
Bilayer photovoltaic devices with PPQ as the electron acceptor layer
Upload
independentCategory
view
0download
0
Perfluorination of tetracene: effects on the optical gap and electron-acceptor properties. An electrochemical,
theoretical DFT, and Raman spectroscopic study.
Rocío Ponce Ortiza, Reyes Malavé Osunaa, Mari Carmen Ruiz Delgadoa, Juan Casadoa, Víctor Hernándeza, Juan Teodomiro López Navarretea, Youichi Sakamotob , Toshiyasu Suzukib.
aDepartment of Physical Chemistry, University of Málaga, 29071-Málaga, Spain. bInstitute for Molecular Science, Myodaiji, Okazaki 444-8585, Japan.
ABSTRACT
We report the synthesis and characterization of perfluorinated tetracene; a material with potential applications in organic electronics. The electrochemical behaviour of the compound is analyzed by differential pulse voltammetry, and compared with that of tetracene. The structure of perfluorotetracene is planar as observed for pentacene. We also report a comparative Raman spectroscopic study of tetracene and perfluorotetracene in relation to their π-conjugational properties. Density functional theory (DFT) calculations have been also performed, at the B3LYP/6-31G** level, to assess information regarding the topologies and energies of the frontier molecular orbitals (MOs) around the gap, and about the vibrational normal modes associated with the Raman features selectively enhanced by the π-conjugation. Keywords: Raman spectroscopy, tetracene, perfluorination, π–conjugation, DFT calculations.
1. INTRODUCTION Organic thin-film transistors (OTFTs) using organic semiconductors as an active layer are of great interest for their use in lightweight, low-cost, large-area and flexible electronic products such as flat-panel displays, smart cards, and radio-frequency (rf) tags. OTFTs are more compatible with polymeric substrates than conventional silicon-based transistors because they can be fabricated by a low-temperature process. The performance of OTFTs has been much improved in the last two decades through various innovations such as the development of new organic semiconductors and the optimization of the deposition conditions to control the ordering of the molecules.1 Acene materials, which consist of n-linked benzene rings (n=3: anthracene; n=4: tetracene; n=5: pentacene), are promising candidates for organic semiconductors because their planar shapes facilitate crystal packing and the extended π-system over molecules enables the intermolecular overlap of π-systems. Tetracence OTFTs have shown a field effect mobility of 0.01 cm2/V s on an untreated silicon dioxide (SiO2) gate insulator and 0.1 cm2/V s on chemically treated SiO2. Of all the OTFT materials reported so far, pentacene OTFTS fabricated on the octadecylchlorosilane (OTS) treated substrates have achieved a maximum field effect mobility of 1.7 cm2/V s and a current on/off ratio of 108,2 values which are comparable to those of amorphous silicon. When one considers producing bipolar transistors and complementary circuits with acenes, the n-type organic semiconductor should have similar physical and electrical properties than the p-type semiconductor except for the type of carriers.3 Some of us have previously demonstrated that aromatic perfluorocarbons such as perfluoro-p-sexiphenyl (C36F26) were efficient n-type semiconductors for the electron-transport layer of organic-light-emitting diodes.4 Because fluorine is the most electronegative of all the elements and relatively small (hydrogen < fluorine < carbon), perfluorination is a straightforward way to convert a p-type organic semiconductor to a n-type one, without changing the molecular size greatly. Molecular spectroscopy is a fundamental tool to establish structure-property relationships guiding the design of new and improved molecular materials. In particular, Raman spectroscopy is very well suited for the study of conjugated systems. Raman frequencies and intensities are experimental observables emerging directly from the π-conjugated backbone which account for the most important electronic signature of oligothiophenes and related materials. The Effective Conjugation Coordinate (ECC) model predicts two main trends for the Raman spectral profiles of conjugated molecules: (a) selective enhancement of particular scatterings associated with collective C=C/C–C stretching vibrations of the conjugated path.5 (this phenomenon relates to the occurrence of an electron-phonon mechanism which is at the origin of
Organic Optoelectronics and Photonics II, edited by Paul L. Heremans, Michele Muccini, Eric A. Meulenkamp,Proc. of SPIE Vol. 6192, 61922V, (2006) · 0277-786X/06/$15 · doi: 10.1117/12.662526
Proc. of SPIE Vol. 6192 61922V-1

their outstanding optical and electrical features) and (b) frequency downshift of these intense bands upon relaxation of the skeletal structure as the consequence of either greater π-electron conjugation in the neutral state or quinoidization induced by ionization. When these spectroscopic data are combined with quantum chemical calculations it is possible to assess precisely relevant molecular parameters which would be very difficult to evaluate by conventional experimental techniques. First principles quantum chemical calculations in the framework of DFT theory are very well suited to model extended π-conjugated systems due to implemented electron-correlation effects. We report herein the synthesis of perfluorotetracene (C18F12) as a potential n-type semiconductor for OFETs. The conjugational properties of this new material are analyzed by means of FT-Raman spectroscopy. We also perform a comparative study of the optical and electrochemical properties of perfluorotetracene and tetracene by using differential pulse voltammetry and UV-Vis-NIR absorption spectroscopy. The whole set of experimental data are interpreted with the help of DFT and TDDFT quantum chemical calculations, at the B3LYP/6-31G** level, to assess information about the optimized molecular structure, equilibrium atomic charges distribution, energies and topologies of the frontier Molecular Orbitals (MO) around the gap, vibrational normal modes associated to the most outstanding Raman scatterings, and vertical one-electron excitations which give rise to the main optical absorptions.
2. EXPERIMENTAL AND THEORETICAL DETAILS Perfluorotetracene (C18F12) was synthesized, as a potential n-type semiconductor, according to Scheme 1. The Diels-Alder reaction of tetrafluorobenzyne and 2-methoxyfuran followed by methylation with dimethyl sulfate gave 1,2,3,4-tetrafluoro-5,8-dimethoxynaphthalene (1) in 20% yield. The Friedel-Crafts reaction of tetrafluorophthalic anhydride with 1 in the presence of aluminum chloride and sodium chloride at 200 °C provided 5,12-tetracenedione 2 in 79% yield. Fluorination of 2 with sulfur tetrafluoride in hydrogen fluoride at 150 °C afforded perfluoro-(5,6,11,12-tetrahydrotetracene) (3) in 38% yield. Defluorination of 3 with zinc at 280 °C gave perfluorotetracene in 54 % yield. Perfluorotetracene was purified by train sublimation and used for characterization. It is soluble in organic solvents such as chloroform and toluene. The structure was determined by 19F NMR, mass spectrometry and elemental analysis. Melting points were obtained on a Büchi melting point apparatus B-540. 1H and 19F NMR spectra were recorded on a JEOL JNM-LA 500 at 500 and 470.4 MHz, respectively. Chemical shifts were reported as δ values (ppm) relative to internal tetramethylsilane (1H NMR) or hexafluorobenzene (19F NMR). EI mass spectra were collected on a Shimadzu GCMS-QP5050A at 70 eV. UV-vis spectra were recorded on a JASCO V-570 spectrophotometer. Emission spectra were collected on a JASCO FP-6600 spectrofluorophotometer. The calorimetric data were obtained on a TA Instruments DSC 2920 at a scanning rate 10 °C min-1 under a continous flow (50 mL min-1) of argon. Analytical TLCs were performed on commercial Merck plates coated with silica gel 60 F254. Flash chromatographic separations were carried out on Fuji Silysia FL60D. FT-Raman scattering spectra were collected on a Bruker FRA106/S apparatus and a Nd:YAG laser source (λexc = 1064 nm), in a back-scattering configuration. The operating power for the exciting laser radiation was kept to 100 mW in all the experiments. Samples were analyzed as pure solids in sealed capillaries. Typically, 1000 scans with 2 cm-1 spectral resolution were averaged to optimize the signal-to-noise ratio. Cyclic (CV) and differential pulse (DPV) voltammetries were recorded on a BAS-100B/W electrochemical analyzer, by using a three-electrode configuration. All measurements were performed at room temperature under an argon atmosphere in 0.1 M 1,2-dichlorobenzene solution of (n-Bu)4NPF6. Concentrations in electrochemical experiments ranged from 10-4 to 10-3 M. A Pt disk working electrode (1-mm diameter) was polished with 0.05-µm alumina before measurements. A Pt wire was used as the counter electrode. The reference electrode was an Ag/0.01 M AgNO3 electrode filled with 0.1 M (n-Bu)4NClO4 in CH3CN. All potentials quoted in the paper are referenced to the ferrocene/ferrocenium couple (Fc/Fc+) as the internal standard. Density Functional Theory (DFT) calculations were carried out by means of the Gaussian 03 program6 running on SGI Origin 2000 supercomputer. We used the Becke's three-parameter exchange functional combined with the LYP correlation functional (B3LYP).7 It has already been shown that the B3LYP functional yields similar geometries for medium–sized molecules as MP2 calculations do with the same basis sets.8,9 Moreover, the DFT force fields calculated using the B3LYP functional yield infrared spectra in very good agreement with experiments.10,11 We also made use of
Proc. of SPIE Vol. 6192 61922V-2

F MeO F OMe
F-LBr +1) n-BuLi, ether, -78 00 F
FF 2) K2003 Me2SO4 reflux FF F OMe
F 0 1 (20%)
OH 0F o SF4
F HF, 150°CA1013 NaCI, 200 °C FF OHO F
(f3U/o) F F F FF F FF F FZn
280 00FFFFFF F F F F3 (38%) (54%)
the standard 6-31G* basis set.12 Optimal geometries were determined on isolated entities. All geometrical parameters were allowed to vary independently apart from planarity of the rings. On the resulting ground-state optimized geometries, harmonic vibrational frequencies and IR and Raman intensities were calculated with the B3LYP functional. We used the often-practiced adjustment of the theoretical force fields in which calculated harmonic vibrational frequencies are uniformly scaled down by a factor of 0.96 for the 6-31G** calculations, as recommended by Scott and Radom.9 This scaling procedure is often accurate enough to disentangle serious experimental misassignments. All quoted vibrational frequencies reported along the manuscript are thus scaled values. The theoretical spectra were obtaining by convoluting the scaled frequencies with Gaussian functions (10 cm-1 width at the half-height). The relative heights of the Gaussians were determined from the theoretical Raman scattering activities. Vertical one-electron excitations were computed by using the time-dependent DFT (TDDFT) approach.13,14 The twenty lowest-energy electronic excited states were computed for all the molecules. The computational cost of TDDFT is roughly comparable to that of single-excitation theories based on an HF ground state, such as single-excitation configuration interactions (CIS). Numerical applications reported so far indicate that TDDFT formalism employing current exchange-correlation functionals performs significantly better than HF-based single excitation theories for the low-lying valence excited states of both closed-shell and open-shell molecules.15,16 TDDFT calculations were carried out using the B3LYP functional and the 6-31G** basis set on the previously optimized molecular geometries obtained at the same level of calculation. Molecular orbital contours were plotted using Molekel 4.3.17
Scheme 1. Synthesis of perfluotetracene
3. RESULTS AND DISCUSSION 3.1 UV-Vis absorption data Figure 1 displays the UV-Vis absorption spectra of tetracene and perfluorotetracene in 1,2-dichlorobenzene. The HOMO-LUMO gaps obtained from the onset of absorption are 2.54 eV for tetracene and 2.39 eV for perfluorotetracene. Vibronic components are clearly distinguishable for the two oligoacenes, consistent with electromagnetic absorption from a single molecular conformation in solution. On the other hand, the fluorescence emissions in 1,2-dichlorobenzene also exhibit a redshift in passing from tetracene (2.57 eV) to perfluorotetracene (2.38 eV). Finally, we observe that the
Proc. of SPIE Vol. 6192 61922V-3

Stokes shift is rather small for these two fused systems, consistent with a quite close geometric match between the ground and the first singlet excited states for both tetracene and its perfluorinated counterpart.
Figure 1: Absorption (left) and emission (right) spectra of tetracene and perfluorotetracene in 1,2-dichlorobenzene. 3.2 TDDFT calculations on electronic transitions To gain insight into the nature of the UV-Vis absorptions, the twenty lowest-energy electronic excited states of tetracene and perfluorotetracene were computed at the B3LYP/6-31G** level using the TDDFT approach. Calculations predict the appearance of only one visible absorption band for tetracene, but two optical transitions for perfluorotetracene. The UV-Vis bands, measured in 1,2-dichlorobenzene solution, at 2.60 eV (tetracene) and 2.45 eV (perfluorotetracene) correspond to the excitation to the first singlet excited state, which is computed at 2.49 eV for tetracene (with a oscillator strength, f, of 0.05) and at 2.30 eV for perfluorotetracene (f = 0.05). This transition is mainly described by a one-electron vertical excitation from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). Thus, TDDFT model chemistry reproduces, with good accuracy, the experimental wavelengths for the optical transitions of these two oligoacenes, and also nicely accounts for the redshift of the π-π* transition in passing from tetracene to its perfluorinated counterpart. Both the HOMO and LUMO are of π-nature and spread over the whole frame of fused rings (see Figure 2). In addition, from the comparison between the B3LYP/6-31G** energy levels around the band gap region for both acenes (also shown in Figure 2) we learn that perfluorination of tetracene causes an overall stabilization of both the doubly occupied and empty frontier orbitals. Furthermore, we observe that perfluorination downshifts the LUMO more than the HOMO (i.e., by 0.964 and 0.758 eV, respectively), thus leading to a narrowing, by ≈ 0.2 eV, of the HOMO-LUMO gap from 2.77 eV in tetracene to 2.57 eV in perfluorotetracene. As depicted in Figure 2, the topologies of the frontier MOs of perfluorotetracene are almost identical to those of the related MOs in tetracene, except for the small electron density on the peripheral fluorine atoms either in the doubly occupied (i.e., HOMO, HOMO-1, and so on) or empty (i.e., LUMO, LUMO+1, and so on) wavefunctions. Finally, we also observe that upon perfluorination there occurs a crossing between the LUMO+1 and LUMO+2 energy levels, since the former orbital is largely stabilized, by 1.45 eV, with respect to the non-fluorinated acene, whereas the latter orbital is “only” stabilized by 0.93 eV.
0.0
0.2
0.4
0.6
0.8
1.0
350 400 450 500 550
UV-
vis
Abs
orba
nce
Wavelength (nm)
Perfluoro-tetracene
Tetracene
0.0
0.2
0.4
0.6
0.8
1.0
400 450 500 550 600 650 700 750Em
issi
on In
tens
ity (a
rbitr
ary
units
)Wavelength (nm)
Perfluoro-tetracene
Tetracene
Proc. of SPIE Vol. 6192 61922V-4

-0.38 __________
K -044
LUMO+1 -1.37
ur -2.09'N -1.83
-3.06
-4.87
HOMO', -5.63
- /-6.34
--
HOMO-1 ''-- -6.99
(a) (b)
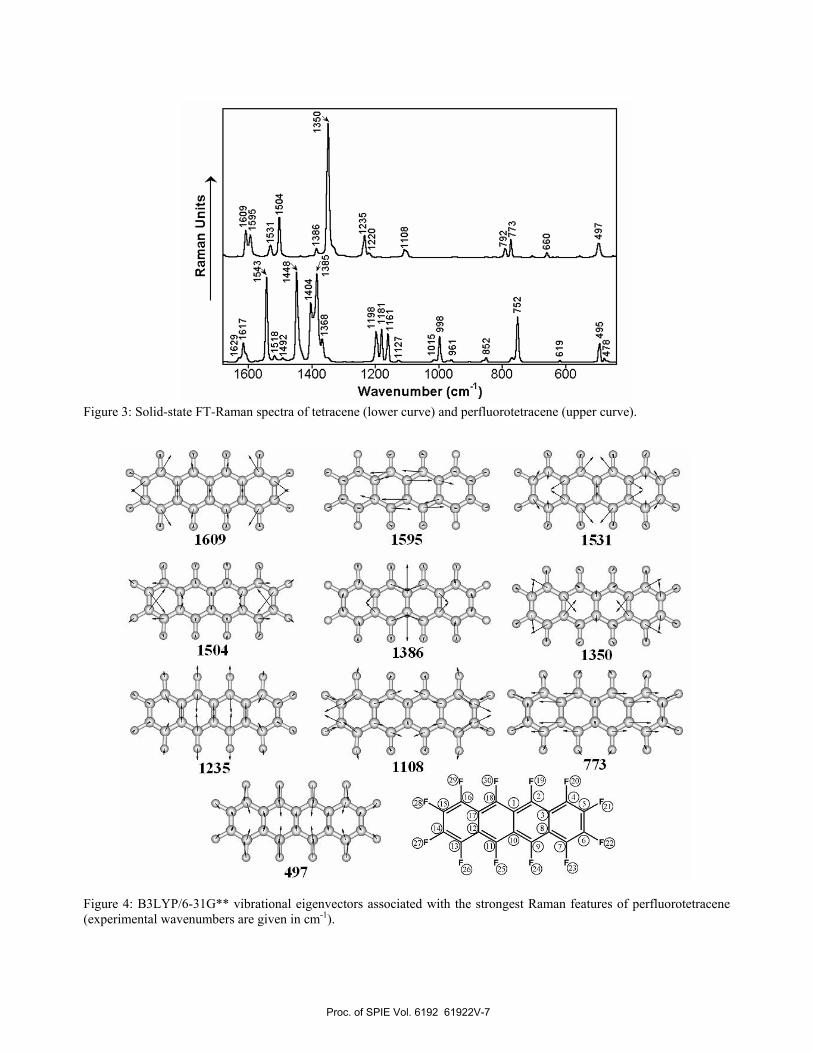
Figure 2: B3LYP/6-31G** electronic density contours (0.03 e/bohr3) and absolute energies (in eV) for some selected frontier MOs around the band gap region for tetracene (a) and perfluorotetracene (b). On the other hand, the sharp electromagnetic absorption of perfluorotetracene measured at 2.97 eV must be correlated with the TDDFT//B3LYP/6-31G** electronic transition at 3.28 eV (with a oscillator strength, f, of 0.19). TDDFT calculations also reveal that the corresponding vertical one-electron excitation can be mainly described as the HOMO → LUMO+1 transition. As a consequence of the aforementioned crossing between the LUMO+1 and LUMO+2 energy levels in passing from tetracene to perfluorotetracene, the electromagnetic absorption due to the related HOMO → LUMO+2 transition is expected to occur at significantly higher energies for the non-perfluorinated acene, as indeed predicted by the TDDFT//B3LYP/6-31G**calculations, which compute this vertical one-electron excitation to appear at 4.75 eV. 3.3 Experimental and theoretical Raman spectra Figure 3 shows the FT-Raman spectra recorded for tetracene and perfluorotetracene, as pure solid samples. It turns out that perfluorotetracene displays a much more simple Raman spectral profile than tetracene, in spite that these two acenes have the same number of atoms and belong to the same symmetry point group (D2h). In this regard, we observe that, while tetracene displays several Raman scatterings with comparable intensities at 1543, 1448, 1404, 1385, 1198, 1181, 1161, 998, 752 and 495 cm-1; the strongest Raman feature for perfluorotetracene is by far that recorded at 1350 cm-1. From the earliest studies on electrically conducting polymers, Raman spectroscopy has been widely used to characterize many different types of π-conjugated systems, both oligomers and polymers, with particular attention to: (i) the analysis of the effectiveness of the π-conjugation along a homologous series of oligomers,18,19 (ii) the characterization of different types of conjugational defects induced by either chemical doping or photoexcitation,20 or (iii) the “quantification” of the degree of intramolecular charge transfer in push-pull π-conjugated chromophores.21,22
Proc. of SPIE Vol. 6192 61922V-5

The intriguing observation of a limited number of overwhelmingly strong Raman scatterings, even for systems having complex chemical structures,19f,20f was definitively accounted for over twenty years ago by G. Zerbi and co-workers through the development of the Effective Conjugation Coordinate (ECC) formalism. This model, which is now well-accepted, postulates the existence of a unique collective C=C/C–C stretching mode strongly involved in the electron-phonon coupling inherent to these one-dimensional π-conjugated chains.5 In α-linked aromatic π-conjugated oligomers, the so-termed collective ECC coordinate has the analytic form of a linear combination of ring C=C/C–C stretchings, which points in the direction from an aromatic structure (usually that of the ground electronic state) to a quinonoid one (usually that corresponding to the first dipole allowed electronic excited state). The ECC formalism states that the totally symmetric C=C/C–C stretching modes entering in the lattice dynamics of the ECC vibrational coordinate, that is those which give rise to the few and selectively enhanced Raman features, should undergo large dispersions both in peak position and intensity on increasing conjugation length along a given set of neutral oligomers. Thus, changes in Raman frequencies and relative intensities with increasing chain length are particularly useful in evaluating the mean conjugation length for a given family of π-conjugated compounds. Furthermore, upon chemical or electrochemical oxidation/reduction of these π-conjugated heteroaromatic systems, various types of quinonoid-like charged defects are created.23 The subsequent quinoidization of the π-conjugated frame gives rise to a further redshift of the strongest Raman lines due to the progressive softening of the conjugated C=C bonds, which is a marker of the type of charged defects created upon oxidation or reduction.19,20 To gain a deeper insight into the FT-Raman spectral profiles of these two acenes, we decided to carry out vibrational analyses at the B3LYP/6-31G** level of theory. Figure 4 depicts the vibrational eigenvectors associated with the strongest Raman features of perfluorotetracene, together with the measured wavenumbers. We can see from this figure that the ν(CC) stretching mode of perfluorotetracene at 1350 cm-1 much more strongly couples to the LUMO than the other Raman-active normal modes of this fused compound. This can be understood as follows. The reduced mass for the Ag vibration at 1350 cm-1 is 12.19, therefore, the displacements of carbon atoms are much larger than those of fluorine atoms along this mode. When perfluorotetracene becomes distorted along this particular Ag vibration, toward the same direction as shown in Figure 4, the bonding (antibonding) interactions between two neighboring carbon atoms become stronger (weaker), and the antibonding interactions between two neighboring carbon and fluorine atoms in the LUMO become weaker. Therefore, the LUMO is largely stabilized in energy by such molecular distortion. This is the reason why the Ag ν(CC) stretching mode of perfluorotetracene at 1350 cm-1 strongly couples to the LUMO. We also learn from Figure 4 that the Raman-active Ag vibrations at 1504 and 1235 cm-1 can also couple somewhat to the LUMO of perfluorotetracene. This can be rationalized in the following terms: when perfluorotetracene becomes distorted along the Ag mode at 1504 cm-1, the bonding interactions between C(5) and C(6) atoms, and between C(14) and C(15) atoms become stronger, whereas the antibonding interactions in the LUMO between C(5) and F(21) atoms, between C(6) and F(22) atoms, between C(14) and F(27) atoms, and between C(15) and F(28) atoms become weaker (atom numbering is also displayed in Figure 4). Nonetheless, the bonding interactions between C(3) and C(4) atoms, between C(7) and C(8) atoms, between C(12) and C(13) atoms, and between C(16) and C(17) atoms, become weaker along the aforementioned Ag mode at 1504 cm-1; and thus the LUMO would be slightly destabilized by such an effect; which is however overcompensated by the aforementioned stabilization, being the reason why the Raman-active Ag vibration at 1504 cm-1 couples to a lower extent to the LUMO than the above Ag mode at 1350 cm-1, even though both Raman-active Ag modes arise from two closely related ν(CC) stretching vibrations, and they have similar reduced masses. Finally, when the π-conjugated backbone of perfluorotetracene becomes distorted along the ν(C–F) stretching vibration at 1235 cm-1, the weak bonding interactions in the LUMO between C(3) and C(8) atoms, and between C(12) and C(17) atoms become somewhat stronger; whereas the antibonding interactions between C(2) and F(19) atoms, between C(9) and F(24) atoms, between C(11) and F(25) atoms, and between C(18) and F(30) atoms, become weaker; therefore, the LUMO is estabilized along such a molecular distortion. However, we see that there are not significant variations along this vibrational mode in the strengths of the main orbital interactions between each pair of neighboring carbon atoms. Hence, the Ag ν(C–F) mode at 1235 cm-1 is much less coupled to the LUMO than the Ag ν(CC) stretching at 1350 cm-1.
Proc. of SPIE Vol. 6192 61922V-6
Ram
an U
nits
1629
C
) >
1617
16
09
o 15
95
o 15
31
cmio
14
48
1404
13
50
o __
____
o
•__S
8 13
85
____
____
____
____
___
F')
1198
12
20
E
1235
hf1
1127
1015
1108
o 99
8 o
961
852
0,
o 79
2
—75
2 66
0 61
9 0
495
497
Figure 3: Solid-state FT-Raman spectra of tetracene (lower curve) and perfluorotetracene (upper curve).
Figure 4: B3LYP/6-31G** vibrational eigenvectors associated with the strongest Raman features of perfluorotetracene (experimental wavenumbers are given in cm-1).
Proc. of SPIE Vol. 6192 61922V-7

w
UI
0 0
3.4 X-ray structure Single crystals of perfluorotetracene were successfully grown by slow sublimation at 190 °C under a flow of 250 Pa of argon. A reddish-orange plate was used for X-ray crystallography.24 The structure of perfluorotetracene is planar as observed for tetracene.25 The C-C bond distances range from 1.34 to 1.45 Å, and the C-F bond distances (1.34-1.35 Å) are typical of aromatic organofluorine compounds. As illustrate in Figure 5, both tetracene and perfluorotetracene adopt herringbone structures with angles of 51.3° and 91.1°, respectively. The shortest C-C intermolecular contacts for perfluorotetracene (3.30-3.31 Å), indicated by arrows in Figure 6, are lower than the sum of van der Waals radii (3.40 Å). The interplanar distance within the π-stacks, 3.28 Å, is also shorter than the layer separation of graphite (3.35 Å). The molecular structure and packing of perfluorotetracene are quite similar to those of perfluoropentacene (C-C bonds: 1.35-1.45 Å; C-F bonds: 1.34-1.35 Å; herringbone angle: 91.2°; short C-C contacts: 3.22-3.25 Å) except for the Z values (perfluorotetracene: 4; perfluoropentace: 2).26
Figure 5: Molecular packing diagrams of (a) tetracene and (b) perfluorotetracene.
Figure 6: Molecular arrangement of perfluorotetracene. 3.5 Electrochemical data Figure 7 displays the electrochemical measurements recorded for tetracene and perfluorotetracene in 1,2-dichlorobenzene. The DPV of tetracene shows a reduction peak at -2.13 V and an oxidation peak at 0.46 V; whereas for perfluorotetracene both the reduction and oxidation peaks are, as expected, positively shifted (i.e., -1.49 and 1.02 V, respectively). Thus, the HOMO-LUMO gaps determined from the redox peaks are 2.59 eV for tetracene and 2.51 eV for perfluorotetracene. In polar solvents such as THF, perfluorotetracene shows a reversible reduction wave at -1.37 V (Figure 7). Thus, the DFT calculations are in agreement with the experimental results (i.e., as mentioned above, the HOMO-LUMO gaps calculated at the B3LYP/6-31G** level of theory are 2.77 eV for tetracene and 2.57 eV for perfluorotetracene).
Proc. of SPIE Vol. 6192 61922V-8

Figure 7: DPV (left) and CV (right) waves of tetracene and perfluorotetracene. 3.6 Field-effect mobilities The n-channel FET activity was observed for OFETs based on perfluorotetracene (electron mobility: 4 x 10-4 cm2 V-1 s-1; on/off ratio: 103).27 Efforts are underway to improve the FET performance of perfluorotetracene.
4. CONCLUSIONS We have report the synthesis and characterization of perfluorinated tetracene; a material with potential applications in organic electronics. The detailed analysis of the electromagnetic absorption and fluorescence emission spectra of tetracene and perfluorotetracene, performed with the guidance of TDDFT quantum chemical calculations, reveals a narrowing of the gap in passing from the former to the latter system. The electrochemical study of these two acenes also indicates that perfluorotetracene has a smaller HOMO-LUMO gap than tetracene. On the other hand, the DFT//B3LYP/6-31G** study of both fused systems shows that, whereas the topologies of the frontier molecular orbitals of perfluorotetracene greatly resemble those of the corresponding MOs in tetracene (i.e., except for the small electron density on the fluorine atoms); both the doubly-occupied and the empty energy levels around the band gap region are significantly more stable for perfluorotetracene than for tetracene, what is a direct consequence of the presence in the former system of very many peripheral fluorine atoms (i.e., the most electronegative of all the elements) surrounding the π-conjugated frame. X-ray diffraction data indicate that perfluotetracene, like perfluoropentacene, displays a fully flat molecular structure and adopt a π-stacked packing motif. The comparative analysis of the Raman spectra of perfluorotetracene and tetracene in relation to their π-conjugational properties (carried our with the help of suited B3LYP/6-31G** vibrational calculations) shows that the strongest by far Raman feature of perfluorotetracene at 1350 cm-1 is due to a skeletal Ag ν(CC) stretching mode strongly coupled to the LUMO of this fused system. Finally, efforts are underway to improve the FET performance of perfluorotetracene.
ACKNOWLEDGMENTS This work was supported by Japan Society for the Promotion of Science (Grant-in-Aid for Scientific Research B14340226 and Grant-in-Aid for Young Scientists B15750161). We thank Dr. M. Akita for help in X-ray structural determination. Research at the University of Málaga was supported by the Ministerio de Educación y Ciencia (MEC) of Spain through project BQU2003-03194, and by the Junta de Andalucía for funding our FQM-0159 scientific group. M.C.R.D, R.P.O and R.M.O. are also grateful to MEC and Junta de Andalucía for their personal doctoral grants.
-2.5-2.0-1.5-1.0-0.50.00.51.01.5E (V) vs Fc/Fc+
4 µA
Perfluoro-tetracene
Tetracene4 µA
-2.0-1.5-1.0-0.50.0E (V) vs Fc/Fc+
2 µA
2 µA
Perfluoro-tetracene
Tetracene
-1.69
-1.34-1.41
-1.33
Proc. of SPIE Vol. 6192 61922V-9

REFERENCES [1] (a) Katz, H. E.; Bao, Z.; Gilant, S. L. Acc. Chem. Res., 2001, 34, 359. (b) Lin, Y. Y.; Gundlach, D. J.; Nelson, S. F.; Jackson, T. N. IEEE Electron Device Lett. 1997, 18, 606. (c) Meyer zu Heringdorf, F. J.; Reuter, M. C.; Tromp, R. M. Nature (London), 2001, 412, 517. [2] Gundlach, D. J.; Klauk, H.; Cheraw, C. D.; Kuo, C. C.; Huang, J. R.; Jackson, T. N. Proc. IEDM 1999, 99, 111. [3] (a) Dodabalapur, A.; Katz, H. E.; Torsi, L.; Haddon, R. C. Science 1995, 269, 1560. (b) Dodabalapur, A.; Katz, H. E.; Torsi, L.; Haddon, R. C. Appl. Phys. Lett. 1996, 68, 1108. (c) Dodabalapur, A.; Laquindanum, J.; Katz, H. E.; Bao, Z. Appl. Phys. Lett. 1996, 69, 4227. (d) Lin, Y. Y.; Dodabalapur, A.; Sarpeshkar, R.; Bao, Z.; Li, W.; Baldwin, K.; Raju, V. R.; Katz, H. E. Appl. Phys. Lett. 1999, 74, 2714. [4] Heidenhain, S. B.; Sakamoto, Y.; Suzuki, T.; Miura, A.; Fujikawa, H.; Mori, T.; Tokito, S.; Taga, Y. J. Am. Chem. Soc. 2000, 122, 10240. [5] (a) Zerbi, G.; Castiglioni, C.; Del Zoppo, M. Electronic Materials: The Oligomer Approach; Wiley–VCH: Weinheim, 1998; p. 345. (b) Castiglioni, C.; Gussoni, M.; Lopez Navarrete, J. T.; Zerbi, G. Solid State Commun. 1988, 65, 625. (c) Lopez Navarrete, J. T.; Zerbi, G. J. Chem. Phys. 1991, 94, 957 and 965 (d) Hernandez, V.; Castiglioni, C.; Del Zoppo, M.; Zerbi, G. Phys. Rev. B 1994, 50, 9815. (e) Agosti, E.; Rivola, M.; Hernandez, V.; Del Zoppo, M.; Zerbi, G. Synth. Met. 1999, 100, 101. G. (f) Zerbi, Handbook of Conducting Polymers; Marcel Dekker: New York, 1998. [6] Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A. Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision B.04; Gaussian Inc.: Pittsburgh PA, 2003. [7] Becke, A. D. J. Chem. Phys. 1993, 98, 1372. [8] Stephens, P. J.; Devlin, F. J.; Chabalowski, F. C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623. [9] Novoa, J. J.; Sosa, C. J. Phys. Chem. 1995, 99, 15837. [10] Scott, A. P.; Radom, L. J. Phys. Chem. 1996, 100, 16502. [11] Rauhut, G.; Pulay, P. J. Phys. Chem. 1995, 99, 3093. [12] Francl, M. M.; Pietro, W. J.; Hehre, W. J.; Binkley, J. S.; Gordon, M. S.; Defrees, D. J.; Pople, J. A. J. Chem. Phys. 1982, 77, 3654. [13] Runge, E..; Gross, E. K. U. Phys.Rev..Lett. 1984, 52, 997. Gross, E. K. U.; Kohn, W. Adv.Quantum Chem 1990, 21, 255. Gross, E. K. U. and Driezler, R.M., Eds.; Plenum Press: New York, 1995; p.149. [14] Casida, M. E. Recent Advances in Density Functional Methods, Part I; Chong, D.P., Ed.; World Scientific: Singapore, 1995; p.115. [15] Koch, W.; Holthausen, M. C. A Chemist's Guide to Density Functional Theory; Wiley-VCH:Weinheim, 2000. [16] Casado, J.; Miller, L.L.; Mann, K.R.; Pappenfus, T.M.; Kanemitsu, Y.; Ortí, E.; Viruela, P.M.; Pou-Amérigo, R.; Hernandez, V.; López Navarrete, J. T. J. Phys. Chem. B 2002, 106, 3872. [17] Portmann, S.; Lüthi, H. P. Chimia 2000, 54, 766–770. [18] (a) Sakamoto, A.; Furukawa, Y.; Tasumi, M. J. Phys. Chem. 1994, 98, 4635.(b) Yokonuma, N.; Furukawa, Y.; Tasumi, M.; Kuroda, M.; Nakayama, J. Chem. Phys. Lett. 1996, 255, 431. (c) Harada, I.; Furukawa, Y. Vibrational Spectra and Structure; Durig, J., Ed.; Elsevier: Amsterdam, 1991, Vol. 19, p. 369. [19] (a) Hernandez, V.; Casado, J.; Ramirez, F.J.; Zotti, G.; Hotta, S.; Lopez Navarrete, J. T. J. Chem. Phys. 1996, 104, 9271. (b) Casado, J.; Hernandez, V.; Hotta, S.; Lopez Navarrete, J. T. J. Chem. Phys. 1998, 109, 10419. (c) Moreno Castro, C.; Ruiz Delgado, M. C.; Hernandez, V.; Hotta, S.; Casado, J.; Lopez Navarrete, J. T. J. Chem. Phys. 2002, 116, 10419. (d) Moreno Castro, C.; Ruiz Delgado, M. C.; Hernandez, V.; Shirota, Y.; Casado, J.; Lopez Navarrete, J. T. J. Phys. Chem. B 2002, 106, 7163. (e) Ruiz Delgado, M. C.; Hernandez, V.; Lopez Navarrete, J. T.; Tanaka, S.; Yamashita, Y. J. Phys. Chem. B 2004, 108, 2516. (f) Ruiz Delgado, M. C.; Casado, J.; Hernandez, V.; Lopez Navarrete, J. T.; Fuhrmann, G.; Bauerle, P. J. Phys. Chem. B 2004, 108, 3158. (g) Casado, J.; Ponce Ortiz, R.; Ruiz Delgado, M. C.; Azumi, R.; Oakley, R. T.; Hernandez, V.; Lopez Navarrete, J. T. J. Phys. Chem. B 2005, 109, 10115.
Proc. of SPIE Vol. 6192 61922V-10

[20] (a) Casado, J.; Hernandez, V.; Hotta, S.; Lopez Navarrete, J. T. J. Chem. Phys. 1998, 109, 10419. (b) Casado, J.; Hernandez, V.; Hotta, S.; Lopez Navarrete, J. T. Adv. Mater. 1998, 10, 1258. (c) Casado, J.; Miller, L.L.; Mann, K.R.; Pappenfus, T.M.; Kanemitsu, Y.; Orti, E.; Viruela, P.M.; Pou-Amerigo, P.; Hernandez, V.; Lopez Navarrete, J. T. J. Phys. Chem. B 2002, 106, 3872. (d) Casado, J.; Miller, L.L.; Mann, K.R.; Pappenfus, T.M.; Hernandez, V.; Lopez Navarrete, J. T. J. Phys. Chem. B 2002, 106, 3597. (e) Casado, J.; Ruiz Delgado, M. C.; Shirota, Y.; Hernandez, V.; Lopez Navarrete, J. T. J. Phys. Chem. B 2003, 107, 2637. (f) Casado, J.; Hernandez, V.; Ponce Ortiz, R.; Ruiz Delgado, M. C.; Lopez Navarrete, J. T.; Fuhrmann, G.; Bauerle, P. J. Raman Spectrosc. 2004, 35, 592. [21] (a) Hernandez, V.; Casado, J.; Effenberger, F.; Lopez Navarrete, J. T. J. Chem. Phys. 2000, 112, 5105. (b) Delgado Ledesma, S.; Ponce Ortiz, R.; Ruiz Delgado, M. C.; Vida, Y.; Perez-Inestrosa, E.; Casado, J.; Hernandez, V.; Kim, O.-K.; Lehn, J.-M.; Lopez Navarrete, J. T. Chem. Eur. J. 2004, 10, 3805. [22] (a) Gonzalez, M.; Segura, J. L.; Seoane, C.; Martin, N.; Garin, J.; Orduna, J.; Alcala, R.; Villacampa, B.; Hernandez, V.; Lopez Navarrete, J. T. J. Org. Chem. 2001, 66, 8872. (b) Casado, J.; Pappenfus, T. M.; Miller, L. L.; Mann, K. R.; Orti, E.; Viruela, P. M.; Pou-Amerigo, P.; Hernandez, V.; Lopez Navarrete, J. T. J. Am. Chem. Soc., 2003, 125, 2534. [23] (a) Ehrendorfer, Ch.; Karpfen, A. J. Phys. Chem. 1994, 98, 7492. (b) Ehrendorfer, Ch.; Karpfen, A. J. Phys. Chem. 1995, 99, 5341. [24] Crystal data for perfluorotetracene: monoclinic, space group P21/c, a = 26.750(5) Å, b = 4.5921(9) Å, c = 11.482(2) Å, β = 101.434(4)°, V = 1382.5(5) Å3, T = 293 K, Z = 4, R = 0.034, GOF = 0.82. [25] Holmes, D.; Kumaraswamy, S.; Matzger, A. J.; Vollhardt, K. P. C. Chem. Eur. J. 1999, 5, 3399. [26] Sakamoto, Y.; Suzuki, T; Kobayashi, M.; Gao, Y.; Fukai, Y.; Inoue, Y.; Sato, F.; Tokito, S. J. Am. Chem. Soc. 2004, 126, 8138. [27] (a) Inoue, Y.; Sakamoto, Y.; Suzuki, T.; Tokito, S. To be submitted for publication. (b) OFETs with tetracene: Gundlach, D. J.; Nichols, J. A.; Zhou, L.; Jackson, T. N. Appl. Phys. Lett. 2002, 80, 2925-2927.
Proc. of SPIE Vol. 6192 61922V-11
Copyright © 2022 FDOKUMEN











 2 studied by infrared and Raman spectroscopies and DFT computations](https://static.fdokumen.com/doc/165x107/6316cce2d16b3722ff0d1094/molecular-structure-and-vibrational-spectrum-of-mgch-3-2so-6clo-4-2-studied.jpg)








