
Investigations of two soy-based adhesives: Soy flour - Oregon ...
Upload
independentCategory
view
1download
0
Cancer Sci | November 2007 | vol. 98 | no. 11 | 1740–1746 doi: 10.1111/j.1349-7006.2007.00595.x© 2007 Japanese Cancer Association
Blackwell Publishing Asia
Soy-derived isoflavones inhibit the growth of adult T-cell leukemia cells in vitro and in vivoMasao Yamasaki,1,2 Satoshi Fujita,1 Erina Ishiyama,1 Ayako Mukai,1 Harishkumar Madhyastha,2 Yoichi Sakakibara,1,2 Masahito Suiko,1,2 Kinta Hatakeyama,3 Takayuki Nemoto,4 Kazuhiro Morishita,2,5 Hiroaki Kataoka,2,6 Hirohito Tsubouchi2,7 and Kazuo Nishiyama1,8
1Department of Biochemistry and Applied Biosciences, Faculty of Agriculture, University of Miyazaki, 1-1 Gakuenkibanadai-nishi, Miyazaki 889-2192; 2Miyazaki Prefectural Industrial Support Foundation, 16500-2 Higashi-Kaminaka, Sadowara-cho, Miyazaki 880-0303; Departments of 3Pathology, 4Pharmacology, and 5Biochemistry, 6Section of Oncopathology and Regenerative Biology, Faculty of Medicine, University of Miyazaki, 5200 Kihara Kiyotake, Miyazaki 889-1692; 7Section of Digestive Disease and Life-style-related Disease, Kagoshima University Graduate School of Medical and Dental Sciences, 8-35-1 Sakuragaoka, Kagoshima 890-8544, Japan
(Received April 16, 2007/Revised July 10, 2007/Accepted July 17, 2007/Online publication August 28, 2007)
Adult T-cell leukemia occurs in human T-lymphotropic virus typeI-infected individuals and is endemic to the south-western area ofKyushu in Japan. In this communication, we examined the effect ofsoy isoflavones on the growth of adult T-cell leukemia cells in vitroand in vivo. In the in vitro study, daidzein and genistein but notglycitein significantly inhibited the proliferation of ED-40515 andHut102 cells in a dose-dependent manner. Among the isoflavonesstudied, genistein had the highest growth-inhibitory effect; however,genistein did not exert an apparent growth-inhibitory effect onJurkat and Molt-4 cells, which were non-adult T-cell leukemiacells. Genistein prevented the G1/S or G2/M transition at 3 and 10 or30 μμμμM, respectively. Genistein upregulated p21 protein expressiontogether with p53 accumulation. In addition, treatment with 30 μμμμMgenistein strongly induced phosphorylation of checkpoint kinase(CHK) 2 and p53 at serines 15, 20 and 37. Caffeine, an inhibitor ofataxia-telangiectasia mutated protein kinase, alleviated the genistein-induced p53 and CHK2 phosphorylation, suggesting the involvementof DNA damage at 30 μμμμM. However, marked phosphorylation ofCHK2 and p53 could not be detected at 3 and 10 μμμμM genistein. Thesedata indicate that genistein has biphasic growth-inhibitory properties.The in vivo studies demonstrated that soy-derived isoflavonessignificantly inhibit ED-40515 cell growth and infiltration into variousorgans in non-obese diabetic severe combined-immunodeficiencycommon γγγγ-chain knockout mice. Taken together, it is evidentthat soy isoflavones might serve as a promising compound for thetreatment of adult T-cell leukemia. (Cancer Sci 2007; 98: 1740–1746)
South-western Kyushu in Japan is known to be an area with ahigh prevalence of HTLV-I. In particular, the prevalence rate
has reached 37% in a small selected population in south-westernKyushu.(1,2) HTLV-I is the etiological agent of the CD4+ T-cellmalignancy ATL and the neurological disorder TSP/HAM.Therefore, it is designated as an endemic disease, particularly incertain pockets of the Japanese population. ATL was first reportedin Japan,(3) and a subsequent study proved the relationshipbetween HTLV-I and ATL.(4) Among infected individuals, only1–5% develop ATL and it is well established that this takes atleast 20–50 years after HTLV-I infection. ATL is categorizedinto four types according to their clinical phenotypes: acute,chronic, smoldering and lymphoma. Among them, the acutetype constitutes 55–75% of all ATL cases,(5,6) but acute ATL isaggressive, malignant and fatal when accompanied by pulmonarycomplications, opportunistic infections and uncontrolled hyper-calcemia. In addition, aggressive forms of ATL are resistant toconventional chemotherapy. Therefore, it is important to findappropriate therapeutic methods to prevent the development ofATL or to prolong survival after its occurrence.
It has been suggested that local cofactors such as food cultureplay an important role in ATL occurrence. Particularly in Japan,
it takes approximately 50 years after infection (most of them areby vertical transmission) for ATL to develop and this duration iscomparatively longer than in other areas of the world. Recentstudies have revealed that some components of natural foodssuch as curcumin, capsaicin and fucoidan are effective for ATLtreatment.(7–10) Soybean isoflavone is known to inhibit the prolifer-ation or to induce cell death in various cancer cells includinghepatoma, melanoma, breast cancer and non-ATL leukemiacells.(11–17) Most soybean isoflavones are glycosylated and aredigested to their aglycon forms, which are effectively absorbedby the intestinal tract. The major isoflavones in soybeans aregenistin, daidzin and glycitin. The cellular uptake and biocom-patibility of genistein, daidzein and glycitein have been studiedextensively and strong chemopreventive effects in other types ofcancer and non-ATL leukemia cells have been revealed. However,scientific understanding is still not clear on the area of ATL pre-vention. Therefore, we examined the effect of these isoflavoneson the proliferation of ATL cells in vitro and in vivo.
Materials and Methods
Materials. Daidzein, genistein and glycitein were obtainedfrom Fujicco (Kobe, Japan). Caffeine (Wako Pure Chemicals,Osaka, Japan) was used as an ATM kinase inhibitor. Peroxidase-conjugated antimouse IgG, peroxidase-conjugated antirabbit IgGand anti-p53 antibodies were purchased from Santa CruzBiotechnology (Santa Cruz, CA, USA). Anti-β-actin cloneAC-15 was from Sigma (St Louis, MO, USA) and other antibodies(against phosphorylated p53 [serines 15, 20 and 37], CHK1,CHK2, and phosphorylated CHK1 and CHK2) were purchasedfrom Cell Signaling Technology (Beverly, MA, USA).
Cell culture. Hut102 cells, Jurkat and Molt-4 cells werepurchased from or provided by Fujisaki Cell Center, HayashibaraBiochemical Laboratories (Okayama, Japan). The ATL cell lineED-40515 is an interleukin-2-independent human T-cell line ofleukemic-cell origin established from an ATL patient.(18) Thesecells were maintained in RPMI-1640 medium supplementedwith 10% fetal bovine serum containing 100 U/mL penicillin Gand 100 μg/mL streptomycin. Cells were subcultured twicea week and in the actual in vitro experiment, the cell numberwas adjusted to 1.0 × 105 cells/mL and cultured with 0–30 μMisoflavones.
8To whom correspondence should be addressed. E-mail: [email protected]: ATL, adult T-cell leukemia; ATM, ataxia-telangiectasia mutated;CBP, CREB binding protein; Cdc25C, cell division cycle 25C; CDK, cyclin-dependentkinase; CHK, checkpoint kinase; GADD45a, growth arrest and DNA damage induciblegene 45a; HTLV-I, human T-lymphotropic virus type I; NOG, non-obese diabetic-severe combined immunodeficiency common γ-chain knockout; PBS, phosphate-buffered saline; PK, protein kinase; SHG, soyaflavone HG; TSP/HAM, tropical spasticparaparesis/HTLV-I-associated myelopathy; TTBS, Tris-buffered saline with 0.1%Tween-20.
Yamasaki et al. Cancer Sci | November 2007 | vol. 98 | no. 11 | 1741© 2007 Japanese Cancer Association
Cell cycle analysis. For cell cycle analysis, nuclei wereisolated from cells using 0.1% citrate after the culture period.Isolated nuclei were fixed using ice-cold methanol for 30 min.Then, nuclei were treated with 10 μg/mL propidium iodide and10 μg/mL RNase. Cell cycle analysis was carried out using anEpics XL (Beckman Coulter, Fullerton, CA, USA) equippedwith MultiCycle software (San Diego, CA, USA).
Western blot analysis. The cell cycle-related proteins p53,phosphorylated p53, CHK1, CHK2 and phosphorylated CHK1and CHK2 were detected by western blot analysis. At the end ofthe culture period, cells were lysed in 50 mM Tris-HCl (pH 7.5)containing 150 mM NaCl, 2% Triton X-100, 2 mM ethylene-diaminetetraacetic acid, 50 mM NaF, 30 mM Na4P2O7 and 2%protease inhibitor cocktail (Nacalai Tesque, Kyoto Japan).Protein concentrations were measured using the BCA proteinassay reagent (Pierce, Rockford, IL, USA). Lysates containing10 μg protein were separated by electrophoresis on a 10%sodium dodecylsulfate–polyacrylamide gel, then transferred to apolyvinylidene fluoride (PVDF) Hybond-P membrane (Amersham-Pharmacia Biotech, Buckinghamshire, UK). Blocking was carriedout using 5% non-fat milk or 5% bovine serum albumin fractionV (for phosphorylated protein detection) in TTBS, and antibodieswere diluted in Can Get Signal solutions 1 and 2 (Toyobo,Tokyo, Japan). The membrane was washed with TTBS aftereach antibody-binding reaction. Detection of each protein wascarried out using the ECL Plus kit (Amersham-Pharmacia).
Mice. Non-obese diabetic-severe combined immunodeficiencycommon γ-chain knockout mice were obtained from the CentralInstitute for Experimental Animals (Kawasaki, Japan). All micewere maintained under specific pathogen-free conditions in theDepartment of Bio-resources, Frontier Science Research Center,University of Miyazaki. The Ethical Review Committee of theFaculty of Medicine, University of Miyazaki, approved theexperimental protocol.
Inoculation of cell lines and isoflavone administration into SCIDmice. Cells were washed twice and resuspended in PBS andwere inoculated into the peritoneal cavity of the mice at a doseof 1 × 107 cells per mouse. SHG, which is 47.3% total isoflavoneform (daidzin 33.76%, genistin 6.89% and glycitin 16.44%), wasobtained from Fuji Oil (Osaka, Japan). Mice were treated witheither PBS (control mice) or drug (100 μg SHG/mouse/day)intraperitoneally every day for 4 weeks. Tumor growth andprogression were monitored by observation and measurementof the tumor, the physical condition of the mice and theirbodyweight during the 1-month follow-up period.
Growth measurement of intraperitoneal tumors. ED-40515 cellsmade tumors in the peritoneum after injection into the peritonealcavity and the tumor growth was evaluated by measurement with acaliper device on a weekly basis. The tumor volume was estimatedusing the formula volume (mm3) = width × length × height. Micewere killed 40 days after inoculation with the ED-40515 cell lines.
Histological and cytological examination. Tumor tissues, peripheralblood and various organs were collected from the mice. The tumortissues and organs were fixed with 4% paraformaldehyde forhematoxylin–eosin staining and examined under a microscope.
MIB-1 index. Sections were deparaffinized and subjected toheat-induced epitope retrieval by autoclave for 15 min. Slides thenwere incubated at room temperature with antibodies directedtoward MIB-1 (mouse monoclonal, 1:160; Dako, Carpentaria,CA, USA). Antibodies were detected using the avidin–biotin–peroxidase complex method using 3,3′-diaminobenzidine as thechromogen. Standard positive controls were used throughout.Normal sera served as the negative control. Sections werecounterstained with hematoxylin. The evaluation of MIB-1 labelingindex was assessed at the periphery of the lesions, where thenumber of positive cells per >1000 cells examined was counted.This procedure was carried out for five representative tumorareas from each slide.
Statistical analysis. Data were analyzed using Student’s t-testto evaluate the significance of difference at P < 0.05.
Results
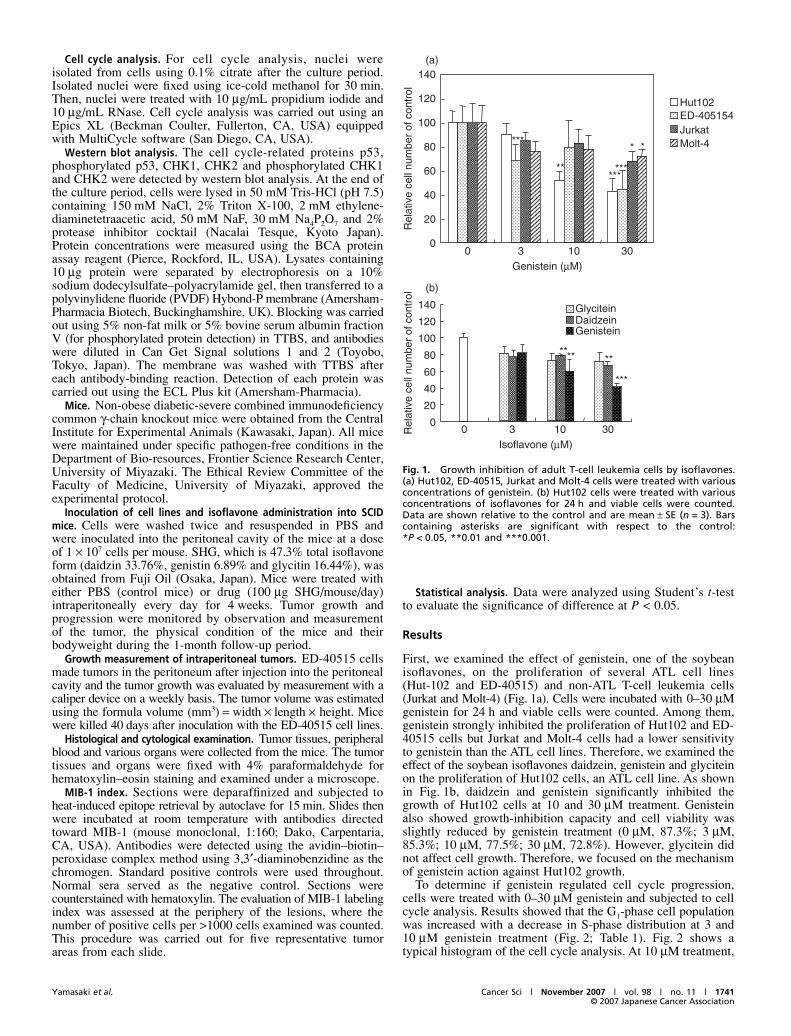
First, we examined the effect of genistein, one of the soybeanisoflavones, on the proliferation of several ATL cell lines(Hut-102 and ED-40515) and non-ATL T-cell leukemia cells(Jurkat and Molt-4) (Fig. 1a). Cells were incubated with 0–30 μMgenistein for 24 h and viable cells were counted. Among them,genistein strongly inhibited the proliferation of Hut102 and ED-40515 cells but Jurkat and Molt-4 cells had a lower sensitivityto genistein than the ATL cell lines. Therefore, we examined theeffect of the soybean isoflavones daidzein, genistein and glyciteinon the proliferation of Hut102 cells, an ATL cell line. As shownin Fig. 1b, daidzein and genistein significantly inhibited thegrowth of Hut102 cells at 10 and 30 μM treatment. Genisteinalso showed growth-inhibition capacity and cell viability wasslightly reduced by genistein treatment (0 μM, 87.3%; 3 μM,85.3%; 10 μM, 77.5%; 30 μM, 72.8%). However, glycitein didnot affect cell growth. Therefore, we focused on the mechanismof genistein action against Hut102 growth.
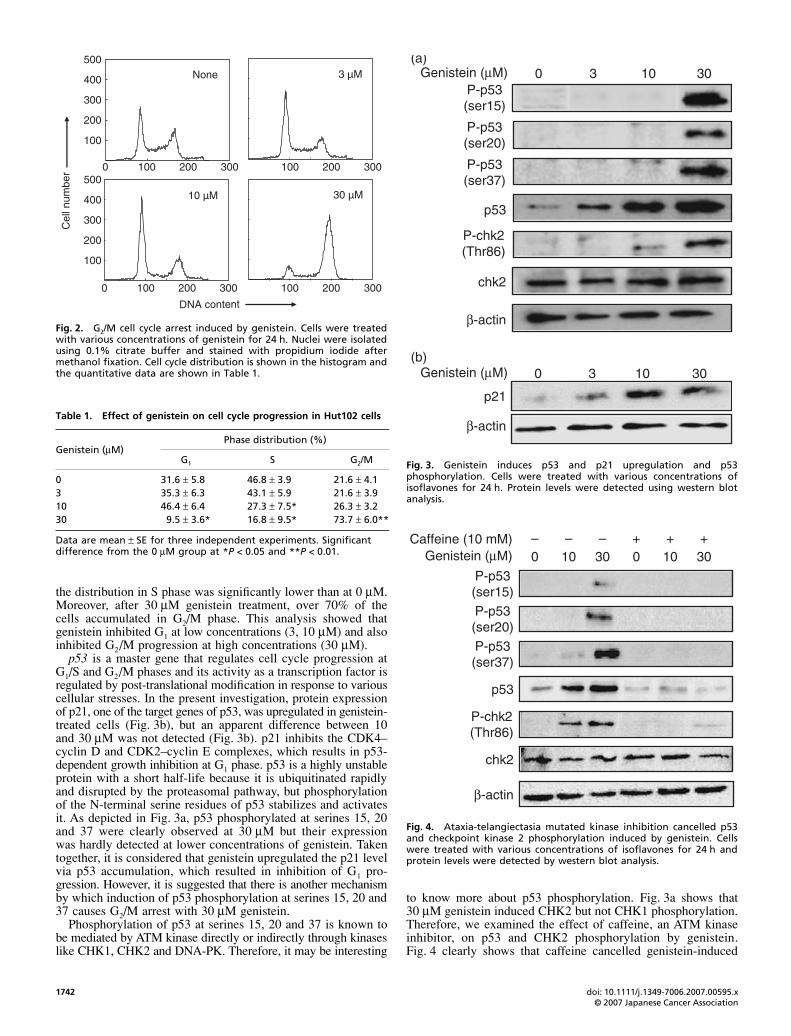
To determine if genistein regulated cell cycle progression,cells were treated with 0–30 μM genistein and subjected to cellcycle analysis. Results showed that the G1-phase cell populationwas increased with a decrease in S-phase distribution at 3 and10 μM genistein treatment (Fig. 2; Table 1). Fig. 2 shows atypical histogram of the cell cycle analysis. At 10 μM treatment,
Fig. 1. Growth inhibition of adult T-cell leukemia cells by isoflavones.(a) Hut102, ED-40515, Jurkat and Molt-4 cells were treated with variousconcentrations of genistein. (b) Hut102 cells were treated with variousconcentrations of isoflavones for 24 h and viable cells were counted.Data are shown relative to the control and are mean ± SE (n = 3). Barscontaining asterisks are significant with respect to the control:*P < 0.05, **0.01 and ***0.001.
1742 doi: 10.1111/j.1349-7006.2007.00595.x© 2007 Japanese Cancer Association
the distribution in S phase was significantly lower than at 0 μM.Moreover, after 30 μM genistein treatment, over 70% of thecells accumulated in G2/M phase. This analysis showed thatgenistein inhibited G1 at low concentrations (3, 10 μM) and alsoinhibited G2/M progression at high concentrations (30 μM).
p53 is a master gene that regulates cell cycle progression atG1/S and G2/M phases and its activity as a transcription factor isregulated by post-translational modification in response to variouscellular stresses. In the present investigation, protein expressionof p21, one of the target genes of p53, was upregulated in genistein-treated cells (Fig. 3b), but an apparent difference between 10and 30 μM was not detected (Fig. 3b). p21 inhibits the CDK4–cyclin D and CDK2–cyclin E complexes, which results in p53-dependent growth inhibition at G1 phase. p53 is a highly unstableprotein with a short half-life because it is ubiquitinated rapidlyand disrupted by the proteasomal pathway, but phosphorylationof the N-terminal serine residues of p53 stabilizes and activatesit. As depicted in Fig. 3a, p53 phosphorylated at serines 15, 20and 37 were clearly observed at 30 μM but their expressionwas hardly detected at lower concentrations of genistein. Takentogether, it is considered that genistein upregulated the p21 levelvia p53 accumulation, which resulted in inhibition of G1 pro-gression. However, it is suggested that there is another mechanismby which induction of p53 phosphorylation at serines 15, 20 and37 causes G2/M arrest with 30 μM genistein.
Phosphorylation of p53 at serines 15, 20 and 37 is known tobe mediated by ATM kinase directly or indirectly through kinaseslike CHK1, CHK2 and DNA-PK. Therefore, it may be interesting
to know more about p53 phosphorylation. Fig. 3a shows that30 μM genistein induced CHK2 but not CHK1 phosphorylation.Therefore, we examined the effect of caffeine, an ATM kinaseinhibitor, on p53 and CHK2 phosphorylation by genistein.Fig. 4 clearly shows that caffeine cancelled genistein-induced
Fig. 2. G2/M cell cycle arrest induced by genistein. Cells were treatedwith various concentrations of genistein for 24 h. Nuclei were isolatedusing 0.1% citrate buffer and stained with propidium iodide aftermethanol fixation. Cell cycle distribution is shown in the histogram andthe quantitative data are shown in Table 1.
Table 1. Effect of genistein on cell cycle progression in Hut102 cells
Genistein (μM)Phase distribution (%)
G1 S G2/M
0 31.6 ± 5.8 46.8 ± 3.9 21.6 ± 4.13 35.3 ± 6.3 43.1 ± 5.9 21.6 ± 3.910 46.4 ± 6.4 27.3 ± 7.5* 26.3 ± 3.230 9.5 ± 3.6* 16.8 ± 9.5* 73.7 ± 6.0**
Data are mean ± SE for three independent experiments. Significant difference from the 0 μM group at *P < 0.05 and **P < 0.01.
Fig. 3. Genistein induces p53 and p21 upregulation and p53phosphorylation. Cells were treated with various concentrations ofisoflavones for 24 h. Protein levels were detected using western blotanalysis.
Fig. 4. Ataxia-telangiectasia mutated kinase inhibition cancelled p53and checkpoint kinase 2 phosphorylation induced by genistein. Cellswere treated with various concentrations of isoflavones for 24 h andprotein levels were detected by western blot analysis.

Yamasaki et al. Cancer Sci | November 2007 | vol. 98 | no. 11 | 1743© 2007 Japanese Cancer Association
phosphorylation of p53, and CHK2 was also completelyblocked by caffeine treatment. Interestingly, the upregulation ofp53 observed with both 10 and 30 μM genistein treatment wascompletely cancelled by caffeine.
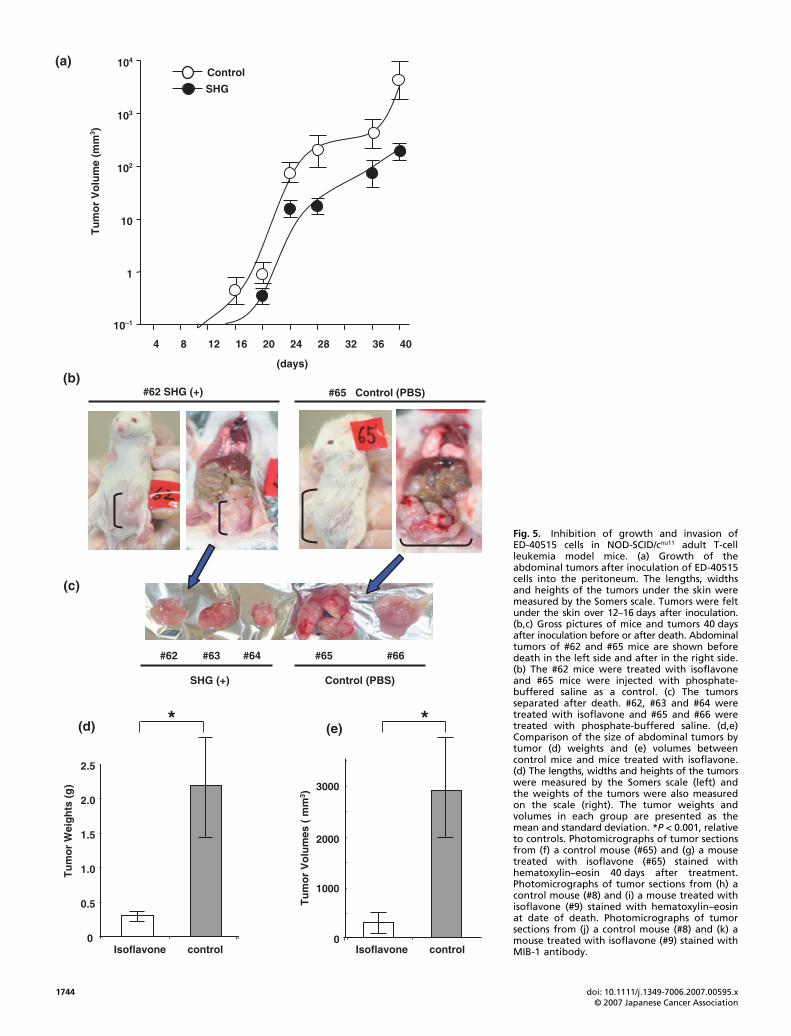
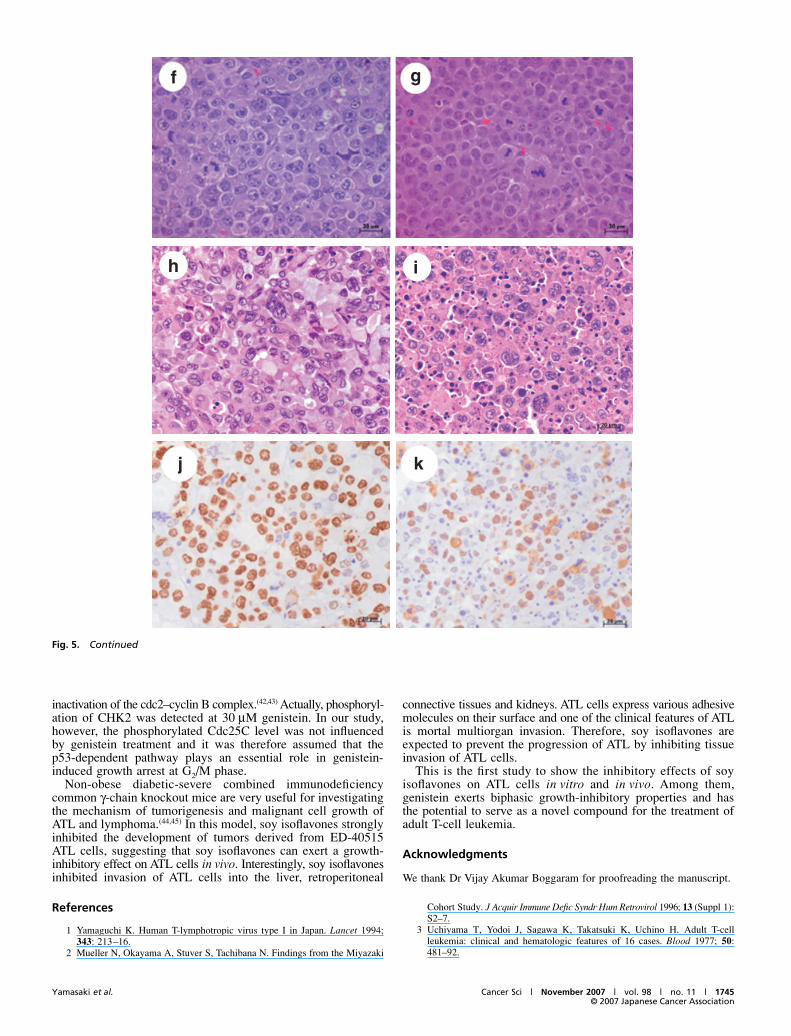
To investigate and further authenticate our in vitro observations,we continued our ATL prevention studies using isoflavones inthe mouse model. We injected ED-40515 cells (1 × 107) into theintraperitoneal cavity of NOG mice. Mice were treated intra-peritoneally with either PBS (as control, #65 and #66 mice) orSHG (100 μg/mouse/day, #62, #63 and #64 mice) daily for 40 days.ED-40515 cell inoculation with PBS injection promoted thedevelopment of abdominal tumors in the abdomen at 12 daysafter inoculation. However, tumors were found in the micetreated with SHG at 16 days after inoculation of ED-40515 cells(Fig. 5a). At 40 days after inoculation, the abdominal tumorsinoculated with PBS (#65) were bigger than those with SHG(#62) (Fig. 5b). After the mice were killed the mass of abdomi-nal tumors that had developed in the peritoneum was separatedeasily from the intestinal tract. The sizes of control tumors wereapproximately five times bigger than those of SHG-treatedtumors (Fig. 5b–e). Along with the abdominal tumors, leukemiacells in control mice invaded the liver or retroperitoneal connec-tive tissues and kidneys. In contrast, SHG-treated mice had asmall enlargement of abdominal tumors in the intraperitonealcavity with very low infiltration into organs (data not shown).By hematoxylin–eosin staining of the tumor sections, theleukemia cells in the tumors proliferated diffusely and showedhigh malignancy with aberrant-sized nuclei and high amountsof chromatin. In contrast, leukemia cells in the SHG-treatedtumors proliferated diffusely but necrotic cells were found eas-ily in the tumors (Fig. 5f,g). Moreover, we treated and observedmice with either PBS or SHG for 3 months. Two mice treatedwith PBS (#8 and #11) died at 35 or 47 days, and two mice treatedwith SHG died at 67 or 84 days (#9 and #10). Pathological find-ings showed that the tumor cells in the SHG-treated mice (#8)showed severe condensed nuclei and eosinophilic cytoplasm, asapoptotic cell death (Fig. 5h,i). We further investigated the cellproliferation activity using the MIB-1 labeling index. The MIB-1labeling index of the tumors from control mice treated with PBSwas 78% on average and the index of the tumors from mice treatedwith SHG was 41% on average, suggesting that tumor cells treatedwith SHG were less proliferative than control tumor cells (P <0.01). We carried out the same experiment three times using a totalof 12 mice and got the same results. Therefore, clinical and patho-logical evaluation of tumor formation after injection of ED-40515cells showed that SHG treatment significantly inhibits ED-40515cell growth and infiltration in various organs of NOG mice.
Discussion
In the present communication, we examined the effect of soybean-derived isoflavones on the growth of Hut102 and ED-40515cells infected with HTLV-I. Accumulated data indicate that thesoybean isoflavones daidzein, genistein and glycitein inhibit thegrowth of various cancer cells.(11–17,19–24) In the present study, ourdata revealed that these isoflavones apparently inhibited thegrowth of Hut102 and ED-40515 cells. Daidzein significantlyinhibited the growth of Hut102 cells at 10 and 30 μM treatment.Daidzein and its metabolite equol are known to inhibit severalcancer cells via inhibition of G1/S-phase progression.(25,26) Xianget al. showed that genistein more strongly inhibited the growthof prostate cancer cells than did daidzein and glycitein.(27)
Interestingly, as shown in Fig. 1a, the growth-inhibitory effect ofgenistein on Hut102 and ED-40515 cells was stronger than thaton Jurkat and Molt-4 cells, the non-ATL T-cell leukemia celllines. This data implies that the target molecule of genistein ismore specifically or strongly expressed in ATL cells than innon-ATL T-cell leukemia cells. Taken together, these data
indicated that genistein was the most potent growth inhibitor ofATL cells. This is the first report to show the growth-inhibitoryeffect of soy isoflavones on ATL cells. Here, we focused onunderstanding the mechanisms underlying genistein-inducedgrowth inhibition.
The product of p21, one of the target genes of p53, acts as aninhibitor of cyclin E–CDK2 or cyclin D–CDK4/6. Inhibition ofthese CDK–cyclin complexes induces cell cycle arrest in G1phase. In the present study, 3 and 10 μM genistein treatmentlead to G1/S arrest accompanied by p21 and p53 upregulation.In a previous report, genistein exhibited an inhibitory effect onG1/S-phase cell cycle progression at low doses in melanomacells.(28) Therefore, upregulation of p21 may be explained byp53 accumulation. However, it has been reported that p53function is decreased in HTLV-I-infected cells irrespective ofthe absence of gene mutation.(29) A recent study revealed that Tax,encoded by the pX region of HTLV-1, strongly activates thenuclear factor-κB pathway and that phosphorylation of thep65/RelA subunit of nuclear factor-κB at serine 536 inhibits p53activity.(30,31) Ariumi et al. suggested that Tax is likely to competewith p53 for binding with CBP, thereby repressing its trans-activation function.(32) Therefore, it was considered that genisteinmight improve p53 function. However, genistein is known toinduce p21 in a p53-independent manner in prostate cancercells.(33) Moreover, genistein induces proteasome inhibition andaccumulation of proteasome target proteins.(34) Therefore, wecould not ignore the possibility of p53-independent inductionof p21 by genistein. Interestingly, genistein did upregulate p21levels but caused G2/M arrest at 30 μM. Taken together, our dataindicate that there are two independent mechanisms involved inthe effect of genistein on Hut102 cells.
Genistein inhibited the proliferation of some cancer lines via G2/Marrest but details of the mechanisms are still unknown.(11–17,20)
Our data also clearly show that genistein inhibited the growth ofHut102 cells via G2/M arrest at 30 μM treatment. p53 plays apivotal role in protecting cells from various endogenous andexogenous stresses and regulates various groups of genes involvedin G2/M cell cycle regulation.(35) In the present study, we showedthat genistein induced phosphorylation of CHK2 and p53 at serines15, 20 and 37. One of the representative pathways responsiblefor the phosphorylation of p53, CHK1 and CHK2 is cellularDNA damage. Tax is known to interact with CHK1 and attenuatesDNA damage-induced G2 arrest;(36) however, the reason whygenistein could not induce CHK1 phosphorylation is still unknown.ATM kinase is activated in response to ionizing radiation andDNA-damaging agents through autophosphorylation at serine1981. p53 is a well-known substrate of ATM and is directly orindirectly (via CHK1 and CHK2) phosphorylated at serines 15,20 and 37 in response to DNA damage.(37–39) Therefore, accumulationof p53 induced by 3 and 10 μM genistein might be induced by a DNAdamage-independent pathway because phosphorylation of CHK2and p53 could not be detected. CHK1 and CHK2 mediate thecellular response to DNA damage and regulate the cell cycle at theG2 checkpoint; they can both phosphorylate serine 20 of p53.(40)
Recently, cumulative data have shown the relationship betweenG2/M arrest and p53 in response to DNA damage. As reviewedby Zhan,(41) GADD45a, one of the genes downstream of p53, isinduced by ATM activation. In the present study, genistein couldnot upregulate GADD45a in Hut102 cells (data not shown). Asshown in Fig. 3, the genistein-induced phosphorylation of CHK2and p53 at serines 15, 20 and 37 was completely cancelled bycaffeine, an ATM inhibitor, indicating induction of DNA damageand ATM activation. These results indicate that genistein inducedp53 phosphorylation via ATM–CHK2 activation in Hut102 cells.
Checkpoint kinase also plays a pivotal role in G2/M arrest inresponse to DNA damage in the p53-independent pathway.In this pathway, CHK2 phosphorylates Cdc25C, which resultsin cdc2 export from the nucleus to the cytosolic fraction and
1744 doi: 10.1111/j.1349-7006.2007.00595.x© 2007 Japanese Cancer Association
Fig. 5. Inhibition of growth and invasion ofED-40515 cells in NOD-SCID/cnu11 adult T-cellleukemia model mice. (a) Growth of theabdominal tumors after inoculation of ED-40515cells into the peritoneum. The lengths, widthsand heights of the tumors under the skin weremeasured by the Somers scale. Tumors were feltunder the skin over 12–16 days after inoculation.(b,c) Gross pictures of mice and tumors 40 daysafter inoculation before or after death. Abdominaltumors of #62 and #65 mice are shown beforedeath in the left side and after in the right side.(b) The #62 mice were treated with isoflavoneand #65 mice were injected with phosphate-buffered saline as a control. (c) The tumorsseparated after death. #62, #63 and #64 weretreated with isoflavone and #65 and #66 weretreated with phosphate-buffered saline. (d,e)Comparison of the size of abdominal tumors bytumor (d) weights and (e) volumes betweencontrol mice and mice treated with isoflavone.(d) The lengths, widths and heights of the tumorswere measured by the Somers scale (left) andthe weights of the tumors were also measuredon the scale (right). The tumor weights andvolumes in each group are presented as themean and standard deviation. *P < 0.001, relativeto controls. Photomicrographs of tumor sectionsfrom (f) a control mouse (#65) and (g) a mousetreated with isoflavone (#65) stained withhematoxylin–eosin 40 days after treatment.Photomicrographs of tumor sections from (h) acontrol mouse (#8) and (i) a mouse treated withisoflavone (#9) stained with hematoxylin–eosinat date of death. Photomicrographs of tumorsections from (j) a control mouse (#8) and (k) amouse treated with isoflavone (#9) stained withMIB-1 antibody.
Yamasaki et al. Cancer Sci | November 2007 | vol. 98 | no. 11 | 1745© 2007 Japanese Cancer Association
inactivation of the cdc2–cyclin B complex.(42,43) Actually, phosphoryl-ation of CHK2 was detected at 30 μM genistein. In our study,however, the phosphorylated Cdc25C level was not influencedby genistein treatment and it was therefore assumed that thep53-dependent pathway plays an essential role in genistein-induced growth arrest at G2/M phase.
Non-obese diabetic-severe combined immunodeficiencycommon γ-chain knockout mice are very useful for investigatingthe mechanism of tumorigenesis and malignant cell growth ofATL and lymphoma.(44,45) In this model, soy isoflavones stronglyinhibited the development of tumors derived from ED-40515ATL cells, suggesting that soy isoflavones can exert a growth-inhibitory effect on ATL cells in vivo. Interestingly, soy isoflavonesinhibited invasion of ATL cells into the liver, retroperitoneal
connective tissues and kidneys. ATL cells express various adhesivemolecules on their surface and one of the clinical features of ATLis mortal multiorgan invasion. Therefore, soy isoflavones areexpected to prevent the progression of ATL by inhibiting tissueinvasion of ATL cells.
This is the first study to show the inhibitory effects of soyisoflavones on ATL cells in vitro and in vivo. Among them,genistein exerts biphasic growth-inhibitory properties and hasthe potential to serve as a novel compound for the treatment ofadult T-cell leukemia.
Acknowledgments
We thank Dr Vijay Akumar Boggaram for proofreading the manuscript.
References
1 Yamaguchi K. Human T-lymphotropic virus type I in Japan. Lancet 1994;343: 213–16.
2 Mueller N, Okayama A, Stuver S, Tachibana N. Findings from the Miyazaki
Cohort Study. J Acquir Immune Defic Syndr Hum Retrovirol 1996; 13 (Suppl 1):S2–7.
3 Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T-cellleukemia: clinical and hematologic features of 16 cases. Blood 1977; 50:481–92.
Fig. 5. Continued

1746 doi: 10.1111/j.1349-7006.2007.00595.x© 2007 Japanese Cancer Association
4 Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC.Detection and isolation of type C retrovirus particles from fresh and culturedlymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl AcadSci USA 1980; 77: 7415–19.
5 Hanchard B, Gibbs WN, Lofters W et al. Adult T-cell leukemia/lymphoma (ATL)in Jamaica. In: Blattner WA, ed. Human retrovirology: HTLV. New York:Raven Press, 1990: 173–83.
6 Yamaguchi K, Kiyokawa T, Futami G, Ishii T, Takatsuki K. Pathogenesis ofadult T-cell leukemia from clinical pathologic features. In: Blattner WA, ed.Human retrovirology: HTLV. New York: Raven Press, 1990; 163–71.
7 Tomita M, Kawakami H, Uchihara JN et al. Curcumin (diferuloylmethane)inhibits constitutively active NF-κB, leading to suppression of cell growthof human T-cell leukemia virus type I-infected T-cell lines and primary adultT-cell leukemia cells. Int J Cancer 2006; 118: 765–72.
8 Tomita M, Kawakami H, Uchihara JN et al. Curcumin suppresses con-stitutive activation of AP-1 by downregulation of Jun D protein in HTLV-1-infected T-cell lines. Leuk Res 2006; 30: 313–21.
9 Zhang J, Nagasaki M, Tanaka Y, Morikawa S. Capsaicin inhibits growth ofadult T-cell leukemia cells. Leuk Res 2003; 27: 275–83.
10 Haneji K, Matsuda T, Tomita M et al. Fucoidan extracted from Cladosiphonokamuranus Tokida induces apoptosis of human T-cell leukemia virus type1-infected T-cell lines and primary adult T-cell leukemia cells. Nutr Cancer2005; 52: 189–201.
11 Liao CH, Pan SL, Guh JH, Teng CM. Genistein inversely affects tubulin-binding agent-induced apoptosis in human breast cancer cells. BiochemPharmacol 2004; 67: 2031–8.
12 Chang KL, Kung ML, Chow NH, Su SJ. Genistein arrests hepatoma cells atG2/M phase: involvement of ATM activation and upregulation of p21waf1/cip1 and Wee1. Biochem Pharmacol 2004; 67: 717–26.
13 Hewitt AL, Singletary KW. Soy extract inhibits mammary adenocarcinomagrowth in a syngeneic mouse model. Cancer Lett 2003; 192: 133–43.
14 Su SJ, Chow NH, Kung ML, Hung TC, Chang KL. Effects of soy isoflavoneson apoptosis induction and G2–M arrest in human hepatoma cells involve-ment of caspase-3 activation, Bcl-2 and Bcl-XL downregulation, and Cdc2kinase activity. Nutr Cancer 2003; 45: 113–23.
15 Casagrande F, Darbon JM. Effects of structurally related flavonoids on cellcycle progression of human melanoma cells: regulation of cyclin-dependentkinases CDK2 and CDK1. Biochem Pharmacol 2001; 61: 1205–15.
16 Frey RS, Li J, Singletary KW. Effects of genistein on cell proliferation andcell cycle arrest in nonneoplastic human mammary epithelial cells:involvement of Cdc2, p21 (waf/cip1), p27 (kip1), and Cdc25C expression.Biochem Pharmacol 2001; 61: 979–89.
17 Cappelletti V, Fioravanti L, Miodini P, Di Fronzo G. Genistein blocks breastcancer cells in the G2 (M) phase of the cell cycle. J Cell Biochem, 2000; 79:594–600.
18 Takatsuki K, Uchiyama T, Sagawa K, Yodoi J. Adult T-cell leukemia inJapan. In: Seno S, Takaku F, Irino S, eds. Topics in Hematology. Amsterdam,the Netherlands: Excerpta Medica, 1977; 73–7.
19 Khanna KK, Keating KE, Kozlov S et al. ATM associates with andphosphorylates p53: mapping the region of interaction. Nat Genet 1998; 20:398–400.
20 Matsukawa Y, Marui N, Sakai T et al. Genistein arrests cell cycleprogression at G2–M. Cancer Res 1993; 53: 1328–31.
21 Ying C, Hsu JT, Shieh SC. Growth inhibition of human endothelial cells bythe phyto-oestrogen biochanin A, a metabolite of genistein. Br J Nutr 2001;85: 615–20.
22 Rice L, Samedi VG, Medrano TA et al. Mechanisms of the growth inhibitoryeffects of the isoflavonoid biochanin A on LNCaP cells and xenografts.Prostate 2002; 52: 201–12.
23 Ying C, Hsu JT, Hung HC, Lin DH, Chen LF, Wang LK. Growth and cellcycle regulation by isoflavones in human breast carcinoma cells. ReprodNutr Dev 2002; 42: 55–64.
24 Yoshida H, Teramoto T, Ikeda K, Yamori Y. Glycitein effect on suppressingthe proliferation and stimulating the differentiation of osteoblastic MC3T3-E1 cells. Biosci Biotechnol Biochem 2001; 65: 1211–13.
25 Pan W, Ikeda K, Takebe M, Yamori Y. Genistein, daidzein and glyciteininhibit growth and DNA synthesis of aortic smooth muscle cells from stroke-prone spontaneously hypertensive rats. J Nutr 2001; 131: 1154–8.
26 Hedlund TE, Johannes WU, Miller GJ. Soy isoflavonoid equol modulates thegrowth of benign and malignant prostatic epithelial cells in vitro. Prostate2003; 54: 68–78.
27 Xiang H, Schevzov G, Gunning P, Williams HM, Silink M. A comparativestudy of growth-inhibitory effects of isoflavones and their metabolites onhuman breast and prostate cancer cell lines. Nutr Cancer 2002; 42: 224–32.
28 Kuzumaki T, Kobayashi T, Ishikawa K. Genistein induces p21 (Cip1/WAF1)expression and blocks the G1 to S phase transition in mouse fibroblast andmelanoma cells. Biochem Biophys Res Commun 1998; 251: 291–5.
29 Reid RL, Lindholm PF, Mireskandari A, Dittmer J, Brady JN. Stabilizationof wild-type p53 in human T-lymphocytes transformed by HTLV-I. Oncogene1993; 8: 3029–36.
30 Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. ActivatedAKT regulates NF-κB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 2005; 24: 6719–28.
31 Jeong SJ, Radonovich JM, Brady JN, Pise-Masison CA. HTLV-I tax inducesa novel interaction between p65/RelA and p53 that results in inhibition ofp53 transcriptional activity. Blood 2004; 104: 1490–7.
32 Ariumi Y, Kaida A, Lin JY, Hirota M, Masui O, Yamaoka S. HTLV-1 taxoncoprotein represses the p53-mediated trans-activation function throughcoactivator CBP sequestration. Oncogene 2000; 19: 1491–9.
33 Choi YH, Lee WH, Park KY, Zhang L. p53-independent induction of p21(WAF1/CIP1), reduction of cyclin B1 and G2/M arrest by the isoflavonegenistein in human prostate carcinoma cells. Jpn J Cancer Res, 2000; 91:164–73.
34 Kazi A, Daniel KG, Smith DM, Kumar NB, Dou QP. Inhibition of theproteasome activity, a novel mechanism associated with the tumor cell apoptosis-inducing ability of genistein. Biochem Pharmacol 2003; 66: 965–76.
35 Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene2001; 20: 180–5.
36 Park HU, Jeong JH, Chung JH, Brady JN. Human T-cell leukemia virus type1 Tax interacts with CHK1 and attenuates DNA-damage induced G2 arrestmediated by CHK1. Oncogene 2004; 23: 4966–74.
37 Canman CE, Lim DS, Cimpri CHKA et al. Activation of the ATM kinase byionizing radiation and phosphorylation of p53. Science 1998; 281: 1677–9.
38 Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation ofSer-20 mediates stabilization of human p53 in response to DNA damage.Proc Natl Acad Sci USA 1999; 96: 13 777–82.
39 Tibbetts RS, Brumbaugh KM, Williams JM et al. A role for ATR in the DNAdamage-induced phosphorylation of p53. Genes Dev 1999; 13: 152–7.
40 Hirao A, Kong YY, Matsuoka S et al. DNA damage-induced activation ofp53 by the checkpoint kinase CHK2. Science 2000; 287: 1824–7.
41 Zhan Q. Gadd45a, a p53- and BRCA1-regulated stress protein, in cellularresponse to DNA damage. Mutation Res 2005; 569: 133–43.
42 Takizawa CG, Morgan DO. Control of mitosis by changes in the subcellularlocation of cyclin-B1–Cdk1 and Cdc25C. Curr Opin Cell Biol 2000; 12:658–65.
43 Lopez-Girona A, Furnari B, Mondesert O, Russell P. Nuclear localization ofCdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 1999; 97:172–5.
44 Ito M, Hiramatsu H, Kobayashi K et al. NOD/SCID/γnull mouse: anexcellent recipient mouse model for engraftment of human cells. Blood2002; 100: 3175–82.
45 Dewan MZ, Terashima K, Taruishi M et al. Rapid tumor formation of humanT-cell leukemia virus type 1-infected cell lines in novel NOD-SCID/cnull
mice: suppression by an inhibitor against NF-κB. J Virol 2003; 77: 5286–94.
Copyright © 2022 FDOKUMEN




















