
Testing drug response in the presence of genetic information
10
Pharmacogenetics 2000, 10:503–510 Original article 0960-314X # 2000 Lippincott Williams & Wilkins Testing drug response in the presence of genetic information: sampling issues for clinical trials Lon R. Cardon a , Ramana M. Idury b , Timothy J.R. Harris c , John S. Witte d and Robert C. Elston d a Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK, b Kiva Genetics, Inc., Mountain View, California, USA, c Structural GenomiX, Inc., Encinitas, California, USA, d Department of Epidemiology and Biostatistics, Case Western Reserve University, Cleveland, Ohio, USA Received 18 August 1999; accepted 19 December 1999 Progress towards construction of a dense map of di-allelic markers across the human genome has generated considerable enthusiasm for pharmacogenomic applications. To date, however, nearly all of the effort on single nucleotide polymorphism (SNP) projects has been focused on marker identification and screening, not on how the SNP genotype data actually can be used in clinical trials to advance medical practice. Here, we explore how different properties of SNPs impact the size, scope and design of clinical trials using a simple trial design. We evaluate the clinical trial sampling requirements under different allele frequencies, gene action, gene effect size and number of markers in a genome screen. Power and sample size calculations suggest that allele frequency and type of gene action can have a dramatic impact on trial sample sizes, in that under some conditions the required sample sizes are too large to be applicable in a costly clinical trial setting. In other situations, however, pharmacogenomic clinical trials can yield significant sampling/cost savings over traditional trials. These properties are discussed with regard to the general usage of genetic information in clinical trial settings. Pharmacogenetics 10:503–510 # 2000 Lippincott Williams & Wilkins Keywords: trial design, SNP, single nucleotide polymorphism, power Introduction Individual differences in response to drugs have been studied for over 65 years (Nebert, 1997), yet modern medicine remains rooted in undifferentiated treat- ment of individuals. A physician’s selection of drug dosage and treatment typically is based upon empiri- cal averages obtained from clinical trials, not upon the individual being administered the drug. This practice of undifferentiated treatment, which is claimed to contribute to a loss of over US$100 billion per year in hospital admissions, productivity, and premature death (Marshall, 1997a), has stimulated substantial interest in drug development strategies that might better predict individual profiles of efficacy and toxicity (Drews & Ryser, 1997). This interest has led to a resurgence in the field of pharmacogenomics, aimed at identifying genetic loci contributing to variability in drug response (Marshall, 1997b). There are now a number of examples of variation in drug metabolism and efficacy attributable to specific DNA polymorphisms (Weber, 1997) and of mutations in receptors of drug target pathways (Meyer, 1997). Recent research has focused on drugs for common diseases such as asthma, Alzheimer’s disease, and coronary artery disease and hyper- tension (Poirier et al., 1995; Tan et al., 1997; Ferrari, 1998; Kuivenhoven et al., 1998). Despite the emer- gence of relevant knowledge, genetic information has not yet been widely integrated into the clinical trial setting. To date, pharmacogenomic research has been firmly entrenched in the candidate gene model and has been largely limited to the study of compounds after they have reached the marketplace. In contrast to the candidate gene approach, a new pharmacogenomic paradigm is emerging in which the entire human genome is screened for single nucleotide polymorphisms (SNPs) that may be asso- ciated with drug response. The genome-screen model of pharmacogenomics is being driven by techno- logical advancements for identifying and rapidly scoring SNPs in and around the 60 000–100 000 Correspondence to Lon R. Cardon, Wellcome Trust Centre for Human Genetics, University of Oxford, Roosevelt Drive, Oxford OX3 7BN, UK Tel: 44 1865 287 587; fax: 44 1865 287 650; e-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Testing drug response in the presence of genetic information

Pharmacogenetics 2000, 10:503±510 Original article
0960-314X # 2000 Lippincott Williams & Wilkins
Testing drug response in the presence of geneticinformation: sampling issues for clinical trials
Lon R. Cardona, Ramana M. Iduryb, Timothy J.R. Harrisc, John S. Witted andRobert C. Elstond
aWellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK, bKiva Genetics, Inc., Mountain View,California, USA, cStructural GenomiX, Inc., Encinitas, California, USA, dDepartment of Epidemiology and Biostatistics,Case Western Reserve University, Cleveland, Ohio, USA
Received 18 August 1999; accepted 19 December 1999
Progress towards construction of a dense map of di-allelic markers across the humangenome has generated considerable enthusiasm for pharmacogenomic applications. To date,however, nearly all of the effort on single nucleotide polymorphism (SNP) projects has beenfocused on marker identi®cation and screening, not on how the SNP genotype data actuallycan be used in clinical trials to advance medical practice. Here, we explore how differentproperties of SNPs impact the size, scope and design of clinical trials using a simple trialdesign. We evaluate the clinical trial sampling requirements under different allelefrequencies, gene action, gene effect size and number of markers in a genome screen. Powerand sample size calculations suggest that allele frequency and type of gene action can have adramatic impact on trial sample sizes, in that under some conditions the required samplesizes are too large to be applicable in a costly clinical trial setting. In other situations,however, pharmacogenomic clinical trials can yield signi®cant sampling/cost savings overtraditional trials. These properties are discussed with regard to the general usage of geneticinformation in clinical trial settings. Pharmacogenetics 10:503±510 # 2000 Lippincott Williams & Wilkins
Keywords: trial design, SNP, single nucleotide polymorphism, power
Introduction
Individual differences in response to drugs have beenstudied for over 65 years (Nebert, 1997), yet modernmedicine remains rooted in undifferentiated treat-ment of individuals. A physician's selection of drugdosage and treatment typically is based upon empiri-cal averages obtained from clinical trials, not uponthe individual being administered the drug. Thispractice of undifferentiated treatment, which isclaimed to contribute to a loss of over US$100 billionper year in hospital admissions, productivity, andpremature death (Marshall, 1997a), has stimulatedsubstantial interest in drug development strategiesthat might better predict individual pro®les of ef®cacyand toxicity (Drews & Ryser, 1997). This interest hasled to a resurgence in the ®eld of pharmacogenomics,aimed at identifying genetic loci contributing tovariability in drug response (Marshall, 1997b).
There are now a number of examples of variationin drug metabolism and ef®cacy attributable tospeci®c DNA polymorphisms (Weber, 1997) and ofmutations in receptors of drug target pathways(Meyer, 1997). Recent research has focused on drugsfor common diseases such as asthma, Alzheimer'sdisease, and coronary artery disease and hyper-tension (Poirier et al., 1995; Tan et al., 1997; Ferrari,1998; Kuivenhoven et al., 1998). Despite the emer-gence of relevant knowledge, genetic information hasnot yet been widely integrated into the clinical trialsetting. To date, pharmacogenomic research has been®rmly entrenched in the candidate gene model andhas been largely limited to the study of compoundsafter they have reached the marketplace.
In contrast to the candidate gene approach, a newpharmacogenomic paradigm is emerging in whichthe entire human genome is screened for singlenucleotide polymorphisms (SNPs) that may be asso-ciated with drug response. The genome-screen modelof pharmacogenomics is being driven by techno-logical advancements for identifying and rapidlyscoring SNPs in and around the 60 000±100 000
Correspondence to Lon R. Cardon, Wellcome Trust Centre for HumanGenetics, University of Oxford, Roosevelt Drive, Oxford OX3 7BN, UKTel: �44 1865 287 587; fax: �44 1865 287 650;e-mail: [email protected]

genes in the human genome (Lander, 1999). Thepotential for proprietary commercial usage of SNPsand SNP maps has led to new public fundinginitiatives and pharmaceutical collaborations aimedat identi®cation and public dissemination of SNPs(Collins et al., 1997; Masood, 1999).
The emergence of an SNP map will present manyopportunities for integrating genetics into drug dis-covery, design, and development (Regalado, 1997).However, the potential to screen many polymorph-isms in many genes in an ef®cient and cost-effectivemanner only partially ful®lls the requirements forpharmaceutical use of genetic information. At pres-ent, it is unknown how to optimally integrate SNPdata with clinical trials and whether or not incor-poration of genetic data will require substantivechanges in the size and cost of clinical trials.
As it has long been known that the simultaneousstudy of multiple factors can lead to economy in thecost of determining the effect of each factor as well asnew insights into how the factors interact (Fisher,1935), one might expect that the synthesis of geneticstudies and clinical trials should make clinical trialsmore economical, increase ef®cacy by knowing howdrugs and genotypes interact to produce a response,and predict side-effects when they have a geneticaetiology. However, these advances will only bepossible when clinical trials are speci®cally designedto capitalize on the underlying genetic effects. As a®rst step towards designing such trials, we need toknow how different properties of the aetiological lociimpact the clinical trial size.
Here, we consider how different properties of SNPsin¯uence the expected size and power of clinicaltrials. Speci®cally, we consider the following issues:What effect do allele frequency and gene action(dominant, recessive, additive) have on clinical trialsampling requirements? How large a genetic effectwill be needed to in¯uence trial outcomes and con-clusions? Under what conditions will genetic informa-tion increase or decrease the size of clinical trials?What is the impact of testing up to 100 000 SNPs onfalse positive rates and therefore sample size require-ments?
Methods
There are numerous variations on study design thatone could consider for this type of power study (e.g.multiple doses, quantitative versus discrete outcomes,various placebo effects, haplotype combinations, epi-static interactions). Here we adopt a simple generictrial design in an attempt to provide general informa-tion about pharmacogenomic sampling requirements.In this regard, for all statistical power and sample
size calculations, we restrict our concern to discreteoutcomes in an hypothesized clinical trial, i.e. sub-jects in the trial either respond to a given compoundor they do not. In this context, `response' can refer toef®cacy, toxicity, or any of the various metabolicoutcomes typically assessed in the course of clinicaltrials.
The statistical method used to calculate power andrequired sample sizes was recently described (Elstonet al., 1999). Brie¯y, this design assumes a di-allelicmarker (e.g. SNP) with genotype frequencies(1 ÿ q)2, 2q(1 ÿ q), and q2 for genotypes A2 A2,A2 A1 and A1 A1, respectively, and D discrete dosegroups of drug administration (group 0 may beadministered a placebo). The number of individualsin each dose group may be ®xed by the experimenter,as in any (non-genetic) clinical trial, but the numberof individuals in each genotype group cannot be ®xedunless selective sampling for a single locus is con-ducted. The response rate in each cell of these three(genotypes) by D (doses, including the placebo) de-sign, ðij, is estimated by pij � Rij/Nij, where Rij andNij represent the number of responders and totalnumber of individuals in each cell, respectively. Thetotal number of individuals required in the trial isthus
N �X2
i�0
XDÿ1
j�0
Nij
For any dose in this design, the overall responserate is:
Kj � (1ÿ q)2ð0j � 2q(1ÿ q)ð1j � q2ð2j
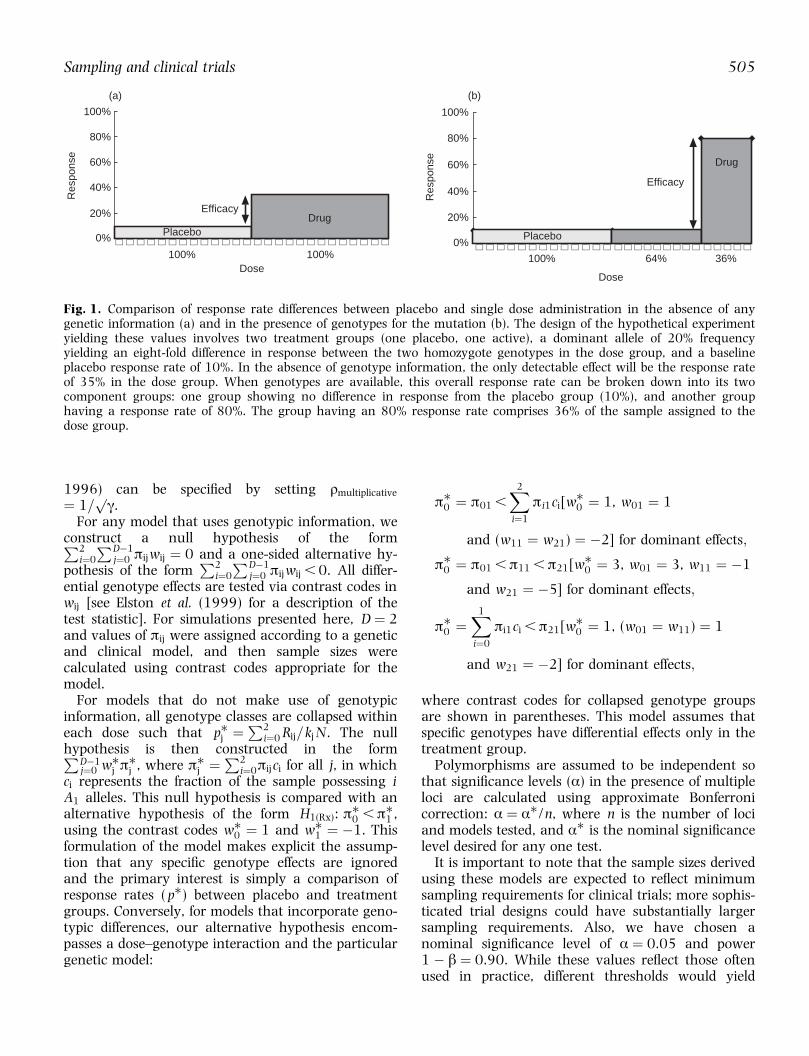
For example, if ð0j , ð1j � ð2j and q � 0.2, thisoverall response rate is made up of (1.0 ÿ0.2)2 � 64% of the population having response rateð0j and the remaining 36% of the population havingresponse rate ð1j � ð2j. When ð0j � 0.1 andð1j � ð2j � 0.8, Kj � 35% (Fig. 1).
Following Risch and Merikangas (1996), we modelthe effect of any polymorphism as genotype relativerisks (GRR), such that the overall dose-speci®c riskrelative to the placebo is:
Kj
ð0j� (1ÿ q)2 � 2q(1ÿ q)ãr� q2ã
where ã � ð2j/ð0j > 1.0 and ð1j � ãrð0j. Additive,dominant, and recessive gene effects can then beencapsulated in r:
radditive � (ã� 1)=2ã
rdominant � 1:0
rrecessive � 1=ã
Also, the multiplicative GRR (Risch & Merikangas,
504 Cardon et al.
1996) can be speci®ed by setting rmultiplicative
� 1=pã.
For any model that uses genotypic information, weconstruct a null hypothesis of the formP2
i�0
PDÿ1j�0 ðijwij � 0 and a one-sided alternative hy-
pothesis of the formP2
i�0
PDÿ1j�0 ðijwij , 0. All differ-
ential genotype effects are tested via contrast codes inwij [see Elston et al. (1999) for a description of thetest statistic]. For simulations presented here, D � 2and values of ðij were assigned according to a geneticand clinical model, and then sample sizes werecalculated using contrast codes appropriate for themodel.
For models that do not make use of genotypicinformation, all genotype classes are collapsed withineach dose such that p�j �
P2i�0 Rij=kj N. The null
hypothesis is then constructed in the formPDÿ1j�0 w�j ð�j , where ð�j �
P2i�0ðijci for all j, in which
ci represents the fraction of the sample possessing iA1 alleles. This null hypothesis is compared with analternative hypothesis of the form H1(Rx): ð�0 ,ð�1 ,using the contrast codes w�0 � 1 and w�1 � ÿ1. Thisformulation of the model makes explicit the assump-tion that any speci®c genotype effects are ignoredand the primary interest is simply a comparison ofresponse rates ( p�) between placebo and treatmentgroups. Conversely, for models that incorporate geno-typic differences, our alternative hypothesis encom-passes a dose±genotype interaction and the particulargenetic model:
ð�0 � ð01 ,X2
i�1
ði1ci[w�0 � 1, w01 � 1
and (w11 � w21) � ÿ2] for dominant effects;
ð�0 � ð01 ,ð11 ,ð21[w�0 � 3, w01 � 3, w11 � ÿ1
and w21 � ÿ5] for dominant effects;
ð�0 �X1
i�0
ði1ci ,ð21[w�0 � 1, (w01 � w11) � 1
and w21 � ÿ2] for dominant effects;
where contrast codes for collapsed genotype groupsare shown in parentheses. This model assumes thatspeci®c genotypes have differential effects only in thetreatment group.
Polymorphisms are assumed to be independent sothat signi®cance levels (á) in the presence of multipleloci are calculated using approximate Bonferronicorrection: á � á�/n, where n is the number of lociand models tested, and á� is the nominal signi®cancelevel desired for any one test.
It is important to note that the sample sizes derivedusing these models are expected to re¯ect minimumsampling requirements for clinical trials; more sophis-ticated trial designs could have substantially largersampling requirements. Also, we have chosen anominal signi®cance level of á � 0.05 and power1 ÿ â � 0.90. While these values re¯ect those oftenused in practice, different thresholds would yield
Fig. 1. Comparison of response rate differences between placebo and single dose administration in the absence of anygenetic information (a) and in the presence of genotypes for the mutation (b). The design of the hypothetical experimentyielding these values involves two treatment groups (one placebo, one active), a dominant allele of 20% frequencyyielding an eight-fold difference in response between the two homozygote genotypes in the dose group, and a baselineplacebo response rate of 10%. In the absence of genotype information, the only detectable effect will be the response rateof 35% in the dose group. When genotypes are available, this overall response rate can be broken down into its twocomponent groups: one group showing no difference in response from the placebo group (10%), and another grouphaving a response rate of 80%. The group having an 80% response rate comprises 36% of the sample assigned to thedose group.
100%
80%
60%
40%
20%
0%
100% 100%Dose
PlaceboDrug
Efficacy
Res
pons
e
(a)
100%
80%
60%
40%
20%
0%
Res
pons
e
100% 64% 36%
Dose
Placebo
Drug
Efficacy
(b)
Sampling and clinical trials 505
different outcomes, especially with respect to theabsolute numbers of samples required (Fig. 2).
Results
Figure 1 shows an illustrative comparison of the typeof pharmacogenomic effect with which we are con-cerned. In this example, a dominant genotype effectconfers an eight-fold increase in response over thebaseline genotype. The increasing allele has a popula-tion frequency of 20%. If the aetiologic genotypeeffect were ignored, as in the case of a traditionalnon-genetic clinical trial (Fig. 1a), then the observeddose response rate would be only 35%. For manyindications, this response rate would not be regardedas strong evidence for ef®cacy in comparison with aplacebo rate of 10% as we have assumed here. Inreality, however, 36% of the trial sample has aresponse rate of 80% while the remainder of thegroup has the same response rate as placebo (Fig.1b). As such a large a genotypic effect may in¯uenceinvestigators' conclusions of ef®cacy and determinethe future development and usage of the compound
(e.g. Richard et al., 1997), it is of some importance todetermine how large a trial should be conducted todetect it.
The effects of allele frequency, genotype relativerisk (GRR), and mode of gene action on the necessarysample size are summarized in Fig. 2. We ®rstdescribe the relative in¯uence of these effects withrespect to a candidate study of a speci®c variant, e.g.a study in which the molecular target of drug actionis known, and one is interested speci®cally in howgenetic variation in the target in¯uences ef®cacyand/or action. We subsequently explore equivalentsituations in the context of genome-screens, i.e. aspeci®c variant in¯uences ef®cacy or action, but thevariant is unknown.
The type of gene action
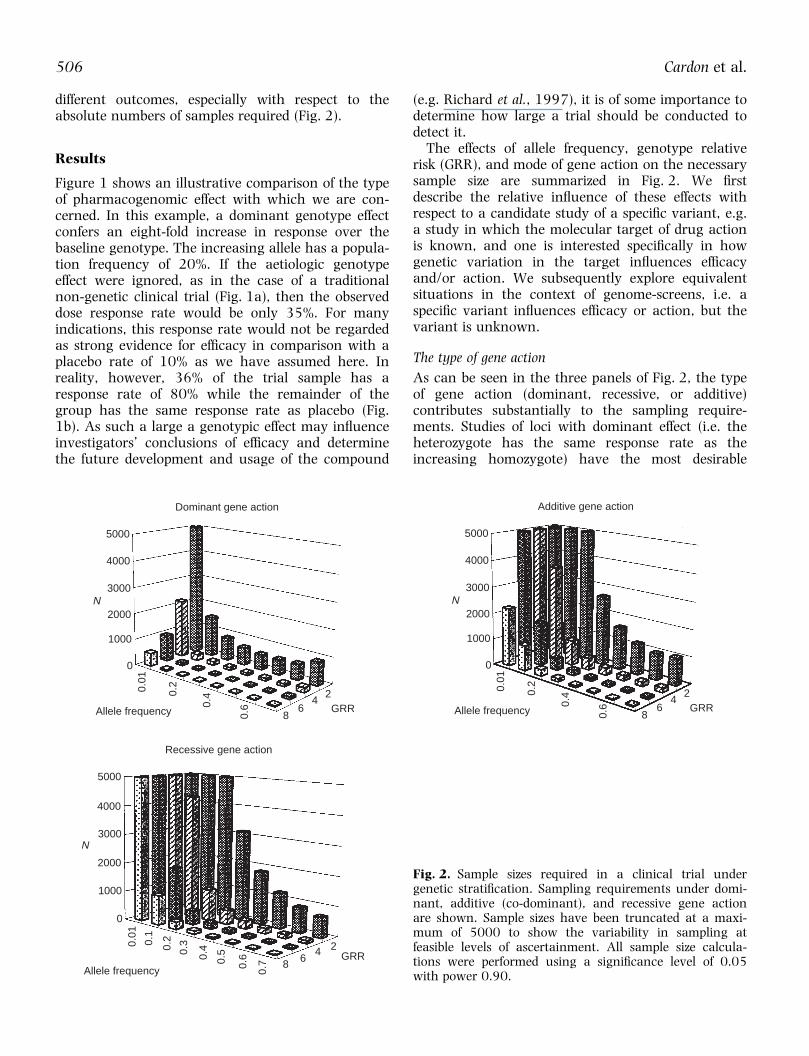
As can be seen in the three panels of Fig. 2, the typeof gene action (dominant, recessive, or additive)contributes substantially to the sampling require-ments. Studies of loci with dominant effect (i.e. theheterozygote has the same response rate as theincreasing homozygote) have the most desirable
Fig. 2. Sample sizes required in a clinical trial undergenetic strati®cation. Sampling requirements under domi-nant, additive (co-dominant), and recessive gene actionare shown. Sample sizes have been truncated at a maxi-mum of 5000 to show the variability in sampling atfeasible levels of ascertainment. All sample size calcula-tions were performed using a signi®cance level of 0.05with power 0.90.
5000
4000
3000
2000
1000
0
0.01
0.2
0.4
0.6
86
42
Dominant gene action
N
Allele frequency GRR
5000
4000
3000
2000
1000
0
0.01
0.2
0.4
0.6
86
42
Additive gene action
N
Allele frequency GRR
5000
4000
3000
2000
1000
0
0.01
0.2
0.4
0.6
8 64 2
Recessive gene action
N
Allele frequencyGRR
0.1
0.3
0.5
0.7
506 Cardon et al.
sampling characteristics, often requiring less than50% of the number of patients needed to detect anadditive or recessive locus. This is essentially thegenetic architecture of the apolipoprotein E locus,which was reported to be correlated with response tothe acetylcholinesterase inhibitor, tacrine, in a studyof a small number of Alzheimer's disease patients(Poirier et al., 1995). However, unless the suscept-ibility allele is relatively common (> 30%) and theeffect of the gene is relatively large (GRR . 4)pharmacogenomic studies of recessive and additiveloci have inherent sampling requirements that aretoo extreme for most early stage clinical trials andsome phase III designs. These data emphasize theneed for knowledge of genetic parameters prior toinitiating candidate mutation ef®cacy trials. In theabsence of any information about allele frequency,distribution of response and effect size, pharmaco-genomic trials may be grossly under- or overpow-ered.
SNP allele frequencies
The frequencies of alleles at the polymorphism ofinterest have a dramatic impact on the likelihood ofdetecting a dosage effect (Fig. 2). For relatively un-common alleles (, 10%), most genetic models re-quire in excess of 1000 samples to detect theunderlying genotypic effect. Studies of rare alleles(1% or less) are essentially impractical, as the num-ber of samples required exceeds the realm of nearlyall clinical trials. However, the situation improvesnoticeably when the frequency of the susceptibilityallele is 30% or greater. In this case, fewer than300±400 individuals are required under nearly anygenetic effect size and model (the exception beingvery modest genotypic effects). Thus, candidate genestudies in which the investigator has prior knowledgeabout the magnitude and pattern of genotypic effectsmay be feasible, but studies involving a large numberof loci of unknown frequency and effect size maysuffer from a lack of statistical power to detect anyeffects.
Gene effect size
Situations in which the genetic variant has a smalleffect require sample sizes that would be unreason-able for all but the very largest phase II trials andsome phase III trials (Fig. 2). For example, when theincreasing genotype confers only a two-fold increasein response over the baseline genotype, pharmaco-genomic sample sizes range from 582 to over28 000. As this simulated example represents a best-case scenario in which the investigator tests only onelocus and knows the type of genotypic effect of thatlocus, most situations will actually require a much
larger sample size than this to detect signi®cance atthe modest level of á � 0.05. Conversely, as ex-pected, situations in which the gene has a muchlarger in¯uence on response rates require relativelysmall samples, particularly when the susceptibilityallele has a frequency of 30% or greater. Theseoutcomes suggest that pharmacogenomic loci thathave a small or moderate effect on the outcomemeasure may be most fruitfully assessed when verylarge trials can be undertaken, such as in a phase IVsetting. In this case, personalization of treatment maycome only after the drug has been put on themarket.
Ignoring aetiologic genetic effects
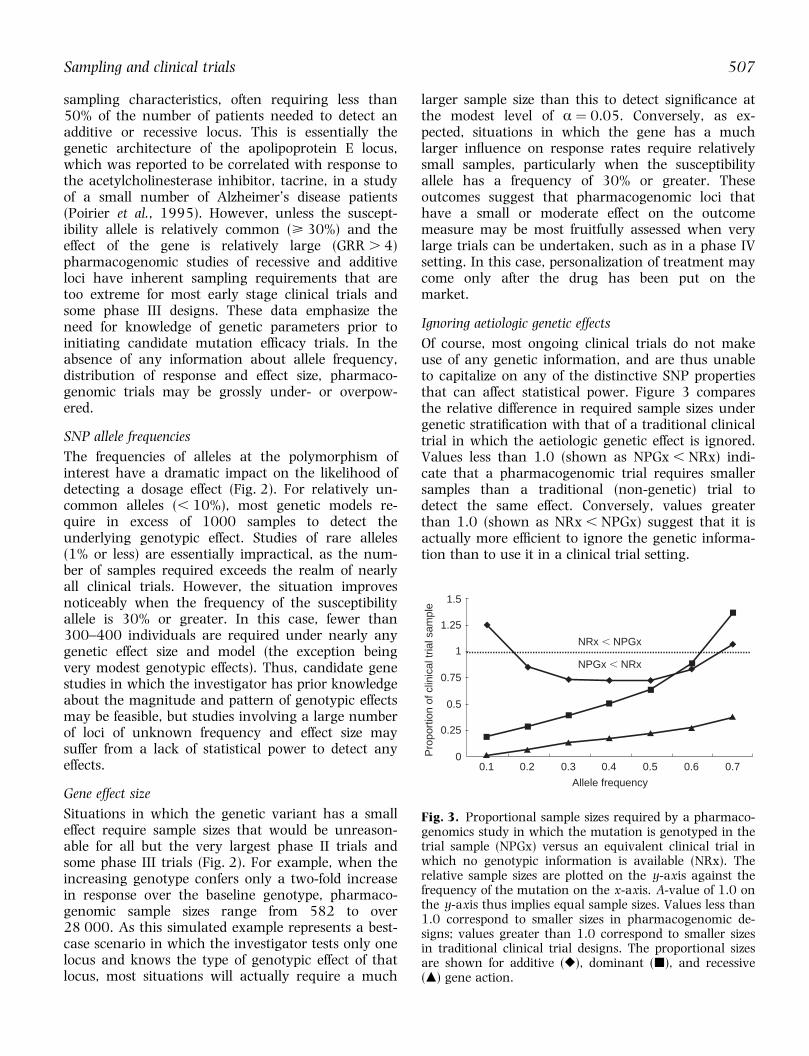
Of course, most ongoing clinical trials do not makeuse of any genetic information, and are thus unableto capitalize on any of the distinctive SNP propertiesthat can affect statistical power. Figure 3 comparesthe relative difference in required sample sizes undergenetic strati®cation with that of a traditional clinicaltrial in which the aetiologic genetic effect is ignored.Values less than 1.0 (shown as NPGx , NRx) indi-cate that a pharmacogenomic trial requires smallersamples than a traditional (non-genetic) trial todetect the same effect. Conversely, values greaterthan 1.0 (shown as NRx , NPGx) suggest that it isactually more ef®cient to ignore the genetic informa-tion than to use it in a clinical trial setting.
1.5
1.25
1
0.75
0.5
0.25
00.1 0.2 0.3 0.4 0.5 0.6 0.7
Allele frequency
Pro
port
ion
of c
linic
al tr
ial s
ampl
e
NPGx , NRx
NRx , NPGx
Fig. 3. Proportional sample sizes required by a pharmaco-genomics study in which the mutation is genotyped in thetrial sample (NPGx) versus an equivalent clinical trial inwhich no genotypic information is available (NRx). Therelative sample sizes are plotted on the y-axis against thefrequency of the mutation on the x-axis. A-value of 1.0 onthe y-axis thus implies equal sample sizes. Values less than1.0 correspond to smaller sizes in pharmacogenomic de-signs; values greater than 1.0 correspond to smaller sizesin traditional clinical trial designs. The proportional sizesare shown for additive (r), dominant (j), and recessive(m) gene action.
Sampling and clinical trials 507

Unsurprisingly, the incorporation of genetic infor-mation into clinical trials can reduce the sample sizesconsiderably, typically by a factor of at least two. Therelative saving in sampling is primarily a function ofthe frequency of the genetic variant, although thetype of gene action is also a salient factor. Interest-ingly, in the allele frequency range of 0.3±0.5,pharmacogenomic trials have substantially smallersampling requirements than traditional clinical trials,regardless of the type of gene action. However,pharmacogenomic trials do not imply a uniformreduction in the size/cost of a given clinical trial. Insome cases, particularly in the extreme ranges ofallele frequencies, an ef®cient trial design would notinvolve any genotyping at all. This again emphasizesthe utility of prior information about gene action andallele frequency, as some mutations will afford hugesavings in sample size, while others afford little ornone at all.
Genome-wide screening
The previous examples have addressed a single di-allelic locus, as might be encountered in a pharmaco-genomic study of a candidate gene mutation. Forgenome-wide screens of SNPs, however, we wish toknow the required sample sizes when testing hun-dreds or thousands of markers. In Fig. 4, we presentthe relative increase in sample size required as afunction of the number of loci tested. We account forincreases in false positive rates due to multiple testingby Bonferroni correction; this may be conservativefor a dense SNP map because of dependencies amongthe SNPs. All values presented in the ®gure arerelative to sample sizes required for a single candidatemutation study (as in Fig. 2). As might be expected,the sampling requirements can be seen to increasewhen more than one locus is tested, as testing 10±100 genes requires samples 1.5±2 times larger thanthose shown in Fig. 2. Testing 100 000 loci, as mightbe conducted in a genome-wide screen (Risch &Merikangas, 1996), requires approximately three-foldgreater sample size than that required for a singlecandidate gene. This relatively small increase mightappear somewhat surprising given that the actualsigni®cance level required in the 100 000 locus caseis 2.5 3 10ÿ7 [nominal á � 0.05/100 000 3 2 (al-leles)] whereas for one locus we have required onlyá � 0.025 [0.05/2 (alleles)], but this result alsofollows from theoretical arguments (Witte et al.,1999). In addition, our simulations indicate that thesample size ratios are largely independent of geneticmodel, allele frequency, and gene effect size, again inagreement with large sample theory (Witte et al.,1999). That is, all of the locus characteristics thatin¯uence the design of the candidate gene study
described above also in¯uence the design of a gen-ome-wide study of the same design. This outcome isof practical utility since it suggests that one maydesign a study using a candidate gene hypothesis,then multiply the sample size by a constant to obtaina broad approximation to the requirements for de-tecting a similar effect anywhere in the genome.
Discussion
The successful integration of pharmacogenomics andclinical trials is predicated on the availability of avery large number of genetic markers distributedthroughout the human genome and on the ability tomake use of the resulting genotype data in a clinicaltrial setting. Given the large-scale initiatives forgenome-wide SNP scanning, it appears very likelythat a suf®cient number of markers will soon beavailable for use in clinical trials. The data presentedhere suggest that frequency of the aetiologic allele,pattern of gene action, and the number of loci testedare key determining factors for establishing geno-typic-drug response correlations.
In general, the use of genetic information cangreatly reduce the number of patients that must beenrolled in any clinical trial; however, unless certainconditions are met the sample sizes remain too largefor most trial settings. As is widely known in thestudy of complex disease genes, the properties of theaetiological variant (frequency, mode of action, effectsize) dictate the sampling requirements of eachparticular study. It is therefore perhaps unsurprisingthat such factors also determine the sampling re-
350%300%250%200%150%100%
50%0%
1 locus 10 loci 100 loci 1000 loci 10000loci
100000loci
Number of loci tested
Incr
ease
in N
Fig. 4. The effect of testing multiple loci in a pharmaco-genomic study. Bars represent the expected relative samplesizes required when 1, 10, 1000, 10 000 and 100 000loci are tested. The signi®cance level desired after approx-imate Bonferroni correction for multiple tests is á � 0.025corresponding to a single di-allelic locus. The relativeincreases are shown using an allele frequency of 0.5 and agenotype relative risk (GRR) of 6. Other models of geneaction, allele frequency and GRR yield similar results(Witte et al., 1999).
508 Cardon et al.

quirements of genetically based clinical trials. Never-theless, this dependence on basic polymorphismfeatures has signi®cant implications for usage ofanonymous SNPs in clinical trials, particularly in thecase of genome-wide studies. Because the number ofsamples required for a speci®c indication can vary byorders of magnitude depending on allele frequency,gene action, and effect size, it is crucial that priorgenetic information be available to ensure appropri-ate study design. In direct contrast, one major appealof genome-wide studies is that they require noassumptions about the underlying genetic architec-ture. The present data suggest that this aspect ofgenome-wide SNP screens does not lend itself toclinical trial settings; in the absence of any under-standing of the genetic factors contributing to out-come, genome-wide clinical trials of anonymousSNPs may be impractical. Indeed, if pharmaco-genomic trials are conducted without any knowledgeof the genetic parameters, they may be grossly under-or overpowered; the former resulting in a misseddetection of genetic effect, the latter resulting in aconsiderable waste of resources.
Because the likelihood of success of pharmaco-genomic studies is largely dependent upon the avail-ability of as much prior information as possible, it isof interest to consider how such information mightbe obtained. One possible solution could be to designlarger phase I and II trials, in which speci®c candi-date loci tests or exploratory genome-screens areconducted to determine the relevant genetic para-meters. This would then lead to a reduction in thesampling requirements for the more costly phase IIItrials. At the genome level, it may also be fruitful toconduct genome-screens in very large phase IV trialsrather than in smaller phase I±III trials or to simplyallow that rare allele effects may be missed in thegenome-screen. Alternatively, if a small number ofcandidate genes are to be tested, one could pre-selectthe trial sample according to genotype(s) to ensureadequate representation of the rare allele in alltreatment cells. Clearly, if genetic information is to bewidely incorporated into the clinical trial setting,further studies are needed to explore how to designtrials most effectively to make use of genotypic data.
At the genome level, the predominant role of allelefrequency presents somewhat of a conundrum. It isclear that studies of rare variants require sample sizesthat are too large and cost-prohibitive for most trialsettings, and the sizes cannot be reduced by pre-selection for thousands of genotypes. Thus, it wouldseem that genome-screen studies would be wellserved to avoid genotyping markers with infrequentalleles, as the likelihood for successful outcomes isvery low anyway. Indeed, several investigators have
argued in favour of focusing on common variants forthis and other reasons (Lander, 1996; Collins et al.,1997; Chakravarti, 1998, 1999). However, if theaetiologic allele(s) are uncommon, as others haveargued is likely to be the case (Weiss, 1996;Terwilliger & Weiss, 1998), this strategy could resultin missing precisely the effect that the study wasdesigned to detect. Solutions to this problem mayinclude conducting genome-screens in very largephase IV trials rather than in smaller phase I±IIItrials or simply allowing that rare allele effects maybe missed in the genome-screen. In either case, ourresults suggest that, in the absence of any priorgenetic information, trials aimed at identifying raremutations (e.g. for side-effects) from a genome-screenare not suitable for pharmacogenomic applications.
Of course, in many situations, the investigator willnot be interested in rare alleles and may have someunderstanding of the putative gene action and effectsize. In such situations a genome-screen for pharma-cogenomic loci may be quite feasible. For the samplesizes presented here, we have assumed a very simpleclinical trial design of two dose groups strati®ed bygenotypes at each marker in a screen. Under thissimple study design, the results suggest that agenome-screen of 100 000 loci only requires approxi-mately three times the number of samples it wouldtake to conduct a candidate gene study of a singlelocus. In some cases, this implies a three-fold increasein a sample size that is already implausibly large fora clinical trial. In other cases, however, this increaseis well within extant sampling expectations. Thisoutcome highlights the importance of the emergingSNP map of the genome, as well as the potential forsigni®cant advances towards personalized medicine.
References
Chakravarti A. It's raining SNPs, hallelujah? Nature Genet 1998;19:216±217.
Chakravarti A. Population genetics ± making sense out ofsequence. Nature Genet 1999; 21:56±60.
Collins FS, Guyer MS, Chakravarti A. Variations on a theme:cataloging human DNA sequence variation. Science 1997;278:1580±1581.
Drews J, Ryser S. The role of innovation in drug development.Nature Biotechnol 1997; 15:1318±1319.
Elston RC, Idury RI, Cardon LR, Lichter JB. The study ofcandidate genes in drug trials: sample size considerationsStat Med 1999; 18:741±751.
Ferrari P. Pharmacogenomics: a new approach to individualtherapy of hypertension. Curr Opin Neph Hypertens 1998;7:217±222.
Fisher RA. The design of experiments. Edinburgh: Oliver and Boyd;1935.
Kuivenhoven JA, Jukema JW, Zwinderman AH, de Knijff P,McPherson R, Bruschke AV, et al. The role of a commonvariant of the cholesteryl ester transfer protein gene in the
Sampling and clinical trials 509

progression of coronary atherosclerosis. The RegressionGrowth Evaluation Statin Study Group. N Engl J Med 1998;338:86±93.
Lander ES. The new genomics: global views of biology. Science1996; 274:536±539.
Lander ES. Array of hope. Nature Genet 1999; 21:3±4.Marshall A. Getting the right drug into the right patient. Nature
Biotechnol 1997a; 15:1249±1251.Marshall A. Laying the foundations for personalized medicines.
Nature Biotechnol 1997b; 15:955±957.Masood E. As consortium plans free SNP map of human genome.
Nature 1999; 398:545±546.Meyer UA. Medically relevant genetic variation of drug effects.
In: Evolution in Health and Disease. Oxford, UK: OxfordUniversity Press; 1997.
Nebert DW. Polymorphisms in drug-metabolizing enzymes: whatis their clinical relevance and why do they exist? Am J HumGenet 1997; 60:265±271.
Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, etal. Apolipoprotein E4 allele as a predictor of cholinergicde®cits and treatment outcome in Alzheimer disease. ProcNatl Acad Sci USA 1995; 92:12260±12264.
Regalado A. Inventing the pharmacogenomics business. In Vivo1997; 9:52±64.
Richard F, Helbecque N, Neuman E, Guez D, Levy R, Amouyel P.APOE genotyping and response to drug treatment inAlzheimer's disease. Lancet 1997; 349:539.
Risch N, Merikangas K. The future of genetic studies of complexhuman diseases. Science 1996; 273:1516±1517.
Tan S, Hall IP, Dewar J, Dow E, Lipworth B. Association betweenbeta 2-adrenoceptor polymorphism and susceptibility tobronchodilator desensitisation in moderately severe stableasthmatics. Lancet 1997; 350:995±999.
Terwilliger JD, Weiss KM. Linkage disequilibrium mapping ofcomplex disease: fantasy or reality? Curr Opin Biotechnol1998; 9:578±594.
Weber WW. Pharmacogenetics. New York: Oxford UniversityPress; 1997.
Weiss KM. Is there a paradigm shift in genetics? Lessons from thestudy of human diseases. Mol Phylogenet Evol 1996; 5:259±265.
Witte JS, Elston RC, Cardon LR. On the relative sample sizerequired for multiple comparisons. Stat Med 1999; 69:369±372.
510 Cardon et al.

Pharmacogenetics 2001, 11:459±460 Correspondence
0960-314X # 2001 Lippincott Williams & Wilkins
Testing drug response in the presence of geneticinformation
Patrice P. Rioux
PMB 214, 405 Waltham Street, Lexington, MA 02421-7934, USA.
In a recent paper, Cardon et al. (2000) have tried togive us some clues for testing drug response in thepresence of genetic information, and I was particu-larly interested in their Fig. 3. From this ®gure, itappears that using a genetic hypothesis in clinicaltrials would almost always be bene®cial in terms ofsample sizes. However, I would have been happy toknow how they built these curves since, in manycases, it is mathematically impossible to have such agenetic hypothesis.
For instance, in Fig. 1, they explain that instead ofhaving a group of patients with a 35% response rate,you can assume that only a smaller group of pa-tients, characterized by genotypic information, iseffectively responding differently from the placebogroup. However, by de®nition, the response rate inthis smaller group cannot be higher than 100%. Byde®nition of a mean, the smaller group has to be bigenough to compensate the fact that the rest of thetreated group is responding similar to the placebo(10% in their example). Different genetic hypothesescan then be chosen to support this breaking intosmaller groups, but they have to be in agreementwith the Hardy±Weinberg equilibrium. The mostinteresting hypothesis would be to have a homo-genous group of homozygous patients with a 100%response rate linked to a recessive single nucleotidepolymorphism (SNP) allele, and so only a SNP allelefrequency of at least 0.52 can support such a case(Rioux, 2000). The authors have chosen anotherhypothesis, i.e. to have a homogenous group ofhomozygous and heterozygous patients with an 80%response rate linked to a dominant SNP allele, whichcorresponds to a 0.2 allele frequency. Despite that, itassumes that having one or two copies of an allelelinked to the ef®cacy of a drug would have the sameimpact in terms of drug metabolism, even with a100% response rate, the allele frequency would haveto be at least 0.15.
Knowing that, from personal experience, mostSNPs occur at very low frequencies, it would beuseful to clarify these points in order that we do not
have to genotype patients in clinical trials whenthere is no potentially plausible genetic hypothesis.
References
Cardon LR, Idury RM, Harris TJ, Witte JS, Elston RC. Testingdrug response in the presence of genetic information:sampling issues for clinical trials. Pharmacogenetics 2000;10:503±510.
Rioux P. Clinical trials in pharmacogenetics and pharmaco-genomics: methodology and applications. Am J Health-SystPharm 2000; 57:887±898.
Authors' reply
Lon R. Cardona, John Witteb and Robert Elstonb
aWellcome Trust Centre for Human Genetics, Universityof Oxford, Roosevelt Drive, Oxford OX3 7BN, UK andbCase Wester Reserve University, 10900 Euclid Avenue,Cleveland, Ohio 44106, USA
We are grateful for the comments of Dr Rioux as theyprovide us an opportunity to further clarify theoutcomes of our study (Cardon et al., 2000). Regard-ing point one, it is perhaps useful to reconsider ourmodel of response. Let q re¯ect the frequency of aresponse-increasing allele within a candidate gene, sothat the wild-type (wt) allele occurs with frequency1 ± q, and the three genotypes have frequencies (1 ±q)2, 2(1 ± q)q and q2. All of the modelling describedin our paper assumes nothing further than the basicassumptions of Mendelian inheritance and Hardy±Weinberg equilibrium. With regard to drug response,we assume that response in a placebo group occursat a ®xed rate with respect to genotype, but thatresponse may differ by genotype in the dose group(s).The relationships in Fig. 1, highlighted by Dr Rioux,follow directly from these principles. In particular,the ®gure re¯ects an hypothetical example in which10% of a placebo group responds to treatment, while35% of a dose group responds to the same treatment.
Correspondence to Patrice P. Rioux, PMB 214, 405 Waltham Street,Lexington, MA 02421-7934, USA. E-mail: [email protected]
Correspondence to Lon R. Cardon, Wellcome Trust Centre for HumanGenetics, University of Oxford, Roosevelt Drive, Oxford OX3 7BN, UKTel: �44 1865 287 587; fax: �44 1865 287 650; e-mail: [email protected]

This simple scenario can occur under a (response-increasing) allele frequency of 0.20, dominant inheri-tance and a genotype relative risk (GRR) of 8, asdenoted in the legend to the ®gure. In this case, thethree genotype frequencies are wt/wt: 0.802 � 0:64;wt/increaser: 2(0.20 3 0.8) � 0.32; and increaser/increaser: 0.202 � 0.04. The genotype relative risk of8 and dominant inheritance implies that the homo-zygote (increaser/increaser) and heterozygote classeshave an eight-fold increase over the response rate ofthe wt/wt homozygote. That is 64% do not differfrom the placebo group, whereas the remaining 36%have an 80% response rate (i.e. a response rate eighttimes that in the placebo group). Ignoring geneticinformation would yield an average response of:0.10 3 0.64 � 0.80 3 0.36 � 35%.
In Fig. 3, also mentioned by Dr Rioux, we made nofurther assumptions or additions to this model. Forsimplicity, we maintained the model of a GRR of 8 asdescribed in Fig. 1. Dr Rioux expressed concern aboutthe genetic plausibility of this model. Indeed, giventhe simple model of 10% placebo and eight-fold GRRjust described, any allele frequency can occur withdominant, recessive or additive inheritance, so noneof the models we described imply Hardy±Weinberg orother undisclosed allele frequency variations. Cer-tainly, there are risk-ratio/allele frequency modelsthat are implausible for pharmacogenetics applica-tions. Under our model, many of these would occurwhenever the GRR exceeded 10. However, we didnot consider any of these situations in our paper.
Regarding point two, Dr Rioux describes a beliefthat most SNPs occur at very low frequencies. It isunarguable that most monogenic diseases are rareand, consequently, are determined by rare alleles.However, polygenic diseases (and possibly variability
in response to many drugs) are thought to be in¯u-enced by many common genes and environmentalfactors. Indeed, this is the prevailing hypothesisguiding worldwide SNP-detection efforts, which haveculminated in an unprecedented degree of opencollaboration within the pharmaceutical industryand academia (Masood, 1999). As a result, there arecurrently over 1.5 million human reference SNPs inthe public domain (dbSNP: http://www.ncbi.nlm.nih.gov/SNP/index.html), many of which were detectedusing methods designed for common variants(Altshuler et al., 2000; Mullikin et al., 2000; TheInternational SNP Working Group, 2001). Withinthe next few years, it is expected that genotypingtechnologies will be available to evaluate large sets ofsuch polymorphisms in clinical samples. Thus, therewill soon be a wealth of data for empirical veri®ca-tion or refutation of Dr Rioux's personal experiencewith rare SNPs.
References
Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J,Linton L, Lander ES. An SNP map of the human genomegenerated by reduced representation shotgun sequencing.Nature 2000; 407:513±516.
Cardon LR, Idury RM, Harris TJ, Witte JS, Elston RC. Testingdrug response in the presence of genetic information:sampling issues for clinical trials. Pharmacogenetics 2000;10:503±510.
Masood E. As consortium plans free SNP map of human genome.Nature 1999; 398:545±546.
Mullikin JC, Hunt SE, Cole CG, Mortimore BJ, Rice CM, Burton J,et al. An SNP map of human chromosome 22. Nature 2000;407:516±520.
The International SNP Working Group. A map of humangenome sequence variation containing 1.42 million singlenucleotide polymorphisms. Nature 2001; 409:928±933.
460 Rioux et al.













![Testing Testing [Read-Only] - Czone - East Sussex](https://static.fdokumen.com/doc/165x107/6327b2a26d480576770d6757/testing-testing-read-only-czone-east-sussex.jpg)






