
Síntese e caracterização de material zeolítico de cinzas de carvão ...
Upload
khangminh22Category
view
1download
0
sid.inpe.br/mtc-m21b/2017/07.20.20.04-TDI
SÍNTESE E CARACTERIZAÇÃO DE HÍBRIDOSCELULÓSICOS MODIFICADOS COM ÓXIDOS
METÁLICOS HIDRATADOS PARA APLICAÇÃO COMOADITIVOS EM ESPUMAS RÍGIDAS DE POLIURETANO
Ester Pinheiro dos Santos
Tese de Doutorado do Cursode Pós-Graduação em Engenhariae Tecnologia Espaciais/Ciência eTecnologia de Materiais e Sensores,orientada pelas Dras. Maria doCarmo de Andrade Nono, e MariaLúcia Caetano Pinto da Silva,aprovada em 23 de junho de 2017.
URL do documento original:<http://urlib.net/8JMKD3MGP3W34P/3PAKJLH>
INPESão José dos Campos
2017

PUBLICADO POR:
Instituto Nacional de Pesquisas Espaciais - INPEGabinete do Diretor (GB)Serviço de Informação e Documentação (SID)Caixa Postal 515 - CEP 12.245-970São José dos Campos - SP - BrasilTel.:(012) 3208-6923/6921E-mail: [email protected]
COMISSÃO DO CONSELHO DE EDITORAÇÃO E PRESERVAÇÃODA PRODUÇÃO INTELECTUAL DO INPE (DE/DIR-544):Presidente:Maria do Carmo de Andrade Nono - Conselho de Pós-Graduação (CPG)Membros:Dr. Plínio Carlos Alvalá - Centro de Ciência do Sistema Terrestre (CST)Dr. André de Castro Milone - Coordenação de Ciências Espaciais e Atmosféricas(CEA)Dra. Carina de Barros Melo - Coordenação de Laboratórios Associados (CTE)Dr. Evandro Marconi Rocco - Coordenação de Engenharia e Tecnologia Espacial(ETE)Dr. Hermann Johann Heinrich Kux - Coordenação de Observação da Terra (OBT)Dr. Marley Cavalcante de Lima Moscati - Centro de Previsão de Tempo e EstudosClimáticos (CPT)Silvia Castro Marcelino - Serviço de Informação e Documentação (SID)BIBLIOTECA DIGITAL:Dr. Gerald Jean Francis BanonClayton Martins Pereira - Serviço de Informação e Documentação (SID)REVISÃO E NORMALIZAÇÃO DOCUMENTÁRIA:Simone Angélica Del Ducca Barbedo - Serviço de Informação e Documentação(SID)Yolanda Ribeiro da Silva Souza - Serviço de Informação e Documentação (SID)EDITORAÇÃO ELETRÔNICA:Marcelo de Castro Pazos - Serviço de Informação e Documentação (SID)André Luis Dias Fernandes - Serviço de Informação e Documentação (SID)

sid.inpe.br/mtc-m21b/2017/07.20.20.04-TDI
SÍNTESE E CARACTERIZAÇÃO DE HÍBRIDOSCELULÓSICOS MODIFICADOS COM ÓXIDOS
METÁLICOS HIDRATADOS PARA APLICAÇÃO COMOADITIVOS EM ESPUMAS RÍGIDAS DE POLIURETANO
Ester Pinheiro dos Santos
Tese de Doutorado do Cursode Pós-Graduação em Engenhariae Tecnologia Espaciais/Ciência eTecnologia de Materiais e Sensores,orientada pelas Dras. Maria doCarmo de Andrade Nono, e MariaLúcia Caetano Pinto da Silva,aprovada em 23 de junho de 2017.
URL do documento original:<http://urlib.net/8JMKD3MGP3W34P/3PAKJLH>
INPESão José dos Campos
2017

Dados Internacionais de Catalogação na Publicação (CIP)
Santos, Ester Pinheiro dos.Sa59s Síntese e caracterização de híbridos celulósicos modificados
com óxidos metálicos hidratados para aplicação como aditivos emespumas rígidas de poliuretano / Ester Pinheiro dos Santos. – SãoJosé dos Campos : INPE, 2017.
xxxiv + 212 p. ; (sid.inpe.br/mtc-m21b/2017/07.20.20.04-TDI)
Tese (Doutorado em Engenharia e TecnologiaEspaciais/Ciência e Tecnologia de Materiais e Sensores) –Instituto Nacional de Pesquisas Espaciais, São José dos Campos,2017.
Orientadoras : Dras. Maria do Carmo de Andrade Nono, eMaria Lúcia Caetano Pinto da Silva.
1. Celulose. 2. Óxidos metálicos hidratados. 3. Híbridosorgânico-inorgânico. 4. Poliuretano. 5. Compósitos. I.Título.
CDU 661.728.7:661.8
Esta obra foi licenciada sob uma Licença Creative Commons Atribuição-NãoComercial 3.0 NãoAdaptada.
This work is licensed under a Creative Commons Attribution-NonCommercial 3.0 UnportedLicense.
ii


iii

iv

v
Dedicatória
A você, pai: toda minha dedicação
para mais uma vitória.
(in memoriam)
A você, mãe, nossa conquista.
A meu eterno amor, Alexandre.

vi

vii
AGRADECIMENTOS
A Deus, pela força, sabedoria, discernimento e por todas as realizações conquistadas
durante esta caminhada.
À Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, CAPES, pelo auxílio
financeiro.
Ao Instituto Nacional de Pesquisas Espaciais, INPE, pelo programa de pós-graduação e
à infraestrutura disponibilizada pelo Laboratório Associados de Sensores Materiais,
LAS.
À Escola de Engenharia de Lorena, EEL/USP, na qual construí grande parte da minha
formação, por toda a infraestrutura, financiamento e empenho dos professores e
funcionários envolvidos de todas as formas possíveis durante minha estada.
À Empresa Dow Brasil e seus integrantes Vinícius e Glória, pelo fornecimento dos
reagentes, atenção e auxílio concedidos para a síntese dos compósitos.
À Prof.a Dr.a Maria do Carmo Nono de Andrade pela orientação, disciplinas ministradas
e pela experiência profissional concedida durante a execução deste trabalho.
À Prof.a Dr.a Maria Lucia Caetano Pinto da Silva pela orientação, disposição, amizade,
incentivo e por sempre acreditar na minha capacidade e dedicação para o
desenvolvimento desta tese.
Ao Prof. Dr. Amilton Martins dos Santos por permitir a realização das análises de
calorimetria exploratória diferencial e aos integrantes de seu laboratório, Rodolfo e
Maria Luiza, pela colaboração.

viii
Ao Prof. Dr. Carlos Antônio Reis Pereira Baptista por disponibilizar a infraestrutura
necessária para a realização dos ensaios de compressão.
À Prof.a Dr.a Neidenei Gomes Ferreira por me contagiar com todo o seu conhecimento
sobre Física de Superfície, pelos conselhos e amparo nos momentos críticos.
Ao Prof. Dr. Antônio Fernando Beloto por todos conselhos, empenho e disposição em
me ajudar a compreender todos os trâmites do curso.
À Maria Lucia Brison pela atenção e solicitude na realização das análises de
microscopia eletrônica de varredura.
Ao João Paulo Barros Machado pelo auxílio nas análises de difratometria de raios x.
À Bárbara Pereira pela atenção ao executar as análises de espectroscopia de
infravermelho.
A minha mãe, Suzette, por ser a minha fortaleza que, com seu amor incondicional, me
guiou em todas as minhas jornadas, estando presente em cada uma de nossas conquistas.
A meu esposo, Alexandre, pelo amor imensurável que compartilhamos há 15 anos, pela
paciência e compreensão, especialmente, por ser o meu maior encorajador e espectador
na concretização dos meus objetivos.
A todos os familiares que contribuíram por meio de orações, apoio e encorajamento
para que pudesse superar as inúmeras atribulações dispersas durante esta jornada.
A meu grande amigo, Rafael, pela colaboração técnico-científica e apoio mas,
principalmente, pela eterna amizade.

ix
À Amanda Cassiano e ao Adriano Cavalca pela amizade que construímos nestes anos
juntos.
Aos meus queridos Brandão e Otávio, não somente por toda ajuda disponibilizada no
laboratório, mas também pela amizade e torcida para que obtivesse o título.
A todos os alunos de iniciação científica e estagiários Roberta, Nathália, Milena,
Nicole, Izabela, Matheus, Júlia, Kaique e Thaísa, Renan e Ian e de pós-graduação
Nícolas, Thaís e Neuana, que encontrei durante esta longa caminhada, obrigada pela
colaboração, companheirismo e por todos os momentos que pudemos partilhar.
E a todos que não foram citados, porém emanaram todas as suas aspirações para que
pudesse atingir meu objetivo, muito obrigada!

x

xi
“Talvez não tenha conseguido fazer o melhor, mas lutei para que o melhor fosse feito.
Não sou o que deveria ser, mas graças a Deus, não sou o que era antes.”
Marthin Luther King

xii

xiii
RESUMO
O sinergismo de propriedades entre diferentes componentes em um único material torna-se cada vez mais atrativo, no que se refere à diversidade de aplicações assim como na utilização de subprodutos agroindustriais. Além disso, a busca por fontes renováveis aliada à elevada disponibilidade evidenciam as fibras lignocelulósicas como precursoras no desenvolvimento de novos materiais. Os híbridos orgânico-inorgânicos apresentam-se promissores, podendo ser aplicados em vários setores, tais como catálise, adsorção e como aditivos em matrizes poliméricas para a produção de membranas e compósitos. A proposta deste trabalho é desenvolver e caracterizar híbridos, provenientes da combinação entre a celulose da folha de bananeira e os óxidos metálicos hidratados de nióbio (Nb2O5.nH2O), de alumínio (Al2O3.nH2O) e de zinco (ZnO.nH2O), visando a produção de espumas rígidas de poliuretano e verificar o efeito destes aditivos híbridos e seus precursores nas propriedades térmicas e mecânicas dos compósitos. Inicialmente, as folhas de bananeira foram submetidas a diferentes tratamentos químicos, a fim de avaliar a modificação mais eficiente para o isolamento da celulose. Posteriormente, os híbridos foram sintetizados via precipitação convencional e um estudo de proporção foi realizado, com diferentes relações mássicas entre a celulose e os óxidos metálicos hidratados. O estabelecimento da melhor proporção de cada aditivo híbrido foi determinado por termogravimetria. Os híbridos escolhidos foram caracterizados por testes de absorção de água, DSC, FTIR, DRX, MEV/EDX e, em seguida, incorporados à matriz de poliuretano para a síntese das espumas rígidas. Os compósitos foram obtidos mediante a adição de 1 a 5 % m/m dos híbridos escolhidos, além dos respectivos precursores (celulose e óxidos metálicos hidratados), e caracterizados por TGA, DSC e ensaio de compressão. Os resultados de termogravimetria indicaram as proporções 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O como as melhores relações celulose/óxido (híbridos). As curvas DSC exibiram o efeito da fração inorgânica na supressão de calor, evidenciando os híbridos 97Celfb/3Nb2O5.nH2O e 94Celfb/6Al2O3.nH2O com propriedades em potencial para atuarem como retardantes de chamas. As análises de EDX confirmaram a incorporação dos metais na matriz celulósica, corroborando com os resultados de DRX e FTIR. Nos testes de absorção de água o híbrido 94Celfb/6Al2O3.nH2O apresentou a maior redução no caráter hidrofílico, comparado à celulose isolada e aos demais híbridos, o que nos induz a esperar por um aditivo que proporcione aumento de adesão na interface fibra/matriz polimérica, resultando em melhores propriedades térmicas e mecânicas. Os resultados térmicos e mecânicos evidenciaram o híbrido 94Celfb/6Al2O3.nH2O como o melhor aditivo na formulação para o compósito PU + 1% m/m 94Celfb/6Al2O3.nH2O.

xiv

xv
SYNTHESIS AND CHARACTERIZATION OF CELLULOSIC HYBRIDS
MODIFIED WITH HYDROUS METALLIC OXIDES FOR APPLICATION AS
ADDITIVES IN RIGID POLYURETHANE FOAMS
ABSTRACT
The synergism properties between different components in an unique material become increasingly attractive, as regards the variety of applications like the use of agro industrial by-products. Furthermore, the search for renewable sources combined with high availability evidence the lignocellulosic fibers as precursor in the development of new materials. Organic-inorganic hybrids have to be promising and can be applied in several sectors, such as catalysis, adsorption and as additives in polymer matrices in the production of membranes and composites. The purpose of this work is to develop and characterize hybrids derived from the combination between the cellulose of leaf banana’s plant and hydrous metallic oxides of niobium (Nb2O5.nH2O), aluminum (Al2O3.nH2O) and zinc (ZnO.nH2O), aiming at the production of rigid polyurethane foams and to verify the effect of these additives hybrid and their precursors on the thermal and mechanical properties of the composites. Initially, the banana leaves were submitted to different chemical treatments, in order to evaluate the most efficient modification for isolation of cellulose. After, the hybrids were synthesized through conventional precipitation and a proportion study was performed, with different mass relations between the cellulose and the hydrous metallic oxides. The establishment of the best proportion of each additive hybrid was determined by thermogravimetric analysis. The selected hybrids were characterized by water absorption tests, DSC, FTIR, DRX, MEV/EDX and then incorporated into the polyurethane matrix for the synthesis of rigid foams. The composites were obtained by adding 1 to 5 % m/m of the chosen hybrids, beyond to the respective precursors (cellulose and hydrous metallic oxides) and characterized by TGA, DSC and compression test. The results of thermogravimetric analysis indicated the proportions 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O and 97Celfb/3ZnO.nH2O as the best cellulose/oxide ratios (hybrids). DSC curves exhibited the effect of oxides on heat suppression, evidencing the hybrids 97Celfb/3Nb2O5.nH2O and 94Celfb/6Al2O3.nH2O with potential properties to act as flame retardants. EDX analyzes confirmed the incorporation of the metals in the cellulosic matrix, corroborating with the results of XRD and FTIR. In the tests of water absorption, the hybrid 94Celfb/6Al2O3.nH2O presented the greatest reduction in hydrophilic character, compared to the isolated cellulose and the other hybrids, which induces us to wait for an additive that provides increased adhesion at the fiber/polymeric matrix interface, resulting in better thermal and mechanical properties. The thermal and mechanical results show the hybrid 94Celfb/6Al2O3.nH2O as the best additive in the formulation for the composite PU + 1% m/m 94Celfb/6Al2O3.nH2O.

xvi

xvii
LISTA DE FIGURAS
Figura 2.1 - Classificação para as fibras naturais e sintéticas. .......................................... 4!Figura 2.2 - Classificação geral para as fibras lignocelulósicas. ....................................... 6!Figura 2.3 - Representação esquemática da estrutura de uma fibra vegetal. .................... 7!Figura 2.4 - Estrutura da lignina. .................................................................................... 11!Figura 2.5 - Principais unidades de fenóis substituídos presentes na lignina: (a) trans-p-
cumarílico, (b) trans-coferílico e (c) trans-sinapílico. ............................................ 12!Figura 2.6 - Estrutura parcial da hemicelulose. ............................................................... 13!Figura 2.7 - Estrutura da celulose. ................................................................................... 14!Figura 2.8 - Representação esquemática das interações (a) intramolecular e (b)
intermolecular na celulose. ...................................................................................... 15!Figura 2.9 - Estrutura supramolecular da celulose para as fibras vegetais. .................... 16!Figura 2.10 - Transformação da celulose nativa em seus respectivos polimorfos. ......... 17!Figura 2.11 - Reação global para o tratamento alcalino. ................................................. 21!Figura 2.12 - Estrutura das fibras (a) sem tratamento e (b) com tratamento alcalino. .... 22!Figura 2.13 - Hidrólise básica da lignina. ....................................................................... 22!Figura 2.14 - Reações de acetilação: (a) com catalisador e (b) sem catalisador. ............ 25!Figura 2.15 - Índice de safra da banana de 2006 a 2016. ................................................ 27!Figura 2.16 - Representação esquemática de uma bananeira. ......................................... 28!Figura 2.17 - Classificação dos compósitos em função do reforço. ................................ 45!Figura 2.18 - Reação global para a síntese de poliuretanos. ........................................... 47!Figura 4.1 - Fluxograma para o isolamento da celulose via tratamento ácido. ............... 57!Figura 4.2 - Fluxograma para o isolamento da celulose via tratamento básico. ............. 58!Figura 4.3 - Fluxograma para a síntese dos óxidos metálicos hidratados via precipitação
convencional (PC). .................................................................................................. 59!Figura 4.4 - Fluxograma para a síntese dos híbridos orgânico-inorgânicos via
precipitação convencional (PC). ............................................................................. 61!Figura 4.5 - Esquema para a síntese do poliuretano. ....................................................... 62!Figura 4.6 - Esquema para a síntese dos compósitos de poliuretano com os aditivos da
formulação, sendo aditivo: Celfb, MyOz.nH2O ou (100-x)Celfb/xMyOz.nH2O. ..... 63!Figura 4.7 - Esquema para identificação e corte para os blocos de compósitos. ............ 64!

xviii
Figura 4.8 - Esquema para amostragem dos compósitos. ............................................... 65!Figura 5.1 - Transformação macroscópica em função dos tratamentos químicos: (a)
folha de bananeira bruta, (b) celulose P1, (c) celulose P2 e (d) celulose P3, em
placas de Petri com 10 cm de diâmetro. .................................................................. 71!Figura 5.2 - Diferenças na remoção da lignina mediante (a) um ataque ácido e (b) dois
ataques ácido. .......................................................................................................... 72!Figura 5.3 - Transformação macroscópica da folha de bananeira in natura: (a) folha de
bananeira bruta e seca e (b) celulose após dois ataques ácido (P2) pulverizada a 20
mesh, em placas de Petri com 10 cm de diâmetro. ................................................. 72!Figura 5.4 - Óxido de zinco hidratado, ZnO.nH2O, em placa de Petri com 10 cm de
diâmetro. .................................................................................................................. 73!Figura 5.5 - Aditivos híbridos escolhidos no estudo de proporção:
(a) 97Celfb/3 Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O e (c) 97Celfb/3ZnO.nH2O,
em placas de Petri com 10 cm de diâmetro. ............................................................ 73!Figura 5.6 - Proporções dos híbridos com separação de fases:
(a) 95Celfb/5Nb2O5.nH2O, (b) 93Celfb/7Al2O3.nH2O e (c) 85Celfb/15ZnO.nH2O,
em placas de Petri com 10 cm de diâmetro. ............................................................ 74!Figura 5.7 - Seções de 10 x 5 cm retiradas do interior dos blocos: (a) poliuretano puro
(PU), (b) PU + 1% Celfb, (c) PU + 2% Celfb, (d) PU + 3% Celfb, (e) PU + 4%
Celfb e (f) PU + 5% Celfb. ...................................................................................... 75!Figura 5.8 - Difratogramas de raios X para (a) folha bruta, (b) celulose P1, (c) celulose
P2 e (d) celulose P3. ................................................................................................ 77!Figura 5.9 - Espectros na região do infravermelho para (a) folha bruta, (b) celulose P1,
(c) celulose P2 e (d) celulose P3. ............................................................................ 80!Figura 5.10 - Proposta de mecanismo para o isolamento da celulose, via ataque ácido
(P2), para a folha de bananeira. ............................................................................... 81!Figura 5.11 - Representação da acetilação parcial da celulose, via tratamento ácido P2.
................................................................................................................................. 82!Figura 5.12 - Proposta de mecanismo para o isolamento da celulose, via ataque básico
(P1 e P3), para a folha de bananeira. ....................................................................... 83!

xix
Figura 5.13 - Representação do isolamento da celulose, via tratamento básico P1 e P3.
................................................................................................................................. 84!Figura 5.14 - Perfis das curvas TGA/DTG para (a) folha bruta, (b) celulose P1, (c)
celulose P2 e (d) celulose P3. .................................................................................. 85!Figura 5.15 - Micrografias para (a) folha bruta, (b) celulose P1, (c) celulose P2 e (d)
celulose P3, com ampliação de 100x. ..................................................................... 89!Figura 5.16 - Gráfico da faixa de pH para precipitação de alguns hidróxidos metálicos.
................................................................................................................................. 91!Figura 5.17 - Perfis das curvas TGA/DTG para os óxidos hidratados: (a) Nb2O5.nH2O,
(b) Al2O3.nH2O e (c) ZnO.nH2O, em atmosfera de ar sintético. ............................. 94!Figura 5.18 - Perfis das curvas DSC para os óxidos hidratados: (a) Nb2O5.nH2O, (b)
Al2O3.nH2O e (c) ZnO.nH2O, em atmosfera de nitrogênio. .................................... 96!Figura 5.19 - Difratogramas para: (a) Nb2O5.nH2O e (b) Nb2O5 calcinado à 600 °C. .... 97!Figura 5.20 - Difratogramas para: (c) Al2O3.nH2O e (d) Al2O3 calcinado à 1000 °C. ... 98!Figura 5.21 - Difratogramas para: (e) ZnO.nH2O e (f) ZnO calcinado à 600 °C. ........... 98!Figura 5.22 - Perfis das curvas TGA para os híbridos (100-x)Celfb/xNb2O5.nH2O, em
atmosfera de ar sintético, para x = 3, 5, 10, 15 e 20. ............................................. 101!Figura 5.23 - Perfis das curvas TGA para os híbridos (100-x)Celfb/xAl2O3.nH2O, em
atmosfera de ar sintético, para x = 3, 4, 5, 6, 7, 10, 15 e 20. ................................. 104!Figura 5.24 - Perfis das curvas TGA para os híbridos (100-x)Celfb/xZnO.nH2O, em
atmosfera de ar sintético, para x = 3, 4, 5, 6, 7, 10, 15 e 20. ................................. 107!Figura 5.25 - Comparação entre as curvas TGA das proporções escolhidas e seus
precursores para os híbridos: (a) 97Celfb/3Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O
e (c) 97Celfb/3ZnO.nH2O. ................................................................................... 110!Figura 5.26 - Perfis das curvas DSC para: (a) celulose, (b) 97Celfb/3Nb2O5.nH2O, (c)
94Celfb/6Al2O3.nH2O e (d) 97Celfb/3ZnO.nH2O em atmosfera de nitrogênio. .. 111!Figura 5.27 - Difratogramas para (a) 97Celfb/3Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O
e (c) 97Celfb/3ZnO.nH2O e seus respectivos precursores. ................................... 115!Figura 5.28 - Espectros na região do infravermelho para (a) 97Celfb/3Nb2O5.nH2O, (b)
94Celfb/6Al2O3.nH2O e (c) 97Celfb/3ZnO.nH2O. ............................................... 117!

xx
Figura 5.29 - Micrografias para (a) celulose, (b) 97Celfb/3Nb2O5.nH2O, (c)
94Celfb/6Al2O3.nH2O e (d) 97Celfb/3ZnO.nH2O, com ampliação de 100x. ....... 118!Figura 5.30 - Efeito da modificação química na capacidade de absorção de água em
função do tempo para a folha bruta, celulose e os aditivos híbridos. .................... 120!Figura 5.31 - Micrografias para (a) PU puro; (b) PU + 1% m/m Celfb; (c) PU + 2% m/m
Celfb; (d) PU + 3% m/m Celfb; (e) PU + 4% m/m Celfb e (f) PU + 5% m/m Celfb,
com ampliação de 23 e 24x. .................................................................................. 122!Figura 5.32 - Perfis das curvas TGA para os precursores PU e Celfb e compósitos PU +
Celfb, em atmosfera de ar sintético, com adição de 1 a 5 % m/m de Celfb. ......... 126!Figura 5.33 - Perfis das curvas TGA para os precursores PU e Nb2O5.nH2O e
compósitos PU + Nb2O5.nH2O, em atmosfera de ar sintético, com adição de 1 a 5
% m/m de Nb2O5.nH2O. ........................................................................................ 129!Figura 5.34 - Perfis das curvas TGA para os precursores PU e Al2O3.nH2O e compósitos
PU + Al2O3.nH2O, em atmosfera de ar sintético, com adição de 1 a 5 % m/m de
Al2O3.nH2O. .......................................................................................................... 131!Figura 5.35 - Perfis das curvas TGA para os precursores PU e ZnO.nH2O e compósitos
PU + ZnO.nH2O, em atmosfera de ar sintético, com adição de 1 a 5 % m/m de
ZnO.nH2O. ............................................................................................................ 133!Figura 5.36 - Perfis das curvas TGA para os precursores PU, Celfb, Nb2O5.nH2O e
97Celfb/3Nb2O5.nH2O e compósitos PU + 97Celfb/3Nb2O5.nH2O, em atmosfera
de ar sintético, com adição de 1 a 5 % m/m de 97Celfb/3Nb2O5.nH2O. ............... 135!Figura 5.37 - Perfis das curvas TGA para os precursores PU, Celfb, Al2O3.nH2O e
94Celfb/6Al2O3.nH2O e compósitos PU + 94Celfb/6Al2O3.nH2O, em atmosfera de
ar sintético, com adição de 1 a 5 % m/m de 94Celfb/6Al2O3.nH2O. .................... 138!Figura 5.38 - Perfis das curvas TGA para os precursores PU, Celfb, ZnO.nH2O e
97Celfb/3ZnO.nH2O e compósitos PU + 97Celfb/3ZnO.nH2O, em atmosfera de ar
sintético, com adição de 1 a 5 % m/m de 97Celfb/3ZnO.nH2O. ........................... 141!Figura 5.39 - Perfis das curvas DSC para: (a) PU, (b) PU + 3% m/m Nb2O5.nH2O, (c)
PU + 4 % m/m Al2O3.nH2O e (d) PU + 2 % m/m ZnO.nH2O em atmosfera de
nitrogênio. ............................................................................................................. 144!

xxi
Figura 5.40 - Perfis das curvas DSC para: (a) PU, (b) PU + 2 % m/m Celfb, (c) PU + 4
% m/m 97Celfb/3Nb2O5.nH2O, (d) PU + 1 % m/m 94Celfb/6Al2O3.nH2O e (e) PU
+ 2 % m/m 97Celfb/3ZnO.nH2O em atmosfera de nitrogênio. ............................. 145!Figura 5.41 - Curva resistência à compressão em função da deformação para o PU puro
até o limite do corpo de prova 1 (CP1). ................................................................ 148!Figura 5.42 - Corpos de prova para o PU puro antes e após o ensaio até o limite de
compressão do CP1. .............................................................................................. 149!Figura 5.43 - Curvas resistência à compressão em função da deformação para os
compósitos contendo óxidos metálicos hidratados. .............................................. 150!Figura 5.44 - Curvas resistência à compressão em função da deformação para os
compósitos contendo celulose e híbridos. ............................................................. 150!

xxii

xxiii
LISTA DE TABELAS
Tabela 2.1 - Principais diferenças entre hemicelulose e celulose. .................................. 13!Tabela 4.1 - Reagentes utilizados para a síntese do poliuretano. .................................... 53!Tabela 4.2 – Tratamentos químicos empregados para o isolamento da celulose. ........... 56!Tabela 4.3 - Condições de síntese e lavagem para os óxidos metálicos hidratados. ....... 59!Tabela 4.4 - Estudo de proporções dos híbridos orgânico-inorgânico. ........................... 60!Tabela 5.1 - Rendimento de celulose para as propostas de tratamento químico da folha
de bananeira. ........................................................................................................... 76!Tabela 5.2 – Valores dos índices de cristalinidade (Ic) para as celuloses extraídas pelas
diferentes propostas de tratamento químico. ........................................................... 78!Tabela 5.3 – Dados das curvas TGA/DTG para a folha bruta e celuloses isoladas em
atmosfera de ar sintético. ......................................................................................... 86!Tabela 5.4 – Dados das curvas TGA/DTG para os óxidos metálicos hidratados em
atmosfera de ar sintético. ......................................................................................... 95!Tabela 5.5 – Dados das curvas DSC para os óxidos metálicos hidratados em atmosfera
de nitrogênio. ........................................................................................................... 97!Tabela 5.6 - Dados das curvas TGA/DTG para os híbridos (100-x)Celfb/xNb2O5.nH2O e
precursores. ........................................................................................................... 102!Tabela 5.7 - Dados das curvas TGA/DTG para os híbridos (100-x)Celfb/xAl2O3.nH2O e
precursores. ........................................................................................................... 105!Tabela 5.8 - Dados das curvas TGA/DTG para os híbridos (100-x)Celfb/xZnO.nH2O e
precursores. ........................................................................................................... 108!Tabela 5.9 – Dados das curvas DSC para a celulose e híbridos escolhidos em atmosfera
de nitrogênio. ......................................................................................................... 113!Tabela 5.10 – Valores dos índices de cristalinidade (Ic) para a celulose e híbridos
97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O. .............. 116!Tabela 5.11 – Valores obtidos nos testes de absorção de água para a folha bruta (Fb),
celulose (Celfb) e híbridos 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e
97Celfb/3ZnO.nH2O. ............................................................................................ 120!

xxiv
Tabela 5.12 - Dados das curvas TGA/DTG para os compósitos PU + Celfb e
precursores. ........................................................................................................... 127!Tabela 5.13 - Dados das curvas TGA/DTG para os compósitos PU/Nb2O5.nH2O e
precursores. ........................................................................................................... 130!Tabela 5.14 - Dados das curvas TGA/DTG para os compósitos PU + Al2O3.nH2O e
precursores, sendo n.d. = não detectado. ............................................................... 132!Tabela 5.15 - Dados das curvas TGA/DTG para os compósitos PU + ZnO.nH2O e
precursores. ........................................................................................................... 134!Tabela 5.16 - Dados das curvas TGA/DTG para os compósitos .................................. 136!Tabela 5.17 - Dados das curvas TGA/DTG para os compósitos .................................. 139!Tabela 5.18 - Dados das curvas TGA/DTG para os compósitos .................................. 142!Tabela 5.19 – Dados das curvas DSC para compósitos contendo os óxidos metálicos
hidratados, celulose e híbridos orgânico-inorgânicos em atmosfera de nitrogênio.
............................................................................................................................... 146!Tabela 5.20 - Propriedades mecânicas para os compósitos PU + Celfb. ...................... 151!Tabela 5.21 - Propriedades mecânicas para os compósitos PU + Nb2O5.nH2O. .......... 151!Tabela 5.22 - Propriedades mecânicas para os compósitos PU + Al2O3.nH2O. ........... 152!Tabela 5.23 - Propriedades mecânicas para os compósitos PU + ZnO.nH2O. .............. 152!Tabela 5.24 - Propriedades mecânicas para os compósitos PU + 97Celfb/3Nb2O5.nH2O.
............................................................................................................................... 152!Tabela 5.25 - Propriedades mecânicas para os compósitos PU + 94Celfb/6Al2O3.nH2O.
............................................................................................................................... 153!Tabela 5.26 - Propriedades mecânicas para os compósitos PU + 97Celfb/3ZnO.nH2O.
............................................................................................................................... 153!

xxv
LISTA DE ABREVIATURAS E SIGLAS
ABAL Associação Brasileira do Alumínio
AM Anidrido maleico
APP Polifosfato de amônio
ASTM American Society for Testing and Materials
CBMM Companhia Brasileira de Metalurgia e Mineração
CMC Celulose microcristalina
CP Corpo de prova
DMCHA N,N-dimetilciclo hexilamina
DRIFT Espectroscopia de Infravermelho por Reflectância Difusa
DRX Difração de Raios X
DSC Calorimetria Exploratória Diferencial
EDX Espectroscopia de Energia Dispersiva de Raios X
EEL/USP Escola de Engenharia de Lorena/Universidade de São Paulo
EVA Poli(etileno-co-acetato de vinila)
FISQP Ficha de Informação de Segurança de Produtos Químicos
FTIR Espectroscopia de Infravermelho com Transformada de Fourier
GZO Óxido de zinco dopado com gálio
IBGE Instituto Brasileiro de Geografia e Estatística
INPE Instituto Nacional de Pesquisas Espaciais e Inovação
ITO Óxido de índio dopado com estanho
JCPDS Joint Committee on Powder Diffraction Standards
LAS Laboratório Associado de Sensores e Materiais
LOQ Departamento de Engenharia Química
LSS Lauril sulfato de sódio
MDI Difenilmetano diisocianato
MEV Microscopia Eletrônica de Varredura
PBS Poli(succinato de butileno)
PC Precipitação convencional
PEAD Polietileno de alta densidade

xxvi
PEBD Polietileno de baixa densidade
PHBV Poli(3-hidroxibutirato-co-3-hidroxivalerato)
PLA Poli(ácido lático)
PP Polipropileno
PU Poliuretano
RE Resíduo Experimental
RT Resíduo Teórico
S1 Parede secundária externa
S2 Parede secundária intermediária
S3 Parede secundária interna
sol. Solução
TCO Óxidos Transparentes Condutores
TDI Tolueno diisocianato
TGA/DTG Termogravimetria e sua derivada
UV Ultravioleta

xxvii
LISTA DE SÍMBOLOS
% m/m Porcentagem massa por massa % m/v Porcentagem massa por volume % v/v Porcentagem volume por volume % vol Porcentagem volumétrica °C Graus Celsius °C.min-1 Graus Celsius por minuto Al Alumínio Al2O3 Óxido de alumínio ou alumina Al2O3.nH2O Óxido de alumínio hidratado C-C Ligação carbono-carbono C4H6O3 Anidrido acético Cd2+ Cátion cádmio Cel Celulose Celfb Celulose da folha da bananeira CH3CO Grupo acetila Cl- Ânion cloreto cm Centímetro cm-1 Por centímetro CO Monóxido de carbono CO2 Dióxido de carbono Cr4+ Cátion cromo D Dextrógira eq.L-1 Equivalente por litro eV elétron-volt Fb Folha de bananeira Fe3O4.nH2O Óxido de ferro hidratado g Grama g/g Grama por grama g.cm-3 Massa específica h Hora H2O Água H2Odeio. Água deionizada H2SO4 Ácido sulfúrico HCl Ácido clorídrico HCN Ácido cianídrico HF Ácido fluorídrico HNO3 Ácido nítrico In2O3 Óxido de índio KBr Brometo de potássio kJ.mol-1 Quilojoule por mol

xxviii
kN Quilonewton kPa Quilopascal Kps Produto de solubilidade kV Quilovolt mg/g Miligrama por grama MgO.nH2O Óxido de magnésio hidratado min Minuto mL Mililitro mL.min-1 Mililitros por minuto mm Milímetro mm.min-1 Milímetro por minuto Mn3O4.nH2O Óxido de manganês hidratado mol.L-1 Mol por litro Mt Megatonelada n Grau de hidratação NaOH Hidróxido de sódio Nb Nióbio Nb-Ti Liga de nióbio-titânio Nb2O5 Óxido de nióbio Nb2O5.nH2O Óxido de nióbio hidratado NbO Monóxido de nióbio NbO2 Dióxido de nióbio NH3 Amônia nm Nanometro o Posição orto OH Grupo hidroxila p Posição para Pa Pascal Pb2+ Cátion chumbo pH Potencial hidrogeniônico rpm Rotações por minuto s Segundo S.m-1 Siemens por metro SnO2 Óxido de estanho T Temperatura t Tempo Tamb Temperatura ambiente Tcal. Tempratura de calcinação Tg Temperatura de transição vítrea Tinicial Temperatura Inicial u.a. Unidade arbitrária Zn Zinco

xxix
Zn(OH)2 Hidróxido de zinco ZnO Óxido de zinco ZnO.nH2O Óxido de zinco hidratado ZrO2.nH2O Óxido de zircônio hidratado α Alfa β Beta γ Gama Δ Aquecimento δ Delta ΔH Entalpia de reação ΔHdec. Entalpia de decomposição ΔHdes. Entalpia de desidratação ΔHdespol. Entalpia de despolimerização ΔT Variação de temperatura η Eta θ Teta κ Kappa χ Chi

xxx

xxxi
SUMÁRIO
1.! INTRODUÇÃO ........................................................................................................ 1!
2.! REVISÃO BIBLIOGRÁFICA ................................................................................ 3!2.1.! FIBRAS NATURAIS ........................................................................................ 3!2.2.! FIBRAS LIGNOCELULÓSICAS ................................................................... 5!
2.2.1.! Composição química das fibras lignocelulósicas ........................................ 9!a)! Pectinas, ceras e extrativos .......................................................................... 10!b)! Lignina ......................................................................................................... 10!c)! Hemicelulose ................................................................................................ 12!d)! Celulose ....................................................................................................... 14!
2.3.! MODIFICAÇÕES EM FIBRAS VEGETAIS .............................................. 18!2.3.1.! Modificações físicas ................................................................................... 19!2.3.2.! Modificações químicas ............................................................................... 20!
a)! Mercerização ............................................................................................... 20!b)! Acetilação .................................................................................................... 24!
2.4.! FIBRA DE BANANEIRA ............................................................................... 27!2.5.! FIBRA DE BANANEIRA E SUAS POTENCIAIS APLICAÇÕES ........... 29!2.6.! HÍBRIDOS ORGÂNICO-INORGÂNICO ................................................... 38!2.7.! ÓXIDOS METÁLICOS HIDRATADOS ..................................................... 40!
2.7.1.! Óxido de nióbio hidratado, Nb2O5.nH2O ................................................... 40!2.7.2.! Óxido de alumínio hidratado, Al2O3.nH2O ................................................ 42!2.7.3.! Óxido de zinco hidratado, ZnO.nH2O ........................................................ 43!
2.8.! COMPÓSITOS ................................................................................................ 44!2.9.! POLIURETANOS ........................................................................................... 46!
3.! OBJETIVOS ........................................................................................................... 51!3.1.! OBJETIVO GERAL ....................................................................................... 51!3.2.! OBJETIVOS ESPECÍFICOS ........................................................................ 51!
4.! MATERIAIS E MÉTODOS .................................................................................. 53!4.1.! MATERIAIS .................................................................................................... 53!
4.1.1.! Folhas de bananeira (Musa sapientum) .................................................... 53!

xxxii
4.1.2.! Reagentes de síntese para a matriz de poliuretano ................................... 53!4.1.3.! Outros reagentes ........................................................................................ 54!
4.2.! EQUIPAMENTOS .......................................................................................... 54!4.2.1.! Laboratório de Novos Materiais, Departamento de Engenharia Química
(LOQ), EEL/USP ..................................................................................................... 54!4.2.2.! Laboratório de Polímeros, Departamento de Engenharia Química (LOQ),
EEL/USP ................................................................................................................. 55!4.2.3.! Laboratório de Infravermelho, Departamento de Biotecnologia (LOT),
EEL/USP ................................................................................................................. 55!4.2.4.! Laboratório de Ensaios Mecânicos, Departamento de Materiais (LOM),
EEL/USP ................................................................................................................. 55!4.2.5.! Laboratório Associado de Sensores e Materiais (LAS), INPE .................. 55!
4.3.! MÉTODOS ...................................................................................................... 55!4.3.1.! Preparação das folhas de bananeira, Fb ................................................... 56!4.3.2.! Isolamento da celulose a partir das folhas de bananeira, Celfb ............... 56!
a)! Ataque ácido ................................................................................................ 56!b)! Ataque básico ............................................................................................... 57!
4.3.3.! Síntese dos óxidos metálicos hidratados, MyOz.nH2O ............................... 58!4.3.4.! Calcinação dos óxidos metálicos hidratados, MyOz.nH2O ........................ 60!4.3.5.! Síntese dos híbridos orgânico-inorgânico, (100-x)Celfb/xMyOz.nH2O ..... 60!4.3.6.! Preparação da matriz de poliuretano, PU ................................................. 61!4.3.7.! Preparação dos compósitos ....................................................................... 62!4.3.8.! Corte e amostragem dos compósitos .......................................................... 63!4.3.9.! Fragmentação e pulverização dos compósitos .......................................... 65!
4.4.! CARACTERIZAÇÕES .................................................................................. 65!4.4.1.! Teste de absorção de água ......................................................................... 65!4.4.2.! Espectroscopia de infravermelho com transformada de Fourier, FTIR .... 66!4.4.3.! Difratometria de raios X, DRX .................................................................. 66!4.4.4.! Termogravimetria e sua derivada, TGA/DTG ........................................... 67!4.4.5.! Calorimetria Exploratória Diferencial, DSC ............................................ 67!

xxxiii
4.4.6.! Microscopia eletrônica de varredura e espectroscopia de energia
dispersiva de raios X, MEV/EDX ............................................................................ 68!4.4.7.! Densidade aparente ................................................................................... 68!4.4.8.! Ensaios de compressão .............................................................................. 69!
5.! RESULTADOS E DISCUSSÕES ......................................................................... 71!5.1.! AVALIAÇÃO MACROSCÓPICA ................................................................ 71!
5.1.1.! Folha de bananeira e celulose ................................................................... 71!5.1.2.! Óxidos metálicos hidratados ...................................................................... 72!5.1.3.! Híbridos orgânico-inorgânicos .................................................................. 73!5.1.4.! Matriz e compósitos ................................................................................... 74!
5.2.! AVALIAÇÃO FÍSICO-QUÍMICA: PRECURSORES E HÍBRIDOS ....... 76!5.2.1.! Isolamento da celulose ............................................................................... 76!5.2.2.! Síntese e caracterização dos óxidos metálicos hidratados ........................ 91!5.2.3.! Síntese dos híbridos (100-x)Celfb/xMyOz.nH2O: estudo de proporção ..... 99!
a)! Híbridos (100-x)Celfb/xNb2O5.nH2O ......................................................... 101!b)! Híbridos (100-x)Celfb/xAl2O3.nH2O .......................................................... 104!c)! Híbridos (100-x)Celfb/xZnO.nH2O ............................................................ 107!
5.3.! HÍBRIDOS: PROPORÇÕES ESCOLHIDAS ........................................... 111!5.4.! ABSORÇÃO DE ÁGUA ............................................................................... 119!5.5.! COMPÓSITOS DE PU: ESTRUTURA CELULAR E MORFOLOGIA 122!5.6.! COMPÓSITOS DE PU: COMPORTAMENTO TÉRMICO E
PROPRIEDADES MECÂNICAS .......................................................................... 124!5.6.1.! Termogravimetria .................................................................................... 125!
a)! Compósitos PU + celulose, PU + Celfb .................................................... 125!b)! Compósitos PU + Nb2O5.nH2O ................................................................. 129!c)! Compósitos PU + Al2O3.nH2O ................................................................... 131!d)! Compósitos PU + ZnO.nH2O .................................................................... 133!e)! Compósitos PU + 97Celfb/3Nb2O5.nH2O .................................................. 135!f)! Compósitos PU + 94Celfb/6Al2O3.nH2O ................................................... 138!g)! Compósitos PU + 97Celfb/3ZnO.nH2O ..................................................... 141!
5.6.2.! Calorimetria Exploratória Diferencial .................................................... 144!

xxxiv
5.6.3.! Comportamento mecânico ....................................................................... 148!
6.! CONCLUSÕES .................................................................................................... 155!
REFERÊNCIAS BIBLIOGRÁFICAS ...................................................................... 157!
APÊNDICE A -! CURVAS TGA/DTG PARA OS HÍBRIDOS (100-
X)Celfb/XMYOZ.nH2O ................................................................................................ 177!
APÊNDICE B -! MICROGRAFIAS PARA OS COMPÓSITOS DE PU +
MYOZ.NH2O E PU + (100-X)CELFB/XMYOZ.NH2O .............................................. 189!
APÊNDICE C -! CURVAS TGA/DTG: MATRIZ DE PU E COMPÓSITOS PU
+ CELFB; PU + MYOZ.NH2O E PU + (100-X)CELFB/XMYOZ.NH2O ................. 195!

1
1. INTRODUÇÃO
A motivação para o desenvolvimento deste projeto baseia-se na utilização de
subprodutos agroindustriais, visando uma adequação tecnológica para materiais
voltados a vários setores da engenharia. A abundância destes resíduos produzidos é de
extrema importância para aplicações industriais de maior valor agregado, pois
caracteriza-se como fonte de material celulósico, sendo empregado principalmente
como reforço em compósitos poliméricos (ALBINANTE; PACHECO; VISCONTE,
2013).
Das inúmeras fibras vegetais disponíveis, a fibra de bananeira destaca-se devido
ao alto índice na produção do fruto no país, com uma safra de 6,96 milhões de toneladas
em 2016 (IBGE, 2017). A elevada safra reflete tanto no mercado financeiro, como na
geração de grandes quantidades de subprodutos, os quais podem ser aplicados em
diferentes segmentos, tais como: artesanal, científico e tecnológico.
Uma vez que as fibras apresentam-se altamente hidrofílicas, in natura, as
modificações químicas são alternativas para a redução desta hidrofilicidade, quando as
respectivas fontes vegetais destinam-se como reforço em matrizes poliméricas
(KHALIL; BHAT; YUSRA, 2012). Devido à diversidade de fibras vegetais disponíveis
no país, a escolha da modificação química adequada é crucial para que a fração
cristalina (celulose) seja preservada e devidamente isolada.
Dentre os tratamentos químicos, o tratamento alcalino (ataque básico) é uma das
modificações mais baratas e utilizadas nas fibras naturais, quando as mesmas são
empregadas como reforço (GURUNATHAN; MOHANTY; NAYAK, 2015). Porém,
para minimizar as agressões ambientais de diversos componentes tóxicos também
utilizados, outros tipos de tratamentos químicos estão sendo estudados, como por
exemplo, os tratamentos superficiais, os quais apresentam-se eficientes tanto no
isolamento quanto na preservação da fração cristalina. Dentre eles, pode-se citar a
acetilação, na qual utiliza-se ácido acético ou anidrido acético, ambos catalisados por
ácidos fortes (ácido nítrico ou ácido sulfúrico) (LI; TABIL; PANIGRAHI, 2007;
KABIR et al., 2012b).

2
A escolha dos óxidos metálicos hidratados mencionados nesta tese baseia-se nas
propriedades de seus respectivos metais, as quais também serão observadas nos
híbridos. O alumínio apresenta baixa massa específica (2,74 g.cm-3) e condutividade
elétrica (3,42 x 107 S.m-1). O óxido de alumínio, por sua vez, é um dos materiais
cerâmicos mais importantes e por exibir alta resistência elétrica é aplicado como
isolante elétrico. Já o nióbio é resistente à oxidação e ao impacto, com excelente
ductilidade. Considerando o setor metalúrgico, é essencial na produção de ligas
resistentes a altas temperaturas e aços inoxidáveis especiais. O óxido de zinco destaca-
se devido à sua elevada atividade óptica e luminescente, além de abundância natural e
baixa toxicidade. É muito atrativo para aplicações em eletrônica e optoeletrônica como,
por exemplo, em semicondutores e sensores.
Assim como os óxidos metálicos hidratados citados, os poliuretanos (PU’s)
ganham evidência devido à sua versatilidade única na obtenção de estruturas
poliméricas, as quais abrangem os termoplásticos, os termorrígidos e os elastômeros.
Considerando a vasta gama de combinações de diferentes tipos de matérias-primas, é
possível obter diversos produtos para atender as necessidades dos vários segmentos de
mercado (ZIA; BHATTI; BHATTI, 2007; KURANSKA; PROCIAK, 2012).
Com base no exposto, este trabalho visa o desenvolvimento de novos materiais
compósitos com valor agregado maior, comparado aos seus precursores, e de
desempenho. Isto é, considerando as características intrínsecas dos metais a serem
estudados e a versatilidade da matriz polimérica, a presença do híbrido modificará as
características do material final, alterando propriedades mecânicas e térmicas, as quais
definirão as aplicações em diferentes setores da engenharia.

3
2. REVISÃO BIBLIOGRÁFICA
2.1. FIBRAS NATURAIS
A crescente preocupação da sociedade sobre impacto ambiental, sustentabilidade,
fontes renováveis de energia, reciclagem e biodegradabilidade assumiram papéis
importantes como funções não estruturais no desenvolvimento de um material,
apresentando-se como incentivos à utilização adequada de subprodutos e resíduos
(GIBSON, 2010; AL-OQLA et al., 2015).
Ao longo dos últimos anos, pesquisas relacionadas à utilização de fibras naturais
no desenvolvimento de materiais multifuncionais tornaram-se atrativas mediante à gama
de aplicações, principalmente na produção de compósitos poliméricos (AL-OQLA et
al., 2015; PEREIRA et al., 2015; GURUNATHAN; MOHANTY; NAYAK, 2015;
YAN; KASAL; HUANG, 2016).
De uma maneira geral, as fibras naturais podem ser classificadas de acordo com
sua origem: vegetal, animal ou mineral. As fibras de origem animal são constituídas
basicamente por proteínas, tais como: cabelo, seda ou lã. Por outro lado, as fibras de
origem mineral são compostas essencialmente por silicatos como, por exemplo, o
amianto. E as fibras de base vegetal ou lignocelulósicas apresentam a celulose como um
de seus componentes principais (JOHN; THOMAS, 2008; JAWAID; KHALIL, 2011;
KHALIL; BHAT; YUSRA, 2012; GURUNATHAN; MOHANTY; NAYAK, 2015). A
Figura 2.1 apresenta um diagrama com a classificação para as diversas fibras e alguns
exemplos.

4
Figura 2.1 - Classificação para as fibras naturais e sintéticas.
Fonte: Adaptada de Gurunathan, Mohanty e Nayak (2015).
A utilização de fibras naturais em compósitos tem sido extensivamente explorada
devido à facilidade de seu processamento, anisotropia e ao aprimoramento das
propriedades térmicas e mecânicas do material final (TERZOPOULOU et al., 2015).
Além disso, proporciona uma diminuição nos problemas relacionados tanto à poluição
ambiental quanto à eliminação de resíduos (AL-OQLA et al., 2015). Devido à vasta
variedade de fibras naturais, inúmeras combinações para a produção de compósitos,
cada qual com propriedades intrínsecas, tornam este nicho científico cada vez mais
atrativo.
Kim; Lin; Bhattacharyya (2015) investigaram o efeito das fibras de lã e do
polifosfato de amônio (APP), como retardantes de chama, em compósitos de
polipropileno (PP). Neste trabalho, os autores avaliaram tanto o comportamento térmico
quanto mecânico dos compósitos PP-fibras curta de lã. Os compósitos foram preparados
por fusão, seguida pela moldagem por compressão, em diferentes formulações, as quais
foram submetidas a testes de flamabilidade (calorimetria e queima vertical). Baseado
FIBRA
NATURAL SINTÉTICA
MINERAL ANIMAL ORGÂNICA INORGÂNICA
CELULÓSICA/LIGNOCELULÓSICA
Seda
Cabelo
Lã
Amianto Aramida/Kevlar
Poliéster aromático
Polietileno
Vidro
Carbono
Boro
Carbeto de silício

5
nos resultados dos testes, um aumento significativo na propriedade de retardância à
chama foi evidenciado no compósito PP-lã com 30 % m/m de lã, juntamente com 20 %
m/m de APP. Nestes testes foram observados um decréscimo significativo na taxa de
libertação de calor e uma auto-extinção de chama nos compósitos. Além disso, a adição
de APP nos materiais influenciou positivamente as propriedades mecânicas, devido ao
aumento dos módulos de tração dos compósitos PP-lã-APP, apesar da diminuição na
resistência à tração. Segundo os autores, os resultados apresentaram-se satisfatórios,
pois tratou-se de um estudo investigativo.
Em outro trabalho, a síntese e a degradação de compósitos contendo fibra de seda
em uma matriz de poli(3-hidroxibutirato-co-3-hidroxivalerato) (PHBV) foram avaliados
por Miroiu et al. (2015). As fibras de seda e o PHBV são biopolímeros naturais com
excelente biocompatibilidade, porém com diferentes biodegradabilidades, foram
escolhidos a fim de melhorar as propriedades como materiais de revestimentos, visando
aplicações biomédicas. Os autores evidenciam que todos os resultados provaram que o
aumento do teor de PHBV confere uma diminuição na hidrofilicidade, uma degradação
mais lenta e um comportamento mais estável dos revestimentos poliméricos. Tais
resultados forneceram informações de suporte para diversas aplicações biomédicas,
incluindo a liberação controlada de drogas.
2.2. FIBRAS LIGNOCELULÓSICAS
A produção de resíduos/subprodutos agroindustriais constitui uma fonte
abundante de fibras lignocelulósicas, cujo principal componente químico é a celulose.
Considerando a abundância dos resíduos produzidos, tais subprodutos são de suma
importância para aplicações de maior valor agregado, principalmente como reforço em
compósitos poliméricos (SILVA et al., 2009; ALBINANTE; PACHECO; VISCONTE,
2013).
As fibras lignocelulósicas podem ser subdividas em outras categorias, em relação
à parte da planta da qual é extraída, tais como: caule, folha, sementes, fruto, madeira,
espiga, dentre outras (YAN; KASAL; HUANG, 2016; ONUAGULUCHI; BANTHIA,
2016). A Figura 2.2 apresenta esta classificação e alguns exemplos de fibras vegetais.

6
Figura 2.2 - Classificação geral para as fibras lignocelulósicas.
Fonte: Adaptada de Gurunathan, Mohanty e Nayak (2015).
As fibras vegetais podem ser definidas como um material compósito constituído
por um arranjo de microfibrilas de celulose, envoltas em uma matriz amorfa de
hemicelulose e lignina, unidas por meio de ligações de hidrogênio, com a formação de
uma rede tridimensional (JOHN; THOMAS, 2008; MERLINI; SOLDI; BARRA, 2011;
KABIR et al., 2013). Esta matriz amorfa tem por finalidade agir como barreira natural à
degradação microbiana, atuando como uma proteção mecânica (SILVA et al., 2009).
A parte mais importante de uma fibra vegetal é a parede celular, que consiste em
uma estrutura complexa, formada por um tubo oco, contendo diversas camadas
distintas. Cada camada é composta por celulose, incorporada em uma matriz de lignina
e hemicelulose. Na estrutura de cada fibra (Figura 2.3) podem ser identificadas uma
cavidade central (lúmen), encarregada pelo transporte de água e nutrientes e pela parede
celular; a lamela média e as paredes primária e secundária. A parede secundária é
constituída por três camadas: parede secundária externa (S1), parede secundária
intermediária (S2) e parede secundária interna (S3). Todas as camadas apresentam
microfibrilas de celulose, em diferentes orientações, de acordo com cada camada (DE
CELULÓSICA/LIGNOCELULÓSICA
CAULE FOLHA SEMENTES CAPIM/CANA MADEIRA FRUTO ESPIGA
Linho
Cânhamo
Juta
Rami
Kenaf
Sisal
Banana
Abacá
Agave
Ráfia
Algodão
Bucha
Coco Conífera
Curauá
“De lei”
Arroz
Aveia
Centeio
Cevada
Milho
Trigo
Bagaço
Bambu
Esparto

7
SIQUEIRA; FILHO, 2010; AKIL et al., 2011; THAKUR; THAKUR, 2014; PEREIRA
et al., 2015).
Figura 2.3 - Representação esquemática da estrutura de uma fibra vegetal.
Fonte: Adaptada de Thakur e Thakur (2014).
A parede primária, formada durante o crescimento das células, consiste em um
arranjo desordenado de microfibrilas de celulose, localizadas em uma matriz de pectina,
hemicelulose, lignina e proteína. Apresenta funções como suporte estrutural e mecânico,
além de proteger contra patógenos e desidratação da fibra. E as paredes secundárias são
formadas por microfibrilas de celulose cristalina organizadas em um arranjo espiral, no
qual a camada intermediária (S2) é responsável pelas propriedades mecânicas da fibra.
Nesta camada há uma série de microfibrilas, compostas por longas cadeias de celulose,
dispostas helicoidalmente, organizadas no sentido da fibra. Tais microfibrilas, dispostas
em uma região formada por lignina e hemicelulose, apresentam um diâmetro de 10 a 30
nm e são resultantes do empacotamento de 30 a 100 cadeias de celulose estendidas. Por
fim, a lamela média, camada exterior da célula, é constituída principalmente por
pectina, atuando como cimento entre as fibras. Parâmetros microestruturais, tais como
Lúmen Parede
secundária S3
Parede secundária S2
Parede secundária S1
Parede primária
Lamela média
Matriz amorfa composta por hemicelulose e
lignina
Rede de microfibrilas de celulose

8
dimensões, distribuição, orientação, empacotamento, defeitos, tensão, cristalinidade e
estrutura são variáveis importantes que determinam as propriedades das fibras.
Consequentemente, tais parâmetros contribuem diretamente nas propriedades finais dos
compósitos e tornam-se relevantes na escolha da fonte vegetal, com a finalidade de
obter propriedades específicas (JOHN; THOMAS, 2008; PAUL et al., 2008; SILVA et
al., 2009; DE SIQUEIRA; FILHO, 2010; KHALIL; BHAT; YUSRA, 2012; MENON;
RAO, 2012; PEREIRA et al., 2015).
A demasiada exploração de recursos não renováveis, principalmente de fontes
petroquímicas, assim como a má gestão dos resíduos agroindustriais impactam
diretamente em desequilíbrios ambientais e ecológicos. Tais fatores estimulam o
interesse das comunidade científica e industrial para a utilização de fibras vegetais no
desenvolvimento de materiais com potencial tecnológico, principalmente em
compósitos poliméricos (THAKUR; THAKUR, 2014; PAPPU et al., 2015).
Características como biodegradabilidade, disponibilidade, baixo custo e
propriedades mecânicas satisfatórias são um dos atrativos para a utilização de fibras
vegetais como substitutas às fibras sintéticas (YAN; KASAL; HUANG, 2016). Fibras
sintéticas como náilon, rayon, vidro, carbono e aramida predominam no mercado
industrial, principalmente nos setores de construção civil, automotivo, aeroespacial e
esportivo. Entretanto, as fibras de vidro, por exemplo, apresentam alta massa específica
(2,5 g.cm-3) e alto custo, além de riscos por inalação, difícil processamento e
reciclagem, alto consumo de energia para obtenção e não são renováveis (PAPPU et al.,
2015; TERZOPOULOU et al., 2015; GURUNATHAN; MOHANTY; NAYAK, 2015).
As fibras vegetais, por sua vez, apresentam diversas vantagens: provém de fontes
renováveis, são atóxicas, não abrasivas, recicláveis, possuem baixo consumo de energia
na produção, apresentam propriedades mecânicas específicas (resistência e módulo de
elasticidade) comparáveis às fibras sintéticas (p.ex., fibra de vidro) e estimulam
empregos na zona rural (NETO; PARDINI, 2006; PAUL et al., 2008; ROSÁRIO et al.,
2011; ALBINANTE; PACHECO; VISCONTE, 2013; FIORE; DI BELLA; VALENZA,
2015; YAN; KASAL; HUANG, 2016). Ademais, quando queimadas, as fibras
apresentam pouco resíduo, emitindo menos dióxido de carbono (CO2) para a atmosfera,
comparado ao removido durante o crescimentos da respectiva planta (JAWAID;

9
KHALIL, 2011). Devido à baixa massa específica, podem satisfazer as exigências do
mercado global, especialmente para as indústrias automotiva e aeroespacial (PAUL et
al., 2008; DITTENBER; GANGARAO, 2012; THAKUR; THAKUR, 2014).
Contudo, as fibras lignocelulósicas exibem algumas limitações, as quais
influenciam diretamente no processamento e nas características finais do compósito.
Dentre estas desvantagens pode-se citar: a natureza hidrofílica, caráter oposto à maioria
das matrizes poliméricas mais utilizadas, diminuindo a adesão na interface fibra/matriz,
a qual conduz à uma dispersão não uniforme do elemento de reforço. Outro contraponto
é a temperatura de processamento, restrita a aproximadamente 200 °C, o que limita a
escolha da matriz polimérica. A absorção de umidade das fibras vegetais também
apresenta-se como fator adverso, pois conduz a formação de vazios na interface,
reduzindo as propriedades mecânicas e estabilidade dimensional dos compósitos.
Apesar das características desfavoráveis, as vantagens permanecem predominantes, uma
vez que a maioria destas limitações pode ser alterada com o emprego de tratamentos
químicos (NETO; PARDINI, 2006; JOHN; THOMAS, 2008; PICKERING; ARUAN
EFENDY; LE, 2016).
Diversas fibras vegetais são utilizadas como reforço em diferentes matrizes
poliméricas, tais como bagaço de cana de açúcar, curauá, linho, cânhamo, juta, sisal,
algodão, palha de arroz, bambu, folha do abacaxi, kenaf, abacá, banana, dentre outras.
2.2.1. Composição química das fibras lignocelulósicas
As fibras vegetais podem ser definidas como materiais compósitos, constituídos
por uma variedade de componentes como celulose, hemicelulose, lignina, pectinas,
ceras e extrativos (gorduras, proteínas e sais inorgânicos), os quais afetam diretamente
suas propriedades e, consequentemente, as propriedades do respectivo compósito.
Fatores como espécie, idade e parte da planta, produção, local da colheita, condições do
solo, intempéries etc interferem diretamente nesta composição (KABIR et al., 2012b;
ALBINANTE; PACHECO; VISCONTE, 2013; PEREIRA et al., 2015; PAPPU et al.,
2015; ONUAGULUCHI; BANTHIA, 2016; YAN; KASAL; HUANG, 2016).

10
a) Pectinas, ceras e extrativos
A pectina é o principal componente da lamela média, constituída por
polissacarídeos ricos em ácido galacturônico (JOHN; THOMAS, 2008; KABIR et al.,
2012b). Confere flexibilidade às fibras vegetais e atua como um “cimento”,
promovendo a adesão entre as fibrilas (YAN; KASAL; HUANG, 2016). Ainda que sua
estrutura exata não seja conhecida, a literatura descreve que seus principais
componentes, polímeros lineares do ácido galacturônico, encontram-se ligados
covalentemente na parede celular primária e na lamela média, formando uma rede
complexa de pectina (TERZOPOULOU et al., 2015).
A maioria das ceras encontrada nas fibras vegetais são misturas de longas cadeias
de hidrocarbonetos alifáticos substituídos. Também podem ser identificados ácidos
graxos, fenólico e esteárico, álcoois primários e secundários, cetonas, aldeídos, dentre
outros. Em geral, situam-se na parte exterior das plantas, impactando na molhabilidade
e adesão das fibras (JOHN; THOMAS, 2008; GURUNATHAN; MOHANTY;
NAYAK, 2015; YAN; KASAL; HUANG, 2016).
Além das pectinas e ceras, pequenas quantidades de extrativos (orgânicos) e
cinzas (inorgânicos) estão presentes na estrutura das fibras vegetais. Estes extrativos
orgânicos são responsáveis pela cor, odor e resistência à deterioração, enquanto os
componentes inorgânicos podem conferir abrasividade às fibras (KABIR et al., 2012b).
b) Lignina
A lignina, posteriormente à celulose, é o componente mais abundante dentre as
biomassas lignocelulósicas. É classificada como um polifenol, de estrutura muito
complexa, constituída por um arranjo irregular de diversas unidades de fenilpropano, as
quais contêm grupos hidroxila e metoxila como substituintes no grupo fenil. Este
arranjo resulta em uma macromolécula tridimensional, amorfa e altamente reticulada
(SILVA et al., 2009; SANTOS et al., 2012; THAKUR; THAKUR, 2014; YAN;
KASAL; HUANG, 2016). Sua composição apresenta, como principais precursores, três
unidades de fenóis substituídos: os álcoois trans-p-cumarílico, trans-coferílico e trans-
sinapílico. Estas unidades dão origem a um vasto número de ligações e grupos
funcionais devido ao acoplamento entre as mesmas, predominantemente por ligações

11
éteres entre as unidades da lignina (SILVA et al., 2009; WATKINS et al., 2015;
PEREIRA et al., 2015). As Figuras 2.4 e 2.5 apresentam a estrutura da lignina e suas
principais unidades, respectivamente.
Figura 2.4 - Estrutura da lignina.
Fonte: Adaptada de Watkins et al. (2015).
j m a t e r r e s t e c h n o l . 2 0 1 5;4(1):26–32 27
CH2OH
H HH
H
H H
HH
H OH H OH
OH OHC
C
C C
C C C
C
C C
O O HH
H
H
H OH
OHC C
C
C C
O
O O O
CH2OH CH 2OHA
H2COH
H2COH
H2COH
H2COH
CH2OH
CH2
H2COH
H2COH
HCOH
OCH
CHO
CHO
H2C
CH2
H2COH
H2COH
H2COH
H2COHH2COH
CH2OH
H2COH
H2COH
H2COH
H2COH
Lignin
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
OCH3
OCH3
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
HOCH2
HOCH2
HOCH2
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH2
OCH3
OCH3
OCH3
CH
HC
HC
HC
HC
HC
HC HC
HC
HCHC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CHOCH
CH
OH
CHCH
CH
OH
CH
CH
CH COCO
CO
CH
CHCO
CHO
CH
OH
CO
O
O
O
O
O
O
O
O
OC
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
B
CH2OH
OH
Coniferylalcohol
Sinapylalcohol
P-coumarylalcohol
OH
OMe OMe
OMe = OCH3methoxyl group
MeO
OH
CH2OH CH 2OHC
Fig. 1 – (a) Schematic representation of the cellulose structure [1] (b) lignin structure [2], and (c) structural units of lignin [2].

12
Figura 2.5 - Principais unidades de fenóis substituídos presentes na lignina: (a) trans-p-cumarílico, (b) trans-coferílico e (c) trans-sinapílico.
Fonte: Adaptada de Gurunathan, Mohanty e Nayak (2015).
Situada na parede celular, este componente amorfo e insolúvel em água atua como
uma “cola” entre as fibrilas de celulose, proporcionando a adesão entre estas fibrilas
devido à presença de ligações covalentes entre as cadeias de lignina e
celulose/hemicelulose (SILVA et al., 2009; THAKUR; THAKUR, 2014; WATKINS et
al., 2015). Por conferir rigidez às fibras vegetais, sua ausência impediria seu alcance em
grandes alturas, afetando diretamente na sustentação das plantas (YAN; KASAL;
HUANG, 2016).
c) Hemicelulose
A hemicelulose é classificada como um heteropolissacarídeo e consiste em uma
combinação de vários monossacarídeos polimerizados, de baixa massa molar, incluindo
carboidratos de cinco carbonos (como xilose e arabinose), carboidratos de seis carbonos
(como galactose, glucose e manose), ácido 4-o-metil glucurônico e resíduos de ácido
galactorônico (SILVA et al., 2009; SANTOS et al., 2012; THAKUR; THAKUR, 2014).
Apresenta uma estrutura totalmente amorfa, contendo grupos hidroxila e acetila,
fortemente ligada às fibrilas de celulose por meio de ligações de hidrogênio (YAN;
KASAL; HUANG, 2016). A Figura 2.6 exibe a estrutura parcial da hemicelulose.
j m a t e r r e s t e c h n o l . 2 0 1 5;4(1):26–32 27
CH2OH
H HH
H
H H
HH
H OH H OH
OH OHC
C
C C
C C C
C
C C
O O HH
H
H
H OH
OHC C
C
C C
O
O O O
CH2OH CH 2OHA
H2COH
H2COH
H2COH
H2COH
CH2OH
CH2
H2COH
H2COH
HCOH
OCH
CHO
CHO
H2C
CH2
H2COH
H2COH
H2COH
H2COHH2COH
CH2OH
H2COH
H2COH
H2COH
H2COH
Lignin
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
OCH3
OCH3
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
HOCH2
HOCH2
HOCH2
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH2
OCH3
OCH3
OCH3
CH
HC
HC
HC
HC
HC
HC HC
HC
HCHC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CHOCH
CH
OH
CHCH
CH
OH
CH
CH
CH COCO
CO
CH
CHCO
CHO
CH
OH
CO
O
O
O
O
O
O
O
O
OC
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
B
CH2OH
OH
Coniferylalcohol
Sinapylalcohol
P-coumarylalcohol
OH
OMe OMe
OMe = OCH3methoxyl group
MeO
OH
CH2OH CH 2OHC
Fig. 1 – (a) Schematic representation of the cellulose structure [1] (b) lignin structure [2], and (c) structural units of lignin [2].
OH
CH CHCH2OH
(a)
OCH3
OH
CH CHCH2OH
(b)
j m a t e r r e s t e c h n o l . 2 0 1 5;4(1):26–32 27
CH2OH
H HH
H
H H
HH
H OH H OH
OH OHC
C
C C
C C C
C
C C
O O HH
H
H
H OH
OHC C
C
C C
O
O O O
CH2OH CH 2OHA
H2COH
H2COH
H2COH
H2COH
CH2OH
CH2
H2COH
H2COH
HCOH
OCH
CHO
CHO
H2C
CH2
H2COH
H2COH
H2COH
H2COHH2COH
CH2OH
H2COH
H2COH
H2COH
H2COH
Lignin
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
OCH3
OCH3
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
HOCH2
HOCH2
HOCH2
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH2
OCH3
OCH3
OCH3
CH
HC
HC
HC
HC
HC
HC HC
HC
HCHC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CHOCH
CH
OH
CHCH
CH
OH
CH
CH
CH COCO
CO
CH
CHCO
CHO
CH
OH
CO
O
O
O
O
O
O
O
O
OC
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
B
CH2OH
OH
Coniferylalcohol
Sinapylalcohol
P-coumarylalcohol
OH
OMe OMe
OMe = OCH3methoxyl group
MeO
OH
CH2OH CH 2OHC
Fig. 1 – (a) Schematic representation of the cellulose structure [1] (b) lignin structure [2], and (c) structural units of lignin [2].
j m a t e r r e s t e c h n o l . 2 0 1 5;4(1):26–32 27
CH2OH
H HH
H
H H
HH
H OH H OH
OH OHC
C
C C
C C C
C
C C
O O HH
H
H
H OH
OHC C
C
C C
O
O O O
CH2OH CH 2OHA
H2COH
H2COH
H2COH
H2COH
CH2OH
CH2
H2COH
H2COH
HCOH
OCH
CHO
CHO
H2C
CH2
H2COH
H2COH
H2COH
H2COHH2COH
CH2OH
H2COH
H2COH
H2COH
H2COH
Lignin
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
OCH3
OCH3
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
HOCH2
HOCH2
HOCH2
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH2
OCH3
OCH3
OCH3
CH
HC
HC
HC
HC
HC
HC HC
HC
HCHC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CHOCH
CH
OH
CHCH
CH
OH
CH
CH
CH COCO
CO
CH
CHCO
CHO
CH
OH
CO
O
O
O
O
O
O
O
O
OC
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
B
CH2OH
OH
Coniferylalcohol
Sinapylalcohol
P-coumarylalcohol
OH
OMe OMe
OMe = OCH3methoxyl group
MeO
OH
CH2OH CH 2OHC
Fig. 1 – (a) Schematic representation of the cellulose structure [1] (b) lignin structure [2], and (c) structural units of lignin [2].
OCH3
OH
CH CHCH2OH
(c)
H3CO

13
Figura 2.6 - Estrutura parcial da hemicelulose.
Fonte: Adaptada de Ferreira, Rocha e Da Silva (2009).
Localizada em todas as camadas da parede celular, a hemicelulose concentra-se
nas paredes primárias e secundárias, em associação com celulose e lignina (KABIR et
al., 2012b; PEREIRA et al., 2015). A partir desta associação, as propriedades estruturais
na parede celular são definidas, desempenhando funções na regulação do crescimento e
desenvolvimento das plantas (FERREIRA; ROCHA; DA SILVA, 2009). A Tabela 2.1
aborda as principais diferenças entre hemicelulose e celulose, uma vez que ambas são
classificadas como polissacarídeos e exibem estruturas similares.
Tabela 2.1 - Principais diferenças entre hemicelulose e celulose. CARACTERÍSTICAS HEMICELULOSE CELULOSE
Composição química
Unidades de pentoses e hexoses
Unidades de glicose
Grau de polimerização
Baixo
Alto
Formação de arranjo fibroso
Não
Sim (microfibrilas)
Cristalinidade Amorfo Semicristalino Reações com ácidos inorgânicos diluídos
Rápida
Lenta
Solubilidade em álcalis Solúvel Insolúvel Fonte: Adaptada de Pereira Jr, Couto e Santa Anna (2008).
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
CH3
C H3C O C H3C
O

14
d) Celulose
A celulose (Figura 2.7) é o polímero natural mais abundante na Terra e o principal
componente dos organismos vegetais, podendo ser sintetizada por bactérias, algas e
animais como os tunicados (KHALIL; BHAT; YUSRA, 2012).
Figura 2.7 - Estrutura da celulose.
Fonte: Adaptada de Ferreira, Rocha e Da Silva (2009).
Este biopolímero é definido como um homopolissacarídeo linear, composto por
unidades de D-anidroglicopiranose, etereficadas por ligações β-1,4-glicosídicas, nos
carbonos 1 e 4 (JOHN; THOMAS, 2008; KHALIL; BHAT; YUSRA, 2012; SANTOS
et al., 2012). A junção de n unidades de D-glicopiranose, seguida pela eliminação de
uma molécula de água, dá origem à macromolécula de celulose. A unidade de repetição
deste polímero é denominada de celobiose, que nada mais é que um dímero de glicose
(GURUNATHAN; MOHANTY; NAYAK, 2015).
A presença de grupos OH livres na molécula de celulose possibilita a formação de
ligações de hidrogênio, bem como forças de Van der Waals. Em cada unidade de
repetição há seis grupos hidroxila, os quais estabelecem interações do tipo ligações de
hidrogênio intra e intermoleculares (OLIVEIRA, 2007; SILVA et al., 2009;
GURUNATHAN; MOHANTY; NAYAK, 2015). A estrutura da celulose também
apresenta ligações acetal, as quais são responsáveis pela formação dos anéis de piranose
(FERREIRA; ROCHA; DA SILVA, 2009).
As ligações de hidrogênio intramoleculares ocorrem entre os grupos OH da
mesma molécula de celulose (Figura 2.8a), conferindo rigidez à cadeia. As ligações
intermoleculares (Figura 2.8b), entre grupos OH de cadeias adjacentes, são responsáveis
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
OH
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
Ferreira et al.624 Quim. Nova
Independente do preço e da disponibilidade do petróleo e do gás natural, a queima de combustíveis fósseis é um fator que não pode (não deve) ser negligenciado por nenhum país, pois seu efeito no aquecimento global não é restrito. Neste cenário, é que as biomassas de fontes renováveis aparecem como alternativas economicamente atrativas para fixação de CO2, produção de biocombustíveis e insumos básicos para suprir as indústrias químicas.8
Além da questão econômica, a transição dos processos de pre-paração de insumos químicos a partir do petróleo para um sistema baseado em biomassas terá que transpor outros obstáculos, sendo o mais importante as diferenças químicas dos dois tipos de matérias-primas. As biomassas são compostas de substâncias mais comple-xas (carboidratos, aminoácidos, proteínas, lipídios e biopolímeros (polissacarídeos)9 como celulose, hemiceluloses, quitina, amido e lignina) que variam consideravelmente de massas moleculares (Esquema 2).
CARBOIDRATOS
Os carboidratos compõem 75% da biomassa da Terra represen-tando a maior fonte renovável do planeta. Anualmente são converti-dos mais de 100 bilhões de m3 de CO2 e H2O em 200 mil toneladas de carboidratos pelas plantas superiores e algas.10-12 Nesta classe, destacam-se a celulose, a hemicelulose, o amido e a sacarose, dentre outros carboidratos de massas moleculares menores. Estes carboi-dratos já são utilizados industrialmente em larga escala para diversos fins, mas alguns são utilizados principalmente para fins alimentares, tendo poucas aplicações nas indústrias químicas.13,14
Por serem materiais renováveis e de baixo custo, esses carboi-dratos deverão ser no futuro as fontes economicamente mais viáveis para substituir os atuais derivados petroquímicos. Em 1995 publi-camos, nesta mesma revista, um artigo relatando a visão da época sobre o uso de carboidratos abundantes e suas aplicabilidades em síntese orgânica.15 Esta nova revisão pretende abordar, além do uso de carboidratos de baixas massas moleculares, as questões relativas às biomassas renováveis oriundas da fotossíntese e suas alternativas para a produção de intermediários e produtos da química fina.
Nestes últimos anos, muitas novas reações e materiais foram criados visando a produção de novos intermediários passíveis de uso pelos setores químicos da indústria, com a perspectiva que estes deveriam ser economicamente viáveis, para substituir aqueles derivados das fontes petroquímicas. Esta mudança baseia-se no fato das matérias-primas fósseis serem irrevogavelmente finitas, além da pressão gerada sobre o nosso meio ambiente com o aumento do CO2 oriundo da queima crescente de combustíveis fósseis. Com esta linha
de atuação, as indústrias químicas vêem despertando seus interesses para fontes de matérias-primas limpas e renováveis, que possam manter o equilíbrio de CO2 no planeta.16
Os carboidratos existem difundidos em todos os seres vivos e são essenciais para a vida.17 Alguns existem praticamente puros, tais como a sacarose, o amido e a celulose, e este último no algodão, na madeira e no papel. Eles são os constituintes estruturais de tecidos de plantas (celulose), de alguns animais (quitina em crustáceos e insetos) e paredes de celulares de bactérias. Como novos açúcares biotransformados fazem parte das membranas celulares e do DNA e RNA, que carregam importantes informações genéticas nas células, glicoconjugados com uma variedade de produtos naturais, tais como antibióticos, glicolipídios, e glicoproteínas, sendo este dois últimos responsáveis por reter informações de reconhecimento supramole-cular que formam a base da glicobiologia.
Apesar do papel principal dos carboidratos relacionado com suas diversas funções na bioquímica dos seres vivos, esta classe de substância tem sido largamente estudada pelos químicos orgânicos nos seus diversos aspectos estereoquímicos, mecanísticos, sintéticos e analíticos. A biomassa renovável de carboidratos é constituída prin-cipalmente de polissacarídeos como a celulose, o amido, a inulina e a hemicelulose que, por sua vez, são constituídos de unidades de menores massas moleculares. Para estes carboidratos utiliza-se o termo genérico monossacarídeo, indicando uma única unidade de açúcar sem conexões glicosídicas com outras unidades.
Os polissacarídeos podem ser utilizados in natura na alimenta-ção, produtos têxteis, papel, madeira para construção, revestimentos industriais, cosméticos etc. Também podem sofrer modificações nas cadeias poliméricas de modo a adaptá-los para usos específicos, por exemplo, o rayon e a quitosana, obtidos respectivamente da celulose e a quitina, que mantêm as estruturas gerais dos polissacarídeos correspondentes. Em outro extremo têm-se as substâncias orgânicas classificadas como comodities e intermediários da química fina que têm baixas massas moleculares. É bem recente o interesse da indústria química nas fontes de matérias-primas renováveis,18 cujo desafio é transformar essa biomassa em carboidratos menores como dissaca-rídeos (celobiose) ou monossacarídeos (glicose, frutose, xilose) e este por sua vez nestas substâncias com maior valor agregado.19 Os carboidratos de massas moleculares mais baixas apresentam maiores vantagens que os polissacarídeos para a preparação de insumos e intermediários da química fina, pois são de baixo custo e abundantes para usos em escala industrial (Esquema 3).
Celulose, amido, quitina e quitosana
Os carboidratos podem possuir outras funcionalidades além dos grupos hidroxilas. Por exemplo, os amino-açúcares são carboidratos que possuem um grupo hidroxila substituído por uma amina. Os mais conhecidos destes carboidratos são os polissacarídeos quitina e quitosana20,21 (Figura 4) e o monossacarídeo D-glicosamina (2-amino-2-desoxi-D-glicose). Uma excelente revisão sobre os derivados
O
OH
HOHO
OHO
OHO
HO OH
OH
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
O
OH
OHO
OHO O
OH
HO
OHO
O
OH
HO
OHO
O
OHHO
HO
OHOH
O
OH
OHO
NHAc
O O
OH
HONHAc
O O
OH
HONHAc
OO
OH
HOHO
NHAc
OH
1
2´1´
D-sacarose2-O-( -D-glicopiranosil)- -D-frutofuranose
Ligação glicosídica(1,2´)
D-Glicose
D-Frutose
Ligação glicosídica(1 4)
1 4
celulose
amilose
Ligação glicosídica(1,4´)
14´
)n
(
D-glicose
n
Ligação glicosídica(1,4´)
14´
quitina
n
(Um dos componentes do amido)
N-Acetil-D-glicosilamina
Esquema 2. As principais biomassas de renováveis baseadas em carboi-dratos
Polissacarídeos Carboidratos com baixas massas moleculares
Materiais poliméricos especiais
Insumos químicos eintermediários da química fina
Esquema 3. Resumo esquemático das abordagens para os usos de polissa-carídeos
Ferreira et al.624 Quim. Nova
Independente do preço e da disponibilidade do petróleo e do gás natural, a queima de combustíveis fósseis é um fator que não pode (não deve) ser negligenciado por nenhum país, pois seu efeito no aquecimento global não é restrito. Neste cenário, é que as biomassas de fontes renováveis aparecem como alternativas economicamente atrativas para fixação de CO2, produção de biocombustíveis e insumos básicos para suprir as indústrias químicas.8
Além da questão econômica, a transição dos processos de pre-paração de insumos químicos a partir do petróleo para um sistema baseado em biomassas terá que transpor outros obstáculos, sendo o mais importante as diferenças químicas dos dois tipos de matérias-primas. As biomassas são compostas de substâncias mais comple-xas (carboidratos, aminoácidos, proteínas, lipídios e biopolímeros (polissacarídeos)9 como celulose, hemiceluloses, quitina, amido e lignina) que variam consideravelmente de massas moleculares (Esquema 2).
CARBOIDRATOS
Os carboidratos compõem 75% da biomassa da Terra represen-tando a maior fonte renovável do planeta. Anualmente são converti-dos mais de 100 bilhões de m3 de CO2 e H2O em 200 mil toneladas de carboidratos pelas plantas superiores e algas.10-12 Nesta classe, destacam-se a celulose, a hemicelulose, o amido e a sacarose, dentre outros carboidratos de massas moleculares menores. Estes carboi-dratos já são utilizados industrialmente em larga escala para diversos fins, mas alguns são utilizados principalmente para fins alimentares, tendo poucas aplicações nas indústrias químicas.13,14
Por serem materiais renováveis e de baixo custo, esses carboi-dratos deverão ser no futuro as fontes economicamente mais viáveis para substituir os atuais derivados petroquímicos. Em 1995 publi-camos, nesta mesma revista, um artigo relatando a visão da época sobre o uso de carboidratos abundantes e suas aplicabilidades em síntese orgânica.15 Esta nova revisão pretende abordar, além do uso de carboidratos de baixas massas moleculares, as questões relativas às biomassas renováveis oriundas da fotossíntese e suas alternativas para a produção de intermediários e produtos da química fina.
Nestes últimos anos, muitas novas reações e materiais foram criados visando a produção de novos intermediários passíveis de uso pelos setores químicos da indústria, com a perspectiva que estes deveriam ser economicamente viáveis, para substituir aqueles derivados das fontes petroquímicas. Esta mudança baseia-se no fato das matérias-primas fósseis serem irrevogavelmente finitas, além da pressão gerada sobre o nosso meio ambiente com o aumento do CO2 oriundo da queima crescente de combustíveis fósseis. Com esta linha
de atuação, as indústrias químicas vêem despertando seus interesses para fontes de matérias-primas limpas e renováveis, que possam manter o equilíbrio de CO2 no planeta.16
Os carboidratos existem difundidos em todos os seres vivos e são essenciais para a vida.17 Alguns existem praticamente puros, tais como a sacarose, o amido e a celulose, e este último no algodão, na madeira e no papel. Eles são os constituintes estruturais de tecidos de plantas (celulose), de alguns animais (quitina em crustáceos e insetos) e paredes de celulares de bactérias. Como novos açúcares biotransformados fazem parte das membranas celulares e do DNA e RNA, que carregam importantes informações genéticas nas células, glicoconjugados com uma variedade de produtos naturais, tais como antibióticos, glicolipídios, e glicoproteínas, sendo este dois últimos responsáveis por reter informações de reconhecimento supramole-cular que formam a base da glicobiologia.
Apesar do papel principal dos carboidratos relacionado com suas diversas funções na bioquímica dos seres vivos, esta classe de substância tem sido largamente estudada pelos químicos orgânicos nos seus diversos aspectos estereoquímicos, mecanísticos, sintéticos e analíticos. A biomassa renovável de carboidratos é constituída prin-cipalmente de polissacarídeos como a celulose, o amido, a inulina e a hemicelulose que, por sua vez, são constituídos de unidades de menores massas moleculares. Para estes carboidratos utiliza-se o termo genérico monossacarídeo, indicando uma única unidade de açúcar sem conexões glicosídicas com outras unidades.
Os polissacarídeos podem ser utilizados in natura na alimenta-ção, produtos têxteis, papel, madeira para construção, revestimentos industriais, cosméticos etc. Também podem sofrer modificações nas cadeias poliméricas de modo a adaptá-los para usos específicos, por exemplo, o rayon e a quitosana, obtidos respectivamente da celulose e a quitina, que mantêm as estruturas gerais dos polissacarídeos correspondentes. Em outro extremo têm-se as substâncias orgânicas classificadas como comodities e intermediários da química fina que têm baixas massas moleculares. É bem recente o interesse da indústria química nas fontes de matérias-primas renováveis,18 cujo desafio é transformar essa biomassa em carboidratos menores como dissaca-rídeos (celobiose) ou monossacarídeos (glicose, frutose, xilose) e este por sua vez nestas substâncias com maior valor agregado.19 Os carboidratos de massas moleculares mais baixas apresentam maiores vantagens que os polissacarídeos para a preparação de insumos e intermediários da química fina, pois são de baixo custo e abundantes para usos em escala industrial (Esquema 3).
Celulose, amido, quitina e quitosana
Os carboidratos podem possuir outras funcionalidades além dos grupos hidroxilas. Por exemplo, os amino-açúcares são carboidratos que possuem um grupo hidroxila substituído por uma amina. Os mais conhecidos destes carboidratos são os polissacarídeos quitina e quitosana20,21 (Figura 4) e o monossacarídeo D-glicosamina (2-amino-2-desoxi-D-glicose). Uma excelente revisão sobre os derivados
O
OH
HOHO
OHO
OHO
HO OH
OH
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
O
OH
OHO
OHO O
OH
HO
OHO
O
OH
HO
OHO
O
OHHO
HO
OHOH
O
OH
OHO
NHAc
O O
OH
HONHAc
O O
OH
HONHAc
OO
OH
HOHO
NHAc
OH
1
2´1´
D-sacarose2-O-( -D-glicopiranosil)- -D-frutofuranose
Ligação glicosídica(1,2´)
D-Glicose
D-Frutose
Ligação glicosídica(1 4)
1 4
celulose
amilose
Ligação glicosídica(1,4´)
14´
)n
(
D-glicose
n
Ligação glicosídica(1,4´)
14´
quitina
n
(Um dos componentes do amido)
N-Acetil-D-glicosilamina
Esquema 2. As principais biomassas de renováveis baseadas em carboi-dratos
Polissacarídeos Carboidratos com baixas massas moleculares
Materiais poliméricos especiais
Insumos químicos eintermediários da química fina
Esquema 3. Resumo esquemático das abordagens para os usos de polissa-carídeos
n
Ligação β-1,4-glicosídica

15
pela formação da estrutura supramolecular (CUNHA, 2011; KABIR et al., 2012b;
TERZOPOULOU et al., 2015). E as interações de Van der Waals, também entre as
cadeias de celulose, permite a formação de regiões cristalinas, bem ordenadas. Devido à
estas interações, que favorecem a formação de estruturas cristalinas altamente
ordenadas, a celulose torna-se completamente insolúvel em água e na maioria dos
solventes orgânicos (SILVA et al., 2009; KHALIL; BHAT; YUSRA, 2012).
Figura 2.8 - Representação esquemática das interações (a) intramolecular e (b) intermolecular na celulose.
Fonte: Adaptada de Cunha (2011).
A partir das interações inter e intramoleculares, são formadas estruturas primárias
organizadas, denominadas fibrilas, originando assim a parede celular vegetal, na qual
estão dispostas as macro e as microfibrilas. A Figura 2.9 apresenta a estrutura
supramolecular da celulose onde ocorre esta formação.
Ferreira et al.624 Quim. Nova
Independente do preço e da disponibilidade do petróleo e do gás natural, a queima de combustíveis fósseis é um fator que não pode (não deve) ser negligenciado por nenhum país, pois seu efeito no aquecimento global não é restrito. Neste cenário, é que as biomassas de fontes renováveis aparecem como alternativas economicamente atrativas para fixação de CO2, produção de biocombustíveis e insumos básicos para suprir as indústrias químicas.8
Além da questão econômica, a transição dos processos de pre-paração de insumos químicos a partir do petróleo para um sistema baseado em biomassas terá que transpor outros obstáculos, sendo o mais importante as diferenças químicas dos dois tipos de matérias-primas. As biomassas são compostas de substâncias mais comple-xas (carboidratos, aminoácidos, proteínas, lipídios e biopolímeros (polissacarídeos)9 como celulose, hemiceluloses, quitina, amido e lignina) que variam consideravelmente de massas moleculares (Esquema 2).
CARBOIDRATOS
Os carboidratos compõem 75% da biomassa da Terra represen-tando a maior fonte renovável do planeta. Anualmente são converti-dos mais de 100 bilhões de m3 de CO2 e H2O em 200 mil toneladas de carboidratos pelas plantas superiores e algas.10-12 Nesta classe, destacam-se a celulose, a hemicelulose, o amido e a sacarose, dentre outros carboidratos de massas moleculares menores. Estes carboi-dratos já são utilizados industrialmente em larga escala para diversos fins, mas alguns são utilizados principalmente para fins alimentares, tendo poucas aplicações nas indústrias químicas.13,14
Por serem materiais renováveis e de baixo custo, esses carboi-dratos deverão ser no futuro as fontes economicamente mais viáveis para substituir os atuais derivados petroquímicos. Em 1995 publi-camos, nesta mesma revista, um artigo relatando a visão da época sobre o uso de carboidratos abundantes e suas aplicabilidades em síntese orgânica.15 Esta nova revisão pretende abordar, além do uso de carboidratos de baixas massas moleculares, as questões relativas às biomassas renováveis oriundas da fotossíntese e suas alternativas para a produção de intermediários e produtos da química fina.
Nestes últimos anos, muitas novas reações e materiais foram criados visando a produção de novos intermediários passíveis de uso pelos setores químicos da indústria, com a perspectiva que estes deveriam ser economicamente viáveis, para substituir aqueles derivados das fontes petroquímicas. Esta mudança baseia-se no fato das matérias-primas fósseis serem irrevogavelmente finitas, além da pressão gerada sobre o nosso meio ambiente com o aumento do CO2 oriundo da queima crescente de combustíveis fósseis. Com esta linha
de atuação, as indústrias químicas vêem despertando seus interesses para fontes de matérias-primas limpas e renováveis, que possam manter o equilíbrio de CO2 no planeta.16
Os carboidratos existem difundidos em todos os seres vivos e são essenciais para a vida.17 Alguns existem praticamente puros, tais como a sacarose, o amido e a celulose, e este último no algodão, na madeira e no papel. Eles são os constituintes estruturais de tecidos de plantas (celulose), de alguns animais (quitina em crustáceos e insetos) e paredes de celulares de bactérias. Como novos açúcares biotransformados fazem parte das membranas celulares e do DNA e RNA, que carregam importantes informações genéticas nas células, glicoconjugados com uma variedade de produtos naturais, tais como antibióticos, glicolipídios, e glicoproteínas, sendo este dois últimos responsáveis por reter informações de reconhecimento supramole-cular que formam a base da glicobiologia.
Apesar do papel principal dos carboidratos relacionado com suas diversas funções na bioquímica dos seres vivos, esta classe de substância tem sido largamente estudada pelos químicos orgânicos nos seus diversos aspectos estereoquímicos, mecanísticos, sintéticos e analíticos. A biomassa renovável de carboidratos é constituída prin-cipalmente de polissacarídeos como a celulose, o amido, a inulina e a hemicelulose que, por sua vez, são constituídos de unidades de menores massas moleculares. Para estes carboidratos utiliza-se o termo genérico monossacarídeo, indicando uma única unidade de açúcar sem conexões glicosídicas com outras unidades.
Os polissacarídeos podem ser utilizados in natura na alimenta-ção, produtos têxteis, papel, madeira para construção, revestimentos industriais, cosméticos etc. Também podem sofrer modificações nas cadeias poliméricas de modo a adaptá-los para usos específicos, por exemplo, o rayon e a quitosana, obtidos respectivamente da celulose e a quitina, que mantêm as estruturas gerais dos polissacarídeos correspondentes. Em outro extremo têm-se as substâncias orgânicas classificadas como comodities e intermediários da química fina que têm baixas massas moleculares. É bem recente o interesse da indústria química nas fontes de matérias-primas renováveis,18 cujo desafio é transformar essa biomassa em carboidratos menores como dissaca-rídeos (celobiose) ou monossacarídeos (glicose, frutose, xilose) e este por sua vez nestas substâncias com maior valor agregado.19 Os carboidratos de massas moleculares mais baixas apresentam maiores vantagens que os polissacarídeos para a preparação de insumos e intermediários da química fina, pois são de baixo custo e abundantes para usos em escala industrial (Esquema 3).
Celulose, amido, quitina e quitosana
Os carboidratos podem possuir outras funcionalidades além dos grupos hidroxilas. Por exemplo, os amino-açúcares são carboidratos que possuem um grupo hidroxila substituído por uma amina. Os mais conhecidos destes carboidratos são os polissacarídeos quitina e quitosana20,21 (Figura 4) e o monossacarídeo D-glicosamina (2-amino-2-desoxi-D-glicose). Uma excelente revisão sobre os derivados
O
OH
HOHO
OHO
OHO
HO OH
OH
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
O
OH
OHO
OHO O
OH
HO
OHO
O
OH
HO
OHO
O
OHHO
HO
OHOH
O
OH
OHO
NHAc
O O
OH
HONHAc
O O
OH
HONHAc
OO
OH
HOHO
NHAc
OH
1
2´1´
D-sacarose2-O-( -D-glicopiranosil)- -D-frutofuranose
Ligação glicosídica(1,2´)
D-Glicose
D-Frutose
Ligação glicosídica(1 4)
1 4
celulose
amilose
Ligação glicosídica(1,4´)
14´
)n
(
D-glicose
n
Ligação glicosídica(1,4´)
14´
quitina
n
(Um dos componentes do amido)
N-Acetil-D-glicosilamina
Esquema 2. As principais biomassas de renováveis baseadas em carboi-dratos
Polissacarídeos Carboidratos com baixas massas moleculares
Materiais poliméricos especiais
Insumos químicos eintermediários da química fina
Esquema 3. Resumo esquemático das abordagens para os usos de polissa-carídeos
H
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
H
H
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
(a)
(b)
Ferreira et al.624 Quim. Nova
Independente do preço e da disponibilidade do petróleo e do gás natural, a queima de combustíveis fósseis é um fator que não pode (não deve) ser negligenciado por nenhum país, pois seu efeito no aquecimento global não é restrito. Neste cenário, é que as biomassas de fontes renováveis aparecem como alternativas economicamente atrativas para fixação de CO2, produção de biocombustíveis e insumos básicos para suprir as indústrias químicas.8
Além da questão econômica, a transição dos processos de pre-paração de insumos químicos a partir do petróleo para um sistema baseado em biomassas terá que transpor outros obstáculos, sendo o mais importante as diferenças químicas dos dois tipos de matérias-primas. As biomassas são compostas de substâncias mais comple-xas (carboidratos, aminoácidos, proteínas, lipídios e biopolímeros (polissacarídeos)9 como celulose, hemiceluloses, quitina, amido e lignina) que variam consideravelmente de massas moleculares (Esquema 2).
CARBOIDRATOS
Os carboidratos compõem 75% da biomassa da Terra represen-tando a maior fonte renovável do planeta. Anualmente são converti-dos mais de 100 bilhões de m3 de CO2 e H2O em 200 mil toneladas de carboidratos pelas plantas superiores e algas.10-12 Nesta classe, destacam-se a celulose, a hemicelulose, o amido e a sacarose, dentre outros carboidratos de massas moleculares menores. Estes carboi-dratos já são utilizados industrialmente em larga escala para diversos fins, mas alguns são utilizados principalmente para fins alimentares, tendo poucas aplicações nas indústrias químicas.13,14
Por serem materiais renováveis e de baixo custo, esses carboi-dratos deverão ser no futuro as fontes economicamente mais viáveis para substituir os atuais derivados petroquímicos. Em 1995 publi-camos, nesta mesma revista, um artigo relatando a visão da época sobre o uso de carboidratos abundantes e suas aplicabilidades em síntese orgânica.15 Esta nova revisão pretende abordar, além do uso de carboidratos de baixas massas moleculares, as questões relativas às biomassas renováveis oriundas da fotossíntese e suas alternativas para a produção de intermediários e produtos da química fina.
Nestes últimos anos, muitas novas reações e materiais foram criados visando a produção de novos intermediários passíveis de uso pelos setores químicos da indústria, com a perspectiva que estes deveriam ser economicamente viáveis, para substituir aqueles derivados das fontes petroquímicas. Esta mudança baseia-se no fato das matérias-primas fósseis serem irrevogavelmente finitas, além da pressão gerada sobre o nosso meio ambiente com o aumento do CO2 oriundo da queima crescente de combustíveis fósseis. Com esta linha
de atuação, as indústrias químicas vêem despertando seus interesses para fontes de matérias-primas limpas e renováveis, que possam manter o equilíbrio de CO2 no planeta.16
Os carboidratos existem difundidos em todos os seres vivos e são essenciais para a vida.17 Alguns existem praticamente puros, tais como a sacarose, o amido e a celulose, e este último no algodão, na madeira e no papel. Eles são os constituintes estruturais de tecidos de plantas (celulose), de alguns animais (quitina em crustáceos e insetos) e paredes de celulares de bactérias. Como novos açúcares biotransformados fazem parte das membranas celulares e do DNA e RNA, que carregam importantes informações genéticas nas células, glicoconjugados com uma variedade de produtos naturais, tais como antibióticos, glicolipídios, e glicoproteínas, sendo este dois últimos responsáveis por reter informações de reconhecimento supramole-cular que formam a base da glicobiologia.
Apesar do papel principal dos carboidratos relacionado com suas diversas funções na bioquímica dos seres vivos, esta classe de substância tem sido largamente estudada pelos químicos orgânicos nos seus diversos aspectos estereoquímicos, mecanísticos, sintéticos e analíticos. A biomassa renovável de carboidratos é constituída prin-cipalmente de polissacarídeos como a celulose, o amido, a inulina e a hemicelulose que, por sua vez, são constituídos de unidades de menores massas moleculares. Para estes carboidratos utiliza-se o termo genérico monossacarídeo, indicando uma única unidade de açúcar sem conexões glicosídicas com outras unidades.
Os polissacarídeos podem ser utilizados in natura na alimenta-ção, produtos têxteis, papel, madeira para construção, revestimentos industriais, cosméticos etc. Também podem sofrer modificações nas cadeias poliméricas de modo a adaptá-los para usos específicos, por exemplo, o rayon e a quitosana, obtidos respectivamente da celulose e a quitina, que mantêm as estruturas gerais dos polissacarídeos correspondentes. Em outro extremo têm-se as substâncias orgânicas classificadas como comodities e intermediários da química fina que têm baixas massas moleculares. É bem recente o interesse da indústria química nas fontes de matérias-primas renováveis,18 cujo desafio é transformar essa biomassa em carboidratos menores como dissaca-rídeos (celobiose) ou monossacarídeos (glicose, frutose, xilose) e este por sua vez nestas substâncias com maior valor agregado.19 Os carboidratos de massas moleculares mais baixas apresentam maiores vantagens que os polissacarídeos para a preparação de insumos e intermediários da química fina, pois são de baixo custo e abundantes para usos em escala industrial (Esquema 3).
Celulose, amido, quitina e quitosana
Os carboidratos podem possuir outras funcionalidades além dos grupos hidroxilas. Por exemplo, os amino-açúcares são carboidratos que possuem um grupo hidroxila substituído por uma amina. Os mais conhecidos destes carboidratos são os polissacarídeos quitina e quitosana20,21 (Figura 4) e o monossacarídeo D-glicosamina (2-amino-2-desoxi-D-glicose). Uma excelente revisão sobre os derivados
O
OH
HOHO
OHO
OHO
HO OH
OH
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
O
OH
OHO
OHO O
OH
HO
OHO
O
OH
HO
OHO
O
OHHO
HO
OHOH
O
OH
OHO
NHAc
O O
OH
HONHAc
O O
OH
HONHAc
OO
OH
HOHO
NHAc
OH
1
2´1´
D-sacarose2-O-( -D-glicopiranosil)- -D-frutofuranose
Ligação glicosídica(1,2´)
D-Glicose
D-Frutose
Ligação glicosídica(1 4)
1 4
celulose
amilose
Ligação glicosídica(1,4´)
14´
)n
(
D-glicose
n
Ligação glicosídica(1,4´)
14´
quitina
n
(Um dos componentes do amido)
N-Acetil-D-glicosilamina
Esquema 2. As principais biomassas de renováveis baseadas em carboi-dratos
Polissacarídeos Carboidratos com baixas massas moleculares
Materiais poliméricos especiais
Insumos químicos eintermediários da química fina
Esquema 3. Resumo esquemático das abordagens para os usos de polissa-carídeos
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
H
H
Ferreira et al.624 Quim. Nova
Independente do preço e da disponibilidade do petróleo e do gás natural, a queima de combustíveis fósseis é um fator que não pode (não deve) ser negligenciado por nenhum país, pois seu efeito no aquecimento global não é restrito. Neste cenário, é que as biomassas de fontes renováveis aparecem como alternativas economicamente atrativas para fixação de CO2, produção de biocombustíveis e insumos básicos para suprir as indústrias químicas.8
Além da questão econômica, a transição dos processos de pre-paração de insumos químicos a partir do petróleo para um sistema baseado em biomassas terá que transpor outros obstáculos, sendo o mais importante as diferenças químicas dos dois tipos de matérias-primas. As biomassas são compostas de substâncias mais comple-xas (carboidratos, aminoácidos, proteínas, lipídios e biopolímeros (polissacarídeos)9 como celulose, hemiceluloses, quitina, amido e lignina) que variam consideravelmente de massas moleculares (Esquema 2).
CARBOIDRATOS
Os carboidratos compõem 75% da biomassa da Terra represen-tando a maior fonte renovável do planeta. Anualmente são converti-dos mais de 100 bilhões de m3 de CO2 e H2O em 200 mil toneladas de carboidratos pelas plantas superiores e algas.10-12 Nesta classe, destacam-se a celulose, a hemicelulose, o amido e a sacarose, dentre outros carboidratos de massas moleculares menores. Estes carboi-dratos já são utilizados industrialmente em larga escala para diversos fins, mas alguns são utilizados principalmente para fins alimentares, tendo poucas aplicações nas indústrias químicas.13,14
Por serem materiais renováveis e de baixo custo, esses carboi-dratos deverão ser no futuro as fontes economicamente mais viáveis para substituir os atuais derivados petroquímicos. Em 1995 publi-camos, nesta mesma revista, um artigo relatando a visão da época sobre o uso de carboidratos abundantes e suas aplicabilidades em síntese orgânica.15 Esta nova revisão pretende abordar, além do uso de carboidratos de baixas massas moleculares, as questões relativas às biomassas renováveis oriundas da fotossíntese e suas alternativas para a produção de intermediários e produtos da química fina.
Nestes últimos anos, muitas novas reações e materiais foram criados visando a produção de novos intermediários passíveis de uso pelos setores químicos da indústria, com a perspectiva que estes deveriam ser economicamente viáveis, para substituir aqueles derivados das fontes petroquímicas. Esta mudança baseia-se no fato das matérias-primas fósseis serem irrevogavelmente finitas, além da pressão gerada sobre o nosso meio ambiente com o aumento do CO2 oriundo da queima crescente de combustíveis fósseis. Com esta linha
de atuação, as indústrias químicas vêem despertando seus interesses para fontes de matérias-primas limpas e renováveis, que possam manter o equilíbrio de CO2 no planeta.16
Os carboidratos existem difundidos em todos os seres vivos e são essenciais para a vida.17 Alguns existem praticamente puros, tais como a sacarose, o amido e a celulose, e este último no algodão, na madeira e no papel. Eles são os constituintes estruturais de tecidos de plantas (celulose), de alguns animais (quitina em crustáceos e insetos) e paredes de celulares de bactérias. Como novos açúcares biotransformados fazem parte das membranas celulares e do DNA e RNA, que carregam importantes informações genéticas nas células, glicoconjugados com uma variedade de produtos naturais, tais como antibióticos, glicolipídios, e glicoproteínas, sendo este dois últimos responsáveis por reter informações de reconhecimento supramole-cular que formam a base da glicobiologia.
Apesar do papel principal dos carboidratos relacionado com suas diversas funções na bioquímica dos seres vivos, esta classe de substância tem sido largamente estudada pelos químicos orgânicos nos seus diversos aspectos estereoquímicos, mecanísticos, sintéticos e analíticos. A biomassa renovável de carboidratos é constituída prin-cipalmente de polissacarídeos como a celulose, o amido, a inulina e a hemicelulose que, por sua vez, são constituídos de unidades de menores massas moleculares. Para estes carboidratos utiliza-se o termo genérico monossacarídeo, indicando uma única unidade de açúcar sem conexões glicosídicas com outras unidades.
Os polissacarídeos podem ser utilizados in natura na alimenta-ção, produtos têxteis, papel, madeira para construção, revestimentos industriais, cosméticos etc. Também podem sofrer modificações nas cadeias poliméricas de modo a adaptá-los para usos específicos, por exemplo, o rayon e a quitosana, obtidos respectivamente da celulose e a quitina, que mantêm as estruturas gerais dos polissacarídeos correspondentes. Em outro extremo têm-se as substâncias orgânicas classificadas como comodities e intermediários da química fina que têm baixas massas moleculares. É bem recente o interesse da indústria química nas fontes de matérias-primas renováveis,18 cujo desafio é transformar essa biomassa em carboidratos menores como dissaca-rídeos (celobiose) ou monossacarídeos (glicose, frutose, xilose) e este por sua vez nestas substâncias com maior valor agregado.19 Os carboidratos de massas moleculares mais baixas apresentam maiores vantagens que os polissacarídeos para a preparação de insumos e intermediários da química fina, pois são de baixo custo e abundantes para usos em escala industrial (Esquema 3).
Celulose, amido, quitina e quitosana
Os carboidratos podem possuir outras funcionalidades além dos grupos hidroxilas. Por exemplo, os amino-açúcares são carboidratos que possuem um grupo hidroxila substituído por uma amina. Os mais conhecidos destes carboidratos são os polissacarídeos quitina e quitosana20,21 (Figura 4) e o monossacarídeo D-glicosamina (2-amino-2-desoxi-D-glicose). Uma excelente revisão sobre os derivados
O
OH
HOHO
OHO
OHO
HO OH
OH
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
O
OH
OHO
OHO O
OH
HO
OHO
O
OH
HO
OHO
O
OHHO
HO
OHOH
O
OH
OHO
NHAc
O O
OH
HONHAc
O O
OH
HONHAc
OO
OH
HOHO
NHAc
OH
1
2´1´
D-sacarose2-O-( -D-glicopiranosil)- -D-frutofuranose
Ligação glicosídica(1,2´)
D-Glicose
D-Frutose
Ligação glicosídica(1 4)
1 4
celulose
amilose
Ligação glicosídica(1,4´)
14´
)n
(
D-glicose
n
Ligação glicosídica(1,4´)
14´
quitina
n
(Um dos componentes do amido)
N-Acetil-D-glicosilamina
Esquema 2. As principais biomassas de renováveis baseadas em carboi-dratos
Polissacarídeos Carboidratos com baixas massas moleculares
Materiais poliméricos especiais
Insumos químicos eintermediários da química fina
Esquema 3. Resumo esquemático das abordagens para os usos de polissa-carídeos
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
H
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
H
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005

16
Figura 2.9 - Estrutura supramolecular da celulose para as fibras vegetais.
Fonte: Adaptada de Rojas, Bedoya e Ciro (2015).
Nas fibras lignocelulósicas, as macrofibrilas são descritas como um feixe de
microfibrilas, as quais apresentam-se ligadas umas à outras por hemicelulose, polímeros
amorfos provenientes de diferente açúcares e pectinas, envoltas por lignina. Já as
microfibrilas são o arranjo de diversas moléculas de celulose empacotadas, compostas
por n unidades de glicose (SILVA; D’ALMEIDA, 2009; TAIPINA, 2012; MENON;
RAO, 2012). Estas microfibrilas são o menor agregado de cadeias de celulose, formadas
por arranjos cristalinos separados por domínios amorfos (JOHN; THOMAS, 2008;
GURUNATHAN; MOHANTY; NAYAK, 2015). As dimensões das microfibrilas são
dependentes da fonte celulósica (ZHAO et al., 2007).
A variação na conformação ou no empacotamento das cadeias de celulose, devido
às interações de hidrogênio, proporciona a formação de diferentes polimorfos, que
podem ser interconvertidos por vários processos de tratamento. Sete formas cristalinas
foram identificadas para celulose, as quais são designadas como Iα, Iβ, II, IIII , IIIII, IVI
e IVII, com repetição das unidades monoméricas a cada 1,03 nm (SILVA;
Macrofibrila
Moléculas de glicose
Fibras de celulose
Microfibrila Parede celular
Fonte vegetal

17
D’ALMEIDA, 2009; OGEDA; PETRI, 2010; PEREIRA et al., 2015). A Figura 2.10
esquematiza a interconversão para os diferentes polimorfos da celulose.
Figura 2.10 - Transformação da celulose nativa em seus respectivos polimorfos.
Fonte: Adaptada de Silva e D’Almeida (2009).
Dentre os diferentes polimorfos identificados na celulose, os mais comuns e,
consequentemente, os mais estudados são os tipo I e II. A celulose I, ou celulose nativa,
é a base da estrutura cristalina na célula unitária, a qual encontra-se sob as formas Iα e
Iβ. A celulose Iα é o componente principal produzido por bactérias e fungos, com uma
célula unitária triclínica. E a celulose Iβ é a forma cristalina majoritária presente em
plantas superiores, com estrutura monoclínica. Em ambas as estruturas, as cadeias de
celulose estão orientadas paralelamente, variando-se somente a intensidade e os planos,
nos quais ocorrem as interações de hidrogênio. A proporção entre as frações dos
polimorfos Iα e Iβ está relacionada com a origem (SILVA; D’ALMEIDA, 2009;
OGEDA; PETRI, 2010; TAIPINA, 2012).
A celulose tipo II é a mais estável termicamente e pode ser formada a partir da
mercerização (tratamento alcalino) ou regeneração (solubilização seguida de
precipitação) da celulose nativa. A maior estabilidade deste polimorfo, em relação às
interações de hidrogênio, está associada à orientação das cadeias de celulose: paralela
(tipo I) e antiparalela (tipo II). A transição da estrutura do tipo I para o tipo II é
irreversível (OGEDA; PETRI, 2010).
CELULOSE IVI
Glicerol
T = 260°C
CELULOSE IIII
NH3(l)
CELULOSE Iα
NH3(l)
CELULOSE Iβ
NaOH
NaOH
NaOH
Δ
Δ
Δ CELULOSE II
Glicerol
T = 260°C
CELULOSE IIIII
CELULOSE IVII

18
O polimorfo III pode ser obtido por um processo reversível, a partir das celuloses
I e II, via tratamento com amônia líquida, dando origem às estruturas IIII e IIIII. E o
polimorfo IV pode ser formado aquecendo-se a celulose III com glicerol à 206 ºC. As
formas IVI e IVII são geradas a partir da celulose IIII e IIIII, respectivamente (KLEMM
et al., 2005; OLIVEIRA, 2007).
Um contraponto na utilização das fibras vegetais em compósitos poliméricos é o
seu caráter altamente hidrofílico, o qual afeta diretamente a interface fibra/matriz
(KHALIL; BHAT; YUSRA, 2012; ALBINANTE; PACHECO; VISCONTE, 2013;
GURUNATHAN; MOHANTY; NAYAK, 2015). Tal natureza hidrofílica promove uma
fraca adesão interfacial, devido à elevada absorção de umidade, conduzindo à alterações
dimensionais das próprias fibras e, consequentemente, no desempenho mecânico do
respectivo compósito (FIORE; DI BELLA; VALENZA, 2015; YAN; KASAL;
HUANG, 2016). Além disso, a presença de pectinas e componentes cerosos, que atuam
como barreira nas fibras, também impedem sua adesão com a matriz. Visando a eficácia
desta adesão, as modificações superficiais apresentam-se como uma alternativa para
reduzir a natureza hidrofílica das fibras vegetais, favorecendo a compatibilidade com
diversas matrizes poliméricas (KABIR et al., 2012b; FARUK et al., 2012; KHALIL;
BHAT; YUSRA, 2012; PEREIRA et al., 2015).
2.3. MODIFICAÇÕES EM FIBRAS VEGETAIS
A interface fibra/matriz é a zona de difusão ou reação, na qual duas fases são
quimicamente e/ou mecanicamente combinadas e desempenha um papel fundamental
nas propriedades dos compósitos (KABIR et al., 2012b).
Diversas metodologias de modificação de superfície têm sido exploradas, a fim de
reduzir a natureza hidrofílica das fibras vegetais. A presença dos grupos hidroxila
ligados à sua estrutura favorece tais modificações, com significativa melhora em
características superficiais, como adesão, molhabilidade, resistência à tração ou
porosidade (BRÍGIDA et al., 2010; GEORGE; CHAE; BRESSLER, 2016). Estes
tratamentos apresentam diferentes mecanismos de atuação, os quais estão diretamente
correlacionados à eficiência, podendo ser classificados em físicos e químicos
(GURUNATHAN; MOHANTY; NAYAK, 2015; GEORGE; CHAE; BRESSLER,

19
2016; ZHOU; FAN; CHEN, 2016). A seguir serão descritos as principais rotas para
cada tipo de modificação.
2.3.1. Modificações físicas
Os tratamentos físicos promovem mudanças nas propriedades estruturais e
superficiais das fibras, sem alterar sua composição (FARUK et al., 2012; FIORE; DI
BELLA; VALENZA, 2015; NG et al., 2015). Estas mudanças estão relacionadas com a
introdução de ligações cruzadas na superfície, modificação da energia de superfície e/ou
geração de radicais livres e grupos reativos, os quais influenciam no ancoramento
mecânico com a matriz polimérica (VÄISÄNEN et al., 2016; ZHOU; FAN; CHEN,
2016).
Dentre as diversas metodologias, as mais empregadas para fibras vegetais são as
que envolvem a utilização de plasma (SARIKANAT et al., 2016), descarga corona
(RAGOUBI et al., 2012), radiações γ (LI et al., 2015) e ultravioleta (UV) (ZAMAN;
KHAN; KHAN, 2009) e explosão de vapor (SONIA; PRIYA DASAN, 2013).
Os tratamentos por plasma e descarga corona têm como princípio a ionização das
moléculas presentes no gás, promovendo a oxidação da superfície e a alteração da
energia superficial das fibras vegetais (STEPCZYÑSKA, 2015; YAN; KASAL;
HUANG, 2016). O resultado consiste em um aumento tanto na polaridade quanto na
rugosidade superficiais (PICKERING; ARUAN EFENDY; LE, 2016).
A utilização de radiações, γ e UV, favorece a produção de radicais nas cadeias de
celulose devido à remoção de hidrogênio e grupos hidroxila, rompimento de algumas
ligações C-C e cisão da cadeia, proporcionando um ganho de polaridade (ZHOU et al.,
2015; ZHOU; FAN; CHEN, 2016).
A modificação por explosão de vapor fundamenta-se no aquecimento da fibra
vegetal, a altas temperatura e pressão, o qual promove uma ruptura mecânica do
material celulósico por meio de uma descarga violenta de vapor (explosão). Os
resultados incluem superfícies mais lisas, com rigidez reduzida, além de uma melhor
distribuição das fibras (SATYANARAYANA; ARIZAGA; WYPYCH, 2009;
GURUNATHAN; MOHANTY; NAYAK, 2015). Assim como os tratamentos por

20
descarga corona e plasma, o efeito desta modificação depende do tempo, temperatura e
composição dos gases envolvidos (PICKERING; ARUAN EFENDY; LE, 2016).
2.3.2. Modificações químicas
As modificações químicas baseiam-se na utilização de reagentes, os quais
promovem uma reação na estrutura das fibras, com a introdução de grupos funcionais e
alteração em sua composição. Tais modificações melhoraram as interações interfaciais,
com redução na absorção de umidade e acréscimo na compatibilidade com a matriz (LA
MANTIA; MORREALE, 2011; KABIR et al., 2012b; DATTA; KOPCZYNSKA, 2015;
FIORE; DI BELLA; VALENZA, 2015; PEREIRA et al., 2015; GEORGE; CHAE;
BRESSLER, 2016). As ligações envolvidas neste tipo de modificação podem ser
covalente, ligação de hidrogênio ou ácido-base (ALBINANTE; PACHECO;
VISCONTE, 2013).
Diversos agentes químicos são empregados para a modificação de superfície das
fibras vegetais, incluindo álcalis (mercerização), ácido/anidrido acético, silanos, ácido
acrílico, isocianatos, permanganato de potássio, peróxidos, éteres, ácido esteárico,
agentes de acoplamento (anidrido maleico), dentre outros (LI; TABIL; PANIGRAHI,
2007; KABIR et al., 2012b; DATTA; KOPCZYNSKA, 2015; GURUNATHAN;
MOHANTY; NAYAK, 2015; PICKERING; ARUAN EFENDY; LE, 2016; YAN;
KASAL; HUANG, 2016). A seguir serão descritos os tratamentos com álcali
(mercerização) e com ácido acético, pois estas foram as modificações químicas
utilizadas no presente trabalho.
a) Mercerização
O tratamento alcalino, ou mercerização, é uma das modificações químicas mais
antigas, baratas e eficazes empregadas em fibras vegetais, quando estas são aplicadas
como reforço em matrizes termoplásticas e termofixas (LI; TABIL; PANIGRAHI,
2007; SANCHEZ et al., 2010; ALBINANTE; PACHECO; VISCONTE, 2013;
GURUNATHAN; MOHANTY; NAYAK, 2015). Segundo ASTM D1695-07, a
mercerização pode ser definida como um processo no qual as fibras vegetais são
submetidas a uma interação com solução aquosa relativamente concentrada de uma base

21
forte, a fim de promover grande intumescimento, resultando em alterações estruturais,
dimensionais, morfológicas e mecânicas das mesmas.
Neste tratamento, usualmente com NaOH, a modificação mais importante
ocorrida é o rompimento das ligações de hidrogênio, devido à ionização dos grupos
hidroxila presentes em vários componentes das fibras (LI; TABIL; PANIGRAHI, 2007;
KABIR et al., 2012b; NG et al., 2015; ZHOU; FAN; CHEN, 2016). A Figura 2.11
apresenta a reação esquemática para a mercerização.
Figura 2.11 - Reação global para o tratamento alcalino.
Sendo: R = celulose.
Fonte: Produção da autora.
A mercerização remove grande parte da lignina, além de hemicelulose, ceras e
óleos que recobrem a superfície externa da parede celular das fibras vegetais (FARUK
et al., 2012; FIORE; DI BELLA; VALENZA, 2015; GURUNATHAN; MOHANTY;
NAYAK, 2015), influenciando na composição química, no grau de polimerização e na
orientação dos cristais de celulose (BOGOEVA-GACEVA et al., 2007; KALIA et al.,
2013; LI; TABIL; PANIGRAHI, 2007). Devido ao rompimento das ligações de
hidrogênio, há mudança na orientação da celulose cristalina altamente empacotada, com
formação de regiões amorfas, as quais proporcionam maior acessibilidade para outros
agentes químicos (KABIR et al., 2012b; ZHOU; FAN; CHEN, 2016). A Figura 2.12
exibe uma representação esquemática para a estrutura da fibra celulósica antes e após o
tratamento alcalino.
R O H + Na+ :-OH R O:- Na+ + H2O + impurezas

22
Figura 2.12 - Estrutura das fibras (a) sem tratamento e (b) com tratamento alcalino.
Fonte: Adaptada de Kabir et al. (2012b).
Durante o processo, a hemicelulose solubiliza-se à baixas concentrações de álcali.
Em tais condições, a lignina sofre hidrólise básica com a quebra das ligações éter entre
as unidades de fenilpropano, formando grupos fenólicos, responsáveis por sua
solubilização (ALBINANTE; PACHECO; VISCONTE, 2013). A Figura 2.13
representa a hidrólise com alguns dos componentes fenólicos formados.
Figura 2.13 - Hidrólise básica da lignina.
Fonte: Adaptada de Albinante, Pacheco e Visconte (2013).
Após o tratamento alcalino, há um aumento da rugosidade com maior exposição
da superfície das fibras, tornando os grupos hidroxila mais acessíveis, favorecendo a
changes the orientation of highly packed crystalline cellulose orderand forming an amorphous region. This provides more access topenetrate chemicals. In the amorphous region, cellulose micro-molecules are separated at large distances and the spaces are filledby water molecules. Alkali sensitive hydroxyl (OH) groups presentamong the molecules are broken down, which then react withwater molecules (HAOH) and move out from the fibre structure.The remaining reactive molecules form fibre–cell–O–Na groups be-tween the cellulose molecular chains [9]. Due to this, hydrophilichydroxyl groups are reduced and increase the fibres moistureresistance property. It also takes out a certain portion of hemicel-luloses, lignin, pectin, wax and oil covering materials [26–28]. Asa result, the fibre surface becomes clean. In other words, the fibresurface becomes more uniform due to the elimination of microv-oids and thus the stress transfer capacity between the ultimatecells improves. In addition to this, it reduces fibre diameter andthereby increases the aspect ratio (length/diameter). This increaseseffective fibre surface area for good adhesion with the matrix [14].Mechanical and thermal behaviors of the composite are improvedsignificantly by this treatment. If the alkali concentration is higherthan the optimum condition, the excess delignification of the fibrecan take place, which results in weakening or damaging the fibres[22,26]. The chemical reaction of the fibre–cell and NaOH is repre-sented in Scheme 1. Fig. 4 presents the schematic view of the cel-lulose fibre structure, before and after an alkali treatment. Treatedfibres have lower lignin content, partial removal of wax and oilcover materials and distension of crystalline cellulose order. Table4 summarizes the recent results on the effect of alkali treatmentson the mechanical and thermal properties of composites.
4.1.2. Silane treatmentSilane is a multifunctional molecule which is used as a coupling
agent to modify fibre surfaces. The composition of silane forms achemical link between the fibre surface and the matrix through asiloxane bridge. It undergoes several stages of hydrolysis, conden-sation and bond formation during the treatment process of the fi-bre. Silanols form in the presence of moisture and hydrolysablealkoxy groups (hydrolysis, see Scheme 2a [36]). During condensa-tion process, one end of silanol reacts with the cellulose hydroxylgroup (Si–O–cellulose, see Scheme 2b [36]) and other end reacts(bond formation) with the matrix (Si-Matrix) functional group.This co-reactivity provides molecular continuity across the inter-face of the composite. It also provides the hydrocarbon chain thatrestrains the fibre swelling into the matrix [22,32]. As a result, fibrematrix adhesion improves and stabilizes the composite properties[26]. Natural fibres exhibit micro-pores on their surface and silanecoupling agents act as a surface coating. This penetrates into thepores and develops mechanically interlocked coatings on fibre sur-face. Silane treated fibre composites provide better tensile strengthproperties than the alkali treated fibre composites [33]. Seki [34]investigated the effect of alkali (5% NaOH for 2 h) and silane (1%oligomeric siloxane with 96% alcohol solution for 1 h) treatmentson flexural properties of jute epoxy and jute polyester composites.For jute epoxy composites silane over alkali treatments showedabout 12% and 7% higher strength and modulus properties com-pared to the alkali treatment alone. Similar treatments reportedaround 20% and 8% improvement for jute polyester composites. Se-ver et al. [35] applied different concentrations of (0.1%, 0.3% and0.5%) silane (c-Methacryloxy-propyl-trimethoxy-silane) treat-ments on jute fabrics polyester composites. Tensile, flexural andinterlaminar shear strength properties were investigated and com-pared with the untreated fibre composites. The results for 0.3% si-lane treated composites showed around 40%, 30% and 55%improvement in tensile, flexural and interlaminar shear strengthrespectively.
4.1.3. Acetylation treatmentAcetylation treatment is known as esterification method for
plasticizing of natural fibres. Acetyl group (CH3CO) reacts withthe hydrophilic hydroxyl groups (OH) of the fibre and takes outthe existed moisture. As a result, hydrophilic nature of the fibreis decreased and improves dimensional stability of the composites[36]. Moreover, this treatment provides rough surface topographywith less number of void contents that give better mechanicalinterlocking with the matrix [4,26]. Fibres are acetylated withand without an acid catalyst to graft acetyl groups onto the cellu-lose structure. In general, acetic acid and acetic anhydride individ-ually does not react sufficiently with the fibres. To accelerate thereaction, fibres are initially soaked in acetic acid and consequentlytreated with acetic anhydride between the time periods of 1–3 h
Fibre-cell-OH + NaOH Fibre cell-O-Na+ + H2O + impurities
Scheme 1.
Lignin
Cellulose
Wax & Oil
(i) (ii) Fig. 4. Typical structure of (i) untreated and (ii) alkalized cellulose fibre [6].
Table 4Recent works on alkali treated fibre reinforced polymer composites.
Fibre matrix composites Applied treatment methods Results on mechanical and thermal properties References
Flax–epoxy Alkali treatment 30% increased in tensile strength and modulus with the removal of pectin [26]Sisal–polyester 0.5%, 1%, 2%, 4%, 10% NaOH
treatment at room temperature4% alkali treatment reported maximum tensile strength properties [26]
Hemp non-woven matwith euphorbia resin
0.16% NaOH for 48 h Tensile strength was increased by 30% and doubled the shear strength propertieswas found compared to the untreated fibre composites
[27]
Jute–vinylester 5% NaOH for 4, 6 and 8 h 4 h alkali treated composite accounted 20% and 19% increased in flexural strengthand interlaminar shear strength properties
[28]
Sisal–polycaprolactonecomposite
10% NaOH for 1, 3, 24 and 48 h Increased in elastic modulus with the increased with reaction time [29]
Hemp fibre 8% NaOH treatment Thermal stability was increased by 4% [30]Coir–polyester 5% NaOH treatment for 72 h Flexural and impact strength was increased by 40% with respect to the untreated
fibre composites[31]
M.M. Kabir et al. / Composites: Part B 43 (2012) 2883–2892 2887
changes the orientation of highly packed crystalline cellulose orderand forming an amorphous region. This provides more access topenetrate chemicals. In the amorphous region, cellulose micro-molecules are separated at large distances and the spaces are filledby water molecules. Alkali sensitive hydroxyl (OH) groups presentamong the molecules are broken down, which then react withwater molecules (HAOH) and move out from the fibre structure.The remaining reactive molecules form fibre–cell–O–Na groups be-tween the cellulose molecular chains [9]. Due to this, hydrophilichydroxyl groups are reduced and increase the fibres moistureresistance property. It also takes out a certain portion of hemicel-luloses, lignin, pectin, wax and oil covering materials [26–28]. Asa result, the fibre surface becomes clean. In other words, the fibresurface becomes more uniform due to the elimination of microv-oids and thus the stress transfer capacity between the ultimatecells improves. In addition to this, it reduces fibre diameter andthereby increases the aspect ratio (length/diameter). This increaseseffective fibre surface area for good adhesion with the matrix [14].Mechanical and thermal behaviors of the composite are improvedsignificantly by this treatment. If the alkali concentration is higherthan the optimum condition, the excess delignification of the fibrecan take place, which results in weakening or damaging the fibres[22,26]. The chemical reaction of the fibre–cell and NaOH is repre-sented in Scheme 1. Fig. 4 presents the schematic view of the cel-lulose fibre structure, before and after an alkali treatment. Treatedfibres have lower lignin content, partial removal of wax and oilcover materials and distension of crystalline cellulose order. Table4 summarizes the recent results on the effect of alkali treatmentson the mechanical and thermal properties of composites.
4.1.2. Silane treatmentSilane is a multifunctional molecule which is used as a coupling
agent to modify fibre surfaces. The composition of silane forms achemical link between the fibre surface and the matrix through asiloxane bridge. It undergoes several stages of hydrolysis, conden-sation and bond formation during the treatment process of the fi-bre. Silanols form in the presence of moisture and hydrolysablealkoxy groups (hydrolysis, see Scheme 2a [36]). During condensa-tion process, one end of silanol reacts with the cellulose hydroxylgroup (Si–O–cellulose, see Scheme 2b [36]) and other end reacts(bond formation) with the matrix (Si-Matrix) functional group.This co-reactivity provides molecular continuity across the inter-face of the composite. It also provides the hydrocarbon chain thatrestrains the fibre swelling into the matrix [22,32]. As a result, fibrematrix adhesion improves and stabilizes the composite properties[26]. Natural fibres exhibit micro-pores on their surface and silanecoupling agents act as a surface coating. This penetrates into thepores and develops mechanically interlocked coatings on fibre sur-face. Silane treated fibre composites provide better tensile strengthproperties than the alkali treated fibre composites [33]. Seki [34]investigated the effect of alkali (5% NaOH for 2 h) and silane (1%oligomeric siloxane with 96% alcohol solution for 1 h) treatmentson flexural properties of jute epoxy and jute polyester composites.For jute epoxy composites silane over alkali treatments showedabout 12% and 7% higher strength and modulus properties com-pared to the alkali treatment alone. Similar treatments reportedaround 20% and 8% improvement for jute polyester composites. Se-ver et al. [35] applied different concentrations of (0.1%, 0.3% and0.5%) silane (c-Methacryloxy-propyl-trimethoxy-silane) treat-ments on jute fabrics polyester composites. Tensile, flexural andinterlaminar shear strength properties were investigated and com-pared with the untreated fibre composites. The results for 0.3% si-lane treated composites showed around 40%, 30% and 55%improvement in tensile, flexural and interlaminar shear strengthrespectively.
4.1.3. Acetylation treatmentAcetylation treatment is known as esterification method for
plasticizing of natural fibres. Acetyl group (CH3CO) reacts withthe hydrophilic hydroxyl groups (OH) of the fibre and takes outthe existed moisture. As a result, hydrophilic nature of the fibreis decreased and improves dimensional stability of the composites[36]. Moreover, this treatment provides rough surface topographywith less number of void contents that give better mechanicalinterlocking with the matrix [4,26]. Fibres are acetylated withand without an acid catalyst to graft acetyl groups onto the cellu-lose structure. In general, acetic acid and acetic anhydride individ-ually does not react sufficiently with the fibres. To accelerate thereaction, fibres are initially soaked in acetic acid and consequentlytreated with acetic anhydride between the time periods of 1–3 h
Fibre-cell-OH + NaOH Fibre cell-O-Na+ + H2O + impurities
Scheme 1.
Lignin
Cellulose
Wax & Oil
(i) (ii) Fig. 4. Typical structure of (i) untreated and (ii) alkalized cellulose fibre [6].
Table 4Recent works on alkali treated fibre reinforced polymer composites.
Fibre matrix composites Applied treatment methods Results on mechanical and thermal properties References
Flax–epoxy Alkali treatment 30% increased in tensile strength and modulus with the removal of pectin [26]Sisal–polyester 0.5%, 1%, 2%, 4%, 10% NaOH
treatment at room temperature4% alkali treatment reported maximum tensile strength properties [26]
Hemp non-woven matwith euphorbia resin
0.16% NaOH for 48 h Tensile strength was increased by 30% and doubled the shear strength propertieswas found compared to the untreated fibre composites
[27]
Jute–vinylester 5% NaOH for 4, 6 and 8 h 4 h alkali treated composite accounted 20% and 19% increased in flexural strengthand interlaminar shear strength properties
[28]
Sisal–polycaprolactonecomposite
10% NaOH for 1, 3, 24 and 48 h Increased in elastic modulus with the increased with reaction time [29]
Hemp fibre 8% NaOH treatment Thermal stability was increased by 4% [30]Coir–polyester 5% NaOH treatment for 72 h Flexural and impact strength was increased by 40% with respect to the untreated
fibre composites[31]
M.M. Kabir et al. / Composites: Part B 43 (2012) 2883–2892 2887
NaOH
Ceras e óleos
Celulose
Lignina
(a) (b)
j m a t e r r e s t e c h n o l . 2 0 1 5;4(1):26–32 27
CH2OH
H HH
H
H H
HH
H OH H OH
OH OHC
C
C C
C C C
C
C C
O O HH
H
H
H OH
OHC C
C
C C
O
O O O
CH2OH CH 2OHA
H2COH
H2COH
H2COH
H2COH
CH2OH
CH2
H2COH
H2COH
HCOH
OCH
CHO
CHO
H2C
CH2
H2COH
H2COH
H2COH
H2COHH2COH
CH2OH
H2COH
H2COH
H2COH
H2COH
Lignin
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
OCH3
OCH3
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
HOCH2
HOCH2
HOCH2
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH2
OCH3
OCH3
OCH3
CH
HC
HC
HC
HC
HC
HC HC
HC
HCHC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CHOCH
CH
OH
CHCH
CH
OH
CH
CH
CH COCO
CO
CH
CHCO
CHO
CH
OH
CO
O
O
O
O
O
O
O
O
OC
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
B
CH2OH
OH
Coniferylalcohol
Sinapylalcohol
P-coumarylalcohol
OH
OMe OMe
OMe = OCH3methoxyl group
MeO
OH
CH2OH CH 2OHC
Fig. 1 – (a) Schematic representation of the cellulose structure [1] (b) lignin structure [2], and (c) structural units of lignin [2].
NaOH
j m a t e r r e s t e c h n o l . 2 0 1 5;4(1):26–32 27
CH2OH
HHH
H
HH
HH
HOHHOH
OHOHC
C
CC
CCC
C
CC
OOHH
H
H
HOH
OH CC
C
CC
O
OO O
CH2OHCH 2OH A
H2COH
H2COH
H2COH
H2COH
CH2OH
CH2
H2COH
H2COH
HCOH
OCH
CHO
CHO
H2C
CH2
H2COH
H2COH
H2COH
H2COH H2COH
CH2OH
H2COH
H2COH
H2COH
H2COH
Lignin
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
OCH3
OCH3
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
H3CO
OCH3
OCH3
HOCH2
HOCH2
HOCH2
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH3
OCH2
OCH3
OCH3
OCH3
CH
HC
HC
HC
HC
HC
HCHC
HC
HCHC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CHO CH
CH
OH
CHCH
CH
OH
CH
CH
CHCO CO
CO
CH
CHCO
CHO
CH
OH
CO
O
O
O
O
O
O
O
O
O C
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
B
CH2OH
OH
Coniferylalcohol
Sinapylalcohol
P-coumarylalcohol
OH
OMeOMe
OMe = OCH3methoxyl group
MeO
OH
CH2OHCH 2OH C
Fig. 1 – (a) Schematic representation of the cellulose structure [1] (b) lignin structure [2], and (c) structural units of lignin [2].
OH
2-metóxi-fenol
OCH3
OH
j
m
a
t
e
r
r
e
s
t
e
c
h
n
o
l
.
2
0
1
5;4
(1):26–32
27
CH
2 OH
HH
HH
HH
HHH
OH
HO
H
OH
OH
C
CCC
CC
C
CCC
OO
HH
H
H
HO
H
OH
CC
CCC O
OO
O
CH
2 OH
CH
2 OH
A
H2 C
OH
H2 C
OH
H2 C
OH
H2 C
OH
CH
2 OH
CH
2
H2 C
OH
H2 C
OH
HC
OH
OC
H
CH
OCH
O
H2 C
CH
2
H2 C
OH
H2 C
OH
H2 C
OHH
2 CO
HH
2 CO
H
CH
2 OH
H2 C
OH
H2 C
OH
H2 C
OH
H2 C
OH
Lignin
H3 C
O
H3 C
O
H3 C
O
H3 C
O
H3 C
O
OC
H3
OC
H3
OC
H3
OC
H3
H3 C
O
H3 C
O
H3 C
O
H3 C
O
H3 C
O
H3 C
O
H3 C
O
H3 C
O
OC
H3OC
H3
HO
CH
2
HO
CH
2
HO
CH
2
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H3
OC
H2
OC
H3
OC
H3
OC
H3
CH
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
HC
CH
CH
CH
CH
CH
OH
CH
CH
CH
CH
CH
OC
H
CH
OH
CH
CH
CH
OH
CH
CH
CH
CO
CO
CO
CH
CH
CO
CH
O
CH
OH
CO
O
O
O
O
O
O
O
O
OC
O
O
O
O
O
O
O
O
OO
O
O
O
O
O
B
CH
2 OH
OH
Coniferyl
alcoholS
inapylalcohol
P-coum
arylalcohol
OH
OM
eO
Me
OM
e = OC
H3
methoxyl group
MeO
OH
CH
2 OH
CH
2 OH
C
Fig.
1
–
(a)
Schem
atic
representation
of
the
cellulose
structu
re
[1]
(b)
lignin
structu
re
[2],
and
(c)
structu
ral
un
its
of
lignin
[2].
OCH2CH3
H3CH2CO
2,6-dietóxi-fenol

23
aderência mecânica com a matriz (LI; TABIL; PANIGRAHI, 2007; FARUK et al.,
2012; FIORE; DI BELLA; VALENZA, 2015). Além disso, com a eliminação da
hemicelulose, a qual apresenta caráter hidrofílico, há uma redução na capacidade de
absorção de umidade das fibras, o que permite uma melhor adesão interfacial
(DITTENBER; GANGARAO, 2012).
Na mercerização, parâmetros como tipo e concentração da solução alcalina,
temperatura e tempo de tratamento afetam diretamente a eficiência da modificação
(MOHANTY; MISRA; DRZAL, 2001; ALBINANTE; PACHECO; VISCONTE, 2013;
PEREIRA et al., 2015). Por exemplo, uma alta concentração de álcali, acima da
condição ótima, pode enfraquecer ou até mesmo danificar as fibras (GURUNATHAN;
MOHANTY; NAYAK, 2015).
Neste contexto, diversos estudos têm sido desenvolvidos, a fim de avaliar quais as
melhores condições deste tratamento aplicadas à gama de fibras vegetais disponíveis.
Sanchez et al. (2010) empregaram o tratamento alcalino em fibras de bagaço de
cana-de-açúcar e verificaram a influência da modificação nas propriedades de
compósitos com resinas de poliéster insaturado. As fibras foram tratadas com sol.
NaOH (10 % m/m), por 24 h, a temperatura ambiente. Os compósitos foram preparados
mediante a mistura manual do reforço com a matriz, na proporção de 15 % m/m de
fibras com e sem tratamento. Os resultados indicaram uma melhora nas propriedades de
impacto, com aumento do módulo de elasticidade em flexão, sem alteração significativa
do módulo de elasticidade em tração dos compósitos, em relação à resina pura. Além de
favorecer a compatibilidade com a matriz, o tratamento aumentou a estabilidade térmica
das fibras de bagaço.
Venkateshwaran; Perumal; Arunsundaranayagam (2013), por sua vez, reportaram
o efeito das diferentes concentrações de NaOH (0,5; 1,0; 2,0; 5,0; 10; 15 e 20 %) sobre
as propriedades mecânicas dos compósitos de epóxi/fibras de bananeira. Segundo os
autores, o aumento da concentração de álcali danificou a superfície das fibras, além de
reduzir as propriedades mecânicas dos compósitos. Os resultados indicaram que NaOH
a 1 % promove um aumento em aproximadamente 50 % nas propriedades mecânicas,
quando comparadas aos demais compósitos, sendo esta a melhor condição de
tratamento alcalino.

24
Mahjoub et al. (2014) estudaram as diferentes condições de tratamento alcalino,
aplicadas às fibras de kenaf e o seu efeito nas propriedades destas fibras. As fibras
foram imersas em solução de NaOH (5 %, 7 %, 10 % e 15 %), por um período de 1, 3 e
24 h. Segundo os autores, os tratamentos com as concentrações de 10 % e 15 %,
danificaram a textura da fibra, a qual apresentou-se muito mais fina e frágil, em relação
à fibra não tratada. Além disso, a resistência à tração diminuiu com o aumento tanto da
concentração de NaOH quanto com o tempo de imersão, sendo a melhor condição 5 % e
3 h de imersão.
Fiore; Di Bella; Valenza (2015) também avaliaram as diferentes condições de
mercerização para as fibras de kenaf e a influência destas modificações nas
propriedades mecânicas dos compósitos com matriz epóxi. As fibras foram tratadas com
sol. NaOH 6 % m/m para os tempos de imersão de 48 e 144 h. Os resultados indicaram
que o tratamento com tempo de imersão de 48 h melhorou as propriedades mecânicas
dos compósitos, devido à completa remoção dos componentes amorfos da superfície,
favorecendo a compatibilidade com a matriz.
A combinação entre diferentes tratamentos químicos também apresenta-se como
alternativa no aumento da adesão interfacial. Neste contexto, Orue et al. (2016) trataram
as fibras de sisal por dois tratamentos diferentes (mercerização e silanização) e a
combinação de ambos, a fim de verificar o resultado de tais tratamentos nas
propriedades de tração das fibras e dos respectivos compósitos à base de poli(ácido
láctico) (PLA). Os valores de resistência à tração e módulo de elasticidade das fibras
diminuíram em 55 % e 30 %, respectivamente, após a combinação entre os tratamentos
de mercerização e silanização. Em todos os compósitos contendo as fibras tratadas com
álcali e álcali/silano, as propriedades mecânicas foram magnificadas em relação aos
compósitos com as fibras sem tratamento e tratadas somente com silano. A presença de
álcali no tratamento aumentou a rugosidade da superfície, favorecendo a aderência
mecânica.
b) Acetilação
A acetilação é um método para modificação de superfície que causa a
plastificação das fibras de celulose, aumentando sua hidrofobicidade (LI; TABIL;

25
PANIGRAHI, 2007; KABIR et al., 2012b; GURUNATHAN; MOHANTY; NAYAK,
2015). Esta modificação baseia-se em uma reação de esterificação na qual os grupos
hidroxila presentes na lignina e hemicelulose (fração amorfa) são substituídos por
grupos acetila (CH3CO). A reação ocorre preferencialmente na região amorfa uma vez
que a estrutura da celulose apresenta-se altamente empacotada, devido às ligações de
hidrogênio, impedindo a difusão do reagente de acetilação (BOGOEVA-GACEVA et
al., 2007; ALBINANTE; PACHECO; VISCONTE, 2013; KALIA et al., 2013; ZHOU;
FAN; CHEN, 2016).
Em geral, a reação ocorre à altas temperaturas, com ácido acético (CH3COOH) ou
anidrido acético (C4H6O3) como reagentes. Entretanto, com a finalidade de acelerar a
reação e maximizar o grau de acetilação, pode-se utilizar catalisadores como os ácidos
sulfúrico (H2SO4) ou nítrico (HNO3) (KABIR et al., 2012b; GURUNATHAN;
MOHANTY; NAYAK, 2015; ZHOU; FAN; CHEN, 2016). A Figura 2.14 apresenta as
reações de acetilação com e sem catalisador.
Figura 2.14 - Reações de acetilação: (a) com catalisador e (b) sem catalisador.
Fonte: Adaptada de Kabir et al. (2012b).
Ao final da acetilação, as fibras apresentam-se hidrofóbicas, com uma superfície
rugosa, porém com menor número de vazios (KABIR et al., 2012b; ZHOU; FAN;
CHEN, 2016). Tais características favorecem a aderência mecânica, bem como
aumentam a dispersão das fibras com a matriz (GURUNATHAN; MOHANTY;
NAYAK, 2015; YAN; KASAL; HUANG, 2016). Em alguns trabalhos, foi relatado que
HNO3
Δ R O H +
+ H2O H3C
O C
O R
(a)
H3C O
C O H
(b)
R O H +
H3C O
C O
H3C C O
Δ
H3C O
C O R
+ H3C O
C O H

26
a acetilação melhora as propriedades térmicas, além de contribuir também na remoção
de pectinas (GEORGE; CHAE; BRESSLER, 2016).
Kabir et al. (2012a) preparam compósitos tipo sanduíche com fibras de cânhamo e
resina de poliéster. Para tal, foram utilizados diferentes tratamentos químicos, tais como
mercerização, silanização e acetilação. De acordo com os resultados de microscopia, as
fibras acetiladas apresentaram sua superfície altamente rugosa e exibiram um aumento
da temperatura de degradação, além de uma melhoria nas propriedades de resistência
para os respectivos compósitos.
Com o objetivo de melhorar a dispersão de celulose microcristalina (CMC) em
uma matriz de poli(ácido lático) (PLA) para a produção de compósitos, Mukherjee et al.
(2013) empregaram a acetilação como metodologia para modificação de superfície.
Neste trabalho, a CMC foi tratada com cloreto de acetila, em meio de piridina em
atmosfera inerte e precipitada com etanol. Os compósitos foram preparados com
diferentes quantidade de CMC (1 a 5 % m/m) e posteriormente caracterizados. De
acordo com os resultados observados, a quantidade de 2,5 % m/m apresentou-se como
valor ótimo, com relação à dispersão, pois acima deste valor há a aglomeração de CMC
na matriz de PLA. O comportamento térmico também foi avaliado e demonstrou que a
cristalinidade de todos os compósitos com CMC acetilada aumentou em relação aos
compósitos com CMC sem tratamento, também com valor ótimo de 2,5 % m/m. Os
autores concluíram que a CMC acetilada é uma forte candidata para atuar como agente
nucleante na preparação de compósitos à base de PLA, os quais podem ser destinados à
aplicações biomédicas.
Em outro estudo, porém desenvolvido por Hu et al. (2015), nanocristais de
celulose foram extraídos, a partir do línter de algodão, via hidrólise ácida e
posteriormente acetilados com anidrido acético. Estes nanocristais foram introduzidos
em matriz de poli(succinato de butileno) (PBS) para o desenvolvimento de espumas
biodegradáveis, com diferentes quantidades do reforço (0, 1, 2, 3, 5, 7, 10 % m/m).
Baseado nos resultados, as alterações nas quantidades de nanocristais acetilados
influenciaram diretamente no tamanho das células e na densidade das espumas. A
adição de 5 % m/m foi a proporção que apresentou os melhores resultados em
propriedades mecânicas, cristalinidade e morfologia celular. Os autores acreditam que

27
estes materiais apresentam grande potencial para a substituição de espumas à base de
petroquímicos.
2.4. FIBRA DE BANANEIRA
Das inúmeras fibras vegetais disponíveis, a fibra de bananeira destaca-se devido
ao alto índice na produção do fruto no país, com uma safra de 6,96 milhões de toneladas
em 2016 (IBGE, 2017). A elevada safra e sua estabilidade ao longo dos anos reflete
tanto no mercado financeiro, como na geração de grandes quantidades de subprodutos,
os quais podem ser aplicados em diversos setores, tais como: artesanal, científico e
tecnológico. A Figura 2.15 apresenta o índice de safra no Brasil, nos últimos 11 anos,
segundo dados do Instituto Brasileiro de Geografia e Estatística (IBGE).
Figura 2.15 - Índice de safra da banana de 2006 a 2016.
Fonte: IBGE (2016).
O alto índice de safra e a facilidade de cultivo devido ao clima tropical,
proporcionam ao Brasil o quinto lugar na produção mundial de banana. Segundo dados
correspondentes ao mês de jan/2017, as regiões Sudeste e Nordeste, somadas, são
responsáveis por 65 % da produção nacional, sendo o estado da Bahia o maior produtor
com 1004000 t, seguido por São Paulo com 985332 t (IBGE, 2017).
7,0 7,1 7,0 6,8 7,0 7,3
6,9 6,9 6,9 6,9 7,0
2006 2007 2008 2009 2010 2011 2012 2013 2014 2015 2016
Índice de safra (milhões de toneladas/ano)

28
As bananeiras são plantas herbáceas, pertencentes à família Musacease, gênero
Musa. A família Musaceae inclui cerca de 40 espécies, as quais são distribuídas entre
três gêneros: Musa, Musella, e Ensete (BENÍTEZ et al., 2013). De origem asiática, são
cultivadas principalmente pelos seus frutos, os quais são um dos mais utilizados na
alimentação humana (TELI; VALIA, 2013; PAPPU et al., 2015). Em geral, as
bananeiras podem ser divididas, segundo a representação esquemática exibida na Figura
2.16.
Figura 2.16 - Representação esquemática de uma bananeira.
Fonte: Produção da autora.
Após a colheita do fruto, o qual constitui apenas 12 % em peso da planta, grandes
quantidades de resíduos são produzidas, tais como ráquis, pseudocaule, folhas e engaço
(ZULUAGA et al., 2007; ELANTHIKKAL et al., 2010; PAPPU et al., 2015). Apesar
do emprego destes rejeitos na adubação dos solos, quando acumulados em abundância
facilitam a proliferação de fungos e insetos devido à umidade local, prejudicando a
plantação (BECKER et al., 2011; ZIMMERMANN et al., 2014). Outras possibilidades
para a utilização de cascas, folhas e pseudocaule, por exemplo, incluem sua
transformação em matéria orgânica para incorporação ao solo; alimentos para os
ruminantes (feno) e como substrato para a produção de biomassa (PAPPU et al., 2015).
Folha
Engaço
Fruto
Ráquis
Coração
Pseudocaule Filho

29
Entretanto, tais subprodutos também constituem uma fonte importante de fibras, com
potencial aplicação em compósitos poliméricos.
As fibras de bananeira podem ser obtidas a partir do pseudocaule, o qual é
constituído pelo conjunto rígido de bainhas foliares sobrepostas, sendo possível extrair
cinco tipos diferentes de fibras, desde a mais áspera até a de textura mais fina
(OLIVEIRA, 2007; ALBINANTE et al., 2012; BENÍTEZ et al., 2013). De acordo com
a literatura, grande parte dos trabalhos utiliza o pseudocaule da bananeira como fonte de
celulose para aplicação em compósitos, além de tratamentos químicos mais agressivos
tanto para a fibra quanto para o meio ambiente. Contudo, poucos trabalhos são relatados
nos quais empregam-se outros componentes da planta, com tratamentos químicos
superficiais.
2.5. FIBRA DE BANANEIRA E SUAS POTENCIAIS APLICAÇÕES
O interesse em compósitos reforçados com fibras vegetais torna-se cada vez mais
atrativo devido às inúmeras pesquisas, aplicações industriais e à variedade de fibras
vegetais disponíveis (RAMACHANDRAN; BANSAL; RAICHURKAR, 2016). Dentre
as diversas fibras, as fibras de bananeira evidenciam-se devido à potencialidade de
aplicações, principalmente em compósitos. Vários setores, tais como, biomédico,
alimentício, eletroeletrônico, construção civil, automobilístico, etc são relatados como
os principais campos de aplicação para as fibras de bananeira (PAPPU et al., 2015).
A utilização de partículas de celulose em escala nanométrica como reforço em
compósitos poliméricos teve início há duas décadas. Trabalhos envolvendo diferentes
metodologias para isolamento do componente celulósico, tanto na forma de microfibras
ou microfibrilas quanto cristais (“whiskers” ou “nanowhiskers”) têm sido reportados.
Zuluaga et al. (2007) descreveram a extração de microfibras de celulose, a partir
do ráquis da bananeira, pela combinação de tratamentos químicos e mecânicos. Os
autores observaram que o tipo de tratamento influenciou diretamente a morfologia das
microfibras. Isto é, com o tratamento químico foram obtidos agregados curtos de
celulose microcristalina, enquanto pelo processo mecânico (homogeneização a alta
pressão) foram obtidas microfibras individualizadas.

30
Em estudo semelhante Elanthikkal et al. (2010) produziram microfibras de
celulose a partir de resíduos de bananeira, visando aplicações biomédicas. O estudo
avaliou as diferentes condições de hidrólise ácida, nas quais estes resíduos foram
submetidos. Inicialmente, os resíduos foram pré-tratados com NaOH (1 mol.L-1, à 80 °C
por 4 h) e, em seguida, branqueados com solução de hipoclorito de sódio (NaOCl, 5 %,
à 80°C, por 3 h), para remoção dos componentes amorfos. Posteriormente, o material
pré-tratado foi hidrolisado com H2SO4 e os efeitos da temperatura, tempo e
concentração do ácido sobre as propriedades das microfibras foram avaliados. Baseado
nos resultados, a estabilidade das suspensões de celulose foi diretamente influenciada
pela temperatura e concentração de ácido. A diminuição nas dimensões das microfibras
também foi afetada pela concentração de ácido, assim como pelos métodos de secagem.
Por outro lado, o tempo de hidrólise não apresentou efeito significativo nas
propriedades das microfibras. A partir da hidrólise ácida os autores obtiveram celulose
com maior cristalinidade, comparada à celulose isolada pós mercerização e
branqueamento.
Ibrahim et al. (2010) avaliaram as propriedades físico-químicas e mecânica das
fibras de bananeira (pseudocaule) e de seus compósitos de polietileno de alta densidade
(PEAD). Para a modificação das fibras foram utilizadas duas rotas de tratamento:
alcalino (NaOH, 10 % m/m, 170 °C) por 3 h e explosão de vapor (220 °C) por 240 s. Os
compósitos foram preparados em diferentes frações mássicas (20, 40 e 60 %), para
ambas as fibras modificadas, com e sem anidrido maleico (AM) como agente
compatibilizante. De acordo com os autores, os compósitos contendo 20 % de fibras
com ambos os tratamentos e com AM apresentaram os melhores resultados de
resistência à tração, devido à melhor adesão entre fibras/matriz, em relação às fibras
sem tratamento. Com o aumento da fração mássica de fibras, também observaram uma
diminuição na flexibilidade, devido à redução na aderência com a matriz polimérica.
Comparando-se os tratamentos utilizados, com a explosão a vapor foram obtidas
partículas menores (microfibrilas) com um maior índice de cristalinidade, resultado
refletido nos compósitos.
Deepa et al. (2011) também utilizaram a metodologia de explosão a vapor
combinada aos tratamentos químicos para extração de nanofibras de bananeira. As

31
fibras foram tratadas com NaOH em autoclave por 1 h, branqueadas e, por fim,
hidrolisadas. Os resultados apresentados evidenciaram uma redução no diâmetro, um
aumento na estabilidade térmica e alto índice de cristalinidade das nanofibras, devido à
combinação de explosão a vapor e hidrólise ácida. De acordo com os autores, tais
propriedades tornam este material adequado para aplicação como reforço em
compósitos.
Em outro trabalho, Merlini; Soldi; Barra (2011) relataram as propriedades térmica
e mecânica de compósitos de poliuretano (PU), proveniente do óleo de mamona e fibras
de bananeira (pseudocaule). Neste trabalho os autores avaliaram o comprimento das
fibras (20 e 30 mm), a fração volumétrica (5, 10 e 15 % vol) e o tratamento químico nas
fibras com solução de hidróxido de sódio (NaOH, 2,5 mol.L-1) por 1 h. Segundo os
autores, as fibras tratadas apresentaram altas resistência à tração e ao cisalhamento, em
comparação às fibras sem tratamento. O comprimento e fração volumétrica ótimos, nos
quais foram obtidos os máximos valores de resistência à tração e módulo de elasticidade
foram de 30 mm e 15 % vol, respectivamente, em ambas as fibras com e sem
tratamento. Outro ponto observado foi o aumento da transição vítrea (Tg) com o
aumento da fração volumétrica de fibras, o qual pode estar associado à diminuição da
mobilidade das cadeias com a incorporação das fibras de bananeira devido às interações
interfaciais. Baseado nos resultados, os autores concluíram que as fibras de bananeira
podem ser utilizadas como eficiente agente de reforço na matriz estudada.
As propriedades mecânicas e térmicas também foram estudas por Shih; Huang
(2011), porém em compósitos de poli(ácido lático) (PLA) e fibras de bananeira. As
fibras, tratadas com NaOH, foram inseridas à matiz após a compatibilização com silano,
nas proporções de 20, 40 e 60 % m/m. A incorporação de 40 % m/m de fibras de
bananeira aumentou a estabilidade térmica e as resistências à tração e flexão dos
compósitos, em aproximadamente duas vezes, em relação à matriz pura. Com estes
resultados, os autores concluíram que a adição de fibras à matriz pode reduzir custos de
fabricação dos compósitos.
Benítez et al. (2013) estudaram o efeito dos tratamentos químicos nas
propriedades mecânicas e térmicas das fibras de bananeira das Canárias (pseudocaule)
para aplicação em compósitos processados por moldagem por injeção. Hidróxido de

32
sódio (0,1, 1,0 e 4,0 eq.L-1) e AM, sob diferentes condições de pressão (atmosférica e
129 kPa) e temperatura (ambiente e 107 °C), foram utilizados para tratar a fibra.
Segundo os autores, a influência da pressão foi mais relevante do que a concentração de
NaOH, pois com o aumento da concentração não foram observadas melhorias tanto no
comportamento térmico, quanto mecânico. Apesar da modificação com AM
proporcionar um aumento na estabilidade térmica da fibra, as propriedades mecânicas
não apresentaram alterações significativas. Os autores concluíram que a concentração
de NaOH de 1,0 eq.L-1 com pressão à 129 kPa exibiu os melhores resultados para as
propriedades térmicas, sem diminuição no comportamento mecânico. Além disso, a
resistência à tração e módulo de elasticidade para esta espécie de fibra de bananeira são
mais elevados, quando comparados à outras fibras, tornando-as hábeis para utilização
como reforço.
Com a finalidade de redução de custos nos tratamentos empregados para as fibras
lignocelulósicas, Pereira et al. (2013) propuseram utilizar somente lavagens com água
destilada. Após o processo de lavagem, as fibras das cascas da banana (5 e 10 % m/m)
foram misturadas com PEAD para avaliação das características físico-químicas dos
materiais obtidos. Segundo os resultados, as fibras lavadas com água exibiram redução
de grande parte dos componentes amorfos (impurezas), menor índice de cristalinidade e
aumento da estabilidade térmica, quando comparadas às fibras não lavadas. Nos
compósitos, a adição de 5 % m/m das fibras lavadas proporcionou um aumento na
resistência à tração e no módulo de elasticidade, comparados à matriz pura,
confirmando sua atuação como agente de reforço. Entretanto, com 10 % de fibras há
diminuição destes valores, devido à redução da adesão na interface fibra/matriz.
Zimmermann et al. (2014) utilizaram diferentes concentrações de NaOH (1, 5 e 10
% m/v) para modificação química das fibras de bananeira, com o objetivo de avaliar seu
efeito nas propriedades de compósitos expandidos de poli(etileno-co-acetato de vinila)
(EVA). Os resultados indicaram que o tratamento alcalino promove a extração dos
componentes amorfos, porém altas concentrações de NaOH (10 % m/v) ocasionaram
efeitos degradativos na fibra, além de perdas nas propriedades mecânicas de resistência
à tração dos compósitos. Com o aumento de fibras nos compósitos expandidos há um
decréscimo das propriedades mecânicas, com a diminuição da densidade devido ao

33
aumento dos espaços vazios no interior dos compósitos. Entretanto, foram observadas
que as propriedades mecânicas específicas de resistência ao rasgo (ASTM D 62400)
exibiram os melhores resultados com 10 % m/m de fibras.
Gonçalves et al. (2015) estudaram as propriedades físico-químicas da banana roxa
(Musa velutina), visando sua aplicação como reforço em compósitos. As fibras,
extraídas do pseudocaule, foram tratadas com NaOH (5 % m/v), por 1 h em temperatura
ambiente. Os resultados evidenciaram a eficiência do tratamento alcalino, com remoção
de 36 % de hemicelulose e 67 % de lignina, em relação à fibra bruta, com aumento na
cristalinidade. As análises de FTIR confirmam o desaparecimento de bandas referentes
aos componentes amorfos e nas micrografias foi observada uma superfície mais lisa,
sem impurezas, com exposição das fibrilas de celulose. Os autores concluíram que esta
espécie de fibra de bananeira tem potencial para aplicação como reforço em matrizes
poliméricas, não só por estes resultados, mas também pelas propriedades mecânicas
magnificadas após a modificação.
Paul; Kanny; Redhi (2015) utilizaram a seiva da bananeira para sintetizar uma
bio-resina, objetivando a preparação de compósitos contendo fibras de bananeira
(pseudocaule) como reforço. A bio-resina foi obtida a partir de uma blenda formada
pela seiva, propilenoglicol e AM. Para efeito de comparação também foi preparada uma
resina de controle, porém sem a seiva. Os compósitos foram confeccionados sob a
forma de painéis, contendo duas camadas de fibra (30 % m/m) e a respectiva resina,
pelo método de moldagem por infusão. Os painéis permaneceram sob cura por 24 h e
pós-curados a 80 °C por 3 h. De acordo com os testes mecânicos, a adição de seiva de
bananeira à resina promoveu um aumento de 15 % da resistência à tração, 12 % no
módulo de elasticidade e 25 % no módulo de flexão nos compósitos, quando
comparados aos painéis produzidos sem seiva. O compósito contendo seiva também
exibiu uma melhora na estabilidade térmica, com um aumento na sua temperatura de
transição vítrea, assim como uma melhor compatibilidade com as fibras. Por fim, os
testes de degradação revelaram um aumento de 21 % no diâmetro da zona de
crescimento de fungos para os compósitos contendo a seiva, confirmando a formação de
um biocompósito. Em estudo anterior, os mesmos autores relataram o desenvolvimento
e as propriedades físico-químicas da bio-resina, visando aplicações destinadas ao

34
interior de veículos, tais como suportes de painel, guarnição da porta interna, etc
(PAUL; KANNY; REDHI, 2013).
Fibras de origem mineral como, por exemplo o asbesto ou amianto, apresentam
propriedades que as tornam atrativas para diversas aplicações, tais como, estabilidade
térmica, resistência ao calor, baixa condutividade elétrica, etc (SHRIVER; ATKINS,
2008). Apesar de amplamente utilizadas em compósitos, atualmente são evitadas e
proibidas em muitos países, por problemas relacionados à saúde, os quais estão
associados desde doenças respiratórias até câncer de pulmão (IDRIS et al., 2015; XIE et
al., 2015; PICKERING; ARUAN EFENDY; LE, 2016).
Neste contexto, Idris et al. (2015) propuseram a substituição de asbestos por
cascas de banana para a produção de pastilhas de freio. Neste trabalho foram utilizados
dois tipos de cascas de banana: não carbonizada e carbonizada à 1200 °C. As amostras
de pastilhas de freio foram preparadas mediante a mistura de resina fenólica com ambos
os tipos de cascas, em diferentes proporções. Os moldes foram obtidos por prensagem a
quente à 150 °C (pressão de 9,81x107 Pa, por 2 min) e curados a 130 °C por 8 h.
Baseado nos resultados, as amostras contendo 25 % m/m de cascas não carbonizadas e
30 % m/m de cascas carbonizadas exibiram os melhores resultados em termos de
resistência à compressão, dureza e gravidade específicas, assim como nos testes de
absorção de óleo e água e taxa de desgaste. Segundo os autores, o resultado desta
pesquisa é um indicativo que as partículas de cascas de banana podem ser utilizadas
como um substituto para o amianto satisfatoriamente.
As fibras de bananeira também apresentaram-se como promissores adsorventes na
remoção de óleos e metais pesados. Cascas de banana foram utilizadas por Memon et al.
(2009) para adsorção de íons Cr4+ em efluentes industriais. Os autores observaram uma
capacidade máxima de adsorção de aproximadamente 132 mg/g, sendo este o maior
valor encontrado, dentre os diversos estudos direcionados à remoção de íons cromo.
Anwar et al. (2010) também utilizaram cascas de banana como adsorvente e
avaliaram sua eficiência, porém na remoção dos íons Cd2+ e Pb2+. Baseado nos
resultados, a capacidade máxima de adsorção para os metais cádmio foi 5,71 mg/g e de
2,18 mg/g para o chumbo. Segundo os autores, este estudo confirma a eficácia das

35
cascas de banana na remoção de metais pesados em águas residuais e efluentes
industriais.
Teli; Valia (2013), por sua vez, relataram a utilização de fibras de bananeira
modificadas quimicamente para promover a absorção de óleo. Inicialmente, as fibras de
bananeira foram esterificadas com anidrido acético, visando a diminuição da
hidrofilicidade e, em seguida, caracterizadas. De acordo com os resultados, a
esterificação apresentou-se eficiente na redução da hidrofilicidade das fibras de
bananeira, as quais exibiram uma capacidade máxima de absorção de 18 g de óleo/g de
fibra, com um reciclo de três vezes. Além de evidenciarem vantagens como baixo custo
e alta capacidade de absorção, os autores concluíram que estas fibras modificadas
poderiam ser utilizadas na recuperação de óleo em derramamentos. Trabalho similar
também foi reportado por Shang et al. (2016), no qual a capacidade máxima de
absorção de óleo foi de 32 g de óleo/g de fibra.
A combinação de dois ou mais materiais possibilita a obtenção de uma série de
propriedades intrínsecas. Este efeito, denominado hibridização, apresenta-se promissor
em compósitos, pela combinação de diferentes tipos de fibras de reforço em uma
mesma matriz (KUMAR et al., 2016). A partir de uma seleção adequada das fibras, as
características individuais destes reforços podem ser exploradas e os custos do
compósito final podem ser reduzidos (IDICULA; JOSEPH; THOMAS, 2010;
PICKERING; ARUAN EFENDY; LE, 2016). Devido às suas propriedades mecânicas
elevadas, as fibras de bananeira podem ser facilmente combinadas a outros tipos de
fibras, tanto naturais quanto sintéticas (RAMACHANDRAN; BANSAL;
RAICHURKAR, 2016).
Idicula; Joseph; Thomas (2010) reportaram um efeito positivo nas propriedades
mecânicas em compósitos de poliéster, reforçados com fibras curtas de banana/sisal,
devido à adição de fibras de bananeira. Segundo os autores, a distribuição das fibras
influenciou as propriedades de tração, principalmente com o aumento do número de
camadas de fibras de bananeira, com os maiores valores de tração e flexão observados
na proporção 3:1 banana/sisal.
Boopalan; Niranjanaa; Umapathy (2013) avaliaram as propriedades mecânicas e
térmicas de compósitos (resina epóxi) reforçados com fibras de juta e bananeira in

36
natura. Os compósitos foram processados por moldagem, com diferentes proporções
mássicas entre as fibras de juta/bananeira (100/0, 75/25, 50/50, 25/75 e 0/100) e
submetidos a testes de tração, flexão, impacto, térmico e absorção de água. Segundo os
autores, a adição de fibra de bananeira promoveu um aumento na resistência à tração
(17 %), flexão (4,3 %) e resistência ao impacto (35,5 %). Entretanto, somente a
composição com 50 % m/m de fibra de bananeira apresentou os melhores resultados nos
testes mecânicos e térmicos, com o menor índice de absorção de umidade.
Alavudeen et al. (2015) relataram o efeito da distribuição das fibras de bananeira e
kenaf, incorporadas randomicamente e sob a forma de tecidos (plano e trançado), em
uma matriz de poliéster. O estudo avaliou as propriedades mecânicas dos compósitos
contendo os reforços individuais e híbridos, em ambas as formas de distribuição.
Paralelamente, este estudo também avaliou o efeito dos tratamentos químicos com
NaOH e lauril sulfato de sódio (LSS), em diferentes concentrações (5, 10 e 15 % m/v) e
tempos (2, 4, 6 e 8 h). Ambas as modificações químicas proporcionaram às fibras maior
resistência mecânica e adesão, porém a condição ótima adotada para a continuidade do
estudo foi 10 % de LSS por 8 h devido aos melhores resultados. Para as diferentes
formas de tecido, o tipo liso demonstrou as melhores propriedades de resistência à
tração, comparado ao tipo trançado. Já a máxima resistência mecânica foi observada nos
tecidos lisos, porém formados pelo reforço híbrido, diferentemente dos compósitos
contendo as fibras distribuídas randomicamente. Os autores concluíram que as
propriedades mecânicas dos compósitos reforçados com tecido híbrido aumentaram
após a adição de fibras de bananeira, evidenciando a superioridade destas fibras em
relação às fibras de kenaf.
Em outro trabalho, Kumar et al. (2016) avaliaram o efeito da porcentagem relativa
e distribuição das fibras de bananeira (B) e coco (C), como reforço híbrido, nas
propriedades vibracionais e mecânicas de compósitos em matriz de poliéster. Neste
estudo, as fibras curtas de bananeira (~ 4 mm de comprimento) e fibras de coco (tecido)
foram tratadas via mercerização (NaOH, 1 eq.L-1). Posteriormente, as fibras foram
dispostas em diferentes padrões de camadas (CBC, CCB, BCB e BBC), mantendo-se a
mesma % m/m total de fibras. Testes de tração, flexão e de impacto, juntamente com as
características vibracionais e de amortecimento foram avaliados, visando aplicações

37
automotivas. Os autores observaram que a distribuição das fibras influenciou
significativamente as propriedades mecânicas e proporcionou alterações na rigidez,
refletidas nos testes vibracionais. O compósito CBC exibiu a maior resistência à flexão
e o maior valor de amortecimento, indicando uma melhor capacidade de absorção de
energia, devido à estrutura porosa da fibra de coco. Já o compósito BBC apresentou a
maior resistência à tração e flexão. Segundo os autores, os compósitos com maior
porcentagem relativa de fibras de bananeira exibiram propriedades mecânicas
magnificadas, destacando as características intrínsecas desta fibra.
Ramachandran; Bansal; Raichurkar (2016) também estudaram a combinação de
fibras curtas (2-4 mm) de bananeira, bambu e linho, adicionadas à resina epóxi,
randomicamente. Neste estudo, foram preparadas diferentes proporções mássicas dos
compósitos, sendo designadas como A (90:10, resina/bambu), B (90:5:5,
resina/bambu/banana) e C (90:5:5,resina/bambu/linho). Os compósitos foram
caracterizados por testes de impacto (Izod e Charpy), de dureza Rockwell e FTIR. Os
autores observaram que o compósito B apresentou os melhores resultados em todos os
testes de impacto, além de uma melhor compatibilidade com a resina, confirmada por
FTIR. Entretanto, o compósito C exibiu o melhor resultado nos testes de dureza. Os
autores afirmaram que este estudo foi preliminar e, futuramente, pretendem realizar
mais testes com uma variedade de composições e diferentes tamanhos de fibras, visando
aplicações industriais.
Trabalhos similares contendo fibras de bananeira com abacá/fibras de vidro
(OHMANTH; KATHIRAVAN; GANDIKOTA, 2016); juta (RAMNATH et al., 2015) e
linho (SRINIVASAN et al., 2014) também reportaram o efeito da hidridização nas
propriedades térmica e mecânicas dos compósitos.
Assim como a junção de diferentes fibras vegetais apresenta-se eficaz no ganho de
propriedades térmicas e mecânicas, a combinação entre componentes orgânicos e
inorgânicos tem se mostrado bastante eficiente como reforço em compósitos
poliméricos, dando origem aos materiais denominados híbridos orgânico-inorgânicos.

38
2.6. HÍBRIDOS ORGÂNICO-INORGÂNICO
Nas duas ultimas décadas, o interesse científico e tecnológico no desenvolvimento
de materiais híbridos orgânico-inorgânicos tornou-se mais significativo devido à
versatilidade de aplicações em diversos setores industriais (KANGO et al., 2013).
Os materiais híbridos orgânico-inorgânicos podem ser definidos como uma
combinação entre componentes inorgânicos (minérios, óxidos metálicos hidratados, sais
e etc.) e matrizes orgânicas (polímeros termoplásticos, termorrígidos, frações
celulósicas etc.). Esta combinação alia as estabilidades térmica e química da fração
inorgânica com a processabilidade e flexibilidade da fração orgânica, fornecendo
características intrínsecas ao material final (JOSÉ; PRADO, 2005; KANGO et al., 2013;
CAMPOS; URBANO; RIVAS, 2014a; FIGUEIRA; SILVA; PEREIRA, 2015).
No que se refere à preparação, três maneiras distintas são descritas: incorporação
física dos constituintes, ligações químicas entre os componentes e a combinação de
ambos os métodos. Destaca-se o segundo método, pela ocorrência de ligações
covalentes entre as frações orgânica e inorgânica (JOSÉ; PRADO, 2005).
Considerando-se a natureza de suas ligações, os híbridos orgânico-inorgânicos podem
ser divididos em Classe I, II e III.
Na Classe I as frações orgânica e inorgânica encontram-se dispersas e interagem
por meio de ligações de hidrogênio, forças de Van der Waals ou ligações iônicas. Nos
materiais da Classe II as frações exibem ligações covalente, iônica-covalente ou
ligações coordenadas (KICKELBICK, 2007; CUNHA et al., 2010; CARRARO;
GROSS, 2014; FIGUEIRA; SILVA; PEREIRA, 2015). E os híbridos pertencentes à
Classe III, exibem uma combinação de interações descritas nas Classe I e II (JOSÉ;
PRADO, 2005).
Estes materiais híbridos representam uma ampla e diversificada classe de
sistemas, provenientes de uma combinação intrínseca, devido à formação da ligação
química entre as frações orgânica e inorgânica (CARRARO; GROSS, 2014). São
macroscopicamente homogêneos, devido à mistura de suas frações em nível molecular,
normalmente em escala de nanômetro a sub-micrômetro, porém suas propriedades
refletem a natureza química dos blocos pelos quais foram formados (JOSÉ; PRADO,
2005; KICKELBICK, 2007).

39
Geralmente, os termos híbridos e compósitos são encontrados na literatura como
sinônimos. Entretanto, estes materiais distinguem-se em relação às interações entre as
fases que os compõem e à processabilidade. Os compósitos são descritos como
materiais que exibem dois ou mais componentes (ou fases) quimicamente diferentes,
combinados em escala macroscópica, os quais apresentam uma interface distinta com
separação destes componentes (MARINUCCI, 2011). Ou seja, são obtidos pela mistura
física de suas fases, resultando em materiais heterogêneos. Por outro lado, os híbridos
são sintetizados a partir de soluções homogêneas formadas por componentes orgânicos,
oligômeros ou polímeros reativos, juntamente com o componente inorgânico. Neste
sistema, a fração inorgânica é obtida in situ, a qual permite o controle da morfologia e,
consequentemente, da homogeneidade do sistema (SILVA et al., 2005). Deve-se
salientar que estes híbridos não exibem somente uma mera soma de características de
seus precursores, e sim um sinergismo devido à interação de ambas as fases, enquanto
novas propriedades são obtidas (BENVENUTTI et al., 2009; CUNHA et al., 2010).
Considerando a ampla variedade de combinações possíveis entre os componentes
orgânico e inorgânico, inúmeros materiais multifuncionais podem ser desenvolvidos,
visando aplicações industriais inovadoras em diversos setores (CUNHA et al., 2010;
MASCHIO; PEREIRA; DA SILVA, 2012). Áreas como a adsorção (ZULFIKAR et al.,
2016), óptica (KUMAR; PARK, 2014), medicina (FRAGAL et al., 2016),
revestimentos (FIGUEIRA; SILVA; PEREIRA, 2015), células solares (WRIGHT;
UDDIN, 2012), catálise e sensores (WANG et al., 2013) são apenas alguns exemplos de
aplicações promissoras para estes materiais híbridos.
A combinação entre a celulose proveniente de subprodutos agroindustriais e
óxidos metálicos hidratados tem se mostrado promissora na obtenção de compósitos
poliméricos e membranas.
Mulinari et al. (2009) avaliaram o efeito da modificação na superfície da celulose
do bagaço de cana com óxido de zircônio hidratado (ZrO2.nH2O) nas propriedades
mecânicas e morfológicas em compósitos de polietileno de alta densidade (PEAD). Os
resultados indicaram que a modificação com ZrO2.nH2O aumentou a resistência à
ruptura dos compósitos, elevando o módulo de elasticidade em 38 %, em relação à

40
celulose não modificada com ZrO2.nH2O. Tais resultados confirmaram que a adição de
10 % m/m no PEAD foi suficiente para a atuação do híbrido como reforço.
Ottoboni (2011), por sua vez, modificou a superfície da celulose do bagaço de
cana, porém com óxido de nióbio hidratado (Nb2O5.nH2O), visando sua aplicação como
reforço em compósitos de polietileno de baixa densidade (PEBD). De acordo com os
resultados, o híbrido 90Cel/10Nb2O5.nH2O apresentou-se como a melhor proporção,
exibindo propriedades de retardância à chamas. Nos compósitos, a substituição da
celulose pelo híbrido aumentou os valores de resistência à tração máxima e módulo
elástico, com diminuição nos valores de deformação.
Maschio; Pereira; Da Silva (2012) também observaram o efeito de retardância à
chama em híbridos provenientes da celulose do bagaço de cana e Nb2O5.nH2O. Segundo
os resultados de calorimetria, a incorporação da fração inorgânica à celulose inibiu os
picos referentes ao processo de combustão do componente orgânico.
Silva (2013) relatou a aplicação de híbridos com celulose do bagaço de cana e
óxido de alumínio hidratado (Al2O3.nH2O) em membranas. Os resultados de
calorimetria também indicaram o mesmo efeito de retardância à chama nos híbridos,
como reportado para os materiais contendo Nb2O5.nH2O. As membranas preparadas a
partir dos híbridos exibiram maior estabilidade térmica e resistência à combustão,
quando comparadas às membranas contendo somente celulose.
Outros óxidos metálicos hidratados tais como, manganês, Mn3O4.nH2O;
magnésio, MgO.nH2O e ferro, Fe3O4.nH2O também podem ser citados como exemplos
para compor a fração inorgânica.
2.7. ÓXIDOS METÁLICOS HIDRATADOS
2.7.1. Óxido de nióbio hidratado, Nb2O5.nH2O
Materiais contendo nióbio destacam-se por apresentarem diversas aplicações
especiais, principalmente nos setores aeroespacial, eletroeletrônico, de catálise e de
adsorção (FOROUGHI-ABARI; CADIEN, 2011; LOPES et al., 2015). Tal interesse é
justificado pois o Brasil detêm as maiores reservas mundiais de nióbio (98,2 %), as
quais encontram-se nos estados de Minas Gerais (411,5 Mt), Amazonas (159,7 Mt),

41
Goiás (106,8 Mt) e Rondônia (42,1 Mt). No cenário mundial, o país destaca-se como o
principal produtor, com participação de 93,7 % no mercado, seguido por Canadá (5,28
%) e demais países (1,05 %) (DNPM, 2015). É encontrado na natureza sob a forma dos
minérios pirocloro, (Na,Ca)2Nb2O6(OH,F), e columbita, (Fe,Mn)Nb2O6 (GIBSON;
KELEBEK; AGHAMIRIAN, 2015).
O nióbio é um material refratário, com massa específica de 8,57 g.cm-3, boa
condutividade térmica, o qual exibe elevados pontos de fusão (2477 °C) e ebulição
(4744 °C) (NICO; MONTEIRO; GRAÇA, 2016). O metal possui cinco grandes classes
de compostos: fosfatos, carbetos, sulfetos, nitretos e óxidos, com estados de oxidação de
+5 (mais estável) até -1 (LEE, 1999; LOPES et al., 2015). Apresenta excelente
ductilidade, resistência à oxidação e resistência ao impacto (YOON, 2010). De acordo
com a Companhia Brasileira de Metalurgia e Mineração (CBMM), no setor metalúrgico
é de extrema importância na produção de ligas resistentes à altas temperaturas (por
exemplo, veículos aeroespaciais) e aços inoxidáveis especiais, além de aplicações no
campo da supercondutividade (ligas Nb-Ti) (NICO; MONTEIRO; GRAÇA, 2016).
Os óxidos de nióbio apresentam-se como materiais versáteis, com potenciais
aplicações tecnológicas, tais como capacitores, dispositivos ópticos, óxidos condutores
transparentes, células solares, catalisadores etc (LIU; XUE; LI, 2011; NICO;
MONTEIRO; GRAÇA, 2016). Devido à reação com o oxigênio, são formados óxidos
em diferentes estequiometrias, como o pentóxido (Nb2O5), dióxido (NbO2) e monóxido
de nióbio (NbO) (LEE, 1999; LOPES et al., 2015).
O pentóxido de nióbio, Nb2O5, é um semicondutor tipo “n”, com band gap de 3,4
eV, sendo um sólido branco, estável ao ar e insolúvel em água (ZHAO et al., 2012;
KAWANO; KAKEMOTO; IRIE, 2015). Possui sítios ácidos de Brönsted e Lewis, com
alta acidez e características anfotéricas, podendo ser dissolvido tanto em ácidos fortes
como em bases fortes (LOPES et al., 2015).
O óxido de nióbio hidratado, Nb2O5.nH2O, também denominado ácido nióbico, é
branco, possui grau de hidratação indeterminado e maior reatividade que o óxido anidro
(RODRIGUES, 2008). Possui alta resistência ao ataque por ácidos, álcalis, oxidantes e
redutores, é praticamente insolúvel em água e apresenta uma estrutura rígida, sofrendo
pouca dilatação ou compressão quando imerso em solução aquosa (RODRIGUES; DA

42
SILVA, 2009a; RODRIGUES; DA SILVA, 2009b). A síntese deste óxido sob a forma
de nanopartículas é uma das rotas mais promissoras para sua aplicação em setores como
catálise, imobilização de enzimas, troca iônica e adsorção (TAGLIAFERRO; DA
SILVA; DA SILVA, 2005; RODRIGUES; DA SILVA, 2010; TAGLIAFERRO et al.,
2011).
2.7.2. Óxido de alumínio hidratado, Al2O3.nH2O
O alumínio, primeiro metal e o terceiro elemento químico mais abundante da
crosta terrestre, evidencia-se como um material de extrema importância devido às suas
diversas propriedades, segundo dados da Associação Brasileira do Alumínio (ABAL).
A baixa massa específica aliada à ductilidade, maleabilidade, condutividades elétrica e
térmica, resistência química à corrosão, além do caráter anfótero tornam-se atrativas em
aplicações de componentes estruturais em transportes e construção civil e no setor de
embalagens (KARIM et al., 2016).
A bauxita é o principal minério para a produção de alumínio e demais compostos,
a qual possui um teor superior a 40 % de Al2O3, sendo composta por gibbsita
[Al(OH)3], diásporo [AlO(OH)] e a boehmita [AlO(OH)]. As reservas mundiais de
bauxita somam-se 28,1 bilhões de toneladas. O Brasil detém o 3º lugar com 2,6 bilhões
de toneladas, reserva concentrada na região Amazônica, perdendo para Guiné (7,4
bilhões) e Austrália (6,5 bilhões) (DNPM, 2015).
O óxido de alumínio (Al2O3), um dos materiais cerâmicos mais importantes, é
conhecido por existir em diferentes formas polimórficas metaestáveis, denominadas
aluminas de transição. Tais polimorfos incluem γ, θ, η, δ, χ, κ e β alumina, antes da
completa transformação para a fase termodinamicamente estável (α-alumina). Estas
aluminas são amplamente utilizadas na indústria química por apresentarem propriedades
físico-químicas específicas como, por exemplo, boa resistência mecânica e estabilidade
térmica (FAVARO et al., 2010; SALEM; CHINELATTO; CHINELATTO, 2014;
NOGUEIRA et al., 2016).
As diferentes estruturas existentes proporcionam ao Al2O3 uma diversidade de
aplicações, tais como: adsorventes, abrasivos, isolante elétrico, dispositivos ópticos e
eletrônicos, camadas protetoras antioxidantes e membranas, sendo o mais utilizado

43
como catalisador e suportes catalíticos (FAVARO et al., 2010; NAWROCKI;
FIJOŁEK, 2013; MALKI et al., 2014).
O óxido de alumínio hidratado (Al2O3.nH2O) é o componente inorgânico mais
extensivamente utilizado como retardante de chamas (XU; YAN; CHEN, 2016). Este
óxido hidratado decompõe-se endotermicamente com a liberação de vapores de água, os
quais diluem os gases combustíveis presentes na chama, favorecendo seu efeito
antichama. Outra característica de grande importância que contribui para sua aplicação
como retardante é a formação de Al2O3 como camada protetora na superfície do
produto, minimizando a difusão de oxigênio para o meio reativo, impedindo a troca de
calor (HULL; WITKOWSKI; HOLLINGBERY, 2011). Dentre as principais vantagens,
destacam-se o baixo custo e baixa toxidez, provenientes da não liberação de gases
tóxicos ou substâncias corrosivas durante a queima, atuando simultaneamente como
retardante de chama e supressor de fumaça (WANG; LIU; WANG, 2010).
Além de vários estudos envolvendo a utilização do Al2O3.nH2O como retardantes
de chama (XU; YAN; CHEN, 2016; HOFFENDAHL et al., 2015; LIANG et al., 2013;
WANG; LIU; WANG, 2010), outros trabalhos reportaram aplicações em adsorção (LIN
et al., 2016) e híbridos orgânico-inorgânicos (LIU et al., 2016a; CAMPOS; URBANO;
RIVAS, 2014a; CAMPOS; URBANO; RIVAS, 2014b; SILVA, 2013).
2.7.3. Óxido de zinco hidratado, ZnO.nH2O
Outro metal de destaque, devido à sua vasta demanda nas indústrias, é o zinco,
sendo o quarto metal mais consumindo no mundo (MOEZZI; MCDONAGH; CORTIE,
2012). Sua principal aplicação é na galvanização do aço ou ferro, como revestimento
anticorrosivo, visando as indústrias automobilística, de construção civil e de
eletrodomésticos, particularmente da linha branca (LEE, 1999; EJTEMAEI;
GHARABAGHI; IRANNAJAD, 2014). Além disso, este metal é essencial para o corpo
humano por participar de muitas reações do metabolismo celular, incluindo processos
fisiológicos, tais como função imune, defesa antioxidante, crescimento e
desenvolvimento (MAFRA; COZZOLINO, 2004; SHRIVER; ATKINS, 2008).
Em 2014, as estimativas divulgadas pelo United States Geological Suvey (USGS),
em relação às reservas mundiais de zinco, em metal contido, atingiram 234,5 Mt. Os

44
cinco maiores produtores, China, Austrália, Peru, Índia e México, respondem por 68,9
% da produção mundial, dos quais apenas 0,9 % das reservas estão localizadas no
Brasil. Cerca de 2,2 Mt deste metal situam-se nos municípios de Vazante e Paracatu, no
Estado de Minas Gerais (DNPM, 2015).
Os óxidos condutores transparentes (TCO) são materiais que apresentam várias
aplicações em eletrônica e optoeletrônica. Tais materiais são classificados como óxidos
semicondutores, com “band gap” entre 3 e 5 eV, adquirindo assim uma alta
transmitância na região do visível. São exemplos de TCO’s: SnO2, In2O3, ITO, ZnO,
GZO, entre outros (OHTA; HOSONO, 2004).
Dentre os óxidos semicondutores mencionados, evidencia-se o ZnO devido à sua
elevada atividade óptica e luminescente, além da baixa toxicidade. É um material muito
atrativo para aplicações em eletrônica e optoeletrônica por ser um semicondutor com
um “bandgap” de 3,37 eV a temperatura ambiente, tornando-o transparente à luz visível
capaz de atuar na faixa do UV. Possui energia de ligação excitônica de 60 meV, a qual
aumenta a eficiência de emissão de luz, exibindo melhor resistência à radiação sendo
ideal para aplicações aeroespaciais (RAJA; RAMESH; GEETHA, 2014;
SORNALATHA; MURUGAKOOTHAN, 2014; LEE et al., 2016). Boa estabilidade
fotoquímica, características piezoelétricas, bem como biocompatibilidade também são
propriedades intrínsecas do ZnO (MIRZAEI et al., 2016).
Devido às suas propriedades distintas, o óxido de zinco é um dos materiais
inorgânicos mais versáteis, com aplicações em dispositivos optoeletrônicos
(GHAFOURI; SHARIATI; EBRAHIMZAD, 2012), sensores (WANG et al., 2015),
células solares (PERIYAT; ULLATTIL, 2015), dentre outras. Sob a forma de
nanopartículas, é utilizado em cosméticos, pigmentos, biossensores, liberação de drogas
e agentes antibacterianos (LIU et al., 2016b).
2.8. COMPÓSITOS
A definição de compósito indica que este é formado por dois ou mais
componentes, o que implica que todo material com dois ou mais componentes (ou
fases) diferentes pode ser classificado como compósito. Entretanto, admite-se que as
fases constituintes exibam nítida diferença em suas propriedades físicas e químicas,

45
apresentando uma fase contínua e uma descontínua. A fase descontínua, ou reforço,
pode ser composta por partículas, fibras vegetais/sintéticas ou lâminas. E a fase
contínua, ou matriz, abrange os metais, as cerâmicas e os polímeros. Logo, define-se um
compósito como um material que exibe dois ou mais componentes quimicamente
diferentes que, em escala macroscópica, exibe uma interface distinta com separação
destes componentes (NETO; PARDINI, 2006; MARINUCCI, 2011). A Figura 2.17
ilustra uma representação geral de classificação, baseada na estrutura/geometria do
reforço.
Figura 2.17 - Classificação dos compósitos em função do reforço.
Fonte: Adaptada de Neto e Pardini (2006).
Analogamente, os compósitos poliméricos são definidos como materiais
multicomponentes, constituídos por elementos de reforço ou enchimento envolvidos por
uma matriz polimérica (CALLISTER Jr., 2008). A matriz polimérica pode ser
classificada como polímeros termoplásticos, termorrígidos e elastômeros.
COMPÓSITOS
PARTÍCULAS
ORIENTADAS ALEATÓRIAS
Unidirecional
Bidirecional (tecidos)
FIBRAS
DESCONTÍNUAS (CURTAS)
CONTÍNUAS (LONGAS)
Aleatórias
Orientadas
MULTICAMADA CAMADA ÚNICA
Híbridos
Laminados

46
Os polímeros termoplásticos são àqueles capazes de amolecer e fluir, quando
sujeitos a um aumento de temperatura e pressão; e endurecer, em um produto de formas
definidas, quando resfriados. Novas aplicações de temperatura e pressão conduzem a
um novo ciclo de fusão e endurecimento, sendo esta alteração uma transformação física
e reversível. Os termorrígidos ou termofixos amolecem uma única vez com o
aquecimento e tornam-se rígidos, após o processo de cura, devido à formação de
ligações cruzadas. Posteriores aquecimentos não modificam seu estado físico, pela
ocorrência de uma transformação química irreversível. Por fim, os elastômeros
abrangem tanto os termoplásticos quantos os termorrígidos. São materiais amorfos que,
à temperatura ambiente, podem ser deformados repetidas vezes em pelo menos duas
vezes o seu comprimento original. Retirado este esforço, adquirem rapidamente o
tamanho original (CANEVAROLO Jr., 2010).
Dentre os polímeros mais comumente empregados em compósitos pode-se citar
como termoplásticos polietileno (PE) e polipropileno (PP), e poliuretanos (PU’s),
poliésteres, epóxi e fenólicos, como termorrígidos (ROSÁRIO et al., 2011; PEREIRA et
al., 2015). Os elastômeros incluem as borrachas natural e sintética.
2.9. POLIURETANOS
Em relação à matriz polimérica, os poliuretanos (PU’s) ganham evidência devido
à sua versatilidade na obtenção de estruturas poliméricas, as quais abrangem os
termoplásticos, os termorrígidos e os elastômeros. Devido ao amplo conjunto de
composições possíveis, os poliuretanos possuem inúmeras aplicações comerciais, tais
como revestimentos, espumas, adesivos, selantes, couro sintético, membranas,
elastômeros e utensílios biomédicos, fazendo esta classe de polímeros uma das mais
largamente utilizadas nos âmbitos industriais e domésticos (ZIA; BHATTI; BHATTI,
2007; KURANSKA; PROCIAK, 2012).
Os PU’s podem ser definidos como copolímeros de bloco, constituídos por blocos
de polióis de baixa massa molecular, ligados covalentemente ao grupo uretano (-
NHCO-O-) (MAHMOOD et al., 2016). Em geral, são obtidos pela reação entre
isocianatos e compostos contendo grupos hidroxila ativos (polióis), bi ou poli-
funcionais, além da adição de catalisadores, extensores de cadeia, tensoativos, agentes

47
de expansão etc (VILAR, 2005; KONG; NARINE, 2007). Cerca de 95 % de todos os
isocianatos consumidos são derivados do tolueno diisocianato (TDI) e do difenilmetano
diisocianato (MDI). E, dentre a grande variedade de polióis, pode-se citar como
exemplos os polióis poliéteres, polióis poliésteres alifáticos e aromáticos, óleo de
mamona etc (VILAR, 2005; CANGEMI; SANTOS; NETO, 2009). A Figura 2.18
apresenta a reação global para a síntese dos PU’s.
Figura 2.18 - Reação global para a síntese de poliuretanos.
Fonte: Produção da autora.
No setor de espumas, no qual os poliuretanos exibem sua maior parcela de
mercado, participam como materiais pouco densos em aplicações estruturais, podendo
ser classificadas como espumas flexíveis, semirrígidas ou rígidas, dependendo dos
desempenhos mecânicos e densidade (MAHMOOD et al., 2016).
As espumas rígidas, utilizadas neste trabalho, tornam-se atrativas devido às
seguintes propriedades: baixa densidade, boas propriedades mecânicas, alta resistência
às intempéries, baixa condutividade térmica e excelente capacidade de amortecimento
(ZIELENIEWSKA et al., 2016; ESTRAVÍS et al., 2016). Tais materiais apresentam
uma variedade de aplicações, atuando como isolantes térmicos ou absorvedores de
impacto e de som, sendo imprescindíveis às indústrias automotiva, moveleira e da
construção civil (IBEH; BUBACZ, 2008; SEPTEVANI et al., 2015). Outra importante
aplicação das espumas rígidas é em painéis sanduíche, os quais são destinados aos
setores aeroespacial, ferroviário e naval (ŞERBAN et al., 2016).
As reações envolvidas na síntese de espumas de poliuretano incluem a formação
do grupo uretano, reações de reticulação e a formação da espuma, com um calor de
reação para a formação do PU de ~ 100-110 kJ.mol-1 de uretano. Durante a
+ R1 OH HO n R2 NCO OCN n
Poliol Diisocianato
O
O
C N
H
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
R2 N C
O
O R1
H
462 MORAIS, S.A.L. et al.
Na década de 1970, o governo brasileiro incentivouos reflorestamentos, e com isso grandes áreas foramutilizadas para a plantação de espécies de Eucalyptuse Pinus, entre estes o Pinus oocarpa. A maioria dosreflorestamentos se localizou no Cerrado por causado preço da terra naquela época. Porém, reflorestamentossituados em Angatuba, SP, e outras regiões alcançaramrendimentos surpreendentes (MOURA et al., 1998).
Estudos recentes têm evidenciado que híbridosdesse Pinus com Pinus caribaea var. hondurensis,Pinus tecunumanii, Pinus patula e outras espéciessão promissores, em virtude da qualidade da madeirae da facilidade de propagação (MOURA et al., 1998).
Durante o incentivo aos programas dereflorestamento, no início dos anos da década de 1970,largas áreas de plantações de Pinus oocarpa foramestabelecidas, principalmente na região do Cerradobrasileiro, que compõe, aproximadamente, 25% doterritório nacional (MOURA et al., 1998).
No Quadro 1, apresenta-se a quantidade aproximadaem que os constituintes macromoleculares estruturais(celulose, hemiceluloses e lignina) e extrativos estãopresentes nas madeiras de coníferas, folhosas e gramíneas(THOMAS, 1977; SJÖSTRÖM,1993; IRANMAHBOOBet al., 2002).
A celulose é um polissacarídeo constituído porunidades monoméricas de b-D-glucose. Na Figura 1,apresenta-se a formação da molécula de celulose viaeliminação de água (SJÖSTRÖM, 1993).
A molécula de celulose do algodão consiste deaproximadamente 15.000 unidades de glucose, já nacelulose de madeiras há cerca de 10.000 unidades. Cadaunidade de glucose possui massa de 162 u (unidadesde massa atômica). Portanto, ao dividir a massa molecularda celulose por 162, obtém-se seu grau de polimerização(GP), equação 1. Na celulose nativa, o GP varia de 7.000a 15.000 (PHILIPP e D’ALMEIDA, 1988; SJÖSTRÖM,1993).
(1)
Componentes Composição (%)
Químicos Madeiras de Madeiras de GramíneasConíferas Folhosas
Celulose 42 ± 2 45 ± 2 36 ± 5Hemiceluloses 27 ± 2 30 ± 5 27 ± 3Lignina 28 ± 3 20 ± 4 11 ± 3Extrativos 3 ± 2 3 ± 2 26 ± 5
Quadro 1 – Composição química aproximada dos constituintesde madeiras de coníferas, folhosas e gramíneas
Table 1 – Average chemical composition of softwoods, hardwoodsand grass
Figura 1 – Formação da cadeia de celulose pela união de unidades β-D-glucose.Figure 1 – Schematic illustration of the cellulose chain formation.
R. Árvore, Viçosa-MG, v.29, n.3, p.461-470, 2005
Grupo uretano n
Poliuretano

48
polimerização dos PU’s, podem ocorrer reações paralelas ou secundárias, envolvendo o
grupo isocianato, como a formação do grupo alofanato, ureia distribuída e biureto
(MAHMOOD et al., 2016). Segundo Antunes et al. (2011), a formação das espumas de
PU ocorre devido à geração in situ do agente de expansão (CO2) durante polimerização
exotérmica, conduzindo a espumas reticuladas com estruturas celulares.
No entanto, as espumas de PU possuem algumas limitações, dentre as quais se
destacam a baixa estabilidade oxidativa e alta flamabilidade, acarretando no uso intenso
e frequente de aditivos para permitir o seu emprego (CHATTOPADHYAY;
WEBSTER, 2009). Após a ignição, estas espumas geram uma grande quantidade de
calor num curto período de tempo, com a liberação de gases tóxicos, como CO, CO2,
HCN, metanol, piridina, acetonitrila, acrilonitrila e demais componente nocivos à saúde
humana (XI et al., 2015; YANG et al., 2015).
Tendo em vista que a formação da espuma de poliuretano decorre principalmente
da formação de CO2 in situ, durante a reação exotérmica de polimerização de polióis e
isocianato ambos sob a forma líquida, a incorporação de aditivos bem como a obtenção
de compósitos é facilitada por essa rota (KURANSKA; PROCIAK, 2012). Entretanto, a
incorporação de retardantes de chama halogenados, principal classe de aditivos
convencionais utilizados para esse fim em espumas de poliuretano, acarretam
geralmente em perdas de propriedades mecânicas em função das altas quantidades e da
baixa interação química entre a carga e a matriz polimérica.
Diferentes aditivos tem sido incorporados ao PU, em pequenas quantidades,
visando potenciais propriedades mecânicas e térmicas (PARK; OH; KIM, 2013). Em
função do baixo custo, do baixo peso específico, do alto módulo, da resistência à tração
e da presença de hidroxilas nas superfícies, as fibras lignocelulósicas tem atraído grande
interesse na busca de espumas/compósitos leves de poliuretano, com propriedades
mecânicas magnificadas em função da grande adesão entre fibra-matriz.
A densidade apresenta-se como um dos parâmetros físicos mais importantes pois
tem influência significativa nas propriedades mecânicas e térmicas das espumas de PU
e, consequentemente, nos respectivos compósitos (PISZCZYK et al., 2012; YANG et
al., 2015). De acordo com Cordero et al. (2015) a adição de componentes celulósicos
implica na reação destes com o isocianato, formando ligações uretânicas adicionais, as

49
quais podem influenciar nas propriedades mecânicas e térmicas dos compósitos. A
estrutura polimérica deve ser firme o suficiente para resistir às tensões, pois a aplicação
de esforços externos pode deformar a estrutura celular, conduzindo ao colapso das
células (VILAR, 2005).
Mosiewicki et al. (2015) observaram que a adição de celulose nanométrica, em
proporções acima de 3 % m/m não influenciaram significativamente nos valores de
densidade aparente nas espumas de PU. Estravís et al. (2016) relataram um aumento na
densidade com a adição de argilas. Segundo os autores, esta tendência poderia estar
relacionada ao aumento da viscosidade da mistura reacional, antes do início da reação
de síntese do PU. Entretanto, as variações nos valores de densidade foram consideradas
mínimas pelos autores.
Por outro lado, Cordero et al. (2015) reportaram que a incorporação de celulose
nanocristalina pode aumentar ou diminuir a densidade aparente das espumas, pois tal
propriedade está diretamente relacionada à formulação de síntese do PU. Segundo os
autores, as alterações observadas nos valores de densidade ocorreram devido à atuação
da celulose nanocristalina como um agente de nucleação durante a fase de expansão;
enquanto o melhoramento das propriedades mecânicas à provável migração da celulose
para os segmentos rígidos, formando ligações adicionais na rede polimérica.
Segundo trabalhos reportados, a adição de compostos derivados do fósforo como
retardantes de chama (ZHANG et al., 2014; LUO et al., 2015; XI et al., 2015; XU;
WANG; ZHENG, 2015; YANG et al., 2015); argilas (PISZCZYK et al., 2012; PAUZI
et al., 2014; NIKKHAH et al., 2015; ESTRAVÍS et al., 2016) e componentes
celulósicos (PARK; OH; KIM, 2013; RIBEIRO DA SILVA et al., 2013a; WU et al.,
2014; CORDERO et al., 2015; HADJADJ et al., 2016; ZHOU; SAIN; OKSMAN,
2016), são alguns exemplos que mostraram-se promissores no que tange a melhoria de
propriedades e na versatilidade de aplicações das espumas de PU.
Conforme mencionado anteriormente neste trabalho, os óxidos metálicos
hidratados demonstram potenciais aplicações como retardantes de chama atóxicos,
permitindo uma maior adesão fibra-matriz por possuírem hidroxilas em suas superfícies,
as quais podem reagir com grupos isocianatos presentes nos precursores da matriz de
PU. Além disto, como já supramencionado, encontram-se estudos da obtenção de

50
híbridos de celulose/óxidos metálicos hidratados, materiais que potencialmente podem
vir a prover um ganho combinado de retardância à chama e de propriedades mecânicas,
sem comprometer a densidade do compósito (OTTOBONI, 2011; SILVA, 2013). No
entanto, até o presente momento não encontram-se menções da avaliação deste tipo de
material voltado para o setor aeroespacial.
Diante do exposto, esta tese visa o desenvolvimento de novos materiais
compósitos utilizando uma matriz polimérica de poliuretano reforçada com híbridos
provenientes da combinação entre a celulose da folha de bananeira e os óxidos
metálicos hidratados de Zn, Al e Nb. Considerando as diversas características
intrínsecas de cada metal, estes aditivos híbridos poderão fornecer propriedades únicas
ao material final, acarretando um alto valor agregado e aplicações mais nobres. Além da
preparação e caracterização físico-química dos compósitos, foram realizados estudos da
melhor proporção tanto dos aditivos híbridos quanto dos compósitos, assim como
ensaios mecânicos.

51
3. OBJETIVOS
3.1. OBJETIVO GERAL
Desenvolver aditivos híbridos provenientes da combinação entre a celulose da
folha de bananeira e os óxidos metálicos hidratados de alumínio (Al2O3.nH2O), de zinco
(ZnO.nH2O) e de nióbio (Nb2O5.nH2O) para a produção de compósitos com matriz de
poliuretano.
3.2. OBJETIVOS ESPECÍFICOS
! Avaliar o tratamento químico mais eficiente para o isolamento da celulose a
partir de folhas de bananeira;
! Caracterizar as diferentes celuloses obtidas e definir o tratamento químico;
! Sintetizar e caracterizar os óxidos metálicos hidratados de alumínio
(Al2O3.nH2O), de zinco (ZnO.nH2O) e de nióbio (Nb2O5.nH2O);
! Sintetizar e caracterizar os híbridos orgânico-inorgânicos, (100-
x)Celfb/xMyOz.nH2O, em diferentes proporções mássicas celulose/óxido
metálico hidratado;
! Avaliar e definir a melhor composição de cada híbrido;
! Sintetizar e caracterizar a matriz polimérica de poliuretano (PU);
! Preparar e caracterizar os compósitos: PU + Celfb, PU + MyOz.nH2O e PU +
(100-x)Celfb/xMyOz.nH2O;
! Estudar e definir a melhor relação matriz polimérica/celulose;
! Estudar e definir a melhor relação matriz polimérica e óxidos: PU/Nb2O5.nH2O,
PU + Al2O3.nH2O, PU + ZnO.nH2O;
! Estudar e definir a melhor relação matriz polimérica e híbridos: PU + (100-
x)Celfb/xNb2O5.nH2O, PU + (100-x)Celfb/xAl2O3.nH2O, PU + (100-
x)Celfb/xZnO.nH2O;
! Estudar os efeitos térmico e mecânico nos compósitos preparados .

52

53
4. MATERIAIS E MÉTODOS
Exceto por algumas caracterizações realizadas no Laboratório Associado de
Sensores e Materiais (LAS) do Instituto Nacional de Pesquisas Espaciais (INPE), todo o
projeto foi desenvolvido no Laboratório de Novos Materiais, alocado no Departamento
de Engenharia Química (LOQ) da Escola de Engenharia de Lorena da Universidade de
São Paulo (EEL/USP), devido à infraestrutura já disponível.
A seguir são apresentados os materiais, equipamentos e métodos utilizados para o
desenvolvimento deste trabalho.
4.1. MATERIAIS
4.1.1. Folhas de bananeira (Musa sapientum)
Para obtenção da fração celulósica foram coletadas folhas verdes e secas, de
bananeiras com mesmas localidade e espécie (prata), na cidade de Lorena-SP.
4.1.2. Reagentes de síntese para a matriz de poliuretano
Os reagentes específicos para a síntese foram gentilmente cedidos pela empresa
DOW, localizada em Jundiaí-SP, e utilizados como recebidos. A Tabela 4.1 apresenta
as informações referentes aos componentes.
Tabela 4.1 - Reagentes utilizados para a síntese do poliuretano. Reagente Nome comercial Constituintes
Isocianato
VoracorTM CE 101
Diisocianato de difenilmetano, isômeros e homólogos
Mistura de Poliol Poliéter e aditivos
VoracorTM CR 873
Poliol poliéter, glicerol óxido de propileno; 1,1-dicloro-1-
fluoretano; propileno glicol; amina alcoxilada e DMCHA
Fonte: Ficha de informação de segurança de produto químico (FISQP), fornecida pela
empresa Dow.

54
4.1.3. Outros reagentes
Os reagentes descritos foram utilizados conforme recebidos:
! Acetona, C3H6O, 99,5% m/m, Synth;
! Ácido acético glacial, CH3COOH, 100 % m/m, Merck;
! Ácido clorídrico, HCl, 36,5% m/m, Synth;
! Ácido fluorídrico, HF, 48 % m/m, Nuclear;
! Ácido nítrico, HNO3, 65 % m/m, Synth;
! Álcool etílico, C2H6O, 95 % v/v, Synth;
! Alumínio metálico, P. D. Indústria e Comércio Ltda;
! Ar sintético grau 5.0, 99,999 %, Air Liquide;
! Clorofórmio, CHCl3, resíduo de coluna cromatográfica, Departamento de
Engenharia Química, LOQ, EEL/USP;
! Diclorometano, CH2Cl2, resíduo de coluna cromatográfica, Departamento de
Engenharia Química, LOQ, EEL/USP;
! Hidróxido de amônio, NH4OH, 28-30 % m/m, Synth;
! Hidróxido de sódio, NaOH, 99 % m/m, Merck;
! Nióbio metálico, Departamento de Engenharia de Materiais, LOM, EEL/USP;
! Nitrato de prata, AgNO3, 99,8% m/m, Cromoline;
! Nitrogênio gasoso, N2, grau 5.0, 99,999 %, Air Liquide;
! Tetraidrofurano, C4H8O, resíduo de coluna cromatográfica, Departamento de
Engenharia Química, LOQ, EEL/USP;
! Zinco metálico, com granulometria de 20 mesh, Pentaflex.
4.2. EQUIPAMENTOS
4.2.1. Laboratório de Novos Materiais, Departamento de Engenharia Química
(LOQ), EEL/USP
! Agitador de peneiras, Bertel;
! Agitador magnético com aquecimento Ika, RCT Basic;
! Agitador manual pressurizado Atlas Copco;
! Balança analítica Chyo Balance Corporation, JK-200;

55
! Compressor Chiaperini, série 10248;
! Estufa elétrica Nova Ética, 402-3D;
! Moinho de facas, Marconi, MA-340;
! Mufla elétrica Quimis, Q 318-24; ! ! Termobalança Shimadzu, TGA-50.
4.2.2. Laboratório de Polímeros, Departamento de Engenharia Química
(LOQ), EEL/USP
! Calorímetro de Varredura Diferencial TA Instruments, Q20.
4.2.3. Laboratório de Infravermelho, Departamento de Biotecnologia (LOT),
EEL/USP
! Espectrômetro de infravermelho Perkin Elmer, Spectrum GX.
4.2.4. Laboratório de Ensaios Mecânicos, Departamento de Materiais (LOM),
EEL/USP
! Máquina Universal de Ensaios Mecânicos EMIC, DL 3000.
4.2.5. Laboratório Associado de Sensores e Materiais (LAS), INPE
! Difratômetro de raios X X PANalytical, X’Pert Powder;
! Espectrômetro de energia dispersiva de raios X Bruker, XFLASH 5010;
! Microscópio eletrônico de varredura Jeol, JMS-5310;
! Microscópio eletrônico de varredura de alta resolução, Tescan, Mira 3.
4.3. MÉTODOS
Neste presente trabalho, inicialmente, foram isolados e sintetizados os materiais
necessários para o desenvolvimento dos compósitos: celulose, óxidos metálicos
hidratados, híbridos orgânico-inorgânico e matriz polimérica. Cada componente obtido
foi caracterizado individualmente. No caso dos materiais multicomponentes (híbridos e
compósitos), os mesmos foram caracterizados e comparados com seus precursores.

56
4.3.1. Preparação das folhas de bananeira, Fb
As folhas coletadas foram lavadas com água corrente e, em seguida, picadas
manualmente, para a remoção de suas nervuras (talo central da folha) e mantidas à
sombra para secagem. As folhas secas foram trituradas em um liquidificador doméstico,
somente para redução de tamanho, não havendo necessidade de uma uniformização.
4.3.2. Isolamento da celulose a partir das folhas de bananeira, Celfb
Para verificar qual a melhor proposta de tratamento químico para o isolamento da
celulose foram avaliadas três diferentes metodologias, de acordo com a Tabela 4.2. As
metodologias propostas baseiam-se em trabalhos anteriores do grupo de pesquisa do
Laboratório de Novos Materiais da EEL/USP.
Tabela 4.2 – Tratamentos químicos empregados para o isolamento da celulose. Metodologia Tratamento Químico
Proposta 1 (P1) 1 ataque ácido + 1 ataque básico
Proposta 2 (P2) 2 ataques ácidos
Proposta 3 (P3) 2 ataques ácidos + 1 ataque básico
Fonte: Produção da autora.
a) Ataque ácido
Neste tratamento químico empregou-se uma adaptação da metodologia proposta
por Brendel; Iannetta; Stewart (2000). Em um erlenmeyer, 10,0 g de folhas de
bananeira, previamente secas e trituradas, foram adicionadas a uma mistura ácida
contendo 200,00 mL de solução 80 % v/v de ácido acético e 23,00 mL de ácido nítrico
65 % m/m. O sistema foi mantido sob agitação, em banho de glicerina, a 120 °C por 20
min. O conteúdo foi resfriado à temperatura ambiente, filtrado a vácuo, lavado com
água deionizada até pH neutro e duas vezes com etanol para a remoção de resíduos
orgânicos.

57
b) Ataque básico
A celulose isolada, proveniente do ataque ácido, foi transferida quantitativamente
para um béquer e imersa em uma mistura de 40,00 mL de solução de hidróxido de sódio
1,5 mol.L-1 e 40,00 mL de água deionizada. O sistema foi mantido sob aquecimento a
40 °C por 30 min. Em seguida, foi filtrado a vácuo, lavado com água deionizada até pH
neutro e duas vezes com etanol para a remoção de resíduos orgânicos (SILVA, 2013).
As celuloses obtidas ao final de cada proposta (Tabela 4.2) foram secas em estufa
a 50 °C até peso constante. Para evitar a proliferação de fungos durante armazenamento
em dessecador, adicionou-se 10,00 mL de acetona. Uma vez definido o tratamento
químico adequado, foram preparados vários lotes a fim de obter a quantidade suficiente
para todo o desenvolvimento do trabalho. Em seguida, toda a celulose adquirida foi
pulverizada e peneirada em 20 mesh. As Figuras 4.1 e 4.2 apresentam os fluxogramas
referentes aos ataques ácido e básico, respectivamente.
Figura 4.1 - Fluxograma para o isolamento da celulose via tratamento ácido.
Fonte: Produção da autora.
FOLHA BRUTA
Ataque ácido CH3COOH/HNO3
10:1
Agitação T = 120°C; t = 20 min
Resfriamento Tamb
Filtração a vácuo
2° Ataque ácido
Secagem T = 50°C
Celfb
Lavagem

58
Figura 4.2 - Fluxograma para o isolamento da celulose via tratamento básico.
Fonte: Produção da autora.
4.3.3. Síntese dos óxidos metálicos hidratados, MyOz.nH2O
Os óxidos metálicos hidratados de nióbio (Nb2O5.nH2O), de alumínio
(Al2O3.nH2O) e de zinco (ZnO.nH2O) foram sintetizados pelo método de precipitação
convencional (PC) (RODRIGUES; MULINARI; DA SILVA, 2009). Primeiramente, o
metal foi dissolvido em 10,00 mL de ácido concentrado e precipitado lentamente com
solução básica, sob constante agitação de 300 rpm, até pH ideal. Este pH foi definido
mediante estudos prévios realizados pelo Grupo de Pesquisas do Laboratório de Novos
Materiais da EEL/USP. Em seguida, o precipitado foi filtrado a vácuo, lavado e seco em
estufa à 50 °C até peso constante. A Tabela 4.3 apresenta as condições de síntese e
lavagem para cada óxido metálico hidratado.
CELULOSE
Ataque básico Sol. NaOH 1,5 mol.L-1
Agitação T = 40°C; t = 30 min
Filtração a vácuo
Lavagem
Secagem T = 50°C
Celfb

59
Tabela 4.3 - Condições de síntese e lavagem para os óxidos metálicos hidratados. Material Ácido Agente
precipitante pH Digestão do
precipitado Lavagem
Nb2O5.nH2O
Mistura de HNO3/HF (1:3) v/v
Solução aquosa de NH4OH (1:3) v/v
8,0
Não
H2Odeio.
até pH neutro
Al2O3.nH2O
HCl
Solução aquosa de NH4OH (1:3) v/v
9,0
Aquecimento até 50°C por
10 min
H2Odeio. até remoção de
íons Cl-
ZnO.nH2O
HCl
Solução aquosa de NaOH
1,5 mol.L-1
10,0
Não
H2Odeio. até remoção de
íons Cl-
Fonte: Produção da autora.
! A Figura 4.3 apresenta o fluxograma para a síntese dos óxidos metálicos
hidratados.
Figura 4.3 - Fluxograma para a síntese dos óxidos metálicos hidratados via precipitação convencional (PC).
Fonte: Produção da autora.
Dissolução ácido concentrado
Agitação Tamb
Precipitação solução básica
Ajuste de pH
Filtração a vácuo
Lavagem H2O deionizada
Secagem T = 50°C
MyOz.nH2O
M = Zn, Al ou Nb
pH neutro Eliminação de
íons Cl-
Nb Al, Zn

60
4.3.4. Calcinação dos óxidos metálicos hidratados, MyOz.nH2O
Os óxidos hidratados foram calcinados, segundo uma adaptação do procedimento
apresentado por Tagliaferro; Da Silva; Da Silva (2005). Este processo tem como
finalidade a remoção das moléculas de água presentes nos materiais, para a
determinação das estruturas cristalinas dos óxidos sintetizados e posterior confirmação a
partir do perfil de difração, obtida pela comparação com as fichas JCPDS (Joint
Committee on Powder Diffraction Standards). Para este procedimento a temperatura de
calcinação (Tcal.) foi obtida a partir das curvas TGA de cada óxido, sendo esta a
temperatura na qual o material não apresenta eventos de perda de massa.
Em cadinhos de porcelana, pesou-se aproximadamente 1,0000 g de cada óxido e
os mesmos foram colocados em mufla com a temperatura inicial registrada em 200 °C.
Em seguida, iniciou-se a rampa de aquecimento, com uma taxa de 100 °C até a Tcal.. Os
cadinhos foram mantidos na Tcal. (definidas pelas curvas TGA) por 6 h e, após este
período, iniciou-se a rampa de resfriamento (sob a mesma taxa) até 200 °C. Os óxidos
anidros foram devidamente armazenados e mantidos em dessecador.
4.3.5. Síntese dos híbridos orgânico-inorgânico, (100-x)Celfb/xMyOz.nH2O
Os híbridos foram sintetizados pelo método PC, como descrito no item 4.3.3,
porém com a adição da celulose, previamente intumescida em água deionizada por 1 h,
à solução ácida contendo o metal dissolvido. O híbrido resultante foi seco em estufa a
50 °C até peso constante.
Para cada híbrido foi realizado um estudo de proporção para avaliar a melhor
relação entre celulose/óxido metálico hidratado. As variações de massas para o
respectivo estudo encontram-se descritas na Tabela 4.4.
Tabela 4.4 - Estudo de proporções dos híbridos orgânico-inorgânico. HÍBRIDO PROPORÇÃO (%m/m)
(100-x)Celfb/xNb2O5.nH2O x = 3, 5, 10, 15 e 20
(100-x)Celfb/xAl2O3.nH2O x = 3, 4, 5, 6, 7, 10, 15 e 20
(100-x)Celfb/xZnO.nH2O x = 3, 4, 5, 6, 7, 10, 15 e 20
Fonte: Produção da autora.

61
A Figura 4.4 demonstra o fluxograma para a síntese dos híbridos orgânico-
inorgânicos.
Figura 4.4 - Fluxograma para a síntese dos híbridos orgânico-inorgânicos via precipitação convencional (PC).
Fonte: Produção da autora.
4.3.6. Preparação da matriz de poliuretano, PU
A matriz polimérica PU foi sintetizada, segundo uma adaptação da metodologia
apresentada por Antunes et al. (2011), pelo processo denominado de “one shot”. Neste
processo, poliol (com surfatantes, aditivos e catalisadores) e isocianato são misturados
sob intensa agitação mecânica, com a formação e a expansão da matriz de maneira
imediata.
Em um copo de polietileno, transferiu-se a quantidade necessária da fase poliol
(fase A) e, em seguida, o isocianato (fase B). Os componentes foram misturados com o
auxílio de um agitador manual pressurizado, a 3000 rpm por 10 s. O “creme” formado
foi vertido em um molde cúbico (18x18x18 cm), forrado com folhas plásticas, no qual
Dissolução ácido concentrado
Adição de Celfb intumescida
Agitação Tamb; t = 5 min
Precipitação solução básica
Ajuste de pH
Filtração a vácuo
(100-x)Celfb/xMyOz.nH2O
M = Zn, Al ou Nb
Lavagem H2O deionizada
Secagem T = 50°C
pH neutro Eliminação de
íons Cl-
Nb Al, Zn

62
ocorreu a expansão do poliuretano. Este molde foi mantido sob exaustão em capela,
devido à liberação de gases, até resfriamento à temperatura ambiente. O bloco de
poliuretano foi retirado do molde e exposto ao ar, sob o abrigo de luz, por 24 h para
total processo de cura. A Figura 4.5 exibe uma representação esquemática para a síntese
do PU.
Figura 4.5 - Esquema para a síntese do poliuretano.
Fonte: Produção da autora.
A quantidade utilizada de isocianato em partes por cem de poliol foi de 115, isto
é, um excesso de 15 partes em relação ao poliol. Entretanto, devido à alta viscosidade
dos componentes, foi calculado um excesso de 10 % em ambas as fases, para minimizar
perdas. A relação entre as fases, o tempo e a velocidade de agitação foram emitidos pela
empresa. Estes parâmetros também foram considerados na preparação dos compósitos.
4.3.7. Preparação dos compósitos
Os compósitos foram preparados de maneira análoga à matriz polimérica, como
descrito no item 4.3.6, porém com a dispersão dos aditivos na fase poliol, realizada
manualmente (Figura 4.6). As quantidades dos materiais adicionados à matriz
polimérica foram baseadas em estudo reportado por Ribeiro da Silva et al. (2013a),
variando de 1 a 5 % m/m em relação à massa da fase poliol, para todos os compósitos
avaliados. Logo, os seguintes compósitos foram sintetizados:
Agitação 3000 rpm
t = 10 s Resfriamento
Tamb
Fase A Poliol
Fase B Isocianato
Copo de polietileno
Caixa de papelão
Bloco de PU

63
! Matriz + celulose, PU + Celfb;
! Matriz + óxido de nióbio hidratado, PU + Nb2O5.nH2O;
! Matriz + óxido de alumínio hidratado, PU + Al2O3.nH2O;
! Matriz + óxido de zinco hidratado, PU + ZnO.nH2O;
! Matriz + melhor proporção híbrido de nióbio, PU + (100-x)Celfb/xNb2O5.nH2O;
! Matriz + melhor proporção híbrido de alumínio, PU + (100-x)Celfb/xAl2O3.nH2O;
! Matriz + melhor proporção híbrido de zinco, PU + (100-x)Celfb/xZnO.nH2O.
Todas as sínteses dos compósitos foram realizadas em copos de polietileno, os
quais foram limpos com solventes provenientes de coluna cromatográfica. Este
procedimento de limpeza foi mantido entre as sínteses para um mesmo aditivo, com a
troca do copo somente na troca de material.
Figura 4.6 - Esquema para a síntese dos compósitos de poliuretano com os aditivos da
formulação, sendo aditivo: Celfb, MyOz.nH2O ou (100-x)Celfb/xMyOz.nH2O.
Fonte: Produção da autora.
4.3.8. Corte e amostragem dos compósitos
Os blocos de compósitos foram cortados com um arco de serra, para a realização
de uma amostragem e devidas caracterizações. Inicialmente, as faces do bloco foram
identificadas como 1, 2 e 3, sendo as faces 1 e 2 paralelas entre si e a face 3 oposta à
etiqueta de identificação do respectivo bloco. Estas faces foram retiradas e descartadas.
Em seguida, cortou-se uma fatia de aproximadamente 1 cm de espessura, do lado oposto
Agitação 3000 rpm
t = 10 s Resfriamento
Tamb
Fase A Poliol Fase B
Isocianato
Copo de polietileno
Caixa de papelão
Bloco de compósito
Aditivo
Agitação manual t = 10 s
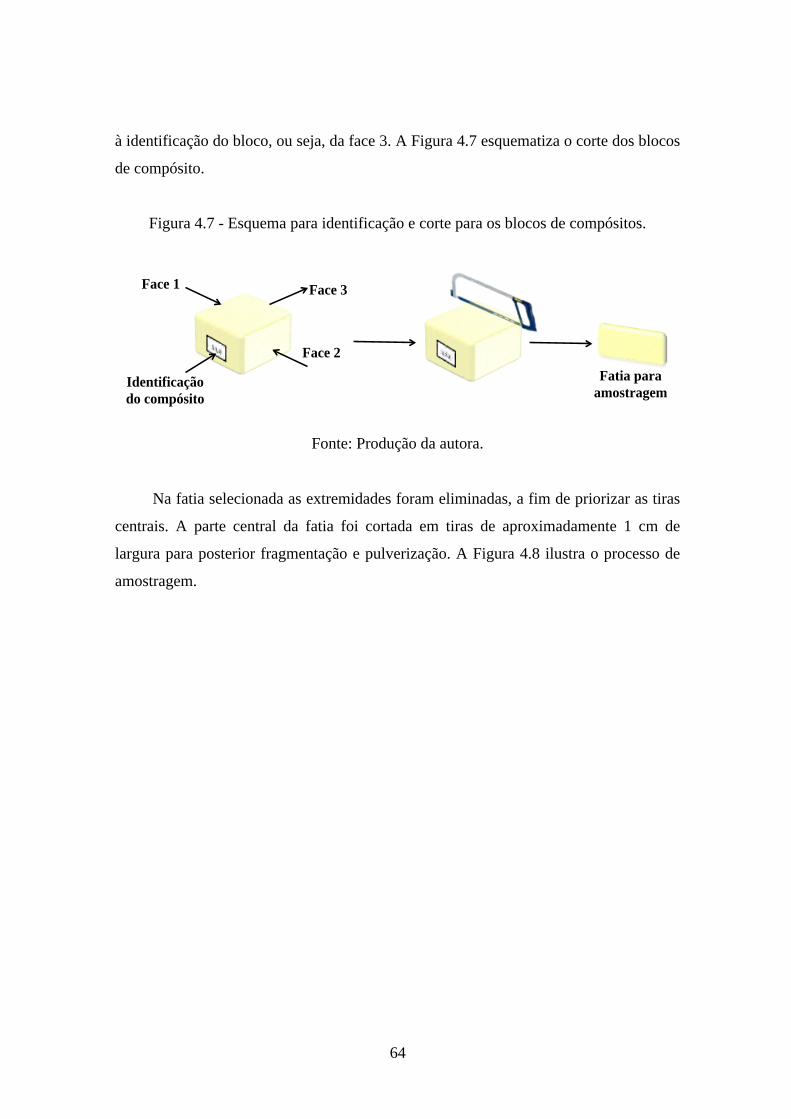
64
à identificação do bloco, ou seja, da face 3. A Figura 4.7 esquematiza o corte dos blocos
de compósito.
Figura 4.7 - Esquema para identificação e corte para os blocos de compósitos.
Fonte: Produção da autora.
Na fatia selecionada as extremidades foram eliminadas, a fim de priorizar as tiras
centrais. A parte central da fatia foi cortada em tiras de aproximadamente 1 cm de
largura para posterior fragmentação e pulverização. A Figura 4.8 ilustra o processo de
amostragem.
Face 2
Face 3 Face 1
Identificação do compósito
Fatia para amostragem

65
Figura 4.8 - Esquema para amostragem dos compósitos.
Fonte: Produção da autora.
4.3.9. Fragmentação e pulverização dos compósitos
Após o processo de amostragem, as tiras centrais foram fragmentadas em
pequenos cubos e transferidas para o copo de acrílico de um processador doméstico. As
amostras foram pulverizadas por 30 s, tempo suficiente para obtê-las sob a forma de pó.
As amostras foram armazenadas em recipientes de polietileno e devidamente
identificadas.
4.4. CARACTERIZAÇÕES
4.4.1. Teste de absorção de água
A quantidade de água absorvida pelas folhas de bananeira (Fb), celulose (Celfb) e
híbridos definidos foi determinada pela imersão dos mesmos (cerca de 1±0,0001 g) em
100,00 mL de água deionizada (LOPES et al., 2011). Os materiais intumescidos foram
Extremidade da fatia
Extremidade da fatia
Tiras centrais

66
filtrados em papel de filtro, secos superficialmente e pesados em balança analítica após
os seguintes tempos: 1, 2, 3, 4, 6, 12, 24 e 48 h.
4.4.2. Espectroscopia de infravermelho com transformada de Fourier, FTIR
A espectroscopia de absorção no infravermelho fundamenta-se na conversão da
radiação absorvida pelas moléculas da amostra em energia de vibração molecular. A
partir das vibrações moleculares, deformações axial ou angular, obtém-se um espectro
com as bandas características (grupos funcionais, insaturações, etc) possibilitando sua
identificação (SILVERSTEIN, 2012).
A presença e a intensidade de grupos funcionais ligados à estrutura dos materiais
foram determinadas por FTIR. As análises foram conduzidas na faixa de comprimento
de onda de 400 a 4000 cm-1, com uma resolução de 4 cm-1, diretamente por refletância
difusa (DRIFT), sem necessidade de preparo com KBr.
4.4.3. Difratometria de raios X, DRX
Os raios X apresentam-se como uma forma de radiação eletromagnética, de altas
energias e curtos comprimentos de onda, na ordem dos espaçamentos atômicos dos
materiais sólidos (CALLISTER Jr., 2008). A incidência destes raios permite a
determinação dos espaçamentos interplanares e parâmetros da rede cristalina do
material, possibilitando a identificação da respectiva estrutura, assim como sua fração
(percentual) cristalina (CANEVAROLO Jr, 2004).
A cristalinidade dos materiais foi medida em um difratômetro, com fonte radiação
CuKα. As amostras foram analisadas a temperatura ambiente, para valores de 2θ entre
10 e 90°, passo de 0,02 e tempo por passo de 10 s, com uma corrente e voltagem de
aceleração de 40 mA e 45 kV, respectivamente.
A partir dos difratogramas foi possível estimar o índice de cristalinidade (Ic) dos
materiais, de acordo com a Equação 4.1 (BUSCHLE-DILLER; ZERONIAN, 1992):
!! = !!!!!!!!
×100 (4.1)

67
Sendo:
I1 = intensidade mínima do vale, referente à fração amorfa;
I2 = intensidade máxima do pico de difração, referente à fração cristalina.
4.4.4. Termogravimetria e sua derivada, TGA/DTG
A termogravimetria pode ser definida como um processo contínuo, no qual mede-
se a variação de massa (perda ou ganho) de um determinado material, em função do
tempo e/ou da temperatura, mediante uma programação controlada de temperatura
(MOTHÉ, 2009). A partir desta técnica é possível acompanhar alterações de massa e
estabelecer os intervalos de temperatura, mediante as transformações físicas
(sublimação, evaporação, condensação) ou químicas (degradação, decomposição,
oxidação) ocorridas durante o aquecimento (CANEVAROLO Jr, 2004).
O comportamento térmico de todos os materiais foi avaliado por
termogravimetria, utilizando cadinho de platina. As análises foram conduzidas sob
atmosfera de ar sintético com vazão de 50 mL.min-1 e taxa de aquecimento de 20
°C.min-1, no intervalo da temperatura ambiente até 900 °C. Para os óxidos metálicos
hidratados, a partir dos dados das curvas TGA/DTG, foi possível calcular o grau de
hidratação (n), segundo a Equação 4.2 (TAGLIAFERRO; DA SILVA; DA SILVA,
2005):
18!! = !!!(!!!!!)!"" (4.2)
Sendo:
X = porcentagem de massa de água liberada;
M = massa molecular do óxido resultante da análise térmica;
n = número de moléculas de água.
4.4.5. Calorimetria Exploratória Diferencial, DSC
A calorimetria exploratória diferencial define-se como uma técnica
termoanalítica, na qual medem-se as temperaturas e o fluxo de calor associado com às
transições da amostra, em relação a um material de referência termicamente inerte,

68
submetidos a uma programação controlada de temperatura. Estas medidas
proporcionam informações qualitativas e quantitativas sobre mudanças físicas e
químicas, as quais incluem processos endotérmicos, exotérmicos ou mudanças na
capacidade calorífica (MOTHÉ, 2009).
Os calores envolvidos e as temperaturas de transição dos materiais definidos
foram avaliados por calorimetria exploratória diferencial. As análises foram realizadas
em cadinho de alumínio selado, sob atmosfera de nitrogênio, com vazão de 50 mL.min-1
e taxa de aquecimento de 10 °C.min-1 , no intervalo da temperatura ambiente até 400
°C.
4.4.6. Microscopia eletrônica de varredura e espectroscopia de energia
dispersiva de raios X, MEV/EDX
As características microestruturais de um material, tais como tamanho e forma de
grão, composição, distribuição de fases, defeitos cristalinos, superfícies, etc, podem ser
determinadas por técnicas de microscopia. Na microscopia eletrônica ocorre a interação
do feixe de elétrons incidente com a superfície da amostra, na qual parte do feixe é
refletida e parte coletada pelo detector (CALLISTER Jr., 2008). Esta interação provoca
a emissão de elétrons secundários, retroespalhados, Auger e raios X característicos.
As superfícies da folha bruta, celulose e híbridos foram investigadas utilizando-se
um microscópio eletrônico de varredura, com uma aceleração de voltagem de 20 kV. As
amostras secas foram fixadas em um suporte, com o auxílio de uma fita de carbono
autocolante dupla face, submetidas ao recobrimento metálico com ouro e analisadas por
elétrons secundários. Os elementos foram detectados por um espectrômetro de energia
dispersiva de raios X, sem a necessidade de recobrimento com ouro.
Para a matriz e compósitos de PU, utilizou-se um microscópio eletrônico de
varredura de alta resolução, com uma aceleração de voltagem de 5 kV. O preparo das
amostras procedeu-se analogamente aos demais materiais.
4.4.7. Densidade aparente
A densidade aparente dos compósitos de PU foi determinada calculando-se a
média dos resultados da razão entre a massa e o volume de seis CP’s por material, nas

69
diferentes proporções de aditivos, segundo Narine et al. (2007). As medições de massa
foram realizadas em balança analítica com precisão de ± 0,0001 g, enquanto as
dimensões das amostras cúbicas foram efetuadas com o auxílio de um paquímetro
analógico com precisão de ± 0,1 mm.
4.4.8. Ensaios de compressão
Os CP’s, sob a forma de cubos com dimensões iguais a 45 mm, foram cortados a
partir dos blocos dos respectivos compósitos PU + Celfb; PU + MyOz.nH2O e PU +
(100-x)Celfb/xMyOz.nH2O. Os ensaios de compressão foram conduzidos a temperatura
ambiente, em uma Máquina Universal de Ensaios Mecânicos, de acordo com a norma
ASTM D1621-10. A força de compressão de 5 kN foi aplicada no sentido contrário ao
da expansão dos compósitos (eixo Z), com uma velocidade de 2,5 mm.min-1. Para o
primeiro CP de cada material a compressão foi realizada até o limite final do ensaio (~ 5
mm), a fim de avaliar a capacidade de absorção de energia na deformação e,
consequentemente, a recuperação do material celular. Para os demais CP’s, a carga
compressiva foi aplicada até 25 % de deformação.
Os ensaios de compressão foram realizados em seis corpos de prova para cada
compósito produzido. Os valores de resistência à compressão foram calculados
baseados no método padrão, para deformações de 10 %; e os respectivos módulos de
Young foram calculados a partir da Lei de Hooke, ambos segundo a norma ASTM
D1621-10.

70

71
5. RESULTADOS E DISCUSSÕES
5.1. AVALIAÇÃO MACROSCÓPICA
5.1.1. Folha de bananeira e celulose
A Figura 5.1 demonstra os aspectos macroscópicos das folhas de bananeira bruta
(Fb) e das celuloses extraídas pelas diferentes propostas de tratamento químico (P1, P2
e P3).
Figura 5.1 - Transformação macroscópica em função dos tratamentos químicos: (a) folha de bananeira bruta, (b) celulose P1, (c) celulose P2 e (d) celulose P3, em placas de
Petri com 10 cm de diâmetro.
Fonte: Produção da autora.
A celulose P1 (Figura 5.1b) apresenta-se mais amarelada, em relação à celulose
P2 (Figura 5.1c) e P3 (Figura 5.1d), devido ao número de ataques ácidos ser menor, o
qual está relacionado à remoção da fração amorfa: lignina e hemicelulose. Segundo
(TSERKI et al., 2005), a mistura ácida reage primeiramente com as frações amorfas, as
quais impedem sua difusão para a fração cristalina, preservando-a. Na proposta P1, 1
ataque ácido/1 ataque básico, a hemicelulose solubiliza-se a baixas concentrações de
álcalis, enquanto a lignina hidrolisa-se em meio básico (KABIR et al., 2012b);
(ALBINANTE; PACHECO; VISCONTE, 2013). Ou seja, tais tratamentos não foram
suficientes para remoção da lignina, a qual apresenta-se marrom, tornando a celulose P1
com tom mais amarelado. A Figura 5.2 demonstra a efetividade do número de ataques
ácido na remoção da lignina.
(a) (b) (c) (d)

72
Figura 5.2 - Diferenças na remoção da lignina mediante (a) um ataque ácido e (b) dois ataques ácido.
Fonte: Produção da autora.
A partir das Figuras 5.2a e 5.2b nota-se o efeito dos tratamentos químicos
efetuados em Fb, com relação à remoção da fração amorfa. A Figura 5.3, por sua vez,
esquematiza a transformação macroscópica do componente vegetal (folha de bananeira
in natura) até a obtenção do componente celulósico, considerando-se os dois ataques
ácido (P2).
Figura 5.3 - Transformação macroscópica da folha de bananeira in natura: (a) folha de bananeira bruta e seca e (b) celulose após dois ataques ácido (P2) pulverizada a 20
mesh, em placas de Petri com 10 cm de diâmetro.
Fonte: Produção da autora.
5.1.2. Óxidos metálicos hidratados
A partir dos metais Nb, Al e Zn foram obtidos os respectivos óxidos hidratados:
Nb2O5.nH2O, Al2O3.nH2O e ZnO.nH2O. Após a secagem, todos os óxidos
apresentaram-se brancos sob a forma de um pó fino.
Segundo Lee (1999), a cor dos compostos está associada às transições eletrônicas
d-d dos metais. Para os metais estudados, o zinco apresenta o orbital d preenchido,
enquanto alumínio não possui tal orbital, com configuração 3s23p1 para a última
(a) (b)
(a) (b)

73
camada. E o nióbio apresenta a configuração 4d45s1, com orbital d parcialmente
preenchido. Entretanto, nos pentóxidos os metais possuem configuração d0 e,
consequentemente, são brancos. A Figura 5.4 apresenta somente o óxido de zinco
hidratado (ZnO.nH2O), uma vez que os óxidos de alumínio hidratado (Al2O3.nH2O) e de
nióbio hidratado (Nb2O5.nH2O) exibiram as mesmas características macroscópicas.
Figura 5.4 - Óxido de zinco hidratado, ZnO.nH2O, em placa de Petri com 10 cm de diâmetro.
Fonte: Produção da autora.
5.1.3. Híbridos orgânico-inorgânicos
Exceto pelas proporções que exibiram separação de fases visível
macroscopicamente, todos os híbridos orgânico-inorgânico deste trabalho apresentaram-
se amarelos, sem qualquer distinção perceptível a olho nu. A Figura 5.5 exibe os
aditivos híbridos escolhidos no estudo de proporção, discutido adiante no item 5.2.3.
Figura 5.5 - Aditivos híbridos escolhidos no estudo de proporção: (a) 97Celfb/3 Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O e (c) 97Celfb/3ZnO.nH2O, em
placas de Petri com 10 cm de diâmetro.
Fonte: Produção da autora.
(a) (b) (c)

74
A Figura 5.6 exibe as proporções que, a partir das quais foram observadas
separação entre as fases celulósica e inorgânica, após a síntese, no momento da
filtração.
Figura 5.6 - Proporções dos híbridos com separação de fases: (a) 95Celfb/5Nb2O5.nH2O, (b) 93Celfb/7Al2O3.nH2O e (c) 85Celfb/15ZnO.nH2O, em
placas de Petri com 10 cm de diâmetro.
Fonte: Produção da autora.
De acordo com a Figura 5.6 nota-se uma pequena mudança na coloração nos
híbridos 95Celfb/5Nb2O5.nH2O (Figura 5.6a) e 85Celfb/15ZnO.nH2O (Figura 5.6c),
apresentando-se esbranquiçados, devido a presença dos respectivos óxidos na superfície
da celulose. Por outro lado, no híbrido 93Celfb/7Al2O3.nH2O (Figura 5.6b) foi
observada a presença de grumos, indicados pelas setas, devido à aglutinação do
Al2O3.nH2O na superfície da celulose. Acredita-se que esta aglutinação tenha ocorrido
devido ao aspecto do respectivo óxido formado, o qual é extremamente gelatinoso, não
influenciando na coloração do material final.
5.1.4. Matriz e compósitos
A Figura 5.7 ilustra a matriz de poliuretano (PU) e os respectivos compósitos
contendo a celulose (Celfb) como aditivo nas diferentes quantidades incorporadas.
(a) (c) (b)

75
Figura 5.7 - Seções de 10 x 5 cm retiradas do interior dos blocos: (a) poliuretano puro (PU), (b) PU + 1% Celfb, (c) PU + 2% Celfb, (d) PU + 3% Celfb, (e) PU + 4% Celfb e
(f) PU + 5% Celfb.
Fonte: Produção da autora.
Todos os compósitos, independente do aditivo incorporado, apresentaram as
mesmas características da matriz pura (Figura 5.7a). A partir das Figuras 5.7b, 5.7c,
5.7d, 5.7e e 5.7f é possível identificar as partículas de celulose no interior da espuma,
além de uma maior distribuição com o aumento da porcentagem adicionada.
Os compósitos contendo os híbridos orgânico-inorgânico exibiram os mesmos
aspectos em termos de cor e distribuição destes aditivos. Nos compósitos contendo a
fração inorgânica esta distribuição não foi perceptível macroscopicamente devido ao
fato de todos os óxidos metálicos hidratados serem brancos.
(a) (b) (c)
(d) (e) (f)

76
5.2. AVALIAÇÃO FÍSICO-QUÍMICA: PRECURSORES E HÍBRIDOS
5.2.1. Isolamento da celulose
Para a determinação das condições de isolamento da celulose a partir da folha de
bananeira foi realizado um estudo, a fim de verificar qual a metodologia de tratamento
químico adequada, avaliando-se as propriedades físico-químicas e o efeito das
modificações na estrutura do material isolado. Ambas as propostas P1, P2 e P3,
descritas na Tabela 5.1, foram caracterizadas individualmente e comparadas ao material
de partida (folha bruta).
Inicialmente, para confirmar o isolamento da celulose a partir dos tratamentos
químicos avaliados, calculou-se o rendimento de cada proposta. Este rendimento foi
calculado em função dos valores da massa inicial de folha bruta e massa final do
material seco. A Tabela 5.1 apresenta os valores obtidos.
Tabela 5.1 - Rendimento de celulose para as propostas de tratamento químico da folha de bananeira.
METODOLOGIA Massa inicial
(g)
Massa final
(g)
Rendimento
(%)
P1 10,0018 2,5503 25,50
P2 10,0006 2,6008 26,01
P3 10,0022 2,3028 23,02
Fonte: Produção da autora.
Baseado nos resultados apresentados por Bilba; Arsene; Ouensanga (2007), a
folha de bananeira apresenta a seguinte composição em massa: 23-28 % de celulose, 15-
19 % de hemicelulose, 22-27 % de lignina, sendo o restante composto por extrativos,
umidade e cinzas. Comparando-se a composição determinada pelos autores com os
rendimentos obtidos, todas as propostas foram eficientes, extraindo praticamente toda a
celulose contida na folha.
Após a confirmação do isolamento, avaliou-se a cristalinidade das celuloses, a fim
de avaliar qual proposta de tratamento foi mais eficaz. Silva et al. (2009) definem a

77
celulose como um homopolissacarídeo linear, formado por unidades de glicose,
eterificadas por ligações β-1,4-glicosídicas. Segundo Canevarolo Jr. (2010), a
cristalinidade em polímeros é definida como o alinhamento dos segmentos das cadeias,
com a formação de um arranjo tridimensionalmente perfeito, denominado domínio
cristalino. Além disso, a linearidade dos polímeros influencia diretamente na
cristalinidade, devido à maior facilidade no empacotamento das cadeias.
Consequentemente, a celulose apresentará domínios amorfos e cristalinos.
A Figura 5.8 apresenta os difratogramas para a folha bruta, em comparação com
as propostas P1, P2 e P3.
Figura 5.8 - Difratogramas de raios X para (a) folha bruta, (b) celulose P1, (c) celulose P2 e (d) celulose P3.
Fonte: Produção da autora.
Em todos os materiais observam-se três picos com valores de 2θ próximos a 15°,
22° e 35°, referentes aos planos cristalográficos (101), (002) e (040), respectivamente,
os quais são intrínsecos de fibras lignocelulósicas (SATYANARAYANA;
GUIMARÃES; WYPYCH, 2007; GUIMARÃES et al., 2009; GUIMARÃES et al.,
2010; PEREIRA et al., 2014; LU et al., 2015).
0 500
1000 1500 2000 2500 3000 3500 4000 4500 5000 5500
10 20 30 40 50 60 70 80 90
Inten
sidad
e (u.a
.)
2θ (graus)
Folha bruta maior 20 mesh
I1
I2
0 500
1000 1500 2000 2500 3000 3500 4000 4500 5000 5500
10 20 30 40 50 60 70 80 90
Inten
sidad
e (u.
a.)
2θ (graus)
Cel 20 mesh
I2
I1
0 500
1000 1500 2000 2500 3000 3500 4000 4500 5000 5500
10 20 30 40 50 60 70 80 90
Inten
sidad
e (u.
a.)
2θ (graus)
Proposta 1
I2
I1
0 500
1000 1500 2000 2500 3000 3500 4000 4500 5000 5500
10 20 30 40 50 60 70 80 90
Inte
nsid
ade (
u.a.)
2θ (graus)
Proposta 3
I2
I1
a) b)
c) d)

78
De acordo com Ass; Belgacem; Frollini (2006), a celulose pode ser classificada
em relação às regiões nas quais ocorrem difração. A celulose tipo I exibe um pico
referente à fração cristalina em 22°≤ 2θ ≤ 23°, enquanto para a celulose tipo II este pico
ocorre em 18°≤ 2θ ≤ 22°. As regiões correspondentes à fração amorfa para a celulose
tipo I e celulose tipo II ocorrem em 18°≤ 2θ ≤ 19° e 13°≤ 2θ ≤ 15°, respectivamente.
Em todas as propostas, o material isolado foi classificado como celulose tipo I.
A partir dos resultados obtidos nos difratogramas foi possível estimar o índice de
cristalinidade (Ic) do material celulósico, definido pela Equação 4.1. A Tabela 5.2
apresenta os valores dos Ic para a folha bruta e celulose isolada para as três propostas.
Tabela 5.2 – Valores dos índices de cristalinidade (Ic) para as celuloses extraídas pelas diferentes propostas de tratamento químico.
MATERIAL I1 (u.a.) I2 (u.a.) Ic (%)
Folha bruta (Fb) 751 1242 39,5
Celulose (P1) 952 4556 79,1
Celulose (P2) 570 3272 82,6
Celulose (P3) 1086 5267 79,4
Fonte: Produção da autora.
Os resultados da Tabela 5.2 exibem um aumento significativo do índice de
cristalinidade, comparado à folha bruta, o qual confirma satisfatoriamente o isolamento
da celulose devido à eliminação das frações amorfas (lignina e hemicelulose). A
proximidade entre os valores de Ic apontam que todas as propostas de tratamento
químico foram eficientes para o isolamento da celulose, estando concordantes com os
resultados de rendimento. Entretanto, a celulose P2 apresentou um valor relativamente
maior, comparada às celulose de P1 e P3. Conforme Silva et al. (2009) e Albinante;
Pacheco; Visconte (2013), as ligações de hidrogênio na celulose proporcionam uma
forte tendência para a formação de cristais, tornando-a completamente insolúvel em
água e na maioria dos solventes orgânicos. Além disso, segundo Tserki et al. (2005), os
grupos hidroxila que reagem com o agente de acetilação, são àqueles referentes à
hemicelulose e lignina (fração amorfa). Já os grupos OH da celulose, por sua vez, sendo

79
a forma mais compactada devido a estas ligações, impedem a difusão do reagente,
resultando na preservação da fração cristalina.
As modificações químicas ocorridas na superfície das celuloses foram avaliadas
por FTIR. A Figura 5.9 apresenta os espectros de infravermelho referentes às propostas
P1, P2 e P3, em comparação à folha bruta.
O espectro da folha de bananeira sem tratamento (Figura 5.9a) confirma que a
fibra vegetal é composta não somente por celulose, mas também por outros materiais
não-celulósicos, tais como lignina, pectina e hemicelulose (BENÍTEZ et al., 2013).
A banda na região de 3600-3200 cm-1 refere-se à vibração do estiramento de
vários grupos hidroxila (–OH). Segundo Ibrahim et al. (2010), dentre os componentes
que contêm grupos hidroxila, pode-se citar água absorvida e álcoois alifáticos primário
e secundário encontrados na celulose, hemicelulose e lignina. Na região de 2900-2800
cm-1, observa-se a vibração associada ao estiramento de grupos –CHn. Na região de
1700-1100 cm-1, há a sobreposição de bandas devido aos grupos -C–C, –C=C, –OH, –
C=O, –CHn, –C–O–C e –CH, referente às ligações aromáticas, presentes na
hemicelulose e lignina. E, de 900-700 cm-1, há absorção de bandas -CHn (alifáticas e
aromáticas) dos carboidratos e lignina presentes no material bruto (BILBA; ARSENE;
OUENSANGA, 2007; MERLINI; SOLDI; BARRA, 2011).

80
Figura 5.9 - Espectros na região do infravermelho para (a) folha bruta, (b) celulose P1, (c) celulose P2 e (d) celulose P3.
Fonte: Produção da autora.
No espectro da proposta P2 (Figura 5.9c) é possível verificar um sinal de alta
intensidade em 1740 cm-1, o qual refere-se à carbonila de um éster, confirmando a
acetilação da folha bruta devido ao tratamento com a mistura ácida CH3COOH/HNO3.
Observa-se também que na região de 3600-3200 cm-1 há um estreitamento da banda,
referente aos grupos –OH, sendo este um indicativo de que a acetilação foi parcial,
ocorrendo somente nas hidroxilas mais externas da cadeia de celulose (D’ALMEIDA et
al., 2005; LOPES et al., 2011; TELI; VALIA, 2013). Em 1271 cm-1, pode-se identificar
a banda relacionada ao estiramento dos acoplamentos –C–O, a qual também confirma a
acetilação da folha. Além disso, variações na região entre 3000 e 2800 cm-1, referentes
aos acoplamentos –C–H, são indícios do tratamento químico utilizado.
A Figura 5.10 apresenta a proposta de mecanismo para P2, conforme resultados
apresentados por FTIR. Este mecanismo foi dividido em três etapas: I) protonação do
ácido acético com o catalisador ácido nítrico, II) ataque ácido na folha de bananeira e
III) isolamento e acetilação da celulose, com regeneração do catalisador ácido nítrico
(HNO3).
0
20
40
60
80
100
120
140
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tran
smitâ
ncia
(%)
Número de ondas (cm-1)
Proposta 3
0
20
40
60
80
100
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tran
smitâ
ncia
(%)
Número de ondas (cm-1)
Proposta 2
0
20
40
60
80
100
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tran
smitâ
ncia
(%)
Número de ondas (cm-1)
Proposta 1
0
20
40
60
80
100
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tran
smitâ
ncia
(%)
Número de ondas (cm-1)
Folha bruta
(a) (b)
(c) (d)

81
Figura 5.10 - Proposta de mecanismo para o isolamento da celulose, via ataque ácido (P2), para a folha de bananeira.
I) Protonação do ácido acético
II) Ataque ácido na folha de bananeira
III) Isolamento e acetilação da celulose
Sendo R = cadeia de celulose.
Fonte: Produção da autora.
A acetilação da folha de bananeira pelo mecanismo proposto também pode ser
representada pela reação global, exibida na Figura 5.11.
H3C O:
C O H
H+ NO3- +
NO3-
H3C O
C O H
+ H
NO3-
H3C O
C O H
+ H
+ H O R .. H3C
O
C O H
H
H O R
NO3- +
H3C
O
C O H
H
H O R
NO3- +
H3C O
C O R
+ H2O + H+ NO3-

82
Figura 5.11 - Representação da acetilação parcial da celulose, via tratamento ácido P2.
Fonte: Produção da autora.
Os espectros para as propostas P1 (Figura 5.10b) e P3 (Figura 5.10d)
apresentaram um perfil similar, pois em ambas efetuou-se um ataque básico com
NaOH, após o isolamento com a mistura ácida. Isto é, apesar da acetilação ter ocorrido
não foi possível identificar a banda referente à carbonila do éster devido à remoção dos
grupos acetila pela substituição por grupos hidroxila, o que pode ser confirmado na
região de 3600-3200 cm-1.
A Figura 5.12 retrata a proposta de mecanismo para P1 e P3, de acordo com os
resultados apresentados por FTIR. Este mecanismo tem início com a celulose já
acetilada, devido aos ataques ácidos (um para P1 e dois para P3) e término com o ataque
básico com NaOH.
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
HNO3 T = 120 °C
+!
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
Ac Ac
Ac Ac
H3C O
C O H
+ H2O + H+ NO3-

83
Figura 5.12 - Proposta de mecanismo para o isolamento da celulose, via ataque básico (P1 e P3), para a folha de bananeira.
I) Introdução de grupos OH
II) Neutralização e eliminação
do grupo acetila
Com os grupos acetila eliminados, a celulose retoma à sua estrutura original,
somente com grupos OH, confirmados pela banda na região de 3600-3200 cm-1.
Entretanto, após a neutralização e eliminação dos grupos acetila, as hidroxilas serão
grupos susceptíveis ao ataque da base. Logo, tem-se que:
III) Isolamento e ataque básico da celulose
Sendo R = cadeia de celulose.
Fonte: Produção da autora.
A Figura 5.13 apresenta a proposta para a reação global do ataque básico.
H3C O
C O R
+ Na+ :-OH H3C
O-
C O R
Na+
O H
H3C O
C O :- Na+
H3C
O-
C O R
Na+
O H
+ H O R
R O H + Na+ :-OH R O:- Na+ + H2O

84
Figura 5.13 - Representação do isolamento da celulose, via tratamento básico P1 e P3.
Fonte: Produção da autora.
Como o presente trabalho visa o desenvolvimento de compósitos poliméricos, a
introdução de grupos hidrofóbicos na estrutura da celulose favorecerão a
compatibilidade com a matriz, aumentando a adesão na interface (KABIR et al., 2012b).
A partir das modificações químicas ocorridas na celulose e considerando-se sua
aplicação na síntese de compósitos, a proposta P2 apresenta-se como a melhor opção
pois, dentre as propostas avaliadas, a acetilação destaca-se por ser uma modificação
química superficial e menos agressiva, contribuindo para a preservação da fração
cristalina e apresentando-se eficiente no isolamento.
Após a confirmação do isolamento, assim como as modificações químicas
ocorridas na estrutura da fração orgânica, o comportamento térmico também foi
avaliado.
O perfil de degradação térmica de um material, em condições não isotérmicas,
fornece informações quanto a sua resistência ou estabilidade térmicas, quando
submetido à variações de temperatura (MOTHÉ; AZEVEDO, 2009).
As diferenças estruturais dos três componentes majoritários presentes nas fibras
lignocelulósicas estão diretamente relacionadas às etapas de perda massa, durante a
decomposição (YANG et al., 2007). A hemicelulose é composta por vários sacarídeos
T = 40 °C
+!
+ H2O
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
Ac
Ac Ac
Ac
NaOH
H3C O
C O :- Na+
+
Potencialidades e oportunidades na química da sacarose e outros açúcares 625Vol. 32, No. 3
hidrossolúveis da quitosana e sua aplicações nas áreas farmacêuticas foi publicada recentemente por Silva e colaboradores.22
Celulose
A madeira é a principal fonte industrial de celulose. Ela é cons-tituída principalmente pelos seguintes materiais lignocelulósicos: hemicelulose (20 a 25%), celulose (40-50%) e lignina (25 a 30%), além de componentes inorgânicos (Esquema 4). Este conjunto de substâncias é responsável pelas propriedades mecânicas da madeira. As hemiceluloses são polissacarídeos ramificados com baixo grau de polimerização contendo uma mistura de polímeros de pentoses, como a D-xilose e a L-arabinose e, em menor quantidade as hexoses D-glicose, D-galactose, D-manose e ácidos glucurônicos. As hemice-luloses não são cristalinas e encontram-se intercaladas às microfibrilas da celulose dando elasticidade e impedindo que elas se toquem. As hemiceluloses variam nas estruturas e nas composições dependendo da fonte natural.
A celulose é encontrada em plantas sob a forma de microfibrilas de 2 a 20 nm de diâmetro e entre 100 a 40.000 nm de comprimento, tendo entre 2.000-25.000 de resíduos de glicose.23 Estas microfi-bras estão em ligações lineares do tipo -(1 4) entre unidades de D-glucopiranose e na conformações 4C1, onde formam agregados supramoleculares estruturalmente bastante resistentes e utilizados nas paredes celulares das plantas.24 Cada resíduo de glicose está orientado em 180º em relação ao próximo resíduo. As ligações de hidrogênio in-tramoleculares entre O3-H O5’, O6H O2’ e O6 HO3’ mantêm a rede mais fixa e com características hidrofóbicas. As interações de van der Waals entre as cadeias poliméricas formam zonas com estruturas cristalinas, em sua maioria, bastante ordenadas que não permitem a água penetrar no seu interior.25 Porém, outras zonas amorfas e secas podem absorver água e tornar a celulose macia e flexível. Estas liga-ções, juntamente com as zonas cristalinas, que alternam com zonas amorfas, correspondem a cerca de dois terços da celulose presente na madeira. Apesar da natureza higroscópica das moléculas individuais de celulose, a absorção de moléculas de água só é possível nas zonas amorfas devido à falta de espaços vazios na estrutura cristalina. A organização cristalina da celulose influencia a sua reatividade ao controlar o acesso de substâncias químicas ou enzimas aos grupos funcionais e às ligações químicas nas regiões cristalinas. As hidroxilas são os grupos mais abundantes na celulose, seguidos pelas ligações acetal que formam o anel das piranoses.26
Cada espécie de árvore possui fibras de celulose de diferentes morfologias. Devido a isso, esse polissacarídeo tem muitas utili-
zações na fabricação de diversos tipos de papel, e também como agente antiaglutinante (anticaking agent), emulsificante, estabilizante, dispersante, espessante, indutor de gelatinização, entre outros, pois em contato com a água pode aumentar o volume e a textura, espe-cialmente em cosméticos. A celulose pode ser modificada nas suas hidroxilas e produzir matéria com propriedades importantes (ex. triacetato de celulose, nitrocelulose, carboximetil celulose e rayon). O rayon ou viscose é uma fibra celulósica semissintética modificada na hidroxila primária. Esta pequena modificação confere um aspecto sedoso e brilhante na fibra celulósica.
No Brasil, a celulose é produzida em grande escala a partir do eucalipto por diversas empresas (ex. Aracruz, Jari, Suzano e Votoran-tin) e em 2004 contava com 220 empresas localizadas em 16 estados e 450 municípios, gerando 100.000 empregos, o que torna o país o maior produtor mundial de celulose de eucalipto.27,28 Esta espécie utilizada tem o dobro da produtividade de outras coníferas plantadas no Brasil e da maioria das árvores nativas.
Hemiceluloses
As hemiceluloses são polissacarídeos que representam um dos componentes da parede celular vegetal e estão intimamente asso-ciadas à celulose, definindo as propriedades estruturais na parede celular além de desempenhar funções na regulação do crescimento e desenvolvimento das plantas. Os xiloglucanos (Xg) e os galac-tomananos são hemiceluloses presentes tanto em parede primária (função estrutural) como em parede de reserva em sementes de algumas espécies (função de reserva). Os resultados mostraram que o grau de ramificação e o tamanho da cadeia afetam a interação entre Xg e a celulose. Enquanto os graus de galactosilação e fucosilação estão relacionados com a força de ligação entre os polissacarídeos, o tamanho da molécula do Xg afeta a sua capacidade (quantidade) de interação à celulose, consequentemente a autointeração.
Amido
É o segundo polissacarídeo mais abundante. Pode ser encontrado em sementes, raízes e fibras de plantas. É a fonte de energia, em termos de glicose, mais abundante para os humanos. Ele é composto basicamente de dois polissacarídeos de D-glicose: amilose e amilo-pectina. A diferença entre estes dois polímeros está no encadeamento das unidades glicosídicas. Na amilose a cadeia é linear e as ligações são -(1 4’). Na amilopectina há uma ramificação na hidroxila de C6 com uma ligação -(1 6’). Porém, as ligações -(1 4’) continuam no polímero. A sua composição varia de acordo com a planta. Por exemplo, o amido da batata difere basicamente do amido do aipim (macaxeira) na relação entre amilose e amilopectina. A maioria dos seres vivos pode utilizar estes polissacarídeos como fonte energéti-ca, pois possuem enzimas -amilases na saliva e no intestino que quebram a ligação -(1 4’) entre as unidade de glicose. O mesmo não acontece com a celulose. A maioria dos animais não consegue utilizá-la como alimento. A razão está na ligação -(1 4’) que não está presente nos animais superiores. Entretanto, cupins conseguem digerir a celulose facilmente, pois têm no seu intestino bactérias sim-bióticas que secretam a enzima celulase que faz a hidrólise da ligação
-(1 4’). Alguns vertebrados herbívoros também conseguem fazer a digestão, pois têm bactérias no estômago que secretam celulase.
Na conformação -(1 4’), mais estável nos polissacarídeos ami-lose, amilopectina e glicogênio, as cadeiras ficam rígidas tornando a cadeia curva ao invés de linear, como na celulose. Este arranjo produz estruturas compactas e helicoidais produzindo grânulos densos que podem ser visualizados em muitas células.
O glicogênio é o principal polissacarídeo de estocagem de energia
O
MeOO
OH
O
O
OH
O
MeO
MeO
OH
O
HO
O
MeO
OH
OMe
O
OMe
O
OHO
OMe
HO O
MeO
OOHO
OH
OO
AcO OHO OHO
O
O O
HO OAc
O OH
HO
OMe
O
CO2H
hemicelulose
lignina
O
OH
HOHO
OH
OO
OH
HO OHO O
OH
HOOH
OH
celulose
n
glicose
etanol
NaOH/CS2
OO
O
HO OHO
O-Na+
S
Rayon
OH
Esquema 4. Estruturas parciais dos componentes lignocelulósicos e deri-vados
:- Na+
Na+ -:
:- Na+
:- Na+

85
de cinco e seis carbonos e exibe uma estrutura amorfa e repleta de ramificações, a qual
decompõe-se facilmente a baixas temperaturas, liberando voláteis como CO, CO2 e
alguns hidrocarbonetos. Por outro lado, a celulose um homopolissacarídeo linear,
constituída por unidades de glicose, sem ramificações, exibe uma alta estabilidade
térmica. E a lignina, por possuir uma estrutura amorfa, altamente ramificada, com
vários anéis aromáticos, decompõe-se lentamente no intervalo da temperatura ambiente
até 900 °C (YANG et al., 2007; SILVA et al., 2009; KABIR et al., 2012b). As Figuras
5.14a a 5.14d apresentam os perfis das curvas TGA/DTG para a folha bruta e celuloses
isoladas.
Figura 5.14 - Perfis das curvas TGA/DTG para (a) folha bruta, (b) celulose P1, (c) celulose P2 e (d) celulose P3.
Fonte: Produção da autora.
De acordo com os perfis térmicos, as curvas TGA/DTG exibiram de três a quatro
eventos térmicos: o primeiro exibe a desidratação da celulose, o segundo a combustão
da hemicelulose, o terceiro a combustão da celulose e o quarto a degradação da lignina
remanescente (MUSTATA et al., 2015; DA SILVA et al., 2015).
-0,014
-0,012
-0,01
-0,008
-0,006
-0,004
-0,002
0
0,002
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
Proposta 3 - P3 DTG
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
Proposta 2 - P2 DTG
-0,016
-0,014
-0,012
-0,01
-0,008
-0,006
-0,004
-0,002
0
0,002
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
Proposta 1 - P1 DTG
-1,60E-02
-1,40E-02
-1,20E-02
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
Folha bruta - Fb DTG
(a) (b)
(c) (d)
323 °C
457 °C 370 °C
346 °C 365 °C

86
Primeiramente, verifica-se uma mudança na inclinação da curva de perda de
massa, indicando que os materiais isolados apresentam diferentes teores de celulose, os
quais são inerentes aos tratamentos químicos efetuados. Tal fato pode ser relacionado ao
comparar os eventos de maior perda de massa das celuloses P1, P2 e P3 com a folha
bruta, nos quais verifica-se um aumento na porcentagem de perda, relativa à fração
celulósica. Entretanto, nota-se que no segundo evento das propostas P1 e P3, não há
separação das frações referentes à hemicelulose e celulose, indicando uma sobreposição
de suas respectivas decomposições, o que compromete a eficiência destes tratamentos
no isolamento da celulose. Diferentemente, a proposta P2 exibe estes dois eventos bem
distintos, indicando que grande parte da quantidade de hemicelulose foi removida,
quando comparada à folha bruta e que a fração de interesse (celulose) foi isolada. A
Tabela 5.3 apresenta os dados obtidos a partir da termogravimetria.
Tabela 5.3 – Dados das curvas TGA/DTG para a folha bruta e celuloses isoladas em atmosfera de ar sintético.
MATERIAL
Intervalos Curvas TGA (°C)
Perda de massa Curvas TGA
(%)
Temperatura Curva DTG
(°C)
Resíduo Experimental
(RE) (%)
Folha de bananeira bruta
25-165 165-413 413-574 574-900
7,81 46,33 32,18 1,63
76 323 457 661
12,05
Celulose P1
25-170 170-426 426-900
4,78 61,36 23,65
63 370
-
10,21
Celulose P2
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
11,26
Celulose P3
25-165 165-461 461-900
6,06 70,46 15,31
60 365 525
8,17
Fonte: Produção da autora.
Segundo os dados da Tabela 5.3, outro fator que evidencia o efeito dos
tratamentos químicos nos materiais isolados é a temperatura da curva DTG do evento de

87
maior perda de massa. Relacionando-se tais temperaturas, nota-se uma diferença em
todas as propostas, quando comparadas à folha bruta (323 °C): 370 °C para P1, 346 °C
para P2 e 365 °C para P3. Contudo, estas temperatura também são um indicativo da
estabilidade térmica dos materiais isolados. Neste caso, a celulose de P2 é o material
menos estável termicamente, em relação à P1 e P3, porém ainda é mais estável em
relação à folha bruta. Esta diminuição na estabilidade térmica também foi observada por
Teli; Valia (2013). Os autores utilizaram fibras de bananeira acetiladas para aplicação
em absorção de óleos, visando a limpeza de derramamentos de petróleo. O
comportamento térmico das fibras bruta e acetiladas foi avaliado por termogravimetria e
os resultados indicaram que o material acetilado apresentou uma estabilidade térmica
menor. De acordo com os autores, a perda de estabilidade térmica pode estar
relacionada à desintegração das interações intermoleculares (ligações de hidrogênio)
devido à acetilação, isto é, a substituição de grupos OH por grupos acetila. Os
resultados são concordantes com as observações feitas por FTIR, pois ambas as
propostas P1 e P3, as quais não possuem acetilação parcial apresentam uma maior
estabilidade térmica. Além disso, os mecanismos propostos para todos os tratamentos
químicos confirmam tais observações.
Avaliando-se o evento de maior perda de massa para as três celuloses, observa-se
que P2 exibe o pico da DTG mais pontiagudo e afunilado, indicando o isolamento da
fração celulósica mais pura, em relação à P1 e P3, corroborando com os resultados de
rendimento e índice de cristalinidade (Ic). Como mencionado, o agente da acetilação
reage primeiramente com as frações amorfas, as quais impedem sua difusão para a
fração cristalina preservando-a (TSERKI et al., 2005).
Os perfis das curvas TGA/DTG para as propostas P1 e P3 também podem ser
correlacionados com os tratamentos químicos realizados. Avaliando-se o primeiro
evento de ambas, P3 apresenta uma maior perda de massa (6,06 %), referente à perda de
grupos hidrofílicos (OH), em relação à P1 (4,18 %). Como em P3 efetuam-se dois
ataques ácidos, é necessária uma quantidade muito maior de álcali para a remoção dos
grupos acetila. Logo, a celulose retoma à sua estrutura original, somente com grupos
OH, como discutido nos resultados de FTIR e mecanismos.

88
Baseado nos resultados das curvas TGA/DTG de P1 e P3, também é possível
afirmar que P3 exibiu uma menor perda de massa no terceiro evento (15,31 %),
comparada com P1 (23,65 %), evidenciando que P3 removeu uma maior fração de
lignina, porém ambas as propostas foram pouco eficientes em relação à P2 (8,75 %).
Segundo Kabir et al. (2012b) e Albinante; Pacheco; Visconte (2013), a hemicelulose é
solúvel a baixas concentrações de álcalis, enquanto a lignina sofre hidrólise básica. Ou
seja, por P1 ser composta por um ataque ácido e um ataque básico, tais tratamentos não
foram suficientes para remoção da lignina. Outra evidência que justifica tal fato é a
perda de massa contínua no quarto evento de P1, também relacionado à degradação da
lignina, que ocorre da temperatura ambiente até 900 °C (MUSTATA et al., 2015).
A partir das observações sobre o comportamento térmico das celuloses obtidas
nas três propostas, P2 ainda apresenta-se como a melhor opção de tratamento. Apesar da
perda de estabilidade térmica, frente às outras propostas, P2 apresenta a fração
celulósica devidamente isolada, com grande parte de sua fração amorfa eliminada
(Figura 5.14 e Tabela 5.3).
As Figuras 5.15a, 5.15b, 5.15c e 5.15d apresentam as micrografias para a folha de
bananeira bruta, P1, P2 e P3, respectivamente.

89
Figura 5.15 - Micrografias para (a) folha bruta, (b) celulose P1, (c) celulose P2 e (d) celulose P3, com ampliação de 100x.
Fonte: Produção da autora.
As fibras lignocelulósicas são formadas a partir das microfibrilas de celulose
cristalina, envoltas em uma matriz amorfa composta por hemicelulose e lignina, as
quais ligam tais microfibrilas por meio de ligações de hidrogênio, com a formação de
uma rede tridimensional (MERLINI; SOLDI; BARRA, 2011; KABIR et al., 2013). Na
micrografia da folha bruta (Figura 5.15a) observam-se tais características, como
reportadas pelos autores, além de um aspecto rugoso.
Kabir et al. (2013) avaliaram os efeitos das modificações químicas nas superfícies
das fibras de cânhamo, tratadas com hidróxido de sódio, ácido acético/anidrido acético e
siloxano oligomérico. Segundo os autores, as micrografias para a fibra acetilada e
tratada com NaOH exibiram em suas superfícies as microfibrilas de celulose, separadas
individualmente, devido à remoção do revestimento amorfo (hemicelulose e lignina).
Entretanto, as microfibrilas de celulose para a fibra acetilada apresentaram-se mais
fragmentadas e individualizadas.
Comparando-se os resultados apresentados pelos autores com a micrografia de P2
(Figura 5.15c), observa-se uma superfície mais limpa e fragilizada, com microfibrilas
(c) (d)
(a) (b)
SE MAG: 100 x HV: 20,0 kV WD: 0,0 mm

90
curtas, devido à remoção do revestimento amorfo, ocasionada pelo tratamento com a
mistura ácida. A acetilação remove o revestimento amorfo de hemicelulose e lignina,
evidenciando as microfibrilas de celulose (KABIR et al., 2013).
Nas micrografias de P1 (Figura 5.15b) e P3 (Figura 5.15d), observa-se uma
superfície mais rugosa, com microfibrilas finas e emaranhadas, como consequência do
tratamento básico, porém mais compactadas, em relação à P2 (GU, 2009; SANCHEZ et
al., 2010; MERLINI; SOLDI; BARRA, 2011).
A partir das análises de EDX da folha bruta foi possível identificar a presença de
potássio, elemento característico da própria bananeira (BORGES et al., 2006).
Entretanto, em P2 há a remoção deste elemento devido ao tratamento químico utilizado.
Em P1 e P3 foi confirmada a presença de sódio, devido ao tratamento básico, como
proposto no mecanismo. Como em P3 foram realizados dois ataques ácidos, a
disponibilidade de sítios para a entrada do sódio foi menor, confirmado pela
porcentagem identificada no EDX (0,29 %). Em P1, com um ataque ácido, a acetilação
ocorreu em menos sítios, sendo necessária pouca quantidade de NaOH para a remoção
destes grupos. Logo, P1 apresenta maior quantidade de sódio (0,50 %). As micrografias
das folha bruta e materiais isolados corroboram com todos os resultados apresentados
confirmando a extração da celulose a partir da folha de bananeira.
As celuloses obtidas nos tratamentos químicos foram submetidas a testes de
intumescimento em água por 1 h, uma vez que o intumescimento é uma etapa crucial
para a síntese dos híbridos. Segundo os testes, somente a celulose obtida em P2
dispersou-se em água, com a liberação das microfibrilas de celulose, o que não ocorreu
para as celuloses de P1 e P3. A dispersão das microfibrilas proporcionou um aumento
na área de contato, que favorecerá a precipitação dos óxidos metálicos hidratados na
síntese dos híbridos orgânico-inorgânicos.
A partir de todos os resultados apresentados para as três celuloses obtidas nas
diferentes propostas de tratamentos químicos, a proposta P2 mostrou-se a melhor
metodologia para o isolamento da celulose proveniente das folhas de bananeira.

91
5.2.2. Síntese e caracterização dos óxidos metálicos hidratados
O método de precipitação possui diversos fatores os quais influenciam não
somente no equilíbrio químico, mas também na pureza e propriedades físicas do
respectivo precipitado. Dentre estes parâmetros, pode-se citar a concentração dos íons
presentes, o pH, o agente precipitante, a temperatura, a lavagem do precipitado, digestão
do precipitado etc (SKOOG et al., 2014).
O primeiro parâmetro a ser considerado foi o pH para a precipitação dos óxidos
metálicos hidratados de zinco, alumínio e nióbio. O gráfico, representado na Figura
5.16, exibe as faixas de pH de alguns hidróxidos metálicos. De acordo com o gráfico, a
área hachurada indica a faixa de pH necessário para atingir seus respectivos produtos de
solubilidade (Kps) e as áreas em branco denotam a faixa em que os íons permanecem
em solução. Como os pH’s para Nb2O5.nH2O, Al2O3.nH2O e ZnO.nH2O foram de 8,0;
9,0 e 10,0, respectivamente, todos encontram-se dentro da faixa necessária para a
precipitação.
Figura 5.16 - Gráfico da faixa de pH para precipitação de alguns hidróxidos metálicos.
Fonte: Adaptado de VOGEL (1981).

92
Outro parâmetro importante foi o agente precipitante. Na Tabela 4.3, das
condições de síntese dos óxidos, somente para a precipitação do ZnO.nH2O utilizou-se
NaOH como agente precipitante, ao invés de hidróxido de amônio (NH4OH). Segundo
Vogel (1981), a não precipitação do ZnO.nH2O, em presença de solução de NH4OH se
justifica pela formação do sal de amônio, pois este reduz a concentração dos íons
hidroxila (-OH) e impede que o Kps seja atingido. Isto é, como a reação é reversível, o
equilíbrio é deslocado no sentido inverso, de formação de reagentes, ocasionando o
chamado efeito do íon comum, representado pela Equação química 5.1. Para a
representação das reações considerou-se o meio reacional descrito na Tabela 4.3.
(5.1)
Além do tipo de agente precipitante, a concentração deste no meio reacional
também é relevante na precipitação dos óxidos, principalmente na síntese do
ZnO.nH2O, devido à utilização de uma base forte (NaOH).
De acordo com Vogel (1981), Zn(OH)2 precipita-se facilmente com NaOH,
entretanto um excesso de NaOH pode provocar a solubilização do óxido, formando
tetrahidroxizincato de sódio, o qual é solúvel, descrita na Equação química 5.2.
!!" !" !(!) + !!"!"(!) !↔ !" (5.2)
A reação apresenta-se sob a forma de equilíbrio, uma vez que o hidróxido de
zinco se reconstituiu devido ao seu caráter anfótero. Logo, a concentração do agente
precipitante é um fator relevante, pois está diretamente relacionada ao controle do pH
no meio reacional, como discutido.
O processo de digestão dos precipitados também se mostra relevante na síntese
dos óxidos, principalmente na síntese do Al2O3.nH2O. A digestão ou envelhecimento
dos precipitados pode ser definida como o conjunto de transformações irreversíveis que
ocorre em um precipitado, após sua formação (BACCAN et al., 2001). Nos precipitados
gelatinosos, as partículas são muito finas e pequenas, tornando-se filtráveis após o
processo de precipitação. Para impedir a perda do material no momento da filtração, o
!"(!" .)!! + !2!!"(!" .)! + 2!!"!!"(!)!!!!!!!!!!!!!!"(!")!(!) !+ !2!!"!!"(!)!!"(!"#.)!! + !2!!"(!"#.)! + 2!!"!!"(!)!!!!!!!!!!!!!!"(!")!(!) !+ !2!!"!!"(!)!!"!! + !2!"! + 2!"!!"!!!!!!!!!!!!!!"(!")! !+ !2!!"!!"!!"!! + !2!"! + 2!"!!"!!!!!!!!!!!!!!"(!")! !+ !2!!"!!"!

93
óxido é aquecido em temperaturas brandas, ou até próximo do ponto de ebulição da
solução (se a solubilidade permitir) para promover a aglomeração das partículas,
favorecendo a filtrabilidade do material.
A lavagem dos óxidos também foi outro fator de suma importância. Para os
óxidos Al2O3.nH2O e ZnO.nH2O, nos quais os metais de partida foram dissolvidos em
HCl concentrado, foi necessária a completa remoção dos íons cloreto (Cl-) do meio
reacional.
Após a abordagem dos parâmetros que influenciam na precipitação dos óxidos
Nb2O5.nH2O, Al2O3.nH2O e ZnO.nH2O, as reações envolvidas nas sínteses podem ser
representadas pelas Equações químicas 5.3, 5.4 e 5.5, respectivamente:
!"(!) + !!"#!(!) + !"(!) → [!"#!!](!".)! + !"!(!) + !!!!(!) (5.3)
[!"#!!](!".)! + !!"!!"(!) → !"!!!.!!!!(!) !+ !!"!(!".)! + !(!".)!
2!!"(!) + 6!!"#(!) → 2!!"(!".)!! + !6!!"(!".)! + !3!!!(!) (5.4)
!"(!".)!! + !!"(!".)! + !!"!!"(!) → !"!!!.!!!!(!) !+ !!"!(!".)! + !!"(!".)!
2!!"(!) + 6!!"#(!) → 2!!"(!".)!! + !6!!"(!".)! + !3!!(!) (5.5)
!"(!".)!! + ! !!"(!".)! + !!"#$(!) → !"#.!!!!(!) !+ !!"(!".)! + !!!(!".)!
A termogravimetria possibilita calcular o número de moléculas de água, isto é, o
grau de hidratação (n), assim como determinar a temperatura necessária para efetuar o
processo de calcinação dos óxidos hidratados. Na curva TGA, a temperatura de
calcinação (Tcal.) pode ser definida como a temperatura na qual o material não exibe
perda de massa. O processo de calcinação é fundamental para a determinação das
estruturas cristalinas dos óxidos sintetizados, a partir da comparação com as fichas
padrão JCPDS.
As Figuras 5.17a, 5.17b e 5.17c apresentam as curvas TGA/DTG para os óxidos
metálicos hidratados Nb2O5.nH2O, Al2O3.nH2O e ZnO.nH2O, respectivamente.

94
Figura 5.17 - Perfis das curvas TGA/DTG para os óxidos hidratados: (a) Nb2O5.nH2O, (b) Al2O3.nH2O e (c) ZnO.nH2O, em atmosfera de ar sintético.
Fonte: Produção da autora.
De acordo com as curvas, todos os óxidos exibem dois eventos, os quais são
identificados como etapas de desidratação. A primeira etapa refere-se à perda de
moléculas de água fracamente ligadas e a segunda, à perda de moléculas de água
fortemente ligadas ou condensação dos grupos hidroxila (TAGLIAFERRO; DA
SILVA; DA SILVA, 2005; RODRIGUES; DA SILVA, 2009a).
A partir dos dados da Tabela 5.4, calculou-se o grau de hidratação, de acordo com
a Equação 4.2, descrita no item 4.4.4. Os dados referentes às curvas TGA/DTG para os
óxidos metálicos hidratados, assim como os valores de n e Tcal. estão apresentados na
Tabela 5.4.
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900 Temperatura (°C)
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
ZnOnH2O DTG
-0,0035
-0,003
-0,0025
-0,002
-0,0015
-0,001
-0,0005
0
0,0005
0
20
40
60
80
100
120
0 300 600 900 Temperatura (°C)
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Al2O3nH2O DTG
-1,60E-03
-1,40E-03
-1,20E-03
-1,00E-03
-8,00E-04
-6,00E-04
-4,00E-04
-2,00E-04
0,00E+00
2,00E-04
0
20
40
60
80
100
120
0 300 600 900 Temperatura (°C)
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Nb2O5nH2O DTG
(a) (b)
(c)
88 °C 356 °C 295 °C
270°C
90 °C
78 °C

95
Tabela 5.4 – Dados das curvas TGA/DTG para os óxidos metálicos hidratados em atmosfera de ar sintético.
MATERIAL
Intervalos Curva TGA (°C)
Perda de massa
Curva TGA (%)
Temperatura Curva DTG (°C)
Resíduo Experimental
(RE) (%)
Grau de
hidratação (n)
Temperatura de calcinação
(Tcal.) (°C)
Nb2O5.nH2O
25-200 200-900
7,37 12,87
88 356
79,76
3,7
600
Al2O3.nH2O
25-196 196-900
10,06 22,19
78 295
67,75
2,7
1000
ZnO.nH2O
25-144 144-900
1,41 21,27
90 270
77,32
1,3
600
Fonte: Produção da autora.
Baseado na curva TGA/DTG e nos dados da Tabela 5.4, nota-se que a
temperatura de calcinação para o Al2O3.nH2O (1000 °C) é superior à temperatura
observada na curva termogravimétrica (750 °C). De acordo com Shriver; Atkins (2008),
a desidratação do Al2O3.nH2O em temperaturas inferiores à 900 °C ocasiona a formação
de estruturas policristalinas metaestáveis. Porém, a partir de 1000 °C é possível obter a
α-alumina, sendo esta a fase mais estável presente no Al2O3.nH2O, com estrutura
cristalina bem definida. Desta forma, salienta-se que a temperatura adotada para o
processo de calcinação do Al2O3.nH2O foi de 1000 °C.
Outra análise térmica essencial é a calorimetria exploratória diferencial (DSC), na
qual são calculadas as entalpias das transformações físico-químicas ocorridas mediante
aquecimento. As Figuras 5.18a, 5.18b e 5.18c apresentam as curvas DSC para os óxidos
hidratados Nb2O5.nH2O, Al2O3.nH2O e ZnO.nH2O, respectivamente.

96
Figura 5.18 - Perfis das curvas DSC para os óxidos hidratados: (a) Nb2O5.nH2O, (b) Al2O3.nH2O e (c) ZnO.nH2O, em atmosfera de nitrogênio.
Fonte: Produção da autora.
Segundo as curvas DSC, todos os óxidos metálicos hidratados exibem picos
endotérmicos, referentes aos eventos de desidratação e transformações de fase. A curva
DSC para o Nb2O5.nH2O (Figura 5.18a) exibe um único pico, o qual denota a
desidratação de moléculas de água fortemente ligadas. O Al2O3.nH2O (Figura 5.18b),
entretanto, apresenta dois picos que referem-se ao mesmo evento descrito para
Nb2O5.nH2O e um terceiro pico que configura a transformação das fases metaestáveis
presente em sua composição, enquanto a curva DSC para o ZnO.nH2O (Figura 5.18c)
apresenta dois picos relativos aos mesmos eventos descritos para o Al2O3.nH2O.
Comparando-se as curvas DSC, nota-se que somente o ZnO.nH2O exibe em seu
primeiro evento um pico largo e bipartido, possivelmente devido à presença de íons
sódio provenientes do agente precipitante. Outro ponto relevante a salientar é que as
alterações nos perfis das curvas DSC, comparadas às curvas TGA/DTG, são decorrentes
à diferença nas atmosferas. A Tabela 5.5 apresenta os dados obtidos pelas curvas DSC.
(a) (b)
-8
-6
-4
-2
0
2
0 50 100 150 200 250 300 350 400
Flux
o de
cal
or (W
/g)
Temperatura (°C)
-10
-8
-6
-4
-2
0
2
0 50 100 150 200 250 300 350 400
Flux
o de
cal
or (W
/g)
Temperatura (°C)
-4
-3
-2
-1
0
1
0 50 100 150 200 250 300 350 400
Flux
o de
cal
or (W
/g)
Temperatura (°C)
184 °C
151 °C
188 °C
319 °C
163 °C
271 °C
(c)
! EXO ! EXO
! EXO

97
Tabela 5.5 – Dados das curvas DSC para os óxidos metálicos hidratados em atmosfera de nitrogênio.
MATERIAL
Intervalos Curva DSC (°C)
Entalpia de reação ΔH
(J.g-1)
Temperatura inicial do evento Tinicial (°C)
Nb2O5.nH2O 167-206 78 183
Al2O3.nH2O
144-160 183-217 250-356
5 137 290
150 186 294
ZnO.nH2O
143-225 238-300
440 85
161 255
Fonte: Produção da autora.
Uma vez avaliado os perfis térmicos das frações inorgânicas, a comparação entre
os difratogramas dos materiais hidratados e calcinados mostra-se necessária, a fim de
determinar a respectiva estrutura cristalina de cada óxido metálico hidratado. As
estruturas hidratada e calcinada estão representadas pelas Figuras 5.19a e 5.19b para o
Nb2O5.nH2O, 5.20c e 5.20d para o Al2O3.nH2O e 5.21e e 5.21f para o ZnO.nH2O.
Figura 5.19 - Difratogramas para: (a) Nb2O5.nH2O e (b) Nb2O5 calcinado à 600 °C.
Fonte: Produção da autora.
0
500
1000
1500
2000
2500
3000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Nb2O5nH2O - estudo proporção
0
1000
2000
3000
4000
5000
6000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Nb2O5 LF calcinado
(a) (b)

98
Figura 5.20 - Difratogramas para: (c) Al2O3.nH2O e (d) Al2O3 calcinado à 1000 °C.
Fonte: Produção da autora.
Figura 5.21 - Difratogramas para: (e) ZnO.nH2O e (f) ZnO calcinado à 600 °C.
Fonte: Produção da autora.
O difratograma para o Nb2O5.nH2O (Figura 5.19a) sintetizado via PC exibe um
perfil com características amorfas, similar à trabalhos anteriores, obtido pela mesma
metodologia de precipitação (OTTOBONI, 2011; MASCHIO; PEREIRA; DA SILVA,
2012). No difratograma para o óxido calcinado (Figura 5.19b) foram identificados picos
característicos da estrutura cristalina ortorrômbica do Nb2O5, segundo comparação com
a ficha padrão JCPDS 271003 e com a literatura (LIU; XUE; LI, 2011).
No difratograma do óxido de alumínio hidratado (Figura 5.20c) observa-se a
presença da gibbsita ou hidróxido de alumínio (γ-Al(OH)3), sendo esta a precursora para
a obtenção das diferentes fases transicionais da alumina (MALKI et al., 2014). Segundo
0
5000
10000
15000
20000
25000
30000
35000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
ZnOnH2O - lote final calcinado
0
1000
2000
3000
4000
5000
6000
7000
8000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Series1
(e) (f)
0
500
1000
1500
2000
2500
3000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Al2O3 LF calcinado 1000
0
1000
2000
3000
4000
5000
6000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Al2O3nH2O - estudo proporção
(c) (d)

99
Bhattacharya et al. (2004), entre 400-900 °C são formadas várias fases metaestáveis
com estruturas polimórficas, tais como χ, κ, γ, δ, θ, ρ etc. À 1000 °C ocorre a completa
transformação da χ a κ-alumina e à 1100 °C há o surgimento da α-alumina juntamente
com a κ-alumina. Comparando-se o difratograma do Al2O3 calcinado até 1000 °C
(Figura 5.20d) com os resultados apresentados pelos autores, pode-se afirmar a
obtenção da mesma mistura de fases. Apesar do surgimento da α-alumina ocorrer
somente a partir de 1100 °C, acredita-se que sua presença seja devido ao tempo de
exposição do óxido na temperatura de 1000 °C (6 h).
As Figuras 5.21e e 5.21f exibem os difratogramas para o óxido de zinco hidratado
e calcinado à 600 °C, respectivamente. O óxido calcinado apresenta picos
característicos da estrutura hexagonal da wurtzita, estando de acordo com a ficha padrão
JCPDS 361451 e com a literatura (MUTHUKUMARAN; GOPALAKRISHNAN, 2012;
SORNALATHA; MURUGAKOOTHAN, 2014; RAJA; RAMESH; GEETHA, 2014).
A abordagem dos parâmetros que influenciam as sínteses dos óxidos metálicos
hidratados, assim como as caracterizações são de suma importância, pois a partir destes
materiais foram obtidos os híbridos orgânico-inorgânicos.
5.2.3. Síntese dos híbridos (100-x)Celfb/xMyOz.nH2O: estudo de proporção
A definição da melhor relação entre a celulose e os óxidos metálicos hidratados
foi realizada a partir da termogravimetria. Para o estudo de proporção todos os híbridos
foram comparados aos seus respectivos precursores celulose e óxidos metálicos
hidratados. A fração celulósica utilizada para a síntese dos híbridos, assim como o seu
comportamento térmico foram discutidos no item 5.2.1. Analogamente à celulose, o
comportamento térmico dos óxidos metálicos hidratados encontra-se descrito no item
5.2.2. Logo, neste item foram relatadas somente as comparações entres as curvas TGA
dos híbridos e seus respectivos precursores. As curvas TGA/DTG individualizadas de
todas as proporções dos aditivos híbridos (100-x)Celfb/xNb2O5.nH2O, (100-
x)Celfb/xAl2O3.nH2O e (100-x)Celfb/xZnO.nH2O encontram-se no Apêndice A.
A partir das curvas TGA/DTG dos híbridos, os seguintes parâmetros foram
avaliados: diferença de temperatura da curva DTG, porcentagem de perda no evento de
maior perda de massa e comparação entre as porcentagens de resíduo experimental

100
(%RE) e resíduo teórico (%RT). A %RE pode ser definida como a quantidade de
material obtida ao final da análise, ou seja, a massa remanescente no cadinho. E a %RT
refere-se à soma dos resíduo experimental da fração celulósica com a fração de óxido
calculada. E, segundo Maschio; Pereira; Da Silva (2012) e Pereira et al. (2014), quanto
maior a diferença de temperatura do híbrido, em comparação com os precursores (Celfb
e MyOz.nH2O), maior a interação entre o inorgânico e a fração celulósica.
Como mencionado na Tabela 4.4, foram sintetizadas várias proporções com
diferentes variações mássicas entre celulose e óxidos metálicos hidratados. De acordo
com a avaliação macroscópica (item 5.1.3), foi observada separação de fases, visível
macroscopicamente, a qual desqualificaria o material como um híbrido. Entretanto,
todos os resultados de termogravimetria foram apresentados e avaliados a fim de
demonstrar como a análise térmica apresenta-se eficaz na definição das proporções
destes híbridos orgânico-inorgânicos.
A seguir, seguem as definições das proporções para os híbridos
(100-x)Celfb/xNb2O5.nH2O, (100-x)Celfb/xAl2O3.nH2O e (100-x)Celfb/xZnO.nH2O.

101
a) Híbridos (100-x)Celfb/xNb2O5.nH2O
A Figura 5.22 apresenta as curvas TGA para os híbridos de nióbio e os seus
respectivos precursores.
Figura 5.22 - Perfis das curvas TGA para os híbridos (100-x)Celfb/xNb2O5.nH2O, em atmosfera de ar sintético, para x = 3, 5, 10, 15 e 20.
Fonte: Produção da autora.
As curvas TGA para as proporções 97/3 e 95/5 exibem dois eventos térmicos: o
primeiro denota a desidratação da celulose e do Nb2O5.nH2O, referente às moléculas de
água fracamente ligadas e o segundo denota a combustão da hemicelulose, celulose, a
segunda etapa de desidratação, relativa às moléculas de água fortemente ligadas, e à
degradação da lignina remanescente. Os demais híbridos, 90/10, 85/15 e 80/20
apresentaram três eventos referentes às mesmas etapas descritas para os híbridos 97/3 e
95/5 e um terceiro evento similar ao ocorrido na celulose isolada, relativo à degradação
da lignina remanescente. Esta mudança no perfil caracteriza-se devido à separação de
fases observada macroscopicamente, o que denota a não formação do híbrido
satisfatoriamente. A Tabela 5.6 exibe os dados obtidos por termogravimetria.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C )
Celfb Nb2O5.nH2O 97/3 95/5 90/10 85/15 80/20

102
Tabela 5.6 - Dados das curvas TGA/DTG para os híbridos (100-x)Celfb/xNb2O5.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva
TGA (%)
Temperatura Curva DTG
(°C)
Resíduo Experimental
RE (%)
Resíduo Teórico RT (%)
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
11,26
-
Nb2O5.3,7H2O
25-200 200-900
7,37 12,87
88 356
79,90
-
97/3
25-170 170-900
4,33 88,68
61 373
6,99
14,26
95/5
25-174 174-900
5,06 85,65
59 363
9,29
16,26
90/10
25-178 178-474 474-900
5,79 72,20 9,00
61 356 582
13,01
21,26
85/15 25-178
178-478 478-900
6,82 70,26 8,60
59 355 572
14,32
26,26
80/20 25-174
174-483 483-900
5,65 68,15 9,28
61 359 606
16,92
31,26
Fonte: Produção da autora.
De acordo com os dados da Tabela 5.6, o híbrido 97Celfb/3Nb2O5.nH2O exprime
uma alta temperatura na curva DTG (373 °C), quando comparado à celulose (346 °C),
com uma diferença de 27 °C indicando uma maior interação entre os materiais isolados.
Além disso, observa-se uma perda de massa contínua até aproximadamente 750 °C,
enquanto que na curva da celulose esta perda de massa ocorre até 600 °C. Com isso,
pode-se afirmar que aproximadamente 3 % m/m de Nb2O5.nH2O promovem uma alta
estabilidade térmica na fração celulósica, prolongando a degradação do híbrido devido à
incorporação do componente inorgânico.
Ottoboni (2011) sintetizou híbridos provenientes da combinação entre a celulose
do bagaço de cana de açúcar e Nb2O5.nH2O para a preparação de compósitos com
matriz de polietileno de alta densidade (PEAD). Neste trabalho foram sintetizadas as
seguintes proporções de celulose/Nb2O5.nH2O: 90/10, 80/20, 70/30 e 60/40. De acordo

103
com os resultados de termogravimetria em ar sintético para os materiais híbridos, a
proporção 90Cel/10Nb2O5.nH2O apresentou a menor temperatura da DTG (333 °C)
comparada à celulose do bagaço (350 °C). Ou seja, a maior variação de temperatura
com um ΔT = 17 °C, indicando a maior interação entre os componentes. A autora
também salienta que o óxido promoveu uma degradação lenta da fração celulósica até
aproximadamente 550 °C em todas as proporções, diferentemente da celulose isolada na
qual o término do processo de degradação ocorre em 480 °C.
Como descrito no item 5.1.3, a partir da proporção 95/5 observou-se separação de
fases, visível macroscopicamente, desqualificando os demais materiais como híbridos.
Avaliando-se os dados da Tabela 5.6, nota-se que as temperaturas da curva DTG para as
proporções 90/10, 85/15 e 80/20 apresentam-se próximas à temperatura da DTG do
Nb2O5.nH2O isolado (356 °C), caracterizando a heterogeneidade do material.
Entretanto, observa-se que %RE e %RT exibem uma grande variação, indicando que a
quantidade de óxido incorporada à celulose foi menor que a estequiométrica.

104
b) Híbridos (100-x)Celfb/xAl2O3.nH2O
As curvas TGA para os híbridos de alumínio e materiais de partida encontram-se
na Figura 5.23.
Figura 5.23 - Perfis das curvas TGA para os híbridos (100-x)Celfb/xAl2O3.nH2O, em atmosfera de ar sintético, para x = 3, 4, 5, 6, 7, 10, 15 e 20.
Fonte: Produção da autora.
Os híbridos (100-x)Celfb/xAl2O3.nH2O exibem três eventos térmicos em suas
curvas TGA. O primeiro refere-se as desidratações da celulose e do Al2O3.nH2O,
relativo às moléculas de água fracamente ligadas; o segundo refere-se a combustão da
hemicelulose, da celulose e a segunda desidratação do óxido, devido à condensação de
grupos hidroxila. O terceiro refere-se à degradação da lignina remanescente e a
desidratação final do Al2O3.nH2O, devido à formação de estruturas metaestáveis
presentes no óxido (BHATTACHARYA et al., 2004). A Tabela 5.7 apresenta os dados
adquiridos pela termogravimetria.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C )
Celfb Al2O3.nH2O 97/3 96/4 95/5 94/6 93/7 90/10 85/15 80/20

105
Tabela 5.7 - Dados das curvas TGA/DTG para os híbridos (100-x)Celfb/xAl2O3.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva
TGA (%)
Temperatura Curva DTG
(°C)
Resíduo Experimental
RE (%)
Resíduo Teórico RT (%)
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
11,26
-
Al2O3.2,7H2O
25-196 196-900
10,06 22,19
78 295
67,75
-
97/3
25-173 173-413 413-900
4,20 65,94 18,92
69 360 433
10,94
14,26
96/4
25-182 182-418 418-900
3,84 68,97 18,17
65 361 435
9,02
15,26
95/5
25-182 182-423 423-900
4,10 68,48 16,76
65 367 561
10,66
16,26
94/6
25-200 200-414 414-900
6,47 59,07 17,86
69 350 420
16,60
17,26
93/7
25-191 191-405 405-900
6,27 62,11 21,41
60 349 423
10,21
18,26
90/10
25-182 182-404 404-900
4,18 63,14 19,66
64 353 425
13,02
21,26
85/15
25-182 182-409 409-900
4,55 65,54 19,16
64 360 430
10,75
26,26
80/20
25-187 187-396 396-900
6,33 47,60 22,06
67 346 419
24,01
31,26
Fonte: Produção da autora.
Analogamente aos híbridos de nióbio e segundo os dados da Tabela 5.7, os
híbridos contendo alumínio também mostram uma diferença de temperatura, em relação
aos seus precursores, confirmando a síntese de novos materiais devido a incorporação
do Al2O3.nH2O à celulose.

106
De acordo com a Tabela 5.7, o híbrido 95Celfb/5Al2O3.nH2O apresenta a maior
diferença de temperatura (ΔT = 21 °C), entretanto sua %RE (10,66 %) está distante da
%RT (16,26 %), desqualificando sua escolha. Apesar do híbrido 94Celfb/6Al2O3.nH2O
exibir uma mínima alteração na temperatura (ΔT = 4 °C), os valores de resíduo estão
muito próximos, %RE (16,60 %) e %RT (17,26 %). Ou seja, praticamente toda a
quantidade estequiométrica calculada para o óxido foi incorporada à matriz celulósica,
sendo esta selecionada como a melhor proporção. A partir da proporção
93Celfb/7Al2O3.nH2O, observou-se separação de fases entre os precursores, visível
macroscopicamente, também justificável pelas grandes diferenças entre as %RE e %RT.
Silva (2013) preparou híbridos a partir da celulose do bagaço de cana de açúcar e
óxido de alumínio hidratado, para posterior aplicação em membranas. Para o estudo de
proporção foram sintetizadas as seguintes frações mássicas celulose/Al2O3.nH2O: 95/5,
90/10, 85/15 e 80/20. A partir da termogravimetria em ar sintético, os híbridos
apresentam comportamento térmico similar, porém com uma variação de temperatura
em relação à celulose entre 24 e 29 °C. Devido à similaridade de comportamento, a
proporção definida pela autora foi a 95Cel/5Al2O3.nH2O devido à menor quantidade de
óxido requerida na síntese. Os resultados descritos corroboram com a literatura.

107
c) Híbridos (100-x)Celfb/xZnO.nH2O
A Figura 5.24 apresenta as curvas TGA para os híbridos de zinco e seus materiais
de partida.
Figura 5.24 - Perfis das curvas TGA para os híbridos (100-x)Celfb/xZnO.nH2O, em atmosfera de ar sintético, para x = 3, 4, 5, 6, 7, 10, 15 e 20.
Fonte: Produção da autora.
De acordo com a Figura 5.24, a curva TGA do híbrido 97Celfb/3ZnO.nH2O exibe
duas etapas de perda de massa: a primeira retrata as desidratações da celulose e do
ZnO.nH2O, relativa a perda de moléculas de H2O fracamente ligadas; e a segunda
retrata a sequência dos eventos de combustão da hemicelulose e celulose, a completa
desidratação do óxido, referente às moléculas de H2O fortemente ligadas, além da
degradação da lignina remanescente. As proporções 96/4, 95/5, 94/6, 93/7, 90/10 e
80/20 apresentaram três eventos referentes às mesmas etapas descritas para o 97/3, com
o terceiro evento similar ao ocorrido na celulose isolada, referente à degradação da
lignina remanescente. Somente a proporção 85Celfb/15ZnO.nH2O apresenta quatro
eventos de perda de massa em sua curva TGA, com um perfil bem similar à celulose
isolada. Como a partir desta observou-se separação de fases, acredita-se que este
comportamento esteja relacionado à heterogeneidade do material, fato também
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
Celfb
ZnO.nH2O
97/3
96/4
95/5
94/6
93/7
90/10
85/15
80/20

108
observado nos híbridos de Nb. Tal inferência também é justificável pela %RE ser bem
menor (20,34 %) que a %RT (26,26 %). A Tabela 5.8 apresenta os dados obtidos pela
termogravimetria.
Tabela 5.8 - Dados das curvas TGA/DTG para os híbridos (100-x)Celfb/xZnO.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva
TGA (%)
Temperatura Curva DTG
(°C)
Resíduo Experimental
(RE) (%)
Resíduo Teórico
(RT) (%)
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
11,26
-
ZnO.1,3H2O
25-144 144-900
1,41 21,27
90 270
77,32
-
97/3
25-170 170-900
3,85 82,6
62 385
13,55
14,26
96/4
25-174 174-400 400-900
5,79 66,75 13,35
61 354 465
14,11
15,26
95/5
25-174 174-409 409-900
5,14 69,42 10,62
68 354 458
14,82
16,26
94/6
25-178 178-400 400-900
4,29 68,33 13,48
65 355 474
13,90
17,26
93/7
25-174 174-400 400-900
4,59 66,91 14,05
63 357 450
14,45
18,26
90/10
25-165 165-426 426-900
4,51 61,08 15,81
67 350 482
18,60
21,26
85/15 25-157
157-244 244-435 435-900
4,06 1,67
57,48 16,45
66 203 347 472
20,34
26,26
80/20
25-178 178-391 391-900
6,08 66,70 6,55
63 343 475
20,67
31,26
Fonte: Produção da autora.

109
Segundo os dados apresentados na Tabela 5.8 e, como supramencionado neste
trabalho, a interação do óxido de zinco hidratado com a celulose é evidenciada pela
diferença das temperaturas do evento de maior perda de massa, entre os materiais
isolados e os híbridos. Isto é, quanto maior esta diferença de temperatura, maior é a
interação (MASCHIO; PEREIRA; DA SILVA, 2012; PEREIRA et al., 2014).
Dentre as relações mássicas avaliadas, a proporção 97Celfb/3ZnO.nH2O apresenta
a maior interação, com um ΔT = 39 °C. Analisando-se as porcentagens %RE e % RT,
observa-se que são próximas, sendo este um indicativo de que quase toda a quantidade
estequiométrica calculada para o óxido foi incorporada à matriz celulósica. De acordo
com a curva TGA do híbrido 97Celfb/3ZnO.nH2O (Figura 5.24), nota-se uma perda de
massa contínua no terceiro evento até 900 °C. Ou seja, esta curva corrobora com o fato
de que aproximadamente 3 % m/m de ZnO.nH2O promovem uma alta estabilidade
térmica na celulose, prolongando sua degradação devido à incorporação do óxido à
estrutura celulósica.
Nos demais híbridos, o comportamento térmico foi semelhante entre eles, exceto
na proporção 85Cel/15ZnO.nH2O, como citado. As diferenças de temperatura para estes
materiais apresentaram-se muito próximas à celulose isolada, confirmando a baixa
interação entre seus componentes. Além disso, as porcentagens %RE e %RT também
mostram-se distantes, sendo este um indício de pequenas quantidades de ZnO.nH2O
ligado quimicamente à celulose.
As Figuras 5.25a, 5.25b e 5.25c apresentam os perfis térmicos para as proporções
escolhidas 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O,
respectivamente, evidenciando as diferenças em relação aos precursores.

110
Figura 5.25 - Comparação entre as curvas TGA das proporções escolhidas e seus precursores para os híbridos: (a) 97Celfb/3Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O e
(c) 97Celfb/3ZnO.nH2O.
Fonte: Produção da autora.
As proporções escolhidas, 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e
97Celfb/3ZnO.nH2O, foram caracterizadas por DSC, FTIR, DRX, MEV/EDX e testes
de absorção de água para confirmar a obtenção de novos materiais, sem separação de
fases, os quais apresentam ligações químicas entre seus precursores. Ressalta-se que
para a preparação dos compósitos foram utilizadas somente as proporções escolhidas
(97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O) como aditivos
híbridos.
0
20
40
60
80
100
120
0 300 600 900
Per
da d
e m
assa
(%
)
Temperatura (°C)
Celfb
ZnO.nH2O
97/3
0
20
40
60
80
100
120
0 300 600 900
Per
da d
e m
assa
(%
)
Temperatura (°C )
Celfb
Al2O3.nH2O
94/6
0
20
40
60
80
100
120
0 300 600 900
Per
da d
e m
assa
(%
)
Temperatura (°C )
Celfb
Nb2O5.nH2O
97/3
(a) (b)
(c)

111
5.3. HÍBRIDOS: PROPORÇÕES ESCOLHIDAS
Com o propósito de complementar o estudo térmico e verificar as entalpias
envolvidas nos processos de transformação de fase, celulose e híbridos escolhidos
foram caracterizados por DSC. As Figuras 5.26a, 5.26b, 5.26c e 5.26d apresentam as
curvas calorimétricas para celulose, 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e
97Celfb/3ZnO.nH2O, respectivamente.
Figura 5.26 - Perfis das curvas DSC para: (a) celulose, (b) 97Celfb/3Nb2O5.nH2O, (c) 94Celfb/6Al2O3.nH2O e (d) 97Celfb/3ZnO.nH2O em atmosfera de nitrogênio.
Fonte: Produção da autora.
A curva DSC para a celulose (Figura 5.26a) apresenta três picos endotérmicos e
um exotérmico. O primeiro, em 81 °C e ∆Hdes. = 20 J.g-1, denota a desidratação da
celulose, referente à volatilização dos grupos hidroxila, devido à acetilação parcial. O
segundo, em 149 °C com ∆Hdec. = 10 J.g-1, denota a decomposição da hemicelulose; o
terceiro em 187 °C com ∆Hdec. = 57 J.g-1 refere-se à decomposição da celulose. O
-6
-4
-2
0
2
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C)
-3
-2
-1
0
1
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C)
-3
-2,5
-2
-1,5
-1
-0,5
0
0,5
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
(a) (b)
187 °C 156 °C
342 °C
189 °C
337 °C
(c)
! EXO ! EXO
! EXO
-4
-3
-2
-1
0
1
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C)
(d)
! EXO
81 °C
149 °C
323 °C
83 °C
86 °C 149 °C
82 °C
146 °C
187 °C
351 °C

112
último pico em 323 °C, denota a despolimerização com a formação de resíduos sólidos,
com ∆Hdespol. = -44 J.g-1.
Segundo Kabir et al. (2013), celulose, hemicelulose e lignina exibem eventos
distintos, através de uma sequência de picos endotérmicos e exotérmicos, mediante as
transformações de fase, em temperaturas definidas. Nestas transformações, a
volatilização de gases é responsável pelos eventos endotérmicos, enquanto a formação
de resíduos sólidos pelos eventos exotérmicos (YANG et al., 2007). Os resultados
apresentados neste trabalho corroboram com os dados discutidos no item 5.2.1.
Comportamento similar também foi relatado em outros trabalhos (SUN et al., 2004;
DEEPA et al., 2011; OLIVEIRA et al., 2012; KABIR et al., 2013).
A curva de DSC para o híbrido 97Celfb/3Nb2O5.nH2O (Figura 5.26b) exibe três
picos endotérmicos: o primeiro ocorrido em 83 °C e ∆Hdes. = 24 J.g-1, refere-se à
desidratação da celulose; o segundo, em 156 °C com ∆Hdec. = 101 J.g-1, denota as
decomposições da hemicelulose e da celulose e a desidratação do Nb2O5.nH2O. Neste
segundo evento, observa-se um alargamento do pico, o qual está relacionado à
sobreposição das transformações ocorridas simultaneamente. E o terceiro pico, em 342
°C com ∆Hdespol. = 102 J.g-1, refere-se à despolimerização da celulose, com formação de
resíduos sólidos e a quebra das ligações relativas ao Nb2O5.nH2O presente na estrutura
da celulose. Esta transformação é decorrente da presença do óxido, resultando em um
efeito endotérmico, com supressão de calor.
O híbrido 94Celfb/6Al2O3.nH2O apresenta quatro picos endotérmicos na curva de
DSC (Figura 5.26c): o primeiro em 86 °C e ∆Hdes. = 17 J.g-1 corresponde à desidratação
da celulose; o segundo em 149 °C com ∆Hdec. = 7 J.g-1, devido a decomposição da
hemicelulose e a primeira desidratação do óxido, relativa às moléculas de água
fracamente ligadas. O terceiro ocorrido em 189 °C com ∆Hdec. = 68 J.g-1 representa a
decomposição da celulose e segunda desidratação do óxido, referente às moléculas de
água fortemente ligadas; e o quarto, em 337 °C com ∆Hdespol. = 120 J.g-1, refere-se à
despolimerização da celulose, com formação de resíduos sólidos e quebra da ligação
entre a estrutura celulósica e o Al2O3.nH2O, com supressão de calor.
A curva DSC para o híbrido 97Celfb/3ZnO.nH2O (Figura 5.26d) exibe três picos
endotérmicos e um exotérmico. Os picos endotérmicos denotam os mesmos eventos

113
mencionados para o híbrido 97Celfb/3Nb2O5.nH2O, porém nas seguintes temperaturas e
entalpias: 82 °C e ∆Hdes. = 16 J.g-1 (1° pico), 146 °C com ∆Hdec. = 11 J.g-1 (2° pico) e
187 °C com ∆Hdec. = 67 J.g-1 (3° pico). O último pico invertido (exotérmico), em 351 °C
com ∆Hdespol. = -28 J.g-1, corresponde à despolimerização da celulose, com formação de
resíduos sólidos e a quebra das ligações relativas ao óxido de zinco hidratado. Nota-se
que a entalpia referente ao último evento para o 97Celfb/3ZnO.nH2O apresenta-se
menor, quando comparada aos híbridos de nióbio e de alumínio, devido à menor energia
necessária para a quebra destas ligações entre os metais e a estrutura orgânica. Vale
salientar que as diferenças observadas entre as temperaturas das curvas TGA/DTG em
comparação às curvas DSC são decorrentes da atmosfera envolvida em cada análise. A
Tabela 5.9 apresenta os dados obtidos pelas análises de DSC.
Tabela 5.9 – Dados das curvas DSC para a celulose e híbridos escolhidos em atmosfera de nitrogênio.
MATERIAL
Intervalos Curva DSC
(°C)
Entalpia de reação ΔH (J.g-1)
Temperatura de início do evento
Tinicial (°C)
Celfb 52-93
140-160 181-205 300-355
20 10 57 -44
73 148 186 309
97Celfb/3Nb2O5.nH2O
54-96 135-219 269-363
24 101 102
73 149 307
94Celfb/6Al2O3.nH2O
69-98 142-160 185-213 275-363
17 7
68 120
75 147 186 310
97Celfb/3ZnO.nH2O
71-93 133-160 181-205 333-375
16 11 67 -28
75 145 186 340
Fonte: Produção da autora.
Analisando-se os dados da Tabela 5.9, observa-se que somente os híbridos
97Celfb/3Nb2O5.nH2O e 94Celfb/6Al2O3.nH2O exibiram picos endotérmicos na região

114
de despolimerização da celulose, com valores das entalpias de ∆Hdespol. = 102 J.g-1 e
∆Hdespol. = 120 J.g-1, respectivamente. Esta inversão de pico, isto é, a mudança na
entalpia de despolimerização da celulose observada nestes híbridos é um indicativo do
efeito dos óxidos de Nb2O5.nH2O e Al2O3.nH2O como agentes retardantes de chamas.
Outro ponto importante é a diferença entre as entalpias de despolimerização da
celulose e do híbrido, ou seja, a quantidade de calor trocada neste processo devido ao
efeito de cada óxido metálico hidratado incorporado à fração orgânica.
Matematicamente, o calor envolvido neste processo pode ser expresso na Equação 5.6:
∆!!"#$%&'(&" = ∆!!"#$%&. !í!"#$% !− ! ∆!!"#$%&. !"#$#%&" ! (5.6)
Sendo:
∆Hresultante = a entalpia referente ao efeito da presença do óxido.
Logo, para os híbridos 97Celfb/3Nb2O5.nH2O e 94Celfb/6Al2O3.nH2O a
quantidade de calor trocada foi de ΔHresultante = 146 J.g-1 e de ΔHresultante = 164 J.g-1,
respectivamente. Nota-se que a maior quantidade de calor trocada encontra-se para o
híbrido 94Celfb/6Al2O3.nH2O, confirmando a eficiência do Al2O3.nH2O como
retardante de chama. De acordo com Hull; Witkowski; Hollingbery (2011), este óxido
hidratado decompõe-se endotermicamente com a liberação de vapores de água, os quais
diluem os gases combustíveis, seguida pela formação de Al2O3 como camada protetora
na superfície do produto, minimizando a difusão de oxigênio para o meio reativo, a qual
impede a troca de calor.
Comparando-se a fração dos óxidos incorporados à celulose e a quantidade de
calor trocada, observa-se que aproximadamente 3 % m/m de Nb2O5.nH2O proporcionou
um efeito similar de retardância à celulose, com uma entalpia total bem próxima ao
híbrido de alumínio com aproximadamente 6 % m/m.
Apesar do híbrido 97Celfb/3ZnO.nH2O ser o mais estável termicamente, dentre as
proporções definidas, por apresentar a maior diferença na temperatura da DTG (ΔT =
39 °C), como discutido em termogravimetria (item 5.2.3), este não apresenta
característica de retardância à chamas. Tal evidência pode ser confirmada pela presença

115
do pico exotérmico, na região de despolimerização da celulose, com uma entalpia de
ΔHdespol. = -28 J.g-1 e pela ínfima quantidade de calor trocada (ΔHresultante = 16 J.g-1).
Todos estes resultados apresentam-se concordantes com trabalhos reportados pelo grupo
(OTTOBONI, 2011; MASCHIO; PEREIRA; DA SILVA, 2012; SILVA, 2013).
As Figuras 5.27a, 5.27b e 5.27c apresentam os difratogramas para os híbridos
97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O, respectivamente,
comparados à suas frações individuais correspondentes.
Figura 5.27 - Difratogramas para (a) 97Celfb/3Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O e (c) 97Celfb/3ZnO.nH2O e seus respectivos precursores.
Fonte: Produção da autora.
Os difratogramas dos híbridos não exibem picos característicos de seus
respectivos óxidos metálicos hidratados, somente os picos referentes à celulose, porém
em diferentes intensidades. A ausência dos picos característicos dos MyOz.nH2O sugere
que não há depósito da fração inorgânica na superfície da celulose. Os difratogramas
0
1000
2000
3000
4000
5000
6000
7000
8000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
)
2θ (graus)
Celfb ZnO.nH2O 97Celfb/3ZnO.nH2O
0
500
1000
1500
2000
2500
3000
3500
4000
4500
5000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Celfb Nb2O5nH2O 97Celfb/3Nb2O5.nH2O
0
1000
2000
3000
4000
5000
6000
7000
10 20 30 40 50 60 70 80 90
Inte
nsid
ade
(u.a
.)
2θ (graus)
Celfb Al2O3.nH2O 94Celfb/6Al2O3.nH2O
(a) (b)
(c)

116
também confirmam a formação de novos materiais, pois cada híbrido exibe um perfil de
difração diferente de suas frações de partida, corroborando com a termogravimetria.
Analogamente à celulose, estimou-se o índice de cristalinidade (Ic) de cada
híbrido, segundo a Equação 4.1. A Tabela 5.10 apresenta os valores dos Ic para as três
proporções definidas, comparadas à celulose isolada.
Tabela 5.10 – Valores dos índices de cristalinidade (Ic) para a celulose e híbridos 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O.
Material I1 (u.a.) I2 (u.a.) Ic (%)
Celulose 570 3272 82,6
97Celfb/3Nb2O5.nH2O 904 4460 79,7
94Celfb/6Al2O3.nH2O 1329 5907 77,5
97Celfb/3ZnO.nH2O 963 4432 78,3
Fonte: Produção da autora.
De acordo com a Tabela 5.10, os índices de cristalinidade (Ic) dos híbridos
apresentam-se menores, quando comparados à celulose, indicando que tal decréscimo
na cristalinidade é devido à incorporação dos óxidos metálicos hidratados à matriz
celulósica. Os resultados obtidos mostram-se concordantes com trabalhos do grupo do
Laboratório de Novos Materiais (OTTOBONI, 2011; MASCHIO; PEREIRA; DA
SILVA, 2012; SILVA, 2013). Segundo Cunha et al. (2010), a permanência do padrão
de difração da celulose sugere que a modificação com os óxidos pouco afetou suas
regiões cristalinas.
Os híbridos escolhidos também foram submetidos à análises de FTIR, a fim de
verificar a presença de grupos funcionais devido às modificações químicas efetuadas.
As Figuras 5.28a, 5.28b e 5.28c apresentam os espectros para os híbridos
97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O, respectivamente.

117
Figura 5.28 - Espectros na região do infravermelho para (a) 97Celfb/3Nb2O5.nH2O, (b) 94Celfb/6Al2O3.nH2O e (c) 97Celfb/3ZnO.nH2O.
Fonte: Produção da autora.
Nos espectros dos híbridos é possível identificar a presença da carbonila de éster,
referente ao grupo acetila, devido à acetilação parcial da celulose, e grupos hidroxila.
Em todos os materiais há um deslocamento da carbonila de 1740 cm-1 (celulose) para
1744 cm-1 (97Celfb/3Nb2O5.nH2O), 1757 cm-1 (94Celfb/6Al2O3.nH2O) e 1763 cm-1
(97Celfb/3ZnO.nH2O), confirmando a permanência deste grupo após a modificação
química com os óxidos metálicos hidratados. Isto é, de acordo com os espectros a
incorporação dos óxidos à celulose ocorreu nas hidroxilas disponíveis, uma vez que esta
foi acetilada parcialmente, como discutido no item 5.2.1. A permanência do grupo
acetila nos híbridos pode favorecer a compatibilidade com a matriz polimérica na
produção dos compósitos.
As Figuras 5.29a, 5.29b, 5.29c e 5.29d exibem as micrografias da celulose, dos
híbridos 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O,
respectivamente, para a avaliação superficial.
0
20
40
60
80
100
120
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tra
smit
ânci
a (%
)
Número de onda (cm-1)
97Celfb/3ZnO.nH2O
0
20
40
60
80
100
120
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tra
nsm
itân
cia
(%)
Número de onda (cm-1)
94Celfb/6Al2O3.nH2O
0
20
40
60
80
100
120
400 800 1200 1600 2000 2400 2800 3200 3600 4000
Tra
nsm
itân
cia
(%)
Número de onda (cm-1)
97Celfb/3Nb2O5.nH2O
(a) (b)
(c)
1744 cm-1 1757 cm-1
1763 cm-1

118
Figura 5.29 - Micrografias para (a) celulose, (b) 97Celfb/3Nb2O5.nH2O, (c) 94Celfb/6Al2O3.nH2O e (d) 97Celfb/3ZnO.nH2O, com ampliação de 100x.
Fonte: Produção da autora.
De acordo com as micrografias, em todos materiais é evidente a ausência de
depósito dos óxidos na superfície da celulose, corroborando com os resultados
apresentados por DRX. As análises de EDX identificaram a presença dos metais Nb, Al
e Zn nos híbridos, confirmando a incorporação dos óxidos metálicos hidratados à
estrutura da celulose.
Para o híbrido 97Celfb/3ZnO.nH2O, a análise de EDX também identificou uma
pequena quantidade de sódio devido ao agente precipitante, corroborando com o
resultado exibido na curva DSC para o ZnO.nH2O, discutido no item 5.2.2. Na síntese
do híbrido 97Celfb/3ZnO.nH2O, como o pH do meio reacional é básico, os grupos
hidroxila disponíveis na celulose serão atacados, fornecendo sítios ativos para a entrada
do ZnO.nH2O. Neste caso, há uma competição entre os metais zinco e sódio devido ao
excesso de agente precipitante (NaOH) para atingir o pH = 10. Entretanto, como a
reatividade do zinco é maior que a do sódio, consequentemente, o zinco ocupará mais
sítios em relação ao sódio e este último permanecerá na estrutura da celulose; assim
(b) (a)
SE MAG: 100 x HV: 20,0 kV WD: 0,0 mm
SE MAG: 100 x HV: 20,0 kV WD: 0,0 mm
(c)
SE MAG: 100 x HV: 20,0 kV WD: 0,0 mm
(d)

119
como discutido no ataque básico da celulose (item 5.2.1). Além disso, o raio iônico do
zinco é muito menor (0,074 nm) comparado ao sódio (0,102 nm), favorecendo sua
entrada em um número maior de sítios ativos.
5.4. ABSORÇÃO DE ÁGUA
Como mencionado, a absorção de umidade caracteriza-se como uma das
principais desvantagens das fibras lignocelulósicas, devido aos grupamentos
hidrofílicos presentes em sua estrutura química (SPINACÉ et al., 2009; ALBINANTE;
PACHECO; VISCONTE, 2013; KABIR et al., 2013). Tal desvantagem enfraquece a
adesão interfacial, afeta a estabilidade dimensional e contribui para o processo de
degradação destas fibras, impedindo sua aplicação como reforço em compósitos
poliméricos. A baixa resistência à absorção de umidade, não só reduz o desempenho
mecânico como também prejudica as propriedades a longo prazo (KABIR et al., 2012b;
PEREIRA et al., 2015; PICKERING; ARUAN EFENDY; LE, 2016).
Neste contexto, as modificações químicas apresentadas neste trabalho tiveram por
objetivo verificar seus efeitos na absorção de H2O, reduzindo a hidrofilicidade dos
materiais celulósicos, com o propósito de favorecer a compatibilidade com a matriz de
PU com caráter hidrofóbico para a produção dos compósitos. Foram realizados testes de
absorção de água em iguais condições para folha bruta, celulose e híbridos
97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O, a fim de avaliar
os efeitos da modificação superficial na fonte vegetal e da incorporação dos
componentes inorgânicos na fração celulósica. Ou seja, o quanto tais modificações
influenciaram na capacidade de absorção de água. A Figura 5.30 apresenta as curvas de
absorção de água e a Tabela 5.11 exibe os dados obtidos para a folha bruta, a celulose e
os aditivos híbridos.

120
Figura 5.30 - Efeito da modificação química na capacidade de absorção de água em função do tempo para a folha bruta, celulose e os aditivos híbridos.
Fonte: Produção da autora.
Tabela 5.11 – Valores obtidos nos testes de absorção de água para a folha bruta (Fb), celulose (Celfb) e híbridos 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e
97Celfb/3ZnO.nH2O. Tempo
(h)
!"##"!!!!"##"!"#$%!!"#$%
(g/g)
!"##"!!!!"##"!"#$#%&"
(g/g)
!"##"!!!!"##"!í!"#$%!!"
(g/g)
!"##"!!!!"##"!í!"#$%!!"
(g/g)
!"##"!!!!"##"!í!"#$%!!"
(g/g) 1 4,64 4,25 2,75 2,39 2,36 2 5,56 4,49 3,15 2,33 2,59 3 5,60 4,45 3,08 2,12 2,86 4 5,87 4,54 3,03 1,85 2,84 6 5,59 5,03 3,23 2,38 2,84
12 6,33 4,01 2,76 2,11 2,26 24 48
6,99 6,96
4,45 4,62
3,30 3,11
2,29 2,34
2,95 2,65
Fonte: Produção da autora.
Segundo Albinante; Pacheco; Visconte (2013), a acetilação é um tratamento
químico que modifica a superfície das fibras, tornando-as mais hidrofóbicas devido à
substituição parcial dos grupos hidroxila, presentes nas fibra lignocelulósicas, por
grupos acetila, como discutido no item 5.2.1. Para a folha bruta observa-se um aumento
na absorção de água até o tempo de 24 h, com saturação após 48 h de imersão. De
0,00
1,00
2,00
3,00
4,00
5,00
6,00
7,00
8,00
1 2 3 4 6 12 24 48
Mas
sa H
2O/M
assa
adi
tivo
Tempo (h)
Folha bruta - Fb
Celfb
97Celfb/3Nb2O5.nH2O
94Celfb/6Al2O3.nH2O
97Celfb/3ZnO.nH2O

121
acordo com Spinacé et al. (2009), a umidade pode causar o inchamento na parede
celular da fibra até que a mesma esteja saturada com água e, acima disto, a umidade
apresenta-se como água livre nas lacunas da fibra, não ocasionando o seu inchamento.
Os dados da Tabela 5.11 indicam que o ataque ácido atuou como redutor na
hidrofilicidade da celulose em todos os testes avaliados, com absorção de água
praticamente constante ao longo do tempo, devido à modificação parcial na superfície.
Os resultados corroboram com os apresentados por FTIR, TGA/DTG e trabalhos
anteriores (D’ALMEIDA et al., 2005; LOPES et al., 2011; MBOUGUENG et al., 2012;
CAI et al., 2013). Avaliando-se os valores, após 24 h de imersão, houve uma redução de
36 % na capacidade de absorção de água da celulose. Comparando-se os resultados
obtidos para a celulose com os híbridos, observa-se uma redução no caráter hidrofílico
devido à presença dos componentes inorgânicos, nos seguintes valores:
97Celfb/3Nb2O5.nH2O = 26 %, 94Celfb/6Al2O3.nH2O = 49 % e 97Celfb/3ZnO.nH2O =
34 %. A partir dos testes de absorção de água, destaca-se o híbrido
94Celfb/6Al2O3.nH2O, pois exibiu a maior redução no caráter hidrofílico comparado
aos demais.
Baseado nos resultados abordados, o ataque com a mistura ácida
CH3COOH/HNO3 e a incorporação dos óxidos metálicos hidratados apresentaram-se
como eficientes modificações químicas na redução do caráter hidrofílico destes
materiais de base celulósica para aplicação como aditivo na matriz hidrofóbica de PU.

122
5.5. COMPÓSITOS DE PU: ESTRUTURA CELULAR E MORFOLOGIA
A Figura 5.31 exibe as micrografias da matriz de PU e dos compósitos PU +
Celfb.
Figura 5.31 - Micrografias para (a) PU puro; (b) PU + 1% m/m Celfb; (c) PU + 2% m/m Celfb; (d) PU + 3% m/m Celfb; (e) PU + 4% m/m Celfb e (f) PU + 5% m/m Celfb, com
ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

123
A partir da micrografia do PU rígido (Figura 5.31a), observam-se células de
formato poliédrico, predominantemente fechadas, com uma distribuição relativamente
uniforme e pequenas regiões com células colapsadas. As micrografias dos compósitos
PU/celulose (Figuras 5.31b-f), por sua vez, indicam alterações na estrutura das células,
tais como, no formato (de poliédrico para esférico), aumento no tamanho médio e maior
número de regiões colapsadas, à medida em que aumenta-se a quantidade de celulose
incorporada à matriz.
A formação das espumas rígidas de PU envolve diferentes reações químicas, as
quais ocorrem desde as etapas de nucleação até o crescimento final, originando espumas
reticuladas com estruturas celulares (VILAR, 2005). Nas espumas, o termo célula é
designado para representar a menor unidade estrutural, a qual está relacionada ao
conteúdo vazio presente em sua estrutura (RIBEIRO DA SILVA et al., 2013b;
CORDERO et al., 2015). Com um formato tipicamente poliédrico, estas células são
obtidas quando as bolhas em crescimento se tocam, devido à geração in situ do agente
de expansão (CO2) (VILAR, 2005; ANTUNES et al., 2011). Diferentes aspectos
morfológicos, tais como geometria, concentração volumétrica, tipo (célula fechada ou
aberta) e teor de gás inerte podem influenciar em propriedades mecânicas, térmicas e de
isolamento da espuma final (CORDERO et al., 2015).
De acordo com Zieleniewska et al. (2016), a adição de partículas sólidas na fase
poliol pode alterar significativamente o processo de crescimento da espuma,
influenciando a fase de nucleação. Parâmetros como o número e a homogeneidade das
microbolhas de gás formadas durante a agitação mecânica desta pré-mistura
determinam a nucleação das espumas. Consequentemente, a incorporação de aditivos
pode intervir no processo de nucleação, afetando o número de células e a espessura da
parede celular.
Comparando-se os compósitos PU + 1% m/m Celfb (Figura 5.31b) e PU + 5%
m/m Celfb (Figura 5.31f), por exemplo, evidencia-se que o aumento de celulose
proporcionou uma maior quantidade de furos com células altamente distorcidas na
estrutura destes compósitos. Segundo Ribeiro da Silva et al. (2013a) o aumento da
viscosidade na pré-mistura, devido à incorporação de aditivos, pode afetar a expansão

124
do PU, ocasionando a formação de células desestruturadas e distribuição de tamanhos
menos uniforme, comparada ao PU puro.
Resultados similares foram observados por Yang et al. (2015), nos quais a
incorporação de quantidades maiores de retardante de chamas ao PU rígido provocou
colapso celular e ruptura das paredes celulares, originando furos na estrutura formada.
Os autores atribuem estes efeitos às tensões internas ou ao crescimento desequilibrado
da espuma induzido pela alta viscosidade, resultante dos altos teores de retardante
adicionado à fase poliol.
A incorporação dos demais aditivos, óxidos metálicos hidratados e híbridos
(Apêndice B), também proporcionou os mesmos efeitos observados nos compósitos
contendo celulose.
5.6. COMPÓSITOS DE PU: COMPORTAMENTO TÉRMICO E
PROPRIEDADES MECÂNICAS
A síntese do PU baseia-se na reação entre o isocianato e compostos contendo
grupos hidroxila ativos, tais como os polióis, além da adição de catalisadores,
extensores de cadeia, tensoativos etc (KONG; NARINE, 2007). Esta reação promove a
formação de uma estrutura complexa, composta por moléculas interpenetradas de
diferentes massas moleculares, funcionalidades e cristalinidade. O resultado é uma
estrutura formada por segmentos rígidos e flexíveis, provenientes de seus precursores
isocianato e poliol, respectivamente. Consequentemente, a distribuição destes
segmentos ao longo da cadeia do PU determinará as propriedades finais do polímero
(CORDERO et al., 2015).
Segundo Kong; Narine (2007), além da distribuição entre os segmentos rígidos e
flexíveis, fatores como a composição química e a fração destes segmentos e o grau de
ligações cruzadas também podem influenciar nas propriedades finais dos PU’s. Tais
fatores podem ser manipulados mediante a variação na razão estequiométrica de
poliol/isocianato ou na taxa entre grupos hidroxila (OH) e grupos isocianato (NCO).
Como a formulação trata-se de um sistema fechado, isto é, sem a especificação de
sua composição química este trabalho visa investigar o efeito dos aditivos inseridos

125
nesta formulação e sua influência nas propriedades térmicas e mecânicas dos
compósitos, com o intuito de direcionar um campo de aplicação.
5.6.1. Termogravimetria
A degradação térmica dos PU’s é um processo muito complexo devido à
ocorrência de vários fenômenos físico-químicos. Diversos fatores podem influenciar no
perfil térmico destes polímeros, tais como o número de grupos uretano, tipo e
funcionalidade do poliol, ramificações, isocianato não reagido etc (JAVNI et al., 2000;
NARINE et al., 2007). Além disso, a estabilidade térmica destes polímeros
correlaciona-se com os tipos de segmentos e sua distribuição ao longo da cadeia
(CORCUERA et al., 2010).
Nesta seção, a termogravimetria foi utilizada para investigar o perfil térmico dos
compósitos e avaliar o efeito na estabilidade destes, mediante a inserção dos diferentes
aditivos à matriz de PU. A seguir são apresentados todos os resultados dos referidos
compósitos, em comparação com a matriz polimérica e os respectivos aditivos. As
curvas TGA/DTG da matriz PU e dos compósitos PU + Celfb, PU + Nb2O5.nH2O, PU +
Al2O3.nH2O, PU + ZnO.nH2O, PU + 97Celfb/3Nb2O5.nH2O, PU + 94Celfb/6
Al2O3.nH2O e 97Celfb/3ZnO.nH2O encontram-se no Apêndice C.
a) Compósitos PU + celulose, PU + Celfb
De uma maneira geral, o processo de degradação térmica das espumas rígidas de
PU ocorre em duas e três etapas, sob atmosfera de nitrogênio e ar sintético,
respectivamente. Em ambas as atmosferas, a primeira etapa denota a volatilização de
pequenas moléculas, com uma perda de massa de ~ 3 %, em temperaturas próximas à
190 °C (JIAO et al., 2013). A segunda etapa pode ser descrita como a decomposição das
ligações do grupo uretano, referente aos segmentos rígidos, com conversão a isocianato
e álcool, seguida pela formação de aminas, pequenos componentes de transição e
dióxido de carbono (CO2). E a terceira etapa, efetuada em altas temperaturas, refere-se
às decomposições dos segmentos flexíveis, ureia substituída, isocianurato e outros
compostos estáveis termicamente; ou ainda, a quebra da ligação C-C (JAVNI et al.,
2000; CHATTOPADHYAY; WEBSTER, 2009; CORCUERA et al., 2010;

126
MEYABADI et al., 2013; RIBEIRO DA SILVA et al., 2013a). A Figura 5.32 apresenta
as curvas TGA.
Figura 5.32 - Perfis das curvas TGA para os precursores PU e Celfb e compósitos PU + Celfb, em atmosfera de ar sintético, com adição de 1 a 5 % m/m de Celfb.
Fonte: Produção da autora.
A curva TGA para o PU puro apresenta quatro eventos de perda de massa: o
primeiro e segundo eventos, relatam a volatilização de pequenas moléculas; o terceiro
denota a decomposição das ligações uretânicas e o quarto evento refere-se às
decomposições dos segmentos flexíveis e demais componentes estáveis termicamente e
quebra da ligação C-C, com completa decomposição em 750 °C. Exceto o compósito
PU + 1% m/m Celfb que exibe três eventos térmicos, os demais apresentam quatro
eventos de perda de massa, os quais correspondem aos mesmos eventos mencionados
para o PU puro.
Comparando-se as curvas dos compósitos com o PU puro, observa-se que com o
aumento do ativo Celfb no meio reacional, há uma mudança significativa no perfil
térmico, a qual pode estar relacionada à maior interação matriz/celulose. Ou seja, além
da reação com os grupos OH presentes no poliol, é possível que os grupos isocianato
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
Celfb
PU + 1%m/m Celfb
PU + 2%m/m Celfb
PU + 3%m/m Celfb
PU + 4%m/m Celfb
PU + 5%m/m Celfb

127
tenham reagido com os grupos hidroxila disponíveis na estrutura da celulose, uma vez
que esta apresenta-se parcialmente acetilada, como discutido no item 5.2.1. A Tabela
5.12 exibe os dados obtidos para os compósitos de PU + Celfb.
Tabela 5.12 - Dados das curvas TGA/DTG para os compósitos PU + Celfb e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
PU
25-136 136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
PU + 1%m/m Cefb
25-268 268- 511 511-900
0,66 53,00 29,10
53 339 682
PU + 2%m/m Cefb
25-166 166-264 264-506 506-900
0,05 0,77
54,17 33,02
61 206 344 661
PU + 3%m/m Cefb
25-123 123-238 238-500 500-900
0,71 1,00
55,10 36,09
60 173 344 684
PU + 4%m/m Cefb
25-123 123-226 226-500 500-900
0,59 0,66
55,52 35,10
61 197 336 850
PU + 5%m/m Cefb
25-136 136-230 230-511 511-900
0,81 1,08
57,82 23,77
63 161 344 710
Fonte: Produção da autora.
A estabilidade térmica dos PU’s, assim como de seus compósitos, é uma
propriedade importante, devido à liberação de voláteis nocivos à saúde humana. Após a

128
ignição, estes polímeros produzem uma grande quantidade de calor num curto período
de tempo, com a liberação de gases tóxicos, tais como CO, CO2, HCN, metanol,
piridina, acetonitrila, acrilonitrila e dentre outros (XI et al., 2015; YANG et al., 2015).
De acordo com Yang et al. (2015), a estabilidade térmica dos PU’s está
relacionada ao grau de reticulação ao invés do seu peso molecular. O alto grau de
reticulação é função do número de ligações cruzadas existentes na estrutura, as quais
interferem na estabilidade da rede. Logo, quanto mais reticulado o PU, maior sua
estabilidade, tanto em ambientes inertes quanto oxidativos, e maior a energia térmica
necessária para que ocorra o rompimento destas ligações adicionais.
Considerando-se a liberação de voláteis do PU e o objetivo geral do trabalho em
verificar a atuação dos aditivos (celulose, óxidos metálicos hidratados e híbridos) como
inibidores no processo de decomposição da matriz polimérica, alguns critérios foram
avaliados mediante o perfil térmico dos materiais. Nas curvas TGA observa-se a
temperatura de DTG e a evolução de massa na etapa de decomposição do PU, os quais
podem influenciar diretamente na estabilidade térmica dos compósitos.
De acordo com os critérios citados, os compósitos com 2, 3 e 5 % m/m de
celulose apresentaram as maiores temperaturas (344 °C). Porém o compósito contendo 2
% m/m de celulose foi escolhido como a melhor proporção pois, além da temperatura,
exibiu a menor perda de massa (54,17 %), em relação às demais proporções citadas. Ou
seja, esta quantidade foi suficiente para reduzir a inibição de voláteis do PU e
proporcionar a maior estabilidade térmica à matriz. Segundo Mosiewicki et al. (2009), a
reação adicional entre o isocianato e os grupos hidroxila presentes na celulose origina
novas ligações, as quais fornecem uma alta estabilidade térmica aos compósitos.
Park; Oh; Kim (2013) preparam biocompósitos de PU elastomérico, a partir do
óleo de mamona, reforçados com nanowhiskers de celulose. Inicialmente, os autores
modificaram quimicamente a celulose, a fim de favorecer uma melhor dispersão e
incorporação na matriz de PU. Os resultados obtidos por TGA mostraram que, para as
quantidades de 0; 0,5; 1,0 e 5,0 % m/m de celulose modificada, as temperaturas de DTG
foram 392, 407, 410 e 412 °C, respectivamente. Segundo os autores, o aumento do
conteúdo de nanowhiskers dificulta o processo de decomposição dos compósitos,
promove a ligação entre a fração celulósica e o isocianato, melhorando a estabilidade

129
térmica, evidenciada pelo aumento da temperatura da DTG. Os resultados exibidos para
os compósitos PU + Celfb apresentam-se concordantes com a literatura.
Os critérios mencionados foram adotados na avaliação de todos os compósitos, a
fim de definir o material com a maior estabilidade térmica.
b) Compósitos PU + Nb2O5.nH2O
A Figura 5.33 apresenta as curvas TGA para os compósitos contendo
Nb2O5.nH2O e a Tabela 5.13 os dados obtidos via termogravimetria.
Figura 5.33 - Perfis das curvas TGA para os precursores PU e Nb2O5.nH2O e compósitos PU + Nb2O5.nH2O, em atmosfera de ar sintético, com adição de 1 a 5 %
m/m de Nb2O5.nH2O.
Fonte: Produção da autora.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
Nb2O5.nH2O
PU + 1%m/m Nb2O5.nH2O
PU + 2%m/m Nb2O5.nH2O
PU + 3%m/m Nb2O5.nH2O
PU + 4%m/m Nb2O5.nH2O
PU + 5%m/m Nb2O5.nH2O

130
Tabela 5.13 - Dados das curvas TGA/DTG para os compósitos PU/Nb2O5.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
PU
25-136 136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
Nb2O5.nH2O
25-200 200-900
7,37 12,87
88 356
PU + 1%m/m Nb2O5.nH2O
25-119 119-221 221-489 489-900
1,22 1,12
54,17 35,71
57 157 337 679
PU + 2%m/m Nb2O5.nH2O
25-100 100-226 226-528 528-900
0,32 1,83
56,40 31,48
63 152 347 725
PU + 3%m/m Nb2O5.nH2O
25-100 100-221 221-523 523-900
0,63 1,79
54,70 35,24
63 142 353 714
PU + 4%m/m Nb2O5.nH2O
25-100 100-213 213-523 523-900
0,71 1,86
55,69 32,79
64 149 351 716
PU + 5%m/m Nb2O5.nH2O
25-100 100-226 226-511 511-900
0,53 2,42
54,50 35,56
63 148 351 691
Fonte: Produção da autora.
Segundo a Figura 5.33 e os dados da Tabela 5.13, todas as curvas TGA dos
compósitos PU + Nb2O5.nH2O apresentam quatro eventos térmicos, os quais denotam
as mesmas etapas descritas para o PU puro (item a). Avaliando-se o perfil térmico dos
compósitos, em todas as proporções observa-se o deslocamento dos terceiro e quarto
eventos, com variações de suas temperaturas máximas de decomposição, em relação à
matriz pura.

131
Avaliando-se os dados da Tabela 5.13, a proporção contendo 3 % m/m de
Nb2O5.nH2O apresentou a maior temperatura da DTG na etapa de decomposição dos
grupos uretânicos (353 °C), comparada ao PU puro (340 °C), e uma perda de massa de
54,70 %, evidenciando o compósito PU + 3% m/m Nb2O5.nH2O como o material de
maior estabilidade térmica dentre as proporções avaliadas para este aditivo inorgânico.
c) Compósitos PU + Al2O3.nH2O
A Figura 5.34 representa as curvas TGA para os compósitos de PU +
Al2O3.nH2O, enquanto a Tabela 5.14 exibe os dados obtidos na termogravimetria.
Figura 5.34 - Perfis das curvas TGA para os precursores PU e Al2O3.nH2O e compósitos PU + Al2O3.nH2O, em atmosfera de ar sintético, com adição de 1 a 5 % m/m de
Al2O3.nH2O.
Fonte: Produção da autora.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
Al2O3.nH2O
PU + 1%m/m Al2O3.nH2O
PU + 2%m/m Al2O3.nH2O
PU + 3%m/m Al2O3.nH2O
PU + 4%m/m Al2O3.nH2O
PU + 5%m/m Al2O3.nH2O

132
Tabela 5.14 - Dados das curvas TGA/DTG para os compósitos PU + Al2O3.nH2O e precursores, sendo n.d. = não detectado.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
PU
25-136 136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
Al2O3.nH2O
25-196 196-900
10,06 22,19
78 295
PU + 1%m/m Al2O3.nH2O
25-85 85-213
213-500 500-900
0,30 1,34
59,27 20,48
63 151 346 nd
PU + 2%m/m Al2O3.nH2O
25-100 100-221 221-500 500-900
0,26 1,59
58,05 20,77
63 146 349 n.d.
PU + 3%m/m Al2O3.nH2O
25-94 94-213
213-494 494-900
1,04 1,55
53,59 34,20
58 142 342 678
PU + 4%m/m Al2O3.nH2O
25-94 94-217
217-506 506-900
0,80 2,26
57,85 14,98
61 134 356 n.d.
PU + 5%m/m Al2O3.nH2O
25-100 100-213 213-496 496-900
0,69 1,53
59,86 14,09
62 144 354 n.d.
Fonte: Produção da autora.
De acordo com a Figura 5.34, todos os compósitos contendo Al2O3.nH2O exibem
os mesmos eventos térmicos descritos para o PU puro. Comparando-se as curvas,
exceto pela proporção de 3 % m/m, podem ser observados perfis similares entre os
demais compósitos, com variações nos valores de perdas de massa e nas temperaturas
da DTG nos terceiro e quarto eventos. Além disso, todos compósitos apresentam o
prolongamento do último evento de decomposição até 900 °C, diferentemente dos
compósitos de PU + Nb2O5.nH2O (Figura 5.33), dos quais somente o material com 1 %

133
m/m do referido óxido exibiu este comportamento. Tal característica evidencia a
atuação do Al2O3.nH2O como um eficiente aditivo, retardando a decomposição final da
matriz.
Considerando-se os critérios adotados para a escolha do material de maior
estabilidade térmica e, de acordo com os dados da Tabela 5.14, o compósito com 4 %
m/m de Al2O3.nH2O exprime um aumento de 16 °C na temperatura da DTG do evento
de decomposição das ligações uretânicas, em relação ao PU puro (340 °C), sendo este o
compósito escolhido para o aditivo Al2O3.nH2O.
d) Compósitos PU + ZnO.nH2O
As curvas TGA e os dados referentes aos compósitos PU + ZnO.nH2O encontram-
se na Figura 5.35 e na Tabela 5.15, respectivamente.
Figura 5.35 - Perfis das curvas TGA para os precursores PU e ZnO.nH2O e compósitos PU + ZnO.nH2O, em atmosfera de ar sintético, com adição de 1 a 5 % m/m de
ZnO.nH2O.
Fonte: Produção da autora.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
ZnO.nH2O
PU + 1%m/m ZnO.nH2O
PU + 2%m/m ZnO.nH2O
PU + 3%m/m ZnO.nH2O
PU + 4%m/m ZnO.nH2O
PU + 5%m/m ZnO.nH2O

134
Tabela 5.15 - Dados das curvas TGA/DTG para os compósitos PU + ZnO.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
PU 25-136
136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
ZnO.nH2O
25-144 144-900
1,41 21,27
90 270
PU + 1%m/m
ZnO.nH2O
25-100 100-226 226-481 481-900
0,74 1,63
51,17 39,98
57 157 343 630
PU + 2%m/m
ZnO.nH2O
25-94 94-226
226-494 494-900
0,44 1,71
52,11 39,21
59 157 346 634
PU + 3%m/m
ZnO.nH2O
25-102 102-213 213-489 489-681 681-900
0,85 2,04
49,83 28,46 7,52
63 203 341 620 708
PU + 4%m/m
ZnO.nH2O
25-100 100-234 234-489 489-685 685-900
0,65 2,34
51,18 30,71 7,34
61 146 342 631 681
PU + 5%m/m
ZnO.nH2O
25-115 115-200 200-498 498-681 681-900
0,47 1,58
55,50 25,50 8,88
61 156 348 631 706
Fonte: Produção da autora.
Segundo a Figura 5.35, os compósitos PU + 1%m/m ZnO.nH2O e PU + 2%m/m
ZnO.nH2O exibem quatro eventos de perda de massa análogas ao PU puro. Por outro
lado, a partir da adição de 3 % m/m de ZnO.nH2O nota-se uma alteração no último
evento, relativo à decomposição dos segmentos flexíveis, o qual apresenta-se bipartido.
Comparando-se com os compósitos PU + Nb2O5.nH2O (Figura 5.33) e PU +

135
Al2O3.nH2O (Figura 5.34), a adição de ZnO.nH2O ao PU não modificou
significativamente o perfil térmico dos respectivos compósitos. Desta forma, pode-se
inferir que o aumento do aditivo ZnO.nH2O incorporado ao PU produz um efeito maior
de interação com os segmentos flexíveis (poliol) ao invés dos segmentos rígidos
(isocianato). Entretanto, segundo os critérios já supramencionados na avaliação da
estabilidade térmica, o compósito PU + 2%m/m ZnO.nH2O (52,11 % e 346 °C) pode
ser definido como o melhor material, dentre os compósitos contendo ZnO.nH2O.
e) Compósitos PU + 97Celfb/3Nb2O5.nH2O
A Figura 5.36 apresenta as curvas TGA para os compósitos PU +
97Celfb/3Nb2O5.nH2O com os respectivos dados expressos na Tabela 5.16.
Figura 5.36 - Perfis das curvas TGA para os precursores PU, Celfb, Nb2O5.nH2O e 97Celfb/3Nb2O5.nH2O e compósitos PU + 97Celfb/3Nb2O5.nH2O, em atmosfera de ar
sintético, com adição de 1 a 5 % m/m de 97Celfb/3Nb2O5.nH2O.
Fonte: Produção da autora.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
Celfb
Nb2O5.nH2O
97Celfb/3Nb2O5.nH2O
PU + 1%m/m 97Celfb/3Nb2O5.nH2O
PU + 2%m/m 97Celfb/3Nb2O5.nH2O
PU + 3%m/m 97Celfb/3Nb2O5.nH2O
PU + 4%m/m 97Celfb/3Nb2O5.nH2O
PU + 5%m/m 97Celfb/3Nb2O5.nH2O

136
Tabela 5.16 - Dados das curvas TGA/DTG para os compósitos PU + 97Celfb/3Nb2O5.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
PU 25-136
136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
Nb2O5.nH2O
25-200 200-900
7,37 12,87
88 356
97Celfb/3Nb2O5.nH2O
25-170 166-900
4,33 88,68
61 373
PU + 1%m/m
97Celfb/3Nb2O5.nH2O
25-100 100-213 213-511 511-900
1,22 1,99
53,39 37,23
58 152 343 754
PU + 2%m/m
97Celfb/3Nb2O5.nH2O
25-89 89-217
217-523 523-900
0,71 2,77
57,65 34,23
63 143 344 723
PU + 3%m/m
97Celfb/3Nb2O5.nH2O
25-100 100-226 226-528 528-900
0,55 2,02
58,92 31,56
63 138 346 851
PU + 4%m/m
97Celfb/3Nb2O5.nH2O
25-106 106-217 217-528 528-900
1,01 1,95
58,83 32,29
62 157 351 865
PU + 5%m/m
97Celfb/3Nb2O5.nH2O
25-100 100-221 221-540 540-900
0,75 2,61
58,42 33,81
61 142 345 736
Fonte: Produção da autora.
Analogamente aos compósitos PU + Nb2O5.nH2O (item b), todos os materiais
contendo o híbrido 97Celfb/3Nb2O5.nH2O exibem quatro eventos térmicos, sendo estes
as mesmas etapas descritas para o PU puro. Segundo as curvas (Figura 5.36) e os dados

137
da Tabela 5.16, a adição do híbrido 97Celfb/3Nb2O5.nH2O resultou na formação de dois
tipos de perfis térmicos, devido à heterogeneidade do material incorporado. Entretanto,
comparando-se com os compósitos contendo somente Nb2O5.nH2O (Figura 5.33 e
Tabela 5.13), observa-se um único perfil com pequenas variações de estabilidade
térmica, diferentemente dos compósitos contendo o híbrido. Acredita-se que esta
alteração no perfil térmico possa estar relacionada à contribuição dos grupos hidroxila
presentes na estrutura do material. Ou seja, no Nb2O5.nH2O os grupos hidroxila são
provenientes somente das águas de hidratação, enquanto que no híbrido esta
contribuição provém tanto da hidratação do óxido metálico hidratado quanto da celulose
parcialmente acetilada. Consequentemente, as reações para síntese dos compósitos
ocorreram de maneira randômica, uma vez que o isocianato reagirá com os grupamentos
OH disponíveis, como supramencionado.
De acordo com os critérios de avaliação, o compósito PU + 4%m/m
97Celfb/3Nb2O5.nH2O exibiu a maior estabilidade térmica, com a maior temperatura da
DTG (351 °C) e menor evolução de massa (58,83 %), além do prolongamento do último
evento até 900 °C.

138
f) Compósitos PU + 94Celfb/6Al2O3.nH2O
As curvas TGA para os compósitos contendo o híbrido 94Celfb/6Al2O3.nH2O são
representadas pela Figura 5.37 e os dados são exibidos na Tabela 5.17.
Figura 5.37 - Perfis das curvas TGA para os precursores PU, Celfb, Al2O3.nH2O e 94Celfb/6Al2O3.nH2O e compósitos PU + 94Celfb/6Al2O3.nH2O, em atmosfera de ar
sintético, com adição de 1 a 5 % m/m de 94Celfb/6Al2O3.nH2O.
Fonte: Produção da autora.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
Celfb
Al2O3.nH2O
94Celfb/6Al2O3.nH2O
PU + 1%m/m 94Celfb/6Al2O3.nH2O
PU + 2%m/m 94Celfb/6Al2O3.nH2O
PU + 3%m/m 94Celfb/6Al2O3.nH2O
PU + 4%m/m 94Celfb/6Al2O3.nH2O
PU + 5%m/m 94Celfb/6Al2O3.nH2O

139
Tabela 5.17 - Dados das curvas TGA/DTG para os compósitos PU + 94Celfb/6Al2O3.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
PU 25-136
136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
Al2O3.nH2O
25-196 196-900
10,06 22,19
78 295
94Celfb/6Al2O3.nH2O
25-200 200-414 414-900
6,47 59,07 17,86
69 350 420
PU + 1%m/m
94Celfb/6Al2O3.nH2O
25-115 115-221 221-519 519-900
1,54 1,96
53,57 38,60
61 152 345 791
PU + 2%m/m
94Celfb/6Al2O3.nH2O
25-106 106-221 221-519 519-900
0,60 1,82
56,23 34,33
61 145 346 826
PU + 3%m/m
94Celfb/6Al2O3.nH2O
25-100 100-221 221-540 540-900
0,58 2,06
60,28 31,16
63 155 346 806
PU + 4%m/m
94Celfb/6Al2O3.nH2O
25-100 100-234 234-540 540-900
0,58 2,03
57,58 33,45
63 147 349 864
PU + 5%m/m
94Celfb/6Al2O3.nH2O
25-100 100-217 217-523 523-900
0,41 2,13
56,57 34,40
63 140 345 729
Fonte: Produção da autora.
De acordo com a Figura 5.37, todos os compósitos PU + 94Celfb/6Al2O3.nH2O
exibiram quatro eventos térmicos, sendo estes os mesmos descritos para o polímero

140
puro. A partir das curvas TGA, observa-se que a adição do híbrido de alumínio à matriz
polimérica alterou significativamente o perfil térmico de todos os compósitos. Dentre as
proporções avaliadas, o compósito com 4 % m/m do híbrido apresentou a maior
estabilidade térmica (349 °C), comparada ao PU puro (340 °C), enquanto as demais
exibiram perfis similares com poucas variações. Apesar do compósito com 1 % m/m
exibir a menor temperatura de DTG (345 °C), o mesmo foi escolhido como o melhor
material com o aditivo híbrido 94Celfb/6Al2O3.nH2O por apresentar a menor perda de
massa (53,57 %), devido à supressão dos voláteis presentes no PU. Em todas as
avaliações do comportamento térmico por termogravimetria o critério evolução de
perda de massa no evento de decomposição do PU prevalece em relação à temperatura
devido à maior sensibilidade da técnica.
Comparando-se com os compósitos contendo somente Al2O3.nH2O, observa-se
que é necessária uma menor quantidade de aditivo híbrido (1 % m/m) em relação ao
aditivo inorgânico isolado (4 % m/m) para promover a estabilidade térmica do
compósito. Independentemente do efeito na temperatura ser maior no compósito PU + 4
% m/m Al2O3.nH2O (356 °C), em relação ao PU + 1% m/m 94Celfb/6Al2O3.nH2O (345
°C), ressalta-se o efeito das modificações químicas efetuadas para a síntese deste
híbrido, as quais proporcionaram similar melhora da propriedade térmica.

141
g) Compósitos PU + 97Celfb/3ZnO.nH2O
A Figura 5.38 apresenta as curvas TGA dos compósitos PU +
97Celfb/3ZnO.nH2O e a Tabela 5.18 exibe os dados referentes à termogravimetria.
Figura 5.38 - Perfis das curvas TGA para os precursores PU, Celfb, ZnO.nH2O e 97Celfb/3ZnO.nH2O e compósitos PU + 97Celfb/3ZnO.nH2O, em atmosfera de ar
sintético, com adição de 1 a 5 % m/m de 97Celfb/3ZnO.nH2O.
Fonte: Produção da autora.
0
20
40
60
80
100
120
0 300 600 900
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU
Celfb
ZnO.nH2O
97Celfb/3ZnO.nH2O
PU + 1%m/m 97Celfb/3ZnO.nH2O
PU + 2%m/m 97Celfb/3ZnO.nH2O
PU + 3%m/m 97Celfb/3ZnO.nH2O
PU + 4%m/m 97Celfb/3ZnO.nH2O
PU + 5%m/m 97Celfb/3ZnO.nH2O

142
Tabela 5.18 - Dados das curvas TGA/DTG para os compósitos PU + 97Celfb/3ZnO.nH2O e precursores.
MATERIAL
Intervalos Curva TGA
(°C)
Perda de massa Curva TGA
(%)
Temperatura Curva DTG
(°C)
PU 25-136
136-243 243-485 485-900
0,03 3,92
49,67 35,34
60 195 340 635
Celfb
25-162 162-300 300-460 460-900
4,77 8,13
67,09 8,75
63 294 346 551
ZnO.nH2O
25-144 144-900
1,41 21,27
90 270
97Celfb/3ZnO.nH2O
25-170 174-900
3,85 82,60
62 385
PU + 1%m/m
97Celfb/3ZnO.nH2O
25-111 111-234 234-485 485-900
1,21 1,43
50,85 40,67
58 157 341 621
PU + 2%m/m
97Celfb/3ZnO.nH2O
25-106 106-230 230-506 506-900
0,77 1,66
52,80 38,85
62 158 347 652
PU + 3%m/m
97Celfb/3ZnO.nH2O
25-106 106-221 221-500 500-900
0,87 1,51
55,14 37,48
63 158 344 655
PU + 4%m/m
97Celfb/3ZnO.nH2O
25-111 111-217 217-511 511-900
0,77 1,55
56,49 36,73
65 159 350 681
PU + 5%m/m
97Celfb/3ZnO.nH2O
25-106 106-217 217-500 500-900
0,82 1,85
54,33 39,05
64 154 348 642
Fonte: Produção da autora.

143
Assim como observado nos compósitos PU + ZnO.nH2O (Figura 5.35), a
incorporação do híbrido 97Celfb/3ZnO.nH2O não exprimiu efeito significativo no perfil
térmico dos materiais. Isto é, a baixa interação tanto nos compósitos contendo o óxido
ZnO.nH2O quanto contendo o híbrido 97Celfb/3ZnO.nH2O está relacionada às sutis
diferenças visualizadas nos perfis das curvas de ambos os materiais. Apesar da presença
de grupos OH contidos na fração orgânica do híbrido, os quais reagiriam com os grupos
isocianato, a adição do híbrido 97Celfb/3ZnO.nH2O pouco favoreceu o aumento da
estabilidade térmica nestes compósitos. De acordo com os critérios adotados, a
proporção contendo 2 % m/m do referido híbrido foi escolhida como a de maior
estabilidade térmica (347 °C), comparada ao PU puro (340 °C).

144
5.6.2. Calorimetria Exploratória Diferencial
As análises de DSC foram realizadas nos compósitos selecionados pelas curvas
TGA/DTG com maior estabilidade térmica, com a finalidade de avaliar o efeito dos
aditivos Celfb, Nb2O5.nH2O, Al2O3.nH2O, ZnO.nH2O, 97Celfb/3Nb2O5.nH2O,
94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O incorporados à matriz polimérica, em
relação ao calor trocado. As Figuras 5.39 e 5.40 apresentam as curvas DSC para os
compósitos com os aditivos inorgânicos e de fração celulósica, respectivamente. E a
Tabela 5.19 exibe os dados referentes a todos os compósitos representados pelas curvas
calorimétricas.
Figura 5.39 - Perfis das curvas DSC para: (a) PU, (b) PU + 3% m/m Nb2O5.nH2O, (c) PU + 4 % m/m Al2O3.nH2O e (d) PU + 2 % m/m ZnO.nH2O em atmosfera de
nitrogênio.
Fonte: Produção da autora.
-3
-2
-1
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
-2,5
-2
-1,5
-1
-0,5
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
-2
-1,5
-1
-0,5
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
-4
-3
-2
-1
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
(a) (b)
307 °C
302 °C
(c)
! EXO ! EXO
! EXO
(d)
! EXO
281 °C
301 °C
152 °C

145
Figura 5.40 - Perfis das curvas DSC para: (a) PU, (b) PU + 2 % m/m Celfb, (c) PU + 4 % m/m 97Celfb/3Nb2O5.nH2O, (d) PU + 1 % m/m 94Celfb/6Al2O3.nH2O e (e) PU + 2
% m/m 97Celfb/3ZnO.nH2O em atmosfera de nitrogênio.
Fonte: Produção da autora.
-1,5
-1
-0,5
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
-1
-0,5
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
-2
-1,5
-1
-0,5
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
-4
-3
-2
-1
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
(a) (b)
305 °C
300 °C
(c)
! EXO ! EXO
! EXO
(d)
! EXO
281 °C
297 °C
-4
-3
-2
-1
0
0 50 100 150 200 250 300 350 400
Flu
xo d
e ca
lor
(W/g
)
Temperatura (°C )
(e) ! EXO
299 °C

146
Tabela 5.19 – Dados das curvas DSC para compósitos contendo os óxidos metálicos hidratados, celulose e híbridos orgânico-inorgânicos em atmosfera de nitrogênio.
MATERIAL
Intervalos Curva DSC
(°C)
Entalpia de reação ΔH (J.g-1)
Temperatura de início do evento
Tinicial (°C) PU 229-310 -97 240
PU + 3 % m/m Nb2O5.nH2O
250-341
125
274
PU + 4 % m/m Al2O3.nH2O
125-181 232-342
6 158
135 268
PU + 2 % m/m
ZnO.nH2O
232-338
164
260
PU + 2 % m/m
Celfb
232-337
187
268
PU + 4 % m/m
97Celfb/3Nb2O5.nH2O
232-339
139
267
PU + 1 % m/m
94Celfb/6Al2O3.nH2O
215-363
231
257
PU + 2 % m/m
97Celfb/3ZnO.nH2O
232-338
122
270
Fonte: Produção da autora.
De acordo com a Tabela 5.19, o PU apresenta um pico exotérmico,
correspondente à entalpia de decomposição das ligações uretânicas (-97 J.g-1), enquanto
que os compósitos contendo os óxidos metálicos hidratados (Figura 5.39) exibiram
somente um pico endotérmico. Observa-se que a presença do aditivo inorgânico na
matriz inibiu a evolução de calor, referente à esta decomposição. Outro indício do efeito
da incorporação dos óxidos metálicos hidratados é a alteração nas temperaturas iniciais
do evento de 240 °C (PU) para 274 °C (Nb2O5.nH2O), 268 °C (Al2O3.nH2O) e 260 °C
(ZnO.nH2O). A partir dos dados das curvas DSC da Tabela 5.19, as entalpias resultantes
foram calculadas, segundo a Equação 5.6, descrita no item 5.3. A entalpia resultante
para os compósitos com aditivos inorgânicos segue a sequência: PU + 2% m/m

147
ZnO.nH2O = 261 J.g-1 > PU + 4% m/m Al2O3.nH2O = 255 J.g-1 > PU + 3% m/m
Nb2O5.nH2O = 222 J.g-1.
Analogamente aos compósitos com os óxidos metálicos hidratados, as curvas
DSC para os compósitos contendo tanto celulose quanto os híbridos orgânico-
inorgânicos (Figura 5.40) apresentaram um único pico endotérmico. Nota-se que a
presença dos componentes de fração celulósica no PU também inibiu a evolução de
calor, referente à decomposição dos grupos uretânicos. Assim como os óxidos, a
incorporação dos aditivos celulósicos também modificaram as temperaturas iniciais do
evento, de 240 °C (PU) para 268 °C (Celfb) 267 °C (97Celfb/3Nb2O5.nH2O), 257 °C
(94Celfb/6Al2O3.nH2O) e 270 °C (97Celfb/3ZnO.nH2O). Considerando-se a equação
5.6, descrita no item 5.3, as entalpias resultantes para os compósitos com os aditivos
celulósicos seguem a sequência: PU + 1% m/m 94Celfb/6Al2O3.nH2O = 328 J.g-1 > PU
+ 2% m/m Celfb = 284 J.g-1 > PU + 4% m/m 97Celfb/3Nb2O5.nH2O = 236 J.g-1 > PU +
2% m/m 97Celfb/3ZnO.nH2O = 219 J.g-1.

148
5.6.3. Comportamento mecânico
As propriedades mecânicas dos compósitos contendo os diferentes aditivos foram
avaliadas a partir das curvas de resistência à compressão em função da deformação.
Como descrito no item 4.4.8, para o primeiro corpo de prova (CP1) de cada compósito o
teste foi realizado até seu limite, a fim de avaliar a capacidade de absorção de energia na
deformação e, consequentemente, a recuperação do material celular. As Figuras 5.41 e
5.42 exibem a curva para o CP1 do PU puro e os CP’s antes e após a máxima
compressão, respectivamente.
Figura 5.41 - Curva resistência à compressão em função da deformação para o PU puro até o limite do corpo de prova 1 (CP1).
Fonte: Produção da autora.
0
5
10
15
20
25
30
0 20 40 60 80 100
Res
istê
ncia
à c
ompr
essã
o (M
Pa)
Deformação (%)

149
Figura 5.42 - Corpos de prova para o PU puro: antes e após o ensaio até o limite de compressão do CP1.
Fonte: Produção da autora.
A curva de resistência à compressão em função da deformação para o CP1 do PU
puro (Figura 5.41) apresenta um perfil característico para materiais celulares, categoria
na qual encontram-se as espumas rígidas, exibindo três estágios (ŞERBAN et al., 2016).
O primeiro refere-se à região elástica linear, na qual determina-se o módulo de
Young (módulo elástico) da espuma. O segundo denota a região plástica linear onde a
espuma é submetida à uma ampla deformação mecânica, sob um mínimo aumento de
carga. Na região plana deste estágio ocorre o regime plástico, devido ao colapso da
parede celular das espumas. E o último estágio representa a densificação da espuma,
com a elevação exponencial da carga aplicada sobre o material, sem deformação
considerável mediante à aplicação desta carga (NARINE et al., 2007; RIBEIRO DA
SILVA et al., 2013b; ŞERBAN et al., 2016). Em todos os CP1 dos diferentes
compósitos foram observados perfis similares, com os três estágio bem definidos.
Para a exibição das respectivas curvas dos compósitos contendo os diferentes
aditivos (Celfb, Nb2O5.nH2O, Al2O3.nH2O, ZnO.nH2O, 97Celfb/3Nb2O5.nH2O,
94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O), adotou-se como parâmetro os materiais
que apresentaram os maiores valores de resistência à compressão. As Figuras 5.43 e
5.44 exibem as curvas tensão-deformação para os compósitos contendo os óxidos
metálicos hidratados e híbridos, respectivamente, comparadas ao poliuretano puro.
Sent
ido
de e
xpan
são
da
espu
ma
CP original CP após compressão

150
Figura 5.43 - Curvas resistência à compressão em função da deformação para os compósitos contendo óxidos metálicos hidratados.
Fonte: Produção da autora.
Figura 5.44 - Curvas resistência à compressão em função da deformação para os compósitos contendo celulose e híbridos.
Fonte: Produção da autora.
0
2
4
6
8
10
12
14
16
0 10 20 30
Res
istên
cia
à co
mpr
essã
o (M
Pa)
Deformação (%)
PU puro
PU + 3% m/m Celfb
PU + 2% m/m 97Celfb/3Nb2O5.nH2O
PU + 1% m/m 94Celfb/6Al2O3.nH2O
PU + 2% m/m 97Celfb/3ZnO.nH2O
0
2
4
6
8
10
12
14
16
0 5 10 15 20 25 30
Res
istên
cia
à co
mpr
essã
o (M
Pa)
Deformação (%)
PU puro
PU + 5% m/m Nb2O5.nH2O
PU +1% m/m Al2O3.nH2O
PU + 2% m/m ZnO.nH2O

151
As Tabelas 5.20, 5.21, 5.22, 5.23, 5.24, 5.25 e 5.26 sintetizam as propriedades
mecânicas e os valores de densidade aparente para os compósitos PU + Celfb, PU +
Nb2O5.nH2O, PU + Al2O3.nH2O, PU + ZnO.nH2O, PU + 97Celfb/3Nb2O5.nH2O, PU +
94Celfb/6Al2O3.nH2O e PU + 97Celfb/3ZnO.nH2O, respectivamente.
Tabela 5.20 - Propriedades mecânicas para os compósitos PU + Celfb.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% Celfb 28,8 ± 0,4 9,9 ± 1,0 174,8 ± 24,3 PU + 2% Celfb PU + 3% Celfb PU + 4% Celfb PU + 5% Celfb
28,8 ± 1,3 29,5 ± 0,6 28,9 ± 0,4 29,5 ± 1,0
11,7 ± 0,8 12,0 ± 0,6 10,8 ± 0,4 11,2 ± 0,6
222,6 ± 21,7 197,3 ± 52,9 196,7 ± 17,3 178,5 ± 24,5
Fonte: Produção da autora.
Tabela 5.21 - Propriedades mecânicas para os compósitos PU + Nb2O5.nH2O.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% Nb2O5.nH2O 28,6 ± 2,1 10,0 ± 1,0 157,0 ± 15,5 PU + 2% Nb2O5.nH2O PU + 3% Nb2O5.nH2O PU + 4% Nb2O5.nH2O PU + 5% Nb2O5.nH2O
29,6 ± 1,0 29,9 ± 0,3 29,6 ± 1,1 32,1 ± 4,2
11,4 ± 0,5 11,5 ± 0,5 11,7 ± 0,4 12,6 ± 0,9
186,4 ± 38,8 214,8 ± 19,9 193,9 ± 26,0 211,3 ± 40,1
Fonte: Produção da autora.

152
Tabela 5.22 - Propriedades mecânicas para os compósitos PU + Al2O3.nH2O.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% Al2O3.nH2O 30,5 ± 1,1 12,2 ± 0,5 189,3 ± 39,6 PU + 2% Al2O3.nH2O PU + 3% Al2O3.nH2O PU + 4% Al2O3.nH2O PU + 5% Al2O3.nH2O
30,2 ± 1,0 30,0 ± 2,0 29,8 ± 1,3 30,5 ± 2,3
11,9 ± 0,4 11,7 ± 0,6 11,3 ± 0,3 11,1 ± 0,5
207,8 ± 41,7 233,1 ± 34,6 223,2 ± 20,8 215,3 ± 23,4
Fonte: Produção da autora.
Tabela 5.23 - Propriedades mecânicas para os compósitos PU + ZnO.nH2O.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% ZnO.nH2O 28,8 ± 1,5 12,0 ± 0,5 239,7 ± 16,3 PU + 2% ZnO.nH2O PU + 3% ZnO.nH2O PU + 4% ZnO.nH2O PU + 5% ZnO.nH2O
30,3 ± 1,6 30,3 ± 1,0 28,9 ± 0,7 30,4 ± 0,8
12,2 ± 0,4 12,1 ± 0,4 11,5 ± 0,5 11,4 ± 0,5
216,7 ± 15,1 238,0 ± 13,5 229,9 ± 14,6 230,1 ± 23,9
Fonte: Produção da autora.
Tabela 5.24 - Propriedades mecânicas para os compósitos PU + 97Celfb/3Nb2O5.nH2O.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% 97Celfb/3Nb2O5.nH2O 29,4 ± 0,7 11,8 ± 0,8 205,9 ± 23,1 PU + 2% 97Celfb/3Nb2O5.nH2O PU + 3% 97Celfb/3Nb2O5.nH2O PU + 4% 97Celfb/3Nb2O5.nH2O PU + 5% 97Celfb/3Nb2O5.nH2O
30,8 ± 0,7 29,9 ± 0,7 30,2 ± 1,2 29,4 ± 0,3
12,0 ± 0,5 11,8 ± 0,7 11,9 ± 0,3 11,6 ± 0,4
232,1 ± 45,0 204,2 ± 50,4 211,5 ± 26,3 218,7 ± 20,7
Fonte: Produção da autora.

153
Tabela 5.25 - Propriedades mecânicas para os compósitos PU + 94Celfb/6Al2O3.nH2O.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% 94Celfb/6Al2O3.nH2O 30,7 ± 1,1 12,9 ± 0,3 245,8 ± 17,6 PU + 2% 94Celfb/6Al2O3.nH2O PU + 3% 94Celfb/6Al2O3.nH2O PU + 4% 94Celfb/6Al2O3.nH2O PU + 5% 94Celfb/6Al2O3.nH2O
31,1 ± 1,2 30,1 ± 1,0 29,5 ± 1,5 29,1 ± 1,3
12,7 ± 0,1 12,6 ± 0,2 11,3 ± 0,3 12,0 ± 0,3
224,3 ± 28,6 248,1 ± 20,8 223,2 ± 20,8 210,9 ± 42,4
Fonte: Produção da autora.
Tabela 5.26 - Propriedades mecânicas para os compósitos PU + 97Celfb/3ZnO.nH2O.
MATERIAL Densidade aparente (kg.m-3)
Resistência à compressão
(MPa)
Módulo de Young (MPa)
PU 27,9 ± 1,1 11,5 ± 0,3 185,6 ± 29,8 PU + 1% 97Celfb/3ZnO.nH2O 29,5 ± 0,3 12,1 ± 0,2 228,2 ± 17,1 PU + 2% 97Celfb/3ZnO.nH2O PU + 3% 97Celfb/3ZnO.nH2O PU + 4% 97Celfb/3ZnO.nH2O PU + 5% 97Celfb/3ZnO.nH2O
29,7 ± 0,9 28,7 ± 0,5 29,0 ± 1,8 29,3 ± 1,1
12,3 ± 0,3 11,9 ± 0,4 11,5 ± 0,3 11,3 ± 0,8
198,5 ± 34,1 221,3 ± 24,6 198,5 ± 30,1 200,7 ± 28,8
Fonte: Produção da autora.
A partir dos dados obtidos nos testes densidade (Tabelas 5.20 a 5.26), nota-se que
a incorporação dos aditivos, no geral, favoreceu um pequeno aumento ( ~ 7 %) na
densidade aparente em todos os compósitos, sendo evidenciado nos materiais contendo
os híbridos 97Celfb/3Nb2O5.nH2O e 94Celfb/6Al2O3.nH2O. No entanto, a incorporação
dos aditivos não promoveu diferença estatística para os demais compósitos, devido às
pequenas quantidades empregadas (1 a 5 % m/m), resultando em um sistema
heterogêneo com pouca variabilidade nos resultados, conforme reportado por Estravís et
al. (2016). De acordo com Ribeiro da Silva et al. (2013b), a alteração na densidade está
relacionada às estruturas celulares mais compactas, isto é, há mais material por unidade
de área podendo implicar também em um aumento nos valores de resistência e módulo.
Avaliando-se os resultados de resistência à compressão (Tabelas 5.20 a 5.26),
observa-se a seguinte sequência: PU + 1% m/m 94Celfb/6Al2O3.nH2O > PU + 5% m/m

154
Nb2O5.nH2O > PU + 2% m/m 97Celfb/3ZnO.nH2O > PU + 1% m/m Al2O3.nH2O ~ PU
+ 2% m/m ZnO.nH2O > PU + 2% m/m 97Celfb/3Nb2O5.nH2O ~ PU + 3% m/m Celfb.
Para os valores de densidade, tem-se: PU + 5% m/m Nb2O5.nH2O > PU + 2%
m/m 94Celfb/6Al2O3.nH2O > PU + 2% m/m 97Celfb/3Nb2O5.nH2O > PU + 1% m/m
Al2O3.nH2O > PU + 5% m/m ZnO.nH2O > PU + 2% m/m 97Celfb/3ZnO.nH2O > PU +
3% m/m Celfb.
Por fim, os valores dos módulos de Young: PU + 3% m/m 94Celfb/6Al2O3.nH2O
> PU + 3% m/m ZnO.nH2O > PU + 3% m/m Al2O3.nH2O > PU + 2% m/m
97Celfb/3Nb2O5.nH2O > PU + 1% m/m 97Celfb/3ZnO.nH2O > PU + 2% m/m Celfb >
PU + 3% m/m Nb2O5.nH2O.
Com relação à propriedade de resistência à compressão, os compósitos com Celfb,
97Celfb/3Nb2O5.nH2O, Nb2O5.nH2O, Al2O3.nH2O e ZnO.nH2O não apresentaram
diferença estatística, quando comparados à matriz pura. Entretanto, nota-se um aumento
no valor da resistência à compressão de 12 % e 7 % para os híbridos
94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O, respectivamente. Os compósitos
contendo o híbrido 97Celfb/3ZnO.nH2O apresentaram-se efetivos no ganho de
propriedade mecânica, diferentemente do ocorrido em relação à estabilidade térmica,
como discutido no item 5.2.3. A partir dos resultados gerais, destaca-se o híbrido
94Celfb/6Al2O3.nH2O devido às suas potenciais propriedades térmicas e mecânicas,
evidenciadas nos respectivos compósitos. Além disso, destaca-se também sua alta
hidrofobicidade devido às modificações químicas efetuadas (item 5.4) corroborando
com os resultados mecânicos. Considerando-se os valores dos módulos de Young, os
aditivos promoveram um aumento no módulo, contudo todos os materiais
apresentaram-se similares estatisticamente. Comportamentos semelhantes com
diferentes aditivos incorporados ao poliuretano também foram reportados por (LI; REN;
RAGAUSKAS, 2010; RIBEIRO DA SILVA et al., 2013b; YANG et al., 2015; XI et al.,
2015).
Desta forma, os resultados de densidade, resistência à compressão e módulo de
Young indicam que a celulose, os óxidos metálicos hidratados de nióbio, alumínio e
zinco, assim como os respectivos híbridos prestam-se para a aplicação como aditivos ao
PU.

155
6. CONCLUSÕES
O tratamento com a mistura ácida (CH3COOH/HNO3) foi efetivo no isolamento
da celulose.
A termogravimetria mostrou-se viável para o estabelecimento das melhores
proporções, indicando os híbridos os 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O e
97Celfb/3ZnO.nH2O.
Os difratogramas para os híbridos 97Celfb/3Nb2O5.nH2O, 94Celfb/6Al2O3.nH2O
e 97Celfb/3ZnO.nH2O exibiram perfis de difração diferentes de seus precursores,
confirmando a formação de novos materiais.
Os espectros de FTIR para os híbridos 97Celfb/3Nb2O5.nH2O,
94Celfb/6Al2O3.nH2O e 97Celfb/3ZnO.nH2O evidenciaram a permanência do grupo
acetila.
As curvas DSC evidenciaram a atuação dos híbridos 97Celfb/3Nb2O5.nH2O e
94Celfb/6Al2O3.nH2O como retardantes de chamas.
As micrografias destacaram a ausência de depósito dos óxidos na superfície da
celulose e as análises de EDX identificaram a presença dos metais nos respectivos
híbridos.
Nos testes de absorção de água o híbrido 94Celfb/6Al2O3.nH2O apresentou a
maior redução no caráter hidrofílico.
As curvas TGA dos compósitos definiram as seguintes proporções: PU + 4% m/m
Al2O3.nH2O > PU + 3% m/m Nb2O5.nH2O > PU + 4% m/m 97Celfb/3Nb2O5.nH2O >
PU + 2% m/m 97Celfb/3ZnO.nH2O > PU + 2% m/m ZnO.nH2O > PU + 1% m/m
94Celfb/6Al2O3.nH2O > PU + 2% m/m Celfb.
As curvas DSC exibiram retardância à chama para todos os compósitos na
seguinte sequência: PU + 1% m/m 94Celfb/6Al2O3.nH2O > PU + 2% m/m Celfb > PU
+ 2% m/m ZnO.nH2O > PU + 4% m/m Al2O3.nH2O > PU + 4% m/m
97Celfb/3Nb2O5.nH2O > PU + 3% m/m Nb2O5.nH2O > PU + 2% m/m
97Celfb/3ZnO.nH2O.
As micrografias para os compósitos de PU exibiram um número maior células
colapsadas com o aumento da quantidade dos aditivos incorporados.

156
Os resultados térmicos e mecânicos evidenciam o híbrido 94Celfb/6Al2O3.nH2O
como o melhor aditivo na formulação para o compósito PU + 1% m/m
94Celfb/6Al2O3.nH2O.

157
REFERÊNCIAS BIBLIOGRÁFICAS
ASSOCIAÇÃO BRASILEIRA DO ALUMÍNIO, ABAL.Aluminio. Disponível em: <http://www.abal.gov.br> Acesso em 18 mar. 2016 AKIL, H. M.; OMAR, M. F.; MAZUKI, A. A. M.; SAFIEE, S.; ISHAK, Z. A. M.; ABU BAKAR, A. Kenaf fiber reinforced composites: A review. Materials and Design, v. 32, n. 8-9, p. 4107-4121, 2011. AL-OQLA, F. M.; SAPUAN, S. M.; ANWER, T.; JAWAID, M.; HOQUE, M. E. Natural fiber reinforced conductive polymer composites as functional materials: A review. Synthetic Metals, v. 206, p. 42-54, 2015. ALAVUDEEN, A.; RAJINI, N.; KARTHIKEYAN, S.; THIRUCHITRAMBALAM, M.; VENKATESHWAREN, N. Mechanical properties of banana/kenaf fiber-reinforced hybrid polyester composites: Effect of woven fabric and random orientation. Materials and Design, v. 66, p. 246-257, 2015. ALBINANTE, S. R.; PACHECO, E. B. A. V.; VISCONTE, L. L. Y.; TAVARES, M. I. B. Caracterização de fibras de bananeira e de coco por ressonância magnética nuclear de alta resolução no estado sólido. Polímeros, v. 22, n. 5, p. 460-466, 2012. ALBINANTE, S. R.; PACHECO, E. B. A. V.; VISCONTE, L. L. Y. Revisão dos tratamentos químicos da fibra natural para mistura com poliolefinas. Química Nova, v. 36, n. 1, p. 114-122, 2013. ANTUNES, M.; Canoa, A.; Haurieb, L.; Velasco, J. I. Esparto wool as reinforcement in hybrid polyurethane composite foams. Industrial Crops and Products, v. 34, p. 1641-1648, 2011. ANWAR, J.; SHAFIQUE, U.; WAHEED-UZ-ZAMAN; SALMAN, M.; DAR, A.; ANWAR, S. Removal of Pb (II) and Cd (II) from water by adsorption on peels of banana. Bioresource Technology, v. 101, p. 1752-1755, 2010. ASS, B. A. P.; BELGACEM, M. N.; FROLLINI, E. Mercerized linters cellulose: characterization and acetylation in N,N-dimethylacetamide/lithium chloride. Carbohydrate Polymers, v. 63, p. 19-29, 2006. AMERICAN SOCIETY FOR TESTING AND MATERIALS (ASTM). D 1695-07: Standard terminology of cellulose and cellulose derivatives. Estados Unidos, ago. 2012. 9 p. AMERICAN SOCIETY FOR TESTING AND MATERIALS (ASTM). D 1621-10: Standard test method for compressive properties of rigid cellular plastics. Estados Unidos, abr. 2010. 5 p.

158
BACCAN, N.; ANDRADE, J. C.; GODINHO, O. E. S.; BARONE, J. S. Química analítica quantitativa elementar. 3a ed. São Paulo: Edgard Blucher Ltda, 2001. 324 p. BECKER, D.; KLEINSCHMIDT, A. C.; BALZER, P. S.; SOLDI, V. Influência da sequência de mistura do pp-ma nas propriedades dos compósitos de pp e fibra de bananeira. Polímeros, v. 21, n. 1, p. 7-12, 2011. BENÍTEZ, A. N.; MONZÓN, M. D.; ANGULO, I.; ORTEGA, Z.; HERNÁNDEZ, P. M.; MARRERO, M. D. Treatment of banana fiber for use in the reinforcement of polymeric matrices. Measurement, v. 46, p. 1065-1073, 2013. BENVENUTTI, E. V.; MORO, C. C.; COSTA, T. M. H.; GALLAS, M. R. Materiais híbridos à base de sílica obtidos pelo método sol-gel. Química Nova, v. 32, n. 7, p. 1926-1933, 2009. BHATTACHARYA, I. N.; DAS, S. C.; MUKHERJEE, P. S.; PAUL, S.; MITRA, P. K. Thermal decomposition of precipitated fine aluminium trihydroxide. Scandinavian Journal of Metallurgy, v. 33, p. 211-219, 2004. BILBA, K.; ARSENE, M. A.; OUENSANGA, A. Study of banana and coconut fibers Botanical composition, thermal degradation and textural observations. Bioresource Technology, v. 98, p. 58-68, 2007. BOGOEVA-GACEVA, G.; AVELLA, M.; MALINCONICO, M.; BUZAROVSKA, A.; GROZDANOV, A.; GENTILE, G.; ERRICO, M. E. Natural fiber eco-composites. Polymer Composites, v. 28, p. 98-107, 2007. BOOPALAN, M.; NIRANJANAA, M.; UMAPATHY, M. J. Study on the mechanical properties and thermal properties of jute and banana fiber reinforced epoxy hybrid composites. Composites Part B: Engineering, v. 51, p. 54-57, 2013. BORGES, A. L.; SILVA, S. O.; CALDAS, R. C.; LEDO, C. A S. Teores foliares de nutrientes em genótipos de bananeira. Revista Brasileira de Fruticultura, v. 28, n. 2, p. 314-318, 2006. BRENDEL, O.; IANNETTA, P. P. M.; STEWART, D. A rapid and simple method to isolate pure alpha-cellulose. Phytochemical Analysis, v. 11, p. 7-10, 2000. BRÍGIDA, A. I. S.; CALADO, V. M. A.; GONÇALVES, L. R. B.; COELHO, M. A. Z. Effect of chemical treatments on properties of green coconut fiber. Carbohydrate Polymers, v. 79, p. 832-838, 2010. BUSCHLE-DILLER, G.; ZERONIAN, S. H. Enhancing the reactivity and strength of cotton fibers. Journal of Applied Polymer Science, v. 45, p. 967-979, 1992.

159
CAI, J.; Fei, P.; XIONG, Z.; SHI, Y.; YAN, K.; XIONG, H. Surface acetylation of bamboo cellulose: Preparation and rheological properties. Carbohydrate polymers, v. 92, p. 11-8, 2013. CALLISTER Jr, W. D. Compósitos. In: ___(7a Ed.). Ciências e Engenharia de Materiais: Uma Introdução. Rio de Janeiro: LTC, 2008. cap. 16, p. 422-454. CAMPOS, C. H.; URBANO, B. F.; RIVAS, B. L. Synthesis and characterization of organic-inorganic hybrid composites from poly(acrylic acid)-[3-(trimethoxysilyl)propyl methacrylate]-Al2O3. Composites Part B: Engineering, v. 57, p. 1-7, 2014a. CAMPOS, C. H.; URBANO, B. F.; RIVAS, B. L. Hybrid composites from poly[(4-vinylbenzyl)trimethylammonium chloride]-metal oxide using simultaneous radical polymerization/sol-gel synthesis. Materials Letters, v. 131, p. 198-202, 2014b. CANEVAROLO Jr., S. V. Técnicas de caracterização de polímeros. 1a ed. São Paulo: Artliber Editora, 2004. 448 p. CANEVAROLO Jr., S. V. Ciências dos polímeros: um texto básico para tecnólogos e engenheiros. 3a ed. São Paulo: Artliber Editora, 2010. 280 p. CANGEMI, J. M.; SANTOS, A. M.; NETO, S. C. Poliuretano: de travesseiros a preservativos, um polímero versátil. Química Nova na Escola, v. 31, n.3., p. 159-164, 2009. CARRARO, M.; GROSS, S. Hybrid materials based on the embedding of organically modified transition metal oxoclusters or polyoxometalates into polymers for functional applications: A review. Materials, v. 7, p. 3956-3989, 2014. COMPANHIA DE METALURGIA E MINERAÇÃO, CBMM. Nióbio. Disponível em <http://www.cbmm.com.br> Acesso em 25 mar. 2016. CHATTOPADHYAY, D. K.; WEBSTER, D. C. Thermal stability and flame retardancy of polyurethanes. Progress in Polymer Science, v. 34, p. 1068-1133, 2009. CORCUERA, M. A.; RUEDA, L.; FERNANDEZ D’ARLAS, B.; ARBELAIZ, A.; MARIETA, C.; MONDRAGON, I.; ECEIZA, A. Microstructure and properties of polyurethanes derived from castor oil. Polymer Degradation and Stability, v. 95, p. 2175-2184, 2010. CORDERO, A. I.; AMALVY, J. I.; FORTUNATI, E.; KENNY, J. M.; CHIACCHIARELLI, L. M. The role of nanocrystalline cellulose on the microstructure of foamed castor-oil polyurethane nanocomposites. Carbohydrate Polymers, v. 134, p. 110-118, 2015.

160
CUNHA, A. G.; FREIRE, C. S. R.; SILVESTRE, A. J. D.; NETO, C. P.; GANDINI, A. Preparation and characterization of novel highly omniphobic cellulose fibers organic-inorganic hybrid materials. Carbohydrate Polymers, v. 80, p. 1048-1056, 2010. CUNHA, A. G. G. N. Preparação de novos materiais por modificação heterogênea de celulose. 2011. 202 p. Tese (Doutorado em Química) - Universidade de Aveiro, 2011. D’ALMEIDA, A. L. F. S.; Calado, V.; Barreto, D. W.; d’Almeida, J. R. M. Acetilação da fibra de bucha (Luffa cylindrica). Polímeros, v. 15, n. 1, p. 59-62, 2005. DATTA, J.; KOPCZYNSKA, P. Effect of kenaf fibre modification on morphology and mechanical properties of thermoplastic polyurethane materials. Industrial Crops and Products, v. 74, p. 566-576, 2015. DE SIQUEIRA, F. G.; FILHO, E. X. F. Plant cell wall as a substrate for the production of enzymes with industrial applications. Mini-Reviews in Organic Chemistry, v. 7, p. 54-60, 2010. DEEPA, B.; ABRAHAM, E.; CHERIAN, B. M.; BISMARCK, A.; BLAKER, J. J.; POTHAN, L. A.; LEAO, A. L. DE SOUZA, S. F.; KOTTAISAMY, M. Structure, morphology and thermal characteristics of banana nano fibers obtained by steam explosion. Bioresource Technology, v. 102, p. 1988-1997, 2011. DITTENBER, D. B.; GANGARAO, H. V. S. Critical review of recent publications on use of natural composites in infrastructure. Composites Part A: Applied Science and Manufacturing, v. 43, p. 1419-1429, 2012. DNPM, Departamento Nacional de Produção Mineral. Sumário mineral, Ano 2015. Disponível em: <http://www.dnpm.gov.br> Acesso em 16 mar. 2016. EJTEMAEI, M.; GHARABAGHI, M.; IRANNAJAD, M. A review of zinc oxide mineral beneficiation using flotation method. Advances in Colloid and Interface Science, v. 206, p. 68-78, 2014. ELANTHIKKAL, S.; GOPALAKRISHNAPANICKER, U.; VARGHESE, S.; GUTHRIE, J. T. Cellulose microfibres produced from banana plant wastes: Isolation and characterization. Carbohydrate Polymers, v. 80, p. 852-859, 2010. ESTRAVÍS, S.; TIRADO-MEDIAVILLA, J.; SANTIAGO-CALVO, M.; RUIZ-HERRERO, J. L.; VILLAFAÑE, F.; RODRÍGUEZ-PÉREZ, M. A. Rigid polyurethane foams with infused nanoclays: Relationship between cellular structure and thermal conductivity. European Polymer Journal, v. 80, p. 1-15, 2016.

161
FARUK, O.; BLEDZKI, A. K.; FINK, H. P.; SAIN, M. Biocomposites reinforced with natural fibers: 2000-2010. Progress in Polymer Science, v. 37, p. 1552-1596, 2012. FAVARO, L.; BOUMAZA, A.; ROY, P.; LÉDION, J.; SATTONNAY, G.; BRUBACH, J. B.; HUNTZ, A. M.; TÉTOT, R. Experimental and ab initio infrared study of χ-, κ- and α-aluminas formed from gibbsite. Journal of Solid State Chemistry, v. 183, p. 901-908, 2010. FERREIRA, V. F.; ROCHA, D. R. DA; DA SILVA, F. DE C. Potencialidades e oportunidades na química da sacarose e outros açúcares. Química Nova, v. 32, n. 3, p. 623-638, 2009. FIGUEIRA, R. B.; SILVA, C. J. R.; PEREIRA, E. V. Organic-inorganic hybrid sol-gel coatings for metal corrosion protection/: a review of recent progress. Journal of Coatings Technology and Research, v. 12, n. 1, p. 1-35, 2015. FIORE, V.; DI BELLA, G.; VALENZA, A. The effect of alkaline treatment on mechanical properties of kenaf fibers and their epoxy composites. Composites Part B: Engineering, v. 68, p. 14-21, 2015. FOROUGHI-ABARI, A.; CADIEN, K. C. Growth, structure and properties of sputtered niobium oxide thin films. Thin Solid Films, v. 519, p. 3068-3073, 2011. FRAGAL, E. H.; CELLET, T. S. P.; FRAGAL, V. H.; COMPANHONI, M. V. P.; UEDA-NAKAMURA, T.; MUNIZ, E. C.; SILVA, R.; RUBIRA, A. F. Hybrid materials for bone tissue engineering from biomimetic growth of hydroxiapatite on cellulose nanowhiskers. Carbohydrate Polymers, v. 152, p. 734-746, 2016. GEORGE, M.; CHAE, M.; BRESSLER, D. C. Composite materials with bast fibres: Structural, technical, and environmental properties. Progress in Materials Science, v. 83, p. 1-23, 2016. GHAFOURI, V.; SHARIATI, M.; EBRAHIMZAD, A. Photoluminescence investigation of crystalline undoped ZnO nanostructures constructed by RF sputtering. Scientia Iranica, v. 19, n. 3, p. 934-942, 2012. GIBSON, C. E.; KELEBEK, S.; AGHAMIRIAN, M. Niobium oxide mineral flotation: A review of relevant literature and the current state of industrial operations. International Journal of Mineral Processing, v. 137, p. 82-97, 2015. GIBSON, R. F. A review of recent research on mechanics of multifunctional composite materials and structures. Composite Structures, v. 92, p. 2793-2810, 2010. GONÇALVES, A. P. B.; MIRANDA, C. S.; GUIMARÃES, D. H.; OLIVEIRA, J. C.; CRUZ, A. M. F.; DA SILVA, F. L. B. M.; LUPORINI, S.; JOSÉ, N. M. Physicochemical, mechanical and morphologic characterization of purple banana fibers. Materials Research, v. 18, n. 2, p. 205-209, 2015.

162
GU, H. Tensile behaviours of the coir fibre and related composites after NaOH treatment. Materials and Design, v. 30, p. 3931-3934, 2009. GUIMARÃES, J. L.; FROLLINI, E.; DA SILVA, C. G.; WYPYCH, F.; SATYANARAYANA, K. G. Characterization of banana, sugarcane bagasse and sponge gourd fibers of Brazil. Industrial Crops and Products, v. 30, p. 407-415, 2009. GUIMARÃES, J. L.; WYPYCH, F.; SAUL, C. K.; RAMOS, L. P.; SATYANARAYANA, K. G. Studies of the processing and characterization of corn starch and its composites with banana and sugarcane fibers from Brazil. Carbohydrate Polymers, v. 80, p. 130-138, 2010. GURUNATHAN, T.; MOHANTY, S.; NAYAK, S. K. A review of the recent developments in biocomposites based on natural fibres and their application perspectives. Composites Part A: Applied Science and Manufacturing, v. 77, p. 1-25, 2015. HADJADJ, A.; JBARA, O.; TARA, A.; GILLIOT, M.; MALECK, F.; MAAFI, E. M.; TIGHZERT, L. Effects of cellulose fiber content on physical properties of polyurethane based composites. Composite Structures, v. 135, p. 217-223, 2016. HOFFENDAHL, C.; FONTAINE, G.; DUQUESNE, S.; TASCHNER, F.; MEZGER, M.; BOURBIGOT, S. The combination of aluminum trihydroxide (ATH) and melamine borate (MB) as fire retardant additives for elastomeric ethylene vinyl acetate (EVA). Polymer Degradation and Stability, v. 115, p. 77-88, 2015. HU, F.; LIN, N. CHANG, P. R.; HUANG, J. Reinforcement and nucleation of acetylated cellulose nanocrystals in foamed polyester composites. Carbohydrate Polymers, v. 129, p. 208-215, 2015. HULL, T. R.; WITKOWSKI, A.; HOLLINGBERY, L. Fire retardant action of mineral fillers. Polymer Degradation and Stability, v. 96, p. 1462-1469, 2011. IBEH, C. C.; BUBACZ, M. Current trends in nanocomposite foams. Journal of Cellular Plastics, v. 44, p. 493-515, 2008. IBGE, Instituto Brasileiro de Geografia e Estatística. Levantamento sistemático da produção agrícola, jan. 2017. Disponível em: <http://www.ibge.gov.br>. Acesso em 17 fev. 2017. IBRAHIM, M. M.; DUFRESNE, A.; EL-ZAWAWY, W. K.; AGBLEVOR, F. A. Banana fibers and microfibrils as lignocellulosic reinforcements in polymer composites. Carbohydrate Polymers, v. 81, p. 811-819, 2010. IDICULA, M.; JOSEPH, K.; THOMAS, S. Mechanical performance of short banana / sisal hybrid fiber reinforced polyester composites. Journal of Reinforced Plastics and Composites, v. 29, n. 1, p. 12-29, 2010.

163
IDRIS, U. D.; AIGBODION, V. S.; ABUBAKAR, I. J.; NWOYE, C. I. Eco-friendly asbestos free brake-pad: Using banana peels. Journal of King Saud University - Engineering Sciences, v. 27, p. 185-192, 2015. JAVNI, I.; PETROVIĆ, Z. S.; GUO, A.; FULLER, R. Thermal stability of polyurethanes based on vegetable oils. Journal of Applied Polymer Science, v. 77, p. 1723-1734, 2000. JAWAID, M.; KHALIL, H. P. S. A. Cellulosic/synthetic fibre reinforced polymer hybrid composites: A review. Carbohydrate Polymers, v. 86, p. 1-18, 2011. JIAO, L. XIAO, H.; WANG, Q.; SUN, J. Thermal degradation characteristics of rigid polyurethane foam and the volatile products analysis with TG-FTIR-MS. Polymer Degradation and Stability, v. 98, p. 2687-2696, 2013. JOHN, M. J.; THOMAS, S. Biofibres and biocomposites. Carbohydrate Polymers, v. 71, p. 343-364, 2008. JOSÉ, N. M.; PRADO, L. A. S. DE A. Materiais híbridos orgânico-inorgânicos: preparação e algumas aplicações. Química Nova, v. 28, n. 2, p. 281-288, 2005. KABIR, M. M.; WANG, H.; LAU, K. T.; CARDONA, F.; ARAVINTHAN, T. Mechanical properties of chemically-treated hemp fibre reinforced sandwich composites. Composites Part B: Engineering, v. 43, p. 159-169, 2012a. KABIR, M. M.; WANG, H.; LAU, K. T.; CARDONA, F. Chemical treatments on plant-based natural fibre reinforced polymer composites: An overview. Composites Part B: Engineering, v. 43, p. 2883-2892, 2012b. KABIR, M. M.; WANG, H.; LAU, K. T.; CARDONA, F. Effects of chemical treatments on hemp fibre structure. Applied Surface Science, v. 276, p. 13-23, 2013. KALIA, S.; THAKUR, K.; CELLI, A.; KIECHEL, M. A.; SCHAUER, C. L. Surface modification of plant fibers using environment friendly methods for their application in polymer composites, textile industry and antimicrobial activities: A review. Journal of Environmental Chemical Engineering, v. 1, p. 97-112, 2013. KANGO, S.; KALIA, S.; CELLI, A.; NJUGUNA, J.; HABIBI, Y.; KUMAR, R. Surface modification of inorganic nanoparticles for development of organic-inorganic nanocomposites - A review. Progress in Polymer Science, v. 38, p. 1232-1261, 2013. KARIM, F.; BORA, T.; CHAUDHARI, M.; HABIB, K.; MOHAMMED, W.; DUTTA, J. Measurement of aluminum oxide film by Fabry-Pérot interferometry and scanning electron microscopy. Aceito pela revista Journal of Saudi Chemical Society. Acesso em: 15 set. 2016.

164
KAWANO, T.; KAKEMOTO, H.; IRIE, H. Niobium (V) oxide with added silver as a thermoelectric material prepared by spark plasma sintering. Materials Letters, v. 156, p. 94-97, 2015. KHALIL, H. P. S. A.; BHAT, A. H.; YUSRA, A. F. I. Green composites from sustainable cellulose nanofibrils: A review. Carbohydrate Polymers, v. 87, p. 963-979, 2012. KICKELBICK, G. Introduction to hybrid materials. In: ___(1a Ed.). Hybrid Materials. Synthesis, Characterization, and Applications. Weinheim: Wiley-VCH Verlag GmbH & Co., 2007. cap. 1, p. 1-48. KIM, N. K.; LIN, R. J. T.; BHATTACHARYYA, D. Effects of wool fibres, ammonium polyphosphate and polymer viscosity on the flammability and mechanical performance of PP/wool composites. Polymer Degradation and Stability, v. 119, p. 167-177, 2015. KLEMM, D.; HEUBLEIN, B.; FINK, H. P.; BOHN, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angewandte Chemie International Edition, v. 44, p. 3358-3393, 2005. KONG, X.; NARINE, S. S. Physical properties of polyurethane plastic sheets produced from polyols from canola oil. Biomacromolecules, v. 8, p. 2203-2209, 2007. KUMAR, G. M.; PARK, J. Structural and optical property studies on indium doped ZnO nanostructures for solution based organic-inorganic hybrid p-n junctions. Journal of Colloid and Interface Science, v. 430, p. 229-233, 2014. KUMAR, K. S., SIVA, I.; RAJINI, N.; JAPPES, J. T. W.; AMICO, S. C. Layering pattern effects on vibrational behavior of coconut sheath/banana fiber hybrid composites. Materials and Design, v. 90, p. 795-803, 2016. KURANSKA, M.; PROCIAK, A. Porous polyurethane composites with natural fibres. Composites Science and Technology, v. 72, p. 299-304, 2012. LA MANTIA, F. P.; MORREALE, M. Green composites: A brief review. Composites Part A: Applied Science and Manufacturing, v. 42, p. 579-588, 2011. LEE, J. D. Química inorgânica não tão concisa. 5 a ed. São Paulo: Edgar Blucher Ltda, 1999. 527 p. LEE, K. M.; LAI, C. W.; NGAI, K. S.; JUAN, J. C. Recent developments of zinc oxide based photocatalyst in water treatment technology: A review. Water Research, v. 88, p. 428-448, 2016.

165
LI, R.; GU, Y.; YANG, Z.; LI, M.; WANG, S.; ZHANG, Z. Effect of γ irradiation on the properties of basalt fiber reinforced epoxy resin matrix composite. Journal of Nuclear Materials, v. 466, p. 100-107, 2015. LI, X.; TABIL, L. G.; PANIGRAHI, S. Chemical treatments of natural fiber for use in natural fiber-reinforced composites: A review. Journal of Polymers and the Environment, v. 15, p. 25-33, 2007. LI, Y.; REN, H.; RAGAUSKAS, A. J. Rigid polyurethane foam reinforced with cellulose whiskers: Synthesis and characterization. Nano-Micro Letters, v. 2, n. 2, p. 89-94, 2010. LIANG, Y.; YU, J.; FENG, Z.; AI, P. Flammability and thermal properties of bitumen with aluminium trihydroxide and expanded vermiculite. Construction and Building Materials, v. 48, p. 1114-1119, 2013. LIN, L.; LIN, Y.; LI, C.; WU, D.; KONG, H. Synthesis of zeolite/hydrous metal oxide composites from coal fly ash as efficient adsorbents for removal of methylene blue from water. International Journal of Mineral Processing, v. 148, p. 32-40, 2016. LIU, J.; XUE, D.; LI, K. Single-crystalline nanoporous Nb2O5 nanotubes. Nanoscale Research Letters, v. 6, p. 138-145, 2011. LIU, Q.; XU, C.; JI, G.; LIU, H.; MO, Y.; TOLLERUD, D. J.; GU, A.; ZHANG, Q. Sublethal effects of zinc oxide nanoparticles on male reproductive cells. Toxicology in Vitro, v. 35, p. 131-138, 2016a. LIU, Y.; LV, C.; DING, J.; QIAN, P.; ZHANG, X.; YU, Y.; YE, S.; CHEN, Y. The use of the organic-inorganic hybrid polymer Al(OH)3-polyacrylamide to flocculate particles in the cyanide tailing suspensions. Minerals Engineering, v. 89, p. 108-117, 2016b. LOPES, F. F. M.; ARAÚLO, G. T.; LUNA, S.; NASCIMENTO, J. W. B.; DA SILVA, V. R. Modificação das propriedades das fibras de curauá por acetilação. Revista Brasileira de Engenharia Agrícola e Ambiental, v. 15, n. 3, p. 316-321, 2011. LOPES, O. F.; MENDONÇA, V. R.; SILVA, F. B. F.; PARIS, E. C.; RIBEIRO, C. Óxidos de nióbio: uma visão sobre a síntese do Nb2O5 e sua aplicação em fotocatálise heterogênea. Química Nova, v. 38, n. 1, p. 106-117, 2015. LU, Q.; LIN, W.; TANG, L.; WANG, S.; CHEN, X.; HUANG, B. A mechanochemical approach to manufacturing bamboo cellulose nanocrystals. Journal of Materials Science, v. 50, p. 611-619, 2015.

166
LUO, F.; WU, K.; GUO, H.; ZHAO, Q.; LIANG, L.; LU, M. Effect of cellulose whisker and ammonium polyphosphate on thermal properties and flammability performance of rigid polyurethane foam. Journal of Thermal Analysis and Calorimetry, v. 122, p. 717-723, 2015. MAFRA, D.; COZZOLINO, S. M. F. Importância do zinco na nutrição humana. Revista de Nutrição, v. 17, n. 1, p. 79-87, 2004. MAHJOUB, R.; YATIM , J. M.; SAM , A. R. M.; HASHEMI , S. H.; Tensile properties of kenaf fiber due to various conditions of chemical fiber surface modifications. Construction and Building Materials, v. 55, p. 103-113, 2014. MAHMOOD, N.; YUAN, Z.; SCHMIDT, J.; XU, C. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renewable and Sustainable Energy Reviews, v. 60, p. 317-329, 2016. MALKI, A.; MEKHALIF, Z.; DETRICHE, S.; FONDER, G.; BOUMAZA, A.; DJELLOUL, A. Calcination products of gibbsite studied by X-ray diffraction, XPS and solid-state NMR. Journal of Solid State Chemistry, v. 215, p. 8-15, 2014. MARINUCCI, G. Materiais Compósitos. In: ___(1a Ed.). Materiais compósitos poliméricos: fundamentos e tecnologia. São Paulo: ArtLiber Editora, 2011. cap. 1, p. 21-32. MASCHIO, L. J.; PEREIRA, P. H. F.; DA SILVA, M. L. C. P. Preparation and characterization of cellulose/hydrous niobium oxide hybrid. Carbohydrate Polymers, v. 89, p. 992-996, 2012. MBOUGUENG, P. D.; TENIN, D.; SCHER, J.; TCHIÉGANG, C. Influence of acetylation on physicochemical, functional and thermal properties of potato and cassava starches. Journal of Food Engineering, v. 108, p. 320-326, 2012. MEMON, J. R.; MEMON, S. Q.; BHANGER, M. I.; EL-TURKI , A.; HALLAM, K. R.; ALLEN, G. C. Banana peel: A green and economical sorbent for the selective removal of Cr (VI) from industrial wastewater. Colloids and Surfaces B: Biointerfaces, v. 70, p. 232-237, 2009. MENON, V.; RAO, M. Trends in bioconversion of lignocellulose: Biofuels, platform chemicals & biorefinery concept. Progress in Energy and Combustion Science, v. 38, p. 522-550, 2012. MERLINI, C.; SOLDI, V.; BARRA, G. M. O. Influence of fiber surface treatment and length on physico-chemical properties of short random banana fiber-reinforced castor oil polyurethane composites. Polymer Testing, v. 30, p. 833-840, 2011. MEYABADI, T. F.; SADEGHI, G. M. M.; DADASHIAN, F.; ASL, H. E. Z. From cellulosic waste to nanocomposites. Part 2: Synthesis and characterization of polyurethane/cellulose nanocomposites. Journal of Materials Science, v. 48, p. 7283-7293, 2013.

167
MIROIU, F. M.; STEFAN, N.; VISAN, A. I.; NITA, C.; LUCULESCU, C. R.; RASOGA, O.; SOCOL, M.; ZGURA, I.; CRISTESCU, R.; RACIUN, D.; SOCOL, G. Composite biodegradable biopolymer coatings of silk fibroin-poly(3-hydroxybutyric-acid-co-3-hydroxyvaleric-acid) for biomedical applications. Applied Surface Science, v. 355, p. 1123-1131, 2015. MIRZAEI, A.; CHEN, Z.; HAGHIGHAT, F.; YERUSHALMI, L. Removal of pharmaceuticals and endocrine disrupting compounds from water by zinc oxide-based photocatalytic degradation: A review. Sustainable Cities and Society, v. 27, p. 407-418, 2016. MOEZZI, A.; MCDONAGH, A. M.; CORTIE, M. B. Zinc oxide particles: Synthesis, properties and applications. Chemical Engineering Journal, v. 185-186, p. 1-22, 2012. MOHANTY, A. K.; MISRA, M.; DRZAL, L. T. Surface modifications of natural fibers and performance of the resulting biocomposites/: An overview. Composite Interfaces, v. 8, n. 5, p. 313-343, 2001. MOSIEWICKI, M. A.; CASADO, U.; MARCOVICH, N. E.; ARANGUREN, M. I. Polyurethanes from tung oil: Polymer characterization and composites. Polymer Engineering and Science, v. 49, p. 685-692, 2009. MOSIEWICKI, M. A.; ROJEK, P.; MICHAŁOWSKI, S.; ARANGUREN, M. I.; PROCIAK, A. Rapeseed oil-based polyurethane foams modified with glycerol and cellulose micro/nanocrystals. Journal of Applied Polymer Science, v. 132, p. 41602, 2015. MOTHÉ, C. G.; DE AZEVEDO, A. D. Análise térmica dos materiais. 1a ed. São Paulo: Artliber Editora, 2009, 324 p. MUKHERJEE, T.; SANI, M.; KAO, N.; GUPTA, R. K.; QUAZI, N.; BHATTACHARYA, S. Improved dispersion of cellulose microcrystals in polylactic acid (PLA) based composites applying surface acetylation. Chemical Engineering Science, v. 101, p. 655-662, 2013. MULINARI, D. R.; Voorwald, H. J. C.; CIOFFI, M. O. H.; DA SILVA, M. L. C. P.; LUZ, S. M. Preparation and properties of HDPE/sugarcane bagasse cellulose composites obtained for thermokinetic mixer. Carbohydrate Polymers, v. 75, p. 317-321, 2009. MUSTATA, F. S. C.; TUDORACHI, N.; MUSTATA, A.; MUSTATA, F. Physical and thermal characterization of some cellulose fabrics as reinforced materials for composite. Journal of Thermal Analysis and Calorimetry, v. 120, p. 1703-1714, 2015. MUTHUKUMARAN, S.; GOPALAKRISHNAN, R. Structural, FTIR and photoluminescence studies of Cu doped ZnO nanopowders by co-precipitation method. Optical Materials, v. 34, p. 1946-1953, 2012.

168
NARINE, S. S.; KONG, X.; BOUZIDI, L.; SPORNS, P. Physical properties of polyurethanes produced from polyols from seed oils: II. Foams. Journal of the American Oil Chemists’ Society, v. 84, p. 65-72, 2007. NAWROCKI, J.; FIJOŁEK, L. Effect of aluminium oxide contaminants on the process of ozone decomposition in water. Applied Catalysis B: Environmental, v. 142-143, p. 533-537, 2013. NETO, L. N.; PARDINI, L. C. Compósitos estruturais: Ciência e tecnologia. 1a ed. São Paulo: Edgar Blucher Ltda, 2006, 313 p. NG, H.-M.; SIN, L. T.; TEE, T-T.; BEE, S-T.; HUI, D.; LOW, C-Y.; RAHMAT, A. R. Extraction of cellulose nanocrystals from plant sources for application as reinforcing agent in polymers. Composites Part B: Engineering, v. 75, p. 176-200, 2015. NICO, C.; MONTEIRO, T.; GRAÇA, M. P. F. Niobium oxides and niobates physical properties: Review and prospects. Progress in Materials Science, v. 80, p. 1-37, 2016. NIKKHAH, A. A.; ZILOUEI, H.; ASADINEZHAD, A.; KESHAVARZ, A. Removal of oil from water using polyurethane foam modified with nanoclay. Chemical Engineering Journal, v. 262, p. 278-285, 2015. NOGUEIRA, F. G. E.; ASENCIOS, Y. J. O.; RODELLA, C. B.; PORTO, A. L. M.; ASSAF, E. M. Alternative route for the synthesis of high surface-area η-Al2O3/Nb2O5 catalyst from aluminum waste. Materials Chemistry and Physics, v. 184, p. 23-30, 2016. OGEDA, T. L.; PETRI, D. F. S. Hidrólise enzimática de biomassa. Química Nova, v. 33, n. 7, p. 1549-1558, 2010. OHMANTH, B.; KATHIRAVAN, R.; GANDIKOTA, S. The effect of chemical treatment on mechanical and thermal properties of abaca-glass-banana hybrid fibre composites. International Journal of ChemTech Research, v. 9, n. 3, p. 665-671, 2016. OHTA, H.; HOSONO, H. Transparent oxide optoelectronics. Materials Today, v. 7, p. 42-51, 2004. OLIVEIRA, A. L. P. Extracção e Caracterização de Constituintes da Bananeira ‘Dwarf Cavendish’. 2007. 250 p. Tese (Doutorado em Química) - Universidade de Aveiro, 2007. OLIVEIRA, F. R.; ERKENS, L.; FANGUEIRO, R.; SOUTO, A. P. Surface Modification of Banana Fibers by DBD Plasma Treatment. Plasma Chemistry and Plasma Processing, v. 32, p. 259-273, 2012. ONUAGULUCHI, O.; BANTHIA, N. Plant-based natural fibre reinforced cement composites: A review. Cement and Concrete Composites, v. 68, p. 96-108, 2016.

169
ORUE, A. et al. The effect of alkaline and silane treatments on mechanical properties and breakage of sisal fibers and poly(lactic acid)/sisal fiber composites. Composites Part A: Applied Science and Manufacturing, v. 84, p. 186-195, 2016. OTTOBONI, F. S. Desenvolvimento de novos compósitos baseados em polietileno e híbridos gerados a partir de resíduos agrícolas modificados com óxido de nióbio. 2011. 121 p. Dissertação (Mestrado em Engenharia Química) - Escola de Engenharia de Lorena, Universidade de São Paulo, 2011. PAPPU, A.; PATIL, V.; JAIN, S.; MAHINDRAKAR, A.; HAQUE, R.; THAKUR, V. K. Advances in industrial prospective of cellulosic macromolecules enriched banana biofibre resources: A review. International Journal of Biological Macromolecules, v. 79, p. 449-458, 2015. PARK, S. H.; OH, K. W.; KIM, S. H. Reinforcement effect of cellulose nanowhisker on bio-based polyurethane. Composites Science and Technology, v. 86, p. 82-88, 2013. PAUL, S. A.; BOUDENNE, A.; IBOS, L.; CANDAU, Y.; JOSEPH, J.; THOMAS, S. Effect of fiber loading and chemical treatments on thermophysical properties of banana fiber/polypropylene commingled composite materials. Composites Part A: Applied Science and Manufacturing, v. 39, p. 1582-1588, 2008. PAUL, V.; KANNY, K.; REDHI, G. G. Formulation of a novel bio-resin from banana sap. Industrial Crops and Products, v. 43, p. 496-505, 2013. PAUL, V.; KANNY, K.; REDHI, G. G. Mechanical, thermal and morphological properties of a bio-based composite derived from banana plant source. Composites Part A: Applied Science and Manufacturing, v. 68, p. 90-100, 2015. PAUZI, N. N. P. N.; MAJID, R. A.; DZULKIFL, M. H.; YAHYA, M. Y. Development of rigid bio-based polyurethane foam reinforced with nanoclay. Composites Part B: Engineering, v. 67, p. 521-526, 2014. PEREIRA, P. H. F.; BENINI, K. C. C. C.; WATASHI, C. Y.; VOORWALD, H. J. C.; CIOFFI, M. O. H. Characterization of high density polyethylene (HDPE) reinforced with banana peel fibers. BioResources, v. 8, n. 2, p. 2351-2365, 2013. PEREIRA, P. H. F.; VOORWALD, H. J. C.; CIOFFI, M. O. H.; DA SILVA, M. L. C. P.; REGO, A. M. B.; FERRARIA, A. M.; DE PINHO, M. N. Sugarcane bagasse cellulose fibres and their hydrous niobium phosphate composites: synthesis and characterization by XPS, XRD and SEM. Cellulose, v. 21, p. 641-652, 2014.

170
PEREIRA, P. H. F.; ROSA, M. F.; CIOFFI, M. O. H.; BENINI, K. C. C. C.; MILANESE, A. C.; VOORWALD, H. J. C.; MULINARI, D. R. Vegetal fibers in polymeric composites: a review. Polímeros, v. 25, n. 1, p. 9-22, 2015. PEREIRA JR, N.; COUTO, M. A. P. G.; SANTA ANNA, L. M. M. Biomass of lignocellulosic compostion for fuel ethanol production and the context of biorefinery. 1a ed. Rio de Janeiro: Series on Biotechnology, 2008. v. 2, 47 p. PERIYAT, P.; ULLATTIL, S. G. Sol-gel derived nanocrystalline ZnO photoanode film for dye sensitized solar cells. Materials Science in Semiconductor Processing, v. 31, p. 139-146, 2015. PICKERING, K. L.; ARUAN EFENDY, M. G.; LE, T. M. A review of recent developments in natural fibre composites and their mechanical performance. Composites Part A: Applied Science and Manufacturing, v. 83, p. 98-112, 2016. PISZCZYK, Ł.; STRANKOWSKI, M.; DANOWSKA, M.; HAPONIUK, J. T.; GAZDA, M. Preparation and characterization of rigid polyurethane-polyglycerol nanocomposite foams. European Polymer Journal, v. 48, p. 1726-1733, 2012. RAGOUBI, M.; GEORGE, B.; MOLINA, S.; BIENAIMÉ, D.; MERLIN, A.; HIVER, J. M.; DAHOUN, A. Effect of corona discharge treatment on mechanical and thermal properties of composites based on miscanthus fibres and polylactic acid or polypropylene matrix. Composites Part A: Applied Science and Manufacturing, v. 43, p. 675-685, 2012. RAJA, K.; RAMESH, P. S.; GEETHA, D. Structural, FTIR and photoluminescence studies of Fe doped ZnO nanopowder by co-precipitation method. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, v. 131, p. 183-188, 2014. RAMACHANDRAN, M.; BANSAL, S.; RAICHURKAR, P. Experimental study of bamboo using banana and linen fibre reinforced polymeric composites. Perspectives in Science, v. 8, p. 313-316, 2016. RAMNATH, B. V.; SHARAVANAN, R.; CHANDRASEKARAN, M.; ELANCHEZHIAN, C.; SATHYANARAYANAN, R.; RAJA, R. N.; KOKAN, S. J. Experimental determination of mechanical properties of banana jute hybrid composite. Fibers and Polymers, v. 16, n. 1, p. 164-172, 2015. RIBEIRO DA SILVA, V.; MOSIEWICKI, M. A.; YOSHIDA, M. I.; DA SILVA, M. C.; STEFANI, P. M.; MARCOVICH, N. E. Polyurethane foams based on modified tung oil and reinforced with rice husk ash I: Synthesis and physical chemical characterization. Polymer Testing, v. 32, p. 438-445, 2013a.

171
RIBEIRO DA SILVA, V.; MOSIEWICKI, M. A.; YOSHIDA, M. I.; DA SILVA, M. C.; STEFANI, P. M.; MARCOVICH, N. E. Polyurethane foams based on modified tung oil and reinforced with rice husk ash II: Mechanical characterization. Polymer Testing, v. 32, p. 665-672, 2013b. RODRIGUES, L. A. Síntese de nanopartículas de óxido de nióbio hidratado via microemulsão inversa. 2008. 122 p. Dissertação (Mestrado em Engenharia Química) Escola de Engenharia de Lorena, Universidade de São Paulo, 2008. RODRIGUES, L. A.; DA SILVA, M. L. C. P. Adsorção de íons fosfato em óxido de nióbio hidratado. Química Nova, v. 32, n. 5, p. 1206-1211, 2009a. RODRIGUES, L. A.; DA SILVA, M. L. C. P. An investigation of phosphate adsorption from aqueous solution onto hydrous niobium oxide prepared by co-precipitation method. Colloids and Surfaces A: Physicochemical and Engineering Aspects, v. 334, p. 191-196, 2009b. RODRIGUES, L. A.; DA SILVA, M. L. C. P. Synthesis of Nb2O5.nH2O nanoparticles by water-in-oil microemulsion. Journal of Non-Crystalline Solids, v. 356, p. 125-128, 2010. RODRIGUES, L. A.; MULINARI, D. R.; DA SILVA, M. L. C. P. Adsorção de íons sulfato em ZrO2.nH2O preparado pelo método da precipitação convencional e da precipitação em solução homogênea. Cerâmica, v. 55, p. 40-45, 2009. ROJAS, J.; BEDOYA, M.; CIRO, Y. Current trends in the production of cellulose nanoparticles and nanocomposites for biomedical applications. In: ___(1a ed.). Cellulose - Fundamental Aspects and Current Trends. InTech, 2015 cap. 8, p. 193-228. ROSÁRIO, F.; PACHEKOSKI, W. M.; SILVEIRA, A. P. J.; DOS SANTOS, S. F.; JÚNIOR, H. S.; CASARIN, S. A. Resíduos de sisal como reforço em compósitos de polipropileno virgem e reciclado. Polímeros, v. 21, n. 2, p. 90-97, 2011. SALEM, R. E. P.; CHINELATTO, A. S. A.; CHINELATTO, A. L. Síntese de pós de alumina por meio de um método Pechini modificado com adição de sementes em diferentes atmosferas de calcinação. Cerâmica, v. 60, p. 108-116, 2014. SANCHEZ, E. M. S.; CAVANI, C. S.; LEAL, C. V.; SANCHEZ, C. G. Compósito de resina de poliéster insaturado com bagaço de cana-de-açúcar: influência do tratamento das fibras nas propriedades. Polímeros, v. 20, n. 3, p. 194-200, 2010. SANTOS, F. A.; DE QUEIRÓZ, J. H.; COLODETTE, J. L.; FERNANDES, S. A.; GUIMARÃES, V. M.; REZENDE, S. T. Potencial da palha de cana-de-açúcar para produção de etanol. Química Nova, v. 35, n. 5, p. 1004-1010, 2012.

172
SARIKANAT, M.; SEKI, Y.; SEVER, K.; BOZACI, E.; DEMIR, A.; OZDOGAN, E. The effect of argon and air plasma treatment of flax fiber on mechanical properties of reinforced polyester composite. Journal of Industrial Textiles, v. 45, n. 6, p. 1252-1267, 2016. SATYANARAYANA, K. G.; ARIZAGA, G. G. C.; WYPYCH, F. Biodegradable composites based on lignocellulosic fibers - An overview. Progress in Polymer Science, v. 34, p. 982-1021, 2009. SATYANARAYANA, K. G.; GUIMARÃES, J. L.; WYPYCH, F. Studies on lignocellulosic fibers of Brazil. Part I: Source, production, morphology, properties and applications. Composites Part A: Applied Science and Manufacturing, v. 38, p. 1694-1709, 2007. SEPTEVANI, A. A.; EVANS, D. A. C.; CHALEAT, C.; MARTIN, D. J.; ANNAMALAI, P. K. A systematic study substituting polyether polyol with palm kernel oil based polyester polyol in rigid polyurethane foam. Industrial Crops and Products, v. 66, p. 16-26, 2015. ŞERBAN, D.-A.; WEISSENBORN, O.; GELLER, S.; MARŞAVINA, L.; GUDE, M. Evaluation of the mechanical and morphological properties of long fibre reinforced polyurethane rigid foams. Polymer Testing, v. 49, p. 121-127, 2016. SHANG, W.; SHENG, Z.; SHEN, Y.; AI, B.; ZHENG, L.; YANG, J.; XU, Z. Study on oil absorbency of succinic anhydride modified banana cellulose in ionic liquid. Carbohydrate Polymers, v. 141, p. 135-142, 2016. SHIH, Y.-F.; HUANG, C.-C. Polylactic acid (PLA)/banana fiber (BF) biodegradable green composites. Journal of Polymer Research, v. 18, p. 2335-2340, 2011. SHRIVER, D. F.; ATKINS, P. W. Química inorgânica. 4a ed. Porto Alegre: Bookman, 2008, 848 p. SILVA, D. J.; D’ALMEIDA, M. L. O. Nanocristais de celulose. O Papel, v. 70, n. 7, p. 34-52, 2009. SILVA, R.; HARAGUCHI, S. K.; MUNIZ, E. C.; RUBIRA, A. F. Aplicações de fibras lignocelulósicas na química de polímeros e em compósitos. Química Nova, v. 32, n. 3, p. 661-671, 2009. SILVA, S. S.; FERREIRA, R. A. S.; FU, L.; CARLOS, L. D.; MANO, J. F.; REIS, R. L.; ROCHA, J. Functional nanostructured chitosan-siloxane hybrids. Journal of Materials Chemistry, v. 15, p. 3952-3961, 2005. SILVA, D. R.; CRESPI, M. S.; CRNKOVIC, P. C. G. M.; RIBEIRO, C. A. Pyrolysis, combustion and oxy-combustion studies of sugarcane industry wastes and its blends. Journal of Thermal Analysis and Calorimetry, v. 121, p. 309-318, 2015.

173
SILVA, L. P. Preparação e caracterização de híbridos de celulose do bagaço de cana-de-açúcar e óxido de alumínio hidratado para aplicação em membranas. 2013. 169 p. Dissertação (Mestrado em Engenharia Química) Escola de Engenharia de Lorena, Universidade de São Paulo, 2013. SILVERSTEIN, R. M.; WEBSTER, F. X.; KIEMLE, D. J. Espectroscopia no infravermelho. In: ___(7a ed.). Identificação espectrométrica de compostos orgânicos. Rio de Janeiro: LTC, 2012. cap. 2, p. 70-122. SKOOG, D. A.; WEST, D. M.; HOLLER, F. J.; CROUCH, S. R. Fundamentos de química analítica. 9a ed. Cengage Learning, 2014, 1070 p. SONIA, A.; PRIYA DASAN, K. Chemical, morphology and thermal evaluation of cellulose microfibers obtained from Hibiscus sabdariffa. Carbohydrate Polymers, v. 92, p. 668-674, 2013. SORNALATHA, D. J.; MURUGAKOOTHAN, P. Characterization of hexagonal ZnO nanostructures prepared by hexamethylenetetramine (HMTA) assisted wet chemical method. Materials Letters, v. 124, p. 219-222, 2014. SPINACÉ, M. A. S.; LAMBERT, C. S.; FERMOSELLI, K. K. G.; DE PAOLI, M. A. Characterization of lignocellulosic curaua fibres. Carbohydrate Polymers, v. 77, p. 47-53, 2009. SRINIVASAN, V. S.; BOOPATHY, S. R.; SANGEETHA, D.; RAMNATH, B. V. Evaluation of mechanical and thermal properties of banana-flax based natural fibre composite. Materials and Design, v. 60, p. 620-627, 2014. STEPCZYÑSKA, M. Analysis of the decay of some effects of modification of polylactide surface layers. Polimery, v. 60, n. 7-8, p. 462-467, 2015. SUN, J. X.; SUN, X. F.; ZHAO, H.; SUN, R. C. Isolation and characterization of cellulose from sugarcane bagasse. Polymer Degradation and Stability, v. 84, p. 331-339, 2004. TAGLIAFERRO, G. V.; DA SILVA, M. L. C. P.; DA SILVA, G. L. J. P. Influência do agente precipitante na preparação do óxido de nióbio (V) hidratado pelo método da precipitação em solução homogênea. Química Nova, v. 28, n. 2, p. 250-254, 2005. TAGLIAFERRO, G. V.; PEREIRA, P. H. F.; RODRIGUES, L. A.; DA SILVA, M. L. C. P. Adsorção de chumbo, cádmio e prata em óxido de nióbio (V) hidratado preparado pelo método da precipitação em solução homogênea. Química Nova, v. 34, n. 1, p. 101-105, 2011.

174
TAIPINA, M. O. Nanocristais de celulose: obtenção, caracterização e modificação de superfície. 2012. 89 p. Dissertação (Mestrado em Química) Instituto de Química, Universidade Estadual de Campinas, 2012. TELI, M. D.; VALIA, S. P. Acetylation of banana fibre to improve oil absorbency. Carbohydrate Polymers, v. 92, p. 328-333, 2013. TERZOPOULOU, Z. N.; PAPAGEORGIOU, G. Z.; PAPADOPOULOU, E.; ATHANASSIADOU, E.; ALEXOPOULOU, E.; BIKIARIS, D. N. Green composites prepared from aliphatic polyesters and bast fibers. Industrial Crops and Products, v. 68, p. 60-79, 2015. THAKUR, V. K.; THAKUR, M. K. Processing and characterization of natural cellulose fibers/thermoset polymer composites. Carbohydrate Polymers, v. 109, p. 102-117, 2014. TSERKI, V.; ZAFEIROPOULOS, N. E.; SIMON, F.; PANAYIOTOU, C. A study of the effect of acetylation and propionylation surface treatments on natural fibres. Composites Part A: Applied Science and Manufacturing, v. 36, p. 1110-1118, 2005. VÄISÄNEN, T.; HAAPALA, A.; LAPPALAINEN, R.; TOMPPO, L. Utilization of agricultural and forest industry waste and residues in natural fiber-polymer composites/: A review. Waste Management, v. 54, p. 62-73, 2016. VENKATESHWARAN, N.; PERUMAL, A. E.; ARUNSUNDARANAYAGAM, D. Fiber surface treatment and its effect on mechanical and visco-elastic behaviour of banana/epoxy composite. Materials and Design, v. 47, p. 151-159, 2013. VILAR, W. D. Química e tecnologia dos poliuretanos. Vilar Consultoria Técnica, 2005. Disponível em: <http://www.poliuretanos.com.br> Acesso em 20 ago. 2016. VOGEL, A. I. Química analítica qualitativa. 1a ed. São Paulo: Mestre Jou, 1981, 664 p. WANG, S.; KANG, Y.; WANG, L.; ZHANG, H.; WANG, Y.; WANG, Y. Organic/inorganic hybrid sensors: A review. Sensors and Actuators B: Chemical, v. 182, p. 467-481, 2013. WANG, Z.; SONG, C.; YIN, H.; ZHANG, H. Capacitive humidity sensors based on zinc oxide nanorods grown on silicon nanowires arrays at room temperature. Sensors and Actuators A: Physical, v. 235, p. 234-239, 2015. WANG, Z.-Y.; LIU, Y.; WANG, Q. Flame retardant polyoxymethylene with aluminium hydroxide/melamine/novolac resin synergistic system. Polymer Degradation and Stability, v. 95, p. 945-954, 2010.

175
WATKINS, D.; NURUDDIN, M.; HOSUR, M.; TCHERBI-NARTEH, A.; JEELANI, S. Extraction and characterization of lignin from different biomass resources. Journal of Materials Research and Technology, v. 4, n. 1, p. 26-32, 2015. WRIGHT, M.; UDDIN, A. Organic-inorganic hybrid solar cells: A comparative review. Solar Energy Materials and Solar Cells, v. 107, p. 87-111, 2012. WU, G-M.; CHEN, J.; HUO, S-P.; LIU, G-F.; KONG, Z-W. Thermoset nanocomposites from two-component waterborne polyurethanes and cellulose whiskers. Carbohydrate Polymers, v. 105, p. 207-213, 2014. XI, W.; QIAN, L.; CHEN, Y.; WANG, J.; LIU, X. Addition flame-retardant behaviors of expandable graphite and [bis(2-hydroxyethyl)amino]-methyl-phosphonic acid dimethyl ester in rigid polyurethane foams. Polymer Degradation and Stability, v. 122, p. 36-43, 2015. XIE, X.; ZHOU, Z.; JIANG, M.; XU, X.; WANG, Z.; HUI, D. Cellulosic fibers from rice straw and bamboo used as reinforcement of cement-based composites for remarkably improving mechanical properties. Composites Part B: Engineering, v. 78, p. 153-161, 2015. XU, W.; WANG, G.; ZHENG, X. Research on highly flame-retardant rigid PU foams by combination of nanostructured additives and phosphorus flame retardants. Polymer Degradation and Stability, v. 111, p. 142-150, 2015. XU, Z.; YAN, L.; CHEN, L. Synergistic flame retardant effects between aluminum hydroxide and halogen-free flame retardants in high density polyethylene composites. Procedia Engineering, v. 135, p. 630-636, 2016. YAN, L.; KASAL, B.; HUANG, L. A review of recent research on the use of cellulosic fibres, their fibre fabric reinforced cementitious, geo-polymer and polymer composites in civil engineering. Composites Part B: Engineering, v. 92, p. 94-132, 2016. YANG, H.; YAN, R.; CHEN, H.; LEE, D. H.; ZHENG, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel, v. 86, p. 1781-1788, 2007. YANG, R.; HU, W.; XU, L.; SONG, Y.; LI, J. Synthesis, mechanical properties and fire behaviors of rigid polyurethane foam with a reactive flame retardant containing phosphazene and phosphate. Polymer Degradation and Stability, v. 122, p. 102-109, 2015. YOON, J-S. The fabrication of niobium powder by sodiothermic reduction process. International Journal of Refractory Metals and Hard Materials, v. 28, p. 265-269, 2010. ZAMAN, H. U.; KHAN, M. A.; KHAN, R. A. Improvement of mechanical properties of jute fibers-polyethylene/polypropylene composites: effect of green dye and UV radiation. Polymer-Plastics Technology and Engineering, v. 48, p. 1130-1138, 2009.

176
ZHANG, M.; PAN, H.; ZHANG, L.; HU, L.; ZHOU, Y. Study of the mechanical, thermal properties and flame retardancy of rigid polyurethane foams prepared from modified castor-oil-based polyols. Industrial Crops and Products, v. 59, p. 135-143, 2014. ZHAO, H.; KWAK, J. H.; ZHANG, Z. C.; BROWN, H. M.; AREY, B. W.; HOLLADAY, J. E. Studying cellulose fiber structure by SEM , XRD , NMR and acid hydrolysis. Carbohydrate Polymers, v. 68, p. 235-241, 2007. ZHAO, Y.; ZHOU, X.; YE, L.; TSANG, S. C. E. Nanostructured Nb2O5 catalysts. Nano Reviews, v. 3, p. 17631-17642, 2012. ZHOU, X.; SAIN, M. M.; OKSMAN, K. Semi-rigid biopolyurethane foams based on palm-oil polyol and reinforced with cellulose nanocrystals. Composites Part A: Applied Science and Manufacturing, v. 83, p. 56-62, 2016. ZHOU, Y.; FAN, M.; CHEN, L.; ZHUANG, J. Lignocellulosic fibre mediated rubber composites: An overview. Composites Part B: Engineering, v. 76, p. 180-191, 2015. ZHOU, Y.; FAN, M.; CHEN, L. Interface and bonding mechanisms of plant fibre composites: An overview. Composites Part B: Engineering, v. 101, p. 31-45, 2016. ZIA, K. M.; BHATTI, H. N.; BHATTI, I. A. Methods for polyurethane and polyurethane composites, recycling and recovery: A review. Reactive and Functional Polymers, v. 67, p. 675-692, 2007. ZIELENIEWSKA, M.; LESZCZYŃSKI, M. K.; SZCZEPKOWSKI, L.; BRYŚKIEWICZ, A.; KRZYŻOWSKA, M.; BIEŃ, K.; RYSZKOWSKA, J. Development and applicational evaluation of the rigid polyurethane foam composites with egg shell waste. Polymer Degradation and Stability, v. 132, p. 78-86, 2016. ZIMMERMANN, M. V.; TURELLA, T. C.; ZATTERA, A. J.; SANTANA, R. M. C. Influência do tratamento químico da fibra de bananeira em compósitos de poli(etileno-co-acetato de vinila) com e sem agente de expansão. Polímeros, v. 24, n. 1, p. 58-64, 2014. ZULFIKAR, M. A.; AFRITA, S.; WAHYUNINGRUM, D.; LEDYASTUTI, M. Preparation of Fe3O4-chitosan hybrid nano-particles used for humic acid adsorption. Environmental Nanotechnology, Monitoring & Management, v. 6, p. 64-75, 2016. ZULUAGA, R.; PUTAUX, J-L.; RESTREPO, A.; MONDRAGON, I.; GAÑÁN, P. Cellulose microfibrils from banana farming residues: isolation and characterization. Cellulose, v. 14, p. 585-592, 2007.
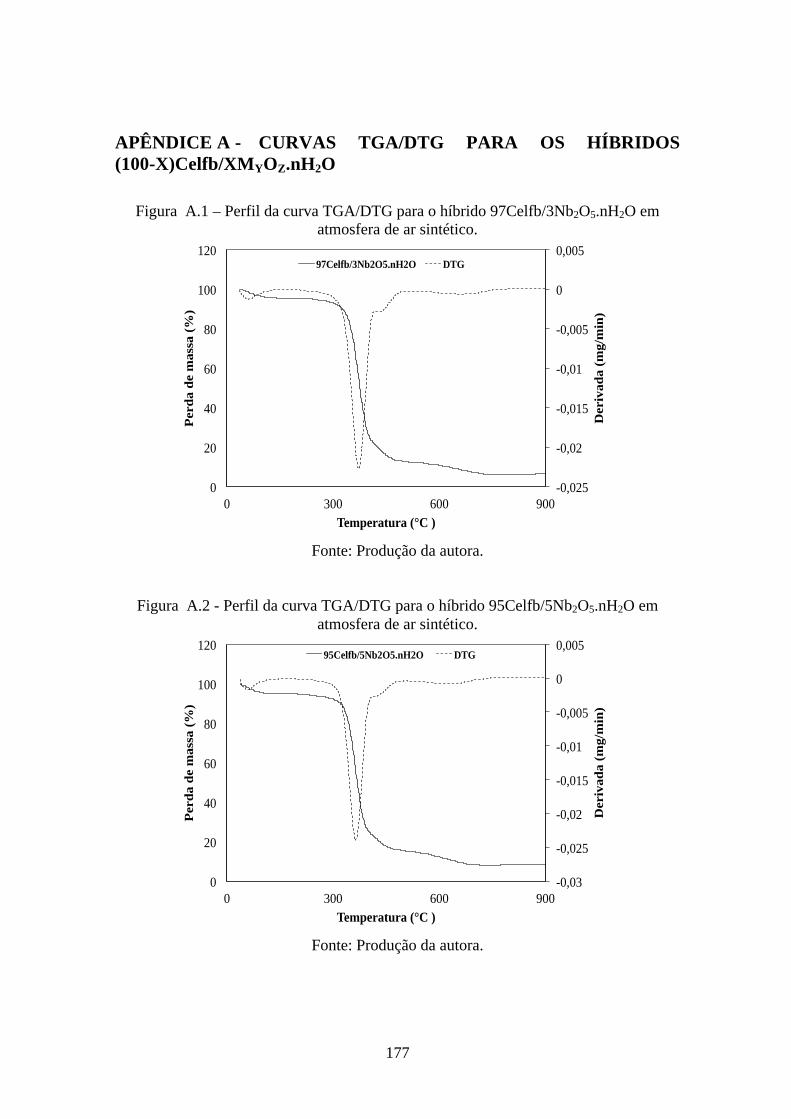
177
APÊNDICE A - CURVAS TGA/DTG PARA OS HÍBRIDOS (100-X)Celfb/XMYOZ.nH2O
Figura A.1 – Perfil da curva TGA/DTG para o híbrido 97Celfb/3Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.2 - Perfil da curva TGA/DTG para o híbrido 95Celfb/5Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C )
97Celfb/3Nb2O5.nH2O DTG
-0,03
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C )
95Celfb/5Nb2O5.nH2O DTG

178
Figura A.3 - Perfil da curva TGA/DTG para o híbrido 90Celfb/10Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.4 - Perfil da curva TGA/DTG para o híbrido 85Celfb/15Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-0,03
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C )
90Celfb/10Nb2O5.nH2O DTG
-3,00E-02
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C )
85Celfb/15Nb2O5.nH2O DTG

179
Figura A.5 - Perfil da curva TGA/DTG para o híbrido 80Celfb/20Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.6 - Perfil da curva TGA/DTG para o híbrido 97Celfb/3Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C )
80Celfb/20Nb2O5.nH2O DTG
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
97Celfb/3Al2O3.nH2O DTG

180
Figura A.7 - Perfil da curva TGA/DTG para o híbrido 96Celfb/4Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.8 - Perfil da curva TGA/DTG para o híbrido 95Celfb/5Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
96Celfb/4Al2O3.nH2O DTG
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
1,00E-02
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
95Celfb/5Al2O3.nH2O DTG

181
Figura A.9 - Perfil da curva TGA/DTG para o híbrido 94Celfb/6Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.10 - Perfil da curva TGA/DTG para o híbrido 93Celfb/7Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
1,00E-02
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
94Celfb/6Al2O3.nH2O DTG
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0,01
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
93Celfb/7Al2O3.nH2O DTG

182
Figura A.11 - Perfil da curva TGA/DTG para o híbrido 90Celfb/10Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.12 - Perfil da curva TGA/DTG para o híbrido 85Celfb/15Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
1,00E-02
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
90Celfb/10Al2O3.nH2O DTG
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0,01
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
85Celfb/15Al2O3.nH2O DTG

183
Figura A.13 - Perfil da curva TGA/DTG para o híbrido 80Celfb/20Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.14 - Perfil da curva TGA/DTG para o híbrido 97Celfb/3ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-0,02
-0,015
-0,01
-0,005
0
0,005
0,01
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C )
80Celfb/20Al2O3.nH2O DTG
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
97Celfb/3ZnO.nH2O DTG

184
Figura A.15 - Perfil da curva TGA/DTG para o híbrido 96Celfb/4ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.16 - Perfil da curva TGA/DTG para o híbrido 95Celfb/5ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-3,00E-02
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
96Celfb/4ZnO.nH2O DTG
-0,03
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
95Celfb/5ZnO.nH2O DTG

185
Figura A.17 - Perfil da curva TGA/DTG para o híbrido 94Celfb/6ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.18 - Perfil da curva TGA/DTG para o híbrido 93Celfb/7ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-0,03
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
94Celfb/6ZnO.nH2O DTG
-0,03
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
93Celfb/7ZnO.nH2O DTG
186
Figura A.19 - Perfil da curva TGA/DTG para o híbrido 90Celfb/10ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura A.20 - Perfil da curva TGA/DTG para o híbrido 85Celfb/15ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-2,50E-02
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
90Celfb/10ZnO.nH2O DTG
-2,00E-02
-1,50E-02
-1,00E-02
-5,00E-03
0,00E+00
5,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
85Celfb/15ZnO.nH2O DTG

187
Figura A.21 - Perfil da curva TGA/DTG para o híbrido 80Celfb/20ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-0,03
-0,025
-0,02
-0,015
-0,01
-0,005
0
0,005
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
80Celfb/20ZnO.nH2O DTG

188

189
APÊNDICE B - MICROGRAFIAS PARA OS COMPÓSITOS DE PU + MYOZ.NH2O E PU + (100-X)CELFB/XMYOZ.NH2O
Figura B.1 - Micrografias para (a) PU puro; (b) PU + 1% m/m Nb2O5.nH2O; (c) PU + 2% m/m Nb2O5.nH2O; (d) PU + 3% m/m Nb2O5.nH2O; (e) PU + 4% m/m Nb2O5.nH2O
e (f) PU + 5% m/m Nb2O5.nH2O, com ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

190
Figura B.2 - Micrografias para (a) PU puro; (b) PU + 1% m/m Al2O3.nH2O; (c) PU + 2% m/m Al2O3.nH2O; (d) PU + 3% m/m Al2O3.nH2O; (e) PU + 4% m/m Al2O3.nH2O e
(f) PU + 5% m/m Al2O3.nH2O, com ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

191
Figura B.3 - Micrografias para (a) PU puro; (b) PU + 1% m/m ZnO.nH2O; (c) PU + 2% m/m ZnO.nH2O; (d) PU + 3% m/m ZnO.nH2O; (e) PU + 4% m/m ZnO.nH2O e (f) PU +
5% m/m ZnO.nH2O, com ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

192
Figura B.4 - Micrografias para (a) PU puro; (b) PU + 1% m/m 97Celfb/3Nb2O5.nH2O; (c) PU + 2% m/m 97Celfb/3Nb2O5.nH2O; (d) PU + 3% m/m 97Celfb/3Nb2O5.nH2O; (e) PU + 4% m/m 97Celfb/3Nb2O5.nH2O e (f) PU + 5% m/m 97Celfb/3Nb2O5.nH2O, com
ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

193
Figura B.5 - Micrografias para (a) PU puro; (b) PU + 1% m/m 94Celfb/6Al2O3.nH2O; (c) PU + 2% m/m 94Celfb/6Al2O3.nH2O; (d) PU + 3% m/m 94Celfb/6Al2O3.nH2O; (e) PU + 4% m/m 94Celfb/6Al2O3.nH2O e (f) PU + 5% m/m 94Celfb/6Al2O3.nH2O, com
ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

194
Figura B.6 - Micrografias para (a) PU puro; (b) PU + 1% m/m 97Celfb/3ZnO.nH2O; (c) PU + 2% m/m 97Celfb/3ZnO.nH2O; (d) PU + 3% m/m 97Celfb/3ZnO.nH2O; (e) PU
+ 4% m/m 97Celfb/3ZnO.nH2O e (f) PU + 5% m/m 97Celfb/3ZnO.nH2O, com ampliação de 23 e 24x.
Fonte: Produção da autora.
a) b) c)
d) e) f)

195
APÊNDICE C - CURVAS TGA/DTG: MATRIZ DE PU E COMPÓSITOS PU + CELFB; PU + MYOZ.NH2O E PU + (100-X)CELFB/XMYOZ.NH2O
Figura C.1 - Perfil da curva TGA/DTG para a matriz de poliuretano PU em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.2 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m Celfb em atmosfera de ar sintético.
Fonte: Produção da autora.
-4,00E-03
-3,50E-03
-3,00E-03
-2,50E-03
-2,00E-03
-1,50E-03
-1,00E-03
-5,00E-04
0,00E+00
5,00E-04
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU DTG
-3,00E-03
-2,50E-03
-2,00E-03
-1,50E-03
-1,00E-03
-5,00E-04
0,00E+00
5,00E-04
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 1%m/m Celfb DTG

196
Figura C.3 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m Celfb em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.4 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m Celfb em atmosfera de ar sintético.
Fonte: Produção da autora.
-4,00E-03
-3,50E-03
-3,00E-03
-2,50E-03
-2,00E-03
-1,50E-03
-1,00E-03
-5,00E-04
0,00E+00
5,00E-04
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 2%m/m Celfb DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 3%m/m Celfb DTG

197
Figura C.5 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m Celfb em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.6 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m Celfb em atmosfera de ar sintético.
Fonte: Produção da autora.
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 4%m/m Celfb DTG
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 5%m/m Celfb DTG

198
Figura C.7 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.8 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 1%m/m Nb2O5.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 2%m/m Nb2O5.nH2O DTG

199
Figura C.9 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.10 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,00E-02 -9,00E-03 -8,00E-03 -7,00E-03 -6,00E-03 -5,00E-03 -4,00E-03 -3,00E-03 -2,00E-03 -1,00E-03 0,00E+00 1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 3%m/m Nb2O5.nH2O DTG
-9,00E-03
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 4%m/m Nb2O5.nH2O DTG

200
Figura C.11 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.12 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-9,00E-03
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 5%m/m Nb2O5.nH2O DTG
-9,00E-03
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 1%m/m Al2O3.nH2O DTG

201
Figura C.13 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.14 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-9,00E-03
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 2%m/m Al2O3.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 3%m/m Al2O3.nH2O DTG

202
Figura C.15 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.16 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,40E-02
-1,20E-02
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 4%m/m Al2O3.nH2O DTG
-1,20E-02
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 5%m/m Al2O3.nH2O DTG

203
Figura C.17 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.18 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 1%m/m ZnO.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 2%m/m ZnO.nH2O DTG

204
Figura C.19 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.20 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 3%m/m ZnO.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 4%m/m ZnO.nH2O DTG

205
Figura C.21 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.22 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m 97Celfb/3Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-9,00E-03
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Per
da d
e m
assa
(%)
Temperatura (°C)
PU + 5%m/m ZnO.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 1%m/m 97Celfb/3Nb2O5.nH2O DTG
206
Figura C.23 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m 97Celfb/3Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.24 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m 97Celfb/3Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,20E-02
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 2%m/m 97Celfb/3Nb2O5.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 3%m/m 97Celfb/3Nb2O5.nH2O DTG

207
Figura C.25 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m 97Celfb/3Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.26 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m 97Celfb/3Nb2O5.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,00E-02 -9,00E-03 -8,00E-03 -7,00E-03 -6,00E-03 -5,00E-03 -4,00E-03 -3,00E-03 -2,00E-03 -1,00E-03 0,00E+00 1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 4%m/m 97Celfb/3Nb2O5.nH2O DTG
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 5%m/m 97Celfb/3Nb2O5.nH2O DTG

208
Figura C.27 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m 94Celfb/6Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.28 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m 94Celfb/6Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,20E-02
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 1%m/m 94Celfb/6Al2O3.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 2%m/m 94Celfb/6Al2O3.nH2O DTG

209
Figura C.29 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m 94Celfb/6Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.30 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m 94Celfb/6Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 3%m/m 94Celfb/6Al2O3.nH2O DTG
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 4%m/m 94Celfb/6Al2O3.nH2O DTG

210
Figura C.31 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m 94Celfb/6Al2O3.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.32 - Perfil da curva TGA/DTG para o compósito PU + 1% m/m 97Celfb/3ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-9,00E-03
-8,00E-03
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 1%m/m 97Celfb/3ZnO.nH2O DTG
-7,00E-03
-6,00E-03
-5,00E-03
-4,00E-03
-3,00E-03
-2,00E-03
-1,00E-03
0,00E+00
1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 5%m/m 94Celfb/6Al2O3.nH2O DTG

211
Figura C.33 - Perfil da curva TGA/DTG para o compósito PU + 2% m/m 97Celfb/3ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.34 - Perfil da curva TGA/DTG para o compósito PU + 3% m/m 97Celfb/3ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,00E-02 -9,00E-03 -8,00E-03 -7,00E-03 -6,00E-03 -5,00E-03 -4,00E-03 -3,00E-03 -2,00E-03 -1,00E-03 0,00E+00 1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 2%m/m 97Celfb/3ZnO.nH2O DTG
-1,00E-02 -9,00E-03 -8,00E-03 -7,00E-03 -6,00E-03 -5,00E-03 -4,00E-03 -3,00E-03 -2,00E-03 -1,00E-03 0,00E+00 1,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 3%m/m 97Celfb/3ZnO.nH2O DTG

212
Figura C.35 - Perfil da curva TGA/DTG para o compósito PU + 4% m/m 97Celfb/3ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
Figura C.36 - Perfil da curva TGA/DTG para o compósito PU + 5% m/m 97Celfb/3ZnO.nH2O em atmosfera de ar sintético.
Fonte: Produção da autora.
-1,20E-02
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 4%m/m 97Celfb/3ZnO.nH2O DTG
-1,00E-02
-8,00E-03
-6,00E-03
-4,00E-03
-2,00E-03
0,00E+00
2,00E-03
0
20
40
60
80
100
120
0 300 600 900
Der
ivad
a (m
g/m
in)
Perd
a de
mas
sa (%
)
Temperatura (°C)
PU + 5%m/m 97Celfb/3ZnO.nH2O DTG
Copyright © 2022 FDOKUMEN




















