
Revised recommendations for the management of Gaucher disease in children
12
REVIEW Revised recommendations for the management of Gaucher disease in children Paige Kaplan & Hagit Baris & Linda De Meirleir & Maja Di Rocco & Amal El-Beshlawy & Martina Huemer & Ana Maria Martins & Ioana Nascu & Marianne Rohrbach & Lynne Steinbach & Ian J. Cohen Received: 18 November 2011 / Accepted: 5 June 2012 / Published online: 8 July 2012 # Springer-Verlag 2012 Abstract Gaucher disease is an inherited pan-ethnic disor- der that commonly begins in childhood and is caused by deficient activity of the lysosomal enzyme glucocerebrosi- dase. Two major phenotypes are recognized: non- neuropathic (type 1) and neuropathic (types 2 and 3). Symp- tomatic children are severely affected and manifest growth retardation, delayed puberty, early-onset osteopenia, signif- icant splenomegaly, hepatomegaly, thrombocytopenia, ane- mia, severe bone pain, acute bone crises, and fractures. Symptomatic children with types 1 or 3 should receive enzyme replacement therapy, which will prevent debilitating and often irreversible disease progression and allow those with non-neuropathic disease to lead normal healthy lives. Children should be monitored every 6 months (physical exam including growth, spleen and liver volume, neurologic exam, hematologic indices) and have one to two yearly skeletal assessments (bone density and imaging, preferably with magnetic resonance, of lumbar vertebrae and lower limbs), with specialized cardiovascular monitoring for some type 3 patients. Response to treatment will determine the frequency of monitoring and optimal dose of enzyme re- placement. Treatment of children with type 2 (most severe) neuropathic Gaucher disease is supportive. Pre-symptomatic children, usually with type 1 Gaucher, increasingly are P. Kaplan (*) Children’ s Hospital of Philadelphia, University of Pennsylvania, 9th Floor, Colket Translational Research Building, Civic Center Blvd, Philadelphia, PA 19104, USA e-mail: [email protected] H. Baris Beilinson-Schneider Gaucher Clinic, Rabin Medical Center, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel L. De Meirleir Pediatric Neurology, UZ Brussels, Brussels, Belgium M. Di Rocco Rare Disease Unit, II Division of Pediatrics, Gaslini Institute, Genoa, Italy A. El-Beshlawy Department of Pediatrics, Cairo University School of Medicine, Cairo, Egypt M. Huemer Department of Pediatrics, LKH Bregenz, Bregenz, Austria A. M. Martins Center of Reference in Inborn Errors of Metabolism, Universidade Federal de Sao Paulo, Sao Paulo, Brazil I. Nascu Center of Genetic Diseases, Children’ s Hospital Emergency Cluj, Cluj, Romania M. Rohrbach Division of Metabolism, Connective Tissue Unit, University Children’ s Hospital, Zurich, Switzerland L. Steinbach Department of Radiology, University of California, San Francisco, CA, USA I. J. Cohen Beilinson-Schneider Gaucher Clinic, Department of Pediatric Hematology Oncology, Schneider Children’ s Medical Center of Israel and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel Eur J Pediatr (2013) 172:447–458 DOI 10.1007/s00431-012-1771-z
Transcript of Revised recommendations for the management of Gaucher disease in children

REVIEW
Revised recommendations for the management of Gaucherdisease in children
Paige Kaplan & Hagit Baris & Linda De Meirleir &
Maja Di Rocco & Amal El-Beshlawy & Martina Huemer &
Ana Maria Martins & Ioana Nascu &
Marianne Rohrbach & Lynne Steinbach & Ian J. Cohen
Received: 18 November 2011 /Accepted: 5 June 2012 /Published online: 8 July 2012# Springer-Verlag 2012
Abstract Gaucher disease is an inherited pan-ethnic disor-der that commonly begins in childhood and is caused bydeficient activity of the lysosomal enzyme glucocerebrosi-dase. Two major phenotypes are recognized: non-neuropathic (type 1) and neuropathic (types 2 and 3). Symp-tomatic children are severely affected and manifest growthretardation, delayed puberty, early-onset osteopenia, signif-icant splenomegaly, hepatomegaly, thrombocytopenia, ane-mia, severe bone pain, acute bone crises, and fractures.Symptomatic children with types 1 or 3 should receiveenzyme replacement therapy, which will prevent debilitatingand often irreversible disease progression and allow those
with non-neuropathic disease to lead normal healthy lives.Children should be monitored every 6 months (physicalexam including growth, spleen and liver volume, neurologicexam, hematologic indices) and have one to two yearlyskeletal assessments (bone density and imaging, preferablywith magnetic resonance, of lumbar vertebrae and lowerlimbs), with specialized cardiovascular monitoring for sometype 3 patients. Response to treatment will determine thefrequency of monitoring and optimal dose of enzyme re-placement. Treatment of children with type 2 (most severe)neuropathic Gaucher disease is supportive. Pre-symptomaticchildren, usually with type 1 Gaucher, increasingly are
P. Kaplan (*)Children’s Hospital of Philadelphia, University of Pennsylvania,9th Floor, Colket Translational Research Building,Civic Center Blvd,Philadelphia, PA 19104, USAe-mail: [email protected]
H. BarisBeilinson-Schneider Gaucher Clinic, Rabin Medical Center,Sackler Faculty of Medicine, Tel Aviv University,Tel Aviv, Israel
L. De MeirleirPediatric Neurology, UZ Brussels,Brussels, Belgium
M. Di RoccoRare Disease Unit, II Division of Pediatrics, Gaslini Institute,Genoa, Italy
A. El-BeshlawyDepartment of Pediatrics, Cairo University School of Medicine,Cairo, Egypt
M. HuemerDepartment of Pediatrics, LKH Bregenz,Bregenz, Austria
A. M. MartinsCenter of Reference in Inborn Errors of Metabolism,Universidade Federal de Sao Paulo,Sao Paulo, Brazil
I. NascuCenter of Genetic Diseases, Children’s Hospital Emergency Cluj,Cluj, Romania
M. RohrbachDivision of Metabolism, Connective Tissue Unit,University Children’s Hospital,Zurich, Switzerland
L. SteinbachDepartment of Radiology, University of California,San Francisco, CA, USA
I. J. CohenBeilinson-Schneider Gaucher Clinic, Department of PediatricHematology Oncology, Schneider Children’s Medical Center ofIsrael and Sackler Faculty of Medicine, Tel Aviv University,Tel Aviv, Israel
Eur J Pediatr (2013) 172:447–458DOI 10.1007/s00431-012-1771-z

being detected because of affected siblings and screening inhigh-prevalence communities. In this group, annual exami-nations (including bone density) are recommended. How-ever, monitoring of asymptomatic children with affectedsiblings should be guided by the age and severity of mani-festations in the first affected sibling. Treatment is necessaryonly if signs and symptoms develop. Conclusion: Earlydetection and treatment of symptomatic types 1 and 3Gaucher disease with regular monitoring will optimize out-come. Pre-symptomatic children require regular monitoring.Genetic counseling is important.
Keywords Gaucher disease type 1 .Gaucher disease type 2 .
Gaucher disease type 3 . Glucocerebrosidase .
Glucocerebroside . Enzyme replacement therapy . Geneticcounseling . Monitoring . Disease management . Treatmentrecommendations
Introduction
Gaucher disease (GD) is a genetic pan-ethnic disorder af-fecting approximately 1:40,000 to 1:60,000 individualsglobally [17, 28, 49]. In Ashkenazi Jews, it is one of themost common genetic disorders, with a carrier frequency of1:17 [35]. It is inherited in an autosomal recessive mannerand is caused by deficiency of the lysosomal enzyme acidβ-glucosidase (glucocerebrosidase). This leads to accumu-lation of the substrate, glucosylceramide (glucocerebroside),in the lysosomes of macrophages and other cells such asosteoblasts [50]. These lipid-laden “Gaucher cells” accumu-late in various tissues and organs, particularly the spleen,liver, bone marrow (Fig. 1a), lung, and brain.
Although Gaucher disease manifests along a spectrum,typically it is classified clinically into three subtypes: type 1(non-neuropathic, MIM 230800), type 2 (acute neuropathic,MIM 230900), and type 3 (chronic neuropathic, MIM23100). In Europe, Canada, and the USA, the most preva-lent form (94 %) is GD type 1, characterized by the absenceof primary central nervous system involvement. In other
parts of the world such as Egypt, Japan, Sweden, andPoland, the neuropathic forms can have a higher prevalencethan GD type 1 [37, 64].
The signs and symptoms of non-neuropathic GD in chil-dren include splenomegaly, hepatomegaly, thrombocyto-penia, anemia, failure to thrive, growth retardation,delayed puberty, osteopenia, severe bone pain (acute bonecrises), and fractures [40]. In non-neuropathic GD, oftendescribed incorrectly as an adult disorder, the majority ofpatients manifest symptoms in childhood and are diagnosedbefore age 20 years [10]. Earlier onset correlates with moresevere disease and a high risk of morbid complications [38,47]. Seventy-five percent of untreated symptomatic childrenhave suboptimal growth [39], with 34–42 % being at orbelow the fifth percentile in height [1, 38]. Splenomegaly,hepatomegaly, and bone disease are each found in more than80 % of children at diagnosis [38]. In untreated children,median spleen volumes are more than 20 times the normalsize and median liver volumes are twice the normal size forthe child’s age and weight [1]. Puberty is significantlydelayed in ~60 % of adolescents with untreated GD [42],which can have a considerable psychological impact. Ane-mia and moderate to severe thrombocytopenia, both foundin approximately 40 % of children [38], are caused byhypersplenism. Anemia is a major cause of fatigue [38]and thrombocytopenia increases the tendency for bleeding(including frequent nose bleeds) and bruising [33].
Bone disease is caused by accumulation of Gaucher cells inthe bone marrow, glucocerebrosides in osteoblasts [50], andeffects of cytokines released from these cells. Approximately80 % of children with GD have at least one skeletal abnor-mality at diagnosis [38]. X-ray of the distal femur may showthe characteristic Erlenmeyer flask deformity (Fig. 1b). Somechildren experience chronic bone pain and/or bone crises(acute-onset, prolonged episodes of pain that are dull initiallybut become excruciating, and usually precede osteonecrosisand fractures). Bone mineral density (BMD), mass, and corti-cal bone thickness are abnormally low [47, 59].
GD types 2 and 3 represent a continuum of diseaseinvolvement distinguished mainly by the rate of neurologic
Fig. 1 a MRI showing marrowinfiltration. b X-ray showingErlenmeyer flask deformity
448 Eur J Pediatr (2013) 172:447–458

degeneration—rapid/acute in type 2 and chronic in type 3.Acute neuropathic GD (type 2) is very rare (1:500,000) [17]and is panethnic. Affected children may appear normal atbirth but manifest systemic and neurologic signs by 2 years(often much earlier) and deteriorate rapidly. Most childrendie in infancy, some prenatally, and others after a few years[31]. GD type 3 manifests in early childhood, often withsimilar features to type 1, and has a slowly progressivecourse often leading to death in the second and third deca-des. Neurological signs such as horizontal saccade-initiationfailure, inability to complete vertical gaze, abnormally slowobject tracking, and convergent squint usually develop earlyin childhood but can also develop later. A disease severityscore has been developed for GD type 3 [18] and at leastthree subtypes have been delineated. Patients with type 3ahave progressive neurologic disease with myoclonus anddementia. Patients with type 3b have significant visceraland skeletal disease, similar to severe type 1, but withneurologic signs (mainly horizontal supranuclear gaze pal-sy) that can present before or after somatic signs and symp-toms. Bone disease usually manifests as chest deformitiesand severe kyphoscoliosis without bone pain and crises[64]. Type 3c is characterized by variable neurologic signs(including hydrocephalus), corneal opacities, and cardiacvalvular and aortic calcification leading to congestive heartfailure and arrhythmias.
In symptomatic children with GD types 1 or 3, earlyenzyme replacement therapy can prevent the evolution ofserious, disabling, and irreversible somatic (but not neuro-logic) complications.
Guidelines on the diagnosis and management of non-neuropathic pediatric GD were published in 2004 [3, 29].Recently, a group of international Gaucher experts fromseveral medical specialities met to update these guidelinesand to expand them to include recommendations for chil-dren who are diagnosed pre-symptomatically and those withneuropathic disease.
Diagnosis of Gaucher disease in children
Symptomatic children
The diagnosis of GD in symptomatic children is based onmedical history, physical examination, and laboratory testsconfirmed by enzyme and/or gene assay.
History should encompass abdominal swelling and dis-comfort, early satiety for food, fatigue, bruising or bleedingtendency, growth deceleration and/or retardation, bone pain,and poor school performance. Family history should includeinquiries about the presence of siblings with the disease,consanguinity, ethnicity, and close relatives with hyper-splenism, splenectomy, and the presence of diseases
associated with GD such as early onset Parkinson-like dis-ease and Alzheimer disease [25, 36, 62].
Physical examinations assess growth, abdomen, skin(pallor, bruises, and petechiae), musculoskeletal and neuro-logical systems, heart, and lungs. Palpation of the abdomenshould start in the inguinal area so as not to overlook theedge of a massively enlarged spleen and liver. The absenceof neurological signs in early childhood does not rule outneurological disease because eye signs can also developlater in childhood.
A blood count should be performed and is likely to showanemia, thrombocytopenia, and, possibly, leucopenia. Un-related factor XI deficiency, common in Ashkenazi Jews,may also be present.
The definitive diagnosis is made by measurement of acidβ-glucosidase activity levels in total white cells, mononu-clear cells, fibroblasts, or dried blood spots on filter paper[6, 53], and/or molecular assay of the gene. Bone marrowaspiration and/or biopsy are invasive and are not requiredfor diagnosis [12]. Determination of the subtype in GD type3 is important to guide monitoring of disease progression.
Asymptomatic children
Increasingly, children are being diagnosed with GD pre-symptomatically (through enzyme testing or molecularanalysis) by genetic screening of their parents, prenatal/neonatal diagnosis in the Ashkenazi Jewish community,or because of relatives with GD. Newborn screening forlysosomal disorders will reveal other asymptomatic children.
Molecular analysis and genetic counseling
Molecular analysis of the acid β-glucosidase (GBA) gene isrecommended in every child with confirmed GD (includingthose diagnosed presymptomatically by enzyme testing) tohelp differentiate non-neuropathic (type 1) from chronicneuropathic (type 3), which can be clinically similar in earlychildhood. Although genotype–phenotype correlations arenot absolute, at least one N370S mutation protects againstneuropathic disease, whereas at least one L444P mutationtends to increase disease severity. The majority of patientsdiagnosed by Ashkenazi Jewish community screening willhave N370S mutations. Homozygosity or compound hetero-zygosity for L444P and D409H is usually associated with aneuropathic phenotype; evidence in Brazilian patients alsosuggests that a G377S mutation can be associated withneuropathic disease [6, 27, 61]. Knowledge of the genotypecan be used for family screening and prenatal diagnosis.
Parents should be offered genetic counseling by a pro-vider with experience in Gaucher disease. For those diag-nosed through community screening, counseling should be
Eur J Pediatr (2013) 172:447–458 449
given prior to pregnancy. Couples who are both carriersshould be advised that every child that they conceive to-gether will have a 25 % risk of having Gaucher disease. Fora couple who both carry N370S mutations, counseling aboutthe wide range of symptoms and ages at onset is extremelyimportant [74].
Baseline assessments in confirmed cases
A thorough baseline assessment is essential for all childrendiagnosed with Gaucher disease, even those diagnosed pre-symptomatically (Table 1). This includes blood tests, meas-urements of spleen and liver volume, and assessment ofbone disease.
Blood tests
Laboratory testing should include a complete blood count,liver and kidney function tests, and other biomarkers.Possible abnormalities include high immunoglobulin levels[70], low cholesterol levels, especially HDL cholesterol[15], prolonged prothrombin time (PT) and partial thrombo-plastin time (PTT) (found in 30 % of patients), and elevated
biomarkers (chitotriosidase, CCL18, tartrate-resistant acidphosphatase [TRAP], and angiotensin converting enzyme[ACE]). About 6 % of individuals do not express chitotrio-sidase (an enzyme secreted by activated macrophages thathydrolyses the trisaccharide derived from chitin) due to anull mutation in the gene; in some parts of the world such asTaiwan, this encompasses approximately 30 % of the pop-ulation [8]. In those GD patients, measurement of the alter-nate biomarkers ACE and TRAP or CCL18 is useful forongoing monitoring of disease manifestations and efficacyof therapeutic intervention [8, 34].
Before starting treatment, it is recommended that a base-line serum sample (3 mL) be obtained for storage in a bank(−70 °C) for future reference. This is invaluable to measureantibodies in case of an adverse reaction to enzyme treat-ment or for other tests/studies that may become relevantlater.
Spleen, liver, and bone assessments
Spleen and liver volume and infiltration should be measuredwith magnetic resonance imaging (MRI). If MRI is notavailable, ultrasonography (less accurate) can give someindication of organ size.
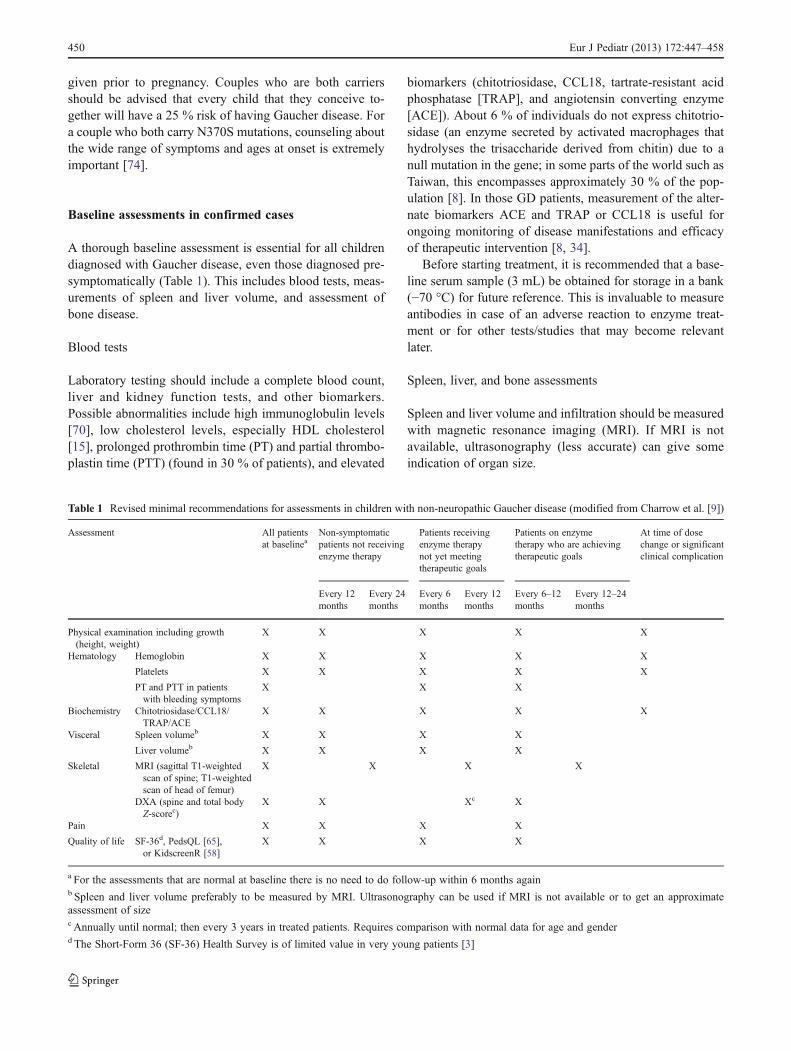
Table 1 Revised minimal recommendations for assessments in children with non-neuropathic Gaucher disease (modified from Charrow et al. [9])
Assessment All patientsat baselinea
Non-symptomaticpatients not receivingenzyme therapy
Patients receivingenzyme therapynot yet meetingtherapeutic goals
Patients on enzymetherapy who are achievingtherapeutic goals
At time of dosechange or significantclinical complication
Every 12months
Every 24months
Every 6months
Every 12months
Every 6–12months
Every 12–24months
Physical examination including growth(height, weight)
X X X X X
Hematology Hemoglobin X X X X X
Platelets X X X X X
PT and PTT in patientswith bleeding symptoms
X X X
Biochemistry Chitotriosidase/CCL18/TRAP/ACE
X X X X X
Visceral Spleen volumeb X X X X
Liver volumeb X X X X
Skeletal MRI (sagittal T1-weightedscan of spine; T1-weightedscan of head of femur)
X X X X
DXA (spine and total bodyZ-scorec)
X X Xc X
Pain X X X X
Quality of life SF-36d, PedsQL [65],or KidscreenR [58]
X X X X
a For the assessments that are normal at baseline there is no need to do follow-up within 6 months againb Spleen and liver volume preferably to be measured by MRI. Ultrasonography can be used if MRI is not available or to get an approximateassessment of sizec Annually until normal; then every 3 years in treated patients. Requires comparison with normal data for age and genderd The Short-Form 36 (SF-36) Health Survey is of limited value in very young patients [3]
450 Eur J Pediatr (2013) 172:447–458
Bone assessments ideally should include bone mineraldensity, best measured with dual-energy X-ray absorption(DXA) of the total body, lumbar spine and/or hips, and MRIof lumbar spine and femurs.
Treatment guidelines
Enzyme replacement therapy
Every child and adolescent with symptomatic GD (Table 2)should be treated with regular intravenous infusions ofenzyme replacement therapy. The dose should be individu-alized according to the clinical and molecular status of thepatient. The recombinant enzyme is available in over 80countries, including all of Europe. As it is very expensive,reimbursement procedures, schedules, and doses for initialtreatment differ from region to region. When possible, se-verely affected children should receive 60 U/kg per 2 weeks.For moderately affected children, at least 30 U/kg should begiven, and if treatment goals are not met, the dose should beincreased (Table 3). The child’s weight should be checked ateach assessment and the appropriate number of units perkilogram prescribed. A permanent indwelling venous cath-eter is not necessary unless there is recurring difficulty inaccessing the vein or if the child is extremely fearful despitepre-treatment with a topical anesthetic such as lidocaine.
Enzyme replacement results in the breakdown of storedglucosylceramide and improvement or normalization ofvisceral, growth, and bone manifestations. Most of the literature
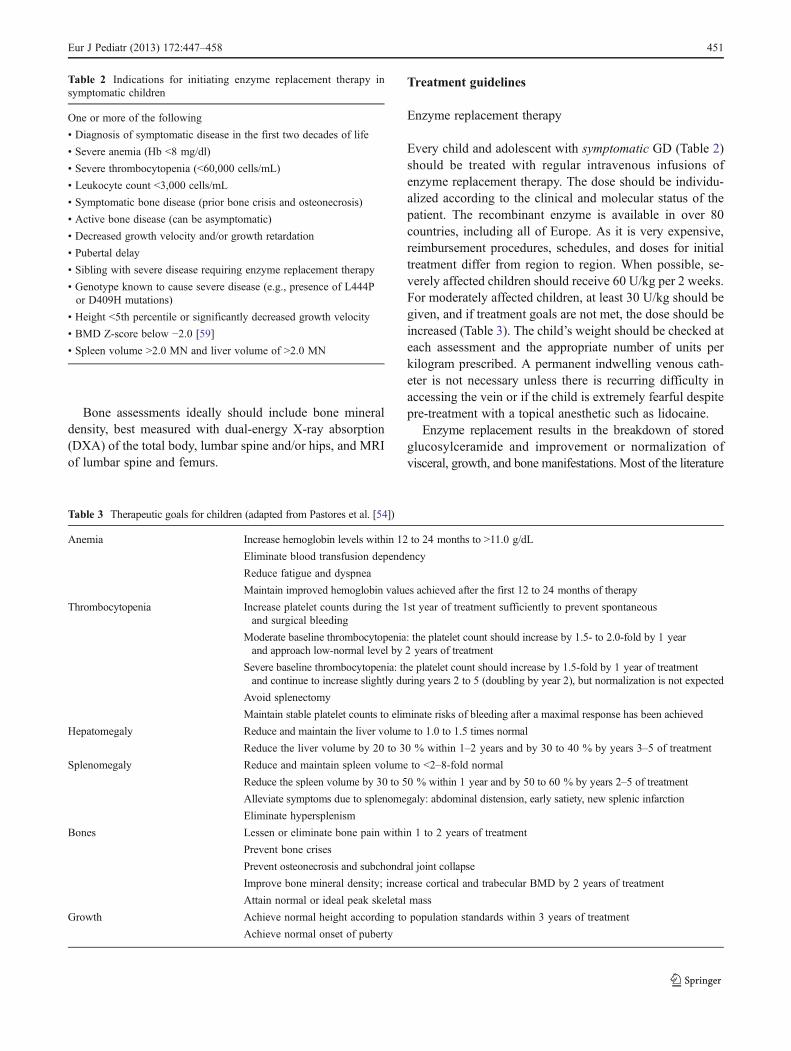
Table 2 Indications for initiating enzyme replacement therapy insymptomatic children
One or more of the following
• Diagnosis of symptomatic disease in the first two decades of life
• Severe anemia (Hb <8 mg/dl)
• Severe thrombocytopenia (<60,000 cells/mL)
• Leukocyte count <3,000 cells/mL
• Symptomatic bone disease (prior bone crisis and osteonecrosis)
• Active bone disease (can be asymptomatic)
• Decreased growth velocity and/or growth retardation
• Pubertal delay
• Sibling with severe disease requiring enzyme replacement therapy
• Genotype known to cause severe disease (e.g., presence of L444Por D409H mutations)
• Height <5th percentile or significantly decreased growth velocity
• BMD Z-score below −2.0 [59]
• Spleen volume >2.0 MN and liver volume of >2.0 MN
Table 3 Therapeutic goals for children (adapted from Pastores et al. [54])
Anemia Increase hemoglobin levels within 12 to 24 months to >11.0 g/dL
Eliminate blood transfusion dependency
Reduce fatigue and dyspnea
Maintain improved hemoglobin values achieved after the first 12 to 24 months of therapy
Thrombocytopenia Increase platelet counts during the 1st year of treatment sufficiently to prevent spontaneousand surgical bleeding
Moderate baseline thrombocytopenia: the platelet count should increase by 1.5- to 2.0-fold by 1 yearand approach low-normal level by 2 years of treatment
Severe baseline thrombocytopenia: the platelet count should increase by 1.5-fold by 1 year of treatmentand continue to increase slightly during years 2 to 5 (doubling by year 2), but normalization is not expected
Avoid splenectomy
Maintain stable platelet counts to eliminate risks of bleeding after a maximal response has been achieved
Hepatomegaly Reduce and maintain the liver volume to 1.0 to 1.5 times normal
Reduce the liver volume by 20 to 30 % within 1–2 years and by 30 to 40 % by years 3–5 of treatment
Splenomegaly Reduce and maintain spleen volume to <2–8-fold normal
Reduce the spleen volume by 30 to 50 % within 1 year and by 50 to 60 % by years 2–5 of treatment
Alleviate symptoms due to splenomegaly: abdominal distension, early satiety, new splenic infarction
Eliminate hypersplenism
Bones Lessen or eliminate bone pain within 1 to 2 years of treatment
Prevent bone crises
Prevent osteonecrosis and subchondral joint collapse
Improve bone mineral density; increase cortical and trabecular BMD by 2 years of treatment
Attain normal or ideal peak skeletal mass
Growth Achieve normal height according to population standards within 3 years of treatment
Achieve normal onset of puberty
Eur J Pediatr (2013) 172:447–458 451
on the effects of enzyme replacement therapy refers to resultsobtained with alglucerase (Ceredase®, Genzyme Corpora-tion)—macrophage-targeted placentally-derived human gluco-cerebrosidase, which became available in 1991—and itsrecombinant successor, imiglucerase (Cerezyme®, GenzymeCorporation), available since 1994. An alternate form of recom-binant human glucocerebrosidase, velaglucerase alfa (VPRIV®,Shire plc), became available in 2010 [22, 71]. A third recombi-nant form of human glucocerebrosidase, taliglucerase alfa(Pfizer/Protalix BioTherapeutics, Inc.) received approval fromthe US FDA in May 2012 [72].
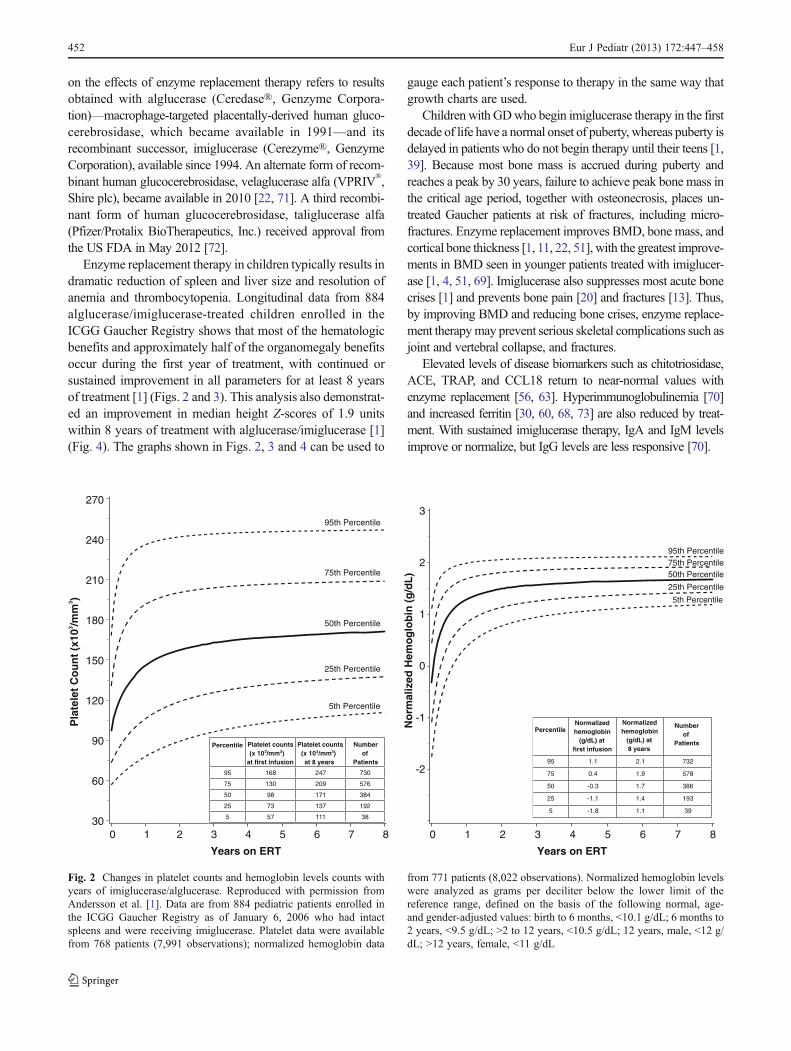
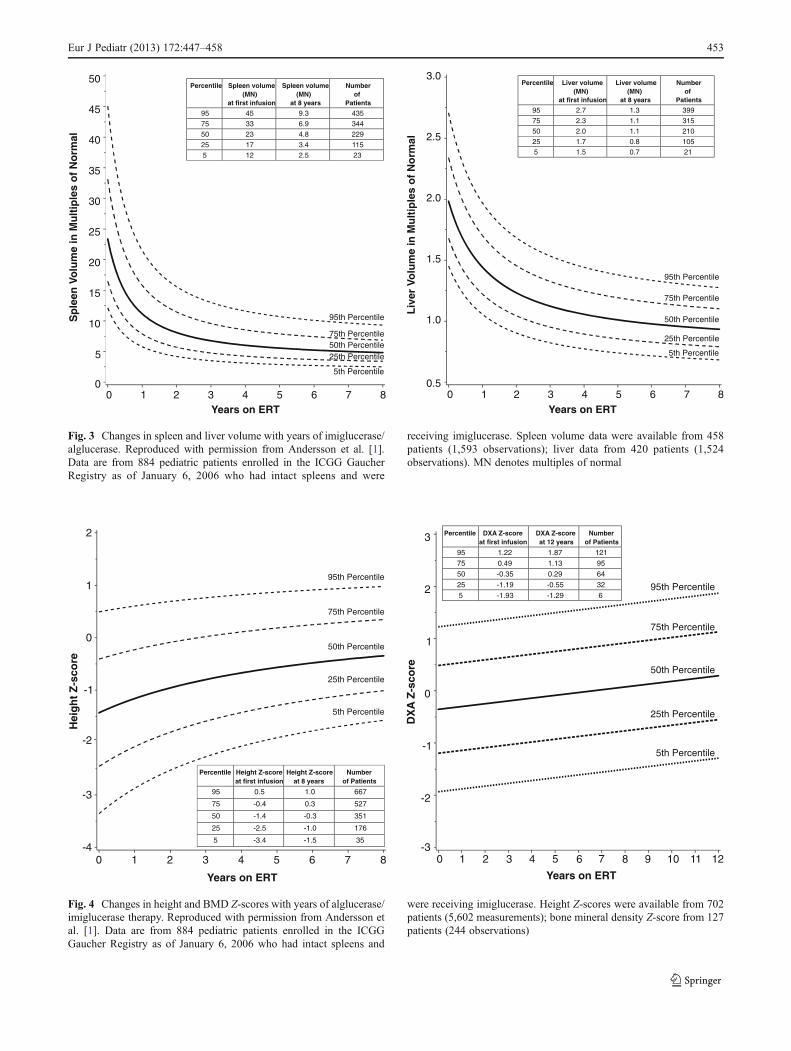
Enzyme replacement therapy in children typically results indramatic reduction of spleen and liver size and resolution ofanemia and thrombocytopenia. Longitudinal data from 884alglucerase/imiglucerase-treated children enrolled in theICGG Gaucher Registry shows that most of the hematologicbenefits and approximately half of the organomegaly benefitsoccur during the first year of treatment, with continued orsustained improvement in all parameters for at least 8 yearsof treatment [1] (Figs. 2 and 3). This analysis also demonstrat-ed an improvement in median height Z-scores of 1.9 unitswithin 8 years of treatment with alglucerase/imiglucerase [1](Fig. 4). The graphs shown in Figs. 2, 3 and 4 can be used to
gauge each patient’s response to therapy in the same way thatgrowth charts are used.
Children with GDwho begin imiglucerase therapy in the firstdecade of life have a normal onset of puberty, whereas puberty isdelayed in patients who do not begin therapy until their teens [1,39]. Because most bone mass is accrued during puberty andreaches a peak by 30 years, failure to achieve peak bone mass inthe critical age period, together with osteonecrosis, places un-treated Gaucher patients at risk of fractures, including micro-fractures. Enzyme replacement improves BMD, bone mass, andcortical bone thickness [1, 11, 22, 51], with the greatest improve-ments in BMD seen in younger patients treated with imiglucer-ase [1, 4, 51, 69]. Imiglucerase also suppresses most acute bonecrises [1] and prevents bone pain [20] and fractures [13]. Thus,by improving BMD and reducing bone crises, enzyme replace-ment therapymay prevent serious skeletal complications such asjoint and vertebral collapse, and fractures.
Elevated levels of disease biomarkers such as chitotriosidase,ACE, TRAP, and CCL18 return to near-normal values withenzyme replacement [56, 63]. Hyperimmunoglobulinemia [70]and increased ferritin [30, 60, 68, 73] are also reduced by treat-ment. With sustained imiglucerase therapy, IgA and IgM levelsimprove or normalize, but IgG levels are less responsive [70].
5th Percentile
50th Percentile
75th Percentile
95th Percentile
90
120
150
180
210
240
270
Years on ERT
60
25th Percentile
3010 32 54 86 7
33
Pla
tele
tC
ou
nt
(x10
/mm
)
Percentile Platelet counts (x 103/mm3)
at first infusion
Platelet counts (x 103/mm3) at 8 years
Numberof
Patients
95 168 247 730
75 130 209 576
50 98 171 384
25 73 137 192
5 57 111 38
No
rmal
ized
Hem
og
lob
in (
g/d
L)
-1
0
1
2
3
Years on ERT
5th Percentile
25th Percentile
50th Percentile
95th Percentile75th Percentile
-2
10 32 54 86 7
PercentileNormalized hemoglobin
(g/dL) at first infusion
Normalized hemoglobin
(g/dL) at 8 years
Numberof
Patients
95 1.1 2.1 732
75 0.4 1.9 578
50 -0.3 1.7 386
25 -1.1 1.4 193
5 -1.8 1.1 39
Fig. 2 Changes in platelet counts and hemoglobin levels counts withyears of imiglucerase/alglucerase. Reproduced with permission fromAndersson et al. [1]. Data are from 884 pediatric patients enrolled inthe ICGG Gaucher Registry as of January 6, 2006 who had intactspleens and were receiving imiglucerase. Platelet data were availablefrom 768 patients (7,991 observations); normalized hemoglobin data
from 771 patients (8,022 observations). Normalized hemoglobin levelswere analyzed as grams per deciliter below the lower limit of thereference range, defined on the basis of the following normal, age-and gender-adjusted values: birth to 6 months, <10.1 g/dL; 6 months to2 years, <9.5 g/dL; >2 to 12 years, <10.5 g/dL; 12 years, male, <12 g/dL; >12 years, female, <11 g/dL
452 Eur J Pediatr (2013) 172:447–458
Years on ERT
Sp
leen
Volu
me
inM
ult
iple
so
fN
orm
al
0
10
15
20
25
30
35
40
45
50
5
5th Percentile
50th Percentile75th Percentile
95th Percentile
25th Percentile
10 32 54 86 7
Percentile Spleen volume (MN)
at first infusion
Spleen volume (MN)
at 8 years
Numberof
Patients
95 45 9.3 435
75 33 6.9 344
50 23 4.8 229
25 17 3.4 115
5 12 2.5 23
0.5
1.5
2.0
2.5
3.0
Years on ERTL
iver
Volu
me
inM
ult
iple
so
fN
orm
al
1.0
5th Percentile
50th Percentile
75th Percentile
95th Percentile
25th Percentile
10 32 54 86 7
Percentile Liver volume (MN)
at first infusion
Liver volume (MN)
at 8 years
Numberof
Patients
95 2.7 1.3 399
75 2.3 1.1 315
50 2.0 1.1 210
25 1.7 0.8 105
5 1.5 0.7 21
Fig. 3 Changes in spleen and liver volume with years of imiglucerase/alglucerase. Reproduced with permission from Andersson et al. [1].Data are from 884 pediatric patients enrolled in the ICGG GaucherRegistry as of January 6, 2006 who had intact spleens and were
receiving imiglucerase. Spleen volume data were available from 458patients (1,593 observations); liver data from 420 patients (1,524observations). MN denotes multiples of normal
25th Percentile
50th Percentile
75th Percentile
5th Percentile
Hei
gh
tZ
-sco
re
-4
-3
-2
-1
0
1
2
Years on ERT
95th Percentile
0 32 54 86 7
1
Percentile Height Z-score at first infusion
Height Z-score at 8 years
Numberof Patients
95 0.5 1.0 667
75 -0.4 0.3 527
50 -1.4 -0.3 351
25 -2.5 -1.0 176
5 -3.4 -1.5 35
DX
AZ
-sco
re
-2
-1
0
1
2
3
-3
Years on ERT
10 11 1296 7 8541 2 30
5th Percentile
50th Percentile
75th Percentile
95th Percentile
25th Percentile
Percentile DXA Z-scoreat first infusion
DXA Z-score at 12 years
Numberof Patients
95 1.22 1.87 121
75 0.49 1.13 95
50 -0.35 0.29 64
25 -1.19 -0.55 32
5 -1.93 -1.29 6
Fig. 4 Changes in height and BMD Z-scores with years of alglucerase/imiglucerase therapy. Reproduced with permission from Andersson etal. [1]. Data are from 884 pediatric patients enrolled in the ICGGGaucher Registry as of January 6, 2006 who had intact spleens and
were receiving imiglucerase. Height Z-scores were available from 702patients (5,602 measurements); bone mineral density Z-score from 127patients (244 observations)
Eur J Pediatr (2013) 172:447–458 453

Approximately 7 % of patients have had recurrent adverseevents during or up to several days following an infusion [5].These hypersensitivity or non-allergic adverse events can beprevented by slowing and extending the infusion time and/orby pretreatment with antihistamines or corticosteroids. Based onextended experience with imiglucerase, a ramping schedule thatstarts with a very slow infusion rate and gradually increases hasprevented adverse reactions. Usually, children with GD areassessed and receive their (initial) courses of treatment in thehospital environment, where there is close monitoring.However, if there have been no adverse reactions to enzymereplacement therapy, treatment in the home environment after6–12 months has become an option in some countries and canincrease compliance and improve quality of life for patients andtheir families. The duration of hospital-based infusions beforeswitching patients to home infusion should be individualized foreach patient, but if no infusion related adverse events haveoccurred for 6–12 months, subsequent development of IgEantibodies is unlikely.
Treatment of acute bone crises
Bone crises can cause excruciating pain for up to severalweeks and are often resistant to intensive narcotic programs.Diagnosis can be made by MRI [16] or Technetium bonescans [41]. High-dose oral prednisone 1 gm/m2 for 2 daysfollowed by smaller doses of prednisone has been shown toalleviate the pain within several hours [14] and has no long-term side effects [16]. Non-steroidal anti-inflammatorydrugs may be used to treat non-specific pain. Radiographsare usually nondiagnostic in an acute crisis but may even-tually show periosteal elevation several weeks later [14].
Treatment of patients with neuropathic disease
There is no evidence that enzyme replacement therapy, even athigh doses, can prevent or slow neurological progression inpatients with type 2 or type 3 GD [7, 57]. Enzyme replacementtherapy is not recommended for type 2 GD. Managementshould be focused on supportive care [66]. Partial splenecto-my (surgical or thromboembolism) can alleviate some symp-toms in type 2 patients [55]. For children with type 3 GD,enzyme replacement therapy is recommended at a minimumdose of 60 U/kg per 2 weeks to ameliorate the severe visceralmanifestations [66]. Enzyme replacement therapy for patientswith type 3 GD is not approved in all countries. In theEuropean Union and Canada, imiglucerase is indicated foruse in patients with a confirmed diagnosis of type 3 GD whoexhibit clinically significant anemia, thrombocytopenia, bonedisease, hepatomegaly, or splenomegaly. In many othercountries, imiglucerase is not specifically indicated for type3 GD. At this time, velaglucerase alfa is approved for use onlyin type 1 GD patients.
Monitoring disease progression and treatment efficacy
Table 1 summarizes the recommended baseline and follow-up assessments for patients on treatment and patients whoare not on treatment. Treatment goals are shown in Table 3.With regard to quality of life assessments, note that these areimportant for parents/caregivers as well as for patients.
Monitoring symptomatic children
Physical examinations and hematologic assessments, includingbiomarkers, should be performedat diagnosis and every 6monthsthereafter. When treatment goals are met, the number and fre-quency of laboratory assessments can be decreased, but historyand physical exams still should be performed every 6 months.
Visceral assessments
In symptomatic children, volumetric MRI should be done every12–24 months, depending on the severity of organomegaly.When stable/normal volumes have been achieved, the MRI canbe done less frequently. Volumetric measures are reported asmultiples of normal (MN) size predicted for body weight. Inyounger children, oral or intravenous sedation is often necessaryduring the MRI. Because of the radiation dose involved, com-puterized tomography (CT) is no longer acceptable for routinemonitoring. Because of lack of sensitivity, ultrasound is notrecommended, unless MRI is unavailable. When ultrasound isused, it should not inform decisions to change drug doses, sinceorgan volumes can only be estimated, even by an experiencedradiologist [9, 21]. Manual measurements of the distance belowcostal margin in centimeters are also not very accurate.
Skeletal assessments
Bone disease can cause significant morbidity and long-termdisability [26]. The extent of bone disease does not alwayscorrelate with the spleen/liver enlargement, anemia, or throm-bocytopenia, so bone evaluations are required regardless ofinvolvement of other systems. Untreated children and youngadults between 10 and 20 years of age have significantly moresevere skeletal disease [38] and lower bone density than youngerchildren [59], reflecting the progressive nature of untreateddisease in childhood. Although not all children experienceosteonecrosis, osteopenia, and/or bone crises, these diseasemanifestations can have significant long-term sequelae, suchas joint collapse, fractures, and chronic pain. Splenectomy, acommon treatment in the pre-enzyme replacement era, has theunfortunate side effect of making the bone disease worse be-cause there is more accumulation of macrophages, chemokines,and cytokines [2].
Evaluating and monitoring the impact of skeletal diseasemust be comprehensive, and the treating physician should
454 Eur J Pediatr (2013) 172:447–458

regularly inquire about bone crises and bone pain. Physicalexamination should assess height, range of motion of joints,scoliosis, and deformities. Bone mineral density is best mea-sured with dual-energy X-ray absorption (DXA) of the totalbody, lumbar spine, and/or hips [69]. In children, bone mineraldensity must be expressed as an age- and gender-matchedZ-score, requiring appropriate software for data analysis.
In general, X-rays are not sufficiently sensitive to moni-tor disease progression, show changes too late and, if usedroutinely, expose the patient to excessive ionizing radiation.
MRI is the most widely used and accepted method formonitoring skeletal and marrow disease in GD and can be donewith sedation in very young children. MRI of the spine andfemur (the “gold standard”) gives information about abnormalremodelling, marrow infiltration, osteonecrosis, and osteoscle-rosis. InterpretingMRI during childhood requires knowledge ofthe conversion of hematopoietic (red) to fatty (yellow) marrowin the femur [52, 67] so that discrepancies from normal devel-opment can be observed. The infiltration of fatty marrow byGaucher cells can be observed with the Dixon quantitativechemical shift imaging (Dixon-QCSI), a special MRI tech-nique; however, norms for children have not been establishedand the technique is not available in most centers. The bonemarrow burden score obtained with MRI is a reproduciblesemiquantitative scoring system [48].
Growth velocity (height and weight) should be assessedevery 6 months in patients on treatment. MRI should bedone at baseline (time of diagnosis) and every subsequent12 months in patients on treatment as well as at the time ofany dose change. DXA should be performed in treatedchildren at baseline and then every 12–24 months until itis normal or if there is a dose change; then every 3 years(preferably in the same center with the same machine).
Monitoring asymptomatic children
Like symptomatic children, asymptomatic children who arediagnosed with GD because of affected siblings should bemonitored at least every 6 months. Since affected siblingsusually have a similar disease course, additional monitoringshould be guided by the age and severity of manifestationsin the older affected sibling(s).
Children diagnosed by community screening pose a man-agement dilemma, especially if they have an unknown ge-notype or one that predicts a more attenuated disease course.Such children could manifest symptoms of GD at any age oreven remain asymptomatic for their entire lives. At mini-mum, annual examinations are recommended. The frequen-cy of monitoring for infants detected in newborn screeningprograms will be determined by the genotype: for those witha “severe” genotype, assessments should be done every6 months; for those with a more benign genotype, includingN370S, assessments should be done annually.
Visceral and skeletal assessments
Ultrasound exams are acceptable for monitoring organome-galy in asymptomatic children only until clinical deteriorationmakes enzyme replacement therapy an option. MRI of bonesshould be done every 12 to 24 months (if available) and as ispractical, considering the need for sedation. DXA should bedone every 12 months, especially if MRI is not done.
Monitoring of neuropathic disease
Monitoring of patients with type 3 GD is dictated by thesubtype. For type 3a, the emphasis is on neurological involve-ment; for type 3b there is more emphasis on internal organs,hematological, and skeletal complications; and for type 3c, closecardiovascular follow-up is required. In addition to the standardtests performed in non-neuropathic GD patients, the followingtests should be performed: neurological examination includingeye movements, additional neuro-ophthalmological investiga-tions, testing of peripheral hearing, brain imaging, EEG, andneuro-psychometry. Avalidatedmodified severity score tomon-itor the clinical course is a useful tool [19]. In addition, type 3cpatients should also have periodic chest X-ray and echocardio-gram for calcification evaluation, cardiac CT angiography orcoronary artery catheterization evaluation for calcifications (andpossible atherosclerosis), and a periodic pulmonary evaluationfor interstitial lung disease.
Alternative treatment modalities
Miglustat (Zavesca®, Actelion Pharmaceuticals US) is anoral treatment for adult patients with GD type 1 that reducesthe production of the substrate glucosylceramide and otherglycosphingolipids. Miglustat is approved for symptomaticadult type 1 GD patients with mild to moderate clinicalmanifestations for whom enzyme therapy is not an option;however, it is not approved to treat children with GD.Isolated case reports in children with type 3 suggest it mighthelp decrease seizure frequency [44]. Side effects includeperipheral neuropathy and diarrhea [32].
An alternative oral substrate reduction therapy, eliglustattartrate (Genzyme, a Sanofi company, Cambridge, USA), spe-cifically inhibits production of glucosylceramide and is beingevaluated in clinical trials of adults with type 1 GD. A phase 2study showed statistically significant improvements in keysigns, including hemoglobin level, platelet count, spleen andliver volumes, and lumbar spine bone mineral density [45, 46].A pediatric investigational plan is under development to estab-lish the safety and efficacy of eliglustat in children with GD.
Significant morbidity and mortality limit the use of hemato-poietic stem cell transplantation (HSCT) in patients with GDtype 1. As enzyme replacement therapy is a highly efficacious
Eur J Pediatr (2013) 172:447–458 455

treatment for the hematological, visceral ,and skeletal manifes-tations of GD, this remains the first line of therapy. However,HSCT could have the theoretic advantage over enzyme therapyin stabilizing the neurological aspects of GD [24]. Studies ofgene therapy in animal models of GD show promise, but thisresearch is still preclinical [23, 43].
Conclusion
Gaucher disease usually manifests in childhood. To preventdevelopment of serious, irreversible disease, children withsymptomatic and pre-symptomatic Gaucher disease requirethorough, regularmulti-modalitymonitoring. Symptomatic chil-dren require enzyme replacement therapy. These revised guide-lines widen the spectrum of children with GD who should bemonitored at regular intervals to include those with neuropathicdisease and those who are diagnosed presymptomatically. En-hanced understanding of the natural history of childhood GDand long-term data on recombinant enzyme therapy outcomeshave led us to recommend more measurements of bone health(bone density measurements because of high prevalence of lowbone density) and growth, and to specify when dose modifica-tion should be considered.
Acknowledgments The meeting to formulate these recommendationswas funded by an unrestricted grant from the Genzyme Corporation. Gen-zyme also provided medical writing support from Frans van Huizen, Ph.D.and Lisa Underhill, MS, of Genzyme Global Medical Affairs. Genzyme hadno formative role in developing these management guidelines or in thedecision to submit the manuscript for publication. All views expressed andtreatment recommendations are solely those of the authors.
Conflict of interest disclosures Paige Kaplan has received grants andhonoraria from Genzyme. Hagit N. Baris has received grants and honorariafrom Genzyme. Linda De Merlier has received honoraria from Genzyme.Maja Di Rocco has received honoraria from Genzyme and Actelion. AmalEl Beshlawy has recieved honoraria from Genzyme. Martina Huemer hasreceived speaker honoraria from Genzyme. AnaMaria Martins has receivedsupport to attend congress and honoraria as a consultant from Genzyme.Ioana Nascu has received honoraria from Genzyme. M. Rohrbach hasreceived unrestricted grants and/or honoraria from Actelion, Genzyme, andShire. Lynne Steinbach has received honoraria fromGenzyme. Ian J. Cohenhas received a grant and honoraria from Genzyme.
References
1. Andersson H, Kaplan P, Kacena K, Yee J (2008) Eight-year clin-ical outcomes of long-term enzyme replacement therapy for 884children with Gaucher disease type 1. Pediatrics 122:1182–1190
2. Ashkenazi A, Zaizov R, Matoth Y (1986) Effect of splenectomyon destructive bone changes in children with chronic (Type I)Gaucher disease. Eur J Pediatr 145:138–141
3. Baldellou A, Andria G, Campbell PE, Charrow J, Cohen IJ,Grabowski GA, Harris CM, Kaplan P, McHugh K, Mengel E,Vellodi A (2004) Paediatric non-neuronopathic Gaucher disease:recommendations for treatment and monitoring. Eur J Pediatr163:67–75
4. Bembi B, Ciana G, Mengel E, Terk MR, Martini C, Wenstrup RJ(2002) Bone complications in children with Gaucher disease. Br JRadiol 75(Suppl 1):A37–A44
5. Beutler E, Grabowski G (2006) Gaucher disease. In: Scriver C,Beaudet A, Sly W, Valle D (eds) The metabolic and molecularbases of inherited disease. McGraw-Hill, New York, pp 3635–3668
6. Bodamer OA, Hung C (2010) Laboratory and genetic evaluationof Gaucher disease. Wien Med Wochenschr 160:600–604
7. Bove KE, Daugherty C, Grabowski GA (1995) Pathological find-ings in Gaucher disease type 2 patients following enzyme therapy.Hum Pathol 26:1040–1045
8. Chang KL, Hwu WL, Yeh HY, Lee NC, Chien YH (2009) CCL18as an alternative marker in Gaucher and Niemann-Pick diseasewith chitotriosidase deficiency. Blood Cells Mol Dis 44:38–40
9. Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P,Pastores G, Prakash-Cheng A, Rosenbloom BE, Scott CR,Wappner RS, Weinreb NJ (2004) Enzyme replacement therapyand monitoring for children with type 1 Gaucher disease: consen-sus recommendations. J Pediatr 144:112–120
10. Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P,Pastores G, Rosenbloom BE, Scott CR, Wappner RS, WeinrebNJ, Zimran A (2000) The Gaucher registry: demographics anddisease characteristics of 1698 patients with Gaucher disease.Arch Intern Med 160:2835–2843
11. Charrow J, Dulisse B, Grabowski GA, Weinreb NJ (2007) Theeffect of enzyme replacement therapy on bone crisis and bone painin patients with type 1 Gaucher disease. Clin Genet 71:205–211
12. Cohen IJ (2003) Gaucher's disease. Haema 6:262–26313. Cohen IJ, Katz K, Kornreich L, Horev G, Frish A, Zaizov R (1998)
Low-dose high-frequency enzyme replacement therapy preventsfractures without complete suppression of painful bone crises inpatients with severe juvenile onset type I Gaucher disease. BloodCells Mol Dis 24:296–302
14. Cohen IJ, Kornreich L, Mekhmandarov S, Katz K, Zaizov R(1996) Effective treatment of painful bone crises in type I gauch-er's disease with high dose prednisolone. Arch Dis Child 75:218–222
15. Cohen IJ, Yaniv I, Baris H (2010) Diagnosis of severe type 1Gaucher's disease before irreversible damage occurs: is HDL cho-lesterol the answer? Br J Haematol 150:118–119
16. Cohen IJ, Zaizov R (1998) Reply to a commentary By Elstein et al.On the paper by Cohen, Ij Et Al - Bcmd 24–296–302, 1998. BloodCells Mol Dis 24:306–308
17. Cox TM, Schofield JP (1997) Gaucher's disease: clinical featuresand natural history. Baillieres Clin Haematol 10:657–689
18. Davies EH, Mengel E, Tylki-Szymanska A, Kleinotiene G, ReinkeJ, Vellodi A (2011) Four-year follow-up of chronic neuronopathicGaucher disease in Europeans using a modified severity scoringtool. J Inherit Metab Dis 34:1053–1059
19. Davies EH, Mengel E, Tylki-Szymanska A, Kleinotiene G, ReinkeJ, Vellodi A (2011) Four-year follow-up of chronic neuronopathicGaucher disease in Europeans using a modified severity scoringtool. J Inherit Metab Dis
20. El-Beshlawy A, Ragab L, Youssry I, Yakout K, El-Kiki H, Eid K,Mansour IM, Abd El-Hamid S, Yang M, Mistry PK (2006)Enzyme replacement therapy and bony changes in Egyptianpaediatric Gaucher disease patients. J Inherit Metab Dis29:92–98
21. Elstein D, Abrahamov A, Hadas-Halpern I, Zimran A (2000)Withdrawal of enzyme replacement therapy in Gaucher's disease.Br J Haematol 110:488–492
22. Elstein D, Cohn GM, Wang N, Djordjevic M, Brutaru C, Zimran A(2011) Early achievement and maintenance of the therapeutic goalsusing velaglucerase alfa in type 1 Gaucher disease. Blood CellsMol Dis 46:119–123
456 Eur J Pediatr (2013) 172:447–458

23. Enquist IB, Nilsson E, Ooka A, Mansson JE, Olsson K, EhingerM, Brady RO, Richter J, Karlsson S (2006) Effective cell and genetherapy in a murine model of Gaucher disease. Proc Natl Acad SciU S A 103:13819–13824
24. Erikson A (2001) Remaining problems in the management ofpatients with Gaucher disease. J Inherit Metab Dis 24(Suppl2):122–126, discussion 187–128
25. Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M,Sidransky E (2008) The spectrum of parkinsonian manifestationsassociated with glucocerebrosidase mutations. Arch Neurol65:1353–1357
26. Grabowski G, Beutler E (2010) Gaucher disease. In: Scriver C,Beaudet A, Sly W et al (eds) The online metabolic and molecularbasis of inherited diseases (wwwommbidcom). McGraw-Hill, NewYork
27. Grabowski G, Kolodny E, Weinreb N, Rosenbloom B, Prakash-Cheng A, Kaplan P, Charrow J, Pastores G, Mistry P (2006)Gaucher disease: phenotypic and genetic variation. In: Scriver C,Beaudet A, Sly W et al (eds) The online metabolic and molecularbasis of inherited diseases (wwwommbidcom). McGraw-Hill, NewYork
28. Grabowski GA (2008) Phenotype, diagnosis, and treatment ofGaucher's disease. Lancet 372:1263–1271
29. Grabowski GA, Andria G, Baldellou A, Campbell PE, Charrow J,Cohen IJ, Harris CM, Kaplan P, Mengel E, Pocovi M, Vellodi A(2004) Pediatric non-neuronopathic Gaucher disease: presentation,diagnosis and assessment. Consensus statements. Eur J Pediatr163:58–66
30. Grabowski GA, Barton NW, Pastores G, Dambrosia JM, BanerjeeTK, McKee MA, Parker C, Schiffmann R, Hill SC, Brady RO(1995) Enzyme therapy in type 1 Gaucher disease: comparativeefficacy of mannose-terminated glucocerebrosidase from naturaland recombinant sources. Ann Intern Med 122:33–39
31. Gupta N, Oppenheim IM, Kauvar EF, Tayebi N, Sidransky E(2011) Type 2 Gaucher disease: phenotypic variation and genotyp-ic heterogeneity. Blood Cells Mol Dis 46:75–84
32. Hollak CE, Hughes D, van Schaik IN, Schwierin B, Bembi B(2009) Miglustat (Zavesca) in type 1 Gaucher disease: 5-yearresults of a post-authorisation safety surveillance programme.Pharmacoepidemiol Drug Saf 18:770–777
33. Hollak CE, Levi M, Berends F, Aerts JM, van Oers MH (1997)Coagulation abnormalities in type 1 Gaucher disease are due tolow- grade activation and can be partly restored by enzyme sup-plementation therapy. Br J Haematol 96:470–476
34. Hollak CE, van Weely S, van Oers MH, Aerts JM (1994) Markedelevation of plasma chitotriosidase activity. A novel hallmark ofGaucher disease. J Clin Invest 93:1288–1292
35. Horowitz M, Pasmanik-Chor M, Borochowitz Z, Falik-Zaccai T,Heldmann K, Carmi R, Parvari R, Beit-Or H, Goldman B, Peleg L,Levy-Lahad E, Renbaum P, Legum S, Shomrat R, Yeger H,Benbenisti D, Navon R, Dror V, Shohat M, Magal N, Navot N,Eyal N (1998) Prevalence of glucocerebrosidase mutations in theIsraeli Ashkenazi Jewish population. Hum Mutat 12:240–244
36. Hruska KS, Goker-Alpan O, Sidransky E (2006) Gaucher diseaseand the synucleinopathies. J Biomed Biotechnol 2006:78549
37. Jeong SY, Park SJ, Kim HJ (2011) Clinical and genetic character-istics of Korean patients with Gaucher disease. Blood Cells MolDis 46:11–14
38. Kaplan P, Andersson HC, Kacena KA, Yee JD (2006) The clinicaland demographic characteristics of nonneuronopathic Gaucherdisease in 887 children at diagnosis. Arch Pediatr Adolesc Med160:603–608
39. Kaplan P, Mazur A, Manor O, Charrow J, Esplin J, Gribble TJ,Wappner RS, Wisch JS, Weinreb NJ (1996) Acceleration of re-tarded growth in children with Gaucher disease after treatmentwith alglucerase. J Pediatr 129:149–153
40. Katz K, Cohen IJ, Ziv N, Grunebaum M, Zaizov R, Yosipovitch Z(1987) Fractures in children who have Gaucher disease. J BoneJoint Surg Am 69:1361–1370
41. Katz K, Mechlis-Frish S, Cohen IJ, Horev G, Zaizov R, Lubin E(1991) Bone scans in the diagnosis of bone crisis in patients whohave Gaucher disease. J Bone Joint Surg Am 73:513–517
42. Kauli R, Zaizov R, Lazar L, Pertzelan A, Laron Z, Galatzer A,Phillip M, Yaniv Y, Cohen IJ (2000) Delayed growth and pubertyin patients with Gaucher disease type 1: natural history and effectof splenectomy and/or enzyme replacement therapy. Isr Med AssocJ 2:158–163
43. Kim EY, Hong YB, Lai Z, Kim HJ, Cho YH, Brady RO, Jung SC(2004) Expression and secretion of human glucocerebrosidasemediated by recombinant lentivirus vectors in vitro and in vivo:implications for gene therapy of Gaucher disease. BiochemBiophys Res Commun 318:381–390
44. Kraoua I, Sedel F, Caillaud C, Froissart R, Stirnemann J, ChaurandG, Flodrops H, Tari S, Gourfinkel-An I, Mathieu S, Belmatoug N,Billette de Villemeur T, Mignot C (2011) A French experience oftype 3 Gaucher disease: phenotypic diversity and neurologicaloutcome of 10 patients. Brain Dev 33:131–139
45. Lukina E, Watman N, Arreguin EA, Banikazemi M, Dragosky M,Iastrebner M, Rosenbaum H, Phillips M, Pastores GM, RosenthalDI, Kaper M, Singh T, Puga AC, Bonate PL, Peterschmitt MJ(2010) A phase 2 study of eliglustat tartrate (Genz-112638), an oralsubstrate reduction therapy for Gaucher disease type 1. Blood116:893–899
46. Lukina E, Watman N, Arreguin EA, Dragosky M, Iastrebner M,Rosenbaum H, Phillips M, Pastores GM, Kamath RS, RosenthalDI, Kaper M, Singh T, Puga AC, Peterschmitt MJ (2010)Improvement in hematological, visceral, and skeletal manifesta-tions of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood116:4095–4098
47. Maas M, Hangartner T, Mariani G, McHugh K, Moore S,Grabowski GA, Kaplan P, Vellodi A, Yee J, Steinbach L (2008)Recommendations for the assessment and monitoring of skeletalmanifestations in children with Gaucher disease. Skeletal Radiol37:185–188
48. Maas M, van Kuijk C, Stoker J, Hollak CE, Akkerman EM, AertsJF, den Heeten GJ (2003) Quantification of bone involvement inGaucher disease: MR imaging bone marrow burden score as analternative to Dixon quantitative chemical shift MR imaging–ini-tial experience. Radiology 229:554–561
49. Meikle PJ, Hopwood JJ, Clague AE, Carey WF (1999) Prevalenceof lysosomal storage disorders. Jama 281:249–254
50. Mistry PK, Liu J, Yang M, Nottoli T, McGrath J, Jain D, Zhang K,Keutzer J, Chuang WL, Mehal WZ, Zhao H, Lin A, Mane S, LiuX, Peng YZ, Li JH, Agrawal M, Zhu LL, Blair HC, Robinson LJ,Iqbal J, Sun L, Zaidi M (2010) Glucocerebrosidase gene-deficientmouse recapitulates Gaucher disease displaying cellular and mo-lecular dysregulation beyond the macrophage. Proc Natl Acad SciU S A 107:19473–19478
51. Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR,Hangartner T (2011) Osteopenia in Gaucher disease develops earlyin life: response to imiglucerase enzyme therapy in children,adolescents and adults. Blood Cells Mol Dis 46:66–72
52. Moore SG, Dawson KL (1990) Red and yellow marrow in thefemur: age-related changes in appearance at MR imaging.Radiology 175:219–223
53. Muller KB, Rodrigues MD, Pereira VG, Martins AM, D'AlmeidaV (2010) Reference values for lysosomal enzymes activities usingdried blood spots samples—a Brazilian experience. Diagn Pathol5:65
54. Pastores GM, Weinreb NJ, Aerts H, Andria G, Cox TM, Giralt M,Grabowski GA, Mistry PK, Tylki-Szymanska A (2004)
Eur J Pediatr (2013) 172:447–458 457

Therapeutic goals in the treatment of Gaucher disease. SeminHematol 41:4–14
55. Pena AH, Kaplan P, Ganesh J, Clevac E, Marie Cahill A (2009)Partial splenic embolization in a child with Gaucher disease, mas-sive splenomegaly and severe thrombocytopenia. Pediatr Radiol39:1006–1009
56. Poll LW, Koch JA, Willers R, Aerts H, Scherer A, Haussinger D,Modder U, vom Dahl S (2002) Correlation of bone marrow re-sponse with hematological, biochemical, and visceral responses toenzyme replacement therapy of nonneuronopathic (type 1)Gaucher disease in 30 adult patients. Blood Cells Mol Dis28:209–220
57. Prows CA, Sanchez N, Daugherty C, Grabowski GA (1997)Gaucher disease: enzyme therapy in the acute neuronopathic var-iant. Am J Med Genet 71:16–21
58. Ravens-Sieberer U, Gosch A, Rajmil L, Erhart M, Bruil J, Duer W,Auquier P, Power M, Abel T, Czemy L, Mazur J, Czimbalmos A,Tountas Y, Hagquist C, Kilroe J, Kidscreen Group E (2005)KIDSCREEN-52 quality-of-life measure for children and adoles-cents. Expert Rev Pharmacoecon Outcomes Res 5:353–364
59. Robertson PL, Maas M, Goldblatt J (2007) Semiquantitative as-sessment of skeletal response to enzyme replacement therapy forGaucher's disease using the bone marrow burden score. AJR Am JRoentgenol 188:1521–1528
60. Rosenthal DI, Doppelt SH, Mankin HJ, Dambrosia JM, Xavier RJ,McKusick KA, Rosen BR, Baker J, Niklason LT, Hill SC et al(1995) Enzyme replacement therapy for Gaucher disease: skeletalresponses to macrophage-targeted glucocerebrosidase. Pediatrics96:629–637
61. Rozenberg R, Araujo FT, Fox DC, Aranda P, Nonino A, MichelettiC, Martins AM, Cravo R, Sobreira E, Pereira LV (2006) Highfrequency of mutation G377S in Brazilian type 3 Gaucher diseasepatients. Braz J Med Biol Res 39:1171–1179
62. Sidransky E (2005) Gaucher disease and parkinsonism. Mol GenetMetab 84:302–304
63. Stirnemann J, Belmatoug N, Vincent C, Fain O, Fantin B, MentreF (2010) Bone events and evolution of biologic markers inGaucher disease before and during treatment. Arthritis Res Ther12:R156
64. Tylki-Szymanska A, Vellodi A, El-Beshlawy A, Cole JA, Kolodny E(2010) Neuronopathic Gaucher disease: demographic and clinicalfeatures of 131 patients enrolled in the International CollaborativeGaucher Group Neurological Outcomes Subregistry. J Inherit MetabDis 33:339–346
65. Varni JW, Burwinkle TM, Seid M (2005) The PedsQL as a pedi-atric patient-reported outcome: reliability and validity of thePedsQL Measurement Model in 25,000 children. Expert RevPharmacoecon Outcomes Res 5:705–719
66. Vellodi A, Tylki-Szymanska A, Davies EH, Kolodny E, Bembi B,Collin-Histed T, Mengel E, Erikson A, Schiffmann R (2009)Management of neuronopathic Gaucher disease: revised recom-mendations. J Inherit Metab Dis 32:660–664
67. Waitches G, Zawin JK, Poznanski AK (1994) Sequence and rate ofbone marrow conversion in the femora of children as seen on MRimaging: are accepted standards accurate? AJR Am J Roentgenol162:1399–1406
68. Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH,Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS,Zimran A (2002) Effectiveness of enzyme replacement therapy in1028 patients with type 1 Gaucher disease after 2 to 5 years oftreatment: a report from the Gaucher Registry. Am J Med 113:112–119
69. Wenstrup RJ, Kacena KA, Kaplan P, Pastores GM, Prakash-ChengA, Zimran A, Hangartner TN (2007) Effect of enzyme replacementtherapy with imiglucerase on BMD in type 1 Gaucher disease. JBone Miner Res 22:119–126
70. Wine E, Yaniv I, Cohen IJ (2007) Hyperimmunoglobulinemia inpediatric-onset type 1 Gaucher disease and effects of enzymereplacement therapy. J Pediatr Hematol Oncol 29:451–457
71. Zimran A, Altarescu G, Philips M, Attias D, Jmoudiak M, DeebM, Wang N, Bhirangi K, Cohn GM, Elstein D (2010) Phase 1/2and extension study of velaglucerase alfa replacement therapy inadults with type 1 Gaucher disease: 48-month experience. Blood115:4651–4656
72. Zimran A, Brill-Almon E, Chertkoff R, Petakov M, Blanco-FavelaF, Munoz ET, Solorio-Meza SE, Amato D, Duran G, Giona F,Heitner R, Rosenbaum H, Giraldo P, Mehta A, Park G, Phillips M,Elstein D, Altarescu G, Szleifer M, Hashmueli S, Aviezer D (2011)Pivotal trial with plant cell-expressed recombinant glucocerebrosi-dase, taliglucerase alfa, a novel enzyme replacement therapy forGaucher disease. Blood 118:5767–5773
73. Zimran A, Elstein D, Levy-Lahad E, Zevin S, Hadas-Halpern I,Bar-Ziv Y, Foldes J, Schwartz AJ, Abrahamov A (1995)Replacement therapy with imiglucerase for type 1 Gaucher's dis-ease. Lancet 345:1479–1480
74. Zuckerman S, Lahad A, Zimran A, Levy-Lahad E, Sagi M (2008)Attitudes of couples identified through screening as carriers ofGaucher disease type 1. Clin Genet 74:566–570
458 Eur J Pediatr (2013) 172:447–458




















