
Serum Withdrawal Potentiates the Toxic Effects of Methamphetamine in Vitro
10
82 Serum Withdrawal Potentiates the Toxic Effects of Methamphetamine in Vitro JEAN LUD CADET a AND SONIA ORDONEZ Molecular Neuropsychiatry Section, NIH/NIDA, Intramural Research Program, Baltimore, Maryland 21224, USA ABSTRACT: Methamphetamine (METH) has been shown to cause neurotoxic damage both in vitro and in vivo. The mechanisms of action are thought to involve the production of pathophysiologic concentration of free radicals. The present study was undertaken to assess the toxic effects of METH caused dose-dependent increased production of reactive oxygen species (ROS) and cell death. Cell death caused by METH was characterized by cytoplasmic vacuolar formation, shrink- age of cytoplasm and nuclear dissolution. Flow cytometric evaluation also re- vealed that this toxin causes changes similar to those observed in cells undergoing apoptosis. When taken together these observations suggest the METH can cause these cells to die via apoptosis. Further experiments indicated that growth of these cells in low (1%) serum or in the absence of serum markedly enhanced the apoptotic effects of METH. These data provide further support for the ideas that METH can cause ROS-mediated apoptosis. INTRODUCTION Methamphetamine (METH) is an illicit drug that is abused throughout the world. The acute administration of this agent can cause neuropsychiatric complications in- cluding psychosis, strokes, coma, and death. 18,23 Abrupt cessation of its use can cause withdrawal symptoms that can lead to suicide. While the acute effects of the drug might be due to increases in the levels of synaptic dopamine (DA), the long- term effects might be secondary to persistent perturbations in monoaminergic systems. 2 For example, the administration of METH can cause marked depletion of dopaminergic or serotonergic markers in rodents, nonhuman primates, as well as hu- man METH users. 4,7,11,14,19,22,26 Several laboratories are actively seeking to decipher the cellular and molecular mechanisms of METH-induced neurotoxicity. The accumulated data suggest that su- peroxide radicals, hydrogen peroxide, hydroxyl radicals, and nitric oxicide might play major roles in its toxicity. 2,12,14–16,24 The production of these reactive oxygen species is thought to be related to the metabolism of METH-induced DA release as well as changes in glutamate metabolism within the striatum. In the case of DA, the production of free radicals is thought to occur via oxidation or metabolic breakdown by monoamine oxidase with subsequent production of reactive oxygen species a Address for correspondence: Jean Lud Cadet, M.D., Molecular Neuropsychiatry Section, NIH/NIDA, Intramural Research Program, P.O. Box 5180, Baltimore, Maryland 21224. Tel.: (410) 550-2953; fax: (410) 550-2745. e-mail: [email protected]
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Serum Withdrawal Potentiates the Toxic Effects of Methamphetamine in Vitro

82
Serum Withdrawal Potentiates the Toxic Effects of Methamphetamine in Vitro
JEAN LUD CADETa AND SONIA ORDONEZ
Molecular Neuropsychiatry Section, NIH/NIDA, Intramural Research Program, Baltimore, Maryland 21224, USA
ABSTRACT: Methamphetamine (METH) has been shown to cause neurotoxicdamage both in vitro and in vivo. The mechanisms of action are thought to involvethe production of pathophysiologic concentration of free radicals. The presentstudy was undertaken to assess the toxic effects of METH caused dose-dependentincreased production of reactive oxygen species (ROS) and cell death. Cell deathcaused by METH was characterized by cytoplasmic vacuolar formation, shrink-age of cytoplasm and nuclear dissolution. Flow cytometric evaluation also re-vealed that this toxin causes changes similar to those observed in cellsundergoing apoptosis. When taken together these observations suggest theMETH can cause these cells to die via apoptosis. Further experiments indicatedthat growth of these cells in low (1%) serum or in the absence of serum markedlyenhanced the apoptotic effects of METH. These data provide further support forthe ideas that METH can cause ROS-mediated apoptosis.
INTRODUCTION
Methamphetamine (METH) is an illicit drug that is abused throughout the world.The acute administration of this agent can cause neuropsychiatric complications in-cluding psychosis, strokes, coma, and death.18,23 Abrupt cessation of its use cancause withdrawal symptoms that can lead to suicide. While the acute effects of thedrug might be due to increases in the levels of synaptic dopamine (DA), the long-term effects might be secondary to persistent perturbations in monoaminergicsystems.2 For example, the administration of METH can cause marked depletion ofdopaminergic or serotonergic markers in rodents, nonhuman primates, as well as hu-man METH users.4,7,11,14,19,22,26
Several laboratories are actively seeking to decipher the cellular and molecularmechanisms of METH-induced neurotoxicity. The accumulated data suggest that su-peroxide radicals, hydrogen peroxide, hydroxyl radicals, and nitric oxicide mightplay major roles in its toxicity.2,12,14–16,24 The production of these reactive oxygenspecies is thought to be related to the metabolism of METH-induced DA release aswell as changes in glutamate metabolism within the striatum. In the case of DA, theproduction of free radicals is thought to occur via oxidation or metabolic breakdownby monoamine oxidase with subsequent production of reactive oxygen species
aAddress for correspondence: Jean Lud Cadet, M.D., Molecular Neuropsychiatry Section,NIH/NIDA, Intramural Research Program, P.O. Box 5180, Baltimore, Maryland 21224.Tel.: (410) 550-2953; fax: (410) 550-2745.
e-mail: [email protected]

83CADET & ORDONEZ: SERUM WITHDRAWAL
(ROS) such as superoxide radicals, hydrogen peroxide, and hydroxyl radicals.2 Thisidea is supported by the recent evidence that the toxic effects of the DA metabolite,6-OHDA, involve the production of superoxide radicals, since transgenic mice thatoverexpress human CuZu superoxide dismutase are protected against the neurode-generative effects of this compound on nigrostriatal dopaminergic neurons.1
The present study further evaluated the toxic effects of METH using an in vitrosystem and also sought to determine if serum withdrawal could cause exacerbationof its toxic effects.
MATERIALS AND METHODS
Cell Cultures
Stock immortalized mesencephalic AF-5 cells were a gift from Dr. W.J Freed(NIDA/NIH). Cells were plated at a concentration of 1.5 × 106 cells/plate, incubatedin Dulbecco’s modified Eagle’s medium/Ham’s F12 (DMEM/F12, 1:1), 10% fetalcalf serum, 2 mM L-glutamine, 100 U/ml penicillin G, and 20 ng/ml human recom-binant basic growth factor (bFGF, Chemicon International, Temecula, California,USA) in an incubator at 37°C, 5% CO2. Cells were subcultured in 6-well plates forMETH treatment as described below.
Treatment of the Cultures and Assessment of Toxicity
Cytotoxicity was measured by the release of the lactate dehydrogenase enzymeinto the culture supernatant upon damage of the plasma membrane or during celldeath. Briefly, cells were plated at a concentration of 1 × 104 cells/well in a 96-wellplate. Cells were allowed to attach overnight. After removal of the media, cells werewashed twice with HBSS. Subsequently, different doses of METH were added to thewells containing DMEM with 10% or 1% FBS or no serum; for controls, cells werecultured with the drugs. Plates were then incubated at 37°C and 5% CO2 for 24 hr.An aliquot of culture supernatant was collected cell free and incubated with the re-action mixture from a kit (cytotoxicity detection, Boehringer). The color intensityformed during the reaction is proportional to the number of lysed cells. Absorbancewas measured using an ELISA reader plate at 490 nm.
Analysis of Apoptosis
Cells were plated at a concentraton of 1 × 106 cells per as described above withvarious concentrations of serum and incubated for 24 hr with METH. Floating cellsas well as adherent cells were pooled, washed with PBS, pelleted, and resuspendedin 0.5 ml PBS. Slides for microscopy were prepared by placing a drop of this cellsuspension on a clean slide, air drying, and fixing with cold acetone. The rest of thecell suspension was fixed with 4% paraformaldehyde for use in flow cytometry aspreviously described.3
84 ANNALS NEW YORK ACADEMY OF SCIENCES
Flow Cytometry
Apoptosis was assessed by flow cytometry using an APO-BrdU Kit (PhoenixFlow Systems Inc., San Diego, CA) as detected in the manufacturer’s protocol. Thiskit uses a two-color staining method that labels DNA strand breaks and total cellularDNA; this approach serves to detect cells undergoing apoptosis using flowcytometry.17 3′-Hydroxyl ends of fragmented DNA are labeled with bromolateddeoxyuridine triphosphate nucleotides (BrdUTP) using deoxyl-nucleotidyltransferase. These sites are then identified by a fluorescent labeled anti-BrdU mono-clonal antibody. Nonapoptotic cells do not incorporate Br-dUTP owing to the lackof exposed 3′-hydoxyl DNA ends.
Flow cytometric data were acquired on a Becton-Dickinson FACScan cytometer(San Jose, CA) using the Consor 40 data acquisition software. Data from 20,000 cellswere acquired and stored in list mode. Data analysis was performed with the PC Lysisanalysis program from Becton-Dickinson. The percentage of apoptotic cells in eachtreatment sample corresponds to the percentage of cells showing the fluorescent sig-nal after subtraction of the control using the histogram subtraction function.
RESULTS
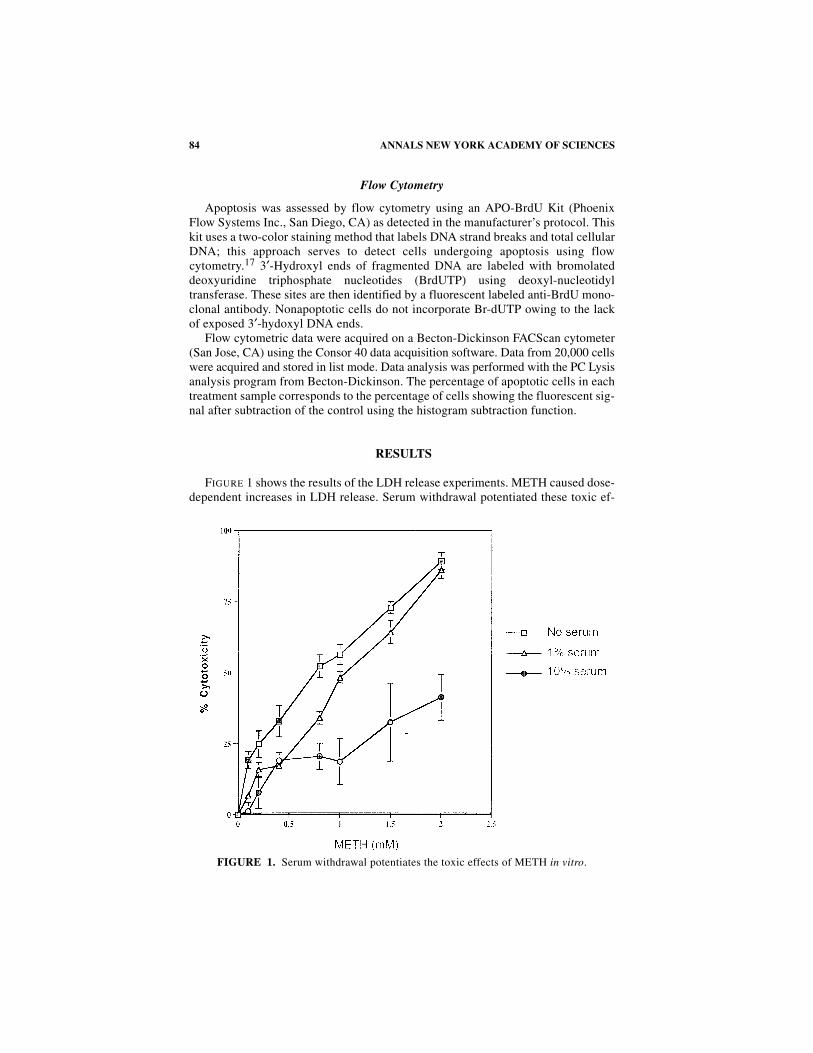
FIGURE 1 shows the results of the LDH release experiments. METH caused dose-dependent increases in LDH release. Serum withdrawal potentiated these toxic ef-
FIGURE 1. Serum withdrawal potentiates the toxic effects of METH in vitro.
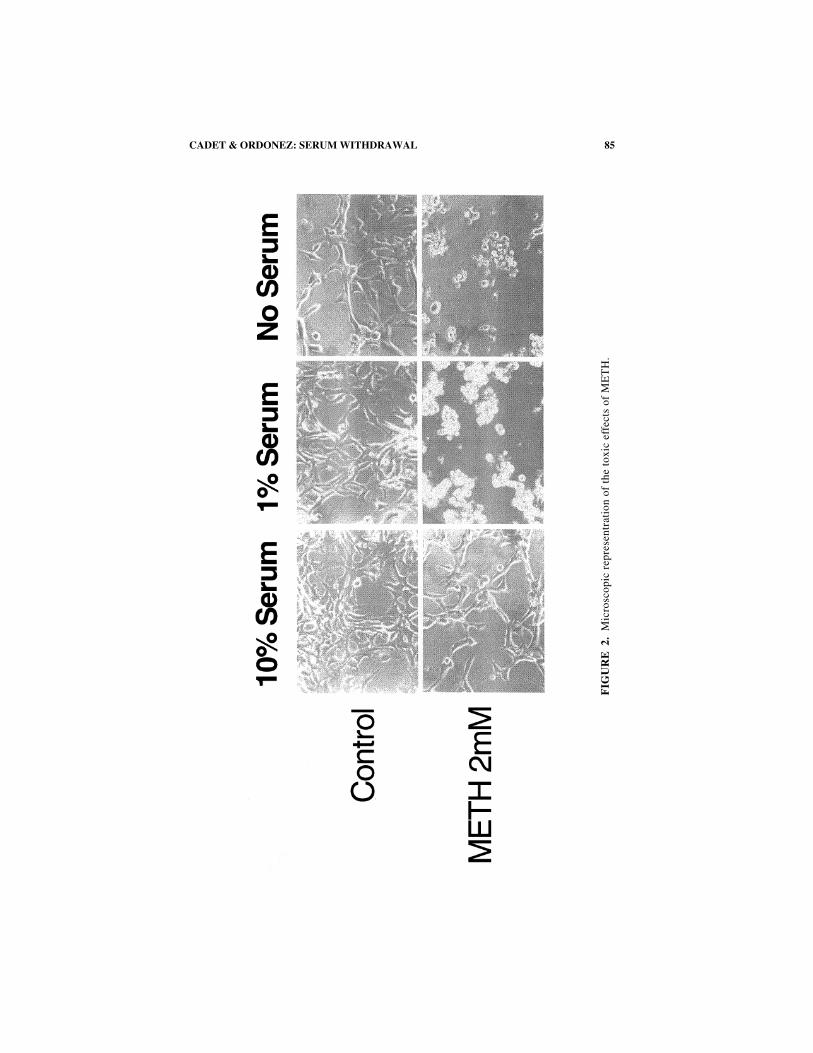
85CADET & ORDONEZ: SERUM WITHDRAWAL
FIG
UR
E2.
Mic
rosc
opic
rep
rese
ntra
tion
of
the
toxi
c ef
fect
s of
ME
TH
.

86 ANNALS NEW YORK ACADEMY OF SCIENCES
FIG
UR
E3.
Flo
w c
ytom
etri
c ev
iden
ce t
hat
ME
TH
cau
ses
apop
toti
c ch
ange
s in
vit
ro.

87CADET & ORDONEZ: SERUM WITHDRAWAL
FIGURE 4. Quantitative analysis of the apoptic effects of METH on AF cells measured by flow cytometry.
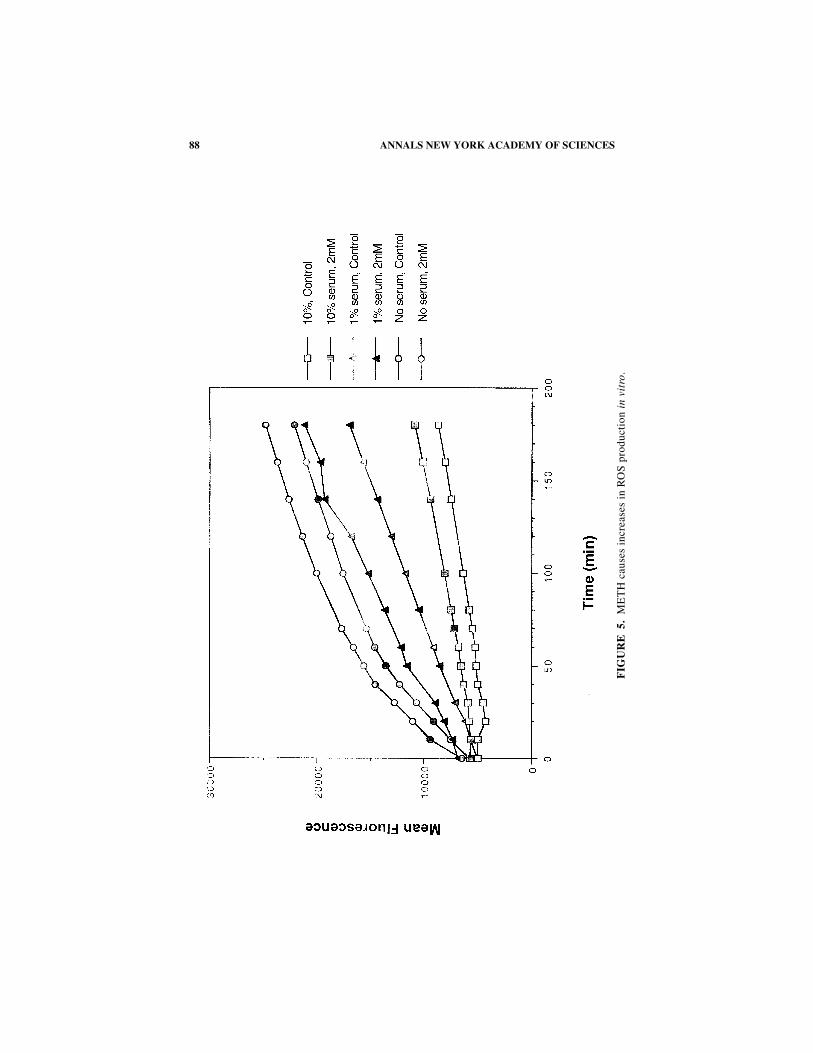
88 ANNALS NEW YORK ACADEMY OF SCIENCES
FIG
UR
E5.
ME
TH
cau
ses
incr
ease
s in
RO
S p
rodu
ctio
n in
vit
ro.

89CADET & ORDONEZ: SERUM WITHDRAWAL
fects of the drug. For example, in 10% serum, the toxic effects of METH seem toplateau, whereas there was a marked shifting of the dose-response curve within the1% and no serum conditions (FIG. 1).
FIGURE 2 shows the results of the microscopic evaluation of the cells. Microscopicanalysis of drug-induced effects revealed that high doses of METH caused very eas-ily observable morphological changes in the cells. The differences between the 1%or no serum in contrast to the 10% serum were very apparent. Specifically, METH-induced toxicity was associated with the presence of vacuoles, cytoplasmicshrinkage, and nuclear dissolution. In order to determine if METH was causing celldeath through an apoptotic process, we used flow cytometry to seek evidence ofinternucleosomal breakdown of DNA. As previously reported using bothimmortalized3 and primarily cortical cultures,25 METH caused dose-dependentDNA breaks (FIGS. 3 and 4). These were exacerbated by serum withdrawal.
FIGURE 5 shows that METH exposure results in significant increases in ROS pro-duction in vitro.
DISCUSSION
The main findings in this paper are that METH can cause apoptosis in another im-mortalized neural cell. These observations support previous observations made byus3,6 and others.21,25 In addition, the present paper shows that these effects can beexacerbated by serum withdrawal. These observations suggest that the mechanismsinvolved in METH-induced toxicity in vivo might be comparable to the deleteriouseffects of serum withdrawal.8,10 For example, serum withdrawal has also beenshown to cause cell death via apoptosis in a number of in vitro systems.10,20
Moreover, among other mechanisms this process might be dependent on the produc-tion of free radicals10,20 in a fashion similar to what is presently being reported fromMETH. Thus, the present study further supports the accumulating evidence that isdocumenting a role for free radicals in the toxic effects of METH (see Ref. 2 for areview).
Because these cells are not known to produce DA, this discussion implicates in-trinsic effects of METH itself or of metabolites in the production of reactive oxygenspecies, vacuoles, and the other METH-induced morphological changes in this im-mortalized cell line. This interpretation is consonant with findings that METH cancause cellular damage in areas of the brain that do not produce much dopamine.6,9,21
It is also consistent with a recent demonstration that amphetamine analogs can causeapoptosis in cultures of primary cortical cells that do not make DA.25 Thus, whentaken together with the convincing evidence that DA is involved in the toxicity ofMETH in some regions of the brain,16,27 the present observations suggest that theremight be both DA-dependent and DA-independent toxic effects of METH. While themechanisms involved remain to be clarified, the present results indicate that any ex-planation of the full effects of METH on the brain will have to take into consider-ation the biochemical pharmacology of METH itself.

90 ANNALS NEW YORK ACADEMY OF SCIENCES
REFERENCES
1. ASANUMA, M., H. HIRATA & J.L. CADET. 1998. Attenuation of 6-hydroxydopamine-induced dopaminergic nigrostriatal lesions in superoxide dismutase transgenic mice.Neuroscience 85: 907–917.
2. CADET, J.L. & C. BRANNOCK. 1998. Free radicals and the pathobiology of braindopamine systems. Neurochem. Int. 32: 117–131.
3. CADET, J.L., S.V. ORDONEZ & J.V. ORDONEZ. 1997. Methamphetamine induces apopto-sis in immortalized neural cells: protection by the proto-oncogene, bcl-2. Synapse25: 176–184.
4. CADET, J.L., P. SHENG, S. ALI, R. ROTHMAN, E. CARLSON & C. EPSTEIN. 1994. Attenuationof methamphetamine-induced neurotoxicity in copper/zinc superoxide dismutasetransgenic mice. J. Neurochem. 62: 380–383.
5. CADET, J.L. & C. BRANNOCK. 1998. Free radicals and the pathobiology of braindopamine systems. Neurochem. Int. 32: 117–131.
6. DENG, X., B. LADENHEIM, L.I. TSAO & J.L. CADET. 1999. Null mutation of c-fos causesexacerbation of methamphetamine-induced neurotoxicity. J. Neurosci. 19: 10107–10115.
7. DE VITO, M.J. & G.C. WAGNER. 1989. Methamphetamine-induced neuronal damage: apossible role for free radicals. Neuropharmacology 28: 1145–1150.
8. EDWARDS, S.N. & A.M. TOLKOVSKY. 1994. Characterization of apoptosis in cultured ratsympathetic neurons after nerve growth factor withdrawal. J. Cell Biol. 124: 537–546.
9. EISCH, A.J., L.C. SCHMUED & J.F. MARSHALL. 1998. Characterizing cortical neuroninjury with Fluoro-Jade labeling after a neurotoxic regimen of methamphetamine.Synapse 30: 329–333.
10. GREENLUND, L.J., T.L. DECKWORTH & E.M. JOHNSON. 1995. Superoxide dismutasedelays neuronal apoptosis: a role for reactive oxygen species in programmed neu-ronal death. Neuron 14: 303–315.
11. HIRATA, H. & J.L. CADET. 1997. p53-knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J.Neurochem. 69: 780–790.
12. HIRATA, H., M. ASANUMA & J.L. CADET. 1998. Melatonin attenuates methamphetamine-induced toxic effects on dopamine and serotonin terminals in mouse brain. Synapse30: 150–155.
13. ITZHAK, Y., J.I. MARTIN, M.D. BLAK & S.F. ALI. 1998. Effect of melatonin on meth-amphetamine- and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotox-icity and methamphetamine-induced behavioral sensitization. Neuropharmacology37: 781–791.
14. ITZHAK, Y., C. GANDIA, P.L. HUANG & S.F. ALI. 1998. Resistance of neuronal nitricoxide synthase-deficient mice to methamphetamine-induced dopaminergic neurotoxicity.J. Pharmacol. Exp. Ther. 284: 1040–1047.
15. JAYANTHI, S., B. LADENHEIM, A.M. ANDREWS & J.L. CADET. 1999. Overexpression ofhuman copper/zinc superoxide dismutase in trangenic mice attenuates oxidative stresscause by methylenedioxymethamphetamine (Ecstasy). Neuroscience 91: 1379–1387.
16. LAVOIE, M.J. & T.G. HASTINGS. 1999. Dopamine quinone formation and protein modi-fication associated with the striatal neurotoxicity of methamphetamine: evidenceagainst a role for extracellular dopamine. J. Neurosci. 19: 1484–1491.
17. LI, X. & Z. DARZYNKIEWICZ. 1995. Labeling DNA strand breaks with BrdUTP:detection of apoptosis and cell proliferation. Cell Proliferation 28: 572–579.
18. MILLER, M.A. 1991. Trends and patterns of methamphetamine smoking in Hawaii.NIDA Res. Monogr. 115: 72–83.
19. O'CALLAGHAN, J.P. & D.B. MILLER. 1994. Neurotoxicity profiles of substitutedamphetamines in the C57BL/6J mouse. J. Pharmacol. Exp. Ther. 270: 741–751.
20. PARK, D.S., E.J. MORRIS, L. STEFANIS, C.M. TROY, M.L. SHELANSKI, H.M. GELLER &L.A. GREENE. 1998. Multiple pathways of neuronal death induced by DNA-damagingagents, NGF deprivation, and oxidative stress. J. Neurosci. 18: 830–840.
21. PU, C., H.W. BROENING & C.V. VORHEES. 1996. Effect of methamphetamine onglutamate-positive neurons in the adult and developing rat somatosensory cortex.Synapse 23: 328–334.

91CADET & ORDONEZ: SERUM WITHDRAWAL
22. RICAURTE, G.A., R.W. GUILLERY, L.S. SEIDEN, C.R. SCHUSTER & R.Y. MOORE. 1982.Dopamine nerve terminal degeneration produced by high doses of methylamphet-amine in the rat brain. Brain Res. 235: 93–103.
23. SHAW, K.P. 1999. Human methamphetamine fatalities in Tawian during 1991–1996. J.Forensic Sci. 44: 27–31.
24. SHENG, P., C. CERRUTI, S. ALI & J.L. CADET. 1996. Nitric oxide is a mediator of meth-amphetamine (METH)-induced neurotoxicity: in vitro evidence from primary cul-tures of mesencephalic cells. Ann. N.Y. Acad. Sci. 801: 174–186.
25. STUMM, G., J. SCHLEGEL, T. SCHAFER, C. WURZ, H.D. MENNEL, J.C. KRIEG & H. VEDDER.1999. Amphetamines induce apoptosis and regulation of bcl-x splice variants in neo-cortical neurons. FASEB J. 13: 1065–1072.
26. WILSON, J.M., K.S. KALASINSKY, A.I. LEVEY, C. BERGERON, G. REIBER, R.M. ANTHONY,G.A. SCHMUNK, K. SHANNAK, J.W. HAYCOCK & S.J. KISH. 1996. Striatal dopaminenerve terminal markers in human, chronic methamphetamine users. Nat. Med. 2:699–703.
27. YAMAMOTO, B.K. & W. ZHU. 1998. The effects of methamphetamine on the productionof free radicals and oxidative stress. J. Pharmacol. Exp. Ther. 287: 107–114.




















