
Phylogenetic Relationships of World Populations of Bemisia tabaci (Gennadius) Using Ribosomal ITS1
8
Phylogenetic Relationships of World Populations of Bemisia tabaci (Gennadius) Using Ribosomal ITS1 Paul J. De Barro, Felice Driver, John W. H. Trueman,* and John Curran CSIRO Entomology, G.P.O. Box 1700, Canberra, ACT 2601, Australia; and *Research School of Biological Sciences, Australian National University, Canberra, ACT 0200, Australia Received June 15, 1999; revised December 13, 1999 A phylogeny of Bemisia tabaci is presented based on the ITS1 region of ribosomal DNA. The monophyly of each biotype of Bemisia is supported, and a strong biogeographic pattern is evident in the data. Popula- tions from the Americas (including the A biotype) form a clade sister to a clade comprising the silverleafing or B biotype and a nonsilverleafing biotype from the North Africa/Mediterranean region. Polymorphisms in rDNA suggest that silverleafing is a recent phenom- enon, while the nonsilverleafing form is the ancestral or plesiomorphic state. Based on this phylogeny, if B. argentifolii is accepted as a separate species then one would have to review the taxonomic status of all bio- types of B. tabaci. In particular, a new name clearly would be needed for an Egypt/Spain/Sudan/Nigeria clade, and the monophyly of the haplotypes remaining in B. tabaci would be open to serious doubt. To make new species assignments in these circumstances seems premature. The phylogenetic relationships of the different populations of B. tabaci and the origins of effective natural enemies of the B biotype suggest that knowing the origin of the B biotype is not essen- tial to finding effective agents and supports the notion that crop management is the key aspect to control. © 2000 Academic Press Key Words: ribosomal DNA; Bemisia tabaci; Bemisia argentifolii; phylogeny. INTRODUCTION Bemisia tabaci (Gennadius) (Hemiptera:Aleyrodi- dae) is a worldwide pest of vegetable, ornamental, and field crops. It is composed of numerous populations that, while morphologically indistinguishable (Rosell et al., 1997), vary considerably in their relative ability to transmit geminiviruses (Bedford et al., 1994; Markham et al., 1994; Brown and Bird, 1995), rate of development (Wang and Tsai, 1996), ability to utilize different hosts (Burban et al., 1992; Brown and Bird, 1995; Bedford et al., 1994), and ability to induce phys- iological changes in some hosts (Costa and Brown, 1991; Cohen et al., 1992; Bedford et al., 1994). They also vary considerably with respect to esterase (Liu et al., 1992; Wool et al., 1993, 1994; Coats et al., 1994; Legg et al., 1994; Brown et al., 1995a,b; Gunning et al., 1997) and RAPD-PCR profiles (Gawel and Bartlett, 1993; De Barro and Driver, 1997; Guirao et al., 1997), and there is considerable morphological variation among their endosymbionts (Costa et al., 1995). To- gether, these differences have been used to character- ize numerous biotypes (Bedford et al., 1992, 1993, 1994; Beitia et al., 1997). On the basis of the above, Bellows et al. (1994) con- cluded that the B. tabaci B biotype was a different species and revised the name to B. argentifolii. How- ever, this reassignment is contentious, given that it was based on a comparison between the United States A biotype and the B biotype and failed to include indi- viduals representing the remaining world fauna. The B biotype was first detected in Australia in 1994 (Gunning et al., 1995) and is the second biotype of B. tabaci to be discovered in that country. The first is a largely innocuous biotype widely distributed across northern Australia (De Barro and Driver, 1997). This biotype occurs on several native plant hosts and is believed to be indigenous to Australia and the Pacific islands of Papua New Guinea, Solomon Islands, New Caledonia, and Vanuatu (De Barro et al., 1998). It is a known vector of several closely related forms of tomato leaf curl geminiviruses (Dry et al., 1993). The presence of an indigenous biotype of B. tabaci with a proven ability to vector geminiviruses raised the possibility that additional viruses also may be present. One of the hallmarks of an incursion of the B biotype is an in- crease in the number of viral diseases occurring in crops. This is due to the B biotype’s broader host range that enables it to move geminiviruses from noncrop hosts into crops (Brown, 1994; Markham et al., 1994). The origin of the B biotype is unknown. It was intro- duced to Australia from the United States where it was also introduced. Not knowing the origin or relation- ships among the different biotypes made it difficult to assess the threat posed by the new introduction. A Molecular Phylogenetics and Evolution Vol. 16, No. 1, July, pp. 29 –36, 2000 doi:10.1006/mpev.1999.0768, available online at http://www.idealibrary.com on 1055-7903/00 $35.00 Copyright © 2000 by Academic Press All rights of reproduction in any form reserved. 29
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Phylogenetic Relationships of World Populations of Bemisia tabaci (Gennadius) Using Ribosomal ITS1

Phylogenetic Relationships of World Populations of Bemisia tabaci
tebtaBNieoawtwcinstottt
dfitatMdd
Molecular Phylogenetics and EvolutionVol. 16, No. 1, July, pp. 29–36, 2000doi:10.1006/mpev.1999.0768, available online at http://www.idealibrary.com on
(Gennadius) Using Ribosomal ITS1Paul J. De Barro, Felice Driver, John W. H. Trueman,* and John Curran
CSIRO Entomology, G.P.O. Box 1700, Canberra, ACT 2601, Australia; and *Research School of Biological Sciences,Australian National University, Canberra, ACT 0200, Australia
Received June 15, 1999; revised December 13, 1999
1991; Cohen et al., 1992; Bedford et al., 1994). They
A phylogeny of Bemisia tabaci is presented based onhe ITS1 region of ribosomal DNA. The monophyly ofach biotype of Bemisia is supported, and a strongiogeographic pattern is evident in the data. Popula-ions from the Americas (including the A biotype) formclade sister to a clade comprising the silverleafing orbiotype and a nonsilverleafing biotype from the
orth Africa/Mediterranean region. Polymorphismsn rDNA suggest that silverleafing is a recent phenom-non, while the nonsilverleafing form is the ancestralr plesiomorphic state. Based on this phylogeny, if B.rgentifolii is accepted as a separate species then oneould have to review the taxonomic status of all bio-
ypes of B. tabaci. In particular, a new name clearlyould be needed for an Egypt/Spain/Sudan/Nigeria
lade, and the monophyly of the haplotypes remainingn B. tabaci would be open to serious doubt. To makeew species assignments in these circumstanceseems premature. The phylogenetic relationships ofhe different populations of B. tabaci and the originsf effective natural enemies of the B biotype suggesthat knowing the origin of the B biotype is not essen-ial to finding effective agents and supports the notionhat crop management is the key aspect to control.
© 2000 Academic Press
Key Words: ribosomal DNA; Bemisia tabaci; Bemisiaargentifolii; phylogeny.
INTRODUCTION
Bemisia tabaci (Gennadius) (Hemiptera:Aleyrodi-ae) is a worldwide pest of vegetable, ornamental, andeld crops. It is composed of numerous populationshat, while morphologically indistinguishable (Rosell etl., 1997), vary considerably in their relative abilityo transmit geminiviruses (Bedford et al., 1994;arkham et al., 1994; Brown and Bird, 1995), rate of
evelopment (Wang and Tsai, 1996), ability to utilizeifferent hosts (Burban et al., 1992; Brown and Bird,
1995; Bedford et al., 1994), and ability to induce phys-iological changes in some hosts (Costa and Brown,
29
also vary considerably with respect to esterase (Liu etal., 1992; Wool et al., 1993, 1994; Coats et al., 1994;Legg et al., 1994; Brown et al., 1995a,b; Gunning et al.,1997) and RAPD-PCR profiles (Gawel and Bartlett,1993; De Barro and Driver, 1997; Guirao et al., 1997),and there is considerable morphological variationamong their endosymbionts (Costa et al., 1995). To-gether, these differences have been used to character-ize numerous biotypes (Bedford et al., 1992, 1993,1994; Beitia et al., 1997).
On the basis of the above, Bellows et al. (1994) con-cluded that the B. tabaci B biotype was a differentspecies and revised the name to B. argentifolii. How-ever, this reassignment is contentious, given that itwas based on a comparison between the United StatesA biotype and the B biotype and failed to include indi-viduals representing the remaining world fauna.
The B biotype was first detected in Australia in 1994(Gunning et al., 1995) and is the second biotype of B.tabaci to be discovered in that country. The first is alargely innocuous biotype widely distributed acrossnorthern Australia (De Barro and Driver, 1997). Thisbiotype occurs on several native plant hosts and isbelieved to be indigenous to Australia and the Pacificislands of Papua New Guinea, Solomon Islands, NewCaledonia, and Vanuatu (De Barro et al., 1998). It is aknown vector of several closely related forms of tomatoleaf curl geminiviruses (Dry et al., 1993). The presenceof an indigenous biotype of B. tabaci with a provenability to vector geminiviruses raised the possibilitythat additional viruses also may be present. One of thehallmarks of an incursion of the B biotype is an in-crease in the number of viral diseases occurring incrops. This is due to the B biotype’s broader host rangethat enables it to move geminiviruses from noncrophosts into crops (Brown, 1994; Markham et al., 1994).
The origin of the B biotype is unknown. It was intro-duced to Australia from the United States where it wasalso introduced. Not knowing the origin or relation-ships among the different biotypes made it difficult toassess the threat posed by the new introduction. A
1055-7903/00 $35.00Copyright © 2000 by Academic PressAll rights of reproduction in any form reserved.

study of the phylogenetic relationships of the different
t
D
tTw
acteristics between those of B. hancocki and B. leakii,
30 DE BARRO ET AL.
biotypes of B. tabaci was therefore undertaken to puthe Australian situation into a world context.
MATERIALS AND METHODS
NA Extraction
DNA was extracted from single, whole specimens, inhe manner described by De Barro and Driver (1997).he crude lysate was boiled for 5 min and samplesere stored at 220°C until required.
Amplification and Sequencing
Polymerase chain reaction (PCR) was used to am-plify the ribosomal ITS1 region. All reaction volumeswere 50 mL, containing 20 pM each primer; 200 mMeach dGTP, dATP, dCTP, and dTTP, 2.5 mM MgCl2,7% dimethyl sulfoxide, 2 mL DNA lysate, 13 suppliedbuffer, and 2.5U Taq polymerase (Fisher Biotech, Aus-tralia). PCR amplifications were done in a Hybaid ther-mocycler and included a precycle denaturation step for5 min at 94°C, followed by the addition of the Taqpolymerase, and then a final postcycle extension stepat 72°C for 5 min. Primers for the ITS1 generegion were TW81 59-GTTTCCGTAGGTGAACCTGC-39(Brust et al., 1998) and B. tabaci 5.8R 59-ATCCGCGAGC-CGAGTGATCC-39. Cycling conditions for the primer setwere denaturation at 94°C for 1 min, annealing at 57°Cfor 1 min 15 s, and extension at 72°C for 1 min 30 s, for 35cycles.
The ITS1 amplicons were purified and prepared forsequencing by electrophoresis in 0.8% TAE agarosegels containing 10 mg ml21 ethidium bromide (Sam-brook et al., 1989). Fragments were excised and trans-ferred to a microfuge tube. The agarose slices weremashed in 30 mL sterile distilled water using a tooth-pick and then incubated at 50°C for 1 h. Samples wereleft at room temperature overnight to allow the DNA toelute from the gel. The samples were stored at 220°Cuntil required. Amplicons were selectively precipitatedand ligated into the pPCR-Script Amp SK(1) cloningvector from Stratagene (La Jolla, CA), according to themanufacturer’s protocol.
Five microliters of the eluted PCR amplicons or ap-proximately 250 ng cloned plasmid DNA and the ap-propriate PCR primers were used for sequencing ac-cording to the ABI PRISM Dye Terminator CycleSequencing Ready Reaction Kit Manual (PE-AppliedBioSystems). Both strands of each fragment were se-quenced and reactions were loaded onto an ABI Model373A Sequencer.
Phylogenetic Analyses
A total of 31 B. tabaci populations were included inthe analysis. Three outgroups were selected: (1) B.hancocki, (2) a species, “BLC,” in the B. leakii complex(ex Rosa sp., from Korea) having morphological char-
and (3) a native Australian whitefly, “NAW,” moreclosely related to B. tabaci than the other two outgroupspecies (Martin, 1999) (Table 1). All whiteflies wereidentified morphologically using the 4th instar. A totalof 104 sequences representing one to three individualsfrom each population were used in the analysis (Table1). Populations containing individuals belonging to theB biotype were identified by their ability to causesquash silverleafing (Costa and Brown, 1991) and byRAPD-PCR (De Barro and Driver, 1997).
Some pairs of ITS1 sequences were identical at allpositions. The duplicates were removed and the re-maining representative sequence was renamed to in-dicate its multiple provenance. The ingroup sequenceswere all very similar and readily aligned to each other,but the relationships of distant outgroups BLC and B.hancocki were not so clear. We aligned the sequencesby first applying Clustal W (Thompson et al., 1994) tothe ingroup (including NAW), after which a few incon-sistent calls on variable length C-repeats were cor-rected by hand, and then aligning BLC and B. hancockito this alignment using the Clustal W profile-align-ment routine. This approach gave the same trees asthose of a conventional, single-step Clustal W align-ment of all taxa, but at a shorter length. An alignmentthat produces a shorter tree is a better alignment(Wheeler and Gladstein, 1994) and on this basis wepreferred the two-step over the conventional align-ment.
Phylogenetic analyses were performed using PAUP*(Swofford, 1998) prerelease version 4.0d64 and betatest version 4.0b2a. In a preliminary step, the questionof possible saturation and loss of phylogenetic signalinvolving BLC and B. hancocki was addressed by cal-culating pairwise distances between selected ingroupand outgroup sequences according to the HKY85 dis-tance model (Hasegawa et al., 1985; Swofford et al.,1996). This was plotted against the transition (ti)/transversion (tv) ratio. A sequence from Acyrthosiphonpisum (Aphididae) (GenBank Accession No. AF024643)was used to provide a yet more distant, unequivocallysaturated reference point for this comparison.
In the main analysis, evolutionary trees were esti-mated using a number of parsimony models, and sub-sets of the taxa were analyzed using likelihood models.The full data set contains too many taxa for existingmaximum-likelihood tree estimation routines; reduceddata sets were therefore created. One data set wascreated by selecting 25 taxa to span the phylogenetictree estimated from parsimony analysis (Table 1). Oth-ers were created (i) by random selection of 25 taxausing the “Randbetween” function in Microsoft Excel4.0 (20 data sets) and (ii) by taking each node whichshowed high (.85%) bootstrap support under all par-simony models and condensing them to a reconstructed
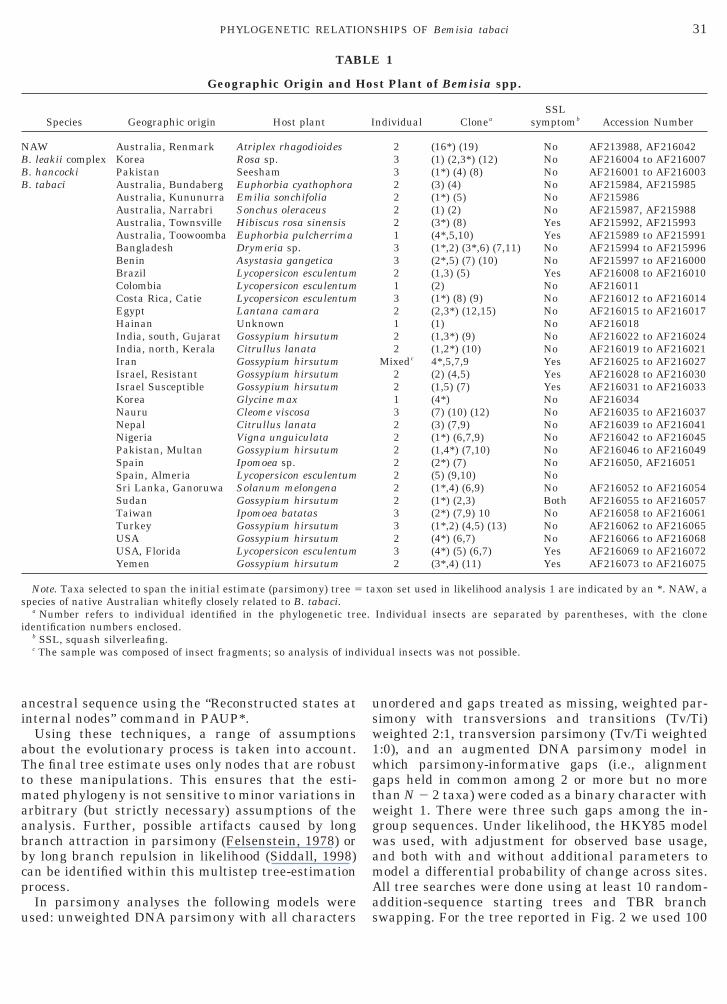
TABLE 1
s
i
31PHYLOGENETIC RELATIONSHIPS OF Bemisia tabaci
ancestral sequence using the “Reconstructed states atinternal nodes” command in PAUP*.
Using these techniques, a range of assumptionsabout the evolutionary process is taken into account.The final tree estimate uses only nodes that are robustto these manipulations. This ensures that the esti-mated phylogeny is not sensitive to minor variations inarbitrary (but strictly necessary) assumptions of theanalysis. Further, possible artifacts caused by longbranch attraction in parsimony (Felsenstein, 1978) orby long branch repulsion in likelihood (Siddall, 1998)can be identified within this multistep tree-estimationprocess.
In parsimony analyses the following models wereused: unweighted DNA parsimony with all characters
Geographic Origin and H
Species Geographic origin Host plant
NAW Australia, Renmark Atriplex rhagodioidesB. leakii complex Korea Rosa sp.B. hancocki Pakistan SeeshamB. tabaci Australia, Bundaberg Euphorbia cyathophora
Australia, Kununurra Emilia sonchifoliaAustralia, Narrabri Sonchus oleraceusAustralia, Townsville Hibiscus rosa sinensisAustralia, Toowoomba Euphorbia pulcherrimaBangladesh Drymeria sp.Benin Asystasia gangeticaBrazil Lycopersicon esculentumColombia Lycopersicon esculentumCosta Rica, Catie Lycopersicon esculentumEgypt Lantana camaraHainan UnknownIndia, south, Gujarat Gossypium hirsutumIndia, north, Kerala Citrullus lanataIran Gossypium hirsutumIsrael, Resistant Gossypium hirsutumIsrael Susceptible Gossypium hirsutumKorea Glycine maxNauru Cleome viscosaNepal Citrullus lanataNigeria Vigna unguiculataPakistan, Multan Gossypium hirsutumSpain Ipomoea sp.Spain, Almeria Lycopersicon esculentumSri Lanka, Ganoruwa Solanum melongenaSudan Gossypium hirsutumTaiwan Ipomoea batatasTurkey Gossypium hirsutumUSA Gossypium hirsutumUSA, Florida Lycopersicon esculentumYemen Gossypium hirsutum
Note. Taxa selected to span the initial estimate (parsimony) tree 5pecies of native Australian whitefly closely related to B. tabaci.
a Number refers to individual identified in the phylogenetic tredentification numbers enclosed.
b SSL, squash silverleafing.c The sample was composed of insect fragments; so analysis of ind
unordered and gaps treated as missing, weighted par-simony with transversions and transitions (Tv/Ti)weighted 2:1, transversion parsimony (Tv/Ti weighted1:0), and an augmented DNA parsimony model inwhich parsimony-informative gaps (i.e., alignmentgaps held in common among 2 or more but no morethan N 2 2 taxa) were coded as a binary character withweight 1. There were three such gaps among the in-group sequences. Under likelihood, the HKY85 modelwas used, with adjustment for observed base usage,and both with and without additional parameters tomodel a differential probability of change across sites.All tree searches were done using at least 10 random-addition-sequence starting trees and TBR branchswapping. For the tree reported in Fig. 2 we used 100
st Plant of Bemisia spp.
ndividual CloneaSSL
symptomb Accession Number
2 (16*) (19) No AF213988, AF2160423 (1) (2,3*) (12) No AF216004 to AF2160073 (1*) (4) (8) No AF216001 to AF2160032 (3) (4) No AF215984, AF2159852 (1*) (5) No AF2159862 (1) (2) No AF215987, AF2159882 (3*) (8) Yes AF215992, AF2159931 (4*,5,10) Yes AF215989 to AF2159913 (1*,2) (3*,6) (7,11) No AF215994 to AF2159963 (2*,5) (7) (10) No AF215997 to AF2160002 (1,3) (5) Yes AF216008 to AF2160101 (2) No AF2160113 (1*) (8) (9) No AF216012 to AF2160142 (2,3*) (12,15) No AF216015 to AF2160171 (1) No AF2160182 (1,3*) (9) No AF216022 to AF2160242 (1,2*) (10) No AF216019 to AF216021
Mixedc 4*,5,7,9 Yes AF216025 to AF2160272 (2) (4,5) Yes AF216028 to AF2160302 (1,5) (7) Yes AF216031 to AF2160331 (4*) No AF2160343 (7) (10) (12) No AF216035 to AF2160372 (3) (7,9) No AF216039 to AF2160412 (1*) (6,7,9) No AF216042 to AF2160452 (1,4*) (7,10) No AF216046 to AF2160492 (2*) (7) No AF216050, AF2160512 (5) (9,10) No2 (1*,4) (6,9) No AF216052 to AF2160542 (1*) (2,3) Both AF216055 to AF2160573 (2*) (7,9) 10 No AF216058 to AF2160613 (1*,2) (4,5) (13) No AF216062 to AF2160652 (4*) (6,7) No AF216066 to AF2160683 (4*) (5) (6,7) Yes AF216069 to AF2160722 (3*,4) (11) Yes AF216073 to AF216075
xon set used in likelihood analysis 1 are indicated by an *. NAW, a
Individual insects are separated by parentheses, with the clone
dual insects was not possible.
o
I
ta
e.
ivi
prt
32 DE BARRO ET AL.
random-addition-sequence starting trees, with thenumber of trees held at any one time constrained to nomore than 1000. The search yielded 9020 trees in nineapparent TBR islands each of 1000 trees plus six smallislands of between 1 and 5 trees. These trees were theninput as starting trees to recover further trees on thelarger island or islands, resulting in 10,328 trees, ofwhich Fig. 2 is the strict consensus.
Nonparametric bootstrap resamplings of the data set(Felsenstein, 1985) were used to evaluate the supportfor groups (nodes) on parsimony trees. The set ofgroups that were robust to manipulations of the evolu-tionary model and tree-estimation technique is coinci-dent with the set of groups that showed bootstrapsupport .60% under weighted parsimony. The treeestimate that we report is confined to these groups.
RESULTS
The full data set of 104 sequences contained 11 iden-tical sequences. The analysis therefore used the 93nonidentical sequences. Sequence length ranged from513 to 530 bases. In the alignment 467 sites wereconstant and there were 60 parsimony-informativesites. The alignment is available from Treebase, http://herbaria.harvard.edu/treebase, Accession No. SN345.
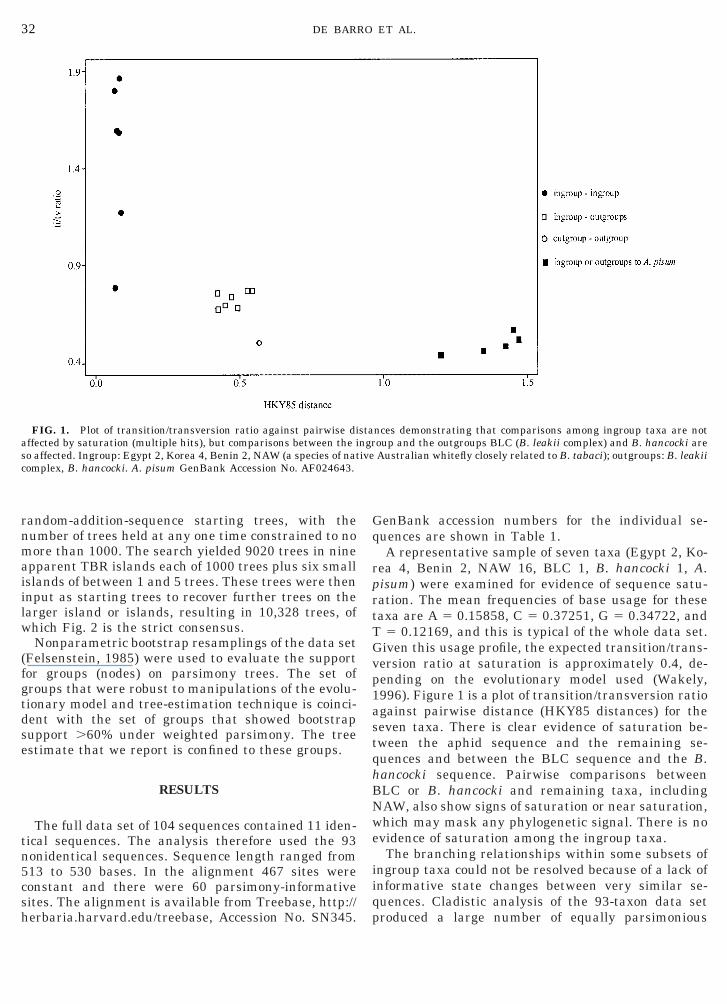
FIG. 1. Plot of transition/transversion ratio against pairwise diaffected by saturation (multiple hits), but comparisons between the iso affected. Ingroup: Egypt 2, Korea 4, Benin 2, NAW (a species of natcomplex, B. hancocki. A. pisum GenBank Accession No. AF024643.
GenBank accession numbers for the individual se-quences are shown in Table 1.
A representative sample of seven taxa (Egypt 2, Ko-rea 4, Benin 2, NAW 16, BLC 1, B. hancocki 1, A.
isum) were examined for evidence of sequence satu-ation. The mean frequencies of base usage for theseaxa are A 5 0.15858, C 5 0.37251, G 5 0.34722, and
T 5 0.12169, and this is typical of the whole data set.Given this usage profile, the expected transition/trans-version ratio at saturation is approximately 0.4, de-pending on the evolutionary model used (Wakely,1996). Figure 1 is a plot of transition/transversion ratioagainst pairwise distance (HKY85 distances) for theseven taxa. There is clear evidence of saturation be-tween the aphid sequence and the remaining se-quences and between the BLC sequence and the B.hancocki sequence. Pairwise comparisons betweenBLC or B. hancocki and remaining taxa, includingNAW, also show signs of saturation or near saturation,which may mask any phylogenetic signal. There is noevidence of saturation among the ingroup taxa.
The branching relationships within some subsets ofingroup taxa could not be resolved because of a lack ofinformative state changes between very similar se-quences. Cladistic analysis of the 93-taxon data setproduced a large number of equally parsimonious
nces demonstrating that comparisons among ingroup taxa are notoup and the outgroups BLC (B. leakii complex) and B. hancocki areAustralian whitefly closely related to B. tabaci); outgroups: B. leakii
stangrive
Fec
s
33PHYLOGENETIC RELATIONSHIPS OF Bemisia tabaci
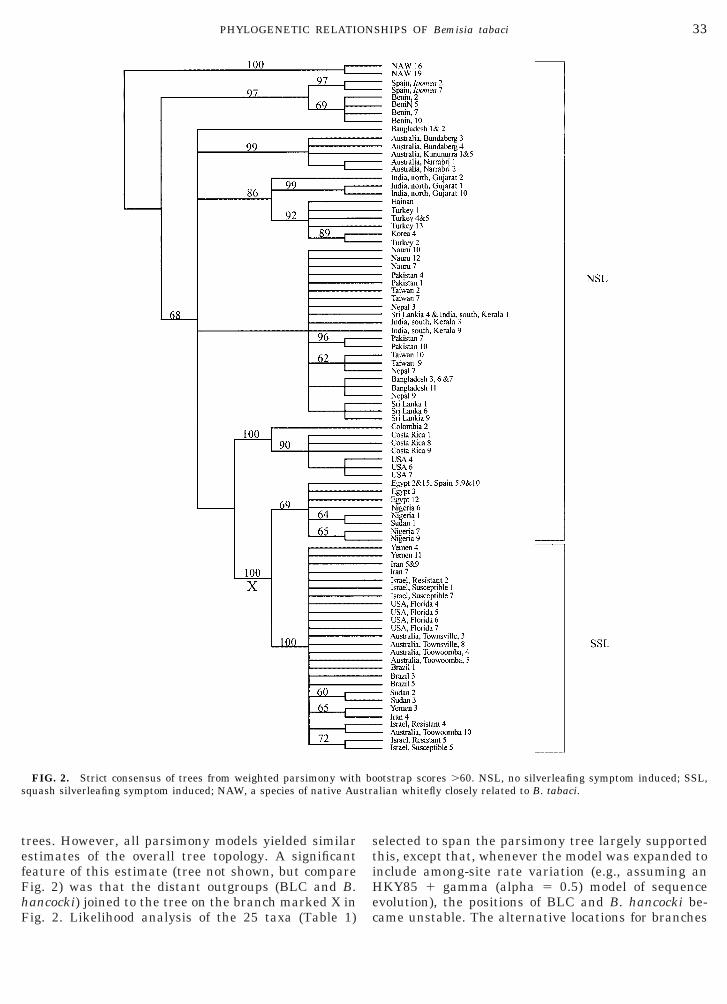
trees. However, all parsimony models yielded similarestimates of the overall tree topology. A significantfeature of this estimate (tree not shown, but compareFig. 2) was that the distant outgroups (BLC and B.hancocki) joined to the tree on the branch marked X in
ig. 2. Likelihood analysis of the 25 taxa (Table 1)
FIG. 2. Strict consensus of trees from weighted parsimony withquash silverleafing symptom induced; NAW, a species of native Au
selected to span the parsimony tree largely supportedthis, except that, whenever the model was expanded toinclude among-site rate variation (e.g., assuming anHKY85 1 gamma (alpha 5 0.5) model of sequencevolution), the positions of BLC and B. hancocki be-ame unstable. The alternative locations for branches
otstrap scores .60. NSL, no silverleafing symptom induced; SSL,alian whitefly closely related to B. tabaci.
bostr

subtending BLC and B. hancocki are near or adjacent is a synapomorphy of the SSL clade (Fig. 2), suggesting
as
s
34 DE BARRO ET AL.
to NAW.This instability, which is probably attributable to
sequence saturation, has the potential to create arti-facts in the remainder of the tree. To avoid making afalse inference of ingroup relationships, BLC and B.hancocki were removed from the analysis. Morpholog-ical evidence indicates that NAW is sister to the re-maining ingroup taxa. Further, the NAW ITS1 se-quences cluster separately from all other sequences onpairwise distance measures. For example, NAW ap-pears as sister group to all other sequences in neigh-bor-joining trees, even when BLC and B. hancocki areincluded. When BLC and B. hancocki are removed, thebranch that subtends NAW is either the longest or thesecond longest branch under all parsimony models. Wereran all tree estimation models omitting BLC and B.hancocki and taking NAW as an outgroup at which toroot the trees.
When applied to this reduced data set, each parsi-mony model gave the same overall tree topology, andthat topology was identical to the topology found usingthe full data set. A representative result from cladisticanalysis, the strict consensus of trees found usingweighted parsimony (10,328 trees at length 413; CI 50.7298, excluding uninformative characters; RI 50.9528) is shown in Fig. 2. Numbers above thebranches are nonparametric bootstrap scores .60% ina PAUP* “fast” bootstrap test (1000 pseudoreplicates)(Felsenstein, 1985; Swofford, 1998). Likelihood analy-ses of the remaining subsets (23 taxa) of the previouslyselected taxa recovered this same topology whether ornot the model included an allowance for among-siterate variation. Likelihood analyses using 20 randomsubsets, each of 25 taxa selected from the 86 taxa inthis analysis, and an HKY85 model adjusted for ob-served base frequencies each returned a set of treesconsistent with this branching structure. We considerthe tree shown in Fig. 2 to be a best estimate of thephylogeny of the sequences, but caution against reli-ance on those nodes for which no bootstrap score isshown.
DISCUSSION
The tree supports the monophyly of an Americangroup which includes the A biotype (sequences USA 4,6, and 7 in Fig. 2). It supports the monophyly of the Bbiotype (SSL in Fig. 2) and a sister relationship be-tween the American group and a clade comprising theB biotype plus a number of nonsilverleafing forms fromnorthern Africa. Several other groups among the non-silverleafing forms are strongly supported and there isa strong geographic pattern to the tree. The anomalousoccurrences of the B biotype in Australia, Brazil, andthe United States are known to result from pest incur-sions since the early 1980s. The silverleafing symptom
that it is a recently evolved phenomenon and thatabsence of silverleafing symptom induction is the ple-siomorphic state.
Some rDNA polymorphisms, i.e., point mutations orsingle-position indels (in one case a two-position indel),exist within single individuals; however, in no casedoes an individual carry rDNA sequences from otherthan its local geographic group. The within-clade struc-ture of the tree is partly incompatible with an assump-tion that relationships among individuals are strictlyhierarchical and this probably reflects reticulationamong populations within each of the well-supportedclades.
The data provide some support for populations fromSpain ex Ipomoea and Benin being the sister group tothe remaining populations of B. tabaci, although thisresult is somewhat equivocal. It provides strong sup-port for the B biotype being Middle Eastern in origin.All closely related populations have a North African–Mediterranean distribution. Asia contains several dis-tinct populations, but the relationships among themare not well established. Australia is represented by asingle, clearly monophyletic group, although a sepa-rate study has demonstrated that the eastern andwestern elements of this group are biologically incom-patible and can be separated using RAPD data (DeBarro and Hart, in press). The Americas make upanother distinct biogeographic region with a sugges-tion that populations from the United States (A bio-type) and Costa Rica may be distinct from those ofColombia.
The ITS region of ribosomal DNA is a fast-evolvingsection of the genome and may be expected to resolvehierarchical relationships only among recently di-verged lineages. Additional compatible evidence for allof the main conclusions in our analysis recently hascome from an independent study of more slowly evolv-ing gene sequences by Frolich et al. (1999). These au-thors analyzed fragments of the mitochondrial 16S andnuclear COI genes from 10 and 21 B. tabaci haplo-types, respectively, using the species T. vaporariorum(greenhouse whitefly) as outgroup. Their parsimonyanalyses give support to an American clade which con-tains the A biotype and a Middle Eastern clade whichcontains the B biotype (represented in the 16S analysisby a single United States sequence and in the COIanalysis by a pair of United States sequences). Likeli-hood methods using our models and their data concur.A Sudanese nonsilverleafing haplotype (haplotype pairin the COI analysis) is sister to the B biotype, andthese clades together are sister to the American clade.Their trees, rooted at T. vaporariorum, have variouslyn Indian haplotype or a group of Benin haplotypes asister group to the remainder.Thus, the evidence from ITS1, 16S, and COI is con-
istent in placing the A and B biotypes and associated

nonsilverleafing and silverleafing forms of B. tabaci as
pbbUthtncinp
guchr
tpsmMrttmeHt1ogbacstepmo(
REFERENCES
B
B
B
C
C
C
C
D
D
35PHYLOGENETIC RELATIONSHIPS OF Bemisia tabaci
shown in Fig. 2. The renaming of the B biotype to B.argentifolii (Bellows et al., 1994) was based on a com-
arison between the B biotype and the indigenous Aiotype found in the southern United States. The Aiotype is represented in our phylogeny by individualsSA 4, 6, and 7. Based on this phylogeny, if B. argen-
ifolii is accepted as a separate species then one wouldave to review the taxonomic status of all biotypes of B.abaci. In particular, a new name clearly would beeeded for the Egypt (and Spain)–Sudan–Nigerialade, and the monophyly of the haplotypes remainingn B. tabaci would be open to serious doubt. To makeew species assignments in these circumstances seemsremature.While this study has presented a broad biogeo-
raphic framework for B. tabaci, it is not possible tose these data at the microgeographic scale. Smallhanges between individuals that lead to changes inost utilization or vector efficacy are unlikely to beeflected at this level of phylogenetic analysis.
This analysis raises an interesting interpretation ofhe approach to biological control of the B biotype, inarticular the search for effective natural enemies. Noturprisingly, some of the most effective natural ene-ies of this biotype are from the northeast Africa/iddle East region, yet nearly all countries in this
egion have current serious problems with the B bio-ype (Goolsby et al., 1998). This suggests that many ofhe problems associated with controlling the B biotypeay not be the result of having ineffective natural
nemies, but are issues instead of crop management.ighly effective agents are also found in countries con-
aining biotypes that are quite distinct (Goolsby et al.,998; De Barro et al., unpublished). This is contrary tone of the tenets of biological control because it sug-ests, in this case, that knowing the origin of the Biotype is not essential in terms of finding effectivegents. Again, these findings support the notion thatrop management is the key aspect to control. A similarituation was recently hypothesized to exist regardinghe cane toad, Bufo marinus. Where a pest organismxists in its natural range as two or more distinctopulations, control organisms may be potentiallyore effective if selected from population clusters
ther than the one from which the pest has dispersedSlade and Moritz, 1998).
ACKNOWLEDGMENTS
The authors thank Gina Banks, Ian Bedford, and Peter Markham(John Innes Institute), Matthew Cahill (ACRI Rothamsted), andSylvia Green (AVRDC) for access to some of the populations used inthis analysis. This study was funded by ACIAR, Centre for Sustain-able Cotton Production, CRDC, HRDC, NIAA, and QVFG.
Bedford, I. D., Briddon, R. W., Markham, P. G., Brown, J. K., andRosell, R. C. (1992). “Bemisia tabaci—Biotype Characterisationand the Threat of This Whitefly Species to Agriculture,” BrightonCrop Control Conference—Pests and Diseases, pp. 1235–1240.
Bedford, I. D., Briddon, R. W., Markham, P. G., Brown, J. K., andRosell, R. C. (1993). A new species of Bemisia or biotype of Bemisiatabaci (Genn.), as a future pest of European agriculture. BCPBMonograph No 54: Plant Health and the European Single Market,pp. 381–386.
Bedford, I. D., Briddon, R. W., Brown, J. K., Rosell, R. C., andMarkham, P. G. (1994). Geminivirus transmission and biologicalcharacterisation of Bemisia tabaci (Gennadius) from different geo-graphic regions. Ann. Appl. Biol. 125: 311–325.
Beitia, F., Mayo, I., Robles-Chillida, E. M., Guira, P., and Cenis, J. L.(1997). Current status of Bemisia tabaci (Gennadius) in Spain: Thepresence of biotypes of this species. In “Integrated Control inProtected Crops “Mediterranean climate”” (R. Albajes and A.Carnero, Eds.), pp. 99–106. IOBC wprs Bulletin OILB srop 20.
Bellows, T. S., Perring, T. M., Gill, R. J., and Headrick, D. H. (1994).Description of a species of Bemisia (Homoptera: Aleyrodidae). Ann.Entomol. Soc. Am. 87: 195–206.
Brown, J. K. (1994). Current status of Bemisia tabaci as a plant pestand virus vector in agroecosystems worldwide. FAO Plant Prot.Bull. 42: 3–32.
Brown, J. K., and Bird, J. (1995). Variability within the Bemisiatabaci species complex and its relation to new epidemics caused bygeminiviruses. CEIBA 36: 73–80.
Brown, J. K., Frohlich, D. R., and Rosell, R. C. (1995a). The sweet-potato or silverleaf whiteflies: Biotypes of Bemisia tabaci or a newspecies complex. Annu. Rev. Entomol. 40: 511–534.rown, J. K., Coats, S. A., Bedford, I. D., Markham, P. G., Bird, J.,and Frohlich, D. R. (1995b). Characterisation and distribution ofesterase electromorphs in the whitefly, Bemisia tabaci (Genn.)(Homoptera: Aleyrodidae). Biochem. Genet. 33: 205–214.rust, R. A., Ballard, J. W. O., Driver, F., Hartley, D. M., Galway,N. J., and Curran J. (1998). Molecular systematics, morphologicalanalysis, and hybrid crossing identify a third taxon, Aedes (Halae-des) wardandensis sp. nov., of the Aedes (Halaedes) australis spe-cies-group (Diptera: Culicidae). Can. J. Zool. 76: 1236–1246.
urban, C., Fishpool, L. D. C., Fauquet, C., Fargette, D., andThouvenel, J. C. (1992). Host-associated biotypes within WestAfrican populations of the whitefly Bemisia tabaci (Genn.), (Hom.,Aleyrodidae). J. Appl. Entomol. 113: 416–423.
oats, S. A., Brown, J. K., and Hendrix, D. L. (1994). Biochemicalcharacterisation of biotype-specific esterases in the whitefly, Be-misia tabaci Genn (Homoptera: Aleyrodidae). Insect Biochem. Mol.Biol. 24: 723–728.ohen, S., Duffus, J. E., and Liu, H. Y. (1992). A new Bemisia tabacibiotype in the southwestern United States and its role in silverleafof squash and transmission of lettuce infectious yellows virus.Phytopathology 82: 86–90.
osta, H. S., and Brown, J. K. (1991). Variation in biological char-acteristics and esterase patterns among populations of Bemisiatabaci, and the association of one population with silverleaf symp-tom induction. Entomol. Exp. Appl. 61: 211–219.osta, H. S., Westcot, D. M., Ullman, D. E., Rosell, R., Brown, J. K.,and Johnson, M. W. (1995). Morphological variation in Bemisiaendosymbionts. Protoplasma 189: 194–202.e Barro, P. J., and Driver, F. (1997). Use of RAPD PCR to distin-guish the B biotype from other biotypes of Bemisia tabaci (Genna-dius) (Hemiptera: Aleyrodidae). Aust. J. Entomol. 36: 149–152.e Barro, P. J., Liebregts W., and Carver, M. (1998). The distributionand identity of biotypes of Bemisia tabaci (Gennadius) (Hemiptera:

Aleyrodidae) in member countries of the Secretariat of the PacificCommunity. Aust. J. Entomol. 37: 214–218.
D
D
F
F
F
G
G
G
G
G
H
L
L
Markham, P. G., Bedford, I. D., Liu, S., and Pinner, M. S. (1994). Thetransmission of geminiviruses by Bemisia tabaci. Pestic. Sci. 42:
M
R
S
S
S
S
S
T
W
W
W
W
W
36 DE BARRO ET AL.
e Barro, P. J., and Hart, P. J. (2000). Mating interactions betweentwo biotypes of the whitefly, Bemisia tabaci (Hemiptera: Aleyrodi-dae) in Australia. Bull. Entomol. Res., in pressry, I. B., Rigden, J. E., Krake, L. R., Mullineaux, P. M., andRezaian, M. A. (1993). Nucleotide sequence and genome organiza-tion of tomato leaf curl geminivirus. J. Gen. Virol. 74: 147–151.
elsenstein, J. (1978). Cases in which parsimony or compatibilitymethods will be positively misleading. Syst. Zool. 27: 401–410.
elsenstein, J. (1985). Confidence limits on phylogenies: An ap-proach using the bootstrap. Evolution 39: 783–791.
rohlich, D. R., Torres-Jerez, I., Bedford, I. D., Markham, P. G., andBrown, J. K. (1999). A phylogeographical analysis of the Bemisiatabaci species complex based on mitochondrial DNA markers. Mol.Ecol. 8: 1683–1691.awell, N. J., and Bartlett, A. C. (1993). Characterization of differ-ences between whiteflies using RAPD-PCR. Insect Mol. Biol. 2:33–38.oolsby, J. A., Ciomperlik, M. A., Legaspi, B. C., Legaspi, J. C., andWendel, L. E. (1998). Laboratory and field evaluation of exoticparasitoids of Bemisia tabaci (Gennadius) (Biotype B) (Homoptera:Aleyrodidae) in the Lower Rio Grande Valley of Texas. Biol. Con-trol 12: 127–135.uirao, P., Beitia, F., and Cenis, J. L. (1997). Biotype determinationof Spanish populations of Bemisia tabaci (Hemiptera: Aleyrodi-dae). Bull. Entomol. Res. 87: 587–593.unning, R. V., Byrne, F. J., Conde, B. D., Connelly, M. I., Herg-strom, K., and Devonshire, A. L. (1995). First report of B-biotypeBemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) in Austra-lia. J. Aust. Entomol. Soc. 34: 116.unning, R. V., Byrne, F. J., and Devonshire, A. L. (1997). Electro-phoretic analysis of non-B and B-biotype Bemisia tabaci (Genna-dius) (Hemiptera: Aleyrodidae) in Australia. Aust. J. Entomol. 36:245–249.asegawa, M., Kishino, H., and Yano, T. (1985). Dating of the hu-man–ape splitting by a molecular clock of mitochondrial DNA. J.Mol. Evol. 21: 160–174.
egg, J. P., Gibson, R. W., and Otim-Nape, G. W. (1994). Geneticpolymorphism amongst Ugandan populations of Bemisia tabaci(Gennadius) (Homoptera: Aleyrodidae), vector of African cassavageminivirus. Trop. Sci. 34: 73–81.
iu, H. Y., Cohen, S., and Duffus, J. E. (1992). The use of isozymepatterns to distinguish sweetpotato whitefly (Bemisia tabaci) bio-types. Phytoparasitica 20: 187–194.
123–128.
artin, J. H. (1999). “The Whitefly Fauna of Australia (Sternorrhyn-cha: Aleyrodidae): A Taxonomic Account and Identification Guide,”CSIRO Entomol. Tech. Paper No. 38, 197 pp.
osell, R. C., Bedford, I. D., Frohlich, D. R., Gill, R. J., Brown, J. K.,and Markham, P. G. (1997). Analysis of morphological variation indistinct populations of Bemisia tabaci (Homoptera: Aleyrodidae).Ann. Entomol. Soc. Am. 90: 575–589.
ambrook, J., Fritsch, E. F., and Maniatis, T. (1989). “MolecularCloning: A Laboratory Manual,” 2nd ed., Cold Spring Harbor Lab-oratory Press, Cold Spring Harbor, NY.
iddall, M. E. (1998). Success of parsimony in the four-taxon case:Long-branch repulsion by likelihood in the Farris Zone. Cladistics14: 209–220.
lade, R. W., and Moritz C. (1998). Phylogeography of Bufo marinusfrom its natural and introduced ranges. Proc. R. Soc. Lond. B 265:769–777.
wofford, D. L., Olsen, G. J., Waddell, P. J., and Hillis, D. M. (1996).Phylogenetic inference. In “Molecular Systematics” (D. M. Hillis,C. Moritz, and B. K. Mable, Eds.), 2nd ed., Sinauer Sunderland,MA.
wofford, D. L. (1998). PAUP*: Phylogenetic analysis using parsi-mony. (and other methods). Sinauer, Sunderland, MA.
hompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). Clustal W:Improving the sensitivity of progressive multiple sequence align-ment through sequence weighting, position-specific gap penaltiesand weight matrix choice. Nucleic Acids Res. 22: 4673–4680.
akely, J. (1996). The excess of transitions among nucleotide sub-stitutions: New methods of estimating transition bias underscoreits significance. Trends Ecol. Evol. 11: 158–163.
ang, K., and Tsai, J. H. (1996). Temperature effect on developmentand reproduction of silverleaf whitefly (Homoptera: Aleyrodidae).Ann. Entomol. Soc. Am. 89: 375–384.
heeler, W. C., and Gladstein, D. (1994). Malign: A multiple se-quence alignment program. J. Hered. 85: 417.
ool, D., Gerling, D., Bellotti, A. C., and Morales, F. J. (1993).Esterase electrophoretic variation in Bemisia tabaci (Genn.)(Hom., Aleyrodidae) among host plants and localities in Israel.J. Appl. Entomol. 115: 185–196.
ool, D., Calvert, L., Constantino, L. M., Bellotti, A. C., and Gerling,D. (1994). Differentiation of Bemisia tabaci (Genn.) (Hom., Aley-rodidae) populations in Colombia. J. Appl. Entomol. 117: 122–134.














