
Cu isotopic fractionation in the supergene environment with and without bacteria

Available online at www.sciencedirect.com
www.elsevier.com/locate/gca
Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
Isotopic fractionation and reaction kinetics between Cr(III)and Cr(VI) in aqueous media
Sonja Zink a,*, Ronny Schoenberg b,1, Michael Staubwasser c
a Institute for Mineralogy, Leibniz University of Hannover, Callinstrasse 3, 30167 Hannover, Germanyb Centre for Geobiology and Department of Earth Science, University of Bergen, Allegaten 41, 3007 Bergen, Norway
c Institute for Geology and Mineralogy, University of Cologne, Zulpicher Strasse 49a, 50674 Cologne, Germany
Received 29 September 2009; accepted in revised form 12 July 2010; available online 24 July 2010
Abstract
The redox-sensitive stable isotope geochemistry of chromium bears the potential to monitor the attenuation of chromate pol-lution and to investigate changes in environmental conditions in the present and the past. The use of stable Cr isotope data as ageo-environmental tracer, however, necessitates an understanding of the reaction kinetics and Cr fractionation behaviour dur-ing redox transition and isotope exchange. Here, we report stable chromium isotope fractionation data for Cr(VI) reduction,Cr(III) oxidation and isotopic exchange between soluble Cr(III) and Cr(VI) in aqueous media. The reduction of Cr(VI) toCr(III) with H2O2 under strongly acidic conditions shows a near-equilibrium isotope fractionation of D53/52Cr(Cr(III)–Cr(VI)) of�3.54 ± 0.35&. At pH neutrality, however, the reduction experiments show a kinetic isotope fractionation D53/52Cr(Cr(III)–Cr(VI))
of �5& for the extent of reduction of up to 85% of the chromium. The oxidation of Cr(III) to Cr(VI) in alkaline media, usingH
2O2 as the oxidant, cannot be explained by a single, unidirectional reaction. Our experiments indicate that the involvement of
the unstable intermediates Cr(IV) and Cr(V) and their disproportionation during redox reactions between Cr(III) and Cr(VI)influence the overall fractionation factor, depending on the prevailing pH conditions and the reaction rates. No detectable iso-tope exchange between soluble Cr(VI) and Cr(III) species at pH values of 5.5 and 7 was revealed over a timescale of days toweeks. This means that, at least within such a time frame, the isotopic composition of Cr(VI) in a natural system will not beinfluenced by equilibration with any Cr(III) and thus reveal the true extent of reduction, given that the Cr isotope compositionof the source Cr(VI) and the fractionation factor for the prevailing conditions are known.� 2010 Elsevier Ltd. All rights reserved.
1. INTRODUCTION
Variations in the stable isotope composition of redoxelements have proven to be powerful tools to investigatechanges in environmental conditions throughout Earth’shistory. For example, systematic changes in the iron isotopecomposition of chemical sediments and diagenetic sulphidesfrom the Archaean to the early Proterozoic were interpretedto show the build up of significant amounts of free oxygenin the atmosphere (Rouxel et al., 2005).
0016-7037/$ - see front matter � 2010 Elsevier Ltd. All rights reserved.
doi:10.1016/j.gca.2010.07.015
* Corresponding author. Tel.: +49 511 762 8970; fax: +49 511762 2110.
E-mail address: [email protected] (S. Zink).1 Present address: Institute for Geoscience, Eberhard Karls
University of Tubingen, Wilhelmstr. 56, 72074 Tubingen, Germany.
Systematic differences in molybdenum isotope composi-tions have been reported for oxic, suboxic, anoxic andeuxinic marine sediments, reflecting different mechanismsof authigenic molybdenum accumulation (Siebert et al.,2003; Poulson et al., 2006). Furthermore, it has been pro-posed that molybdenum isotope compositions in reducingmarine sediments may be used as tracers for organic carboncycling in continental margin sediments (McManus et al.,2006; Siebert et al., 2006). Similarly, systematic stable ura-nium isotope variations between oxic, suboxic and euxinicsediments make this isotope system a useful tracer to depictchanges in marine redox conditions over geological time-scales (Weyer et al., 2008). The large chromium isotopefractionation between different Cr species that were deter-mined by theoretical models and experiments (Ellis et al.,2002; Schauble et al., 2004) render this element to be

Fig. 1. Prevalent Cr(III) and Cr(VI) species in an aquatic system atvarious physicochemical conditions. (a) Eh-pH diagram for thedifferent Cr(III) and Cr(VI) species; (b) the predominant Cr(III)species at different pH values given as their degree of dissociation(Barnowski, 2001); (c) the fraction of the Cr2O7
2�, HCrO4� and
CrO4� at a given Cr(VI) concentration of 5 mmol L�1, depending
on the prevailing pH (Palmer and Puls, 1994).
5730 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
another promising tool to detect redox changes in aque-ous environments. Recently, a characterisation of theCr isotope signatures of solid Earth reservoirs has beencarried out (Schoenberg et al., 2008), however, systematicinvestigations of the chromium isotope cycling in theaquatic environment and marine sediments are not avail-able yet.
In nature chromium occurs mainly in the two oxidationstates Cr(III) and Cr(VI) with trivalent chromium being byfar the predominant species in rocks. In aquatic systemsboth Cr(III) and Cr(VI) species may be dominant, depend-ing on the prevailing reduction potential Eh and the pHconditions of the system (Fig. 1a). In aqueous solutionsCr(III) mainly exists as hydrolysed Cr3+ and its hydroxocomplexes, such as Cr(OH)2
+ and Cr(OH)3. The abundanceof these Cr(III) species is strongly depending on the ambi-ent pH conditions (Kotas and Stasicka, 2000) as shown inFig. 1b. Cr(III) is poorly soluble – the equilibrium constantof Cr(III)-hydroxide (log K) in water is <�6.84 (Rai et al.,1987) – and preferentially adsorbs onto organic and inor-ganic particulate matter. Cr(VI) on the other hand is highlysoluble – the solubility of K2CrO4 in water, for example, is629 g L�1 (Nriagu and Nieboer, 1988) – and occurs asCrO4
2�, HCrO4� and Cr2O7
2� ions in aquatic systems(Rai et al., 1989). Fig. 1c shows that the relative propor-tions of these Cr(VI) species in solutions depend on thepH condition, as well as the Cr(VI) concentration (Palmerand Puls, 1994).
The reduction of Cr(VI) to Cr(III) on the surface ofmagnetite crystals and (Fe(II)-bearing) minerals in sedi-ments in aqueous solution is accompanied by a supposedlykinetic, mass-dependent Cr isotope fractionation of�3.4 ± 0.1& on the 53Cr/52Cr ratio (Ellis et al., 2002). The-oretically, the amount of natural attenuation of toxicCr(VI) in aquifers by reduction to Cr(III) can be tracedand quantified isotopically, as the 53Cr/52Cr ratio of theresidual Cr(VI) compounds in solution increases along aRayleigh-type function (Blowes, 2002; Ellis et al., 2002).Adsorption of Cr(VI) on mineral surfaces does not appearto cause significant equilibrium stable Cr isotope fraction-ation. Ellis et al. (2004) demonstrated that adsorption ofCr(VI) onto c-Al2O3 and goethite causes an initial kineticisotope effect between the soluble and the adsorbed speciesof +0.70& in the 53Cr/52Cr ratio after exposures of lessthan 2 h. However, ongoing isotopic exchange towardsequilibrium decreases the difference in this ratio to less than+0.04& after only 24 h, which is negligible compared to theisotope fractionation caused by Cr reduction. DissimilatoryCr(VI) reduction by Shewanella oneidensis results in a Crisotope fractionation of 4.00 to 4.50& in 53Cr/52Cr, therebyenriching the remaining unreduced Cr(VI) in heavy isotopes(Sikora et al., 2008). Thus, microbially mediated Cr isotopicfractionation appears to be significantly larger than thatresulting from abiotic Cr(VI) reduction by magnetite ornatural estuarine and pond sediments (Ellis et al., 2002).Equilibrium Cr isotope fractionation between Cr(VI) andCr(III) species calculated using vibrational spectra and bothab initio and empirical force-field models qualitatively agreewith experimentally determined ones. However, the calcu-lated equilibrium and the experimentally determined, sup-
posedly kinetic Cr isotope fractionation factors differ by afactor of about 1.5–2 (Schauble et al., 2004). The theoreticalmodels of Schauble et al. (2004) indicate that the largest equi-librium Cr isotope fractionations can be expected to resultfrom redox transformations. Furthermore, these modelspredict much smaller differences in Cr isotope compositionsbetween species of equal oxidation states, but different bond-ing partners.

Cr isotopic fractionation and reaction kinetics in aqueous media 5731
So far, no experimental data have been published thatinvestigate the Cr isotope fractionation during oxidationfrom Cr(III) to Cr(VI), which may take place through oxi-dative weathering on land or within the oxygenated watermasses and sediments of the oceans. However, input ofCr(III) by hydrothermal vent systems at mid-ocean ridgesis a significant source for the reduced Cr species to the mar-ine environment (Sander and Koschinsky, 2000; Sanderet al., 2003). Possible Cr isotope fractionation during oxida-tion of hydrothermal Cr(III) might therefore be an impor-tant process for the overall marine Cr isotope cycling.Similarly, little is known about the reaction kinetics ofcoexisting Cr(III) and Cr(VI) species in aquatic systems.Previous investigations of the reaction kinetics of differentiron species in solution show a rapid isotope exchange be-tween Fe(II) and Fe(III) species towards equilibrium withinonly a few minutes at room temperature (Johnson et al.,2002). No comparable data about the kinetics of equilib-rium chromium isotope exchange of soluble Cr species areavailable yet. Moreover, further systematic investigationsto analyse chromium isotope fractionation during Cr(VI)reduction under various conditions might prove useful sincedifferent combinations of Cr(VI) and Cr(III) species coexistin solutions of variable pH conditions (Fig. 1), which mightinfluence the reduction kinetics and equilibrium behaviourof chromium. Other stable isotope systems like iron (e.g.Dauphas and Rouxel, 2006) and sulphur (Ohmoto andLasaga, 1982; Canfield, 2001; Detmers et al., 2001) reveallarge variations in isotope fractionation factors duringreduction under different conditions.
Here we present an extensive experimental dataset thatinvestigates stable chromium isotope fractionation duringoxidation and reduction. In contrast to previous experi-ments, where Cr(III) was adsorbed onto organic or inor-ganic matter (Ellis et al., 2002, 2004), experimental setupspresented here keep both Cr(III) and Cr(VI) in solution.In addition, the isotope exchange kinetics between solubleCr(III) and Cr(VI) were determined by enriched 50Cr tracerexperiments. All experiments were performed under variouspH conditions to investigate whether the variable, predom-inant Cr(III) and Cr(VI) species influence the Cr isotopefractionation during redox transformations.
2. EXPERIMENTAL AND ANALYTICAL METHODS
2.1. Experimental conditions
The Cr(VI) reduction experiments carried out by Elliset al. (2002) and Sikora et al. (2008) were aimed at reflectingnatural conditions of aquatic systems. In these experimentssolutions were held at near pH neutrality (pH values be-tween 6 and 7) and natural inorganic and organic com-pounds, such as magnetite, sediment from the SanFrancisco Bay, and Shewanella oneidensis were used asreducing agents, which constantly withdrew the reducedCr(III) by adsorption. Here, we chose a simple closed-system experimental setup for our reduction and oxidationexperiments that kept both, trivalent and hexavalentchromium in solution. In such a set up Cr(III) and Cr(VI)are expected to follow a Rayleigh distillation function in
d53/52CrSRM979 versus fraction of Cr(III) diagrams in thecase of a unidirectional, kinetic isotope reaction with a rel-atively fast forward reaction rate (i.e. �constant fraction-ation factor) and a negligible backward reaction, whilefollowing parallel, straight lines in case of equilibrium iso-tope exchange (e.g. Broecker and Oversby, 1971; Younget al., 2002; Anbar, 2004; Weiss et al., 2008).
All oxidising and reducing experiments were performedin 7 mL PFA beakers. The Cr standard solutions used inthis study were purchased from �Merck (Darmstadt, Ger-many) and were isotopically characterised in a former study(Schoenberg et al., 2008). In particular, these chromiumsolutions are (i) a 1000 lg mL�1 Cr(III) standard solutionin 0.5 mol L�1 HNO3 and (ii) a 450 lg mL�1 Cr(VI) stan-dard solution in water. Small volumes for the reaction solu-tions were chosen to ensure rapid and complete mixing ofthe components involved in the chemical reactions andallowing for direct loading of the total sample onto thechromatographic column for separation of Cr(III) andCr(VI). Separation of the total amounts of Cr(III) andCr(VI) also allowed to check the yield of soluble chromiumspecies and therefore to test for precipitation of Cr(III),which could influence the results. After the redox transfor-mation the Cr(III) and the Cr(VI) fractions were separatedby anion exchange chromatography (see Section 2.2) forconcentration and isotope analyses.
2.1.1. Reduction
Different Cr(VI) species, i.e. CrO42�, HCrO4
� andCr2O7
2�, can coexists in the same solution with their rela-tive abundance being dependent on the pH value. The samestatement holds true for the soluble Cr(III) species Cr3+,CrOH2+, Cr(OH)2
+, Cr(OH)3 and Cr(OH)4�. The signifi-
cance here is that the fractionation factor between the oxi-dised and reduced chromium complexes might bedepending on the pH, as a result of their different bondstrengths. This can be expressed by the pH dependent for-ward reaction Eqs. (1) and (2), which were defined in termsof the predominant Cr(VI) and Cr(III) species:
2HCrO4� þ 3H2O2 þ 8Hþ
! 2Cr3þ þ 8H2Oþ 3O2 at pH 1 ð1Þ
2CrO42� þ 3H2O2 þ 2H2O
! 2CrðOHÞ2þ þ 6OH� þ 3O2 at pH 7 ð2Þ
As shown in Eqs. (1) and (2) H2O2 was used as a reduc-tion agent in all our reduction experiments. The experimen-tal volumes given below were too small for in situ pHdeterminations without potentially risking significant con-tamination of the samples. The pH values for all the differ-ent reduction experiments were therefore calculated and, inaddition, verified on solutions of much larger volumes, butwith equal compositions as the experimental ones.
Given the expected complexity of the reduction reactionwith the possible involvement of intermediate unstableCr(V) and Cr(IV) species, a first series of test experiments(designated R1) was performed to investigate the influenceof the reagent handling on the stability of the reaction. Inthese experiments 100 lL of a 450 lg mL�1 Cr(VI) stan-dard solution in H2O (0.9 lmol Cr) were mixed with

5732 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
50 lL 0.6 mol L�1 HCl (30 lmol) as proton donor and10 lL 0.3% H2O2 (0.9 lmol) to cause partial reduction ofCr(VI) to Cr(III) at a pH of �0.7 (i.e. according to Eq.(1)). The stability tests included (i) testing the sequence inwhich the different agents were added to the reagent vessel,(ii) checking whether surplus H2O2 and/or boiling of thesample solution to evaporate surplus H2O2 after the reac-tion was completed influenced the Cr(III):Cr(VI) concen-tration ratio and therefore the isotopic composition of thetwo chromium pools. Thereby, H2O2 was dissociated insome, but not all experiments, and (iii) testing whetherthe reaction time influenced the results, by performing thechromatographic separation immediately after the mixingof the reactants in one experiment and waiting for 2 h be-fore separation in another experiment.
In a second series of experiments we tested the influenceof the pH conditions and hence the different combinationsof soluble Cr(III) and Cr(VI) species on the Cr isotope frac-tionation during chromium reduction. In one set of experi-ments (designated R2-a) 100 lL of the 450 lg mL�1 Cr(VI)standard solution (0.9 lmol) were mixed with 50 lL6 mol L�1 HCl (300 lmol) to obtain a pH of �1. Increas-ing amounts of H2O2 up to 13.2 lmol were admixed to par-allel experiments to obtain varying amounts of partialCr(VI) reduction. In a second set of experiments (desig-nated R2-b) near-neutral pH conditions were aimed at.Thereby, the same increasing amounts of H2O2 as used inthe first set of experiments were mixed to parallel reactionvessels containing 100 lL of 450 lg mL�1 Cr(VI) solution(0.9 lmol), but no accessory acid was added here. SurplusH2O2 of both sets of experiments was boiled away byshortly heating the beakers on a hotplate. The samples werethen diluted with 900 lL H2O and ready for chromato-graphic separation of trivalent from hexavalent chromium.
2.1.2. Oxidation
Oxidation experiments were carried out under alkalineconditions, using H2O2 as oxidising agent. Under these con-ditions the sole stable Cr(VI) complex is CrO4
2�, whileCr(III) mainly exists as Cr(OH)3 and the forward reactioncan be described by Eq. (3):
2CrðOHÞ3 þ 3H2O2 þ 4OH�
! 2CrO42� þ 8H2O at pH 10:5 ð3Þ
Increasing amounts of H2O2 (0.5–150 lmol in 5–15 lL)were added to two parallel sets of samples (designated O1-aand O1-b), each containing 50 lL of Cr(III) (�1 lmol) in10 lL NH3 (133 lmol), resulting in immediate partial oxi-dation of Cr(III) to Cr(VI), according to Eq. (3). The highionic strength keeps the Cr(III) in solution, at least for theduration of the experiments, which is in the order of sec-onds to minutes. In this time frame no precipitates formedand no losses of the total Cr amount were detected. SurplusH2O2 was again boiled away by heating the solutions. Asecond series of tests (designated O2) was carried out, toinvestigate whether further redox transformations takeplace over extended periods of time. Here, each experimentcontained 20 lL 25% NH3 (266 lmol) and 12 lL 3% H2O2
(10.6 lmol), to which again 50 lL Cr(III) solution was
added. The reaction times for the different experiments, be-fore separation of Cr(III) from Cr(VI), were 0, 2, 6, 24 and48 h. All these experiments were repeated to investigate thereproducibility of the oxidation reaction under the givenconditions. All samples were then diluted with 900 lLH2O and ready for chromatographic separation of trivalentfrom hexavalent chromium.
2.1.3. Isotope exchange
The isotope exchange kinetics between soluble Cr(III)and Cr(VI) were investigated by means of enriched isotopetracer experiments. In these experiments 50Cr-enrichedCr(III) solutions and Cr(VI) solutions of natural isotopeabundance were mixed in 250 mL PE bottles, in which dif-ferent pH conditions were adjusted and maintained usingphosphate buffers. Four parallel experiments were arranged(designated E1 to E4). For two different pH values of 5.5and 7, solutions with Cr(III):Cr(VI) ratios of 1:2 and 2:1were prepared. Well-defined amounts of Cr(III) standardand enriched 50Cr isotope tracer solutions were mixed toyield a d50/52CrSRM979 value of approximately 800&. Afterevaporating to dryness these mixtures were taken up inphosphate buffer solutions of the above-mentioned pH val-ues. To these solutions a Merck Cr(VI) standard (in H2O)used in an earlier study (Schoenberg et al., 2008) withd50/52CrSRM979 = 0.052& was admixed to obtain a totalchromium concentration of 50 lg g�1 in each experiment.0.5 mL aliquots were taken from the solutions at certaintime intervals and directly loaded onto anion exchange col-umns to separate Cr(III) from Cr(VI).
2.2. Separation of Cr(III) and Cr(VI)
Cr(III) and Cr(VI) fractions were separated by an anionexchange chromatography that was slightly modified fromthe method described in Schoenberg et al. (2008). Two mil-lilitres of Dowex AG 1X8, 100–200 mesh, anion exchangeresin were transferred to Spectrum� PP columns 104704.The resin was successively cleaned with 5 mL of 5 mol L�1
HNO3, ultrapure H2O, 6 mol L�1 HCl and ultrapure H2O.Where necessary, the columns were preconditioned withHCl to yield the same pH values as the samples. The sam-ples were loaded onto the resin and the Cr(III) fractionswere eluted with 8 mL of 0.2 mol L�1 HCl and 8 mL ultra-pure H2O. The resin was then washed with 8 mL ultrapureH2O to obtain pH neutrality. The remaining Cr(VI) frac-tions were reduced to Cr(III) (Goetz and Heumann, 1988;Ball and Bassett, 2000) and eluted with a total of 7 mL of2 mol L�1 HNO3. The purification yields of the Cr(III)and Cr(VI) fractions during separation by anion exchangechromatography were determined in independent experi-ments. Although this method allows to separate Cr(III)and Cr(VI) from each other and thus to directly comparethe isotopic composition of these two chromium pools foreach sample, it bears the drawback of a small cross-contam-ination between the two pools. From our extensive experi-ence of purifying Cr(VI) from a variety of sample matriceswith this method (Schoenberg et al., 2008) and independentexperiments carried out for this study, we know that thecolumn yield for Cr(VI) reproducibly lies between 92%

Cr isotopic fractionation and reaction kinetics in aqueous media 5733
and 96%. We observed that the loss of 4–8% of Cr(VI) oc-curs directly during the loading of the sample onto the resinand will thus be collected in the Cr(III) fraction if both spe-cies (i.e. Cr(VI) and Cr(III)) are present. The yield for theCr(III) elution from the resin with 8 mL of 0.2 mol L�1
HCl and 8 mL of H2O was determined to be 96–99%.The residual 1–4% of the Cr(III) is released from the resintogether with the Cr(VI), upon reduction and elution ofthe latter to Cr(III) with 7 mL of 2 mol L�1 HNO3. AllCr isotope data were thus corrected for this cross-contam-ination, but both uncorrected and corrected values are re-ported. For the corrected Cr(III) and Cr(VI) data weassumed an average contamination of Cr(VI) in the Cr(III)fraction of 6% and an average Cr(III) contamination in theCr(VI) fraction of 2.5%. The upper and lower boundaries ofthe variability in the cross-contamination are expressed asthe asymmetric uncertainty limits of the corrected data-points given in the tables and figures. However, the correc-tion of the cross-contamination becomes only significantwhen the isotopic difference between the Cr(III) and Cr(VI)pools becomes very large and one of the fractions verysmall in comparison to the other.
2.3. Cr isotope analysis
All chromium isotope measurements were performed ona ThermoFinnigan Neptune MC–ICP mass spectrometer.The chromium signals 50Cr+, 52Cr+, 53Cr+ and 54Cr+ weredetected simultaneously together with the monitor signals48Ti+, 51V+, and 56Fe+ to correct for minor isobaric inter-ferences on 50Cr+ and 54Cr+ when necessary. Further poly-atomic interferences like 40Ar12C+ on 52Cr+, 40Ar14N+ on54Cr+ and 40Ar16O+ on 56Fe+ were completely resolvedby operating the instrument in medium-resolution mode(Halicz et al., 2008; Schoenberg et al., 2008). In this workCr isotope data are given in the d-notation followingEq. (4):
d53=52CrSRM979 ¼53Cr=52Crsample
53Cr=52CrSRM979
� �� 1
d50=52CrSRM979 ¼50Cr=52Crsample
50Cr=52CrSRM979
� �� 1 ð4Þ
For reasons of simplification the d values of samples arereported as the per mill deviation of a particular Cr isotoperatio relative to that of the certified Cr isotope standardNIST SRM 979, which was also used in previous works(Ball and Bassett, 2000; Ellis et al., 2002, 2004; Haliczet al., 2008; Schoenberg et al., 2008; Sikora et al., 2008).The isotopic fractionation that occurs during redox reac-tions can be expressed by the fractionation factor a:
a ¼ Rproduct
Rreactant
ð5Þ
with R being the 53Cr/52Cr ratio of the respective oxidisedor reduced chromium pool. The relative isotopic differenceD between two substances can be calculated according toEq. (6) or it can be approximated through the isotopic frac-tionation factor a as shown in Eq. (7), given that the isoto-pic difference between the two substances is fairly small:
D53=52Crðsample1�sample2Þ ¼ d53=52Crsample1;SRM979
� d53=52Crsample2;SRM979 ð6Þ
and
ln a � D53=52Crðsample1�sample2Þ ð7Þ
Aliquots of all Cr(III) and Cr(VI) fractions resulting fromthe reduction, oxidation, and isotope exchange experimentswere taken for Cr concentration determination by opticalemission spectroscopy (ICP-OES). An adequate amount ofa 50Cr–54Cr double spike solution was added to all chromiumfractions resulting from the reduction and oxidation experi-ments in order to correct for the instrumental mass bias dur-ing the measurements (Schoenberg et al., 2008). For eachanalytical session between March and August 2008 an ali-quot of a NIST SRM 979 stock solution, premixed with a de-fined amount of the 50Cr–54Cr double spike, was subjected tothe same Cr separation method as the samples and used asinternal control standard during the Cr isotope measure-ments. The external reproducibility of all these NISTSRM 979 data (N = 81) is 0.003 ± 0.053& (2 SD) ond53/52CrSRM979. Due to reasons lined out in Schoenberget al. (2008) the average d53/52CrSRM979 values of each singlemeasurement session was normalised to the nominald53/52CrSRM979 value of zero and the d53/52CrSRM979 valuesof the samples were corrected accordingly. By doing so, theinstrumental reproducibility of the NIST SRM 979 measure-ments decreases to 0.033& (2 SD) on d53/52CrSRM979 a valuethat is similar to the reproducibility of 0.024& for standardsolutions reported in Schoenberg et al. (2008).
For the isotope exchange experiments, the use of the50Cr–54Cr double spike method to correct for the instru-mental mass bias, however, was not possible. In this exper-imental setup the 50Cr tracer, that was added to the Cr(III)standard solution, is expected to exchange between Cr(III)and Cr(VI) to an unknown degree with time. Here, the stan-dard-sample bracketing method, which is widely used forstable isotope measurements with MC–ICP-MS (e.g.Mason et al., 2004; Schoenberg and von Blanckenburg,2005; Peel et al., 2008), was applied to correct for the instru-mental mass bias. NIST SRM 979 was used as the bracket-ing standard throughout the Cr isotope measurements forthe isotope exchange experiments. The Cr(III) standardsolution in 0.5 mol L�1 HNO3 by �Merck, which was usedfor the experiments, was routinely measured several timeswithin each analytical session to assess the accuracy andreproducibility of the isotope analyses. During this studythe measured Cr isotope composition of the �MerckCr(III) standard was d53/52CrSRM979 = �0.435 ± 0.041&
(2 SD, N = 108), which is in good agreement with previousmeasurements using the double spike method for mass biascorrection yielding d53/52CrSRM979 = �0.443 ± 0.022&
(2 SD, N = 4) (Schoenberg et al., 2008).
3. RESULTS AND DISCUSSION
3.1. Reduction experiments
All data for the reduction experiments are given inTable 1 and 2 and are illustrated in Figs. 2 and 3. The first

Table 1Cr isotope data of the first set of reduction experiments – influence of reagent handling.
Series Sample pHValue
Procedure* fCr(III) d53/52CrSRM979 d53/52CrSRM979** fCr(VI) d53/52CrSRM979 d53/52CrSRM979
** D53/52Cr(Cr(III)–Cr(VI))
a b c d Cr(III) uncorrectedCr(III)
correctedCr(III)
Cr(VI) uncorrectedCr(VI)
correctedCr(VI)
corrected
R1 1 0.7 x 0.70 �2.06 �2.24+0.06
0.30 4.65 5.07+0.06 �7.31
�0.06 �0.06
R1 2 0.7 x x 0.70 �1.98 �2.15+0.06
0.30 4.58 4.98+0.06 �7.13
�0.06 �0.06
R1 3 0.7 x 2 0.70 �2.04 �2.22+0.06
0.30 4.69 5.11+0.06 �7.32
�0.06 �0.06
R1 4 0.7 x 0.69 �2.09 �2.27+0.06
0.31 4.60 5.00+0.06 �7.27
�0.06 �0.06
R1 5 0.7 x 2 0.69 �2.06 �2.24+0.06
0.31 4.48 4.87+0.06 �7.11
�0.06 �0.06Mean �7.23
* a: Mixing of HCl with H2O2, then addition of Cr(VI); b: mixing of HCl with Cr(VI), then addition of H2O2; c: boiling on hotplate; d:reaction time in hours before separation of Cr(III) and Cr(VI).** Uncertainties are either the external reproducibility (2SD) or the calculated uncertainty limits, resulting from the variability of the cross-contamination between Cr(III) and Cr(VI) pools, whichever was larger.
Table 2Cr isotope data of the second set of reduction experiments – influence of pH.
Series Sample pHValue
fCr(III) d53/52CrSRM979 d53/52CrSRM979** fCr(VI) d53/52CrSRM979 d53/52CrSRM979
** D53/52Cr(Cr(III)–Cr(VI))
Cr(III) uncorrectedCr(III)
correctedCr(III)
Cr(VI) uncorrectedCr(VI)
correctedCr(VI)
corrected
R2-a 1 �1 0.21 �2.18 �3.00+0.32
0.79 0.55 0.57+0.05 �3.57
�0.40 �0.05
R2-a 2 �1 0.47 �1.52 �1.72+0.07
0.53 1.31 1.37+0.05 �3.09
�0.07 �0.05
R2-a 3 �1 0.67 �1.12 �1.22+0.05
0.33 2.15 2.32+0.11 �3.55
�0.05 �0.11
R2-a 4 �1 0.83 �0.64 �0.68+0.05
0.17 2.80 3.26+0.34 �3.95
�0.05 �0.29
R2-a 5* �1 1.00 �0.04 �0.04+0.05�0.05
R2-a 6* �1 1.00 �0.06 �0.06+0.05 Mean �3.54�0.05
R2-b 1 �7 1.00 0.01 0.01+0.05 0.27�0.05
R2-b 2 �7 0.34 �4.06 �4.86+0.29
0.66 2.15 2.23+0.05 �7.10
�0.31 �0.05
R2-b 3 �7 0.64 �2.77 �3.04+0.09
0.36 4.81 5.16+0.22 �8.20
�0.09 �0.21
R2-b 4 �7 0.85 �1.32 �1.42+0.05
0.15 7.29 8.67+1.06 �10.08
�0.05 �0.88
R2-b 5 �7 0.94 �0.31 �0.33+0.05
0.06 4.63 7.58+4.38 �7.92
�0.05 �2.08
R2-b 6 �7 0.94 �0.16 �0.18+0.05
0.06 2.81 4.87+3.54 �5.05
�0.05 �1.47
* Samples 5 and 6 were completely reduced.** Uncertainties are either the external reproducibility (2SD) or the calculated uncertainty limits resulting from the variability of the cross-contamination between Cr(III) and Cr(VI) pools, whichever was larger.
5734 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
series of experiments (R1) reveals that reagent handlingdoes not influence the stability of the reduction reactions.All experiments show the same extent of reduction withfCr(III) of 0.69–0.70 and the same difference between Cr(III)and Cr(VI) in D53/52Cr(Cr(III)–Cr(VI)) of �7.11 to �7.32&,independent from the order of reagent mixing, the presence
of surplus H2O2 after the reaction and the time that passedbefore separation of Cr(III) from Cr(VI) by anion chroma-tography (Table 1 and Fig. 2).
The chromium isotopic difference between Cr(III) andCr(VI) in these experiments is much larger than the isotopefractionation of �3.4& in D53/52Cr(Cr(III)–Cr(VI)) determined

Fig. 2. Results from the first set of Cr(VI) reduction experiments (R1): d53/52CrSRM979 values of Cr(VI) plotted as diamonds andd53/52CrSRM979 values of Cr(III) plotted as circles versus the fraction of Cr(III). Uncorrected values are plotted as open symbols, the correctedvalues as filled symbols. The solid lines correspond to a Rayleigh fractionation with 103 � lna (�D53/52Cr(Cr(III)–Cr(VI))) of �4.2& and thestippled parallel lines to an equilibrium isotope fractionation with a constant difference of D53/52Cr(Cr(III)–Cr(VI)) of �7.2&. All uncertainties onthe d53/52CrSRM979 values are smaller than the symbols.
Fig. 3. Results from the second set of Cr(VI) reduction experiments (R2) under acidic conditions with pH�1 (a) and near-neutral conditionswith pH �7 (b): corrected d53/52CrSRM979 values of unreduced Cr(VI) plotted as black diamonds, and corrected d53/52CrSRM979 values oforiginated Cr(III) plotted as black circles versus the fraction of Cr(III). The open symbols show the uncorrected d53/52CrSRM979 values. In (a)the remaining Cr(VI) and reduced Cr(III) plot along two parallel lines (stippled lines) with a constant difference of D53/52Cr(Cr(III)–Cr(VI)) of�3.54 ± 0.35& (1 SD, N = 4) and the solid lines show a Rayleigh fractionation with 103 � lna (�D53/52Cr(Cr(III)–Cr(VI))) of �3.54&. Thecurves in (b) (solid lines) correspond to a Rayleigh fractionation with 103 � lna (�D53/52Cr(Cr(III)–Cr(VI))) of �5.0&. All uncertainties on thed53/52CrSRM979 values are given in the figure unless they are smaller than the symbols.
Cr isotopic fractionation and reaction kinetics in aqueous media 5735

5736 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
by Ellis et al. (2002) and confirmed by Schoenberg et al.(2008). Fig. 2 shows that the Cr(III) and Cr(VI) pools ofthese experiments with a reproducible D53/52Cr(Cr(III)–Cr(VI))
of �7.11 to �7.32& can either be fitted onto an equilibriumisotope fractionation model with an according fraction-ation factor a of ca. 0.9928 on the 53Cr/52Cr ratio (stippledlines) or onto a Rayleigh-type model with an isotopic frac-tionation a of 0.9958 on the 53Cr/52Cr ratio (solid lines).This fractionation factor a of 0.9958, representing a kineticreduction reaction, is still somewhat higher than that deter-mined by abiotic Cr(VI) reduction of 0.9966 (Ellis et al.,2002; Schoenberg et al., 2008), but within the range of frac-tionation factors of 0.9955–0.9960 determined for biogenicCr(VI) reduction (Sikora et al., 2008).
In the second series of reduction experiments we investi-gated the influence of the pH on the chromium isotope frac-tionation. At a pH�1 (R2-a) the Cr isotope compositionsof the remaining Cr(VI) and reduced Cr(III) fractions ap-pear to plot along two parallel lines in a d53/52CrSRM979
vs. fCr(III) diagram (Table 2 and Fig. 3a).The constant difference D53/52Cr(Cr(III)–Cr(VI)) of
�3.54 ± 0.35& (1 SD, N = 4) seems to indicate a near-equilibrium Cr isotope fractionation between these two dis-solved oxidation states of chromium under the prevailingconditions. The isotopic difference D53/52Cr(Cr(III)–Cr(VI)) of�3.54 ± 0.35&, obtained in this set of reduction experi-ments, is within uncertainty equal to the value of�3.4 ± 0.1& that was published for Cr(VI) reduction atpH 6–7 using magnetite and estuarine and pond sedimentsas reducing agents (Ellis et al., 2002). However, our experi-ments at pH�1 are the first to indicate that the Cr isotopefractionation between soluble Cr(III) and Cr(VI) duringreduction may take place under near-equilibrium conditions.
Theoretical calculations predict equilibrium chromiumisotope fractionations of 1–6& on the 53Cr/52Cr ratio be-tween different Cr(III) and Cr(VI) complexes at tempera-tures of 300–0 �C (Schauble et al., 2004). Cr(III) is mainlyoctahedrally coordinated, while Cr(VI) is almost exclusivelytetrahedrally coordinated. In low temperature aquatic envi-ronments the most common bonding partners of chromiumare oxygen and hydroxyl. The empirical and ab initio force-field models of Schauble et al. (2004), which are based onthe bonding environment of various Cr complexes, suggestthat natural inorganic Cr isotope fractionation at theEarth’s surface may be driven largely by reduction and oxi-dation processes, whereas the effect of ligand-exchangewithin the same chromium oxidation state on the Cr iso-tope fractionation is, in comparison, modest. The experi-mental results of our reduction series R2-a presented hereand earlier findings by Ellis et al. (2002) appear to supportthe models of Schauble et al. (2004). The chromium isotopefractionation during Cr(VI) reduction to Cr(III) appears tobe independent from the predominant chromium species,which are CrO4
2� and Cr(OH)2+ at pH 7 (Ellis et al.,
2002) and HCrO4� and Cr3+ at pH�1 (this study).
Interestingly, the Cr(III) and Cr(VI) fractions of ourreduction experiments at pH 7 (R2-b) plot along a Ray-leigh-type function in a d53/52CrSRM979 vs. fCr(III) diagramwith a fractionation factor a of 0.995 for up to 85% ofCr(VI) reduction (i.e. fCr(III) = 0.85). Two of these Cr(VI)
reduction experiments at pH 7 with fCr(III) of around 0.94significantly deviate from this Rayleigh function (Table 2and Fig. 3b). However, the very low Cr content of theseCr(VI) fractions and the large isotopic differences to theirrespective Cr(III) pools (see Table 2) cause significantcross-contamination corrections on the d53/52CrSRM979 val-ues of 2.95& and 2.06&, respectively. The sizes of theasymmetrical uncertainties calculated for these residualCr(VI) pools (Table 2 and Fig. 3b) are a quantitative mea-sure for the precision and qualitative indicator for the accu-racy of these data-points. We conclude that the deviation ofthese Cr(VI) fractions from the Rayleigh function of theother data-points in the d53/52CrSRM979 vs. fCr(III) diagramis to some, but most likely not to the full, extent causedby the inaccurate correction of the cross-contaminationwith Cr(III). The kinetic fractionation along a Rayleigh-type function with a fractionation of ca. �5& in the53Cr/52Cr ratio between the Cr(VI) and the immediateCr(III) reduction product is much larger than the fraction-ation of �3.4 ± 0.1& at pH 6–7 determined by Ellis et al.(2002) and �3.54 ± 0.35& at pH�1 (this study), the lattereven indicating Cr isotope fractionation under near-equilib-rium conditions. A similar observation has already beenmade by the Cr(VI) reduction experiments at pH �1 ofthe first series (R1) that yielded a D53/52Cr(Cr(III)–Cr(VI)) of�4.2& (see above) assuming a unidirectional process alonga Rayleigh function.
In a strong acidic environment the formal potential E�0(i.e. E� modified for pH – 0; calculated employing theNernst equation for the predominant species at the givenpH value) for the reduction of Cr(VI) to Cr(III) is 1.38 V.At a pH value of 7 the species dependent potential E�0 isonly approximately 0.36 V (Nriagu and Nieboer, 1988),pointing to slower reduction rates at near-neutral condi-tions than at highly acidic ones. Furthermore, the modeof Cr(VI) reduction with H2O2 via peroxochromium com-plexes is different at a pH of 7 than it is at a pH�1 (Fig. 4).
Under highly acidic conditions equilibrium betweenH2CrO4 and HCrO4
� develops very quickly (Van Niekerket al., 2007). The formation of the blue peroxochromi-um(VI) complex [CrO(O2)2(OH2)] from H2CrO4 orHCrO4
� requires the addition of two H2O2 molecules. Itis generally agreed that the addition of the first H2O2 mol-ecule to form an intermediate Cr(VI)/H2O2 adduct is therate-limiting step for this reaction pathway (Moore et al.,1966; Orhanovic and Wilkins, 1967; Funahashi et al.,1978; Van Niekerk et al., 2007). The decomposition of theblue peroxochromium(VI) complex [CrO(O2)2(OH2)] toCr(III) and oxygen is very quick again, due to its instabilityin aqueous solutions (Moore et al., 1966; Orhanovic andWilkins, 1967; Funahashi et al., 1978; Zhang and Lay,1998; Gili et al., 2002; Vander Griend et al., 2002; VanNiekerk et al., 2007). Thus, under highly acidic conditionsthe reduction of Cr(VI) to Cr(III) with H2O2 is a first-orderreaction, solely being dependent on the availability of H2O2
and HCrO4� (Pettine et al., 2002). Under less acidic or
near-neutral conditions and a strong surplus of H2O2,however, the reduction process is a second- to third-orderreaction that involves unstable intermediate Cr(V) species(Vander Griend et al., 2002). Here, chromate rapidly

Fig. 4. Reaction pathways for Cr reduction in acidic and Cr oxidation in alkaline media, using hydrogen peroxide. Solid-lined double arrowsrepresent equilibrium reactions with very different rate constants, described in the literature. The stippled-lined double arrow represents areaction pathway, whose existence is still under debate.
Cr isotopic fractionation and reaction kinetics in aqueous media 5737
transforms to the violet peroxochromium(VI) complex[CrO(O2)2(OH)]�, which decomposes to a series of differentperoxochromium(V) complexes (i.e. mono-, di-, tri- and tet-ra-peroxochromium(V)), depending on the pH value andthe relative concentrations of the reactants (Gili et al.,2002; Vander Griend et al., 2002; Zhang and Lay, 1998).This reaction pathway goes along with a high consumptionof hydrogen peroxide. The violet peroxochromium(VI)complex can also directly reduce to Cr(OH)2
+. However,it’s still debated whether a direct reduction of the differentperoxochromium(V) complexes to Cr(OH)2
+ takes place(Pettine et al., 2002; Vander Griend et al., 2002). In bothcases the reaction pathways inhibit an effective reductionof all chromate to Cr(III) by producing the peroxochromi-um(VI) and peroxochromium(V) complexes, while consum-ing large amounts of the available hydrogen peroxide.
The effective fractionation of a multi-step reaction de-pends on the relative rates of the single reaction steps. Ina simplified model, with one dominating rate-limiting stepof the overall reaction, which takes place late in the chainof reaction steps, the overall fractionation factor can be de-fined by the product of the fractionation factors of all singlereactions involved (Anbar, 2004). However, the complexityof the reaction pathways of Cr(VI) reduction (Fig. 4), whichinvolves at last two rate-limiting reaction steps, requires amore complex model to describe the overall isotopic frac-tionation. Rees (1973), for example, developed differentmodels for complex bacterial sulphur isotope fractionationand Hayes (2001) describes a detailed model for biosyn-thetic processes involving a network of different chemicalreactions. However, the information about fractionationfactors and reaction rates for each of the various Cr(VI)reduction steps that are available from the literature arecontroversial (Pettine et al., 2002; Vander Griend et al.,2002). It is therefore difficult to apply one of these existingmodels to Cr(VI) reduction at near-neutral pH conditions.All these differences in reaction rates and variability of thefractionation factors for the various pathways, when reduc-ing Cr(VI) to Cr(III) with H2O2, might explain (i) why a
near-equilibrium Cr isotope fractionation trend betweenCr(VI) and Cr(III) is observed at pH�1, while at pH neu-tral conditions a kinetic process with larger isotopic frac-tionation is observed and (ii) why at pH neutrality apossible deviation from a Rayleigh fractionation is ob-served at extents of Cr reduction higher than 85% (seeFig. 3b).
Our experiments with H2O2 as reducing agent support apredominantly kinetic Cr isotope fractionation for Cr(VI)reduction by natural sediments at a pH of 6–7, as was sug-gested earlier (Ellis et al., 2002). Interestingly, the fraction-ation factor of our reduction experiments is much largerthan that described for Cr reduction at pH 6–7 (Elliset al., 2002), while the similarity between the equilibriumfractionation factor obtained from our Cr reduction exper-iments at pH�1 and the fractionation factor reported byEllis et al. (2002) is conspicuous. We hypothesise that thepresence of H2O2 at near-neutral pH conditions, whichleads to the formation of peroxochromium(VI) and espe-cially peroxochromium(V) complexes strongly influencesthe overall reaction kinetics of Cr(VI) reduction to Cr(III)towards a larger isotopic fractionation than is observedfor a pH �1. Clearly, further experiments are necessaryto address the question to what extent equilibrium and ki-netic Cr isotope fractionation and the formation of peroxospecies and their decomposition processes that accompanyCr(VI) reduction to Cr(III) at pH 7 overlap each other.
3.2. Oxidation experiments
All data for oxidation experiments are given in Tables 3and 4 and are illustrated in Figs. 5 and 6. The oxidationexperiments of Cr(III) to Cr(VI) in alkaline conditions re-veal a much smaller Cr isotope fractionation than the Crreduction experiments in acidic or near-neutral conditions.Both sets of the first Cr oxidation series (O1-a and O1-b),that were carried out independently, show the same rateof oxidation accompanied with the same chromium isotopefractionation, which verifies the reproducibility of the
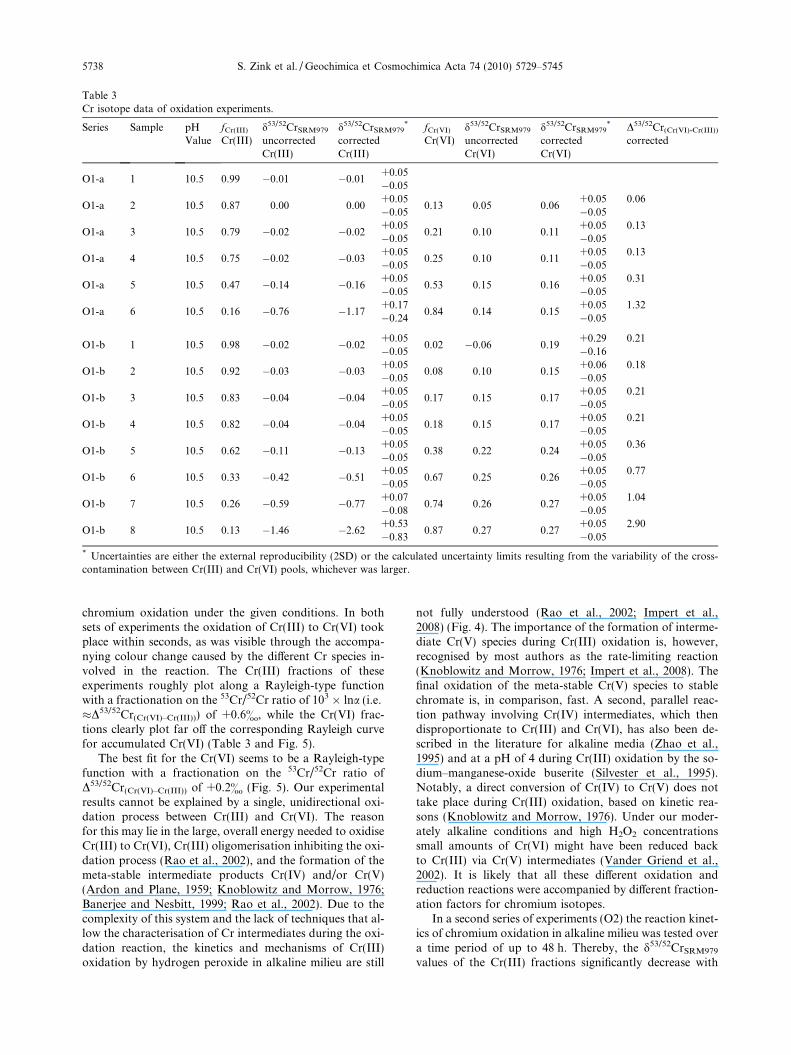
Table 3Cr isotope data of oxidation experiments.
Series Sample pHValue
fCr(III) d53/52CrSRM979 d53/52CrSRM979* fCr(VI) d53/52CrSRM979 d53/52CrSRM979
* D53/52Cr(Cr(VI)-Cr(III))
Cr(III) uncorrectedCr(III)
correctedCr(III)
Cr(VI) uncorrectedCr(VI)
correctedCr(VI)
corrected
O1-a 1 10.5 0.99 �0.01 �0.01+0.05�0.05
O1-a 2 10.5 0.87 0.00 0.00+0.05
0.13 0.05 0.06+0.05 0.06
�0.05 �0.05
O1-a 3 10.5 0.79 �0.02 �0.02+0.05
0.21 0.10 0.11+0.05 0.13
�0.05 �0.05
O1-a 4 10.5 0.75 �0.02 �0.03+0.05
0.25 0.10 0.11+0.05 0.13
�0.05 �0.05
O1-a 5 10.5 0.47 �0.14 �0.16+0.05
0.53 0.15 0.16+0.05 0.31
�0.05 �0.05
O1-a 6 10.5 0.16 �0.76 �1.17+0.17
0.84 0.14 0.15+0.05 1.32
�0.24 �0.05
O1-b 1 10.5 0.98 �0.02 �0.02+0.05
0.02 �0.06 0.19+0.29 0.21
�0.05 �0.16
O1-b 2 10.5 0.92 �0.03 �0.03+0.05
0.08 0.10 0.15+0.06 0.18
�0.05 �0.05
O1-b 3 10.5 0.83 �0.04 �0.04+0.05
0.17 0.15 0.17+0.05 0.21
�0.05 �0.05
O1-b 4 10.5 0.82 �0.04 �0.04+0.05
0.18 0.15 0.17+0.05 0.21
�0.05 �0.05
O1-b 5 10.5 0.62 �0.11 �0.13+0.05
0.38 0.22 0.24+0.05 0.36
�0.05 �0.05
O1-b 6 10.5 0.33 �0.42 �0.51+0.05
0.67 0.25 0.26+0.05 0.77
�0.05 �0.05
O1-b 7 10.5 0.26 �0.59 �0.77+0.07
0.74 0.26 0.27+0.05 1.04
�0.08 �0.05
O1-b 8 10.5 0.13 �1.46 �2.62+0.53
0.87 0.27 0.27+0.05 2.90
�0.83 �0.05
* Uncertainties are either the external reproducibility (2SD) or the calculated uncertainty limits resulting from the variability of the cross-contamination between Cr(III) and Cr(VI) pools, whichever was larger.
5738 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
chromium oxidation under the given conditions. In bothsets of experiments the oxidation of Cr(III) to Cr(VI) tookplace within seconds, as was visible through the accompa-nying colour change caused by the different Cr species in-volved in the reaction. The Cr(III) fractions of theseexperiments roughly plot along a Rayleigh-type functionwith a fractionation on the 53Cr/52Cr ratio of 103 � lna (i.e.�D53/52Cr(Cr(VI)–Cr(III))) of +0.6&, while the Cr(VI) frac-tions clearly plot far off the corresponding Rayleigh curvefor accumulated Cr(VI) (Table 3 and Fig. 5).
The best fit for the Cr(VI) seems to be a Rayleigh-typefunction with a fractionation on the 53Cr/52Cr ratio ofD53/52Cr(Cr(VI)–Cr(III)) of +0.2& (Fig. 5). Our experimentalresults cannot be explained by a single, unidirectional oxi-dation process between Cr(III) and Cr(VI). The reasonfor this may lie in the large, overall energy needed to oxidiseCr(III) to Cr(VI), Cr(III) oligomerisation inhibiting the oxi-dation process (Rao et al., 2002), and the formation of themeta-stable intermediate products Cr(IV) and/or Cr(V)(Ardon and Plane, 1959; Knoblowitz and Morrow, 1976;Banerjee and Nesbitt, 1999; Rao et al., 2002). Due to thecomplexity of this system and the lack of techniques that al-low the characterisation of Cr intermediates during the oxi-dation reaction, the kinetics and mechanisms of Cr(III)oxidation by hydrogen peroxide in alkaline milieu are still
not fully understood (Rao et al., 2002; Impert et al.,2008) (Fig. 4). The importance of the formation of interme-diate Cr(V) species during Cr(III) oxidation is, however,recognised by most authors as the rate-limiting reaction(Knoblowitz and Morrow, 1976; Impert et al., 2008). Thefinal oxidation of the meta-stable Cr(V) species to stablechromate is, in comparison, fast. A second, parallel reac-tion pathway involving Cr(IV) intermediates, which thendisproportionate to Cr(III) and Cr(VI), has also been de-scribed in the literature for alkaline media (Zhao et al.,1995) and at a pH of 4 during Cr(III) oxidation by the so-dium–manganese-oxide buserite (Silvester et al., 1995).Notably, a direct conversion of Cr(IV) to Cr(V) does nottake place during Cr(III) oxidation, based on kinetic rea-sons (Knoblowitz and Morrow, 1976). Under our moder-ately alkaline conditions and high H2O2 concentrationssmall amounts of Cr(VI) might have been reduced backto Cr(III) via Cr(V) intermediates (Vander Griend et al.,2002). It is likely that all these different oxidation andreduction reactions were accompanied by different fraction-ation factors for chromium isotopes.
In a second series of experiments (O2) the reaction kinet-ics of chromium oxidation in alkaline milieu was tested overa time period of up to 48 h. Thereby, the d53/52CrSRM979
values of the Cr(III) fractions significantly decrease with

Table 4Cr isotope data of oxidation experiments – influence of reaction time.
Series Sample pHValue
Time [h] fCr(III) d53/52CrSRM979 d53/52CrSRM979* fCr(VI) d53/52CrSRM979 d53/52CrSRM979
* D53/52Cr(Cr(VI)–Cr(III))
Cr(III) uncorrectedCr(III)
correctedCr(III)
Cr(VI) uncorrectedCr(VI)
correctedCr(VI)
corrected
O2 1a 10.5 0.1 0.33 �0.86 �1.04+0.07
0.67 0.45 0.46+0.05 1.51
�0.07 �0.05
O2 1b 10.5 0.1 0.32 �1.00 �1.21+0.08
0.68 0.47 0.49+0.05 1.70
�0.09 �0.05
O2 2a 10.5 2 0.23 �1.00 �1.37+0.14
0.77 0.46 0.47+0.05 1.84
�0.17 �0.05
O2 2b 10.5 2 0.27 �0.86 �1.08+0.08
0.73 0.29 0.30+0.05 1.38
�0.10 �0.05
O2 3a 10.5 6 0.19 �1.00 �1.41+0.16
0.81 0.18 0.18+0.05 1.59
�0.20 �0.05
O2 3b 10.5 6 0.19 �1.02 �1.39+0.15
0.81 0.05 0.06+0.05 1.44
�0.19 �0.05
O2 4a 10.5 24 0.30 �1.30 �1.60+0.11
0.70 0.59 0.61+0.05 2.21
�0.13 �0.05
O2 4b 10.5 24 0.28 �1.69 �2.11+0.16
0.72 0.65 0.67+0.05 2.78
�0.18 �0.05
O2 5a 10.5 48 0.21 �1.64 �2.27+0.25
0.79 0.53 0.54+0.05 2.81
�0.30 �0.05
O2 5b 10.5 48 0.22 �2.48 �3.36+0.34
0.78 0.78 0.80+0.05 4.16
�0.41 �0.05
* Uncertainties are either the external reproducibility (2SD) or the calculated uncertainty limits resulting from the variability of the cross-contamination between Cr(III) and Cr(VI) pools, whichever was larger.
Fig. 5. Results of the first set of two parallel Cr(III) oxidation experiments: corrected d53/52CrSRM979 values of both series (O1-a and O1-b)plotted as filled symbols. The open symbols show the uncorrected d53/52CrSRM979 values. The Cr(III) fractions of these experiments roughlyplot along a Rayleigh-type function with a fractionation on the 53Cr/52Cr ratio of 103 � lna (i.e. �D53/52Cr(Cr(VI)–Cr(III))) of +0.6& (stippledlines). The best fit for the Cr(VI) seems to be a Rayleigh-type function with a fractionation on the 53Cr/52Cr ratio of D53/52Cr(Cr(VI)–Cr(III)) of+0.2& (solid lines). All uncertainties on the d53/52CrSRM979 values are given in the figure unless they are smaller than the symbols.
Cr isotopic fractionation and reaction kinetics in aqueous media 5739
time whereas the d53/52CrSRM979 values of the correspond-ing Cr(VI) fractions reveal a small time-dependent increase(Table 4, Fig. 6).
This increase in chromium isotope fractionation withtime can be explained by slow, ongoing oxidation of Cr(III)to Cr(VI) and a possible back reduction of a small propor-tion of Cr(VI) to Cr(V) and/or Cr(III). Unfortunately, therelatively large uncertainties in the Cr(III) and Cr(VI) con-centration determinations of these experiments due to thecross-contamination during species separation (see Section
2.2.) do not allow the detection of a small, systematicincrease in Cr(VI) concentrations over time, as would be ex-pected under such circumstances. Therefore, an additionalCr(III) oxidation experiment in alkaline milieu was per-formed, but this time the Cr(VI) concentration was deter-mined photometrically by UV–VIS without previousseparation of Cr(VI) from Cr(III). In this experiment smallaliquots of 2.5 mL were taken from a single reaction solu-tion of 25 mL (60 lL H2O2 were added to 25 mL of 5 lmolCr(III) solution with a pH of 10.5) over a time period of

Fig. 6. Results of the second set of Cr(III) oxidation experiments (O2): (a) gives the d53/52CrSRM979 values of Cr(III) and Cr(VI) versus thereaction time. The two stippled lines describe the average reaction trends of the measured data-points. (b) Shows the Cr(VI) concentration inthe experimental dissolution, determined by photometric measurements in an additional experiment, versus reaction time. All uncertainties onthe d53/52CrSRM979 values are given in the figure unless they are smaller than the symbols.
5740 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
285 h and the Cr(VI) concentrations were directly measuredon equal factor (0.2 mmol L�1 Cr) dilutions of thesealiquots by UV–VIS. The results in Fig. 6b clearly demon-strate a significant increase in Cr(VI) concentration over areaction time of 285 h. This observation appears to be incontrast to the reaction kinetics of Cr reduction experi-ments (experiment R1, Table 1 and Fig. 2) where no timedependence of the reaction rates was observed. However,the results from these experiments support our assumptionthat the increasing Cr isotope fractionation between Cr(VI)and Cr(III) with time described above (experiments O2)might be the result of ongoing redox reactions of meta-sta-ble Cr(IV) and Cr(V) with Cr isotope fractionationsthrough both, oxidation with the respective a expected tobe >1 and small amounts of back-reduction with a expectedto be <1.
In a recent study, Frei et al. (2009) presented Cr isotopedata of banded iron formations (BIFs) that were depositedthroughout large parts of the Archaean, the Early Protero-zoic and the Late Neoproterozoic in order to investigate therise in atmospheric oxygen levels during these eras. All BIFsreported in this study have Cr isotope compositions that areeither within the range of high-temperature solid Earth res-ervoirs (Schoenberg et al., 2008) or heavier. Frei et al.(2009) suggest that the positive d53/52CrSRM979 values storedin some of the BIFs are the result of Cr isotope fraction-
ation caused by partial Cr(III) oxidation to Cr(VI) in ero-sion products and soils on land, using manganese oxidesas a reaction catalyst. The isotopically heavy Cr(VI) wassubsequently transported into the oceans. The fact thatthe Cr(VI) pools of our oxidation experiments are onlyslightly heavier in their Cr isotopic compositions than theoriginal Cr(III) source (see Tables 3 and 4 and Fig. 5) doesnot contradict the much larger isotope fractionations dur-ing Cr(III) oxidation by manganese oxides suggested bythe the BIF data reported by Frei et al. (2009), where LateNeoproterozoic samples have d53/52CrSRM979 values of upto +4.9&. Given the extreme conditions of our oxidationexperiments – a purely aquatic system with a pH of 10.5,using H2O2 as oxidising agent – the extent to which our re-sults can be directly translated into natural systems is atleast questionable. Preliminary Cr(III) oxidation experi-ments using the d-MnO2 birnessite revealed significant Crisotope fractionation (Bain and Bullen, 2005). However,the variability in d53/52CrSRM979 values of the developingCr(VI) pool between �2.5 and +0.7& of these experimentsalso points to the complexity of the Cr(III) oxidation pro-cess, involving the formation of unstable intermediatesCr(IV) and Cr(V) and their disproportionation, reportedin this study. We agree, in principle, with Frei et al.’s(2009) assessment of the global Cr cycling and the isotopicshifts involved with it. However, apart from the Cr isotope

Table 5Cr isotope data of isotope exchange experiments at a pH of 7.
Sample Time [h] d50/52CrCr(III),SRM979* d50/52CrCr(VI),SRM979
*
Series E1 at pH 7 with Cr(III):Cr(VI) of 1:2
1 0 913+46
2.9+5.7
�42 �5.7
2 2 918+48
11.4+5.3
�44 �5.4
3 4 922+50
16.0+5.1
�45 �5.2
4 7 916+47
12.8+5.4
�43 �5.5
5 10 925+51
18.1+5.0
�46 �5.1
6 13 929+53
14.3+5.0
�48 �5.0
7 24 915+47
7.6+5.5
�42 �5.6
8 36 923+51
8.3+5.2
�46 �5.3
9 48 913+47
12.4+5.4
�43 �5.4
10 72 914+48
11.5+5.4
�43 �5.4
11 96 923+52
21.3+4.9
�47 �4.9
12 120 923+52
14.3+5.0
�47 �5.0
13 530 870+31
24.2+7.1
�29 �7.2
14 1370 876+35
40.7+6.2
�32 �6.3
Series E3 at pH 7 with Cr(III):Cr(VI) of 2:1
1 0 868+10
8.0+22.2
�10 �23.4
2 2 876+10
48.4+19.1
�11 �20.0
3 4 876+11
67.1+17.5
�11 �18.3
4 7 874+10
42.7+20.0
�10 �21.0
5 10 868+11
72.5+17.3
�11 �18.1
6 13 877+11
55.3+17.9
�11 �18.6
7 24 876+10
25.9+20.8
�10 �21.8
8 36 877+10
23.3+20.3
�11 �21.3
9 48 866+10
38.4+20.2
�10 �21.2
10 72 873+10
35.5+20.2
�10 �21.2
11 96 877+11
69.1+17.8
�11 �18.5
12 120 876+11
109.2+15.0
�12 �15.6
13 530 810+8
124.6+16.8
�8 �17.6
14 1370 807+9
170.2+13.4
�9 �14.4
* Uncertainties are the calculated uncertainty limits resulting fromthe variability of the cross-contamination between Cr(III) andCr(VI) pools.
Table 6Cr isotope data of isotope exchange experiments at a pH of 5.5.
Sample Time [h] d50/52CrCr(III),SRM979* d50/52CrCr(VI),SRM979
*
Series E2 at pH 5.5 with Cr(III):Cr(VI) of 1:2
1 0 878+40 �0.6
+6.0�37 �6.1
2 2 876+39 �6.5
+6.2�36 �6.3
3 4 882+42 �3.2
+5.9�38 �5.9
4 7 881+42 �1.4
+5.9�38 �5.9
5 10 886+44
6.0+5.5
�40 �5.6
6 13 891+46
6.0+5.3
�42 �5.4
7 24 898+49
3.4+5.1
�44 �5.2
8 36 891+46
3.3+5.4
�42 �5.4
9 48 883+42
3.9+5.7
�39 �5.8
10 72 887+44
3.8+5.5
�40 �5.6
11 96 892+47
9.4+5.2
�42 �5.3
12 120 901+51
6.5+4.9
�46 �5.0
13 530 847+27 �8.9
+8.5�25 �8.6
14 1370 725+26 �4.0
+6.4�24 �6.5
Series E4 at pH 5.5 with Cr(III):Cr(VI) of 2:1
1 0 861+9 �8.1
+24.3�9 �25.7
2 2 863+9 �24.4
+26.3�9 �27.9
3 4 863+9 �15.6
+25.4�9 �26.9
4 7 863+9 �14.1
+25.3�9 �26.8
5 10 860+9
22.0+21.9
�9 �23.1
6 13 869+10
24.3+20.5
�10 �21.4
7 24 865+9
8.7+23.3
�9 �24.5
8 36 868+10
12.0+22.2
�10 �23.3
9 48 859+9
4.9+23.7
�9 �25.0
10 72 863+9
7.6+23.4
�9 �24.7
11 96 871+11
20.8+18.5
�12 �19.3
12 120 867+10
12.1+20.2
�11 �21.1
13 530 805+7 �29.4
+29.6�7 �31.8
14 1370 810+8
68.4+19.7
�8 �20.8
* Uncertainties are the calculated uncertainty limits resulting fromthe variability of the cross-contamination between Cr(III) andCr(VI) pools.
Cr isotopic fractionation and reaction kinetics in aqueous media 5741
5742 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
fractionation during oxidation with MnO2 in soils westrongly suggest that partial back reduction of the solubleCr(VI) to insoluble Cr(III) in soils and river beds duringtransport is a significant factor in enriching the residualCr(VI) that finally reaches the oceans in heavy Cr isotopes.
3.3. Isotopic exchange experiments
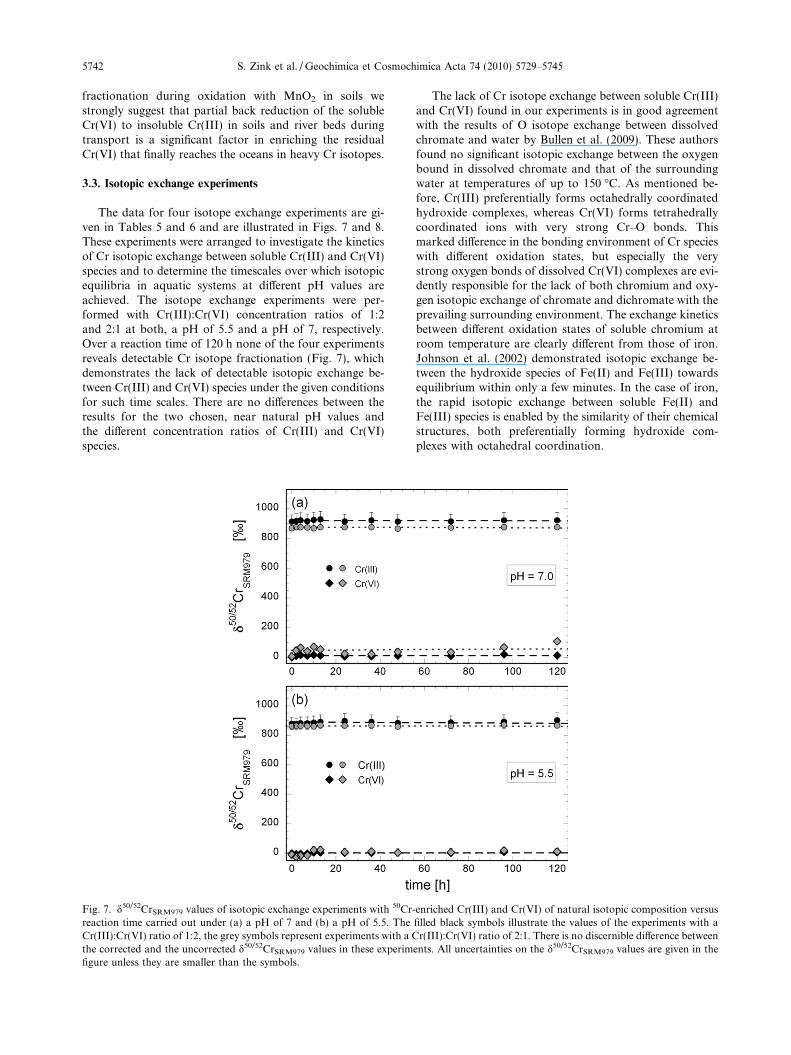
The data for four isotope exchange experiments are gi-ven in Tables 5 and 6 and are illustrated in Figs. 7 and 8.These experiments were arranged to investigate the kineticsof Cr isotopic exchange between soluble Cr(III) and Cr(VI)species and to determine the timescales over which isotopicequilibria in aquatic systems at different pH values areachieved. The isotope exchange experiments were per-formed with Cr(III):Cr(VI) concentration ratios of 1:2and 2:1 at both, a pH of 5.5 and a pH of 7, respectively.Over a reaction time of 120 h none of the four experimentsreveals detectable Cr isotope fractionation (Fig. 7), whichdemonstrates the lack of detectable isotopic exchange be-tween Cr(III) and Cr(VI) species under the given conditionsfor such time scales. There are no differences between theresults for the two chosen, near natural pH values andthe different concentration ratios of Cr(III) and Cr(VI)species.
Fig. 7. d50/52CrSRM979 values of isotopic exchange experiments with 50Cr-reaction time carried out under (a) a pH of 7 and (b) a pH of 5.5. TheCr(III):Cr(VI) ratio of 1:2, the grey symbols represent experiments with a Cthe corrected and the uncorrected d50/52CrSRM979 values in these experimfigure unless they are smaller than the symbols.
The lack of Cr isotope exchange between soluble Cr(III)and Cr(VI) found in our experiments is in good agreementwith the results of O isotope exchange between dissolvedchromate and water by Bullen et al. (2009). These authorsfound no significant isotopic exchange between the oxygenbound in dissolved chromate and that of the surroundingwater at temperatures of up to 150 �C. As mentioned be-fore, Cr(III) preferentially forms octahedrally coordinatedhydroxide complexes, whereas Cr(VI) forms tetrahedrallycoordinated ions with very strong Cr–O bonds. Thismarked difference in the bonding environment of Cr specieswith different oxidation states, but especially the verystrong oxygen bonds of dissolved Cr(VI) complexes are evi-dently responsible for the lack of both chromium and oxy-gen isotopic exchange of chromate and dichromate with theprevailing surrounding environment. The exchange kineticsbetween different oxidation states of soluble chromium atroom temperature are clearly different from those of iron.Johnson et al. (2002) demonstrated isotopic exchange be-tween the hydroxide species of Fe(II) and Fe(III) towardsequilibrium within only a few minutes. In the case of iron,the rapid isotopic exchange between soluble Fe(II) andFe(III) species is enabled by the similarity of their chemicalstructures, both preferentially forming hydroxide com-plexes with octahedral coordination.
enriched Cr(III) and Cr(VI) of natural isotopic composition versusfilled black symbols illustrate the values of the experiments with ar(III):Cr(VI) ratio of 2:1. There is no discernible difference between
ents. All uncertainties on the d50/52CrSRM979 values are given in the

Fig. 8. Results of the same isotopic exchange experiments as shown in Fig. 7, but with an extended time scale of up to eight weeks. Thechanges in the Cr isotopic composition visible for the steps with much longer reaction times are most likely due to precipitation of Cr(III) andadsorption effects of Cr(III) and Cr(VI).
Cr isotopic fractionation and reaction kinetics in aqueous media 5743
It is noteworthy that after several weeks visible coagula-tions of Cr(III)-hydroxides precipitated which results insmall, but significant changes in the Cr isotope composi-tions of the Cr(III) and Cr(VI) species.
The high Cr3+ concentrations, which were 5000 and10,000 times higher than the solubility of Cr(III) in water,were necessary to arrange the experiments in a simple setupwith manageable volumes and to allow the separation ofenough Cr(III) and Cr(VI) from a small aliquot of theexperimental solution for accurate Cr isotope analyses.Although precipitation of Cr(OH)3 at these moderate pHvalues takes place rapidly (Fricke and Windhausen, 1923;Rai et al., 1987), no visible coagulations were observedfor the first 120 h. We speculate that small isotope fraction-ation during precipitation of Cr(OH)3, adsorption effectsand contamination of the separated Cr(VI) with particulateCr(III), which did not wash out from the anion exchangeresin together with soluble Cr(III) during species separa-tion, are responsible for the small change in isotopic com-positions of Cr(III) and Cr(VI) complexes over extendedexperimental run times.
In natural settings like groundwater plumes and aquifersthe isotope exchange rates between Cr(III) and Cr(VI) willdepend on several parameters, such as the form in whichthe different Cr species are present, their concentration,the ambient temperature and the prevailing pH conditions.In a natural system contact times between Cr(III) andCr(VI) are certainly much longer than the timescale of days
to weeks tested in our experiments. Significant isotope ex-change may therefore take place in groundwater plumesand aquifers over the course of months to years.
4. SUMMARY AND CONCLUSIONS
The present study of isotope fractionation and reactionkinetics during chromium reduction and chromium oxida-tion with hydrogen peroxide and isotopic exchange of dis-solved Cr(VI) and Cr(III) has revealed following results:
– Reduction of Cr(VI) to Cr(III) with H2O2 at pH�1 is accompanied by a near-equilibrium isotopefractionation trend with a D53/52Cr(Cr(III)–Cr(VI)) of�3.54 ± 0.35&. This value is consistent with an earlierdetermination under pH neutral conditions, using naturalsediment and magnetite as reducing agents (Ellis et al.,2002). At pH neutrality, however, our experimental setup with peroxide as reducing agent reveals a kinetic iso-tope fractionation D53/52Cr(Cr(III)–Cr(VI)) of approximately�5& for reduction up to 85% Cr. The deviation from thiskinetic isotope fractionation for the extent of reactionhigher than 85% Cr might be the result of the formationand decomposition of peroxochromium(V) complexes.
– Isotope fractionation during oxidation of Cr(III) toCr(VI) in an alkaline milieu using H2O2 appears to besmall, favouring heavy Cr isotopes in the higher oxida-tion state. Our experimental results do not fit a simple

5744 S. Zink et al. / Geochimica et Cosmochimica Acta 74 (2010) 5729–5745
unidirectional Cr isotope fractionation model. We inter-pret our data of the oxidation experiments as the resultof the formation and dissimilation of one or both of theintermediates Cr(IV) and Cr(V).
– Our experiments reveal that there is no significant isoto-pic exchange between dissolved Cr(VI) and Cr(III) at apH between 5.5 and 7, at least over a reaction time ofdays to weeks. This result is consistent with the lack ofisotope exchange between oxygen bound in dissolvedchromate CrO4
2� and that of the surrounding water(Bullen et al., 2009).
Our results clearly confirm that the Cr isotope composi-tion of dissolved chromate in groundwater is a suitable tracerto assess the natural attenuation of anthropogenic Cr(VI)pollution along groundwater aquifers (Blowes, 2002; Elliset al., 2002; Izbicki et al., 2008; Berna et al., 2010). Thestrength of this tracer lies in the specific isotopic fractionationbehaviour during redox changes combined with the reactionkinetics between dissolved Cr(VI) and Cr(III). Large changesin the Cr isotopic composition of a specific anthropogenicchromate pollutant in the groundwater plume will almostexclusively be due to partial reduction of Cr(VI) to Cr(III),resulting in an isotope fractionation D53/52Cr(Cr(III)–Cr(VI))
of �3.4& or bigger. Partial back-oxidation of the dissolvedCr(III)-hydroxides by Mn-oxides in the soil is not expectedto cause large changes in the Cr isotope composition of theprevailing chromates (Izbicki et al., 2008). The very slow iso-tope exchange kinetics between dissolved Cr(III) and Cr(VI)species at near-neutral pH conditions, as determined in thisstudy, are also unlikely to cause significant changes in theCr isotope composition of the dissolved chromates ingroundwater, at least over time intervals of several weeks.
ACKNOWLEDGMENTS
We thank Alexandra Tangen for her support in the laboratory.Ingo Horn is thanked for maintaining the MC–ICP-MS during thecourse of this work. Robert Frei, two anonymous reviewers and theassociate editor James Farquhar are thanked for providingthoughtful and constructive comments that helped to significantlyimprove the quality of this manuscript. This project was financiallysupported by German Research Foundation (DFG) GrantSCHO1071/3-1 to R.S.
REFERENCES
Anbar A. (2004) Iron stable isotopes: beyond biosignatures. Earth
Planet. Sci. Lett. 217, 223–236.
Ardon M. and Plane R. A. (1959) The formation of a dinuclearCr(III) species by oxidation of chromous solutions. J. Am.
Chem. Soc. 81, 3197–3200.
Bain D. J. and Bullen T. D. (2005) Chromium isotope fractionationduring oxidation of Cr(III) by manganese oxides. Geochim.
Cosmochim. Acta 69, A212.
Ball J. and Bassett R. (2000) Ion exchange separation of chromiumfrom natural water matrix for stable isotope mass spectrometricanalysis. Chem. Geol. 168, 123–134.
Banerjee D. and Nesbitt H. (1999) Oxidation of aqueous Cr(III) atbirnessite surfaces: constraints on reaction mechanism. Geo-
chim. Cosmochim. Acta 63, 1671–1687.
Barnowski C (2001) Entwicklung und Untersuchung von Metho-den zur Speziation von Chrom in Umgebungsaerosolen, Uni-
versitat Dortmund, Ph.D. Thesis.Berna E. C., Johnson T. M., Makdisi R. S. and Basu A. (2010) Cr
stable isotopes as indicator of Cr(VI) reduction in groundwater:a detailed time-series study of a point-source plume. Environ.
Sci. Technol. 44, 1043–1048.
Blowes D. (2002) Tracking hexavalent Cr in groundwater. Science
295, 2024–2025.
Bullen T., Widory D. and Petelet-Giraud E. (2009) Multi-isotopeapproaches for identification of metal contamination sources inenvironmental systems. Geochim. Cosmochim. Acta 73, A173.
Broecker W. and Oversby V. (1971) Chemical Equilibria in the
Earth. McGraw-Hill, New York, 318 pp.Canfield D. E. (2001) Biogeochemistry of sulfur isotopes. Rev. Min.
Geochem. 43, 607–636.
Dauphas N. and Rouxel O. (2006) Mass spectrometry and naturalvariations of iron isotopes. Mass Spectron. Rev. 25, 831–832.
Detmers J., Bruchert V., Habicht K. S. and Kuever J. (2001)Diversity of sulfur isotope fractionations by sulfate-reducingprokaryotes. Appl. Environ. Microbiol. 67, 888–894.
Ellis A., Johnson T. and Bullen T. (2002) Chromium isotopes andthe fate of hexavalent chromium in the environment. Science
295, 2060–2062.
Ellis A. S., Johnson T. M. and Bullen T. D. (2004) Using chromiumstable isotope ratios to quantify Cr(VI) reduction: lack ofsorption effects. Environ. Sci. Technol. 38, 3604–3607.
Frei R., Gaucher C., Poulton S. W. and Canfield D. E. (2009)Fluctuations in Precambrian atmospheric oxygenation recordedby chromium isotopes. Nature 461, 250–254.
Fricke R. and Windhausen O. (1923) Uber die Alterung desChromhydroxydes, sowie uber Alkalichromite und ihre Losun-gen. Z. Anorg. Allg. Chem. 132, 273–288.
Funahashi S., Uchida F. and Tanaka M. (1978) Reactions ofhydrogen peroxide with metal complexes. 3. Thermodynamicand kinetic studies on the formation, dissociation, and decom-position of peroxochromium(VI) complexes in acid media.Inorg. Chem. 17, 2784–2789.
Gili P., Mederos A., Lorenzo-Luis P., De La Rosa E. and Muoz A.(2002) On the interaction of compounds of chromium(VI) withhydrogen peroxide. A study of chromium(VI) and (V) peroxidesin the acid–basic pH range. Inorg. Chim. Acta 331, 16–24.
Goetz A. and Heumann K. (1988) Chromium trace determinationin inorganic, organic and aqueous samples with isotope dilutionmass spectrometry. Fresenius Z. Anal. Chem. 331, 123–128.
Halicz L., Yang L., Teplyakov N., Burg A., Sturgeon R. andKolodny Y. (2008) High precision determination of chromiumisotope ratios in geological samples by MC–ICP-MS. J. Anal.
At. Spectrom. 23, 1622–1627.
Hayes J. M. (2001) Fractionation of carbon and hydrogen isotopesin biosynthetic processes. Rev. Min. Geochem. 43, 225–277.
Impert O., Katafias A., Kita P. and Woroniecka M. (2008) Kineticsof chromate(III) oxidation by hydrogen peroxide in alkalinesolutions – revisited. Polish J. Chem. 82, 1121–1126.
Izbicki J. A., Ball J. W., Bullen T. D. and Sutley S. J. (2008)Chromium, chromium isotopes and selected trace elements,western Mojave Desert, USA. Appl. Geochem. 23, 1325–1352.
Johnson C., Skulan J., Beard B., Sun H., Nealson K. and BratermanP. (2002) Isotopic fractionation between Fe(III) and Fe(II) inaqueous solutions. Earth Planet. Sci. Lett. 195, 141–153.
Knoblowitz M. and Morrow J. (1976) Kinetic study of anintermediate present in the hydrogen-peroxide oxidation ofchromium(III) to chromium(VI). Inorg. Chem. 15, 1674–1677.
Kotas J. and Stasicka Z. (2000) Chromium occurrence in theenvironment and methods of its speciation. Environ. Pollut. 107,
263–283.

Cr isotopic fractionation and reaction kinetics in aqueous media 5745
Mason T., Weiss D., Horstwood M., Parrish R., Russell S.,Mullane E. and Coles B. (2004) High-precision Cu and Znisotope analysis by plasma source mass spectrometry – Part 2.Correcting for mass discrimination effects. J. Anal. At. Spec-
trom. 19, 218–226.
McManus J., Berelson W., Severmann S., Poulson R., HammondD., Klinkhammer G. and Holm C. (2006) Molybdenum anduranium geochemistry in continental margin sediments:paleoproxy potential. Geochim. Cosmochim. Acta 70, 4643–
4662.
Moore P., Kettle S. and Wilkins R. (1966) The kinetics offormation of blue peroxychromic acid in aqueous solution.Inorg. Chem. 5, 466–467.
Nriagu J. and Nieboer E. (1988) Chromium in Natural and Human
Environments. Wiley Interscience, New York.Ohmoto H. and Lasaga A. C. (1982) Kinetics of reactions between
aqueous sulfates and sulfides in hydrothermal systems. Geo-
chim. Cosmochim. Acta 46, 1727–1745.
Orhanovic M. and Wilkins R. (1967) Kinetic studies of thereactions of peroxy compounds of chromium (VI), vanadium(V), and titanium (IV) in acid media. J. Am. Chem. Soc. 89,
278–282.
Palmer C and Puls R (1994) Natural attenuation of hexavalentchromium in groundwater and soils. EPA Ground Water Issue
EPA/540/5-94/505. pp. 1–12.Peel K., Weiss D., Chapman J., Arnold T. and Coles B. (2008) A
simple combined sample-standard bracketing and inter-elementcorrection procedure for accurate mass bias correction andprecise Zn and Cu isotope ratio measurements. J. Anal. At.
Spectrom. 23, 103–110.
Pettine M., Campanella L. and Millero F. (2002) Reduction ofhexavalent chromium by H2O2 in acidic solutions. Environ. Sci.
Technol. 36, 901–907.
Poulson R., Siebert C., McManus J. and Berelson W. (2006)Authigenic molybdenum isotope signatures in marine sedi-ments. Geology 34, 617–620.
Rai D., Eary L. and Zachara J. (1989) Environmental chemistry ofchromium. Sci. Total Environ. 86, 15–23.
Rai D., Sass B. M. and Moore D. A. (1987) Chromium(III)hydrolysis constants and solubility of chromium(III) hydroxide.Inorg. Chem. 26, 345–349.
Rao L., Zhang Z., Friese J., Ritherdon B., Clark S., Hess N. andRai D. (2002) Oligomerization of chromium(III) and its impacton the oxidation of chromium(III) by hydrogen peroxide inalkaline solutions. J. Chem. Soc. Dalton Trans. 36, 267–274.
Rees C. E. (1973) A steady-state model for sulphur isotopefractionation in bacterial reduction processes. Geochim. Cos-
mochim. Acta 37, 1141–1162.
Rouxel O., Bekker A. and Edwards K. (2005) Iron isotopeconstraints on the Archean and Paleoproterozoic ocean redoxstate. Science 307, 1088–1091.
Sander S. and Koschinsky A. (2000) Onboard-ship redox specia-tion of chromium in diffuse hydrothermal fluids from the NorthFiji Basin. Mar. Chem. 71, 83–102.
Sander S., Koschinsky A. and Halbach P. (2003) Redox speciationof chromium in the oceanic water column of the Lesser Antilles
and offshore Otago Peninsula, New Zealand. J. Mar. Freshwa-
ter Res. 54, 745–754.
Schauble E., Rossman G. and Taylor H. (2004) Theoreticalestimates of equilibrium chromium-isotope fractionations.Chem. Geol. 205, 99–114.
Schoenberg R. and von Blanckenburg F. (2005) An assessment ofthe accuracy of stable Fe isotope ratio measurements onsamples with organic and inorganic matrices by high-resolutionmulticollector ICP-MS. Int. J. Mass Spectrom. 242, 257–
272.
Schoenberg R., Zink S., Staubwasser M. and von Blanckenburg F.(2008) The stable Cr isotope inventory of solid Earth reservoirsdetermined by double spike MC–ICP-MS. Chem. Geol. 249,
294–306.
Siebert C., McManus J., Bice A., Poulson R. and Berelson W.(2006) Molybdenum isotope signatures in continental marginmarine sediments. Earth Planet. Sci. Lett. 241, 723–733.
Siebert C., Nagler T., von Blanckenburg F. and Kramers J. (2003)Molybdenum isotope records as a potential new proxy forpaleoceanography. Earth Planet. Sci. Lett. 211, 159–171.
Sikora E., Johnson T. and Bullen T. (2008) Microbial mass-dependent fractionation of chromium isotopes. Geochim. Cos-
mochim. Acta 72, 3631–3641.
Silvester E., Charlet L. and Manceau A. (1995) Mechanism ofChromium(III) Oxidation by Na-Buserite. J. Phys. Chem. 99,
16662–16669.
Van Niekerk W., Pienaar J., Lachmann G., Van Eldik R. andHamza M. (2007) A kinetic and mechanistic study of thechromium (VI) reduction by hydrogen peroxide in acidicaqueous solutions. Water SA 33, 619–625.
Vander Griend D., Golden J. and Arrington, Jr., C. (2002) Kineticsand mechanism of chromate reduction with hydrogen peroxidein base. Inorg. Chem. 41, 7042–7048.
Weiss D. J., Rehkdmper M., Schoenberg R., McLaughlin M.,Kirby J., Campbell P. G. C., Arnold T., Chapman J., Peel K.and Gioia A. S. (2008) Application of nontraditional stable-isotope systems to the study of sources and fate of metals in theenvironment. Environ. Sci. Technol. 42, 655–664.
Weyer S., Anbar A., Gerdes A., Gordon G., Algeo T. and Boyle E.(2008) Natural fractionation of U-238/U-235. Geochim. Cos-
mochim. Acta 72, 345–359.
Young E., Galy A. and Nagahara H. (2002) Kinetic and equilib-rium mass-dependent isotope fractionation laws in nature andtheir geochemical and cosmochemical significance. Geochim.
Cosmochim. Acta 66, 1095–1104.
Zhang L. and Lay P. (1998) EPR spectroscopic studies on theformation of chromium(v) peroxo complexes in the reaction ofchromium(vi) with hydrogen peroxide. Inorg. Chem. 37, 1729–
1733.
Zhao Z., Rush J., Holcman J. and Bielski B. (1995) The oxidationof chromium(III) by hydroxyl radical in alkaline solution – astopped-flow and pre-mix pulse radiolysis study. Radiat. Phys.
Chem. 45, 257–263.
Associate editor: James Farquhar
Copyright © 2022 FDOKUMEN




















