
Photoinitiation of peroxyoxalate chemiluminescence: application to flow injection analysis of...
6
1050 Anal. Chem. 1990. 62. 1050-1055 quent removal of organic constituents and easily ionizable elements like the alkali and alkaline-earth metals should allow a more accurate measurement to be made. The preconcen- tration achieved with the electrodeposition improves the detection limits of both techniques significantly relative to levels that might have been obtained for direct determination. Much work remains to be done to further establish the ac- curacy and precision of the techniques. ACKNOWLEDGMENT We gratefully acknowledge the use of electron microscopy facilities at the Ultrastructure Center at the University of Georgia and the ICP-MS results provided by S. J. Jiang and R. S. Houk of Iowa State University. Registry No. Zn (element), 7440-66-6; Mn (element), 7439- 96-5; Co (element), 7440-48-4; Cu (element), 7440-50-8; Cr (ele- ment), 7440-47-3; Ni (element), 7440-02-0; Fe (element), 7439-89-6; Cd (element), 7440-43-9; Pb (element), 7439-92-1; Hg (element), LITERATURE CITED 7439-97-6; H20, 7732-18-5; Nb, 7440-03-1. Akagi, T.; Fuwa, K.; Haraguchi, H. Anal. Chim. Acta 1985, 777, 139. Hudnlk, V.; Gomiscek, S.; Gorenc. Anal. Chim. Acta 1978, 98, 39-46. Danielsson. L. G.: Magnusson, 6.; Westerlund, S. Anal. Chkn. Acta 1978. 98, 47-57. Bruland, K. W.; Franks, R. P.; Knauer, G. A,; Martin, J. H. Anal. Chim. Acta 1979, 105, 233-245. Bond, A. M.; Wallace, G. G. Anal. Chim. Acta 1984, 164, 223-232. Isshiki, K.: Tsujl, F.; Kuwamoto, T.; Nakayama, E. Anal. Chem. 1987, 59, 2491-2495. (7) Batley, G. E.; Matousek, J. P. Anal. Chem. 1977, 49, 2031-2034. (8) Abdullah, M.: Fuwa, K.; Haraguchi, H. Appl. Spectrosc. 1987, 41, (9) Matusiewicz, H.; Fish, J.; Malinski, T. Anal. Chem. 1987, 59, (10) Manthey, M.; Riley, P. J.; Wallace, G. G. Am. Lab. 1988, July, 21-29. (11) Cox, J. A.; Twardowski, 2. Anal. Chlm. Acta 1980, 779, 39-45. (12) Koropchak, J. A.: Zlotorzynska, E. D. Anal. Chem. 1988, 60, 328-331. (13) O'Halloran, R. J. Anal. Chim. Acta 1982, 740, 51-58. (14) Hoyer, 6.: Florence, T. M.; Batley, G. E. Anal. Chem. 1987, 59, (15) Van den Berg, C. M. G. Anal. Chim. Acta 1984, 764, 195-207. (16) Donat, J. R.; Bruiand, K. W. Anal. Chem. 1988, 60, 240-244. (17) Long, S. E.; Snook, R. D. Ana/yst 1983, 708, 1331-1338. (18) King, J. N.; Fritz, J. S. Anal. Chem. 1987. 59, 703-708. (19) Bond, A. M. 26th Eastern Analytical Symposium Official Program, N.Y., 1987; Paper NO. 202, p 179. (20) Sioda. R. E. Anal. Chem. 1988, 60, 1177-1179. (21) Ciszewski. A.; Fish, J. R.; Malinski, T.; Sioda, R. E. Anal. Chem. 1989, 67, 856. (22) Tan, S. H.; Horlick, G. Appl. Spetrosc. 1988, 40, 446-464. (23) Vaughan, M. A.; Horlick, G. Appl. Spectrosc. 1986, 40, 434-445. (24) Olivares, J. A.; Houk, R. S. Anal. Chem. 1988, 58, 20. (25) Gregoire, C. Spectochim. Acta, Part B 1987, 42, 895. (26) Kester, D. R.; Duedall, I. W.; Connors, D. N.; Ptykowicz, R. M. Limnol. Oceanogr. 1967, 72, 176. (27) Chong, N. S.: Norton, M. L.; Anderson, J. L., unpublished work. (28) Wittry, D. B. European Congress on Electron Microscopy, 3rd Leiden; Elsevier: New York, 1980; p 14. (29) Driscoll, J. N.; Jacobus, N. Am. Lab. 1988, August, 68-75. (30) McLaren, J. W.; Mykytluk, A. P.; Willie, S. N.; Berman, S. S. Anal. Chem 1985, 57, 2907-2911. 715-720. 2264-2269. 1608- 161 4. RECEIVED for review August 1,1989. Accepted February 15, 1990. We gratefully acknowledge the financial support of NSF Grant CHE-8600224. Photoinitiation of Peroxyoxalate Chemiluminescence: Application to Flow Injection Analysis of Chemilumophores Robert E. Milofsky and John W. Birks* Department of Chemistry and Biochemistry and Cooperative Institute for Research in Environmental Sciences (CIRES), Campus Box 216, University of Colorado, Boulder, Colorado 80309 Photolnltlatlon of peroxyoxalate chemllumlnescence Is re- ported for the first tlme and applied to the detectlon of am- ino-substituted polycycllc aromatic hydrocarbons in flow in- jection analysis. Thls novel detection system Is slmpwfled by ellmlnatlon of the reagent hydrogen peroxide used In most IIqukCphase chemllwnlnescence detection schemes. Like the H,O,-Inltlated reaction, the sensltlvlty of detection in pho- tolnltiated chemilumlnescence (PICL) Is enhanced by use of a base catalyst such as Imidazole. Lhnlts of detection are in the low- to mld-picogram range and are comparable to those obtalned by fluorescence detection using the same apparatus. It is proposed that the PICL reaction begins wlth hydrogen abstraction by the triplet-excited state of the oxalate ester, followed by addition of 02. Subsequent steps In the mecha- nlsm proceed to form high-energy Intermediates that transfer energy to the chemllumophore. INTRODUCTION Since the discovery of peroxyoxalate chemiluminescence in the 1960s and its characterization in subsequent years (1-4), the phenomenon has been applied to the detection of a variety of analytes in both static (5-9) and flowing systems (10-38). The chemical reactions involved in these detection schemes can be summarized by three basic steps, written as follows: oxalate ester + H202 - intermediates chemilumophore + intermediates - chemilumophore* chemilumophore* - chemilumophore + hu These reactions are extremely complicated and have numerous side reactions, making them very sensitive to factors such as solvent composition, pH, temperature, and base catalyst (38). Rauhut postulated dioxetanedione as the intermediate (2). McCapra suggested that dioxetanedione transfers its energy to a fluorophore via the chemically initiated electron exchange luminescence (CIEEL) mechanism (39,40). The role of di- oxetanedione in peroxyoxalate chemiluminescence recently has been questioned and other high-energy intermediates proposed (41, 42). In this paper we report photoinitiation of peroxyoxalate chemiluminescence for the first time and demonstrate its application to the sensitive detection of amino-PAH, which serve as model chemilumophores. This unique method of analysis, although not as sensitive as hydrogen peroxide 0003-2700/90/0362-1050$02.50/0 8 1990 American Chemical Society
Transcript of Photoinitiation of peroxyoxalate chemiluminescence: application to flow injection analysis of...

1050 Anal. Chem. 1990. 62. 1050-1055
quent removal of organic constituents and easily ionizable elements like the alkali and alkaline-earth metals should allow a more accurate measurement to be made. The preconcen- tration achieved with the electrodeposition improves the detection limits of both techniques significantly relative to levels that might have been obtained for direct determination. Much work remains to be done to further establish the ac- curacy and precision of the techniques.
ACKNOWLEDGMENT We gratefully acknowledge the use of electron microscopy
facilities at the Ultrastructure Center at the University of Georgia and the ICP-MS results provided by S. J. Jiang and R. S. Houk of Iowa State University.
Registry No. Zn (element), 7440-66-6; Mn (element), 7439- 96-5; Co (element), 7440-48-4; Cu (element), 7440-50-8; Cr (ele- ment), 7440-47-3; Ni (element), 7440-02-0; Fe (element), 7439-89-6; Cd (element), 7440-43-9; Pb (element), 7439-92-1; Hg (element),
LITERATURE CITED
7439-97-6; H20, 7732-18-5; Nb, 7440-03-1.
Akagi, T.; Fuwa, K.; Haraguchi, H. Anal. Chim. Acta 1985, 777, 139. Hudnlk, V.; Gomiscek, S.; Gorenc. Anal. Chim. Acta 1978, 98, 39-46. Danielsson. L. G.: Magnusson, 6.; Westerlund, S. Anal. Chkn. Acta 1978. 98, 47-57. Bruland, K. W.; Franks, R. P.; Knauer, G. A,; Martin, J. H. Anal. Chim. Acta 1979, 105, 233-245. Bond, A. M.; Wallace, G. G. Anal. Chim. Acta 1984, 164, 223-232. Isshiki, K.: Tsujl, F.; Kuwamoto, T.; Nakayama, E. Anal. Chem. 1987, 59, 2491-2495.
(7) Batley, G. E.; Matousek, J. P. Anal. Chem. 1977, 49, 2031-2034. (8) Abdullah, M.: Fuwa, K.; Haraguchi, H. Appl. Spectrosc. 1987, 41,
(9) Matusiewicz, H.; Fish, J.; Malinski, T. Anal. Chem. 1987, 59,
(10) Manthey, M.; Riley, P. J.; Wallace, G. G. Am. Lab. 1988, July, 21-29. (11) Cox, J. A.; Twardowski, 2. Anal. Chlm. Acta 1980, 779, 39-45. (12) Koropchak, J. A.: Zlotorzynska, E. D. Anal. Chem. 1988, 6 0 ,
328-331. (13) O'Halloran, R. J. Anal. Chim. Acta 1982, 740, 51-58. (14) Hoyer, 6.: Florence, T. M.; Batley, G. E. Anal. Chem. 1987, 59,
(15) Van den Berg, C. M. G. Anal. Chim. Acta 1984, 764, 195-207. (16) Donat, J. R.; Bruiand, K. W. Anal. Chem. 1988, 60, 240-244. (17) Long, S. E.; Snook, R. D. Ana/yst 1983, 708, 1331-1338. (18) King, J. N.; Fritz, J. S. Anal. Chem. 1987. 59, 703-708. (19) Bond, A. M. 26th Eastern Analytical Symposium Official Program,
N.Y., 1987; Paper NO. 202, p 179. (20) Sioda. R. E. Anal. Chem. 1988, 60, 1177-1179. (21) Ciszewski. A.; Fish, J. R.; Malinski, T.; Sioda, R . E. Anal. Chem. 1989,
67, 856. (22) Tan, S. H.; Horlick, G. Appl. Spetrosc. 1988, 40, 446-464. (23) Vaughan, M. A.; Horlick, G. Appl. Spectrosc. 1986, 40, 434-445. (24) Olivares, J. A.; Houk, R. S. Anal. Chem. 1988, 58, 20. (25) Gregoire, C. Spectochim. Acta, Part B 1987, 42, 895. (26) Kester, D. R.; Duedall, I. W.; Connors, D. N.; Ptykowicz, R. M. Limnol.
Oceanogr. 1967, 72, 176. (27) Chong, N. S.: Norton, M. L.; Anderson, J. L., unpublished work. (28) Wittry, D. B. European Congress on Electron Microscopy, 3rd Leiden;
Elsevier: New York, 1980; p 14. (29) Driscoll, J. N.; Jacobus, N. Am. Lab. 1988, August, 68-75. (30) McLaren, J. W.; Mykytluk, A. P.; Willie, S. N.; Berman, S. S. Anal.
Chem 1985, 57, 2907-2911.
715-720.
2264-2269.
1608- 161 4.
RECEIVED for review August 1,1989. Accepted February 15, 1990. We gratefully acknowledge the financial support of NSF Grant CHE-8600224.
Photoinitiation of Peroxyoxalate Chemiluminescence: Application to Flow Injection Analysis of Chemilumophores
Robert E. Milofsky and John W. Birks* Department of Chemistry and Biochemistry and Cooperative Institute for Research i n Environmental Sciences (CIRES) , Campus Box 216, University of Colorado, Boulder, Colorado 80309
Photolnltlatlon of peroxyoxalate chemllumlnescence Is re- ported for the first tlme and applied to the detectlon of am- ino-substituted polycycllc aromatic hydrocarbons in flow in- jection analysis. Thls novel detection system Is slmpwfled by ellmlnatlon of the reagent hydrogen peroxide used In most IIqukCphase chemllwnlnescence detection schemes. Like the H,O,-Inltlated reaction, the sensltlvlty of detection in pho- tolnltiated chemilumlnescence (PICL) Is enhanced by use of a base catalyst such as Imidazole. Lhnlts of detection are in the low- to mld-picogram range and are comparable to those obtalned by fluorescence detection using the same apparatus. I t is proposed that the PICL reaction begins wlth hydrogen abstraction by the triplet-excited state of the oxalate ester, followed by addition of 02. Subsequent steps In the mecha- nlsm proceed to form high-energy Intermediates that transfer energy to the chemllumophore.
INTRODUCTION Since the discovery of peroxyoxalate chemiluminescence
in the 1960s and its characterization in subsequent years (1-4), the phenomenon has been applied to the detection of a variety
of analytes in both static (5-9) and flowing systems (10-38). The chemical reactions involved in these detection schemes can be summarized by three basic steps, written as follows:
oxalate ester + H202 - intermediates
chemilumophore + intermediates - chemilumophore*
chemilumophore* - chemilumophore + hu
These reactions are extremely complicated and have numerous side reactions, making them very sensitive to factors such as solvent composition, pH, temperature, and base catalyst (38). Rauhut postulated dioxetanedione as the intermediate (2). McCapra suggested that dioxetanedione transfers its energy to a fluorophore via the chemically initiated electron exchange luminescence (CIEEL) mechanism (39,40). The role of di- oxetanedione in peroxyoxalate chemiluminescence recently has been questioned and other high-energy intermediates proposed (41, 42).
In this paper we report photoinitiation of peroxyoxalate chemiluminescence for the first time and demonstrate its application to the sensitive detection of amino-PAH, which serve as model chemilumophores. This unique method of analysis, although not as sensitive as hydrogen peroxide
0003-2700/90/0362-1050$02.50/0 8 1990 American Chemical Society

ANALYTICAL CHEMISTRY. VOL. 62, NO. 10. MAY 15. 1990 - 1051
Sample Mixing n e
Pholochemlcal
Oxalals Eslw
Hydropen Donor
Scheme I 3 TSPO 1 hr - I-* - -*
c,d , , 0-c-c-0 6 0 , , u +a* - c l ~ o . d o ~ c l
CI CI I I!
generated peroxyoxalate chemiluminescence, is comparable to fluorescence detection and could possibly he further op-. timized to provide more sensitive and selective detection than fluorescence for many compounds. The eight amino-PAH investigated showed low- to mid-picogram detection limits in the presence of imidazole as a base catalyst.
The mechanism of peroxyoxalate chemiluminescence is still highly uncertain, and it has been shown that at least two high-energy intermediates are involved (43). Our hypothesized mechanism for photoinitiated chemiluminescence is given in Scheme I. The mechanism begins with A one-electron pho- toreduction of the oxalate ester and is followed hy the addition of molecular oxygen. The remainder of the mechanism, al- though speculative, shows that photoinitiation and initiation hy HzOz can, via reasonable reaction steps, lead to the same intermediates.
EXPERIMENTAL SECTION Chemicals. All PAH and amino-PAH were obtained from
either Aldrich Chemical Co. or Analabs and used without further purification. Bis(2,4,6-trichlorophenyl) oxalate (TCPO) and im- idazole were purchased from Fluka Chemical Co. and Sigma Chemical Co., respectively. Ethyl acetate, obtained from Burdick and Jackson as a high-purity solvent, and isopropyl alcohol (IPA), obtained from Mallinckrodt, were used without further purifi- cation.
Static Experiments. Luminescence resulting from photolysis of solutions in cuvettes was detected by photon counting. The photocathode of a RCA 1P28 photomultiplier tube was biased at -1000 V hy a Hewlett-Packard Model 6516A high-voltage power supply, and photons were counted with an Ortec Model 454 pulse amplifier, Model 346 discriminator, and Model 9315 counter, Photolysis sources included a 5M)-W tungsten-halogen lamp, an 8-W fluorescent black lamp (Sylvania Model E 8TS IBLB, Am, = 366 nm), a 4.5-mW Pen Ray low-pressure Hg lamp (254 nm), and a Lumnnics Model 500 KrF excimer laser (X = 249 nm) having a maximum power of 140 mJ/pulse and operated at 10 Hz.
Flow Injection System. The flow injection apparatus, shown schematically in Figure 1, consists of a Waten Model 6000A HPLC pump operated at 0.5 mL/min to provide the hulk flow of ethyl
Figure 1. Schematic diagram of the FIA apparatus.
acetate carrier, a Rhecdyne 7125 injector with a 20-WL loop to introduce the analyte, and an Isco Model 314 high-pressure syringe pump to deliver the oxalate ester/IPA/ethyl acetate reagent through a photochemical reador. A Valco low dead volume mixing tee was used to mix the mobile phase carrying the analyte with the photolyzed TCPO solution. The reagent stream was pumped through a coiled PTFE photoreactor (3.5 m of 30-gauge tubing obtained from Small Parts, Inc.) at a flow rate of 0.5 mL/min, resulting in a photochemical residence time of approximately 30 s. A 4.5-mW Pen-Ray low-pressure mercury lamp (254 nm) ENHI as the excitation source. The photon flux to the photoreactor was enhanced by wrapping it with aluminum foil. For investi- gations utilizing a catalyst, the imidazole was added to the ethyl acetate carrier. A Kratos FS 910 fluorescence detector with its light source turned off and no emission filter was used for chemiluminescence detection. The signal was recorded by an Omniscribe chart recorder.
For detection limit determinations, the reagent stream consisted of 0.16 M IPA and 3 mM TCPO in ethyl acetate, and the carrier stream was either neat ethyl acetate or 10 mM imidazole/ethyl acetate. The photochemical reagent solution was saturated with oxygen by bubbling oxygen through the solution prior to filling the syringe pump. Analytes (PAH and amino-PAH) were made up in ethyl acetate.
RESULTS AND DISCUSSION Mechanistic Studies. Upon irradiation of TCPO, using
ruhrene as the chemilumophore, chemiluminescence was ob- served only when both a hydrogen-atom-donating (HAD) species, such as isopropyl alcohol, and dissolved oxygen were present. Compounds capable of donating hydrogen atoms to triplet-excited carbonyl compounds include alcohols, ethers, aldehydes. and ketones (44). No photoinitiated chemilu- minescence was observed when TCPO was photolyzed in carbon tetrachloride or solvents that are poor hydrogen donors such as acetonitrile. I t was found that the PICL reaction may he initiated by either a low-pressure mercury lamp or a KrF excimer laser (249 nm). The extinction coefficient of TCPO is fairly strong from 240 to 320 nm with c = 1000 L cm-l at a Amax of 254 nm. The photoinitiated chemiluminescence signal was also found to he characteristic of the fluorophore present in solution, and no signal was observed in the absence of a fluorescent analyte.
That the reacti,on is not initiated by singlet oxygen is ev- idenced hy the fact that white light, which is strongly absorbed by rubrene (a good singlet oxygen sensitizer), does not initiate the reaction. No chemiluminescence is observed if rubrene is photolyzed with 254 nm, 366 nm, or white light and then mixed with TCPO. Although rubrene absorbs strongly in these wavelength regions, TCPO has negligible absorption beyond 320 nm. Chemiluminescence is observed when a TCPO solution containing IPA and dissolved oxygen is first

1052 ANALYTICAL CHEMISTRY, VOL. 62, NO. 10, MAY 15, 1990
5000 e 4000
w . 2 3000 a 0 C
0 r 2 2000
n.
I
loool k 0 200 400 600 800
12
Time, s Flgure 2. Chemiluminescence emission intensity as a function of time for PICL and for H,O, initiation.
photolyzed with 249 (excimer laser) or 254 nm (Hg lamp) light and then mixed with a rubrene solution. Thus, we conclude that the reaction is initiated by the absorption of light by TCPO.
It could be postulated that absorption of light by TCPO simply sensitizes the formation of hydrogen peroxide via the mechanism
TCPO + hu -+ TCPO*
TCPO* + RH - HTCPO + R HTCPO + 02 + H02 + TCPO
HOz + HOz -+ H2Oz + 0 2
An upper limit to the concentration of HzOz formed by this mechanism may be calculated from the lamp intensity ( I = 5.8 X 1015 photons cm-2 s-l), area of cuvette irradiated (A = 2 cm2), time of irradiation ( t = 30 s), extinction coefficient of TCPO a t 254 nm ( 6 = 1000 L mol-' cm-I), path length ( 1 = 1 cm), concentration of TCPO (c = 1.5 X mol L-I), volume of solution (V = 2 X L), maximum possible quantum yield (@ = 1) for hydrogen abstraction by excited- state TCPO, and Avagadro's number (NA) using the expression
[H,OZ] = IAta(1 - 10-"'))/2NAV
where the factor of 2 in the denominator takes into account the fact that two H o p radicals must be formed in order to produce one H 2 0 2 molecule. For the specified conditions, an upper limit to the concentration of HzOz formed by photo- sensitization is calculated to be 1.4 X M. Figure 2 shows the chemiluminescence intensity as a function of time actually obtained under these conditions. Also shown in Figure 2 are the emission profiles obtained in dark solutions having the same concentrations of TCPO and rubrene but spiked to and M in H202 It can be seen that not only is PICL much more intense than the maximum possible signal from the HzOz-initiated reaction, but the kinetics of the two reactions are entirely different. Over a period of 600 s, PICL undergoes rapid decay, while emission from the HzOz reaction slowly builds up. I t is clear from these results that the PICL mechanism is not that of HzO, sensitization. It is interesting to note that under certain reaction conditions, such as those of Figure 2, a second maximum occurs in the PICL reaction. A similar phenomenon was observed by Alvarez et al. (43) for the H202-initiated reaction under conditions of nearly equal molar concentration of the oxalate ester and HzOz.
Brina and Bard (45) have demonstrated that peroxyoxalate chemiluminescence can be initiated by an electrochemical
Flow Rate, mllmin Figure 3. Effect of the flow rate of photochemical reagent on the PICL S/N ratio. Each point was measured a minimum of 3 times. Error bars are f 1 u.
reduction of the oxalate ester. The requirement that a good hydrogen atom donor such as IPA be present for photoini- tiated chemiluminescence is strong evidence that the first step of the reaction is a photoreduction.
PICL is not limited to TCPO as the oxalate ester; for ex- ample, we found that most of the oxalate ester/chemi- lumophore systems used in the commercially available Cya- lume Light Sticks could be photoinitiated by 254-nm light. Also, as in HzOz-initiated chemiluminescence, it was found that a base catalyst greatly enhances the chemiluminescence signal for PICL (see Table I and discussion below).
A possible mechanism for photoinitiated chemiluminescence is given in Scheme I. In this mechanism, TCPO absorbs light to form an excited singlet state, 'TCPO*, which undergoes rapid intersystem crossing to the lowest triplet state, TCPO*, consistent with the well-known photochemistry of carbonyl compounds (44) . The second step of the reaction is a one- electron reduction (alternatively, abstraction of H) of trip- let-state excited TCPO (a strong oxidant) by a weakly reducing species such as IPA. A likely fate of the free radical (I) formed is the addition of oxygen to form 11. This could be followed by the elimination of the trichlorophenolate anion to form another radical, species 111. I t is possible that species I11 is directly involved in excitation of the chemilumophore. Al- ternatively, a second one-electron reduction leads to species IV, the high-energy intermediate postulated for HzOz-initiated chemiluminescence by Catherall et al. (41). This final re- duction may occur as the result of absorption of another photon, or, more likely, by encountering reducing agents in solution such as other free radicals, the chemilumophore, or the imidazole catalyst. Intermediate IV could directly excite the chemilumophore by the CIEEL mechanism or (perhaps less likely) rearrange to form dioxetanedione (V) as the energy transfer species.
Application to Flow Injection Analysis. Although not fully optimized, flow injection analysis with PICL detection exhibits high sensitivity toward the model compounds tested. Figure 3 shows the effect of TCPO reagent flow rate on the signal-to-noise ratio. For the reagent concentrations chosen (3 mM TCPO, 0.16 M IPA), the optimal flow rate was found to be 0.5 mL/min. The shoulder occurring at low flow rates in the flow-rate optimization curve given in Figure 3 is re- producible. Increasing the reagent flow rate decreases the residence time in the photochemical reactor, reduces the concentration of the analyte in the detection cell, and shortens the delay time between mixing of the photoproducts with the
ANALYTICAL CHEMISTRY, VOL. 62, NO. 10, MAY 15, 1990 1053
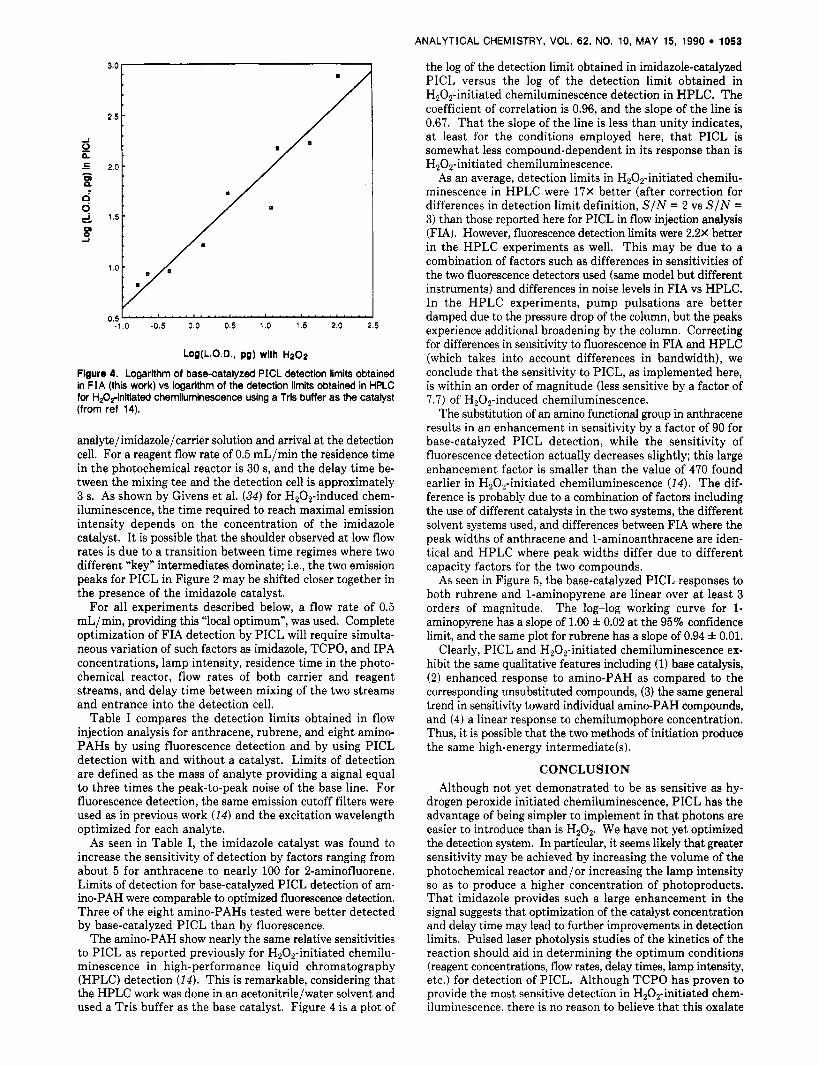
the log of the detection limit obtained in imidazole-catalyzed PICL versus the log of the detection limit obtained in H,O,-initiated chemiluminescence detection in HPLC. The coefficient of correlation is 0.96, and the slope of the line is 0.67. That the slope of the line is less than unity indicates, at least for the conditions employed here, that PICL is somewhat less compound-dependent in its response than is HzOz-initiated chemiluminescence.
As an average, detection limits in H20z-initiated chemilu- minescence in HPLC were 17X better (after correction for differences in detection limit definition, SIN = 2 vs SIN = 3) than those reported here for PICL in flow injection analysis (FIA). However, fluorescence detection limits were 2.2X better in the HPLC experiments as well. This may be due to a combination of factors such as differences in sensitivities of the two fluorescence detectors used (same model but different instruments) and differences in noise levels in FIA vs HPLC. In the HPLC experiments, pump pulsations are better damped due to the pressure drop of the column, but the peaks experience additional broadening by the column. Correcting for differences in sensitivity to fluorescence in FIA and HPLC (which takes into account differences in bandwidth), we conclude that the sensitivity to PICL, as implemented here, is within an order of magnitude (less sensitive by a factor of 7.7) of H202-induced chemiluminescence.
The substitution of an amino functional group in anthracene results in an enhancement in sensitivity by a factor of 90 for base-catalyzed PICL detection, while the sensitivity of fluorescence detection actually decreases slightly; this large enhancement factor is smaller than the value of 470 found earlier in H,Oz-initiated chemiluminescence (14). The dif- ference is probably due to a combination of factors including the use of different catalysts in the two systems, the different solvent systems used, and differences between FIA where the peak widths of anthracene and 1-aminoanthracene are iden- tical and HPLC where peak widths differ due to different capacity factors for the two compounds.
As seen in Figure 5 , the base-catalyzed PICL responses to both rubrene and 1-aminopyrene are linear over a t least 3 orders of magnitude. The log-log working curve for 1- aminopyrene has a slope of 1.00 f 0.02 at the 95% confidence limit, and the same plot for rubrene has a slope of 0.94 f 0.01.
Clearly, PICL and HzOz-initiated chemiluminescence ex- hibit the same qualitative features including (1) base catalysis, (2) enhanced response to amino-PAH as compared to the corresponding unsubstituted compounds, (3) the same general trend in sensitivity toward individual amino-PAH compounds, and (4) a linear response to chemilumophore concentration. Thus, it is possible that the two methods of initiation produce the same high-energy intermediate(s).
I"" t
1 .o
V 0.5' ' ' ' " ' ' " ' ' . ' " ' ' ' " ' ' . " ' ' ' " ' ' ' . I
-1.0 -0.5 0.0 0.5 1.0 1.5 2.0 2.5
Log(L.O.D., pg) with H202
Figure 4. Logarithm of base-catalyzed PICL detection limits obtained in FIA (this work) vs logarithm of the detection limits obtained in HPLC for H,O,-initiated Chemiluminescence using a Tris buffer as the catalyst (from ref 14).
analyte/imidazole/carrier solution and arrival at the detection cell. For a reagent flow rate of 0.5 mL/min the residence time in the photochemical reactor is 30 s, and the delay time be- tween the mixing tee and the detection cell is approximately 3 s. As shown by Givens et al. (34) for HzOz-induced chem- iluminescence, the time required to reach maximal emission intensity depends on the concentration of the imidazole catalyst. It is possible that the shoulder observed at low flow rates is due to a transition between time regimes where two different "key" intermediates dominate; i.e., the two emission peaks for PICL in Figure 2 may be shifted closer together in the presence of the imidazole catalyst.
For all experiments described below, a flow rate of 0.5 mL/min, providing this "local optimum", was used. Complete optimization of FIA detection by PICL will require simulta- neous variation of such factors as imidazole, TCPO, and IPA concentrations, lamp intensity, residence time in the photo- chemical reactor, flow rates of both carrier and reagent streams, and delay time between mixing of the two streams and entrance into the detection cell.
Table I compares the detection limits obtained in flow injection analysis for anthracene, rubrene, and eight amino- PAHs by using fluorescence detection and by using PICL detection with and without a catalyst. Limits of detection are defined as the mass of analyte providing a signal equal to three times the peak-to-peak noise of the base line. For fluorescence detection, the same emission cutoff filters were used as in previous work (14) and the excitation wavelength optimized for each analyte.
As seen in Table I, the imidazole catalyst was found to increase the sensitivity of detection by factors ranging from about 5 for anthracene to nearly 100 for 2-aminofluorene. Limits of detection for base-catalyzed PICL detection of am- ino-PAH were comparable to optimized fluorescence detection. Three of the eight amino-PAHs tested were better detected by base-catalyzed PICL than by fluorescence.
The amino-PAH show nearly the same relative sensitivities to PICL as reported previously for H,O,-initiated chemilu- minescence in high-performance liquid chromatography (HPLC) detection (14). This is remarkable, considering that the HPLC work was done in an acetonitrile/water solvent and used a Tris buffer as the base catalyst. Figure 4 is a plot of
CONCLUSION Although not yet demonstrated to be as sensitive as hy-
drogen peroxide initiated chemiluminescence, PICL has the advantage of being simpler to implement in that photons are easier to introduce than is H202. We have not yet optimized the detection system. In particular, it seems likely that greater sensitivity may be achieved by increasing the volume of the photochemical reactor and/or increasing the lamp intensity so as to produce a higher concentration of photoproducts. That imidazole provides such a large enhancement in the signal suggests that optimization of the catalyst concentration and delay time may lead to further improvements in detection limits. Pulsed laser photolysis studies of the kinetics of the reaction should aid in determining the optimum conditions (reagent concentrations, flow rates, delay times, lamp intensity, etc.) for detection of PICL. Although TCPO has proven to provide the most sensitive detection in Hz02-initiated chem- iluminescence, there is no reason to believe that this oxalate

1054 ANALYTICAL CHEMISTRY, VOL. 62, NO. 10, MAY 15, 1990
T a b l e I . L i m i t s of D e t e c t i o n for Flow I n j e c t i o n A n a l y s i s
compound
6-aminochrysene 1-aminopyrene anthracene 1-aminoanthracene 9-aminophenanthrene 1-aminonaphthalene 2-aminofluorene 2,7-diaminofluorene 1,8-diamino-
rubrene naphthalene
PICL without catalyst,
Pg
65 53
3600 59
340 460
3700 2000 5000
520
PICL with
catalyst, Pg
9.0 6.6
8.5 760
16 53 38
150 170
32
fluorescence detection limit,
Pg (L, Aem) 1.6 (266, >389) 2.8 (240, >370)
69 (240, >370) 84 (200, >37O) 21 (221, >370) 24 (238, >370) 8 (200, >389)
33 (200, >389) 180 1248, >389)
15 (210, >370)
I -9 -8 .7 - 6 - 5
Log (Concentration, M)
Figure 5. Working curves for rubrene and 1-aminopyrene obtained in F I A with base-catalyzed PICL. Error bars represent one standard deviation. Minimum of three measurements per data point.
ester is optimal for PICL. Indeed, an oxalate ester having different photophysical and photochemical properties may be better suited to PICL detection.
Photoinitiated chemiluminescence would be especially useful for detection in gel electrophoresis where addition of HzOz would be problematic. In fact, it was with this appli- cation in mind that the PICL experiments were first initiated. For example, the four fluorophores used in nucleic acid se- quencing (one for each of the four bases) (46) could be replaced with chemilumophores. PICL has the potential of providing much more sensitive detection than that of fluorescence in this application.
There has been a great deal of interest recently in single molecule detection by laser-induced fluorescence (LIF) (47) . PICL has the potential to improve on LIF by delaying the luminescence until long after the laser pulse, thus reducing scattered light and allowing for optimal collection of the lu- minescence. Here, an oxalate ester would be converted to high-energy intermediates by the laser pulse, and chemilu- minescence would follow.
Because PICL requires a hydrogen-atom donor such as IPA, it should be possible to develop a scheme for detecting hy- drogen-atom-donating compounds instead of fluorophores. In this case the analyte would be mixed with an oxalate ester before entering a photochemical reactor and the fluorophore added just prior to the luminescence detector. Alternatively, the fluorophore could be immobilized on silica and held within the detection cell, as in our previous work with immobilized 3-aminofluoranthene (28).
Since many of the side reactions occurring in peroxyoxalate chemiluminescence probably involve hydrogen peroxide, PICL provides a means of investigating the kinetics and mechanism of the peroxyoxalate reaction in a less complicated system. Such studies currently are underway in our laboratory.
ACKNOWLEDGMENT We thank Tad Koch for helpful discussions concerning
possible mechanisms for photoinitiation of peroxyoxalate chemiluminescence.
Registry No. TCPO, 1165-91-9; 02, 7782-44-7; imidazole, 288-32-4; peroxyoxalate, 119666-18-1.
LITERATURE CITED (1) Chandross, E. A. Tetrahedron Lett. 1963, 761-766. (2) Rauhut, M. M.; Boilyky, L. J.; Roberts, B. G.; Loy, M.; Whitman, R. H.;
Ianotta. A. V.; Semsel, A. M.; Ciark, R. A. J. Am. Chem. SOC. 1987, 89, 6515-6522.
(3) Rauhut, M. M. Acc. Chem. Res. 1969, 2 , 80. (4) Tseng, S.-S.; Mohan, A. G.; Haines, L. G.; Vizcarra, L. S.; Rauhut, M.
M. J. Ora. Chem. 1979. 44. 4113-4116. (5) Williams,-D. C.; Huff, G. F.; Seitz. W. R. Anal. Chem. 1976, 48,
1003- 1006. (6) Scott, G.; Seitz. W. R.; Ambrose, J. Anal. Chim. Acta 1980, 775,
221-228. (7) Williams, D. C.; Seitz, W. R. Anal. Chem. 1976, 48, 1478. (8) Curtis. T. G.; Seitz, W. R. J. Chromatogr. 1977, 134, 343-350. (9) Sherman, P. A.; Holzbecher, J.; Ryan, D. E. Ami. Chim. Acta 1978,
97, 21-27. (10) Kobayashi, S.; Imai, K. Anal. Chem. 1980, 52, 424-427. (11) Kobayashi, S.; Sekino, J.; Honda, K.; Imai, K. Anal. Biochem. 1981,
772. 99-104. (12) Sigvardson, K. W.; Birks, J. W. Anal. Chem. 1983, 55, 432-435. (13) Melbin, G. J. Liq. Chromatogr. 1983, 6 , 1603-1616. (14) Sigvardson, K. W.; Kennish, J. M.; Birks, J. W. Anal. Chem. 1984. 56,
1096-1102. (15) Sigvardson, K . W.; Birks, J. W. J. Chromatogr. 1984, 316, 507-518. (16) Weinberger, R. J. Chromatogr. 1984, 374, 155-165. (17) dedong, G. J.; Lammers, N.; Spruit, F. J.; Brinkman, U. A. Th.; Frei, R.
W. Chromatographia 1984, 18, 129-133. (18) Miyaguchi, K.; Honda, K.; Imai, K. J . Chromatogr. 1984, 303,
(19) Melbin, G.; Smith, B. E. F. J. Chromatogr. 1984, 372. 203-210. (20) Koziol, T.; &ayeski, M. L.; Weinberger, R. J. Chromatogr. 1984, 377,
(21) Honda, K.; Miyaguchi, K.; Imai, K. Anal. Chim. Acta 1985, 777, 103- 1 10.
(22) Honda, K.; Miyaguchi, K.; Imai. K. Anal. Chim. Acta 1985, 777, 11 1-120.
(23) van Zoonen. P.; Kamminga, D. A,; Gooijer, C.; Velthorst, N. H.; Frei, R. W. Anal. Chim. Acta 1985, 767, 249-256.
(24) Grayeski, M. L. I n Chemi- and8ioiuminescence; Burr, J. G., Ed.; Dek- ker: New York, 1985; pp 469-473.
(25) van Zoonen, P.; Kamminga, D. A,; Gooijer, C.; Velthorst, N. H.: Frei. R. W.; Gubitz, G. Anal. Chem. 1986, 58, 1245-1248.
(26) Honda, K.; Miyaguchi, K.; Nishino, H.; Tanaka, H.; Yao, T.; Imai, K. Anal. Biochem. 1986, 753, 50-53.
(27) Poulsen, J. R.; Birks, J. W.; van Zoonen, P.; Gooijer, C.; Velthorst, N. H.; Frei. R. W. Chromatographia 1988, 2 1 , 587-595.
(28) Poulsen, J. R.; Birks, J. W.; Gubitz, G.; van Zoonen. P.; Gooijer, C.; Verthorst, N. H.; Frei, R. W. J. Chromatogr. 1986, 360, 371-383.
(29) van Zoonen, P.; de Herder, I.; Gooijer, C.; Verthorst, N. H.; Frei, R. W.; Kuntzberg. E.; Gubitz, G. Anal. Lett. 1986, 79, 1949-1961.
(30) Imai. K.; Matsunaga, Y.; Tsukamoto, Y.; Nishitani. A. J. Chromatogr. 1987, 303, 173-176.
(31) Mann, B.; Grayeski, M. L. J. Chromatogr. 1987, 386, 149-158. (32) Grayeski, M. L.; Devasto. J. K. Anal. Chem. 1987, 59, 1203-1206. (33) Kwakman, P. J. M.; Brinkman, U. A. Th.; Frei, R. W.; DeJong, G. J.;
Spruit, F. J.; Lammers, G. F. M.; van den Berg, J. H. M. Chromato-
173-176.
355-366.
graphia 1987, 24, 395-399. (34) Hanaoka, N.; Givens, R. S.; Schowen, R. L.; Kuwana. T. Anal. Chem.
1988. 60. 2193-2197. (35) Nozaki, 0.; Ohba, Y ; Imai, K. Anai. Chim. Acta 1988. 205,
255-260. (36) Jansen. H.; Brinkman, U. A. Th.; Frei, R. W. J. Chromatogr. 1988,
440, 217-223. (37) Nieman, T. A. I n Chemiluminescence and Photochemical Reaction
Detection in chromatography; Brks, J. W., Ed.; VCH Publishers: New York, 1989; pp 99-147.
(38) Givens. R. S.; Schowen, R. L. I n Chemiluminescence andPhotochem- ical Reaction Detection in Chromatography; Birks, J. W., Ed.; VCH Publishers: New York, 1989; pp 125-147.
(39) Schuster, G. B. Acc. Chem. Res. 1979, 72, 366-373. (40) McCapra, F.; Perring, K.; Hart, R. J.; Hann, R. A . Tetrahedron Lett.
1981, 5087-5090. (41) Catherall, C. L. R.; Palmer, T. F.; Cundall. R. B. J. Chem. Soc.. Fara-
day Trans. 2 1984, 80, 823-036, 837-849. (42) Orlovic. M.; Schowen, R. L.; Givens, R. S.; Alvarez, F.; Matuszewski,
B.; Parekh, N J Org. Chem. 1989, 54, 3606-3610.

Anal. ch8m. 1990, 62, 1055-1060 1055
(43) Aivarer, F. J.; Parekh, N. J.; Matusquewski, 8.; Givens, R. S.; Higveh. 229-237. T.; Schowen, R. L. J . Am. Chem. SOC. 1986, 708, 6435-6437.
(44) Turro, N. J. Modern Molecular Photochemistty; Benjamin: New York,
(45) Brina, I?.; Bard, A. J. J . Electroanal. Chem. Interfacial Electrochem.
(46) Landegren, U.; Kaiser, R.: Caskey. C. T.; Hood, L. Science 1988, 242,
(47) Worthy, W. Chem. Eng. News 1989, 67(41), 21-23.
1981; pp 362-392.
1987, 238, 277-295. RECEIVED for review November 3, 1989. Accepted February 5, 1990.
Isoprene Measurement by Ozone-Induced Chemiluminescence
Alan J. Hills* and Patrick R. Zimmerman National Center for Atmospheric Research (NCAR), P.O. Box 3000, Boulder, Colorado 80307-3000
An instrument has been constructed that monitors gaseous Isoprene contlnuously. The basis for detection is chemliu- minescence wlth ozone. The lsoprene/ozone reactlon pro- duces electronically excited formaldehyde whose subsequent emission to the ground state is vlewed with a blue-sensitive photomuitlplier tube. The instrument has a response time of 0.1 8, Is llnear over 3 orders of magnitude, and has a detec- tlon limit for isoprene of 400 pptv (at S I N = 2 and 5-s eiec- tronic thne constant). Seiectivltles over various alkenes and other compounds are presented. The first real-time Isoprene fluxes from oak leaves, using a single living leaf, are mea- sured as a function of light modulation.
Atmospheric concentrations of isoprene have been of in- terest for many years. Isoprene (2-methyl-1,3-butadiene) is emitted by woody plants and deciduous trees in large quan- tities and its emission rate is species dependent, proportional to solar intensity, and leaf temperature ( 1 ) . It is often the most abundant non-methane hydrocarbon in forest canopies (2-4). Large emission rates combined with an extremely high reactivity toward the hydroxyl radical, OH (kisoprene+OH = 1.01 x cm3 molecule-' 5-l) (5), allow isoprene to dominate the daytime chemistry of atmospheric OH in many areas such as tropical forests (6). The atmospheric oxidation of isoprene is also of importance since the reaction products are important precursors for the formation of O3 in rural atmospheres ( 4 ) . In order to gain a better knowledge of natural hydrocarbon emissions and their impact on atmospheric quality, especially in relation to the anthropogenically modified, polluted at- mosphere, studies have sought to measure ambient isoprene concentrations and profiles. Atmospheric isoprene concen- trations have been measured by using grab sampling into stainless steel cannisters followed by gas chromatography analysis (2-4). Difficulties with this method are: (1) Insta- bility of isoprene is such containers can lead to gradual loss with respect to time (2 , 7 , 8 ) , especially at sub-part-per-bil- lion-by-volume (ppbv) levels and low humidity or in the presence of ambient oxidants, which are also sampled into the cannisters. (2) Chromatographic analysis is relatively labor
technique precludes important eddy correlation flux mea- surements.
Biological interest in isoprene is also very strong since isoprene emission has been linked to primary photosynthetic processes in many types of vegetation (9-11). The photo- synthesis/isoprene emission relationship is thought to be on the order of seconds, but present analytical techniques require several minutes of sampling followed by longer periods of chromatographic analysis time. Thus, fundamental infor- mation regarding the basic mechanistic isoprene/photosyn- thesis relationship is obscured.
Chemiluminescence (CL) is a technique that is amenable to real-time monitoring of certain atmospheric species. I t is selective, owing to the relatively small number of compounds that chemiluminesce upon reaction with a given reactant, and very sensitive since the chemiluminescence appears out of a near zero light background. In principle, a single photon generated from a chemiluminescent reaction can be detected. Useful chemiluminescence systems have been described for NO reacted with O3 (12-14), reduced sulfur and selenium compounds with Fz (15,16) , iodinated hydrocarbons with F2 (17), H2S and CH3SH with OClO (18), and halogenated or- ganics with sodium vapor (19) . The chemiluminescence of alkene/03 reactions has been explored as an ozone monitor (20) , as GC detectors (8, 21, 22) , and as ambient alkene monitors (23,24). Becker et al. (24) did not report a detection limit but Schurath (23) lists a detection limit of 3 ppb for the most sensitively detected alkenes.
We describe here a real time detection system, sensitive to isoprene, based on the chemiluminescence reaction between a primary alkene and ozone
(1)
Olefin/03 mechanistic studies have been made by Pitts et al. (25-29) and others (30). Emission from HCHO* (lA2) occurs in the region 450-550 nm. As a first step toward development of a sub-ppbv ambient atmospheric instrument, an application is presented in which isoprene flux is monitored from 7.1 cm2 of a single oak leaf, in real time. Leaf fluxes were measured as part of a biochemical study relating photosynthetic pro- cesses to isoprene production.
alkene + O3 - HCHO* + products
intensive, costly, and slow. Therefore only a limited number of samples can be collected and analyzed per day. This low
diurnal variability and adds difficulty in relating emission rates to environmental variables. In addition, data are not available in real time, thus prohibiting interactive refinement of ex- perimental procedures. (3) The inherent slowness of the
EXPERIMENTAL SECTION Instrumentation. To determine the best photomultiplier tube
the wavelength range 200-800 nm on a discharge-flow system described earlier (31, 32) coupled to an intensified diode array scanning spectrometer (IDARSS, TN 1710, Tracor-Northern). The IDARSS instrument was wavelength calibrated using a
frequency precludes characterization Of (PMT)/filter combination, O,/dkene spectra were collected Over
0003-2700/90/0362-1055$02.50/0 0 1990 American Chemical Society




















