
Incorporation of primary patient-derived glycoproteins into authentic infectious hepatitis C virus...
13
Incorporation of Primary Patient-Derived Glycoproteins Into Authentic Infectious Hepatitis C Virus Particles Juliane Doerrbecker, 1 Martina Friesland, 1 Nina Riebesehl, 1 Corinne Ginkel, 1 Patrick Behrendt, 1,2 Richard J.P. Brown, 1 Sandra Ciesek, 1,2 Heiner Wedemeyer, 2 Christoph Sarrazin, 3 Lars Kaderali, 4 Thomas Pietschmann, 1 * and Eike Steinmann 1 * The Japanese fulminant hepatitis-1 (JFH1)-based hepatitis C virus (HCV) infection sys- tem has permitted analysis of the complete viral replication cycle in vitro. However, lack of robust infection systems for primary, patient-derived isolates limits systematic func- tional studies of viral intrahost variation and vaccine development. Therefore, we aimed at developing cell culture models for incorporation of primary patient-derived glycopro- teins into infectious HCV particles for in-depth mechanistic studies of envelope gene func- tion. To this end, we first constructed a packaging cell line expressing core, p7, and NS2 based on the highly infectious Jc1 genotype (GT) 2a chimeric genome. We show that this packaging cell line can be transfected with HCV replicons encoding cognate Jc1-derived glycoprotein genes for production of single-round infectious particles by way of trans- complementation. Testing replicons expressing representative envelope protein genes from all major HCV genotypes, we observed that virus production occurred in a genotype- and isolate-dependent fashion. Importantly, primary GT 2 patient-derived glycoproteins were efficiently incorporated into infectious particles. Moreover, replacement of J6 (GT 2a) core, p7, and NS2 with GT 1a-derived H77 proteins allowed production of infectious HCV particles with GT 1 patient-derived glycoproteins. Notably, adaptive mutations known to enhance virus production from GT 1a-2a chimeric genomes further increased virus release. Finally, virus particles with primary patient-derived E1-E2 proteins possessed biophysical properties comparable to Jc1 HCVcc particles, used CD81 for cell entry, were associated with ApoE and could be neutralized by immune sera. Conclusion: This work describes cell culture systems for production of infectious HCV particles with primary envelope protein genes from GT 1 and GT 2-infected patients, thus opening up new opportunities to dissect envelope gene function in an individualized fashion. (HEPATOLOGY 2014;60:508-520) G lobally, an estimated 160 million people are chronically infected with hepatitis C virus (HCV) 1 and are therefore at a high risk for developing severe liver disease including hepatic steatosis, fibrosis, cirrhosis, and hepatocellular carcinoma. 2 Due to error-prone RNA replication, HCV is a genetically highly variable virus and based on phylogenetic analyses viral isolates are grouped into seven genotypes, which differ between each other by more than 30% at the nucleotide level. 3 This genetic variability facilitates immune evasion and contributes to viral persistence. Importantly, genotype (GT)-dependent viral determi- nants influence both treatment response and the natural course of hepatitis C, since GTs 1 and 4 are more diffi- cult to treat with interferons compared to GTs 2 and 3 and as GT 3 is associated with liver steatosis. 4 Abbreviations: E-I, encephalomyocarditisvirus internal ribosomal entry site; HCV, hepatitis C virus; HCVcc, hepatitis C virus cell culture; HCVpp, hepatitis C virus pseudoparticles; HCV TCP , hepatitis C virus trans-complemented; GT, genotype; JFH1, Japanese fulminant hepatitis-1; P-I, poliovirus internal ribosomal entry site; qRT-PCR, quantitative real time-polymerase chain reaction; TCID 50 , tissue culture infectious dose 50. From the 1 Institute for Experimental Virology, Twincore, and Hannover Medical School Hannover, Germany, and Helmholtz Centre for Infection Research, Braunschweig, Germany; 2 Department of Gastroenterology, Hepatology and Endocrinology, Medical School Hannover, Germany; 3 Klinikum der Johann Wolfgang Goethe-Universit€ at, Medizinische Klinik 1, Germany; 4 Institute for Medical Informatics and Biometry, Medical Faculty, Technische Universit€ at Dresden, Germany. Received January 7, 2014; accepted April 23, 2014. E.S. was supported by the DFG (STE 1954/1-1) and an intramural young investigator award of the Helmholtz Centre for Infection Research. T.P. was sup- ported by grants from the DFG (PI 734/2-1) and the Helmholtz Association (SO-024) and the i-Med initiative. *These authors contributed equally. 508
-
Upload
mh-hannover -
Category
Documents
-
view
6 -
download
0
Transcript of Incorporation of primary patient-derived glycoproteins into authentic infectious hepatitis C virus...

Incorporation of Primary Patient-Derived GlycoproteinsInto Authentic Infectious Hepatitis C Virus Particles
Juliane Doerrbecker,1 Martina Friesland,1 Nina Riebesehl,1 Corinne Ginkel,1 Patrick Behrendt,1,2
Richard J.P. Brown,1 Sandra Ciesek,1,2 Heiner Wedemeyer,2 Christoph Sarrazin,3 Lars Kaderali,4
Thomas Pietschmann,1* and Eike Steinmann1*
The Japanese fulminant hepatitis-1 (JFH1)-based hepatitis C virus (HCV) infection sys-tem has permitted analysis of the complete viral replication cycle in vitro. However, lackof robust infection systems for primary, patient-derived isolates limits systematic func-tional studies of viral intrahost variation and vaccine development. Therefore, we aimedat developing cell culture models for incorporation of primary patient-derived glycopro-teins into infectious HCV particles for in-depth mechanistic studies of envelope gene func-tion. To this end, we first constructed a packaging cell line expressing core, p7, and NS2based on the highly infectious Jc1 genotype (GT) 2a chimeric genome. We show that thispackaging cell line can be transfected with HCV replicons encoding cognate Jc1-derivedglycoprotein genes for production of single-round infectious particles by way of trans-complementation. Testing replicons expressing representative envelope protein genes fromall major HCV genotypes, we observed that virus production occurred in a genotype- andisolate-dependent fashion. Importantly, primary GT 2 patient-derived glycoproteins wereefficiently incorporated into infectious particles. Moreover, replacement of J6 (GT 2a)core, p7, and NS2 with GT 1a-derived H77 proteins allowed production of infectiousHCV particles with GT 1 patient-derived glycoproteins. Notably, adaptive mutationsknown to enhance virus production from GT 1a-2a chimeric genomes further increasedvirus release. Finally, virus particles with primary patient-derived E1-E2 proteins possessedbiophysical properties comparable to Jc1 HCVcc particles, used CD81 for cell entry, wereassociated with ApoE and could be neutralized by immune sera. Conclusion: This workdescribes cell culture systems for production of infectious HCV particles with primaryenvelope protein genes from GT 1 and GT 2-infected patients, thus opening up newopportunities to dissect envelope gene function in an individualized fashion. (HEPATOLOGY
2014;60:508-520)
Globally, an estimated 160 million people arechronically infected with hepatitis C virus(HCV)1 and are therefore at a high risk for
developing severe liver disease including hepatic steatosis,fibrosis, cirrhosis, and hepatocellular carcinoma.2 Due toerror-prone RNA replication, HCV is a geneticallyhighly variable virus and based on phylogenetic analysesviral isolates are grouped into seven genotypes, which
differ between each other by more than 30% at thenucleotide level.3 This genetic variability facilitatesimmune evasion and contributes to viral persistence.Importantly, genotype (GT)-dependent viral determi-nants influence both treatment response and the naturalcourse of hepatitis C, since GTs 1 and 4 are more diffi-cult to treat with interferons compared to GTs 2 and 3and as GT 3 is associated with liver steatosis.4
Abbreviations: E-I, encephalomyocarditisvirus internal ribosomal entry site; HCV, hepatitis C virus; HCVcc, hepatitis C virus cell culture; HCVpp, hepatitis Cvirus pseudoparticles; HCVTCP, hepatitis C virus trans-complemented; GT, genotype; JFH1, Japanese fulminant hepatitis-1; P-I, poliovirus internal ribosomal entrysite; qRT-PCR, quantitative real time-polymerase chain reaction; TCID50, tissue culture infectious dose 50.
From the 1Institute for Experimental Virology, Twincore, and Hannover Medical School Hannover, Germany, and Helmholtz Centre for Infection Research,Braunschweig, Germany; 2Department of Gastroenterology, Hepatology and Endocrinology, Medical School Hannover, Germany; 3Klinikum der Johann WolfgangGoethe-Universit€at, Medizinische Klinik 1, Germany; 4Institute for Medical Informatics and Biometry, Medical Faculty, Technische Universit€at Dresden, Germany.
Received January 7, 2014; accepted April 23, 2014.E.S. was supported by the DFG (STE 1954/1-1) and an intramural young investigator award of the Helmholtz Centre for Infection Research. T.P. was sup-
ported by grants from the DFG (PI 734/2-1) and the Helmholtz Association (SO-024) and the i-Med initiative.*These authors contributed equally.
508

Unfortunately, primary patient-derived HCV replicatespoorly in cell culture, precluding in-depth assessment ofviral functions causing these differences.
The development of in vitro cell-culture systems,including subgenomic replicons for GT 1, 2 and morerecently for GT 3 and 4,5-8 HCV pseudoparticles,9,10
and ultimately the Japanese fulminant hepatitis-1(JFH1)-based infection system11-13 have alloweddetailed investigation of many aspects of the viral life-cycle and facilitated evaluation of antiviral com-pounds.14 More recently, additional GT 2 and GT 1infectious culture systems including JFH-2,15 J6cc,J8cc,16 H77-S,17 and TN18 have been reported. Finally,construction of JFH1-chimeras has permitted analysis ofvirus assembly and HCV entry across all HCV geno-types.11,19,20 However, these models are limited to dis-tinct, more or less representative clonal HCV sequenceswhich may not adequately reflect the entire spectrum offunctional traits of primary HCV in vivo.
Nevertheless, using these systems we and others haverecently reported HCV isolate-dependent use of claudinand occludin family members for infection, thus high-lighting strain-specific functional differences betweenHCV isolates during cell entry.21,22 These differences arecaused by the viral glycoproteins E1 and E2 that mediatereceptor interactions critical for cell entry and infectionof hepatocytes. In parallel, these viral proteins are themain targets for neutralizing antibodies that exert strongimmune pressure, thus rendering the glycoproteins mostvariable among all HCV proteins.3 However, to whatextent divergent glycoprotein function and HCV receptoruse contributes to viral immune escape and differentialcourse of infection between individuals is unknown.Therefore, to facilitate individualized studies of HCVglycoprotein function, in this work we aimed at develop-ing cell culture systems for assessment of functional traitsof patient-derived primary glycoproteins in the contextof authentic liver-cell derived HCV particles.
Materials and Methods
Ethics Statement. The study was approved by theEthics Committee of the Hannover Medical School.Patients provided written informed consent.
Cell Culture and Cell Lines. Huh-7.5, Huh-7.5.1,and 293T cells were cultured in Dulbecco’s modifiedminimal essential medium (DMEM, Life Technolo-gies) supplemented with 13 nonessential amino acids(Invitrogen), 100 lg/mL streptomycin (Invitrogen),100 IU/mL penicillin (Invitrogen), and 10% fetalbovine serum (DMEM complete) at 37�C and 5%CO2. Packaging cell lines Huh-7.5[C][p7NS2]J6,Huh-7.5[C][p7NS2]H77, Huh-7.5[C][p7NS2]H77/V787A, Huh-7.5.1[C][p7NS2]J6, and Huh-7.5.1[C][p7NS2]H77/V787A ectopically expressing viral pro-teins were created by lentiviral gene transfer asdescribed recently.23 Gene transfer involved vectorsencoding a blasticidin-S deaminase resistance gene,and therefore cells were selected by addition of blastici-din at a dose of 5 lg/mL.
Phylogenetic Analysis. E1E2 nucleotide alignmentswere generated using the ClustalX algorithm imple-mented in MEGA5.24 Nucleotide sequences werealigned according to encoded amino acid sequences, toensure maintenance of the overlying open readingframe. Alignments were manually adjusted and gap-stripped prior to phylogenetic reconstruction to avoiderroneous comparison of nonhomologous sites. Thegenotypes of recovered E1E2 sequences were con-firmed by phylogenetic analyses, using the maximumlikelihood method implemented in PhyML 3.0.25
Robustness of major phylogenetic groupings wasassessed by way of the bootstrap approach using 1,000replicates, and presented as percentages.
Plasmids and Viral Genomes. The pWPI derivatespWPI-core-J6_BLR, pWPI-core-H77_BLR, pWPI-p7NS2-J6_BLR, pWPI-p7NS2-H77_BLR pWPI-p7NS2-H77/V787A_BLR were generated as previouslydescribed.26 The plasmids pFK PI-EI-NS3-NS5B/JFH1and pFK PI-G-Luc-EI-NS3-NS5B/JFH1 have beendescribed elsewhere.23,26 The basic helper replicon vectorwas engineered with a poliovirus-derived internal ribo-some entry site (IRES) (designated P-I) downstream ofthe JFH1-derived 5-nontranslated region (50NTR)(nucleotides 1 to 341 of JFH1) and separated from theHCV 50NTR by a spacer region of 72 nucleotides. Thedesign of the Renilla luciferase reporter replicon isaccording to the R2a Jc1 construct described before.27 In
Address reprint requests to: PD Dr. Eike Steinmann or Prof. Dr. Thomas Pietschmann, Institute for Experimental Virology, TWINCORE, Centre for Experi-mental and Clinical Infection Research; a joint venture between the Medical School Hannover (MHH) and the Helmholtz Centre for Infection Research (HZI),Feodor-Lynen-Str. 7, 30625 Hannover, Germany. E-mail: [email protected] or [email protected]; fax: 149 2200 27139.
Copyright VC 2014 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.27190Potential conflict of interest: Dr. Pietschmann consults for Biotest and Janssen.
HEPATOLOGY, Vol. 60, No. 2, 2014 DOERRBECKER ET AL. 509

brief, the Renilla luciferase is fused to the NS3-NS5Bcoding region of JFH1 by way of the foot-and-mouthdisease virus 2A peptide coding region that liberates thereporter from the downstream HCV proteins. TheDCE1E2 genes of isolates H77 (GT 1a), Con1 (GT1b), J4 (GT 1b), JFH1 (GT 2a), J6CF (GT 2a), J8(GT 2b), S52 (GT 3a), ED43 (GT 4a), SA13 (GT 5a),Hk6a (GT 6a), QC69 (GT 7a), and plasma derived iso-lates were transferred into the first cistron of pFK PI-EI-NS3-NS5B/JFH1 by polymerase chain reaction (PCR)-based insertion. Further details regarding the cloningstrategies and exact nucleotide sequences can be obtainedupon request.
In Vitro Transcription, Transfection, and Produc-tion/Titration of Trans-Complemented HCV Parti-cles. HCV trans-complemented particles wereproduced as described previously.23 For infectious parti-cle quantification, virus-containing culture fluids wereharvested at indicated timepoints and filtered through a0.45-lm pore size filter. Viral infectivity of cell-freesupernatants was determined by infection of na€ıve Huh-7.5 target cells. Infectious particle amounts were quanti-fied by a slightly modified limiting dilution assay onna€ıve Huh-7.5 cells with the NS5A-specific antibodyE9E10 as described11 and tissue culture infectious dose50 (TCID50) was calculated.28 Determination of Renillaluciferase was performed as described before.29
Quantification of HCV RNA and Core Protein.Quantification of HCV RNA was performed asdescribed previously.30 HCV core protein content incell culture supernatants was measured with a commer-cially available diagnostic kit (Architect Anti-HCV,Abbott).
Western Blot Analysis. Immunoblot analysis wasperformed as described elsewhere.26 Primary antibodiesagainst core (C7-50), E2 (AP33), NS2 (6H6), andactin were used in a 1:1,000 dilution, except for theE2-specific antibody which was diluted 1:500.
Separation of Trans-Complemented HCV Particleson Density Gradient. Trans-complemented HCVparticles were analyzed by iodixanol step gradient asdescribed previously.29
Neutralization Assays. For quantification ofantibody-mediated neutralization, inhibition levels oftrans-complemented particle infection were analyzedby neutralization assay as described previously.13,30
Immunoglobulins were purified from sera of HCVpatients infected with GT 1 (Ab1a_1 and Ab1a_2) or2 (Ab2a_1) or healthy controls using the Thermo Sci-entific NAb Protein G Spin Kit. In addition, neutrali-zation assays were performed with the monoclonalantibody AR4A and the control antibody RO4.
Results
Construction of a Jc1-Based Packaging Cell Linefor Incorporation of Various Glycoproteins IntoSingle-Round Infectious HCV Particles. The HCVglycoproteins E1 and E2 are critically involved in virusassembly and cell entry. Retroviral pseudotypes carry-ing various E1-E2 proteins from cloned HCV consen-sus genomes or primary patient-derived isolates inplace of retroviral envelope proteins (HCVpp) havebecome a popular model to investigate HCV cellentry.9,10,31 However, HCVpp are produced in humannonliver cells and therefore lack a lipoprotein coat typ-ical for authentic, cell culture-derived HCV particles(HCVcc) originating from human liver cells. More-over, there are fundamental differences between cellentry of HCVpp and authentic HCVcc as, forinstance, the Niemann Pick C1-like 1 cholesterolreceptor (NPC1L1) is critical for HCVcc infection invitro and in vivo but dispensable for HCVpp infec-tion.32 On the other hand, currently available HCVccsystems do not permit functional testing of multipleE1-E2 genes and therefore are not suited for in-depthanalysis of functional traits of patient-derived envelopeprotein genes. To overcome these limitations we con-structed an HCV packaging cell line constitutivelyexpressing core, p7, and NS2 of the highly infectiousGT 2a chimera Jc1 and confirmed protein expressionby western blot19 (Fig. 1A). Once these cells are trans-fected with a subgenomic JFH1 replicon expressingE1-E2 genes in the first cistron, all viral factors areavailable for production of infectious viral progeny.Notably, the resulting particles which are formed byway of trans-complementation (HCV trans-comple-mented particles, HCVTCP
23), incorporate the repliconwhich lacks the coding regions for core, p7, and NS2.As a consequence, these HCVTCP are infectious foronly a single round since the aforementioned criticalviral assembly cofactors are not transduced to theinfected cells, thus precluding further viral spread.
To test if this cell line allows production of infec-tious HCVTCP with glycoproteins from all knownHCV genotypes, we cloned replicons encoding the E1-E2 genes of representative isolates from each viralgenotype and transfected these into the packaging cellline. As expected, all replicons propagated efficiently inthe transfected cell, as evidenced by the accumulationof abundant NS5A protein (Fig. 1B). Different levelsof E2 protein were detected, which likely reflects varia-tion of the epitope targeted by the AP33 monoclonalantibody used for protein analysis. In parallel, wedetermined the release of HCV core protein into the
510 DOERRBECKER ET AL. HEPATOLOGY, August 2014

culture fluid as marker for export of HCV particles(Fig. 1C). Notably, a modest level of core protein wasdetected upon transfection of the control repliconencoding a gaussia luciferase protein (G-Luc) insteadof E1-E2 (Fig. 1C). However, these core structuresreleased in the absence of viral glycoproteins were non-infectious, as evidenced by the limiting dilution assaydisplayed in Fig. 1D. In contrast, when repliconsencoding GT 1 through GT 7-derived E1-E2 proteins
were transfected, infectious progeny was producedexcept for the ED43 strain (GT 4a). J6 (GT 2a),JFH1 (GT 2a), and QC69 (GT 7a) E1-E2 proteinsyielded peak infectious titers above 105 TCID50/mLcorrelating with maximal release of core protein (Fig.1C,D). However, GT 1-derived E1-E2 proteins sup-ported only modest virus production, with peak virustiters more than 1,000-fold lower. Similarly, virus pro-duction in the presence of J8 (GT 2b), S52 (GT 3a),SA13 (GT 5a), and HK6a (GT 6a) was comparativelylow (Fig. 1D). Since all E1-E2 genes tested in the Jc1packaging cell line are derived from viral consensusgenomes proven to produce infectious HCV in cellculture and for some also in vivo, we can rule out thatdysfunctional E1-E2 proteins limited virus produc-tion.14 Notably, using chimeric HCV full-lengthgenomes expressing E1-E2 genes from different viralgenotypes we have reported that HCV envelope pro-teins support virus production in a genotype-dependent fashion.33 This is likely because E1-E2 pro-teins interact with other viral proteins in a genotype-specific manner.33 Therefore, incompatibility of E1-E2proteins with the remaining viral factors in the contextof the GT 2a Jc1-packaging cell line is likely due tolimited virus production from replicons expressing E1-E2 sequences highly divergent from the cognate J6-derived E1-E2 proteins.
Nevertheless, a modest level of infectious progenywas produced for most tested E1-E2 genes and highlyefficient virus release was observed for both GT 2astrains (JFH1 and J6) as well as a GT 7a isolate(QC69).
Fig. 1. Construction of a Jc1-based packaging cell line for incorpo-ration of various E1-E2 proteins into single-round infectious HCV par-ticles (HCVTCP). (A) Schematic representation of the Jc1 genome andthe bicistronic JFH1 replicon expressing E1-E2 genes in the first cis-tron. Viral sequences derived from the J6 and JFH1 strains aredepicted in dark and light gray, respectively and poliovirus-IRES (P-I)and encephalomyocarditisvirus-IRES (E-I) are given as black bars.Gene cassettes inserted into the packaging cell line by lentiviral genetransfer are indicated above the Jc1 genome. Expression of core andNS2 proteins in the packaging cell line was monitored by westernblot. Note that the packaging cell line does not express functional E1-E2 genes. These are introduced by transfection of replicons encodingE1-E2 genes derived from given molecular clones of HCV20 which rep-resent all HCV genotypes. A replicon expressing the gene encodinggaussia luciferase (G-Luc) was used as negative control for virus pro-duction. E2 and NS5A protein expression at 48 hours posttransfectionwas monitored by western blot (B). Release of core protein at thistimepoint was determined by a core-specific ELISA (C), while produc-tion of infectious progeny was determined 24, 48 and 72 hours post-transfection by using a limiting dilution assay (D). Mean values withstandard error for particle production and core release are given fromthree and two independent experiments, respectively. Background levelof the assay is shown in a dotted line.
HEPATOLOGY, Vol. 60, No. 2, 2014 DOERRBECKER ET AL. 511

Efficient Production of Infectious Particles WithPrimary GT 2-Infected Patient-Derived Glycopro-teins. These findings raised the hope that at least GT2a-derived E1-E2 proteins may be readily incorporatedonto infectious progeny in the background of the Jc1packaging cell line. To test this, E1-E2 sequences frompatient’s plasma were obtained by RT-PCR, clonedinto the bicistronic JFH1 helper replicon (Fig. 1A)and subsequently transfected into the Jc1 packagingcell line. Remarkably, four of five GT 2b-derived pri-mary E1-E2 genes produced infectious progeny withpeak titers ranging between 103 to 2 3 104 TCID50/mL (Fig. 2C). Moreover, three of five tested GT 2a-E1-E2 sequences yielded infectious virus between 2 3
104 to 2 3 105 TCID50/mL (Fig. 2C). A representa-tive immunofluorescence picture of infection efficiencywith H2a-4 and J6-derived glycoproteins is shown inSupporting Fig. S1. For those three sequences that didnot produce detectable infectious virus (H2b-3, H2a-1,and H2a-5), we observed intracellular E2 proteinexpression (Fig. 2A) and at least for two of themincreased liberation of HCV core protein, suggestingthat for these two, primarily noninfectious viruses werereleased (Fig. 2B). In contrast, core release upon trans-fection of H2a-1 was similar to the G-Luc control, sug-gesting that for this strain release of virus was notsupported. If failure to produce infectious virus in thesethree strains is due to amplification of nonfunctionalE1-E2 sequences circulating in vivo or due to introduc-tion of mutations by PCR is unknown. Nevertheless,our results show that the Jc1 packaging cell line permitsefficient incorporation of primary GT 2a- and GT 2b-derived E1-E2 protein sequences onto single-roundinfectious HCVTCP particles for the majority of primaryisolates (70%), rendering this model a powerful systemto explore functional traits of primary GT 2-derivedE1-E2 proteins in cell culture.
Construction and Characterization of an E1-E2Packaging System for GT 1 Patient-Derived Glyco-proteins. As depicted in Fig. 1D, the trans-comple-mentation efficiency of GT 1 glycoproteins was lowwith viral titers of about 102 TCID50/mL in the Jc1-based packaging system. To overcome this limitationand to also incorporate GT 1-derived patient isolatesin infectious particles, we aimed to develop a packag-ing system optimized for GT 1 E1-E2 sequences.Since virus production of H77 (GT 1a)-JFH1 full-length chimeric genomes was most efficient with ajunction between H77 and JFH1 after residue 842 ofthe H77 polyprotein directly downstream of the firsttransmembrane domain of NS2 was chosen, our GT1-packaging cells were constructed with this optimized
junction for intergenotypic chimeras.19 Moreover,long-term passage of H77-JFH1 full-length viruses hasled to identification of adaptive mutations that increasevirus production from these viruses. These are locatedwithin p734 (H77 V787A), and within NS334 (H77Q1247L). Finally, Kaul et al.35 reported that theV2440L exchange within the NS5A protein of JFH1facilitates virus release of several intergenotypic JFH1
Fig. 2. Efficient production of infectious particles with genotype 2patient-derived glycoproteins. JFH1-based replicons encoding givenpatient-derived E1-E2 genes were transfected into the Jc1-based pack-aging cell line lacking endogenous E1-E2 expression. A repliconencoding the J6-E1-E2 proteins or the gene encoding gaussia lucifer-ase (G-Luc) was transfected as positive or negative control, respec-tively. E2 and NS5A expression were monitored by western blot (A)and core release was quantified by a core-specific ELISA (B). Produc-tion of infectious progeny was determined by a limiting dilution assay(C). Mean values with standard error for particle production and corerelease are given from three and two independent experiments,respectively. Background level of the assay is shown in a dotted line.
512 DOERRBECKER ET AL. HEPATOLOGY, August 2014

chimeras. To find out which adaptive mutations wouldpermit optimal virus production for primary GT 1E1-E2 proteins, we created two distinct packaging celllines expressing H77 core, p7 with or without theadaptive mutation in p7 (Huh-7.5[C][p7NS2]H77/V787A and Huh-7.5[C][p7NS2]H77, respectively)and a chimeric H77/JFH1 NS2 protein (Fig. 3A).Moreover, three distinct replicons were constructedwhich all expressed the H77 E1-E2 proteins in thefirst cistron but encoded either a wild-type JFH1 repli-case (NS3-NS5B), a replicase with the NS3 Q1247Lmutation (NS3-NS5B/Q1247L), or a replicase withboth the NS3 and the NS5A mutation (NS3-NS5B/Q1247l1V2440L). Subsequently, viral protein expres-sion in the two different packaging cell lines was con-firmed by western blot (Fig. 3B) and the efficiency ofvirus production upon transfection of these repliconswas assessed by use of the limiting dilution assay (Fig.3C). Notably, transfection of the wild-type repliconwith H77 E1-E2 proteins into the packaging cell lineexpressing wild-type p7 yielded only a low virus titer(Fig. 3C, lane 1). In contrast, when the Q1247Lmutation was encoded by the replicon (Fig. 3C, lane3), �30-fold higher virus titers reaching up to 1 3
103 TCID50/mL were attained in both packaging celllines (with or without p7 mutation; lanes 3 and 5,respectively), indicating that the Q1247L mutation iscritical for production of HCVTCP with H77 envelopeproteins. In line with this notion, use of the repliconencoding both the NS3 and NS5A mutations did notfurther improve virus production (Fig. 3C, lane 8).Thus, in conclusion, at least for H77, the aforemen-tioned adaptive mutation in NS3 strongly increasesassembly of infectious HCVTCP production in H77-JFH1 packaging cells.
To explore if these mutations also facilitate produc-tion of infectious HCVTCP particles carrying primaryE1-E2 genes from GT 1a infected individuals wecloned six distinct E1-E2 sequences from GT 1a-infected patients and one from a GT 1b patient intoJFH1-based helper replicons. Note that we created E1-E2-encoding replicons without adaptive mutations andwith the Q1247L and V2440L mutations to explorethe impact of adaptive mutations on incorporation ofGT 1-derived E1-E2 proteins onto HCVTCP. Subse-quently, wild-type replicons were transfected into theJc1-based packaging cell line (Fig. 4A) or into thewild-type H77-packaging cell line (Fig. 4B), whereasthe adapted replicon was transfected into the adaptedH77-packaging cell line (Fig. 4C). Protein expressionas well as virus production was monitored as describedabove. Irrespective of which cell line was transfected
Fig. 3. Construction and characterization of E1-E2 protein packag-ing systems for GT 1 patient-derived glycoproteins. (A) Schematic rep-resentation of the chimeric H77/C3 genome19 and the bicistronicJFH1 replicon are given at the top. H77-derived sequences aredepicted in white and JFH1-derived sequences in light gray. Adaptivemutations are indicated by asterisks. H77 E1-E2-encoding repliconswith wild-type JFH1 sequence or with the Q1247L mutation, theV2440L mutation or with both adaptive changes were transfected intothe packaging cell lines encoding either wild-type H77 genes (Huh-7.5[C]p7NS2]H77) or V787A adapted H77 genes (Huh-7.5[C]p7NS2]H77/V787A). H77 transgene expression in the packag-ing cell lines upon lentiviral gene transfer was monitored by westernblot (B). Production of infectious progeny was monitored using the lim-iting dilution assay (C). Presence of adaptive mutations during virusproduction is indicated below each lane. Mean values with standarderror are given from three independent experiments. Background levelof the assay is shown in a dotted line.
HEPATOLOGY, Vol. 60, No. 2, 2014 DOERRBECKER ET AL. 513

and which replicon context was chosen, we observedsimilar expression of E1-E2 protein genes and NS5Aprotein, suggesting that the cellular and the repliconbackground neither grossly affected E1-E2 proteinexpression nor replicon propagation (Fig. 4, upperpanel). Notably, detection of E2 proteins was variablebetween patient isolates, which likely reflects variabilityof the epitope recognized by the AP33 antibody usedfor protein detection. Interestingly, release of core pro-tein observed upon transfection of each E1-E2 encod-ing adapted replicon into the adapted H77 packagingcell line was clearly higher when compared to the G-Luc-encoding control replicon, suggesting that each ofthese replicons supported production and release ofHCVTCP (Fig. 4C, middle panel). Moreover, in eachcase core release was higher compared to when therespective wild-type replicon was transfected into wild-type H77 packaging cells, suggesting that the adaptivechanges facilitated virus production for all tested GT1-derived E1-E2 sequences (Fig. 4, middle panel).Most important, production of infectious progeny wasachieved for 8 of 10 tested GT 1-derived E1-E2
sequences in the adapted GT 1a packaging system andwas between 10- to 100-fold higher compared to thewild-type H77 packaging and the Jc1-packaging sys-tem (Fig. 4, lower panel). A representative immunoflu-orescence picture of infection efficiency with H1a-6and H77-derived glycoproteins is shown in SupportingFig. S1.
Thus, in conclusion, by creating a custom-madeH77-JFH1-based packaging system including a fewadaptive mutations we were able to develop a robustcell culture system for production of authentic HCVparticles carrying functional GT 1 patient-derived E1-E2 sequences.
Phylogenetic Analysis of HCV Glycoproteins forProductive Trans-Complementation. E1E2 sequencesderived from standard laboratory strains representinggenotypes 1-7 were aligned with patient-derived E1E2glycoprotein sequences and used to generate a phyloge-netic reconstruction using the best-fit nucleotide substi-tution model (GTR1I1C) under the maximumlikelihood criterion (Fig. 5). As expected, patient-derived E1E2s clustered with reference sequences
Fig. 4. GT 1a-specific packaging system with adaptive mutations facilitates virus production with GT 1-patient-derived E1-E2 glycoproteins. (A)Wild-type JFH1-derived replicons expressing given E1-E2 sequences or gaussia luciferase were transfected into the Jc1 packaging cell line. Viralprotein expression was determined by western blot and ELISA assay (upper and middle panel, respectively), whereas virus production 72 hoursposttransfection was measured by a limiting dilution assay (lower panel). Alternatively, given wild-type JFH1 replicons were transfected into theGT 1a wild-type packaging cell line (B) or adapted JFH1 replicons were transfected into adapted GT 1a packaging cells (C). In both cases proteindetection and virus production was monitored as described in (A). Mean values with standard error for particle production and core release aregiven from three and two independent experiments, respectively. Background level of the assay is shown in a dotted line.
514 DOERRBECKER ET AL. HEPATOLOGY, August 2014

according to the viral genotype and therefore correlatedwith the observed genotype- and isolate-dependent effi-ciency in trans-complementation. Of note, the position-ing of GT 7 in the E1E2 phylogeny indicates it isgenetically more closely related to GT 2 than all othergenotypes (Fig. 5).
HCV Particles Bearing Patient-Derived Glycopro-teins Have Authentic Buoyant Densities and EnterTarget Cells in a CD81- and ApoE-Dependent Man-ner. To firmly establish that infectious particles carry-ing primary, patient-derived E1-E2 genes areauthentic, we compared CD81- and ApoE-dependenceof cell entry as well as buoyant density of released par-ticles between H77, J6 laboratory strain-derived andH1a-6 and H2a-4 patient-derived HCVTCP particles(Figs. 6, 7). As depicted in Fig. 6A,B, anti-CD81 anti-bodies reduced infectivity of all tested HCVTCP par-ticles to a similar degree. Moreover, ApoE-specificantibodies partially neutralized infection of all fourtested HCVTCP types (Fig. 6C,D). Finally, theseHCVTCP particles all shared the broad distribution ofviral RNA, core, and infectivity including low densityfractions that is typical for HCVcc particles andreflects their association with human lipoproteins (Fig.7). In summary, these results show that HCVTCP car-rying primary, patient-derived glycoproteins possesssimilar properties as authentic HCV infectious virions,
including use of CD81 for target cell entry, close asso-ciation with ApoE, and comparable buoyant densityprofiles.
Neutralization of Luciferase-Based ReporterHCVTCP With Patient-Derived Immunoglobulinsand Monoclonal Antibodies. To facilitate the detec-tion of infection events, we next generated reporterreplicons for incorporation of patient-derived GT 1-and GT 2- E1-E2 genes into reporter virus particles(Supporting Fig. S2A). To this end, we fused a Renillaluciferase gene to the N-terminus of the NS3-NS5Bcoding region by way of a foot-and-mouth diseasevirus 2A peptide sequence so that the reporter is liber-ated from the HCV polyprotein after translation.These novel reporter genomes replicated to high levelsin transfected Huh-7.5.1-based GT 2a (SupportingFig. S2B) and GT 1a (Supporting Fig. S2C) packagingcell lines, which were generated to enhance the effi-ciency of trans-complementation. Moreover, produc-tion of infectious particles was determined by a highlysensitive luciferase assay and was more efficient in theGT 2a packaging system compared to the GT 1aadapted packaging system, as observed before (Sup-porting Fig. S2D,E). Next, we analyzed the neutraliza-tion susceptibility of HCVTCP incorporating GT 2a(Fig. 8A-D) and GT 1a-derived glycoproteins (Fig.8E,F) to antibodies derived from genotype-matched
Fig. 5. Positioning of patient-derivedisolates within the E1E2 phylogeny.Unrooted radial tree for HCV E1E2 isdepicted. Bootstrap values assigned tointernal nodes indicate well-supportedbranches leading to the major HCVgenotypes/subtypes. Isolate namesare given at terminal nodes and geno-type 1 and 2 sequences are high-lighted in gray. E1E2 sequences, whichproduced functional virions in GT 1 orGT 2 trans-complementation assays,are denoted by � or *, respectively.Branch lengths are relative to the scalebar and are proportional to nucleotidesubstitutions per site.
HEPATOLOGY, Vol. 60, No. 2, 2014 DOERRBECKER ET AL. 515

patient sera and in case of GT 2a glycoproteins also tothe monoclonal antibody AR4A which broadly neutral-izes several HCV genotypes.36 Purified immunoglobu-lins from an HCV GT 2-infected patient (Ab2a_1)neutralized J6- as well as GT 2a-patient derived glyco-proteins in a dose-dependent manner (Fig. 8A). More-over, the AR4A monoclonal antibody inhibitedinfection of GT 2a reporter viruses, with the H2a-4-derived glycoproteins being somewhat more sensitiveto neutralization by this antibody compared to the J6-derived envelope proteins (Fig. 8C). In the same line,GT 1a-derived glycoproteins were neutralized by puri-fied antibodies from GT 1-infected patients (Ab1a_1and Ab1a_2) with slight differences between H77- andpatient isolate-based trans-complemented particles (Fig.8E,F), with no cytotoxicity observed (data not shown).In conclusion, primary patient-derived glycoproteinsincorporated into infectious particles can be neutral-ized by patient-derived immunoglobulins and mono-clonal antibodies and analyzed by a highly sensitivereporter assay.
Discussion
By exploiting the ability of HCV to incorporatesubgenomic replicons into single-round infectious par-ticles, in this study we developed cell culture systemsfor production of authentic, infectious HCV incorpo-rating primary patient-derived glycoproteins from GT1- and GT 2-infected individuals. Due to expressionof E1-E2 genes from a single gene cassette insertedinto autonomously replicating HCV replicons, poten-tial issues resulting from aberrant polyprotein process-ing are excluded and simple cloning of a large numberof patient-derived E1-E2 genes is facilitated. We feelthat these novel systems encompassing also reporter-based infection readouts are attractive models forstudying E1-E2 protein function in an individualizedfashion.
Importantly, several lines of evidence provided inthis study highlight that these particles recapitulate keyfeatures of authentic E1-E2 protein function both interms of virus assembly and cell entry. Specifically, we
Fig. 6. HCV particles carrying primary patient-derived E1-E2 proteins enter target cells in a CD81-dependent manner and are neutralized byApoE-specific antibodies. (A,B) HCVTCP harboring primary GT 1a- (A) or GT 2a-derived glycoproteins (B) were incubated with CD81-antibodies for1 hour at 37�C before inoculation of Huh-7.5 for 4 hours at 37�C. Infectivity was determined by focus forming unit assay. Mean values withstandard error are given from three independent experiments. (C,D) Experimental procedure as above in the presence of ApoE antibodies.
516 DOERRBECKER ET AL. HEPATOLOGY, August 2014

show that these particles infect Huh-7.5 cells in aCD81-dependent fashion and can be neutralized bypatient-derived antibodies or monoclonal antibodies.Moreover, neutralization of infection by ApoE-specificantibodies as well as the heterogeneous density distri-bution of cell-free HCVTCP indicate that these virusesincorporate host-derived lipoproteins. This latter find-ing is particularly relevant since HCVpp, which havebeen used previously for assessment of primarypatient-derived glycoprotein function, are produced in
a human kidney cell line (293T) that lacks endogenousexpression of human lipoproteins, thus confoundingthe usefulness of HCVpp for analysis of the interplaybetween E1-E2 proteins with lipoproteins. Moreover,HCVpp do not permit studies of glycoprotein func-tions during virus assembly since these particles areassembled in the absence of other HCV factors andlipoproteins. In contrast, HCVTCP, like full-lengthHCV, require expression of all viral structural andnonstructural proteins as well as apolipoproteins for
Fig. 7. Density gradient analysis of HCV particles carrying primary patient-derived E1-E2 proteins. Given HCVTCP with either GT 1a (H77 andH1a-6) or GT 2a (J6 and H2a-4) E1-E2 sequences were harvested 72 hours after transfection of Huh-7.5 packaging cells and were resolvedusing an iodixanol step gradient. Nine to 11 fractions were harvested from bottom to top and HCV infectivity (A,D), core protein levels (B,E), andRNA copy numbers (C,F) were determined for each fraction. Values are plotted against the density of the respective fraction measured by refrac-tometry. One representative experiment out of three independent experiments is shown.
HEPATOLOGY, Vol. 60, No. 2, 2014 DOERRBECKER ET AL. 517
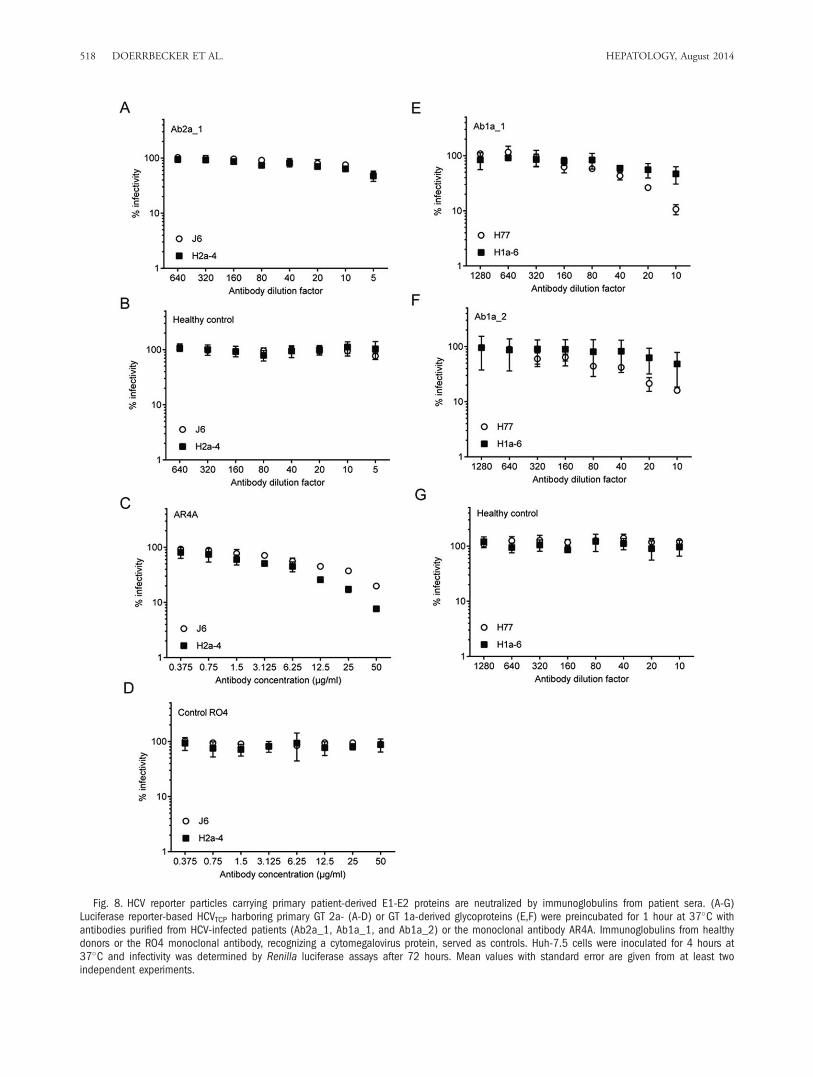
Fig. 8. HCV reporter particles carrying primary patient-derived E1-E2 proteins are neutralized by immunoglobulins from patient sera. (A-G)Luciferase reporter-based HCVTCP harboring primary GT 2a- (A-D) or GT 1a-derived glycoproteins (E,F) were preincubated for 1 hour at 37�C withantibodies purified from HCV-infected patients (Ab2a_1, Ab1a_1, and Ab1a_2) or the monoclonal antibody AR4A. Immunoglobulins from healthydonors or the RO4 monoclonal antibody, recognizing a cytomegalovirus protein, served as controls. Huh-7.5 cells were inoculated for 4 hours at37�C and infectivity was determined by Renilla luciferase assays after 72 hours. Mean values with standard error are given from at least twoindependent experiments.
518 DOERRBECKER ET AL. HEPATOLOGY, August 2014

release of infectious progeny.23,37 Moreover, we showthat E1-E2 genes are incorporated into HCVTCP in anisolate- and genotype-dependent fashion. This findingis consistent with our previous observation with chi-meric full-length genomes carrying E1-E2 genes fromisolates deviating from the other viral genes.33 In thisprevious work we could show that genetic incompati-bility of E1-E2 proteins with remaining viral factorsduring assembly limits envelopment of HCV nucleo-capsids likely due to inefficient protein-protein interac-tions between the glycoprotein complex and otherviral proteins.33 The results displayed in this presentstudy show that genetic compatibility of E1-E2 alsogoverns production of HCVTCP confirming that thissystem recapitulates authentic viral assembly pathways.
Clearly, genetic incompatibility of glycoproteins withviral factors derived from other HCV genomes restrictsuse of a single packaging cell line for incorporation ofE1-E2 proteins across all viral genotypes. Nevertheless,our data indicate that within a given HCV genotypeinsertion of E1-E2 genes from various isolates is welltolerated, as 70 to 80% of tested GT 2 and GT 1 iso-lates produced robust infectious titers in their cognatepackaging systems. Notably, the packaging system basedon the Jc1 chimera,19 which grows to very high titers incell culture,33 is particularly suited to produce highlyinfectious HCVTCP. This is likely attributable to specificfeatures of J6-derived core and p7 protein functionssupporting a high level of virus production.33 However,these beneficial properties of the J6 strain do not over-rule the requirement for intragenotypic compatibility ofthe E1-E2 sequences since the GT 1a packaging systemwith adaptive mutation rescues a greater proportion ofGT 1-derived E1-E2 isolates (80% instead of 70% forthe Jc1 packaging system) and consistently yields higherinfectious titers. Therefore, construction of custom-made packaging systems for each HCV genotype shouldbe a reasonable strategy to ultimately create cell culturesystems that permit assessment of functional E1-E2 pro-tein traits across all naturally occurring HCV strains.The infectious full-length JFH1-based chimeric HCVgenomes including distinct adaptive changes should bea reasonable starting point for these efforts.20
Ultimately, in-depth functional comparison betweenHCV E1-E2 protein sequences should shed furtherlight on the requirements for E1-E2 function andprotein-protein interactions during virus assembly.Moreover, these experiments may reveal unique fea-tures of the interplay between E1-E2 and lipoproteinsand host cell entry factors. Finally, these models shouldbe instrumental for exploring viral resistance to HCVentry inhibitors and ultimately for development of
vaccine candidates that elicit broadly protective cross-neutralizing antibodies.
Acknowledgment: We thank Takaji Wakita(National Institute of Infectious Diseases, Tokyo), JensBukh and Judith Gottwein (Copenhagen UniversityHospital, Copenhagen) for JFH1 and infectious chi-meric HCV constructs, respectively; Charles Rice(Rockefeller University, New York City) for Huh-7.5cells, 6H6, and E9E10 monoclonal antibody; DariusMoradpour (Centre Hospitalier Universitaire Vaudois,Lausanne) for the core-specific C7-50 antibody and toGENENTECH; Arvind Patel (University of Glasgow,Glasgow) for providing AP33; Frank Chisari (ScrippsResearch Institute, La Jolla, CA) for Huh-7.5.1 cells;Mansun Law (Scripps Research Institute, La Jolla, CA)for the AR4A antibody; Steven Foung (Stanford Uni-versity School of Medicine, Stanford, CA) for theRO4 antibody; Francois Penin (Institut de Biologie etChimie des Proteins) for helpful discussion; and allmembers of the Institute of Experimental Virology,Twincore, for helpful suggestions and discussions.
References
1. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis Cvirus infection. Lancet Infect Dis 2005;5:558-567.
2. Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. HEPATO-
LOGY 1997;26:15S-20S.3. Simmonds P. Genetic diversity and evolution of hepatitis C virus—15
years on. J Gen Virol 2004;85:3173-3188.4. Negro F. Hepatitis C virus-induced steatosis: an overview. Dig Dis
2010;28:294-299.5. Lohmann V, Korner F, Koch J, Herian U, Theilmann L,
Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs ina hepatoma cell line. Science 1999;285:110-113.
6. Saeed M, Gondeau C, Hmwe S, Yokokawa H, Date T, Suzuki T, et al.Replication of hepatitis C virus genotype 3a in cultured cells. Gastroen-terology 2013;144:56-58 e57.
7. Peng B, Yu M, Xu S, Lee YJ, Tian Y, Yang H, et al. Development ofrobust hepatitis C virus genotype 4 subgenomic replicons. Gastroenter-ology 2013;144:59-61 e56.
8. Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, MizokamiM, et al. Efficient replication of the genotype 2a hepatitis C virus sub-genomic replicon. Gastroenterology 2003;125:1808-1817.
9. Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C viruspseudo-particles containing functional E1-E2 envelope protein com-plexes. J Exp Med 2003;197:633-642.
10. Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM,et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entryof pseudotyped retroviral particles. Proc Natl Acad Sci U S A 2003;100:7271-7276.
11. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, LiuCC, et al. Complete replication of hepatitis C virus in cell culture. Sci-ence 2005;309:623-626.
12. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z,et al. Production of infectious hepatitis C virus in tissue culture from acloned viral genome. Nat Med 2005;11:791-796.
13. Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR,et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci US A 2005;102:9294-9299.
HEPATOLOGY, Vol. 60, No. 2, 2014 DOERRBECKER ET AL. 519

14. Steinmann E, Pietschmann T. Cell culture systems for hepatitis C virus.Curr Top Microbiol Immunol 2013;369:17-48.
15. Date T, Kato T, Kato J, Takahashi H, Morikawa K, Akazawa D, et al.Novel cell culture-adapted genotype 2a hepatitis C virus infectiousclone. J Virol 2012;86:10805-10820.
16. Li YP, Ramirez S, Gottwein JM, Scheel TK, Mikkelsen L, Purcell RH,et al. Robust full-length hepatitis C virus genotype 2a and 2b infectiouscultures using mutations identified by a systematic approach applicableto patient strains. Proc Natl Acad Sci U S A 2012;109:E1101-1110.
17. Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. Production ofinfectious genotype 1a hepatitis C virus (Hutchinson strain) in culturedhuman hepatoma cells. Proc Natl Acad Sci U S A 2006;103:2310-2315.
18. Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. Highlyefficient full-length hepatitis C virus genotype 1 (strain TN) infectiousculture system. Proc Natl Acad Sci U S A 2013;109:19757-19762.
19. Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S,Steinmann E, et al. Construction and characterization of infectiousintragenotypic and intergenotypic hepatitis C virus chimeras. Proc NatlAcad Sci U S A 2006;103:7408-7413.
20. Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC,Knudsen ML, et al. Development and characterization of hepatitis Cvirus genotype 1-7 cell culture systems: role of CD81 and scavengerreceptor class B type I and effect of antiviral drugs. HEPATOLOGY 2009;49:364-377.
21. Haid S, Grethe C, Dill MT, Heim M, Kaderali L, Pietschmann T. Iso-late-dependent use of Claudins for cell entry by hepatitis C virus.HEPATOLOGY 2014;59:24-34.
22. Sourisseau M, Michta ML, Zony C, Israelow B, Hopcraft SE, NarbusCM, et al. Temporal analysis of hepatitis C virus cell entry with occlu-din directed blocking antibodies. PLoS Pathog 2013;9:e1003244.
23. Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T.Efficient trans-encapsidation of hepatitis C virus RNAs into infectiousvirus-like particles. J Virol 2008;82:7034-7046.
24. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S.MEGA5: molecular evolutionary genetics analysis using maximum like-lihood, evolutionary distance, and maximum parsimony methods. MolBiol Evol 2011;28:2731-2739.
25. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, GascuelO. New algorithms and methods to estimate maximum-likelihood phy-logenies: assessing the performance of PhyML 3.0. Syst Biol 2010;59:307-321.
26. Brohm C, Steinmann E, Friesland M, Lorenz IC, Patel A, Penin F,et al. Characterization of determinants important for hepatitis C virusp7 function in morphogenesis by using trans-complementation. J Virol2009;83:11682-11693.
27. Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, et al.Recruitment and activation of a lipid kinase by hepatitis C virus NS5A
is essential for integrity of the membranous replication compartment.Cell Host Microbe 2011;9:32-45.
28. Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M,Steinmann J, et al. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. HEPATOLOGY 2011;54:1947-1955.
29. Anggakusuma, Colpitts CC, Schang LM, Rachmawati H, Frentzen A,Pfaender S, et al. Turmeric curcumin inhibits entry of all hepatitis Cvirus genotypes into human liver cells. Gut 2013 [Epub ahead ofprint].
30. Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M,Herrmann E, et al. Hepatitis C virus hypervariable region 1 modulatesreceptor interactions, conceals the CD81 binding site, and protectsconserved neutralizing epitopes. J Virol 2010;84:5751-5763.
31. Maurin G, Fresquet J, Granio O, Wychowski C, Cosset FL, LavilletteD. Identification of interactions in the E1E2 heterodimer of hepatitisC virus important for cell entry. J Biol Chem 2011;286:23865-23876.
32. Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, HussainS, et al. Identification of the Niemann-Pick C1-like 1 cholesterolabsorption receptor as a new hepatitis C virus entry factor. Nat Med2012;18:281-285.
33. Steinmann E, Doerrbecker J, Friesland M, Riebesehl N, Ginkel C,Hillung J, et al. Characterization of hepatitis C virus intra- and inter-genotypic chimeras reveals a role of the glycoproteins in virus envelop-ment. J Virol 2013;87:13297-13306.
34. Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, AlterHJ, et al. Development of JFH1-based cell culture systems for hepatitisC virus genotype 4a and evidence for cross-genotype neutralization.Proc Natl Acad Sci U S A 2008;105:997-1002.
35. Kaul A, Woerz I, Meuleman P, Leroux-Roels G, Bartenschlager R. Cellculture adaptation of hepatitis C virus and in vivo viability of anadapted variant. J Virol 2007;81:13168-13179.
36. Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, et al.Human broadly neutralizing antibodies to the envelope glycoproteincomplex of hepatitis C virus. Proc Natl Acad Sci U S A 2012;109:6205-6210.
37. Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A,Doerrbecker J, et al. Apolipoprotein E co-determines tissue-tropism ofhepatitis C virus and it is crucial for viral cell-to-cell transmission bycontributing to a post-envelopment step of assembly. J Virol 2013.
Supporting Information
Additional Supporting Information may be foundin the online version of this article at the publisher’swebsite.
520 DOERRBECKER ET AL. HEPATOLOGY, August 2014




















