
OPUS-PSP: An Orientation-dependent Statistical All-atom Potential Derived from Side-chain Packing
14
OPUS-PSP: An Orientation-dependent Statistical All-atom Potential Derived from Side-chain Packing Mingyang Lu 1 , Athanasios D. Dousis 2 and Jianpeng Ma 1,2 ⁎ 1 Verna and Marrs McLean Department of Biochemistry and Molecular Biology, Baylor College of Medicine, One Baylor Plaza, Houston, TX77030, USA 2 Department of Bioengineering, Rice University, Houston, TX 77005, USA Received 24 September 2007; received in revised form 6 November 2007; accepted 13 November 2007 Available online 19 November 2007 Here we report an orientation-dependent statistical all-atom _ potential derived from side-chain _ packing, named OPUS-PSP. It features a basis set of 19 rigid-body blocks extracted from the chemical structures of all 20 amino acid residues. The potential is generated from the orientation-specific packing statistics of pairs of those blocks in a non-redundant structural database. The purpose of such an approach is to capture the essential elements of orientation dependence in molecular packing interactions. Tests of OPUS-PSP on commonly used decoy sets demonstrate that it sig- nificantly outperforms most of the existing knowledge-based potentials in terms of both its ability to recognize native structures and consistency in achieving high Z-scores across decoy sets. As OPUS-PSP excludes inter- actions among main-chain atoms, its success highlights the crucial impor- tance of side-chain packing in forming native protein structures. Moreover, OPUS-PSP does not explicitly include solvation terms, and thus the poten- tial should perform well when the solvation effect is difficult to determine, such as in membrane proteins. Overall, OPUS-PSP is a generally applicable potential for protein structure modeling, especially for handling side-chain conformations, one of the most difficult steps in high-accuracy protein structure prediction and refinement. © 2007 Elsevier Ltd. All rights reserved. Edited by M. Levitt Keywords: knowledge-based potential function; decoy recognition/ structure prediction; protein folding; side-chain packing; orientation dependence Introduction Despite major advances in high-throughput pro- tein crystallization and other tools, modern experi- mental techniques in structural biology have not kept up with the rapid pace of genome sequencing. Consequently, the substantial sequence-structure gap is a driving force for research in protein structure prediction. However, accurately predict- ing the structure of a protein directly from its amino acid sequence is far from routine due to various technical difficulties, one of which is the lack of accurate empirical potential functions. In general, there are two types of potential functions for protein structure modeling: physics- based and knowledge-based. Physics-based poten- tial functions, such as the CHARMM force field, 1 are developed from ab initio quantum chemical calculations, whereas knowledge-based potential functions are developed from statistical analysis of known protein structures. In many applications, knowledge-based potential functions outperform the physics-based potentials. 2,3 Knowledge-based potentials may be further divided into two cate- gories: all-atom potentials and coarse-grained (semi)-residue-based potentials. For more than two decades, developing various potential func- tions has been an extremely active field. Many comprehensive review articles are available in the literature. 2,4–12 One of the most important determinants of protein structure is side-chain packing, since the *Corresponding author. E-mail address: [email protected]. Present address: J. Ma, One Baylor Plaza, BCM-125, Houston, TX 77030, USA. Abbreviations used: PSP, potential based on side-chain packing; LJ, Lennard-Jones; HPMF, hydrophobic potential of mean force. doi:10.1016/j.jmb.2007.11.033 J. Mol. Biol. (2008) 376, 288–301 Available online at www.sciencedirect.com 0022-2836/$ - see front matter © 2007 Elsevier Ltd. All rights reserved.
Transcript of OPUS-PSP: An Orientation-dependent Statistical All-atom Potential Derived from Side-chain Packing

doi:10.1016/j.jmb.2007.11.033 J. Mol. Biol. (2008) 376, 288–301
Available online at www.sciencedirect.com
OPUS-PSP: An Orientation-dependent StatisticalAll-atom Potential Derived from Side-chain Packing
Mingyang Lu1, Athanasios D. Dousis2 and Jianpeng Ma1,2⁎
1Verna and Marrs McLeanDepartment of Biochemistryand Molecular Biology, BaylorCollege of Medicine, One BaylorPlaza, Houston, TX77030, USA2Department of Bioengineering,Rice University, Houston,TX 77005, USA
Received 24 September 2007;received in revised form6 November 2007;accepted 13 November 2007Available online19 November 2007
*Corresponding author. E-mail [email protected] address: J. Ma, One Baylo
Houston, TX 77030, USA.Abbreviations used: PSP, potentia
packing; LJ, Lennard-Jones; HPMF, hof mean force.
0022-2836/$ - see front matter © 2007 E
Here we report an orientation-dependent statistical all-atom _potentialderived from side-chain _packing, named OPUS-PSP. It features a basis set of19 rigid-body blocks extracted from the chemical structures of all 20 aminoacid residues. The potential is generated from the orientation-specificpacking statistics of pairs of those blocks in a non-redundant structuraldatabase. The purpose of such an approach is to capture the essentialelements of orientation dependence in molecular packing interactions. Testsof OPUS-PSP on commonly used decoy sets demonstrate that it sig-nificantly outperforms most of the existing knowledge-based potentials interms of both its ability to recognize native structures and consistency inachieving high Z-scores across decoy sets. As OPUS-PSP excludes inter-actions among main-chain atoms, its success highlights the crucial impor-tance of side-chain packing in forming native protein structures. Moreover,OPUS-PSP does not explicitly include solvation terms, and thus the poten-tial should perform well when the solvation effect is difficult to determine,such as in membrane proteins. Overall, OPUS-PSP is a generally applicablepotential for protein structure modeling, especially for handling side-chainconformations, one of the most difficult steps in high-accuracy proteinstructure prediction and refinement.
© 2007 Elsevier Ltd. All rights reserved.
Keywords: knowledge-based potential function; decoy recognition/structure prediction; protein folding; side-chain packing; orientationdependence
Edited by M. LevittIntroduction
Despite major advances in high-throughput pro-tein crystallization and other tools, modern experi-mental techniques in structural biology have notkept up with the rapid pace of genome sequencing.Consequently, the substantial sequence-structuregap is a driving force for research in proteinstructure prediction. However, accurately predict-ing the structure of a protein directly from its aminoacid sequence is far from routine due to various
ess:
r Plaza, BCM-125,
l based on side-chainydrophobic potential
lsevier Ltd. All rights reserve
technical difficulties, one of which is the lack ofaccurate empirical potential functions.In general, there are two types of potential
functions for protein structure modeling: physics-based and knowledge-based. Physics-based poten-tial functions, such as the CHARMM force field,1
are developed from ab initio quantum chemicalcalculations, whereas knowledge-based potentialfunctions are developed from statistical analysis ofknown protein structures. In many applications,knowledge-based potential functions outperformthe physics-based potentials.2,3 Knowledge-basedpotentials may be further divided into two cate-gories: all-atom potentials and coarse-grained(semi)-residue-based potentials. For more thantwo decades, developing various potential func-tions has been an extremely active field. Manycomprehensive review articles are available in theliterature.2,4–12
One of the most important determinants ofprotein structure is side-chain packing, since the
d.

289Orientation-dependent All-atom Statistical Potential from Side-chain Packing
sequence identities of all polypeptide chains aresolely designated by side-chains. In the literature,however, methods for generating statistical poten-tials are limited in their ability to describe side-chain packing. On the one hand, all-atom energyfunctions typically ignore the heterogeneous che-mical bond connectivity.13 For example, betweentwo favorably packed side-chain groups, theproximity of individual atoms is substantiallyinfluenced by chemical bonding interactions. Inother words, certain unfavorably interacting atomscan be near each other. Another issue is that,without proper description of chemical bonds, theorientational heterogeneity among packing groupsis not well represented. On the other hand, typical(semi)-residue-based potentials insufficientlydescribe side-chain packing due to coarse-graining.The literature documents extensive efforts to ex-ploit orientation preference for improving the de-scription of side-chain packing,14–18 but muchremains to be done to fully capture the orientationof side-chains when statistics are derived fromknown structures.Here we have developed an orientation-dependent
statistical _potential based on side-chain _packing(PSP), named OPUS-PSP. It is designed to overcomethe aforementioned drawbacks and bridge the gapbetween all-atom and residue-based potentials. Thefoundation of OPUS-PSP hinges solely on side-chainpacking interactions described by a unique basisset of rigid-body building blocks. This basis set isformed by decomposing the chemical structures of20 amino acid residues into 19 block types. Thepotential is then generated from the orientation-specific packing statistics of pairs of those blocks ina non-redundant structural database. OPUS-PSPcaptures the essential details of side-chain flexibilityand packing orientation without compromising theconciseness of a coarse-grained approach. Anotherfeature of OPUS-PSP is that it does not considermain-chain-main-chain interactions, especiallymain-chain hydrogen-bonding interactions. Thepotential also does not explicitly include the solva-tion effect.OPUS-PSP was tested on several commonly used
decoy sets. The results demonstrate that it signifi-cantly outperforms most of the knowledge-basedpotentials in the literature, e.g. DFIRE,19 DOPE20/DOPE-Cβ45, and HPMF.21 Due to the exclusion ofinteractions among main-chain atoms, the success ofthe potential highlights the crucial importance ofside-chain packing in building native protein struc-tures. Furthermore, OPUS-PSP is widely applicabledue to the generality of side-chain packing. In aforthcoming publication, we will show the effec-tiveness of the potential in improving side-chainmodeling, one of the most important and difficultsteps in high-accuracy protein structure predictionand refinement.For clarity, the theoretical framework of OPUS-
PSP is briefly outlined before the description of themain results, while the methodological details aregiven in Methods. Some lengthy technical issues
that are of less importance to the general audienceare explained in Supplementary Data.
Theoretical framework
In this section, we outline the theoretical frame-work of OPUS-PSP. Additional details of theimplementation are given in the Methods and inSupplementary Data.Two major components are involved in construct-
ing OPUS-PSP: (a) the definition and geometry of 19rigid-body blocks; (b) a knowledge-based energyfunction derived from packing statistics of theseblocks, combined with a repulsive Lennard-Jonesterm to deter steric clashes. We also apply coarse-graining and symmetry to improve the statistics.
Definitions of rigid-body blocks and relativeorientation
To begin, we decompose the 20 residues into astructural basis of 19 rigid-body blocks. Theseblocks, shown in Figure 1(a), share three importantcharacteristics: (a) all atoms in a block are bondedand in the same residue; (b) the bonded atoms areassumed to form a rigid body; (c) all heavy atomsare assumed to be in the same plane. Note thatalthough assumptions (b) and (c) are invalid for theproline ring of block type 19, these assumptions arefound to be reasonable for our potential. Further-more, we ignore the alpha carbon atoms of allresidues except Pro and Gly because we assume thatshielding by bonded atoms around alpha carbonsreduces the influence of those alpha carbons onside-chain packing (our results support this assump-tion). Each residue contains more than one block,but each block appears only once in a single residue.The block compositions of the 20 residue typesare summarized in Figure 1(b). For notationalconsistency, we shall denote residue types (20total) with m and n, block types (19 total) with aand b, block indices with α and β, and atomic indiceswith i and j.OPUS-PSP requires a coordinate system to define
the relative orientation of a pair of blocks. In order tomake it permutationally invariant (i.e. order ofblocks does not matter), we express the relativeorientation of block types a and b using threevariables: two relative direction vectors ra→b andrb→a, and an inter-rotation angle ψab along the axisconnecting the origins of the two blocks in theirrespective molecular reference frames, as illustratedin Figure 2. For a pair of blocks, these three variables(computed in the laboratory reference frame),coupled with the molecular reference frame foreach block, completely define the relative orienta-tion of that pair of blocks, i.e. the axial rotationaround the line linking the origins of the two blocksand the pivot motion around the origin of eachblock. The origin and molecular reference frame foreach block type and the inter-rotation angle arespecified in Supplementary Data.

290 Orientation-dependent All-atom Statistical Potential from Side-chain Packing
Energy function
OPUS-PSP is composed of an orientation-depen-dent packing energy term Eorient and a repulsiveenergy term Erepul:
EPSP ¼ Eorient þ wrepulErepul ð1Þwhere wrepul is a weight parameter optimizedagainst a small subset of decoy sets (see Methods).To calculate the total orientation-dependent pack-
ing energy, Eorient, we first define the packing energyfor a pair of blocks in relative orientation space usingthe Boltzmann formula:
E Vab,a,bð Þ ¼ �kBT logpobsðVab,a,bÞprefðVab,a,bÞ ð2Þ
Here, pobs is the probability of a particularorientation state for block types a and b in contact(“contact probability”; see Methods for the defini-tion of block contact) with respect to all observedcontact states for any block pair extracted from thenon-redundant structure database, pref is the con-tact probability of all possible occurrences of thatstate without packing interactions (the referencestate), Ωab=(ra→b, rb→a, ψab) specifies the relativeorientation of a and b, and kBT is the Boltzmannconstant (set to unity). Thus, to derive the packingenergy function, we must: (a) count the number ofnative contacts for all relative orientations of allpairs of blocks using a non-redundant proteinstructure database (pobs); (b) estimate the referencestate that encapsulates all possible packing orienta-tions (pref). These two steps are explained in detailin Methods.The total orientation-dependent packing energy is
calculated by summing the packing energies of allpairs of blocks in contact (“block contact pairs”)between all pairs of non-consecutive residues:
Eorient ¼Xa,h
yða,hÞEðBðaÞ,BðhÞÞ ð3Þ
Here, δ(α,β) is one when blocks α and β are incontact and zero otherwise, B(α)=a maps block α toits block type a, and Ê(a,b)=n(a,b)E(Ωab,a,b), wheren(a,b) is the average number of pairs of heavy atomsin contact (“atom contact pairs”; two atoms are incontact when their pairwise distance is less than 5 Å)between block types a and b, used as a weightingterm for block size. The weighting term is calculatedby random sampling in the manner of the referencestate probability calculation (see Methods), and it isnecessary because larger blocks contribute moreatom contact pairs and therefore more energy. Incalculating Eorient, we restrict our calculation to side-chain-side-chain and main-chain-side-chain interac-tions only. The main-chain-main-chain hydrogenbonding and other short-range interactions are notconsidered. Thus, we ignore all contact pairs ofblock types {1,5,6,7}, as types 1 and 7 are main-chainfunctional groups and types 5 and 6 have contactinteractions that are highly correlated to those of the
main-chain atoms (unpublished). The sensitivity ofEorient to the features above is analyzed in Results.The repulsive term Erepul is necessary because a
purely orientation-dependent packing function can-not deter steric clashes, and it is defined as:
Erepul ¼Xi,j
ELJði; jÞ ð4Þ
where ELJ(i,j) is a repulsive Lennard-Jones (LJ)potential (no attractive term) for two atoms i and j.Like Eorient, the summation in the LJ term excludesinteractions between pairs of main-chain atomsand between two atoms in the same residue. Notethat OPUS-PSP minimizes over-counting of similarinteractions because Eorient and Erepul are typicallyorthogonal.
Coarse-graining of orientation bins andsymmetry
Given the limited amount of non-homologousprotein data available for building the energyfunction, it is necessary to coarse-grain the orienta-tion space and exploit the symmetry of the 19 blocks.These blocks are classified into nine symmetryclasses that belong to two basic groups: planeshapes (IV, V, VII-IX) and line shapes (I-III, VI), asshown in Figure 1(a). Note that VI is considered aline shape because of the 6-fold axial symmetry ofthe phenyl ring.For each plane-shaped block, the relative direction
with respect to the molecular reference frame of theblock is coarse-grained into 26 bins. These bins areillustrated in Figure 3(a). For each line-shaped block,the cylindrical symmetry permits usage of fivelatitudinal bins as shown in Figure 3(b). Figure 3(c)illustrates the θ and ϕ ranges of each relativedirection bin. The inter-rotation angle is coarse-grained into four bins spanning π/2 radians each.Thus, for two blocks in contact, the maximal
number of bins is 26×4×26=2704. However, inpractice, we combine certain redundant bins basedon the intrinsic molecular symmetry of the blocks,such that the effective number of bins is muchsmaller. For details of this symmetry treatment, seeSupplementary Data.To balance the trade-off between the number of
bins and the available structure data for statisticalanalysis, we find that a choice of 26 directional binsis appropriate for plane-shaped blocks. In a coarserapproximation, we tried six bins (±x, ±y, ±z) and theresults were much worse. Using more than 26 binswould severely deplete the structure data.
Results
Decoy set recognition
The predictive power of OPUS-PSP was tested inbenchmark studies using the popular decoy setcollections: Decoys ‘R’ Us,22 HR,23 Rosetta (and
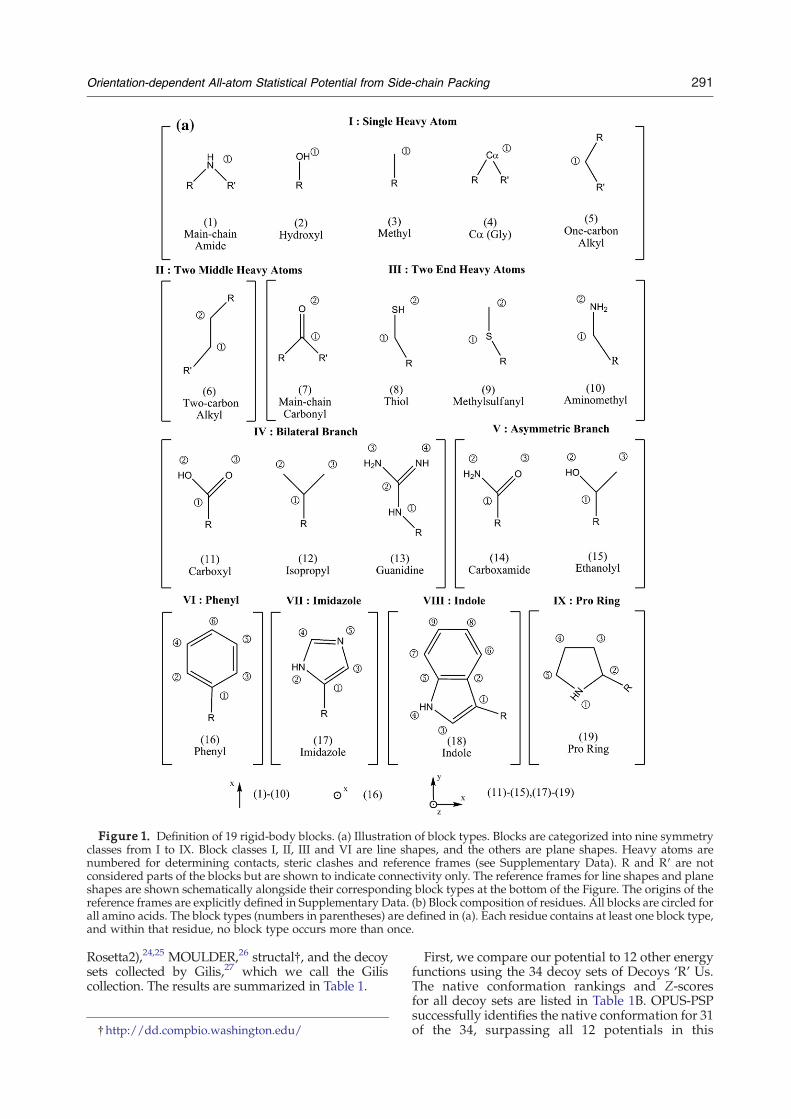
Figure 1. Definition of 19 rigid-body blocks. (a) Illustration of block types. Blocks are categorized into nine symmetryclasses from I to IX. Block classes I, II, III and VI are line shapes, and the others are plane shapes. Heavy atoms arenumbered for determining contacts, steric clashes and reference frames (see Supplementary Data). R and R′ are notconsidered parts of the blocks but are shown to indicate connectivity only. The reference frames for line shapes and planeshapes are shown schematically alongside their corresponding block types at the bottom of the Figure. The origins of thereference frames are explicitly defined in Supplementary Data. (b) Block composition of residues. All blocks are circled forall amino acids. The block types (numbers in parentheses) are defined in (a). Each residue contains at least one block type,and within that residue, no block type occurs more than once.
291Orientation-dependent All-atom Statistical Potential from Side-chain Packing
Rosetta2),24,25 MOULDER,26 structal†, and the decoysets collected by Gilis,27 which we call the Giliscollection. The results are summarized in Table 1.
†http://dd.compbio.washington.edu/
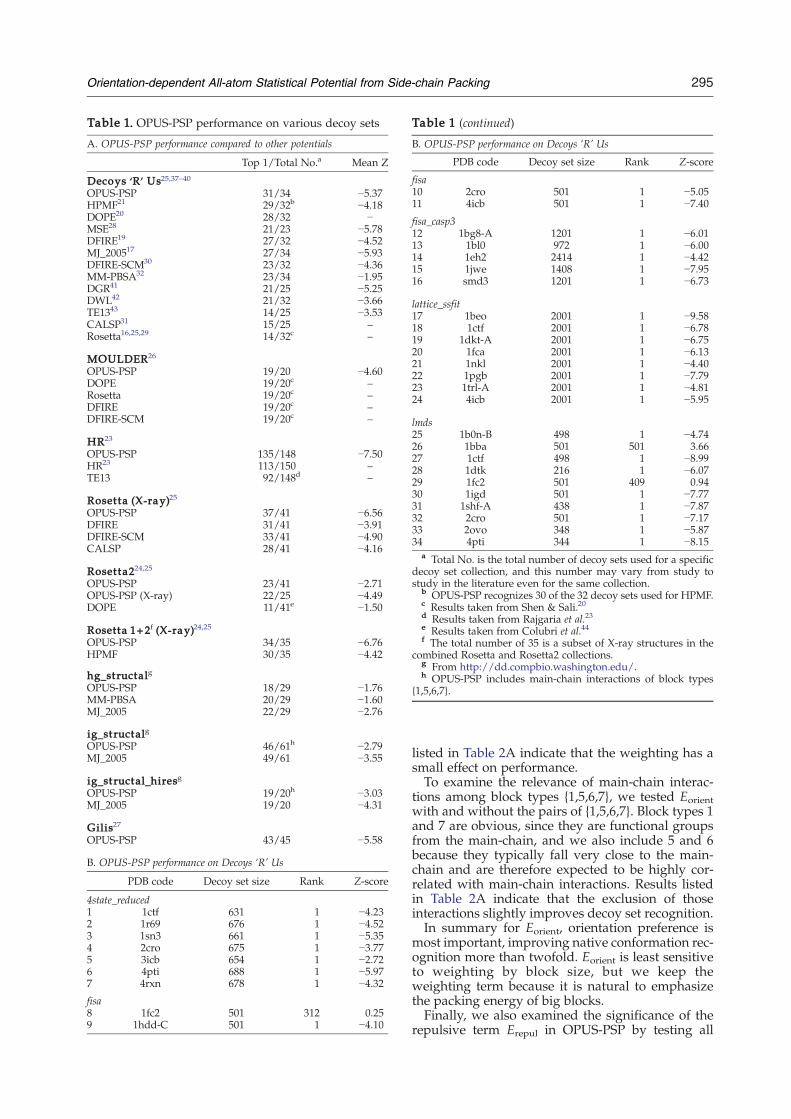
First, we compare our potential to 12 other energyfunctions using the 34 decoy sets of Decoys ‘R’ Us.The native conformation rankings and Z-scoresfor all decoy sets are listed in Table 1B. OPUS-PSPsuccessfully identifies the native conformation for 31of the 34, surpassing all 12 potentials in this
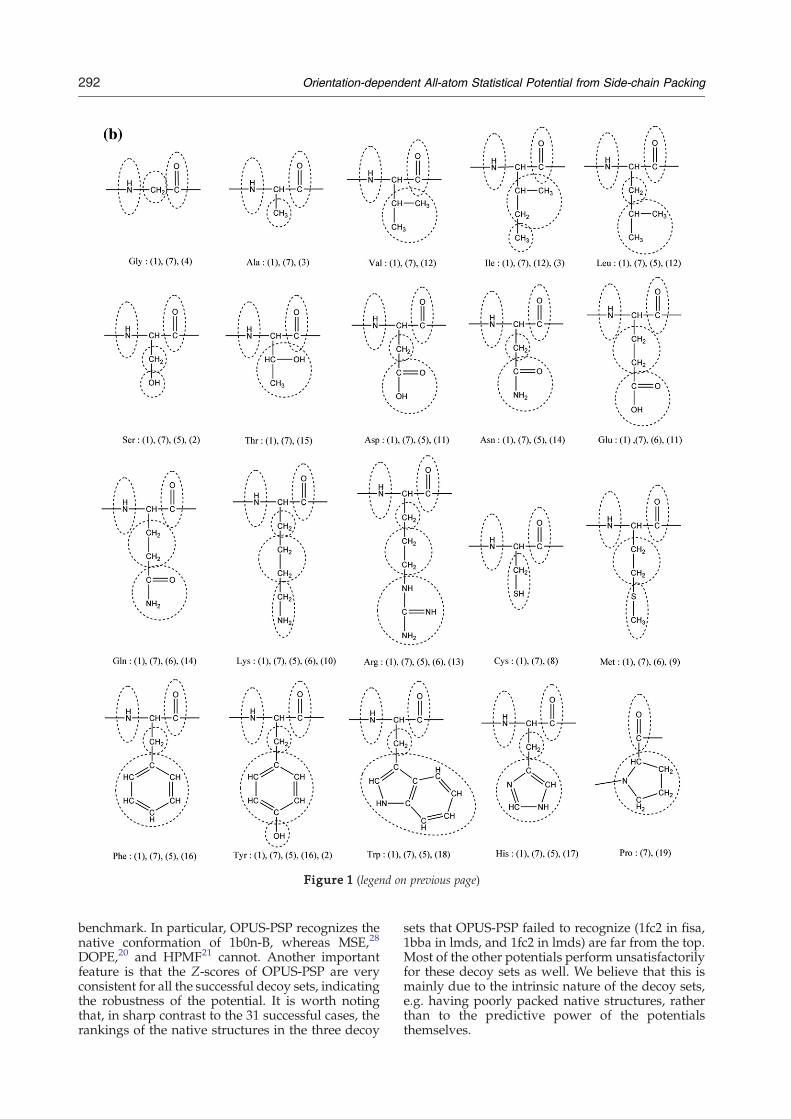
Figure 1 (legend on previous page)
292 Orientation-dependent All-atom Statistical Potential from Side-chain Packing
benchmark. In particular, OPUS-PSP recognizes thenative conformation of 1b0n-B, whereas MSE,28
DOPE,20 and HPMF21 cannot. Another importantfeature is that the Z-scores of OPUS-PSP are veryconsistent for all the successful decoy sets, indicatingthe robustness of the potential. It is worth notingthat, in sharp contrast to the 31 successful cases, therankings of the native structures in the three decoy
sets that OPUS-PSP failed to recognize (1fc2 in fisa,1bba in lmds, and 1fc2 in lmds) are far from the top.Most of the other potentials perform unsatisfactorilyfor these decoy sets as well. We believe that this ismainly due to the intrinsic nature of the decoy sets,e.g. having poorly packed native structures, ratherthan to the predictive power of the potentialsthemselves.

Figure 2. The definition of rela-tive orientation. If block types a andb are in contact, then ra→b and rb→aare the relative direction vectors andψab is the inter-rotation angle alongthe axis connecting the origins oaand ob of the two blocks.
293Orientation-dependent All-atom Statistical Potential from Side-chain Packing
OPUS-PSP's performance on the MOULDERdecoy set collection is similar to that of DOPE,20
Rosetta,16,25,29 DFIRE,19 and DFIRE-SCM.30 Thesingle structure that all of the tested energy func-tions fail to correctly identify is an NMR structure(PDB code 2pna).On the HR decoy set collection, which contains
many high-resolution near-native decoys, OPUS-PSP performs very well. It recognizes 135 of 148native conformations, and of the thirteen missed, sixare still ranked within the top five.On the Rosetta and Rosetta2 decoy set collections,
OPUS-PSP performs very well on structures solvedby X-ray crystallography, whereas it is less success-ful on the broader set that includes structures solvedby NMR spectroscopy.20 This is reasonable giventhat our potential is trained using X-ray structures.OPUS-PSP considerably outperforms DFIRE,19
DFIRE-SCM,30 and CALSP31 on the X-ray structuresubset of the Rosetta decoys. For the more challen-ging Rosetta2 collection, which contains a largenumber of decoys that are biased toward the nativestructure, OPUS-PSP recognizes more than twice thenative conformations that DOPE recognizes.Out of all the benchmarks, OPUS-PSP is out-
performed only on the structal decoy sets by theMM-PBSA32 and MJ_2005 potentials.17 These decoysets contain decoys generated by comparativemodeling of globins and immunoglobulins, ∼60%of which have a Cα RMSD (from the native con-formation) less than 2.5 Å. For the ig_structal andig_structal_hires sets, we find that OPUS-PSP per-forms better if main-chain interactions betweenpairs of block types {1,5,6,7} are also included in thetotal energy calculation (see Theoretical Framework).The potential with the closest performance to
OPUS-PSP is the very recent hydrophobic potentialof mean force (HPMF) model.21 OPUS-PSP outper-forms HPMF in the number of native conformationsidentified and in mean Z-score for both Decoys ’R’
Us and the combined set of X-ray structures fromthe Rosetta and Rosetta2 collections (“Rosetta1+2”in Table 1A).21 Moreover, HPMF contains manyenergy parameters that are trained from a subset ofdecoys, whereas OPUS-PSP has only one adjustableweight for two nearly independent terms, and thusOPUS-PSP's performance is less dependent uponthe choice of training set.
Sensitivity analysis of energy function features
We conducted a sensitivity analysis to identify thefeatures of OPUS-PSP that are most important forcorrectly identifying native conformations withina set of decoys. First, we tested the orientation-dependent packing energy term Eorient of OPUS-PSPin three aspects: (a) orientation preference; (b)weighting by block size; (c) main-chain interactionsamong blocks types {1,5,6,7}. We generated energyfunctions for all combinations of these three aspects,and we tested the resulting eight energy functions(including the energy function described by equa-tion (3)) on the 34 decoy sets in Decoys ‘R’ Us.To test the importance of orientation preference
for two block types a and b that are in contact, wecompared Eorient to a simple contact energy functionwith no preference for orientation:
E a,bð Þ ¼ �kBTlogpobsða,bÞprefða,bÞ
Here, pobs(a,b)=Nobs(a,b)/∑a,b Nobs(a,b) and pref
(a,b)≈χ(a)χ(b), where χ(a)=Nobs (a)/∑a Nobs (a) is
the mole fraction of block type a. Results listed inTable 2A indicate the overwhelming importance ofthe orientation preference.To test the effect of weighting the pairwise
packing energies by block size, we compared thedecoy recognition results with and without the size-weighting term n(a,b) from equation (3). Results
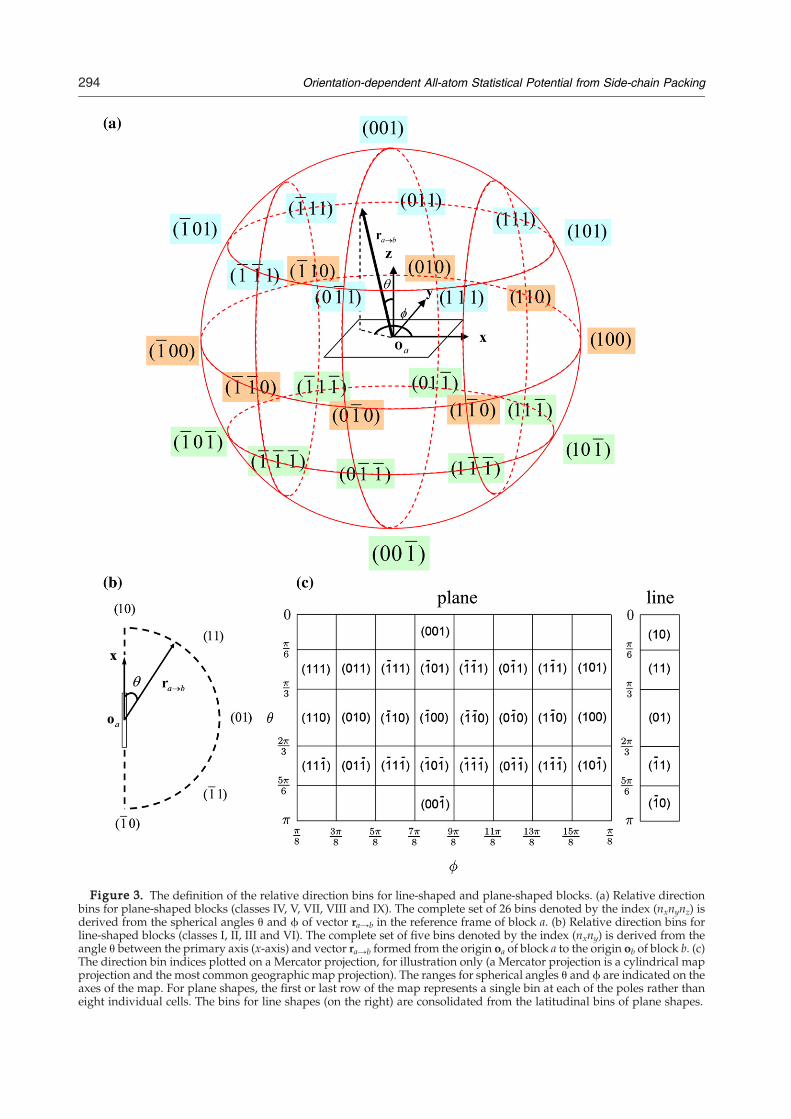
Figure 3. The definition of the relative direction bins for line-shaped and plane-shaped blocks. (a) Relative directionbins for plane-shaped blocks (classes IV, V, VII, VIII and IX). The complete set of 26 bins denoted by the index (nxnynz) isderived from the spherical angles θ and ϕ of vector ra→b in the reference frame of block a. (b) Relative direction bins forline-shaped blocks (classes I, II, III and VI). The complete set of five bins denoted by the index (nxny) is derived from theangle θ between the primary axis (x-axis) and vector ra→b formed from the origin oa of block a to the origin ob of block b. (c)The direction bin indices plotted on a Mercator projection, for illustration only (a Mercator projection is a cylindrical mapprojection and the most common geographic map projection). The ranges for spherical angles θ and ϕ are indicated on theaxes of the map. For plane shapes, the first or last row of the map represents a single bin at each of the poles rather thaneight individual cells. The bins for line shapes (on the right) are consolidated from the latitudinal bins of plane shapes.
294 Orientation-dependent All-atom Statistical Potential from Side-chain Packing
able 1. OPUS-PSP performance on various decoy sets
. OPUS-PSP performance compared to other potentials
Top 1/Total No.a Mean Z
ecoys ‘R’ Us25,37–40PUS-PSP 31/34 −5.37PMF21 29/32b −4.18OPE20 28/32 −SE28 21/23 −5.78FIRE19 27/32 −4.52J_200517 27/34 −5.93FIRE-SCM30 23/32 −4.36M-PBSA32 23/34 −1.95GR41 21/25 −5.25WL42 21/32 −3.66E1343 14/25 −3.53ALSP31 15/25 –osetta16,25,29 14/32c –
OULDER26
PUS-PSP 19/20 −4.60OPE 19/20c –osetta 19/20c –FIRE 19/20c –FIRE-SCM 19/20c –
R23
PUS-PSP 135/148 −7.50R23 113/150 –E13 92/148d –
osetta (X-ray)25PUS-PSP 37/41 −6.56FIRE 31/41 −3.91FIRE-SCM 33/41 −4.90ALSP 28/41 −4.16
osetta224,25PUS-PSP 23/41 −2.71PUS-PSP (X-ray) 22/25 −4.49OPE 11/41e −1.50
osetta 1+2f (X-ray)24,25PUS-PSP 34/35 −6.76PMF 30/35 −4.42
g_structalgPUS-PSP 18/29 −1.76M-PBSA 20/29 −1.60J_2005 22/29 −2.76
g_structalgPUS-PSP 46/61h −2.79J_2005 49/61 −3.55
g_structal_hiresgPUS-PSP 19/20h −3.03J_2005 19/20 −4.31
ilis27PUS-PSP 43/45 −5.58
. OPUS-PSP performance on Decoys ‘R’ Us
PDB code Decoy set size Rank Z-score
state_reduced1ctf 631 1 −4.231r69 676 1 −4.521sn3 661 1 −5.352cro 675 1 −3.773icb 654 1 −2.724pti 688 1 −5.974rxn 678 1 −4.32
sa1fc2 501 312 0.25
1hdd-C 501 1 −4.10
B. OPUS-PSP performance on Decoys ‘R’ Us
PDB code Decoy set size Rank Z-score
fisa10 2cro 501 1 −5.0511 4icb 501 1 −7.40
fisa_casp312 1bg8-A 1201 1 −6.0113 1bl0 972 1 −6.0014 1eh2 2414 1 −4.4215 1jwe 1408 1 −7.9516 smd3 1201 1 −6.73
lattice_ssfit17 1beo 2001 1 −9.5818 1ctf 2001 1 −6.7819 1dkt-A 2001 1 −6.7520 1fca 2001 1 −6.1321 1nkl 2001 1 −4.4022 1pgb 2001 1 −7.7923 1trl-A 2001 1 −4.8124 4icb 2001 1 −5.95
lmds25 1b0n-B 498 1 −4.7426 1bba 501 501 3.6627 1ctf 498 1 −8.9928 1dtk 216 1 −6.0729 1fc2 501 409 0.9430 1igd 501 1 −7.7731 1shf-A 438 1 −7.8732 2cro 501 1 −7.1733 2ovo 348 1 −5.8734 4pti 344 1 −8.15
a Total No. is the total number of decoy sets used for a specificdecoy set collection, and this number may vary from study tostudy in the literature even for the same collection.
b OPUS-PSP recognizes 30 of the 32 decoy sets used for HPMF.c Results taken from Shen & Sali.20d Results taken from Rajgaria et al.23e Results taken from Colubri et al.44f The total number of 35 is a subset of X-ray structures in the
combined Rosetta and Rosetta2 collections.g From http://dd.compbio.washington.edu/.h OPUS-PSP includes main-chain interactions of block types
{1,5,6,7}.
Table 1 (continued)
295Orientation-dependent All-atom Statistical Potential from Side-chain Packing
T
A
DOHDMDMDMDDTCR
MODRDD
HOHT
RODDC
ROOD
ROH
hOMM
iOM
iOM
GO
B
41234567
fi89
(continued on next page)
listed in Table 2A indicate that the weighting has asmall effect on performance.To examine the relevance of main-chain interac-
tions among block types {1,5,6,7}, we tested Eorientwith and without the pairs of {1,5,6,7}. Block types 1and 7 are obvious, since they are functional groupsfrom the main-chain, and we also include 5 and 6because they typically fall very close to the main-chain and are therefore expected to be highly cor-related with main-chain interactions. Results listedin Table 2A indicate that the exclusion of thoseinteractions slightly improves decoy set recognition.In summary for Eorient, orientation preference is
most important, improving native conformation rec-ognition more than twofold. Eorient is least sensitiveto weighting by block size, but we keep theweighting term because it is natural to emphasizethe packing energy of big blocks.Finally, we also examined the significance of the
repulsive term Erepul in OPUS-PSP by testing all

Table 2. Sensitivity analysis of OPUS-PSP features
A. Sensitivity analysis of Eorient features using Decoy ‘R’ Us
Weight {1,5,6,7}
Orientation preference
+ –
+ – 30 (−5.36) 12 (−2.43)– – 30 (−5.31) 13 (−2.46)+ + 24 (−4.63) 14 (−2.27)– + 24 (−4.14) 12 (−1.81)
B. The comparison of OPUS-PSP with and without the repulsive termErepul using various decoy sets
Decoy sets
Repulsive term
– +
Decoy ’R’ Us 30/34 (−5.36) 31/34 (−5.37)Rosetta 45/92 (−3.25) 55/92 (−4.17)Rosetta (X-ray) 33/41 (−5.25) 37/41 (−6.56)Rosetta2 17/41 (−2.43) 23/41 (−2.71)Rosetta2 (X-ray) 16/25 (−3.93) 22/25 (−4.49)Rosetta 1+2 (X-ray) 30/35 (−5.75) 34/35 (−6.76)MOULDER 19/20 (−4.40) 19/20 (−4.60)HR 142/148 (−7.10) 135/148 (−7.50)hg_structal 18/29 (−1.91) 18/29 (−1.76)ig_structal 49/61 (−2.92) 46/61 (−2.79)ig_structal_hires 20/20 (−3.09) 19/20 (−3.03)Gilis Collection 38/45 (−5.19) 43/45 (−5.58)
The notation +/− denotes inclusion/exclusion of a particularfeature. The column labeled {1,5,6,7} represents the interactionsamong block types {1,5,6,7}. The integer values are the number ofnative conformations ranked first by OPUS-PSP (i.e. correctlyrecognized), and the mean Z-scores are in parentheses.Note that the version of OPUS-PSP with Erepul is used for Table 1.
296 Orientation-dependent All-atom Statistical Potential from Side-chain Packing
decoy sets with and without this term. The resultsare shown in Table 2B. With Erepul, the performanceof OPUS-PSP increases for all decoy sets except forthe HR and structal decoy sets. In the case of HR,OPUS-PSP without Erepul recognizes seven morenative conformations than with Erepul, but of thoseseven missed with Erepul, six native conformationsare still ranked among the top five. In the ig_structal_hires set, OPUS-PSP without Erepul outper-forms all other potentials, recognizing all 20 nativeconformations.
Table 3. Examples of the most-favored contact patterns
Interaction Contact pattern Notes Inter
π–π 17–17i Offset orthogonal Hydro17–17ii
Cation-π 18–18i Offset orthogonal18–18ii Offset orthogonal17–18i Offset parallel17–18ii Offset parallel16–18i Orthogonal16–18ii Orthogonal16–16 Orthogonal Elect13–18 Parallel13–16 Parallel
Cyclic side-chain 18–19 Offset orthogonal16–19 Parallel
Disulfide bond 8–8i8–8ii
The indices of contact block pairs (illustrated in Figure 4) are shown i
Most-favored interaction patterns
Here we found that the packing between certainblock types present clear patterns consistent with aprevious analysis of anisotropic side-chainpacking.16 In Table 3 and Figure 4, we show the 30most-probable and unique packing patternsextracted from the non-redundant structure data-base. They are presented in terms of their chemicalnature of interaction, e.g. hydrophobic, electrostatic,π-π and cation-π interactions.Several universal patterns may be identified from
these examples. For instance, aromatic side-chainstend to form certain patterns of π-π and cation-πinteractions. The packing patterns for hydrophobicinteractions favor contact at the branching pointsmore than at the tips of the branches, as in cases 12-12ii, 12-15, and 15-15. The presence of nitrogen andoxygen atoms also noticeably influences the packingpatterns.
Discussion
Here we report an orientation-dependent statis-tical all-atom potential. Amino acid residues areconstructed from a basis set of 19 rigid-body blocks,and the potential is then established by statisticalanalysis of the orientation-specific packing patternsamong those blocks in a non-redundant structuraldatabase. The purpose of this scheme is to capturethe essential details of side-chain flexibility andpacking orientation. Tests of OPUS-PSP on com-monly used decoy sets show that it significantlyoutperforms most of the knowledge-based poten-tials in the literature, especially the recent DFIRE,19
DOPE20/DOPE-Cβ45, and HPMF21 potentials.OPUS-PSP is expected to be particularly useful inmodeling side-chain conformations, one of the mostimportant and difficult steps in high-accuracy pro-tein structure prediction and refinement.OPUS-PSP has many advantages in terms of its
coarse-graining scheme, interaction parameters andreference state. In terms of OPUS-PSP's coarse-
action Contact pattern Notes
phobic 15–15 Contact at rear, two Os apart12–1912–12i Offset parallel12–12ii Contact at rear10–18 (10) rear in contact w/ (18) side12–18 Offset parallel12–15 Contact at rear12–16 Parallel
rostatic 11–13i (13) side in contact w/ (11) head11–13ii (13) head points to (11) head14–18 N–N15–19 Parallel14–15 O–O2–17 Hydrogen bond10–11 N points to midpoint of two Os
n the column Contact pattern.

Figure 4. Examples of most-probable packing patterns (30 in total). For each pattern, the numbers represent theindices of the block types in contact, and the lowercase Roman numerals specify different contact patterns for a given pairof block types. For most block pairs, the consistent clustering of contact patterns provides partial validation of OPUS-PSP.Note that examples such as 17–17i and 12–12i have multiple clusters due to symmetry. Table 3 briefly summarizes thecontact patterns illustrated here.
297Orientation-dependent All-atom Statistical Potential from Side-chain Packing
graining scheme, the unique 19 rigid-body basis setcaptures the essential elements of orientation depen-dence in molecular packing interactions. For exam-ple, OPUS-PSP can sense subtle changes in even asingle rotamer conformation because the orienta-tions of the constituent blocks are significantlychanged as a consequence.In terms of the orientational parameters, a key
trade-off exists between the number of parametersand their sensitivity in describing the orientation.Choosing too many parameters risks over-fitting the
limited data, yet the packing pattern sensitivity of thepotential suffers if the orientational parameters aretoo coarse. To achieve a better balance,we extensivelyuse block symmetry to consolidate redundant bins.We also employ Bayesian statistics to compensatefor the cases in which the available data are sparse(see Methods and Supplemental Data).The reference state is also important in con-
structing a statistical potential. In OPUS-PSP, thereference state is calculated by random sampling ofblocks (see Methods), so the excluded-volume

298 Orientation-dependent All-atom Statistical Potential from Side-chain Packing
effects and block-specific geometries are naturallytaken into account. To the best of our knowledge,this is the first case that the geometry of interactinggroups is explicitly considered in constructing thereference state.We want to emphasize that OPUS-PSP is built
almost exclusively on the orientation-specific pack-ing interactions of side-chains. Only a repulsiveenergy term is added to prevent steric clash. In fact,we found that the potential without the repulsiveterm performs very well by itself in decoy setrecognition. The repulsive term is required for themore general purpose of structure prediction.No main-chain hydrogen bonding interactions are
explicitly considered in OPUS-PSP. One reason isthat the rigid-body basis set used for constructingOPUS-PSP is not suited for optimizing main-chainhydrogen bonding interactions. Another reason isthat main-chain hydrogen bonding interactionsalone are not very specific in terms of pairingbetween two residues, i.e. any pair of residues canform main-chain hydrogen bonds, while not allpairs of side-chains can pack against each otherfavorably. As clearly demonstrated in our results,OPUS-PSP, which excludes the interactions amongmain-chain atoms, performs very well.In addition, no explicit solvation terms are
included in OPUS-PSP. Although the side-chainpacking interactions are not completely orthogonalto the solvation effect, e.g. hydrophobic side-chainsprefer to pack together, it is advantageous toexplicitly avoid the solvation effect if doing sodoes not impair performance. One reason is thatOPUS-PSP is statistically formulated from a struc-ture database of soluble proteins. If the solvationeffect is explicitly included, the potential would beless reliable for systems such as membrane proteinswith a very different solvation environment. Weexpect the current form of OPUS-PSP to performwell in modeling membrane proteins, as their side-chain packing interactions should be very similar tothose of soluble proteins.
Methods
As already outlined in Theoretical Framework, thepacking energy function requires: (a) the propor-tions of observed native block-pair contacts for allrelative orientations; (b) the reference state thatencapsulates the idealized preference of all possiblepacking orientations. In addition, we apply Baye-sian inference to estimate the statistics of block-pairorientations that occur infrequently in the proteinstructure database.
Counting pair contacts from a non-homologousstructure database
To determine pobs, the observed fraction ofoccurrences of a particular pair-orientation statewith respect to all observed states (see equation (2)),we tally the number of contacts Nobs between all
pairs of blocks at all relative orientations that existwithin a representative, non-homologous structuredatabase, i.e. pobs(Ωab,a,b)=N
obs(Ωab,a,b)/Ntotalobs , and
Ntotalobs =∑a,b Nobs(Ωab,a,b). Contact occurs when any
heavy atom from one block falls within a 5Å radiusof any heavy atom from another block. Our non-homologous structure database of 3211 proteinchains is generated from the PISCES server33using a percent identity cutoff of 40%, a resolutioncutoff of 1.8Å, and an R-factor cutoff of 0.25. The40% identity cutoff is a looser criterion than those ofmost other knowledge-based potentials in theliterature, which increases the available set ofstructures and thereby improves the statistics. Weargue that side-chain packing preference is lesssensitive to the percent identity. In addition, our testresults on decoy set recognition are very similarwhen comparing our current version and a versionbuilt from the structure database with 30% identitycutoff.
Calculating the reference state probability
The reference state probability pref(Ωab,a,b) inequation (2) is calculated as the product of thecontact-pair-orientation statistic pref(Ωab|a,b) andthe contact probability pref(a,b):
prefðVab,a,bÞ ¼ prefðVabja,bÞprefða,bÞ ð5Þ
We use random sampling to compute pref(Ωab|a,b), since no obvious analytical solution exists. Twoblocks are randomly placed in space withoutconsideration for chemical interactions. Block pairsare discarded if they are out of contact range (N5Å)or clashing (b2� 2:0=
ffiffiffi26
pÅ, where the atomic radius
of a carbon atom is 2.0 Å). Otherwise, the relativeorientation of the block pair is determined andtabulated. A large number of such contact pairs (anaverage of 105 counts/bin) are generated to mini-mize the sampling error.The calculation of the block contact probability
pref(a,b) is not straightforward, in contrast to theestimation of contact probability for two residues ortwo atoms, which is intensively discussed in theliterature.13,19,34,35 Typically, residues are assumedto be of similar size, and thus the quasi-chemicalapproximation based on mole fractions of theresidues is reasonable.34 We use this approximationas the starting point for estimating the contactprobability for two blocks. First, let the mole fractionof block type a be:
mðaÞ ¼Xm
mðmÞyða,mÞ,
where χ(m)=Nobs(m)/Ntotalobs is the mole fraction of
residue type m, δ(a,m) is one if a is in m and zerootherwise, and the summation is over all 20 residuetypes. To represent the connectivity of the blockswithin a residue, we assume that each block type ain a residue type m has an equal and constantprobability ξ of being in contact with any block type

299Orientation-dependent All-atom Statistical Potential from Side-chain Packing
b of residue type n if m and n are in contact. Hence,from the quasi-chemical approximation:
prefða,bÞcXm,n
mðmÞmðnÞðsdyða,mÞyðb,nÞÞ ¼ smðaÞmðbÞ
Next, since ∑a,b pref(a,b)=∑a,b χ(a)χ(b)=1, then ξmust be unity, and:
prefða; bÞcmðaÞmðbÞ ð6ÞEquation (6) implies that the quasi-chemical ap-proximation is appropriate for estimating thecontact probabilities of two blocks. Note that theequation above does not explicitly consider thesize differences of residues and blocks. Presumably,larger blocks or residues have more contacts thansmaller ones. However, the energy functions de-rived from reference states that incorporate contactnumber are found to be less powerful than thosethat do not.34 Our current study also supports thisconclusion, as our energy function using the quasi-chemical approximation so far produces the bestresults.
Rare data estimation
Given the relatively small size (3211 chains) of thenon-homologous protein dataset used to build ourpotential, many pair-orientations occur too infre-quently for the data to be statistically significant.Thus, we use Bayesian statistics to solve this raredata problem, the details of which are described inSupplementary Data.
The repulsive Lennard-Jones (LJ) potential term
Similar to that used in the literature,36 the re-pulsive LJ potential function for two atoms i and jtakes the form:
ELJði; jÞ ¼49:69� 40:06dij*; dij*a½0; 1=1:33�
eij½ðdij*Þ�12 � 2ðdij*Þ�6�; dij*að1=1:33; 1=1:12�0; dij*að1=1:12;lÞ
8><>:
ð7Þwhere dij
* =dij/aij, dij is the distance between atoms iand j, aij=ai+aj, eij ¼ ffiffiffiffiffiffieiej
p , and ai, aj and ei, ej areatomic radii and well depths for the LJ calcula-tions, respectively. Thus, ELJ (i,j) is linear in theregion of severe steric clash, and it is zero in (i.e.excludes) the attractive region of the LJ potential. Inorder to allow hydrogen and disulfide bonds, aij isspecifically chosen for atoms involving hydrogen ordisulfide bonds. The detailed LJ parameters aredescribed in Supplementary Data.The total LJ energy is calculated by summing over
all possible pairs of heavy atoms except in thefollowing three cases: (a) pairs of main-chain heavyatoms including Cβ (the Cβ position is largelydetermined by the main-chain); (b) two atoms inthe same residue; (c) Pro Cδ and C of the previousresidue, as they are separated by only two bonds.
Weight optimization for the combined energyfunction
To determine the optimal weight parameter wrepul,we ran multiple decoy recognition trials using wrepulranging from 0.0 to 2.0 at 0.1 intervals. The trainingset includes all seven decoy sets in the 4state_reduced subset of Decoys ‘R’ Us and the 13 decoysets in the Gilis collection that are not included inother benchmarks in this work. The decoys in4state_reduced have fewer steric clashes than thedecoys in the Gillis collection. Thus, we can use bothcollections in the optimization to find a balancebetween orientation-dependence and steric-clash-avoidance. From our results, wrepul=1.2 is the opti-mal weight, as it produces the best native conforma-tion rankings and Z-scores. In any case, the decoyrecognition performance of OPUS-PSP is not sensi-tive to the weight value.As an added benefit, this weight optimization is
fairly straightforward with few disadvantages. First,the risk of over-fitting is reduced because there isonly one weight parameter to optimize. Second,Eorient and Erepul are nearly independent, such thatthe combined energy is unlikely to suffer from over-counting. Third, our training set contains decoysgenerated from various methods, thereby balancingthe contributions of the energy terms. This weightoptimizationmethod, which is very similar to that ofHPMF, is supported by our tests of OPUS-PSP onthe other decoy sets.
Acknowledgements
J.M. acknowledges financial support by a grantfrom the National Institutes of Health (R01-GM067801) and a grant from the National ScienceFoundation CAREER Award (MCB-0237796). M.L.is partially supported by a pre-doctoral fellowshipfrom the Keck Center for Interdisciplinary Bio-science Training of the Gulf Coast Consortia. A.D.is supported by a pre-doctoral fellowship from theKeck Center through the National Library ofMedicine Computational Biology and MedicineTraining Program (NLM grant no. 5T15LM07093).
Supplementary Data
Supplementary data associated with this articlecan be found, in the online version, at doi:10.1016/j.jmb.2007.11.033
References
1. MacKerell, A. D., Bashford, D., Jr, Bellott, M.,Dunbrack, R. L., Jr, Evanseck, J. D., Field, M. J. et al.(1998). All-atom empirical potential for molecularmodeling and dynamics studies of proteins. J. Phys.Chem. B102, 3586–3616.

300 Orientation-dependent All-atom Statistical Potential from Side-chain Packing
2. Skolnick, J. (2006). In quest of an empirical potentialfor protein structure prediction. Curr. Opin. Struct.Biol. 16, 166–171.
3. Bradley, P., Malmstrom, L., Qian, B., Schonbrun, J.,Chivian, D., Kim, D. E. et al. (2005). Free modelingwith Rosetta in CASP6. Proteins: Struct. Funct. Genet.61(Suppl. 7), 128–134.
4. Sippl, M. J. (1995). Knowledge-based potentials forproteins. Curr. Opin. Struct. Biol. 5, 229–235.
5. Jernigan, R. L. & Bahar, I. (1996). Structure-derivedpotentials and protein simulations. Curr. Opin. Struct.Biol. 6, 195–209.
6. Moult, J. (1997). Comparison of database potentialsand molecular mechanics force fields. Curr. Opin.Struct. Biol. 7, 194–199.
7. Lazaridis, T. & Karplus, M. (2000). Effective energyfunctions for protein structure prediction. Curr. Opin.Struct. Biol. 10, 139–145.
8. Gohlke, H. & Klebe, G. (2001). Statistical potentialsand scoring functions applied to protein-ligand bind-ing. Curr. Opin. Struct. Biol. 11, 231–235.
9. Russ, W. P. & Ranganathan, R. (2002). Knowledge-based potential functions in protein design. Curr.Opin. Struct. Biol. 12, 447–452.
10. Buchete, N. V., Straub, J. E. & Thirumalai, D. (2004).Development of novel statistical potentials for proteinfold recognition. Curr. Opin. Struct. Biol. 14, 225–232.
11. Poole, A. M. & Ranganathan, R. (2006). Knowledge-based potentials in protein design. Curr. Opin. Struct.Biol. 16, 508–513.
12. Zhou, Y., Zhou, H., Zhang, C. & Liu, S. (2006). Whatis a desirable statistical energy function for proteinsand how can it be obtained? Cell Biochem. Biophys. 46,165–174.
13. Chen, W.W. & Shakhnovich, E. I. (2005). Lessons fromthe design of a novel atomic potential for proteinfolding. Protein Sci. 14, 1741–1752.
14. Buchete, N. V., Straub, J. E. & Thirumalai, D. (2004).Orientational potentials extracted from protein struc-tures improve native fold recognition. Protein Sci. 13,862–874.
15. Mukherjee, A., Bhimalapuram, P. & Bagchi, B. (2005).Orientation-dependent potential of mean force forprotein folding. J. Chem. Phys. 123, 014901.
16. Misura, K. M., Morozov, A. V. & Baker, D. (2004).Analysis of anisotropic side-chain packing in proteinsand application to high-resolution structure predic-tion. J. Mol. Biol. 342, 651–664.
17. Miyazawa, S. & Jernigan, R. L. (2005). How effectivefor fold recognition is a potential of mean force thatincludes relative orientations between contactingresidues in proteins? J. Chem. Phys. 122, 024901.
18. Wu, Y., Lu, M., Chen, M., Li, J. & Ma, J. (2007). OPUS-Ca: a knowledge-based potential function requiringonly Cα positions. Protein Sci. 16, 1449–1463.
19. Zhou, H. & Zhou, Y. (2002). Distance-scaled, finiteideal-gas reference state improves structure-derivedpotentials of mean force for structure selection andstability prediction. Protein Sci. 11, 2714–2726.
20. Shen, M. Y. & Sali, A. (2006). Statistical potential forassessment and prediction of protein structures.Protein Sci. 15, 2507–2524.
21. Lin, M. S., Fawzi, N. L. & Head-Gordon, T. (2007).Hydrophobic potential of mean force as a solvationfunction for protein structure prediction. Structure, 15,727–740.
22. Samudrala, R. & Levitt, M. (2000). Decoys ‘R’ Us: adatabase of incorrect conformations to improve pro-tein structure prediction. Protein Sci. 9, 1399–1401.
23. Rajgaria, R., McAllister, S. R. & Floudas, C. A. (2006).A novel high resolution Calpha–Calpha distancedependent force field based on a high quality decoyset. Proteins: Struct. Funct. Genet. 65, 726–741.
24. Tsai, J., Bonneau, R., Morozov, A. V., Kuhlman, B.,Rohl, C. A. & Baker, D. (2003). An improved proteindecoy set for testing energy functions for proteinstructure prediction. Proteins: Struct. Funct. Genet. 53,76–87.
25. Simons, K. T., Kooperberg, C., Huang, E. & Baker, D.(1997). Assembly of protein tertiary structures fromfragments with similar local sequences using simu-lated annealing and Bayesian scoring functions. J. Mol.Biol. 268, 209–225.
26. John, B. & Sali, A. (2003). Comparative protein struc-ture modeling by iterative alignment, model buildingand model assessment. Nucl. Acids Res. 31, 3982–3992.
27. Gilis, D. (2004). Protein decoy sets for evaluatingenergy functions. J. Biomol. Struct. Dynam. 21, 725–736.
28. McConkey, B. J., Sobolev, V. & Edelman, M. (2003).Discrimination of native protein structures usingatom-atom contact scoring. Proc. Natl Acad. Sci. USA,100, 3215–3220.
29. Simons, K. T., Ruczinski, I., Kooperberg, C., Fox, B. A.,Bystroff, C. & Baker, D. (1999). Improved recognition ofnative-like protein structures using a combination ofsequence-dependent and sequence-independent fea-tures of proteins.Proteins: Struct. Funct. Genet. 34, 82–95.
30. Zhang, C., Liu, S., Zhou, H. & Zhou, Y. (2004). Anaccurate, residue-level, pair potential of mean forcefor folding and binding based on the distance-scaled,ideal-gas reference state. Protein Sci. 13, 400–411.
31. Zhang, J., Chen, R. & Liang, J. (2006). Empiricalpotential function for simplified protein models: com-bining contact and local sequence-structure descrip-tors. Proteins: Struct. Funct. Genet. 63, 949–960.
32. Lee, M. C., Yang, R. & Duan, Y. (2005). Comparisonbetween Generalized-Born and Poisson-Boltzmannmethods in physics-based scoring functions forprotein structure prediction. J. Mol. Model, 12, 101–110.
33. Wang, G. & Dunbrack, R. L., Jr (2003). PISCES: aprotein sequence culling server. Bioinformatics, 19,1589–1591.
34. Skolnick, J., Jaroszewski, L., Kolinski, A. & Godzik, A.(1997). Derivation and testing of pair potentials forprotein folding. When is the quasichemical approx-imation correct? Protein Sci. 6, 676–688.
35. Miyazawa, S. & Jernigan, R. L. (1996). Residue-residuepotentials with a favorable contact pair term and anunfavorable high packing density term, for simulationand threading. J. Mol. Biol. 256, 623–644.
36. Kuhlman, B., Dantas, G., Ireton, G. C., Varani, G.,Stoddard, B. L. & Baker, D. (2003). Design of a novelglobular protein fold with atomic-level accuracy.Science, 302, 1364–1368.
37. Park, B. & Levitt, M. (1996). Energy functions thatdiscriminate X-ray and near native folds from well-constructed decoys. J. Mol. Biol. 258, 367–392.
38. Samudrala, R., Xia, Y., Levitt, M. & Huang, E. S.(1999). A combined approach for ab initio construc-tion of low resolution protein tertiary structures fromsequence. Pac. Symp. Biocomput. 505–516.
39. Xia, Y., Huang, E. S., Levitt, M. & Samudrala, R. (2000).Ab initio construction of protein tertiary structuresusing a hierarchical approach. J. Mol. Biol. 300, 171–185.
40. Keasar, C. & Levitt, M. (2003). A novel approach todecoy set generation: designing a physical energyfunction having local minima with native structurecharacteristics. J. Mol. Biol. 329, 159–174.

301Orientation-dependent All-atom Statistical Potential from Side-chain Packing
41. Dehouck, Y., Gilis, D. & Rooman, M. (2006). A newgeneration of statistical potentials for proteins. Biophys.J. 90, 4010–4017.
42. Dong, Q., Wang, X. & Lin, L. (2006). Novel knowl-edge-based mean force potential at the profile level.BMC Bioinformat. 7, 324.
43. Tobi, D. & Elber, R. (2000). Distance-dependent, pairpotential for protein folding: results from linearoptimization. Proteins: Struct. Funct. Genet. 41, 40–46.
44. Colubri, A., Jha, A. K., Shen, M. Y., Sali, A., Berry, R. S.,Sosnick, T. R. & Freed, K. F. (2006). Minimalistrepresentations and the importance of nearest neigh-bor effects in protein folding simulations. J. Mol. Biol.363, 835–857.
45. Fitzgerald, J. E., Jha, A. K., Colubri, A., Sosnick, T. R. &Freed, K. F. (2007). Reduced Cβ statistical potentialscan outperform all-atom potentials in decoy identifi-cation. Protein Sci. 16, 2123–2139.




















