
PAGE: A Framework for Easy PArallelization of GEnomic Applications
10
PAGE: A Framework for Easy PArallelization of GEnomic Applications Mucahid Kutlu Department of Computer Science and Engineering Ohio State University Columbus, OH, 43210 Email: [email protected] Gagan Agrawal Department of Computer Science and Engineering Ohio State University Columbus, OH, 43210 Email: [email protected] Abstract—With the availability of high-throughput and low- cost sequencing technologies, an increasing amount of genetic data is becoming available to researchers. There is clearly a potential for significant new scientific and medical advances by analysis of such data, however, it is imperative to exploit paral- lelism and achieve effective utilization of the computing resources to be able to handle massive datasets. Thus, frameworks that can help researchers develop parallel applications without dealing with low-level details of parallel coding are very important for advances in genetic research. In this study, we develop a middleware, PAGE, which supports ‘mapreduce-like’ processing, but with significant differences from a system like Hadoop, to be useful and effective for parallelizing analysis of genomic data. Particularly, it can work with map functions written in any language, thus allowing utilization of existing serial tools (even those for which only an executable is available) as map functions. Thus, it can greatly simplify parallel application development for scenarios where complex data formats and/or nuanced serial algorithms are involved, as is often the case for genomic data. It allows parallelization by partitioning by-locus or partitioning by-chromosome, provides different scheduling schemes, and execution models, to match the nature of algorithms common in genetic research. We have evaluated the middleware system using four popular genomic applications, including VarScan, Unified Genotyper, Realigner Target Creator, and Indel Realigner, and compared the achieved performance against with two popular frameworks (Hadoop and GATK). We show that our middleware outperforms GATK and Hadoop and it is able to achieve high parallel efficiency and scalability. I. I NTRODUCTION Analysis of genetic data is becoming increasingly critical for medical research and even practice. Trends in sequenc- ing technologies have drastically reduced the cost of, and increased the speed of, collecting gene sequences [1], [6]. This data is now being shared aggressively through various projects, and researchers at different institutions can download and analyze the data. An example of such efforts is the 1000 Human Genome Project 1 , which has already produced 200TB of genome data across 1700 samples, and made it available on the Amazon Cloud Storage 2 . As a large number of researchers have access to the data, the focus is beginning to shift to analysis of this data. There exist many tools [15], [24], [30], [34] and libraries [4] to 1 www.1000genomes.org 2 http://aws.amazon.com/1000genomes/ ease the implementation of new data analysis programs on genetic data. One of the complexities in this area is that data formats in which sequences are stored are very specialized. Existing tools, in most cases, alleviate the need for scientists understanding these formats and coding complex algorithms. Consistent with the overall trend in data analytics, use of parallelism is inevitable in analysis of genomic data also. Low- ered response time, including facilitating interactive analysis of large-scale data, can open us many novel opportunities for researchers. Again, just like all other fields, the difficulty of carrying-out parallel implementations, especially for domain experts, is a large obstacle to widespread use of parallelism for analysis of genomic data. The current state-of-the-art in parallel analysis of genomic data is very limited. There are many serial software suites that lack any parallelization capability[19], [23], [24], [26], [30]. General MapReduce frameworks such as Hadoop [36] can potentially be used, but even parallelization using such a framework can be a hard problem for the community [20], [29]. Particularly, developing a parallel implementation with the use of MapReduce will require knowledge of data formats and reprogramming of an existing serial program, neither of which is desirable. Load imbalance is another problem while processing genomic data, which requires nuanced solutions. Genome Analysis Tool Kit (GATK)[27] provides developers a MapReduce-like framework specific for analysis of genomic data. However, GATK does not provide distributed memory parallelization for most of its programs and its scalability is low, as we will show in Section VI. Thus, we can see that easing parallelization of applications that process genomic data, while also achieving high parallel efficiency, is a challenging problem. In this study, we are proposing PAGE, which is a MapReduce-like middleware that allows users to continue to utilize their existing programs, but achieve parallelization with a modest additional effort. The map and reduce programs can be separate executables written in any (and even two distinct) programming languages. We can also work with any of the popular genomic data formats. The middleware divides the genome structure into regions/intervals and runs each map task for a different region, performing load balancing in the process. Subsequently, these partial results are reduced to obtain the final result(s). In order to improve efficiency, our middleware provides streaming task scheduling, in which the tasks can be scheduled dynamically with the help of a master node, and the reduce tasks do not have to wait for all map tasks to be finished. We evaluate PAGE with 4 existing applications, which are !oƼ ! ෝ !ȫōÞȫ Þ!Ƽ ෝ !!!ϧෝÞ!Ƽ!ϧ Þ
Transcript of PAGE: A Framework for Easy PArallelization of GEnomic Applications

PAGE: A Framework for Easy PArallelization ofGEnomic Applications
Mucahid KutluDepartment of Computer Science
and EngineeringOhio State UniversityColumbus, OH, 43210
Email: [email protected]
Gagan AgrawalDepartment of Computer Science
and EngineeringOhio State UniversityColumbus, OH, 43210
Email: [email protected]
Abstract—With the availability of high-throughput and low-cost sequencing technologies, an increasing amount of geneticdata is becoming available to researchers. There is clearly apotential for significant new scientific and medical advances byanalysis of such data, however, it is imperative to exploit paral-lelism and achieve effective utilization of the computing resourcesto be able to handle massive datasets. Thus, frameworks that canhelp researchers develop parallel applications without dealingwith low-level details of parallel coding are very important foradvances in genetic research.
In this study, we develop a middleware, PAGE, which supports‘mapreduce-like’ processing, but with significant differences froma system like Hadoop, to be useful and effective for parallelizinganalysis of genomic data. Particularly, it can work with mapfunctions written in any language, thus allowing utilization ofexisting serial tools (even those for which only an executableis available) as map functions. Thus, it can greatly simplifyparallel application development for scenarios where complexdata formats and/or nuanced serial algorithms are involved, asis often the case for genomic data. It allows parallelization bypartitioning by-locus or partitioning by-chromosome, providesdifferent scheduling schemes, and execution models, to matchthe nature of algorithms common in genetic research.
We have evaluated the middleware system using four populargenomic applications, including VarScan, Unified Genotyper,Realigner Target Creator, and Indel Realigner, and comparedthe achieved performance against with two popular frameworks(Hadoop and GATK). We show that our middleware outperformsGATK and Hadoop and it is able to achieve high parallelefficiency and scalability.
I. INTRODUCTION
Analysis of genetic data is becoming increasingly criticalfor medical research and even practice. Trends in sequenc-ing technologies have drastically reduced the cost of, andincreased the speed of, collecting gene sequences [1], [6].This data is now being shared aggressively through variousprojects, and researchers at different institutions can downloadand analyze the data. An example of such efforts is the 1000Human Genome Project1, which has already produced 200TBof genome data across 1700 samples, and made it availableon the Amazon Cloud Storage2.
As a large number of researchers have access to the data,the focus is beginning to shift to analysis of this data. Thereexist many tools [15], [24], [30], [34] and libraries [4] to
1www.1000genomes.org2http://aws.amazon.com/1000genomes/
ease the implementation of new data analysis programs ongenetic data. One of the complexities in this area is that dataformats in which sequences are stored are very specialized.Existing tools, in most cases, alleviate the need for scientistsunderstanding these formats and coding complex algorithms.
Consistent with the overall trend in data analytics, use ofparallelism is inevitable in analysis of genomic data also. Low-ered response time, including facilitating interactive analysisof large-scale data, can open us many novel opportunities forresearchers. Again, just like all other fields, the difficulty ofcarrying-out parallel implementations, especially for domainexperts, is a large obstacle to widespread use of parallelismfor analysis of genomic data.
The current state-of-the-art in parallel analysis of genomicdata is very limited. There are many serial software suitesthat lack any parallelization capability[19], [23], [24], [26],[30]. General MapReduce frameworks such as Hadoop [36]can potentially be used, but even parallelization using such aframework can be a hard problem for the community [20],[29]. Particularly, developing a parallel implementation withthe use of MapReduce will require knowledge of data formatsand reprogramming of an existing serial program, neither ofwhich is desirable. Load imbalance is another problem whileprocessing genomic data, which requires nuanced solutions.Genome Analysis Tool Kit (GATK)[27] provides developers aMapReduce-like framework specific for analysis of genomicdata. However, GATK does not provide distributed memoryparallelization for most of its programs and its scalability islow, as we will show in Section VI.
Thus, we can see that easing parallelization of applicationsthat process genomic data, while also achieving high parallelefficiency, is a challenging problem. In this study, we areproposing PAGE, which is a MapReduce-like middleware thatallows users to continue to utilize their existing programs, butachieve parallelization with a modest additional effort. Themap and reduce programs can be separate executables writtenin any (and even two distinct) programming languages. We canalso work with any of the popular genomic data formats. Themiddleware divides the genome structure into regions/intervalsand runs each map task for a different region, performing loadbalancing in the process. Subsequently, these partial resultsare reduced to obtain the final result(s). In order to improveefficiency, our middleware provides streaming task scheduling,in which the tasks can be scheduled dynamically with the helpof a master node, and the reduce tasks do not have to wait forall map tasks to be finished.
We evaluate PAGE with 4 existing applications, which are
!000111444 IIIEEEEEEEEE !888ttthhh IIInnnttteeerrrnnnaaatttiiiooonnnaaalll PPPaaarrraaalllllleeelll &&& DDDiiissstttrrriiibbbuuuttteeeddd PPPrrroooccceeessssssiiinnnggg SSSyyymmmpppooosssiiiuuummm
!555333000---222000777555 222000!444
UUU...SSS... GGGooovvveeerrrnnnmmmeeennnttt WWWooorrrkkk NNNooottt PPPrrrooottteeecccttteeeddd bbbyyy UUU...SSS... CCCooopppyyyrrriiiggghhhttt
DDDOOOIII !000...!!000999///IIIPPPDDDPPPSSS...222000!444...!999
777222

Locations 1 2 3 4 5 6 7 8 9Reference Bases A A C G T A C C CRead-1 A A C GRead-2 A C G GRead-3 A C G T ARead-4 C G T T C CRead-5 T A T C G
Fig. 1. Representation of an alignment file with reference bases.
VarScan [19], and three applications currently supported byGATK, which are Unified Genotyper [7], Realigner TargerCreator [9], and Indel Realigner [8]. We compare the per-formance of PAGE against GATK for the three applicationscurrently available as part of GATK, and against Hadoop forVarScan. In both cases, we show that PAGE performs signifi-cantly better. The scalability of PAGE for these applications is12.7x, 12.8x, 14.1x and 9x, respectively, when the computingpower is increased by 16 times.
II. GENOMIC DATASETS, APPLICATIONS, ANDPARALLELIZATION
A. Genetic DataUsing sequencing technologies such as Illumina/Solexa3,
we obtain genomic data, which comprises nucleotide base se-quences (sequences of bases, i.e., A, T, G, or C). Subsequently,these sequences can be mapped to particular regions of thegenome by a sequence alignment program [21], [25], [23].Fasta4 is a common (and simple) format, where nucleotidesequences are stored in the text form. The results of a sequencealignment program is more complex, as the file comprisesof reads, where each read is a sequence of nucleotides ofa varying length. Each read is mapped to a region of thegenome, with each mapping having features like location,bases in the sequence, strand (direction) of the sequence,quality of the bases in the sequence, existence of insertionor deletions, and others. A representation of alignment ofreads with reference bases is shown in Figure 1. As can beseen from the Figure, two or more different reads can bemapped to the same or intersecting regions, i.e., the numberof alignments for a particular locus can be more than one andvary among different locations, causing what is referred to ascoverage variance. Most common alignment file format is theSequence/Alignment MAP (SAM) [24] format, which keepsalignment information in a text format. To save space, it is acommon way to keep these alignment files in binary format,specifically, the BAM files, as is the case for the publiclyavailable data from the 1000 Human Genome Project. How-ever, we cannot process BAM files directly because of theirbinary nature. Instead, we can use the SamTools software [24]or the Picard Java API [4].
B. Representative Applications for Processing Genomic DataA variety of analysis tasks are applied on genomic data
stored in FASTA, SAM, BAM, or other related formats. Suchanalysis can include simple diagnostic tools that obtain basicstatistical information, perform quality controls for analyzingerrors on the genetic data, aligning read sequences, variantidentification, comparison of genetic data of different samples,or others. We explain three representative data analysis tasksbelow.
3http://www.illumina.com/technology/solexa technology.ilmn4http://zhanglab.ccmb.med.umich.edu/FASTA/
SNP Calling: In comparing the genomes of two individualsof the same species, at some locations only a single nucleotidemay vary. These single variations in the genes are called SingleNucleotide Polymorphism (SNP), which can have a significanteffect on the biological difference between members of thesame species. For example, it is known that only one SNP isresponsible for the earwax type [38]. Therefore, detection ofSNPs and analyzing their effects for diseases is crucial, withapplications including personalized medicine [13], [19], [26],[35].
Process of identifying SNPs may seem like a simple datascan problem (though over a massive dataset, because of thelength of each genome). However the errors during digiti-zation of a sample’s genetic data and the alignment files’nature complicates their detection. Specifically, we need tofind differences in nucleotides across different samples bylooking at potentially unreliable or unmatched informationacross different reads while also trying to eliminate falsepositives.Detecting Problematic Regions: During the process of align-ment, some regions may require special attention due toproblems like low quality, or misaligned or duplicate reads.Identifying these problematic regions is important for accu-racy of the later analyses. Detection of indels (insertion ordeletion) [9] and duplicates5 are examples of this type ofapplications.Quality Improvement: As mentioned above, issues like mis-aligned or low quality read sequences impact the correctness ofthe further analyses. Therefore, the alignment files may needto be re-processed to increase their overall quality. Eliminatingthe duplicates6 or applying realignment for the suspiciousintervals [8] are examples of these applications.
C. Parallelization of AlgorithmsWe examine the parallelization of the algorithms we have
introduced from two perspectives: 1) parallelization of pro-cessing a single genome, and 2) parallelization when mul-tiple genomes (from different members of the species) areprocessed.Parallelization of Processing a Single Genome: We cangroup algorithms in 3 groups according to how they process asingle genome, and how it affects their correct parallelization.Locus-based Algorithms: Each single location is consideredseparately and can be processed independently. Therefore, thegenome can be divided into separate blocks by splitting it atany point, and the parallel execution will still be correct. SNPcalling can be considered in this group because the existenceof SNP in a particular location depends on only the nucleotidesat that location and is independent from nucleotides in otherlocations. Diagnostic tools like counting number of bases [10],[30] are other examples of this group.Region-based algorithms: Interval(s) in genome comprising ofmultiple consecutive locations need to be processed together.Pattern matching algorithms in which a certain sequence ofnucleotides is searched across genome and processed forfurther analysis is an example of this group [11], [12]. Ifthe genome is divided into chunks arbitrarily and each chunkis processed independently, the processing may be incorrect.However, each chromosome, and in some cases, even certainparts of chromosomes, can be processed independently.Genome-based algorithm: The entire genome should be pro-cessed together in this group. An example will be an alignment
5MarkDuplicates feature of Picard [4]6For example, rmdup feature of SamTools
777333
problem, where a read sequence can be mapped to any regionof the genome [8], [21], [23], [25].Parallelization of Multiple Genome Processing: Most ge-nomic data analysis tasks today involve processing of multiplegenomes (i.e.. from different samples or members of the samespecies). The algorithms involved may do so in two differentways:Independent Processing of Genomes: The same algorithm maybe applied to each sample separately to obtain a differentoutput for each of them. These outputs could optionally becombined together in the end. Alignment [21], determininglow quality regions [9], realigning [9] and improving thequality of the files [11] are all examples of these.
For the parallelization of this group of applications, theapplication can be executed separately in different computingnodes. However, the challenge is load balancing. Particularly,input file sizes may vary drastically. For example, the size ofthe files for different samples ranges from 50 MB to 5 GB inthe pilot dataset of the 1000 Human Genome Project, whichis sequenced by MOSAIK7. Thus, a parallelization frameworkneeds to address load balance problem, and hide the solutionsfrom the user.Inter-dependent Processing of Genomes: In algorithms forSNP calling, or those for obtaining statistical informationsummarizing all inputs, or finding similarities/differences ofgenomes8, data from multiple genomes is accessed at the sametime. In other words, these applications produce a single outputwhile processing multiple genomes.
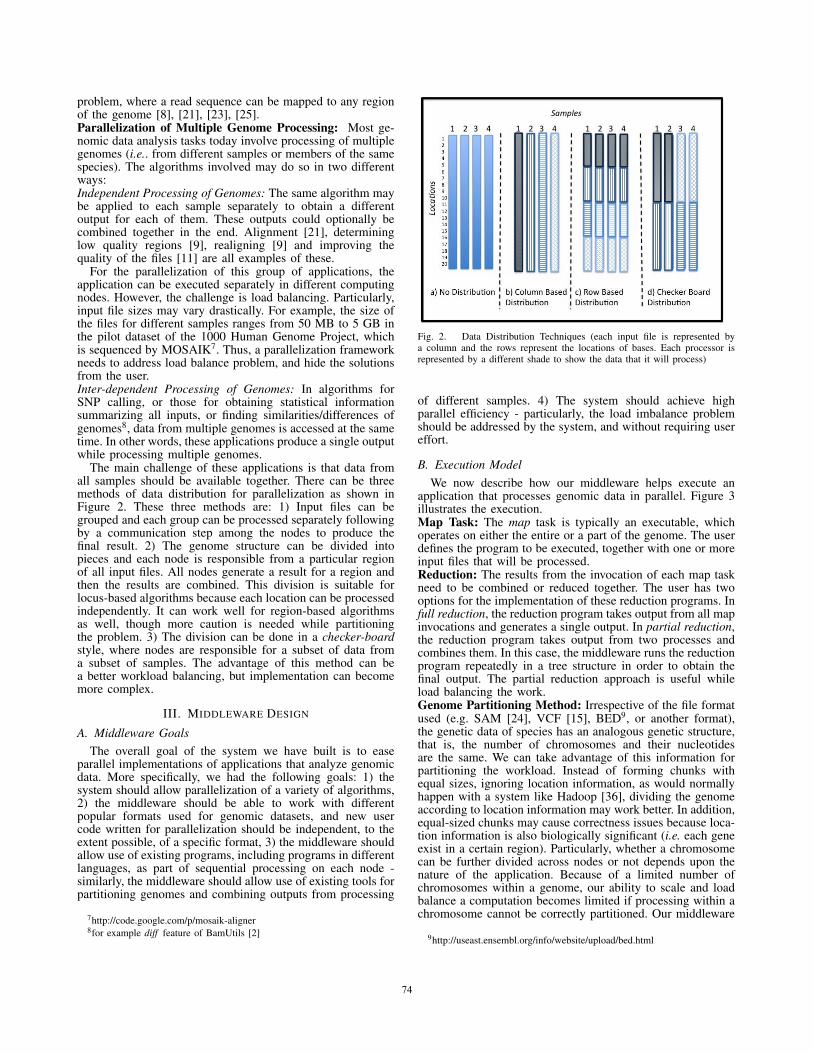
The main challenge of these applications is that data fromall samples should be available together. There can be threemethods of data distribution for parallelization as shown inFigure 2. These three methods are: 1) Input files can begrouped and each group can be processed separately followingby a communication step among the nodes to produce thefinal result. 2) The genome structure can be divided intopieces and each node is responsible from a particular regionof all input files. All nodes generate a result for a region andthen the results are combined. This division is suitable forlocus-based algorithms because each location can be processedindependently. It can work well for region-based algorithmsas well, though more caution is needed while partitioningthe problem. 3) The division can be done in a checker-boardstyle, where nodes are responsible for a subset of data froma subset of samples. The advantage of this method can bea better workload balancing, but implementation can becomemore complex.
III. MIDDLEWARE DESIGN
A. Middleware GoalsThe overall goal of the system we have built is to ease
parallel implementations of applications that analyze genomicdata. More specifically, we had the following goals: 1) thesystem should allow parallelization of a variety of algorithms,2) the middleware should be able to work with differentpopular formats used for genomic datasets, and new usercode written for parallelization should be independent, to theextent possible, of a specific format, 3) the middleware shouldallow use of existing programs, including programs in differentlanguages, as part of sequential processing on each node -similarly, the middleware should allow use of existing tools forpartitioning genomes and combining outputs from processing
7http://code.google.com/p/mosaik-aligner8for example diff feature of BamUtils [2]
Fig. 2. Data Distribution Techniques (each input file is represented bya column and the rows represent the locations of bases. Each processor isrepresented by a different shade to show the data that it will process)
of different samples. 4) The system should achieve highparallel efficiency - particularly, the load imbalance problemshould be addressed by the system, and without requiring usereffort.
B. Execution ModelWe now describe how our middleware helps execute an
application that processes genomic data in parallel. Figure 3illustrates the execution.Map Task: The map task is typically an executable, whichoperates on either the entire or a part of the genome. The userdefines the program to be executed, together with one or moreinput files that will be processed.Reduction: The results from the invocation of each map taskneed to be combined or reduced together. The user has twooptions for the implementation of these reduction programs. Infull reduction, the reduction program takes output from all mapinvocations and generates a single output. In partial reduction,the reduction program takes output from two processes andcombines them. In this case, the middleware runs the reductionprogram repeatedly in a tree structure in order to obtain thefinal output. The partial reduction approach is useful whileload balancing the work.Genome Partitioning Method: Irrespective of the file formatused (e.g. SAM [24], VCF [15], BED9, or another format),the genetic data of species has an analogous genetic structure,that is, the number of chromosomes and their nucleotidesare the same. We can take advantage of this information forpartitioning the workload. Instead of forming chunks withequal sizes, ignoring location information, as would normallyhappen with a system like Hadoop [36], dividing the genomeaccording to location information may work better. In addition,equal-sized chunks may cause correctness issues because loca-tion information is also biologically significant (i.e. each geneexist in a certain region). Particularly, whether a chromosomecan be further divided across nodes or not depends upon thenature of the application. Because of a limited number ofchromosomes within a genome, our ability to scale and loadbalance a computation becomes limited if processing within achromosome cannot be correctly partitioned. Our middleware
9http://useast.ensembl.org/info/website/upload/bed.html
777444

(a) Inter-dependent Processing of Genomes (b) Independent Processing of Genomes
Fig. 3. Execution Models of PAGE
asks whether the partitioning can be by-locus, and if that isnot the case, a by-chromosome partitioning is performed. Sincethe number of bases is different for each chromosome, ourmiddleware tries to decrease the imbalance among regions bytaking into account the chromosome lengths.
Another factor for partitioning is whether the processingacross samples is independent or inter-dependent. If the for-mer, we have greater flexibility in achieving load balance,whereas in the latter case, we have to divide the genomelocations for a single genome.Load Balancing Method: The genome needs to be dividedinto small regions according to the partitioning method. Thenall of these regions should be assigned to computing nodes.Scheduling of the processing of regions, i.e., the tasks, is oneof the challenges for the middleware. Our middleware providestwo types of scheduling schemes, which are the static anddynamic schemes. In static scheduling, the number of regionsis set to number of processors and regions are determinedaccording to partitioning method. Each of these regions areassigned to a different processor. After all map tasks finish, thereduction phase can begin. The problem with such schedulingis that it may not work well for all cases, because of filesize differences among the inputs and coverage variance oflocations, as described in the previous section. Therefore, ourmiddleware also provides dynamic scheduling. This schemeemploys one of the processors as the master node and operatesin a streaming fashion. The details of this scheduling schemeare explained in the next section.
C. Middleware Applicability
The execution model described above creates certain restric-tions on the kind of processing that can be parallelized. First,the map tasks must be able to take interval/region parameter asinput or must be able to work together with programs that areable to partition genomes into pieces such as SamTools [24].Next, the reduction should have one of the formats we haddiscussed above. Finally, the algorithm should be safe tobe parallelized by processing different areas of the genomeindependently. Formally, let P be the map function, !, !!,and !" be three regions, such that concatenation of !! and!" gives us !. Then the following property should hold:
" !!" # " !!!"! " !!""
where, ! is the reduction function. It is easy to see that alllocus-based algorithms can be parallelized by this system. Forregion-based and genome-based algorithms, the correctnessdepends upon the specific algorithm and the partitioningfunction used. For example, for the region-based algorithms ifwe divide the data only by chromosomes, the algorithm canbe parallelized correctly, though with some limitations on itsscalability.
IV. MIDDLEWARE IMPLEMENTATION
Our system involves a carefully optimized implementationof basic design described in the previous section. Particularly,we use the streaming idea, i.e.. the reduce tasks can start eventhough there are map tasks still executing, with the goal ofimproving workload balance and increasing performance. Thepseudo-code for this scheme is given as Algorithm 1.
In order to schedule tasks dynamically, we assign one ofthe nodes as the master node, i.e., to manage the flow oftasks while the other nodes do the actual computations. Themaster node first initializes two queues # and !, which keepmap and reduce tasks to be executed, respectively. Note thatif there will be separate outputs for each input, there will bea separate ! queue for each output file. For simplification,we assume that there will be single output. The master alsoinitializes the "$%&'()( array, which keeps workers’ statusinformation (Line 2). A worker can be in #*" , !+,-.+,or /,0+ status. The master node also calculates the regionsof the genome according to genome division method (Line 3)and inserts these regions into the queue # as map tasks (Line4). Note that if we apply the by-chromosome partitioning, eachregion will be a different chromosome. Instead, if we applythe by-locus partitioning, we have 1 equal length regions. Ifthere will be multiple outputs, we sort the input files accordingto their size and put the regions of the larger inputs first intothe queue in order to increase the performance.
The master node starts waiting a task request from theworker nodes (Line 6). When it receives a request, it firstchecks what type of task the requesting node has finished. Ifthe requesting worker finished a map-task, the master nodeinserts address of the intermediate result files of that finishedmap task to the queue ! (Lines 7-8). Note that a map-taskcan produce multiple results to be inserted into the queue !.If requesting worker finished a reduce task, and if it is not the
777555

Algorithm 1 PAGE’s Streaming Dynamic Scheduling Algo-rithm
1: Master Node2: Initialize map-tasks queue # , reduce-tasks queue ! and
processor status array "$%&'()(3: Divide the genome(s) into 1 chunks4: Enqueue N map tasks into #5: repeat6: Wait for a request from workers7: if Requesting node finished a map task (2! ) then8: enqueue result(s) of 2! to R9: else if Requesting node finished a reduce task (2") &
2" is not the final reduce task then10: enqueue result of 2" to R11: end if12: if M is not empty then13: 2! # 3456464!#"14: send 2! to the requesting node15: "$%&'()($$45647(89:9%34% # #*"16: else if final output is not produced yet then17: "$%&'()($$45647(89:9%34% # /,0+18: for all IDLE nodes / do19: if R has sufficient entries then20: 2" = entries dequeued from R21: send 2" to /22: "$%&'()($/% # !+,-.+23: else24: break25: end if26: end for27: else28: break29: end if30: until all tasks are done31: Send end message to all nodes32: Worker Node33: repeat34: Request a task from master node35: if task is a #*" then36: intervals = intervals received from master node37: map( intervals )38: else if task is a !+,-.+ then39: files = files received from master node40: reduce( files )41: else42: break43: end if44: until true
final reduce task, it enqueues the output of the finished reducetask to the queue ! (Lines 9-10). Then, if there is a map-taskin queue M, master node assigns it to the requesting node(Lines 12-15). If all the map tasks are assigned, and the finalresult has not yet been produced, the master node first setsthe task requesting node’s status to /,0+ (Line 17). Next, itchecks the queue ! if there are a sufficient number of entriesto be reduced. If so, it assigns them to idle nodes (Lines 18-25). Note that if the reduction type is full reduction, all themap tasks for the corresponding output must be finished andall the intermediate results of that output should be assigned
to a single node. If it is partial reduction, each idle node getsonly 2 intermediate results (if available) dequeued from thequeue !. If all the map and reduce tasks have finished, theexecution is over and master node sends the end message toall nodes.
During this process, worker nodes simply execute a loop.At the beginning of each iteration, they request a task fromthe master node (Line 34). They can run map task (Lines 35-37) or reduce task (Lines 38-40) depending on the messagethey received from the master node. Once they receive the endmessage, they finish the execution.
V. APPLICATIONS DEVELOPED AND PROGRAMMABILITY
We implemented our middleware in C programming lan-guage with the MPI library. We implemented four applicationsthat have different properties, leading to differences in theway they use the middleware and are parallelized. Three ofthese applications are from the Genome Analysis ToolKit(GATK) [27], which is another framework for parallelizinggenomic applications. One of the advantages of using GATKbased applications is that we can compare performance againstparallelization performed by GATK. We now explain theseapplications.Realigner Target Creator (RTC): RTC [9] is one of pro-grams available from GATK. RTC finds suspicious indels inthe genome that may need realignment. The execution modelfor RTC is independent processing since each indel intervallist is special for each input file. The reduction is simpleconcatenation of all output files, which are the list of intervals.Indel Realigner (IR): IR [8] is another program availablefrom GATK. IR takes the output of RTC, which is a list ofindels to be realigned, as input, and applies a local realign-ment. Its execution model is independent processing, sincerealignment is specific operation for each alignment file. Weused merge feature of SamTools for reduction of intermediateresults, which are basically alignment files.Unified Genotyper (UG): UG [7] is yet another programfrom GATK that finds SNPs and indels by applying Bayesiangenotype likelihood model and generates a single VCF file.We used concatenate function of VCFTools for the reductionof the intermediate results.VarScan(VS): VarScan[19] is a commonly-used serial Java-based variant detection program, which finds variants likeSNPs and indels among multiple genomes. It takes multiplealignment files and a reference file to detect variants andgenerates a single output file. It is a locus-based algorithm.One important drawback of the software is that it cannotprocess the alignment files which are in SAM or BAM formats,instead, it requires a pre-processing step in which all thealignment files and the reference genome data are piled up intoa single text-based file that is called the mpileup file. Normally,VarScan does not have any parameters to process a specificregion. However, we can take advantage of SamTools, whichcan generate mpileup files and take a parameter to processonly specific regions. For the reduction, we used cat bash shellcommand to concatenate the output files, which are basicallylists of SNPs.
In Table I, the map task commands for using these appli-cations with PAGE, their execution models, and the reductionmethods we used are shown. The bolded regionloc, inputloc,extrainputloc, outputloc texts in map task commands arereserved words of PAGE, helping to use the genomic applica-tions and showing where to insert region, input and outputfile parameters in the corresponding genomic application’sexecution command.
777666

TABLE IAPPLICATIONS PARALLELIZED USING PAGE
Application Map-Task Command Execution Model ReductionRTC java -jar GenomeAnalysisTK.jar -nt 1 -I inputloc -L
regionloc -R reference file -T RealignerTargetCreator -ooutputloc
Independent cat bash shell command
IR java -jar GenomeAnalysisTK.jar -nt 1 -I inputloc -L re-gionloc -R reference file -targetIntervals extrainputloc-T IndelRealigner -o outputloc
Independent merge feature of SamTools
UG java jar GenomeAnalysisTK.jar -nt 1 -nct 1 -I in-put file 1 -I input file 2 ... -I input file n -L regionloc-R reference file -T UnifiedGenotyper -o outputloc
Inter-dependent concatenate feature of VCFTools
VarScan samtools mpileup -b alignment file list -r regionloc-f reference file ! java -jar VarScan.jar mpileup2snp!outputloc
Inter-dependent cat bash shell command
TABLE IIPARALLELIZATION CAPABILITY OF GATK FOR ITS OWN TOOLS.
Application Data Thread CPU Thread GATK-QueueRealigner TargetCreator (RTC)
! " "
Indel Realigner(IR)
" " !
UnifiedGenotyper (UG)
! ! !
A. Programmability and Parallelization ComparisonTwo closest frameworks to PAGE are Hadoop Streaming[3]
and GATK. Hadoop Streaming is a utility based on the popularframework Hadoop [36]. The key feature of Hadoop Streamingis that it allows users to use executable programs and scriptsas map and reduce tasks. As we have stated earlier, GATK is acomparable framework that provides parallelization at differentlevels. It has 2 types of threads, i.e., data and CPU threadsfor shared memory parallelization. Particularly, with respectto the three tools we have used, RTC can be parallelizedby employing multiple data threads while UG can use bothtypes of threads. On the other hand, there is no shared-memory parallelization for IR. In order to maintain distributedmemory parallelization, GATK can work together with an-other software called GATK-Queue10, which is a scriptingframework for genomic analysis. GATK-Queue runs GATKon different nodes for different regions of the genome, andthen gathers the intermediate results to form the final output.GATK-Queue can work together with GATK’s own shared-memory parallelization. However, GATK-Queue can be usedonly for UG and IR, not for RTC. The parallelization capabilityof GATK for RTC, IR, and UG is summarized in Table II.
In the next section, we will compare performance of PAGEagainst Hadoop Streaming (for VarScan) and GATK (for theother three applications). Before that, however, we comparethe programming effort involved with these systems.
In parallelization using GATK, one does not need to writeany code for map and reduce tasks. In other words, par-allelization with GATK and PAGE can be considered verycomparable.
Now, focusing on Hadoop Streaming, which we used toparallelize VarScan - in order to run VarScan in parallel, weneed to perform 3 operations: 1) Conversion of alignment filesin the BAM format and reference file in the Fasta format intothe mpileup file format 2) Running VarScan to detect SNPs,3) Concatenating lists of SNPs to form the final output. A
10http://www.broadinstitute.org/gatk/guide/article?id=1306
comparison of how these 3 operations are performed withHadoop Streaming and PAGE is shown in Table III. As canbe seen from the Table, PAGE does not require much effortbecause of its ability to employ SamTools (and take availableoptions for partitioning in SamTools). In comparison, HadoopStreaming needs to deal with data formats explicitly. Wehave shown two independent approaches for parallelizing anapplication like VarScan with Hadoop Streaming in Table III.The two approaches require comparable programming effort.In the execution times we report, the scripting method wasused.
VI. EXPERIMENTAL RESULTS
In this section, we report results from a series of experimentswe conducted to examine the performance of our middleware,using four genomic applications we had described earlier, andcompare PAGE with Hadoop Streaming and GATK.
A. Experimental Setup and Parameter ConfigurationsIn our experiments, we used a cluster where each node has
8 cores 2.53 GHz Intel (R) Xeon (R) processor and 12 GBmemory. We used the genomic data from the 1000 HumanGenome Project, which was downloaded from the AmazonCloud Storage Systems. We selected 40 random samples fromthe pilot data set and used their alignment files sequencedby MOSAIK on different applications. In order to set numberof threads for shared memory parallelization of GATK forUG, we run it with different number of parameters for atraining dataset and selected the parameter set that gave thebest result. We performed the same method to set the numberof regions for dynamic scheduling scheme (while using by-locus partitioning with PAGE).
B. Experimental ResultsWe now evaluate the performance of PAGE, with two
sets of experiments focusing on scalability while increasingcomputing power, and performance as the data volume isincreased.Parallel Scalability Evaluation: We evaluated the paral-lel scalability of applications with PAGE by varying num-ber of nodes (and cores) we allocated, while also varyingthe scheduling schemes and data partitioning methods. Weused by-locus partitioning with all 4 applications while by-chromosome partitioning is applied only for RTC and IR. Thisis because the execution model of the other two applications,VarScan and UG, is inter-dependent processing in which themaximum number of data chunks is limited by the numberof chromosomes in the genome (24 in our case). We used 10
777777

TABLE IIIPROGRAMMABILITY COMPARISON OF HADOOP STREAMING AND PAGE
Hadoop Streaming PAGE
Data Conversion
Bash Scripting! Manually divide genome into equal regions! Run SamTools for each region in parallel! Upload the data to HDFS
OR Hadoop Implementation! Define BAM and Fasta file formats to Hadoop! Run SamTools as a map task! Merge results in mpileup format as a reduce task
Run SamTools &VarScan together as amap task
Data Processing Run VarScan as a map taskReduction cat bash shell command cat bash shell command
files for RTC and IR, 30 files for UG and 40 files for VarScan.The results are shown in Figure 4.
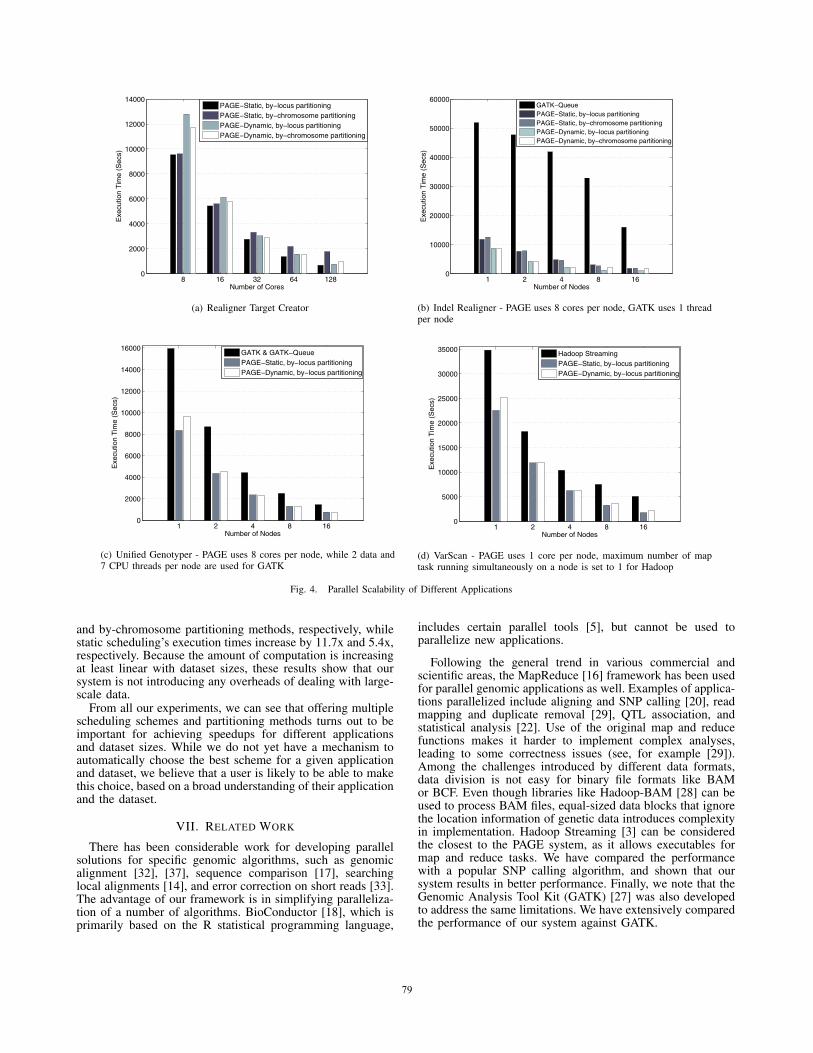
In scalability experiments with RTC, we are not able to useGATK-Queue, and thus no comparison of distributed memoryscalability is feasible. In order to see its shared-memoryparallelization capability, we run RTC for 10 files with 8threads and the execution took 78012 seconds. Subsequently,we performed scalability experiments with RTC with differentparameters of PAGE. As seen from the Figure 4(a), PAGE’sall executions with 8 cores (within a single node) outperformGATK. In observing the performance, as the number of nodesincreases, static scheduling has higher performance with lowernumber of nodes, which is because with dynamic scheduling,one of the cores is allocated as the master process. Staticscheduling with by-locus partitioning has better performancethan dynamic scheduling with by-locus partitioning. However,when we apply by-chromosome partitioning and use more than16 cores, dynamic scheduling outperforms static scheduling.By increasing number of cores from 8 to 128 and usingby-locus partitioning, the speed-ups obtained for static anddynamic scheduling schemes are 14.1x and 17x (number ofworkers increased from 7 to 127), respectively. When weapply by-chromosome partitioning, the speed-ups for staticand dynamic scheduling schemes decreased to 5.5x and 12.3x,respectively, as expected. Overall, these results show thatdynamic scheduling is better than static scheduling in terms ofmaintaining workload balance among nodes when the chunksare not of equal size.
RTC can be considered as a region-based algorithm, sinceits output contains a list of regions. However, a by-locus par-titioning does result in correct parallelization, and particularly,we can see that PAGE produced the same output with GATKfor both partitioning types.
In scalability evaluation of IR, we compared PAGE withthe GATK-Queue framework. As noted in the title of theFigure 4(b), GATK is unable to use multiple cores within eachnode. Thus, it is not surprising that the absolute performancefor PAGE is much better. But, it is interested to note thatgoing from 1 node to 16 nodes, GATK’s speed increasedby 3.25x while PAGE’s speed increased by 9x with dynamicscheduling and by-locus partitioning. We can again see thatusing our dynamic scheduling has better performance thanstatic scheduling when we apply by-chromosome partitioning.
In scalability evaluation of UG, we compared PAGE withparallelization of GATK and GATK-Queue. We increasednumber of nodes from 1 to 16. In each node, we employed allcores for PAGE’s execution while we used 2 data threads and7 CPU threads for GATK’s execution. As seen from the Fig-ure 4(c), PAGE’s both scheduling schemes outperform GATK.
In terms of actual execution time, static scheduling givesbetter results for lower scalability while dynamic schedulingperforms better for higher number of nodes. When the numberof cores is increased by 16x, execution time of PAGE-dynamic,PAGE-static and GATK increased by 12.8x, 11x and 10.9x,respectively.
VarScan is not supported on GATK, and our comparisonfocused on using Hadoop Streaming. In previous experiments,we increased number of nodes from 1 to 16. However, usingsingle node was not enough to keep all converted data (roughlyaround 510 GB) in HDFS. Therefore, we increased number ofnodes from 8 to 128 while keeping the computing power atthe same level by allocating single core on each node. In orderto achieve this in Hadoop’s execution, we set the maximumnumber of map tasks running simultaneously by a task trackerto 1. As seen from Figure 4(d), PAGE has better performancein terms of actual execution time for all cases. PAGE achieves12.7x speedup with static scheduling and 11.8x speedup withdynamic scheduling while the execution with Hadoop achieves6.9x speedup.Scalability With Respect to Dataset Sizes: In this set ofexperiments, we varied the number of files to be processed inorder to see the performance of PAGE, Hadoop and GATKwith different dataset sizes. We used 128 processors forPAGE’s executions and used the equivalent parameter settingsfor GATK and Hadoop. We run our 4 applications with thesesystems, and increased the load from 1 to 16 alignment files(all of similar sizes). The results are shown in Figure 5.
For RTC experiments, we are unable to make any compar-ison against another platform. However, we can see that theincrease in execution time for the best version is at most linear.The by-chromosome partitioning has lower performance thanby-locus partitioning, as expected. For the low number of files,its performance is worse because of not being able to use allthe nodes due to insufficient number of chromosomes. Withby-chromosome partitioning, static scheduling gives betterresults than dynamic scheduling in this experiment, in contrastwith the previous experiment. This is because the sizes of filesused in previous experiment had a higher variance than thefiles used in this experiment. Overall, we can conclude thatstatic scheduling is preferable when the data can be dividedinto equal chunks; on the other hand, dynamic scheduling doesbetter managing unequal sized chunks.
In experiments with IR, recall that GATK-Queue can onlyuse 1 of the 8 cores. Still, we can see that performance ofour system is better by more than a factor of 8. Dynamicscheduling also outperforms static scheduling for all cases.When the data set size increased by 16x, dynamic scheduling’sexecution times increase only by 4.7x and 2.7x for by-locus
777888
8 16 32 64 1280
2000
4000
6000
8000
10000
12000
14000
Number of Cores
Exe
cutio
n T
ime
(Sec
s)
PAGE!Static, by!locus partitioningPAGE!Static, by!chromosome partitioningPAGE!Dynamic, by!locus partitioningPAGE!Dynamic, by!chromosome partitioning
(a) Realigner Target Creator
1 2 4 8 160
10000
20000
30000
40000
50000
60000
Number of Nodes
Exe
cutio
n T
ime
(Sec
s)
GATK!QueuePAGE!Static, by!locus partitioningPAGE!Static, by!chromosome partitioningPAGE!Dynamic, by!locus partitioningPAGE!Dynamic, by!chromosome partitioning
(b) Indel Realigner - PAGE uses 8 cores per node, GATK uses 1 threadper node
1 2 4 8 160
2000
4000
6000
8000
10000
12000
14000
16000
Number of Nodes
Exe
cutio
n T
ime
(Sec
s)
GATK & GATK!QueuePAGE!Static, by!locus partitioningPAGE!Dynamic, by!locus partitioning
(c) Unified Genotyper - PAGE uses 8 cores per node, while 2 data and7 CPU threads per node are used for GATK
1 2 4 8 160
5000
10000
15000
20000
25000
30000
35000
Number of Nodes
Exe
cutio
n T
ime
(Sec
s)
Hadoop StreamingPAGE!Static, by!locus partitioningPAGE!Dynamic, by!locus partitioning
(d) VarScan - PAGE uses 1 core per node, maximum number of maptask running simultaneously on a node is set to 1 for Hadoop
Fig. 4. Parallel Scalability of Different Applications
and by-chromosome partitioning methods, respectively, whilestatic scheduling’s execution times increase by 11.7x and 5.4x,respectively. Because the amount of computation is increasingat least linear with dataset sizes, these results show that oursystem is not introducing any overheads of dealing with large-scale data.
From all our experiments, we can see that offering multiplescheduling schemes and partitioning methods turns out to beimportant for achieving speedups for different applicationsand dataset sizes. While we do not yet have a mechanism toautomatically choose the best scheme for a given applicationand dataset, we believe that a user is likely to be able to makethis choice, based on a broad understanding of their applicationand the dataset.
VII. RELATED WORK
There has been considerable work for developing parallelsolutions for specific genomic algorithms, such as genomicalignment [32], [37], sequence comparison [17], searchinglocal alignments [14], and error correction on short reads [33].The advantage of our framework is in simplifying paralleliza-tion of a number of algorithms. BioConductor [18], which isprimarily based on the R statistical programming language,
includes certain parallel tools [5], but cannot be used toparallelize new applications.
Following the general trend in various commercial andscientific areas, the MapReduce [16] framework has been usedfor parallel genomic applications as well. Examples of applica-tions parallelized include aligning and SNP calling [20], readmapping and duplicate removal [29], QTL association, andstatistical analysis [22]. Use of the original map and reducefunctions makes it harder to implement complex analyses,leading to some correctness issues (see, for example [29]).Among the challenges introduced by different data formats,data division is not easy for binary file formats like BAMor BCF. Even though libraries like Hadoop-BAM [28] can beused to process BAM files, equal-sized data blocks that ignorethe location information of genetic data introduces complexityin implementation. Hadoop Streaming [3] can be consideredthe closest to the PAGE system, as it allows executables formap and reduce tasks. We have compared the performancewith a popular SNP calling algorithm, and shown that oursystem results in better performance. Finally, we note that theGenomic Analysis Tool Kit (GATK) [27] was also developedto address the same limitations. We have extensively comparedthe performance of our system against GATK.
777999

1.3 2.7 5.3 10.4 21.10
200
400
600
800
1000
1200
1400
Data Size(GB)
Exe
cutio
n T
ime
(Sec
s)
PAGE!Static, by!locus partitioningPAGE!Static, by!chromosome partitioningPAGE!Dynamic, by!locus partitioningPAGE!Dynamic, by!chromosome partitioning
(a) Realigner Target Creator
1.3 2.7 5.3 10.4 21.10
5000
10000
15000
20000
25000
30000
Data Size(GB)
Exe
cutio
n T
ime
(Sec
s)
GATK!QueuePAGE!Static, by!locus partitioningPAGE!Static, by!chromosome partitioningPAGE!Dynamic, by!locus partitioningPAGE!Dynamic, by!chromosome partitioning
(b) Indel Realigner
1.3 2.7 5.3 10.4 21.10
100
200
300
400
500
600
700
800
900
Data Size(GB)
Exe
cutio
n T
ime
(Sec
s)
GATK & GATK!QueuePAGE!Static, by!locus partitioningPAGE!Dynamic, by!locus partitioning
(c) Unified Genotyper
1.3 2.7 5.3 10.4 21.10
100
200
300
400
500
600
700
800
900
1000
Data Size(GB)
Exe
cutio
n T
ime
(Sec
s)
Hadoop StreamingPAGE!Static, by!locus partitioningPAGE!Dynamic, by!locus partitioning
(d) VarScan
Fig. 5. Impact of Dataset Size (Each alignment file is approximately 1.3 GB)
Other tools have been developed in context of genomicdata. SamTools [24] allows user to do operations such asmerging, indexing, sorting and so on, and also provides alibrary in C that can be used for developing other tools.BEDTools [30] is another program which provides variousgenomic data operations, for example, intersecting 2 BEDfiles, computing coverage in BAM files, merging overlappingfeatures and so on. However, BedTools does not provide alibrary to be used for developing tools. GenomicTools [34] isa flexible computational platform that allows users to performoperations on GFF, SAM/BAM and BED formats. Besides itscommand-line tools, it also provides API in C++. However,none of them support parallel application development.
Our work is also related to the problem of task scheduling inparallel systems. Because of a large volume of existing work inthis area, we restrict ourself to the most similar system, whichis MapReduce. In the conventional MapReduce approach, eachtask is assigned dynamically and reduce tasks cannot be startedbefore all map tasks finish. A combiner function can be usedto reduce the amount of intermediate results, and the taskscheduling works in a first-in, first-out fashion, assuming thateach task will require similar execution time. Because thissimple approach may not work well for many cases, a numberof studies have developed other schemes. This includes workon execution in a heterogeneous environment [31], clusters
with multiple users [39] and improving data locality [40].PAGE’s streaming dynamic scheduling differs from the con-ventional MapReduce approach, because reduce tasks do nothave to wait for all map tasks to finish. Our partial reductionmechanism is also different from the use of a combinerfunction, because unlike combiner function, partial reductioncan be applied to intermediate results produced by differentnodes.
VIII. CONCLUSION
In this paper, we have a novel middleware, PAGE, which al-lows users to parallelize genomic applications in a convenientfashion. To maintain performance on different applications anddatasets, PAGE supports multiple scheduling schemes, parti-tioning methods, and execution models. We have evaluatedPAGE with four different programs, which are VarScan, IndelRealigner, Realigner Target Creator, and Unified Genotyper,and compared PAGE against GATK and Hadoop Streaming.
The main observations from our extensive evaluation areas follows. First, PAGE is able to parallelize different ap-plications, maintaining high parallel efficiency. Second, eachscheduling scheme can be useful for a certain type of envi-ronment and application - for example, dynamic schedulingis better when chunks are of unequal sizes, while staticscheduling can be better when the number of nodes is smaller.
888000

Finally, in comparison of PAGE with Hadoop Streaming andGATK, we show that it has better performance and scalability.
Acknowledgements
This work was supported by NSF grants IIS-0916196, CCF-1319420, and ACI-1339757.
REFERENCES
[1] Human genome at ten: the sequence explosion. Nature, 464:670–671,2010.
[2] http://genome.sph.umich.edu/wiki/bamutil.[3] http://hadoop.apache.org/docs/stable/streaming.html.[4] http://picard.sourceforge.net.[5] http://www.bioconductor.org/packages/2.13/bioc/html/seqarray.html.[6] http://www.genome.gov/sequencingcosts.[7] www.broadinstitute.org/gatk/gatkdocs/org broadinstitute sting gatk wal
kers genotyper unifiedgenotyper.html.[8] www.broadinstitute.org/gatk/gatkdocs/org broadinstitute sting gatk wal
kers indels indelrealigner.html.[9] www.broadinstitute.org/gatk/gatkdocs/org broadinstitute sting gatk wal
kers indels realignertargetcreator.html.[10] www.broadinstitute.org/gatk/gatkdocs/org broadinstitute sting gatk wal
kers qc countbases.html.[11] www.broadinstitute.org/gatk/gatkdocs/org broadinstitute sting gatk wal
kers readutils clipreads.html.[12] S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. Basic
local alignment search tool. Journal of molecular biology, 215(3):403–410, October 1990.
[13] Vikas Bansal. A statistical method for the detection of variants fromnext-generation resequencing of dna pools. Bioinformatics, 26(12):i318–i324, June 2010.
[14] R. D. Bjornson, A. H. Sherman, S. B. Weston, N. Willard, and J. Wing.Turboblast : a parallel implementation of blast built on the turbohub. InParallel and Distributed Processing Symposium., Proceedings Interna-tional, IPDPS 2002, Abstracts and CD-ROM, pages 8 pp–, 2002.
[15] Petr Danecek, Adam Auton, Goncalo Abecasis, Cornelis A. Albers, EricBanks, Mark A. DePristo, Robert E. Handsaker, Gerton Lunter, Gabor T.Marth, Stephen T. Sherry, Gilean McVean, Richard Durbin, and 1000Genomes Project Analysis Group. The variant call format and VCFtools.Bioinformatics, 27(15):2156–2158, August 2011.
[16] Jeffrey Dean and Sanjay Ghemawat. Mapreduce: simplified dataprocessing on large clusters. Commun. ACM, 51(1):107–113, January2008.
[17] Elizabeth W. Edmiston, Nolan G. Core, Joel H. Saltz, and Roger M.Smith. Parallel processing of biological sequence comparison algo-rithms. International Journal of Parallel Programming, 17(3):259–275,1988.
[18] RobertC Gentleman, VincentJ Carey, DouglasM Bates, Ben Bol-stad, Marcel Dettling, Sandrine Dudoit, Byron Ellis, Laurent Gautier,Yongchao Ge, Jeff Gentry, Kurt Hornik, Torsten Hothorn, WolfgangHuber, Stefano Iacus, Rafael Irizarry, Friedrich Leisch, Cheng Li, MartinMaechler, AnthonyJ Rossini, Gunther Sawitzki, Colin Smith, GordonSmyth, Luke Tierney, JeanYH Yang, and Jianhua Zhang. Bioconductor:open software development for computational biology and bioinformat-ics. Genome biology, 5(10):1–16, 2004.
[19] Daniel C. Koboldt, Ken Chen, Todd Wylie, David E. Larson, Michael D.McLellan, Elaine R. Mardis, George M. Weinstock, Richard K. Wilson,and Li Ding. Varscan: variant detection in massively parallel sequencingof individual and pooled samples. Bioinformatics, 25(17):2283–2285,September 2009.
[20] Ben Langmead, Michael C. Schatz, Jimmy Lin, Mihai Pop, andSteven L. Salzberg. Searching for snps with cloud computing. Genomebiology, 10(11):R134+, November 2009.
[21] Ben Langmead, Cole Trapnell, Mihai Pop, and Steven L. Salzberg.Ultrafast and memory-efficient alignment of short DNA sequences tothe human genome. Genome biology, 10(3):R25–10, March 2009.
[22] Simone Leo, Federico Santoni, and Gianluigi Zanetti. Biodoop: Bioin-formatics on hadoop. Parallel Processing Workshops, InternationalConference on, 0:415–422, 2009.
[23] Heng Li and Richard Durbin. Fast and accurate short read alignmentwith BurrowsWheeler transform. Bioinformatics, 25(14):1754–1760,July 2009.
[24] Heng Li, Bob Handsaker, Alec Wysoker, Tim Fennell, Jue Ruan, NilsHomer, Gabor Marth, Goncalo Abecasis, Richard Durbin, and 1000Genome Project Data Processing Subgroup. The sequence align-ment/map format and samtools. Bioinformatics (Oxford, England),25(16):2078–2079, August 2009.
[25] Heng Li, Jue Ruan, and Richard Durbin. Mapping short DNA sequenc-ing reads and calling variants using mapping quality scores. GenomeResearch, 18(11):1851–1858, November 2008.
[26] Ruiqiang Li, Yingrui Li, Xiaodong Fang, Huanming Yang, Jian Wang,Karsten Kristiansen, and Jun Wang. Snp detection for massively parallelwhole-genome resequencing. Genome Research, 19(6):1124–1132, June2009.
[27] Aaron McKenna, Matthew Hanna, Eric Banks, Andrey Sivachenko,Kristian Cibulskis, Andrew Kernytsky, Kiran Garimella, David Alt-shuler, Stacey Gabriel, Mark Daly, and Mark A. DePristo. The genomeanalysis toolkit: A mapreduce framework for analyzing next-generationdna sequencing data. Genome Research, 20(9):1297–1303, September2010.
[28] Matti Niemenmaa, Aleksi Kallio, Andre Schumacher, Petri Klemela,Eija Korpelainen, and Keijo Heljanko. Hadoop-BAM: Directly manip-ulating next generation sequencing data in the cloud. Bioinformatics,28(6):876–877, February 2012.
[29] Luca Pireddu, Simone Leo, and Gianluigi Zanetti. SEAL: a dis-tributed short read mapping and duplicate removal tool. Bioinformatics,27(15):2159–2160, August 2011.
[30] Aaron R. Quinlan and Ira M. Hall. BEDTools: a flexible suite of utilitiesfor comparing genomic features. Bioinformatics (Oxford, England),26(6):841–842, March 2010.
[31] Aysan Rasooli and Douglas G. Down. An adaptive scheduling algorithmfor dynamic heterogeneous hadoop systems. In Proceedings of the2011 Conference of the Center for Advanced Studies on CollaborativeResearch, CASCON ’11, pages 30–44, Riverton, NJ, USA, 2011. IBMCorp.
[32] Abhinav Sarje and Srinivas Aluru. Parallel genomic alignments on thecell broadband engine. IEEE Transactions on Parallel and DistributedSystems, 20(11), 2009.
[33] Ankit R. Shah, Sriram Chockalingam, and Srinivas Aluru. A parallelalgorithm for spectrum-based short read error correction. Parallel andDistributed Processing Symposium, International, pages 60–70, 2012.
[34] Aristotelis Tsirigos, Niina Haiminen, Erhan Bilal, and Filippo Utro.GenomicTools: a computational platform for developing high-throughputanalytics in genomics. Bioinformatics (Oxford, England), 28(2):282–283, January 2012.
[35] Zhi Wei, Wei Wang, Pingzhao Hu, Gholson J. Lyon, and HakonHakonarson. Snver: a statistical tool for variant calling in analysis ofpooled or individual next-generation sequencing data. Nucleic acidsresearch, 39(19):e132, October 2011.
[36] Tom White. Hadoop: The Definitive Guide. O’Reilly, first editionedition, June 2009.
[37] Adrianto Wirawan, Kwoh Keong, and Bertil Schmidt. Parallel DNAsequence alignment on the cell broadband engine. In Parallel Processingand Applied Mathematics, pages 1249–1256. 2008.
[38] Koh-Ichiro Yoshiura, Akira Kinoshita, Takafumi Ishida, Aya Ninokata,Toshihisa Ishikawa, Tadashi Kaname, Makoto Bannai, Katsushi Toku-naga, Shunro Sonoda, Ryoichi Komaki, Makoto Ihara, Vladimir A.Saenko, Gabit K. Alipov, Ichiro Sekine, Kazuki Komatsu, Haruo Taka-hashi, Mitsuko Nakashima, Nadiya Sosonkina, Christophe K. Mapen-dano, Mohsen Ghadami, Masayo Nomura, De-Sheng Liang, NobutomoMiwa, Dae-Kwang Kim, Ariuntuul Garidkhuu, Nagato Natsume, TohruOhta, Hiroaki Tomita, Akira Kaneko, Mihoko Kikuchi, Graciela Rus-somando, Kenji Hirayama, Minaka Ishibashi, Aya Takahashi, NaruyaSaitou, Jeffery C. Murray, Susumu Saito, Yusuke Nakamura, and NorioNiikawa. A SNP in the ABCC11 gene is the determinant of humanearwax type. Nature Genetics, 38(3):324–330, January 2006.
[39] Matei Zaharia, Dhruba Borthakur, Joydeep Sen Sarma, Khaled Elmele-egy, Scott Shenker, and Ion Stoica. Job scheduling for multi-usermapreduce clusters. Technical Report UCB/EECS-2009-55, EECSDepartment, University of California, Berkeley, Apr 2009.
[40] Matei Zaharia, Dhruba Borthakur, Joydeep Sen Sarma, Khaled Elmele-egy, Scott Shenker, and Ion Stoica. Delay scheduling: a simple techniquefor achieving locality and fairness in cluster scheduling. In Proceedingsof the 5th European conference on Computer systems, EuroSys ’10,pages 265–278, New York, NY, USA, 2010. ACM.
888!




















