
The Complete Mitochondrial DNA Sequence of the Green Alga Pseudendoclonium akinetum (Ulvophyceae)...
14
The Complete Mitochondrial DNA Sequence of the Green Alga Pseudendoclonium akinetum (Ulvophyceae) Highlights Distinctive Evolutionary Trends in the Chlorophyta and Suggests a Sister-Group Relationship Between the Ulvophyceae and Chlorophyceae Jean-Franc ¸ois Pombert, Christian Otis, Claude Lemieux, and Monique Turmel De ´partement de Biochimie et de Microbiologie, Universite ´ Laval, Que ´bec, Que ´bec, Canada The mitochondrial genome has undergone radical changes in both the Chlorophyta and Streptophyta, yet little is known about the dynamics of mtDNA evolution in either of these lineages. In the Chlorophyta, which comprises four of the five recognized classes of green algae (Prasinophyceae, Trebouxiophyceae, Ulvophyceae, and Chlorophyceae), the mitochondrial genome varies from 16 to 55 kb. This genome has retained a compact gene organization and a relatively complex gene repertoire (‘‘ancestral’’ pattern) in the basal lineages represented by the Trebouxiophyceae and Prasinophyceae, whereas it has been reduced in size and gene complement and tends to evolve much more rapidly at the sequence level (‘‘reduced-derived’’ pattern of evolution) in the Chlorophyceae and the lineage leading to the enigmatic chlorophyte Pedinomonas. To gain information about the evolutionary trends of mtDNA in the Ulvophyceae and also to gain insights into the phylogenetic relationships between ulvophytes and other chlorophytes, we have determined the mtDNA sequence of Pseudendoclonium akinetum. At 95,880 bp, Pseudendoclonium mtDNA is the largest green-algal mitochondrial genome sequenced to date and has the lowest gene density. These derived features are reminiscent of the ‘‘expanded’’ pattern exhibited by embryophyte mtDNAs, indicating that convergent evolution towards genome expansion has occurred independently in the Chlorophyta and Streptophyta. With 57 conserved genes, the gene repertoire of Pseudendoclonium mtDNA is slightly smaller than those of the prasinophyte Nephroselmis olivacea and the trebouxiophyte Prototheca wickerhamii. This ulvophyte mtDNA contains seven group I introns, four of which have homologs in green-algal mtDNAs displaying an ‘‘ancestral’’ or a ‘‘reduced-derived’’ pattern of evolution. Like its counterpart in the chlorophycean green alga Scenedesmus obliquus, it features numerous small, dispersed repeats in intergenic regions and introns. Its overall rate of sequence evolution appears to be accelerated to an intermediary level as compared with the rates observed in ‘‘ancestral’’ and ‘‘reduced-derived’’ mtDNAs. In agreement with the finding that Pseudendoclonium mtDNA exhibits features typical of both the ‘‘ancestral’’ and ‘‘reduced-derived’’ patterns of evolution, phylogenetic analyses of seven mtDNA-encoded proteins revealed a sister-group relationship between this ulvophyte and chlorophytes displaying ‘‘reduced-derived’’ mtDNAs. Introduction The mitochondrial genomes of green plants are highly variable in size, gene content, and organization and show divergent evolutionary trends in some lineages, as revealed by the complete mtDNA sequences that have been reported thus far for 13 members of this group. Representatives of the two main phyla of green plants, the Streptophyta and Chlorophyta, have been examined in these genome analyses. The Streptophyta (Bremer 1985) comprises all embryophytes (land plants) and their closest green-algal relatives, the members of the class Charophyceae sensu Mattox and Stewart (1984). In this phylum, the mitochon- drial genomes of the bryophyte Marchantia polymorpha (Oda et al. 1992), of three angiosperms (Arabidopsis thaliana [Unseld et al. 1997], Beta vulgaris [Kubo et al. 2000], and Oryza sativa [Notsu et al. 2002]), and of the charophycean green alga Chaetosphaeridium globosum (Turmel, Otis, and Lemieux 2002b) have been entirely sequenced. The Chlorophyta (Sluiman 1985) comprises the four other classes of green algae: the Prasinophyceae, Ulvophyceae, Trebouxiophyceae, and Chlorophyceae. The seven chlorophyte mitochondrial DNA (mtDNA) se- quences that are currently available include those of four chlorophycean green algae (Chlamydomonas reinhardtii [Michaelis, Vahrenholz, and Pratje 1990], Chlamydomo- nas eugametos [Denovan-Wright, Nedelcu, and Lee 1998], Chlorogonium elongatum [Kroymann and Zetsche 1998], and Scenedesmus obliquus [Ku ¨ck, Jekosch, and Holzamer 2000; Nedelcu et al. 2000]), the nonphotosynthetic trebouxiophyte Prototheca wickerhamii (Wolff et al. 1994), the prasinophyte Nephroselmis olivacea (Turmel et al. 1999), and a chlorophyte of uncertain phylogenetic affinity, Pedinomonas minor (Turmel et al. 1999). The remaining green alga whose mitochondrial genome has been scrutinized, Mesostigma viride (Turmel, Otis, and Lemieux 2002a), belongs to the Streptophyta (Bhatta- charya et al. 1998; Marin and Melkonian 1999; Karol et al. 2001) or to a lineage that emerged before the divergence of the Streptophyta and Chlorophyta (Lemieux, Otis, and Turmel 2000; Turmel et al. 2002; Turmel, Otis, and Lemieux 2002a). Because of their large size and their tendency to incorporate foreign DNA (from the nucleus and the chloroplast), land-plant mitochondrial genomes have been reported to feature an ‘‘expanded’’ pattern of evolution (Turmel et al. 1999). These mitochondrial genomes are the largest (187 kb in Marchantia to 2,000 kb in muskmelon) among green plants, and they also show the greatest structural complexity. Most of the increased size of land- plant mtDNAs relative to their green-algal homologs is accounted for by noncoding sequences that reside either in intergenic regions or introns (Oda et al. 1992; Unseld et al. Key words: Green algae, Ulvophyceae, Pseudendoclonium akine- tum, mitochondrial DNA, group I introns, repeated sequences. E-mail: [email protected]. Mol. Biol. Evol. 21(5):922–935. 2004 DOI:10.1093/molbev/msh099 Advance Access publication March 10, 2004 Molecular Biology and Evolution vol. 21 no. 5 Ó Society for Molecular Biology and Evolution 2004; all rights reserved. by guest on November 26, 2015 http://mbe.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of The Complete Mitochondrial DNA Sequence of the Green Alga Pseudendoclonium akinetum (Ulvophyceae)...

The Complete Mitochondrial DNA Sequence of the Green AlgaPseudendoclonium akinetum (Ulvophyceae) Highlights DistinctiveEvolutionary Trends in the Chlorophyta and Suggests a Sister-GroupRelationship Between the Ulvophyceae and Chlorophyceae
Jean-Francois Pombert, Christian Otis, Claude Lemieux, and Monique TurmelDepartement de Biochimie et de Microbiologie, Universite Laval, Quebec, Quebec, Canada
The mitochondrial genome has undergone radical changes in both the Chlorophyta and Streptophyta, yet little is knownabout the dynamics of mtDNA evolution in either of these lineages. In the Chlorophyta, which comprises four of the fiverecognized classes of green algae (Prasinophyceae, Trebouxiophyceae, Ulvophyceae, and Chlorophyceae), themitochondrial genome varies from 16 to 55 kb. This genome has retained a compact gene organization and a relativelycomplex gene repertoire (‘‘ancestral’’ pattern) in the basal lineages represented by the Trebouxiophyceae andPrasinophyceae, whereas it has been reduced in size and gene complement and tends to evolve much more rapidly at thesequence level (‘‘reduced-derived’’ pattern of evolution) in the Chlorophyceae and the lineage leading to the enigmaticchlorophyte Pedinomonas. To gain information about the evolutionary trends of mtDNA in the Ulvophyceae and also togain insights into the phylogenetic relationships between ulvophytes and other chlorophytes, we have determined themtDNA sequence of Pseudendoclonium akinetum. At 95,880 bp, Pseudendoclonium mtDNA is the largest green-algalmitochondrial genome sequenced to date and has the lowest gene density. These derived features are reminiscent of the‘‘expanded’’ pattern exhibited by embryophyte mtDNAs, indicating that convergent evolution towards genomeexpansion has occurred independently in the Chlorophyta and Streptophyta. With 57 conserved genes, the generepertoire of Pseudendoclonium mtDNA is slightly smaller than those of the prasinophyte Nephroselmis olivacea and thetrebouxiophyte Prototheca wickerhamii. This ulvophyte mtDNA contains seven group I introns, four of which havehomologs in green-algal mtDNAs displaying an ‘‘ancestral’’ or a ‘‘reduced-derived’’ pattern of evolution. Like itscounterpart in the chlorophycean green alga Scenedesmus obliquus, it features numerous small, dispersed repeats inintergenic regions and introns. Its overall rate of sequence evolution appears to be accelerated to an intermediary level ascompared with the rates observed in ‘‘ancestral’’ and ‘‘reduced-derived’’ mtDNAs. In agreement with the finding thatPseudendoclonium mtDNA exhibits features typical of both the ‘‘ancestral’’ and ‘‘reduced-derived’’ patterns of evolution,phylogenetic analyses of seven mtDNA-encoded proteins revealed a sister-group relationship between this ulvophyte andchlorophytes displaying ‘‘reduced-derived’’ mtDNAs.
Introduction
The mitochondrial genomes of green plants are highlyvariable in size, gene content, and organization and showdivergent evolutionary trends in some lineages, as revealedby the complete mtDNA sequences that have been reportedthus far for 13 members of this group. Representatives ofthe two main phyla of green plants, the Streptophyta andChlorophyta, have been examined in these genomeanalyses. The Streptophyta (Bremer 1985) comprises allembryophytes (land plants) and their closest green-algalrelatives, the members of the class Charophyceae sensuMattox and Stewart (1984). In this phylum, the mitochon-drial genomes of the bryophyte Marchantia polymorpha(Oda et al. 1992), of three angiosperms (Arabidopsisthaliana [Unseld et al. 1997], Beta vulgaris [Kubo et al.2000], and Oryza sativa [Notsu et al. 2002]), and of thecharophycean green alga Chaetosphaeridium globosum(Turmel, Otis, and Lemieux 2002b) have been entirelysequenced. The Chlorophyta (Sluiman 1985) comprisesthe four other classes of green algae: the Prasinophyceae,Ulvophyceae, Trebouxiophyceae, and Chlorophyceae. Theseven chlorophyte mitochondrial DNA (mtDNA) se-quences that are currently available include those of four
chlorophycean green algae (Chlamydomonas reinhardtii[Michaelis, Vahrenholz, and Pratje 1990], Chlamydomo-nas eugametos [Denovan-Wright, Nedelcu, and Lee 1998],Chlorogonium elongatum [Kroymann and Zetsche 1998],and Scenedesmus obliquus [Kuck, Jekosch, and Holzamer2000; Nedelcu et al. 2000]), the nonphotosynthetictrebouxiophyte Prototheca wickerhamii (Wolff et al.1994), the prasinophyte Nephroselmis olivacea (Turmelet al. 1999), and a chlorophyte of uncertain phylogeneticaffinity, Pedinomonas minor (Turmel et al. 1999). Theremaining green alga whose mitochondrial genome hasbeen scrutinized, Mesostigma viride (Turmel, Otis, andLemieux 2002a), belongs to the Streptophyta (Bhatta-charya et al. 1998; Marin and Melkonian 1999; Karol et al.2001) or to a lineage that emerged before the divergence ofthe Streptophyta and Chlorophyta (Lemieux, Otis, andTurmel 2000; Turmel et al. 2002; Turmel, Otis, andLemieux 2002a).
Because of their large size and their tendency toincorporate foreign DNA (from the nucleus and thechloroplast), land-plant mitochondrial genomes have beenreported to feature an ‘‘expanded’’ pattern of evolution(Turmel et al. 1999). These mitochondrial genomes are thelargest (187 kb in Marchantia to 2,000 kb in muskmelon)among green plants, and they also show the greateststructural complexity. Most of the increased size of land-plant mtDNAs relative to their green-algal homologs isaccounted for by noncoding sequences that reside either inintergenic regions or introns (Oda et al. 1992; Unseld et al.
Key words: Green algae, Ulvophyceae, Pseudendoclonium akine-tum, mitochondrial DNA, group I introns, repeated sequences.
E-mail: [email protected].
Mol. Biol. Evol. 21(5):922–935. 2004DOI:10.1093/molbev/msh099Advance Access publication March 10, 2004
Molecular Biology and Evolution vol. 21 no. 5 � Society for Molecular Biology and Evolution 2004; all rights reserved.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

1997; Kubo et al. 2000; Notsu et al. 2002). Sixty-ninemitochondrial genes have been identified in Marchantia(Oda et al. 1992; Gray et al. 1998), whereas about 50 havebeen found in angiosperms (Unseld et al. 1997; Kubo et al.2000; Notsu et al. 2002). This substantial difference incoding capacity is attributed to gene transfer to thenucleus, a widespread and ongoing phenomenon (Schusterand Brennicke 1994; Palmer et al. 2000; Adams et al.2002). In embryophyte mitochondria, unicircular genome-sized molecules coexist in a dynamic equilibrium withsubgenomic circles (Palmer and Shields 1984; Mackenzie,He, and Lyznik 1994). In Marchantia mitochondria,unicircular-genome sized molecules apparently coexistwith linear molecules and complex branched structures ofmultigenomic sizes (Oldenburg and Bendich 2001). Incontrast to their fluid structure, land-plant mitochondrialgenomes evolve extremely slowly at the sequence level; inangiosperm mitochondria, point mutations can occur ata frequency up to 100 times lower than in vertebratemitochondria (Wolfe, Li, and Sharp 1987; Palmer andHerbon 1988).
The more compact green-algal mitochondrial ge-nomes display distinctive evolutionary patterns. Theyrange in size from 16 kb (in C. reinhardtii) to 67.8 kb(in Mesostigma) and encode 12 (in C. reinhardtii, C.eugametos, and Chlorogonium) to 67 (in Chaetosphaeri-dium) genes. An ‘‘ancestral’’ (minimally derived) evolu-tionary pattern (Turmel et al. 1999) has been ascribed tothe circular-mapping mtDNAs of Mesostigma, Chaetos-phaeridium, Nephroselmis, and Prototheca, because oftheir large number of conserved genes (.60), their highgene density, and their important sequence conservation.Not only fewer genes but also a greater variability in genecontent (12 to 42 genes) and structural organization (linearor circular-mapping molecules, or even multimericmolecules, as reported for Polytomella [Fan and Lee2002]) have been found in chlorophycean green-algalmtDNAs. The coding sequences of these genomes arehighly divergent from those of other green plants andfeature numerous deletions/additions; moreover, the rRNAgene-coding regions are fragmented into pieces that aredispersed throughout the genomes. As a consequence ofthis high sequence divergence, chlorophycean taxa exhibitvery long branches in mitochondrial trees, and, mostprobably because of long-branch attraction artifacts,usually lie outside the green-plant clade when other greenplants and non–green-plant taxa are included in theanalyses. A ‘‘reduced-derived pattern’’ (Turmel et al.1999) of evolution has been assigned to the three smallestand gene-poorest chlorophycean mtDNAs (i.e., to C.reinhardtii, C. eugametos, and Chlorogonium mtDNAs).Because of its less-derived characters, the ScenedesmusmtDNA sequence has been considered to display an‘‘intermediate’’ pattern of evolution (Nedelcu et al. 2000).
The present study was undertaken to gather informa-tion about the evolutionary trends of the mitochondrialgenome in the Ulvophyceae and also to gain insights intothe phylogenetic relationships between ulvophytes andother chlorophytes. Various hypotheses have been pro-posed concerning the phylogenetic position of the Ulvo-phyceae within the Chlorophyta, but none is strongly
supported by statistical analyses. On the basis of ultrastruc-tural studies (Mattox and Stewart 1984; O’Kelly and Floyd1984) and some phylogenetic analyses of nuclear smallsubunit (SSU) rDNA sequences (Friedl 1995; Bhatta-charya, Friedl, and Damberger 1996; Nakayama, Wata-nabe, and Inouye 1996; Chapman et al. 1998; Watanabe etal. 2000; Wolf et al. 2002), it has been proposed that theUlvophyceae are a monophyletic assemblage that emergedbefore the divergence of the Trebouxiophyceae andChlorophyceae. Independent inferences from nuclear SSUrDNA sequences (Friedl 1997; Marin and Melkonian 1999)and from concatenated chloroplast SSU and large subunit(LSU) (Turmel et al. 2002) rDNA sequences suggesta possible sister-group relationship between the Ulvophy-ceae and Chlorophyceae, with the Trebouxiophyceaeoccupying a basal position. On the other hand, separatenuclear SSU rDNA trees (Bhattacharya and Medlin 1998)favor the hypothesis that the Chlorophyceae appearedbefore the divergence of the Ulvophyceae and Treboux-iophyceae, whereas recent trees, including a wider diversityof ulvophytes (Friedl and O’Kelly 2002) failed to revolvethe branching order of the Trebouxiophyceae, Chlorophy-ceae, and Ulvophyceae. Moreover, other nuclear SSUrDNA trees, including several ulvophytes (Watanabe,Kuroda, and Maiwa 2001) are in agreement with an earlierreport (Zechman et al. 1990) and with the concept ofthe Ulvophyceae sensu Sluiman (1989) in supporting thenotion that the ulvophytes are nonmonophyletic. In theUlvophyceae sensu Sluiman, ulvophytes and trebouxio-phytes are merged to form a green-algal group witha counterclockwise arrangement of kinetid components.The analyses supporting the monophyly of ulvophytessuffer from a relatively poor and/or biased taxon sampling(all five orders recognized in this class were not rep-resented), whereas those supporting their nonmonophylymay be plagued with long-branch attraction artifacts.
In this study, we report the complete mtDNAsequence of Pseudendoclonium akinetum, a unicellularmember of the Ulvophyceae that belongs to a putativelydeep-branching lineage (Floyd and O’Kelly 1990). At95,880 bp, this ulvophyte mtDNA is the largest green-algal mtDNA analyzed thus far. Our phylogenetic analysesprovide support for a sister-group relationship between theUlvophyceae and Chlorophyceae.
Materials and MethodsStrain and Culture Conditions
Pseudendoclonium akinetum (Tupa 1974) was ob-tained from the University of Texas Algal CultureCollection (UTEX 1912) and grown in modified Volvoxmedium (McCracken, Nadakavukaren, and Cain 1980)under 12 h light/dark cycles.
Isolation and Sequencing of mtDNA
A1T-rich organellar DNA was separated fromnuclear DNA by CsCl-bisbenzimide isopycnic centrifuga-tion (Turmel et al. 1999). After nebulization of this A1T-rich fraction, a plasmid library of DNA fragments (1200 to2500 bp) was prepared (Lemieux, Otis, and Turmel 2000).
Mitochondrial DNA Sequence of Pseudendoclonium 923
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

Plasmid DNA templates and PCR fragments spanninguncloned regions of Pseudendoclonium mtDNA weresequenced using ABI Prism 373XL and 377 (AppliedBiosystems, Foster City, Calif.) DNA sequencers and theABI Prism Big Dye terminator sequencing kit (AppliedBiosystems) as described previously (Lemieux, Otis, andTurmel 2000). Templates that yielded poor sequencesunder these conditions were subjected to sequencing usingthe DYEnamic ET (Amersham Pharmacia Biotech, Baied’Urfe, Canada) and/or the ABI Prism dGTP Big Dye(Applied Biosystems) dye terminator sequencing kits.Sequences were edited and assembled with AUTOAS-SEMBLER version 2.1.1 (Applied Biosystems).
Genome Analyses
Gene content was determined by Blast homologysearches (Altschul et al. 1990) against the nonredundantdatabase of NCBI. Protein-coding genes and open readingframes (ORFs) were localized precisely using ORF-FINDER at NCBI and various programs of the GCGWisconsin version 10.2 package (Accelrys, Burlington,Mass.), whereas sequences coding for tRNAs wereidentified with tRNAscan-SE 1.23 (Lowe and Eddy1997). Patterns of codon usage for protein-coding genesand ORFs were compared using CORRESPOND andCODONPREFERENCE from the Wisconsin package andCAI from the EMBOSS version 2.6.0 package (http://www.hgmp.mrc.ac.uk/Software/EMBOSS/). Repeated se-quence elements were identified with PIPMAKER(Schwartz et al. 2000) and REPUTER version 2.74 (Kurtzet al. 2001) and classified with REPEATFINDER(Volfovsky, Haas, and Salzberg 2001).
Phylogenetic Analyses
Mitochondrial genome sequences were retrievedfrom GenBank: Pseudendoclonium akinetum (accessionnumber AY359242 [this study]), Mesostigma viride(accession number AF353999), Nephroselmis olivacea(accessionnumberAF110138),Protothecawickerhamii (ac-cession number NC_001613), Pedinomonas minor (ac-cession number NC_000892), Scenedesmus obliquus(accession number AF204057), Chlamydomonas eugame-tos (accession number NC_001872), Chlamydomonasreinhardtii (accession number NC_001638), Chlorogo-nium elongatum (accession numbers Y13643 andY13644), Chaetosphaeridium globosum (accession num-ber NC_004118), Marchantia polymorpha (accessionnumber NC_001660), Arabidopsis thaliana (accessionnumber NC_001284), Beta vulgaris (accession numberNC_002511), Chondrus crispus (accession numberNC_001677), and Porphyra purpurea (accessionnumber NC_002007). Deduced amino acid sequencesfrom individual genes were aligned using ClustalWversion 1.81 (Thompson, Higgins, and Gibson 1994).Data sets were prepared by concatenating the alignmentsof individual proteins and removing the ambiguouslyaligned regions with GBLOCKS version 0.73b(Castresana 2000). Phylogenetic trees were inferred usingmaximum-likelihood (ML) and ML-distance methods. ML
trees were computed with PROTML in MOLPHY version2.3b3 (Adachi and Hasegawa 1996) and CODEML inPAML version 3.11 (Yang 1997) using the amino acidsubstitution models JTT-F, mtREV24-F, and WAG-F(Whelan and Goldman 2001). �-distributed rates ofsubstitutions across sites (eight categories) and/or multiplegene (Mgene option) corrections were applied in someCODEML analyses. Local bootstrap probabilities wereestimated by resampling of the estimated log-likelihood(RELL) (Adachi and Hasegawa 1996). Confidence assess-ments (P-values) of tree selections were evaluated by theApproximately Unbiased, Kishino-Hasegawa, and Shimo-daira-Hasegawa tests as implemented in CONSEL version0.1d (Shimodaira and Hasegawa 2001). The effect ofinvariable sites on topologies was determined by analysisof a trimmed data set of 1,555 positions containing onlythe variable sites. �-corrected ML distances werecalculated with TREE-PUZZLE version 5.0.2 (Strimmerand von Haeseler 1996) under the WAG-F model, anddistance trees were computed by weighted neighbor-joining as implemented in WEIGHBOR version 1.2(Bruno, Socci, and Halpern 2000). Support for ML-distance trees was obtained by bootstrapping (100replications) with PUZZLEBOOT version 1.03 (A. Rogerand M. Holder, http://www.tree-puzzle.de).
Comparisons of Amino Acid Substitution Rates inDifferent Lineages
We used the data set of 2,107 amino acid positionsthat was employed for the ML and ML-distance analyses.Differences in the rates of amino acid substitutions amonglineages were assessed using the binomial test of Gu andLi (1992).
ResultsGeneral Features
Pseudendoclonium mtDNA is a circular DNAmolecule of 95,880 bp (fig. 1). Its 57 conserved genesand 36 free-standing ORFs (. 60 codons) represent 47.4%and 11.3% of the genome sequence, respectively. A totalof seven introns interrupt four genes (atp1, cob, cox1, andrnl). Intergenic sequences range from 5 to 2,663 bp in size,with an average of about 600 bp, and feature similar pro-portions of A1T (61.4%) as compared with codingregions (60.2%).
In terms of coding capacity, the mitochondrial genomeof Pseudendoclonium closely resembles those of Nephro-selmis and Prototheca. Pseudendoclonium mtDNA lackssix of the protein-coding genes encoded by itsNephroselmiscounterpart and only three of the protein-coding genesencoded by Prototheca mtDNA (table 1). It encodes tworRNAs, 25 tRNAs, 12 ribosomal proteins, 17 ATP synthaseand respiratory chain components, and also a proteininvolved in the Sec-independent translocation pathway(MttB). The encoded tRNAs each feature a conventionalcloverleaf secondary structure and together are sufficient totranslate all of the codons in Pseudendoclonium mtDNAusing the standard genetic code (table 2). In contrast to
924 Pombert et al.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

Nephroselmis and Prototheca mtDNAs, no gene for 5SrRNA (rrn5) is present in Pseudendoclonium mtDNA.
Six intron ORFs in Pseudendoclonium mtDNAencode proteins that are homologous to endonucleases/maturases of the LAGLIDADG family (table 3); each ofthese intron-encoded proteins contains two copies of theLAGLIDADG motif. Two of the 36 free-standing ORFs,orf289 and orf307, also code for putative LAGLIDADGendonucleases/maturases with two copies of the LAGLI-DADG motif. The endonuclease potentially encoded byorf289 shows 48% sequence identity over 282 alignedamino acid positions with the protein specified by orf298in the fourth intron of cox1 (Pacox1.4), whereas theendonuclease potentially encoded by orf307 has nosubstantial homology with any of the Pseudendocloniumintron-encoded endonucleases/maturases. Of the 34 re-maining free-standing ORFs, only the four harboring morethan 300 codons, as well as orf61 and orf90, featurea pattern of codon usage that is similar to that of conservedgenes (table 2). This observation suggests that these fewORFs may be expressed at the protein level; however, we
have not been able to detect any similarity with knowngene sequences.
Pseudendoclonium mtDNA shares only two smallgene clusters (rps12-rps10 and trnE(uuc)-trnW(cca) withNephroselmis mtDNA) and a single cluster (atp1-nad1)with Prototheca mtDNA. The large cluster of ribosomalprotein genes found in Nephroselmis mtDNA has beensegmented into several clusters in PseudendocloniummtDNA, and as observed for the Nephroselmis andPrototheca mtDNAs, the genes for the SSU and LSUrRNAs have retained their continuous structure.
Introns
All seven introns in Pseudendoclonium mtDNAbelong to the group I family (table 3). All of these introns,with the exception of Paatp1.1, show similarities withgroup I introns inserted at equivalent positions in othergreen-plant and/or fungal mtDNAs (table 4). The lattergreen-plant mtDNAs include those of the streptophyteMarchantia and of various chlorophytes exhibiting an
FIG. 1.—Gene map of Pseudendoclonium mtDNA. Genes on the outside of the map are transcribed in a clockwise direction; those on the inside ofthe map are transcribed counterclockwise. tRNA genes are indicated by the one-letter amino acid code followed by the anticodon in parentheses (Me,elongator methionine; Mf, initiator methionine). Seven group I introns (open boxes) interrupt conserved genes; six of these introns contain an internalORF (gray boxes). Only the free-standing ORFs larger than 60 codons are shown.
Mitochondrial DNA Sequence of Pseudendoclonium 925
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from
‘‘ancestral’’ or a ‘‘reduced-derived’’ pattern of mtDNAevolution. Sequence conservation between the corestructures of four Pseudendoclonium introns (Pacox1.1,Pacox1.2, Pacox1.4, and Parnl.1) and their green-algalhomologs is substantial (fig. 2).
Homologs of the Parnl.1 intron have also beenidentified in the chloroplast rrl genes of several greenalgae (Turmel et al. 1993). One of these chloroplast introns(Chrnl.1) contains an ORF in L9.1 (as in Parnl.1) thatcodes for the DNA-homing endonuclease I-ChuI (Cote et
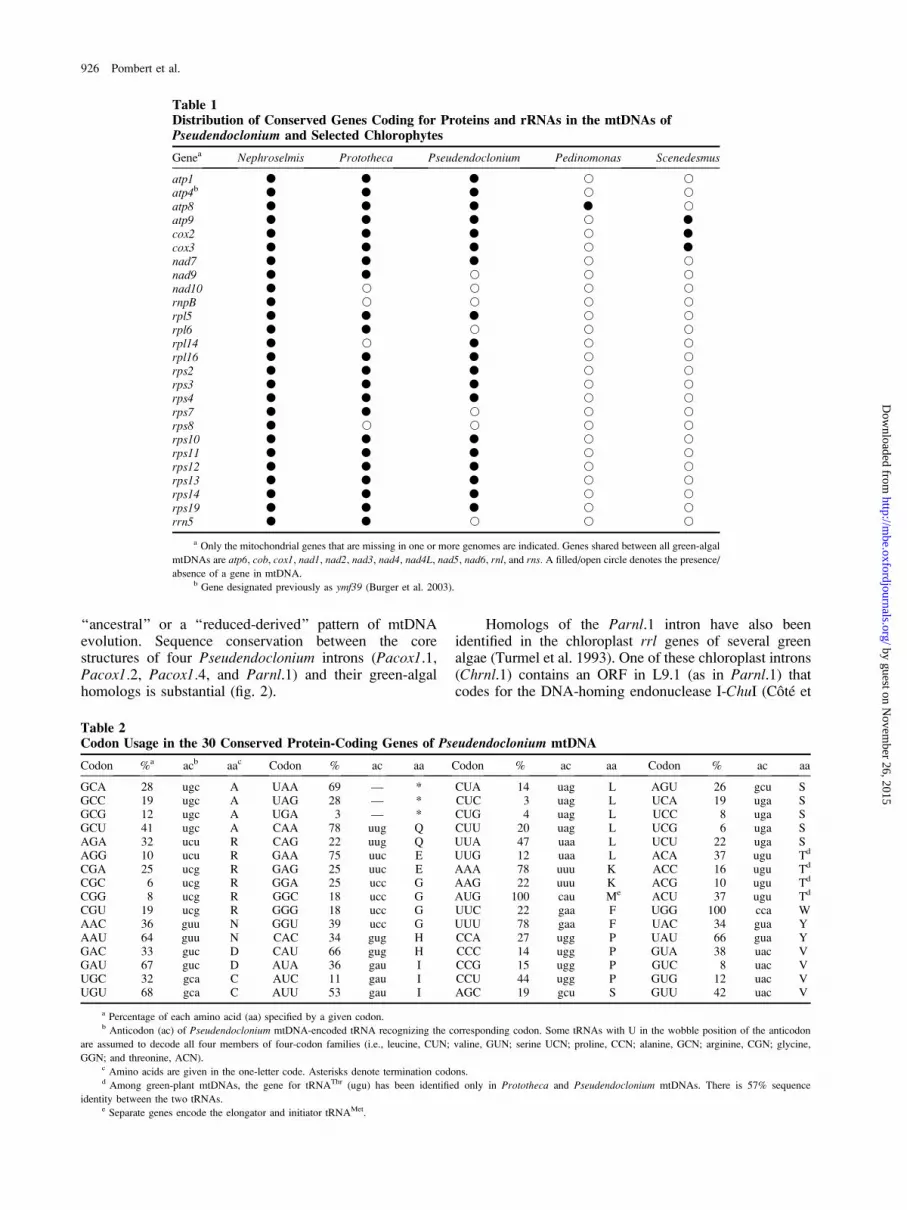
Table 1Distribution of Conserved Genes Coding for Proteins and rRNAs in the mtDNAs ofPseudendoclonium and Selected Chlorophytes
Genea Nephroselmis Prototheca Pseudendoclonium Pedinomonas Scenedesmus
atp1 � � � s satp4b � � � s satp8 � � � � satp9 � � � s �cox2 � � � s �cox3 � � � s �nad7 � � � s snad9 � � s s snad10 � s s s srnpB � s s s srpl5 � � � s srpl6 � � s s srpl14 � s � s srpl16 � � � s srps2 � � � s srps3 � � � s srps4 � � � s srps7 � � s s srps8 � s s s srps10 � � � s srps11 � � � s srps12 � � � s srps13 � � � s srps14 � � � s srps19 � � � s srrn5 � � s s s
a Only the mitochondrial genes that are missing in one or more genomes are indicated. Genes shared between all green-algal
mtDNAs are atp6, cob, cox1, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, rnl, and rns. A filled/open circle denotes the presence/
absence of a gene in mtDNA.b Gene designated previously as ymf39 (Burger et al. 2003).
Table 2Codon Usage in the 30 Conserved Protein-Coding Genes of Pseudendoclonium mtDNA
Codon %a acb aac Codon % ac aa Codon % ac aa Codon % ac aa
GCA 28 ugc A UAA 69 — * CUA 14 uag L AGU 26 gcu SGCC 19 ugc A UAG 28 — * CUC 3 uag L UCA 19 uga SGCG 12 ugc A UGA 3 — * CUG 4 uag L UCC 8 uga SGCU 41 ugc A CAA 78 uug Q CUU 20 uag L UCG 6 uga SAGA 32 ucu R CAG 22 uug Q UUA 47 uaa L UCU 22 uga SAGG 10 ucu R GAA 75 uuc E UUG 12 uaa L ACA 37 ugu Td
CGA 25 ucg R GAG 25 uuc E AAA 78 uuu K ACC 16 ugu Td
CGC 6 ucg R GGA 25 ucc G AAG 22 uuu K ACG 10 ugu Td
CGG 8 ucg R GGC 18 ucc G AUG 100 cau Me ACU 37 ugu Td
CGU 19 ucg R GGG 18 ucc G UUC 22 gaa F UGG 100 cca WAAC 36 guu N GGU 39 ucc G UUU 78 gaa F UAC 34 gua YAAU 64 guu N CAC 34 gug H CCA 27 ugg P UAU 66 gua YGAC 33 guc D CAU 66 gug H CCC 14 ugg P GUA 38 uac VGAU 67 guc D AUA 36 gau I CCG 15 ugg P GUC 8 uac VUGC 32 gca C AUC 11 gau I CCU 44 ugg P GUG 12 uac VUGU 68 gca C AUU 53 gau I AGC 19 gcu S GUU 42 uac V
a Percentage of each amino acid (aa) specified by a given codon.b Anticodon (ac) of Pseudendoclonium mtDNA-encoded tRNA recognizing the corresponding codon. Some tRNAs with U in the wobble position of the anticodon
are assumed to decode all four members of four-codon families (i.e., leucine, CUN; valine, GUN; serine UCN; proline, CCN; alanine, GCN; arginine, CGN; glycine,
GGN; and threonine, ACN).c Amino acids are given in the one-letter code. Asterisks denote termination codons.d Among green-plant mtDNAs, the gene for tRNAThr (ugu) has been identified only in Prototheca and Pseudendoclonium mtDNAs. There is 57% sequence
identity between the two tRNAs.e Separate genes encode the elongator and initiator tRNAMet.
926 Pombert et al.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

al. 1993). The Chrnl.1 and Parnl.1 introns share 69%sequence identity over 169 aligned nucleotides in theircore structures, and their encoded proteins show 65.3%amino acid similarities over 219 aligned amino acidpositions. This important level of sequence conservation isconsistent with our previous finding of closely relatedgroup I introns in the chloroplast and mitochondrialgenomes of green algae (Turmel et al. 1995; Turmel et al.1999; Lucas et al. 2001; Turmel, Otis, and Lemieux2002a) and further supports the idea that lateral transfersof introns have occurred between different organellarcompartments during the evolution of green plants.
Repeated Sequence Elements
A comparison of the Pseudendoclonium mtDNAsequence against itself using PIPMAKER (Schwartz et al.2000) revealed the presence of many degeneratedrepeated elements within intergenic regions and introns(fig. 3). We identified these repeated sequences using theprogram REPUTER and found that they are up to 350 ntin size and do not differ substantially from the rest of thePseudendoclonium genome in terms of nucleotidecomposition. The repeats that were 50 nt or more wereclassified using REPEATFINDER (Volfovsky, Haas, andSalzberg 2001); they represent about 10 kb of thegenome and form four distinct classes, designated C1 toC4 (fig. 3 and table 5). The members of a given classshare no sequence similarity with those of other classes.The first class of repeats (C1) is the largest. Its membersmap to at least 16 different loci (fig. 3) and can be furtherclassified into 14 subclasses, designated C1R1 throughC1R14. The longest repeat within each subclass, definedas the prototype, is made up of repeated units(subrepeats) that also constitute the shorter repeatedelements found in the same subclass. Distinct subclassesfeature different arrangements of repeated units. Thecoordinates of the prototypes and number of repeatedelements within each subclass are given in table 5. Theabundance of subclasses in class 1 suggests thatnumerous recombination and shuffling events betweenrepeated units took place during the evolution of themitochondrial genome. On the other hand, classes 2, 3,and 4 are much less complex than class 1. Each exhibitsonly one subclass of repeats, with two identical copies ofthe prototype sequence that are far apart from one
another. It is likely that these elements arose from singleduplication events. In addition to the repeats describedabove, Pseudendoclonium mtDNA features numerousrepeated elements less than 50 nt that span about 4 kbof this genome (fig. 3).
Phylogenetic Analyses
The amino acid sequences derived from the sevenprotein-coding genes (cob, cox1, nad1, nad2, nad4, nad5,and nad6) that are common to the mtDNAs of Pseuden-doclonium and 11 other green plants were concatenatedand analyzed with ML and ML-distance methods ofphylogenetic inference, using as outgroup the homologoussequences from Mesostigma. Note that when the red algaePorphyra purpurea and Chondrus crispus were used asoutgroup, we observed no change in the position of
Table 3Main Features of Pseudendoclonium Mitochondrial Introns
Intron Classa Size (bp) ORF Size (bp) ORF Locationb
Paatp1.1 IB4 1425 993 L8Pacob.1 IB 911 — —Pacox1.1 IB4 2049 1335 L8Pacox1.2 ID 2148 789 L1Pacox1.3 IB4 1661 534 L8Pacox1.4 IB3 1732 897 L1Parnl.1 IB4 1524 777 L9.1
a Classification of group I introns was according to Michel and Westhof
(1990). Pacob.1 could not be assigned unambiguously to a subclass of IB introns.
Table 4Group I Introns at Identical Gene Locations in Pseudendo-clonium and Other mtDNAs
PseudendocloniumIntron Homologous Introna
AccessionNumber
Pacob.1 Podospora anserina cob i3a (L9.1) NC_001329Podospora curvicolla cob i2 (L9.1) Z69894
Pacox1.1 Prototheca wickerhamii cox1 i1 (L8) NC_001613Chaetosphaeridium globosum cox1
i2 (L8)NC_004118
Marchantia polymorpha cox1i4 (L8)
NC_001660
Agrocybe aegerita cox1 i1 (L8) AF010257Allomyces macrogynus cox1 i5 NC_001715Saccharomyces douglasii cox1 i1 M97514Schizosaccharomyces pombe cox1
i1 (L8)X00886
Pacox1.2 Prototheca wickerhamii cox1 i2 NC_001613Scenedesmus quadricauda cox1
i1 (L1)AB011524
Kluyveromyces lactis cox1 i2 X57546Saccharomyces douglasii cox1 i2 M97514Podospora anserina cox1 i8 (L1) NC_001329
Pacox1.3 Agrocybe aegerita cox1 i2 (L8) AF010257Emericella nidulans cox1 i3 X00790Neurospora crassa cox1 i4 X14669Schizosaccharomyces pombe cox1
i2 (L8)X00886
Pacox1.4 Chlamydomonas eugametos cox1i1 (L9.1)
NC_001872
Marchantia polymorpha cox1i8 (L1)
NC_001660
Mesostigma viride cox1 i2 (L1) AF353999Allomyces macrogynus cox1 i10 NC_001715Dictyostelium discoideum cox1/2
i3 (L1)NC_000895
Kluyveromyces lactis cox1 i4 (L1) X57546Podospora anserina cox1 i15 (L1) NC_001329Saccharomyces cerevisiae cox1
i5b (L9.1)NC_001224
Saccharomyces douglasii cox1i4 (L9.1)
M97514
Schizosaccharomyces pombe cox1 i4 M15671
Parnl.1 Chlamydomonas eugametos rnli2 (L9.1)
NC_001872
Nephroselmis olivacea rnl i2 (L9.1) AF110138
a When an ORF is present in the homologous intron, its position is indicated in
parentheses. L followed by a number refers to the loop extending the base-paired
region identified by the number.
Mitochondrial DNA Sequence of Pseudendoclonium 927
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

Pseudendoclonium. The WAG-F model of amino acidreplacement was selected for all phylogenetic analyses,because it gave higher likelihood values than the JTT-Fand mtREV24-F models (table 6). The ML and ML-distance trees inferred by assuming �-distributed rates ofsubstitutions across sites revealed a strong affiliation ofPseudendoclonium with the highly supported cladecontaining Pedinomonas and the four chlorophyceangreen algae whose mtDNA sequences has been determinedto date (fig. 4). As shown by their very long branches, themembers of this clade, designated here ‘‘reduced-derived’’clade, display a higher rate of mtDNA sequence evolutioncompared to the other green plants examined. Phyloge-netic relationships among members of the ‘‘reduced-derived’’ clade were found to be well resolved in allanalyses, and the affiliation of Pseudendoclonium with thisclade remained strongly supported when we excludedinvariable sites and/or when we included an additionalparameter (Mgene option in CODEML) to take into
account the different evolutionary rates of the selectedmitochondrial proteins.
To our surprise, we found that under certainconditions of phylogenetic analyses the ‘‘reduced-derived’’clade groups with the rest of green algae and land plants.For instance, in the best ML tree shown in figure 4, thecluster formed by Pseudendoclonium and the ‘‘reduced-derived’’ clade occupies the Chlorophyta lineage. Thisresult contrasts with previously reported phylogenies inwhich members of the ‘‘reduced-derived’’ clade branchconsistently outside the monophyletic group formed by therest of green plants, most certainly as a result of long-branch attraction artifacts (Turmel, Otis, and Lemieux1999; Nedelcu et al. 2000). We explored the phylogeneticconditions that contribute to the recovery of the correctbranching pattern in our best ML tree and found that theselected inference method, evolutionary model, and taxonsampling all play an important role in the analyses.Analyses based on ML-distances and performed under the
FIG. 2.—Structural similarities between group I introns in Pseudendoclonium mtDNA and their closest homologs in other green algal mtDNAs.Pacox1.1 and Pacox1.2 were compared with the homologous introns in Prototheca mtDNA, Pacox1.4 was aligned with the homologous intron inMesostigma mtDNA, and Parnl.1 was compared with the homologous intron in Nephroselmis mtDNA. Introns were modeled according to thenomenclature proposed by Burke et al. (1987). Splice sites between exon and intron residues are denoted by arrows. Identical residues found at the samepositions in the compared introns are shown in uppercase characters, whereas positions displaying different nucleotides are denoted by dots. Conservedbase-pairings are denoted by dashes. The numbers inside the variable loops indicate the sizes of these loops in the compared introns, with the uppernumber referring to the Pseudendoclonium sequence.
928 Pombert et al.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

JTT-F and mtREV24-F models failed to recover thecorrect topology as the best tree. Moreover, the inclusionof the Pseudendoclonium taxon in the concatenated dataset and the addition of a � correction for amino acidsubstitutions across sites also represent important factors inthe ML analyses. However, even under the most favorableML conditions for recovering the correct tree, alternatetopologies in which the ‘‘reduced-derived’’ clusterbranches outside the clade formed by the rest of greenplants were detected relatively frequently among theRELL bootstrap replicates. These alternate topologies
proved to be not significantly different (P , 0.1) from thebest tree in the Kishino-Hasegawa and the Shimodaira-Hasegawa tests.
Rates of Amino Acid Substitutions
Considering the branch lengths of the best ML tree(fig. 4), it appears that Pseudendoclonium mtDNA evolvesat a slower rate than the mtDNAs of taxa from the‘‘reduced-derived’’ clade. To determine whether thisinterpretation is correct, we assessed the relative rates of
FIG. 3.—Positions of repeated sequence elements in Pseudendoclonium mtDNA as revealed by PIPMAKER. Repeats of 50 nt or more were sortedinto four classes by REPEATFINDER. Repeats less than 50 nt were not classified. Genes and their polarities are denoted by horizontal arrows, andexons are represented by filled boxes. Similarities between repeated sequence elements are shown as average percentage identity (between 50% to100% identity).
Mitochondrial DNA Sequence of Pseudendoclonium 929
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

amino acid substitutions of mtDNA-encoded proteinsamong the different lineages using the binomial test ofGu and Li (1992) (table 7). The substitution rates ofPseudendoclonium proteins were found to be significantlysmaller (P , 0.001) than those observed for Scenedesmusand other green algae from the ‘‘reduced-derived’’ clade.Pseudendoclonium mtDNA–encoded proteins, however,evolve significantly faster (P , 0.001) than theirNephroselmis and Prototheca counterparts.
DiscussionDistinctive Features of Pseudendoclonium mtDNA
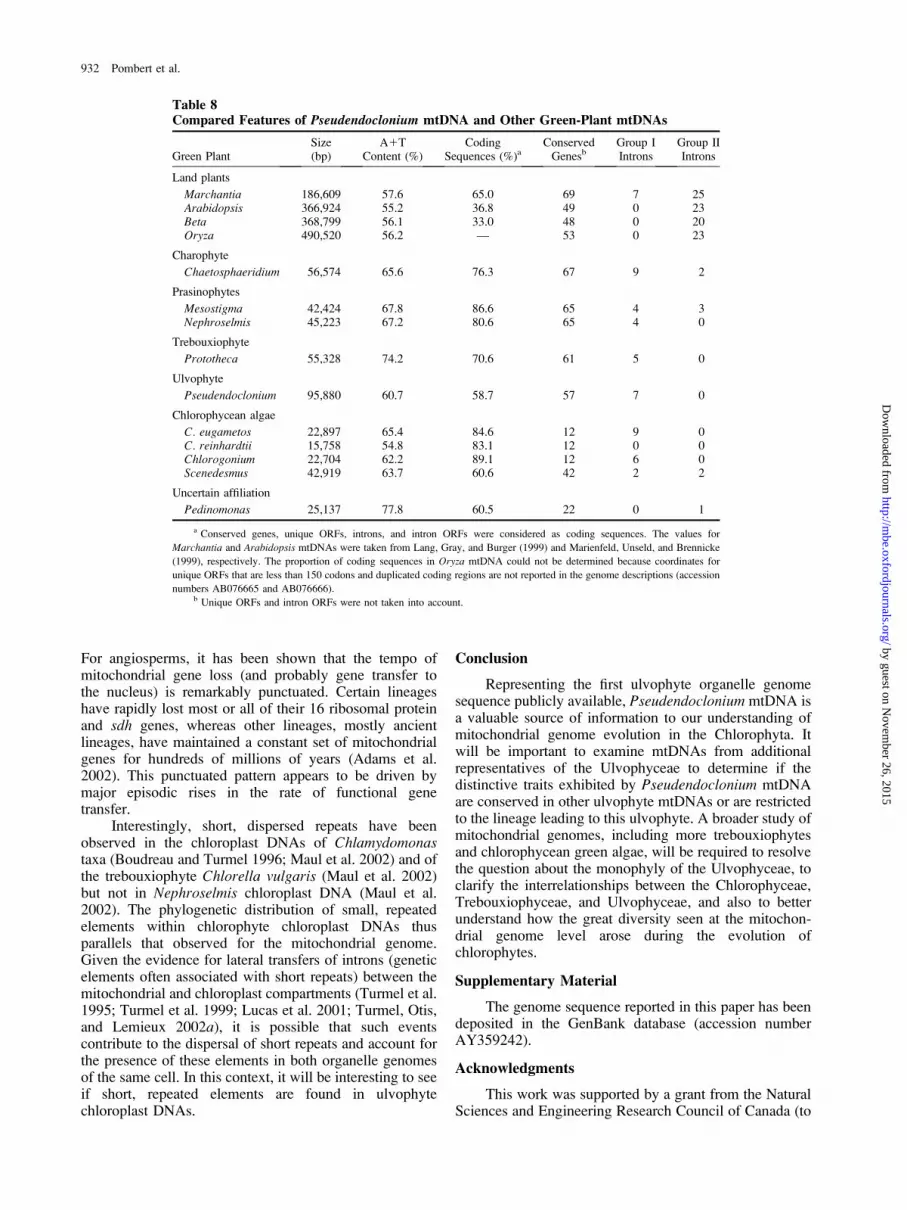
Of all the green-algal mtDNAs sequenced to date,Pseudendoclonium mtDNA is the genome with the largestsize and the lowest gene density (table 8). At 95,880 bp,the size of this genome is almost twofold larger than thoseof the two chlorophytes displaying an ancestral pattern ofevolution (Nephroselmis and Prototheca). Despite thissubstantial expansion in size, Pseudendoclonium mtDNAdisplays a smaller repertoire of conserved genes comparedwith its Nephroselmis and Prototheca counterparts (table8). Most of its increased size is accounted for by dispersedrepeats and sequences of unknown nature/origin that residemainly within the intergenic regions.
The unusually large size and low gene density ofPseudendoclonium mtDNA, together with the presence ofdispersed repeats, is reminiscent of the ‘‘expanded’’ patternof evolution exhibited by embryophyte mtDNAs. Both theexpanded Pseudendoclonium and the embryophytemtDNAs encode a similar number of conserved genes,although they differ in their gene repertoire; moreover,most of their extraneous DNA sequences are of unknownorigin and/or function (Unseld et al. 1997; Kubo et al.2000; Notsu et al. 2002). The finding that Pseudendoclo-nium mtDNA is much larger than its chlorophytecounterparts suggests that mitochondrial genome expan-
sion occurred independently in the Chlorophyta andStreptophyta. Analyses of additional ulvophyte mtDNAswill be required to determine whether the Ulvophyceaedisplays the pattern of progressive genome expansionobserved in the Streptophyta.
On one hand, the gene content (a relatively complexgene repertoire) and gene structure (lack of fragmented andscrambled rRNA genes) of Pseudendoclonium mtDNA arecharacteristic of ‘‘ancestral’’ mtDNAs. On the other hand,its low gene density, abundant repeats, and the absence ofcertain genes (nad9, rpl6, rps7, and rrn5) are typical ofScenedesmus and Pedinomonas mtDNAs. The finding ofgenomic features that are typical of the ‘‘reduced-derived’’pattern of mtDNA evolution, together with the presence ofancestral features, suggests that Pseudendoclonium be-longs to a lineage that appeared after the emergence of theTrebouxiophyceae but before the divergence of theChlorophyceae. Further supporting this notion are threeindependent observations. First, ML and ML-distancetrees inferred from mtDNA-encoded proteins alwayscluster Pseudendoclonium with chlorophycean green algaeand consistently place the trebouxiophyte Prototheca atthe base of this clade (see next section). Second, theoverall rate of sequence evolution appears to be acceler-ated to an intermediary level in PseudendocloniummtDNA as compared with the slow rates observed in‘‘ancestral’’ mtDNAs and the very fast rates detected inPedinomonas, Scenedesmus and chlorophycean green-algal mtDNAs (fig. 4). Third, close relatives of thePacox1.2, Pacox1.4, and Parnl.1 introns are present inmtDNAs displaying both the ‘‘ancestral’’ and ‘‘reduced-derived’’ patterns of evolution.
Phylogenetic Niche of the Ulvophyceae
Our phylogenetic analyses of concatenated mtDNA-encoded protein sequences reveal a close relationshipbetween Pseudendoclonium and chlorophycean greenalgae, with the trebouxiophyte Prototheca occupyinga basal position (fig. 4). These analyses included all thegreen-algal mtDNA sequences available in public data-bases to minimize possible undesirable effects of smalltaxon sampling and used Mesostigma viride as outgroup tomaximize the amount of phylogenetic information. Theposition of Pseudendoclonium relative to the Chlorophy-ceae and Trebouxiophyceae may be the result of genuinephylogenetic signal because it is consistent with our
Table 5Prototypes of Repeated Sequence Elements of 50 nt orMore in Pseudendoclonium mtDNA
Class/Subclass Starta Enda Size (nt)
Number ofOccurrencesb
C1R1 7829 8178 350 8C1R2 22728 22833 106 3C1R3 27871 28104 234 9C1R4 29398 29461 64 2C1R5 35292 35372 81 3C1R6 41072 41233 162 4C1R7 71594 71764 171 5C1R8 80073 80204 132 4C1R9 83421 83505 85 6C1R10 83605 83742 138 4C1R11 84050 84176 127 3C1R12 86491 86566 76 3C1R13 93473 93685 213 7C1R14 94722 94839 118 4C2R1 11683 11792 110 2C3R1 30366 30429 64 2C4R1 74522 74606 85 2
a Coordinates in accession number AY359242.b Number of occurrences of the prototype and subsets of this sequence with-
in the genome.
Table 6Tree-Related Statistics and Log-Likelihood Values for theMitochondrial Data Set of 2,107 Amino Acid Positions
Model of Evolution
mtREV24-F JTT-F WAG-F
Tree length 4.804 4.084 3.905Longest branch 1.360 1.126 1.060Shortest branch 0.011 0.010 0.010Median branch 0.112 0.101 0.092Average branch 0.209 0.178 0.170Log-likelihood 229224.44 229114.30 228984.66
930 Pombert et al.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

finding that Pseudendoclonium mtDNA shares derivedstructural features with its homologs in chlorophyceangreen algae. However, the branching order of theChlorophyceae/Ulvophyceae/Trebouxiophyceae remainsan unresolved issue. The tree recovered in our analysesmay reflect variations in the evolutionary rates of mtDNAsequences rather than the underlying phylogenetic signal,and although we have included all currently availablegreen-algal mtDNAs in our analyses, taxon sampling isstill very low, with only one representative of theUlvophyceae and of the Trebouxiophyceae.
Pseudendoclonium appears to be closely related toPedinomonas, a green alga of uncertain affiliation. Inmitochondrial trees, Pedinomonas branches immediatelyafter the emergence of the Pseudendoclonium lineage.Given the very long branch displayed by Pedinomonas,the phylogenetic position of this chlorophyte might beattributed to long-branch artifacts. However, the existenceof a close relationship between Pedinomonas andulvophytes is supported by the observation that the basalbodies of Pedinomonas and ulvophytes display an absolutecounterclockwise orientation that contrasts with the di-rectly opposed or clockwise orientation found in chloro-phycean green algae (Melkonian 1990). Considering thatthe 25-kb mtDNA of Pedinomonas is highly reduced insize and gene content relative to its Pseudendoclonium andScenedesmus relatives and also considering the muchlonger branch displayed by Pedinomonas mtDNA, itappears that the mitochondrial genome evolved at a moreaccelerated rate in the lineage leading to Pedinomonasthan in the Pseudendoclonium and Scenedesmus lineages.
Repeated Sequence Elements As an EvolutionaryForce in the Chlorophyta
Small repeats have been previously identified inchlorophyte mtDNAs. The few that are found in Proto-
theca mtDNA vary from 30 to 200 nt, are mostly arrangedin tandem, and the recurring motifs are rich in A1T (Wolffet al. 1994). Repeats in Scenedesmus mtDNA range from16 to 118 nt, and, like their Pseudendoclonium counter-parts, account for at least 15% of the genome, flank manyindividual genes, are composed of various subrepeats, andshow a low bias in base composition (Nedelcu et al. 2000).The repeats harbored by chlamydomonad mtDNAs areshorter (9 to 14 nt) and richer in G1C (Boer and Gray1991; Nedelcu and Lee 1998). Unlike all their knownchlorophyte counterparts, the numerous repeated sequencesfound in Pedinomonas mtDNA are densely packed withina discrete, noncoding region of the genome (Turmel et al.1999). The low abundance of repeats in Nephroselmis andPrototheca mtDNAs, together with the increased occur-rence of these elements in the more derived lineagesleading to Pseudendoclonium and Scenedesmus, raise thequestion on their origin. Repeats might have been presentin the mitochondrial genome of the last common ancestorof the ulvophytes and chlorophycean green algae, and havediverged after the split of these lineages. Alternatively, theymight have arisen independently in the Ulvophyceae andChlorophyceae.
Considering that recombination between dispersedrepeats can lead to genome rearrangements (gene lossesor inversions) and that gene content became reduced asfamilies of dispersed repeats emerged and grew in sizeduring the evolution of chlorophyte mtDNAs (at least inderived lineages), we speculate that repeated elementshave played an important role in generating the greatdiversity of size and gene arrangement seen in thesemtDNAs. In chlamydomonad mtDNAs, where genecontent is the poorest of the Chlorophyta lineage,excision of coding regions via recombination betweenflanking short repeats has been invoked to explain theirreduced gene content (Nedelcu 1997; Nedelcu 1998).Dispersed repeats are also thought to act as hot spots forrecombination in land-plant (Palmer and Herbon 1988;Mackenzie, He, and Lyznik 1994), fungal (Jamet-Vierny,Boulay, and Briand 1997), and animal (Lunt and Hyman1997) mtDNAs. Aside from dispersed repeats, otherfactors most probably determine the tempo of gene loss.
FIG. 4.—Phylogenetic position of Pseudendoclonium as inferredfrom the deduced amino acid sequences of seven mitochondrial genes.ML and ML-distances analyses of the data set (2,107 amino acidpositions) were carried out under the WAG-F model of amino acidreplacement, assuming �-distributed rates of substitutions across sites.The best-supported ML tree computed with CODEML is shown.Numbers above the nodes indicate support values; upper and lowervalues are from the ML and ML-distance analyses, respectively. Thechlorophyte lineages displaying a ‘‘reduced-derived’’ pattern of mtDNAevolution are represented as thick lines.
Table 7Differences in the Number of Amino Acid Substitutions inSeven Mitochondrial Proteins and Test for Equal Rates ofSubstitutions in Selected Pairs of Green Algal Lineages
Compared Green Algae(Taxon 1/Taxon 2) N1
a N2b
N1/( N1 1 N2)c Pc
Pseudendoclonium/Nephroselmis 393 142 0.735 3.0 E-28Pseudendoclonium/Prototheca 374 151 0.712 7.4 E-23Pseudendoclonium/Scenedesmus 237 373 0.389 4.1 E-8Pseudendoclonium/Pedinomonas 173 525 0.248 4.4 E-42
a Number of informative substitutions attributable to taxon 1; that is, the
number of amino acid positions at which taxa 1 and 2 are different but taxa 2 and 3
are identical. Taxon 3, the outgroup, was Mesostigma.b Number of informative substitutions attributable to taxon 2; that is the
number of positions at which taxa 1 and 2 are different but taxa 1 and 3 are iden-
tical.c P values were obtained for the binomial tests of the null hypothesis
N1/(N1 1 N2) ¼ 0.5.
Mitochondrial DNA Sequence of Pseudendoclonium 931
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from
For angiosperms, it has been shown that the tempo ofmitochondrial gene loss (and probably gene transfer tothe nucleus) is remarkably punctuated. Certain lineageshave rapidly lost most or all of their 16 ribosomal proteinand sdh genes, whereas other lineages, mostly ancientlineages, have maintained a constant set of mitochondrialgenes for hundreds of millions of years (Adams et al.2002). This punctuated pattern appears to be driven bymajor episodic rises in the rate of functional genetransfer.
Interestingly, short, dispersed repeats have beenobserved in the chloroplast DNAs of Chlamydomonastaxa (Boudreau and Turmel 1996; Maul et al. 2002) and ofthe trebouxiophyte Chlorella vulgaris (Maul et al. 2002)but not in Nephroselmis chloroplast DNA (Maul et al.2002). The phylogenetic distribution of small, repeatedelements within chlorophyte chloroplast DNAs thusparallels that observed for the mitochondrial genome.Given the evidence for lateral transfers of introns (geneticelements often associated with short repeats) between themitochondrial and chloroplast compartments (Turmel et al.1995; Turmel et al. 1999; Lucas et al. 2001; Turmel, Otis,and Lemieux 2002a), it is possible that such eventscontribute to the dispersal of short repeats and account forthe presence of these elements in both organelle genomesof the same cell. In this context, it will be interesting to seeif short, repeated elements are found in ulvophytechloroplast DNAs.
Conclusion
Representing the first ulvophyte organelle genomesequence publicly available, Pseudendoclonium mtDNA isa valuable source of information to our understanding ofmitochondrial genome evolution in the Chlorophyta. Itwill be important to examine mtDNAs from additionalrepresentatives of the Ulvophyceae to determine if thedistinctive traits exhibited by Pseudendoclonium mtDNAare conserved in other ulvophyte mtDNAs or are restrictedto the lineage leading to this ulvophyte. A broader study ofmitochondrial genomes, including more trebouxiophytesand chlorophycean green algae, will be required to resolvethe question about the monophyly of the Ulvophyceae, toclarify the interrelationships between the Chlorophyceae,Trebouxiophyceae, and Ulvophyceae, and also to betterunderstand how the great diversity seen at the mitochon-drial genome level arose during the evolution ofchlorophytes.
Supplementary Material
The genome sequence reported in this paper has beendeposited in the GenBank database (accession numberAY359242).
Acknowledgments
This work was supported by a grant from the NaturalSciences and Engineering Research Council of Canada (to
Table 8Compared Features of Pseudendoclonium mtDNA and Other Green-Plant mtDNAs
Green PlantSize(bp)
A1TContent (%)
CodingSequences (%)a
ConservedGenesb
Group IIntrons
Group IIIntrons
Land plants
Marchantia 186,609 57.6 65.0 69 7 25Arabidopsis 366,924 55.2 36.8 49 0 23Beta 368,799 56.1 33.0 48 0 20Oryza 490,520 56.2 — 53 0 23
Charophyte
Chaetosphaeridium 56,574 65.6 76.3 67 9 2
Prasinophytes
Mesostigma 42,424 67.8 86.6 65 4 3Nephroselmis 45,223 67.2 80.6 65 4 0
Trebouxiophyte
Prototheca 55,328 74.2 70.6 61 5 0
Ulvophyte
Pseudendoclonium 95,880 60.7 58.7 57 7 0
Chlorophycean algae
C. eugametos 22,897 65.4 84.6 12 9 0C. reinhardtii 15,758 54.8 83.1 12 0 0Chlorogonium 22,704 62.2 89.1 12 6 0Scenedesmus 42,919 63.7 60.6 42 2 2
Uncertain affiliation
Pedinomonas 25,137 77.8 60.5 22 0 1
a Conserved genes, unique ORFs, introns, and intron ORFs were considered as coding sequences. The values for
Marchantia and Arabidopsis mtDNAs were taken from Lang, Gray, and Burger (1999) and Marienfeld, Unseld, and Brennicke
(1999), respectively. The proportion of coding sequences in Oryza mtDNA could not be determined because coordinates for
unique ORFs that are less than 150 codons and duplicated coding regions are not reported in the genome descriptions (accession
numbers AB076665 and AB076666).b Unique ORFs and intron ORFs were not taken into account.
932 Pombert et al.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

M.T. and C.L.). J.-F.P. gratefully acknowledges a scholar-ship from CREFSIP (Centre de Recherche sur la Fonction,la Structure et l’Ingenierie des Proteines).
Literature Cited
Adachi, J., and M. Hasegawa. 1996. MOLPHY Version 2.3:programs for molecular phylogenetics based on maximumlikelihood method. Comput. Sci. Monogr. 28:1–150.
Adams, K. L., D. O. Daley, J. Whelan, and J. D. Palmer. 2002.Genes for two mitochondrial ribosomal proteins in floweringplants are derived from their chloroplast or cytosoliccounterparts. Plant Cell 14:931–943.
Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J.Lipman. 1990. Basic local alignment search tool. J. Mol. Biol.215:403–410.
Bhattacharya, D., T. Friedl, and S. Damberger. 1996. Nuclear-encoded rDNA group I introns: origin and phylogeneticrelationships of insertion site lineages in the green algae. Mol.Biol. Evol. 13:978–989.
Bhattacharya, D., and L. Medlin. 1998. Algal phylogeny and theorigin of land plants. Plant Physiol. 116:9–15.
Bhattacharya, D., K. Weber, S. S. An, and W. Berning-Koch.1998. Actin phylogeny identifies Mesostigma viride asa flagellate ancestor of the land plants. J. Mol. Evol.47:544–550.
Boer, P. H., and M. W. Gray. 1991. Short dispersed repeatslocalized in spacer regions of Chlamydomonas reinhardtiimitochondrial DNA. Curr. Genet. 19:309–312.
Boudreau, E., and M. Turmel. 1996. Extensive gene rearrange-ments in the chloroplast DNAs of Chlamydomonas speciesfeaturing multiple dispersed repeats. Mol. Biol. Evol. 13:233–243.
Bremer, K. 1985. Summary of green plant phylogeny andclassification. Cladistics 1:369–385.
Bruno, W. J., N. D. Socci, and A. L. Halpern. 2000. Weightedneighbor joining: a likelihood-based approach to distance-based phylogeny reconstruction. Mol. Biol. Evol. 17:189–197.
Burger, G., B. F. Lang, H.-P. Braun, and S. Marx. 2003. Theenigmatic mitochondrial ORF ymf39 codes for ATP synthasechain b. Nucleic Acids Res. 31:2353–2360.
Burke, J. M., M. Belfort, T. R. Cech, R. W. Davies, R. J.Schweyen, D. A. Shub, J. W. Szostak, and H. F. Tabak. 1987.Structural conventions for group I introns. Nucleic Acids Res.15:7217–7221.
Castresana, J. 2000. Selection of conserved blocks from multiplealignments for their use in phylogenetic analysis. Mol. Biol.Evol. 17:540–552.
Chapman, R. L., M. A. Buchheim, C. F. Delwiche et al. (11 co-authors). 1998. Molecular systematics of the green algae. Pp.508–540 in D. E. Soltis, P. S. Soltis, and J. J. Doyle, eds.Molecular systematics of plants II DNA sequencing. KluwerAcademic Press, Norwell, Mass.
Cote, V., J.-P. Mercier, C. Lemieux, and M. Turmel. 1993. Thesingle group-I intron in the chloroplast rrnL gene ofChlamydomonas humicola encodes a site-specific DNAendonuclease (I-ChuI). Gene 129:69–76.
Denovan-Wright, E. M., A. M. Nedelcu, and R. W. Lee. 1998.Complete sequence of the mitochondrial DNA of Chlamydo-monas eugametos. Plant Mol. Biol. 36:285–295.
Fan, J., and R. W. Lee. 2002. Mitochondrial genome of thecolorless green alga Polytomella parva: two linear DNAmolecules with homologous inverted repeat termini. Mol.Biol. Evol. 19:999–1007.
Floyd, G. L., and C. J. O’Kelly. 1990. Phylum Chlorophyta: classUlvophyceae. Pp. 617–635 in L. Margulis, J. O. Corliss, M.Melkonian, and D. J. Chapman, eds. Handbook of Protoctista.Jones and Bartlett Publishers, Boston.
Friedl, T. 1995. Inferring taxonomic positions and testing genuslevel assignments in coccoid green lichen algae: a phyloge-netic analysis of 18S ribosomal RNA sequences fromDictyochloropsis reticulata and from members of the genusMyrmecia (Chlorophyta, Trebouxiophyceae Cl. Nov.). J.Phycol. 31:632–639.
———. 1997. The evolution of the green algae. Plant Syst. Evol.11(suppl.):87–101.
Friedl, T., and C. J. O’Kelly. 2002. Phylogenetic relationships ofgreen algae assigned to the genus Planophila (Chlorophyta):evidence from 18S rDNA sequence data and ultrastructure.Eur. J. Phycol. 37:373–384.
Gray, M. W., B. F. Lang, R. Cedergren et al. (15 co-authors).1998. Genome structure and gene content in protistmitochondrial DNAs. Nucleic Acids Res. 26:865–878.
Gu, X., and W.-H. Li. 1992. Higher rates of amino acidsubstitution in rodents than in humans. Mol. Phylogenet.Evol. 1:211–214.
Jamet-Vierny, C., J. Boulay, and J.-F. Briand. 1997. Intra-molecular cross-overs generate deleted mitochondrial DNAmolecules in Podospora anserina. Curr. Genet. 31:162–170.
Karol, K. G., R. M. McCourt, M. T. Cimino, and C. F. Delwiche.2001. The closest living relatives of land plants. Science294:2351–2353.
Kroymann, J., and K. Zetsche. 1998. The mitochondrial genomeof Chlorogonium elongatum inferred from the completesequence. J. Mol. Evol. 47:431–440.
Kubo, T., S. Nishizawa, A. Sugawara, N. Itchoda, A. Estiati, andT. Mikami. 2000. The complete nucleotide sequence of themitochondrial genome of sugar beet (Beta vulgaris L.) revealsa novel gene for tRNACys(GCA). Nucleic Acids Res.28:2571–2576.
Kuck, U., K. Jekosch, and P. Holzamer. 2000. DNA sequenceanalysis of the complete mitochondrial genome of the greenalga Scenedesmus obliquus: evidence for UAG being a leucineand UCA being a non-sense codon. Gene 253:13–18.
Kurtz, S., J. V. Choudhuri, E. Ohlebusch, C. Schleiermacher, J.Stoye, and R. Giegerich. 2001. REPuter: the manifoldapplications of repeat analysis on a genomic scale. NucleicAcids Res. 29:4633–4642.
Lang, B. F., M. W. Gray, and G. Burger. 1999. Mitochondrialgenome evolution and the origin of eukaryotes. Annu. Rev.Genet. 33:351–397.
Lemieux, C., C. Otis, and M. Turmel. 2000. Ancestralchloroplast genome in Mesostigma viride reveals an earlybranch of green plant evolution. Nature 403:649–652.
Lowe, T. M., and S. R. Eddy. 1997. tRNAscan-SE: a program forimproved detection of transfer RNA genes in genomicsequence. Nucleic Acids Res. 25:955–964.
Lucas, P., C. Otis, J.-P. Mercier, M. Turmel, and C. Lemieux.2001. Rapid evolution of the DNA-binding site in LAGLI-DADG homing endonucleases. Nucleic Acids Res. 29:960–969.
Lunt, D. H., and B. C. Hyman. 1997. Animal mitochondrialDNA recombination. Nature 387:247.
Mackenzie, S., S. He, and A. Lyznik. 1994. The elusive plantmitochondrion as a genetic system. Plant Physiol. 105:775–780.
Marienfeld, J., M. Unseld, and A. Brennicke. 1999. Themitochondrial genome of Arabidopsis is composed of bothnative and immigrant information. Trends Plant Sci. 4:495–502.
Mitochondrial DNA Sequence of Pseudendoclonium 933
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

Marin, B., and M. Melkonian. 1999. Mesostigmatophyceae,a new class of streptophyte green algae revealed by SSUrRNA sequence comparisons. Protist 150:399–417.
Mattox, K. R., and K. D. Stewart. 1984. Classification of thegreen algae: a concept based on comparative cytology. Pp.29–72 in D. E. G. Irvine and D. M. John, eds. The systematicsof the green algae. Academic Press, London.
Maul, J. E., J. W. Lilly, L. Cui, C. W. dePamphilis, W. Miller, E.H. Harris, and D. B. Stern. 2002. The Chlamydomonasreinhardtii plastid chromosome: islands of genes in a sea ofrepeats. Plant Cell 14:2659–2679.
McCracken, D. A., M. J. Nadakavukaren, and J. R. Cain. 1980. Abiochemical and ultrastructural evaluation of the taxonomicposition of Glaucosphaera vacuolata Korsch. New Phytol.86:39–44.
Melkonian, M. 1990. Chlorophyte orders of uncertain affinities:Order Pedinomonales. Pp. 649–651 in L. Margulis, J. O.Corliss, M. Melkonian, and D. J. Chapman, eds. Handbook ofProtoctista. Jones and Bartlett Publishers, Boston.
Michaelis, G., C. Vahrenholz, and E. Pratje. 1990. MitochondrialDNA of Chlamydomonas reinhardtii: the gene for apocyto-chrome b and the complete functional map of the 15.8 kbDNA. Mol. Gen. Genet. 223:211–216.
Michel, F., and E. Westhof. 1990. Modelling of the three-dimensional architecture of group I catalytic introns based oncomparative sequence analysis. J. Mol. Biol. 216:585–610.
Nakayama, T., S. Watanabe, and I. Inouye. 1996. Phylogeny ofwall-less green flagellates inferred from 18S rDNA sequencedata. Phycol. Res. 44:151–161.
Nedelcu, A. M. 1997. Fragmented and scrambled mitochondrialribosomal RNA coding regions among green algae: a modelfor their origin and evolution. Mol. Biol. Evol. 14:506–517.
———. 1998. Contrasting mitochondrial genome organizationsand sequence affiliations among green algae: potential factors,mechanisms, and evolutionary scenarios. J. Phycol. 34:16–28.
Nedelcu, A. M., and R. W. Lee. 1998. Short repetitive sequencesin green algal mitochondrial genomes: potential roles inmitochondrial genome evolution. Mol. Biol. Evol. 15:690–701.
Nedelcu, A. M., R. W. Lee, C. Lemieux, M. W. Gray, and G.Burger. 2000. The complete mitochondrial DNA sequence ofScenedesmus obliquus reflects an intermediate stage in theevolution of the green algal mitochondrial genome. GenomeRes. 10:819–831.
Notsu, Y., S. Masood, T. Nishikawa, N. Kubo, G. Akiduki, M.Nakazono, A. Hirai, and K. Kadowaki. 2002. The completesequence of the rice (Oryza sativa L.) mitochondrial genome:frequent DNA sequence acquisition and loss during theevolution of flowering plants. Mol. Genet. Genomics268:434–445.
Oda, K., K. Yamato, E. Ohta et al. (11 co-authors). 1992. Geneorganization deduced from the complete sequence of liverwortMarchantia polymorpha mitochondrial DNA. A primitiveform of plant mitochondrial genome. J. Mol. Biol. 223:1–7.
O’Kelly, C. J., and G. L. Floyd. 1984. Flagellar apparatusabsolute orientations and the phylogeny of the green algae.BioSystems 16:227–251.
Oldenburg, D. J., and A. J. Bendich. 2001. Mitochondrial DNAfrom the liverwort Marchantia polymorpha: circularlypermuted linear molecules, head-to-tail concatemers, and a 59protein. J. Mol. Biol. 310:549–562.
Palmer, J. D., K. L. Adams, Y. Cho, C. L. Parkinson, Y.-L. Qiu,and K. Song. 2000. Dynamic evolution of plant mitochondrialgenomes: mobile genes and introns and highly variablemutation rates. Proc. Natl. Acad. Sci. USA 97:6960–6966.
Palmer, J. D., and L. A. Herbon. 1988. Plant mitochondrial DNAevolves rapidly in structure, but slowly in sequence. J. Mol.Evol. 28:87–97.
Palmer, J. D., and C. R. Shields. 1984. Tripartite structure of theBrassica campestris mitochondrial genome. Nature 307:437–440.
Schuster, W., and A. Brennicke. 1994. The plant mitochondrialgenome: physical structure, information content, RNA edit-ing, and gene migration to the nucleus. Annu. Rev. PlantPhysiol. Plant Mol. Biol. 45:61–78.
Schwartz, S., Z. Zhang, K. A. Frazer, A. Smit, C. Riemer, J.Bouck, R. Gibbs, R. Hardison, and W. Miller. 2000.PipMaker—a web server for aligning two genomic DNAsequences. Genome Res. 10:577–586.
Shimodaira, H., and M. Hasegawa. 2001. CONSEL: forassessing the confidence of phylogenetic tree selection.Bioinformatics 17:1246–1247.
Sluiman, H. J. 1985. A cladistic evaluation of the lower andhigher green plants (Viridiplantae). Plant Syst. Evol.149:217–232.
———. 1989. The green algal class Ulvophyceae: an ultrastruc-tural survey and classification. Crypt. Bot. 1:83–94.
Strimmer, K., and A. von Haeseler. 1996. Quartet puzzling:a quartet maximum-likelihood method for reconstructing treetopologies. Mol. Biol. Evol. 16:964–969.
Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994.CLUSTAL W: improving the sensitivity of progressivemultiple sequence alignment through sequence weighting,position-specific gap penalties and weight matrix choice.Nucleic Acids Res. 22:4673–4680.
Tupa, D. D. 1974. An investigation of certain chaetophoraleanalgae. Beihefte zur Nova Hedwigia 46(suppl.):64–67.
Turmel, M., V. Cote, C. Otis, J.-P. Mercier, M. W. Gray, K. M.Lonergan, and C. Lemieux. 1995. Evolutionary transfer ofORF-containing group I introns between different subcellularcompartments (chloroplast and mitochondrion). Mol. Biol.Evol. 12:533–545.
Turmel, M., M. Ehara, C. Otis, and C. Lemieux. 2002.Phylogenetic relationships among Streptophytes as inferredfrom chloroplast small and large subunit rRNA genesequences. J. Phycol. 38:364–375.
Turmel, M., R. R. Gutell, J.-P. Mercier, C. Otis, and C. Lemieux.1993. Analysis of the chloroplast large subunit ribosomalRNA gene from 17 Chlamydomonas taxa: three internaltranscribed spacers and 12 group I intron insertion sites. J.Mol. Biol. 232:446–467.
Turmel, M., C. Lemieux, G. Burger, B. F. Lang, C. Otis, I.Plante, and M. W. Gray. 1999. The complete mitochondrialDNA sequences of Nephroselmis olivacea and Pedinomonasminor. Two radically different evolutionary patterns withingreen algae. Plant Cell 11:1717–1729.
Turmel, M., C. Otis, and C. Lemieux. 1999. The completechloroplast DNA sequence of the green alga Nephroselmisolivacea: insights into the architecture of ancestral chloro-plast genomes. Proc. Natl. Acad. Sci. USA 96:10248–10253.
———. 2002a. The complete mitochondrial DNA sequence ofMesostigma viride identifies this green alga as the earliestgreen plant divergence and predicts a highly compactmitochondrial genome in the ancestor of all green plants.Mol. Biol. Evol. 19:24–38.
———. 2002b. The chloroplast and mitochondrial genomesequences of the charophyte Chaetosphaeridium globosum:insights into the timing of the events that restructuredorganelle DNAs within the green algal lineage that led toland plants. Proc. Natl. Acad. Sci. USA 99:11275–11280.
934 Pombert et al.
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from

Unseld, M., J. R. Marienfeld, P. Brandt, and A. Brennicke. 1997.The mitochondrial genome of Arabidopsis thalianacontains 57 genes in 366,924 nucleotides. Nat. Genet. 15:57–61.
Volfovsky, N., B. J. Haas, and S. L. Salzberg. 2001. A clusteringmethod for repeat analysis in DNA sequences. Genome Biol.2:0027.0021–0027.0011.
Watanabe, S., A. Himizu, L. A. Lewis, G. L. Floyd, and P. A.Fuerst. 2000. Pseudoneochloris marina (Chlorophyta), a newcoccoid ulvophycean alga, and its phylogenetic positioninferred from morphological and molecular data. J. Phycol.36:596–604.
Watanabe, S., N. Kuroda, and F. Maiwa. 2001. Phylogeneticstatus of Helicodictyon planctonicum and Desmochlorishalophila gen. et comb. nov. and the definition of the classUlvophyceae (Chlorophyta). Phycologia 40:421–434.
Whelan, S., and N. Goldman. 2001. A general empirical model ofprotein evolution derived from multiple protein families usinga maximum-likelihood approach. Mol. Biol. Evol. 18:691–699.
Wolf, M., M. Buchheim, E. Hegewald, L. Krienitz, and D.Hepperle. 2002. Phylogenetic position of the Sphaeropleaceae(Chlorophyta). Plant Syst. Evol. 230:161–171.
Wolfe, K. H., W.-H. Li, and P. M. Sharp. 1987. Rates ofnucleotide substitution vary greatly among plant mitochon-drial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci.USA 84:9054–9058.
Wolff, G., I. Plante, B. F. Lang, U. Kuck, and G. Burger. 1994.Complete sequence of the mitochondrial DNA of the chloro-phyte alga Prototheca wickerhamii. J. Mol. Biol. 237:75–86.
Yang, Z. 1997. PAML: a program package for phylogeneticanalysis by maximum likelihood. CABIOS 13:555–556.
Zechman, F. W., E. C. Theriot, E. A. Zimmer, and R. L.Chapman. 1990. Phylogeny of the Ulvophyceae (Chlorophy-ta): cladistic analysis of nuclear-encoded rRNA sequence data.J. Phycol. 26:700–710.
Mark Ragan, Associate Editor
Accepted January 8, 2004
Mitochondrial DNA Sequence of Pseudendoclonium 935
by guest on Novem
ber 26, 2015http://m
be.oxfordjournals.org/D
ownloaded from




















