
Neuropsychiatric Disease and Treatment - MedSpec Publishing
56
dove medical press Neuropsychiatric Disease and Treatment (South African Excerpts Edition) volume 5 · number 4 · 2011 2IÀFLDO -RXUQDO RI WKH International Neuropsychiatric Association
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Neuropsychiatric Disease and Treatment - MedSpec Publishing

dove medical press
Neuropsychiatric Diseaseand Treatment
(South African Excerpts Edition)
volume 5 · number 4 · 2011
International Neuropsychiatric Association
Neuro vo5 no4 2011.indd 1 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 2 2011/10/28 10:13 AM

PublisherReni RouncivellTel: (012) 661 3294 / Fax: 086 561 5122Cell: 082 441 [email protected]
AdvertisingSue-Anne SmookCell: 082 856 [email protected]
Lelani AdendorffCell: 079 512 [email protected]
Subscriptions & AccountsElizabeth VersteegCell: 072 189 [email protected]
Private Bag X1036, LytteltonSouth Africa 0140
FOR ADDRESS CHANGES PLEASE CONTACT:
TRADE ENQUIRIES: Cally Lamprecht [email protected]
ALL OTHERS: Linda vanderberg [email protected]
of theInternational
Neuropsychiatric Association
Neuropsychiatric Disease
and Treatment(South African Excerpts Edition)
EDITORIAL
This quarter’s journal contains articles on a variety of topics.
Treatment of chronic enduring serious mental disorders such as schizophrenia
remains unsatisfactory, with medications that are partially effective and that have
considerable unpleasant and even dangerous side-effects. Clozapine has long been
held to be the drug of choice in “treatment-resistant” schizophrenia. But what can
be done for people with schizophrenia who do not respond to clozapine, or who
experience dangerous adverse effects? A review article on pharmacotherapy for
treatment-resistant schizophrenia outlines the evidence for alternative strategies. The
strategies reviewed include the addition of a range of medications, electro-convulsive
therapy and repetitive transcranial magnetic stimulation. The best evidence is for the
addition of lamotrigine to clozapine, while there is less evidence for other strategies,
and clearly more research is needed in this area.
There is considerable interest in the literature in the neurobiology of depressive
again on the role of cortisol and the HPA-axis. This article reviews the evidence
may cause tryptophan depletion and the release of neurotoxic metabolites of the
kynurenine pathway, both of which have been hypothesized to cause depression.
Possibilities for new antidepressant treatments are suggested.
“Narcolepsy: a review” – outlines the epidemiology, pathogenesis, clinical
symptomatology and treatment of this disorder, which affects about 0.5% of the
population. Current established treatment is available for symptomatic relief, but
there is a need for investigation of treatments that tackle the cause of narcolepsy.
structural and functional neuro-imaging studies. Most of the abnormalities that
have been found to be related to starvation and appear to be reversible following
weight restoration. However, some of the changes appear to persist after recovery,
which may explain persistent cognitive and behaviour patterns. There do appear
to be differences between various categories of eating disorders and different sub-
types and involvement of tempero-pareital areas seems to be related to body image
disturbances.
Adjunct Professor RGM Thom, Division of Psychiatry, Faculty of Health Sciences, University of the Witwatersrand
1
Neuro vo5 no4 2011.indd 1 2011/10/28 10:13 AM

of the
International Neuropsychiatric
Association
dove medical press
CONTENTSvolume 5 · number 4 · 2011
REVIEWS
Pharmacotherapy for treatment-resistant schizophreniaMeghan E Mcilwain, Jeff Harrison, Amanda J Wheeler, Bruce R Russell
of indoleamine 2,3-dioxygenaseDavid M Christmas, JP Potokar, Simon JC Davies
Narcolepsy: a reviewGbolagade Sunmaila, Akintomide, Hugh Rickards
Neuroimaging in eating disordersIgnacio Jáuregui-Lobera
5
23
33
45
2
Neuro vo5 no4 2011.indd 2 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 3 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 4 2011/10/28 10:13 AM

Neuropsychiatric Disease and Treatment Dovepress
Dovepress
open access to scientific and medical research
Schizophrenia is a disabling mental illness with a lifetime prevalence of
0.7% worldwide and significant, often devastating, consequences on social and occupational
functioning. A range of antipsychotic medications are available; however, suboptimal therapeutic
response in terms of psychotic symptoms is common and affects up to one-third of people with
schizophrenia. Negative symptoms are generally less amenable to treatment. Because of the
consequences of inadequate symptom control, effective treatment strategies are required for
people with treatment-resistant schizophrenia. Clozapine has been shown to be more effective
than other antipsychotics in treatment-resistant populations in several studies; however, the
occurrence of adverse effects, some of which are potentially life-threatening, are important
limitations. In addition to those who are intolerant to clozapine, only 30% to 50% experience
clinically significant symptom improvement. This review describes the recent evidence for
treatment strategies for people not responding to nonclozapine antipsychotic agents and people
not responding or only partially responding to clozapine.
antipsychotic, refractory, clozapine
Schizophrenia is a disabling mental illness with a lifetime prevalence of
0.7% worldwide.1 Typically beginning in early adolescence, outcomes for patients
are variable but the course of illness is chronic, often marked with periods of relapse
despite treatment. Schizophrenia has a significant and often devastating impact on
social and occupational functioning for patients, often due to residual negative
symptoms and cognitive deficits.2 This may manifest as the decreased likelihood of
living independently, being in an intimate relationship, achieving formal education,
or being in paid employment.3–6 A range of antipsychotic medications is available,
including f irst-generation antipsychotics (FGAs) and second-generation
antipsychotics (SGAs).7,8 However, suboptimal therapeutic response in terms of
psychotic symptoms is common and affects up to one-third of people.9 Negative
symptoms may be classified as primary (part of the disease process itself ) or second-
ary (to factors such as depression, drug-induced akinesia, or a suspicious
withdrawal)10 and are generally less amenable to treatment.11,12 Antipsychotic agents
have no demonstrable efficacy for primary enduring or “deficit” negative symptoms.13
Improvements in this symptom domain are largely a consequence of a reduction in
positive symptoms.14,15 While antipsychotic agents improve attention in people with
schizophrenia,16,17 the effects observed for other cognitive impairments are incon-
sistent18 and may include worsening.19,20 The net impact of an antipsychotic agent
5
Neuro vo5 no4 2011.indd 5 2011/10/28 10:13 AM

Dovepress
Dovepress
on cognitive function is determined by the beneficial effect
on attention and adverse effects related to anticholinergic
activity and extrapyramidal side effects (EPSE).21
Furthermore, it has been postulated that a practice effect
may account for beneficial effects observed.22 There are no
apparent consistent differences between antipsychotic
agents with respect to their effect on cognition.23–25 Because
of the consequences of inadequate symptom control, effec-
tive treatment strategies are required for people with
treatment-resistant schizophrenia (TRS).
Several definitions of treatment-resistant schizophrenia
exist and vary in their specificity. The criteria employed by
Kane et al to define treatment-resistant (or treatment-
refractory) schizophrenia in the pivotal trial comparing
clozapine to chlorpromazine is used frequently in clinical
trials and audit settings.26,27 Kane et al classified participants
as treatment-resistant if: improvement had not been demon-
strated after 3 periods of treatment with antipsychotics (from
2 or more different chemical classes) in the previous 5 years
equivalent to 1000 mg/day of chlorpromazine (CPZ) for
6 weeks and participants had had no episodes of good func-
tioning in the previous 5 years, Brief Psychiatric Rating Scale
(BPRS) total score 45, Clinical Global Impressions (CGI)
score 4, and score 4 on 2 or 4 positive symptoms items.26
Conley and Kelly presented a modified version of these
criteria to reflect clinical practice patterns and a better under-
standing of optimal dosing: 2 antipsychotic trials (400–600 mg
CPZ equivalents per day) for 4 to 6 weeks with no clinical
improvement, no period of good social or occupational
functioning for 5 years, BPRS total score 45, and a score
of 4 on 2 of 4 positive items.28
Clozapine has been shown to be more effective than other
antipsychotics in treatment-resistant populations in several
studies; however, the occurrence of adverse effects, some of
which are potentially life-threatening, are important limitations.
In addition to those who are intolerant to clozapine, only
30% to 50% experience clinically significant symptom
improvement.29,30 This has prompted unlicensed prescribing
and antipsychotic combination strategies (with or without
clozapine) for which there is the potential for increased side
effects and little robust evidence to support this practice.
This review will summarize key studies and recent evi-
dence for treatment strategies for people not responding to
nonclozapine antipsychotic agents and people not responding
or only partially responding to clozapine. The literature
reviewed was identified by a systematic search of Ovid
Medline & Medline In-Process, Embase (combined file
1947 to present), Cochrane Central Register of Controlled
Trials (CENTRAL/CCTR), and PsycINFO, supplemented
by hand searches of reference lists. The evidence is pre-
sented in 3 sections: clozapine monotherapy versus other
antipsychotics, clozapine augmentation strategies, and
options for clozapine-intolerant or clozapine-resistant people.
The first section is divided into 2 parts comparing clozapine
monotherapy to FGAs and SGAs; each part is stratified by
the level of evidence presented. The clozapine augmentation
section is first stratified by level of evidence (meta-analysis
or randomized controlled trial) then by specific treatment
strategy. The structure of this section reflects the relative
availablity of evidence for the treatment combinations
considered. The third section, treatment options for those
who are intolerant or resistant to clozapine, discusses alterna-
tive antipsychotic monotherapy and nonpharmacological
treatments.
The World Psychiatric Association Section on Pharmaco-
psychiatry utilized data from approximately 1600 random-
ized controlled trials of 51 FGAs and 11 SGAs in the
treatment of schizophrenia.31 Modest benefits were observed
for the use of SGAs compared to FGAs for negative, cogni-
tive, and depressive symptoms, and with a lower risk of
tardive dyskinesia. These benefits were mainly attributed to
the ability of SGAs to provide improvement in positive
symptoms, equivalent to that of FGAs, with a lower risk of
EPSE. There were no consistent differences between SGAs
in terms of efficacy with the exception of clozapine, which
was found to be more efficacious than other antipsychotics
in people who had not responded to 1 or more other antip-
sychotics. Adequate trials of adequate doses of FGAs and
SGAs were found to be key variables in optimizing effective-
ness of antipsychotic agents. Substantial individual vari-
ability was observed in treatment response and adverse
effects. SGAs offer the advantage of fewer acute extrapyra-
midal symptoms and less likelihood of tardive dyskinesia
but produce greater metabolic side effects. Meta-analyses
published subsequent to this summary statement and key
trials on the use of clozapine are presented below.
Leucht et al compared treatment outcomes between SGAs and
FGAs in people with schizophrenia in general in a meta-
analysis of 150 double-blind randomized studies including
21,533 participants.32 The meta-analysis by Essali et al also
compared treatment outcomes between those taking FGAs
6
Neuro vo5 no4 2011.indd 6 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 7 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 8 2011/10/28 10:13 AM

Dovepress
Dovepress
versus SGAs and was largely based on the same data.33
Four SGA agents emerged as superior to FGA agents:
clozapine, amisulpride, olanzapine, and risperidone.32 The
majority of studies (121) were of 12 weeks’ duration, 17 were
of 6 months’ duration, and 12 were longer than 12 months.
It has been postulated that EPSE associated with FGAs may
mimic the symptoms of schizophrenia and in early randomized
controlled trials (RCTs) falsely suggested that SGAs are
superior.34–36 In order to avoid this potential problem, only
participants taking 12 mg/day haloperidol (or 600 mg/day
chlorpromazine equivalents for low- potency FGAs) were
included in this meta-analysis. Positive and Negative Symp-
tom Scale (PANSS) and BPRS scores were used to assess
overall efficacy and specific symptoms domains all of which
were found to be more amenable to treatment with clozapine,
olanzapine, amisulpride, or risperidone versus FGAs.
Treatment with clozapine produced medium effect sizes:
overall symptoms 0.52 (95% confidence intervals [CI]: 0.75
to 0.29, P 0.0001), positive symptoms 0.36 (CI: 0.56 to
0.16, P 0.0001), negative symptoms 0.27 (CI: 0.42 to
0.13, P 0.0001), depression 0.51 (CI: 0.87 to 0.14,
P 0.006). Amisulpride and olanzapine produced similar
improvements compared to FGAs: overall symptoms 0.31
(CI: 0.44 to 0.19, P 0.0001) and 0.28 (CI: 0.38 to 0.18,
P 0.0001), respectively, positive symptoms 0.22 (CI: 0.37
to 0.06, P 0.005) and 0.15 (CI: 0.21 to 0.09, P 0.0001),
negative symptoms 0.27 (CI: 0.40 to 0.14, P 0.0001)
and 0.32 (CI: 0.47 to 0.16, P 0.0001), depression 0.37
(CI: 0.51 to 0.24, P 0.0001) and 0.27 (CI: 0.35 to 0.19,
P 0.0001). The effect sizes associated with risperidone were
small and the improvement observed on the depression subscale
was not significant: overall symptoms 0.13 (CI: 0.22 to 0.05,
P 0.002), positive symptoms 0.13 (CI: 0.20 to
0.05, P 0.001), negative symptoms 0.13 (CI: 0.21 to
0.06, P 0.0001), depression 0.10 (CI: 0.23 to 0.03,
P 0.145). Industry sponsorship, comparator dose, and pro-
phylactic EPSE medication were assessed as moderator vari-
ables but did not yield any consistent effects. Leucht et al
concluded that this reflects the fact that FGAs and SGAs are
heterogeneous classes of compounds and argued that such
categorization can lead to improper generalization and
confusion.32
Meltzer et al investigated the use of clozapine versus FGAs
in treatment responsive participants during a 24-month study.37
Significant improvements in psychopathology, quality of
life and global functioning were observed in both the
clozapine (n 40) and FGA group (n 45) after taking a
range of antipsychotic agents; most commonly haloperidol
but also perphenazine, fluphenazine, loxapine, thioridazine,
thiothixene, molindone, and amoxapine. While a similar
improvement in psychopathology was observed, signifi-
cantly more relapse/rehospitalization drop-outs occurred in
those taking FGAs (19 relapse related hospitalizations
in 10 participants versus 11 relapse related hospitalizations
in 4 participants treated with clozapine). There were no
differences in the occurrence of EPSEs between clozapine
and the FGA groups; however, clozapine was associated
with more weight gain.
In a 12-week double-blind trial, Krakowski et al randomly
assigned participants with schizophrenia or schizoaffective
disorder to receive clozapine (n 33), olanzapine (n 34),
or haloperidol (n 33).38 People with a history of nonresponse
or intolerance to any of the 3 study medications were
excluded. Aggression was assessed using the Modified Overt
Aggression Scale (MOAS) and a cognitive task battery tested
general executive function, visuospatial ability, psychomotor
function, and visual and verbal memory. In the general cogni-
tive index (GCI) no significant improvement was observed
in the haloperidol or clozapine group while clozapine was the
most efficient medication in reducing aggression. An impor-
tant limitation was the concomitant, prophylactic use of
benztropine 4 mg/day for EPSE in the group taking haloperi-
dol, which may increase anticholinergic cognitive impairment.
Participants taking haloperidol showed no increase in body
weight, blood lipids, or glucose.39
The Cochrane Schizophrenia Group performed a meta-
analysis in order to compare several commonly used SGA
agents in terms of efficacy and tolerability in people with
schizophrenia or schizophrenia-like psychoses.40 The pri-
mary outcome measure selected to assess this was change in
total PANSS score, with positive and negative subscores as
secondary outcomes. Outcomes were reported using
weighted mean difference (WMD) in terms of PANSS scores
and the dropout rate due to poor efficacy was included as a
further outcome measure. Seventy-eight randomized, double-
blind studies were included for analysis of which 28 included
treatment with clozapine.
The results relating to clozapine were different to those
anticipated based on previous reports. No significant differ-
ences were found when comparing the total PANSS scores
9
Neuro vo5 no4 2011.indd 9 2011/10/28 10:13 AM

Dovepress
Dovepress
between clozapine and olanzapine (N 619), quetiapine
(N 232), risperidone (N 466), or ziprasidone (N 146);
however, clozapine was found to be significantly more effica-
cious than zotepine (N 59, WMD 6.0, P 0.002).
The results for a decrease in positive symptoms reflected those
found for overall symptoms while quetiapine was found to be
more efficacious than clozapine on the negative symptom sub-
score (N 142, WMD 2.2, P 0.001). Clozapine was
favored over risperidone when comparing dropout rates due
to poor efficacy (N 627, relative risk [RR] 0.40 95% con-
fidence interval [CI] 0.23–0.70, P 0.001). These unexpected
results may be due to the low or very low doses of clozapine
that were used in many of the studies included; several had an
upper limit of 400 mg/day and 5 used dosages under
210 mg/day. In the pivotal studies that established clozapine’s
effectiveness, the average daily dose of clozapine was
600 mg/day and 523 mg/day.26,41 Furthermore the participants
included in these trials may not have been as treatment refrac-
tory as those in other studies demonstrating clozapine’s supe-
riority over other SGA agents.
Substantial concerns about the side effects induced by
SGA agents such as weight gain and metabolic syndrome
may offset modest differences in their effectiveness. In a
meta-analysis of head-to-head comparisons of the metabolic
effects between SGA agents, Rummel-Kluge et al assessed
weight gain and changes in cholesterol and glucose over
48 studies.42 There were 3 main clusters in terms of these
outcomes: olanzapine and clozapine produced the greatest
elevation in weight, cholesterol and glucose (with no signifi-
cant difference between the 2 agents) followed by quetiapine,
risperidone, and sertindole with intermediate elevations.
Aripiprazole and amisulpride showed lower elevations and
ziprasidone the lowest. The authors noted that the dose of
antipsychotic influenced some of the results in meta-
regressions; for example a high dose of olanzapine tended
to produce a greater difference in the outcome measure in
favor of the comparator drug. Another important caveat is
that data on prior antipsychotic treatment for the participants
in the selected studies were not available for analysis.
Phase II of the Clinical Antipsychotic Trials of Intervention
Effectiveness (CATIE) recruited 99 participants who discon-
tinued treatment with olanzapine, quetiapine, risperidone,
or ziprasidone in phase I or IB of the trial primarily due to
inadequate efficacy.43 Participants were randomized to
blinded treatment with another newer SGA not previously
received in the trial (olanzapine n 19, quetiapine n 15, or
risperidone n 16) or open label treatment with clozapine
(n 49). At 3-month assessments, participants treated with
clozapine experienced a greater reduction in PANSS total
score (mean 11.7, standard error [SE] 3.2) than partici-
pants treated with quetiapine (mean 2.5, SE 4.8) or ris-
peridone (mean 4.1, SE 1.9) but not olanzapine
(mean 3.2, SE 2.3). Clozapine was significantly better
only than quetiapine on the PANSS general psychopathology
subscale (mean 4.7, SE 1.5 versus mean 2.3, SE 2.5,
P 0.006). Time to discontinuation for any reason was sig-
nificantly longer for clozapine (median 10.5 months) than
for risperidone (2.8 months) or quetiapine (median 3.3 months)
but not olanzapine (median 2.7 months). Time to
discontinuation is subject to bias in this phase of the study.
Because treatment allocation was known to both clinicians
and participants there may have been reluctance to discon-
tinue clozapine, it being widely considered the best option
for treatment-resistant schizophrenia. The data from this study
support the conclusion that, for participants who prospectively
failed to improve with an SGA, treatment with clozapine was
more effective than switching to another SGA.
Phase III of CATIE allowed 270 participants who had
discontinued antipsychotics in Phases I and II to select
treatment from 9 antipsychotic regimens with the help of their
study doctor.7 Approximately equal numbers of participants
chose 7 of the 9 antipsychotics including clozapine
(33–41 participants each agent). The study used a double-
blind design with the exception of those treated with clozap-
ine, which was open label. The blinding of treatment with
clozapine would have required additional monitoring of all
treatment groups for clozapine specific safety issues, and in
doing so may have affected the ecological validity of the other
agents. All of the commonly used treatments were associated
with substantial symptom improvement at 3 months
and 6 months, with the exception of aripiprazole at 3 months
and ziprasidone and quetiapine at 6 months. A total of 106 par-
ticipants discontinued treatment; there were no significant
differences in the proportions of participants who discontin-
ued the commonly selected medicines (range 33%–46%).
However, discontinuation due to lack of efficacy was lower
for clozapine (5%), risperidone, quetiapine, and ziprasidone
(0%–5%) than olanzapine, aripiprazole, and combination
treatment (13%–18%). Adverse effects were problematic in
the group taking clozapine; the rates of adverse events
classified as moderate or severe were highest for clozapine
(35%), quetiapine (45%), and combination antipsychotic treat-
10
Neuro vo5 no4 2011.indd 10 2011/10/28 10:13 AM

Dovepress
Dovepress
ment (30%). Clinically significant weight gain of at least 7%
was common with clozapine (32%), combination antipsychotic
treatment (39%), and olanzapine (23%). All other SGA agents
were associated with weight loss, in particular aripiprazole
and ziprasidone, which produced the greatest monthly weight
loss of 0.64 kg and 0.59 kg, respectively; clozapine produced
a gain of 0.59 kg/month.
Krakowski et al reported that olanzapine outperformed
clozapine in terms of neurocognitive function in a study of
100 physically aggressive inpatients with schizophrenia or
schizoaffective disorder.38 For metabolic parameters, partici-
pants taking olanzapine gained the most weight compared with
clozapine or haloperidol, but clozapine was associated with
the greatest increases in serum cholesterol, triglycerides and
glucose.39 In the GCI olanzapine was found to be superior
(improvement was approximately 0.5 standard deviations
[SD]) to both clozapine and haloperidol; this was also associ-
ated with a decrease in aggression which was assessed using
the MOAS. Rather than concluding that olanzapine has a
procognitive effect it is perhaps more likely that olanzapine
has less cognitive liability; clozapine has strong intrinsic anti-
cholinergic activity compared with olanzapine.44 Nonetheless,
treatment with clozapine markedly reduced aggression, sug-
gesting that the antiaggressive effects of olanzapine may be
mediated by different neuronal pathways.
It has been suggested that a decrease in serum choles-
terol may result in aggression due to the subsequent
decrease in brain serotonergic activity, given that choles-
terol determines the availability of serotonin receptors and
transporters.45 In a post-hoc analysis of the relationship
between serum cholesterol levels and aggression in these
groups, Krakowski and Czobor found a negative correlation
at baseline.46 Based on changes in total cholesterol (TC)
over the 12-week study period, the investigators used a
Glimmix regression model to predict changes in aggression
(Krakowski, pers comm). For those taking haloperidol it
was predicted that a 141.9% increase in physical aggression
was associated with a decrease of 1 SD unit in TC levels.
Participants whose cholesterol increased by 1 SD in the
clozapine group were predicted to be 67.6% (P 0.001)
less physically aggressive than those whose cholesterol did
not change. It was then postulated that the antiaggressive
effects of clozapine may have been further enhanced by an
increase in cholesterol.
The UK Cost Utility of the Latest Antipsychotic Drugs
in Schizophrenia Study 2 (CUtLASS 2) included 136 people
with schizophrenia and related disorders whose medication
was being changed due to suboptimal response to 2 or more
previous antipsychotic agents.47 Participants were randomly
allocated to receive clozapine or another SGA agent (risperi-
done, olanzapine, quetiapine, or amisulpride) selected by the
treating clinician. The trial was rater-blind and outcome
assessments were carried out for 87% of the participants at
12, 26, and 52 weeks following randomization. No significant
advantage was observed for those taking clozapine compared
with other SGA agents in the Quality of Life score
(3.36 points, 95% CI 0.46–7.71); however, a significant
improvement was seen in the PANSS total score ( 4.93, 95%
CI 8.82 to 1.05). At 12 weeks the group taking clozapine
reported that their mental health was significantly better than
those taking other SGA agents. There were no significant
differences between the treatment groups in the rate of
adverse effects including weight gain.
Suicide has been identified as the leading cause of pre-
mature death among people with schizophrenia.48 The
International Suicide Prevention Trial (InterSePT) assessed
the risk for suicidal behavior in 980 participants with
schizophrenia or schizoaffective disorder treated with clo-
zapine compared to olanzapine over a 2-year period.49 Par-
ticipants in this study, 26.8% of whom were refractory to
previous treatment, were considered at high risk for suicide
because of previous attempts or the presence of suicidal
ideation. The study was conducted as an open-label trial
with masked ratings. Suicidal behavior, defined as suicide
attempts and hospitalizations to prevent suicide, was
observed less frequently in those taking clozapine versus
olanzapine (hazard ratio [HR] 0.76, 95% CI 0.58–0.97).
Worsening on the CGI-Suicide Severity or implicit worsen-
ing as demonstrated by occurrence of suicidal behavior was
also less frequent in those taking clozapine (HR 0.78, 95%
CI 0.61–0.99). Fewer clozapine treated participants
attempted suicide, required hospitalizations or rescue inter-
ventions to prevent suicide (34 versus 55, P 0.03, 82
versus 107, P 0.05 and 118 versus 155, P 0.01, respec-
tively). The need for concomitant antidepressants or anxi-
olytics/soporifics was also less frequent in those taking
clozapine compared with olanzapine (221 versus 258,
P 0.01 and 301 versus 331, P 0.03). Although the num-
ber of completed suicides was greater in the clozapine
group (5 clozapine-treated participants versus 3 olanzapine-
treated participants, P 0.73), this was not significant and
the study was not powered to evaluate this as an endpoint.
It was recognized by the investigators at the outset that the
study would need to include 20,000 participants to detect
11
Neuro vo5 no4 2011.indd 11 2011/10/28 10:13 AM

Dovepress
Dovepress
a decreased relative risk for suicide deaths with clozapine
therapy by 20%.
In a randomized double-blind trial, Harvey et al compared
the cognitive performance of 130 people with schizophrenia
after 12 weeks of treatment with clozapine (n 69) or
ziprasidone (n 61).50 All participants were either resistant
or intolerant to previous antipsychotic treatment. Clozapine-
treated participants showed improvement on the Rey Audi-
tory Verbal Learning Test (RAVLT; episodic memory) and
the Stroop interference test (executive function) but not the
Trail-Making Test (TMT; parts A and B; processing speed)
compared with those taking ziprasidone. None of the indi-
vidual items were observed to improve at 12 weeks between
the treatment groups; however the composite score improved
significantly in those taking ziprasidone compared with
clozapine (effect size D 0.54, P 0.029). One possible
explanation for these results is that clozapine may interfere
with the performance benefits of practice effects. Although
it appears that ziprasidone is superior in reducing cognitive
deficits in this short-term trial, clinical efficacy in terms of
symptom control was not reported.
Davies et al compared clozapine to available SGA agents
in a UK multi-center, rater-blind RCT in people with
psychosis eligible for clozapine to assess cost- effectiveness.51
Over a 1-year period, it was found that clozapine was associ-
ated with higher quality-adjusted life years (QALYs) than
other SGA agents, but at an additional cost. The probability
that clozapine is cost-effective reached 50% if in order to
gain 1 QALY the decision-makers were willing to pay
£33,000. In other words, if the decision-makers were willing
to pay less than £33,000 to gain 1 QALY, other SGA agents
may be more cost- effective than clozapine. However, this
trial was conducted with a relatively small number of
participants (n 67 clozapine; n 69 other SGA agents) and
post-hoc calculations indicated that the power to detect sig-
nificant differences in net money benefit was low (50% if
important differences in costs and QALYs were defined as
£1600 and one-twentieth of a QALY, respectively). Further-
more, it may not be possible to extrapolate the results to
longer-term clozapine treatment or to a population of primar-
ily treatment-resistant people. The authors also noted that
clozapine may be more cost-effective if fewer participants
had clozapine initiated as an inpatient than in this RCT.
The present review found 2 RCTs comparing clozapine
monotherapy with treatment with high-dose olanzapine52,53
and a further study examining treatment with ziprasidone
with treatment-resistant participants.54 These studies will be
discussed in detail below.
Despite proven efficacy in people with schizophrenia show-
ing sub-optimal response to other antipsychotics, only
30% to 50% of people will experience clinically significant
symptom improvement with clozapine treatment.29,30
One-third to two-thirds of people will continue to experience
positive symptoms with adequate doses of clozapine or will
be unable to reach adequate levels due to side effects that
prevent further dose increases.30 Antipsychotic monotherapy
is preferred over augmentation according to schizophrenia
treatment algorithms; for people who do not respond to
first-line antipsychotics, clozapine is recommended. There-
fore clozapine augmentation strategies should be imple-
mented only for those who experience insufficient response
to clozapine monotherapy. An operational definition of
nonresponse to clozapine or ‘ultraresistant’ schizophrenia
is: BPRS improvement of 20% despite a trial with clozap-
ine for 8 weeks and plasma levels 350 g/L, no stable
period of good social and/or occupational functioning
for 5 years, Global Assessment of Functioning (GAF)
40, BPRS total score 45, CGI score 4, and a score
of 4 on 2 of 4 positive symptom items.55
The present review found 4 meta-analyses on the augmenta-
tion of clozapine treatment with another antipsychotic for
people with an inadequate response to clozapine
monotherapy.56–59 These meta-analyses were based on essen-
tially the same data, the largest of which was conducted by
Barbui et al and arrived at similar conclusions with the excep-
tion of Correll et al.56,58
Barbui et al selected 21 studies to determine the efficacy
of a second antipsychotic in combination with clozapine.56
The number of trials evaluating each augmentation agent
was chlorpromazine n 1, pipothiazine n 2, amisulpride
n 1, sulpiride n 7, and the remainder used risperidone
(n 10). The mean length of follow up was 13.8 weeks
(SD 19.6) and the trials were divided into either short-term
studies of less than 10 weeks’ duration or long-term studies.
Clozapine combination strategies were favored in 14 open
(nonblind), randomized studies in terms of effect size or
standardized mean difference (SMD) from various outcome
scales (SMD 0.80, 95% CI 1.14 to 0.46). However,
this trend was not apparent in 6 of the RCTs (SMD 0.12,
95% CI 0.57 to 0.32). Subgroup analysis by trial duration
revealed a similar trend: the open studies favored clozapine
combinations in both long- and short-term trials, the blinded
12
Neuro vo5 no4 2011.indd 12 2011/10/28 10:13 AM

Dovepress
Dovepress
studies showed no advantage for clozapine combinations of
either duration.
Correll et al found antipsychotic combinations in general
to be advantageous over monotherapy in a meta-analysis of
19 studies (1229 participants) in terms of all cause discon-
tinuation (n 1052, RR 0.65, 95% CI 0.54–0.78) and less
study-specific inefficacy (n 1202, RR 0.76, 95% CI
0.63–0.90).58 The mean trial duration was 12.1 weeks (range
4–52 weeks). The most commonly used antipsychotic was
clozapine, though a variety of antipsychotic combinations
were used. In terms of lack of efficacy as defined by each
study, co- treatment including clozapine was superior to
antipsychotic monotherapy (n 764, RR 0.75, 95% CI
0.61–0.93); however, the specific augmenting agents were
not presented separately within the results. Meaningful
results regarding specific psychopathology and adverse
events could not be calculated due to insufficient data.
Sensitivity analyses identified 5 efficacy moderators: clozap-
ine combinations, concurrent polypharmacy initiation,
Chinese trials, trial duration 10 weeks, and SGA–FGA
combinations. Meta- regression of variables from sensitivity
analyses identified 3 significant moderators associated with
superior efficacy of antipsychotic combinations: similar
doses in the mono- and polytherapy arm (P 0.006,
coeff 0.48), SGA FGA combinations (P 0.027,
coeff 0.39) and concurrent polypharmacy initiation
(P 0.050, coeff 0.35). The findings of this study differ
from those of other meta-analyses of antipsychotic combina-
tion treatment and it is important to note that the positive
results for antipsychotic combinations observed were primar-
ily from Chinese studies not included in the other meta-
analyses. A high degree of heterogeneity within the database
and possible publication bias further obscured the signifi-
cance of these findings.
Overall, it appears that the evidence considered for
clozapine augmentation with another antipsychotic in these
meta-analyses is weak and observed benefits are moderate at
best. One consideration to take into account is that these
reviews combined results of all antipsychotic augmentation
irrespective of mechanism of action.
Dysfunctional glutamatergic neurotransmission is postulated
to be an important component underlying the pathophysiology
of schizophrenia.60 Lamotrigine is an anticonvulsant drug
that inhibits excessive glutamate release in the brain by
antagonism of sodium channels and increases gamma-zyric
acid (GABA) release. It has been used as an augmenting
agent on this basis.61,62 Tiihonen et al examined the
advantages of combining clozapine with lamotrigine in 5
randomized placebo-controlled trials (161 participants) of
10 to 24 weeks’ duration.63 On the primary outcome measure
the total score for symptoms of psychosis, the clozapine–
lamotrigine combination was superior to the clozapine–
placebo combination (SMD 0.57, 95% CI 0.25–0.89;
number needed to treat [NNT] 4, 95% CI 3–6). The second-
ary outcome measures also favored this combination (SMD
0.34, 95% CI 0.02–0.65 for decreasing positive symptoms
and SMD 0.43, 95% CI 0.11–0.75 for improving negative
symptoms). The incidence of severe adverse effects or drop-
out rate did not differ between the treatment groups. No
significant heterogeneity was observed in the meta-analysis.
Importantly, this is the first evidence to date of efficacy for
any pharmacological treatment in clozapine-resistant schizo-
phrenia and it is noted by the authors that similar benefits
may not be observed with lamotrigine and other antipsy-
chotic agents apart from clozapine. The effect size for total
score for symptoms of psychosis was 0.57, suggesting ben-
eficial effects for general symptoms which are known to be
robust predictors of functional outcomes; however, scores
were not available for all studies.64
Like anticonvulsants, the use of N-methyl-D-aspartate
(NMDA) -enhancing agents is predicated on the glutamate
hypothesis of schizophrenia, specifically NMDA receptor
hypofunction. Antagonists of NMDA receptors such as
phencyclidine and ketamine produce psychotic symptoms
and neurocognitive deficits in human subjects and exacerbate
psychotic symptoms in people with schizophrenia.65–67 Agonists
at the obligatory NMDA- glycine binding site are glycine,
D-serine, and D-alanine and the partial agonist D-cycloser-
ine, as opposed to agonists at the NMDA recognition site,
which are excitotoxic. These agents, in addition to sarcosine
which increases the availability of glycine in the synapse
by inhibiting the glycine transporter-1 (GlyT-1), have been
investigated as potential therapeutic agents for schizophrenia.
Tsai and Lin performed a meta-analysis of 26 double-blind,
placebo-controlled trials in approximately 800 people taking
an NMDA agonist in addition to stable doses of antipsy-
chotic medication for at least 4 weeks.68 Almost all studies
used the PANSS to assess symptom severity. The pooled
effect size of clinical efficacy of NMDA agonist augmentation
compared with placebo for total psychopathology was 0.40
(95% CI 0.22–0.58) and significant improvement was noted
for depressive, negative, cognitive, positive, and general
13
Neuro vo5 no4 2011.indd 13 2011/10/28 10:13 AM

Dovepress
Dovepress
symptoms. Treatment with glycine, D- serine, and sarcosine
was associated with improvement in multiple symptom
domains while D-cycloserine was not. The concomitant
antipsychotic used appeared to affect the efficacy of the
NMDA-enhancing agent; those treated with risperidone or
olanzapine improved, but those treated with clozapine did
not. Gastrointestinal (GI) upset and nausea were noted more
often in some glycine trials while other side effects were
equivalent for NMDA-enhancing agents and placebo.
Despite a moderate effect size, the efficacy of these agents
may have been overstated due to limitations within the study.
For instance, studies were included only if they provided
“enough data to calculate the effect size” and a test for
homogeneity revealed that there may have been systematic
differences among the included studies. Another important
caveat is that D-cycloserine, D-serine, D-alanine, and sar-
cosine are protected by US patents for which the study
author is a patent holder.
Topiramate is a GABAergic anticonvulsant drug indicated
as add-on pharmacotherapy for adults and children with
primary generalized tonic–clonic and partial-onset seizures.
It has been used for people with schizophrenia to correct a
postulated glutamate deregulation due to NMDA receptor
hypofunction. Topiramate is thought to potentiate inhibitory
GABAergic transmission (probably through a nonbenzodi-
azepine mechanism) and inhibit the activity of kainite on the
AMPA/kainate receptor subtype.69–73
Two studies have examined the use of topiramate as an
adjunct to treatment to clozapine with contrasting results.
Afshar et al conducted a double-blind trial over 8 weeks
with 32 people receiving clozapine treatment for at least
2 months.74 Participants were randomized to receive up to
300 mg/day of topiramate (n 16) or placebo in addition to
clozapine (n 16). Total PANSS scores at baseline were
similar between the groups, indicating a suboptimal response
to clozapine monotherapy (topiramate group 96.87 21.98;
placebo group 101.87 23.05, P 0.53). Clinically signifi-
cant improvement was defined as a 20% decrease in
total PANSS score and was observed in 8 participants
(50%) in the topiramate group and 2 in the placebo group
(12.5%; P 0.05). The differences in the groups’ total
PANSS mean scores were reported at both 3 and 8 weeks
and favored topiramate augmentation: 11.18 8.72
versus 1.56 9.23, P 0.005 and 20.00 11.96
versus 1.31 11.13, P 0.001, respectively. At 8 weeks
a number of side effects were more prevalent in the topira-
mate group such as hypersalivation (75.0% versus 34.7%,
P 0.05) (although this was reported to be present in some
participants prior to the study), psychomotor retardation
(50.0% versus 6.2%, P 0.01), and paresthesia (37.5%
versus 6.2%, P 0.05). Weight loss was also reported more
commonly in the topiramate group (37.5% versus 6.2%,
P 0.05). However, the authors reported that there were
no differences observed in body mass index (BMI) between
the groups or within each group over the trial period. None
of the participants dropped out of the trial due to drug-
induced adverse effects. While the results of this small trial
appear to favor topiramate augmentation, the follow-up
period is relatively short. Furthermore, the investigators did
not assess cognitive impairment, a well-documented, dose-
dependent adverse effect of topiramate that is particularly
relevant to people with schizophrenia.75–78
The double-blind RCT by Muscatello et al79 was a meth-
odologically robust 24-week study that failed to replicate the
benefits of topiramate add-on pharmacotherapy reported by
Afshar et al.74 People receiving clozapine for at least 1 year,
at a stable dose for at least 1 month, with a BPRS score
of 25 were eligible to participate. The clozapine dose
remained unchanged throughout the study and participants
noncompliant with all 10 study visits were excluded. No last
observations were carried forward since this introduces
assumptions which can under- or overestimate the effects of
treatment.80 Participants did not receive any antidepressants
or anticonvulsants for a period of 2 months prior to the study.
A maximum dose of 200 mg/day topiramate was added to
clozapine treatment (n 19; placebo n 24). No significant
improvement in positive, negative, affective, or overall
symptomatology from baseline to week 24 was observed.
In the topiramate group a significant reduction was observed
using the scale for the assessment of positive symptoms
(SAPS) subscale for bizarre behavior (including clothing and
appearance, aggressive behavior, stereotyped behavior and
social, and sexual behavior).81 No significant effects on
cognitive functioning were observed as measured by the
Stroop test, verbal fluency, and the Wisconsin Card Sorting
Test (WCST). No serious adverse events were reported;
however, adjunctive topiramate was more frequently
associated with asthenia, sedation, and paresthesia while
constipation and hypersalivation were reported in the placebo
group. There was no significant change in body weight from
baseline to the end of the trial for the topiramate group.
14
Neuro vo5 no4 2011.indd 14 2011/10/28 10:13 AM

Dovepress
Dovepress
It is possible that this trial did not prove topiramate to be
as useful for clinical symptomatology as the previous study
because a lower dose of topiramate was used (200 mg/day
versus 300 mg/day). Yet this dose was chosen based on find-
ings by Deutsch et al82 in order to avoid cognitive impairment
which was not assessed by Afshar et al.74 Furthermore, the
very small topiramate group (n 19) means that only a large
change in SAPS or WCST would produce a statistically
significant difference. From these studies it appears that at
doses low enough to preserve cognitive function, topiramate
is of little benefit for clinical symptoms.
Memantine is a weak, nonselective NMDA receptor antago-
nist approved for use in the treatment of moderate to severe
Alzheimer’s disease. De Lucena et al studied the effects of
20 mg/day memantine combined with clozapine treatment
for negative symptoms over 12 weeks.83 This double-blind
trial was small (memantine n 10, placebo n 11) and
consisted of those taking clozapine for at least 10 years for
TRS. Significant improvements were seen at week 12 in the
active treatment group for the total BPRS score (19.00 versus
43.18, P 0.001) and on the positive and negative symptom
subscales (4.10 versus 9.18, P 0.007 and 6.10 versus 13.55,
P 0.001). Those taking memantine also showed an
6.12-point (95% CI 4.45–7.79) increase in mean score on
the Mini-Mental State Examination (MMSE), although this
is not the most sensitive measure of cognitive functioning.84
Simpson-Angus Scale (SAS) score and body weight were
not significantly different between the groups. Based on
results from animal studies, it has been postulated that
memantine may improve cognitive function by upregulating
the expression of brain-derived neurotrophic factor (BDNF)
in humans.85 In this study, however, de Lucena did not detect
an association between memantine treatment and increased
serum BDNF levels, which have been highly correlated with
cerebrospinal fluid BDNF levels.86 This may be due to the
small sample size or clozapine treatment prior to randomiza-
tion, which may also have increased serum BDNF levels.87
From this small trial, it appears that memantine may have
beneficial effects in treatment-resistant people taking clo-
zapine in particular; previous studies have not reported
this effect in people taking atypical antipsychotics apart
from clozapine.88 Other cognitive enhancing agents such as
CX516 (an ampakine) and modafinil (a wakefulness-
promoting agent) have shown less promising results in recent
randomized controlled trials.89,90 CX516 did not improve
PANSS scores after 4 weeks of co-administration with
clozapine (n 24), olanzapine (n 18), or risperidone (n 9)
and was associated with fatigue, insomnia, and epigastric
upset compared with placebo.90 In an 8-week trial, modafinil
did not worsen psychosis in 35 people taking clozapine
concurrently but also failed to reduce fatigue, negative
symptoms, or cognitive deficits.89
As a partial D2 agonist, aripiprazole’s mechanism of action
is distinct from that of other antipsychotics. It is a partial
agonist at 5-HT1A
receptors, an agonist at 5-HT2 receptors,
and has been described as the prototype of a new generation
of antipsychotic agents, the dopamine-serotonin system
stabilizers.91 Partial agonism may be a beneficial property by
allowing optimal neurotransmission, for instance, by acting
as an antagonist in areas where there is an abundance of
dopamine causing psychosis while acting as an agonist at
receptor sites where low dopaminergic tone would produce
adverse effects such as EPSE or hyperprolactinemia.92 Adverse
effects associated with this drug such as somnolence, head-
ache, light-headedness, and GI upset may be explained by
its affinity for several other receptors including D3, D
4,
5-HT2C
, 5-HT7,
1, and H
1.
Millar et al studied aripiprazole or placebo in combina-
tion with clozapine in suboptimally controlled outpatients
over a period of 16 weeks.93 Participants in this double-blind,
randomized study were on a stable dose of clozapine for at
least 3 months and had gained at least 2.5 kg since starting
clozapine. At week 16, co-treatment with aripiprazole was
associated with a significant decrease in mean weight com-
pared with placebo (aripiprazole 2.53 kg, placebo 0.018 kg;
P 0.001) and waist circumference (aripiprazole 2.00 cm,
placebo 0 cm; P 0.001). Both treatment groups showed
similar improvement in the GAF. Improvements on the
Epworth Sleepiness Scale and Fatigue Syndrome Inventory
were observed in both groups; a significant difference in
favor of aripiprazole was seen only in week 1.
In an open-label extension of a 16 week double-blind
placebo controlled trial (reviewed in the meta-analysis by
Taylor and Smith 2009),59 Fleischhacker et al administered
aripiprazole (5–15 mg/day) in combination with clozapine
to all participants.94 For participants previously randomized
to adjunctive placebo then treated with adjunctive
aripiprazole for 12 weeks, the weight loss from the end of
the double-blind phase was greater (1.74 kg versus adjunc-
tive aripiprazole 0.47 kg). This finding suggests that while
15
Neuro vo5 no4 2011.indd 15 2011/10/28 10:13 AM

Dovepress
Dovepress
the weight loss was maintained in the initial aripiprazole
group, this effect may plateau after a period of time. Clini-
cally relevant weight loss from baseline was seen in 13%
of those previously in the placebo group and in 21% of
those taking aripiprazole for 28 weeks. Differences in
PANSS scores were not significant between treatment
groups in either phase of the study. The authors reported
that symptom improvements were maintained; however,
only the week 16 PANSS results were reported. Similarly,
it was reported that participants who switched from placebo
to aripiprazole at week 12 had reduced TC, low-density
lipoprotein (LDL) cholesterol, and triglycerides but data
illustrating this were not supplied.
Phase III of the CATIE study included only 2 participants
receiving this combination of antipsychotics and therefore it
was not meaningful to report these separately. However, the
positive outcomes for weight loss in these randomized con-
trolled trials correspond with findings in CATIE III; treatment
with aripiprazole was associated with the most monthly
weight loss (0.64 kg).7 It appears from these trials that the
addition of aripiprazole counteracts or at least decelerates
the weight gain as a result of clozapine treatment without
causing clinical deterioration or improvement.
Two RCTs focused specifically on clozapine versus high
dose olanzapine in TRS.53,95 Olanzapine is structurally simi-
lar to clozapine but has a different receptor affinity profile,
being a weaker agonist for 1 and
2 receptors relative to
its D2, D
4, and 5HT
2A antagonism. In a 6-month, double-
blind RCT Meltzer et al examined the efficacy and tolerabil-
ity of high-dose olanzapine (target dose 25–45 mg/day;
mean dose 34 mg/day; n 19) versus clozapine (target
dose 300–900 mg/day; mean dose 564 mg/day; n 21) in
treatment-resistant participants with schizophrenia or
schizoaffective disorder.53 Between 6 weeks and 6 months
of treatment, significant and robust improvements were
observed in both groups using multiple measures of
psychopathology. The GAF significantly favored clozapine
(P 0.01); however, there were no other significant differ-
ences between each group. While it appears in this small
trial that high-dose olanzapine was as effective as clozapine,
significantly more weight gain in the olanzapine group may
limit its use. At 6 months, the mean increase in BMI for
those taking olanzapine was 2.2 versus 0.3 for those taking
clozapine (P 0.006).
Kumra et al52 concluded in a 12-week controlled
comparison of 39 adolescents with TRS that clozapine was
superior to high-dose olanzapine (included in meta-analysis
by Rummel-Kluge et al).42 In an open-label extension of this
study, the authors investigated the metabolic side effects of
these treatments at 24 weeks and the clinical response at
12 weeks of 10 of the 19 olanzapine-treated participants who
were switched to clozapine due to nonresponse.95 Clinical
response was defined as a decrease of at least 30% on the
BPRS and a CGI-Improvement rating of 1 (very much
improved) or 2 (much improved). On this basis, 7 of the 10
participants switched to clozapine were found to respond to
clozapine. Metabolic side effects were similarly problematic
in both treatment groups but direct comparisons between the
groups were difficult to make due to the large proportion of
participants switched to clozapine. It should also be noted
that the mean weight of the participants at the beginning of
this trial corresponded to a mean BMI percentile of 91.3
(SD 10.0), which may be accounted for by exposure to
SGA agents prior to study entry.
With a much higher affinity for 5-HT2 receptors than
D2 receptors, ziprasidone has one of the highest serotonin/
dopamine binding ratios of the SGA group and a low affin-
ity for H1 and
1 receptors. Sacchetti et al investigated the
use of ziprasidone compared to clozapine over an 18-week
period in acutely unwell people (mean PANSS total
score 107) with a history of multiple refractoriness to
antipsychotics using a double-blind design.54 Decreases in
the PANSS score were similar in each group; clozapine 24.5
(95% CI 29.7 to 19.2) and ziprasidone 25.0 (95%
CI 30.2 to 19.8). Discontinuation rates due to adverse
effects were similar however, ziprasidone offered the
advantage of a more favorable metabolic profile (in terms
of weight, fasting glucose, TC, LDL cholesterol, and
triglycerides). Reductions in movement disorders assessed
by the SAS and Abnormal Involuntary Movement Scale
scores were also observed with ziprasidone but not
clozapine. Clozapine-intolerant and clozapine-resistant
participants were not distinguished from one another in this
study which may have affected the results. The investigators
also acknowledge that the mean dosage of clozapine
(346 mg/day) was within the therapeutic range but lower
than may be used in clinical practice.
A thorough appraisal of the value of nonpharmacological
treatment options is beyond the scope of this review, however
in the context of treatment resistance it is important to
16
Neuro vo5 no4 2011.indd 16 2011/10/28 10:13 AM

Dovepress
Dovepress
acknowledge the potential role of cognitive behavioral
therapy (CBT) and electroconvulsive therapy (ECT).
Recent studies have shown that CBT may be beneficial for
those resistant to clozapine. Barretto et al compared CBT for
psychosis (n 12) to nonspecific social support also termed
“befriending” (BF, n 9) over 20 individualized therapy
sessions over 3 weeks and at 6 months.96 The clozapine dose
remained the same for all participants throughout the trial
and the rater was blinded for the participants’ intervention.
At 6 months modest improvements were observed in the
BPRS total score (CBT mean 25.00, SD 6.85 versus BF
mean 19.00, SD 8.38, P 0.0092), PANSS total score
(CBT mean 74.11, SD 8.76 versus BF mean 66.54
SD 13.95, P 0.0447), and PANSS general symptom sub-
scale (CBT mean 38.44, SD 6.63 versus BF mean 33.45
SD 7.27, P 0.0147). Participants with residual negative
symptoms such as conceptual disorganization, emotional
withdrawal, and blunted affect were excluded from the study.
Although this approach is rational, since such people may
not be able to engage in and benefit from CBT, this limits
the generalizability of the findings; many people with TRS
have residual negative symptoms.
Turkington et al compared CBT (n 31) and BF (n 28)
over a 5-year period in individuals with schizophrenia and
persistent positive symptoms despite adequate trials of antip-
sychotic medication.97 Improvements were observed with
CBT in overall symptoms severity (NNT 10.36, 95%
CI 10.21 to 10.51) and level of negative symptoms
(NNT 5.22, 95% CI 5.06–5.37). While these results suggest
that CBT may improve outcomes for participants, there was
a significant break between the intervention which was
completed at 9 months and follow-up at 18 months and
5 years. Intermediate follow-up assessments and booster
sessions may have revealed greater benefits for CBT.
Matheson et al performed a systematic meta-review to deter-
mine the benefits and adverse outcomes associated with
ECT and repetitive transcranial magnetic stimulation (rTMS)
for people with schizophrenia.98 In contrast to ECT which
produces global central nervous system excitation and
generalized seizures, rTMS allows for targeted stimulation
of superficial layers of the brain which may be effective for
specific symptoms of schizophrenia.99 Furthermore, rTMS is
subconvulsive and does not require an anesthetic or muscle
relaxant. Five systematic reviews with meta-analysis were
included in this meta-review (2 ECT, 3 rTMS) and graded in
terms of the quality of evidence. High quality evidence sug-
gested a short-term, small effect with ECT for the improve-
ment of global symptoms in participants with or without
concurrent antipsychotics (RR 0.76, 95% CI 0.63–0.92).98,100
For rTMS, high quality evidence suggests a moderate to large
decrease in auditory hallucinations (D 0.88, 95% CI
0.52–1.23).98,101 No evidence was found for long-term thera-
peutic or adverse effects of either treatment.
Lévy-Rueff et al conducted a retrospective chart review
of 19 participants with schizophrenia or schizoaffective
disorder nonresponsive or only partially responsive to phar-
macological agents.102 In addition to antipsychotic medication,
participants received maintenance ECT (M-ECT) beyond
acute episodes of psychosis. Participants received an average
of 47 sessions of bilateral M-ECT at 1- to 8-week intervals
for a mean period of 43 months. Improvements in mood,
delusions, anorexia, suicidal ideation, and anxiety were
observed but symptom scores were not reported. With
M-ECT the mean duration of yearly hospitalizations
decreased by 80% within this cohort from 10.5 months
(SD 17 months) in the year preceding M-ECT to 2.1 months
(SD 2.04 months). The mean duration of each hospitalization
decreased by 40%, from 4.13 months (SD 4.0 months) prior
to M-ECT to 2.53 months (SD 3.47). An improvement in
daily functioning was also reported for most participants;
2 participants were discharged from full-time hospitalization
and 1 returned to employment.
The results of the large trials CATIE and CUtLASS chal-
lenged the widely held belief that SGAs are superior to FGAs
in treatment-responsive schizophrenia. One concept that
remains unchanged is clozapine’s superiority over both SGAs
and FGAs in treatment-resistant schizophrenia; a finding
reinforced by the second phase of each of these studies (and
in the case of CATIE the third phase also) and the recent
meta-analyses and RCTs presented in this review. In addition
to people with treatment-resistant schizophrenia, studies sug-
gest that clozapine may be useful for those at high risk of
suicide or aggression. The adverse effects of clozapine are
significant, ranging from acute events such as agranulocytosis
to insidious weight gain and the onset of the metabolic
syndrome. Many studies reported that clozapine treatment
produced the greatest increase in BMI and/or body weight,
closely followed by olanzapine.
The evidence supporting clozapine augmentation is weak
and the benefits observed were moderate at best with the
17
Neuro vo5 no4 2011.indd 17 2011/10/28 10:13 AM

Dovepress
Dovepress
exception of lamotrigine. In the meta-analysis by Tiihonen
et al63 lamotrigine produced significant improvements in the
total PANSS or BPRS score and positive and negative symp-
tom subscales. The use of the NMDA receptor antagonist
memantine was supported by a recent RCT, which reported
improvements in the MMSE, total BPRS, and positive and
negative symptom subscales. Limited evidence suggests that
NMDA agonists may produce clinical improvements in
participants taking olanzapine or risperidone, but not clozap-
ine, while the addition of topiramate to clozapine was of little
benefit at doses low enough to preserve cognitive function.
Clozapine augmentation with aripiprazole resulted in weight
loss or at least halted further weight gain without causing
clinical deterioration or improvement.
Recent RCTs suggest that high-dose olanzapine may be an
important alternative for people intolerant or resistant to clo-
zapine; evidence for the use of ziprasidone in these conditions
is limited. CBT in addition to a nonclozapine antipsychotic for
people not responding or intolerant to clozapine is supported
by small trials. ECT (with or without concurrent antipsychotic
medication) was found to produce small, short-term improve-
ments in global functioning, while significant improvements
specifically in auditory hallucinations were observed with
rTMS. However, more studies are required to determine the
long-term and adverse effects of these treatments.
In terms of clinical practice recommendations where
there is a lack of evidence from RCTs to guide treatment,
clinicians should review single case reports or case series,
which are beyond the scope of this review. When implement-
ing a treatment strategy for which there is limited evidence
clinicians should ensure that the treatment trial is adequate
with objective outcome measures, for example the PANSS.
Larger trials with prospective data using clinically important
outcomes measured by well-validated, approved instruments
are needed to accurately compare the agents available for the
treatment of schizophrenia. Future trials on clozapine aug-
mentation strategies should aim to distinguish between
augmenting agents rather than comparing the results of all
antipsychotic augmentation irrespective of mechanism of
action.
The authors declare no conflicts of interest.
1. Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2(5):e141.
2. Marder SR. Initiatives to promote the discovery of drugs to improve cognitive function in severe mental illness. J Clin Psychiatry. 2006; 67(7):e03.
3. Gureje O, Herrman H, Harvey C, Morgan V, Jablensky A. The Australian national survey of psychotic disorders: Profile of psychosocial disability and its risk factors. Psychol Med. 2002;32(4):639–647.
4. Jablensky A, McGrath J, Herrman H, Castle D, Gureje O, Evans M, et al. Psychotic disorders in urban areas: An overview of the study on low prevalence disorders. Aust N Z J Psychiatry. 2000;34(2): 221–236.
5. Thornicroft G, Tansella M, Becker T, Knapp M, Leese M, Schene A, et al. The personal impact of schizophrenia in europe. Schizophr Res. 2004;69(2–3):125–132.
6. Wheeler A. Sociodemographic, functional and clinical correlates in outpatients with schizophrenia: Comparison with affective disorders. Aust N Z J Psychiatry. 2007;41(10):809–818.
7. Stroup TS, Lieberman JA, McEvoy JP, Davis SM, Swartz MS, Keefe RS, et al. Results of phase 3 of the catie schizophrenia trial. Schizophr Res. 2009;107(1):1–12.
8. Jones PB, Barnes TR, Davies L, Dunn G, Lloyd H, Hayhurst KP, et al. Randomized controlled trial of the effect on quality of life of second- vs first-generation antipsychotic drugs in schizophrenia: Cost utility of the latest antipsychotic drugs in schizophrenia study (cutlass 1). Arch Gen Psychiatry. 2006;63(10):1079–1087.
9. Lehman A LJ, Dixon L. APA practice guidelines: Schizophrenia. Am J Psychiatry. 2004;161(Suppl 2):1–56.
10. Carpenter W Jr, Heinrichs D, Wagman A. Deficit and nondeficit forms of schizophrenia: The concept. Am J Psychiatry. 1988;145(5): 578–583.
11. Erhart SM, Marder SR, Carpenter WT. Treatment of schizophrenia negative symptoms: Future prospects. Schizophr Bull. 32(2): 234–237.
12. Stahl SM, Buckley PF. Negative symptoms of schizophrenia: A problem that will not go away. Acta Psychiatr Scand. 2007;115(1):4–11.
13. Kirkpatrick B, Fenton WS, Carpenter WT, Marder SR. The nimh-matrics consensus statement on negative symptoms. Schizophr Bull. 2006;32(2):214–219.
14. Breier A, Schreiber JL, Dyer J, Pickar D. National institute of mental health longitudinal study of chronic schizophrenia: Prognosis and predictors of outcome. Arch Gen Psychiatry . 1991;48(3): 239–246.
15. Tandon R, Ribeiro SCM, DeQuardo JR, Goldman RS, Goodson J, Greden JF. Covariance of positive and negative symptoms during neuroleptic treatment in schizophrenia: A replication. Biol Psychiatry. 1993;34(7):495–497.
16. Harvey PD, Keefe RSE. Studies of cognitive change in patients with schizophrenia following novel antipsychotic treatment. Am J Psychiatry. 2001;158(2):176–184.
17. Mishara AL, Goldberg TE. A meta-analysis and critical review of the effects of conventional neuroleptic treatment on cognition in schizo-phrenia: Opening a closed book. Biol Psychiatry. 2004;55(10): 1013–1022.
18. Mortimer AM. Cognitive function in schizophrenia – do neuroleptics make a difference? Pharmacol Biochem Behav. 1997;56(4):789–795.
19. Bilder RM. Neurocognitive impairment in schizophrenia and how it affects treatment options. Can J Psychiatry. 1997;42(3):255–264.
20. Green MF, Braff DL. Translating the basic and clinical cognitive neuroscience of schizophrenia to drug development and clinical trials of antipsychotic medications. Biol Psychiatry. 2001;49(4): 374–384.
21. Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 5. Treatment and prevention past, present, and future. Schizophr Res. 2010;122(1–3):1–23.
22. Goldberg TE, Goldman RS, Burdick KE, Malhotra AK, Lencz T, Patel RC, et al. Cognitive improvement after treatment with second-generation antipsychotic medications in first-episode schizophrenia: Is it a practice effect? Arch Gen Psychiatry. 2007;64(10):1115–1122.
18
Neuro vo5 no4 2011.indd 18 2011/10/28 10:13 AM

Dovepress
Dovepress
23. Davidson M, Galderisi S, Weiser M, Werbeloff N, Fleischhacker WW, Keefe RS, et al. Cognitive effects of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: A randomized, open-label clinical trial (eufest). Am J Psychiatry. 2009;166(6):675–682.
24. Hill SK, Bishop JR, Palumbo D, Sweeney JA. Effect of second- generation antipsychotics on cognition: Current issues and future chal-lenges. Expert Rev Neurother. 2009;10(1):43–57.
25. Keefe RSE, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the catie trial. Arch Gen Psychiatry. 2007; 64(6):633–647.
26. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic: A double-blind comparison with chlorprom-azine. Arch Gen Psychiatry. 1988;45(9):789–796.
27. Kerwin RW, Bolonna A. Management of clozapine-resistant schizo-phrenia. Advances in Psychiatric Treatment. 2005;11(2):101–106.
28. Conley RR, Kelly DL. Management of treatment resistance in schizo-phrenia. Biol Psychiatry. 2001;50(11):898–911.
29. Buckley PF, Krowinski AC, Miller DD, Friedman L, Eaton Y, Tronetti M. Clinical and biochemical correlates of ‘high-dose’ clozapine therapy for treatment – refractory schizophrenia. Schizophr Res. 2001; 49(1–2):225–227.
30. Chakos M, Lieberman J, Hoffman E, Bradford D, Sheitman B. Effec-tiveness of second-generation antipsychotics in patients with treatment-resistant schizophrenia: A review and meta-analysis of randomized trials. Am J Psychiatry. 2001;158(4):518–526.
31. Tandon R, Belmaker RH, Gattaz WF, Lopez-Ibor JJ Jr, Okasha A, Singh B, et al. World psychiatric association pharmacopsychiatry section statement on comparative effectiveness of antipsychotics in the treatment of schizophrenia. Schizophr Res. 2008;100(1–3):20–38.
32. Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: A meta-analysis. Lancet. 2009;373(9657):31–41.
33. Essali A, Al-Haj Haasan N, Li C, Rathbone J. Clozapine versus typical neuroleptic medication for schizophrenia. Cochrane Database Syst Rev. 2009;1:CD000059.
34. Geddes J, Freemantle N, Harrison P, Bebbington P. Atypical antipsy-chotics in the treatment of schizophrenia: Systematic overview and meta-regression analysis. BMJ. 2000;321(7273):1371–1376.
35. Leucht S, Wahlbeck K, Hamann J, Kissling W. New generation antipsychotics versus low-potency conventional antipsychotics: A sys-tematic review and meta-analysis. Lancet. 2003;361(9369):1581–1589.
36. Rosenheck RA. Open forum: Effectiveness versus efficacy of second-generation antipsychotics: Haloperidol without anticholinergics as a comparator. Psychiatr Serv. 2005;56(1):85–92.
37. Meltzer HY, Bobo WV, Lee MA, Cola P, Jayathilake K. A randomized trial comparing clozapine and typical neuroleptic drugs in non- treatment-resistant schizophrenia. Psychiatry Res. 2010;177(3): 286–293.
38. Krakowski MI, Czobor P, Nolan KA. Atypical antipsychotics, neu-rocognitive deficits, and aggression in schizophrenic patients. J Clin Psychopharmacol. 2008;28(5):485–493.
39. Krakowski M, Czobor P, Citrome L. Weight gain, metabolic parameters, and the impact of race in aggressive inpatients randomized to double-blind clozapine, olanzapine or haloperidol. Schizophr Res. 2009; 110(1–3):95–102.
40. Leucht S, Komossa K, Rummel-Kluge C, Corves C, Hunger H, Schmid F, et al. A meta-analysis of head-to-head comparisons of second- generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry. 2009;166(2):152–163.
41. Rosenheck R, Cramer J, Xu W, Thomas J, Henderson W, Frisman L, et al. A comparison of clozapine and haloperidol in hospitalized patients with refractory schizophrenia. N Engl J Med. 1997;337(12):809–815.
42. Rummel-Kluge C, Komossa K, Schwarz S, Hunger H, Schmid F, Lobos CA, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: A systematic review and meta-analysis. Schizophr Res. 2010;123:225–233.
43. McEvoy JP, Lieberman JA, Stroup TS, Davis SM, Meltzer HY, Rosenheck RA, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600–610.
44. Chengappa KN, Pollock BG, Parepally H, Levine J, Kirshner MA, Brar JS, et al. Anticholinergic differences among patients receiving standard clinical doses of olanzapine or clozapine. J Clin Psychopharmacol. 2000;20(3):311–316.
45. Engelberg H. Low serum cholesterol and suicide. Lancet. 1992; 339(8795):727–729.
46. Krakowski MI, Czobor P. A prospective longitudinal study of choles-terol and aggression in patients randomized to clozapine, olanzapine, and haloperidol. J Clin Psychopharmacol. 2010;30(2):198–200.
47. Lewis SW, Barnes TR, Davies L, Murray RM, Dunn G, Hayhurst KP, et al. Randomized controlled trial of effect of prescription of clozapine versus other second-generation antipsychotic drugs in resistant schizophrenia. Schizophr Bull. 2006;32(4):715–723.
48. Cohen LJ, Test MA, Brown RL. Suicide and schizophrenia: Data from a prospective community treatment study. Am J Psychiatry. 1990; 147(5):602–607.
49. Meltzer HY, Alphs L, Green AI, Altamura AC, Anand R, Bertoldi A, et al. Clozapine treatment for suicidality in schizophrenia: International suicide prevention trial (intersept). Arch Gen Psychiatry. 2003;60(1): 82–91.
50. Harvey PD, Sacchetti E, Galluzzo A, Romeo F, Gorini B, Bilder RM, et al. A randomized double-blind comparison of ziprasidone vs clozapine for cognition in patients with schizophrenia selected for resistance or intolerance to previous treatment. Schizophr Res. 2008; 105(1–3):138–143.
51. Davies LM, Barnes TR, Jones PB, Lewis S, Gaughran F, Hayhurst K, et al. A randomized controlled trial of the cost-utility of second- generation antipsychotics in people with psychosis and eligible for clozapine. Value Health. 2008;11(4):549–562.
52. Kumra S, Kranzler H, Gerbino-Rosen G, Kester HM, De Thomas C, Kafantaris V, et al. Clozapine and “high-dose” olanzapine in refractory early-onset schizophrenia: A 12-week randomized and double-blind comparison. Biol Psychiatry. 2008;63(5):524–529.
53. Meltzer HY, Bobo WV, Roy A, Jayathilake K, Chen Y, Ertugrul A, et al. A randomized, double-blind comparison of clozapine and high-dose olanzapine in treatment-resistant patients with schizophrenia. J Clin Psychiatry. 2008;69(2):274–285.
54. Sacchetti E, Galluzzo A, Valsecchi P, Romeo F, Gorini B, Warrington L, et al. Ziprasidone vs clozapine in schizophrenia patients refractory to multiple antipsychotic treatments: The mozart study. Schizophr Res. 2009;113(1):112–21.
55. Mouaffak F, Tranulis C, Gourevitch R, Poirier MF, Douki S, Olie JP, et al. Augmentation strategies of clozapine with antipsychotics in the treatment of ultraresistant schizophrenia. Clin Neuropharmacol. 2006; 29:28–33.
56. Barbui C, Signoretti A, Mule S, Boso M, Cipriani A. Does the addition of a second antipsychotic drug improve clozapine treatment? Schizophr Bull. 2009;35(2):458–468.
57. Cipriani A, Boso M, Barbui C. Clozapine combined with different antipsychotic drugs for treatment resistant schizophrenia. Cochrane Database Syst Rev. 2009;3:CD006324.
58. Correll CU, Rummel-Kluge C, Corves C, Kane JM, Leucht S. Antipsychotic combinations vs monotherapy in schizophrenia: A meta-analysis of random-ized controlled trials. Schizophr Bull. 2009;35(2): 443–457.
59. Taylor DM, Smith L. Augmentation of clozapine with a second antipsychotic – a meta-analysis of randomized, placebo-controlled studies. Acta Psychiatr Scand. 2009;119(6):419–425.
60. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9): 1367–1377.
61. Cunningham MO, Jones RSG. The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous gaba release in the rat entorhinal cortex in vitro. Neuropharmacology. 2000;39(11):2139–2146.
19
Neuro vo5 no4 2011.indd 19 2011/10/28 10:13 AM

Dovepress
Dovepress
62. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioural aspects of lamotrigine. Epilepsia. 1991;32(Suppl 2):S4–S8.
63. Tiihonen J, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine-resistant schizophrenia: A systematic review and meta-analysis. Schizophr Res. 2009;109(1–3):10–14.
64. Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153(3):321–330.
65. Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive nmda antagonist, ketamine, in humans: Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3): 199–214.
66. Malhotra AK, Pinals DA, Adler CM, Elman I, Clifton A, Pickar D, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141–150.
67. Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport. 1995;6(6):869–872.
68. Tsai GE, Lin PY. Strategies to enhance n-methyl-d-aspartate receptor-mediated neurotransmission in schizophrenia, a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522–537.
69. White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate enhances gaba-mediated chloride flux and gaba-evoked chloride cur-rents in murine brain neurons and increases seizure threshold. Epilepsy Research. 1997;28(3):167–179.
70. Gibbs JW, Sombati S, DeLorenzo RJ, Coulter DA. Cellular actions of topiramate: Blockade of kainate-evoked inward currents in cultured hippocampal neurons. Epilepsia. 2000;41(Suppl 1):S10–S16.
71. Ängehagen M, Shank R, Hansson E, Rönnbäck L, Ben-Menachem E. Topiramate affects the ability of protein kinase to phosphorylate glutamate receptors activated by kainate Epilepsia. 2001;42 Suppl 7:S10.
72. Arnone D. Review of the use of topiramate for treatment of psychiatric disorders. Ann Gen Psychiatry. 2005;4(1):5.
73. Shank RP, Gardocki JF, Streeter AJ, Maryanoff BE. An overview of the preclinical aspects of topiramate: Pharmacology, pharmacokinetics, and mechanism of action. Epilepsia. 2000;41 Suppl 1:S3–S9.
74. Afshar H, Roohafza H, Mousavi G, Golchin S, Toghianifar N, Sadeghi M, et al. Topiramate add-on treatment in schizophrenia: A randomised, double-blind, placebo-controlled clinical trial. J Psychopharmacol. 2009;23(2):157–162.
75. Arif H, Buchsbaum R, Weintraub D, Pierro J, Resor SR Jr, Hirsch LJ. Patient-reported cognitive side effects of antiepileptic drugs: Predictors and comparison of all commonly used antiepileptic drugs. Epilepsy Behav. 2009;14(1):202–209.
76. Gilliam FG, Veloso F, Bomhof MA, Gazda SK, Biton V, Ter Bruggen JP, et al. A dose-comparison trial of topiramate as monotherapy in recently diagnosed partial epilepsy. Neurology. 2003;60(2):196–1202.
77. Lee S, Sziklas V, Andermann F, Farnham S, Risse G, Gustafson M, et al. The effects of adjunctive topiramate on cognitive function in patients with epilepsy. Epilepsia. 2003;44(3):339–347.
78. Thompson PJ, Baxendale SA, Duncan JS, Sander JW. Effects of topiramate on cognitive function. J Neurol Neurosurg Psychiatry. 2000; 69(5):636–641.
79. Muscatello MRA, Bruno A, Pandolfo G, Micò U, Bellinghieri PM, Scimeca G, et al. Topiramate augmentation of clozapine in schizophrenia: A double-blind, placebo-controlled study. J Psychopharmacology. 2010 Jul 8. [Epub ahead of print].
80. Streiner DL. The case of the missing data: Methods of dealing with dropouts and other research vagaries. Can J Psychiatry. 2002;47(1): 68–75.
81. Andreasen NC. The Scale for Assessment of Positive Symptoms (SAPS). Iowa City, IA; 1984. http://www.movementdisorders.org/UserFiles/file/Long_SAPS_2000_publish(1).pdf.
82. Deutsch SI, Schwartz BL, Rosse RB, Mastropaolo J, Marvel CL, Drapalski AL. Adjuvant topiramate administration: A pharmacologic strategy for addressing nmda receptor hypofunction in schizophrenia. Clin Neuropharmacol. 2003;26(4):199–206.
83. De Lucena D, Fernandes BS, Berk M, Dodd S, Medeiros DW, Pedrini M, et al. Improvement of negative and positive symptoms in treatment-refractory schizophrenia: A double-blind, randomized, placebo- controlled trial with memantine as add-on therapy to clozapine. J Clin Psychiatry. 2009;70(10):1416–1423.
84. Lancu I, Olmer A. The minimental state examination – an up-to-date review. Harefuah. 2006;145(9):687–690, 701.
85. Meisner F, Scheller C, Kneitz S, Sopper S, Neuen-Jacob E, Riederer P, et al. Memantine upregulates bdnf and prevents dopamine deficits in siv-infected macaques: A novel pharmacological action of memantine. Neuropsychopharmacology. 2008;33(9):2228–2236.
86. Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neurophar-macology. 1998;37(12):1553–1561.
87. Gama CS, Andreazza AC, Kunz M, Berk M, Belmonte-de-Abreu PS, Kapczinski F. Serum levels of brain-derived neurotrophic factor in patients with schizophrenia and bipolar disorder. Neurosci Lett. 2007; 420(1):45–48.
88. Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD, et al. A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychop-harmacology. 2009;34(5):1322–1329.
89. Freudenreich O, Henderson DC, Macklin EA, Evins AE, Fan X, Cather C, et al. Modafinil for clozapine-treated schizophrenia patients: A double-blind, placebo-controlled pilot trial. J Clin Psychiatry. 2009;70(12):1674–1680.
90. Goff DC, Lamberti JS, Leon AC, Green MF, Miller AL, Patel J, et al. A placebo-controlled add-on trial of the ampakine, cx516, for cognitive deficits in schizophrenia. Neuropsychopharmacology. 2008;33(3): 465–472.
91. Tamminga CA, Carlsson A. Partial dopamine agonists and dopamin-ergic stabilizers, in the treatment of psychosis. Curr Drug Targets CNS Neurol Disord. 2002;1(2):141.
92. Rivas-Vasquez RA. Aripiprazole: A novel antipsychotic with dopamine stabilising properties. Prof Psychol Res Pr. 2003;34(1):108–111.
93. Millar H, Felter C, Landsberg W. The effects of aripiprazole in combi-nation with clozapine: Patient functioning results from a double-blind, 16-week study in patients with schizophrenia (cn138-170). J Psychop-harmacol. 2008;22(5):A17.
94. Fleischhacker WW, Heikkinen ME, Olie JP, Landsberg W, Dewaele P, McQuade RD, et al. Effects of adjunctive treatment with aripiprazole on body weight and clinical efficacy in schizophrenia patients treated with clozapine: A randomized, double-blind, placebo-controlled trial. Int J Neuropsychopharmcol. 2010;13(8):1115–1125.
95. Kumra S, Kranzler H, Gerbino-Rosen G, Kester HM, DeThomas C, Cullen K, et al. Clozapine versus “high-dose” olanzapine in refractory early-onset schizophrenia: An open-label extension study. J Child Adolesc Psychopharmacol. 2008;18(4):307–316.
96. Barretto EM, Kayo M, Avrichir BS, Sa AR, Camargo MG, Napolitano IC, et al. A preliminary controlled trial of cognitive behavioral therapy in clo-zapine-resistant schizophrenia. J Nerv Ment Dis. 2009;197(11): 865–868.
97. Turkington D, Sensky T, Scott J, Barnes TRE, Nur U, Siddle R, et al. A randomized controlled trial of cognitive-behavior therapy for persistent symptoms in schizophrenia: A five-year follow-up. Schizophr Res. 2008;98(1–3):1–7.
98. Matheson SL, Green MJ, Loo C, Carr VJ. Quality assessment and comparison of evidence for electroconvulsive therapy and repetitive transcranial magnetic stimulation for schizophrenia: A systematic meta-review. Schizophr Res. 2010;118(1–3):201–210.
20
Neuro vo5 no4 2011.indd 20 2011/10/28 10:13 AM

Dovepress
Dovepress
99. Burt T, Lisanby SH, Sackeim HA. Neuropsychiatric applications of transcranial magnetic stimulation: A meta analysis. Int J Neuropsy-chopharmacol. 2002;5(1):73–103.
100. Tharyan P, Adams CE. Electroconvulsive therapy for schizophrenia. Cochrane Database Syst Rev. 2005;18(2):CD000076.
101. Aleman A, Sommer IE, Kahn RS. Efficacy of slow repetitive transcranial magnetic stimulation in the treatment of resistant auditory hallucinations in schizophrenia: A meta-analysis. J Clin Psychiatry. 2007;68(3):416–421.
102. Lévy-Rueff M, Gourevitch R, Lôo H, Olié J-P, Amado I. Maintenance electroconvulsive therapy: An alternative treatment for refractory schizophrenia and schizoaffective disorders. Psychiatry Res. 2010; 175(3):280–283.
21
Neuro vo5 no4 2011.indd 21 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 22 2011/10/28 10:13 AM

Neuropsychiatric Disease and Treatment Dovepress
Dovepress
open access to scientific and medical research
This article highlights the evidence linking depression to increased inflammatory drive
and explores putative mechanisms for the association by reviewing both preclinical and clinical
literature. The enzyme indoleamine 2,3-dioxygenase is induced by proinflammatory cytokines
and may form a link between immune functioning and altered neurotransmission, which results
in depression. Increased indoleamine 2,3-dioxygenase activity may cause both tryptophan deple-
tion and increased neurotoxic metabolites of the kynurenine pathway, two alterations which have
been hypothesized to cause depression. The tryptophan-kynurenine pathway is comprehensively
described with a focus on the evidence linking metabolite alterations to depression. The use of
immune-activated groups at high risk of depression have been used to explore these hypotheses;
we focus on the studies involving chronic hepatitis C patients receiving interferon-alpha, an
immune activating cytokine. Findings from this work have led to novel strategies for the future
development of antidepressants including inhibition of indoleamine 2,3-dioxygenase, moderating
the cytokines which activate it, or addressing other targets in the kynurenine pathway.
depression, inflammation, indoleamine 2,3-dioxygenase, kynurenine, serotonin,
tryptophan
Clinical depression is extremely common and debilitating. It is ranked by the World
Health Organization as the fourth largest cause of burden amongst all diseases and
the leading nonfatal disease burden.1 Current treatments have only moderate efficacy,
with around 35% remission after initial treatment and approximately 70% remission
after four cumulative treatment trials.2 Therefore it is necessary to look beyond cur-
rently characterized neurotransmitter systems to understand the pathophysiology of
depression in order to produce more effective treatments in the long-term.
Emerging evidence demonstrates that: a) major depression is associated with
increased inflammatory drive;3–5 and b) provoking an acute inflammatory response in
healthy humans can result in depression-like behaviors and symptoms.6,7 The nature
of these associations has yet to be delineated with respect to causality. Determining a
plausible biological mechanism remains an important step. In this article we review
a putative mechanism by which increased inflammation may affect mood, by altering
activity of the enzyme indoleamine 2,3-dioxygenase (IDO).
There is a growing body of literature that suggests that major depression is associ-
ated with an increased inflammatory drive. People with depression display increased
23
Neuro vo5 no4 2011.indd 23 2011/10/28 10:13 AM

Dovepress
Dovepress
plasma concentrations of pro-inflammatory cytokines such
as: interleukin-1 (IL-1)3,8 (also increased in cerebrospinal
fluid [CSF]9), interleukin-6 (IL-6),3,4,9–11 tumor necrosis
factor (TNF)4,12 and other acute phase proteins, such as
C-reactive protein (CRP),3 haptoglobin11 and neopterin.13
There have been some negative findings,14,15 but the over-
all picture is sufficient to support both a positive meta-
analysis exploring the associations of CRP, IL-1, IL-6
and depression3 and the suggestion that plasma IL-6 and
soluble IL-2-receptor should be considered biomarkers of
depression.16
Treatment of depression with antidepressants may
reverse derangements in these inflammatory markers.17
Fluoxetine treatment for depression reduces serum IL-6 in
patients.18 Imipramine, clomipramine, venlafaxine, fluox-
etine, sertraline and trazodone have been shown to reduce
the interferon-gamma (INF- /IL-10 ratio of in vitro
human blood samples (a ratio of pro-inflammatory/anti-
inflammatory drive), consistent with an anti-inflammatory
effect.19–21 In addition, nonresponders to selective serotonin
reuptake inhibitor (SSRI) medication continue to exhibit
raised IL-6 levels, raising the possibility that response to
treatment is linked to a reduction of IL-6.22 Preliminary
evidence also exists that an increased body temperature
may also be present in depression and reversed by suc-
cessful treatment.23
Abnormalities of plasma cytokines may occur in various
psychiatric disorders. In bipolar disorder, increased IL-1,
IL-6 and TNF have been reported at differing stages of
the illness.24 In schizophrenia, less consistent results have
been found, but a recent meta-analysis reported increased
plasma IL-6 and IL-1 receptor antagonist levels.25 However,
exploring these illnesses in detail is beyond the scope of
this review, which focuses upon the changes seen in major
depression.
“Sickness behavior” is a characteristic constellation
of symptoms (hypomotility, hyperthermia, hypophagia,
hyperalgesia, decreased interest in exploration, decreased
sexual activity, increased sleep) observed in animals fol-
lowing immune activation26–28 that has been proposed to be
a model of depression.29 Activating an immune response
by injecting lipopolysaccharide (LPS),30 IL-1,31 or IFN32
results in characteristic sickness behavior. In addition, an
acute inflammatory challenge has been reported to produce
depression-like responses in two other animal models of
depression, the tail suppression and sucrose consumption
tests, after the initial illness behaviors have subsided.33
The biochemical and behavioral effects of challenges like
these may also be augmented by social stress,34 analogous
to social risk factors for depression.35 Pretreatment with
the antidepressant imipramine has been found to attenuate
LPS-induced sickness behavior.30
Provoking an acute inflammatory response in healthy
humans, for example via injection of endotoxin,6,7 IL-6,36
or IFN- ,37 also produces symptoms similar to those seen
in depression (such as fatigue, lack of motivation, anorexia,
poor sleep). Although these symptoms are short-lived, subtle
cognitive symptoms similar to those seen in depression are
also present. These include feelings of social isolation6,38
and psychomotor slowing.37 The symptoms produced by
challenge tests such as these resolve quickly and are not
prolonged as is seen in depression.
Indoleamine 2,3-dioxygenase (IDO) and its hepatic
equivalent tryptophan 2,3-dioxygenase (TDO) oxygenate
tryptophan to form kynurenine39 (Figure 1, tryptophan
metabolic pathway). The majority of dietary tryptophan
is metabolized through this pathway with less than 1%
eventually being available for conversion (via hydroxyla-
tion by tryptophan hydroxylase, TPH, and decarboxylation)
into 5HT in the brain.40 Under normal circumstances,
TDO is the dominant enzyme, but IDO is subject to
induction during immune activation. At such times the
effect of increasing the combined availability of IDO and
TDO means that the overall capacity of the kynurenine
pathway is much increased. Therefore serum tryptophan
concentration can be reduced by 25%–50%, leaving pro-
portionally less tryptophan available for conversion to
serotonin.41–43
IDO is ubiquitous throughout the organs and present in
human immune cells including macrophages and microglia.44
Interferons are important in the induction of IDO. The sites
of action are two IFN-stimulated response elements (ISREs)
and IFN- activated site (GAS) element sequences found in
the 5 promoter region of the IDO gene.45,46 IDO can be stimu-
lated by INF- in macrophages and microglia.47,48 However,
other cytokines such as TNF in combination with IL-6 or
IL-1 can induce IDO via signal transducer and activator of
transcription protein (STAT)-independent pathways involv-
ing p38 mitogen-activated protein kinase (p38 MAPK) and
nuclear factor-kappa B (NF- B).49
A proxy measure for in vivo IDO activity, like many
enzymes, is the ratio of product:substrate (in this case
kynurenine:tryptophan). Thus, an increase in the ratio reflects
24
Neuro vo5 no4 2011.indd 24 2011/10/28 10:13 AM

Dovepress
Dovepress
greater enzyme activity, a decrease indicates lower activity
and no change implies the same activity.50
Animal models support the hypothesis that immune-related
sickness behavior may be related to increased activity of
IDO. IDO activity, measured by either the plasma concen-
tration of kynurenine pathway metabolites or IDO mRNA
expression, is increased in animal sickness behavior.51 This
activation is partly mediated by IFN- and TNF, since IFN-
knockout mice, and animals with prior treatment with the
TNF antagonist etanercept, both show reduced IDO activa-
tion and depressive behaviors (in the forced swim and tail
suspension tests).52 IDO knock-out mice lack the expected
depressive behaviors secondary to an immune challenge,
despite normal cytokine responses.51 In addition, inhibition
of IDO blocks the depressive behavior in these models51,53 and
administration of kynurenine induces depressive behavior in
a dose-dependent manner.53
As described above, acute inflammatory challenges reproduce
sickness behavior and depressive cognitions in healthy
humans. Clearly, it is ethically difficult to continue challenges
like these for prolonged periods due to the high degree of
morbidity they cause. Therefore, an alternative is to study
patient cohorts who require long-term pro-inflammatory
treatments for an underlying condition.
An ideal high-risk population that can be used to assess
the effects of increased inflammatory drive is chronic
hepatitis C (HCV) patients being treated with IFN-
therapy. HCV is a common illness, affecting approximately
170 million people worldwide.54,55 Without treatment, it
causes considerable morbidity and mortality; it leads to
chronic infection in approximately 85% of cases, cirrhosis
in 15%–20%, and in cirrhosis patients, 1%–4% progress
to hepatocellular carcinoma.56 HCV patients undergo an
IFN- -based treatment regime for between 6 and 12 months.
Tryptophan
Kynurenine
TDO
TPH
KAT I
KAT II
Kynureninase*
Serotonin pathway
Kynurenine pathway
NMDA agonist
NMDA antagonist
Putative neurotoxin
IDO induced by cytokines
Potentially induced by cytokines
3-hydroxyanthranilic
acid oxygenase*
QPRT
Kynurenine
hydroxylase*
Kynureninase*
IDO ¶
5-HTP 5-HTMelatonin
Kynurenic acid
Xanthurenic acid
3-Hydroxyanthranilic acid
NAD
3-Hydroxykynurenine
Quinolinic acid
Anthranilic acid
*¶
25
Neuro vo5 no4 2011.indd 25 2011/10/28 10:13 AM

Dovepress
Dovepress
During this time there are high rates of depression, estimated
at approximately 25% 57,58 and 33%59,60 (although some stud-
ies report higher prevalences, a precise figure is difficult to
determine due to methodological differences between studies;
those reporting higher rates report self-rated symptoms rather
than utilizing standardized objective depression scales61).
In HCV, IFN- increases inflammatory drive with eleva-
tions in pro-inflammatory cytokines (eg, IL-1, IL-6, IL-8 and
TNF62,63) similar to that observed in depression.3 Although
IFN- is peripherally administered, increases in IFN- , IL-6
and monocyte chemoattractant protein-1 have been observed
in the CSF of this group, providing evidence of central
immunomodulatory effects. In conjunction with the increase
in pro-inflammatory cytokines, the kynurenine:tryptophan
ratio is increased (reflecting increased IDO activity) in both
the blood and CSF.64,65
There are two current hypotheses regarding the mecha-
nism of how increased inflammatory drive and IDO activa-
tion may cause depression in the HCV group – tryptophan
depletion and kynurenine toxicity.
Increased IDO activity should reduce the availabil-
ity of its substrate, the dietary essential amino acid
tryptophan.66 Serotonin (5HT) is produced from tryptophan
via 5- hydroxytryptophan (5HTP). Under normal conditions
the rate-limiting enzyme tryptophan hydroxylase is only
about 50% saturated. Therefore 5HT synthesis varies with
tryptophan availability.67 The evidence linking 5HT dysfunc-
tion to depressive illness has been well described. Many
effective antidepressants (such as SSRIs) work primarily on
increasing serotonin availability in the synaptic cleft.68 This
antidepressant effect can be temporarily reversed using the
acute tryptophan depletion (ATD) technique, which acutely
lowers 5HT by lowering the brain availability of its precursor
tryptophan.69,70 Lower concentrations of plasma tryptophan
have also been reported in depression.71 Imaging studies have
also reported central changes in the 5HT system in depres-
sion including reduced 5HT transporters,72,73 reduced 5HT-1a
receptors,74,75 and reduced 5HT-2a receptors.76
In the HCV cohort, reductions in plasma tryptophan
(and 5HT, although this is an inexact measure, as much
plasma 5HT is stored by platelets and released when they are
stimulated, such as in venepuncture, resulting in inconsistent
results77) have been observed.78 SSRIs are highly effective
at treating or preventing IFN- associated depression.79,80
However, tryptophan does not have clear access to the
brain from the plasma: 95% is protein-bound in the plasma,
leaving 5% free to access the CNS.81 It is transported across
the blood–brain barrier via active transport in competition
with the other large neutral amino acids (LNAAs): valine,
leucine, isoleucine, methionine, phenylalanine and tyrosine.82
Plasma tryptophan concentrations correlate poorly with
those of the CSF.83 Thus, a more accurate measure of brain
tryptophan availability is the tryptophan:LNAA ratio.84 This
ratio remains unchanged in IFN- therapy for hepatitis C
and does not appear to vary with depressive symptoms.64
In keeping with this, CSF levels of tryptophan do not change
during interferon treatment.65 However, this does not entirely
disprove the 5HT reduction theory, as the 5HT metabolite
5-hydroxyindoleacetic acid (5HIAA) is reduced and this
reduction correlates with depressive symptoms.85 Therefore
although absolute tryptophan levels appear not to be altered,
an overall reduction in brain 5HT turnover may still be related
to depression.
Different measures of brain 5HT functioning are required
to further delineate these changes. A sensitive method is
polysomnography. 5HT is important in the regulation of
sleep86 and sleep disturbances prior to interferon treat-
ment have been suggested to predict later depression.87
Serotonergic compounds (such as SSRIs) increase time
until onset of rapid eye movement (REM) sleep (increased
REM latency). Decreasing serotonin availability, by ATD,
has the opposite effect, decreased REM latency.88–90 Altera-
tions in REM latency have been utilized by our group to
detect differences in physiological potency between different
SSRIs,91 proving this technique’s sensitivity to alterations
in central 5HT functioning. Using a within-subjects design
we observed no significant alteration in REM latency after
6 weeks IFN- treatment (Pers comm, David N Christmas,
2011). Our finding of no decrease in REM latency is similar
to an independent study by a different group that observed
an REM latency increase during IFN- treatment.92 The dif-
ference between these studies has yet to be explained, but
importantly neither observed the decrease in REM latency
predicted by the 5HT depletion hypothesis.
An alternative explanation for the lack of decrease in
REM latency is that IDO activation may also affect REM
sleep via a different mechanism to 5HT depletion. Pre-
clinical evidence suggests that glutamatergic neurons are
important in the genesis of REM sleep: indeed kynurenic
acid (which is produced from kynurenine and is an N-methyl-
D-aspartate [NMDA] antagonist) can abolish experimentally
induced REM sleep.93 The control of REM sleep is complex,
with 5HT, acetylcholine, glutamate and gamma-amino
hydroxybutyric acid (GABA) all playing important roles.94
26
Neuro vo5 no4 2011.indd 26 2011/10/28 10:13 AM

Dovepress
Dovepress
At risk of oversimplification, REM-on neurons appear to
be glutamatergic (under tonic inhibition by GABA neu-
rons) and REM-off neurons serotonergic or noradrenergic.
Therefore, altering the balance between kynurenic acid and
quinolinic acid (also on the kynurenine metabolic pathway
and an NMDA agonist [Figure 1]) may also alter both REM
latency and duration. However, IFN- does not alter the
CSF kynurenic acid:quinolinic acid ratio.65 Therefore the
preliminary conclusion is that neither central 5HT function-
ing nor NMDA activation is altered during IFN- treatment.
However, further research is required to form a conclusive
picture.
The products of IDO activation have also been hypothesized
to cause depression. Under normal circumstances the liver
enzyme TDO95 metabolizes tryptophan into kynurenine. TDO
is not induced by immune activation, but is constitutively
active and is induced by tryptophan, tyrosine, histidine,
glucocorticoids and kynurenine. TDO primarily serves
nicotinamide adenine dinucleotide synthesis (Figure 1) and
is the rate-limiting enzyme of the pathway. Under circum-
stances of immune activation, IDO activity is increased,
causing detectable increases in kynurenine and decreases
in tryptophan.41,43,96 Kynurenine is mostly hydroxylated
(kynurenine hydroxylase) into 3-hydroxykynurenine (3-HK).
Kynureninase acts upon both 3-HK and kynurenine; on 3-HK
to form 3-hydroxyanthranilic acid (3-HAA); and on kynure-
nine to form anthranilic acid (although the latter conversion
accounts for only a minority of kynureninase activity).
3-HAA is converted into quinolinic acid by 3-hydroxyanthra-
nilic acid oxygenase. Kynurenine can also be converted into
kynurenic acid by kynurenine aminotransferase I and 3-HK
into xanthurenic acid by kynurenine aminotransferase II.
Some of these kynurenine metabolites modulate neu-
rotransmission and some may be directly neurotoxic. As
mentioned above, quinolinic acid is an NMDA agonist and
kynurenic acid an NMDA antagonist. 3-HK is believed to
be neurotoxic due to increased formation of reactive oxygen
species involved in neuronal apoptosis.97,98 Quinolinic
acid may also be neurotoxic due to increased oxidative
stress,99,100 whereas kynurenic acid has been postulated to be
neuroprotective.101 Under conditions of immune activation,
preclinical evidence suggests kynurenine aminotransferase
activity is unchanged whereas, in addition to IDO, kynure-
nine 3-hydroxylase, kynureninase and 3-hydroxyanthranilic
acid oxygenase activity may be increased51,102 (although the
evidence for induction of the last enzyme is contradictory,
possibly due to species differences). Therefore, kynurenine
metabolism is shifted toward the 3-HK/quinolinic acid
pathway and away from the kynurenic acid pathway, which
should result in greater neurotoxic and reduced neuropro-
tective metabolites. The relative balance of neurotoxic and
neuroprotective pathways of kynurenine metabolism can
be assessed indirectly in vivo by the kynurenine:kynurenic
acid ratio.64,103
During IFN- treatment, the plasma kynurenine:kynurenic
acid ratio is increased and this correlates with depressive
symptoms.64 CSF kynurenine and quinolinic acid also
increase and these increases correlate with increases in
depressive symptoms. However, the kynurenine:kynurenic
acid ratio does not alter as CSF kynurenic acid also rises.65
One cross-sectional study observed increased IDO activ-
ity and a decreased kynurenic acid:kynurenine ratio (reflect-
ing a shift towards neurotoxicity) in otherwise healthy major
depression sufferers compared to controls.104 However, as yet
no studies have been undertaken to identify whether these
differences resolve once the depressive episode has been
successfully treated.
Some preliminary studies have already reported possible
efficacy of anti-inflammatory drugs in depression. A double-
blind, randomized clinical trial reported an advantage of
reboxetine and the cyclo-oxygenase-2 inhibitor celecoxib
over reboxetine and placebo.105 In addition, an open pilot
study reported a benefit of augmentation with aspirin in
depressed patients with no early response to an SSRI.106
However, both these cases require larger more robust trials
to prove their efficacy. A further problem may occur with
this route; there is a large increased risk of gastrointestinal
bleeding when SSRIs and nonsteroidal anti-inflammatory
drugs are combined107 and cyclo-oxygenase-2 inhibitors alone
have been associated with increased cardiovascular and all-
cause mortality above other anti-inflammatory drugs.108,109
Therefore alternative strategies may be required to maintain
a favorable risk–benefit ratio.
Utilizing the above evidence, it is possible to identify
future novel pharmacological targets for antidepressants. The
first target could be antagonizing or reducing IDO activity.
The drug 1-methlytryptophan can inhibit IDO and has been
successful in reducing depressive behaviors following
inflammatory challenges in animal models.51 Clinical trials
using 1-methyltryptophan have commenced in humans as
a putative anticancer agent (trial identifier NCT00567931,
http://clinicaltrials.gov). However, there is some debate as
27
Neuro vo5 no4 2011.indd 27 2011/10/28 10:13 AM

Dovepress
Dovepress
to whether it inhibits human IDO in vivo.110 In addition, IDO
may have immunosuppressive actions in itself, highlighting
the complexity of immune functioning.110
A different avenue may be to block the pro-inflammatory
cytokines that are raised in depression and known to induce
IDO. Monoclonal antibodies are available for human use, to
treat rheumatoid arthritis or inflammatory bowel disease, to
block both TNF (such as infliximab or etanercept) and IL-6
(tocilizumab). Indeed, a clinical trial at Emory University
evaluating the efficacy of infliximab in treatment resis-
tant depression is approaching completion (trial identifier
NCT00463580, http://clinicaltrials.gov).
A third approach may be to block the downstream
actions of excess kynurenine metabolites. As the ratio of
NMDA receptor agonism:antagonism appears to be shifted
towards agonism in depression, NMDA antagonists may
have antidepressant effects. Unfortunately human subjects
exposed to direct NMDA antagonists have experienced
serious side effects such as sedation, memory impairment
and psychosis.111,112 Thus design of NMDA manipulating
compounds may require novel strategies, such as targeting
NMDA cotransmitters. Despite this, several small stud-
ies have shown promising results for the use of ketamine,
an NMDA receptor antagonist, for treatment-resistant
depression.113–117
In summary, major depression appears to be accompanied
by increases in some pro-inflammatory cytokines. In keep-
ing with this, inducing increased inflammation in animals
or humans results in characteristic sickness behavior, or
full-blown major depression in the high-risk HCV cohort.
In tandem with markers of increased inflammation, IDO
is activated, both peripherally and centrally. Although the
evidence falls short of proving a causative link between
inflammation, IDO and mood, the diversity and congruence
of evidence suggests this pathway is a promising field for
future drug targets.
The authors report no conflicts of interest in this work.
1. Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–392.
2. Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917.
3. Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71(12):171–186.
4. Kim YK, Na KS, Shin KH, Jung HY, Choi SH, Kim JB. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(5):1044–1053.
5. Maes M, Bosmans E, Meltzer HY. Immunoendocrine aspects of major depression. Relationships between plasma interleukin-6 and soluble interleukin-2 receptor, prolactin and cortisol. Eur Arch Psychiatry Clin Neurosci. 1995;245(3):172–178.
6. Eisenberger NI, Inagaki TK, Mashal NM, Irwin MR. Inflammation and social experience: an inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain Behav Immun. 2010;24(4):558–563.
7. Reichenberg A, Yirmiya R, Schuld A, et al. Cytokine-associated emo-tional and cognitive disturbances in humans. Arch Gen Psychiatry. 2001;585(5):445–452.
8. Owen BM, Eccleston D, Ferrier IN, Young AH. Raised levels of plasma interleukin-1beta in major and postviral depression. Acta Psychiatr Scand. 2001;103(3):226–228.
9. Levine J, Barak Y, Chengappa KN, Rapoport A, Rebey M, Barak V. Cerebrospinal cytokine levels in patients with acute depression. Neuropsychobiology. 1999;40(4):171–176.
10. Maes M, Meltzer HY, Bosmans E, et al. Increased plasma concentra-tions of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J Affect Disord. 1995;34(4):301–309.
11. Zorrilla EP, Luborsky L, McKay JR, et al. The relationship of depression and stressors to immunological assays: a meta-analytic review. Brain Behav Immun. 2001;15(3):199–226.
12. Hestad KA, Tonseth S, Stoen CD, Ueland T, Aukrust P. Raised plasma levels of tumor necrosis factor alpha in patients with depression: normal-ization during electroconvulsive therapy. J ECT. 2003;19(4):183–188.
13. Maes M, Scharpe S, Meltzer HY, et al. Increased neopterin and interferon-gamma secretion and lower availability of L-tryptophan in major depression: further evidence for an immune response. Psychiatry Res. 1994;54(2):143–160.
14. Brambilla F, Maggioni M. Blood levels of cytokines in elderly patients with major depressive disorder. Acta Psychiatr Scand. 1998;97(4):309–313.
15. Carpenter LL, Heninger GR, Malison RT, Tyrka AR, Price LH. Cere-brospinal fluid interleukin (IL)-6 in unipolar major depression. J Affect Disord. 2004;79(1–3):285–289.
16. Mossner R, Mikova O, Koutsilieri E, et al. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J Biol Psychiatry. 2007;8(3):141–174.
17. Maes M, Yirmyia R, Noraberg J, et al. The inflammatory and neu-rodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. 2009;24(1):27–53.
18. Sluzewska A, Rybakowski JK, Laciak M, Mackiewicz A, Sobieska M, Wiktorowicz K. Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine. Ann N Y Acad Sci. 1995;762:474–476.
19. Lin A, Song C, Kenis G, et al. The in vitro immunosuppressive effects of moclobemide in healthy volunteers. J Affect Disord. 2000;58(1):69–74.
20. Maes M, Song C, Lin AH, et al. Negative immunoregulatory effects of antidepressants: inhibition of interferon-gamma and stimulation of interleukin-10 secretion. Neuropsychopharmacology. 1999;20(4):370–379.
21. Kubera M, Lin AH, Kenis G, Bosmans E, van BD, Maes M. Anti-Inflammatory effects of antidepressants through suppression of the interferon-gamma/interleukin-10 production ratio. J Clin Psychopharmacol. 2001;21(2):199–206.
22. O’Brien SM, Scully P, Fitzgerald P, Scott LV, Dinan TG. Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J Psychiatr Res. 2007;41(3–4):326–331.
28
Neuro vo5 no4 2011.indd 28 2011/10/28 10:13 AM

Dovepress
Dovepress
23. Szuba MP, Guze BH, Baxter LR Jr. Electroconvulsive therapy increases circadian amplitude and lowers core body temperature in depressed subjects. Biol Psychiatry. 1997;42(12):1130–1137.
24. Berk M, Kapczinski F, Andreazza AC, et al. Pathways underly-ing neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev. 2011;35(3):804–817.
25. Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry. 2008;63(8):801–808.
26. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123–137.
27. Dantzer R. Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci. 2001;933:222–234.
28. Kent S, Bluthe RM, Kelley KW, Dantzer R. Sickness behavior as a new target for drug development. Trends Pharmacol Sci. 1992;13(1):24–28.
29. Smith RS. The macrophage theory of depression. Med Hypotheses. 1991;35(4):298–306.
30. Yirmiya R. Endotoxin produces a depressive-like episode in rats. Brain Res. 1996;711(1–2):163–174.
31. Kent S, Rodriguez F, Kelley KW, Dantzer R. Reduction in food and water intake induced by microinjection of interleukin-1 beta in the ventromedial hypothalamus of the rat. Physiol Behav. 1994;56(5): 1031–1036.
32. Fahey B, Hickey B, Kelleher D, O’Dwyer AM, O’Mara SM. The widely-used anti-viral drug interferon-alpha induces depressive- and anxiogenic-like effects in healthy rats. Behav Brain Res. 2007;182(1): 80–87.
33. Frenois F, Moreau M, O’Connor J, et al. Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expres-sion of depressive-like behavior. Psychoneuroendocrinology. 2007;32(5):516–531.
34. Anisman H, Poulter MO, Gandhi R, Merali Z, Hayley S. Interferon-alpha effects are exaggerated when administered on a psychosocial stressor backdrop: cytokine, corticosterone and brain monoamine variations. J Neuroimmunol. 2007;186(1–2):45–53.
35. Righetti-Veltema M, Conne-Perreard E, Bousquet A, Manzano J. Risk factors and predictive signs of postpartum depression. J Affect Disord. 1998;49(3):167–180.
36. Spath-Schwalbe E, Hansen K, Schmidt F, et al. Acute effects of recombinant human interleukin-6 on endocrine and central ner-vous sleep functions in healthy men. J Clin Endocrinol Metab. 1998;83(5):1573–1579.
37. Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol Psychiatry. 2008;63(11):1022–1029.
38. Eisenberger NI, Inagaki TK, Rameson LT, Mashal NM, Irwin MR. An fMRI study of cytokine-induced depressed mood and social pain: the role of sex differences. Neuroimage. 2009;47(3):881–890.
39. Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303(1):1–10.
40. Bender DA. Biochemistry of tryptophan in health and disease. Mol Aspects Med. 1983;6(2):101–197.
41. Werner ER, Fuchs D, Hausen A, et al. Tryptophan degradation in patients infected by human immunodeficiency virus. Biol Chem Hoppe Seyler. 1988;369(5):337–340.
42. Fuchs D, Forsman A, Hagberg L, et al. Immune activation and decreased tryptophan in patients with HIV-1 infection. J Interferon Res. 1990;10(6):599–603.
43. Fuchs D, Moller AA, Reibnegger G, Stockle E, Werner ER, Wachter H. Decreased serum tryptophan in patients with HIV-1 infection corre-lates with increased serum neopterin and with neurologic/psychiatric symptoms. J Acquir Immune Defic Syndr. 1990;39(9):873–876.
44. Dale WE, Dang Y, Brown OR. Tryptophan metabolism through the kynurenine pathway in rat brain and liver slices. Free Radic Biol Med. 2000;29(2):191–198.
45. Konan KV, Taylor MW. Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2,3-dioxygenase gene. J Biol Chem. 1996;271(32):19140–19145.
46. Chon SY, Hassanain HH, Pine R, Gupta SL. Involvement of two regulatory elements in interferon-gamma-regulated expression of human indoleamine 2,3-dioxygenase gene. J Interferon Cytokine Res. 1995;15(6):517–526.
47. Heyes MP, Saito K, Markey SP. Human macrophages convert L- tryptophan into the neurotoxin quinolinic acid. Biochem J. 1992;283(Pt 3):633–635.
48. Aberati-Giani D, Cesura AM. Expression of the kynurenine enzymes in macrophages and microglial cells: regulation by immune modulators. Amino Acids. 1998;14(1–3):251–255.
49. Fujigaki H, Saito K, Fujigaki S, et al. The signal transducer and acti-vator of transcription 1alpha and interferon regulatory factor 1 are not essential for the induction of indoleamine 2,3-dioxygenase by lipopoly-saccharide: involvement of p38 mitogen-activated protein kinase and nuclear factor-kappaB pathways, and synergistic effect of several proinflammatory cytokines. J Biochem. 2006;139(4):655–662.
50. Schrocksnadel K, Wirleitner B, Winkler C, Fuchs D. Monitoring tryptophan metabolism in chronic immune activation. Clin Chim Acta. 2006;364(1–2):82–90.
51. O’Connor JC, Lawson MA, Andre C, et al. Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior. J Immunol. 2009;182(5):3202–3212.
52. O’Connor JC, Andre C, Wang Y, et al. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3- dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. 2009;29(13): 4200–4209.
53. O’Connor JC, Lawson MA, Andre C, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2009;14(5):511–522.
54. Alter MJ. Epidemiology of hepatitis C. Hepatology. 1997;26(3 Suppl 1): 62S–65S.
55. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(9):558–567.
56. Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345(1):41–52.
57. Dieperink E, Ho SB, Thuras P, Willenbring ML. A prospective study of neuropsychiatric symptoms associated with interferon-alpha-2b and ribavirin therapy for patients with chronic hepatitis C. Psychosomatics. 2003;44(2):104–112.
58. Horikawa N, Yamazaki T, Izumi N, Uchihara M. Incidence and clinical course of major depression in patients with chronic hepatitis type C undergoing interferon-alpha therapy: a prospective study. Gen Hosp Psychiatry. 2003;25(1):34–38.
59. Hauser P, Khosla J, Aurora H, et al. A prospective study of the inci-dence and open-label treatment of interferon-induced major depressive disorder in patients with hepatitis C. Mol Psychiatry. 2002;7(9): 942–947.
60. Kraus MR, Schafer A, Faller H, Csef H, Scheurlen M. Psychiatric symptoms in patients with chronic hepatitis C receiving interferon alfa-2b therapy. J Clin Psychiatry. 2003;64(6):708–714.
61. Schafer A, Wittchen HU, Seufert J, Kraus MR. Methodological approaches in the assessment of interferon-alfa-induced depression in patients with chronic hepatitis C – a critical review. Int J Methods Psychiatr Res. 2007;16(4):186–201.
62. Bonaccorso S, Puzella A, Marino V, et al. Immunotherapy with interferon-alpha in patients affected by chronic hepatitis C induces an intercorrelated stimulation of the cytokine network and an increase in depressive and anxiety symptoms. Psychiatry Res. 2001;105(1–2): 45–55.
29
Neuro vo5 no4 2011.indd 29 2011/10/28 10:13 AM

Dovepress
Dovepress
63. Wichers MC, Kenis G, Koek GH, Robaeys G, Nicolson NA, Maes M. Interferon-alpha-induced depressive symptoms are related to changes in the cytokine network but not to cortisol. J Psychosom Res. 2007;62(2):207–214.
64. Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M. IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry. 2005;10(6):538–544.
65. Raison CL, Dantzer R, Kelley KW, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry. 2010;15(4):393–403.
66. Rose WC, Haines WJ, Warner DT. The amino acid requirement of man. V. The role of lysine, arginine and tryptophan. Journal of Biological Chemistry. 1950;206(1):421–430.
67. Schaechter JD, Wurtman RJ. Serotonin release varies with brain tryp-tophan levels. Brain Res. 1990;532(1–2):203–210.
68. Anderson IM, Ferrier IN, Baldwin RC, et al. Evidence-based guide-lines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2008;22(4):343–396.
69. Delgado PL, Char ney DS, Pr ice LH, Aghajanian GK, Landis H, Heninger GR. Serotonin function and the mechanism of antidepressant action. Reversal of antidepressant-induced remission by rapid depletion of plasma tryptophan. Arch Gen Psychiatry. 1990; 47(5):411–418.
70. Hood SD, Bell CJ, Nutt DJ. Acute tryptophan depletion. Part I: rationale and methodology. Aust N Z J Psychiatry. 2005;39(7):558–564.
71. Maes M, Meltzer HY, Scharpe S, et al. Relationships between lower plasma L-tryptophan levels and immune-inflammatory variables in depression. Psychiatry Res. 1993;49(2):151–165.
72. Reimold M, Batra A, Knobel A, et al. Anxiety is associated with reduced central serotonin transporter availability in unmedicated patients with unipolar major depression: a [11C]DASB PET study. Mol Psychiatry. 2008;13(6):606–613, 557.
73. Joensuu M, Tolmunen T, Saarinen PI, et al. Reduced midbrain serotonin transporter availability in drug-naive patients with depression measured by SERT-specific [(123)I] nor-beta-CIT SPECT imaging. Psychiatry Res. 2007;154(2):125–131.
74. Sargent PA, Kjaer KH, Bench CJ, et al. Brain serotonin1 A recep-tor binding measured by positron emission tomography with [11C]WAY–100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry. 2000;57(2):174–180.
75. Drevets WC, Thase ME, Moses-Kolko EL, et al. Serotonin-1 A receptor imaging in recurrent depression: replication and literature review. Nucl Med Biol. 2007;34(7):865–877.
76. Yatham LN, Liddle PF, Shiah IS, et al. Brain serotonin2 receptors in major depression: a positron emission tomography study. Arch Gen Psychiatry. 2000;57(9):850–858.
77. Doggrell SA. The role of 5-HT on the cardiovascular and renal systems and the clinical potential of 5-HT modulation. Expert Opin Investig Drugs. 2003;12(5):805–823.
78. Bonaccorso S, Marino V, Puzella A, et al. Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol. 2002;22(1): 86–90.
79. Kraus MR, Schafer A, Faller H, Csef H, Scheurlen M. Paroxetine for the treatment of interferon-alpha-induced depression in chronic hepatitis C. Aliment Pharmacol Ther. 2002;16(6):1091–1099.
80. Musselman DL, Lawson DH, Gumnick JF, et al. Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med. 2001;344(13):961–966.
81. McMenamy RH. Binding of indole analogues to human serum albumin. Effects of fatty acids. J Biol Chem. 1965;240(11):4235–4243.
82. Oldendorf WH, Szabo J. Amino acid assignment to one of three blood-brain barrier amino acid carriers. Am J Physiol. 1976;230(1):94–98.
83. Salomon RM, Kennedy JS, Johnson BW, et al. Association of a critical CSF tryptophan threshold level with depressive relapse. Neuropsy-chopharmacology. 2003;28(5):956–960.
84. Fernstrom JD. Diet-induced changes in plasma amino acid pattern: effects on the brain uptake of large neutral amino acids, and on brain serotonin synthesis. J Neural Transm Suppl. 1979;(15): 55–67.
85. Raison CL, Borisov AS, Majer M, et al. Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry. 2009;65(4):296–303.
86. Jouvet M. Sleep and serotonin: an unfinished story. Neuropsychop-harmacology. 1999;21(2 Suppl):24S–27S.
87. Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry. 2004;56(11):819–824.
88. Bhatti T, Gillin JC, Seifritz E, et al. Effects of a tryptophan-free amino acid drink challenge on normal human sleep electroencephalogram and mood. Biol Psychiatry. 1998;43(1):52–59.
89. Moore P, Gillin C, Bhatti T, et al. Rapid tryptophan depletion, sleep electroencephalogram, and mood in men with remitted depression on serotonin reuptake inhibitors. Arch Gen Psychiatry. 1998;55(6):534–539.
90. Carhart-Harris RL, Nutt DJ, Munafo MR, Christmas DM, Wilson SJ. Equivalent effects of acute tryptophan depletion on REM sleep in ecstasy users and controls. Psychopharmacology (Berl). 2009;206(2):187–196.
91. Wilson SJ, Bailey JE, Rich AS, Adrover M, Potokar J, Nutt DJ. Using sleep to evaluate comparative serotonergic effects of paroxetine and citalopram. Eur Neuropsychopharmacol. 2004;14(15):367–372.
92. Raison CL, Rye DB, Woolwine BJ, et al. Chronic interferon-alpha administration disrupts sleep continuity and depth in patients with hepatitis c: association with fatigue, motor slowing, and increased evening cortisol. Biol Psychiatry. 2010;68(10):942–949.
93. Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16(10):1959–1973.
94. Fuller PM, Saper CB, Lu J. The pontine REM switch: past and present. J Physiol. 2007;584(Pt 3):735–741.
95. Knox WE, Mehler AH. The conversion of tryptophan to kynurenine in liver. I. The coupled tryptophan peroxidase-oxidase system forming formylkynurenine. J Biol Chem. 1950;187(1):419–430.
96. Silva NM, Rodrigues CV, Santoro MM, Reis LF, Alvarez-Leite JI, Gazzinelli RT. Expression of indoleamine 2,3-dioxygenase, trypto-phan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect Immun. 2002;70(2):859–868.
97. Okuda S, Nishiyama N, Saito H, Katsuki H. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J Neurochem. 1998;70(1):299–307.
98. Stone TW. Endogenous neurotoxins from tryptophan. Toxicon. 2001;39(1):61–73.
99. Santamaria A, Galvan-Arzate S, Lisy V, et al. Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport. 2001;12(4):871–874.
100. Behan WM, McDonald M, Darlington LG, Stone TW. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: protection by melatonin and deprenyl. Br J Pharmacol. 1999;128(8):1754–1760.
101. Stone TW, Addae JI. The pharmacological manipulation of glutamate receptors and neuroprotection. Eur J Pharmacol. 2002;447(2–3):285–296.
102. Saito K, Crowley JS, Markey SP, Heyes MP. A mechanism for increased quinolinic acid formation following acute systemic immune stimulation. J Biol Chem. 1993;268(21):15496–15503.
30
Neuro vo5 no4 2011.indd 30 2011/10/28 10:13 AM

Dovepress
Dovepress
103. Wu HQ, Guidetti P, Goodman JH, et al. Kynurenergic manipulations influence excitatory synaptic function and excitotoxic vulnerability in the rat hippocampus in vivo. Neuroscience. 2000;97(2):243–251.
104. Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: evidence of impaired neuroprotection. J Affect Disord. 2007;98(1–2):143–151.
105. Muller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibi-tor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680–684.
106. Mendlewicz J, Kriwin P, Oswald P, Souery D, Alboni S, Brunello N. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227–231.
107. Loke YK, Trivedi AN, Singh S. Meta-analysis: gastrointestinal bleed-ing due to interaction between selective serotonin uptake inhibitors and non-steroidal anti-inflammatory drugs. Aliment Pharmacol Ther. 2008;27(1):31–40.
108. Kerr SJ, Sayer GP, Whicker SD, Rowett DS, Saltman DC, Mant A. All-cause mortality of elderly Australian veterans using COX-2 selec-tive or non-selective NSAIDs: a longitudinal study. Br J Clin Phar-macol. 2011;71(6):936–942.
109. Abraham NS, El-Serag HB, Hartman C, Richardson P, Deswal A. Cyclooxygenase-2 selectivity of non-steroidal anti-inflammatory drugs and the risk of myocardial infarction and cerebrovascular accident. Aliment Pharmacol Ther. 2007;25(8):913–924.
110. Lob S, Konigsrainer A, Rammensee HG, Opelz G, Terness P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer. 2009;9(6):445–452.
111. Bergink V, van Megen HJ, Westenberg HG. Glutamate and anxiety. Eur Neuropsychopharmacol. 2004;14(3):175–183.
112. Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. Metabotropic glutamate receptors as novel tar-gets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4(2):131–144.
113. Aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depres-sion. Biol Psychiatry. 2010;67(2):139–145.
114. Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ket-amine in depressed patients. Biol Psychiatry. 2000;47(4):351–354.
115. Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placeb-controlled continuation trial. Int J Neuropsychopharmacol. 2010;13(1):71–82.
116. Machado-Vieira R, Yuan P, Brutsche N, et al. Brain-derived neu-rotrophic factor and initial antidepressant response to an N-methyl-D-aspartate antagonist. J Clin Psychiatry. 2009;70(12):1662–1666.
117. Zarate CA Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–864.
31
Neuro vo5 no4 2011.indd 31 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 32 2011/10/28 10:13 AM

Neuropsychiatric Disease and Treatment
Narcolepsy is a lifelong sleep disorder characterized by a classic tetrad of excessive
daytime sleepiness with irresistible sleep attacks, cataplexy (sudden bilateral loss of muscle
tone), hypnagogic hallucination, and sleep paralysis. There are two distinct groups of patients,
ie, those having narcolepsy with cataplexy and those having narcolepsy without cataplexy.
Narcolepsy affects 0.05% of the population. It has a negative effect on the quality of life of its
sufferers and can restrict them from certain careers and activities. There have been advances in
the understanding of the pathogenesis of narcolepsy. It is thought that narcolepsy with cataplexy
is secondary to loss of hypothalamic hypocretin neurons in those genetically predisposed to the
disorder by possession of human leukocyte antigen DQB1*0602. The diagnostic criteria for
narcolepsy are based on symptoms, laboratory sleep tests, and serum levels of hypocretin. There
is no cure for narcolepsy, and the present mainstay of treatment is pharmacological treatment
along with lifestyle changes. Some novel treatments are also being developed and tried. This
article critically appraises the evidence for diagnosis and treatment of narcolepsy.
narcolepsy, cataplexy, hypocretin, modafinil, gamma hydroxybutyrate
Narcolepsy is a debilitating lifelong rapid eye movement (REM) sleep disorder. It is
characterized by the classic tetrad of excessive daytime sleepiness with irresistible
sleep attacks, cataplexy (sudden bilateral loss of muscle tone), hypnagogic hallucina-
tion, and sleep paralysis.1 Other features include fragmented night sleep and automatic
behavior, loss of concentration and memory, and blurry vision.2–5 The presentation is
variable in terms of symptoms and intensity over time, and only about 10% of patients
concurrently exhibit all components of the tetrad.6 There are two distinct groups of
patients, ie, those having narcolepsy with cataplexy and those having narcolepsy
without cataplexy.
It often coexists with other sleep disorders, like obstructive sleep apnea syndrome,
periodic limb movements in sleep, REM sleep behavior disorder, and nocturnal eat-
ing disorder.7–12
The prevalence of narcolepsy in European countries varies from 0.02% to 0.05%.13–15 It
shows marked ethnic variation in prevalence rate, being 0.0002% in Israel and 0.16%
in Japan.16,17 The varying prevalence rates may be as a result of varying disease defini-
tions, varying study designs, varying age group inclusion in studies, or an actual varying
disease prevalence due to other factors.18,19 It has been suggested that the differences in
Dovepress
Dovepress
open access to scientific and medical research
33
Neuro vo5 no4 2011.indd 33 2011/10/28 10:13 AM

prevalence may be partly related to the association between
narcolepsy and the prevalence of the human leukocyte anti-
gen (HLA) DQB1*0602 phenotype.14 The subgroup having
narcolepsy without cataplexy may represent 10%–50% of
the narcolepsy population.20 The estimated incidence rate
is 0.74/100,000 per year for narcolepsy with cataplexy and
1.37/100,000 per year for both narcolepsy with cataplexy and
narcolepsy without cataplexy.21 The incidence rate is highest in
the second decade, and the disorder is more common in men.21
Although most cases are sporadic, there are definite cases
with familial clustering. The risk of narcolepsy in first-degree
relatives of patients is 10–40 times higher than in the general
population.22 There is an environmental contribution as well,
as shown by reported concordance rates of 25%–31% in
monozygotic twins.23 The nature of possible environmental
triggers is unknown. Nevertheless, the onset is frequently asso-
ciated with nonspecific environmental factors, such as head
trauma, stroke, and change in sleep-wake cycle.23 Moreover,
recent studies have shown an association with streptococcal
infection,24,25 HIN1 vaccination or infection,26 and exposure
to heavy metals, insecticides, and weed killers.27
More than 85% of patients having narcolepsy with cataplexy
have HLA DQB1*0602, often in combination with HLA
DR2 (DRB1*1501), while only half of patients having
atypical, mild, or narcolepsy without cataplexy have HLA
DQB1*0602.28 Other alleles of HLA also affect the predis-
position to narcolepsy with cataplexy.29,30 Occurrence of the
HLA DQB*0602 allele is not limited to narcolepsy with
cataplexy, and is found in 12%–38% of the general popula-
tion.23 Moreover, there are some rare patients with definite
cataplexy who do not have HLA DQB1.31 Overall, the poor
discriminatory ability of HLA typing limits its usefulness
and makes it unsuitable as a routine diagnostic test.
Hypocretins 1 and 2, also called orexins A and B, are two
dorsolateral hypothalamic neuropeptides that function in reg-
ulating sleep-wake cycles, food intake, and pleasure-seeking
behavior.32 Amongst the areas of the brain that the neurons
producing hypocretins project to are the locus ceruleus,
tuberomammillary nucleus, raphe nucleus, and ventral
tegmental areas.32 These areas correspond to norepinephrine,
histamine, serotonin, and dopamine secretion, respectively.
Deficiency of hypocretin could lead to malfunctioning of
these systems and therefore abnormalities of REM sleep
and excessive daytime sleepiness.33 Hypocretin neurons
also project to other areas of the hypothalamus, olfactory
bulb, cerebral cortex, and thalamus.34,35 The evidence for
hypocretin deficiency is as follows.
In 1979, Foutz et al showed that narcolepsy was inher-
ited in a single autosomal recessive pattern in Doberman
Pinschers.36 Lin et al later identified this as a mutated
hypocretin receptor 2 gene.37 One of the earliest reports of
hypocretin deficiency in narcolepsy with cataplexy was that
of Nishino et al in 2000. Their study showed that whilst seven
of nine patients having narcolepsy with cataplexy had no
detectable hypocretin, the neuropeptides were detectable in
all their eight matched controls.38 Further studies have sup-
ported hypocretin neurotransmission deficiency in narcolepsy
with cataplexy.31,39,40
It was subsequently found that, unlike in animals, hypo-
cretin deficiency in humans having narcolepsy with cataplexy
was not due to mutation in hypocretin genes but rather
secondary to loss of hypocretin neurons in the dorsolateral
hypothalamus.39,40 Peyron et al found only one patient (an
atypical case with onset of narcolepsy with cataplexy at the
age of 6 months) with hypocretin mutation among 74 patients
screened for mutation. She also found global loss of hypo-
cretin neurons in the brains of six deceased patients who
had suffered from narcolepsy with cataplexy.40 Furthermore,
Thannickal et al found the number of hypocretin neurons to
be reduced by 85% to 95% in association with evidence of
gliosis.39 These studies collectively suggested that the loss
of hypocretin neurons in patients having narcolepsy with
cataplexy might be inflammatory in nature.
The combination of HLA antigens, hypocretin deficiency,
hypocretin neuron loss, the rarity of hypocretin gene muta-
tions, and onset in the second decade of life points strongly
towards an autoimmune etiology.38–40 Initial efforts at isolat-
ing an autoantibody proved unsuccessful.41,42 However, there
was indirect evidence for an autoimmune nature of narco-
lepsy, such as its temporary response to steroids.42,43 Recently,
methods have been developed to demonstrate the presence
of autoantibodies. There has been a demonstration of an
autoantibody which disrupts colonic migrating motor com-
plexes.44 A Tribbles homolog 2 (Trib2) transcript (an autoan-
tigen in autoimmune uveitis45) has been shown to be enriched
in hypocretin neurons from genetically engineered mice.46
Enzyme-linked immunosorbent analysis was used in turn to
Dovepress
Dovepress
34
Neuro vo5 no4 2011.indd 34 2011/10/28 10:13 AM

show that sera from patients having narcolepsy with cataplexy
had higher Trib2-specific antibody titers compared with
those in normal controls or patients with other neurological
diseases.46 Moreover, serum from a patient having narcolepsy
with cataplexy showed specific immunoreactivity with over
86% of hypocretin neurons in the mouse hypothalamus.46 This
finding was replicated in another study that found autoanti-
bodies against Trib2 in 26.1% of patients having narcolepsy
with cataplexy compared with 2.3% of healthy controls.47
Thus, a subgroup of patients having narcolepsy with cata-
plexy might be suffering from anti-Trib2 autoimmune dis-
order. Furthermore, another study has found an association
between narcolepsy with cataplexy and polymorphism in
the T cell receptor alpha genetic locus (encoding the major
receptor for HLA peptide presentation in any disease) in three
ethnic groups compared with normal matched controls.48
Further support for an autoimmune etiology comes from
cytokine studies showing higher interleukin-6, tumor necro-
sis factor- , and tumor necrosis factor receptor p75 levels
in patients having narcolepsy with cataplexy.49–51 However,
Fontana et al have suggested that immune-mediated destruc-
tion of hypocretin cells might occur independent of T cells.52
Moreover, a lack of hypocretin is not specific to narcolepsy
with cataplexy. It has also been reported in patients with
Guillain–Barré and Miller Fisher syndromes.53,54
Loss of hypocretin-producing neurons definitely causes
narcolepsy with cataplexy.31,39,40,55 Other neurological insults,
lesions of the hypothalamus or nearby structures, and global
traumatic, vascular, or inflammatory insults to the brain could
also cause narcolepsy.56–58 All this could affect the levels of
hypocretin either transiently or permanently (see Table 1).55
The first step in the diagnosis of narcolepsy is history-taking
from the patient and from partners, relatives, and friends. Apart
from atonia and areflexia in patients having active cataplexy,
the physical examination should be normal.5 The current
International Classification of Sleep Disorders (ICSD-2)
definition for narcolepsy is shown in Table 2.59 It is based on
history, polysomnography, multiple sleep latency tests (MSLT),
and measurement of hypocretin levels in cerebrospinal fluid.
It classifies narcolepsy into three types (see Table 2). Exces-
sive daytime sleepiness is the most constant feature of narco-
lepsy6 and measuring it accurately is important. There are a
number of subjective and objective scales to measure this.
The main advantages of subjective measures of excessive
daytime sleepiness are that they are less cumbersome, less
time-consuming, and less expensive compared with objective
tests.60 Their main drawback is their inconsistent reliability
in correlating with objective tests.61
The Epworth Sleepiness scale was developed by Murray
Johns in Australia in 1991 and was validated using 150
consecutive clinic patients with a range of sleep disor-
ders and 30 hospital worker controls.60 Thus, the study
sample was from neither the normal population nor from
the same population. The Epworth Sleepiness scale is a
self- administered questionnaire in which patients rate their
likelihood of falling asleep in eight different life situations.
Dovepress
Dovepress
35
Neuro vo5 no4 2011.indd 35 2011/10/28 10:13 AM

Each situation is scored from 0 to 3 and the total score varies
from 0 to 24. It has been validated in different clinical situa-
tions and languages.62–65 It correlates best with the objective
tests in comparison with other subjective tests.60
Other subjective measures include the Stanford Sleepi-
ness scale which rates excessive daytime sleepiness accord-
ing to the subjects’ perception of their sleepiness/alertness
at a particular time,60 and the visual analog scale, with which
subjects indicate their levels of sleepiness along a continu-
ous line between two points. These two tests only measure
sleepiness at a particular time rather than general daytime
sleepiness. Thus, their main application is in research rather
than in the clinic, when they can only be used for point-in-
time estimation of sleepiness.61 Based on the above consid-
erations, our recommended clinical subjective measure is the
Epworth Sleepiness scale.
Polysomnography is a method which is usually performed
overnight in a sleep laboratory before MSLT. Data such
as breathing, movement, time spent asleep, heart rate, and
electroencephalography are obtained.18,66,67 Patients may
have audio and video monitoring that could reveal snoring,
sleep talking, movement, and complex behavior during
sleep.18 Thus, it can reveal the etiology of excessive daytime
sleepiness and other sleep pathologies. However, perform-
ing polysomnography itself can cause disruption to sleep,
potentially affecting MSLT on the following day.18 Thus, it
is recommended that at least 6 hours of sleep is recorded on
the night before MSLT.67
MSLT is the accepted standard objective measure for
excessive daytime sleepiness and has good inter-rater and
intrarater reliability.18,60,66 It consists of five scheduled naps
during the day, each lasting 20 minutes and 2 hours apart.
Ideally, it is carried out under conditions that best help
patients to fall asleep, eg, appropriate temperature and
limited stimulation. Patients are required to stay awake
between each nap opportunity. During the tests, physiologi-
cal data are gathered, such as time taken to fall asleep and
the presence or absence of REM sleep.18 When REM sleep
occurred within 15 minutes of onset, it is termed sleep-
onset REM sleep (SOREM). It is common in narcolepsy
but rare in normal individuals.18 Patients should spend at
least 8 hours in bed per night in the preceding week in order
to avoid erroneous results.68 This can be confirmed using
a documented sleep log or, more objectively, by the use of
wrist actigraphy,69 which entails wearing a motion-sensitive
device that uses lack of movement as a surrogate for the
period spent asleep.68
The current ICSD-2 MSLT criteria shown in Table 2 might
not be specific for narcolepsy, as shown by Allen70 who car-
ried out polysomnography and MSLT in 289 normal males
and 267 normal females, and analyzed subject variables
such as age, gender, body mass index, and HLA typing
against their results on MSLT. Allen found that 4.2% and
0.4% of males and females, respectively, had two SOREMS
and an excessive daytime sleepiness score of 11 on the
Epworth Sleepiness scale (ICSD-2 criteria for narcolepsy
without cataplexy, see Table 2). Furthermore, shift work,
use of non-REM-suppressing antidepressants, a positive
HLA DQB1*0602, decreased oxygen saturation, and a sleep
diary showing a 1-hour decrease in sleep the night before
polysomnography, are all related to two or more SOREMs.
Therefore, several other factors and sleep disorders could
result in two SOREMs, and two SOREMs are not as specific
for narcolepsy as suggested by ICSD-2 criteria.
Moreover, a recent case series showed five patients diag-
nosed as having narcolepsy with cataplexy based on history
alone (consistent with ICSD-2 criteria) but whose diagnoses
were later found not to be corroborated by MSLT, and all
were HLADQB1*0602 negative.71 Thus, the diagnosis of
narcolepsy irrespective of whether it is narcolepsy with
cataplexy or narcolepsy without cataplexy should be based
both on clinical symptoms and MSLT, and reliance on either
alone may be insufficient.
The maintenance of wakefulness test is a variant of the
MSLT, and measures the ability to stay awake. It consists of
4–5 trials of trying to remain awake while in the recumbent
position in a dark room. This is repeated every 2 hours. Each
trial is terminated if no sleep occurs after 40 minutes.45 It
is subject to the same caveats as the MSLT.60 It has been
found to measure alertness rather than sleepiness.72 Thus, it
is used to measure the ability to stay awake in individuals
with jobs that require a high level of alertness and also to
assess response to treatment by those with excessive daytime
sleepiness in pharmacological trials,61 rather than to make
a diagnosis. The correlations between the Epworth Sleepi-
ness scale, MSLT, and maintenance of wakefulness test are
generally not impressive.61,73
Laboratory testing for HLA typing adds little to the
diagnostic evaluation because its sensitivity is highest
in patients having narcolepsy with cataplexy, a group in
which additional diagnosis is rarely necessary.18,19,28,31
Testing for hypocretin suffers from the same problem. In
addition, it needs a lumbar puncture, which is an invasive
Dovepress
Dovepress
36
Neuro vo5 no4 2011.indd 36 2011/10/28 10:13 AM

procedure with potential complications. Nevertheless,
hypocretin measurement should be considered in the fol-
lowing situations: following an equivocal MSLT result;
in patients who cannot follow the instructions for MSLT;
individuals who are unable to stop medications that could
affect the result of MSLT; those with complex psychiatric,
neurological, or medical disorders that could compromise
the result of MSLT; in the patient who gives an excel-
lent history of narcolepsy with cataplexy but who has
normal polysomnography and MSLT. In these patients, a
hypocretin-1 level below 110 pg/mL in cerebrospinal fluid
is highly indicative of narcolepsy, but a higher level does
not necessarily exclude the diagnosis.55
In terms of neuroimaging, magnetic resonance studies
are presently inconclusive in their findings, while magnetic
resonance spectroscopy has revealed abnormal metabolism
in the hypothalamus. Functional neuroimaging has revealed
hypoperfusion and hypermetabolism in the hypothalamus,
limbic cortex, and cerebrum.45,74–81 Overall, neuroimaging is
not useful in the diagnosis of narcolepsy.
Sleep deprivation, sleep apnea, idiopathic hypersomnia,
recurrent hypersomnia (Kleine–Levin syndrome), restless
legs syndrome, and periodic limb movement disorder.82
Neurological conditions commonly associated with
sleepiness and REM sleep behavioral disorder, eg,
Parkinson’s disease, Alzheimer’s disease, and other neu-
rodegenerative conditions, including multiple sclerosis,
stroke, epilepsy, neuromuscular disorders, and structural
brain disorders (bithalamic or bicortical lesions affecting
midline projecting systems).82–84
Medical conditions commonly associated with sleepiness,
such as respiratory disorders (eg, chronic obstructive
pulmonary disease and asthma), cardiac disorders (eg,
congestive heart failure), renal disorders (eg, chronic
renal failure), rheumatologic disorders (eg, arthritis),
inflammatory disorders (eg, lupus), hepatic disorders
(eg, liver failure), and malignancy.82
Major psychiatric disorders sharing symptoms of func-
tional impairment, insomnia, and hypersomnia with
narcolepsy;85 reduced REM latency is common to major
episodes of depression and narcolepsy;4,86,87 REM sleep-
related disorders in narcolepsy share some psychotic
features with schizophrenia, eg, hypnagogic/hypnopom-
pic hallucination;88–90 some features of narcolepsy, such
as sleep paralysis and sleep behavior disorder, could be
misinterpreted as psychosis.91
Stimulants used in the treatment of narcolepsy can give
rise to psychotic symptoms.92
Differences between the core symptoms of psychosis and
symptoms of narcolepsy have been elaborated elsewhere93
in a study comparing 148 narcolepsy patients, 21 schizo-
phrenic patients, and 128 healthy subjects, which found
that episodes of hallucination in narcolepsy were sleep-
related and posture-related, and more likely to be visual and
kinetic (83% and 71% in narcolepsy, respectively, compared
with 29% and 5% in schizophrenia);93 the drawback of
the study included discordant sample sizes for the groups,
and the groups were also from different populations.
In children, narcolepsy may present with only excessive
daytime sleepiness, and the behavioral problem associated
with excessive daytime sleepiness might be misdiagnosed
as ADHD.94
Malingering
Cataplexy has been misdiagnosed as epilepsy or recurrent
syncope.95–97
Conventional treatments are essentially symptomatic, given
that no cure has been found for narcolepsy. American Acad-
emy of Sleep Medicine classification of the levels of evidence
that the authors follow in this paper are shown in Table 3.98
There is no randomized controlled trial to support the
efficacy of nonpharmacological therapy. There is Level III
evidence from a nonblinded controlled study that taking naps
reduces excessive daytime sleepiness both subjectively and
objectively,99 while hypnotherapy has only Level IV evi-
dence from a case series.100 Furthermore, there is Level IV
evidence from an open-label study that a low-carbohydrate,
high-protein diet can improve wakefulness.101 Overall,
Dovepress
Dovepress
37
Neuro vo5 no4 2011.indd 37 2011/10/28 10:13 AM

nonpharmacological therapies are not adequate alone as
primary therapy for narcolepsy. According to an American
Academy of Sleep Medicine report: “Scheduled naps can be
beneficial to combat sleepiness but seldom suffice as primary
therapy for narcolepsy.”102
Pharmacological therapy is the mainstay of treatment for
excessive daytime sleepiness. There is only Level III and IV
evidence of efficacy from trials using earlier medications. The
earliest medication used in the treatment of narcolepsy was
caffeine, but its cardiovascular side effects prohibited its use
at higher doses.103 Other treatments included insulin-induced
hypoglycemia (reported in 1952)104 and trazodone used to
supplement methylphenidate.105 Both were case reports
that led to temporary improvement. Other older treatments
include mazindol, which was studied in a nonrandomized
controlled trial with a short follow-up,106 and propranolol,
which was found to be effective in a case report107 but not
in an open-label study.108 Overall, these earlier medications
are no longer used routinely in the treatment of narcolepsy.
Ritanserin is a serotonin antagonist with Level II evidence
from two studies. One study demonstrated an improvement
in subjective daytime sleepiness but not in sleep latency
on MSLT when ritanserin 5 mg/day was added to usual
medication in 28 patients.109 The second study compared
ritanserin 5 mg and 10 mg with placebo in 134 patients, and
did not find improvement in any of the objective measures,
but did report an improvement in subjective sleep quality.110
According to an American Academy of Sleep Medicine
report: “Ritanserin may be effective treatment of daytime
sleepiness due to narcolepsy.”102 Its most appropriate use is
as an add-on medication.
Selegiline, a monoamine oxidase type B inhibitor, has a
Level II study that demonstrated its efficacy for excessive
daytime sleepiness and cataplexy.111 The American Academy
of Sleep Medicine report has expressed some reservations
about its use as the preferred initial choice for treatment of
excessive daytime sleepiness,102 on account of its potential
for drug and diet interactions.102
Traditional stimulants have been in use since the 1930s,
initially caffeine and ephedrine, then amphetamines,
methamphetamine, and dexamphetamine, and thereafter,
their derivatives pemoline briefly and methylphenidate
later.33,103,112 At low doses, stimulants release dopamine and
noradrenaline through reversal of action of their presynaptic
transporters. At higher doses, amphetamine inhibits vesicu-
lar monoamine transporter 2.113 The side effects of stimulants
include irritability, headache, nervousness, palpitations,
insomnia, and less often, orofacial dyskinesia, anorexia,
nausea, excessive sweating, psychosis, and myocardial
infarction.113–115 Tolerance may develop in up to one-third
of patients, and these have a potential for addiction.113 There
is limited evidence to support their use. The only Level III
evidence available for the efficacy of traditional stimulants
was provided by Mitler et al116 who compared eight patients
with narcolepsy and eight healthy matched controls. The
patients received 0 mg, 20 mg, or 40–60 mg, while the con-
trols received 0 mg, 5 mg, or 10 mg of methamphetamine.
MSLT sleep latency increased in both groups, from 4.3 to
9.3 minutes in patients and from 10.4 to 17.1 minutes in
controls.
Pemoline selectively blocks dopamine reuptake. It has
the potential to cause fatal hepatotoxicity.113 On account of
this, it has been withdrawn from the market and is no longer
recommended.82
Methylphenidate, an N-methyl derivative of amphet-
amine, has a shorter half-life, milder side effects, and low
abuse potential.33 It has Level II and Level IV evidence for
its use. A nonrandomized controlled study compared the
response of narcoleptic patients allocated to four treatment
groups (methylphenidate, pemoline, protriptyline, and
dextroamphetamine) and one placebo group. Each group
had three dose regimens. Improvement in the maintenance
of wakefulness test was used as the objective measure. All
doses of methylphenidate and dextroamphetamine were
efficacious, while doses of pemoline 112.5 mg and doses
of protriptyline up to 60 mg/day were not efficacious.117
Honda et al reported a dose-related improvement in 92% of
106 patients treated with methylphenidate (50 patients had
more than 5 years of treatment).118 However, the improve-
ment might not have been solely due to methylphenidate
because many of the patients were also receiving concurrent
hypnotics or tricyclic antidepressants. Stimulants including
amphetamine, methamphetamine, dextroamphetamine, and
methylphenidate are effective for the treatment of daytime
sleepiness due to narcolepsy.102 However, treatment should
be individualized, and patients should be closely followed
up to ensure efficacy and detection of side effects.102
Modafinil and its R enantiomer, armodafinil, are wake-
promoting agents that have an additional phenyl group and an
amide instead of an amine group in their chemical structure
when compared with amphetamine.113 Although the mecha-
nism of action of modafinil is unclear, it is believed to work
through the dopaminergic, adrenergic, and histaminergic
systems.33 Recently, a functional neuroimaging study has
shown that modafinil increases extracellular levels of dop-
amine in the human brain.119 Furthermore, another study has
Dovepress
Dovepress
38
Neuro vo5 no4 2011.indd 38 2011/10/28 10:13 AM
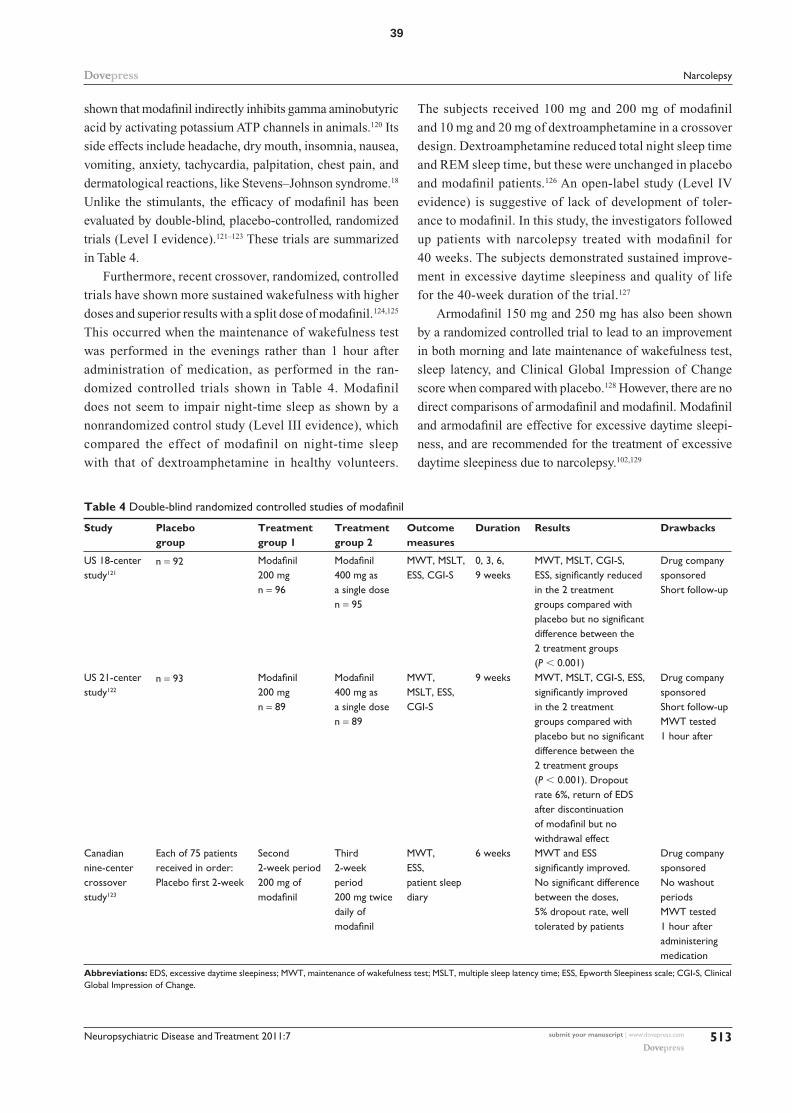
shown that modafinil indirectly inhibits gamma aminobutyric
acid by activating potassium ATP channels in animals.120 Its
side effects include headache, dry mouth, insomnia, nausea,
vomiting, anxiety, tachycardia, palpitation, chest pain, and
dermatological reactions, like Stevens–Johnson syndrome.18
Unlike the stimulants, the efficacy of modafinil has been
evaluated by double-blind, placebo-controlled, randomized
trials (Level I evidence).121–123 These trials are summarized
in Table 4.
Furthermore, recent crossover, randomized, controlled
trials have shown more sustained wakefulness with higher
doses and superior results with a split dose of modafinil.124,125
This occurred when the maintenance of wakefulness test
was performed in the evenings rather than 1 hour after
administration of medication, as performed in the ran-
domized controlled trials shown in Table 4. Modafinil
does not seem to impair night-time sleep as shown by a
nonrandomized control study (Level III evidence), which
compared the effect of modafinil on night-time sleep
with that of dextroamphetamine in healthy volunteers.
The subjects received 100 mg and 200 mg of modafinil
and 10 mg and 20 mg of dextroamphetamine in a crossover
design. Dextroamphetamine reduced total night sleep time
and REM sleep time, but these were unchanged in placebo
and modafinil patients.126 An open-label study (Level IV
evidence) is suggestive of lack of development of toler-
ance to modafinil. In this study, the investigators followed
up patients with narcolepsy treated with modafinil for
40 weeks. The subjects demonstrated sustained improve-
ment in excessive daytime sleepiness and quality of life
for the 40-week duration of the trial.127
Armodafinil 150 mg and 250 mg has also been shown
by a randomized controlled trial to lead to an improvement
in both morning and late maintenance of wakefulness test,
sleep latency, and Clinical Global Impression of Change
score when compared with placebo.128 However, there are no
direct comparisons of armodafinil and modafinil. Modafinil
and armodafinil are effective for excessive daytime sleepi-
ness, and are recommended for the treatment of excessive
daytime sleepiness due to narcolepsy.102,129
Dovepress
Dovepress
39
Neuro vo5 no4 2011.indd 39 2011/10/28 10:13 AM

A report of the effectiveness of imipramine in the treat-
ment of cataplexy in 1960 led to widespread use of tricyclic
antidepressants.130 Their effect could be due to an ability
to inhibit the reuptake of catecholamines, increase muscle
tone, or suppress REM sleep.131–133 Side effects include nau-
sea, anorexia, dry mouth, urinary retention, constipation,
and sexual dysfunction. Rebound cataplexy, which may be
severe and prolonged (status cataplecticus), may occur on
sudden withdrawal from tricyclic antidepressants.33 There
are only case reports and case series (level IV evidence) for
their efficacy.134
Selective serotonin reuptake inhibitors inhibit presyn-
aptic serotonin reuptake and also nocturnal REM sleep.
Femoxetine has Level II evidence for treating cataplexy from
a crossover, placebo-controlled study involving 10 patients.
Only case series and reports (Level IV evidence) are available
for other selective serotonin reuptake inhibitors.135,136
Other antidepressants, including noradrenergic reuptake
inhibitors, such as venlafaxine, duloxetine, reboxetine, and
viloxazine, are being used.137,138 There is a Level III single-
blind, crossover, placebo-controlled study of viloxazine
involving 23 patients, with one dropout.139 There are only
case series (Level V evidence) available for venlafaxine and
duloxetine.137,140
Overall, there is a lack of good evidence for the use of
antidepressants in treating cataplexy. Nevertheless, according
to the American Academy of Sleep Medicine report: “Based
on consensus and clinical experience ... they may be an
effective treatment for cataplexy, hypnagogic hallucination
and sleep paralysis.”102
Gamma hydroxybutyrate, marketed as sodium oxybate, is a
natural metabolite of gamma aminobutyrate (GABA), and
is a GABAB receptor agonist at a pharmacological dose.18
Its exact mechanism of action is not known, but it increases
slow wave sleep, decreases arousals, and has a variable
effect on latency and amount of REM sleep.141,142 Level I
evidence for the efficacy of gamma hydroxybutyrate was
provided by three multicenter, randomized, double-blind,
placebo-controlled studies of the short-term and long-term
efficacy of gamma hydroxybutyrate.143–145 In the first trial, 136
narcolepsy patients with 3–249 cataplexy attacks weekly were
enrolled to receive 3.6 g or 9 g of gamma hydroxybutyrate
or placebo taken in equal divided doses on retiring to bed
and 2.5–4.0 hours later for 4 weeks. The primary measure of
efficacy was a change in baseline weekly cataplectic attacks,
while secondary measures included excessive daytime
sleepiness using the Epworth Sleepiness scale, inadvertent
daytime naps, and night-time awakenings. In total, 88% of
participants completed the trials. Weekly cataplectic attacks
started to decrease at a 6 g dose and became significant at a
9 g dose, and excessive daytime sleepiness was reduced at all
doses, becoming significant at the 9 g dose. The frequency of
inadvertent daytime naps and night-time awakenings reduced
at all doses, becoming significant at the 9 g dose.143 The
second trial included 55 patients stabilized on nightly doses
of gamma hydroxybutyrate for at least 6 months. They were
then randomized into two arms, ie, one abruptly changing to
placebo and the other continuing on gamma hydroxybutyrate
for 2 weeks. Cataplexy gradually returned in those patients
that were changed onto placebo over 2 weeks, but none of the
patients suffered withdrawal symptoms.144 This suggests lack
of development of tolerance to gamma hydroxybutyrate.
The third trial compared the effect of nightly gamma
hydroxybutyrate 4.5 g, 6 g, or 9 g with placebo in 228 adult
patients over 8 weeks.145 The patients showed a significant
dose-related increase in duration of stage 3 and 4 sleep,
reaching a median duration of 52.5 minutes in patients
receiving gamma hydroxybutyrate 9 g nightly. Frequency of
nocturnal awakening and stage 1 sleep were each significantly
decreased at nightly doses of gamma hydroxybutyrate 6 g
and 9 g. This study showed that gamma hydroxybutyrate
decreased the sleep disruption and fragmentation that are
usually associated with narcolepsy. Moreover, two Level IV
(open-label) studies have demonstrated the tolerability and
sustained efficacy of gamma hydroxybutyrate.146,147
From the gamma hydroxybutyrate trials, common side
effects have included dizziness, headache, nausea, pain, som-
nolence, sleep disorder, confusion, infection, vomiting, and
enuresis.33 Illicit use in high doses could result in addiction
and withdrawal. Sudden deaths have been reported in patients
who have risk factors for obstructive sleep apnea.148 Overall,
gamma hydroxybutyrate is effective, and is recommended
for treatment of cataplexy, excessive daytime sleepiness, and
disrupted sleep due to narcolepsy.102
Introduction of hypocretin-1 into the cerebral ventricular system
was found to be useful in mice but not in hypocretin-2 mutated
dogs.149 Intranasal administration is promising, as well as
transplantation of neonatal hypothalamic stem cells into
the brainstem.149,150 Narcolepsy with cataplexy is strongly
Dovepress
Dovepress
40
Neuro vo5 no4 2011.indd 40 2011/10/28 10:13 AM

suspected to be an autoimmune disorder. However, attempts
to modify immune processes, including use of steroids,
plasmapheresis, and intravenous immunoglobulin, have been
met with limited and short-term success.151–153 Histamine
3 receptors regulate the release of histamine. Antagonism of
the histamine 3 receptor enhances wakefulness, while stimula-
tion causes sedation.154 Histamine 3 receptor antagonists have
been shown to be effective in canines and mice.155,156 Other
promising novel treatments include thyrotrophin-releasing
hormone and the nicotine patch.156,157
Our understanding of the pathogenesis of narcolepsy contin-
ues to advance, with substantial evidence that autoimmune
loss of hypocretin neurons is the main cause of narcolepsy
with cataplexy. The standardized criteria for narcolepsy and
diagnostic measures are generally accepted, but might need
to be reviewed in the future. There is established pharma-
cotherapy for symptomatic treatment of narcolepsy. Future
treatment modalities, such as hypocretin analogs and hista-
mine receptor antagonists, should aim to tackle the cause of
narcolepsy.
The authors report no conflicts of interest in this work.
1. Daniels LE. Narcolepsy. Medicine. 1934;13:1–122. 2. Zorick FJ, Salis PJ, Roth T, Kramer M. Narcolepsy and automatic
behavior: A case report. J Clin Psychiatry. 1979;40(4):194–197. 3. Parkes JD, Baraitser M, Marsden CD, Asselman P. Natural history,
symptoms and treatment of the narcoleptic syndrome. Acta Neurol Scand. 1975;52(5):337–353.
4. Broughton R, Ghanem Q, Hishikawa Y, Sugita Y, Nevsimalova S, Roth B. Life effects of narcolepsy in 180 patients from North America, Asia and Europe compared to matched controls. Can J Neurol Sci. 1981;8(4):299–304.
5. Parkes JD, Fenton G, Struthers G, et al. Narcolepsy and cataplexy. Clinical features, treatment and cerebrospinal fluid findings. Q J Med. 1974;43(172):525–536.
6. Morrish E, King MA, Smith IE, Shneerson JM. Factors associated with a delay in the diagnosis of narcolepsy. Sleep Med. 2004;5(1):37–41.
7. Sansa G, Iranzo A, Santamaria J. Obstructive sleep apnea in narcolepsy. Sleep Med. 2010;11(1):93–95.
8. Ferri R, Franceschini C, Zucconi M, et al. Sleep polygraphic study of children and adolescents with narcolepsy/cataplexy. Dev Neuropsychol. 2009;34(5):523–538.
9. Knudsen S, Gammeltoft S, Jennum PJ. Rapid eye movement sleep behaviour disorder in patients with narcolepsy is associated with hypocretin-1 deficiency. Brain. 2010;133(Pt 2):568–579.
10. Schenck CH, Mahowald MW. Motor dyscontrol in narcolepsy: Rapid-eye-movement (REM) sleep without atonia and REM sleep behavior disorder. Ann Neurol. 1992;32(1):3–10.
11. Schenck CH, Hurwitz TD, Bundlie SR, Mahowald MW. Sleep-related eating disorders: Polysomnographic correlates of a heterogeneous syndrome dis-tinct from daytime eating disorders. Sleep. 1991;14(5):419–431.
12. Spaggiari MC, Granella F, Parrino L, Marchesi C, Melli I, Terzano MG. Nocturnal eating syndrome in adults. Sleep. 1994;17(4): 339–344.
13. Ohayon MM, Priest RG, Zulley J, Smirne S, Paiva T. Prevalence of narcolepsy symptomatology and diagnosis in the European general population. Neurology. 2002;58(12):1826–1833.
14. Heier MS, Evsiukova T, Wilson J, Abdelnoor M, Hublin C, Ervik S. Prevalence of narcolepsy with cataplexy in Norway. Acta Neurol Scand. 2009;120(4):276–280.
15. Doherty L, Crowe C, Sweeney B. National narcolepsy survey. Ir Med J. 2010;103(4):112–113.
16. Wilner A, Steinman L, Lavie P, Peled R, Friedmann A, Brautbar C. Narcolepsy-cataplexy in Israeli Jews is associated exclusively with the HLA DR2 haplotype. A study at the serological and genomic level. Hum Immunol. 1988;21(1):15–22.
17. Tashiro T, Kanbayashi T, Iijima S, Hishikawa Y. An epidemiological study of narcolepsy in Japanese. J Sleep Res. 1992;1:228.
18. Peacock J, Benca RM. Narcolepsy: Clinical features, co-morbidities and treatment. Indian J Med Res. 2010;131:338–349.
19. Peterson PC, Husain AM. Pediatric narcolepsy. Brain Dev. 2008; 30(10):609–623.
20. Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C. HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients. Sleep. 1997;20(11):1012–1020.
21. Silber MH, Krahn LE, Olson EJ, Pankratz VS. The epidemiology of narcolepsy in Olmsted County, Minnesota: A population-based study. Sleep. 2002;25(2):197–202.
22. Nishino S, Okura M, Mignot E. Narcolepsy: Genetic predisposition and neuropharmacological mechanisms. Sleep Med Rev. 2000;4(1):57–99.
23. Mignot E. Genetic and familial aspects of narcolepsy. Neurology. 1998;50(2 Suppl 1):S16–S22.
24. Aran A, Lin L, Nevsimalova S, et al. Elevated anti-streptococcal antibodies in patients with recent narcolepsy onset. Sleep. 2009;32(8):979–983.
25. Longstreth WT Jr, Ton TG, Koepsell TD. Narcolepsy and streptococcal infections. Sleep. 2009;32(12):1548.
26. Dauvilliers Y, Montplaisir J, Cochen V, et al. Post-H1N1 narcolepsy-cataplexy. Sleep. 2010;33(11):1428–1430.
27. Ton TG, Longstreth WT Jr, Koepsell TD. Environmental toxins and risk of narcolepsy among people with HLA DQB1*0602. Environ Res. 2010;110(6):565–570.
28. Tafti M. Genetic aspects of normal and disturbed sleep. Sleep Med. 2009;10 Suppl 1:S17–S21.
29. Mignot E, Lin L, Rogers W, et al. Complex HLA-DR and -DQ interac-tions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet. 2001;68(3):686–699.
30. Hor H, Kutalik Z, Dauvilliers Y, et al. Genome-wide association study identifies new HLA class II haplotypes strongly protective against narcolepsy. Nat Genet. 2010;42(9):786–789.
31. Hong SC, Lin L, Jeong JH, et al. A study of the diagnostic utility of HLA typing, CSF hypocretin-1 measurements, and MSLT testing for the diagnosis of narcolepsy in 163 Korean patients with unexplained excessive daytime sleepiness. Sleep. 2006;29(11):1429–1438.
32. Sutcliffe JG, de Lecea L. The hypocretins: Excitatory neuromodulatory peptides for multiple homeostatic systems, including sleep and feeding. J Neurosci Res. 2000;62(2):161–168.
33. Thorpy M. Therapeutic advances in narcolepsy. Sleep Med. 2007; 8(4):427–440.
34. Shibata M, Mondal MS, Date Y, Nakazato M, Suzuki H, Ueta Y. Dis-tribution of orexins-containing fibers and contents of orexins in the rat olfactory bulb. Neurosci Res. 2008;61(1):99–105.
35. Jones BE. Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann N Y Acad Sci. 2008;1129:26–34.
36. Foutz AS, Mitler MM, Cavalli-Sforza LL, Dement WC. Genetic factors in canine narcolepsy. Sleep. 1979;1(4):413–421.
37. Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98(3):365–376.
Dovepress
Dovepress
41
Neuro vo5 no4 2011.indd 41 2011/10/28 10:13 AM

38. Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355(9197): 39–40.
39. Thannickal TC, Moore RY, Nienhuis R, et al. Reduced number of hypo-cretin neurons in human narcolepsy. Neuron. 2000;27(3):469–474.
40. Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6(9):991–997.
41. Black JL 3rd, Silber MH, Krahn LE, et al. Studies of humoral immu-nity to preprohypocretin in human leukocyte antigen DQB1*0602-positive narcoleptic subjects with cataplexy. Biol Psychiatry. 2005; 58(6):504–509.
42. Black JL 3rd, Silber MH, Krahn LE, et al. Analysis of hypocretin (orexin) antibodies in patients with narcolepsy. Sleep. 2005;28(4):427–431.
43. Gledhill RF, Bartel PR, Yoshida Y, Nishino S, Scammell TE. Narcolepsy caused by acute disseminated encephalomyelitis. Arch Neurol. 2004;61(5):758–760.
44. Jackson MW, Reed JH, Smith AJ, Gordon TP. An autoantibody in narcolepsy disrupts colonic migrating motor complexes. J Neurosci. 2008;28(49):13303–13309.
45. Zhang Y, Davis JL, Li W. Identification of Tribbles homolog 2 as an autoantigen in autoimmune uveitis by phage display. Mol Immunol. 2005;42(11):1275–1281.
46. Cvetkovic-Lopes V, Bayer L, Dorsaz S, et al. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J Clin Invest. 2010;120(3):713–719.
47. Toyoda H, Tanaka S, Miyagawa T, Honda Y, Tokunaga K, Honda M. Anti-Tribbles homolog 2 autoantibodies in Japanese patients with narcolepsy. Sleep. 2010;33(7):875–878.
48. Hallmayer J, Faraco J, Lin L, et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet. 2009;41(6):708–711.
49. Hinze-Selch D, Wetter TC, Zhang Y, et al. In vivo and in vitro immune variables in patients with narcolepsy and HLA-DR2 matched controls. Neurology. 1998;50(4):1149–1152.
50. Okun ML, Giese S, Lin L, Einen M, Mignot E, Coussons-Read ME. Exploring the cytokine and endocrine involvement in narcolepsy. Brain Behav Immun. 2004;18(4):326–332.
51. Himmerich H, Beitinger PA, Fulda S, et al. Plasma levels of tumor necro-sis factor alpha and soluble tumor necrosis factor receptors in patients with narcolepsy. Arch Intern Med. 2006;166(16):1739–1743.
52. Fontana A, Gast H, Reith W, Recher M, Birchler T, Bassetti CL. Nar-colepsy: Autoimmunity, effector T cell activation due to infection, or T cell independent, major histocompatibility complex class II induced neuronal loss? Brain. 2010;133(Pt 5):1300–1311.
53. Kanbayashi T, Ishiguro H, Aizawa R, et al. Hypocretin-1 (orexin-A) concentrations in cerebrospinal fluid are low in patients with Guillain-Barre syndrome. Psychiatry Clin Neurosci. 2002;56(3):273–274.
54. Nishino S, Kanbayashi T, Fujiki N, et al. CSF hypocretin levels in Guillain-Barre syndrome and other inflammatory neuropathies. Neurology. 2003;61(6):823–825.
55. Bourgin P, Zeitzer JM, Mignot E. CSF hypocretin-1 assessment in sleep and neurological disorders. Lancet Neurol. 2008;7(7):649–662.
56. Von Economo C. Encephalitis lethargica. Weiner Medizinische Wochenschrift. 1923;73:777–782.
57. Aldrich MS, Naylor MW. Narcolepsy associated with lesions of the diencephalon. Neurology. 1989;39(11):1505–1508.
58. Nishino S, Kanbayashi T. Symptomatic narcolepsy, cataplexy and hypersomnia, and their implications in the hypothalamic hypocretin/orexin system. Sleep Med Rev. 2005;9(4):269–310.
59. American Academy of Sleep Medicine. The International Classifi-cation of Sleep Disorders, Diagnostic and Coding Manual. 2nd ed. Westchester, IL: American Academy of Sleep Medicine; 2005.
60. Johns MW. A new method for measuring daytime sleepiness: The Epworth sleepiness scale. Sleep. 1991;14(6):540–545.
61. Sullivan SS, Kushida CA. Multiple sleep latency test and maintenance of wakefulness test. Chest. 2008;134(4):854–861.
62. Nguyen AT, Baltzan MA, Small D, Wolkove N, Guillon S, Palayew M. Clinical reproducibility of the Epworth Sleepiness Scale. J Clin Sleep Med. 2006;2(2):170–174.
63. DeZee KJ, Jackson JL, Hatzigeorgiou C, Kristo D. The Epworth sleepi-ness scale: Relationship to sleep and mental disorders in a sleep clinic. Sleep Med. 2006;7(4):327–332.
64. Izci B, Ardic S, Firat H, Sahin A, Altinors M, Karacan I. Reliability and validity studies of the Turkish version of the Epworth Sleepiness Scale. Sleep Breath. 2008;12(2):161–168.
65. Chen NH, Johns MW, Li HY, et al. Validation of a Chinese version of the Epworth sleepiness scale. Qual Life Res. 2002;11(8):817–821.
66. Kok SW, Meinders AE, Overeem S, et al. Reduction of plasma leptin levels and loss of its circadian rhythmicity in hypocretin (orexin)-deficient narcoleptic humans. J Clin Endocrinol Metab. 2002;87(2):805–809.
67. Littner MR, Kushida C, Wise M, et al. Practice parameters for clinical use of the multiple sleep latency test and the maintenance of wakeful-ness test. Sleep. 2005;28(1):113–121.
68. Carskadon MA, Dement WC, Mitler MM, Roth T, Westbrook PR, Keenan S. Guidelines for the multiple sleep latency test (MSLT): A standard measure of sleepiness. Sleep. 1986;9(4):519–524.
69. Rack M, Davis J, Roffwarg HP, Richert A, Baran AS. The Multiple Sleep Latency Test in the diagnosis of narcolepsy. Am J Psychiatry. 2005;162(11):2198–2199.
70. Allen RP. When, if ever, can we use REM-onset naps on the MSLT for the diagnosis of narcolepsy? Sleep Med. 2006;7(8):657–659.
71. Morrison I, Buskova J, Nevsimalova S, Douglas NJ, Riha RL. Diagnosing narcolepsy with cataplexy on history alone: Challenging the International Classification of Sleep Disorders (ICSD-2) criteria. Eur J Neurol. 2011;18(7):1017–1020.
72. Sangal RB, Thomas L, Mitler MM. Maintenance of wakefulness test and multiple sleep latency test. Measurement of different abilities in patients with sleep disorders. Chest. 1992;101(4):898–902.
73. Benbadis SR, Mascha E, Perry MC, Wolgamuth BR, Smolley LA, Dinner DS. Association between the Epworth sleepiness scale and the multiple sleep latency test in a clinical population. Ann Intern Med. 1999;130(4 Pt 1):289–292.
74. Plazzi G, Montagna P, Provini F, Bizzi A, Cohen M, Lugaresi E. Pontine lesions in idiopathic narcolepsy. Neurology. 1996;46(5):1250–1254.
75. Bassetti C, Aldrich MS, Quint DJ. MRI findings in narcolepsy. Sleep. 1997;20(8):630–631.
76. Frey JL, Heiserman JE. Absence of pontine lesions in narcolepsy. Neurology. 1997;48(4):1097–1099.
77. Stepien A, Staszewski J, Domzal TM, Tomczykiewicz K, Skrobowska E, Durka-Kesy M. Degenerative pontine lesions in patients with familial narcolepsy. Neurol Neurochir Pol. 2010;44(1):21–27. Polish.
78. Ellis CM, Simmons A, Lemmens G, Williams SC, Parkes JD. Proton spectroscopy in the narcoleptic syndrome. Is there evidence of a brain-stem lesion? Neurology. 1998;50(2 Suppl 1):S23–S26.
79. Lodi R, Tonon C, Vignatelli L, et al. In vivo evidence of neuronal loss in the hypothalamus of narcoleptic patients. Neurology. 2004;63(8): 1513–1515.
80. Joo EY, Hong SB, Tae WS, et al. Cerebral perfusion abnormality in narcolepsy with cataplexy. NeuroImage. 2005;28(2):410–416.
81. Hong SB, Tae WS, Joo EY. Cerebral perfusion changes dur-ing cataplexy in narcolepsy patients. Neurology. 2006;66(11): 1747–1749.
82. Boulos MI, Murray BJ. Current evaluation and management of exces-sive daytime sleepiness. Can J Neurol Sci. 2010;37(2):167–176.
83. Arnulf I. Excessive daytime sleepiness in parkinsonism. Sleep Med Rev. 2005;9(3):185–200.
84. Lee JH, Bliwise DL, Ansari FP, et al. Daytime sleepiness and func-tional impairment in Alzheimer disease. Am J Geriatr Psychiatry. 2007;15(7):620–626.
85. Ervik S, Abdelnoor M, Heier MS, Ramberg M, Strand G. Health-related quality of life in narcolepsy. Acta Neurol Scand. 2006;114(3): 198–204.
Dovepress
Dovepress
42
Neuro vo5 no4 2011.indd 42 2011/10/28 10:13 AM

86. Riemann D. Insomnia and comorbid psychiatric disorders. Sleep Med. 2007;8 Suppl 4:S15–S20.
87. Naumann A, Bellebaum C, Daum I. Cognitive deficits in narcolepsy. J Sleep Res. 2006;15(3):329–338.
88. Bhat SK, Galang R. Narcolepsy presenting as schizophrenia. Am J Psychiatry. 2002;159(7):1245.
89. Kondziella D, Arlien-Soborg P. Diagnostic and therapeutic challenges in narcolepsy-related psychosis. J Clin Psychiatry. 2006;67(11): 1817–1819.
90. Kishi Y, Konishi S, Koizumi S, Kudo Y, Kurosawa H, Kathol RG. Schizophrenia and narcolepsy: A review with a case report. Psychiatry Clin Neurosci. 2004;58(2):117–124.
91. Moturi S, Ivanenko A. Complex diagnostic and treatment issues in psychotic symptoms associated with narcolepsy. Psychiatry (Edgmont). 2009;6(6):38–44.
92. Yasui-Furukori N, Kusunoki M, Kaneko S. Hallucinations associated with modafinil treatment for narcolepsy. J Clin Psychopharmacol. 2009;29(4):408.
93. Dahmen N, Kasten M, Mittag K, Muller MJ. Narcoleptic and schizo-phrenic hallucinations. Implications for differential diagnosis and pathophysiology. Eur J Health Econ. 2002;3 Suppl 2:S94–S98.
94. Nevsimalova S. Narcolepsy in childhood. Sleep Med Rev. 2009; 13(2):169–180.
95. Fejerman N. Nonepileptic disorders imitating generalized idiopathic epilepsies. Epilepsia. 2005;46 Suppl 9:80–83.
96. Calabro RS, Savica R, Lagana A, et al. Status cataplecticus misdiag-nosed as recurrent syncope. Neurol Sci. 2007;28(6):336–338.
97. Macleod S, Ferrie C, Zuberi SM. Symptoms of narcolepsy in children misinterpreted as epilepsy. Epileptic Disord. 2005;7(1):13–17.
98. Centre for Evidence-based Medicine. Levels of evidence 2009 [web page on the Internet]. Oxford: Centre for Evidence-based Medicine; 2009 [updated April 15, 2011]. Available from: http://www.cebm.net/index.aspx?o 1025. Accessed April 20, 2011.
99. Littner M, Johnson SF, McCall WV, et al. Practice parameters for the treatment of narcolepsy: An update for 2000. Sleep. 2001; 24(4):451–466.
100. Wang W, Wang F, Zhao Y, Lv M, Lv X. Two patients with narcolepsy treated by hypnotic psychotherapy. Sleep Med. 2009;10(10):1167.
101. Husain AM, Yancy WS Jr, Carwile ST, Miller PP, Westman EC. Diet therapy for narcolepsy. Neurology. 2004;62(12):2300–2302.
102. Morgenthaler TI, Kapur VK, Brown T, et al. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30(12):1705–1711.
103. Murray TJ, Foley A. Narcolepsy. Can Med Assoc J. 1974;110(1): 63–66.
104. Weitzner HA. Insulin hypoglycemia in treatment of narcolepsy with tem-porary improvement. Perm Found Med Bull. 1952;10(1–4):153–156.
105. Sandyk R. Efficacy of trazodone in narcolepsy. Eur Neurol. 1985; 24(5):335–337.
106. Iijima S, Sugita Y, Teshima Y, Hishikawa Y. Therapeutic effects of mazindol on narcolepsy. Sleep. 1986;9(1 Pt 2):265–268.
107. Kales A, Cadieux R, Soldatos CR, Tan TL. Successful treat-ment of narcolepsy with propranolol: A case report. Arch Neurol. 1979;36(10):650–651.
108. Meier-Ewert K, Matsubayashi K, Benter L. Propranolol: Long-term treatment in narcolepsy-cataplexy. Sleep. 1985;8(2):95–104.
109. Lammers GJ, Arends J, Declerck AC, Kamphuisen HA, Schouwink G, Troost J. Ritanserin, a 5-HT2 receptor blocker, as add-on treatment in narcolepsy. Sleep. 1991;14(2):130–132.
110. Mayer G. Ritanserin improves sleep quality in narcolepsy. Pharma-copsychiatry. 2003;36(4):150–155.
111. Mayer G, Ewert Meier K, Hephata K. Selegeline hydrochloride treat-ment in narcolepsy. A double-blind, placebo-controlled study. Clin Neuropharmacol. 1995;18(4):306–319.
112. Prinzmetal M, Bloomberg W. The use of benzedrine for the treatment of narcolepsy. JAMA. 1935;105(25):2051–2054.
113. Billiard M. Narcolepsy: Current treatment options and future approaches. Neuropsychiatr Dis Treat. 2008;4(3):557–566.
114. Berman SM, Kuczenski R, McCracken JT, London ED. Potential adverse effects of amphetamine treatment on brain and behavior: A review. Mol Psychiatry. 2009;14(2):123–142.
115. Jiao X, Velez S, Ringstad J, Eyma V, Miller D, Bleiberg M. Myocardial infarction associated with adderall XR and alcohol use in a young man. J Am Board Fam Med. 2009;22(2):197–201.
116. Mitler MM, Hajdukovic R, Erman MK. Treatment of narcolepsy with methamphetamine. Sleep. 1993;16(4):306–317.
117. Mitler MM, Hajdukovic R, Erman M, Koziol JA. Narcolepsy. J Clin Neurophysiol. 1990;7(1):93–118.
118. Honda Y, Hishikawa Y, Takahashi Y. Long-term treatment of nar-colepsy with methylphenidate (Ritalin). Curr Ther Res. 1979;25: 288–298.
119. Volkow ND, Fowler JS, Logan J, et al. Effects of modafinil on dop-amine and dopamine transporters in the male human brain: Clinical implications. JAMA. 2009;301(11):1148–1154.
120. Huang Q, Zhang L, Tang H, Wang L, Wang Y. Modafinil modulates GABA-activated currents in rat hippocampal pyramidal neurons. Brain Res. 2008;1208:74–78.
121. [No authors listed]. Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. US Modafinil in Narcolepsy Multicenter Study Group. Ann Neurol. 1998;43(1):88–97.
122. [No authors listed]. Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy: US Modafinil in Narco-lepsy Multicenter Study Group. Neurology. 2000;54(5):1166–1175.
123. Broughton RJ, Fleming JA, George CF, et al. Randomized, double-blind, placebo-controlled crossover trial of modafinil in the treat-ment of excessive daytime sleepiness in narcolepsy. Neurology. 1997;49(2):444–451.
124. Schwartz JR, Feldman NT, Bogan RK, Nelson MT, Hughes RJ. Dosing regimen effects of modafinil for improving daytime wake-fulness in patients with narcolepsy. Clin Neuropharmacol. 2003; 26(5):252–257.
125. Schwartz JR, Feldman NT, Bogan RK. Dose effects of modafinil in sustaining wakefulness in narcolepsy patients with residual evening sleepiness. J Neuropsychiatry Clin Neurosci. 2005;17(3): 405–412.
126. Saletu B, Frey R, Krupka M, Anderer P, Grunberger J, Barbanoj MJ. Differential effects of a new central adrenergic agonist – modafinil – and D-amphetamine on sleep and early morning behaviour in young healthy volunteers. Int J Clin Pharmacol Res. 1989;9(3):183–195.
127. Mitler MM, Harsh J, Hirshkowitz M, Guilleminault C. Long-term efficacy and safety of modafinil (PROVIGIL R) for the treatment of excessive daytime sleepiness associated with narcolepsy. Sleep Med. 2000;1(3):231–243.
128. Harsh JR, Hayduk R, Rosenberg R, et al. The efficacy and safety of armodafinil as treatment for adults with excessive sleepiness associated with narcolepsy. Curr Med Res Opin. 2006;22(4):761–774.
129. Golicki D, Bala MM, Niewada M, Wierzbicka A. Modafinil for narcolepsy: Systematic review and meta-analysis. Med Sci Monit. 2010;16(8):RA177–RA186.
130. Akimoto H, Honda Y, Takahashi Y. Pharmacotherapy in narcolepsy. Dis Nerv Syst. 1960;21:704–706.
131. Guilleminault C, Raynal D, Takahashi S, Carskadon M, Dement W. Evaluation of short-term and long-term treatment of the narcolepsy syndrome with clomipramine hydrochloride. Acta Neurol Scand. 1976;54(1):71–87.
132. Hishikawa Y, Ida H, Nakai K, Kaneko Z. Treatment of narcolepsy with imipramine (tofranil) and desmethylimipramine (Pertofran). J Neurol Sci. 1966;3(5):453–461.
133. Guilleminault C, Wilson RA, Dement WC. A study on cataplexy. Arch Neurol. 1974;31(4):255–261.
134. Houghton WC, Scammell TE, Thorpy M. Pharmacotherapy for cataplexy. Sleep Med Rev. 2004;8(5):355–366.
Dovepress
Dovepress
43
Neuro vo5 no4 2011.indd 43 2011/10/28 10:13 AM

135. Frey J, Darbonne C. Fluoxetine suppresses human cataplexy: A pilot study. Neurology. 1994;44(4):707–709.
136. Langdon N, Shindler J, Parkes JD, Bandak S. Fluoxetine in the treat-ment of cataplexy. Sleep. 1986;9(2):371–373.
137. Izzi F, Placidi F, Marciani MG, et al. Effective treatment of narcolepsy-cataplexy with duloxetine: A report of three cases. Sleep Med. 2009; 10(1):153–154.
138. Moller LR, Ostergaard JR. Treatment with venlafaxine in six cases of chil-dren with narcolepsy and with cataplexy and hypnagogic hallucinations. J Child Adolesc Psychopharmacol. 2009;19(2):197–201.
139. Guilleminault C, Mancuso J, Salva MA, et al. Viloxazine hydrochloride in narcolepsy: A preliminary report. Sleep. 1986;9(1 Pt 2):275–279.
140. Schrader H, Kayed K, Bendixen Markset AC, Treidene HE. The treat-ment of accessory symptoms in narcolepsy: A double-blind cross-over study of a selective serotonin re-uptake inhibitor (femoxetine) versus placebo. Acta Neurol Scand. 1986;74(4):297–303.
141. Lapierre O, Montplaisir J, Lamarre M, Bedard MA. The effect of gamma-hydroxybutyrate on nocturnal and diurnal sleep of normal subjects: Further considerations on REM sleep-triggering mechanisms. Sleep. 1990;13(1):24–30.
142. Mamelak M, Black J, Montplaisir J, Ristanovic R. A pilot study on the effects of sodium oxybate on sleep architecture and daytime alertness in narcolepsy. Sleep. 2004;27(7):1327–1334.
143. [No authors listed]. A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally admin-istered sodium oxybate with placebo for the treatment of narcolepsy. Sleep. 2002;25(1):42–49.
144. [No authors listed]. Sodium oxybate demonstrates long-term efficacy for the treatment of cataplexy in patients with narcolepsy. Sleep Med. 2004;5(2):119–123.
145. Black J, Pardi D, Hornfeldt CS, Inhaber N. The nightly use of sodium oxybate is associated with a reduction in nocturnal sleep disruption: A double-blind, placebo-controlled study in patients with narcolepsy. J Clin Sleep Med. 2010;6(6):596–602.
146. [No authors listed]. A 12-month, open-label, multicenter extension trial of orally administered sodium oxybate for the treatment of narcolepsy. Sleep. 2003;26(1):31–35.
147. Poryazova R, Tartarotti S, Khatami R, et al. Sodium oxybate in narcolepsy with cataplexy: Zurich sleep center experience. Eur Neurol. 2011;65(3):175–182.
148. Zvosec DL, Smith SW, Hall BJ. Three deaths associated with use of Xyrem. Sleep Med. 2009;10(4):490–493.
149. Hanson LR, Martinez P, Taheri S, Kamsheh L, Mignot E, Frey W II. Intranasal administration of hypocretin 1 (orexin A) bypasses the blood-brain barrier and targets the brain: A new strategy for the treat-ment of narcolepsy. Drug Delivery Technol. 2004;4:65–71.
150. Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M. Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci U S A. 2004;101(13):4649–4654.
151. Plazzi G, Poli F, Franceschini C, et al. Intravenous high-dose immuno-globulin treatment in recent onset childhood narcolepsy with cataplexy. J Neurol. 2008;255(10):1549–1554.
152. Valko PO, Khatami R, Baumann CR, Bassetti CL. No persistent effect of intravenous immunoglobulins in patients with narcolepsy with cataplexy. J Neurol. 2008;255(12):1900–1903.
153. Knudsen S, Mikkelsen JD, Bang B, Gammeltoft S, Jennum PJ. Intravenous immunoglobulin treatment and screening for hypocretin neuron-specific autoantibodies in recent onset childhood narcolepsy with cataplexy. Neuropediatrics. 2010;41(5):217–222.
154. Barbier AJ, Berridge C, Dugovic C, et al. Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol. 2004;143(5):649–661.
155. Guo RX, Anaclet C, Roberts JC, et al. Differential effects of acute and repeat dosing with the H3 antagonist GSK189254 on the sleep-wake cycle and narcoleptic episodes in Ox-/- mice. Br J Pharmacol. 2009;157(1):104–117.
156. Tedford CE, Phillips JG, Gregory R, et al. Development of trans-2-[1H-imidazol-4-yl] cyclopropane derivatives as new high-affinity histamine H3 receptor ligands. J Pharmacol Exp Ther. 1999;289(2):1160–1168.
157. Bagai K, Malow BA. A novel approach to treating morning sleep inertia in narcolepsy. J Clin Sleep Med. 2010;6(1):77–78.
Dovepress
Dovepress
44
Neuro vo5 no4 2011.indd 44 2011/10/28 10:13 AM

Neuropsychiatric Disease and Treatment
Neuroimaging techniques have been useful tools for accurate investigation of brain
structure and function in eating disorders. Computed tomography, magnetic resonance imaging,
positron emission tomography, single photon emission computed tomography, magnetic reso-
nance spectroscopy, and voxel-based morphometry have been the most relevant technologies in
this regard. The purpose of this review is to update the existing data on neuroimaging in eating
disorders. The main brain changes seem to be reversible to some extent after adequate weight
restoration. Brain changes in bulimia nervosa seem to be less pronounced than in anorexia ner-
vosa and are mainly due to chronic dietary restrictions. Different subtypes of eating disorders
might be correlated with specific brain functional changes. Moreover, anorectic patients who
binge/purge may have different functional brain changes compared with those who do not binge/
purge. Functional changes in the brain might have prognostic value, and different changes with
respect to the binding potential of 5-HT1A
, 5-HT2A
, and D2/D
3 receptors may be persistent after
recovering from an eating disorder.
neuroimaging, brain changes, brain receptors, anorexia nervosa, bulimia nervosa,
eating disorders
Neuroimaging techniques have been useful tools for accurate investigation of brain
structure and function in eating disorders, mainly in anorexia nervosa.1 The first studies,
by means of structural neuroimaging, ie, computed tomography (CT) and magnetic
resonance imaging (MRI), focused on the brain anatomy in patients with anorexia ner-
vosa and consistently showed sulcal widening and ventricular enlargement that usually
decreased with refeeding.2,3 Other specific findings have been a reduction in total gray
and white matter volumes compared with healthy controls or the persistence of the gray
matter volume changes when weight is restored.4,5 Some of these findings suggest that
the changes are most likely to be due to neuronal damage secondary to malnutrition,
with possible regeneration of myelin accounting for the general reversibility.4 However,
these findings have not improved our understanding of the pathogenesis of anorexia
nervosa.1 More recently, studies have highlighted the use of functional neuroimaging,
which refers to techniques that obtain images of the brain according to its physiology
and biochemistry. This technique provides information on the dynamics of the neural
machinery involved in cognitive systems and emotional states,6 and thus offers far
more information than does structural imaging. Functional neuroimaging studies have
utilized positron emission tomography (PET) and single photon emission computed
tomography (SPECT), the latter used mainly in studies of regional cerebral blood
Dovepress
Dovepress
open access to scientific and medical research
45
Neuro vo5 no4 2011.indd 45 2011/10/28 10:13 AM

flow (rCBF).7 Other studies have used magnetic resonance
spectroscopy8 or voxel-based morphometry.9 With respect
to PET and SPECT, studies can use fluoro-deoxy-glucose
to analyze glucose metabolism or ligands specific for the
serotonin receptor. Thus, these studies provide information
for a neural system (eg, the 5HT2A
receptor). The purpose of
this review is to update the existing data on the main findings
with respect to neuroimaging in eating disorders by means
of a search conducted in PubMed.
The first study of early-onset anorexia nervosa, by means
of SPECT,10 reported unilateral hypoperfusion in 87% of
patients (children and adolescents aged 8–15 years), in which
unilateral hypoperfusion was found on the left in eight of
13 patients, and on the right in five of 13 patients. In that
study, the temporal lobe was hypoperfused in all cases. Years
later, following the same line of research,7 SPECT findings
showed that 73% of children and adolescents with anorexia
nervosa had asymmetry (hypoperfusion) of blood flow in at
least one area (temporal lobe, 9/15; parietal lobe, 5/15; frontal
lobe, 3/15; thalamus, 3/15; and caudate nuclei, 1/15) of the
brain. Considering the relationship between rCBF and several
relevant variables (eg, cerebral dominance, nutritional status,
length of illness, mood, eating disorder psychopathology,
and cognitive profile), another study1 found that, in addi-
tion to unilateral reduction of blood flow in the temporal
lobe and/or associated areas, approximately 75% of patients
with early-onset anorexia nervosa showed no association
between that reduction and cerebral dominance, nutritional
status, length of illness, mood, or eating psychopathology.
Nevertheless, there was a significant association between
the abovementioned reduction and impaired visuospatial
ability, impaired complex visual memory, and enhanced
information processing.
In another study, conducted by Rastam et al11 in anorectic
patients (mean age 22 years), seven years after onset of the
illness, 66% showed reduced blood flow in the temporal or
associated regions, without a significant correlation between
the reduction and body mass index, the lowest body mass
index, residual eating disorder psychopathology, or intel-
ligence quotient.
In a sample of adolescents with restrictive-type anorexia
nervosa, in the early stages of the illness, morphometric gray
matter changes were characterized by means of preprocessed
MRI according to optimized voxel-based morphometry.
The analyses revealed a significant decrease in global gray
matter, and a significant region-specific decrease in gray
matter volume was found bilaterally in the middle cingulate
cortex, the precuneus, and the inferior and superior parietal
lobules.12
In general, the majority of the studies on anorexia
nervosa have reported brain volume deficits and increased
cerebrospinal fluid, suggesting starvation of the brain.4,13–18 In
this regard, another study reported that right dorsal anterior
cingulate cortex volume was significantly reduced in active
patients with anorexia nervosa versus controls, and was cor-
related with lower performance intelligence quotients.19
There is some debate about the abnormalities of cerebral
blood flow in anorexia nervosa as to whether these abnor-
malities are secondary to starvation or indicative of a primary
abnormality predating the illness, representing an underlying
biological substrate. In a recent study, changes in rCBF were
found at both baseline and follow-up (at more than 4 years).
The main affected cerebral area was the medial temporal
region, the data suggesting that rCBF does not return to
normal following weight restoration.20
A previous study21 in female anorectic children (mean age
13.2 years), in which SPECT was performed at the begin-
ning of treatment and after weight gain, reported relatively
increased rCBF in the bilateral parietal and limbic lobes,
including the posterior cingulate cortex, after weight gain.
There was no significant decrease in the rCBF after weight
gain. On the other hand, a significant positive correlation
was observed between body mass index and rCBF in the
right thalamus, right parietal lobe, and right cerebellum.
These results were said to suggest that weight gain during
the process of recovery from early-onset anorexia nervosa
might activate specific brain regions that are possibly relevant
to the pathophysiological aspects of the disorder.
In a pioneer study by Gordon et al,10 temporal lobe
hypoperfusion persisted in three of four patients who had
regained their normal weight. Furthermore, although two of
the four patients had recovered a normal weight/height ratio
after refeeding, the cognitive distortions of anorexia nervosa
persisted, as well as the abnormal rCBF. This suggests that
the hypoperfusion is not related directly to weight loss.
Another study using voxel-based morphometry showed
that several temporal and parietal gray matter regions were
reduced. During follow-up, there was a greater global
increase in gray matter in anorectic patients, and this
increase correlated with a decrease in cortisol. At follow-up
(7 months), there were no differences in global gray matter
Dovepress
Dovepress
46
Neuro vo5 no4 2011.indd 46 2011/10/28 10:13 AM

and white matter volumes between anorectic patients and
controls. The authors concluded that, in adolescent anorectic
patients, gray matter is more affected than white matter and
mainly involves the posterior regions of the brain. Overall,
gray matter alterations are reversible after nutritional
recovery.22 With regards to adult patients with anorexia
nervosa, a decrease in cerebrospinal fluid and an increase in
brain matter, as well as a reversal of ventricular enlargement,
have been observed after weight restoration.13,16,17
The majority of the studies have been developed tak-
ing into account samples of adolescents and adults with
restrictive-type anorexia nervosa. Considering the fact that
it may be possible to find differences between patients at
normal weight, patients after weight restoration, and controls,
a recent study showed that patients with anorexia nervosa had
a significant increase in gray and white matter volume after
weight restoration. In addition, this study showed that patients
had lower levels of gray matter at low weight compared with
controls, which increased with weight restoration.23
Despite some differences, it seems that gray matter
and white matter increase significantly following weight
restoration. With respect to possible differences between
adolescents and adults, it has been stated that gray matter
is more affected than white matter in adolescents.22 The
interaction among low weight, duration of illness, and brain
changes remains controversial to some extent. Thus, while
some authors have reported an inverse correlation between
duration of illness and lower volume of gray matter at low
body weight, but no correlation between low body weight and
measures of brain volume, other authors have found a cor-
relation between body mass index and brain volume, but not
with duration of illness.18,23 Studying patients with anorexia
nervosa who had a different duration of illness, Boghi et al24
found a significant reduction in total white matter volume and
focal gray matter atrophy in the cerebellum, hypothalamus,
caudate nucleus, and frontal, parietal, and temporal areas.
The cerebellum was more affected in patients with a longer
disease duration, whereas the hypothalamic alterations were
more pronounced in patients with a shorter period of food
restriction. A correlation between body mass index and
gray matter was found in the hypothalamus. These authors
suggested that atrophy of cerebellar gray matter could play
a role in the chronic phase of the disease.
Frank et al25 studied groups of women with recovered
restricting-type anorexia nervosa, recovered anorexia ner-
vosa with a binging history, recovered bulimia nervosa
without a history of anorexia nervosa, and controls, by means
of PET and [15O] water in order to assess rCBF. Partial
volume-corrected rCBF values in cortical and subcortical
brain regions were similar between the groups. Neither cur-
rent body mass index nor age correlated with rCBF values.
The authors concluded that rCBF normalizes with long-term
recovery, and stated that altered rCBF is unlikely to confound
functional imaging studies in anorexia nervosa or bulimia
nervosa after recovery. McCormick et al19 found that while
anterior cingulate cortex normalization occurred with weight
restoration, a smaller change in right dorsal anterior cingulate
cortex volume prospectively predicted relapse after treatment.
Another study using functional MRI reported that recovered
patients with anorexia nervosa showed altered task-related
activation in the medial prefrontal cortex, a critical node
of the inhibitory control network. Specifically, whereas
recovered patients with anorexia nervosa and control women
showed similar medial prefrontal cortex activity during trials
when inhibitory demand was low (ie, easy trials), recovered
patients with anorexia nervosa showed significantly less
medial prefrontal cortex activation than control women as
inhibition trials became more difficult (ie, hard trials), sug-
gesting a demand-specific modulation of inhibitory control
circuitry in recovered patients with anorexia nervosa.26
Structural brain changes on CT reported in cases of bulimia
nervosa are similar to those described for anorexia nervosa,
but less pronounced, and they have been related to chronic
dietary restriction.27 Fewer studies of bulimia nervosa have
been performed with MRI than for anorexia nervosa, and they
have usually reported a decreased cortical mass.28–30
The neural bases of eating disorders have been explored by
means of neuroimaging studies employing different stimuli,
such as food or body image. Functional MRI studies have
reported that when malnourished patients with anorexia
nervosa are shown pictures of food, they display abnormal
activity in the insula and orbitofrontal cortex, as well as in
other regions, like the mesial temporal and parietal regions,
and anterior cingulate cortex.31–36 Using PET, SPECT, and
functional MRI, patients with anorexia nervosa eating food
or being exposed to food show activation of the temporal
regions.31,33–36
Comparing chronic anorectic patients, recovered ano-
rectic patients, and controls, Uher et al37 found increased
medial prefrontal and anterior cingulate activation in
response to food stimuli, as well as a lack of activity in the
Dovepress
Dovepress
47
Neuro vo5 no4 2011.indd 47 2011/10/28 10:13 AM

inferior parietal lobule, differentiating the recovered group
from the healthy control subjects. Increased activation of the
right lateral prefrontal, apical prefrontal, and dorsal anterior
cingulate cortices differentiated recovered subjects from
chronically ill patients. Group differences were specific to
food stimuli, whereas processing of emotional stimuli did
not differ between the groups. The authors concluded that
separate neural correlates underlie trait and state character-
istics of anorexia nervosa. The medial prefrontal response to
disease-specific stimuli may be related to trait vulnerability.
Lateral and apical prefrontal involvement is associated with
a good outcome.
Functional MRI studies in women with full or sub-
threshold bulimia nervosa have reported less activation than
healthy controls in the right anterior insula in response to
anticipated receipt of a chocolate milkshake (versus tasteless
solution) and in the left middle frontal gyrus, right posterior
insula, right precentral gyrus, and right mid dorsal insula in
response to consumption of a milkshake (versus tasteless
solution).38
Other studies have sought brain correlates for severe
body image distortion. Boghi et al24 have suggested that
involvement of the temporoparietal areas in brain atrophy
could account for body image distortion. High responsive-
ness in the frontal visual system of the brain and the attention
network, as well as the inferior parietal lobe, including the
anterior part of the intraparietal sulcus, has been described
in patients with anorexia nervosa when confronted with their
own digitally distorted body image as well as an image of a
different person. Moreover, patients with anorexia nervosa
were reported to show only an increase in activation in
response to their own pictures and not to those of others,
indicating different visuospatial processing, while controls
did not differentiate.39 In another study, PET showed a nega-
tive relationship between 5HT2A
receptor activity in the left
parietal cortex, right occipital cortex, and left subgenual
cingulate and the subscale of drive for thinness on the Eating
Disorders Inventory.40 It is well known that the parietal cortex
mediates perceptions of the body, and disturbances in the left
hemisphere may contribute to body image distortion.
Different components of body image (satisfaction
rating and size estimation) have been studied in patients
with anorexia nervosa using functional MRI. Anorectic
patients were less satisfied with their current body shape
than controls. Patients showed stronger activation of the
insula and lateral prefrontal cortex during the satisfaction
rating of thin self-image, indicating a stronger emotional
involvement when presented with distorted images close to
their own ideal body size. In addition, patients overestimated
their own body size.41
Brain involvement in body image satisfaction has been
studied, not only in patients but also in healthy people without
an eating disorder. In a sample of healthy young women, the
impact of images of slim female fashion models was analyzed
by functional neuroimaging. The level of reported anxiety
during exposure to slim bodies correlated with established
measures of shape and weight concern and brain activation in
the bilateral basal ganglia, left amygdala, bilateral dorsal ante-
rior cingulate, and left inferior lateral prefrontal cortex.42
Similar studies have highlighted the relevance of analysis
of the correlation between body image therapy and activa-
tion of brain regions involved in body image representation.
Vocks et al43 analyzed neuronal responses to viewing pho-
tographs of one’s own body before and after treatment in
patients with anorexia nervosa by MRI. Decreases in activa-
tion emerged in a distributed network, and increases were
observed in the extrastriate body area, possibly reflecting
more intense body image processing.
A controlled near-infrared spectroscopy study was developed
to identify brain correlates with different types of eating
disorders. Regional hemodynamic changes in the bilateral
orbitofrontal and right frontotemporal regions were signifi-
cantly smaller in the eating disorder group than in the control
group, and negatively correlated with dieting tendency scores
in the Eating Attitudes Test-26 in the right frontotemporal
regions, and correlated with eating restriction and binge eat-
ing scores in the left orbitofrontal regions. In conclusion, the
authors stated that the tendency towards dieting correlates
with the right frontotemporal cortex, and that dieting behavior
problems correlate with the left orbitofrontal cortex.44
In addition to investigations for brain correlations with
different eating disorder symptoms, other studies have tried to
explore possible and specific brain correlations with different
subtypes of eating disorder. Using SPECT, Beato-Fernández
et al45 found that patients with anorexia nervosa showed
hyperactivation of the left parietal and right superior frontal
areas in response to a positive video stimulus (the patient’s
filmed body image) compared with a neutral (landscape)
stimulus, whereas patients with bulimia nervosa showed
hyperactivation of the right temporal and right occipital areas.
The authors concluded that functional brain abnormalities in
patients with anorexia nervosa might be related to storage of
a distorted prototypic image of the body in the left parietal
lobe, and the activation of the right temporal area after
Dovepress
Dovepress
48
Neuro vo5 no4 2011.indd 48 2011/10/28 10:13 AM

exposure to images of their own body might be consistent
with the adverse response. Following this study, the same
researchers46 used SPECT to investigate the discriminatory
capacity of these psychopathological and neurobiological
variables with regards to different eating disorders. Patients
with nonpurging-type bulimia nervosa showed less eating
and general psychopathology. Furthermore, unlike patients
with restrictive-type anorexia nervosa and purging-type
bulimia nervosa, they did not demonstrate an increase in
rCBF when confronted with their own body image. Temporal
right hyperactivation was one of the discriminatory variables
tested. In this study, the authors concluded that the subgroup
with nonpurging-type bulimia nervosa showed less emotional
alteration and less emotional response when shown their own
body image than the patients with other eating disorders.
Another study34 analyzed the effect of imagining food on
rCBF in patients with anorexia nervosa with and without binge/
purging behavior. The patients with anorexia nervosa accom-
panied by habitual binge/purge behavior showed a significantly
higher percent change in the inferior, superior, prefrontal, and
parietal regions of the right brain than healthy volunteers and
patients with restrictive-type anorexia nervosa. The patients
with habitual binge/purge behavior also had the highest level of
apprehension concerning food intake. Using SPECT, Karhunen
et al47 found similarly high rCBF in the right parietal and
temporal cortical areas in obese women looking at food than
in normal weight women looking at food. Recognition of the
parietal cortex as a mediator of body image perceptions has
led to the suggestion that the parietal lobe contributes to the
awareness of being responsible for one’s own actions.48
Comparing the results of basal SPECT and clinical outcome
in patients with eating disorders, it has been found that tem-
poral hypoperfusion in the acute phase is correlated with the
long-term clinical outcome, suggesting a prognostic value of
temporal hypoperfusion.49 This finding confirms the results of
another study developed by the same research team in which
hypoperfusion of the anterior inferior region of the temporal
lobe, predominantly in the left hemisphere, was found in the
acute phase of the illness.50 Some brain changes, found among
different subtypes of eating disorder, have been suggested to
be possible prognostic factors.46
Brain imaging using PET and development of selective
tracers for the 5-HT system have enabled in vivo analysis of
5-HT receptor function. With respect to the 5-HT1A
receptor,
anorectic patients showed a marked increase (30%–70%) in
[11C] WAY-100635BP, a selective ligand in the prefrontal
and lateral orbital frontal regions, mesial and lateral temporal
lobes, parietal cortex, and dorsal raphe nuclei.51 PET and [11C]
WAY-100635 studies showed that binding potential values
in all brain regions investigated were greater in patients
with bulimia nervosa than in control subjects. The most
robust differences were observed at the angular gyrus, the
medial prefrontal cortex, and the posterior cingulate cortex.
The authors suggested that brain 5-HT1A
receptor binding is
increased in several cortical areas in patients with bulimia
nervosa during bouts of impulsive binge eating.52
With respect to 5-HT2A
receptors, it has been found using
PET that anorectic patients had normal values of the [18F]
altanserin radioligand.51 Using SPECT and 123I-5-I-R91150,
a significantly reduced 5-HT2A
binding index was shown in
the left frontal cortex, left and right parietal cortex, and left
and right occipital cortex in patients with anorexia nervosa
compared with healthy volunteers. A significant left-right
asymmetry was noted in the frontal cortex (left less than
right).53
Bailer et al54 studied different subtypes of eating disorder after
recovery (normal body weight maintained for more than one
year, regular menstrual cycles, no binging or purging) using
PET and [11C] McN5652. They found that both controls and
patients with restrictive anorexia nervosa, purging anorexia
nervosa, and bulimia nervosa showed significant differ-
ences in [11C] McN5652 BP values in the dorsal raphe and
anteroventral striatum. Post hoc analysis revealed that patients
with recovered restrictive anorexia nervosa had significantly
increased [11C] McN5652 BP compared with those having
recovered purging anorexia nervosa in these regions. With
respect to bulimia nervosa, Bailer et al55 used PET and [11C]
WAY100635 to investigate a sample of recovered bulimic
patients and found that they had a 23%–34% elevation of
[11C] WAY binding potential in the subgenual cingulate,
mesial temporal, and parietal regions, and confirmed these
results using 5-HT transporter measures. Pichika et al56 found
that recovered bulimic patients had significantly lower [11C]
DASB binding potential (nondisplaceable) in the midbrain
and superior and inferior cingulate, as well as a significantly
higher [11C] DASB binding potential (nondisplaceable)
in the anterior cingulate and superior temporal gyrus on
voxel-based analysis. Region of interest analysis indicated
Dovepress
Dovepress
49
Neuro vo5 no4 2011.indd 49 2011/10/28 10:13 AM

a lower [11C] DASB binding potential (nondisplaceable) in
the midbrain, including in the dorsal raphe, in patients with
recovered bulimia nervosa, consistent with earlier studies.
Individuals with recovered restrictive anorexia nervosa
have shown a reduced [18F] altanserin binding potential in
the mesial temporal and parietal cortical areas, as well as in
the subgenual and pregenual cingulate cortex.57 Similarly,
women with recovered purging anorexia nervosa have
shown a reduced [18F] altanserin binding potential relative
to controls in the left subgenual cingulate, left parietal, and
right occipital cortex.58 Further, women with recovered
bulimia nervosa have shown a reduced [18F] altanserin
binding potential relative to controls in the orbital frontal
region. In summary, it is suggested that the altered 5-HT1A
and 5-HT2A
receptor binding potential in recovered patients
is reflected in persistent alterations in the frontal, subgenual
cingulate, and mesial temporal regions that are part of the
ventral limbic system.48
In a PET investigation of dopamine D2/D
3 receptor bind-
ing in recovered patients, using PET with [11C] raclopride,
Frank et al59 found that women who were recovered from
anorexia nervosa had a significantly higher [11C] raclopride
binding potential in the anteroventral striatum than controls.
The [11C] raclopride binding potential was positively corre-
lated with harm avoidance in the dorsal caudate and dorsal
putamen in subjects with recovered anorexia nervosa.
PET imaging studies in both ill patients and those with a
recovered eating disorder have found significant correlations
between harm avoidance and 5-HT1A
, 5-HT2A
, DA D2/D
3
receptor binding in the mesial temporal and other limbic
regions.48 It has been suggested that premorbid onset and
the persistence of anxiety and harm avoidance symptoms
after recovery are traits that contribute to the pathogenesis
of anorexia nervosa and bulimia nervosa. The PET imaging
data suggest that such behaviors are related to disturbances of
5-HT and dopamine neurotransmitter function in the limbic
and executive pathways.48
It must be noted that neuroimaging studies in eating disor-
ders have varied widely in terms of sample size, imaging
technology used, age of participants, brain regions assessed,
and duration of illness. Sample sizes have usually been
small, which potentially affects the statistical power of
these studies. To date, the main limitation of this type of
research is that the studies have not consistently identified
brain regions, pathways, or clear behavioral correlates. As
has been noted,48 functional neuroimaging studies in mixed
populations (children, adolescents, and adults) have yielded
inconsistent results due to the wide age range, use of differ-
ent equipment, the different interpretation methods used,
failure to control for emotional arousal, comorbidity, whether
imaging was done before or after a meal, and a number of
other methodological problems. In addition, the actual neu-
roimaging techniques may produce varying results, because
each technique measures something slightly different.7 Also,
different results can be obtained depending on the isotope
used, the type of camera, the scanning parameters, and the
method of data analysis.60
With respect to the neurocircuitry involved in the psy-
chopathology of eating disorders, it is worth mentioning that
the brain is neither an undifferentiated whole nor a collection
of parts or areas.61 In this regard, its overall function is the
result of a complex interaction between different brain areas
and their interconnections. Following this principle, an eat-
ing disorder might emerge as a consequence of disturbance
of a system of interconnecting pathways or circuits in the
brain which regulate cognition, emotion, appetite, and visual
perception.7
In the future, neuroimaging might become a useful tool
for follow-up of the recovery process, enabling clinicians
to differentiate between patients who are recovering
successfully and those who remain chronically ill. Among
these future trends, the study of brain correlates with body
image dissatisfaction as well as body image distortion might
be useful from a diagnostic point of view and, moreover,
serve as a tool to confirm the efficacy of treatment focused
on body image disturbance. The study of clinical subtypes of
eating disorders, usually based on the presence or absence of
binge/purging behavior, is another area of research in which
neuroimaging might contribute to our knowledge about dif-
ferent types of eating disorders from a biological perspective.
In addition, use of functional neuroimaging in the search
for specific neurotransmitter imbalances in the brain could
contribute to improved use of current medications widely
used in eating disorders.
In general, studies of anorexia nervosa have reported brain
volume deficits and increased cerebrospinal fluid, indicating
the effects of starvation in the brain. Despite some controver-
sial results, it appears that the gray and white matter increase
significantly following weight restoration. Brain changes in
bulimia nervosa seem to be less pronounced and are mainly
due to chronic dietary restriction. In response to different
food stimuli, patients with anorexia nervosa show activation
Dovepress
Dovepress
50
Neuro vo5 no4 2011.indd 50 2011/10/28 10:13 AM

in the temporal regions, and patients with bulimia nervosa
show less activation than healthy controls in the right anterior
insula, left middle frontal gyrus, right posterior insula, right
precentral gyrus, and right mid dorsal insula. Body image
distortion has been used to look for correlations between
eating disorders and changes in the brain. Involvement of
the temporoparietal areas with respect to brain atrophy might
account for body image disturbances. A tendency towards
dieting seems to correlate with the right frontotemporal
cortex, and dieting behavior seems to correlate with the left
orbitofrontal cortex.
A number of studies have attempted to identify specific
brain correlates, taking into account the subtypes of eat-
ing disorders. Patients with anorexia nervosa have shown
hyperactivation of the left parietal and right superior frontal
areas in response to a neutral or positive stimulus, whereas
patients with bulimia nervosa have shown hyperactivation of
the right temporal and right occipital areas. Moreover, unlike
patients with restrictive anorexia nervosa and purging bulimia
nervosa, patients with nonpurging bulimia nervosa do not
experience increased rCBF when confronted with their own
body image. Considering the difference between patients with
anorexia nervosa with and without binge/purging behavior,
it has been reported that patients with anorexia nervosa with
habitual binge/purge behavior have a significantly higher per-
cent change in the inferior, superior, prefrontal, and parietal
regions of the right brain. The outcome of an eating disorder
has been related to certain brain changes, suggesting that
temporal hypoperfusion may have a prognostic value. Changes
in 5-HT1A
and 5-HT2A
receptor binding potential may be
persistent after recovery from an eating disorder. The same
applies to some changes in D2/D
3 receptor binding.
The author reports no conflicts of interest in this work.
1. Lask B, Gordon I, Christie D, Frampton I, Chowdhury U, Watkins B. Functional neuroimaging in early-onset anorexia nervosa. Int J Eat Disord. 2005;37 Suppl:S49–S51.
2. Palazidou E, Robinson P, Lishman WA. Neuroradiological and neuropsychological assessment in anorexia nervosa. Psychol Med. 1990; 20(3):521–527.
3. Katzmann DK, Zipursky RB, Lambe EK, Mikulis D. A longitudinal magnetic resonance imaging study of brain changes in adolescents with anorexia nervosa. Arch Pediatr Adolesc Med. 1997;151(8):793–797.
4. Artmann H, Grau H, Adelmann T, Schleiffer R. Reversible and non-reversible enlargement of cerebrospinal fluid space in anorexia nervosa. Neuroradiology. 1985;27(4):304–312.
5. Krieg JC, Pirke KM, Lauer C, Backmund H. Endocrine, metabolic, and cranial computed tomographic findings in anorexia nervosa. Biol Psychiatry. 1988;23(4):377–387.
6. Coyle J. Foreword. In: Ernst M, Rumsey J, editors. Functional Neuroimaging in Child Psychiatry. Cambridge, MA: Cambridge University Press; 2000:13.
7. Chowdhury U, Gordon I, Lask B, Watkins B, Watt H, Christie D. Early-onset anorexia nervosa: is there evidence of limbic system imbalance? Int J Eat Disord. 2003;33(4):388–396.
8. Rost B, Roser W, Bubl R, Radue EW, Buergin D. MRS of the brain in patients with anorexia or bulimia nervosa. Hosp Med. 1999;60(7):474–476.
9. Mühlau M, Gaser C, Ilg R, et al. Gray matter decrease of the anterior cingulate cortex in anorexia nervosa. Am J Psychiatry. 2007;164(12):1850–1857.
10. Gordon I, Lask B, Bryant-Waugh R, Christie D, Timimi S. Childhood-onset anorexia nervosa: towards identifying a biological substrate. Int J Eat Disord. 1997;22(2):159–165.
11. Rastam M, Bjure J, Vestegren E, et al. Regional cerebral blood flow in weight-restored anorexia nervosa: a preliminary study. Dev Med Child Neurol. 2001;43(4):239–242.
12. Gaudio S, Nocchi F, Franchin T, et al. Gray matter decrease distribution in the early stages of Anorexia Nervosa restrictive type in adolescents. Psychiatry Res. 2011;191(1):24–30.
13. Dolan RJ, Mitchell J, Wakeling A. Structural brain changes in patients with anorexia nervosa. Psychol Med. 1988;18(2):349–353.
14. Golden NH, Ashtari M, Kohn MR, et al. Reversibility of cerebral ven-tricular enlargement in anorexia nervosa, demonstrated by quantitative magnetic resonance imaging. J Pediatr. 1996;128(2):296–301.
15. Swayze VW, Andersen A, Arndt S, et al. Reversibility of brain tissue loss in anorexia nervosa assessed with a computerized Talairach 3-D proportional grid. Psychol Med. 1996;26(2):381–390.
16. Kohlmeyer K, Lehmkuhl G, Poustka F. Computed tomography of anorexia nervosa. AJNR Am J Neuroradiol. 1983;4(3):437–438.
17. Neumarker KJ, Bzufka WM, Dudeck U, Hein J, Neumarker U. Are there specific disabilities of number processing in adolescent patients with anorexia nervosa? Evidence from clinical and neuropsychological data when compared to morphometric measures from magnetic resonance imaging. Eur Child Adolesc Psychiatry. 2000;9 Suppl 2:II111–II121.
18. Katzman DK, Lambe EK, Mikulis DJ, Ridgley JN, Goldbloom DS, Zipursky RB. Cerebral gray matter and white matter volume deficits in adolescent girls with anorexia nervosa. J Pediatr 1996;129(6): 794–803.
19. McCormick LM, Ziebell S, Nopoulos P, Cassell M, Andreasen NC, Brumm M. Anterior cingulate cortex: an MRI-based parcellation method. Neuroimage. 2006;32(3):1167–1175.
20. Frampton I, Watkins B, Gordon I, Lask B. Do abnormalities in regional cerebral blood flow in anorexia nervosa resolve after weight restoration? Eur Eat Disord Rev. 2011;19(1):55–58.
21. Komatsu H, Nagamitsu S, Ozono S, Yamashita Y, Ishibashi M, Matsuishi T. Regional cerebral blood flow changes in early-onset anorexia nervosa before and after weight gain. Brain Dev. 2010;32(8): 625–630.
22. Castro-Fornieles J, Caldú X, Andrés-Perpiñá S, et al. A cross-sectional and follow-up functional MRI study with a working memory task in adolescent anorexia nervosa. Neuropsychologia. 2010;48(14): 4111–4116.
23. Roberto CA, Mayer LE, Brickman AM, et al. Brain tissue volume changes following weight gain in adults with anorexia nervosa. Int J Eat Disord. 2011;44(5):406–411.
24. Boghi A, Sterpone S, Sales S, et al. In vivo evidence of global and focal brain alterations in anorexia nervosa. Psychiatry Res. 2011;192(3): 154–159.
25. Frank GK, Bailer UF, Meltzer CC, et al. Regional cerebral blood flow after recovery from anorexia or bulimia nervosa. Int J Eat Disord. 2007;40(6):488–492.
26. Oberndorfer TA, Kaye WH, Simmons AN, Stigo IA, Matthews SC. Demand-specific alteration of medial prefrontal cortex response during an inhibition task in recovered anorexic women. Int J Eat Disord. 2011;44(1):1–8.
Dovepress
Dovepress
51
Neuro vo5 no4 2011.indd 51 2011/10/28 10:13 AM

27. Krieg JC, Lauer C, Pirke KM. Structural brain abnormalities in patients with bulimia nervosa. Psychiatry Res. 1989;27(1):39–48.
28. Laessle RG, Krieg JC, Fichter MM, Pirke KM. Cerebral atrophy and vigilance performance in patients with anorexia and bulimia nervosa. Neuropsychobiology. 1989;21(4):187–191.
29. Hoffman GW, Ellinwood EH Jr, Rockwell WJ, Herfkens RJ, Nishita JK, Guthrie LF. Cerebral atrophy in bulimia. Biol Psychiatry. 1989;25(7):894–902.
30. Husain MM, Black KJ, Doraiswamy PM, et al. Subcortical brain anat-omy in anorexia and bulimia. Biol Psychiatry. 1992;31(7):735–738.
31. Nozoe S, Naruo T, Nakabeppu Y, Soejima Y, Nakajo M, Tanaka H. Changes in regional cerebral blood flow in patients with anorexia nervosa detected through single photon emission tomography imaging. Biol Psychiatry. 1993;34(8):578–580.
32. Nozoe S, Naruo T, Yonekura R, et al. Comparison of regional cere-bral blood flow in patients with eating disorders. Brain Res Bull. 1995;36(3):251–255.
33. Ellison Z, Foong J, Howard R, Bullmore E, Williams S, Treasure J. Functional anatomy of calorie fear in anorexia nervosa. Lancet. 1998; 352(9135):1192.
34. Naruo T, Nakabeppu Y, Sagiyama K, et al. Characteristic regional cerebral blood flow patterns in anorexia nervosa patients with binge/purge behavior. Am J Psychiatry. 2000;157(9):1520–1522.
35. Gordon CM, Dougherty DD, Fischman AJ, et al. Neural substrates of anorexia nervosa: a behavioral challenge study with positrón emission tomography. J Pediatr. 2001;139(1):51–57.
36. Uher R, Murphy T, Brammer M, et al. Medial prefrontal cortex activ-ity associated with symptom provocation in eating disorders. Am J Psychiatry. 2004;161(7):1238–1246.
37. Uher R, Brammer MJ, Murphy T, et al. Recovery and chronicity in anorexia nervosa: brain activity associated with differential outcomes. Biol Psychiatry. 2003;54(9):934–942.
38. Bohon C, Stice E. Reward abnormalities among women with full and subthreshold bulimia nervosa: A functional magnetic resonance imaging study. Int J Eat Disord. 2010. [Epub ahead of print.]
39. Wagner A, Ruf M, Braus DF, Schmidt MH. Neuronal activity changes and body image distortion in anorexia nervosa. Neuro Report. 2003;14(17):2193–2197.
40. Bailer UF, Price JC, Meltzer CC, et al. Altered 5-HT(2A) receptor bind-ing after recovery from bulimia-type anorexia nervosa: relationships to harm avoidance and drive for thinness. Neuropsychopharmacology. 2004;29(6):1143–1155.
41. Mohr HM, Zimmermann J, Röder C, Lenz C, Overbeck G, Grabhorn R. Separating two components of body image in anorexia nervosa using fMRI. Psychol Med. 2010;40(9):1519–1529.
42. Friederich HC, Uher R, Brooks S, et al. I’m not as slim as that girl: neural bases of body shape self-comparison to media images. Neuroimage. 2007;37(2):674–681.
43. Vocks S, Busch M, Schulte D, Grönermeyer D, Herpertz S, Suchan B. Effects of body image therapy on the activation of the extrastriate body area in anorexia nervosa: an fMRI study. Psychiatry Res. 2010;183(2): 114–118.
44. Suda M, Uehara T, Fukuda M, Sato T, Kameyama M, Mikuni M. Dieting tendency and eating behavior problems in eating disorder correlate with right frontotemporal and left orbitofrontal cortex: a near-infrared spectroscopy study. J Psychiatr Res. 2010;44(8):547–555.
45. Beato-Fernández L, Rodríguez-Cano T, García-Vilches, et al. Changes in regional cerebral blood flow after body image exposure in eating disorders. Psychiatry Res. 2009;171(2):129–137.
46. Beato-Fernández L, Rodríguez-Cano T, García-Vilches I. Psychopatho-logical alterations and neuroimaging findings with discriminant value in eating behavior disorders. Actas Esp Psiquiatr. 2011;39(4):203–210.
47. Karhunen LJ, Lappalainen RI, Vanninen EJ, Kuikka JT, Uusitupa MI. Regional cerebral blood flow during food exposure in obese and normal-weight women. Brain. 1997;120 (Pt 9):1675–1684.
48. Fox EA. Purdue Ingestive Behavior Research Center symposium 2007: influences on eating and body weight over the lifespan – childhood and adolescence. Physiol Behav. 2008;94(1):1–7.
49. Jiménez-Bonilla JF, Quirce R, Banzo I, et al. Temporal hypop-erfusion assessed by cerebral blood flow SPECT and long-term clinical outcome in patients with eating disorders. Clin Nucl Med. 2009;34(11):768–772.
50. Jiménez-Bonilla JF, Carril Carril JM, Quirce Pisano R, et al. Assessment of cerebral blood flow in patients with eating disorders in the acute clinical phase using Tc99m-HMPAO spect. Rev Esp Med Nucl. 2008;27(5):350–354.
51. Bailer UF, Frank GK, Henry SE, et al. Exaggerated 5-HT1A but normal 5-HT2 A receptor activity in individuals ill with anorexia nervosa. Biol Psychiatry. 2007;61(9):1090–1099.
52. Tiihonen J, Keski-Rahkonen A, Löppönen M, et al. Brain serotonin 1A receptor binding in bulimia nervosa. Biol Psychiatry. 2004;55(8): 871–873.
53. Audenaert K, Van Laere K, Dumont F, et al. Decreased 5-HT2A receptor binding in patients with anorexia nervosa. J Nucl Med. 2003;44(2):163–169.
54. Bailer UF, Frank GK, Henry SE, et al. Serotonin transporter binding after recovery from eating disorders. Psychopharmacology (Berl). 2007;195(3):315–324.
55. Bailer UF, Bloss CS, Frank GK, et al. 5-HT(1A) receptor binding is increased after recovery from bulimia nervosa compared to control women and is associated with behavioral inhibition in both groups. Int J Eat Disord. 2011;44(6):477–487.
56. Pichika R, Buchsbaum MS, Bailer U, et al. Serotonin transporter bind-ing after recovery from bulimia nervosa. Int J Eat Disord. 2011. [Epub ahead of print.]
57. Frank GK, Kaye WH, Meltzer CC, et al. Reduced 5-HT2A recep-tor binding after recovery from anorexia nervosa. Biol Psychiatry. 2002;52(9):896–906.
58. Bailer UF, Price JC, Meltzer CC, et al. Altered 5-HT(2A) receptor bind-ing after recovery from bulimia-type anorexia nervosa: relationships to harm avoidance and drive for thinness. Neuropsychopharmacology. 2004;29(6):1143–1155.
59. Frank G, Bailer UF, Henry S, et al. Increased dopamine D2/D3 receptor binding after recovery from anorexia nervosa measured by positron emission tomography and [11C] raclopride. Biol Psychiatry. 2005;58(11):908–912.
60. Drevets W. Neuroimaging studies of major depression. In: Morisha J, editor. Advances in Brain Imaging. Washington, DC: American Psychiatric Publishing Inc; 2001.
61. Kalat J. Biological Psychology. 7th ed. Belmont, CA: Wadsworth; 2001.
Dovepress
Dovepress
52
Neuro vo5 no4 2011.indd 52 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 1 2011/10/28 10:13 AM

Neuro vo5 no4 2011.indd 2 2011/10/28 10:13 AM




















