
Calluna vulgaris as a Valuable Source of Bioactive Compounds
Upload
khangminh22Category
view
0download
0
Note: Within nine months of the publication of the mention of the grant of the European patent in the European PatentBulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with theImplementing Regulations. Notice of opposition shall not be deemed to have been filed until the opposition fee has beenpaid. (Art. 99(1) European Patent Convention).
Printed by Jouve, 75001 PARIS (FR)
(19)E
P1
976
870
B1
TEPZZ_97687ZB_T(11) EP 1 976 870 B1
(12) EUROPEAN PATENT SPECIFICATION
(45) Date of publication and mention of the grant of the patent: 05.12.2012 Bulletin 2012/49
(21) Application number: 07700383.8
(22) Date of filing: 17.01.2007
(51) Int Cl.:C07K 14/47 (2006.01) A61K 31/7036 (2006.01)
A61K 48/00 (2006.01) C12Q 1/68 (2006.01)
(86) International application number: PCT/GB2007/000109
(87) International publication number: WO 2007/083094 (26.07.2007 Gazette 2007/30)
(54) PREVENTION/TREATMENT OF ICHTHYOSIS VULGARIS, ATOPY AND OTHER DISORDERS
PRÄVENTION/BEHANDLUNG VON ICHTHYOSIS VULGARIS, ATOPIE UND ANDEREN ERKRANKUNGEN
PREVENTION/TRAITEMENT DE L’ICHTHYOSE VULGAIRE, DE L’ATOPIE ET D’AUTRES TROUBLES
(84) Designated Contracting States: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR
(30) Priority: 18.01.2006 GB 0600948
(43) Date of publication of application: 08.10.2008 Bulletin 2008/41
(73) Proprietor: The University Court of the University of DundeeDundee DD1 4HN (GB)
(72) Inventors: • MCLEAN, William Henry Irwin
Dundee DD1 9SY (GB)• SMITH, Frances Jan Dorothy
Dundee DD1 9SY (GB)
(74) Representative: Chapman, Paul Gilmour et alMarks & Clerk LLP Aurora 120 Bothwell StreetGlasgow G2 7JS (GB)
(56) References cited: WO-A-01/44516 WO-A-2004/010106WO-A-2005/063261 US-A- 6 143 502US-A1- 2005 014 835 US-B1- 6 482 839
• HEWETT DUNCAN R ET AL: "Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome." HUMAN MOLECULAR GENETICS 15 JAN 2005, vol. 14, no. 2, 15 January 2005 (2005-01-15), pages 335-346, XP002434224 ISSN: 0964-6906
• PANCHAL R G ET AL: "Partial functional correction of xeroderma pigmentosum group A cells by suppressor tRNA." HUMAN GENE THERAPY 1 SEP 1999, vol. 10, no. 13, 1 September 1999 (1999-09-01), pages 2209-2219, XP002434225 ISSN: 1043-0342 cited in the application
• SMITH FRANCES J D ET AL: "Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris" NATURE GENETICS, vol. 38, no. 3, March 2006 (2006-03), pages 337-342, XP002434226 ISSN: 1061-4036
• NIRUNSUKSIRI W ET AL: "Decreased profilaggrin expression in ichthyosis vulgaris is a result of selectively impaired posttranscriptional control." THE JOURNAL OF BIOLOGICAL CHEMISTRY 13 JAN 1995, vol. 270, no. 2, 13 January 1995 (1995-01-13), pages 871-876, XP002434227 ISSN: 0021-9258

EP 1 976 870 B1
2
5
10
15
20
25
30
35
40
45
50
55
Description
Field of the Invention
[0001] The present invention relates to the prevention/treatment of ichthyosis vulgaris (IV), atopy and potentiallyother disorders associated with loss-of-function muta-tions in the filaggrin gene sequence. The prevention/ther-apy is based on the use of agents which enable the host’stranslational machinery to read through a nonsense mu-tation found in a mutant allele of the filaggrin gene.
Background of the Invention
[0002] Ichthyosis vulgaris (IV; OMIM# 146700) is themost common inherited disorder of keratinisation andone of the most frequent single gene disorders in hu-mans. The most widely cited incidence figure is 1 in 250based on a survey of 6051 healthy Englishschoolchildren1. The association of IV with the atopic dia-thesis is well established; 37-50% of people with IV haveatopic diseases, in particular atopic dermatitis (ecze-ma)1,15 and conversely around 8% of atopic dermatitispatients have classical features of IV1,16.[0003] The phenotypic characteristics of IV include pal-mar hyperlinearity, keratosis pilaris and a fine scale mostmarkedly seen over the lower abdomen, arms and legs2.Filaggrin (filament aggregating protein) is important inthe formation of the stratum corneum3-5. Keratohyalingranules in the granular layer of interfollicular epidermisare predominantly composed of the 400 kDa protein pro-filaggrin. Following a short, unique N-terminal domain,most of the profilaggrin molecule consists of 10-12 re-peats of the 324 amino acid filaggrin sequence6. Uponterminal differentiation of granular cells, profilaggrin isproteolytically cleaved into ~37 kDa filaggrin peptidesand the N-terminal domain containing an S100-like cal-cium binding domain. Filaggrin rapidly aggregates thekeratin cytoskeleton, causing collapse of the granularcells into flattened anuclear squames. This condensedcytoskeleton is cross-linked by transglutaminases duringformation of the cornified cell envelope (CCE). The CCEis the outermost barrier layer of the skin which not onlyprevents water loss but also impedes the entry of aller-gens and infectious agents7. Filaggrin is therefore a keyprotein in facilitating epidermal differentiation and main-taining barrier function.[0004] Immunoblotting studies have shown that filag-grin protein was absent or markedly reduced in IV pa-tients’ skin and/or keratinocytes8-10. In addition, de-creased filaggrin mRNA has been demonstrated in someindividuals with IV11. A recessive mouse mutant, flakytail (ft), bears the histological and ultrastructural hall-marks of human IV12 and strong genetic linkage has beenobtained to the murine filaggrin locus (FLG)17,18. Al-though biochemical analysis has shown defective pro-filaggrin processing in ft/ft homozygotes12, any genomicmutation in the FLG gene has not hitherto been identified.
[0005] The present inventors have discovered certainloss of function mutations in the gene encoding filaggrinand that the consequence reduction/loss of filaggrin isassociated with the development of IV and other disor-ders such as atopic dermatitis (eczema), asthma, pso-riasis and/or allergies. This work is the subject of severalpapers which are in press (Smith F. J. D. et al., 2006;Palmer C. N. A. et al., 2006) and a co-pending patentapplication GB0525492.5.[0006] Apart from potential treatment by gene therapyapplications, it would be desirable if IV could be treatedusing small molecule drugs designed to overcome atleast some of the identified filaggrin mutations.[0007] Thus, it is amongst the objects of the presentinvention to provide means for preventing and/or treatingIV and other associated disorders.
Summary of the Invention
[0008] The present invention is based in part on workby the inventors in relation to the ability of certain agentsto allow read through of loss-of-function mutations in thefilaggrin gene.[0009] In a first aspect the present invention providesuse of an aminoglycoside antibiotic, negamycin or a mu-tant tRNA capable of enabling read through of a loss-of-function mutation in a filaggrin gene for the manufactureof a medicament for treating IV and/or associated dis-eases.[0010] A method of treating IV and/or other associateddiseases may comprising the step of administrating to asubject an agent which is capable of enabling read-through of a loss-of-function mutation in a filaggrin gene.[0011] It will be appreciated that the present inventionenables treatment of IV and/or to treating an animal sub-ject, especially a human subject who is predisposed todeveloping IV. IV, in severe or mild forms, may be asso-ciated with other diseases, for example atopic dermatitis(eczema), asthma, psoriasis or allergies, such as of acontact type allergy and food allergies (for example, pea-nut allergy). With regards to skin conditions, low levelsof filaggrin expression may lead to development of mildand/or sub-clinical disease. Indeed, many skin conditionsgo undiagnosed and as such treatments may be consid-ered more as a cosmetic treatment. Thus the presentinvention also extends to any such cosmetic therapies.[0012] The loss of function mutations which may beovercome are generally nonsense mutations. Such mu-tations are typically single base modifications which re-sult in the generation of a premature stop codon (i.e. TGA,TAG or TAA). Although the present inventors have iden-tified a number of mutations in the filaggrin gene, whichlead to a loss-of-function, one in particular leads to an in-frame generation of a premature stop codon. This muta-tion is a 1-base substitution at position 1501 of the FLGgene. The mutation is 1501C>T (numbering from initiat-ing ATG), which results in the substitution of a cytidineby a thymidine and a corresponding amino acid change
1 2

EP 1 976 870 B1
3
5
10
15
20
25
30
35
40
45
50
55
at position 501 of an arginine to a stop codon (R501X).As this mutation occurs in the first filaggrin repeat andresults in the generation of a stop codon, no functionalcopies of the filaggrin peptide are produced. Although,the inventors have identified other mutations in the filag-grin gene, this has so far been observed as the mostcommon in European Caucasian populations and it isovercoming of this mutation which is the preferred aspectof the present invention.[0013] The subject may be any subject requiring to betreated, prophylactically or therapeutically and may suit-ably be a newborn or even a foetus. The subject mayhowever be at any stage of life, and therefore includesneonates, children and adults.[0014] There are a number of known agents which areable to induce the read-through of nonsense mutations.One class of agents are certain aminoglycoside antibiot-ics, including gentamicin, paromomycin, neomycin andtobramycin (Bidou L. et al., Gene Therapy 11:619-627,2004; Howard MT., et al. Ann Neurol, 55:422-426, 2004).[0015] Another class of agents including negamycin,is described in US2005/0014835 which is hereby incor-porated by reference. Finally mutated tRNAs may begenerated as nonsense mutation suppressors. Theseare generated from mutant tRNA genes that result in thegeneration of tRNAs that have anticodons altered so thatthey have the ability to read through codons producedby nonsense mutations. This is described, for example,in Panchal RG et al., Human Gene Therapy, 10:2209-2219 (1999).[0016] In addition to the identified agents mentionedabove, there may be other suitable mutation suppressorsand the present invention also provides a method of iden-tifying agents for treating IV.[0017] Thus, in a further aspect there is provided amethod of identifying an agent for treating IV comprisingthe steps of:
a) providing a construct comprising a loss-of-func-tion mutant filaggrin gene joined in-frame to a report-er gene, wherein the loss-of-function mutant filaggringene comprises a stop codon;b) contacting a test agent with said construct; andc) detecting whether or not the test agent is capableof effecting read-through of stop codon in the mutantfilaggrin gene and expression of a reporter genewherein agents capable of effecting read-throughmay be used to treat IV.
[0018] It will be appreciated that the construct will beunder control of appropriate transcription control ele-ments such as promoter and terminator sequences.Moreover, a filaggrin nucleic acid sequence comprisingan internal nonsense sequence will be present in the con-struct. It is not necessary to use the entire filaggrin nucleicacid sequence, only a portion is required. Conveniently,the filaggrin nucleic acid sequence may be 10-1000 bpin length, more preferably 15-200 bp in length.
[0019] A typical mutant filaggrin gene/reporter geneconstruct comprises a 5’ mutant filaggrin nucleic acid se-quence joined in-frame to a 3’ reporter gene sequence.In this manner in order for any expression of the reportergene sequence to occur, there must be read-throughnonsense/stop codon located within the 5’ filaggrin se-quence.[0020] A preferred construct further comprises an ad-ditional positive control reporter gene 5’ of the filaggrinsequence. As will be appreciated all sequences arejoined in-frame with one another. For such a construct,the 5’ positive control reporter is provided so that a usercan ensure the construct is functioning appropriately. Ifno read-through of the filaggrin gene occurs, only thepositive reporter will provide a detectable signal. Howev-er, if read-through of the filaggrin sequence occurs, boththe positive control reporter and the reporter gene 3’ ofthe filaggrin sequence will provide detectable signals. Al-ternatively, the positive reporter may present in the con-struct under control of a separate promoter or, the posi-tive reporter may be encoded by a separate plasmidwhich is co-transfected[0021] Suitable reporter genes for use of the reportergene or positive control reporter gene are well known tothe skilled addressee and include, for example, a luci-ferase gene, β-galactosidase gene, fluorescent genes,such as green fluorescent protein or the like, chloram-phenicol acetyltransferase, β-glucuronidase and the like.Moreover, different versions of a particular gene may beobtained from different species of organism.[0022] A particularly preferred construct comprises therenilla luciferase gene as a positive control reporter, amutant filaggrin nucleic acid sequence and the firefly lu-ciferase gene as the reporter gene, under appropriatetranscriptional control using, for example, the HSV-TKpromoter and SV40-poly A terminator signal, as sche-matically shown in Figure 1. However, many other suit-able constructs can be envisaged and the reporters maybe reporters which only allow cells to survive in certaingrowth medium if the reporter is expressed.[0023] Detection of expression of any particular report-er can be carried out by techniques well known to theskilled reader.[0024] Following a first round of screening, it may beappropriate to test any possible useful agent, by testingthe agent on a cell or cell-line obtained from a patientsuffering from a disease associated with a nonsense mu-tation in order to ascertain/confirm that the agent is ableto cause read-through of the mutation.[0025] It will be appreciated that the method may becarried out in cell based or cell-free systems known inthe art.[0026] The present invention will now be further de-scribed by way of example and with reference to the Fig-ures which show:
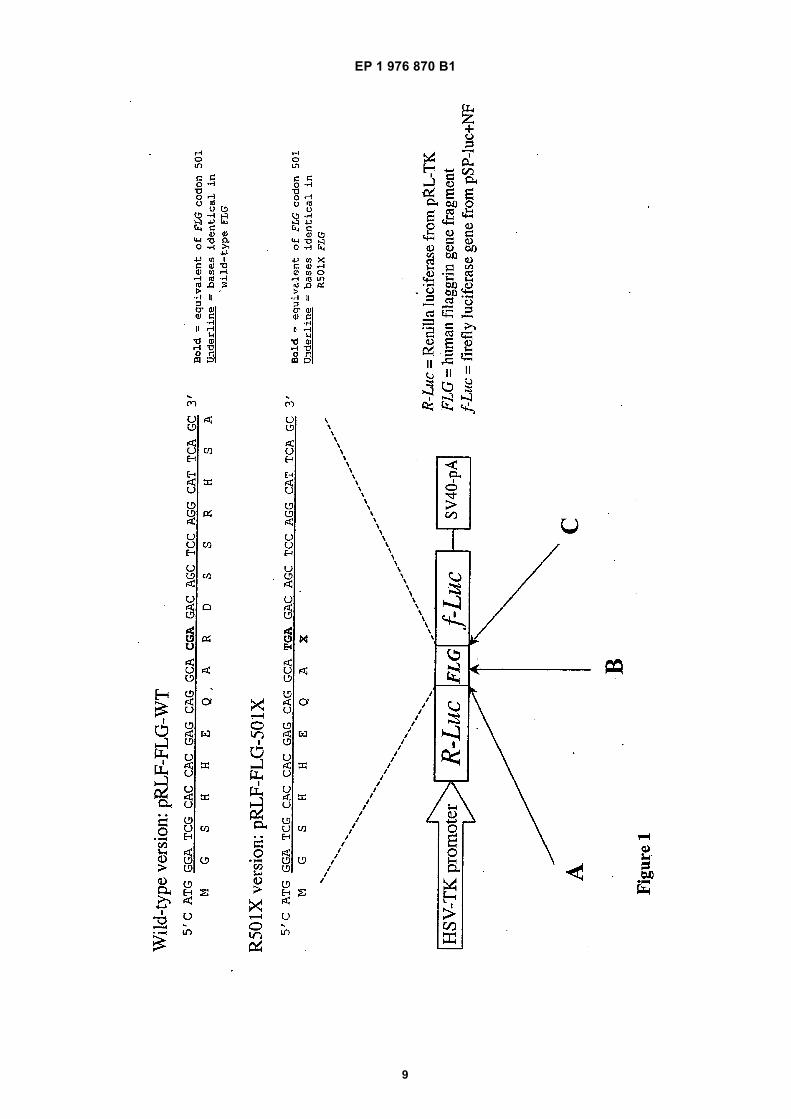
Figure 1 shows a construct for assay of filaggrin non-sense mutation read-through agents. A: TAA stop
3 4

EP 1 976 870 B1
4
5
10
15
20
25
30
35
40
45
50
55
codon mutated out of R-Luc in pRL-TK by 2bp dele-tion & Xba I site put in-frame with FLG and f-Luc; B:FLG oligo cassette cloned into Nco I site of pSP-luc+NF; C: ATG codon mutated out of f-Luc to reduce"leaky" expression.Figure 2 shows FLG oligonucleotide cassettes forcloning into pSP-luc+;Figure. 3 shows TR3 construct: human Arg(CGA)tRNA gene from chromosome 6p22.1;Figure 4 shows TR4B construct: human tRNA-Arg(TCG) gene from chromosome 15q26.1;Figure 5 shows the predicted secondary structuresof TR3 and TR4B suppressor tRNAs showing mu-tated anticodon loops. The anticodon loop is shownin bold and the G>A mutation which allows this topair with TGA codons is marked. Bases shown arepredicted from DNA and do not account for post-transcriptional modiciations;Figure 6 shows read-through of the filaggrin R501Xmutation (a) Epithelial cell line 293 was transientlytransfected with pRLF-FLG-WT or pRLF-FLG-501Xreporter plasmids. 48 hours after transfection, cellswere lysed and luciferase was assayed using thePromega Dual-Luciferase assay system, accordingto the manufacturer’s protocols.
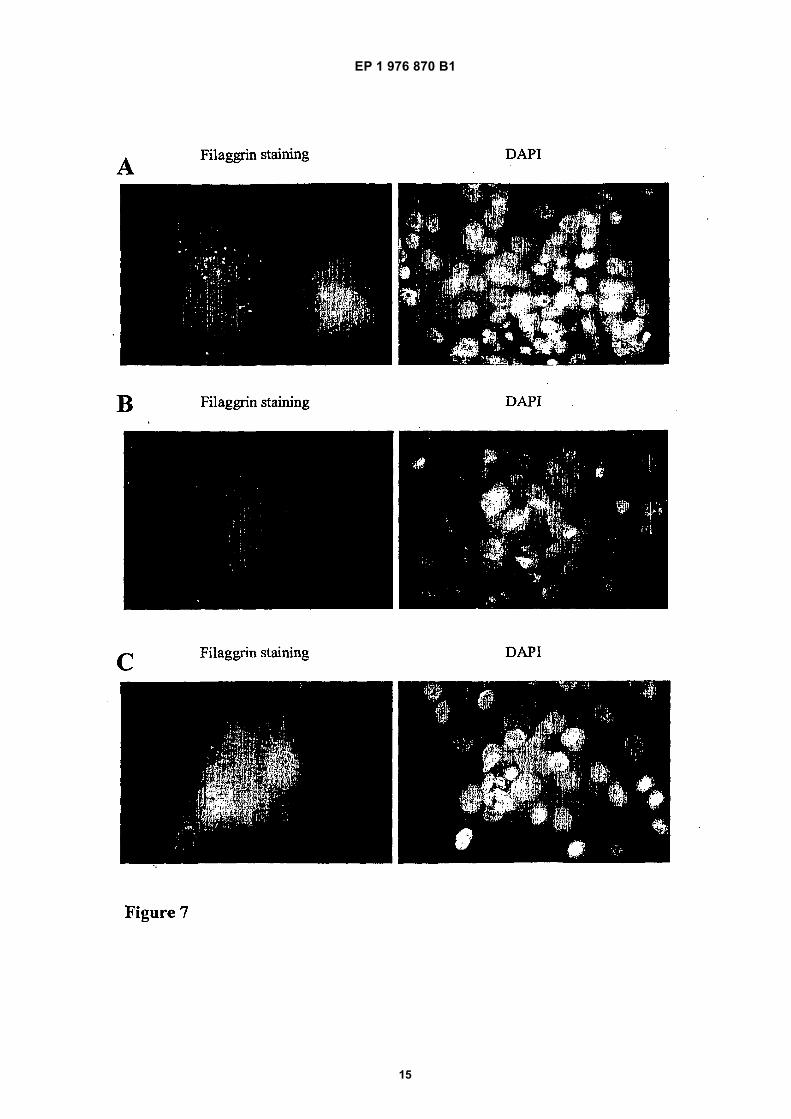
[0027] Untransfected cells gave neither Renilla or Fire-fly signal. The positive control construct pRLF-FLG-WT,containing the wild-type filaggrin sequence, gave positiveRenilla and Firefly signals. In contrast, the pRL-FLG-501X construct gave an equivalent Renilla signal but inthe absence of any read-through agents, gave no detect-able Firefly signal. This demonstrates that the pRLF-FLG-501X construct is not "leaky" and is therefore suit-able for assay or read-through agents.[0028] (b) The 293 cell-line was transiently transfectedwith pRLF-FLG-501X co-reporter containing a fragmentof the human filaggrin gene carrying the R501X mutationcloned in frame between the Renilla and Firefly luciferasegenes.[0029] Read-through activity was measured as Fireflyluciferase activity normalised against Renilla luciferaseactivity. Readings were done in quadruplicate and aver-aged.[0030] Suppressor tRNAs TR3 and TR4BsupTGA al-low read-through of filaggrin R501X nonsense mutation.Gentamicin allows read-through of the filaggrin R501Xmutation in a dose-dependent manner;[0031] Figure 7 shows that gentamicin induces re-ex-pression of filaggrin in keratinocytes from an ichthyosisvulgaris individual (FLG R501X homozygote).[0032] Primary keratinocytes were grown in serum-free KGM and were induced to stratify (differentiate) byshifting from low (0.09 mM) to high-calcium medium (1.89mM). Cultures were fixed using methanol/acetone andstained by indirect immunofluorescence using Novocas-tra monoclonal antibody 15C10 against the human filag-grin repeat sequence. Nucleic were counterstained using
1 mg/ml DAPI (4,6-diamidino-2-phenylindole); (a) normalcontrol keratinocytes express filaggrin upon differentia-tion, (b) R501X homozygote keratinocytes do not ex-press filaggrin upon differentiation, (c) R501X homozy-gote keratinocytes express filaggrin upon differentiationin presence of 600 mg/ml gentamicin (96 hrs incubationwith drug); and[0033] Figure 8 shows filaggrin expression from anR501X patient following gentamycin treatment. Incuba-tion of skin biopsy material from an R501X homozygotetpatient with marked ichthyosis vulgaris with 600 mg/mlgentamicin for 96 hours in organ culture restores filaggrinexpression (arrows). Following incubation, biopsies wereformalin-fixed, paraffin-embedded and processed for im-munohistochemistry using Novocastra anti-human filag-grin repeat monoclonal antibody 15C10 with immunoper-oxidase detection; A: Untreated biopsy; B: Gentamicintreatment
Methods and results
Example 1: Co-reporter gene construct for assay of readthrough agents
[0034] Oligonucleotide cassettes corresponding tobase numbers 1480 - 1523 of the human filaggrin gene,(FLG; numbering the coding sequence from the ATG in-itiation codon; Genbank accession number NM_002016.1), were cloned into the unique Nco I restrictionsite that spans the ATG codon of the firefly luciferasegene (f-Luc) in plasmid vector pSP-luc+NF (Promega).The oligo cassettes, shown in Figure 2, had overhangsadded corresponding to the cohesive ends of Nco I.These cassettes correspond to the region of the humanFLG gene containing codon 501, the site of the commonR501X mutation. Both wild-type and R501X mutant ver-sions were made. Clones with the insert in the correctorientation were identified and verified by DNA sequenc-ing. The ATG codon following insertion of the FLG cas-sette, i.e. the original initiation codon of the f-Luc gene,was mutated to an AGG arginine codon by site-directedmutagenesis with the following primers: FLmut1 5’ AGGCAT TCA GCC AGG GTC ACC GAC GCC 3’ and FLmut25’ GGC GTC GGT GAC CCT GGC TGA ATG CCT 3’(Stratagene QuikChange system). This was to preventpossible use of the original initiation codon, which mightlead to "leaky" expression of firefly luciferase even in thepresence of the FLG cassette containing a TGA or otherstop codon. Clones were verified by DNA sequencing.These clones were designated pSP-R501 and pSP-X501, corresponding to the wild-type and mutant ver-sions, respectively.[0035] The TAA termination codon of the Renilla luci-ferase gene (R-Luc) in plasmid vector pRL-TK (Promega)was mutated out by deletion of 2 bp (TA). This was doneusing by site-directed mutagenesis with the followingprimers: pRL.M1 5’ TCA AAA ATG AAC AAA TTC TAGAGC GGC C 3’ and pRL.M2 5’ GGC CGC TCT AGA ATT
5 6

EP 1 976 870 B1
5
5
10
15
20
25
30
35
40
45
50
55
TGT TCA TTT TTG A 3’ (Stratagene QuikChange sys-tem). The mutated clone (designated pRL-M1) was fullysequenced.[0036] To make the finished co-reporter constructs,Nhe I - Xba I fragments comprising the entire FLG-f-Lucfusion gene were excised from pSP-R501 and pSP-X501. These were cloned into the unique Xba I site ofpRL-M1, which was possible since Nhe I and Xba I havecompatible cohesive ends. Wild-type and R501X mutantclones with the correct orientation were selected by DNAsequencing. The wild-type and R501X mutant co-report-er constructs were designated as pRLF-FLG-WT andpRLF-FLG-501X, respectively (Figure 1).
Example 2: Generation of human suppressor tRNA genes for TGA nonsense mutations.
[0037] The human genome contains a number genesfor CGA-arginine t-RNAs. These are very compact genesand contain intragenic promoters for RNA polymerase III(Lewin B., In: Genes VII, Oxford University Press, 2000,pp.624-626). Two of these genes, which for convenience,were designated TR3 and TR4B, were identified in thecurrent assembly of the human genome (HG17; http://genome.ucsc.edu) and were amplified from normal hu-man control DNA. TR3 is located on chromosome 6p22.1and TR4B is located on chromosome 15q26.1. Specifi-cally, the TR3 gene was amplified as a 668 bp fragmentusing primers TR3.F 5’ CGA TGC AGA CAA TAT GCAGA 3’ and TR3.R 5’ CTA AGC CTA CAA AAC CGA AA3’, corresponding to base numbers chr6:26,407,555-26,408,284 in the HG17 assembly of the hu-man genome. The TR4B gene was amplified as a 465bp fragment using primers TR4B.F 5’ GAC TTC TGGGTG GGC TCT CC 3’ and TR4B.R 5’ CCC TCC CTCTCC ACT TTC CT 3’. This fragment corresponds to basenumbers chr15:87,679,164-87,679,628 in the HG17 as-sembly of the human genome. PCR was performed usingthe High-fidelity PCR system (Roche) and the followingconditions: (94°C 2 min)x1; (94°C 30 sec, X°C 30 sec,72°C 60 sec)x35; (72°C 5 min) x1. For TR3, annealingtemperature X = 55°C; for TR4B, X = 58°C. These frag-ments were cloned into the pCR2.1 vector (InVitrogen)and several clones sequenced to obtain a clone of eachconstruct free from PCR cloning artefacts. Annotated se-quences of the complete constructs are shown in Fig-ures 3 & 4. The predicted secondary structures for theresultant tRNA molecules are shown in Figure 5. Theanticodon loops of these tRNA genes were mutated sothat these would recognise the TGA stop codon insteadof the CGA arginine codon, i.e. the anticodon loop in thegene was mutated from 5’ TCG 3’ to 5’ TCA 3’. In theTR3 gene, this corresponded to the mutation G351A, ar-bitrarily numbering from the first base of primer TR3.F(above). In TR4B, this mutation corresponded to G180A,numbering from the first base of primer TR4B.F (above).
Example 3: Dual luciferase assay for FLG readthrough
[0038] Co-reporter constructs pRLF-FLG-WT andpRLF-FLG-501X were tested by transient transfection in-to 293 cells (transformed human kidney epithelial cellline) using the Fugene-6 system (Roche) in 96-wellplates. At 48 hours post-transfection, cells were subject-ed to the Dual luciferase assay system (Promega) whichdetects specific luciferase signals corresponding to bothRenilla and Firefly luciferase reporter genes. With thewild-type co-reporter, pRLF-FLG-WT, a strong signalwas obtained for both Renilla and Firefly luciferases, asshown in Figure 6a. In contrast, only a Renilla signal wasobtained with the pRLF-FLG-501X construct, showingthat the R501X premature termination codon mutationpresent within the FLG cassette in this construct leadsto complete loss of Firefly luciferase expression (Figure6a).
Example 4: Readthrough assay for FLG nonsense mutations
[0039] Transient transfections of 293 cells were donein 96-well plates as described above, using pRLF-FLG-501X with a range of gentamicin concentrations from 0to 2000 mg/ml. Positive control transfections were donein parallel using pRLF-FLG-WT. Filaggrin nonsense mu-tation readthrough activity was measured using the Dual-luciferase assay system (Promega), as described above.Figure 6b shows that gentamicin allows read-though ofthe R501X premature termination codon mutation in adose-dependent manner, with maximum readthrough ata concentration of 2000 mg/ml in this experiment. As analternative to using aminoglycosides for readthrough ofthe R501X mutation, human suppressor transfer-RNA(tRNA) species TR3supTGA and TR4BsupTGA weretested (see Example 2). These were co-transfected into293 cells with the pRLF-FLG-501X reporter plasmid de-scribed above using the Fugene-6 transient transfectionsystem (Roche). This showed that both suppressor tRNAspecies tested gave a strong read-through signal (Figure6b).
Example 5: Reactivation of filaggrin expression in keratinocyte culture.
[0040] Primary keratinocyte cultures were establishedfrom an individual with severe IV, who had been shownto be homozygous for the filaggrin R501X mutation, asdescribed in Smith FJD et al., Nature Genetics 2006, inpress. Normal primary keratinocytes do not express filag-grin to appreciable levels since the latter is a late-differ-entiation specific protein. However, these cells can beinduced to express filaggrin by shifting the cultures tohigh-calcium medium, which causes stratification and dif-ferentiation of keratinocytes, leading to expression offilaggrin (Smith FJD et al., 2006 in press). Differentiation
7 8

EP 1 976 870 B1
6
5
10
15
20
25
30
35
40
45
50
55
of normal control keratinocytes lead to profilaggrin ex-pression in colonies of cells that were well-stratified (Fig-ure 7). The protein was stained using indirect immun-ofluorescence with monoclonal antibody 15C10 (Novo-castra) against an epitope in the human filaggrin repeatpeptide. In contrast, well-stratified cultures from theR501X homozygote patient failed to express profilaggrin(Figure 7, see also Smith FJD et al., 2006, in press).However, when the differentiated cultures from theR501X homozygote patient were treated with 600 mg/mlgentamicin, well-stratified colonies of cells were seen toexpress profilaggrin at levels comparable to the normalcontrol, at 96 hours (Figure 7). Furthermore, the profilag-grin was present in the form of cytoplasmic granules,comparable to those seen in control keratinocytes. Thus,gentamicin is able to facilitate read-through of the R501Xfilaggrin mutation in cultured cells.
Example 6: Reactivation of epidermal filaggrin ex-pression in organ culture.
[0041] 3 mm cubes of skin biopsy material from a filag-grin R501X/R501X homozygous patient with marked IVwere incubated in Dulbecco’s modified Eagle mediumfor 4 days in culture, plus or minus 600 mg/ml gentamicin.Following incubation, the biopsy material was fixed andembedded for histology and immunohistochemistry. Theuntreated biopsy was completely negative when stainedwith monoclonal antibody 15C10 (Novacastra) againstan epitope in the human filaggrin repeat peptide (Figure8), consistent with homozygosity for filaggrin null muta-tions (Smith FJD et al., Nature Genetics paper, 2006, inpress). In contrast, the biopsy material incubated withgentamicin showed intense staining of the granular celllayers (Figure 8). Absence of histologically identifiablekeratohyalin granules is the hallmark of severe IV due tohomozygous or compound heterozygous filaggrin nullmutations (Smith FJD et al., Nature Genetics paper,2006, in press). Significantly, the recovery of filaggrinstaining seen here with gentamicin treatment, leads tothe de novo appearance of keratohyalin granules (Figure8). Furthermore, in some cells high in the granular layer,where filaggrin staining is particularly intense, the cellmorphology is seen to become more flattened (Figure8). Thus, the recovered protein expression appears tofacilitate the correct terminal differentiation of the epider-mis, consistent with full recovery of filaggrin protein func-tion.
REFERENCES
[0042]
1. Wells, R.S. and Kerr CB, Br Med J., 1:947-949(1966).
2. Judge, M.R., McLean, W.H.I. & Munro, C.S. Dis-orders of keratinization. in Rook’s Textbook of Der-
matology, Vol. 2 (eds. Burns, T., Breathnach, S.,Cox, C. & Griffiths, C.) 34.54-34.56 (Blackwell Sci-entific Publishing, Oxford, 2004).
3. Steinert, P.M., Cantieri, J.S., Teller, D.C., Lons-dale-Eccles, J.D. & Dale, B.A. Characterization of aclass of cationic proteins that specifically interactwith intermediate filaments. Proc Natl Acad Sci 78,4097-4101 (1981).
4. Dale, B.A., Resing, K.A. & Lonsdale-Ecccles, J.D.Filaggrin : a keratin filament associated protein. Ann.NY Acad. Sci. 455, 330-342 (1985).
5. Listwan, P. & Rothnagel, J.A. Keratin bundling pro-teins. Methods Cell Biol 78, 817-27 (2004).
6. Gan, S.Q., McBride, O.W., Idler, W.W., Markova,N. & Steinert, P.M. Organization, structure, and pol-ymorphisms of the human profilaggrin gene. Bio-chemistry 29, 9432-40 (1990).
7. Candi, E., Schmidt, R. & Melino, G. The cornifiedenvelope: a model of cell death in the skin. Nat RevMol Cell Biol 6, 328-40 (2005).
8. Fleckman, P., Holbrook, K.A., Dale, B.A. & Sybert,V.P. Keratinocytes cultured from subjects with ich-thyosis vulgaris are phenotypically abnormal. J In-vest Dermatol 88, 640-5 (1987).
9. Pena Penabad, C. et al. Differential patterns offilaggrin expression in lamellar ichthyosis. Br J Der-matol 139, 958-64 (1998).
10. Sybert, V.P., Dale, B.A. & Holbrook, K.A. Ichthy-osis vulgaris: identification of a defect in synthesisof filaggrin correlated with an absence of keratohy-aline granules. J Invest Dermatol 84, 191-4 (1985).
11. Nirunsuksiri, W., Zhang, S.H. & Fleckman, P.Reduced stability and bi-allelic, coequal expressionof profilaggrin mRNA in keratinocytes cultured fromsubjects with ichthyosis vulgaris. J Invest Dermatol110, 854-61 (1998).
12. Presland, R.B. et al. Loss of normal profilaggrinand filaggrin in flaky tail (ft/ft) mice: an animal modelfor the filaggrin-deficient skin disease ichthyosis vul-garis. J Invest Dermatol 115, 1072-81 (2000).
13. Smith, F.J.D. et al. "Loss-of-function mutationsin the filaggrin gene cause ichthyosis vulgaris", Na-ture Genetics, 2006, in press.
14. Palmer C.N.A. et al. "Haploinsufficiency for theepithelial barrier protein filaggrin is a major predis-posing factor for asthma and atopic dermatitis", sub-
9 10

EP 1 976 870 B1
7
5
10
15
20
25
30
35
40
45
50
55
mitted.
15. Presland, R.B. et al. Evidence for specific prote-olytic cleavage of the N-terminal domain of humanprofilaggrin during epidermal differentiation. J InvestDermatol 108, 170-8 (1997).
16. Ishida-Yamamoto, A., Takahashi, H., Presland,R.B., Dale, B.A. & Iizuka, H. Translocation of pro-filaggrin N-terminal domain into keratinocyte nucleiwith fragmented DNA in normal human skin and lor-icrin keratoderma. Lab Invest 78, 1245-53 (1998).
17. Lane, P.W. Two new mutations in linkage groupXVI of the house mouse. Flaky tail and varitint-wad-dler-J. J Hered 63, 135-40 (1972).
18. Rothnagel, J.A. et al. Characterization of themouse loricrin gene: linkage with profilaggrin and theflaky tail and soft coat mutant loci on chromosome3. Genomics 23, 450-6 (1994).
Claims
1. Use of an aminoglycoside antibiotic, negamycin ora mutant tRNA capable of enabling read through ofa loss-of-function mutation in a filaggrin gene for themanufacture of a medicament for treating ichthyosisvulgaris (IV).
2. The use of claim 1, wherein the loss of function mu-tation is a nonsense mutation.
3. The use of claim 2, wherein the mutation is 1501C>T(numbering from initiating ATG), which results in thesubstitution of a cytidine by a thymidine and a cor-responding amino acid change at position 501 of anarginine to a stop codon (R501X).
4. The use according to any preceding claim, whereinthe subject is a neonate, child or adults.
5. The use according to any preceding claim, whereinthe aminoglycoside is gentamicin, paromomycin, ne-omycin or tobramycin.
6. A method of identifying an agent for treating IV com-prising the steps of:
a) providing a construct comprising a loss-of-function mutant filaggrin gene joined in-frame toa reporter gene, wherein the loss-of-functionmutant filaggrin gene comprises a stop codon;b) contacting a test agent with said construct;andc) detecting whether or not the test agent is ca-pable of effecting read-through of the stop codon
in the mutant filaggrin gene and expression of areporter gene, wherein agents capable of effect-ing read-though may be used to treat IV.
7. The method according to claim 8, wherein the filag-grin nucleic acid sequence is 10-1000 bp in length.
8. The method according to claim 6 or 7, wherein themutant filaggrin gene/reporter gene construct com-prises a 5’ mutant filaggrin nucleic acid sequencejoined in-frame to a 3’ reporter gene sequence.
9. The method according to claim 8, wherein the con-struct further comprises an additional positive controlreporter gene 5’ of the filaggrin sequence.
10. The method according to claim 9, wherein the re-porter gene is luciferase gene, β-galactosidasegene, fluorescent genes, such as green fluorescentprotein, chloramphenicol acetyltransferase or β-glu-curonidase.
11. A method according to claim 10, wherein the con-struct is the pRLF-FLG-501X construct schematical-ly shown in Figure 1.
Patentansprüche
1. Verwendung eines Aminoglycosid-Antibiotikums,Negamycin, oder einer mutanten tRNA, die dasÜberlesen einer Funktionsverlust-Mutation in einemFilaggrin-Gen für die Herstellung eines Medika-ments zur Behandlung von Ichthyosis vulgaris (IV)ermöglichen kann.
2. Verwendung nach Anspruch 1, wobei die Funktions-verlust-Mutation einen Nonsense-Mutation ist.
3. Verwendung nach Anspruch 2, wobei die Mutation1501C>T ist (Nummerierung vom Starter-ATG aus),die zur Substitution eines Cytidins durch ein Thymi-din und zu einer entsprechenden Aminosäureände-rung in Position 501 eines Arginins zu einem Stopp-codon (R501X) führt.
4. Verwendung nach einem der vorstehenden Ansprü-che, wobei die Versuchsperson ein Neugeborenes,ein Kind oder Erwachsener ist.
5. Verwendung nach einem der vorstehenden Ansprü-che, wobei das Aminoglycosid Gentamicin, Paromo-mycin, Neomycin oder Tobramycin ist.
6. Verfahren zur Identifikation eines Wirkstoffs zur Be-handlung von Ichthyosis vulgaris (IV), wobei dasVerfahren die folgenden Schritte aufweist:
11 12

EP 1 976 870 B1
8
5
10
15
20
25
30
35
40
45
50
55
a) Bereitstellen eines Konstrukts, das ein unterFunktionsverlust mutiertes Filaggrin-Gen auf-weist, das unter Erhaltung des Leserasters miteinem Reportergen verbunden ist, wobei dasunter Funktionsverlust mutierte Filaggrin-Genein Stoppcodon aufweist;b) Inkontaktbringen eines Prüfmittels mit demKonstrukt; undc) Nachweisen, ob oder nicht das Prüfmittel einÜberlesen des Stoppcodons in dem mutantenFilaggrin-Gen und die Expression eines Repor-tergens bewirken kann, wobei Mittel, die dasÜberlesen bewirken können, zur Behandlungvon Ichthyosis vulgaris (IV) verwendet werdenkönnen.
7. Verfahren nach Anspruch 6, wobei die Filaggrin-Nu-cleinsäuresequenz eine Länge von 10-1000 bp auf-weist.
8. Verfahren nach Anspruch 6 oder 7, wobei das mu-tante Filaggrin-Gen/Reportergen-Konstrukt eine 5’-mutante Filaggrin-Nucleinsäuresequenz aufweist,die unter Erhaltung des Leserasters mit einer 3’-Re-portergensequenz verbunden ist.
9. Verfahren nach Anspruch 8, wobei das Konstruktferner ein zusätzliches Reportergen als Positivkon-trolle aufweist, das 5’-ständig zu der Filaggrin-Se-quenz ist.
10. Verfahren nach Anspruch 9, wobei das Reportergenein Luciferase-Gen, β-Galactosidase-Gen, Fluores-zenz-Gen, wie z. B. grün fluoreszierendes Protein,Chloramphenicolacetyltransferase oder β-Glucuro-nidase ist.
11. Verfahren nach Anspruch 10, wobei das Konstruktdas schematisch in Fig. 1 dargestellte pRLF-FLG-501X-Konstrukt ist.
Revendications
1. Utilisation d’un antibiotique d’aminoglycoside, de né-gamycine ou d’un ΔRNt mutant capable de permettreune lecture d’une mutation de perte de fonction dansun gène de filaggrine pour la fabrication d’un médi-cament pour le traitement d’une ichtyose vulgaire(IV).
2. Utilisation selon la revendication 1, dans laquelle lamutation de perte de fonction est une mutation nonsens.
3. Utilisation selon la revendication 2, dans laquelle lamutation est 1501C>T (numérotation à partir d’ATGd’initiation), qui conduit à la substitution d’une cyti-
dine par une thymidine et un changement d’acideaminé correspondant à la position 501 d’une arginineà un codon d’arrêt (R501X).
4. Utilisation selon l’une quelconque des revendica-tions précédentes, dans laquelle le sujet est un nou-veau-né, un enfant ou un adulte.
5. Utilisation selon l’une quelconque des revendica-tions précédentes, dans laquelle l’aminoglycosideest la gentamycine, la paromomycine, la néomycineou la tobramycine.
6. Procédé pour l’identification d’un agent pour le trai-tement d’une IV comprenant les étapes:
a) de fourniture d’une construction comprenantun gène de filaggrine mutant de perte de fonc-tion joint en cadre à un gène rapporteur, où legène de filaggrine mutant de perte de fonctioncomprend un codon d’arrêt;b) de mise en contact d’un agent d’essai avecladite construction; etc) de détection si oui ou non l’agent d’essai estcapable d’effectuer une lecture du codon d’arrêtdans le gène de filaggrine mutant et l’expressiond’un gène rapporteur, où des agents capablesd’effectuer une lecture peuvent être utilisés pourtraiter une IV.
7. Procédé selon la revendication 6, dans lequel la sé-quence d’acide nucléique de filaggrine est de10-1000 bp de longueur.
8. Procédé selon la revendication 6 ou 7, dans lequella construction de gène de filaggrine mutant/gènerapporteur comprend une séquence d’acide nucléi-que de filaggrine mutant 5’ jointe en cadre à uneséquence de gène rapporteur 3’.
9. Procédé selon la revendication 8, dans lequel laconstruction comprend en outre un gène rapporteurtémoin positif supplémentaire 5’ de la séquence defilaggrine.
10. Procédé selon la revendication 9, dans lequel le gè-ne rapporteur est un gène de luciférase, un gène deβ-galactosidase, des gènes fluorescents, tels qu’uneprotéine fluorescente verte, la chloramphénicol-acé-tyltransférase ou la β-glucuronidase.
11. Procédé selon la revendication 10, dans lequel laconstruction est la construction pRLF-FLG-501Xmontrée schématiquement dans la Figure 1.
13 14
EP 1 976 870 B1
9

EP 1 976 870 B1
10

EP 1 976 870 B1
11

EP 1 976 870 B1
12

EP 1 976 870 B1
13

EP 1 976 870 B1
14
EP 1 976 870 B1
15

EP 1 976 870 B1
16

EP 1 976 870 B1
17
REFERENCES CITED IN THE DESCRIPTION
This list of references cited by the applicant is for the reader’s convenience only. It does not form part of the Europeanpatent document. Even though great care has been taken in compiling the references, errors or omissions cannot beexcluded and the EPO disclaims all liability in this regard.
Patent documents cited in the description
• GB 0525492 A [0005] • US 20050014835 A [0015]
Non-patent literature cited in the description
• BIDOU L. et al. Gene Therapy, 2004, vol. 11,619-627 [0014]
• HOWARD MT. et al. Ann Neurol, 2004, vol. 55,422-426 [0014]
• PANCHAL RG et al. Human Gene Therapy, 1999,vol. 10, 2209-2219 [0015]
• LEWIN B. Genes. Oxford University Press, 2000, vol.VII, 624-626 [0037]
• SMITH FJD et al. Nature Genetics, 2006 [0040][0041]
• WELLS, R.S. ; KERR CB. Br Med J., 1966, vol. 1,947-949 [0042]
• Disorders of keratinization. JUDGE, M.R. ;MCLEAN, W.H.I. ; MUNRO, C.S. Rook’s Textbookof Dermatology. Blackwell Scientific Publishing,2004, vol. 2, 34.54-34.56 [0042]
• STEINERT, P.M. ; CANTIERI, J.S. ; TELLER, D.C. ;LONSDALE-ECCLES, J.D. ; DALE, B.A. Character-ization of a class of cationic proteins that specificallyinteract with intermediate filaments. Proc Natl AcadSci, 1981, vol. 78, 4097-4101 [0042]
• DALE, B.A. ; RESING, K.A. ; LONSDALE-EC-CCLES, J.D. Filaggrin : a keratin filament associatedprotein. Ann. NY Acad. Sci., 1985, vol. 455, 330-342[0042]
• LISTWAN, P. ; ROTHNAGEL, J.A. Keratin bundlingproteins. Methods Cell Biol, 2004, vol. 78, 817-27[0042]
• GAN, S.Q. ; MCBRIDE, O.W. ; IDLER, W.W. ;MARKOVA, N. ; STEINERT, P.M. Organization,structure, and polymorphisms of the human profilag-grin gene. Biochemistry, 1990, vol. 29, 9432-40[0042]
• CANDI, E., SCHMIDT, R. ; MELINO, G. The cornifiedenvelope: a model of cell death in the skin. Nat RevMol Cell Biol, 2005, vol. 6, 328-40 [0042]
• FLECKMAN, P. ; HOLBROOK, K.A. ; DALE, B.A. ;SYBERT, V.P. Keratinocytes cultured from subjectswith ichthyosis vulgaris are phenotypically abnormal.J Invest Dermatol, 1987, vol. 88, 640-5 [0042]
• PENA PENABAD, C. et al. Differential patterns offilaggrin expression in lamellar ichthyosis. Br J Der-matol, 1998, vol. 139, 958-64 [0042]
• SYBERT, V.P. ; DALE, B.A. ; HOLBROOK, K.A.Ichthyosis vulgaris: identification of a defect in syn-thesis of filaggrin correlated with an absence of kera-tohyaline granules. J Invest Dermatol, 1985, vol. 84,191-4 [0042]
• NIRUNSUKSIRI, W. ; ZHANG, S.H. ; FLECKMAN,P. Reduced stability and bi-allelic, coequal expres-sion of profilaggrin mRNA in keratinocytes culturedfrom subjects with ichthyosis vulgaris. J Invest Der-matol, 1998, vol. 110, 854-61 [0042]
• PRESLAND, R.B. et al. Loss of normal profilaggrinand filaggrin in flaky tail (ft/ft) mice: an animal modelfor the filaggrin-deficient skin disease ichthyosis vul-garis. J Invest Dermatol, 2000, vol. 115, 1072-81[0042]
• SMITH, F.J.D. et al. Loss-of-function mutations in thefilaggrin gene cause ichthyosis vulgaris. Nature Ge-netics, 2006 [0042]
• PALMER C.N.A. et al. Haploinsufficiency for the ep-ithelial barrier protein filaggrin is a major predisposingfactor for asthma and atopic dermatitis [0042]
• PRESLAND, R.B. et al. Evidence for specific prote-olytic cleavage of the N-terminal domain of humanprofilaggrin during epidermal differentiation. J InvestDermatol, 1997, vol. 108, 170-8 [0042]
• ISHIDA-YAMAMOTO, A. ; TAKAHASHI, H. ;PRESLAND, R.B. ; DALE, B.A. ; IIZUKA, H. Trans-location of profilaggrin N-terminal domain into kerat-inocyte nuclei with fragmented DNA in normal humanskin and loricrin keratoderma. Lab Invest, 1998, vol.78, 1245-53 [0042]
• LANE, P.W. Two new mutations in linkage group XVIof the house mouse. Flaky tail and varitint-waddler-J.J Hered, 1972, vol. 63, 135-40 [0042]
• ROTHNAGEL, J.A. et al. Characterization of themouse loricrin gene: linkage with profilaggrin and theflaky tail and soft coat mutant loci on chromosome 3.Genomics, 1994, vol. 23, 450-6 [0042]
Copyright © 2022 FDOKUMEN




















