
Síndromes de falla medular hereditarios - Medigraphic
16
192 www.actapediatrica.org.mx ARTÍCULO DE REVISIÓN Acta Pediatr Mex. 2021; 42 (4): 192-207. Síndromes de falla medular hereditarios: etiología, fisiopatología, diagnóstico y tratamiento Inherited Bone Marrow Failure Syndromes: etiology, pathophysiology, diagnosis, and management Moisés Fiesco-Roa, 1,2 Angélica Monsivais-Orozco, 3 Alfredo Rodríguez,4 Sara Frias, 1,4 Benilde García-de Teresa 1 1 Laboratorio de Citogenéca, Instuto Nacional de Pediatría, CDMX, México. 2 Programa de Maestría y Doctorado en Ciencias Médicas, Odontológicas y de la Salud, Universidad Nacional Autónoma de México, CDMX, México. 3 Servicio de Hematología, Instituto Nacional de Pediatría, CDMX, México. 4 Instuto de Invesgaciones Biomé- dicas, Departamento de Medicina Genómica y Toxicología Ambiental, Universidad Nacional Autónoma de México, CDMX, México. https: //orcid.org/0000-0002-9378- 1007 Recibido: 22 de julio de 2020 Aceptado: 7 de abril de 2021 Correspondencia: Benilde García-de Teresa [email protected] Este arculo debe citarse como: Fiesco- Roa M, Monsivais-Orozco A, Rodríguez A, Frías S, García-de Teresa B. Síndro- mes de falla medular hereditarios: eología, fisiopatología, diagnósco y tratamiento. Acta Pediatr Méx 2021; 42 (4): 192-207. Resumen Los síndromes de falla medular hereditarios son un grupo heterogéneo de enfermedades genéticas debidas a variantes patogénicas en genes relacionados con la hematopoyesis. Los estudios de análisis genómico han permitido delimitar, al menos, 13 síndromes de falla medular hereditarios debidamente caracterizados. El fenotipo de estas entidades es un espectro que va desde críptico, hasta padecimientos con un cuadro clínico muy evidente. Además, pueden cursar con manifestaciones extramedulares, como cáncer o alteraciones funcionales y del desarrollo. Este tipo de padecimientos requiere un alto índice de sospecha, que debe plantearse ante cualquier paciente con alteraciones en la hematopoyesis, incluso si no hay manifestaciones extramedulares o éstas no son evidentes. Los síndromes de falla medular hereditarios son enfermedades complejas cuyo proceso diagnóstico y tratamiento requiere de un equipo interdisciplinario de especialistas, como los que se encuentran en centros de atención de tercer nivel. En esta revisión se exponen las características etiológicas, fisiopatológicas, clínicas y paraclínicas de los principales síndromes de falla medular hereditarios. PALABRAS CLAVE: Anemia de Diamond-Blackfan, Anemia de Fanconi, Disqueratosis congénita, Neutropenia congénita grave, Síndrome de Shwachman-Diamond, Trom- bocitopenia amegacariocítica congénita, Trombocitopenia con ausencia de radio. Abstract Inherited bone marrow failure syndromes (IBMFS) are a heterogeneous group of ge- netic diseases due to pathogenic variants in genes related to hematopoiesis. Genomic analysis techniques have delimited at least 13 well-characterized syndromes. The IBMFS are clinically heterogeneous, the phenotypic spectrum varies from cryptic features to patients with evident manifestations. Furthermore, patients can have extramedullar manifestations, such as cancer and/or functional and structural abnormalities. These diseases require a high index of suspicion and should be considered in any patient with alterations in hematopoiesis, even if extramedullary manifestations are not seen. The IBMFS are complex pathologies whose approach and management require an interdisciplinary team of specialists. This review gathers the etiology, pathophysiology, and phenotype of the main IBMFS. KEYWORDS: Diamond-Blackfan anemia; Fanconi anemia; Dyskeratosis congenita; Severe congenital neutropenia; Shwachman-Diamond syndrome; Congenital amega- karyocytic thrombocytopenia; Thrombocytopenia-absent radii.
-
Upload
khangminh22 -
Category
Documents
-
view
7 -
download
0
Transcript of Síndromes de falla medular hereditarios - Medigraphic

192 www.actapediatrica.org.mx
Artículo de revisión
Acta Pediatr Mex. 2021; 42 (4): 192-207.
Síndromes de falla medular hereditarios: etiología, fisiopatología, diagnóstico y tratamiento
Inherited Bone Marrow Failure Syndromes: etiology, pathophysiology, diagnosis, and management
Moisés Fiesco-Roa,1,2 Angélica Monsivais-Orozco,3 Alfredo Rodríguez,4 Sara Frias,1,4 Benilde García-de Teresa1
1 Laboratorio de Citogenética, Instituto Nacional de Pediatría, CDMX, México.2 Programa de Maestría y Doctorado en Ciencias Médicas, Odontológicas y de la Salud, Universidad Nacional Autónoma de México, CDMX, México.3 Servicio de Hematología, Instituto Nacional de Pediatría, CDMX, México.4 Instituto de Investigaciones Biomé-dicas, Departamento de Medicina Genómica y Toxicología Ambiental, Universidad Nacional Autónoma de México, CDMX, México.https: //orcid.org/0000-0002-9378-1007
Recibido: 22 de julio de 2020
Aceptado: 7 de abril de 2021
Correspondencia: Benilde García-de [email protected]
Este artículo debe citarse como: Fiesco-Roa M, Monsivais-Orozco A, Rodríguez A, Frías S, García-de Teresa B. Síndro-mes de falla medular hereditarios: etiología, fisiopatología, diagnóstico y tratamiento. Acta Pediatr Méx 2021; 42 (4): 192-207.
Resumen
Los síndromes de falla medular hereditarios son un grupo heterogéneo de enfermedades genéticas debidas a variantes patogénicas en genes relacionados con la hematopoyesis. Los estudios de análisis genómico han permitido delimitar, al menos, 13 síndromes de falla medular hereditarios debidamente caracterizados. El fenotipo de estas entidades es un espectro que va desde críptico, hasta padecimientos con un cuadro clínico muy evidente. Además, pueden cursar con manifestaciones extramedulares, como cáncer o alteraciones funcionales y del desarrollo. Este tipo de padecimientos requiere un alto índice de sospecha, que debe plantearse ante cualquier paciente con alteraciones en la hematopoyesis, incluso si no hay manifestaciones extramedulares o éstas no son evidentes. Los síndromes de falla medular hereditarios son enfermedades complejas cuyo proceso diagnóstico y tratamiento requiere de un equipo interdisciplinario de especialistas, como los que se encuentran en centros de atención de tercer nivel. En esta revisión se exponen las características etiológicas, fisiopatológicas, clínicas y paraclínicas de los principales síndromes de falla medular hereditarios.PALABRAS CLAVE: Anemia de Diamond-Blackfan, Anemia de Fanconi, Disqueratosis congénita, Neutropenia congénita grave, Síndrome de Shwachman-Diamond, Trom-bocitopenia amegacariocítica congénita, Trombocitopenia con ausencia de radio.
Abstract
Inherited bone marrow failure syndromes (IBMFS) are a heterogeneous group of ge-netic diseases due to pathogenic variants in genes related to hematopoiesis. Genomic analysis techniques have delimited at least 13 well-characterized syndromes. The IBMFS are clinically heterogeneous, the phenotypic spectrum varies from cryptic features to patients with evident manifestations. Furthermore, patients can have extramedullar manifestations, such as cancer and/or functional and structural abnormalities. These diseases require a high index of suspicion and should be considered in any patient with alterations in hematopoiesis, even if extramedullary manifestations are not seen. The IBMFS are complex pathologies whose approach and management require an interdisciplinary team of specialists. This review gathers the etiology, pathophysiology, and phenotype of the main IBMFS.KEYWORDS: Diamond-Blackfan anemia; Fanconi anemia; Dyskeratosis congenita; Severe congenital neutropenia; Shwachman-Diamond syndrome; Congenital amega-karyocytic thrombocytopenia; Thrombocytopenia-absent radii.

193
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
ANTECEDENTES
Los síndromes de falla medular se caracterizan por la coexistencia de: 1) alteraciones en la hematopoyesis; 2) citopenias que pueden evo-lucionar a pancitopenia; 3) riesgo aumentado de neoplasias.1 Se clasifican en síndromes de falla medular adquiridos (85%) y hereditarios (15%).
Determinar si un paciente tiene un síndrome de falla medular adquirido o hereditario es funda-mental, pues de ello dependerán el tratamiento médico y los regímenes de acondicionamiento en caso de trasplante y seguimiento. Los sín-dromes de falla medular hereditarios se asocian con anomalías extramedulares, tienen un riesgo incrementado de neoplasias y requieren aseso-ramiento genético que incluya la búsqueda de otros familiares afectados.1-4 (Figura 1) La dis-criminación entre un síndrome de falla medular hereditario y uno adquirido suele ser compleja y requiere un seguimiento exhaustivo y metódico. Para esto hace falta que la historia clínica reúna la mayor información posible, sobre todo de los antecedentes heredofamiliares que incluyan la consanguinidad o endogamia y personales patológicos de alteraciones hematológicas, anomalías del desarrollo y procesos oncológi-cos. También es indispensable la exploración física completa y dirigida a la búsqueda de las anormalidades asociadas con un síndrome de falla medular hereditario. La hemoglobina fetal es un parámetro de laboratorio que se encuentra elevado en el 70% de los síndromes de falla medular hereditarios y apoya este diagnóstico.5
Los síndromes de falla medular hereditarios son enfermedades genéticas en las que existe una alteración de los mecanismos hematopoyéticos normales, debido a variantes patogénicas germi-nales que afectan la función de genes asociados con la hematopoyesis.3 El espectro de presen-tación es vasto, puede ir desde pacientes con fenotipo críptico hasta aquellos con un cuadro clínico muy grave.6 Las características hemato-
lógicas pueden manifestarse desde el periodo neonatal o iniciar hasta entrada la edad adulta.1,7 Los estudios recientes demuestran que algunos síndromes de falla medular hereditarios pueden coexistir con falla medular y con pocos o ningún dato acompañante, una presentación que podría confundirse con un síndrome de falla medular adquirido. Por esto es importante que en todos los pacientes con falla medular se descarte una variedad de hereditario.8
Características clínicas, paraclínicas y de clasificación de los síndromes de falla medular hereditarios
Gracias a los estudios de genotipificación, la cantidad de síndromes de falla medular here-
SFMA sin causa identificada 70%
SFMH 15%
SFMA secundario 15%
Figura 1. Clasificación de los síndromes de falla medu-lar. Aproximadamente el 85% de las fallas medulares son adquiridas; en el 70% no logra identificarse una causa, aunque se presume un mecanismo inmune, mientras que en el 15% se identifica etiología infeccio-sa o por intoxicación (secundarias). En el 15% restante logra identificarse una causa hereditaria. Abreviaturas: SFMA: síndromes de falla medular adquiridos; SFMH: síndromes de falla medular hereditarios: síndromes de falla medular hereditarios.1,4

194
Acta Pediátrica de México 2021; 42 (4)
ditarios se ha expandido hasta, al menos, 13 entidades debidamente caracterizadas.3,8 Ense-guida se describen, brevemente, los síndromes de falla medular hereditarios medular heredita-rios clásicos y se contrastan sus manifestaciones hematológicas (Cuadro 1)9-19 y del desarrollo y oncológicas (Cuadro 2).7,20-32 Es necesario reconocer las características fisiopatológicas y moleculares, los estudios clínicos y paraclínicos que confirman el diagnóstico de los síndromes de falla medular hereditarios mencionados. (Cuadro 3)1,3,20,26,33,34 El médico tratante puede apoyarse en estas herramientas y en el algoritmo diagnóstico de la Figura 24,35 para establecer el diagnóstico diferencial de su paciente con falla medular.
Anemia de Fanconi
La anemia de Fanconi es un síndromes de falla medular hereditario cuyo fenotipo hematológico se acompaña, en aproximadamente 80% de los casos, de alteraciones del desarrollo (las más frecuentes son: la talla baja, las malformaciones renales y del eje radial, los cambios en la pig-mentación de la piel y la microcefalia).36 Poco más del 20% de los pacientes con anemia de Fanconi pueden iniciar procesos neoplásicos.37 La fisiopatología recae en una alteración en la reparación del ADN que conduce a la dismi-nución de la cantidad de células progenitoras hematopoyéticas e incrementa el riesgo de cáncer;38 sin embargo, el origen de las alteracio-nes del desarrollo aún no está suficientemente esclarecido.
Disqueratosis congénita
La triada fenotípica clásica de la disqueratosis congénita incluye distrofia ungueal, piel con un patrón de pigmentación reticular y leucoplaquia oral.28 Estas características pueden coexistir antes de la falla medular.19,28 El 50% de los pacientes con disqueratosis congénita tiene alteraciones del desarrollo y el 10% cursa con algún tipo de
cáncer.28 En la disqueratosis congénita la hema-topoyesis alterada se explica por la deficiencia en el mantenimiento y reparación de los telóme-ros, que son estructuras encargadas de proteger los extremos cromosómicos y regular la duración de la vida de las células a través de su acorta-miento luego de las divisiones celulares.39,40 El acortamiento telomérico es un biomarcador de daño hematopoyético en los síndromes de falla medular adquiridos y hereditarios.41 El acorta-miento telomérico es tan marcado (debajo del percentil 1 poblacional para edad) en la disque-ratosis congénita que éste se usa para establecer este diagnóstico.41,42
Síndrome de Shwachman-Diamond
El síndrome de Shwachman-Diamond se caracte-riza por falla medular, insuficiencia pancreática exocrina y alteraciones funcionales y del de-sarrollo sistémicas, sobre todo esqueléticas y hepáticas.43 Lo común es que los pacientes inicien con neutropenia y esteatorrea, el 40% evoluciona a citopenias bilineales y el 20% a pancitopenia.44 El síndrome de Shwachman-Diamond tiene alteraciones en la maduración ribosomal, mitosis, proliferación y diferenciación mieloide.45-48 También hay un aumento en la producción de interleucina 6 (IL-6), estable-ciendo un asa de activación mTOR-STAT3-IL6; esta activación afecta la hematopoyesis a nivel mieloide y linfoide, y contribuye a un alto riesgo de neoplasias de tipo mieloide.49
La trombocitopenia amegacariocítica congénita
La trombocitopenia amegacariocítica congénitan es una enfermedad que cursa con disminución en la cuenta plaquetaria al nacimiento y sue-le evolucionar a pancitopenia antes de los 5 años.20 No suele cursar con manifestaciones extra-medulares,20,50 lo que puede facilitar el diagnóstico diferencial. El fenotipo de la trom-bocitopenia amegacariocítica congénita es consecuencia de la ausencia o disfunción del

195
Fiesco-Roa M, et al. Síndromes de falla medular hereditariosC
uadr
o 1.
Hal
lazg
os H
emat
ológ
icos
de
los
sínd
rom
es d
e fa
lla m
edul
ar h
ered
itari
os
Panc
itop
enia
Ane
mia
Neu
trop
enia
Trom
boci
tope
nia
Ane
mia
de
Fanc
oni
Dis
quer
atos
is
cong
énita
Sínd
rom
e de
Shw
a-ch
man
- D
iam
ond
Trom
boci
tope
nia
ameg
acar
ioci
tica
cong
énita
Ane
mia
de
Dia
mon
d-B
lack
fan
Neu
trop
enia
co
ngén
ita g
rave
Trom
boci
tope
nia
con
ause
ncia
de
radi
o
Al
naci
mie
nto
Leuc
ocito
sis
No
No
No
Pued
e en
cont
rars
e,
prob
able
men
te
rela
cion
ada
con
infe
cció
n
No
No
Sí
Al
diag
nós-
tico
Edad
M
edia
na:
7.5
años
Med
iana
: 15
año
sM
edia
na: 1
.6 a
ños
2-3
años
Med
ia:
4.5
mes
esM
edia
na:
32
mes
es1e
r añ
o de
vid
a
Neu
trop
enia
Mod
erad
a-Se
vera
Fr
ecue
nte
Mod
erad
a-Se
vera
Fr
ecue
nte
Mod
erad
a-Se
vera
Fr
ecue
nte
No
Gen
eral
men
te n
o*Se
vera
No
Ane
mia
Seve
ra
Frec
uent
eM
oder
ada
Frec
uent
eG
ener
alm
ente
no*
No
Seve
ra
Fr
ecue
nte
Gen
eral
men
te
no*
Leve
-Mod
erad
a
Poco
frec
uent
e
Trom
boci
tope
nia
Mod
erad
a-Se
vera
M
uy fr
ecue
nte
Mod
erad
a-Se
vera
M
uy fr
ecue
nte
Gen
eral
men
te n
o*Se
vera
M
uy fr
ecue
nte
Gen
eral
men
te n
o*N
oSe
vera
M
uy fr
ecue
nte
VC
MEl
evad
oEl
evad
oN
orm
alN
orm
alEl
evad
oN
orm
alN
orm
al
Neu
trop
enia
ai
slad
a +
ele
vaci
ón
VC
M
No
No
Sí (
20%
)N
oN
oSí
(9%
)N
o
Neu
trop
enia
ai
slad
a +
ele
vaci
ón
HbF
No
No
Sí (
28%
)N
oN
oN
oN
o
Asp
irad
o de
MO
Mar
cada
men
te h
i-po
celu
lar
afec
taci
ón
trili
neal
Hip
ocel
ular
Nor
moc
elul
ar
al d
iagn
óstic
o e
hipo
celu
lar
con
meg
acar
iopo
iesi
s no
ta
n af
ecta
da u
na v
ez
que
se e
stab
leci
ó la
fa
lla m
edul
ar
Nor
moc
elul
ar,
redu
cció
n ai
slad
a o
ause
ncia
de
meg
acar
ioci
tos
Nor
moc
elul
ar c
on
eritr
obla
stop
enia
Nor
moc
elul
ar
con
arre
sto
mad
urac
ión
en e
stad
io
prom
ielo
cito
a
mie
loci
to
Hip
omeg
acar
iocí
tica
Prog
resi
ónFa
lla m
edul
ar p
ro-
gres
iva.
Inic
ia c
on
trom
boci
tope
nia
y pr
ogre
sa c
on a
nem
ia
y ne
utro
peni
a ha
sta
panc
itope
nia
Falla
med
ular
pro
-gr
esiv
a. T
rom
boci
to-
peni
a es
la c
itope
nia
más
frec
uent
e
Falla
med
ular
pr
ogre
siva
, 20%
tie
nen
neut
rope
nia
inte
rmite
nte
seve
ra
segu
ida
por
cito
-pe
nias
bili
neal
es o
tr
iline
ales
Trom
boci
tope
nia
grav
e al
nac
imie
nto
que
pued
en te
ner
un in
crem
ento
tr
ansi
tori
o de
pla
-qu
etas
y p
rogr
esa
con
anem
ia s
egui
-da
de
leuc
open
ia
llega
ndo
a fa
lla
med
ular
Ane
mia
car
acte
riza
-da
por
ret
icul
ocito
-pe
nia
Neu
trop
enia
se
vera
Leuc
ocito
sis
corr
ige
en
las
prim
eras
sem
anas
de
vida
. Ane
mia
que
cor
ri-
ge e
n lo
s pr
imer
os a
ños
a ra
ngos
nor
mal
es. L
a tr
ombo
cito
peni
a m
ejor
a de
spué
s de
los
2 añ
os
a ni
vele
s po
r de
bajo
de
rang
os n
orm
ales
*Se
pres
enta
rar
a ve
z y
cuan
do ll
ega
a en
cont
rars
e es
mod
erad
a;
Se
pued
e as
ocia
r a
insu
ficie
ncia
car
diac
a gr
ave
e hí
drop
s fe
tal.
Abr
evia
tura
s: H
bF:
hem
oglo
bina
feta
l; M
O:
méd
ula
ósea
; V
CM
: vo
lum
en c
orpu
scul
ar m
edio
. Ref
eren
cias
: (9–
19)

196
Acta Pediátrica de México 2021; 42 (4)
Cua
dro
2. E
pide
mio
logí
a, F
recu
enci
a de
Alte
raci
ones
del
Des
arro
llo y
Man
ifest
acio
nes
Onc
ológ
icas
de
los
sínd
rom
es d
e fa
lla m
edul
ar h
ered
itari
os
A
nem
ia d
e Fa
ncon
iD
isqu
erat
osis
co
ngén
ita
Sínd
rom
e de
Sh
wac
hman
- D
iam
ond
Trom
boci
tope
nia
ameg
a-ca
rioc
itic
a co
ngén
ita
Ane
mia
de
Dia
-m
ond-
Bla
ckfa
nN
eutr
open
ia
cong
énit
a gr
ave
Trom
boci
tope
nia
con
ause
ncia
de
radi
o
Epid
emio
-lo
gía
Prev
alen
cia:
1-5
po
r m
illón
de
pers
onas
Prev
alen
cia:
1-
9 po
r m
illón
de
per
sona
s
Inci
denc
ia e
stim
ada:
1.
3 en
100
,000
Men
os d
e 20
0 ca
sos
repo
rtad
osIn
cide
ncia
est
i-m
ada:
0.2
-0.8
po
r 10
0,00
0
Prev
alen
cia:
3-
8.5
por
mill
ón
de p
erso
nas
Inci
denc
ia e
stim
ada:
0.
4 en
100
,000
Al m
enos
al
guna
alte
-ra
ción
del
de
sarr
ollo
Sí (
79%
), la
m
ayor
ía d
el ti
po
VA
CTE
RL-
H y
/o
PHEN
OS
Sí (
90%
), la
tr
íada
clá
sica
, le
ucop
laqu
ia
oral
, pie
l con
pa
trón
ret
icul
ar
y di
stro
fia
ungu
eal,
está
pr
esen
te e
n 46
%
Sí (
55%
)Sí
(po
co fr
ecue
ntes
)Sí
(50
% u
na
anom
alía
, 25%
do
s o
más
alte
ra-
cion
es)
Sí (
poco
frec
uen-
tes
y vi
stas
en
las
AR
)
Sí (
100%
)
Talla
baj
aSí
(61
%)
Sí (
20%
)Sí
(50
%)
Sí
(13
%)
Sí
(95
%)
Peso
baj
oSí
(20
-40%
)Sí
(8%
)Sí
(10
%)
Sí
(30
%)
Cab
eza
y SN
CM
icro
cefa
lia
(47%
) M
acro
cefa
lia (
en
algu
nos
caso
de
hidr
ocef
alia
, la
cual
ocu
rre
en
5%)
Estr
uctu
rale
s de
l SN
C (
10%
, al
tera
cion
es
hipo
fisia
rias
) C
ogni
tivas
(10
%)
Mic
roce
falia
Es
truc
tura
les
del S
NC
(54
%,
hipo
plas
ia c
ere-
bela
r en
39%
, ca
lcifi
caci
ones
in
trac
rane
ales
) A
taxi
a (7
%)
Cog
nitiv
as
(48%
)
Mic
roce
falia
M
acro
cefa
lia (
en a
lgu-
nos
caso
de
hidr
oce-
falia
) Es
truc
tura
les
del S
NC
(m
alfo
rmac
ión
de
Arn
old-
Chi
ari t
ipo
I) C
ogni
tivas
(20
%)
M
icro
cefa
lia
Mac
roce
falia
(en
al
guno
s ca
so d
e hi
droc
efal
ia)
Estr
uctu
rale
s de
l SN
C (
1%,
alte
raci
ones
hi
pofis
iari
as,
Arn
old-
Chi
ari)
Cog
nitiv
as (
2%)
M
acro
cefa
lia (
76%
) Es
truc
tura
les
del
SNC
(oc
asio
nale
s,
hipo
plas
ia c
ereb
elar
y
verm
iana
) C
ogni
tivas
(7%
)
Car
aO
ftalm
ológ
icas
(1
6%, fi
sura
s pa
l-pe
bral
es c
orta
s,
ptos
is, m
icro
ftal-
mia
, epi
cant
o)
Alte
raci
ones
de
pabe
llone
s au
ri-
cula
res
Faci
es F
anco
ni
(4%
, car
a tr
ian-
gula
r, fis
uras
pal
-pe
bral
es c
orta
s,
ojos
de
impl
an-
taci
ón p
rofu
nda,
m
icro
gnat
ia)
Ofta
lmol
ógic
as
(49%
, est
enos
is
del c
ondu
cto
lacr
imal
, epi
-fo
ra, b
lefa
ritis
, re
tinop
atía
ex
udat
iva/
vas-
cula
r, es
trab
is-
mo,
cat
arat
as,
úlce
ras)
Ofta
lmol
ógic
as (
2%,
hipe
rtel
oris
mo)
O
ftalm
ológ
icas
(5
%, h
iper
terl
o-ri
smo,
epi
cant
o,
estr
abis
mo,
gla
u-co
ma,
cat
arat
a)
Puen
te n
asal
de
prim
ido
Sí
(53
%, f
rent
e pr
o-m
inen
te, p
abel
lone
s au
ricu
lare
s de
impl
an-
taci
ón b
aja)

197
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
Cav
idad
ora
lC
arie
s R
etra
so e
n la
er
upci
ón d
enta
l Pa
lada
r he
ndid
o
Leuc
opla
quia
(7
8%)
Peri
odon
titis
C
arie
s (1
7%)
Hip
opla
sia
del
esm
alte
Ta
urod
ontis
mo
Den
tale
s
Pala
dar
hend
ido
(4
%)
Labi
o he
ndid
o
(0.2
%)
Peri
odon
titis
, es
tom
atiti
s, g
in-
givi
tis
Car
diac
as y
pu
lmon
ares
Mal
form
acio
-ne
s ca
rdia
cas
(1
3.5%
)
Fibr
osis
pul
mo-
nar
(20%
)M
alfo
rmac
ione
s ca
r-di
acas
(11
%)
Mal
form
acio
nes
card
iaca
sM
alfo
rmac
ione
s ca
rdia
cas
(3%
)Fu
nció
n pu
lmo-
nar
Mal
form
acio
nes
car-
diac
as (
15%
)
Gas
troi
ntes
-tin
ales
Ana
les
y tr
aque
-es
ofág
icas
(12
%)
Hep
átic
as (
fun-
cion
ales
, gen
eral
-m
ente
aso
ciad
as
al t
rata
mie
nto
con
andr
ógen
os)
Este
nosi
s es
ofá-
gica
(17
%)
Hep
átic
as (
5-10
%, c
irro
sis
hepá
tica,
fu
ncio
nale
s,
gene
ralm
ente
as
ocia
das
al
trat
amie
nto
con
andr
ógen
os)
Hep
átic
as (
elev
ació
n de
tran
sam
inas
as
hepa
tom
egal
ia)
Panc
reát
icas
(an
o-m
alía
s ex
ocri
nas
que
suel
e m
ejor
ar c
on e
l tie
mpo
)
Gen
itour
i-na
rias
Mal
form
acio
nes
rena
les
(25%
) M
alfo
rmac
ione
s ge
nita
les
(9%
en
varo
nes
y 3%
en
muj
eres
)
Este
nosi
s ur
etra
l (5
%)
Mal
form
acio
nes
rena
les
(3%
) M
alfo
rmac
ione
s ge
nita
les
(3%
, cr
ipto
rqui
dia,
hi-
posp
adia
s, h
erni
a in
guin
al)
Sí
(23
%)
Extr
emi-
dade
s su
peri
ores
Eje
radi
al
(40.
5%, s
i hay
af
ecta
ción
del
ra
dio
tam
bién
lo
hay
del p
ulga
r)
Ej
e ra
dial
(8%
)
Alte
raci
ones
del
eje
ra-
dial
(10
0% a
usen
cia
o hi
popl
asia
bila
tera
l de
radi
o [9
8% b
ilate
ral]
pe
ro c
on p
rese
ncia
de
pulg
ares
) Fo
com
elia
Otr
as e
sque
-lé
ticas
Ver
tebr
ales
(4%
) K
lippe
l Fei
l Sp
reng
el
Nec
rosi
s av
ascu
lar
de
la c
abez
a de
l fé
mur
Dis
osto
sis
met
afisi
aria
pr
ogre
siva
(25
%)
Torá
cica
s (8
%, t
órax
pe
queñ
o)
Hue
sos
wor
mia
nos
Ver
tebr
ales
Ver
tebr
ales
K
lippe
l Fei
l Sp
reng
el
V
erte
bral
es
Torá
cica
s

198
Acta Pediátrica de México 2021; 42 (4)
Piel
y a
ne-
xos
Pigm
enta
ción
de
la p
iel
(39%
, m
anch
as c
afé
con
lech
e, z
onas
de
hip
o/hi
per
pigm
enta
ción
)
Piel
con
pat
rón
retic
ular
(89
%)
Dis
trofi
a un
-gu
eal
(88%
) H
iper
hidr
osis
(1
5%)
Alo
peci
a y
enca
neci
mie
n-to
pre
mat
uro
(1
6%)
Ecze
ma
Ecze
ma
Hem
angi
oma
capi
lar
(2
4%)
Aud
ioló
gi-
cas
Sí (
11.5
%)
Sí (
1%)
Sí
Endo
crin
o-ló
gica
sTi
roid
eas
(50%
) H
ipog
onad
ism
o
(40%
) M
etab
olis
mo
de
carb
ohid
rato
s
(40%
)
Hip
ogon
adis
mo
(6%
) M
etab
olis
mo
óseo
(os
teo-
peni
a/os
teop
o-ro
sis)
Tiro
idea
s M
etab
olis
mo
óseo
(o
steo
peni
a/os
teop
o-ro
sis)
Met
abol
ism
o ós
eo (
oste
ope-
nia/
oste
opor
osis
)
Inm
unol
ó-gi
cas
Infe
ccio
nes
aso-
ciad
as a
neu
tro-
peni
a
Infe
ccio
nes
asoc
iada
s a
neut
rope
nia
Infe
ccio
nes
asoc
iada
s a
neut
rope
nia
Infe
ccio
nes
asoc
iada
s a
neut
rope
nia
In
fecc
ione
s as
ocia
das
a ne
u-tr
open
ia
APL
V: 2
0%
Frec
uenc
ia
de c
ánce
r30
%50
%8-
20%
3-
4%22
%
Proc
esos
on
coló
gico
s as
ocia
dos
SMD
(11
-34%
) LM
A (
10-3
7%)
LLA
Li
nfom
a M
ama
Ost
eosa
rcom
a C
CE
(cab
eza
y cu
ello
, eso
fági
co,
anal
y g
enita
l) N
euro
blas
tom
a*
Nef
robl
asto
ma*
SMD
LM
A
LLA
C
CE
(cab
e-za
y c
uello
, es
ofág
ico,
ana
l y
geni
tal)
SMD
LM
A (
30%
son
LM
A-6
) LL
A
Rep
orte
s an
ecdó
ticos
de
leuc
emia
SMD
LM
A
LLA
O
steo
sarc
oma
Col
on
Trac
to g
enita
l fe
men
ino
SMD
LM
ALM
A
LLA

199
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
Dia
gnós
tico
dife
renc
ial
TAR
, sín
drom
e po
r de
leci
ón
11q2
3 (P
aris
-Tr
ouss
eau)
AF,
fibr
osis
pu
lmon
ar
inte
rstic
ial
NC
G, F
ibro
sis
quís
tica,
sí
ndro
me
Pear
son
TASR
, sín
drom
e po
r de
leci
ón 1
1q23
(Pa
ris-
Trou
ssea
u) P
re-e
clam
psia
, in
sufic
ienc
ia p
lace
ntar
ia
RC
IU, i
nfec
cion
es (
ej.:
TO
RC
H),
TAIN
Eritr
obla
stop
e-ni
a tr
ansi
tori
a de
la in
fanc
ia,
infe
cció
n po
r pa
rvov
irus
B19
, de
ficie
ncia
de
desa
min
asa
de
aden
osin
a po
r va
rian
tes
pato
gé-
nica
s en
CEC
R1
SSD
, Inm
unod
efi-
cien
cias
her
edita
-ri
as,
infe
ccio
nes
(ej.:
TO
RC
H),
Neu
trop
enia
s as
ocia
das
a A
OC
P, s
índr
ome
Bar
th, D
efi-
cien
cia
del g
en
GA
TA2,
Glu
coge
-no
sis
tipo
Ib
AF,
AD
B, T
ASR
, sín
dro-
me
Rob
erts
, Sín
drom
e H
olt-
Ora
m
*Aso
ciad
os a
var
iant
es p
atog
énic
as e
n lo
s ge
nes
FAN
CD
1 y
FAN
CN
A
brev
iatu
ras:
AD
B:
Ane
mia
de
Dia
mon
d-B
lack
fan;
AF:
ane
mia
de
Fanc
oni;
AO
CP:
alb
inis
mo
ocul
ocut
áneo
par
cial
; A
PLV:
Ale
rgia
a la
s Pr
oteí
nas
de la
Lec
he d
e V
aca;
AR
: au
tosó
mic
as r
eces
ivas
; C
CE:
car
cino
ma
de c
élul
as e
scam
osas
; LL
A:
leuc
emia
linf
oblá
stic
a ag
uda;
LM
A:
leuc
emia
mie
loid
e ag
uda;
NC
G:
neut
rope
nia
cong
énita
gra
ve;
PHEN
OS:
Pig
men
taci
ón a
norm
al d
e la
pie
l, H
ead
(mic
roce
falia
), Ey
es (
fisur
as p
alpe
bral
es c
orta
s, m
icro
ftalm
ia),
Neu
roló
gico
, Oto
logí
a, S
hort
Sta
ture
(ta
lla b
aja)
; R
CIU
: re
tras
o en
el c
reci
mie
nto
intr
aute
rino
; SM
D:
sínd
rom
e m
ielo
disp
lási
co;
SNC
: si
stem
a ne
rvio
so c
entr
al;
SSD
: Sí
ndro
me
de S
hwac
hman
- D
ia-
mon
d; T
AIN
: tr
ombo
cito
peni
a al
oinm
une
neon
atal
; TA
R;
trom
boci
tope
nia
con
ause
ncia
de
radi
o; T
ASR
; tr
ombo
cito
peni
a am
egac
ario
cític
a co
n si
nost
osis
rad
ioul
nar;
TO
RC
H:
Toxo
plas
mos
is, O
tros
, Rub
eola
, Cito
meg
alov
irus
, Her
pes;
VA
CTE
RL-
H:
Ver
tebr
al, A
nal,
Car
diac
o, T
raqu
eo-e
sofá
gico
, Est
enos
is e
sofá
gica
/duo
dena
l, R
enal
, Li
mbs
(ex
trem
idad
es),
Hid
roce
falia
. Ref
eren
cias
: (7
,20–
32)
receptor de la trombopoyetina y la consecuente reducción de megacariocitos.51 Los pacientes con trombocitopenia amegacariocítica congé-nita tienen concentraciones de trombopoyetina muy elevadas (hasta 10 veces su valor normal),51 debido a la falta de unión al receptor. Este dato paraclínico resulta muy útil para el diagnóstico diferencial con trombocitopenia por destrucción de plaquetas.52
Anemia de Diamond-Blackfan
La anemia de Diamond-Blackfan es una aplasia pura de serie roja, crónica, macrocítica-nor-mocrómica y con reticulocitopenia debida a una falla de la eritropoyesis. En la médula ósea de estos pacientes los precursores eritroides están disminuidos o ausentes, mientras que se observa una producción normal de leucocitos y plaquetas.1,34 El 25% de los pacientes con ane-mia de Diamond-Blackfan tiene anomalías del desarrollo24 y de 3 a 4% sufre alguna neoplasia.28 La anemia de Diamond-Blackfan se debe a la afectación de la biogénesis y procesamiento de los ribosomas.34 Se ha observado que ocurre acumulación de proteínas ribosomales en el nucleoplasma, lo que inhibe la actividad de la proteína MDM2 (un regulador negativo del factor transcripcional TP53). Si p53 no se inhibe, como en este caso, la tasa de apoptosis en la médula ósea se incrementa, lo que conduce a falla medular.34
Neutropenia congénita grave
Las neutropenias congénitas graves son un conjunto de alteraciones genéticas hetero-géneas, que cursan con una alteración en la diferenciación de los neutrófilos, con un conteo absoluto de neutrófilos menor a 0.5 x 109/L que es persistente por más de tres meses; concen-traciones que, con frecuencia, son inferiores a 0.2 x 109/L y se correlacionan con la gravedad del cuadro clínico.26,53 El fenotipo de las neutro-penias congénitas graves se caracteriza por una

200
Acta Pediátrica de México 2021; 42 (4)
predisposición a las infecciones bacterianas y fúngicas que pueden acompañarse de altera-ciones neurológicas y endocrinas, en particular osteopenia-osteoporosis54 y un riesgo aumen-tado de síndrome mielodisplásico y leucemia mieloide aguda.55 La mitad de los casos se debe a un arresto en la maduración de la serie mie-loide por una producción alterada de elastasa de neutrófilos, que se acumula en el retículo endoplásmico rugoso y provoca una “respuesta a proteína mal plegada”, caracterizada por estrés celular y apoptosis del neutrófilo.26
Trombocitopenia con ausencia de radio
La trombocitopenia con ausencia de radio es una enfermedad genética que cursa con disminución del recuento plaquetario y anomalías esquelé-ticas; específicamente ausencia o hipoplasia bilateral del radio, pero con pulgares.50 Se ha reportado una razón mujer: hombre de 3.8: 1.30 En esta enfermedad se observa una disminución de los megacariocitos en la médula ósea y un aumento de las concentraciones del receptor de la trombopoyetina.56,57 La trombocitopenia, con ausencia de radio, tiene un patrón de he-rencia particular, que requiere la combinación de una variante rara en uno de los alelos (una microdeleción del gen RMB8A o una variante patogénica en este gen que produzca una au-sencia de la proteína) y, en el otro alelo de este mismo gen, una variante de nucleótido único (variante en la región 5’ UTR o en el intrón 1, que se observan, respectivamente, en 3 y 0.4% en población de origen europeo).58,59,60 Cuando coexiste la combinación de ambas variantes se produce una reducción de la proteína Y14, producto del gen RBM8A, que es importante en la maduración plaquetaria.58 A pesar de que hay afectación bialélica del gen RMB8A y que el ries-go de recurrencia es del 25% en estas familias, el mecanismo genético no corresponde a una herencia autosómica recesiva clásica.
TRATAMIENTO Y SEGUIMIENTO DE LOS SÍNDROMES DE FALLA MEDULAR HEREDITARIOS
Los pacientes con síndromes de falla medular hereditarios requieren atención especializada e interdisciplinaria, por lo que su seguimiento debe llevarse en un centro de tercer nivel de atención.
Los medicamentos que aumentan los conteos periféricos se han indicado con éxito parcial en pacientes con síndromes de falla medular here-ditarios.61 En el caso de la anemia de Fanconi y la disqueratosis congénita, los andrógenos han demostrado respuestas hematológicas en la mayoría de los pacientes; sin embargo, casi siempre esas respuestas cesan y aumentan el riesgo de tumores hepáticos.61 Los corticoeste-roides, como la prednisona, se han indicado en la anemia de Diamond-Blackfan con tasas de respuesta incluso de 80%, pero una proporción significativa experimenta los efectos no desea-dos del tratamiento, incluida la toxicidad.22 En las neutropenias congénitas graves se indican factores estimuladores de colonias (FEC-G), que aumentan los conteos de neutrófilos.62 En otros síndromes de falla medular hereditarios los fac-tores estimuladores de colonias de granulocitos solo deben indicarse en casos de infecciones gra-ves, porque la respuesta es pobre e incrementa el riesgo de evolución a trastornos clonales.61,63
El tratamiento de soporte incluye: transfusiones, antifibrinolíticos y quelantes de hierro en caso de sobrecarga, además de tratamiento sintomático personalizado de acuerdo con las manifestacio-nes de cada paciente.61
El trasplante de células progenitoras hema-topoyéticas es el tratamiento curativo de las alteraciones hematológicas. A pesar de las mejoras en las técnicas y resultados de estos tras-plantes aún hay pacientes que no son elegibles
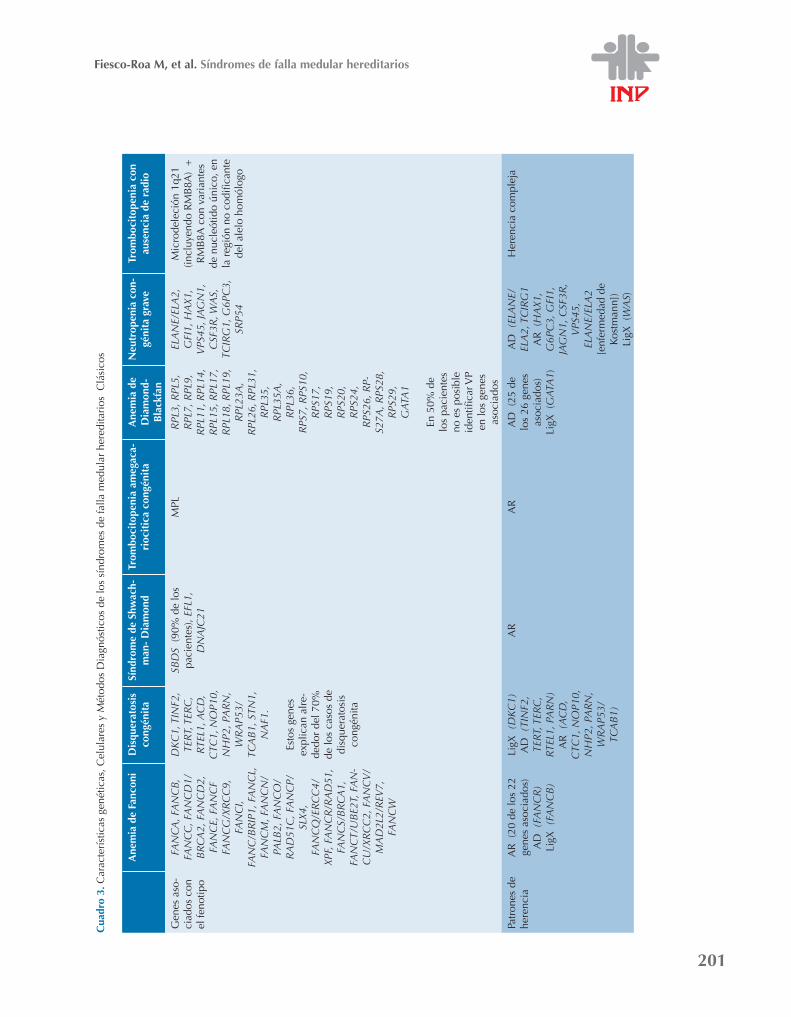
201
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
Cua
dro
3. C
arac
terí
stic
as g
enét
icas
, Cel
ular
es y
Mét
odos
Dia
gnós
ticos
de
los
sínd
rom
es d
e fa
lla m
edul
ar h
ered
itari
os C
lási
cos
A
nem
ia d
e Fa
ncon
iD
isqu
erat
osis
co
ngén
ita
Sínd
rom
e de
Shw
ach-
man
- D
iam
ond
Trom
boci
tope
nia
ameg
aca-
rioc
itic
a co
ngén
ita
Ane
mia
de
Dia
mon
d-B
lack
fan
Neu
trop
enia
con
-gé
nita
gra
veTr
ombo
cito
peni
a co
n au
senc
ia d
e ra
dio
Gen
es a
so-
ciad
os c
on
el fe
notip
o
FAN
CA
, FA
NC
B,
FAN
CC
, FA
NC
D1/
BR
CA
2, F
AN
CD
2,
FAN
CE,
FA
NC
F FA
NC
G/X
RC
C9,
FA
NC
I, FA
NC
/BR
IP1,
FA
NC
L,
FAN
CM
, FA
NC
N/
PALB
2, F
AN
CO
/R
AD
51C
, FA
NC
P/SL
X4,
FAN
CQ
/ER
CC
4/XP
F, F
AN
CR
/RA
D51
, FA
NC
S/B
RC
A1,
FA
NC
T/U
BE2
T, F
AN
-C
U/X
RC
C2,
FA
NC
V/
MA
D2L
2/R
EV7,
FA
NC
W
DK
C1,
TIN
F2,
TER
T, T
ERC
, R
TEL1
, AC
D,
CTC
1, N
OP1
0,
NH
P2, P
AR
N,
WR
AP5
3/TC
AB
1, S
TN1,
N
AF1
.
Esto
s ge
nes
expl
ican
alr
e-de
dor
del 7
0%
de lo
s ca
sos
de
disq
uera
tosi
s co
ngén
ita
SBD
S (9
0% d
e lo
s pa
cien
tes)
, EFL
1,
DN
AJC
21
MPL
RPL
3, R
PL5,
R
PL7,
RPL
9,
RPL
11, R
PL14
, R
PL15
, RPL
17,
RPL
18, R
PL19
, R
PL23
A,
RPL
26, R
PL31
, R
PL35
, R
PL35
A,
RPL
36,
RPS
7, R
PS10
, R
PS17
, R
PS19
, R
PS20
, R
PS24
, R
PS26
, RP-
S27A
, RPS
28,
RPS
29,
GA
TA1
En
50%
de
los
paci
ente
s no
es
posi
ble
iden
tifica
r VP
en lo
s ge
nes
asoc
iado
s
ELA
NE/
ELA
2,
GFI
1, H
AX1
, V
PS45
, JA
GN
1,
CSF
3R, W
AS,
TC
IRG
1, G
6PC
3,
SRP5
4
Mic
rode
leci
ón 1
q21
(in
cluy
endo
RM
B8A
) +
R
MB
8A c
on v
aria
ntes
de
nuc
leót
ido
únic
o, e
n la
reg
ión
no c
odifi
cant
e de
l ale
lo h
omól
ogo
Patr
ones
de
here
ncia
AR
(20
de
los
22
gene
s as
ocia
dos)
A
D (
FAN
CR
) Li
gX (
FAN
CB
)
LigX
(D
KC
1)
AD
(TI
NF2
, TE
RT,
TER
C,
RTE
L1, P
AR
N)
AR
(A
CD
, C
TC1,
NO
P10,
N
HP2
, PA
RN
, W
RA
P53/
TCA
B1)
AR
AR
AD
(25
de
los
26 g
enes
as
ocia
dos)
Li
gX (
GA
TA1)
AD
(EL
AN
E/EL
A2,
TC
IRG
1 A
R (
HA
X1,
G6P
C3,
GFI
1,
JAG
N1,
CSF
3R,
VPS
45,
ELA
NE/
ELA
2 [e
nfer
med
ad d
e Ko
stm
ann]
) Li
gX (
WA
S)
Her
enci
a co
mpl
eja

202
Acta Pediátrica de México 2021; 42 (4)
Mec
anis
mo
celu
lar
alte
rado
Rep
arac
ión
de IC
Ls,
vía
FA/B
RC
AM
ante
ni-
mie
nto
de lo
s te
lóm
eros
Bio
géne
sis
y pr
oces
a-m
ient
o de
rib
osom
as.
Hip
erac
tivac
ión
de la
ví
a m
TOR
-STA
T3
Estim
ulac
ión
de la
pro
-du
cció
n y
dife
renc
iaci
ón
de m
egac
ario
cito
s po
r la
uni
ón d
e TP
O c
on s
u re
cept
or c
elul
ar
Bio
géne
sis
y pr
oces
a-m
ient
o de
ri
boso
mas
Mad
urac
ión
del
linaj
e m
ielo
ide
Mad
urac
ión
de m
egac
a-ri
ocito
s
Mét
odo
de
diag
nóst
ico
Prue
ba d
e ab
erra
-ci
ones
cro
mos
ómi-
cas
indu
cida
s po
r D
iepo
xibu
tano
o
Mito
mic
ina-
C (
Está
n-da
r de
oro
)
Det
ecci
ón d
e va
rian
-te
s pa
togé
nica
s po
r se
cuen
ciac
ión
de lo
s ge
nes
invo
lucr
ados
Med
ició
n de
la
long
itud
telo
mér
ica
flow
-FIS
H
(val
idad
o pa
ra d
iagn
ós-
tico
clín
ico)
, So
uher
n bl
ot,
o qP
CR
Det
ecci
ón d
e va
rian
tes
pa-
togé
nica
s po
r se
cuen
ciac
ión
de lo
s ge
nes
invo
lucr
ados
Ev
alua
ción
de
func
ión
panc
reát
ica
exóc
rina
po
r m
edio
de
dete
rmi-
naci
ón d
e tr
ipsi
nóge
-no
sér
ico
y el
asta
sa
feca
l
Det
ecci
ón d
e va
rian
-te
s pa
togé
nica
s po
r se
cuen
ciac
ión
de lo
s ge
nes
invo
lucr
ados
Det
ecci
ón d
e va
rian
tes
pato
géni
cas
por
secu
enci
a-ci
ón d
el g
en M
PL
D
eter
min
a-ci
ón A
DA
de
eritr
ocito
s.
Se e
ncue
ntra
el
evad
a en
85
% d
e lo
s pa
cien
tes
D
etec
ción
de
vari
ante
s pa
-to
géni
cas
por
secu
enci
ació
n de
los
gene
s in
volu
crad
os
Det
ecci
ón
de v
aria
ntes
pa
togé
nica
s po
r se
cuen
ciac
ión
de
los
gene
s in
volu
-cr
ados
Det
ecci
ón p
or q
PCR
de
la m
icro
dele
ción
1q2
1 +
sec
uenc
iaci
ón S
ange
r (u
sual
men
te) p
ara
dete
ctar
en
RM
B8A
, la
s va
rian
tes
SNPs
(5’
UTR
-SN
P C
hr1:
145
5076
46
[G/A
], o
intr
ónic
o-SN
P C
hr1:
145
5077
65
[G/C
])
Abr
evia
tura
s: A
D:
auto
sóm
ico
dom
inan
te;
AD
A:
aden
osin
a de
sam
inas
a; A
R:
auto
sóm
ico
rece
sivo
; EP
O:
eritr
opoy
etin
a; I
CLs
: un
ione
s co
vale
ntes
inte
rcat
enar
ias
del
DN
A;
LigX
: lig
ado
al X
; qP
CR
: si
glas
en
ingl
és d
e re
acci
ón e
n ca
dena
de
la p
olim
eras
a cu
antit
ativ
a; s
índr
omes
de
falla
med
ular
her
edita
rios
: s
índr
omes
de
falla
med
ular
he
redi
tari
os;
SNP:
sig
las
en in
glés
de
polim
orfis
mo
de n
ucle
ótid
o ún
ico;
TPO
: tr
ombo
poye
tina;
UTR
: si
glas
en
ingl
és d
e re
gión
no
trad
ucid
a; V
P: v
aria
ntes
pat
ogén
ica.
Ref
eren
cias
: (1
,3,2
0,26
,33,
34)

203
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
Sosp
echa
de
SFM
bas
ado
en:
• C
itope
nias
sin
cau
sa a
pare
nte:
H
b <
10 g
/dL
± p
laqu
etas
• <
100K
/uL
± A
NC
<1,
500/
uL•
SM
D
Dat
os s
uges
tivos
de
algú
n SF
MH
esp
ecífi
co
Alte
raci
ones
del
tipo
de
VA
CTE
RL-
H y
/o P
HEN
OS
Leuc
opla
quia
, dis
trofi
a un
guea
l, pi
el c
on p
atró
n re
ticul
ar y
/o fi
bros
is
pulm
onar
Abo
rdaj
e de
cau
sas
infe
ccio
sas,
inm
unol
ógic
as,
nutr
icio
nale
s, H
PN, p
rueb
a ci
toge
nétic
a de
ret
o co
n D
EB, l
ongi
tud
telo
mér
ica
Info
rmat
ivo:
dia
gnós
tico
confi
rmad
o de
SF
MA
sec
unda
ria
(15%
de
los
caso
s de
FM
)*
Info
rmat
ivo:
dia
gnós
tico
confi
rmad
o de
SF
MH
(15%
de
los
caso
s de
FM
)*
• S
ecue
ncia
ción
de
pane
l de
gene
s de
FM
• M
icro
arre
glo
de S
NP
Prue
ba c
itoge
nétic
a de
re
to c
on D
EB
No
info
rmat
ivo
No
info
rmat
ivo
SFM
A d
e ca
usa
no id
entifi
cada
(70%
de
los
caso
s de
FM
)*
No
info
rmat
ivo
Con
tinúa
la s
ospe
-ch
a SF
MH
Long
itud
telo
mér
ica
Bús
qued
a de
VP
en g
e-ne
s as
ocia
dos
al S
SD
Bús
qued
a de
mic
rode
l 1q
21.1
+ S
NP
en R
MB
8A
Insu
ficie
ncia
pan
creá
tica
exóc
rina
con
neu
trop
enia
Trom
boci
tope
nia
con
ause
ncia
/hip
opla
sia
bila
-te
ral d
e ra
dio
con
pulg
ar
pres
ente
Sí
Sí
No
No
WG
S
AF
DC
SSD
TAR
Eval
uaci
ón in
icia
l1.
Con
sang
uini
dad
y en
doga
mia
2. A
ntec
eden
tes
here
dofa
mili
ares
y/o
per
sona
les
pato
-ló
gico
s
a. C
itope
nias
: de
bilid
ad,
palid
ez,
infe
ccio
nes,
san
-gr
ados
b. T
rans
fusi
ones
c. C
ánce
r (le
ucem
ia y
tum
ores
sól
idos
)
d. E
xpos
ició
n a
tóxi
cos
e. E
stea
torr
eaEx
plor
ació
n fís
ica
a. S
omat
omet
ría
b. A
ltera
cion
es d
el d
esar
rollo
del
tipo
de
VA
CTE
RL-
H y
/o P
HEN
OS,
dis
mor
fias
fac
iale
s, d
istr
ofia
un
guea
l, pi
el c
on p
atró
n re
ticul
ar a
ltera
cion
es
pulm
onar
es
*Eva
luac
ión
y se
guim
ient
o in
terd
isci
plin
ario
de
acue
rdo
con
guía
s de
man
ejo
AF:
ane
mia
de
Fanc
oni;
AN
C:
recu
ento
abs
olut
o de
neu
trófi
los
(sig
las
en i
nglé
s de
abs
olut
e ne
utro
phil
coun
t); D
C:
disq
uera
tosi
s co
ngén
ita;
DEB
: di
epox
ibut
ano;
Hb:
hem
oglo
bina
; H
PN:
hem
oglo
binu
ria
paro
xíst
ica
noct
urna
; LM
A: l
euce
mia
mie
loid
e ag
uda;
PH
ENO
S: P
igm
enta
ción
ano
rmal
de
la p
iel,
Hea
d (m
icro
cefa
lia),
Eyes
(fisu
ras
palp
ebra
les
cort
as, m
icro
ftalm
ia),
Neu
roló
gico
, O
tolo
gía,
Sho
rt S
tatu
re (t
alla
baj
a); S
FM: s
índr
ome
de fa
lla m
edul
ar; S
FMA
: sín
drom
e de
falla
med
ular
adq
uiri
do; S
FMH
: sín
drom
e de
falla
med
ular
her
edita
rio;
SM
D: s
índr
ome
mie
lodi
splá
sico
; SN
P: p
olim
orfis
mo
de n
ucle
ótid
o ún
ico;
SSD
: sí
ndro
me
de S
hwac
hman
-Dia
mon
d; T
AR
: tr
ombo
cito
peni
a co
n au
senc
ia d
e ra
dio;
VA
CTE
RL-
H:
Ver
tebr
al,
Ana
l, C
ardi
aco,
Tra
queo
-eso
fági
co,
Este
nosi
s es
ofág
ica/
duod
enal
, Ren
al, L
imbs
(ext
rem
idad
es),
Hid
roce
falia
; VP:
var
iant
e pa
togé
nica
; WG
S: s
ecue
ncia
ción
del
gen
oma
com
plet
o (p
or s
us s
igla
s en
ingl
és w
hole
gen
ome
sequ
enci
ng).
Ref
eren
cias
: 4,6
7
Figu
ra 2
. Alg
oritm
o de
pro
cedi
mie
ntos
par
a el
dia
gnós
tico
etio
lógi
co d
e un
sín
drom
e de
falla
med
ular
. *Ev
alua
ción
y s
egui
mie
nto
inte
rdis
cipl
inar
io d
e ac
uerd
o co
n gu
ías
de m
anej
o. A
F: a
nem
ia
de F
anco
ni;
AN
C:
recu
ento
abs
olut
o de
neu
trófi
los
(sig
las
en in
glés
de
abso
lute
neu
trop
hil c
ount
); D
C:
disq
uera
tosi
s co
ngén
ita;
DEB
: di
epox
ibut
ano;
Hb:
hem
oglo
bina
; H
PN:
hem
oglo
binu
ria
paro
xíst
ica
noct
urna
; LM
A:
leuc
emia
mie
loid
e ag
uda;
PH
ENO
S: P
igm
enta
ción
ano
rmal
de
la p
iel,
Hea
d (m
icro
cefa
lia),
Eyes
(fis
uras
pal
pebr
ales
cor
tas,
mic
rofta
lmia
), N
euro
lógi
co, O
tolo
gía,
Sh
ort S
tatu
re (
talla
baj
a);
SFM
: sí
ndro
me
de fa
lla m
edul
ar;
SFM
A:
sínd
rom
e de
falla
med
ular
adq
uiri
do;
sínd
rom
es d
e fa
lla m
edul
ar h
ered
itari
os :
sín
drom
e de
falla
med
ular
her
edita
rio;
SM
D:
sínd
rom
e m
ielo
disp
lási
co;
SN
P:
polim
orfis
mo
de n
ucle
ótid
o ún
ico;
SS
D:
sín
drom
e de
Shw
achm
an-D
iam
ond;
TA
R:
tro
mbo
cito
peni
a co
n au
senc
ia d
e ra
dio;
V
AC
TER
L-H
: V
erte
bral
, Ana
l, C
ardi
aco,
Tra
queo
-eso
fági
co, E
sten
osis
eso
fági
ca/d
uode
nal,
Ren
al, L
imbs
(ex
trem
idad
es),
Hid
roce
falia
; V
P: v
aria
nte
pato
géni
ca;
WG
S: s
ecue
ncia
ción
del
gen
oma
com
plet
o (p
or s
us s
igla
s en
in
glés
who
le g
enom
e se
quen
cing
). R
efer
enci
as:
(4,3
5)

204
Acta Pediátrica de México 2021; 42 (4)
para estos (por falta de un donador o exclusión por comorbilidades), además de ser un procedi-miento costoso y no accesible a todos.
Debido al riesgo de cáncer hematológico, los pacientes con síndromes de falla medular he-reditarios deben vigilarse de forma constante y continua para poder detectar a tiempo cual-quier indicio de malignidad.19 Se recomienda el estudio de aspirado de médula ósea con análisis citogenético de forma anual, y vigilancia con biometrías hemáticas al menos cada seis meses, aumentando la frecuencia en caso de citopenias.19 Algunos síndromes de falla me-dular hereditarios tienen un riesgo aumentado de tumores sólidos, que puede ser aún más alto después del trasplante de células progenitoras hematopoyéticas, por lo que la vigilancia en la búsqueda de estas alteraciones resulta indispen-sable en el seguimiento del paciente.64
SÍNDROMES DE FALLA MEDULAR HEREDITARIOS EMERGENTES
Los análisis genéticos de secuenciación masiva en paralelo del exoma y del genoma completos en pacientes con pancitopenia han identificado nuevos genes asociados con síndromes de falla medular hereditarios.8 También se ha comproba-do que los pacientes con variantes patogénicas en genes clásicos de síndromes de falla medular hereditarios pueden no tener el fenotipo espera-do. Algunos de los nuevos genes asociados con los síndromes de falla medular hereditarios son:
• GATA2: la haploinsuficiencia de este gen produce pancitopenia sin alteraciones extra-medulares.65
• SAMD9L: el síndrome de ataxia-panci-topenia es producida por una variante patogénica en este gen en el que el signo pivote pueden ser las alteraciones neu-rológicas.66
• SAMD9: variante patogénica en este gen producen el síndrome MIRAGE (Myelo-dysplasia, Infection, Restriction of growth, Adrenal hypoplasia, Genital phenotypes, and Enteropathy).67
• MECOM (complejo MDS1 y EVI1) y HOXA11: se asocian con trombocito-penia amegacariocítica con sinostosis radioulnar.68,69
CONSIDERACIONES FINALES Y PUNTOS PRÁCTICOS
Los síndromes de falla medular hereditarios son enfermedades poco frecuentes y complejas en las que se requiere un alto índice de sospecha, basado en el conocimiento de la enfermedad.
Los pacientes con síndromes de falla medular hereditarios deben atenderse en instituciones de tercer nivel de atención, lo más temprano posible, incluso sin un diagnóstico etiológico específico, para que ahí se confirme y reciban el tratamiento correcto.
Los síndromes de falla medular hereditarios son enfermedades genéticas con un fenotipo que incluye una hematopoyesis alterada, con cito-penias resultantes y predisposición a neoplasias hematológicas y no hematológicas.
Las manifestaciones hematológicas en los sín-dromes de falla medular hereditarios pueden asociarse con diversas alteraciones funcionales y del desarrollo que van de muy leves a graves; se manifiestan con fenotipos extramedulares crípticos, lo que dificulta la distinción de un síndrome de falla medular adquirido.
La ausencia, combinación y sobreposición de las alteraciones funcionales y del desarrollo pueden dificultar el diagnóstico diferencial entre los síndromes de falla medular hereditarios.

205
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
Agradecimientos
Agradecemos a las estudiantes de Medicina Paulina Gómez, Gabriela Baltazar y Jennifer Bal-deras y a la Dra. Adriana Ruiz la lectura crítica de este manuscrito y sus valiosos comentarios.
Este trabajo se benefició del apoyo de los si-guientes fondos: fondos federales del proyecto 41/2014registrado en el INP, PAPIIT205120 y CONA-CYT-FOSISS 233721
REFERENCIAS
1. Wegman-Ostrosky T, Savage SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol 2017; 177 (4): 526-42.
2. Shimamura A. Clinical approach to marrow failure. He-matology Am Soc Hematol Educ Program 2009; 329-37.
3. Kallen ME, Dulau-Florea A, Wang W, Calvo KR. Acquired and germline predisposition to bone marrow failure: Diagnostic features and clinical implications. Semin He-matol 2019; 56 (1): 69-82. https: //doi.org/10.1053/j.seminhematol.2018.05.016
4. Sieff CA. Introduction to Acquired and Inherited Bone Marrow Failure. Hematol Oncol Clin North Am 2018; 32 (4): 569-80. https: //linkinghub.elsevier.com/retrieve/pii/S0889858818307160
5. Alter BP, Rosenberg PS, Day T, Menzel S, Giri N, Savage SA, et al. Genetic regulation of fetal haemoglobin in inherited bone marrow failure syndromes. Br J Haematol 2013; 162 (4): 542-6.
6. Bogliolo M, Bluteau D, Lespinasse J, Pujol R, Vasquez N, D’Enghien CD, et al. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet Med 2018; 20 (4): 458-63.
7. Khincha PP, Savage SA. Neonatal manifestations of inheri-ted bone marrow failure syndromes. Semin Fetal Neonatal Med 2016; 21 (1): 57-65. http: //dx.doi.org/10.1016/j.siny.2015.12.003
8. Bluteau O, Sebert M, Leblanc T, De Latour RP, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood 2018; 131 (7): 717-32.
9. Hashmi S, Allen C, Klaassen R, Fernandez C, Yanofsky R, She-reck E, et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndro-mes and genotype-phenotype correlation. Clin Genet 2017; 79 (5): 448-58. http: //doi.wiley.com/10.1111/j.1399-0004.2010.01468.x
10. Risitano AM, Marotta S, Calzone R, Grimaldi F, Zatterale A. Twenty years of the Italian Fanconi Anemia Registry:
Where we stand and what remains to be learned. Haema-tologica 2016; 101 (3): 319-27.
11. Geddis AE. Congenital Amegakaryocytic Thrombocytopenia and Thrombocytopenia with Absent Radii. Hematol Oncol Clin North Am 2009; 23 (2): 321-31. https: //linkinghub.elsevier.com/retrieve/pii/S0889858809000136
12. Vulliamy T, Dokal I. Dyskeratosis Congenita. Semin Hematol 2006; 43 (3): 157-66. https: //linkinghub.elsevier.com/retrieve/pii/S0037196306000758
13. Ikuse T, Kudo T, Arai K, Fujii Y, Ida S, Ishii T, et al. Shwachman-Diamond syndrome: Nationwide survey and systematic review in Japan. Pediatr Int 2018; 60 (8): 719-26. http: //doi.wiley.com/10.1111/ped.13601
14. King S, Germeshausen M, Strauss G, Welte K, Ballmaier M. Congenital amegakaryocytic thrombocytopenia: a retros-pective clinical analysis of 20 patients. Br J Haematol 2005; 131 (5): 636-44. http: //doi.wiley.com/10.1111/j.1365-2141.2005.05819.x
15. Savoia A, Dufour C, Locatelli F, Noris P, Ambaglio C, Rosti V, et al. Congenital amegakaryocytic thrombocytopenia: clinical and biological consequences of five novel muta-tions. Haematologica 2007; 92 (9): 1186-93. http: //www.haematologica.org/cgi/doi/10.3324/haematol.11425
16. Gong R-L, Wu J, Chen T-X. Clinical, Laboratory, and Mo-lecular Characteristics and Remission Status in Children With Severe Congenital and Non-congenital Neutropenia. Front Pediatr 2018; 6. https: //www.frontiersin.org/arti-cle/10.3389/fped.2018.00305/full
17. DBA Foundation. Learn more. Diagnosis. https: //dbafoun-dation.org/learn-more/diagnosis/
18. National Organization for Rare Disorders. Thrombocytope-nia Absent Radius Syndrome. https: //rarediseases.org/ra-re-diseases/thrombocytopenia-absent-radius-syndrome/
19. Savage SA, Dufour C. Classical inherited bone marrow failu-re syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin Hematol 2017; 54 (2): 105-14. http: //dx.doi.org/10.1053/j.seminhema-tol.2017.04.004
20. Geddis AE. Congenital amegakaryocytic thrombocytopenia. Pediatr Blood Cancer 2011; 57 (2): 199-203.
21. Martínez-Frías ML, Bermejo Sánchez E, García García A, Pérez Fernández JL, Cucalón Manzanos F, Calvo Aguilar MJ, et al. An epidemiological study of the thrombocyto-penia with radial aplasia syndrome (TAR) in Spain. An Esp Pediatr 1998; 49 (6): 619-23. http: //www.ncbi.nlm.nih.gov/pubmed/9972626
22. Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood 2010; 116 (19): 3715-23.
23. Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: Dyskeratosis congenita -update 2015. Eur J Hum Genet 2015; 23 (4): 558. http: //www.nature.com/articles/ejhg2014170
24. Shimamura A, Alter BP. Pathophysiology and manage-ment of inherited bone marrow failure syndromes. Blood

206
Acta Pediátrica de México 2021; 42 (4)
Rev 2010; 24 (3): 101-22. http: //dx.doi.org/10.1016/j.blre.2010.03.002
25. Dokal I. Dyskeratosis congenita in all its forms. Br J Haema-tol 2000; 110 (4): 768-79.
26. Spoor J, Farajifard H, Rezaei N. Congenital neutropenia and primary immunodeficiency diseases. Crit Rev Oncol Hematol 2019; 133: 149-62. https: //doi.org/10.1016/j.critrevonc.2018.10.003
27. Calado RT, Regal JA, Kleiner DE, Schrump DS, Peterson NR, Pons V, et al. A Spectrum of Severe Familial Liver Disorders Associate with Telomerase Mutations. Klein R, editor. PLoS One 2009; 4 (11): e7926. https: //dx.plos.org/10.1371/journal.pone.0007926
28. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the national cancer institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haema-tologica 2018; 103 (1): 30-9.
29. Skórka A, Bielicka-Cymermann J, Gieruszczak-Białek D, Korniszewski L. Thrombocytopenia-absent radius (tar) syndrome: a case with agenesis of corpus callosum, hypo-plasia of cerebellar vermis and horseshoe kidney. Genet Couns 2005; 16 (4): 377-82. http: //www.ncbi.nlm.nih.gov/pubmed/16440880
30. Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, Verschuuren-Bemelmans CC, et al. Thrombocytopenia-absent radius syndrome: A clinical genetic study. J Med Genet 2002; 39 (12): 876-81.
31. Kerr EN, Ellis L, Dupuis A, Rommens JM, Durie PR. The Behavioral Phenotype of School-Age Children with Shwachman Diamond Syndrome Indicates Neurocognitive Dysfunction with Loss of Shwachman-Bodian-Diamond Syndrome Gene Function. J Pediatr 2010; 156 (3): 433-438.e1. https: //linkinghub.elsevier.com/retrieve/pii/S0022347609009019
32. Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: A report from the Diamond Blackfan anemia registry. Blood 2012; 119 (16): 3815-9.
33. Manukjan G, Bösing H, Schmugge M, Strauß G, Schulze H. Impact of genetic variants on haematopoiesis in patients with thrombocytopenia absent radii (TAR) syndrome. Br J Haematol 2017; 179 (4): 606-17. http: //doi.wiley.com/10.1111/bjh.14913
34. Engidaye G, Melku M, Enawgaw B. Diamond blackfan anemia: Genetics, pathogenesis, diagnosis and treatment. Electron J Int Fed Clin Chem Lab Med 2019; 30 (1): 67-81.
35. West AH, Churpek JE. Old and new tools in the clinical diagnosis of inherited bone marrow failure syndromes. Hematology 2017; 2017 (1): 79-87. https: //ashpublica-tions.org/hematology/article/2017/1/79/21056/Old-and-new-tools-in-the-clinical-diagnosis-of
36. Fiesco-Roa MO, Giri N, McReynolds LJ, Best AF, Alter BP. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev 2019; 37.
37. Kutler DI, Singh B, Satagopan J, Batish D, Berwick M, Giampietro PF, et al. A 20-year perspective on the Inter-national Fanconi Anemia Registry (IFAR). Blood 2003; 101: 1249-56.
38. Rodríguez A, D’Andrea A. Fanconi anemia pathway. Curr Biol 2017; 27 (18): R986-8. https: //linkinghub.elsevier.com/retrieve/pii/S0960982217309478
39. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet 2012; 13 (10): 693-704. http: //www.nature.com/articles/nrg3246
40. Wallace DJ. Telomere diseases. N Engl J Med 2010; 362 (12): 1150.
41. Alter BP, Giri N, Savage SA, Rosenberg PS. Telomere length in inherited bone marrow failure syndromes. Haematolo-gica 2015; 100 (1): 49-54.
42. Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telo-mere biology disorders. Expert Rev Hematol 2019; 12 (12): 1037-52. https: //www.tandfonline.com/doi/full/10.1080/17474086.2019.1662720
43. Nelson AS, Myers KC. Diagnosis, Treatment, and Molecular Pathology of Shwachman-Diamond Syndrome. Hematol Oncol Clin North Am 2018; 32 (4): 687-700. https: //linkinghub.elsevier.com/retrieve/pii/S0889858818307147
44. Myers KC, Bolyard AA, Otto B, Wong TE, Jones AT, Harris RE, et al. Variable Clinical Presentation of Shwachman-Diamond Syndrome: Update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr 2014; 164 (4): 866-70. https: //linkinghub.elsevier.com/retrieve/pii/S0022347613014741
45. In K, Zaini MA, Müller C, Warren AJ, von Lindern M, Calkho-ven CF. Shwachman-Bodian-Diamond syndrome (SBDS) protein deficiency impairs translation re-initiation from C/EBPα and C/EBPβ mRNAs. Nucleic Acids Res 2016; 44 (9): 4134-46. https: //academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkw005
46. Nihrane A, Sezgin G, Dsilva S, Dellorusso P, Yamamoto K, Ellis S, et al. Depletion of the Shwachman-Diamond syndrome gene product, SBDS, leads to growth inhibition and increased expression of OPG and VEGF-A. Blood Cells Mol Dis 2009; 42 (1): 85–91. https: //linkinghub.elsevier.com/retrieve/pii/S1079979608002039
47. Austin KM, Gupta ML, Coats SA, Tulpule A, Mostoslavsky G, Balazs AB, et al. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J Clin Invest 2008; 118 (4): 1511-8. http: //www.jci.org/articles/view/33764
48. Orelio C, Verkuijlen P, Geissler J, van den Berg TK, Kuijpers TW. SBDS Expression and Localization at the Mitotic Spindle in Human Myeloid Progenitors. Ben-Jacob E, editor. PLoS One 2009; 4 (9): e7084. https: //dx.plos.org/10.1371/journal.pone.0007084
49. Vella A, D’aversa E, Api M, Breveglieri G, Allegri M, Giaco-mazzi A, et al. MTOR and STAT3 pathway hyper-activation

207
Fiesco-Roa M, et al. Síndromes de falla medular hereditarios
is associated with elevated interleukin-6 levels in patients with shwachman-diamond syndrome: Further evidence of lymphoid lineage impairment. Cancers (Basel) 2020; 12 (3).
50. Geddis AE. Inherited Thrombocytopenia: Congenital Ame-gakaryocytic Thrombocytopenia and Thrombocytopenia With Absent Radii. Semin Hematol 2006; 43 (3): 196-203.
51. Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, et al. C-Mpl Mutations Are the Cause of Congenital Amegakaryocytic Thrombocytopenia. Blood 2001; 97 (1): 139-46.
52. Mukai HY, Kojima H, Todokoro K, Tahara T, Kato T, Hasegawa Y, et al. Serum thrombopoietin (TPO) levels in patients with amegakaryocytic thrombocytopenia are much higher than those with immune thrombocytopenic purpura. Thromb Haemost 1996; 76 (5): 675-8. http: //www.ncbi.nlm.nih.gov/pubmed/8950771
53. Welte K, Zeidler C, Dale DC. Severe Congenital Neutropenia. Semin Hematol 2006; 43 (3): 189-95.
54. Yakisan E, Schirg E, Zeidler C, Bishop NJ, Reiter A, Hirt A, et al. High incidence of significant bone loss in patients with severe congenital neutropenia (Kostmann’s syndrome). J Pediatr 1997; 131 (4): 592-7. https: //linkinghub.elsevier.com/retrieve/pii/S0022347697700684
55. Donadieu J, Beaupain B, Fenneteau O, Bellanné-Chantelot C. Congenital neutropenia in the era of genomics: classifi-cation, diagnosis, and natural history. Br J Haematol 2017; 179 (4): 557-74.
56. Ballmaier M, Schulze H, Strauss G, Cherkaoui K, Wittner N, Lynen S, et al. Thrombopoietin in patients with congenital thrombocytopenia and absent radii: elevated serum levels, normal receptor expression, but defective reactivity to thrombopoietin. Blood 1997; 90 (2): 612-9. http: //www.ncbi.nlm.nih.gov/pubmed/9226161
57. Ballmaier M, Germeshausen M. Advances in the unders-tanding of congenital amegakaryocytic thrombocytopenia. Br J Haematol 2009; 146 (1): 3-16.
58. Albers CA, Newbury-Ecob R, Ouwehand WH, Ghevaert C. New insights into the genetic basis of TAR (thrombocytope-nia-absent radii) syndrome. Curr Opin Genet Dev 2013; 23 (3): 316-23. http: //dx.doi.org/10.1016/j.gde.2013.02.015
59. Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-
frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 2012; 44 (4): 435-9.
60. Soranzo N, Spector TD, Mangino M, Kühnel B, Rendon A, Teumer A, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 2009; 41 (11): 1182-90.
61. Calado RT, Clé D V. Treatment of inherited bone marrow failure syndromes beyond transplantation. Hematology. 2017; 2017 (1): 96-101.
62. Dale DC. Hematopoietic growth factors for the treatment of severe chronic neutropenia. Stem Cells 1995; 13 (2): 94-100. http: //doi.wiley.com/10.1002/stem.5530130201
63. Scagni P, Saracco P, Timeus F, Farinasso L, Dall’Aglio M, Bosa EM, et al. Use of recombinant granulocyte colony-stimulating factor in Fanconi’s anemia. Haematologica 1998; 83 (5): 432-7. http: //www.ncbi.nlm.nih.gov/pubmed/9658728
64. 64. Alter BP. Inherited bone marrow failure syndromes: Considerations pre- and posttransplant. Hematology 2017; 2017 (1): 88-95.
65. McReynolds LJ, Yang Y, Yuen Wong H, Tang J, Zhang Y, Mulé MP, et al. MDS-associated mutations in germline GATA2 mutated patients with hematologic manifestations. Leuk Res 2019; 76: 70-5. https: //doi.org/10.1016/j.leukres.2018.11.013
66. Chen DH, Below JE, Shimamura A, Keel SB, Matsushita M, Wolff J, et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet 2016; 98 (6): 1146-58. http: //dx.doi.org/10.1016/j.ajhg.2016.04.009
67. Veitia RA. MIRAGE Syndrome: Phenotypic Rescue by Soma-tic Mutation and Selection. Trends Mol Med 2019; 25 (11): 937-40. https: //doi.org/10.1016/j.molmed.2019.08.008
68. Thompson AA, Nguyen LT. Amegakaryocytic thrombo-cytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet 2000; 26 (4): 397-8. http: //www.nature.com/articles/ng1200_397
69. Niihori T, Ouchi-Uchiyama M, Sasahara Y, Kaneko T, Hashii Y, Irie M, et al. Mutations in MECOM, Encoding Oncoprotein EVI1, Cause Radioulnar Synostosis with Amegakaryocytic Throm-bocytopenia. Am J Hum Genet 2015; 97 (6): 848-54. https: //linkinghub.elsevier.com/retrieve/pii/S0002929715004127




















