
Patterns of polymorphism and gene flow of gender-associated mitochondrial DNA lineages in European...
11
Introduction A major aim in evolutionary biology is to distinguish the effects of present and past evolutionary pressures on cur- rent patterns of variation (e.g. Avise 1992; Ellsworth et al. 1994; Orti et al. 1994; Thomaz et al. 1996). Analysis of mito- chondrial DNA polymorphism has opened up a phyloge- netic perspective (Avise et al. 1987, 1994) on this work. Recent studies have demonstrated the usefulness of this approach by showing how discordant patterns of varia- tion for organelle and nuclear genomes can suggest differ- ences in historical factors affecting population structure, gene flow and selection intensities (e.g. Karl & Avise 1992; Strauss et al. 1993; Burton & Lee 1994; Quesada et al. 1995b; Dowling et al. 1997). Mussels of the genus Mytilus have an exceptional mode of mtDNA inheritance that provides a novel model system for testing hypotheses relating to the effects of historical and contemporary events on speciation and intraspecific genetic differentiation. They have two distinct mtDNA genomes called F and M (Fisher & Skibinski 1990; Hoeh et al. 1991). The F genome is passed from mothers to their sons and daughters and the M genome is passed from fathers to their sons alone, thus the F genome is inherited maternally and the M genome is inherited paternally (Skibinski et al. 1994a,b; Zouros et al. 1994a,b). This unusual mode of inheritance can account for the high levels of F/M heteroplasmy in males and the high sequence divergence (exceeding 20%) between these two genomes, whose sepa- ration predates speciation in the genus Mytilus (Rawson & Hilbish 1995a; Stewart et al. 1995; Quesada et al. 1996). Taxa from the Mytilus edulis species complex (M. edulis, M. trossulus and M. galloprovincialis) are distributed Molecular Ecology (1998) 7, 1041—1051 ' 1998 Blackwell Science Ltd Patterns of polymorphism and gene flow of gender- associated mitochondrial DNA lineages in European mussel populations H. QUESADA, C. GALLAGHER, D. A. G. SKIBINSKI and D. O. F. SKIBINSKI School of Biological Sciences, University of Wales, Swansea SA2 8PP, UK Abstract Mussels of the genus Mytilus have two types of mitochondrial DNA (mtDNA). The M type is transmitted paternally and the F type is transmitted maternally. RFLP analysis is used to assess phylogenetic relationships and nucleotide diversity and divergence for both mtDNA genomes in European populations of M. edulis and Atlantic and Mediterranean forms of M. galloprovincialis. Ten restriction endonucleases were used to assay variation in regions of the ND2 and COIII genes for a total of 77 individuals. F and M genomes show a concordant phylogenetic split into two major divergent clades, one specific to Mediterranean M. galloprovincialis and the other containing haplotypes from the three taxa. For both genomes, the geographical distribution of mtDNA variation suggests: (i) extensive levels of mtDNA introgression; (ii) asymmetric mtDNA gene flow from Atlantic to Mediterranean populations; and (iii) recurrent historical hybridization events. Significantly higher mtDNA diversity and divergence are observed for the M than F genome in all three Mytilus taxa, although the evolutionary forces responsible for these differences cannot be resolved. The extensive mtDNA gene flow between European Mytilus taxa conflicts with the restricted mtDNA introgression observed in American mussels, implying geographical variation in the nature of nuclear/mtDNA interactions regulating biparental inheritance. Keywords: asymmetric introgression, diversity, heteroplasmy, mitochondrial DNA, Mytilus, paternal inheritance Received 15 December 1997; revision received 16 February 1998; accepted 25 February 1998 Correspondence: Prof. D. O. F. Skibinski. Fax: +44 (0) 1792295447; E-mail: [email protected]
Transcript of Patterns of polymorphism and gene flow of gender-associated mitochondrial DNA lineages in European...

Introduction
A major aim in evolutionary biology is to distinguish theeffects of present and past evolutionary pressures on cur-rent patterns of variation (e.g. Avise 1992; Ellsworth et al.1994; Orti et al. 1994; Thomaz et al. 1996). Analysis of mito-chondrial DNA polymorphism has opened up a phyloge-netic perspective (Avise et al. 1987, 1994) on this work.Recent studies have demonstrated the usefulness of thisapproach by showing how discordant patterns of varia-tion for organelle and nuclear genomes can suggest differ-ences in historical factors affecting population structure,gene flow and selection intensities (e.g. Karl & Avise 1992;Strauss et al. 1993; Burton & Lee 1994; Quesada et al.1995b; Dowling et al. 1997).
Mussels of the genus Mytilus have an exceptional modeof mtDNA inheritance that provides a novel model systemfor testing hypotheses relating to the effects of historicaland contemporary events on speciation and intraspecificgenetic differentiation. They have two distinct mtDNAgenomes called F and M (Fisher & Skibinski 1990; Hoehet al. 1991). The F genome is passed from mothers to theirsons and daughters and the M genome is passed fromfathers to their sons alone, thus the F genome is inheritedmaternally and the M genome is inherited paternally(Skibinski et al. 1994a,b; Zouros et al. 1994a,b). This unusualmode of inheritance can account for the high levels of F/Mheteroplasmy in males and the high sequence divergence(exceeding 20%) between these two genomes, whose sepa-ration predates speciation in the genus Mytilus (Rawson &Hilbish 1995a; Stewart et al. 1995; Quesada et al. 1996).
Taxa from the Mytilus edulis species complex (M. edulis,M. trossulus and M. galloprovincialis) are distributed
Molecular Ecology (1998) 7, 1041Ð1051
© 1998 Blackwell Science Ltd
Patterns of polymorphism and gene flow of gender-associated mitochondrial DNA lineages in Europeanmussel populations
H. QUESADA, C . GALLAGHER, D . A . G . SKIBINSKI and D. O . F. SKIBINSKISchool of Biological Sciences, University of Wales, Swansea SA2 8PP, UK
Abstract
Mussels of the genus Mytilus have two types of mitochondrial DNA (mtDNA). The Mtype is transmitted paternally and the F type is transmitted maternally. RFLP analysis isused to assess phylogenetic relationships and nucleotide diversity and divergence forboth mtDNA genomes in European populations of M. edulis and Atlantic andMediterranean forms of M. galloprovincialis. Ten restriction endonucleases were used toassay variation in regions of the ND2 and COIII genes for a total of 77 individuals. F andM genomes show a concordant phylogenetic split into two major divergent clades, onespecific to Mediterranean M. galloprovincialis and the other containing haplotypes fromthe three taxa. For both genomes, the geographical distribution of mtDNA variationsuggests: (i) extensive levels of mtDNA introgression; (ii) asymmetric mtDNA gene flowfrom Atlantic to Mediterranean populations; and (iii) recurrent historical hybridizationevents. Significantly higher mtDNA diversity and divergence are observed for the M thanF genome in all three Mytilus taxa, although the evolutionary forces responsible for thesedifferences cannot be resolved. The extensive mtDNA gene flow between EuropeanMytilus taxa conflicts with the restricted mtDNA introgression observed in Americanmussels, implying geographical variation in the nature of nuclear/mtDNA interactionsregulating biparental inheritance.
Keywords: asymmetric introgression, diversity, heteroplasmy, mitochondrial DNA, Mytilus,paternal inheritance
Received 15 December 1997; revision received 16 February 1998; accepted 25 February 1998
Correspondence: Prof. D. O. F. Skibinski. Fax: +44 (0) 1792295447;E-mail: [email protected]

world-wide in the littoral zones of temperate and coldoceans. In the Northern hemisphere, M. edulis is found inwestern Europe and eastern North America, M. gallo-provincialis is found in the Mediterranean, westernEurope, California and eastern Asia, and M. trossulus isfound in the Baltic sea, Siberia, eastern Canada and west-ern North America (Koehn 1991; McDonald et al. 1991).Mytilus taxa are closely related and contact zones differ inthe extent of hybridization (Gosling 1992). In Americanpopulations, whenever hybridization among differenttaxa involves M. trossulus, biparental mtDNA inheritanceis disrupted and blocks mtDNA introgression (Zouroset al. 1992; Rawson et al. 1996; Saavedra et al. 1996). Theseobservations suggest that the constraints on compatibilitybetween introgressing nuclear and mitochondrial DNAare stronger than is normally found in species having theusual uniparental maternal mtDNA inheritance(Saavedra et al. 1996; Rawson et al. 1996).
The patterns of mtDNA variation in Europe show strik-ing differences to those in America. One intriguing fea-ture of the F genome in Europe is the apparent lack ofcorrespondence between patterns of mtDNA variationand species boundaries as defined by morphological andallozyme data (Quesada et al. 1995b). In evolutionarytrees, F haplotypes specific to M. edulis and M. galloprovin-cialis cluster together, whereas M. trossulus and M. edulisshare the same frequent mtDNA haplotype. By contrast, amajor F mtDNA phylogeographic break coincident withan abrupt allozyme cline and associated with a sharpoceanographic front in southeast Spain distinguishes con-specific Atlantic and Mediterranean forms of M. gallo-provincialis (Quesada et al. 1995a,c). These observationssuggest extensive levels of introgression, probably influ-enced greatly by Pleistocene glaciations and contempo-rary hydrographic factors (Quesada et al. 1995b). Fewercomparative data are available for the M genome butthese are also intriguing. First, results based on RsaI andDdeI mtDNA restriction patterns suggest larger differ-ences for the M than F genome between Atlantic andMediterranean forms of M. galloprovincialis (Quesada et al.1996). Second, there is no evidence for the occurrence ofthe M genome in Baltic M. trossulus (Wenne & Skibinski1995). These observations invite the speculation that pat-terns of introgression differ between the male and femalemtDNA lineages in European taxa. Thus, differences ofcompatibility between introgressed nuclear backgroundsand each mtDNA type, and/or differences of compatibil-ity between F and M genomes within hybrid males inrelation to the species from which each mtDNA genome isinherited, could lead to asymmetric introgressionbetween F and M genomes. Evidence supporting strongnuclearÐmtDNA interactions governing biparental inheri-tance (Saavedra et al. 1997) might facilitate this possibility.
In the present study, RFLP analysis is used to assess
phylogenetic relationships and patterns of nucleotidediversity and divergence in a homologous segment of theF and M genomes of European M. edulis and the Atlanticand Mediterranean forms of M. galloprovincialis (Quesada1993). We test whether patterns of mtDNA variation andgene flow in hybridizing European taxa are similar in theF and M lineages.
Materials and methods
Mussels were collected during 1994 from sites identifiedpreviously (Skibinski et al. 1983; Quesada 1993) as havingonly Mytilus edulis (Monkton Point, southwest Wales),Atlantic M. galloprovincialis (Rande, northwest Spain) andMediterranean M. galloprovincialis (Pe�iscola, easternSpain). Thirteen mussels of each sex from each locality(although there were 12 M. edulis males) were analysed.Mussels were sexed by microscopic examination ofreproductive tissue from the mantle for the presence ofeggs or sperm. Mitochondria were isolated from wholebody tissue by differential centrifugation and mtDNAwas purified using a CTAB procedure (Fisher & Skibinski1990).
Genome-specific primers were used to amplify a 1.5 kbfragment of M mtDNA (primers 5′-AAACCCTTCGTCCA-CAAGG-Ô3 and 5′-AGCCTTTTTGTCATCATTCTGT-Ô3)and a 1.3 kb fragment of F mtDNA (primers 5′-TCTTG-GTACAACTGCGGGAA-3′ and 5′-ACCAAGAAACG-GAGGCATC-3′) covering homologous regions of the ND2and COIII genes (Skibinski et al. 1994b; Quesada et al. 1996).Conditions for PCR assay were as described in Quesadaet al. (1996). Aliquots from each PCR amplification weredigested with the 10 restriction endonucleases AluI, AvaII,BclI, CfoI, DdeI, MboI, MspI, RsaI, Sau96I and TaqI. PCRamplifications and digestion products were separated on6% polyacrylamide gels and visualized by silver staining(Skibinski et al. 1994b). The M PCR product is larger by153 bp at one end (in the COIII gene) and 122 bp at the otherend (in the ND2 gene) as a result of the M primer-bindingsites flanking the F primer-binding sites. The base sequencedivergence in these nonoverlapping regions is not signifi-cantly different from that for sequence more internal to theprimers, nor do the ND2 and COIII genes differ substan-tially in sequence divergence (Skibinski et al. 1994b). Thusthe small region of nonoverlap between F and M PCRproducts should not lead to any systematic error. However,most importantly, all analyses contained a correction forfragment length by using diversity or divergence pernucleotide site between haplotypes.
For each genome, the number of nucleotide substitu-tions between mtDNA profiles (dij for profiles i and j) wascomputed by the fragment approach of Nei & Li (1979).Estimates of nucleotide diversity (dX) and uncorrecteddivergence (dXY) were computed from the dij values
1042 H. QUESADA ET AL .
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051

according to the method of Nei & Miller (1990). The cor-rected divergence between two samples (dA) was calcu-lated as dA = dXY - (dX + dY)/2, where dX and dY are thenucleotide diversity values for each sample. Assumingneutrality, there are three sources of sampling error in theestimation of nucleotide diversity (Lynch & Crease 1990):the sampling of individuals from each population; thesampling of nucleotide or restriction sites; and the sam-pling of replicate populations evolving independently(drift variance). Felsenstein (1992) and Fu (1994) haveshown that the most efficient estimators (i.e. having mini-mal variance) of nucleotide diversity that include all theabove sources of error are those that take into account thegenealogical relationships of the sample of haplotypes.Unfortunately, these methods are not available for frag-ment data at present. In this study, differences in diversityand divergence between F and M mtDNA genomes weretested using a robust bootstrap approach (Efron 1979;Manly 1994). Two different procedures were used to takeinto account different sources of estimation errors. Formethod I, individuals were resampled with replacementto generate bootstrap samples of individuals for eachpopulation. This method takes account only of samplingerror of individuals. Values of dij and then subsequentlyvalues of nucleotide diversity and divergence were calcu-lated for the bootstrap samples. For diversity, wheneveran individual was included more than once within a boot-strap sample, the dij of zero corresponding to the compari-son of the individual with itself was excluded to minimizeexcessive bias downwards of the bootstrap diversityvalue. This resampling process was carried out for the MmtDNA, and for the F mtDNA in females and males.From distributions of 30 000 bootstrap replicates, confi-dence intervals for estimates of the difference between Fand M genomes were obtained by the bias-corrected per-centile method (Efron 1982; pp. 82Ð84). The level of signif-icance for the difference was set to the largest bootstrapconfidence interval that failed to overlap zero (Manly1994). For method II, the variances and standard errors ofthe estimates were obtained by bootstrapping overrestriction enzymes as indicated by Nei & Miller (1990)using the R E S T S I T E computer package (Miller 1991).Differences among estimates were evaluated using the Z-test (Sokal & Rohlf 1981). In this method, variances aredue to estimation errors of nucleotide substitutions andalso include some but not all of the stochastic (drift) vari-ance that is observed among loci evolving independently(Nei & Miller 1990). By comparing the results of methods Iand II it is possible to gauge the added effect of error dueto sampling of restriction sites and of populations abovethat due simply to the sampling of individuals.
For each genome, molecular variance was partitionedwithin and between taxa using the AMOVA approach ofExcoffier et al. (1992), as implemented in the program
A R L E Q U I N 1.0. This analysis produces estimates of vari-ance components and F-statistic analogues (Φ-statistics)by incorporating information on DNA haplotype diver-gence within an A N O VA format. Values for the F genomewere calculated separately for data from both males andfemales as explained above. The distance data used in thecalculation of Φ-statistics were the dij values computedaccording to the method of Nei & Li (1979). Significancelevels of Φ-statistics were determined via 1000 permuta-tions using the program A R L E Q U I N. Heterogeneity inhaplotype frequencies was also examined by the MonteCarlo method of Roff & Bentzen (1989) using the REAP 4.0program (McElroy et al. 1992).
Phylogenetic trees of the haplotypes from the differentMytilus taxa were constructed using both distance andparsimony methods as implemented in PHYLIP 3.5(Felsenstein 1993). The neighbour-joining method wasused to infer phylogenetic relationships from thesequence divergence estimates among haplotypes. Datamatrices of fragment presence and absence were used togenerate Wagner parsimony phylogenies. The level ofsupport for branching patterns in phylogenetic trees wasdetermined by 100 bootstrap replications of the originaldata using the S E Q B O O T program of PHYLIP. Becauserestriction fragment data are partially correlated and mayinflate bootstrap values in phylogenetic trees (Felsenstein1985), bootstrap estimates were regarded as relative mea-sures of robustness. Similar topologies were obtainedusing the neighbour-joining and Wagner methods, thusfor brevity the neighbour-joining method is presented.
Results
On average, 33.1 and 33.3 restriction fragments werescored for each individual for the F and M genomes,respectively, representing ≈ 132 (F genome) and 133 (Mgenome) bases of endonuclease recognition sequence.Sixty-seven haplotypes were observed in the three taxa,38 for the M and 29 for the F genome (Tables 1 and 2).Pairwise genetic distances between haplotypes are pre-sented in Appendix 1 and 2. Each M haplotype wasdetected once only. By contrast, 11 F haplotypes occurredin more than one individual, with a different haplotypeoccurring at high frequency (0.42Ð0.58) in each of the taxa.Haplotypes 45Ð48 were shared by Atlantic andMediterranean Mytilus galloprovincialis. All 38 males werefound to be heteroplasmic for both F and M genomes,while the 39 females were homoplasmic for the F genome.No significant differences in F haplotype frequencieswere observed between sexes for any taxon as revealed bythe chi-squared test of Roff & Bentzen (1989).
Analysis of molecular variance shows a substantial andsignificant (P < 0.001) level of differentiation among taxa(Table 3). Variance components were higher for the M
MITOCHONDRIAL DNA VARIATION IN MYTILUS 1043
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051

than F genome both within taxa and among taxa.However, both genomes displayed similar levels of differ-entiation as revealed by Φ-statistics. Thus, about 40% ofthe total molecular variance of each F and M genome isdue to differences among taxa. Pairwise analysis amongtaxa showed that all combinations were significantly dif-ferentiated (P < 0.001) for each genome.
Estimates of nucleotide diversity and divergencebetween taxa with standard errors calculated according to
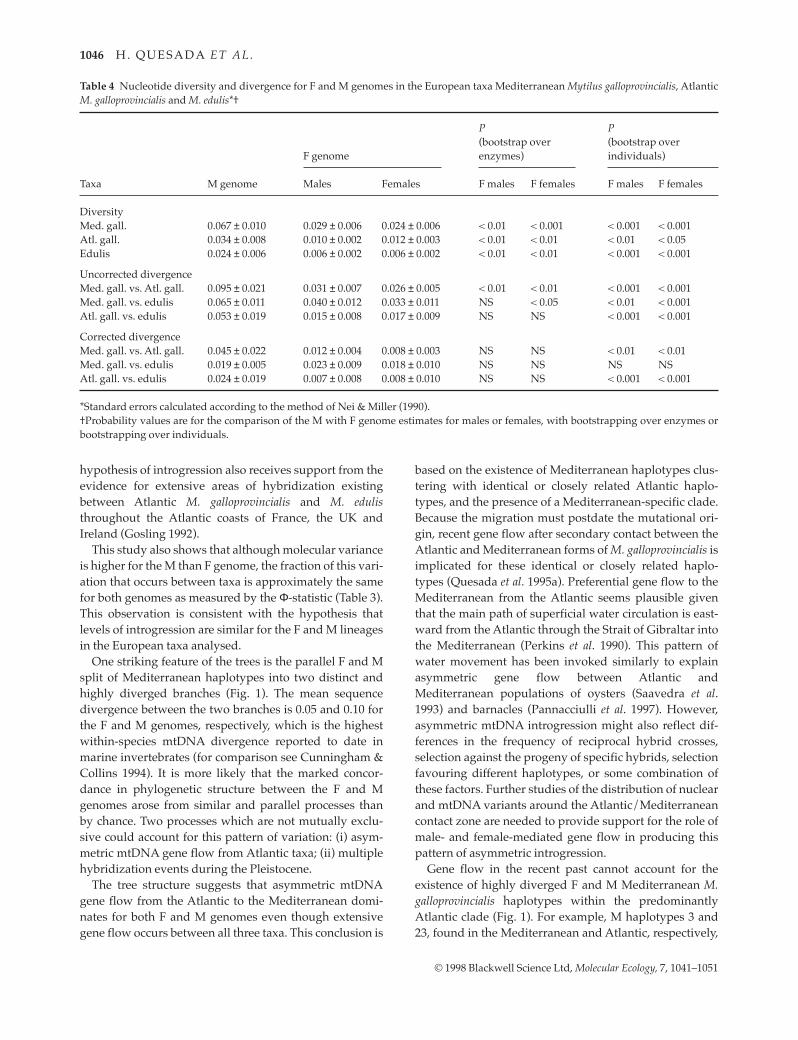
the method of Nei & Miller (1990) are given in Table 4.Estimates of nucleotide diversity were always signifi-cantly higher for the M than the F genome in all threeMytilus taxa. The value for uncorrected divergence for theM genome is greater than that for the F genome in everycomparison. The differences are significant with boot-strap resampling over individuals. However, with boot-strapping over enzymes, when estimation errors ofnucleotide substitution and part of the drift variance aretaken into account, the probability values are higher.Three comparisons become nonsignificant. A similar pat-tern is observed for the corrected divergence, which isgreater for the M than the F genome in all comparisonsexcept one. Comparisons that are significant with boot-strapping over individuals become nonsignificant withbootstrapping over enzymes. The discrepancy betweenboth bootstrap methods is not an unexpected result,because sampling error is higher when samplingnucleotides than individuals. Lynch & Crease (1990) haveshown that the standard errors of diversity and diver-gence estimates for mtDNA surveys involving severalspecies are usually quite large when sampling nucleotidesor individuals, but that only a very small fraction of thetotal sampling variance is caused by sampling of individ-uals. Therefore, drift variance is often of greater impor-tance. Thus, for this study, the exclusive reliance onbootstrap over individuals would have led to substantialunderestimates of standard errors of diversity and diver-gence estimates.
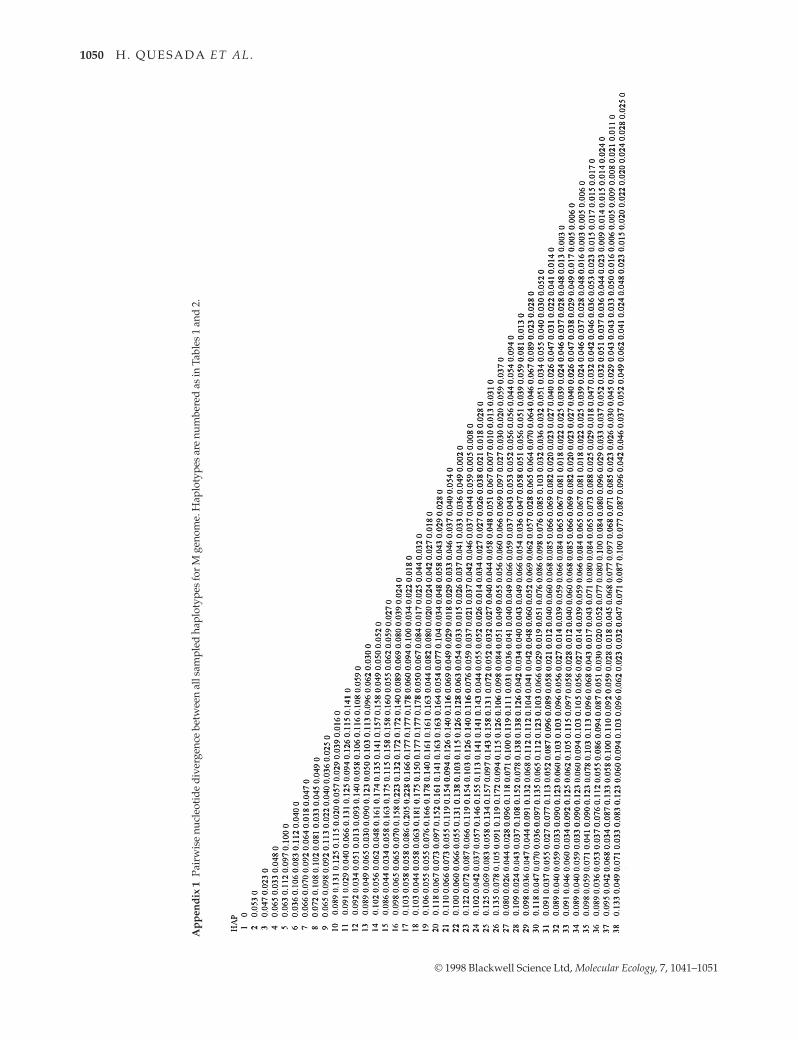
The evolutionary relationships among haplotypes aregiven separately for each genome in Fig. 1. Neighbour-joining trees show a similar topology for both F and Mgenomes, with a division of haplotypes in two majorbranches that is well supported (> 95%) by bootstrap anal-ysis. One of these branches has haplotypes that occuralmost exclusively in Mediterranean M. galloprovincialis.The other has haplotypes from all three taxa. Within thissecond branch, haplotypes from the same taxon tend tocluster together forming distinct sub-branches. The occur-rence of Mediterranean M. galloprovincialis haplotypes inboth of the two major branches is reflected in a substan-tially higher nucleotide diversity for this taxon (Table 4).The proportion of sampled haplotypes in MediterraneanM. galloprovincialis (Tables 1,2) that cluster within the pre-dominantly Atlantic clade does not differ significantlybetween the F (57%) and M (46%) genomes (P = 0.52,FisherÕs exact test). Neighbour-joining trees also showthat branch lengths are considerably longer for the M thanF genome.
Discussion
The results presented in this study demonstrate extensivelevels of introgression for the M genome, and confirm the
1044 H. QUESADA ET AL .
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
Table 1 M genome composite haplotype frequencies in males forMediterranean and Atlantic Mytilus galloprovincialis and M.edulis. Individual enzyme genotypes are represented in the com-posite genotype in the order (from left to right): AluI, AvaII, BclI,CfoI, DdeI, MboI, MspI, RsaI, Sau96I, TaqI
M. galloprovincialis
Haplotype Mediterranean Atlantic M. edulis
1 abaaibeaha 12 adabiagdaa 13 aeaagaabga 14 ahabgbdfaa 15 bbaajeeeka 16 bbaalbebhb 17 bbabkceeca 18 bcaajbfcia 19 bcaajdedja 110 bcaajfehla 111 cdacbaaaaa 112 dhabgadeaa 113 eaabaabgca 114 aaadbadkaa 115 aaadfaajca 116 afadgaciba 117 afadhaajba 118 ahadeagkaa 119 aiadbaakda 120 dgadbagmea 121 eaadaabiaa 122 eaadaabjaa 123 eaaddabiaa 124 eaadmaalaa 125 eaaeaabiac 126 edafcabifa 127 aaacbbgeaa 128 aaagpageaa 129 aiacoaaeda 130 eaabnaboaa 131 eaagaabeaa 132 eaagaabqaa 133 eaagaabraa 134 eaagaabsaa 135 eaahdabgaa 136 edaiaabeaa 137 elagaabpaa 138 faagqadeaa 1
Total 13 13 12

occurrence of a similar pattern for the F genome. Twoobservations support this conclusion. First, for bothgenomes, branches on the evolutionary trees of haplo-types do not correspond exactly with the taxonomic iden-tification of the individuals in which these haplotypes arefound (Fig. 1). Individuals from different taxa, separatedby thousands of kilometres, possess identical or nearlyidentical haplotypes, whereas individuals from the sametaxon can show up to 7% (F genome) and 14% (Mgenome) divergence (Appendix 1 and 2). Second, for bothgenomes, the divergence between Atlantic M. galloprovin-cialis and M. edulis is approximately 50% of that betweenMediterranean M. galloprovincialis and M. edulis (Table 4).This contrasts with morphological and allozymic studiesindicating that M. edulis is more distantly related toAtlantic and Mediterranean M. galloprovincialis than thelatter two are to each other (McDonald et al. 1991; Gosling1992). The alternative hypothesis that these patterns ofvariation are the result of incomplete lineage sorting after
descent from a polymorphic common ancestor ratherthan introgression requires that mtDNA lineages splitprior to the splitting of populations. This cannot accountfor individuals from different taxa having nearly identicalhaplotypes that clearly must have split from each othermore recently than the taxa themselves diverged. The
MITOCHONDRIAL DNA VARIATION IN MYTILUS 1045
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
M. galloprovincialis
Mediterranean Atlantic M. edulis
Haplotype Male Female Male Female Male Female
39 CAAABAAFAA 140 AAAACAADAA 141 BABAEBBBBB 142 BAAAAABCBB 1 143 BABAABBBBB 1 144 BBAABABBCB 1 145 AAAAAAAAAB 4 7 146 BAAAAABBBB 3 1 147 AAAADAAEAA 1 148 AAAACAAEAA 1 7 849 AAAACAAFAA 150 AAAACAAGAA 151 AAAACCAEBA 152 DAAACAAGAA 153 AAAACDAEAA 154 BAAACAAEAA 155 BAAACAAGAA 156 EABAABBHBB 157 AAAACAAEDA 4 758 AAAACAAEDB 1 159 AABACAAEDA 1 160 AAAADAAEDA 361 AAAACAAEEA 162 AAAACAAEFA 163 AAAACAAIEA 164 AAAABAAADA 165 AAAACAAADB 166 AAAAFAAEDA 167 FAAACAAEDA 1
Total 13 13 13 13 12 13
Table 2 F genome composite haplotypefrequencies in females and males forMediterranean and Atlantic Mytilusgalloprovincialis and M. edulis. Individualenzyme genotypes are represented in thecomposite genotype in the order (from leftto right): AluI, AvaII, BclI, CfoI, DdeI, MboI,MspI, RsaI, Sau96I, TaqI
Table 3 Analysis of molecular variance (AMOVA)
Within taxa Among taxa
Genome d.f. Variance d.f. Variance Φ∗ P-value
M 35 0.022 2 0.017 0.431 < 0.001F males 35 0.008 2 0.007 0.460 < 0.001F females 36 0.008 2 0.006 0.416 < 0.001
∗Φ statistics calculated incorporating Nei & Li (1979) distanceamong haplotypes.
hypothesis of introgression also receives support from theevidence for extensive areas of hybridization existingbetween Atlantic M. galloprovincialis and M. edulisthroughout the Atlantic coasts of France, the UK andIreland (Gosling 1992).
This study also shows that although molecular varianceis higher for the M than F genome, the fraction of this vari-ation that occurs between taxa is approximately the samefor both genomes as measured by the Φ-statistic (Table 3).This observation is consistent with the hypothesis thatlevels of introgression are similar for the F and M lineagesin the European taxa analysed.
One striking feature of the trees is the parallel F and Msplit of Mediterranean haplotypes into two distinct andhighly diverged branches (Fig. 1). The mean sequencedivergence between the two branches is 0.05 and 0.10 forthe F and M genomes, respectively, which is the highestwithin-species mtDNA divergence reported to date inmarine invertebrates (for comparison see Cunningham &Collins 1994). It is more likely that the marked concor-dance in phylogenetic structure between the F and Mgenomes arose from similar and parallel processes thanby chance. Two processes which are not mutually exclu-sive could account for this pattern of variation: (i) asym-metric mtDNA gene flow from Atlantic taxa; (ii) multiplehybridization events during the Pleistocene.
The tree structure suggests that asymmetric mtDNAgene flow from the Atlantic to the Mediterranean domi-nates for both F and M genomes even though extensivegene flow occurs between all three taxa. This conclusion is
based on the existence of Mediterranean haplotypes clus-tering with identical or closely related Atlantic haplo-types, and the presence of a Mediterranean-specific clade.Because the migration must postdate the mutational ori-gin, recent gene flow after secondary contact between theAtlantic and Mediterranean forms of M. galloprovincialis isimplicated for these identical or closely related haplo-types (Quesada et al. 1995a). Preferential gene flow to theMediterranean from the Atlantic seems plausible giventhat the main path of superficial water circulation is east-ward from the Atlantic through the Strait of Gibraltar intothe Mediterranean (Perkins et al. 1990). This pattern ofwater movement has been invoked similarly to explainasymmetric gene flow between Atlantic andMediterranean populations of oysters (Saavedra et al.1993) and barnacles (Pannacciulli et al. 1997). However,asymmetric mtDNA introgression might also reflect dif-ferences in the frequency of reciprocal hybrid crosses,selection against the progeny of specific hybrids, selectionfavouring different haplotypes, or some combination ofthese factors. Further studies of the distribution of nuclearand mtDNA variants around the Atlantic/Mediterraneancontact zone are needed to provide support for the role ofmale- and female-mediated gene flow in producing thispattern of asymmetric introgression.
Gene flow in the recent past cannot account for theexistence of highly diverged F and M Mediterranean M.galloprovincialis haplotypes within the predominantlyAtlantic clade (Fig. 1). For example, M haplotypes 3 and23, found in the Mediterranean and Atlantic, respectively,
1046 H. QUESADA ET AL .
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
Table 4 Nucleotide diversity and divergence for F and M genomes in the European taxa Mediterranean Mytilus galloprovincialis, AtlanticM. galloprovincialis and M. edulis*
P P(bootstrap over (bootstrap over
F genome enzymes) individuals)
Taxa M genome Males Females F males F females F males F females
DiversityMed. gall. 0.067 ± 0.010 0.029 ± 0.006 0.024 ± 0.006 < 0.01 < 0.001 < 0.001 < 0.001Atl. gall. 0.034 ± 0.008 0.010 ± 0.002 0.012 ± 0.003 < 0.01 < 0.01 < 0.01 < 0.05Edulis 0.024 ± 0.006 0.006 ± 0.002 0.006 ± 0.002 < 0.01 < 0.01 < 0.001 < 0.001
Uncorrected divergenceMed. gall. vs. Atl. gall. 0.095 ± 0.021 0.031 ± 0.007 0.026 ± 0.005 < 0.01 < 0.01 < 0.001 < 0.001Med. gall. vs. edulis 0.065 ± 0.011 0.040 ± 0.012 0.033 ± 0.011 NS < 0.05 < 0.01 < 0.001Atl. gall. vs. edulis 0.053 ± 0.019 0.015 ± 0.008 0.017 ± 0.009 NS NS < 0.001 < 0.001
Corrected divergenceMed. gall. vs. Atl. gall. 0.045 ± 0.022 0.012 ± 0.004 0.008 ± 0.003 NS NS < 0.01 < 0.01Med. gall. vs. edulis 0.019 ± 0.005 0.023 ± 0.009 0.018 ± 0.010 NS NS NS NSAtl. gall. vs. edulis 0.024 ± 0.019 0.007 ± 0.008 0.008 ± 0.010 NS NS < 0.001 < 0.001
*Standard errors calculated according to the method of Nei & Miller (1990). Probability values are for the comparison of the M with F genome estimates for males or females, with bootstrapping over enzymes orbootstrapping over individuals.

are separated by 9% sequence divergence, and F haplo-types 39 (Mediterranean) and 65 (Atlantic) are separatedby 5% sequence divergence (Appendices 1 and 2). Thepossibility that these haplotypes represent relictMediterranean mtDNA lineages that predate recentmtDNA introgression from Atlantic taxa is not consistentwith their clustering within the Atlantic clade. It is proba-ble that these divergent lineages introgressed from theAtlantic to the Mediterranean as a result of hybridizationevents during the Pleistocene coupled with an historicallyrestricted gene flow between both areas (Quesada et al.1995b). Larger population sizes in the Mediterraneancompared with Atlantic and/or historically more struc-tured populations in the Mediterranean could have pre-vented the extinction of these lineages. At the presenttime, contemporary ecological and hydrographic factors,for example the Almer�aÐOran front, will contribute to themaintenance of the narrow contact zone betweenMediterranean and Atlantic M. galloprovincialis in south-east Spain (Quesada et al. 1995a,c), and to the maintenanceof the phylogeographic structure and increased diversityarising from these historical processes.
This study provides evidence, for the first time, of sig-nificantly higher nucleotide diversity for the M than Fgenome in Mytilus. This is true for all three Mytilus taxaanalysed, confirming earlier claims (that, however, lackedstatistical support) suggesting a higher diversity for the Mgenome (Skibinski et al. 1994b; Stewart et al. 1995, 1996;Quesada et al. 1996). Several factors could account for theobserved difference in diversity between genomes. First,in neutral theory, the expectation of diversity for mtDNAis θ = 2Neu (e.g. Takahata & Nei 1985), where Ne is theeffective population size and u the mutation rate. Thus,differences in θ could be the result of differences in Ne or ubetween F and M genomes, assuming neutral evolution.However, it should be noted that the presence of the Fgenome in both males and females does not imply ahigher Ne for the F lineage, as males do not transmit it.Second, for a given value of θ, the actual level ofnucleotide diversity observed will be subject to stochasticvariation (drift variance) and thus may differ at differentpoints in time. Thus, even if the two genomes have identi-cal values of θ, they can have different values ofnucleotide diversity at one moment in time because ofthis drift variance. As the Nei & Miller (1990) estimates oferror take account of some but not all of the drift variancein diversity, the possibility that the observed differencesin diversity are due to stochastic variation cannot be com-pletely rejected.
The present study also shows a significantly higheruncorrected divergence and longer major branches for theM than F genome (Table 4, Fig. 1), in line with earlierreports (Skibinski et al. 1994b; Rawson & Hilbish 1995a;Stewart et al. 1995, 1996; Quesada et al. 1996). However, as
with diversity, the causes of the differences betweengenomes are difficult to establish. The expectation ofuncorrected divergence in a neutral model is given byθ + 2ut (Takahata & Nei 1985), where u is mutation rate andt is divergence time of the populations. Thus, a significantdifference in uncorrected divergence could be due to a dif-ference in Ne or u. The expectation of corrected divergenceis 2ut, thus the observation of nonsignificant differencesbetween genomes in the present study fails to provide evi-dence of a difference in mutation rate between thegenomes. Another consideration is that if the F and Mgenomes are influenced by selection then the standarderrors derived from neutral theory might not be applicable.Stewart et al. (1996) have shown that relaxed selection on
MITOCHONDRIAL DNA VARIATION IN MYTILUS 1047
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
Fig. 1 Neighbour-joining trees of F and M mtDNA haplotypesbased on nucleotide sequence divergence estimates. The haplo-types are numbered as in Tables 1 and 2. MG corresponds toMediterranean M. galloprovincialis, AG to Atlantic M. galloprovin-cialis, and ED to M. edulis. Numbers along branches indicate thesupport of each of the clades out of 100 bootstrap replicates (onlyvalues higher than 50 are shown). Trees are rooted at the midpoint.

the M genome can explain a higher sequence divergencefor the M genome in American populations of M. edulis andM. trossulus. This explanation could also account, at leastpartly, for the higher diversity and divergence for the Mthan F genome in European mussel populations. However,because the present study cannot take account completelyof the drift variance, the evolutionary forces causing theobserved differences in diversity and divergence inEuropean populations have yet to be identified.
The extensive levels of mtDNA introgression observedin European mussel populations (Quesada et al. 1995band this study) are in clear contrast with the observationof restricted mtDNA gene flow between American taxa(Rawson & Hilbish 1995b; Rawson et al. 1996; Saavedraet al. 1996). Thus, it appears unlikely that the high inci-dence of mtDNA exchange within the western Europehybrid zone is being selected against strongly. Our datafor the F and M genomes suggest that historical and con-temporary factors have had similar effects on the lineagestructure of both genomes at both the intra- and interspe-cific level. For example, the higher level of nucleotidediversity for both genomes in Mediterranean thanAtlantic populations (Table 4) could have accumulated bythe action of asymmetric introgression, coupled with ahistorically restricted gene flow between Atlantic andMediterranean basins (Quesada et al. 1995a). Therefore,when populations diverge following isolation, variationin genetic background might lead to differences betweenpopulations in the mechanisms responsible for the main-tenance of their genetic integrity. Such differences couldaccount for the different patterns of mtDNA introgressionobserved in American and European populations, sug-gesting that routes to reproductive isolation and thenature of nuclear/mitochondral DNA interactions regu-lating biparental inheritance might be different. This sug-gests that it will not be possible to make globalgeneralizations about the evolutionary forces acting onmtDNA in Mytilus through consideration of populationsonly from restricted geographical areas.
Acknowledgements
We thank the Natural Environment Research Council of the UKfor financial support. We are grateful to A. Quesada and F.Rodr�guez for help in the collection of mussels, and to two anony-mous referees for their useful suggestions on this manuscript.H.Q. gratefully acknowledges the support of a postdoctoral fel-lowship from the Ministerio de Educaci�n y Ciencia (Spain).
References
Avise JC (1992) Molecular population structure and the biogeo-graphic history of a regional fauna: a case history with lessonsfor conservation biology. Oikos, 63, 62Ð76.
Avise JC (1994) Molecular Markers, Natural History and Evolution.Chapman & Hall, London.
Avise JC, Arnold J, Ball RM, Bermingham E, Lamb T, Neigel JE,Reeb CA, Saunders NC (1987) Intraspecific phylogeography:the mitochondrial DNA bridge between genetics and systemat-ics. Annual Review of Ecology and Systematics, 18, 489Ð522.
Burton RS, Lee B-N (1994). Nuclear and mitochondrial DNA genegenealogies and allozyme polymorphism across a major phy-logeographic break in the copepod Tigriopus californicus.Proceedings of the National Academy of Sciences USA, 91,5197Ð5201.
Cunningham CW, Collins TM (1994) Developing model systemsfor molecular biogeography: vicariance and interchange inmarine invertebrates. In: Molecular Ecology and Evolution,Approaches and Applications (eds Schierwarer B, Streit B, WagnerGP, DeSalle R), pp. 405Ð433. Birkhauser Verlag, Basel,Switzerland.
Dowling TE, Broughton PE, DeMarais BD (1997) Significant rolefor historical effects in the evolution of reproductive isolation:evidence from patterns of introgression between cyprinidfishes, Luxilus cornutus and Luxilus Chrysocephalus. Evolution,51, 1574Ð1583.
Efron B (1979) Bootstrap methods: Another look at the jackknife.Annals of Statistics, 7, 1Ð26.
Efron B (1982) The jackknife, the bootstrap, and other resamplingplans. In: CBMS-NSF Regional Conference Series in AppliedMathematics, N1 38, Society for Industrial and AppliedMathematics, Philadelphia, PA.
Ellsworth DL, Honeycutt RL, Silvy NJ, Bickham JW, Klimstra WD(1994) Historical biogeography and contemporary patterns ofmitochondrial DNA variation in white-tailed deer from thesoutheastern United States. Evolution, 48, 122Ð136.
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecularvariance inferred from metric distances among DNA haplo-types: application to human mitochondrial DNA restrictiondata. Genetics, 131, 479Ð491.
Felsenstein J (1985) Confidence limits on phylogenies: an approachusing the bootstrap. Evolution, 39, 783Ð791.
Felsenstein J (1992) Estimating effective population size from sam-ples of sequences: inefficiency of pairwise and segregating sitesas compared to phylogenetic estimates. Genetical Research, 59,139Ð147.
Felsenstein J (1993) Phylogenetic Analysis Package (P H Y L I P ).University of Washington.
Fisher C, Skibinski DOF (1990) Sex-biased mitochondrial DNAheteroplasmy in the marine mussel Mytilus. Proceedings of theRoyal Society of London B, 242, 149Ð156.
Fu Y-X (1994) A phylogenetic estimator of effective population sizeor mutation rate. Genetics, 136, 685Ð692.
Gosling EM (1992) Systematics and geographical distribution ofMytilus. In: The Mussel Mytilus: Ecology, Physiology, Genetics andCulture (ed. Gosling EM), pp. 1Ð20. Elsevier, Amsterdam.
Hoeh WR, Blakley KH, Brown WM (1991) Heteroplasmy suggestslimited biparental inheritance of Mytilus mitochondrial DNA.Science, 251, 1488Ð1498.
Karl SA, Avise JC (1992) Balancing selection at allozyme loci in oys-ters: Implications from nuclear RFLPs. Science, 256, 100Ð102.
Koehn RK (1991) The genetics and taxonomy of species in thegenus Mytilus. Aquaqulture, 94, 125Ð147.
Lynch M, Crease TJ (1990) The analysis of population survey dataon DNA sequence variation. Molecular Biology and Evolution, 7,377Ð394.
Manly BF (1994) Randomization and Monte Carlo Methods in Biology.Chapman & Hall, London.
1048 H. QUESADA ET AL .
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051

McDonald JH, Seed R, Koehn RK (1991) Allozymes and morpho-metric characters of three species of Mytilus in the Northernand Southern Hemispheres. Marine Biology, 111, 323Ð333.
McElroy D, Moran P, Bermingham E, Kornfield I (1992) R E A P : anintegrated environment for the manipulation and phylogeneticanalysis of restriction data. Journal of Heredity, 83, 157Ð158.
Miller JC (1991) R E S T S I T E . A phylogenetic program that sorts rawrestriction data. Journal of Heredity, 82, 262Ð263.
Nei M, Li W-H (1979) Mathematical model for studying geneticvariation in terms of restriction endonucleases. Proceedings ofthe National Academy of Sciences USA, 76, 5269Ð5273.
Nei M, Miller JC (1990) A simple method for estimating averagenumber of nucleotide substitutions within and between popu-lations from restriction data. Genetics, 125, 873Ð879.
Orti G, Bell MA, Reimchen TE, Meyer A (1994) Global survey ofmitochondrial DNA sequences in the threespine stickleback:evidence fron recent migrations. Evolution, 48, 608Ð622.
Pannacciulli FG, Bishop JDD, Hawkins SJ (1997) Genetic structureof populations of two species of chthamalus (crustacea:Cirripeda) in the north-east Atlantic and Mediterranean.Marine Biology, 128, 73Ð82.
Perkins H, Kinder T, La Violette P (1990) The Atlantic inflow in theWestern Mediterranean. Journal of Physical Oceanography, 20,242Ð263.
Quesada H (1993) Estructura gen�tica de poblaciones naturales delmejill�n Mytilus galloprovincialis Lmk. Servicio de Publicacionesde la Universidad de Santiago de Compostela, Santiago deCompostela.
Quesada H, Beynon CM, Skibinski DOF (1995a) A mitochondrialDNA discontinuity in the mussel Mytilus galloprovincialis Lmk:Pleistocene vicariance biogeography and secondary intergra-dation. Molecular Biology and Evolution, 12, 521Ð524.
Quesada H, Wenne R, Skibinski DOF (1995b) Differential intro-gression of mitochondrial DNA across species boundarieswithin the marine mussel genus Mytilus. Proceedings of the RoyalSociety of London B, 262, 57Ð64.
Quesada H, Zapata C, Alvarez G (1995c) A multilocus allozymediscontinuity in the mussel Mytilus galloprovincialis: the interac-tion of ecological and life-history factors. Marine EcologyProgress Series, 116, 99Ð115.
Quesada H, Skibinski DAG, Skibinski DOF (1996) Sex-biased het-eroplasmy and mitochondrial DNA inheritance in the musselMytilus galloprovincialis Lmk. Current Genetics, 29, 423Ð426.
Rawson PD, Hilbish TJ (1995a) Evolutionary relationships amongthe male and female mitochondrial DNA lineages in theMytilus edulis species complex. Molecular Biology and Evolution,12, 893Ð901.
Rawson PD, Hilbish TJ (1995b) Distribution of male and femalemtDNA lineages in populations of blue mussels, Mytilus trossu-lus and M. galloprovincialis, along the Pacific coast of NorthAmerica. Marine Biology, 124, 245Ð250.
Rawson PD, Secor CL, Hilbish TJ (1996) The effects of naturalhybridization on the regulation of doubly uniparental mtDNAinheritance in blue mussels (Mytilus spp.). Genetics, 144,241Ð248.
Roff DA, Bentzen P (1989) The statistical analysis of mitochondrialDNA polymorphisms: χ2 and the problem of small samples.Molecular Biology and Evolution, 6, 539Ð545.
Saavedra C, Zapata C, Alvarez G (1993) Allozyme variation inEuropean populations of the oyster Ostrea edulis. MarineBiology, 115, 85Ð95.
Saavedra C, Stewart DT, Stanwood RR, Zouros E (1996) Species-
specific segregation of gender-associated mitochondrial DNAtypes in an area where two mussel species (Mytilus edulis andMytilus trossulus) hybridize. Genetics, 143, 1359Ð1367.
Saavedra C, Reyero M-I, Zouros E (1997) Male-dependent doublyuniparental inheritance of mitochondrial DNA and female-dependent sex-ratio in the mussel Mytilus galloprovincialis.Genetics, 145, 1073Ð1082.
Skibinski DOF, Beardmore JA, Cross TF (1983) Aspects of the pop-ulation genetics of Mytilus (Mytilidae; Mollusca) in the BritishIsles. Biological Journal of the Linnean Society, 19, 137Ð183.
Skibinski DOF, Gallagher C, Beynon CM (1994a) MitochondrialDNA inheritance. Nature, 368, 817Ð818.
Skibinski DOF, Gallagher C, Beynon CM (1994b) Sex-limited mito-chondrial DNA transmission in the marine mussel Mytilusedulis. Genetics, 138, 801Ð809.
Sokal RR, Rohlf FJ (1981) Biometry. W.H. Freeman & Co., SanFrancisco.
Stewart DT, Saavedra C, Stanwood RR, Ball AO, Zouros E (1995)Male and female mitochondrial DNA lineages in the blue mus-sel (Mytilus edulis) species group. Molecular Biology andEvolution, 12, 735Ð747.
Stewart DT, Kenchington ER, Singh RK, Zouros E (1996) Degree ofselective constraint as an explanation of the different rates ofevolution of gender-specific mitochondrial DNA lineages inthe mussel Mytilus. Genetics, 143, 1349Ð1357.
Strauss SH, Hong Y-P, Hipkins VD (1993) High levels of popula-tion differentiation for mitochondrial DNA haplotypes in Pinusradiata, muricata, and attenuata. Theoretical and Applied Genetics,85, 6065Ð6071.
Takahata N, Nei M (1985) Gene genealogy and variance of inter-populational nucleotide differences. Genetics, 110, 325Ð344.
Thomaz D, Guiller A, Clarke B (1996) Extreme divergence of mito-chondrial DNA within species of pulmonate land snails.Proceedings of the Royal Society of London B, 263, 363Ð368.
Wenne R, Skibinski DOF (1995) Mitochondrial DNA heteroplasmyin European populations of the mussels Mytilus trossulus andMytilus edulis. Marine Biology, 122, 619Ð624.
Zouros E, Freeman KR, Ball AO, Pogson GH (1992) Direct evi-dence for extensive paternal mitochondrial DNA inheritance inthe marine mussel Mytilus. Nature, 359, 412Ð414.
Zouros E, Ball AO, Saavedra C, Freeman KR (1994a) MitochondrialDNA inheritance. Nature, 368, 818.
Zouros E, Ball AO, Saavedra C, Freeman KR (1994b) An unusualtype of mitochondrial DNA inheritance in the blue musselMytilus. Proceedings of the National Academy of Sciences USA, 91,7463Ð7467.
David O. F. Skibinski has been working on the population andecological genetics of marine mussels for the last 23 years. Atpresent, his group is investigating the consequences of the excitingdiscovery that Mytilus has two highly diverged mtDNA genomes,with separate male and female routes of inheritance. HumbertoQuesada is a MEC postdoctoral research fellow with broad experi-ence of mussel population genetics, whose current interests includemolecular approaches to studies of ecological genetics and theevolutionary forces acting on mtDNA. Catherine Gallagher is aresearch technician with many years experience of mtDNAanalysis. David A. G. Skibinski is currently carrying out PhDstudies at the University of Sheffield on hydrogenase 4 (Hyf) ofEscherichia coli.
MITOCHONDRIAL DNA VARIATION IN MYTILUS 1049
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
1050 H. QUESADA ET AL .
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
Ap
pen
dix
1Pa
irw
ise
nucl
eotid
e d
iver
genc
e be
twee
n al
l sam
pled
hap
loty
pes
for
M g
enom
e. H
aplo
type
s ar
e nu
mbe
red
as
in T
able
s 1
and
2.

MITOCHONDRIAL DNA VARIATION IN MYTILUS 1051
© 1998 Blackwell Science Ltd, Molecular Ecology, 7, 1041Ð1051
Appendix 2 Pairwise nucleotide divergence between all sampled haplotypes for F genome. Haplotypes are numbered as in Tables 1 and 2.




















