
Nanoprocessability of a one-dimensional oxalato-bridged cobalt(II) complex with 1,2,4-triazole
7
Nanoprocessability of a one-dimensional oxalato-bridged cobalt(II) complex with 1,2,4-triazole David Olea a , Urko Garcı ´a-Couceiro b , Oscar Castillo b , Julio Go ´ mez-Herrero a , Fe ´lix Zamora c, * a Universidad Auto ´ noma de Madrid, Departamento de Fı ´sica de la Materia Condensada, E-28049 Madrid, Spain b Universidad del Paı ´s Vasco, Departamento de Quı ´mica Inorga ´ nica, Apartado 644, E-48080 Bilbao, Spain c Universidad Auto ´ noma de Madrid, Departamento de Quı ´mica Inorga ´ nica (C-VIII), Facultad de Ciencias, E-28049 Madrid, Spain Received 6 June 2006; received in revised form 18 July 2006; accepted 19 July 2006 Available online 3 August 2006 Inorganic Chemistry – The Next Generation. Abstract The polymer {[Co(ox)(Htr) 2 ] 2H 2 O} n (ox = oxalate dianion; Htr = 1,2,4-triazole) (1) has been synthesized and characterized by FT-IR spectroscopy, thermal analysis, variable-temperature magnetic measurements and X-ray diffraction methods. The physical anal- ysis allows us to propose a one-dimensional structure in which [Co(Htr) 2 ] 2+ units are bridged by bis-bidentate oxalato ligands. Magnetic measurements at variable temperature show an overall antiferromagnetic behavior of the compound. Isolated chains of this polymer have been obtained by sonication of 1 in water and deposition on mica or on mica treated with poly-L-lysine. Circular molecules and nano-fibres have been isolated on Highly Oriented Pyrolitic Graphite (HOPG) by casting deposition of sonicated solutions of 1 in eth- anol. The direct reaction on HOPG surface between Co II ,H 2 ox and Htr has proved a useful route to isolate one-dimensional systems on surfaces. The development of new strategies to characterize these types of polymers on surfaces opens the possibility to perform nano- scale studies on their properties and their potential use as nano-materials. Ó 2006 Elsevier B.V. All rights reserved. Keywords: One-dimensional coordination polymers; AFM; Surface chemistry 1. Introduction Supramolecular coordination chemistry has been widely studied during the last few years due to their interesting architectures, their amazing properties and potential tech- nological applications [1–6]. The structural studies of coor- dination supramolecules have been carried out in solution and/or solid state and the studies of their properties mea- sured on a macroscopic scale. The potential use of these new materials in nanotechnology requires the isolation of individual supramolecules on surfaces with arrangements on nanometer scale [7,8] allowing to characterize the indi- vidual properties of each isolated molecule. Among the architectures described, one- and two-dimensional coordi- nation polymers are especially suitable in the search of organizations on surfaces. Nevertheless, studies on surfaces with coordination polymers are very scarce and mainly have been centered on two-dimensional systems [9–13].A typical feature of the coordination polymers is their low or none solubility in most of the solvents, making these materials difficult to be processed [3]. The isolation and characterization of well-ordered one-dimensional coordi- nation polymers on surfaces, has been restricted to a few examples [14–16]. Recently, we have been able to isolate single chains of the one-dimensional coordination poly- mers [Cd(6-MP) 2 ] (6-MP = 6-mercaptopurinate) [14] and 0020-1693/$ - see front matter Ó 2006 Elsevier B.V. All rights reserved. doi:10.1016/j.ica.2006.07.065 * Corresponding author. Tel.: +34 914973962; fax: +34 914974833. E-mail address: [email protected] (F. Zamora). www.elsevier.com/locate/ica Inorganica Chimica Acta 360 (2007) 48–54
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Nanoprocessability of a one-dimensional oxalato-bridged cobalt(II) complex with 1,2,4-triazole

www.elsevier.com/locate/ica
Inorganica Chimica Acta 360 (2007) 48–54
Nanoprocessability of a one-dimensional oxalato-bridgedcobalt(II) complex with 1,2,4-triazole
David Olea a, Urko Garcıa-Couceiro b, Oscar Castillo b,Julio Gomez-Herrero a, Felix Zamora c,*
a Universidad Autonoma de Madrid, Departamento de Fısica de la Materia Condensada, E-28049 Madrid, Spainb Universidad del Paıs Vasco, Departamento de Quımica Inorganica, Apartado 644, E-48080 Bilbao, Spain
c Universidad Autonoma de Madrid, Departamento de Quımica Inorganica (C-VIII), Facultad de Ciencias, E-28049 Madrid, Spain
Received 6 June 2006; received in revised form 18 July 2006; accepted 19 July 2006Available online 3 August 2006
Inorganic Chemistry – The Next Generation.
Abstract
The polymer {[Co(ox)(Htr)2] Æ 2H2O}n (ox = oxalate dianion; Htr = 1,2,4-triazole) (1) has been synthesized and characterized byFT-IR spectroscopy, thermal analysis, variable-temperature magnetic measurements and X-ray diffraction methods. The physical anal-ysis allows us to propose a one-dimensional structure in which [Co(Htr)2]2+ units are bridged by bis-bidentate oxalato ligands. Magneticmeasurements at variable temperature show an overall antiferromagnetic behavior of the compound. Isolated chains of this polymerhave been obtained by sonication of 1 in water and deposition on mica or on mica treated with poly-L-lysine. Circular molecules andnano-fibres have been isolated on Highly Oriented Pyrolitic Graphite (HOPG) by casting deposition of sonicated solutions of 1 in eth-anol. The direct reaction on HOPG surface between CoII, H2ox and Htr has proved a useful route to isolate one-dimensional systems onsurfaces. The development of new strategies to characterize these types of polymers on surfaces opens the possibility to perform nano-scale studies on their properties and their potential use as nano-materials.� 2006 Elsevier B.V. All rights reserved.
Keywords: One-dimensional coordination polymers; AFM; Surface chemistry
1. Introduction
Supramolecular coordination chemistry has been widelystudied during the last few years due to their interestingarchitectures, their amazing properties and potential tech-nological applications [1–6]. The structural studies of coor-dination supramolecules have been carried out in solutionand/or solid state and the studies of their properties mea-sured on a macroscopic scale. The potential use of thesenew materials in nanotechnology requires the isolation ofindividual supramolecules on surfaces with arrangements
0020-1693/$ - see front matter � 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.ica.2006.07.065
* Corresponding author. Tel.: +34 914973962; fax: +34 914974833.E-mail address: [email protected] (F. Zamora).
on nanometer scale [7,8] allowing to characterize the indi-vidual properties of each isolated molecule. Among thearchitectures described, one- and two-dimensional coordi-nation polymers are especially suitable in the search oforganizations on surfaces. Nevertheless, studies on surfaceswith coordination polymers are very scarce and mainlyhave been centered on two-dimensional systems [9–13]. Atypical feature of the coordination polymers is their lowor none solubility in most of the solvents, making thesematerials difficult to be processed [3]. The isolation andcharacterization of well-ordered one-dimensional coordi-nation polymers on surfaces, has been restricted to a fewexamples [14–16]. Recently, we have been able to isolatesingle chains of the one-dimensional coordination poly-mers [Cd(6-MP)2] (6-MP = 6-mercaptopurinate) [14] and

D. Olea et al. / Inorganica Chimica Acta 360 (2007) 48–54 49
[Mn(l-ox)(4atr)2]n (ox = oxalato and 4atr = 4-amine-1,2,4-triazole) on different surfaces [15].
Herein, we report on the search of different ways tocharacterize organizations of the one-dimensional coordi-nation polymer {[Co(ox)(Htr)2] Æ 2H2O}n (ox = oxalatedianion; Htr = 1,2,4-triazole) on a mica and Highly Ori-ented Pyrolitic Graphite (HOPG) surface. The isolationof these architectures together with the magnetic propertiesof this material suggests its potential use in nanotechnolog-ical applications.
2. Experimental
All chemicals were of reagent grade and were used ascommercially obtained. Elemental analyses (C, H, N) wereperformed on a Perkin Elmer 2400 microanalytical ana-lyzer. Metal content was determined by absorption spec-trometry. The IR spectra (KBr pellets) were recorded ona FTIR Mattson 1000 spectrometer in the 4000–400 cm�1
spectral region. Thermal analyses (TG/DTG/DTA) wereperformed on a TA Instruments STD 2960 thermal ana-lyzer in a synthetic air atmosphere (79%N2/21%O2) witha heating rate of 5 �C min�1. Magnetic measurements wereperformed on polycrystalline samples of the compoundswith a Quantum Design SQUID susceptometer coveringthe temperature range of 5–300 K at a magnetic field of1000 G. The susceptibility data were corrected for the dia-magnetism estimated from Pascal’s tables [17] temperature-independent paramagnetism, and the magnetization of thesample holder.
2.1. Synthesis of {[Co(ox)(Htr)2] Æ 2H2O}n
Polycrystalline sample as quantitative yield of com-pound 1 can be obtained by reacting Co(NO3)2 Æ 6H2O,H2C2O4 Æ H2O and Htr in the molar ratio 1:1:6 in an aque-ous media. Yield: 95% (based on Co). Anal. Calc. forC6H10CoN6O6: C, 22.44; H, 3.14; N, 26.17; Co, 18.35.Found: C, 22.45; H, 3.10; N, 26.21; Co, 18.33%. Main IRfeatures (cm�1, KBr pellet): 3415vs for m(O–H); 3110s,2955m, 2855m, 2778m, 2738m, 2680m, 2645m, 2593m,2490m for m(N–H) + m(C–H); 1650vs, 1612vs for mas(O–C–O); 1528m, 1510s for mas(C@N); 1425m for m(C–N);1360m, 1315m for ms(O–C–O); 1300m, 1270m, 1180w,1135w for dip(C–H); 1036m for ms(CO); 993h, 980m;938w, 900m for dring + dop(C–H); 810s for d(O–C–O);638m for sring; 555m, 490m for m(M–O + M–N) + d(C–C–O).
2.2. AFM studies
2.2.1. AFM device
AFM images were acquired in dynamic mode at roomtemperature and ambient pressure using a Nanotec Elect-ronica system (www.nanotec.es). Olympus cantilevers wereused with a nominal force constant of 0.75 N/m and a res-onance frequency of about 70 kHz.
2.2.2. Sample preparation for AFM
Procedure (a): {[Co(ox)(Htr)2] Æ 2H2O}n (1 mg) was sus-pended in ethanol (1 mL) and sonicated (540 W, 40 kHz)for 30 min at 20 �C. The suspension was centrifuged at10000 rpm for 10 min and 10 lL of the clear solutionwas deposited on recently cleaved HOPG. Then, the sam-ple was left in air to allow the complete evaporation ofthe solvent.
Procedure (b): It follows the same procedure as in (a)
but 0.5 mg of {[Co(ox)(Htr)2] Æ 2H2O}n was used and thesuspension was sonicated at 680 W for 1 h.
Procedure (c): {[Co(ox)(Htr)2] Æ 2H2O}n (0.5 mg) wassuspended in water (1 mL) and sonicated (680 W,40 kHz) for 1 h at 20 �C. The suspension was centrifugedat 10000 rpm for 10 min and 10 lL of the clear solutionwas deposited on a cleaved mica sheet, left for 10 min,washed with water and dried with a gentle stream ofnitrogen.
Procedure (d): It follows the same procedure as in (c)
but the solution was deposited on a cleaved mica sheet pre-viously treated with poly-L-lysine.
Procedure (e). A recently cleaved HOPG was dipped for20 min in a water solution (heated at 70 �C) containing1 mL of Co(NO3)2 Æ 6H2O (10�6 mol L�1) mixed with1 mL of a water solution of H2C2O4 Æ H2O (10�6 mol L�1)and 2 mL of a water solution of 1,2,4-triazole(6 Æ 10�6 mol L�1). The HOPG disk was then washed withwater and dried with a gentle stream of nitrogen.
2.2.3. Preparation of the surfacesIn order to obtain reproducible results, very flat sub-
strates with precisely controlled chemical functionalities,freshly prepared just before the chemical deposition, wereused. Two different commercially available supports wereused: Muscovite Mica and Highly Oriented PyroliticGraphite (HOPG). Both of them were cleaved with adhe-sive tape. Freshly exfoliated mica sheets were immersedon a poly-L-lysine solution (0.01% solution; from Sigma)for 15 min and then intensively washed with water.
2.3. X-ray crystallographic studies
The X-ray powder diffraction (XRPD) pattern of com-pound 1 was collected on a Phillips X’PERT powder dif-fractometer with Cu-Ka radiation over the range10 < 2h < 60� with a fixed-time counting of 5 s and a scanstep of 0.02� at 25 �C. The angular position of the first 13peaks of the XRPD pattern were obtained with the WIN-
PLOTR program [18]. Then, the indexing was performedby using DICVOL program [19]. A monoclinic solution withsatisfactory figures of merit, M(13) = 19.8 andF(13) = 20.7, and unit cell parameters of a = 14.95 A,b = 7.50 A, c = 5.51 A and b = 95.38� were found. Thepattern matching analysis of the diffractogram was carriedout by the FULLPROF program [20], using the unit cellparameters obtained in the indexing and a pseudo-voigtfunction for modelling the peak shape. The analysis of
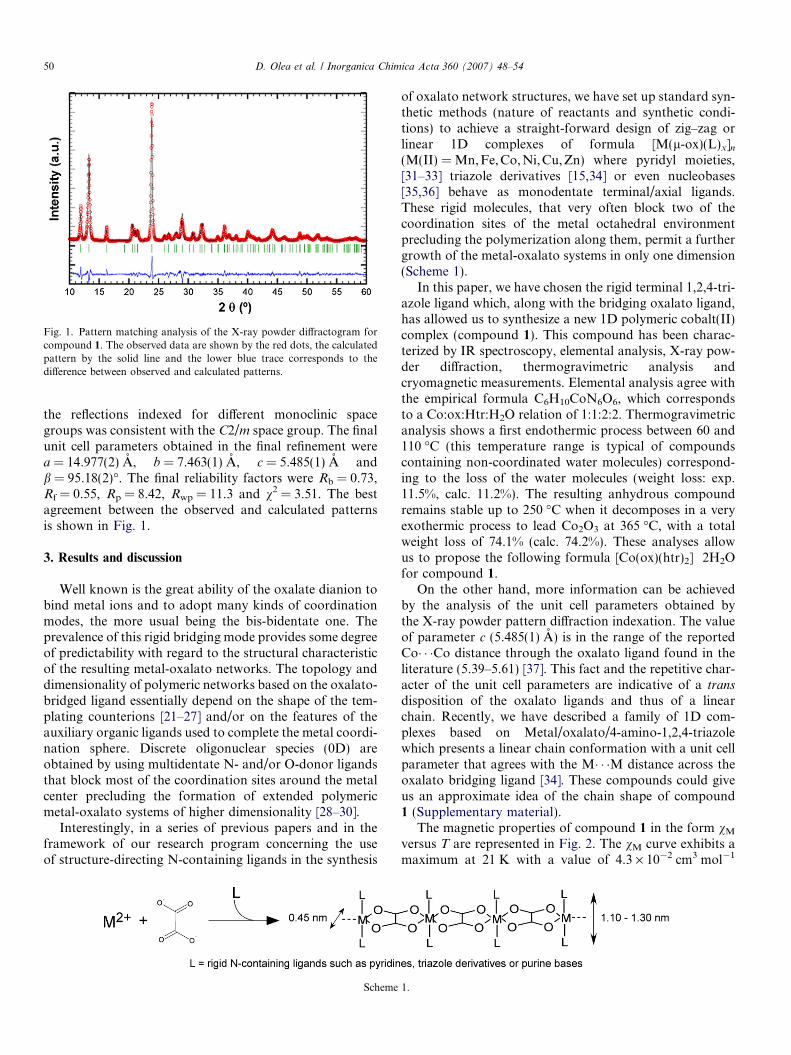
Fig. 1. Pattern matching analysis of the X-ray powder diffractogram forcompound 1. The observed data are shown by the red dots, the calculatedpattern by the solid line and the lower blue trace corresponds to thedifference between observed and calculated patterns.
50 D. Olea et al. / Inorganica Chimica Acta 360 (2007) 48–54
the reflections indexed for different monoclinic spacegroups was consistent with the C2/m space group. The finalunit cell parameters obtained in the final refinement werea = 14.977(2) A, b = 7.463(1) A, c = 5.485(1) A andb = 95.18(2)�. The final reliability factors were Rb = 0.73,Rf = 0.55, Rp = 8.42, Rwp = 11.3 and v2 = 3.51. The bestagreement between the observed and calculated patternsis shown in Fig. 1.
3. Results and discussion
Well known is the great ability of the oxalate dianion tobind metal ions and to adopt many kinds of coordinationmodes, the more usual being the bis-bidentate one. Theprevalence of this rigid bridging mode provides some degreeof predictability with regard to the structural characteristicof the resulting metal-oxalato networks. The topology anddimensionality of polymeric networks based on the oxalato-bridged ligand essentially depend on the shape of the tem-plating counterions [21–27] and/or on the features of theauxiliary organic ligands used to complete the metal coordi-nation sphere. Discrete oligonuclear species (0D) areobtained by using multidentate N- and/or O-donor ligandsthat block most of the coordination sites around the metalcenter precluding the formation of extended polymericmetal-oxalato systems of higher dimensionality [28–30].
Interestingly, in a series of previous papers and in theframework of our research program concerning the useof structure-directing N-containing ligands in the synthesis
Scheme
of oxalato network structures, we have set up standard syn-thetic methods (nature of reactants and synthetic condi-tions) to achieve a straight-forward design of zig–zag orlinear 1D complexes of formula [M(l-ox)(L)x]n(M(II) = Mn,Fe, Co,Ni,Cu, Zn) where pyridyl moieties,[31–33] triazole derivatives [15,34] or even nucleobases[35,36] behave as monodentate terminal/axial ligands.These rigid molecules, that very often block two of thecoordination sites of the metal octahedral environmentprecluding the polymerization along them, permit a furthergrowth of the metal-oxalato systems in only one dimension(Scheme 1).
In this paper, we have chosen the rigid terminal 1,2,4-tri-azole ligand which, along with the bridging oxalato ligand,has allowed us to synthesize a new 1D polymeric cobalt(II)complex (compound 1). This compound has been charac-terized by IR spectroscopy, elemental analysis, X-ray pow-der diffraction, thermogravimetric analysis andcryomagnetic measurements. Elemental analysis agree withthe empirical formula C6H10CoN6O6, which correspondsto a Co:ox:Htr:H2O relation of 1:1:2:2. Thermogravimetricanalysis shows a first endothermic process between 60 and110 �C (this temperature range is typical of compoundscontaining non-coordinated water molecules) correspond-ing to the loss of the water molecules (weight loss: exp.11.5%, calc. 11.2%). The resulting anhydrous compoundremains stable up to 250 �C when it decomposes in a veryexothermic process to lead Co2O3 at 365 �C, with a totalweight loss of 74.1% (calc. 74.2%). These analyses allowus to propose the following formula [Co(ox)(htr)2] Æ 2H2Ofor compound 1.
On the other hand, more information can be achievedby the analysis of the unit cell parameters obtained bythe X-ray powder pattern diffraction indexation. The valueof parameter c (5.485(1) A) is in the range of the reportedCo� � �Co distance through the oxalato ligand found in theliterature (5.39–5.61) [37]. This fact and the repetitive char-acter of the unit cell parameters are indicative of a transdisposition of the oxalato ligands and thus of a linearchain. Recently, we have described a family of 1D com-plexes based on Metal/oxalato/4-amino-1,2,4-triazolewhich presents a linear chain conformation with a unit cellparameter that agrees with the M� � �M distance across theoxalato bridging ligand [34]. These compounds could giveus an approximate idea of the chain shape of compound1 (Supplementary material).
The magnetic properties of compound 1 in the form vM
versus T are represented in Fig. 2. The vM curve exhibits amaximum at 21 K with a value of 4.3 · 10�2 cm3 mol�1
1.

Fig. 2. Magnetic susceptibility (vM) vs. temperature plots for compound 1
showing the Heisenberg (- - -) and Rueff (—) best fits. Inset: fit at low-temperature region by the Ising model (see text).
D. Olea et al. / Inorganica Chimica Acta 360 (2007) 48–54 51
and then decreases very quickly. The vMT value at roomtemperature (2.74 cm3 mol�1 K) is significantly greaterthan the spin only value for a high-spin Co(II) center(1.875 cm3 mol�1 K for S = 3/2 and g = 2.0), but it agreeswith the values observed for octahedral cobalt(II) com-plexes with a significant first-order orbital contribution tothe magnetic moment [38–40]. The inverse magnetic sus-ceptibility follows a Curie-Weiss law above 130 K withC = 2.95 cm3 mol�1 K and h = �19.7 K. In a firstapproach, we have attempted to fit the experimental sus-ceptibility in the entire temperature range using the classi-cal spin Heisenberg model [41] for a one-dimensional chainof SCo = 3/2. A reasonable agreement was obtained withthe parameters J = �9.3 cm�1 and g = 2.64 at T > 40 K,but the results are not very satisfactory in the low temper-ature region owing to fact that the model does not take intoaccount the effects of the spin–orbit coupling for octahe-dral Co(II) complexes (4T1g ground state). Previous mag-netic reports [42] showed that some 1D systems ofcobalt(II) are associated with anisotropic Ising systemswith an effective spin S 0 = 1/2, and they can be fitted at alow temperature range (T < 40 K) where only the groundKramer’s doublet is thermally populated [43,44]. At highertemperatures, a crossover to the Heisenberg type behavioris expected. The best fit (shown in the inset of Fig. 2) gavethe parameters gx = 4.06, gz = 5.96, J = �12.9 cm�1 whichare quite close to those obtained for the one-dimensional[Co(l-ox)(H2O)2]n (J = �13.4 cm�1) [45,46] and {[Co-(l-ox)(H2O)2] Æ 2H2O}n (J = �14.2 cm�1) [38] compoundsthrough the same magnetic model. More recently, Rueffet al. [47,48] have proposed a phenomenological approachfor some low-dimensional Co(II) systems that allow one tohave an estimate of the strength of the antiferromagneticexchange interactions by means of Eq. (1)
vMT ¼ A expð�E1=kBT Þ þ B expð�E2=kBT Þ ð1Þin which A + B equals the Curie constant and E1 and E2 rep-resent the ‘‘activation energies’’ corresponding to spin–orbit
coupling and the antiferromagnetic exchange interaction.The A + B value of 3.11 cm3 mol�1 K obtained with theRueff procedure perfectly agrees with those given in the lit-erature for the Curie constant (C = 2.8–3.5 cm3 mol�1 K).Regarding the value found for the antiferromagnetic ex-change interaction, E2/kB = 3.8 K, J = �7.14 cm�1. Thevalues obtained with the different models are comparableto those found for other oxalato-bridged cobalt(II) com-plexes [38,42,49] (Supplementary material). All theseattempts to simulate the magnetic data should be taken intoaccount only as approximations due to the previously men-tioned limitations of the models, but give us an approximateidea about the strength of the antiferromagnetic interaction.
It is known that most of the coordination polymers arenot soluble in common solvents as such [3]. However, someof these polymers may present, among other properties,reversibility. This concept of dynamic combinatorial chem-istry marks off the possibility to establish reversible connec-tion between the components of the organic and inorganicmaterials [50]. Based on this idea, some of the metal–ligandbonds of coordination polymers can be broken, in an initialstep by thermal or mechanical forces, giving smaller oligo-mers in solution. The use of ultrasound has been proven tobe a suitable source for the scission of the metal–ligandbond of coordination polymers [51]. In following steps,the new fragments generated could be reassembled in solu-tion [51] and/or on surface [14,15] affording the originalarrangement or new architectures, if they are for instanceaffected by new effects as the interaction with the surfaceand/or the solvent.
The AFM images of sonicated suspensions of com-pound 1 in ethanol (1 mg/mL) show the formation ofunspecific aggregates and formation of some chains of 3–6 nm height on HOPG. Less concentrated solutions leadin the same surface to well-ordered one-dimensional fibreswith heights ranging from 0.60 nm to 35 nm (Fig. 3). Theheight of 0.60 nm (Fig. 3a) fits well with the expected valueof the diameter, for the theoretical 1D chain of metal-oxa-late framework (ca. 0.5 nm, see Supplementary material).As it can be seen in the Fig. 3b the higher fibres are longerthan 20 lm and are located in the steps of graphite. Thetemplate effect of the step edges of HOPG to align one-dimensional coordination polymers has already beenreported [15,16]. Fig. 3d shows that some protuberancesare present in these fibres and a large number of lowerfibres coming out from them. The process of the formationof such bundles is reminiscent with the crystallization thattakes place at a higher scale. The protuberances observedcan be considered as an aggregation of non well-orderedoligomers acting as the nucleus growth of fibres. As itcan be seen in Fig. 4, a large number of ordered structuresare present on HOPG, among others, rings, rings with atail and dots. Fig. 4b shows a circular structure of 3 nmheight, suggesting that it is a bundle of at least seven mol-ecules in height. Probably these circular molecules areformed as a consequence of the much lower concentrationof the solution that makes more difficult the recombination

Fig. 4. AFM topogaphic image showing (A) different structures obtainedby deposition of a solution of compound 1 on a HOPG surface. Zoom (B)and height profile (C) of the selected area.
Fig. 5. Topographic image of a molecule obtained by depositing a drop ofwater dispersion of compound 1 onto mica (A). Topographic profile (B)across the line, showing that the height of the structure corresponds to asingle molecule.
Fig. 3. AFM topogaphic image showing (A) a single molecule ofcompound 1 adsorbed on HOPG and topographic profile across the line.High pass filtered AFM image of a higher area of HOPG, showingdifferent structures obtained (B) and (C) topographic profile across theline. Zoomed area of (D) showing a detail of the fibre of (B).
52 D. Olea et al. / Inorganica Chimica Acta 360 (2007) 48–54
of the oligomeric fragments towards the original one-dimensional architecture. Formation of cyclic oligomershas been suggested in the propagation step once the scis-sion and redistribution of the linear oligomers takes place[51]. This is the first time that cyclic oligomers derived fromthe scission of a one-dimensional coordination polymerhave been identified on surface and supports the abovementioned observations in solution.
Recently, we have observed a strong effect of the sur-face on the organization of an analogous one-dimensionalcoordination polymer [15]. In this case, the deposition ona mica surface of sonicated suspensions of 1 in ethanoldoes not give any organization. When water is used as sol-vent, the deposition by casting solution on mica (Fig. 5)and mica treated with poly-L-lysine (Fig. 6) give fibres ofseveral microns long and heights ranging from 5 to13 A. The lower heights of 5 A, are in good agreementwith the diameter expected for isolated single chains ofcompound 1. Mica surface was treated with poly-L-lysinein order to change the surface properties of the substrateand study its effect on the adsorption. The higher densityof fibres deposited on the mica treated with poly-L-lysineindicates a stronger interaction of compound 1 with thepoly-L-lysine surface than with the mica surface. Theincrement in the number of molecules adsorbed on thetreated surface (Fig. 6) may be explained by the changeof the surface properties of the substrate by the poly-L-lysine coating, since the surface becomes more hydropho-bic [52]. This observation is in agreement with the goodadsorption properties observed for 1 in HOPG (hydro-phobic surface) indicating a higher affinity for hydropho-bic substrates. A similar behavior for the analogouspolymer [Mn(l-ox)(4atr)2]n has been described by us[15]. These facts are a clear indication that the nature ofthe substrate and its interaction with the coordination

Fig. 6. AFM topographic image (A) and profile across the line (B) of themolecules adsorbed on mica coated with poly-L-lysine.
D. Olea et al. / Inorganica Chimica Acta 360 (2007) 48–54 53
polymers play an important role in the formation of nano-structures derived from these supramolecules.
Another method employed to obtain isolated fibres hasbeen the in situ reaction of the three building blocks of theone-dimensional coordination polymer 1. Thus, when a
Fig. 7. AFM topographic image of fibres obtained on HOPG by in situ
chemical reaction (A) and topographic profile across the line (B).
HOPG surface was dipped for 20 min, at 70 �C, in a lowconcentrated solution containing stoichiometric quantitiesof their three building blocks Co(II), ox and Htr (1:1:2),long fibrous structures with heights ranging from 0.6 to10 nm have been obtained (Fig. 7). Careful measurementsof the angles between the fibres indicate that most of themare formed following the steps on the graphite surface,since the angles are always close to 120� or 60� [15,16].The aggregates of different heights observed in the sub-strate are probably due to deposits of unreacted ligandsand the Co(II) salt.
4. Conclusions
We report herein the synthesis, chemical characteriza-tion, and crystal structure of the one-dimensional coordina-tion polymer {[Co(ox)(Htr)2] Æ 2H2O}n. Its processability,as nano-rings, fibres and single chains, has been achievedby scission of part of the metal–ligand bonds with ultra-sound and posterior re-organization of the oligomers onsurface. A new feature is the observation for the first timeof circular structures of a coordination polymer that is lin-ear in the solid state. Reaction formation of this polymer atlow concentration of the reactants has been also shown as asuitable way for the isolation of nano-fibres on HOPG. Thishas permitted the morphological characterization of thepolymer by AFM. The results obtained open a new routefor future nanotechnological applications of this type ofmaterials.
Acknowledgements
This work was supported by the Spanish Ministerio deCiencia y Tecnologıa (MAT2005–03047 and MAT2004-05589-C02-01/02), NISAN/IST-2001-38052 and the Uni-versidad del Paıs Vasco/Euskal Herriko Unibertsitatea(9/UPV 00169.310-15329/2003). U. G.-C. thanks the latterinstitution for a predoctoral fellowship.
Appendix A. Supplementary data
IR spectrum, thermoanalytical (TGA/DTA) curves,magnetic data of oxalato-bridged Co(II) complexes andview of the polymeric chain of the analogous compounds[M(ox)(4atr)2]. Supplementary data associated with thisarticle can be found, in the online version, atdoi:10.1016/j.ica.2006.07.065.
References
[1] B. Olenyuk, A. Fechtenkotter, P.J. Stang, Dalton Trans. (1998) 1707.[2] P.J. Stang, B. Olenyuk, Acc. Chem. Res. 30 (1997) 502.[3] C. Janiak, Dalton Trans. (2003) 2781.[4] S. Kitagawa, S. Noro, in: T.J. McCleverty (Ed.), Comprehensive
Coordination Chemistry II, vol. 7, Elsevier, Amsterdam, 2004.[5] J.A.R. Navarro, E. Barea, M.A. Galindo, J.M. Salas, M.A. Romero,
M. Quiros, N. Masciocchi, S. Galli, A. Sironi, B. Lippert, J. Sol. St.Chem. 178 (2005) 2436.

54 D. Olea et al. / Inorganica Chimica Acta 360 (2007) 48–54
[6] J.A.R. Navarro, E. Barea, J.M. Salas, N. Masciocchi, S. Galli, A.Sironi, C.O. Ania, J.B. Parra, Inorg. Chem. 45 (2006) 2397.
[7] J.M. Lehn, Science 295 (2002) 2400.[8] J.M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995.[9] A. Semenov, J.P. Spatz, M. Moller, J.M. Lehn, B. Sell, D. Schubert,
C.H. Weidl, U.S. Schubert, Angew. Chem., Int. Ed. Engl. 38 (1999)2547.
[10] M. Ruben, J. Rojo, F.J. Romero-Salguero, L.H. Uppadine, J.M.Lehn, Angew. Chem., Int. Ed. Engl. 43 (2004) 3644.
[11] U. Ziener, J.M. Lehn, A. Mourran, M. Moller, Chem. Eur. J. 8(2002) 951.
[12] S. Stepanow, M. Lingenfelder, A. Dmitriev, H. Spillmann, E.Delvigne, N. Lin, X.B. Deng, C.Z. Cai, J.V. Barth, K. Kern, NatureMat. 3 (2004) 229.
[13] J.V. Barth, G. Costantini, K. Kern, Nature 437 (2005) 671.[14] D. Olea, S.S. Alexandre, P. Amo-Ochoa, A. Guijarro, F. de Jesus,
J.M. Soler, P.J. de Pablo, F. Zamora, J. Gomez-Herrero, Adv.Mat. 17 (2005) 1761.
[15] U. Garcia-Couceiro, D. Olea, O. Castillo, A. Luque, P. Roman, P.J. dePablo, J. Gomez-Herrero, F. Zamora, Inorg. Chem. 44 (2005) 8343.
[16] S. Novokmet, M.S. Alam, V. Dremov, F.W. Heinemann, P. Muller,R. Alsfasser, Angew. Chem., Int. Ed. Engl. 44 (2005) 803.
[17] A. Earnshaw, Introduction to Magnetochemistry, Academic Press,London, 1968.
[18] J. Roisnel, J. Rodrıguez-Carvajal, WINPLOTR, a New Tool forPowder Diffraction, Laboratoire Leon Brillouin (CEA-CNRS) Centred’Etudes de Saclay, France, 2000.
[19] A. Boultif, D. Louer, J. Appl. Crystallogr. 24 (1991) 987.[20] J. Rodrıguez-Carvajal, FULLPROF, Program Rietveld Pattern
Matching Analysis of Powder Patterns, 1997.[21] E. Coronado, J.R. Galan-Mascaros, C.J. Gomez-Garcia, V. Laukhin,
Nature 408 (2000) 447.[22] E. Coronado, M. Clemente-Leon, J.R. Galan-Mascaros, C. Gime-
nez-Saiz, C.J. Gomez-Garcia, E. Martinez-Ferrero, Dalton Trans.(2000) 3955.
[23] E. Coronado, J.R. Galan-Mascaros, C.J. Gomez-Garcia, J.M.Martinez-Agudo, Inorg. Chem. 40 (2001) 113.
[24] S. Decurtins, H.W. Schmalle, P. Schneuwly, H.R. Oswald, Inorg.Chem. 32 (1993) 1888.
[25] L.O. Atovmyan, G.V. Shilov, R.N. Lyubovskaya, E.I. Zhilyaeva,N.S. Ovanesyan, S.I. Pirumova, I.G. Gusakovskaya, Y.G. Morozov,Jetp. Lett. 58 (1993) 766.
[26] M. Pilkinton, S. Decurtins, in: T.J. Meyer (Ed.), ComprehensiveCoordination Chemistry II, vol. 7, Elsevier, University of Berne,Switzerland, 2004.
[27] H. Tamaki, Z.J. Zhong, N. Matsumoto, S. Kida, M. Koikawa, N.Achiwa, Y. Hashimoto, H. Okawa, J. Am. Chem. Soc. 114 (1992)6974.
[28] D. Cangussu, H.O. Stumpf, H. Adams, J.A. Thomas, F. Lloret, M.Julve, Inorg. Chim. Acta 358 (2005) 2292.
[29] A.L. Fuller, R.W. Watkins, K.R. Dunbar, A.V. Prosvirin, A.M.Arif, L.M. Berreau, Dalton Trans. (2005) 1891.
[30] P. Roman, C. GuzmanMiralles, A. Luque, J.I. Beitia, J.Cano, F. Lloret, M. Julve, S. Alvarez, Inorg. Chem. 35(1996) 3741.
[31] O. Castillo, A. Luque, M. Julve, F. Lloret, P. Roman, Inorg. Chim.Acta 315 (2001) 9.
[32] O. Castillo, A. Luque, F. Lloret, P. Roman, Inorg. Chim. Acta 324(2001) 141.
[33] O. Castillo, A. Luque, P. Roman, F. Lloret, M. Julve, Inorg. Chem.40 (2001) 5526.
[34] U. Garcia-Couceiro, O. Castillo, A. Luque, J.P. Garcia-Teran, G.Beobide, P. Roman, Eur. J. Inorg. Chem. (2005) 4280.
[35] J.P. Garcia-Teran, O. Castillo, A. Luque, U. Garcia-Couceiro, P.Roman, F. Lloret, Inorg. Chem. 43 (2004) 5761.
[36] J.P. Garcia-Teran, O. Castillo, A. Luque, U. Garcia-Couceiro, G.Beobide, P. Roman, Dalton Trans. (2006) 902.
[37] Cambridge Crystallographic Data Centre, CCDC ConQuest 1.8.2006.
[38] U. Garcia-Couceiro, O. Castillo, A. Luque, G. Beobide, P. Roman,Inorg. Chim. Acta 357 (2004) 339.
[39] J.Y. Lu, M.A. Lawandy, J. Li, T. Yuen, C.L. Lin, Inorg. Chem. 38(1999) 2695.
[40] L.M. Zheng, X. Fang, K.H. Lii, H.H. Song, X.Q. Xin, H.K. Fun,K. Chinnakali, I.A. Razak, Dalton Trans. (1999) 2311.
[41] M.E. Fisher, Am. J. Phys. 32 (1964) 343.[42] R. Feyerherm, A. Loose, M.A. Lawandy, J. Li, J. Phys. Chem. Solids
63 (2002) 71.[43] M.E. Fisher, J. Math. Phys. 4 (1963) 124.[44] J.S. Miller (Ed.), Extended Linear Chain Compounds, Plenum, New
York, 1983.[45] I. Sledzinska, A. Murasik, P. Fischer, J. Phys. C Solid State Phys. 21
(1988) 5273.[46] J.A. Lukin, S. Simizu, N.S. Vanderven, S.A. Friedberg, J. Magn.
Magn. Mater. 140 (1995) 1669.[47] J.M. Rueff, N. Masciocchi, P. Rabu, A. Sironi, A. Skoulios, Chem.
Eur. J. 8 (2002) 1813.[48] J.M. Rueff, N. Masciocchi, P. Rabu, A. Sironi, A. Skoulios, Eur. J.
Inorg. Chem. (2001) 2843.[49] J. Glerup, P.A. Goodson, D.J. Hodgson, K. Michelsen, Inorg. Chem.
34 (1995) 6255.[50] J.M. Lehn, Chem. Eur. J. 5 (1999) 2455.[51] J.M.J. Paulusse, R.P. Sijbesma, Angew. Chem., Int. Ed. Engl. 43
(2004) 4460.[52] M. Bussiek, N. Mucke, J. Langowski, Nucleic Acid Res. 31 (2003)
e137.


![3-(Adamantan-1-yl)-4-[( E )-(2,6-difluorobenzylidene)amino]-1-[(4-phenylpiperazin-1-yl)methyl]-1 H -1,2,4-triazole-5(4 H )-thione](https://static.fdokumen.com/doc/165x107/6324d4b3c9c7f5721c01c4ad/3-adamantan-1-yl-4-e-26-difluorobenzylideneamino-1-4-phenylpiperazin-1-ylmethyl-1.jpg)











![Synthesis, Characterization and Antimicrobial activity of 2-(5-Mercapto-3-subsituted-1,5-dihydro-[1,2,4]Triazole’](https://static.fdokumen.com/doc/165x107/6317d6eab6c3e3926d0e1092/synthesis-characterization-and-antimicrobial-activity-of-2-5-mercapto-3-subsituted-15-dihydro-124triazole.jpg)





