
Origin of Lipid A Species Modified with 4-Amino-4-deoxy-L-arabinose in Polymyxin-resistant Mutants...
10
Origin of Lipid A Species Modified with 4-Amino-4-deoxy-L- arabinose in Polymyxin-resistant Mutants of Escherichia coli AN AMINOTRANSFERASE (ArnB) THAT GENERATES UDP-4-AMINO-4-DEOXY-L-ARABINOSE* Received for publication, April 17, 2003 Published, JBC Papers in Press, April 18, 2003, DOI 10.1074/jbc.M304043200 Steven D. Breazeale‡, Anthony A. Ribeiro‡§, and Christian R. H. Raetz‡¶ From the ‡Department of Biochemistry, Duke University Medical Center and the §Department of Radiology and the Duke NMR Spectroscopy Center, Duke University Medical Center, Durham, North Carolina 27710 In Escherichia coli and Salmonella typhimurium, ad- dition of the 4-amino-4-deoxy-L-arabinose (L-Ara4N) moi- ety to the phosphate group(s) of lipid A is required for resistance to polymyxin and cationic antimicrobial pep- tides. We have proposed previously (Breazeale, S. D., Ribeiro, A. A., and Raetz, C. R. H. (2002) J. Biol. Chem. 277, 2886 –2896) a pathway for L-Ara4N biosynthesis that begins with the ArnA-catalyzed C-4 oxidation and C-6 decarboxylation of UDP-glucuronic acid, followed by the C-4 transamination of the product to generate the novel sugar nucleotide UDP-L-Ara4N. We now show that ArnB (PmrH) encodes the relevant aminotransferase. ArnB was overexpressed using a T7lac promoter-driven construct and shown to catalyze the reversible transfer of the amino group from glutamate to the acceptor, uri- dine 5-(-L-threo-pentapyranosyl-4-ulose diphosphate), the intermediate that is synthesized by ArnA from UDP- glucuronic acid. A 1.7-mg sample of the putative UDP-L- Ara4N product generated in vitro was purified by ion exchange chromatography, and its structure was con- firmed by 1 H and 13 C NMR spectroscopy. ArnB, which is a cytoplasmic protein, was purified to homogeneity from an overproducing strain of E. coli and shown to contain a pyridoxal phosphate cofactor, as judged by ultraviolet/visible spectrophotometry. The pyridoxal phosphate was converted to the pyridoxamine form in the presence of excess glutamate. A simple quantitative radiochemical assay was developed for ArnB, which can be used to assay the enzyme either in the forward or the reverse direction. The enzyme is highly selective for glutamate as the amine donor, but the equilibrium con- stant in the direction of UDP-L-Ara4N formation is un- favorable (0.1). ArnB is a member of a very large family of aminotransferases, but closely related ArnB or- thologs are present only in those bacteria capable of synthesizing lipid A species modified with the L-Ara4N moiety. Lipopolysaccharide (LPS) 1 is an immunogenic glycolipid that is the major component of the outer leaflet of the outer mem- branes of Gram-negative bacteria (1–3). LPS consists of three domains, which are the O-specific antigen, the core oligosac- charide, and the lipid A moiety (1–3). The latter provides a hydrophobic membrane anchor and is also the active compo- nent (or endotoxin) of LPS that is responsible for many of the pathophysiological effects associated with Gram-negative sep- sis (1–3). Lipid A potently activates the innate immune system, inducing a host response that includes the production of cati- onic antimicrobial peptides, cytokines, clotting factors, and var- ious immunostimulatory molecules (4 – 6). In severe infections, high levels of cytokines and pro-coagulant activity lead to Gram-negative septic shock, a syndrome characterized by in- creased vascular permeability, hypotension, multiple organ failure, and death (7). The Kdo-lipid A portion of LPS, which is relatively conserved among diverse Gram-negative bacteria, is sufficient to support the growth of Escherichia coli or Salmonella typhimurium (2, 8). Certain covalent modifications to Kdo-lipid A are induced by environmental stimuli, such as low magnesium ion concentra- tions and/or low pH, as would be encountered by bacteria in phagolysosomes of macrophages (9 –11). As shown in Fig. 1 for S. typhimurium, these modifications include the incorporation of palmitate (12, 13), the formation of S-2-hydroxymyristate (14), the addition of phosphoethanolamine (pEtN) (15), and/or the addition 4-amino-4-deoxy-L-arabinose (L-Ara4N) moieties (Fig. 1) (15–17). Addition of the L-Ara4N and pEtN moieties to the phosphate groups of lipid A confers resistance to polymyxin and various cationic antimicrobial peptides (15–17). Resistance is thought to occur by reduction of the net negative charge of lipid A, resulting in a reduced affinity for cationic peptides or polymyxin, thereby preventing these substances from penetrat- ing the outer membrane. The L-Ara4N modification of lipid A is governed by the PmrA/ PmrB two-component regulatory system (10, 18 –21), which is switched on by low pH. The transcription factor PmrA activates genes at the pmrE(ugd) and pmrHFIJKLM loci (10, 22). Con- stitutive mutants of pmrA (pmrA c ) have been isolated in E. coli and S. typhimurium; these mutants modify their lipid A with L-Ara4N and pEtN groups under all growth conditions, and they are polymyxin-resistant (17, 20, 23). However, inactiva- tion of pmrE or of any of the genes in the pmrHFIJKL operon in a pmrA c S. typhimurium mutant results in loss of polymyxin resistance and loss of the L-Ara4N modification of lipid A (10). Sequence analyses of the various genes in the pmr loci have led us to propose a pathway for the biosynthesis of the L-Ara4N moiety and its attachment to lipid A (22). Our hypothesis (Fig. * This work was supported in part by National Institutes of Health Grant GM-51310 (to C. R. H. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. ¶ To whom correspondence should be addressed. Tel.: 919-684-5326; Fax: 919-684-8885; E-mail: [email protected]. 1 The abbreviations used are: LPS, lipopolysaccharide; L-Ara4N, 4-amino-4-deoxy-L-arabinose; UDP-L-Ara4N, uridine-5-diphospho-- (4-amino-4-deoxy-L-arabinose); UDP-Ara4O, uridine 5-(-L-threo-pen- tapyranosyl-4-ulose diphosphate); UDP-GlcUA, UDP-glucuronic acid; pEtN, phosphoethanolamine; IPTG, isopropyl-1-thio--D-galactopy- ranoside; BisTris, bis(2-hydroxyethyl)iminotris(hydroxymethyl)- methane; Kdo, 3-deoxy-D-manno-2-octulosonic acid; NOE, nuclear Overhauser effect. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 27, Issue of July 4, pp. 24731–24739, 2003 © 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. This paper is available on line at http://www.jbc.org 24731 by guest on March 4, 2016 http://www.jbc.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Origin of Lipid A Species Modified with 4-Amino-4-deoxy-L-arabinose in Polymyxin-resistant Mutants...

Origin of Lipid A Species Modified with 4-Amino-4-deoxy-L-arabinose in Polymyxin-resistant Mutants of Escherichia coliAN AMINOTRANSFERASE (ArnB) THAT GENERATES UDP-4-AMINO-4-DEOXY-L-ARABINOSE*
Received for publication, April 17, 2003Published, JBC Papers in Press, April 18, 2003, DOI 10.1074/jbc.M304043200
Steven D. Breazeale‡, Anthony A. Ribeiro‡§, and Christian R. H. Raetz‡¶
From the ‡Department of Biochemistry, Duke University Medical Center and the §Department of Radiology and the DukeNMR Spectroscopy Center, Duke University Medical Center, Durham, North Carolina 27710
In Escherichia coli and Salmonella typhimurium, ad-dition of the 4-amino-4-deoxy-L-arabinose (L-Ara4N) moi-ety to the phosphate group(s) of lipid A is required forresistance to polymyxin and cationic antimicrobial pep-tides. We have proposed previously (Breazeale, S. D.,Ribeiro, A. A., and Raetz, C. R. H. (2002) J. Biol. Chem.277, 2886–2896) a pathway for L-Ara4N biosynthesis thatbegins with the ArnA-catalyzed C-4� oxidation and C-6�decarboxylation of UDP-glucuronic acid, followed bythe C-4� transamination of the product to generate thenovel sugar nucleotide UDP-L-Ara4N. We now show thatArnB (PmrH) encodes the relevant aminotransferase.ArnB was overexpressed using a T7lac promoter-drivenconstruct and shown to catalyze the reversible transferof the amino group from glutamate to the acceptor, uri-dine 5�-(�-L-threo-pentapyranosyl-4�-ulose diphosphate),the intermediate that is synthesized by ArnA from UDP-glucuronic acid. A 1.7-mg sample of the putative UDP-L-Ara4N product generated in vitro was purified by ionexchange chromatography, and its structure was con-firmed by 1H and 13C NMR spectroscopy. ArnB, which isa cytoplasmic protein, was purified to homogeneityfrom an overproducing strain of E. coli and shown tocontain a pyridoxal phosphate cofactor, as judged byultraviolet/visible spectrophotometry. The pyridoxalphosphate was converted to the pyridoxamine form inthe presence of excess glutamate. A simple quantitativeradiochemical assay was developed for ArnB, which canbe used to assay the enzyme either in the forward or thereverse direction. The enzyme is highly selective forglutamate as the amine donor, but the equilibrium con-stant in the direction of UDP-L-Ara4N formation is un-favorable (�0.1). ArnB is a member of a very large familyof aminotransferases, but closely related ArnB or-thologs are present only in those bacteria capable ofsynthesizing lipid A species modified with the L-Ara4Nmoiety.
Lipopolysaccharide (LPS)1 is an immunogenic glycolipid thatis the major component of the outer leaflet of the outer mem-
branes of Gram-negative bacteria (1–3). LPS consists of threedomains, which are the O-specific antigen, the core oligosac-charide, and the lipid A moiety (1–3). The latter provides ahydrophobic membrane anchor and is also the active compo-nent (or endotoxin) of LPS that is responsible for many of thepathophysiological effects associated with Gram-negative sep-sis (1–3). Lipid A potently activates the innate immune system,inducing a host response that includes the production of cati-onic antimicrobial peptides, cytokines, clotting factors, and var-ious immunostimulatory molecules (4–6). In severe infections,high levels of cytokines and pro-coagulant activity lead toGram-negative septic shock, a syndrome characterized by in-creased vascular permeability, hypotension, multiple organfailure, and death (7).
The Kdo-lipid A portion of LPS, which is relatively conservedamong diverse Gram-negative bacteria, is sufficient to supportthe growth of Escherichia coli or Salmonella typhimurium (2,8). Certain covalent modifications to Kdo-lipid A are induced byenvironmental stimuli, such as low magnesium ion concentra-tions and/or low pH, as would be encountered by bacteria inphagolysosomes of macrophages (9–11). As shown in Fig. 1 forS. typhimurium, these modifications include the incorporationof palmitate (12, 13), the formation of S-2-hydroxymyristate(14), the addition of phosphoethanolamine (pEtN) (15), and/orthe addition 4-amino-4-deoxy-L-arabinose (L-Ara4N) moieties(Fig. 1) (15–17). Addition of the L-Ara4N and pEtN moieties tothe phosphate groups of lipid A confers resistance to polymyxinand various cationic antimicrobial peptides (15–17). Resistanceis thought to occur by reduction of the net negative charge oflipid A, resulting in a reduced affinity for cationic peptides orpolymyxin, thereby preventing these substances from penetrat-ing the outer membrane.
The L-Ara4N modification of lipid A is governed by the PmrA/PmrB two-component regulatory system (10, 18–21), which isswitched on by low pH. The transcription factor PmrA activatesgenes at the pmrE(ugd) and pmrHFIJKLM loci (10, 22). Con-stitutive mutants of pmrA (pmrAc) have been isolated in E. coliand S. typhimurium; these mutants modify their lipid A withL-Ara4N and pEtN groups under all growth conditions, andthey are polymyxin-resistant (17, 20, 23). However, inactiva-tion of pmrE or of any of the genes in the pmrHFIJKL operonin a pmrAc S. typhimurium mutant results in loss of polymyxinresistance and loss of the L-Ara4N modification of lipid A (10).
Sequence analyses of the various genes in the pmr loci haveled us to propose a pathway for the biosynthesis of the L-Ara4Nmoiety and its attachment to lipid A (22). Our hypothesis (Fig.
* This work was supported in part by National Institutes of HealthGrant GM-51310 (to C. R. H. R.). The costs of publication of this articlewere defrayed in part by the payment of page charges. This article musttherefore be hereby marked “advertisement” in accordance with 18U.S.C. Section 1734 solely to indicate this fact.
¶ To whom correspondence should be addressed. Tel.: 919-684-5326;Fax: 919-684-8885; E-mail: [email protected].
1 The abbreviations used are: LPS, lipopolysaccharide; L-Ara4N,4-amino-4-deoxy-L-arabinose; UDP-L-Ara4N, uridine-5�-diphospho-�-(4-amino-4-deoxy-L-arabinose); UDP-Ara4O, uridine 5�-(�-L-threo-pen-tapyranosyl-4�-ulose diphosphate); UDP-GlcUA, UDP-glucuronic acid;pEtN, phosphoethanolamine; IPTG, isopropyl-1-thio-�-D-galactopy-
ranoside; BisTris, bis(2-hydroxyethyl)iminotris(hydroxymethyl)-methane; Kdo, 3-deoxy-D-manno-2-octulosonic acid; NOE, nuclearOverhauser effect.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 27, Issue of July 4, pp. 24731–24739, 2003© 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 24731
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

2) is supported by the following observations. 1) ArnT (PmrK)is an inner membrane enzyme that utilizes the novel glycolipidundecaprenyl phosphate-�-L-Ara4N as the donor of theL-Ara4N moiety to modify lipid A (16, 17). 2) ArnA (PmrI) is adehydrogenase/decarboxylase that converts UDP-glucuronicacid to a novel 4�-keto-pentose, designated UDP-Ara4O, whichcan be isolated in milligram quantities as the hydrated ketone(22). 3) Cell extracts of polymyxin-resistant (but not polymyxin-sensitive) E. coli cells convert UDP-Ara4O to the putativesugar nucleotide UDP-L-Ara4N in the presence of L-glutamate(22). 4) Cell extracts of polymyxin-resistant E. coli cells canfurther convert the proposed UDP-L-Ara4N to a putativeformyl-amino derivative in the presence of N-10-formyltetra-hydrofolate (22).
We have now cloned, overexpressed, and purified to homo-geneity the aminotransferase ArnB (PmrH). We demonstratethat ArnB (GenBank™ accession number AAM92146) containsa pyridoxal phosphate cofactor and catalyzes the reversibletransfer of the �-amine moiety from L-glutamate to the C-4�position of UDP-Ara4O, generating UDP-L-Ara4N. The struc-ture of UDP-L-Ara4N was validated by one- and two-dimen-sional NMR spectroscopy. The transamination is highly spe-cific for glutamate as the amine donor. Our biochemical dataestablish ArnB as the key aminotransferase required for thebiosynthesis of the L-Ara4N moiety present on lipid A mole-cules of polymyxin-resistant Gram-negative bacteria. Obvious
ArnB orthologs are present in Yersinia pestis, Pseudomonasaeruginosa, and Burkholderia cepacia, all of which are capableof modifying their lipid A with L-Ara4N (24). The purification ofArnB permits the enzymatic synthesis of milligram quantitiesof UDP-L-Ara4N, which can now be used to probe for theremaining steps in the transfer of L-Ara4N to lipid A. A crystalstructure of ArnB (25), recently obtained as part of a structuralgenomics program, is in accord with our enzymatic and bio-chemical studies.
EXPERIMENTAL PROCEDURES
Buffers, Reagents, and Materials—UDP-glucuronic acid, NAD�, 1.0M triethylammonium bicarbonate, pH 8.5, L-glutamate, polymyxin Bsulfate, kanamycin, and HEPES were purchased from Sigma. TheUDP-[U-14C]glucuronic acid was purchased from PerkinElmer Life Sci-ences. Restriction enzymes (NdeI, NcoI, and XhoI) were obtained fromNew England Biolabs. T4 DNA ligase, various oligonucleotides, andisopropyl-1-thio-�-D-galactopyranoside were purchased from Invitro-gen. The Pfu polymerase was from Stratagene. Polyethyleneimine-cellulose TLC plates were purchased from Merck. DEAE-SepharoseTM
CL-6B was obtained from Amersham Biosciences AB. Protein concen-trations were determined by the bicinchoninic acid method (26) usingthe BCA protein assay reagent (Pierce) with bovine serum albumin asthe standard.
Strains and Recombinant DNA Techniques—Construction ofWD101, a polymyxin-resistant mutant of E. coli W3110, was describedpreviously (17). Preparation and transformation of competent cells bythe CaCl2 method, genomic DNA purification, and gel electrophoresis ofrestriction fragments were performed according to published proce-dures (27, 28). Plasmids were purified using the QIAprep spin miniprepkit (Qiagen). Restriction enzymes, Pfu DNA polymerase, and T4 DNAligase were used as recommended by their manufacturers. DNA se-quencing was performed on an ABIprism 377 instrument at the DukeUniversity DNA Analysis Facility.
Construction of pETArnB, pETArnB-C6H, and pETArnB-N6H—Thepredicted coding region for ArnB (GenBank™ accession numberAAM92146) was amplified by PCR from E. coli W3110 genomic DNAwith primers to the arnB 5� end (5�-CGGGATCCATGGCGGAAGGAA-AAGCAATG-3�) and to the arnB 3� end (5�-CGGGATCCTCGAGTTAT-TGTCCTGCGAGTTGCTG-3�), containing engineered NcoI and XhoIrestriction sites, respectively. The Opti-PrimeTM PCR Optimization kit(Stratagene) was used as described by the manufacturer. The amplifi-cation reaction (50 �l) contained the following components: 5 �l of 10�Opti-Prime Buffer 9 (100 mM Tris-HCl, pH 9.2, 15 mM MgCl2, 250 mM
KCl), 0.2 mM of each dNTP, 250 ng of genomic DNA, 0.5 �M of eachprimer, and 2.5 units of Pfu DNA polymerase. After 30 cycles (94 °C for1 min, 50 °C for 1 min, and 72 °C for 1.5 min) followed by 1 cycle (94 °Cfor 1 min, 50 °C for 1 min, and 72 °C for 10 min), a single product of thepredicted size was obtained. This was isolated with the QIAquick PCRPurification kit (Qiagen), digested with XhoI and NcoI, and then ligatedinto the T7lac expression vector, pET28b (Novagen), previously di-gested with the same enzymes. The presence of the arnB gene (29) andappropriate flanking DNA was confirmed by sequencing of final con-struct, which was designated pETArnB.
Amplification of DNA for cloning a modified arnB gene expressing aC-terminal hexa-histidine fusion protein of ArnB (designated ArnB-C6H) utilized the arnB 5� primer described above for the native proteinand the oligonucleotide 5�-CGGGATCCTCGAGTTGTCCTGCGAGTT-GCTGAAG-3� as the 3� primer. The latter contained an engineered XhoIrestriction site. The PCR (50 �l) contained the following: 5 �l of 10�Opti-Prime Buffer 11 (100 mM Tris-HCl, pH 9.2, 35 mM MgCl2, 250 mM
KCl), 0.2 mM of each dNTP, 250 ng of genomic DNA, 0.5 �M of eachprimer, and 2.5 units of Pfu DNA polymerase. The same temperatureparameters described above were used for the PCR. The resultingproduct was cloned into the expression vector pET28b, as describedabove, to generate pETArnB-C6H. The final construct was confirmed byDNA sequencing.
Amplification of DNA for cloning a modified arnB gene encoding anN-terminal hexa-histidine fusion protein of ArnB (designated ArnB-N6H) was accomplished using the same reaction conditions as forpETArnB-C6H. The arnB 3� primer described above for the nativeprotein was used together with the oligonucleotide 5�-CGGATCCATAT-GGCGGAAGGAAAAGCAATG-3� as the 5� primer, which also con-tained an engineered NdeI restriction site. The PCR product was iso-lated with the QIAquick PCR Purification kit (Qiagen), digested withXhoI and NdeI, and ligated into the pET28b expression vector, previ-
FIG. 1. Regulated covalent modifications of lipid A. The Kdo2-lipid A substructure of lipopolysaccharide is sufficient to support thegrowth of S. typhimurium and E. coli (2). However, as indicated by thedashed bonds, covalent modifications of the phosphate groups and/oracyl chains of lipid A may be induced under certain growth conditions(2). For instance, the phosphates may be substituted with L-Ara4Nand/or pEtN groups (blue); both of these modifications are controlled bythe PmrA transcription factor, which is activated by growth at low pHand is constitutively activated in polymyxin-resistant mutants (2). Ad-ditional minor lipid A derivatives are present in which the locations ofthe L-Ara4N and pEtN groups are reversed (not shown) or in which bothphosphates are modified with the same group (15, 36). Incorporation ofa secondary palmitoyl chain at position 2 in the proximal unit is cata-lyzed by the outer membrane enzyme PagP (13, 48), whereas removal ofthe hydroxymyristoyl chain at position 3 is carried out by PagL (49).Formation of S-2-hydroxymyristate is O2-dependent and requires ahydroxylase encoded by lpxO (14). PagL and LpxO are normally presentin S. typhimurium (50) but not E. coli K-12 (29). With the possibleexception of LpxO, enzymes that modify the acyl chains of lipid A (red)are under the control of the PhoP/PhoQ two-component system, whichis activated by growth in the presence of low Mg2� concentrations(9, 11, 42).
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis24732
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

ously digested with the same enzymes, to generate pETArnB-N6H. Thefinal construct was confirmed by DNA sequencing.
Overexpression of the arnB Gene Product and Purification of ArnB-N6H and ArnB-C6H—Plasmids pETArnB, pETArnB-N6H, orpETArnB-C6H were transformed into E. coli NovaBlue(DE3) cells (No-vagen). Cultures were grown overnight at 37 °C from single colonies inLB broth (30) supplemented with 30 �g/ml kanamycin. The overnightculture was then used to inoculate a fresh 100-ml culture of the samebroth at A600 �0.01. Cells were grown at 37 °C to A600 of �0.6. Expres-sion of ArnB was induced by addition of isopropyl-�-D-thiogalactopyr-anoside to a final concentration of 1 mM, and growth was continued at30 °C for 3.5 h. Cells were harvested by centrifugation at 6500 � g for15 min at 4 °C. The cell pellet was resuspended in 3 ml of buffer A,consisting of 25 mM HEPES, pH 7.5, 10% glycerol, and 300 mM NaCl.Cells were broken by passage through a French pressure cell at 18,000pounds/square inch. Cell debris was removed by centrifugation at16,500 � g for 15 min at 4 °C to give crude cell-free extracts.
For the hexa-histidine fusion proteins, the crude cell-free extractswere loaded onto a 1-ml nickel-nitrilotriacetic acid column (Qiagen),which was pre-equilibrated in buffer A. The column was washed with 15column volumes of buffer A and 15 column volumes of buffer A contain-ing 50 mM imidazole; it was then eluted with 15 column volumes ofbuffer A containing 100 mM imidazole. Proteins were concentrated, andthe imidazole was removed by exchanging for buffer B (consisting of 50mM HEPES, pH 7.5, and 10% glycerol) with the use of a Centricon�(YM-10) membrane, which was subjected to several cycles of low speedcentrifugation and dilution in buffer B. Protein concentrations weredetermined using the bicinchoninic acid procedure (26).
The overexpression of ArnB, ArnB-N6H, and ArnB-C6H, as well asthe initial purification of the hexa-histidine fusion proteins, was mon-itored by SDS-PAGE (4% stacking gel and 8% resolving gel), followed byCoomassie Blue staining (31).
Enzymatic Synthesis and Isolation of UDP-L-Ara4N—A 10-ml reac-tion mixture, containing 0.05 mg/ml pure ArnB-N6H, 1 mM UDP-Ara4O, and 10 mM L-glutamate in 50 mM HEPES, pH 7.5, was incu-bated at 30 °C for 5.5 h. Protein was removed by passing the reactionmixture through a Centricon� (YM-10) membrane. The enzymatic prod-uct was then loaded onto a 10-ml column of DEAE-SepharoseTM CL-6B,pre-equilibrated with 10 mM BisTris, pH 5.9. The column was washedwith 50 ml of deionized water, followed by elution of the product with a0–100 mM BisTris, pH 5.9, gradient (200-ml total volume). Chromatog-raphy was monitored by absorbance of the effluent at 254 nm. Fractions(12 ml) containing the desired product, which eluted at about 30 mM
BisTris, were pooled and diluted 10-fold with H2O. The diluted materialwas then loaded onto a 10-ml column of DEAE-SepharoseTM CL-6B,pre-equilibrated with 10 mM triethylammonium bicarbonate, pH 8.5.The column was washed with 50 ml of deionized water, followed byelution of the product with a 0–100 mM gradient of triethylammoniumbicarbonate, pH 8.5 (200-ml total volume). Chromatography was againmonitored by absorbance at 254 nm. Fractions containing the product(which eluted at about 60 mM triethylammonium bicarbonate) werepooled and concentrated by lyophilization to give the triethylammo-nium salt of the sugar nucleotide. Conversion to the sodium salt wasaccomplished by passage over a 1-ml column of AG�50W-X8 resin(sodium form), followed by lyophilization to give the pure compound (1.7mg, 31% yield).
Enzymatic Synthesis and Purification of UDP-L-[U-14C]Ara4N—Con-version of UDP-[U-14C]glucuronic acid (380 mCi/mmol) to UDP-L-[U-14C]Ara4N was performed in 400-�l reaction mixtures containing 50mM HEPES, pH 7.5, 2 mM dithiothreitol, 3 mM NAD�, 1 mM L-gluta-mate, 0.05 mg/ml ArnA overproducing cell-free extract (22), 0.05 mg/mlpure ArnB-C6H, and 13 �M UDP-[U-14C]glucuronic acid (2 �Ci), previ-ously dried under nitrogen and re-dissolved in a small volume of deion-ized water. The reaction proceeded for 220 min at 30 °C and was thendiluted to 1.5 ml with deionized water before loading onto a 2-ml columnof DEAE-SepharoseTM CL-6B, pre-equilibrated with 10 mM BisTris, pH6. The column was washed with 5 ml of deionized water, followed byelution of the nucleotides with a 0–100 mM gradient of BisTris, pH 6(40-ml total volume). Fractions containing UDP-L-[U-14C]Ara4N werepooled, diluted 10-fold with deionized water, and loaded onto a 2-mlcolumn of DEAE-SepharoseTM CL-6B, pre-equilibrated with 10 mM
triethylammonium bicarbonate, pH 8.5. The column was washed with 5ml of deionized water. The NAD� was resolved from the desired sugarnucleotide with a 0–100 mM gradient of triethylammonium bicarbon-ate, pH 8.5 (40-ml total volume). The radioactive fractions were concen-trated using a Speed-Vac centrifuge and dissolved in 250 �l of deionizedwater. The yield of UDP-L-[U-14C]Ara4N was 74% based on the inputradioactivity. The product was stored frozen at �20 °C.
Nuclear Magnetic Resonance Spectroscopy—The sodium salt of theputative UDP-L-Ara4N (1.7 mg prepared as described above) was dis-solved in 0.6 ml of 99% D2O and placed into a 5 mm NMR tube.Measurements with a 3-mm pH electrode revealed a pD value of 8.00.Proton and 13C NMR chemical shifts were referenced relative to 2,2-dimethylsilapentane-5-sulfonic acid at 0.00 ppm.
1H NMR spectra were obtained on a Varian Inova 800 spectrometerequipped with a Sun Ultra 10 computer and a 5-mm Varian tripleresonance probe. 1H spectra were obtained with a spectral width of 8.0kHz, a 76o pulse flip angle (7 �s), a 4.0-s acquisition time, and a 1.5-srelaxation delay, and were digitized using 96,000 points to obtain adigital resolution of 0.16 Hz/point. Two-dimensional NMR experimentswere performed as described previously (17, 22, 32).
ArnB Enzymatic Assays—The rate of formation of UDP-L-[U-14C]Ara4N by ArnB was quantified using a coupled assay in whichoverexpressed ArnA was first used to generate UDP-[U-14C]Ara4O insitu from UDP-[U-14C]glucuronic acid. The UDP-[U-14C]Ara4O is rela-tively unstable at the low chemical concentrations needed for radioac-tive assays. Briefly, the initial ArnA reaction mixture (10 �l) contained90 mM HEPES, pH 7.5, 3 mM NAD�, and 300 �M UDP-[U-14C]glucu-ronic acid (0.02 �Ci), and 0.1 mg/ml ArnA overproducing cell-free ex-tract from E. coli NovaBlue(DE3)/pETArnA (22). Complete conversionof UDP-[U-14C]glucuronic acid to UDP-[U-14C]Ara4O was achieved af-ter 120 min at 30 °C. To assay for UDP-L-[U-14C]Ara4N synthesis, theamine donor (usually glutamate) and ArnB were added to bring thefinal volume to 20 �l; this was set up to yield final concentrations of 50mM HEPES, pH 7.5, 150 �M UDP-[U-14C]Ara4O (unless otherwiseindicated), and 7.5 mM amine donor. Reactions were incubated furtherat 30 °C. At the desired time points, a 1-�l portion was withdrawn andspotted onto a polyethyleneimine-cellulose TLC plate. The spots wereallowed to dry, and the plate was washed in methanol for 5 min. Afterdrying, the plate was developed using 0.4 M LiCl in 0.25 M aqueousacetic acid. The plate was dried and exposed to a PhosphorImagerscreen. Remaining substrate and radioactive product were quantifiedwith a PhosphorImager (ImageQuant software, Amersham Bio-sciences). ArnB specific enzymatic activity was calculated in unitsof nmol/min/mg.
Ultraviolet-visible Spectroscopy of ArnB—Purified ArnB-C6H (25�M) in 50 mM HEPES, pH 7.5, containing 10% glycerol was incubated at30 °C alone or in the presence of 1 mM L-glutamate in a total volume of100 �l. Spectra were recorded on a BeckmanCoulter DU� 640B UV-visible scanning spectrophotometer. Experimental data were exportedinto Kaleidagraph (Synergy Software, Reading, PA) and plotted. Thereactions were blanked against 50 mM HEPES, pH 7.5, containing thebuffer alone (10% glycerol in 50 mM HEPES, pH 7.5).
RESULTS
Cloning, Overexpression, and Purification of ArnB as a Hexa-histidine-tagged Fusion Protein—The L-Ara4N moiety found onthe phosphate groups of lipid A in polymyxin-resistant Gram-negative bacteria (like pmrA constitutive strains of E. coli or S.typhimurium) is proposed to arise via the novel sugar nucleo-tide, UDP-L-Ara4N (22). The transamination of the precursorUDP-Ara4O (Fig. 2), a nucleotide synthesized in polymyxin-resistant (but not polymyxin-sensitive) E. coli (22), could gen-erate UDP-L-Ara4N (Fig. 2). We have shown previously thatthe E. coli gene arnA encodes a protein that catalyzes theoxidative decarboxylation of UDP-GlcUA to give UDP-Ara4O(22). Furthermore, in the presence of L-glutamate, cell extractsof polymyxin-resistant E. coli can convert UDP-Ara4O to aspecies with the RF expected for such an amino-sugar nucleo-tide at the rate of �2 nmol/min/mg (22). These preliminarydataareconsistentwiththeinvolvementofapyridoxalphosphate-dependent enzyme that utilizes L-glutamate as the amine do-nor for converting UDP-Ara4O to UDP-L-Ara4N.
Upstream of arnA resides arnB (pmrH), another gene re-quired for polymyxin resistance in S. typhimurium (10), whichis predicted to encode an aminotransferase (22, 33). The geneticlocation of arnB and the predicted sequence of its encodedprotein made it a likely candidate for the proposed transami-nase involved in the UDP-L-Ara4N formation (Fig. 2). To vali-date the function of ArnB, the full-length arnB gene was clonedinto the expression vector pET28b behind an inducible T7lac
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis 24733
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

promoter. Expression was carried out in E. coli NovaB-lue(DE3), a polymyxin-sensitive strain that does not modify itsendogenous lipid A with the L-Ara4N moiety (16). A protein ofthe predicted molecular mass of ArnB (�43 kDa) was observedby SDS-gel electrophoresis in cell-free extracts of induced cellsbut was absent in uninduced controls (Fig. 3, lanes 2 and 3,respectively). Extracts of induced cells containing pETArnBcatalyzed the rapid L-glutamate-dependent conversion of UDP-Ara4O to a species with the RF expected for UDP-L-Ara4N(data not shown).
The relatively low level of ArnB expression, as judged by gelelectrophoresis (Fig. 3), led us to engineer hexa-histidine se-quences into the protein to facilitate purification. To this end,arnB was cloned into the pET28b expression vector to expresseither an N-terminal hexa-histidine fusion protein (ArnB-N6H)containing an additional 20 amino acids, or a C-terminal hexa-histidine fusion protein (ArnB-C6H) containing an additional 7amino acids. Similar to the wild-type ArnB, the hexa-histidinefusion proteins were expressed in induced E. coli NovaB-lue(DE3) but were absent in uninduced controls (Fig. 3, lanes 5and 4 and lanes 8 and 9, respectively). The ArnB-specific ac-tivity of a cell extract of induced E. coli NovaBlue(DE3)/pETArnB-C6H is 130 nmol/min/mg. Approximately the same
specific activity is seen with cell extracts overexpressing ArnB-N6H or wild-type ArnB. By using nickel chelation chromatog-raphy, the hexa-histidine ArnB fusion proteins were purified tohomogeneity, as judged by SDS gel electrophoresis (Fig. 3,lanes 6 and 7). The final specific activity of the pure proteins is1.3 � 103 nmol/min/mg.
Analysis of the Covalent Structure of the ArnB Product byOne-dimensional 1H NMR Spectroscopy—By using pure ArnB-N6H, 10 �mol of UDP-Ara4O was converted to the putativeUDP-L-Ara4N in the presence of 100 �mol of L-glutamate, asdescribed under “Experimental Procedures.” The substrate andproduct were separated using anion exchange chromatographyat pH 5.9 (0–100 mM BisTris). The fractions containing theArnB product were then separated from the BisTris, usinganion exchange chromatography at pH 8.5 in the volatilebuffer, triethylammonium bicarbonate (0–100 mM). Fractionscontaining the desired product were lyophilized to give thetriethylammonium salt of the sugar nucleotide, which wasconverted to the sodium salt by passage through a DowexAG®50W-X8 column (sodium form).
The sodium salt (1.7 mg) of the product was dissolved in D2O,and its 1H NMR spectrum was recorded, Fig. 4 (panels A andB). The uracil and ribose moieties of the substrate, UDP-
FIG. 2. Biosynthesis of UDP-L-Ara4N and transfer of the L-Ara4N unit to lipid A. In polymyxin-resistant mutants of E. coli andS. typhimurium, UDP-glucose is first converted to UDP-GlcUA by the dehydrogenase Ugd (PmrE) (2, 10, 33). ArnA then catalyzes the oxidativedecarboxylation of UDP-GlcUA to form the novel sugar nucleotide, UDP-Ara4O (22). As shown in the present study (boxed reaction), ArnBtransaminates the resulting 4�-keto moiety in a reversible reaction to generate UDP-L-Ara4N. The ArnA protein contains a second catalytic domainwith strong homology to methionyl-tRNA formyltransferase (22), which transfers the formyl group from N-10-formyltetrahydrofolate to the 4�amine of UDP-L-Ara4N. Based upon its homology to the yeast dolichoyl phosphate-mannose synthase (51) and recent enzymatic studies in ourlaboratory,3 we suggest that ArnC (formerly PmrF) (10) transfers the L-Ara4-formyl-N unit (indicated by the black rectangle) to undecaprenylphosphate. Following transport of the periplasmic surface of the inner membrane, we speculate that deformylation occurs prior to L-Ara4N additionto lipid A. It is well established by in vitro studies that the polytopic membrane protein ArnT transfers the L-Ara4N moiety (indicated by the bluerectangle) of undecaprenyl phosphate-�-L-Ara4N to lipid A (16, 17). Undecaprenyl phosphate-�-L-Ara4N can be isolated from polymyxin-resistantmutants (17). The proteins involved in the proposed transport and deformylation of undecaprenyl phosphate-�-L-Ara4-formyl-N of have not yetbeen identified.
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis24734
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Ara4O, and of the ArnB product possess virtually identicalchemical shifts and coupling constants (Table I). However, thewell resolved signals arising from the pyranose moiety of theUDP-Ara4O and the putative UDP-L-Ara4N show significantdifferences (Fig. 4, panels B versus C, and Table I). The one-dimensional 1H NMR analysis therefore suggests that ArnBalters only the pyranose moiety of UDP-Ara4O.
Characterization of the ArnB Product by Two-dimensional1H NMR Spectroscopy—The two-dimensional 1H-1H COSYNMR spectrum of the putative UDP-L-Ara4N product (Fig. 5)was used to assign the protons in the ribose, uracil, and pyr-anose rings. As suggested by the one-dimensional spectra, theuridine nucleoside moiety was not altered by ArnB (Table I).Close similarities between the putative UDP-L-Ara4N and theUDP-Ara4O spectra include the strong cross-peak connectivitybetween the uracil H-6 and H-5 doublet resonances (not
shown), the H-1� connectivity to H-2� (4.39 ppm), the H-3�connectivity to H-4� (�4.30 ppm), and the connectivities of H-4�to H-5b� and H-5a� (Table I).
The assignments for the protons of the pyranose moiety ofthe ArnB product (Fig. 4) were unambiguously obtained fromthe COSY analysis (Fig. 5). The anomeric H-1� proton signal, adouble-doublet at 5.63 ppm (JH1�,2� � 3.4, JH1�,P � 7.3 Hz),shows a cross-peak to the H-2� double-triplet at 3.77 ppmJ1�,2� � 3.4, J2�,3� � 9.9 Hz). A second cross-peak from H-2�correlates to H-3� at 4.15 ppm (dd, J2�,3� � 9.9, J3�,4� � 4.6 Hz).H-3� then connects to H-4� at 3.59 ppm (unresolved multiplet).Further tracing of the two remaining cross-peaks from H-4�locate the H-5b� (4.29 ppm; dd, J4�,5� � 2.1, J5b�,5a� � 13.3 Hz)and H-5a� (�3.80 ppm; dd, J4�,5� � 2.0, J5a�,5b� � 13.3 Hz)proton signals.2
The small J1�,2� coupling (3.4 Hz) and the large J2�,3� coupling(9.9 Hz) indicate that the pyranose ring is in a configurationwith an equatorially disposed H-1�, and with axial H-2� andH-3� protons, which would be classified as the � anomer, giventhe L stereochemistry of the sugar (34). The two-dimensionalNOE spectroscopy data (not shown) revealed a strong intra-molecular cross-peak between H-1� and H-2�, in accord with anequatorial H-1� and axial H-2� on the same face of the sugar(Fig. 2) (35). The large trans-diaxial J2�,3� coupling then sug-gests that H-3� is likewise axial. The intermediate value of theJ3�,4� coupling (4.6 Hz) indicates an axial-equatorial coupling,showing that H-4� is in the equatorial position (Fig. 2). A strongintra-molecular cross-peak is observed (not shown) betweenH-3� and H-4� further confirming that they are on the same face(Fig. 2).
The measured values of 2.1 and 2.0 Hz for the two J4�,5�
couplings (equatorial-equatorial and equatorial-axial) do notallow distinction of the axial or equatorial H-5� protons. At-tempts to distinguish these protons for this pyranose ring di-rectly through two-dimensional NOE experiments were unsuc-cessful, as no clear NOE cross-peak was observed between theaxially disposed H-3� (Fig. 2) and either H-5� proton, at both600- and 800-MHz fields. The equatorial H-4� showed a stron-ger NOE cross-peak to H-5b� (�4.29-ppm signal) and a weakerNOE cross-peak to H-5a� (3.80-ppm signal), but those resultsdo not allow firm assignments. However, the H-5a� (3.80 ppm)and H-5b� (4.29 ppm) signals are assigned to the axial andequatorial positions following previous literature values withsynthetic standards (34, 36), based upon the chemical shiftconsideration that a proton in an axial position at a givencarbon atom on a pyranose ring is expected to resonate at �0.5ppm higher field than the corresponding equatorial protonattached to the same carbon (37). Furthermore, the two-dimen-sional NOE spectroscopy data for the �-anomeric L-Ara4N ringof the natural product undecaprenyl phosphate-L-�-Ara4N (17)yielded the expected NOE cross-peaks between H-1�, H-3� andthe upfield H-5� proton resonance, in agreement with the lit-erature assignments, and distinctly different from what is ob-served with the ArnB product generated in vitro.
Evaluation of the Carbon Structure of the ArnB Product byHMQC/HMBC Spectroscopy—13C NMR data for the ArnB en-zymatic product were obtained indirectly through 1H-detectedHMQC and HMBC two-dimensional NMR experiments. Thepartial two-dimensional HMQC 1H-13C correlation map (Fig. 6,panel A) reveals three direct 1H-13C single-bond correlations in
2 The 1H chemical shifts given in the text and in Table I refer to theexperiment shown in Fig. 4. The actual chemical shifts in some of theother experiments (such as the COSY analysis in Fig. 5) may varyslightly (by �0.02 ppm). The analysis in Fig. 5 was done several weeksafter that shown in Fig 4, possibly allowing for small changes in thevolume or pD of the sample.
FIG. 3. Overexpression and purification of two hexa-histidine-tagged derivatives of ArnB. The level of overexpression in crudeextracts and the purity of the tagged ArnB fusion proteins followingaffinity chromatography was evaluated by electrophoresis on a 10%SDS-polyacrylamide gel. In each case either 60 �g of crude extract or 5�g of pure enzyme was loaded per lane, as indicated. The gel wasstained with Coomassie Blue. Lane 1, the molecular mass standards ofphosphorylase b (106 kDa), bovine serum albumin (77 kDa), ovalbumin(51 kDa), carbonic anhydrase (36 kDa), and soybean trypsin inhibitor(28 kDa). Lane 2, uninduced E. coli NovaBlue(DE3)/pETArnB. Lane 3,E. coli NovaBlue(DE3)/pETArnB induced for 3.5 h at 30 °C with 1 mM
IPTG. Lane 4, uninduced E. coli NovaBlue(DE3)/pETArnB-N6H. Lane5, E. coli NovaBlue(DE3)/pETArnB-N6H induced for 3.5 h at 30 °C with1 mM IPTG. Lane 6, ArnB-N6H purified by nickel chelation chromatog-raphy. Lane 7, ArnB-C6H purified by nickel chelation chromatography.Lane 8, E. coli NovaBlue(DE3)/pETArnB-C6H induced for 3.5 h at 30 °Cwith 1 mM IPTG. Lane 9, uninduced E. coli NovaBlue(DE3)/pETArnB-C6H. Lane 10, protein molecular mass standards, as in lane 1.
FIG. 4. Partial 1H NMR spectra of UDP-L-Ara4N and its precur-sor UDP-Ara4O. Panel A shows the aromatic and anomeric protonresonances of UDP-L-Ara4N in D2O at 800 MHz. Panel B shows theribose and pyranose proton resonances of the same sample of UDP-L-Ara4N. Panel C shows the corresponding ribose and pyranose protonresonances of the substrate UDP-Ara4O in D2O at 800 MHz. All spectrawere recorded at 25 °C.
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis 24735
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

the anomeric region (90–105 ppm). The anomeric H-1� near5.63 ppm correlates to the carbon resonance of the pyranose at98.2 ppm (C-1�). The C-1 atom of a pyranose ring that isglycosidically linked with an axial oxygen is expected to reso-nate in the 98–103-ppm range, whereas a C-1 atom that isglycosidically linked with an equatorial oxygen is expected toresonate in the 103–106-ppm region (35). The 98-ppm shift ofC-1� for the 4-amino-4-deoxy-arabinose is thus consistent withan axially disposed oxygen atom and an equatorially disposedH-1�, as deduced from the 1H NMR data. The ribose H-1� protonsignal at 6.00 ppm correlates to the anomeric carbon resonanceat 90.9 ppm (C-1�). The uracil H-5 near 5.99 ppm correlates tothe 105.3-ppm carbon signal, and the uracil H-6 at 7.97 ppmcorrelates to a carbon signal at 144.2 ppm (not shown).
Examination of the HMQC data for the other proton reso-nances of the pyranose moiety shows that the H-5b� and H-5a�proton multiplets correlate to a carbon signal at 62.4 ppm(C-5�), whereas the H-2� and H-3� multiplets connect to carbonresonances at 70.1 and 68.5 ppm, respectively. These carbonshift positions correspond to oxygen-substituted carbons of sug-ars. The nitrogen-substituted carbons of amino sugars are re-ported to resonate near 52–55 ppm (35, 38). The H-4� multipletshows a prominent cross-peak near 55 ppm. This C-4� chemical
TABLE INMR spectroscopy of UDP-L-Ara4N versus UDP-Ara4O
Position13C NMR signals 1H NMR signals
UDP-L-Ara4N UDP-Ara4O UDP-L-Ara4N UDP-Ara4O
� (ppm) � (ppm) Hz � (ppm) HzUridine
2 154.4 154.44 168.8 168.85 105.3 105.2 5.99 (d) J5,6 8.1 6.00 (d) J5,6 8.16 144.2 144.2 7.97 (d) 7.99 (d)
Ribose1� 90.9 90.9 6.00 (d) 6.02 (d)2� 75.6 76.4 �4.39 �4.43� 71.5 72.2 �4.39 �4.44� 85.6 85.8 4.30 �4.35� 66.8 67.5 4.26 (ddd) 3J5�a,4� 2.6 4.27 (ddd) 3J5�a,4� 2.8
J5�a,P 4.5 J5�a,P 5.4J5�a,5�b 11.8 J5�a,5�b 11.8
4.21 3J5�b,4� 2.8 4.22 (ddd) 3J5�b,4� 2.5J5�b,P 5.6 J5�b,P 4.4
Pyranose1� 98.2 98.5 5.63 (dd) J1�,2� 3.4 5.61 (dd) J1�,2� 3.3
3J1�,P 7.3 3J1�,P 7.12� 70.1 73.1 3.77 (dt) J2�,3� 9.9 3.69 (dt) J2�,3� 9.8
4J2�,P 3.4 4J2�,P 3.33� 68.5 75.1 4.15 (dd) J3�,4� 4.6 3.84 (d)4� 54.6 95.2 3.59 (m)5� 62.4 68.0 3.80 (dd) J4�,5�a 2.0 3.58 (d) J5�a,5�b 12.0
J5�a,5�b 13.34.29 (dd) J4�,5�b 2.1 3.96 (d)
FIG. 5. 1H-1H COSY analysis of UDP-L-Ara4N at 800 MHz. Theconnectivities in the ribose and pyranose moieties of UDP-L-Ara4N (1.7mg in D2O at 25 °C) are shown. Key cross-peaks are labeled. The full 1Hand 13C assignments are listed in Table I.
FIG. 6. Partial two-dimensional HMQC and HMBC spectra ofUDP-L-Ara4N. Panel A is a partial two-dimensional HMQC spectrumof UDP-Ara4N in D2O showing the single bond 1H-13C correlationsarising from the uracil H-5, the ribose protons, and the pyranose pro-tons to their respective carbons. Key cross-peaks are labeled. Panel B isthe corresponding partial two-dimensional HMBC spectrum of thesame UDP-Ara4N sample. Of particular importance are the multibondcorrelations from H-5b� and H-5a� to the 54.6-ppm carbon signal of thepyranose, verifying its assignment as C-4�. X is the residual HOD signalremaining after water suppression. The full 1H and 13C assignmentsare listed in Table I.
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis24736
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from
shift is diagnostic evidence confirming C-4� as the site of theamino group substitution. The remaining sugar HMQC cross-peaks arise from protonated carbons of the ribose ring. Theribose assignments derived (Table I) agree with the literaturevalues (35, 39).
The multibond HMBC two-dimensional map (Fig. 6, panel B)allows identification of the quaternary carbons leading to fullassignments (Table I) for the enzyme product. Analyses of themultibond correlations from the uracil H-6 (7.97 ppm) and H-5(5.99 ppm) yielded assignment of the 154 and 168 ppm quater-nary carbon signals to C-2 and C-4, respectively (not shown inFig. 6, panel B), consistent with literature assignments (39).The multibond correlation from the uracil H-6 to C-1� was alsodetected (not shown), thus verifying the linkage of the basegroup to the ribose. The most salient feature in the sugarregion is the detection of the multibond correlations from bothH-5� protons to the 54.6-ppm carbon resonance, thus furthervalidating C-4� as the nitrogen-substituted carbon (Fig. 6). Theremaining multibond correlations in the sugar region verify theassignments of the arabinose and ribose carbon resonances.The 1H and 13C NMR assignments are summarized in Table I.
Comparison of the 13C NMR data for the ArnB product withthe starting substrate, UDP-Ara4O (22) (Table I), reveals thatthe uracil and ribose carbons of both compounds resonate atvery similar positions, consistent with the 1H NMR evidencethat the enzymatic reaction did not alter the uridine moiety.The 13C NMR data dramatically show that the enzyme-cata-lyzed amino group substitution is localized to C-4�, which shiftsupfield by �40.6 ppm, from 95.2 ppm for UDP-Ara4O in D2O(corresponding to the hydrated form of UDP-Ara4O) (22) to54.6 ppm for UDP-L-Ara4N in D2O. The C-3� and C-5� reso-nances show much smaller upfield chemical shifts of 6.6 and5.6 ppm, respectively. The chemical shift change decreases byanother factor of 2 at C-2� to 3 ppm and by a further factor of 10at C-1� to 0.3 ppm.
ArnB-catalyzed Transamination of UDP-Ara4O—PurifiedArnB-C6H in the presence of various amine donors at 7.5 mM
preferentially utilizes L-glutamate for the synthesis of UDP-L-Ara4N from 150 �M UDP-Ara4O. The rate of transaminationwith L-methionine, L-glutamine, and L-alanine is measurable at5, 2, and 1%, respectively, of the rate observed with L-gluta-mate (data not shown). No activity was seen with L isomers ofasparagine, aspartate, glycine, lysine, ornithine, phenylala-nine, or tryptophan. However, the analog L-aminoadipic acid(25) supported the transamination of UDP-Ara4O at about 50%the rate of L-glutamate (not shown).
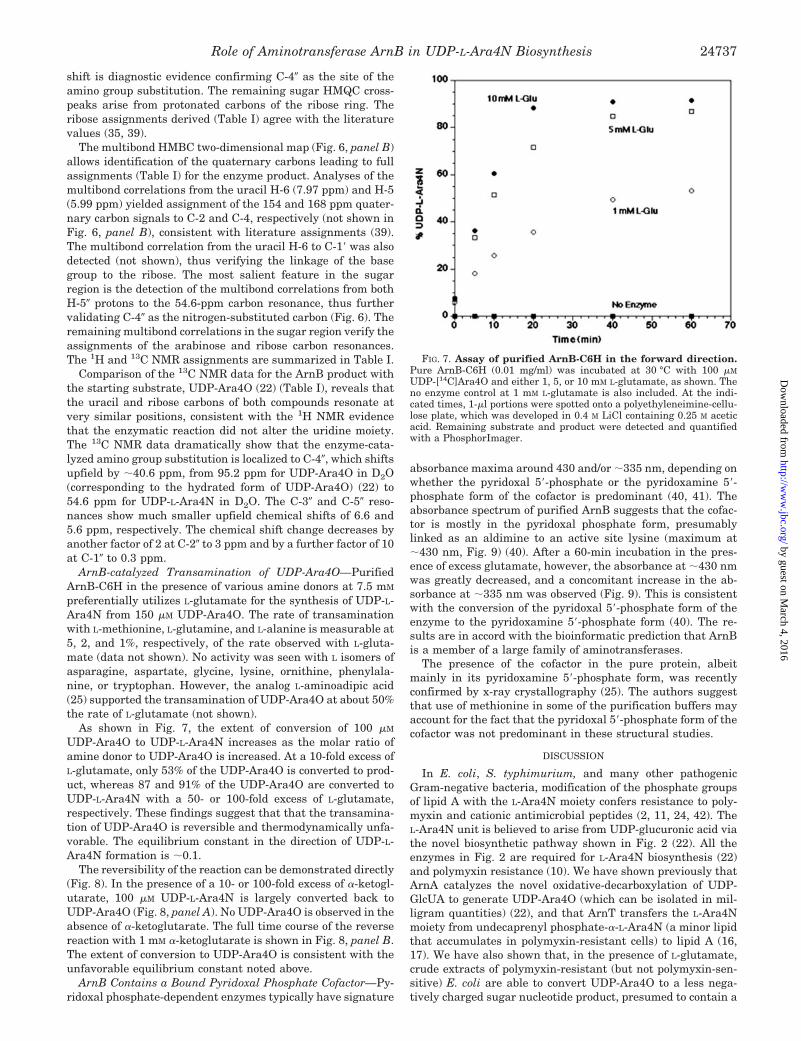
As shown in Fig. 7, the extent of conversion of 100 �M
UDP-Ara4O to UDP-L-Ara4N increases as the molar ratio ofamine donor to UDP-Ara4O is increased. At a 10-fold excess ofL-glutamate, only 53% of the UDP-Ara4O is converted to prod-uct, whereas 87 and 91% of the UDP-Ara4O are converted toUDP-L-Ara4N with a 50- or 100-fold excess of L-glutamate,respectively. These findings suggest that that the transamina-tion of UDP-Ara4O is reversible and thermodynamically unfa-vorable. The equilibrium constant in the direction of UDP-L-Ara4N formation is �0.1.
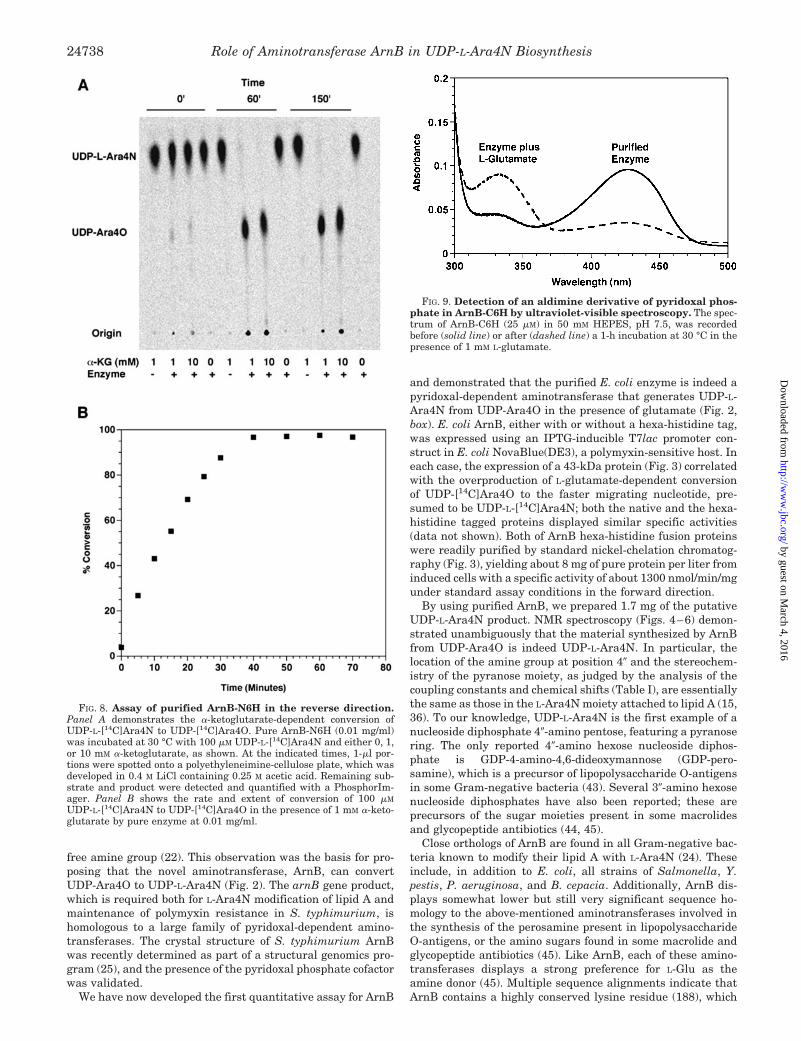
The reversibility of the reaction can be demonstrated directly(Fig. 8). In the presence of a 10- or 100-fold excess of �-ketogl-utarate, 100 �M UDP-L-Ara4N is largely converted back toUDP-Ara4O (Fig. 8, panel A). No UDP-Ara4O is observed in theabsence of �-ketoglutarate. The full time course of the reversereaction with 1 mM �-ketoglutarate is shown in Fig. 8, panel B.The extent of conversion to UDP-Ara4O is consistent with theunfavorable equilibrium constant noted above.
ArnB Contains a Bound Pyridoxal Phosphate Cofactor—Py-ridoxal phosphate-dependent enzymes typically have signature
absorbance maxima around 430 and/or �335 nm, depending onwhether the pyridoxal 5�-phosphate or the pyridoxamine 5�-phosphate form of the cofactor is predominant (40, 41). Theabsorbance spectrum of purified ArnB suggests that the cofac-tor is mostly in the pyridoxal phosphate form, presumablylinked as an aldimine to an active site lysine (maximum at�430 nm, Fig. 9) (40). After a 60-min incubation in the pres-ence of excess glutamate, however, the absorbance at �430 nmwas greatly decreased, and a concomitant increase in the ab-sorbance at �335 nm was observed (Fig. 9). This is consistentwith the conversion of the pyridoxal 5�-phosphate form of theenzyme to the pyridoxamine 5�-phosphate form (40). The re-sults are in accord with the bioinformatic prediction that ArnBis a member of a large family of aminotransferases.
The presence of the cofactor in the pure protein, albeitmainly in its pyridoxamine 5�-phosphate form, was recentlyconfirmed by x-ray crystallography (25). The authors suggestthat use of methionine in some of the purification buffers mayaccount for the fact that the pyridoxal 5�-phosphate form of thecofactor was not predominant in these structural studies.
DISCUSSION
In E. coli, S. typhimurium, and many other pathogenicGram-negative bacteria, modification of the phosphate groupsof lipid A with the L-Ara4N moiety confers resistance to poly-myxin and cationic antimicrobial peptides (2, 11, 24, 42). TheL-Ara4N unit is believed to arise from UDP-glucuronic acid viathe novel biosynthetic pathway shown in Fig. 2 (22). All theenzymes in Fig. 2 are required for L-Ara4N biosynthesis (22)and polymyxin resistance (10). We have shown previously thatArnA catalyzes the novel oxidative-decarboxylation of UDP-GlcUA to generate UDP-Ara4O (which can be isolated in mil-ligram quantities) (22), and that ArnT transfers the L-Ara4Nmoiety from undecaprenyl phosphate-�-L-Ara4N (a minor lipidthat accumulates in polymyxin-resistant cells) to lipid A (16,17). We have also shown that, in the presence of L-glutamate,crude extracts of polymyxin-resistant (but not polymyxin-sen-sitive) E. coli are able to convert UDP-Ara4O to a less nega-tively charged sugar nucleotide product, presumed to contain a
FIG. 7. Assay of purified ArnB-C6H in the forward direction.Pure ArnB-C6H (0.01 mg/ml) was incubated at 30 °C with 100 �M
UDP-[14C]Ara4O and either 1, 5, or 10 mM L-glutamate, as shown. Theno enzyme control at 1 mM L-glutamate is also included. At the indi-cated times, 1-�l portions were spotted onto a polyethyleneimine-cellu-lose plate, which was developed in 0.4 M LiCl containing 0.25 M aceticacid. Remaining substrate and product were detected and quantifiedwith a PhosphorImager.
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis 24737
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from
free amine group (22). This observation was the basis for pro-posing that the novel aminotransferase, ArnB, can convertUDP-Ara4O to UDP-L-Ara4N (Fig. 2). The arnB gene product,which is required both for L-Ara4N modification of lipid A andmaintenance of polymyxin resistance in S. typhimurium, ishomologous to a large family of pyridoxal-dependent amino-transferases. The crystal structure of S. typhimurium ArnBwas recently determined as part of a structural genomics pro-gram (25), and the presence of the pyridoxal phosphate cofactorwas validated.
We have now developed the first quantitative assay for ArnB
and demonstrated that the purified E. coli enzyme is indeed apyridoxal-dependent aminotransferase that generates UDP-L-Ara4N from UDP-Ara4O in the presence of glutamate (Fig. 2,box). E. coli ArnB, either with or without a hexa-histidine tag,was expressed using an IPTG-inducible T7lac promoter con-struct in E. coli NovaBlue(DE3), a polymyxin-sensitive host. Ineach case, the expression of a 43-kDa protein (Fig. 3) correlatedwith the overproduction of L-glutamate-dependent conversionof UDP-[14C]Ara4O to the faster migrating nucleotide, pre-sumed to be UDP-L-[14C]Ara4N; both the native and the hexa-histidine tagged proteins displayed similar specific activities(data not shown). Both of ArnB hexa-histidine fusion proteinswere readily purified by standard nickel-chelation chromatog-raphy (Fig. 3), yielding about 8 mg of pure protein per liter frominduced cells with a specific activity of about 1300 nmol/min/mgunder standard assay conditions in the forward direction.
By using purified ArnB, we prepared 1.7 mg of the putativeUDP-L-Ara4N product. NMR spectroscopy (Figs. 4–6) demon-strated unambiguously that the material synthesized by ArnBfrom UDP-Ara4O is indeed UDP-L-Ara4N. In particular, thelocation of the amine group at position 4� and the stereochem-istry of the pyranose moiety, as judged by the analysis of thecoupling constants and chemical shifts (Table I), are essentiallythe same as those in the L-Ara4N moiety attached to lipid A (15,36). To our knowledge, UDP-L-Ara4N is the first example of anucleoside diphosphate 4�-amino pentose, featuring a pyranosering. The only reported 4�-amino hexose nucleoside diphos-phate is GDP-4-amino-4,6-dideoxymannose (GDP-pero-samine), which is a precursor of lipopolysaccharide O-antigensin some Gram-negative bacteria (43). Several 3�-amino hexosenucleoside diphosphates have also been reported; these areprecursors of the sugar moieties present in some macrolidesand glycopeptide antibiotics (44, 45).
Close orthologs of ArnB are found in all Gram-negative bac-teria known to modify their lipid A with L-Ara4N (24). Theseinclude, in addition to E. coli, all strains of Salmonella, Y.pestis, P. aeruginosa, and B. cepacia. Additionally, ArnB dis-plays somewhat lower but still very significant sequence ho-mology to the above-mentioned aminotransferases involved inthe synthesis of the perosamine present in lipopolysaccharideO-antigens, or the amino sugars found in some macrolide andglycopeptide antibiotics (45). Like ArnB, each of these amino-transferases displays a strong preference for L-Glu as theamine donor (45). Multiple sequence alignments indicate thatArnB contains a highly conserved lysine residue (188), which
FIG. 8. Assay of purified ArnB-N6H in the reverse direction.Panel A demonstrates the �-ketoglutarate-dependent conversion ofUDP-L-[14C]Ara4N to UDP-[14C]Ara4O. Pure ArnB-N6H (0.01 mg/ml)was incubated at 30 °C with 100 �M UDP-L-[14C]Ara4N and either 0, 1,or 10 mM �-ketoglutarate, as shown. At the indicated times, 1-�l por-tions were spotted onto a polyethyleneimine-cellulose plate, which wasdeveloped in 0.4 M LiCl containing 0.25 M acetic acid. Remaining sub-strate and product were detected and quantified with a PhosphorIm-ager. Panel B shows the rate and extent of conversion of 100 �M
UDP-L-[14C]Ara4N to UDP-[14C]Ara4O in the presence of 1 mM �-keto-glutarate by pure enzyme at 0.01 mg/ml.
FIG. 9. Detection of an aldimine derivative of pyridoxal phos-phate in ArnB-C6H by ultraviolet-visible spectroscopy. The spec-trum of ArnB-C6H (25 �M) in 50 mM HEPES, pH 7.5, was recordedbefore (solid line) or after (dashed line) a 1-h incubation at 30 °C in thepresence of 1 mM L-glutamate.
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis24738
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

presumably forms a Schiff’s base with the pyridoxal phosphatecofactor that is detected in the ultraviolet visible absorptionspectrum at 430 nm (Fig. 9). The recently reported crystalstructure of 3-amino-5-hydroxybenzoic acid synthase likewiseshows the cofactor covalently bound to the analogous Lys (188)through the expected internal aldimine linkage (46). Our spec-troscopic data (Fig. 9) are consistent with ArnB being isolatedmainly in the pyridoxal phosphate (aldimine) form of the co-factor, presumably linked to Lys-188. In the presence of excessL-Glu (Fig. 9), the bound pyridoxal phosphate cofactor is con-verted to the pyridoxamine phosphate derivative. The latterwould be used directly to transfer the amine group to UDP-Ara4O (Fig. 2).
The UDP-L-Ara4N (Figs. 4–6) generated by ArnB in vitroprovides a new tool with which to explore the later steps of thepathway for the modification of lipid A with L-Ara4N (Fig. 2). Incell extracts derived from polymyxin-resistant E. coli, UDP-L-Ara4N can be metabolized to what appears to be a 4�-formyl-amino derivative (22). A separate catalytic domain of ArnAcatalyzes formyl group transfer from N-10-formyltetrahydrofo-late to the 4�-amine of UDP-L-Ara4N to make the proposednucleotide UDP-Ara4-formyl-N (Fig. 2).3 The role of this formy-lated nucleotide in the modification of lipid A with L-Ara4N isnot immediately obvious, given that undecaprenyl phosphate-�-L-Ara4N (16, 17) is the Ara4N donor used by ArnT for trans-ferring the L-Ara4N moiety to lipid A.
Biosynthesis of the UDP-L-Ara4-formyl-N intermediate inthe cytoplasm (Fig. 2) might compensate for the relativelyunfavorable equilibrium constant for the conversion of UDP-Ara4O to UDP-L-Ara4N, which we estimate to be about 0.1(Figs. 7 and 8). Preliminary results suggest that ArnC mightactually be selective for UDP-L-Ara4-formyl-N, as proposed inFig. 2.3 However, deformylation would then be necessary priorto L-Ara4N transfer to lipid A from undecaprenyl phosphate-�-L-Ara4N (16, 17). It would make sense for the proposed de-formylation to occur in the periplasm in order to avoid a futilecycle in the cytoplasm (Fig. 2). Furthermore, if the putativeinner membrane transporter (flippase) for undecaprenyl phos-phate-�-L-Ara4-formyl-N (Fig. 2) were unable to transport un-decaprenyl phosphate-�-L-Ara4N, accumulation of the latter onthe outer surface of the inner membrane would take place,thereby ensuring that ArnT has an adequate supply of itsdonor substrate with which to modify lipid A prior to its exportto the outer membrane. The latter process occurs very rapidly(within a minute) in wild-type cells (47). The biochemical char-acterization of the formyltransferase activity of ArnA (22), to-gether with the development of assays for ArnC and the puta-tive deformylase (Fig. 2), should reveal fundamental newinsights into the compartmentalization of lipid A modificationenzymes and the mechanisms of lipid transport across theinner membrane.
Acknowledgments—The Duke NMR Center is supported in part byNCI Grant P30-CA-14236 from the National Institutes of Health (toA. A. R). NMR instrumentation in the Duke NMR Center was funded bythe National Science Foundation, the National Institutes of Health, theNorth Carolina Biotechnology Center and Duke University.
REFERENCES
1. Raetz, C. R. H. (1990) Annu. Rev. Biochem. 59, 129–1702. Raetz, C. R. H., and Whitfield, C. (2002) Annu. Rev. Biochem. 71, 635–7003. Brade, H., Opal, S. M., Vogel, S. N., and Morrison, D. C. (eds) (1999) Endotoxin
in Health and Disease, Marcel Dekker, Inc., New York4. Aderem, A., and Ulevitch, R. J. (2000) Nature 406, 782–7875. Medzhitov, R., and Janeway, C., Jr. (2000) N. Engl. J. Med. 343, 338–3446. Lien, E., and Ingalls, R. R. (2002) Crit. Care Med. 30, S1–S117. Parillo, J. E. (1993) N. Engl. J. Med. 328, 1471–1477
8. Raetz, C. R. H. (1996) in Escherichia coli and Salmonella: Cellular andMolecular Biology (Neidhardt, F. C., ed) Vol. 1, 2nd Ed., pp. 1035–1063,American Society for Microbiology, Washington, D. C.
9. Guo, L., Lim, K. B., Gunn, J. S., Bainbridge, B., Darveau, R. P., Hackett, M.,and Miller, S. I. (1997) Science 276, 250–253
10. Gunn, J. S., Lim, K. B., Krueger, J., Kim, K., Guo, L., Hackett, M., and Miller,S. I. (1998) Mol. Microbiol. 27, 1171–1182
11. Groisman, E. A. (2001) J. Bacteriol. 183, 1835–184212. Guo, L., Lim, K. B., Poduje, C. M., Daniel, M., Gunn, J. S., Hackett, M., and
Miller, S. I. (1998) Cell 95, 189–19813. Bishop, R. E., Gibbons, H. S., Guina, T., Trent, M. S., Miller, S. I., and Raetz,
C. R. H. (2000) EMBO J. 19, 5071–508014. Gibbons, H. S., Lin, S., Cotter, R. J., and Raetz, C. R. H. (2000) J. Biol. Chem.
275, 32940–3294915. Zhou, Z., Ribeiro, A. A., Lin, S., Cotter, R. J., Miller, S. I., and Raetz, C. R. H.
(2001) J. Biol. Chem. 276, 43111–4312116. Trent, M. S., Ribeiro, A. A., Lin, S., Cotter, R. J., and Raetz, C. R. H. (2001)
J. Biol. Chem. 276, 43122–4313117. Trent, M. S., Ribeiro, A. A., Doerrler, W. T., Lin, S., Cotter, R. J., and Raetz,
C. R. H. (2001) J. Biol. Chem. 276, 43132–4314418. Vaara, M., Vaara, T., Jensen, M., Helander, I., Nurminen, M., Rietschel, E. T.,
and Makela, P. H. (1981) FEBS Lett. 129, 145–14919. Roland, K. L., Martin, L. E., Esther, C. R., and Spitznagel, J. K. (1993) J.
Bacteriol. 175, 4154–416420. Helander, I. M., Kilpelainen, I., and Vaara, M. (1994) Mol. Microbiol. 11,
481–48721. Groisman, E. A., Kayser, J., and Soncini, F. C. (1997) J. Bacteriol. 179,
7040–704522. Breazeale, S. D., Ribeiro, A. A., and Raetz, C. R. H. (2002) J. Biol. Chem. 277,
2886–289623. Nummila, K., Kilpelainen, I., Zahringer, U., Vaara, M., and Helander, I. M.
(1995) Mol. Microbiol. 16, 271–27824. Zahringer, U., Lindner, B., and Rietschel, E. T. (1999) in Endotoxin in Health
and Disease (Brade, H., Opal, S. M., Vogel, S. N., and Morrison, D. C., eds)pp. 93–114, Marcel Dekker, Inc., New York
25. Noland, B. W., Newman, J. M., Hendle, J., Badger, J., Christopher, J. A.,Tresser, J., Buchanan, M. D., Wright, T. A., Rutter, M. E., Sanderson, W. E.,Muller-Dieckmann, H. J., Gajiwala, K. S., and Buchanan, S. G. (2002)Structure 10, 1569–1580
26. Smith, P. K., Krohn, R. I., Hermanson, G. T., Mallia, A. K., Gartner, F. H.,Provenzano, M. D., Fujimoto, E. K., Goeke, N. M., Olson, B. J., and Klenk,D. C. (1985) Anal. Biochem. 150, 76–85
27. Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith,J. A., and Struhl, K. (eds) (1989) Current Protocols in Molecular Biology,John Wiley & Sons, Inc., New York
28. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: ALaboratory Manual, 2nd Ed., Cold Spring Harbor, Cold Spring Harbor, NY
29. Blattner, F. R., Plunkett, G., Bloch, C. A., Perna, N. T., Burland, V., Riley, M.,Collado-Vides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F., Gregor, J.,Davis, N. W., Kirkpatrick, H. A., Goeden, M. A., Rose, D. J., Mau, B., andShao, Y. (1997) Science 277, 1453–1474
30. Miller, J. R. (1972) Experiments in Molecular Genetics, Cold Spring HarborLaboratory, Cold Spring Harbor, NY
31. Laemmli, U. K. (1970) Nature 227, 680–68532. Ribeiro, A. A., Zhou, Z., and Raetz, C. R. H. (1999) Magn. Res. Chem. 37,
620–63033. Zhou, Z., Lin, S., Cotter, R. J., and Raetz, C. R. H. (1999) J. Biol. Chem. 274,
18503–1851434. Naleway, J. J., Raetz, C. R. H., and Anderson, L. (1988) Carbohydr. Res. 179,
199–20935. Agrawal, P. K. (1992) Phytochemistry 31, 3307–333036. Zhou, Z., Ribeiro, A. A., and Raetz, C. R. H. (2000) J. Biol. Chem. 275,
13542–1355137. van Halbeek, H. (1996) in Encyclopedia of NMR (Grant, D. M., and Harris,
R. K., eds) Vol. 2, pp. 1107–1137, Wiley, New York38. Agrawal, P. K., Bush, C. A., Qureshi, N., and Takayama, K. (1994) Adv.
Biophys. Chem. 4, 179–23639. Rosenthal, S. N., and Fendler, J. H. (1976) Adv. Phys. Org. Chem. 13, 280–42440. Heinert, D., and Martell, A. E. (1963) J. Am. Chem. Soc. 85, 183–18741. Raetz, C. R. H., and Auld, D. S. (1972) Biochemistry 11, 2229–223642. Ohl, M. E., and Miller, S. I. (2001) Annu. Rev. Med. 52, 259–27443. Albermann, C., and Piepersberg, W. (2001) Glycobiology 11, 655–66144. Chen, H., Thomas, M. G., Hubbard, B. K., Losey, H. C., Walsh, C. T., and
Burkart, M. D. (2000) Proc. Natl. Acad. Sci. U. S. A. 97, 11942–1194745. He, X. M., and Liu, H. W. (2002) Annu. Rev. Biochem. 71, 701–75446. Eads, J. C., Beeby, M., Scapin, G., Yu, T. W., and Floss, H. G. (1999) Biochem-
istry 38, 9840–984947. Doerrler, W. T., Reedy, M. C., and Raetz, C. R. H. (2001) J. Biol. Chem. 276,
11461–1146448. Hwang, P. M., Choy, W. Y., Lo, E. I., Chen, L., Forman-Kay, J. D., Raetz,
C. R. H., Prive, G. G., Bishop, R. E., and Kay, L. E. (2002) Proc. Natl. Acad.Sci. U. S. A. 99, 13560–13565
49. Trent, M. S., Pabich, W., Raetz, C. R. H., and Miller, S. I. (2001) J. Biol. Chem.276, 9083–9092
50. McClelland, M., Sanderson, K. E., Spieth, J., Clifton, S. W., Latreille, P.,Courtney, L., Porwollik, S., Ali, J., Dante, M., Du, F., Hou, S., Layman, D.,Leonard, S., Nguyen, C., Scott, K., Holmes, A., Grewal, N., Mulvaney, E.,Ryan, E., Sun, H., Florea, L., Miller, W., Stoneking, T., Nhan, M.,Waterston, R., and Wilson, R. K. (2001) Nature 413, 852–856
51. Orlean, P., Albright, C., and Robbins, P. W. (1988) J. Biol. Chem. 263,17499–175073 S. D. Breazeale and C. R. H. Raetz, unpublished observations.
Role of Aminotransferase ArnB in UDP-L-Ara4N Biosynthesis 24739
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Steven D. Breazeale, Anthony A. Ribeiro and Christian R. H. Raetz(ArnB) THAT GENERATES UDP-4-AMINO-4-DEOXY-l-ARABINOSE
: AN AMINOTRANSFERASEEscherichia coliPolymyxin-resistant Mutants of Origin of Lipid A Species Modified with 4-Amino-4-deoxy-l-arabinose in
doi: 10.1074/jbc.M304043200 originally published online April 18, 20032003, 278:24731-24739.J. Biol. Chem.
10.1074/jbc.M304043200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/278/27/24731.full.html#ref-list-1
This article cites 46 references, 19 of which can be accessed free at
by guest on March 4, 2016
http://ww
w.jbc.org/
Dow
nloaded from




![Molecular Imaging of Murine Intestinal Inflammation With 2-Deoxy-2-[18F]Fluoro-d-Glucose and Positron Emission Tomography](https://static.fdokumen.com/doc/165x107/6344fffc596bdb97a908b96f/molecular-imaging-of-murine-intestinal-inflammation-with-2-deoxy-2-18ffluoro-d-glucose.jpg)














