
MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0
14
and Evolution. All rights reserved. For permissions, please e-mail: [email protected] The Author 2007. Published by Oxford University Press on behalf of the Society for Molecular Biology 1 Revised April 25, 2007 Letters (software) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0 Koichiro Tamura 1,2 , Joel Dudley 1 , Masatoshi Nei 3 and Sudhir Kumar 1,4* 1 Center for Evolutionary Functional Genomics, The Biodesign Institute, Arizona State University, Tempe, AZ 85287-5301, USA 2 Department of Biological Sciences, Tokyo Metropolitan University, 1-1 Minami-ohsawa, Hachioji, Tokyo 192-0397, Japan 3 Department of Biology and the Institute of Molecular Evolutionary Genetics, The Pennsylvania State University, University Park, PA 16802, USA 4 School of Life Sciences, Arizona State University, Tempe, AZ 85287-4501, USA *Address for Correspondence: Sudhir Kumar Biodesign Institute Building A240 Arizona State University 1001 S. McAllister Avenue Tempe, AZ 85287-5301 Tel: 480-727-6949 E-mail: [email protected] MBE Advance Access published May 7, 2007 by guest on August 18, 2015 http://mbe.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0

and Evolution. All rights reserved. For permissions, please e-mail: [email protected] The Author 2007. Published by Oxford University Press on behalf of the Society for Molecular Biology
1
Revised April 25, 2007
Letters (software)
MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0
Koichiro Tamura1,2, Joel Dudley1, Masatoshi Nei3 and Sudhir Kumar1,4*
1Center for Evolutionary Functional Genomics, The Biodesign Institute, Arizona State
University, Tempe, AZ 85287-5301, USA 2Department of Biological Sciences, Tokyo Metropolitan University, 1-1 Minami-ohsawa,
Hachioji, Tokyo 192-0397, Japan 3Department of Biology and the Institute of Molecular Evolutionary Genetics, The Pennsylvania
State University, University Park, PA 16802, USA 4School of Life Sciences, Arizona State University, Tempe, AZ 85287-4501, USA
*Address for Correspondence:
Sudhir Kumar Biodesign Institute Building A240 Arizona State University
1001 S. McAllister Avenue Tempe, AZ 85287-5301
Tel: 480-727-6949 E-mail: [email protected]
MBE Advance Access published May 7, 2007 by guest on A
ugust 18, 2015http://m
be.oxfordjournals.org/D
ownloaded from

2
Abstract
We announce the release of the fourth version of MEGA software, which expands
on the existing facilities for editing DNA sequence data from auto-sequencers, mining web-
databases, performing automatic and manual sequence alignment, analyzing sequence
alignments to estimate evolutionary distances, inferring phylogenetic trees, and testing
evolutionary hypotheses. Version 4 includes a unique facility to generate captions, written
in figure legend format, in order to provide natural language descriptions of the models
and methods used in the analyses. This facility aims to promote a better understanding of
the underlying assumptions used in analyses, and of the results generated. Another new
feature is the Maximum Composite Likelihood (MCL) method for estimating evolutionary
distances between all pair of sequences simultaneously, with and without incorporating
rate variation among sites and substitution pattern heterogeneities among lineages. This
MCL method also can be used to estimate transition/transversion bias and nucleotide
substitution pattern without the knowledge of the phylogenetic tree. This new version is a
native 32-bit Windows application with multi-threading and multi-user supports, and it is
also available to run in a Linux desktop environment (via the Wine compatibility layer)
and on Intel-based Macintosh computers under the Parallels program. The current test
version of MEGA is available free of charge at http://www.megasoftware.net.
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

3
Since the early 1990s, MEGA software functionality has evolved to include the creation and
exploration of sequence alignments, the estimation of sequence divergence, the reconstruction
and visualization of phylogenetic trees, and the testing of molecular evolutionary hypotheses.
The three versions of MEGA have been released, and they integrate web-based sequence data
acquisition and alignment capabilities (Figure 1) with the evolutionary analyses (Figure 2),
making it much easier to conduct comparative analyses in a single computing environment
(Kumar, Tamura, and Nei 2004). Over time, MEGA has come to enhance the classroom
learning experience as its use by researchers, educators, and students in diverse disciplines has
expanded (Kumar and Dudley 2007). The fourth version (MEGA4) contains three distinct
newly-developed functionalities, which are outlined below.
First, we have developed a Caption Expert software module that generates descriptions
for every result obtained by MEGA4. This description informs the user of all of the options used
in the analysis, including the data subset actually used (e.g., codon positions included), the
chosen option for the handling of sites with gaps or missing data, the evolutionary model of
substitution (e.g., DNA substitution pattern, uniformity of evolutionary rates among sites, and
homogeneity assumption among lineages), and the methods applied for estimating pairwise
distances and for inferring and testing phylogeny. The caption also includes specific citations for
any method, algorithm, and software used in the given analysis. Two examples of descriptions
generated by the Caption Expert are shown in Figure 3.
The availability of these descriptions is intended to promote a better understanding of the
underlying assumptions used in analyses, and of the results produced. This is needed because
MEGA's intuitive graphical interface makes it easy for both novice and expert users to conduct a
variety of computational and statistical analyses. However, some users may not immediately
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

4
realize the underlying assumptions and data-handling options involved in each analysis. Even
expert molecular and population geneticists may not be able to discern all of the assumptions
implied. In general, we expect a written description of methods and results to be useful for
students and researchers when preparing tables and figures for presentation and publication.
Second, we have now added a Maximum Composite Likelihood (MCL) method for
estimating evolutionary distances (dij) between DNA sequences, which MEGA users frequently
employ for inferring phylogenetic trees, divergence times, and average sequence divergences
between and within groups of sequences. In this approach, the Composite Log Likelihood (CL)
obtained as the sum of log likelihood for all sequence pairs in an alignment is maximized by
fitting the common parameters for nucleotide substitution pattern ( � ) to every sequence pair (i,j):
, ln ( , )i j ijCL l d!=" (Tamura, Nei, and Kumar 2004). This approach was previously referred to
as the “Simultaneous Estimation” (SE) method, because all dij’s are simultaneously estimated
(Tamura, Nei, and Kumar 2004). The MCL approach differs from current approaches for
evolutionary distance estimation, wherein each distance is estimated independently of others
either by analytical formulas or by likelihood methods (independent estimation [IE] approach).
The MCL method has many advantages over the IE approach. To begin with, the IE
method for estimating evolutionary distance for each pair of sequences will often cause rather
large errors unless very long sequences are used. The use of the MCL method reduces these
errors considerably, as a single set of parameters estimated from all sequence pairs is applied to
each distance estimation. When distances are estimated with lower errors, distance-based
methods for inferring phylogenies are expected to be more accurate. This is indeed the case for
the Neighbor-Joining method (Saitou and Nei 1987), as the use of the MCL distances leads to a
much higher accuracy (Tamura, Nei, and Kumar 2004). Even when the topologies estimated are
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

5
the same, the use of the MCL distances often gives higher bootstrap values for the estimated
phylogenetic tree compared to the use of IE distances, as is evident from the example given in
Figure 4 A (MCL: bold, IE: italics).
In addition, the IE distances are not always estimable when pairwise distances are
calculated between very distantly related sequences, because the arguments of logarithms in the
analytical formulas may become negative by chance. The probability of occurrence of such
inapplicable cases increases as the number of sequences in the data increases, the evolutionary
distances become larger, and the substitution pattern becomes more complex (Tamura, Nei, and
Kumar 2004). The use of the MCL method eliminates this problem effectively and allows for
the use of sophisticated models in inferring phylogenies from an increasingly larger number of
diverse sequences.
MEGA4 implements the MCL approach for estimating distances between sequence pairs,
average distances between and within groups, and average pairs overall with their variances
estimated by a bootstrap approach. Our implementation of the MCL method allows for the
consideration of substitution rate variation from site-to-site, using an approximation of the
gamma distribution of evolutionary rates, and the incorporation of heterogeneity of base
composition in different species/sequences. The user also has the flexibility to estimate the
numbers of transition and transversion type substitutions per site separately. Naturally, the MCL
distances can be utilized for inferring phylogenies by the distance-based methods, along with the
bootstrap tests of phylogenies.
MEGA4 implements the MCL approach under the Tamura-Nei (1993) substitution
model, in which the rates of two types of transitional substitutions (between purines [a1] and
between pyrimidines [a2]) and the rate of transversional substitutions (b) are considered
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

6
separately by taking into account the unequal frequencies of four nucleotides (base composition
bias). The MCL estimates of transition/transversion rate ratio have been found to be close to the
true values in previous simulation experiments (Tamura, Nei, and Kumar 2004). We have
utilized this feature to provide users with a facility to compute the relative rates of substitutions
between nucleotides based on the MCL estimates of a1, a2, b, and on the observed frequencies of
the four nucleotides under the Tamura-Nei (1993) model (Figure 3C). For ease of comparison,
we have expressed these substitution rates as relative frequencies of substitutions between
nucleotides such that the sum of all frequencies is 100 (see also Gojobori, Li, and Graur 1982).
Third, we have now programmed MEGA4 to run on some versions of Linux through the
Wine software compatibility layer (www.winehq.org). The first advancement alleviates the
problem of performance degradation (and the need to purchase Windows emulation software)
when using MEGA on Linux. Wine is neither a hardware nor a software emulator, but an open
source tool that allows for the native execution of Windows applications on Linux. Our tests of
MEGA4 running on Linux show the display, stability, and performance to be highly satisfactory
and comparable to the native Windows system (Figure 4 B). Furthermore, investigators now
report MEGA4 running on Intel-based Macintosh computers under the Parallels program as well
as it does on Windows-native personal computers (see Hall 2007). The Parallels program is a
native solution for Macs that permits them to simultaneously run Windows and Macintosh
software.
We have also built support for a multi-user environment, which will allow each user of
the same computer to keep his/her customized settings, including file locations, window sizes,
choice of genetic code table, and previously used analysis options. This feature will facilitate
educational and laboratory usage where a single computer is shared by multiple users.
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

7
In conclusion, MEGA4 now contains a wide array of functionalities for the molecular
evolutionary analysis of data (http://www.megasoftware.net/features.html). It is useful to note
that while we are continuously adding new methods and functions to MEGA, we do not intend to
make it a catalogue of all evolutionary analysis methods available. Rather, it is anticipated to
become a workbench for the exploration of sequence data from evolutionary perspectives.
Acknowledgements
We thank the colleagues, students, and volunteers who spent countless hours testing the
early release versions of MEGA; almost all facets of MEGA’s design and implementation
benefited from these comments. We thank Ms. Linwei Wu for assistance with MEGA website
and handling bugs, and Ms. Kristi Garboushian for editorial support. We thank the two
reviewers for suggesting many useful text additions, which have been included in the Figure 1
legend and in the text. We also thank Drs. Masafumi Nozawa and Barry Hall for comments on
an earlier version of this manuscript. The MEGA software project is supported by research
grants from National Institutes of Health (S. K. and M. N.) and from Japan Society for
Promotion of Sciences (K.T.).
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

8
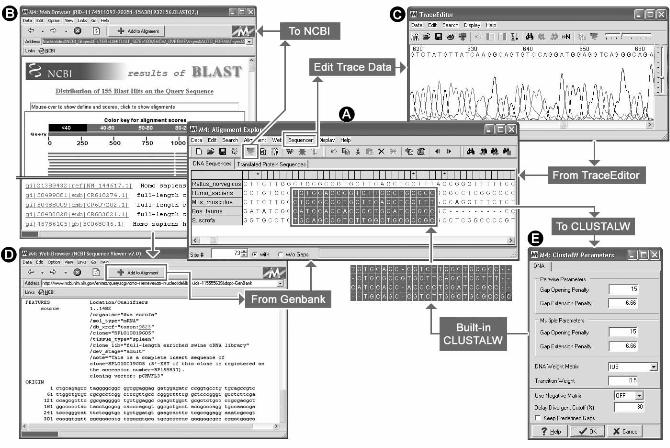
Figure 1. Sequence alignment editor and web-data mining features in MEGA4. In the
Alignment Explorer (panel A), the integrated web browser (pane B) permits downloading
sequences from online databases directly into the current alignment, without the need for manual
cutting-and-pasting and reformatting. The DNA sequences can be translated to the
corresponding protein sequences by a single mouse click, and the protein sequences can be
aligned by ClustalW (panel E) (Thompson, Higgins, and Gibson 1994) and adjusted manually by
eye. Returning to the nucleotide view automatically aligns the nucleotide sequences according to
the protein alignments, and DNA and protein sequence alignments can be exported in a variety
of formats for use with other programs. Alignment Editor also contains facilities for editing and
importing of trace data files output from DNA sequencers (panel C).
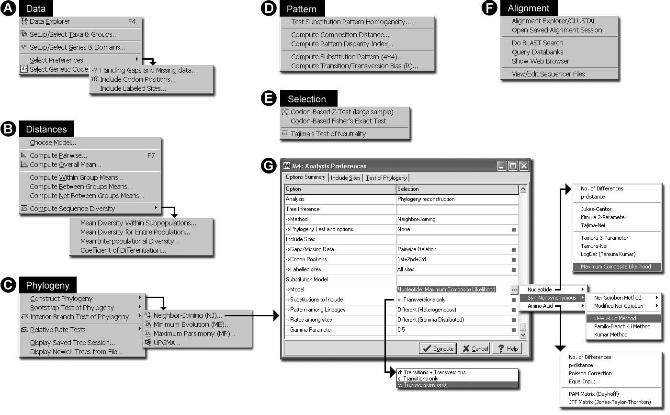
Figure 2. A collection of menus that provide access to many different data analysis options in
MEGA4, including exploration of input data set (A), estimation of evolutionary distances (B),
inferring and testing phylogenetic trees (C), tests of homogeneity of substitution patterns and its
estimation (D), tests of selection (E), alignment of DNA and protein sequences (F), and the
dialog box that provides users with options to select model of substitution and data sub-setting
options (G).
Figure 3. The Tree-Explorer displaying a Neighbor-Joining tree of mitochondrial 16S rRNA
sequences (panel A), and the description generated by the Caption Expert (panel B). Estimates
of the relative probabilities of nucleotide substitutions for 70 control-region sequences of human
mitochondrial DNA sequences are shown in panel C. The gamma shape parameter (a = 0.35)
was estimated using the Yang and Kumar (1996) method, and the rest of the analysis details are
given in panel B. It is worth noting that the Tree Explorer shown in panel A includes a high-
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

9
resolution tree drawing facility that includes displaying trees in a variety of formats, with options
to display/hide branch lengths as well as clade confidence labels, and re-rooting and rearranging
trees, among other functionalities. MEGA4 can export the drawings to graphics programs, and
can export trees in Newick format for use by other programs. Furthermore, MEGA can import
and draw trees from Newick format files that have been estimated by other programs (see panel
2C).
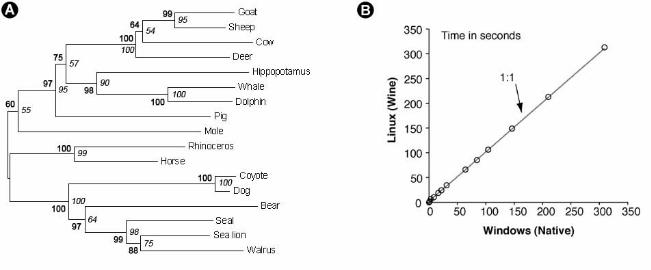
Figure 4. (A) Bootstrap support for the branching order of 16 Laurasiatheria species
reconstructed with MCL approach (bold) and without MCL approach (italics) under the Tamura-
Nei (1993) model (see Figure 3B for rest of the analysis details). The 16S rRNA sequences used
were downloaded from GenBank and were aligned in MEGA4 using CLUSTALW (accession
numbers: AJ428578, NC004029, X72004, AF303109, NC008093, DQ480502, X97336, X79547,
DQ534707, AJ554051, AJ554061, NC000889, NC007704, AB074968, NC005044 and
NC001941). (B) Comparison of MEGA4 performance benchmarks on Windows and Linux
(with Wine application compatibility layer). Identical hardware configuration was used, and
example data sets included in the MEGA4 installation were employed. The results show that
computations executed under Wine are penalized by about 2 seconds, which is attributable to the
need for Wine’s initialization.
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

10
References
Gojobori T.,Li W. H.,Graur D. 1982. Patterns of nucleotide substitution in pseudogenes and
functional genes. J Mol Evol 18:360-369.
Kumar S.,Dudley J. 2007. Bioinformatics for biologists in the genomics era. Bioinformatics (In
press).
Kumar S.,Tamura K.,Nei M. 2004. MEGA3: An integrated software for Molecular Evolutionary
Genetics Analysis and sequence alignment. Brief Bioinform 5:150-163.
Saitou N.,Nei M. 1987. The Neighbor-Joining Method - a New Method for Reconstructing
Phylogenetic Trees. Mol. Biol. Evol. 4:406-425.
Tamura K.,Nei M. 1993. Estimation of the Number of Nucleotide Substitutions in the Control
Region of Mitochondrial-DNA in Humans and Chimpanzees. Mol. Biol. Evol. 10:512-
526.
Tamura K.,Nei M.,Kumar S. 2004. Prospects for inferring very large phylogenies by using the
neighbor-joining method. Proc Natl Acad Sci U S A 101:11030-11035.
Thompson J. D.,Higgins D. G.,Gibson T. J. 1994. Clustal-W - Improving the Sensitivity of
Progressive Multiple Sequence Alignment through Sequence Weighting, Position-
Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 22:4673-4680.
Yang Z.,Kumar S. 1996. Approximate methods for estimating the pattern of nucleotide
substitution and the variation of substitution rates among sites. Mol Biol Evol 13:650-659.
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from

by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from
by guest on August 18, 2015
http://mbe.oxfordjournals.org/
Dow
nloaded from




















