
How accurate are electronic structure methods for actinoid chemistry
37
REGULAR ARTICLE How accurate are electronic structure methods for actinoid chemistry? Boris B. Averkiev • Manjeera Mantina • Rosendo Valero • Ivan Infante • Attila Kovacs • Donald G. Truhlar • Laura Gagliardi Received: 2 December 2010 / Accepted: 16 February 2011 / Published online: 8 March 2011 Ó Springer-Verlag 2011 Abstract The CASPT2, CCSD, and CCSD(T) levels of wave function theory and seven density functionals were tested against experiment for predicting the ionization potentials and bond dissociation energies of actinoid monoxides and dioxides with their cations. The goal is to guide future work by enabling the choice of an appropriate method when performing calculations on actinoid-con- taining systems. We found that four density functionals, namely MPW3LYP, B3LYP, M05, and M06, and three levels of wave function theory, namely CASPT2, CCSD, and CCSD(T), give similar mean unsigned errors for acti- noid–oxygen bond energies and for ionization potentials of actinoid oxides and their cations. Keywords Actinoids Á Electronic structure Á Density functional theory Á CASSCF Á CASPT2 1 Introduction Actinoid chemistry is important for reactor waste man- agement [1–3], and actinoids serve as catalysts for a variety of reactions [4–7], but understanding actinoid chemistry poses formidable challenges to both experimental and computational chemists. From a computational point of view, in order to be able to treat actinoid chemistry, we need electronic structure methods capable of properly describing both electron correlation and relativistic effects in systems that often have large multireference character. Many electronic structure studies have appeared in the literature, involving both wave function theory (WFT) and density functional theory (DFT). The complete active space (CAS) self-consistent field (SCF) method [8] followed by multireference second-order perturbation theory (CASPT2) [9] and DFT with the original B3LYP [10–13] density functional (which is based on the VWN3 approximation for the local spin density component) were tested against experiment for the prediction of bond distances and vibrational frequencies in uranium triatomic molecules and cations [14, 15]. Successively, the CASPT2 [9], relativistic coupled cluster calculations with single and double exci- tations (CCSD) [17, 18], and relativistic coupled cluster calculations with single and double excitations and Dedicated to Professor Pekka Pyykko ¨ on the occasion of his 70th birthday and published as part of the Pyykko ¨ Festschrift Issue. Electronic supplementary material The online version of this article (doi:10.1007/s00214-011-0913-0) contains supplementary material, which is available to authorized users. B. B. Averkiev Á M. Mantina Á R. Valero Á D. G. Truhlar Á L. Gagliardi (&) Department of Chemistry and Supercomputing Institute, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455-0431, USA e-mail: [email protected] D. G. Truhlar e-mail: [email protected] I. Infante Kimika Fakultatea, Euskal Herriko Unibertsitatea and Donostia International Physics Center (DIPC), P.K. 1072, 20080 Donostia, Euskadi, Spain A. Kovacs European Commission, Joint Research Centre, Institute for Transuranium Elements, P.O. Box 2340, 76125 Karlsruhe, Germany A. Kovacs Research Group for Materials Structure and Modeling of the Hungarian Academy of Sciences, Budapest University of Technology and Economics, Szt. Gelle ´rt te ´r 4, 1111 Budapest, Hungary 123 Theor Chem Acc (2011) 129:657–666 DOI 10.1007/s00214-011-0913-0
-
Upload
metaaprendizaje -
Category
Documents
-
view
1 -
download
0
Transcript of How accurate are electronic structure methods for actinoid chemistry

REGULAR ARTICLE
How accurate are electronic structure methods for actinoidchemistry?
Boris B. Averkiev • Manjeera Mantina •
Rosendo Valero • Ivan Infante • Attila Kovacs •
Donald G. Truhlar • Laura Gagliardi
Received: 2 December 2010 / Accepted: 16 February 2011 / Published online: 8 March 2011
� Springer-Verlag 2011
Abstract The CASPT2, CCSD, and CCSD(T) levels of
wave function theory and seven density functionals were
tested against experiment for predicting the ionization
potentials and bond dissociation energies of actinoid
monoxides and dioxides with their cations. The goal is to
guide future work by enabling the choice of an appropriate
method when performing calculations on actinoid-con-
taining systems. We found that four density functionals,
namely MPW3LYP, B3LYP, M05, and M06, and three
levels of wave function theory, namely CASPT2, CCSD,
and CCSD(T), give similar mean unsigned errors for acti-
noid–oxygen bond energies and for ionization potentials of
actinoid oxides and their cations.
Keywords Actinoids � Electronic structure � Density
functional theory � CASSCF � CASPT2
1 Introduction
Actinoid chemistry is important for reactor waste man-
agement [1–3], and actinoids serve as catalysts for a variety
of reactions [4–7], but understanding actinoid chemistry
poses formidable challenges to both experimental and
computational chemists. From a computational point of
view, in order to be able to treat actinoid chemistry, we
need electronic structure methods capable of properly
describing both electron correlation and relativistic effects
in systems that often have large multireference character.
Many electronic structure studies have appeared in the
literature, involving both wave function theory (WFT) and
density functional theory (DFT). The complete active space
(CAS) self-consistent field (SCF) method [8] followed by
multireference second-order perturbation theory (CASPT2)
[9] and DFT with the original B3LYP [10–13] density
functional (which is based on the VWN3 approximation for
the local spin density component) were tested against
experiment for the prediction of bond distances and
vibrational frequencies in uranium triatomic molecules and
cations [14, 15]. Successively, the CASPT2 [9], relativistic
coupled cluster calculations with single and double exci-
tations (CCSD) [17, 18], and relativistic coupled cluster
calculations with single and double excitations and
Dedicated to Professor Pekka Pyykko on the occasion of his 70th
birthday and published as part of the Pyykko Festschrift Issue.
Electronic supplementary material The online version of thisarticle (doi:10.1007/s00214-011-0913-0) contains supplementarymaterial, which is available to authorized users.
B. B. Averkiev � M. Mantina � R. Valero �D. G. Truhlar � L. Gagliardi (&)
Department of Chemistry and Supercomputing Institute,
University of Minnesota, 207 Pleasant St. SE, Minneapolis,
MN 55455-0431, USA
e-mail: [email protected]
D. G. Truhlar
e-mail: [email protected]
I. Infante
Kimika Fakultatea, Euskal Herriko Unibertsitatea
and Donostia International Physics Center (DIPC),
P.K. 1072, 20080 Donostia, Euskadi, Spain
A. Kovacs
European Commission, Joint Research Centre,
Institute for Transuranium Elements,
P.O. Box 2340, 76125 Karlsruhe, Germany
A. Kovacs
Research Group for Materials Structure and Modeling
of the Hungarian Academy of Sciences,
Budapest University of Technology and Economics,
Szt. Gellert ter 4, 1111 Budapest, Hungary
123
Theor Chem Acc (2011) 129:657–666
DOI 10.1007/s00214-011-0913-0

quasiperturbative treatment of connected triple excitations
(CCSD(T)) [19, 20] levels of WFT were tested against
experiment for ionization potentials of actinoid monoxides
and dioxides [16]. In the current paper, we present tests for
a larger set of experimental bond dissociation energies in
which we include actinoid dioxides and their cations and
dications, and, in addition, we test newer density func-
tionals. In particular, the MPW3LYP [21], MOHLYP [22],
M05 [23], M06-L [24], and M06 [25] density functionals
are tested against both some of the previous [14, 16] data
and all of the new test sets. We also checked the perfor-
mance of an additional older functional, namely the PW91
[26] functional, for these systems, because it was shown
[27] that it performs well for vibrational frequencies of the
triatomic molecules NUN, NUO?, and CUO. In related
work, Austin et al. [28] tested the B3LYP, M05, M06-L,
and M06 density functionals and two others against
experiment and compared them with CCSD(T) and CAS-
PT2 results for actinoid reaction energies; they found that
‘‘the M06 functional definitely stands out as being more
successful than the other five functionals tested’’ and ‘‘is
competitive with high-level ab initio methods’’ for these
reaction energies. The present tests should be particularly
useful in that all density functionals and WFT methods
mentioned so far are compared to the same sets of data,
allowing a consistent comparison.
2 Methods
In the following, we will describe the details of the WFT-
based calculations followed by the details of the DFT
calculations.
2.1 WFT calculations of bond dissociation energies
Calculations were performed using CASSCF and CASPT2.
Scalar relativistic effects were included using the Douglas-
Kroll-Hess Hamiltonian to second order [29, 30]. For these
calculations, we used a relativistic ANO-RCC basis set of
triple zeta quality [31, 32], as implemented in the MOL-
CAS 7.4 package [33].
For actinoid monoxides AnO (where An denotes an
actinoid, in particular, Th, Pa, U, Np, Pu, Am, or Cm), a
full valence active space includes 17 orbitals, in particular
13 from the actinoid and four from the oxygen. In the
CASSCF calculations, we used an active space of 16
orbitals obtained by omitting the 2s orbitals of oxygen. For
monoxides, the largest number of electrons in these active
space orbitals is 14 for CmO. In the subsequent CASPT2
calculations, the orbitals up to the 5d of the actinoid and the
1s of oxygen were kept frozen, while the remaining
valence and semi-valence orbitals, now including the
6s and 6p of the actinoid and the 2s of oxygen, were cor-
related. The full valence active space of actinoid dioxides
AnO2 contains 21 orbitals, but calculations with a complete
active space this large are not feasible, so the 2s orbitals of
oxygen, the two lowest 6dpg bonding orbitals, the corre-
sponding antibonding orbitals, and one rg* antibonding
orbital are treated as inactive. Thus, the active space is
composed of 14 orbitals, occupied primarily by the 5f, 6d,
and 7s orbitals of the actinoid atom and the 2p orbitals of
the oxygen atoms. By applying the same truncation along
the whole series, the largest active space in all our calcu-
lations contains 14 electrons in 14 orbitals for CmO2.
Spin–orbit coupling was included using the complete-
active-space state-interaction method, CASSI [34, 35],
which employs an effective one-electron spin–orbit (SO)
Hamiltonian, based on a mean field approximation to the
two-electron contribution [36]. Note that a CASSI calcu-
lation, like a CASSCF calculation, does not include
dynamical correlation. To include this, the spin-free
CASPT2 energies are used as diagonal matrix elements in
the CASSI treatment; this is called spin–orbit-corrected
CASPT2 (SO-CASPT2).
To assist in judging the validity of the CASPT2 results,
we performed exact 2-component (X2C) [37, 38] relativ-
istic calculations at the CCSD and CCSD(T) levels using
the DIRAC08 program [39]. Note that X2C is also called
infinite-order two-component (IO2C). In these coupled
cluster calculations, we correlated the electrons in the 6s,
6p, 5f, 6d, and 7s orbitals of the actinoid atom and the four
valence electrons of the 2p orbitals of the oxygen. We then
restricted the set of virtual orbitals to those most important
for valence and sub-valence correlation by deleting virtual
orbitals with an orbital energy above 10 Eh.
Note that Dyall’s transformation [38] allows one to
separate a four-component relativistic treatment into spin-
free and spin–orbit contributions. The X2C transformation
[37] to a two-component treatment that is used here is
based only on the spin-free part, i.e., we use the Dirac-
Coulomb operator without the Breit spin–orbit term.
Therefore, we add the spin–orbit contribution to the close
coupling calculations as the difference between the
SO-CASPT2 result and the spin-free CASPT2 result.
For the CCSD and CCSD(T) calculations, we used an
uncontracted double-zeta 26s23p17d13f2g basis set [40]
for actinoid atoms and the cc-pVTZ basis set [41] for
oxygen atoms.
The geometries for all WFT calculations are SO-CAS-
PT2 geometries. (Please note that the statement in [16] that
some geometries are spin-free CASPT2 is a typo.) The
zero-point vibrational energy (ZPE) change for the WFT
calculations was taken to be the same as in the M06 cal-
culations (calculated from scaled M06 frequencies as
described in Sect. 2.3).
658 Theor Chem Acc (2011) 129:657–666
123
2.2 WFT calculations of ionization potentials
The WFT calculations of ionization potentials (IPs) are
updated in two respects from those published [16] previ-
ously. Previously, PuO2, PuO2?, and PuO2
2? were com-
puted by RASPT2 [42, 43]; in the present article, all PT2
results are CAS, not RAS. Previously, the change in ZPE
was limited to the symmetric stretching mode; here, a full
ZPE correction is made, based on DFT, as mentioned at the
end of Sect 2.1.
2.3 DFT calculations
For ionization potentials and bond dissociation energies of
actinoid monoxides and dioxides, DFT calculations were
performed with the relativistic MWB60 small-core effec-
tive core potential [44] and the Stuttgart/Dresden-seg-
mented ECP60MWB_SEG basis set [45] for actinoids and
the 6-311?G(2df) basis set [46] for oxygen atoms. All
calculations were carried out with Gaussian09 [47].
Geometries for these calculations were optimized by
DFT for each functional tested. (For additional compar-
isons, we also carried out single-point energy calcula-
tions at SO-CASPT2 geometries, but these results are
given only in supporting information). For all DFT cal-
culations, we used an integration grid that has 400 radial
shells and 974 angular points per shell. All DFT calcu-
lations were performed without enforcing symmetry on
the orbitals. All calculations are spin unrestricted,
although for some of the singlets, the optimized wave
functions retain doubly occupied orbitals as in the
restricted theory even when we started with a guess that
destroys spin symmetry.
Harmonic ZPEs were scaled using the following scale
factors: M05, 0.977; M06, 0.981; M06-L, 0.978; B3LYP,
0.983; MOHLYP, 1.022; MPW3LYP, 0.983, and PW91,
1.007 [48, 49].
We added the spin–orbit contribution to all DFT cal-
culations as the difference between the SO-CASPT2 result
and the spin-free CASPT2 result (although readers who are
interested in the results without such additions may find
them in the supporting information).
3 Results
All numerical results in the article proper include spin–
orbit and ZPE contributions, and all DFT results in the
article proper are at DFT geometries. For the specialist,
the supporting information gives results without spin–
orbit, without ZPE, and without both. The supporting
information also gives DFT results at spin-free CASPT2
geometries.
Tables 1, 2, 3, and 4 report the first and second ioni-
zation potentials (IPs) of AnO and AnO2 obtained with
WFT and DFT, and Tables 5 and 6 give bond dissociation
energies for breaking An–O bonds. Because the values in
Tables 1, 2, 3, 4, 5, and 6 include ZPEs, they are enthalpy
changes at 0 K.
In tables, MUE(7) means the mean unsigned error for
the seven previous rows. MUE(x) in the last row of a table
means the mean unsigned for all x rows of data in the table.
By error, we mean the deviation from experiment. In this
regard, the reader should be aware that most of the
experimental data have an error bar large enough that it
must be taken into account in drawing conclusions about
the comparison of theory to experiment; we cannot state
that a method is better than another when their difference is
within the experimental error bar. We will therefore con-
centrate our attention mainly on analyzing which theoret-
ical models predict results that agree with experiment
within the error bars.
4 Discussion
In discussing numerical results, we will discuss only those
including spin–orbit and ZPE contributions, and we will
discuss DFT results only for calculations at DFT
Table 1 First and Second Ionization Potentials (in eV) of AnO by
WFT
CASPT2 CCSD CCSD(T) Expa
ThO 6.56 6.39 6.53 6.6035 ± 0.0008
PaO 6.28 6.18 6.31 5.9 ± 0.2
UO 6.05 5.92 5.99 6.0313 ± 0.0006
NpO 5.97 5.97 6.04 6.1 ± 0.2
PuO 6.17 5.95 6.00 6.1 ± 0.2
AmO 6.22 6.02 6.04 6.2 ± 0.2
CmO 6.68 6.42 6.46 6.4 ± 0.2
MUE(7) 0.13 0.15 0.13 0.14b
ThO? 11.94 11.92 11.99 11.8 ± 0.7
PaO? 12.10 12.36 12.46 11.8 ± 0.7
UO? 13.08 12.41 12.39 12.4 ± 0.6
NpO? 13.44 13.28 13.23 14.0 ± 0.6
PuO? 14.36 14.11 14.00 14.0 ± 0.6
AmO? 15.04 15.83 15.71 14.0 ± 0.6
CmO? 15.88 15.08 15.08 15.8 ± 0.4
MUE(7) 0.45 0.58 0.58 0.63b
MUE(14) 0.29 0.37 0.36 0.39b
a Most of the experimental values were obtained through indirect
measurements [57]; Values for ThO and UO were accurately mea-
sured by Heaven and co-workers using REMPI method [50, 51]b The MUE(x) for the experiment is referred to the average error bar
Theor Chem Acc (2011) 129:657–666 659
123

geometries. Before discussing the tables, though, we will
briefly discuss the characteristics of the ground states and
the Kohn–Sham determinants.
4.1 Ground states and Kohn–Sham determinants
The ground states and dominant electron configurations
in the CASSCF calculations for the molecules in Tables 1,
2, 3, 4, 5, and 6 are summarized in [16] and that infor-
mation will not be repeated here. For the CCSD and
CCSD(T) calculations, we have converged the Hartree–
Fock single-configuration reference wave functions to
the dominant determinant identified by the CASSCF
calculations.
For the DFT calculations on the molecules in Tables 1,
2, 3, 4, 5, and 6, in every case, we checked all possible
values of the spin projection quantum number MS to find
the lowest-energy spin state and orbital composition. We
compared our findings for the lowest-energy spin state in
DFT to the lowest spin state in spin-free CASPT2, as given
in [16]. We found that it agreed in every case except for
M05, M06, M06-L, and MOHLYP calculations of neutral
UO, for which it turned out that a triplet state is lower than
the quintet state that is predicted to be the ground state by
WFT calculations.
Spin contamination is not severe (see Table S35 of
Supporting Information for a detailed table). For example,
consider the dioxides. For neutral AnO2, the expectation
value of S2 is within 1% of MS (MS ? 1) in all cases, where
MS, as usual, denotes the projection quantum number of
electron spin. The AnO2? monocations satisfy this condi-
tion within 1% for MS B 3/2 and within 2% for other MS.
Table 2 First and second ionization potentials (eV) of AnO from DFT Calculations
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Expa
ThO 6.61 6.84 6.45 6.52 6.17 6.57 6.47 6.6035 ± 0.0008
PaO 6.31 6.67 5.31 6.30 5.78 6.32 8.40 5.9 ± 0.2
UO 5.71 6.09 5.55 6.18 5.84 6.21 6.22 6.0313 ± 0.0006
NpO 5.63 5.79 5.42 6.26 5.85 6.29 6.28 6.1 ± 0.2
PuO 5.75 5.88 5.54 6.38 5.97 6.42 6.39 6.1 ± 0.2
AmO 6.04 6.08 5.77 6.50 6.21 6.54 6.59 6.2 ± 0.2
CmO 6.14 6.27 5.87 6.67 6.19 6.71 6.63 6.4 ± 0.2
MUE(7) 0.28 0.26 0.49 0.23 0.19 0.25 0.56 0.14b
ThO? 11.63 11.84 11.36 12.21 11.82 12.24 12.18 11.8 ± 0.7
PaO? 12.36 12.42 12.86 12.63 14.81 12.67 12.69 11.8 ± 0.7
UO? 13.01 12.95 12.75 13.08 12.63 13.11 13.01 12.4 ± 0.6
NpO? 13.54 13.60 13.29 13.75 13.14 13.78 13.56 14.0 ± 0.6
PuO? 14.19 14.38 14.05 14.42 13.80 14.44 14.22 14.0 ± 0.6
AmO? 15.29 15.37 14.93 15.33 14.69 15.36 15.12 14.0 ± 0.6
CmO? 15.28 15.40 15.15 15.44 15.11 15.47 15.49 15.8 ± 0.4
MUE(7) 0.54 0.54 0.60 0.61 0.81 0.63 0.57 0.63b
MUE(14) 0.41 0.40 0.54 0.42 0.50 0.44 0.56 0.39b
a Most of the experimental values were obtained through indirect measurements [57]; Values for ThO and UO were accurately measured by
Heaven and co-workers using REMPI method [50, 51]b The MUE(x) for the experiment is referred to the average error bar
Table 3 First and second ionization potentials (in eV) of AnO2 from
WFT calculations
CASPT2 CCSD CCSD(T) Expa
ThO2 8.49 8.58 8.58 8.9 ± 0.4
PaO2 5.71 5.93 6.04 5.9 ± 0.2
UO2 6.24 6.05 6.07 6.128 ± 0.003
NpO2 6.30 5.91 6.01 6.33 ± 0.18
PuO2 6.23 5.62 5.73 7.03 ± 0.12
AmO2 6.80 6.66 6.55 7.23 ± 0.15
CmO2 8.31 7.77 7.60 8.5 ± 1.0
MUE(7) 0.31 0.51 0.53 0.29b
ThO2? 15.09 15.86 15.39 16.6 ± 1.0
PaO2? 16.98 16.93 16.87 16.6 ± 0.4
UO2? 14.38 14.13 14.01 14.6 ± 0.4
NpO2? 15.59 15.44 15.26 15.1 ± 0.4
PuO2? 15.38 15.84 15.62 15.1 ± 0.4
AmO2? 16.29 16.45 16.33 15.7 ± 0.6
CmO2? 16.17 16.10 15.95 17.9 ± 1.0
MUE(7) 0.74 0.74 0.76 0.60b
MUE(14) 0.53 0.62 0.65 0.45b
a Most of the experimental values were obtained through indirectmeasurements [57]; Value for UO2 was accurately determined by Hanet al. [52] using REMPI methodb The MUE(x) for the experiment is referred to the average error bar
660 Theor Chem Acc (2011) 129:657–666
123
For the AnO22? dications, the condition is satisfied within
1% for MS B �, within 2% for MS = 1, within 4% for
MS = 3/2, and within 6% for MS = 2. The only exception
is singlet ThO22? molecule, for which S2 has high values
(0.52–0.70) except for the MOHLYP functional
(S2 = 0.09). For the B3LYP functional, the optimization of
the ThO22? as closed-shell singlet (i.e. with constrained
S2 = 0.0) results in Th–O distance 1.851 A, while the
optimization as an open-shell singlet with S2 = 0.7 results
in Th–O distance 1.909 A. The latter structure is 7.6 kcal/
mol lower than the former one. For analogous M06 cal-
culations of ThO22? the distance of closed-shell singlet is
1.800 A, for open-shell singlet is 1.867 A. The latter
structure is 2.2 kcal/mol lower than the former one.
Comparing the orbital character of the DFT determi-
nants to the orbital composition of the CASSCF wave
function is harder because in most cases where the con-
tribution of the Hartree–Fock determinant in the CASSCF
wave function is less than 80% (see Tables 3 and 4 of
[16]), the Kohn–Sham orbitals break symmetry. In several
cases, even when the system can be described to better than
90% by a single configuration, two singly occupied orbitals
(for example, the d and / orbitals in the cases of NpO2?
and the isoelectronic PuO22?) have very similar orbital
energies, and the Kohn–Sham orbitals are mixtures of these
orbitals. When the orbitals do not break symmetry, we can
compare the DFT orbital character to the dominant orbital
composition case of the CASSCF wave function. As an
example, consider the case of M06 calculations for AnO2
and their cations. In most cases, for M06 method, the DFT
orbitals correspond to the CASPT2 orbitals of AnO2 and
their cations. The exceptions are UO2, NpO2, and PuO2
molecules. For NpO2, M06 predicts a 4Uu ground state
when compared with 4Hg for CASPT2, and for PuO2, M06
predicts a 5Rg ground state when compared with 5Uu for
CASPT2. For the UO2 molecule, the M05, M06, and M06-
L functionals give the ground state as 3Hg, while other DFT
methods and WFT methods give the 3Uu state as the ground
state. The difference is due to the HOMO orbital, which is
the 7s orbital of U for the 3Uu state and 5fd orbital of U for
the 3Hg state. In the latter case, singly occupied d and /orbitals are mixed, and their linear combinations constitute
two broken-symmetry orbitals with the same energy.
4.2 Ionization potentials
When comparing WFT results with DFT results, we have to
contextualize the values with respect to available experi-
mental data. The most accurate experiments are for ThO
[50], UO [51], and UO2 [52], where the error on the IP is less
than or equal to 0.003 eV. CASPT2 and CCSD(T) perform
rather well for these cases, with an MUE(3) of 0.06 eV
(where MUE(x) denotes mean unsigned error averaged over
x molecules), while CCSD performs worse—the MUE(3) is
0.13 eV. CASPT2 overcomes this deficiency by a multi-
configurational treatment of static correlation [8, 9], and
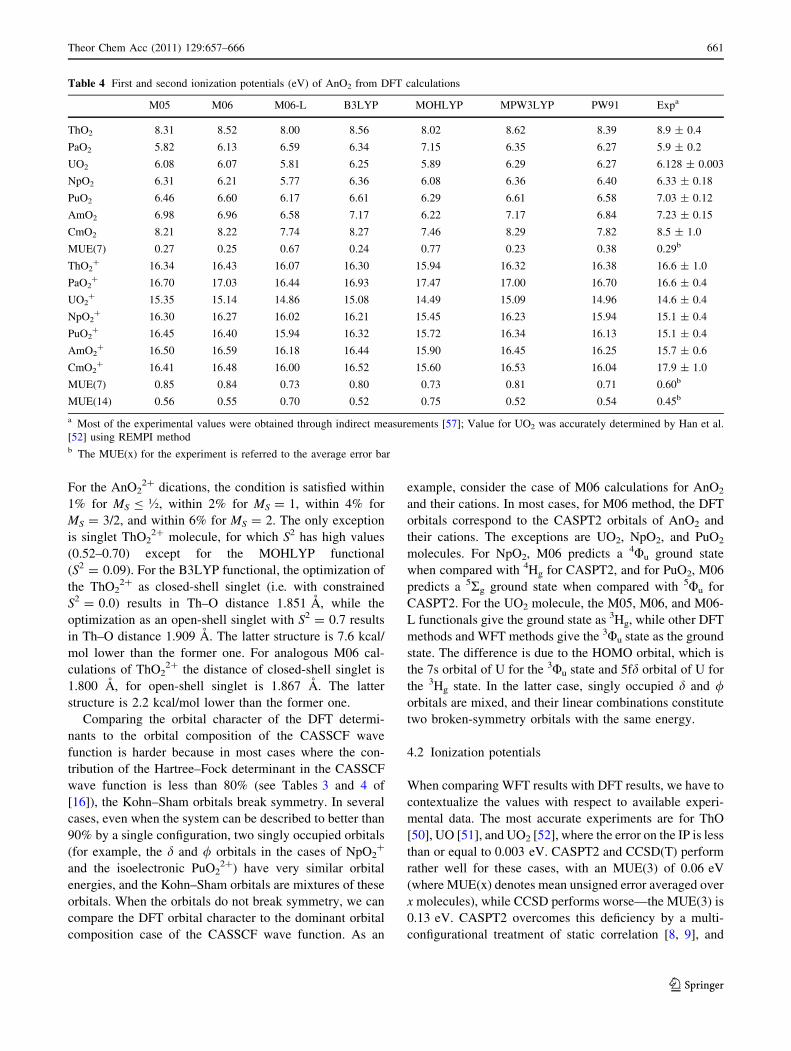
Table 4 First and second ionization potentials (eV) of AnO2 from DFT calculations
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Expa
ThO2 8.31 8.52 8.00 8.56 8.02 8.62 8.39 8.9 ± 0.4
PaO2 5.82 6.13 6.59 6.34 7.15 6.35 6.27 5.9 ± 0.2
UO2 6.08 6.07 5.81 6.25 5.89 6.29 6.27 6.128 ± 0.003
NpO2 6.31 6.21 5.77 6.36 6.08 6.36 6.40 6.33 ± 0.18
PuO2 6.46 6.60 6.17 6.61 6.29 6.61 6.58 7.03 ± 0.12
AmO2 6.98 6.96 6.58 7.17 6.22 7.17 6.84 7.23 ± 0.15
CmO2 8.21 8.22 7.74 8.27 7.46 8.29 7.82 8.5 ± 1.0
MUE(7) 0.27 0.25 0.67 0.24 0.77 0.23 0.38 0.29b
ThO2? 16.34 16.43 16.07 16.30 15.94 16.32 16.38 16.6 ± 1.0
PaO2? 16.70 17.03 16.44 16.93 17.47 17.00 16.70 16.6 ± 0.4
UO2? 15.35 15.14 14.86 15.08 14.49 15.09 14.96 14.6 ± 0.4
NpO2? 16.30 16.27 16.02 16.21 15.45 16.23 15.94 15.1 ± 0.4
PuO2? 16.45 16.40 15.94 16.32 15.72 16.34 16.13 15.1 ± 0.4
AmO2? 16.50 16.59 16.18 16.44 15.90 16.45 16.25 15.7 ± 0.6
CmO2? 16.41 16.48 16.00 16.52 15.60 16.53 16.04 17.9 ± 1.0
MUE(7) 0.85 0.84 0.73 0.80 0.73 0.81 0.71 0.60b
MUE(14) 0.56 0.55 0.70 0.52 0.75 0.52 0.54 0.45b
a Most of the experimental values were obtained through indirect measurements [57]; Value for UO2 was accurately determined by Han et al.
[52] using REMPI methodb The MUE(x) for the experiment is referred to the average error bar
Theor Chem Acc (2011) 129:657–666 661
123

density functional theory includes some static correlation in
the exchange potential [53–56]. On the same molecules, the
M05, M06, MPW3LYP, and B3LYP functionals give
MUE(3) = 0.12 eV, PW91 gives 0.16 eV, MOHLYP gives
0.29 eV, and M06-L gives 0.32 eV.
For the other set of AnO molecules, there is no direct
measurement of the IP, and the experimental error bar of
the first ionization potential is rather large, about 0.2 eV
[57]. Inspection of Table 1 shows that the mean unsigned
error (MUE) for CASPT2, CCSD, and CCSD(T) for the
first IP of AnO is, respectively, 0.13, 0.15, and 0.13 eV.
Thus, among the wave function methods, all three perform
within the experimental uncertainty. The DFT results for
IPs are reported in Table 2. For first IPs, the MUE is 0.28,
0.26, 0.49, 0.23, 0.19, 0.25, and 0.56 eV for M05, M06,
M06-L, B3LYP, MOHLYP, MPW3LYP, and PW91,
respectively.
For the second IP, the experiments show even larger
error bars, ranging from 0.4 eV for CmO? to 0.6–0.7 eV
for the remaining AnO monocationic species [57]. Almost
all WFT and DFT methods agree with experiment within
these limits (the only exception being MOHLYP), in par-
ticular the WFT methods give MUE values of 0.45
(CASPT2), 0.58 (CCSD), and 0.58 eV (CCSD(T)). The
MUEs are 0.54, 0.54, and 0.60 eV for M05, M06, M06-L,
respectively, and 0.61, 0.81, 0.63, and 0.57 eV for B3LYP,
MOHLYP, MPW3LYP, and PW91, respectively.
Now we consider the AnO2 systems. The IPs of these
molecules have been measured with three different meth-
ods, each of them bearing a different error bar. The only
very accurate direct measurement (using the REMPI
method) has been carried out for UO2 [52]. The second
type of experiment employed to measure the first ionization
potential of some of the actinoid species is a well-estab-
lished electron-transfer bracketing technique, which is an
indirect approach that carries an error in the range of
0.1–0.2 eV. In particular, this method has been employed
by Marcalo and Gibson [57] to obtain the IPs of NpO2,
PuO2, and AmO2, with error bars of 0.18, 0.12, and
0.15 eV, respectively. Table 4 shows that WFT methods
tend to give results lower than experiment by more than the
experimental error bar, more prominently for the CCSD
approaches. This has been discussed previously [16], in
particular for the PuO2 molecule, where the first IP was
computed to be considerably lower than the experiment.
The first IP of PuO2 is computed by DFT to range from a
minimum of 6.17 eV for M06-L to a maximum of 6.61 eV
for MPW3LYP and B3LYP. We notice that these values
still fall far from the experimental error bar, and quantum
chemical methods therefore suggest that the actual value of
the first IP of PuO2 is closer to 6–6.5 eV than to 7 eV.
CASPT2 and five of the density functionals give very
similar values for the IP of NpO2, all in a small window of
energies, between 6.21 and 6.36 eV. The 6.21–6.36 eV
window agrees well with the experiment. The CCSD,
CCSD(T), MOHLYP, and M06-L calculations give a lower
IP, outside the experimental error bar.
For AmO2, we encounter a similar situation to PuO2,
with all computed first ionization potentials below the
experimental error bar; however, they are closer to exper-
iment than in the case of PuO2. WFT shows the largest
differences, while B3LYP and MPW3LYP give IPs inside
the experimental error bar. Further experimental work to
confirm that the actual ionization potentials are lower than
the one measured will be difficult due to the hazardous
nature of the materials, and the bracketing technique will
probably remain the most reliable experimental method for
some time.
The third method used to measure the IP of actinoid
species is an indirect method based on the reactions of
actinoid molecules with species of known IPs [57]. In this
case, the error bars are quite large, 0.2 eV for PaO2,
Table 5 Bond Energies (in kcal/mol) for AnO2 AnO2? and AnO2
2?
from WFT Calculations
CASPT2 CCSD CCSD(T) Exp.a
ThO2 147 139 144 164 ± 3
PaO2 190 172 181 187 ± 11
UO2 146 170 183 180 ± 3
NpO2 169 143 158 152 ± 10
PuO2 134 128 144 144 ± 5
AmO2 119 96 110 122 ± 16
CmO2 108 81 95 97 ± 17
MUE(7) 14 17 7 9b
ThO2? 103 88 96 111 ± 9
PaO2? 203 177 185 187 ± 7
UO2? 181 171 182 178 ± 3
NpO2? 162 144 156 146 ± 5
PuO2? 131 130 143 122 ± 9
AmO2? 117 84 98 98 ± 13
CmO2? 73 51 88 49 ± 14
MUE(7) 13 9 13 9b
ThO22? 30 -4 16 0 ± 41
PaO22? 92 73 85 76 ± 26
UO22? 143 127 140 127 ± 7
NpO22? 113 94 109 121 ± 2
PuO22? 104 92 107 97 ± 23
AmO22? 67 67 83 61 ± 31
CmO22? 41 28 47 0 ± 36
MUE(7) 18 10 19 24
MUE(21) 15 12 13 14
a The experimental values from Marcalo and Gibson work [57]
obtained using Born cycle utilizing the measured ionization potentialsb The MUE(x) for the experiment is referred to the average error bar
662 Theor Chem Acc (2011) 129:657–666
123

increasing to 0.4 for ThO2 and to 1.0 eV for CmO2. For
these cases, WFT calculations yield values falling within
the experimental limits. For PaO2, M05 and M06 methods
give values falling within the experimental limits, while
other DFT functionals overestimate the experimental value
of 5.9 eV, especially MOHLYP (7.15 eV). For ThO2, M06,
B3LYP, and MPW3LYP give values falling within the
experimental limits, while other functionals underestimate
the experimental value 8.9 eV.
For the second IPs, the experimental error bars are rather
large, ranging from 0.4 to 1.0 eV [57]. As in the case of the
monoxides, the calculations show significant discrepancies
from the middle experimental value. On average, WFT and
DFT have roughly the same MUE(7); however, in specific
cases, they may differ from each other by a significant
amount. For example, looking at the second IP of UO2, the
CASPT2 calculation gives 14.38 eV, CCSD(T) gives
14.01 eV, and M05, which has performed well in cases
discussed above, yields a value 1 eV higher, 15.35 eV.
Such discrepancies occur quite often in Tables 3 and 4.
Unfortunately, due to the large error bars, the experimental
values do not help much in deciding which method is most
reliable.
The overall performance of methods for the first and
second ionization potentials of AnO and AnO2 is summa-
rized by MUE(AIP28) in Table 7 (The other rows of this
table are discussed below).
4.3 Bond energies
We now consider the performance of the various meth-
ods for bond dissociation energies (BDEs), bearing in
mind that their experimental values are much more
uncertain than those for ionization potentials. This is due
to the fact that the BDEs are computed by a Born cycle
using the measured ionization potentials [57], hence
inheriting their errors along with the errors for other
steps in the cycle. Furthermore, we must consider the
following issue regarding the CASPT2 calculations.
When calculating the reaction AnO2 ? AnO ? O, it is
difficult to keep a balanced active space between the
AnO2 and its dissociation products because a full valence
Table 6 Bond Energies (kcal/mol) for AnO2, AnO2?, and AnO2
2? from DFT Calculations
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp.a
ThO2 159 155 166 157 163 158 176 164 ± 3
PaO2 165 169 199 179 186 181 146 187 ± 11
UO2 172 170 179 171 182 172 193 180 ± 3
NpO2 163 167 172 154 168 155 178 152 ± 10
PuO2 148 153 159 141 155 142 165 144 ± 5
AmO2 109 115 122 108 112 109 128 122 ± 16
CmO2 104 106 116 101 114 102 125 97 ± 17
MUE(7) 10 11 10 7 8 6 21 9b
ThO2? 121 116 130 111 121 112 132 111 ± 9
PaO2? 176 181 169 178 154 179 195 187 ± 7
UO2? 163 171 173 169 180 170 191 178 ± 3
NpO2? 146 156 163 150 161 151 173 146 ± 5
PuO2? 126 131 139 130 141 131 154 122 ± 9
AmO2? 88 95 104 93 112 94 123 98 ± 13
CmO2? 58 62 74 65 86 67 98 49 ± 14
MUE(7) 8 8 15 7 18 7 25 9b
ThO22? 12 11 22 17 25 18 35 0 ± 41
PaO22? 72 76 88 79 94 81 103 76 ± 26
UO22? 102 115 119 118 132 119 141 127 ± 7
NpO22? 84 94 100 93 107 94 117 121 ± 2
PuO22? 81 87 98 88 99 90 112 97 ± 23
AmO22? 61 67 74 67 83 69 97 61 ± 31
CmO22? 28 34 51 37 71 39 82 0 ± 36
MUE(7) 18 14 18 16 23 16 30 24b
MUE(21) 12 11 14 10 16 10 25 14b
a The experimental values from Marcalo and Gibson work [57] obtained using Born cycle utilizing the measured ionization potentialsb The MUE(x) for the experiment is referred to the average error bar
Theor Chem Acc (2011) 129:657–666 663
123

active space is impractical for AnO2, and we need to
truncate the space (see Sect. 2). This lack of balance at
the CASSCF level could be reflected in the description
of the correlation energy, and the error at the CASPT2
level can be larger than what is expected on the basis
of calculations where a full valence active space is
employed. Inspection of Table 5 confirms this by
showing that the MUE for CASPT2 is slightly larger
than that for CCSD(T), especially for the dissociation of
the neutral AnO2 (14 vs. 7 kcal/mol).
DFT methods perform well, especially B3LYP and
MPW3LYP. The M06 functional shows slightly larger
differences from the experiment, but still performs well.
The overall performance of methods for dissociation
energies of AnO2 is shown by MUE(ABDE21) in Table 7.
The bad performance of PW91 is related to systematic
overestimation of the dissociation energies of AnO2 by this
method, with the exception of PaO2, for which PW91
underestimates the bond dissociation energy by 41 kcal/
mol.
4.4 Bond distances
The Ac–O bond distance was optimized for all oxides with
the various density functionals. In Fig. 1, we report the
deviations of DFT bond distances from the CASPT2 bond
distances (A). The absolute values are reported in Sup-
porting Information. The chart shows a nearly systematic
deviation of bond distances predicted by the various
exchange–correlation functionals from those predicted by
CASPT2.
4.5 Overall assessment
The overall performance for both ionization potentials and
bond dissociation energies is given by MUE(AC49) in
Table 7. Four of the density functionals (those containing a
nonzero fraction of Hartree–Fock exchange) and all three
WFT methods show very similar average performance.
Among the three functionals without Hartree–Fock
exchange, the M06-L and MOHLYP functionals perform
better than PW91.
5 Conclusions and outlook
The systems discussed in this paper pose non-trivial chal-
lenges because of their complex electronic structure and
relativistic effects. We have compared the performance of
different methods with the hope of making future users
more aware of their strengths and limitations.
The CASPT2 wave function method used here has been
used successfully in studying many actinoid-containing
systems [58–63]. Recent developments of the Cholesky
decomposition (CD) approach (they have not been
employed in the present study), in combination with the
Table 7 Performance of Tested Methods for 28 Actinoid Ionization Potentials MUE(AIP28) for 21 Actinoid Bond Dissociation Energies
MUE(ABDE21) and for the Actinoid Composite Energy Database of all 49 Ionization Potential and Bond Dissociation Energy Data
MUE(ACE49)
CASPT2 CCSD CCSD(T) M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91
In kcal/mol
MUE(ABDE21) 15 12 13 12 11 14 10 16 10 25
In eV
MUE(AIP28) 0.41 0.50 0.50 0.49 0.47 0.62 0.47 0.63 0.48 0.55
MUE(ABDE21) 0.65 0.53 0.56 0.52 0.47 0.63 0.43 0.71 0.43 1.10
MUE(AC49) 0.51 0.51 0.52 0.50 0.47 0.63 0.45 0.66 0.46 0.79
Fig. 1 Deviations of DFT bond
distances from the CASPT2
bond distances (A)
664 Theor Chem Acc (2011) 129:657–666
123

CASPT2 method [64], have extended the applicability of
the CASPT2 method to larger molecular systems. A CD-
CASPT2 calculation with 1000 basis functions is nowadays
affordable [65]. In addition, CD-CASPT2 calculations can
be performed at even lower computational costs and on
large systems by invoking the frozen natural orbital (FNO)
approximation [66, 67] to effectively reduce the size of the
virtual orbital space with no loss of accuracy. However,
increasing the molecular size is not sufficient. There are
many quantum-chemical problems whose accurate solution
is out of reach for CASPT2 because the adequate number
of MOs cannot be included in the active space [42]. Wave
function methods such as CCSD and CCSD(T) are even
more expensive for large systems. Density functional the-
ory on the other hand remains quite affordable for large
systems, and it has had many successes for transition metal
chemistry [56, 68]. Here, we find that DFT, at least with
well-chosen functionals such as MPW3LYP, B3LYP, M06,
and M05, is, on average, as accurate as or more accurate
than CASPT2, CCSD, and CCSD(T) methods for bond
energies to actinoids. These four functionals also provide
comparable accuracy for ionization potentials of actinoid
compounds.
Acknowledgments This work was supported by the Director, Office
of Basic Energy Sciences, U.S. Department of Energy under Contract
No. USDOE/DE-SC002183, by the Air force Office of Scientific
Research under grant no. FA9550-08-1-0183, by the National Science
Foundation under grant No. CHE09-56776, by 7th Framework Pro-
gramme of the European Commission (Collaboration Project No.
211690), by the Hungarian Scientific Research Foundation (OTKA
No. 75972), and by the University of Minnesota Supercomputing
Institute. We would like to thank John K. Gibson, Berkeley National
Laboratory, for useful discussion.
References
1. Hanschke JM, Stakebake JL (2006) In: Morss LR, Edelstein NM,
Fuger NM, Katz JJ (eds) The chemistry of the actinide and
transactinide elements, vol 5. Springer, Dordrecht
2. Choppin GR, Jensen MP (2006) In: Morss LR, Edelstein NM,
Fuger NM, Katz JJ (eds) Chemistry of the actinides and trans-
actinide elements. Springer, Dordrecht
3. Choppin GR (2007) J Radioanal Nucl Chem 273:695
4. Colmenares CA (1984) Prog Solid State Chem 15:257
5. Barnea E, Eisen MS (2006) Coord Chem Rev 250:855
6. de Almeida KJ, Cesar A (2006) Organometallics 25:3407
7. Stubbert BD, Marks TJ (2007) J Am Chem Soc 129:4253
8. Roos BO, Taylor PR, Siegbahn PEM (1980) Chem Phys 48:157
9. Andersson K, Malmqvist P-A, Roos BO (1992) J Chem Phys
96:1218
10. Becke AD (1988) Phys Rev A 38:3098
11. Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785
12. Becke AD (1993) J Chem Phys 98:5648
13. Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ (1994) J
Phys Chem 98:11623
14. Gagliardi L, Roos BO (2000) Chem Phys Lett 331:229
15. Gagliardi L, Heaven MC, Krogh JW, Roos BO (2005) J Am
Chem Soc 127:86
16. Infante I, Kovacs A, La Macchia G, Shahi ARM, Gibson JK,
Gagliardi L (2010) J Phys Chem A 114:6007
17. Purvis GD, Bartlett RJ (1982) J Chem Phys 76:1910
18. Visscher L, Eliav E, Kaldor U (2001) J Chem Phys 115:9720
19. Raghavachari K, Trucks GW, Pople JA, Head-Gordon M (1989)
Chem Phys Lett 157:479
20. Visscher L, Lee TJ, Dyall KG (1996) J Chem Phys 105:8769
21. Zhao Y, Truhlar DG (2004) J Phys Chem A 108:6908
22. Schultz NE, Zhao Y, Truhlar DG (2005) J Phys Chem A
109:11127
23. Zhao Y, Schultz NE, Truhlar DG (2005) J Chem Phys
123:161103
24. Zhao Y, Truhlar DG (2006) J Chem Phys 125:194101
25. Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
26. Perdew JP (1991) In: Ziesche P, Eschrig H (eds) Electronic
structure of solids ‘91. Akademie, Berlin
27. Clavaguera-Sarrio C, Ismail N, Marsden CJ, Begue D, Pouchan C
(2004) Chem Phys 302:1
28. Austin JP, Burton NA, Hillier IH, Sundararajan M, Vincent MA
(2009) Phys Chem Chem Phys 11:1143
29. Douglas N, Kroll NM (1974) Ann Phys 82:89
30. Hess BA (1986) Phys Rev A 33:3742
31. Roos BO, Lindh R, Malmqvist P-A, Veryazov V, Widmark PO
(2005) Chem Phys Lett 409:295
32. Roos BO, Lindh R, Malmqvist P-A, Veryazov V, Widmark PO,
Borin AC (2008) J Phys Chem A 112:11431
33. Aquilante F, De Vico L, Ferre N, Ghigo G, Malmqvist P-A,
Neogrady P, Pedersen TB, Pitonak M, Reiher M, Roos BO,
Serrano-Andres L, Urban M, Veryazov V, Lindh R (2010) J
Comp Chem 31:224
34. Malmqvist P-A, Roos BO, Schimmelpfennig B (2002) Chem
Phys Lett 357:230
35. Roos BO, Malmqvist P-A (2004) Phys Chem Chem Phys 6:2919
36. Hess BA, Marian CM, Wahlgren U, Gropen O (1996) Chem Phys
Lett 251:365
37. Ilias M, Saue T (2007) J Chem Phys 126:064102
38. Dyall KG (1994) J Chem Phys 100:2118
39. Visscher L, Jensen HJ Aa, Saue T, with new contributions from
Bast R, Dubillard S, Dyall KG, Ekstrom U, Eliav E, Fleig T,
Gomes ASP, Helgaker TU, Henriksson J, Ilias M, Jacob ChR,
Knecht S, Norman P, Olsen J, Pernpointner M, Ruud K, Sałek P,
Sikkema J (2008) DIRAC, a relativistic ab initio electronic
structure program, Release DIRAC08 (see http://dirac.chem.
sdu.dk)
40. Dyall KG (2004) Theor Chem Acc 112:403
41. Dunning TH (1989) J Chem Phys 90:1007
42. Malmqvist P-A, Pierloot K, Shahi ARM, Cramer CJ, Gagliardi L
(2008) J Chem Phys 128:204109
43. Rehaman Moughal Shahi A, Cramer CJ, Gagliardi L (2009) Phys
Chem Chem Phys 11:10964
44. Kuchle W, Dolg M, Stoll H, Preuss H (1994) J Chem Phys
100:7535
45. Cao XY, Dolg M (2004) J Mol Struct (Theochem) 673:203; (see
also the Stuttgart-Cologne basis set site: http://www.theochem.
uni-stuttgart.de/pseudopotentials/clickpse.en.html)
46. Krishnan R, Binkley JS, Seeger R, Pople JA (1980) J Chem Phys
72:650
47. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA,
Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson
GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF,
Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K,
Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao
O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F,
Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN,
Theor Chem Acc (2011) 129:657–666 665
123

Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC,
Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M,
Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts
R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C,
Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth
GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas
O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009)
GAUSSIAN 09 (Revision A.1). Gaussian Inc, Wallingford
48. Alecu IM, Zheng J, Zhao Y, Truhlar DG (2010) J Chem Theory
Comput 6:2872
49. Zheng J, Alecu IM, Lynch BJ, Zhao Y, Truhlar DG (2010)
Database of frequency scale factors for electronic structure
methods. University of Minnesota, Minneapolis. http://comp.
chem.umn.edu/freqscale/index.html. Accessed 26 Nov 2010
50. Goncharov V, Heaven MC (2006) J Chem Phys 124:064312
51. Goncharov V, Kaledin LA, Heaven MC (2006) J Chem Phys
125:133202
52. Han JD, Goncharov V, Kaledin LA, Komissarov AV, Heaven
MC (2004) J Chem Phys 120:5155
53. Becke AD (2000) J Chem Phys 112:4020
54. Gritsenko OV, Schipper PRT, Baerends EJ (1997) J Chem Phys
107:5007
55. Polo V, Grafenstein J, Kraka E, Cremer D (2003) Theor Chem
Acc 109:22
56. Cramer CJ, Truhlar DG (2009) Phys Chem Chem Phys 11:10757
57. Marcalo J, Gibson JK (2009) J Phys Chem A 113:12599
58. Gagliardi L, Pyykko P, Roos BO (2005) Phys Chem Chem Phys
7:2415
59. Gagliardi L, Pyykko P (2004) Angew Chem Int Ed 43:1573
60. Gagliardi L (2006) Theor Chem Acc 116:307
61. Gagliardi L, Roos BO (2007) Chem Soc Rev 36:893
62. Infante I, Gagliardi L, Scuseria GE (2008) J Am Chem Soc
130:7459
63. Infante I, Gagliardi L, Wang XF, Andrews L (2009) J Phys Chem
A 113:2446
64. Aquilante F, Pedersen TB, Lindh R, Roos BO, De Meras AS,
Koch H (2008) J Chem Phys 129:024113
65. Aquilante F, Malmqvist P-A, Pedersen TB, Ghosh A, Roos BO
(2008) J Chem Theory Comput 4:694
66. Aquilante F, Gagliardi L, Pedersen TB, Lindh RJ (2009) Chem
Phys 130:154107
67. La Macchia G, Manni GL, Todorova TK, Brynda M, Aquilante F,
Roos BO, Gagliardi L (2010) Inorg Chem 49:5216
68. Zhao Y, Truhlar DG (2010) Chem Phys Lett. doi:10.1016/j.cplett.
2010.11.060
666 Theor Chem Acc (2011) 129:657–666
123

S1
Supporting Information for
How accurate are electronic structure methods for actinoid chemistry?
Boris Averkiev, Manjeera Mantina, Rosendo Valero, Ivan Infante, Attila Kovacs, Donald G. Truhlar, and Laura Gagliardi
Date of preparation of supporting information: Feb. 11, 2011
Number of pages, including this page: 27
Contents
Tables S1–S2. Calculated bond distances
Tables S3–S4. Calculated zero point vibrational energies
Table S5–S34. Ionization potentials and bond energies
In Tables S5–S34 the MUE for the theoretical columns is the mean unsigned deviation
from the experimental values, whereas the MUE entry for the experimental column is
the average error bar. All numerical results in the article proper include spin–orbit
coupling and zero point vibrational energy (ZPE) contributions, and all DFT results in
the article proper are at DFT geometries. For the specialist, this supporting information
gives results without spin-orbit coupling, without ZPE, and without both. The
supporting information also gives DFT results at spin-free CASPT2 geometries.
Table S35. Calculated expectation values of the square of the total electron spin
Table S36. Bond distances for AnO2 calculated with the aug-cc-pVTZ basis set for oxygen
S2
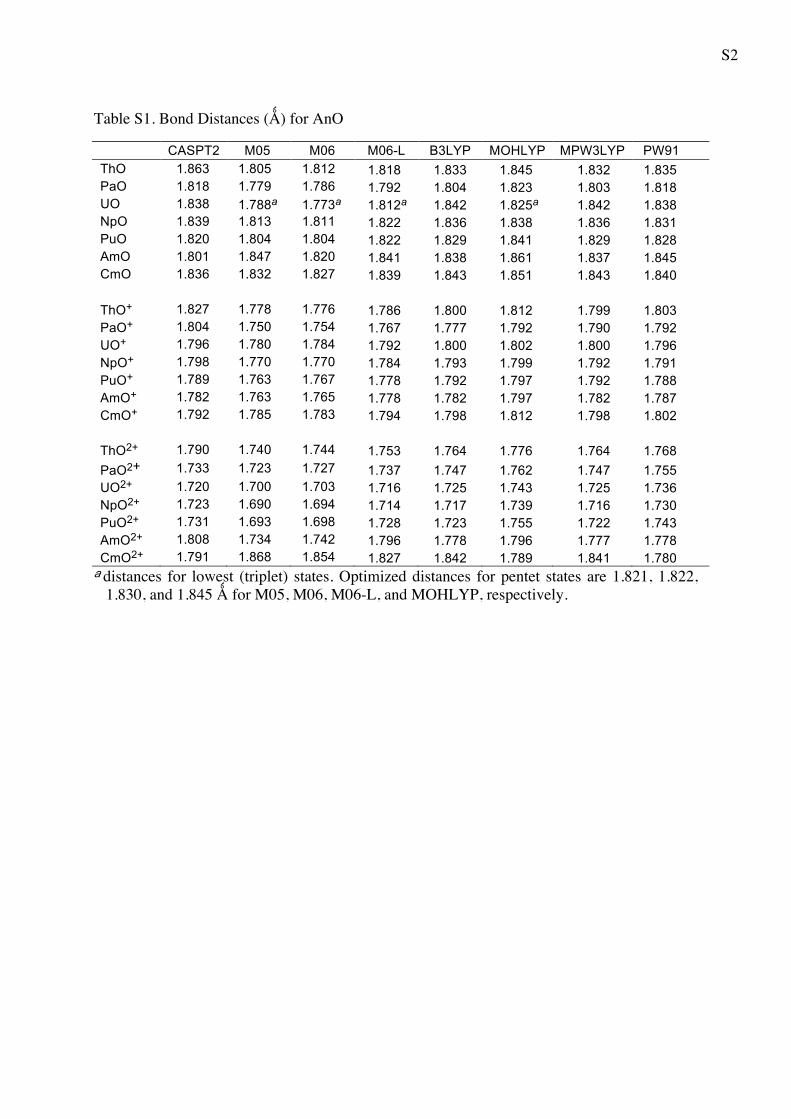
Table S1. Bond Distances (Ǻ) for AnO
CASPT2 M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 ThO 1.863 1.805 1.812 1.818 1.833 1.845 1.832 1.835 PaO 1.818 1.779 1.786 1.792 1.804 1.823 1.803 1.818 UO 1.838 1.788a 1.773a 1.812a 1.842 1.825a 1.842 1.838 NpO 1.839 1.813 1.811 1.822 1.836 1.838 1.836 1.831 PuO 1.820 1.804 1.804 1.822 1.829 1.841 1.829 1.828 AmO 1.801 1.847 1.820 1.841 1.838 1.861 1.837 1.845 CmO 1.836 1.832 1.827 1.839 1.843 1.851 1.843 1.840 ThO+ 1.827 1.778 1.776 1.786 1.800 1.812 1.799 1.803 PaO+ 1.804 1.750 1.754 1.767 1.777 1.792 1.790 1.792 UO+ 1.796 1.780 1.784 1.792 1.800 1.802 1.800 1.796 NpO+ 1.798 1.770 1.770 1.784 1.793 1.799 1.792 1.791 PuO+ 1.789 1.763 1.767 1.778 1.792 1.797 1.792 1.788 AmO+ 1.782 1.763 1.765 1.778 1.782 1.797 1.782 1.787 CmO+ 1.792 1.785 1.783 1.794 1.798 1.812 1.798 1.802 ThO2+ 1.790 1.740 1.744 1.753 1.764 1.776 1.764 1.768 PaO2+ 1.733 1.723 1.727 1.737 1.747 1.762 1.747 1.755 UO2+ 1.720 1.700 1.703 1.716 1.725 1.743 1.725 1.736 NpO2+ 1.723 1.690 1.694 1.714 1.717 1.739 1.716 1.730 PuO2+ 1.731 1.693 1.698 1.728 1.723 1.755 1.722 1.743 AmO2+ 1.808 1.734 1.742 1.796 1.778 1.796 1.777 1.778 CmO2+ 1.791 1.868 1.854 1.827 1.842 1.789 1.841 1.780 a distances for lowest (triplet) states. Optimized distances for pentet states are 1.821, 1.822,
1.830, and 1.845 Ǻ for M05, M06, M06-L, and MOHLYP, respectively.

S3
Table S2. Bond Distances (Ǻ) for AnO2 and Angle (degrees) for ThO2
CASPT2 M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 ThO2 1.923 1.868 1.872 1.883 1.898 1.909 1.897 1.898 ThO2
a 111.9 121.0 120.8 118.3 119.0 119.0 119.0 118.5 PaO2 1.816 1.779 1.786 1.806 1.806 1.833 1.805 1.813 UO2 1.827 1.813 1.816 1.830 1.789 1.818 1.789 1.808 NpO2 1.761 1.791 1.795 1.809 1.821 1.815 1.821 1.803 PuO2 1.744 1.782 1.786 1.805 1.814 1.813 1.813 1.802 AmO2 1.807 1.793 1.800 1.828 1.827 1.856 1.827 1.827b CmO2 1.832 1.809 1.815 1.839 1.839 1.852 1.838 1.840 ThO2
+ 1.832 1.814 1.827 1.847 1.868 1.879 1.868 1.870 PaO2
+ 1.767 1.740 1.745 1.759 1.767 1.812 1.767 1.777 UO2
+ 1.745 1.725 1.730 1.745 1.753 1.773 1.753 1.764 NpO2
+ 1.723 1.704 1.708 1.724 1.732 1.754 1.731 1.745 PuO2
+ 1.704 1.686 1.692 1.709 1.714 1.737 1.714 1.728 AmO2
+ 1.721 1.684 1.690 1.714 1.716 1.743 1.716 1.733 CmO2
+ 1.746 1.709 1.716 1.748 1.748 1.770 1.747 1.759 ThO2
2+ 1.903 1.781 1.867 1.848 1.909 1.864 1.909 1.856
PaO22+ 1.726 1.728 1.738 1.756 1.772 1.792 1.772 1.785
UO22+ 1.710 1.667 1.672 1.688 1.693 1.717 1.693 1.710
NpO22+ 1.700 1.659 1.665 1.684 1.688 1.715 1.688 1.708
PuO22+ 1.675 1.646 1.650 1.670 1.674 1.704 1.673 1.696
AmO22+ 1.679 1.636 1.642 1.664 1.666 1.696 1.666 1.688
CmO22+ 1.674 1.647 1.653 1.687 1.688 1.718 1.688 1.707
a angle values for ThO2 molecule, which has a bent geometry With C2v symmetry bMolecule has a bent geometry with C2v symmetry and a bond angle of 175.1 deg.
S4
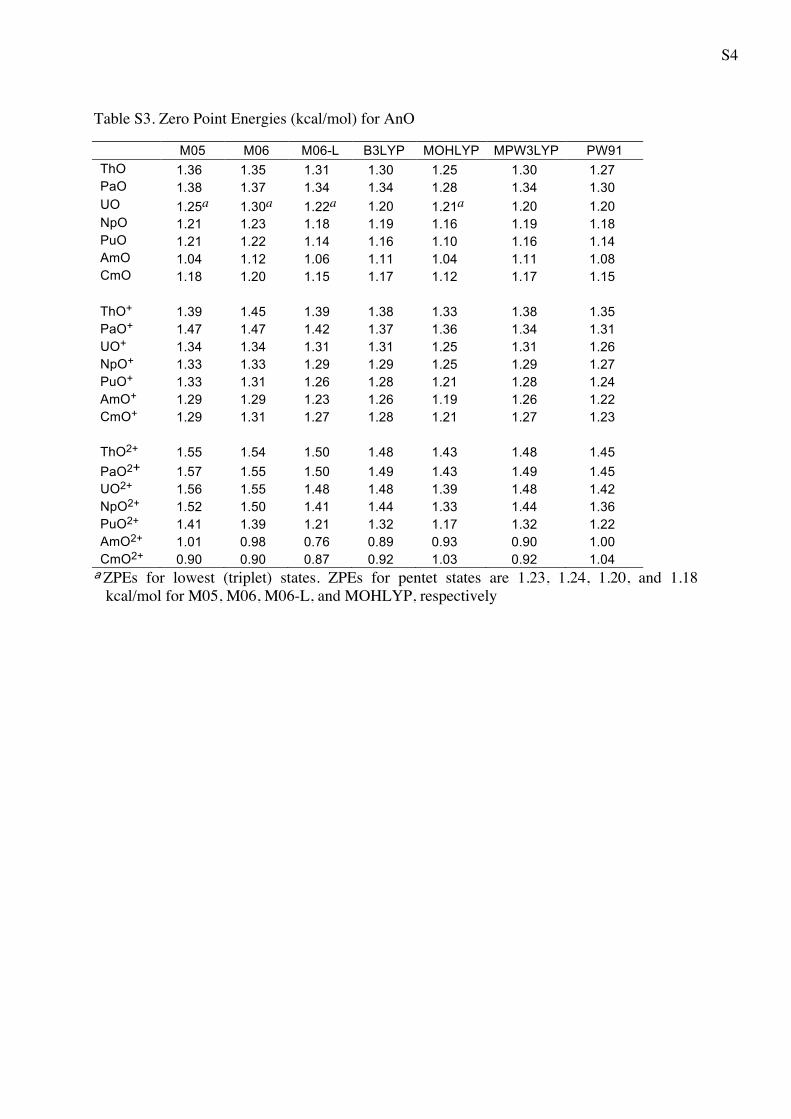
Table S3. Zero Point Energies (kcal/mol) for AnO
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 ThO 1.36 1.35 1.31 1.30 1.25 1.30 1.27 PaO 1.38 1.37 1.34 1.34 1.28 1.34 1.30 UO 1.25a 1.30a 1.22a 1.20 1.21a 1.20 1.20 NpO 1.21 1.23 1.18 1.19 1.16 1.19 1.18 PuO 1.21 1.22 1.14 1.16 1.10 1.16 1.14 AmO 1.04 1.12 1.06 1.11 1.04 1.11 1.08 CmO 1.18 1.20 1.15 1.17 1.12 1.17 1.15 ThO+ 1.39 1.45 1.39 1.38 1.33 1.38 1.35 PaO+ 1.47 1.47 1.42 1.37 1.36 1.34 1.31 UO+ 1.34 1.34 1.31 1.31 1.25 1.31 1.26 NpO+ 1.33 1.33 1.29 1.29 1.25 1.29 1.27 PuO+ 1.33 1.31 1.26 1.28 1.21 1.28 1.24 AmO+ 1.29 1.29 1.23 1.26 1.19 1.26 1.22 CmO+ 1.29 1.31 1.27 1.28 1.21 1.27 1.23 ThO2+ 1.55 1.54 1.50 1.48 1.43 1.48 1.45 PaO2+ 1.57 1.55 1.50 1.49 1.43 1.49 1.45 UO2+ 1.56 1.55 1.48 1.48 1.39 1.48 1.42 NpO2+ 1.52 1.50 1.41 1.44 1.33 1.44 1.36 PuO2+ 1.41 1.39 1.21 1.32 1.17 1.32 1.22 AmO2+ 1.01 0.98 0.76 0.89 0.93 0.90 1.00 CmO2+ 0.90 0.90 0.87 0.92 1.03 0.92 1.04 aZPEs for lowest (triplet) states. ZPEs for pentet states are 1.23, 1.24, 1.20, and 1.18
kcal/mol for M05, M06, M06-L, and MOHLYP, respectively

S5
Table S4. Zero Point Energies (kcal/mol) for AnO2
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 ThO2 2.58 2.56 2.50 2.49 2.41 2.49 2.45 PaO2 2.97 2.96 2.74 2.86 2.62 2.86 2.78 UO2 2.68 2.69 2.56 2.99 2.69 3.00 2.76 NpO2 2.89 2.88 2.75 2.69 2.47 2.69 2.51 PuO2 2.99 2.98 2.79 2.78 2.71 2.78 2.77 AmO2 2.81 2.76 2.50 2.53 2.31 2.54 2.24 CmO2 2.70 2.42 2.43 2.43 2.37 2.44 2.39 ThO2
+ 2.55 2.36 2.24 2.06 2.19 2.05 2.23 PaO2
+ 3.24 3.23 3.08 3.09 2.77 3.08 2.97 UO2
+ 3.42 3.39 3.21 3.22 3.04 3.22 3.08 NpO2
+ 3.64 3.61 3.44 3.40 3.19 3.40 3.24 PuO2
+ 3.77 3.73 3.54 3.53 3.31 3.52 3.37 AmO2
+ 3.77 3.72 3.44 3.47 3.22 3.47 3.30 CmO2
+ 3.40 3.31 2.97 3.00 2.90 3.00 2.97 ThO2
2+ 3.06 2.09 2.24 2.20 2.53 2.20 2.41 PaO2
2+ 3.12 2.88 2.72 2.45 2.57 2.45 2.59 UO2
2+ 3.83 3.78 3.54 3.60 3.28 3.60 3.32 NpO2
2+ 3.87 3.81 3.53 3.60 3.25 3.60 3.29 PuO2
2+ 4.03 3.99 3.72 3.75 3.38 3.75 3.45 AmO2
2+ 4.12 4.06 3.80 3.82 3.52 3.82 3.59 CmO2
2+ 3.79 3.75 3.35 3.36 3.20 3.36 3.30
S6
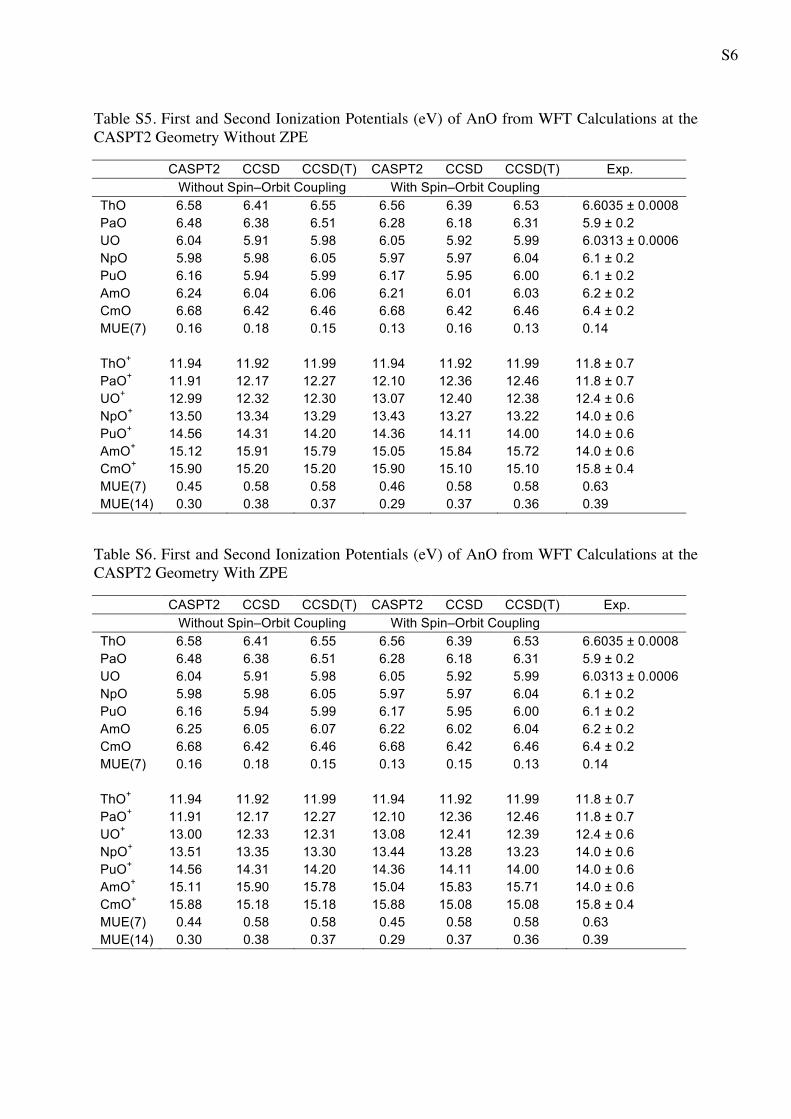
Table S5. First and Second Ionization Potentials (eV) of AnO from WFT Calculations at the CASPT2 Geometry Without ZPE
CASPT2 CCSD CCSD(T) CASPT2 CCSD CCSD(T) Exp. Without Spin–Orbit Coupling With Spin–Orbit Coupling
ThO 6.58 6.41 6.55 6.56 6.39 6.53 6.6035 ± 0.0008 PaO 6.48 6.38 6.51 6.28 6.18 6.31 5.9 ± 0.2 UO 6.04 5.91 5.98 6.05 5.92 5.99 6.0313 ± 0.0006 NpO 5.98 5.98 6.05 5.97 5.97 6.04 6.1 ± 0.2 PuO 6.16 5.94 5.99 6.17 5.95 6.00 6.1 ± 0.2 AmO 6.24 6.04 6.06 6.21 6.01 6.03 6.2 ± 0.2 CmO 6.68 6.42 6.46 6.68 6.42 6.46 6.4 ± 0.2 MUE(7) 0.16 0.18 0.15 0.13 0.16 0.13 0.14 ThO+ 11.94 11.92 11.99 11.94 11.92 11.99 11.8 ± 0.7 PaO+ 11.91 12.17 12.27 12.10 12.36 12.46 11.8 ± 0.7 UO+ 12.99 12.32 12.30 13.07 12.40 12.38 12.4 ± 0.6 NpO+ 13.50 13.34 13.29 13.43 13.27 13.22 14.0 ± 0.6 PuO+ 14.56 14.31 14.20 14.36 14.11 14.00 14.0 ± 0.6 AmO+ 15.12 15.91 15.79 15.05 15.84 15.72 14.0 ± 0.6 CmO+ 15.90 15.20 15.20 15.90 15.10 15.10 15.8 ± 0.4 MUE(7) 0.45 0.58 0.58 0.46 0.58 0.58 0.63 MUE(14) 0.30 0.38 0.37 0.29 0.37 0.36 0.39
Table S6. First and Second Ionization Potentials (eV) of AnO from WFT Calculations at the CASPT2 Geometry With ZPE
CASPT2 CCSD CCSD(T) CASPT2 CCSD CCSD(T) Exp. Without Spin–Orbit Coupling With Spin–Orbit Coupling
ThO 6.58 6.41 6.55 6.56 6.39 6.53 6.6035 ± 0.0008 PaO 6.48 6.38 6.51 6.28 6.18 6.31 5.9 ± 0.2 UO 6.04 5.91 5.98 6.05 5.92 5.99 6.0313 ± 0.0006 NpO 5.98 5.98 6.05 5.97 5.97 6.04 6.1 ± 0.2 PuO 6.16 5.94 5.99 6.17 5.95 6.00 6.1 ± 0.2 AmO 6.25 6.05 6.07 6.22 6.02 6.04 6.2 ± 0.2 CmO 6.68 6.42 6.46 6.68 6.42 6.46 6.4 ± 0.2 MUE(7) 0.16 0.18 0.15 0.13 0.15 0.13 0.14 ThO+ 11.94 11.92 11.99 11.94 11.92 11.99 11.8 ± 0.7 PaO+ 11.91 12.17 12.27 12.10 12.36 12.46 11.8 ± 0.7 UO+ 13.00 12.33 12.31 13.08 12.41 12.39 12.4 ± 0.6 NpO+ 13.51 13.35 13.30 13.44 13.28 13.23 14.0 ± 0.6 PuO+ 14.56 14.31 14.20 14.36 14.11 14.00 14.0 ± 0.6 AmO+ 15.11 15.90 15.78 15.04 15.83 15.71 14.0 ± 0.6 CmO+ 15.88 15.18 15.18 15.88 15.08 15.08 15.8 ± 0.4 MUE(7) 0.44 0.58 0.58 0.45 0.58 0.58 0.63 MUE(14) 0.30 0.38 0.37 0.29 0.37 0.36 0.39
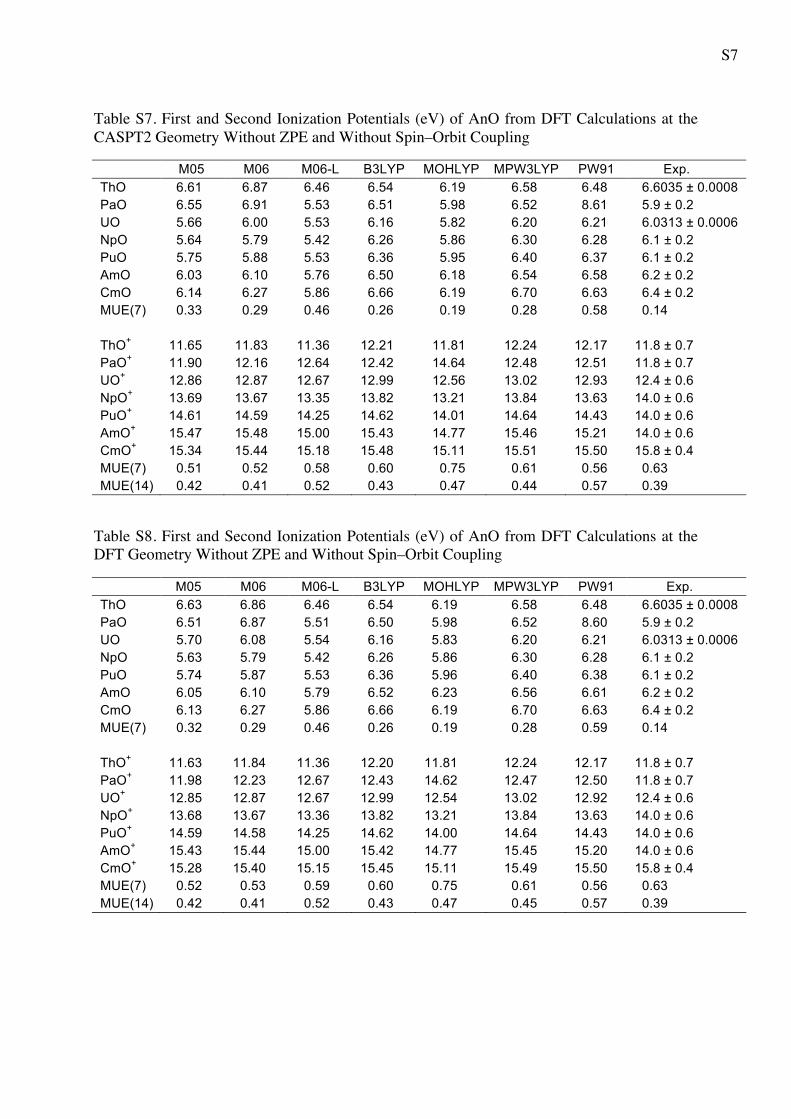
S7
Table S7. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the CASPT2 Geometry Without ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.61 6.87 6.46 6.54 6.19 6.58 6.48 6.6035 ± 0.0008 PaO 6.55 6.91 5.53 6.51 5.98 6.52 8.61 5.9 ± 0.2 UO 5.66 6.00 5.53 6.16 5.82 6.20 6.21 6.0313 ± 0.0006 NpO 5.64 5.79 5.42 6.26 5.86 6.30 6.28 6.1 ± 0.2 PuO 5.75 5.88 5.53 6.36 5.95 6.40 6.37 6.1 ± 0.2 AmO 6.03 6.10 5.76 6.50 6.18 6.54 6.58 6.2 ± 0.2 CmO 6.14 6.27 5.86 6.66 6.19 6.70 6.63 6.4 ± 0.2 MUE(7) 0.33 0.29 0.46 0.26 0.19 0.28 0.58 0.14 ThO+ 11.65 11.83 11.36 12.21 11.81 12.24 12.17 11.8 ± 0.7 PaO+ 11.90 12.16 12.64 12.42 14.64 12.48 12.51 11.8 ± 0.7 UO+ 12.86 12.87 12.67 12.99 12.56 13.02 12.93 12.4 ± 0.6 NpO+ 13.69 13.67 13.35 13.82 13.21 13.84 13.63 14.0 ± 0.6 PuO+ 14.61 14.59 14.25 14.62 14.01 14.64 14.43 14.0 ± 0.6 AmO+ 15.47 15.48 15.00 15.43 14.77 15.46 15.21 14.0 ± 0.6 CmO+ 15.34 15.44 15.18 15.48 15.11 15.51 15.50 15.8 ± 0.4 MUE(7) 0.51 0.52 0.58 0.60 0.75 0.61 0.56 0.63 MUE(14) 0.42 0.41 0.52 0.43 0.47 0.44 0.57 0.39
Table S8. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the DFT Geometry Without ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.63 6.86 6.46 6.54 6.19 6.58 6.48 6.6035 ± 0.0008 PaO 6.51 6.87 5.51 6.50 5.98 6.52 8.60 5.9 ± 0.2 UO 5.70 6.08 5.54 6.16 5.83 6.20 6.21 6.0313 ± 0.0006 NpO 5.63 5.79 5.42 6.26 5.86 6.30 6.28 6.1 ± 0.2 PuO 5.74 5.87 5.53 6.36 5.96 6.40 6.38 6.1 ± 0.2 AmO 6.05 6.10 5.79 6.52 6.23 6.56 6.61 6.2 ± 0.2 CmO 6.13 6.27 5.86 6.66 6.19 6.70 6.63 6.4 ± 0.2 MUE(7) 0.32 0.29 0.46 0.26 0.19 0.28 0.59 0.14 ThO+ 11.63 11.84 11.36 12.20 11.81 12.24 12.17 11.8 ± 0.7 PaO+ 11.98 12.23 12.67 12.43 14.62 12.47 12.50 11.8 ± 0.7 UO+ 12.85 12.87 12.67 12.99 12.54 13.02 12.92 12.4 ± 0.6 NpO+ 13.68 13.67 13.36 13.82 13.21 13.84 13.63 14.0 ± 0.6 PuO+ 14.59 14.58 14.25 14.62 14.00 14.64 14.43 14.0 ± 0.6 AmO+ 15.43 15.44 15.00 15.42 14.77 15.45 15.20 14.0 ± 0.6 CmO+ 15.28 15.40 15.15 15.45 15.11 15.49 15.50 15.8 ± 0.4 MUE(7) 0.52 0.53 0.59 0.60 0.75 0.61 0.56 0.63 MUE(14) 0.42 0.41 0.52 0.43 0.47 0.45 0.57 0.39
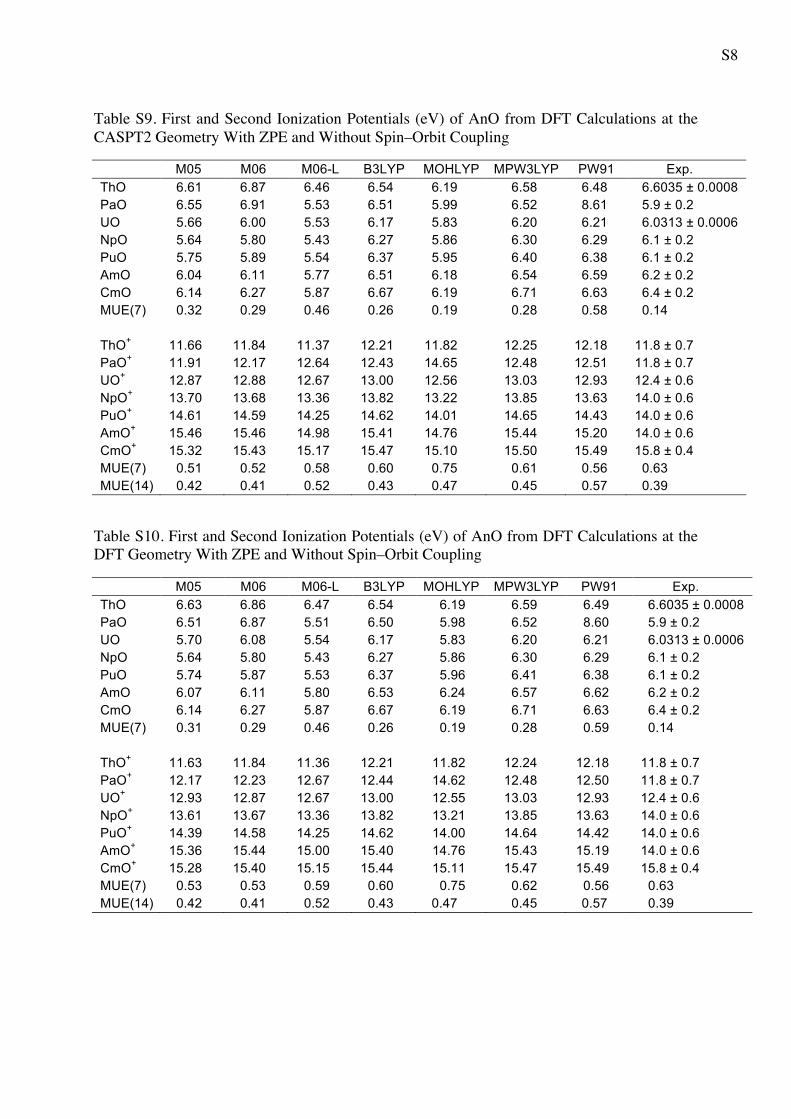
S8
Table S9. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the CASPT2 Geometry With ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.61 6.87 6.46 6.54 6.19 6.58 6.48 6.6035 ± 0.0008 PaO 6.55 6.91 5.53 6.51 5.99 6.52 8.61 5.9 ± 0.2 UO 5.66 6.00 5.53 6.17 5.83 6.20 6.21 6.0313 ± 0.0006 NpO 5.64 5.80 5.43 6.27 5.86 6.30 6.29 6.1 ± 0.2 PuO 5.75 5.89 5.54 6.37 5.95 6.40 6.38 6.1 ± 0.2 AmO 6.04 6.11 5.77 6.51 6.18 6.54 6.59 6.2 ± 0.2 CmO 6.14 6.27 5.87 6.67 6.19 6.71 6.63 6.4 ± 0.2 MUE(7) 0.32 0.29 0.46 0.26 0.19 0.28 0.58 0.14 ThO+ 11.66 11.84 11.37 12.21 11.82 12.25 12.18 11.8 ± 0.7 PaO+ 11.91 12.17 12.64 12.43 14.65 12.48 12.51 11.8 ± 0.7 UO+ 12.87 12.88 12.67 13.00 12.56 13.03 12.93 12.4 ± 0.6 NpO+ 13.70 13.68 13.36 13.82 13.22 13.85 13.63 14.0 ± 0.6 PuO+ 14.61 14.59 14.25 14.62 14.01 14.65 14.43 14.0 ± 0.6 AmO+ 15.46 15.46 14.98 15.41 14.76 15.44 15.20 14.0 ± 0.6 CmO+ 15.32 15.43 15.17 15.47 15.10 15.50 15.49 15.8 ± 0.4 MUE(7) 0.51 0.52 0.58 0.60 0.75 0.61 0.56 0.63 MUE(14) 0.42 0.41 0.52 0.43 0.47 0.45 0.57 0.39
Table S10. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the DFT Geometry With ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.63 6.86 6.47 6.54 6.19 6.59 6.49 6.6035 ± 0.0008 PaO 6.51 6.87 5.51 6.50 5.98 6.52 8.60 5.9 ± 0.2 UO 5.70 6.08 5.54 6.17 5.83 6.20 6.21 6.0313 ± 0.0006 NpO 5.64 5.80 5.43 6.27 5.86 6.30 6.29 6.1 ± 0.2 PuO 5.74 5.87 5.53 6.37 5.96 6.41 6.38 6.1 ± 0.2 AmO 6.07 6.11 5.80 6.53 6.24 6.57 6.62 6.2 ± 0.2 CmO 6.14 6.27 5.87 6.67 6.19 6.71 6.63 6.4 ± 0.2 MUE(7) 0.31 0.29 0.46 0.26 0.19 0.28 0.59 0.14 ThO+ 11.63 11.84 11.36 12.21 11.82 12.24 12.18 11.8 ± 0.7 PaO+ 12.17 12.23 12.67 12.44 14.62 12.48 12.50 11.8 ± 0.7 UO+ 12.93 12.87 12.67 13.00 12.55 13.03 12.93 12.4 ± 0.6 NpO+ 13.61 13.67 13.36 13.82 13.21 13.85 13.63 14.0 ± 0.6 PuO+ 14.39 14.58 14.25 14.62 14.00 14.64 14.42 14.0 ± 0.6 AmO+ 15.36 15.44 15.00 15.40 14.76 15.43 15.19 14.0 ± 0.6 CmO+ 15.28 15.40 15.15 15.44 15.11 15.47 15.49 15.8 ± 0.4 MUE(7) 0.53 0.53 0.59 0.60 0.75 0.62 0.56 0.63 MUE(14) 0.42 0.41 0.52 0.43 0.47 0.45 0.57 0.39
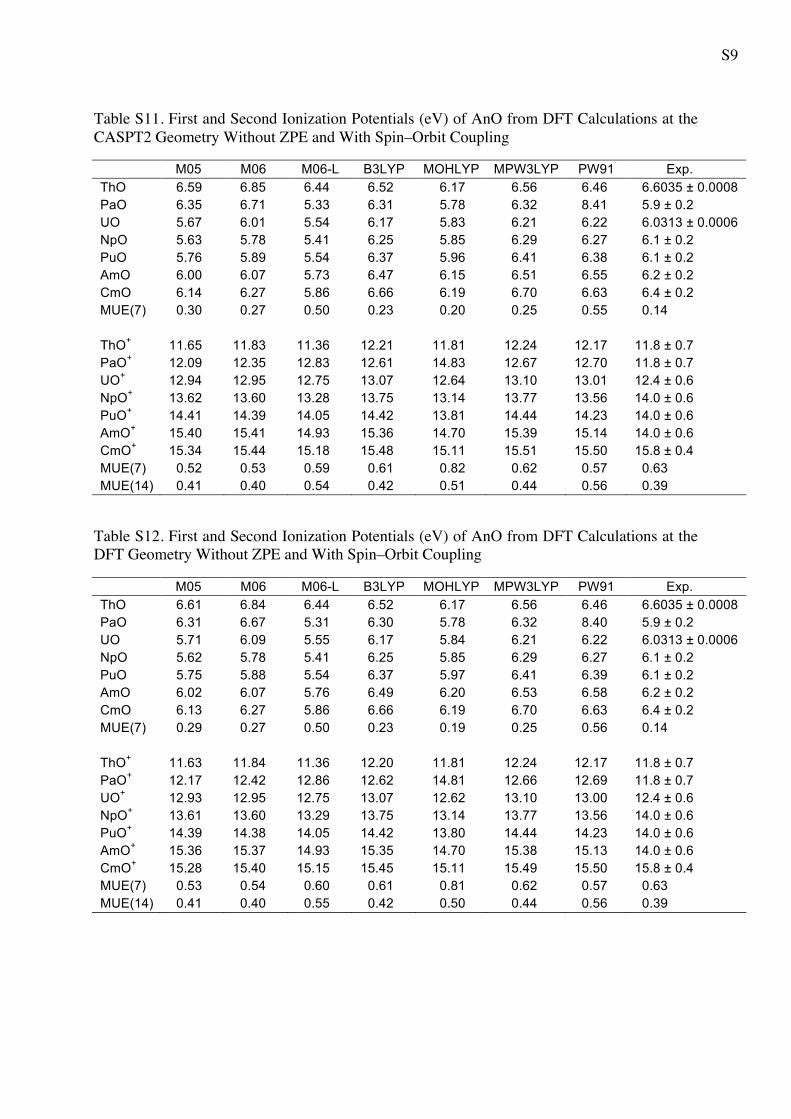
S9
Table S11. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the CASPT2 Geometry Without ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.59 6.85 6.44 6.52 6.17 6.56 6.46 6.6035 ± 0.0008 PaO 6.35 6.71 5.33 6.31 5.78 6.32 8.41 5.9 ± 0.2 UO 5.67 6.01 5.54 6.17 5.83 6.21 6.22 6.0313 ± 0.0006 NpO 5.63 5.78 5.41 6.25 5.85 6.29 6.27 6.1 ± 0.2 PuO 5.76 5.89 5.54 6.37 5.96 6.41 6.38 6.1 ± 0.2 AmO 6.00 6.07 5.73 6.47 6.15 6.51 6.55 6.2 ± 0.2 CmO 6.14 6.27 5.86 6.66 6.19 6.70 6.63 6.4 ± 0.2 MUE(7) 0.30 0.27 0.50 0.23 0.20 0.25 0.55 0.14 ThO+ 11.65 11.83 11.36 12.21 11.81 12.24 12.17 11.8 ± 0.7 PaO+ 12.09 12.35 12.83 12.61 14.83 12.67 12.70 11.8 ± 0.7 UO+ 12.94 12.95 12.75 13.07 12.64 13.10 13.01 12.4 ± 0.6 NpO+ 13.62 13.60 13.28 13.75 13.14 13.77 13.56 14.0 ± 0.6 PuO+ 14.41 14.39 14.05 14.42 13.81 14.44 14.23 14.0 ± 0.6 AmO+ 15.40 15.41 14.93 15.36 14.70 15.39 15.14 14.0 ± 0.6 CmO+ 15.34 15.44 15.18 15.48 15.11 15.51 15.50 15.8 ± 0.4 MUE(7) 0.52 0.53 0.59 0.61 0.82 0.62 0.57 0.63 MUE(14) 0.41 0.40 0.54 0.42 0.51 0.44 0.56 0.39
Table S12. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the DFT Geometry Without ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.61 6.84 6.44 6.52 6.17 6.56 6.46 6.6035 ± 0.0008 PaO 6.31 6.67 5.31 6.30 5.78 6.32 8.40 5.9 ± 0.2 UO 5.71 6.09 5.55 6.17 5.84 6.21 6.22 6.0313 ± 0.0006 NpO 5.62 5.78 5.41 6.25 5.85 6.29 6.27 6.1 ± 0.2 PuO 5.75 5.88 5.54 6.37 5.97 6.41 6.39 6.1 ± 0.2 AmO 6.02 6.07 5.76 6.49 6.20 6.53 6.58 6.2 ± 0.2 CmO 6.13 6.27 5.86 6.66 6.19 6.70 6.63 6.4 ± 0.2 MUE(7) 0.29 0.27 0.50 0.23 0.19 0.25 0.56 0.14 ThO+ 11.63 11.84 11.36 12.20 11.81 12.24 12.17 11.8 ± 0.7 PaO+ 12.17 12.42 12.86 12.62 14.81 12.66 12.69 11.8 ± 0.7 UO+ 12.93 12.95 12.75 13.07 12.62 13.10 13.00 12.4 ± 0.6 NpO+ 13.61 13.60 13.29 13.75 13.14 13.77 13.56 14.0 ± 0.6 PuO+ 14.39 14.38 14.05 14.42 13.80 14.44 14.23 14.0 ± 0.6 AmO+ 15.36 15.37 14.93 15.35 14.70 15.38 15.13 14.0 ± 0.6 CmO+ 15.28 15.40 15.15 15.45 15.11 15.49 15.50 15.8 ± 0.4 MUE(7) 0.53 0.54 0.60 0.61 0.81 0.62 0.57 0.63 MUE(14) 0.41 0.40 0.55 0.42 0.50 0.44 0.56 0.39
S10
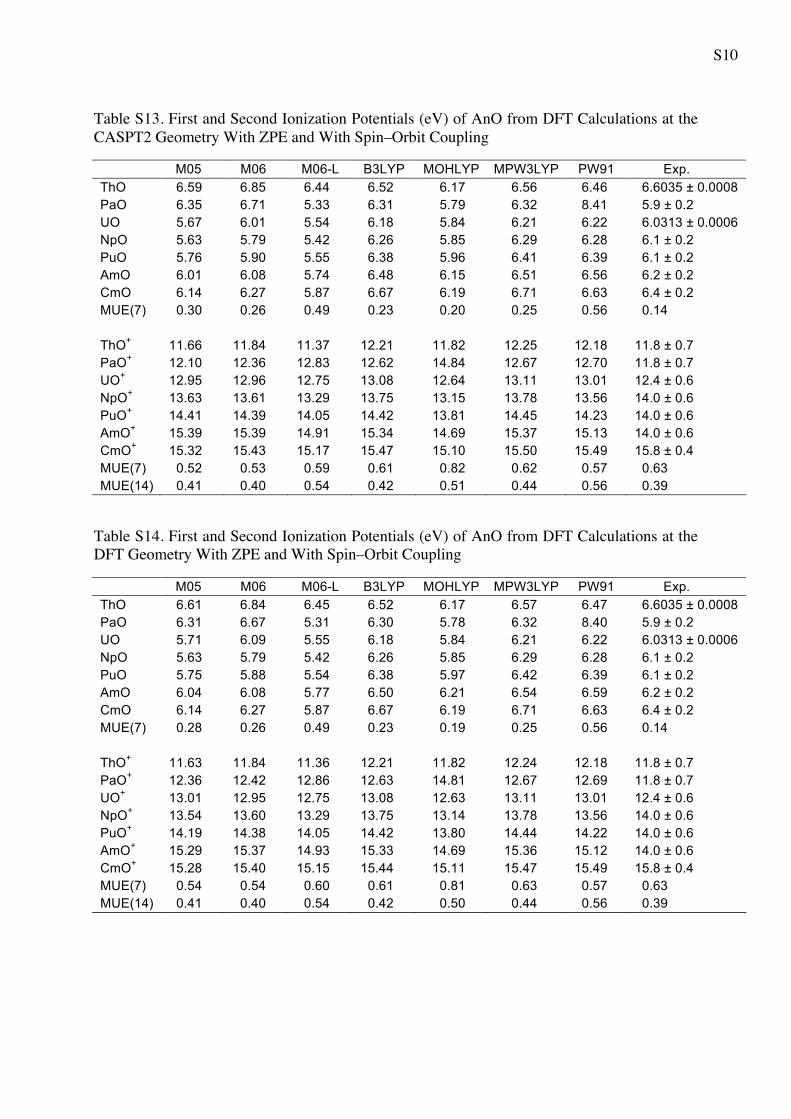
Table S13. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the CASPT2 Geometry With ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.59 6.85 6.44 6.52 6.17 6.56 6.46 6.6035 ± 0.0008 PaO 6.35 6.71 5.33 6.31 5.79 6.32 8.41 5.9 ± 0.2 UO 5.67 6.01 5.54 6.18 5.84 6.21 6.22 6.0313 ± 0.0006 NpO 5.63 5.79 5.42 6.26 5.85 6.29 6.28 6.1 ± 0.2 PuO 5.76 5.90 5.55 6.38 5.96 6.41 6.39 6.1 ± 0.2 AmO 6.01 6.08 5.74 6.48 6.15 6.51 6.56 6.2 ± 0.2 CmO 6.14 6.27 5.87 6.67 6.19 6.71 6.63 6.4 ± 0.2 MUE(7) 0.30 0.26 0.49 0.23 0.20 0.25 0.56 0.14 ThO+ 11.66 11.84 11.37 12.21 11.82 12.25 12.18 11.8 ± 0.7 PaO+ 12.10 12.36 12.83 12.62 14.84 12.67 12.70 11.8 ± 0.7 UO+ 12.95 12.96 12.75 13.08 12.64 13.11 13.01 12.4 ± 0.6 NpO+ 13.63 13.61 13.29 13.75 13.15 13.78 13.56 14.0 ± 0.6 PuO+ 14.41 14.39 14.05 14.42 13.81 14.45 14.23 14.0 ± 0.6 AmO+ 15.39 15.39 14.91 15.34 14.69 15.37 15.13 14.0 ± 0.6 CmO+ 15.32 15.43 15.17 15.47 15.10 15.50 15.49 15.8 ± 0.4 MUE(7) 0.52 0.53 0.59 0.61 0.82 0.62 0.57 0.63 MUE(14) 0.41 0.40 0.54 0.42 0.51 0.44 0.56 0.39
Table S14. First and Second Ionization Potentials (eV) of AnO from DFT Calculations at the DFT Geometry With ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO 6.61 6.84 6.45 6.52 6.17 6.57 6.47 6.6035 ± 0.0008 PaO 6.31 6.67 5.31 6.30 5.78 6.32 8.40 5.9 ± 0.2 UO 5.71 6.09 5.55 6.18 5.84 6.21 6.22 6.0313 ± 0.0006 NpO 5.63 5.79 5.42 6.26 5.85 6.29 6.28 6.1 ± 0.2 PuO 5.75 5.88 5.54 6.38 5.97 6.42 6.39 6.1 ± 0.2 AmO 6.04 6.08 5.77 6.50 6.21 6.54 6.59 6.2 ± 0.2 CmO 6.14 6.27 5.87 6.67 6.19 6.71 6.63 6.4 ± 0.2 MUE(7) 0.28 0.26 0.49 0.23 0.19 0.25 0.56 0.14 ThO+ 11.63 11.84 11.36 12.21 11.82 12.24 12.18 11.8 ± 0.7 PaO+ 12.36 12.42 12.86 12.63 14.81 12.67 12.69 11.8 ± 0.7 UO+ 13.01 12.95 12.75 13.08 12.63 13.11 13.01 12.4 ± 0.6 NpO+ 13.54 13.60 13.29 13.75 13.14 13.78 13.56 14.0 ± 0.6 PuO+ 14.19 14.38 14.05 14.42 13.80 14.44 14.22 14.0 ± 0.6 AmO+ 15.29 15.37 14.93 15.33 14.69 15.36 15.12 14.0 ± 0.6 CmO+ 15.28 15.40 15.15 15.44 15.11 15.47 15.49 15.8 ± 0.4 MUE(7) 0.54 0.54 0.60 0.61 0.81 0.63 0.57 0.63 MUE(14) 0.41 0.40 0.54 0.42 0.50 0.44 0.56 0.39
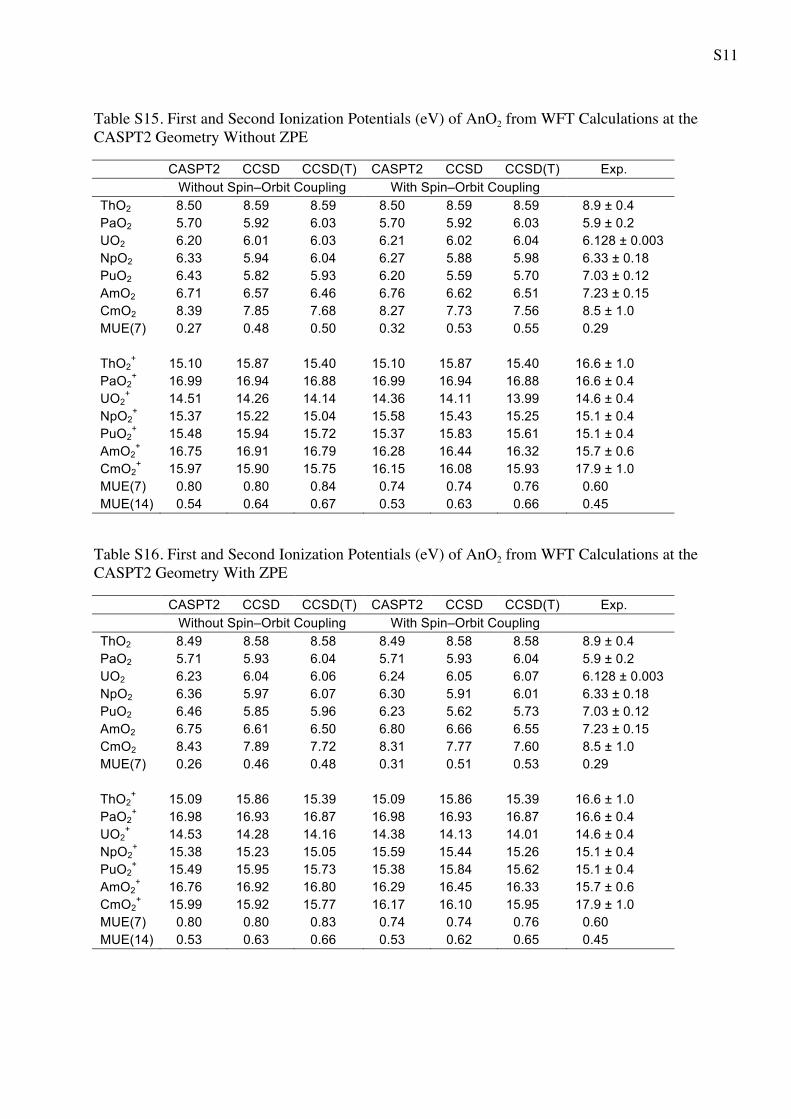
S11
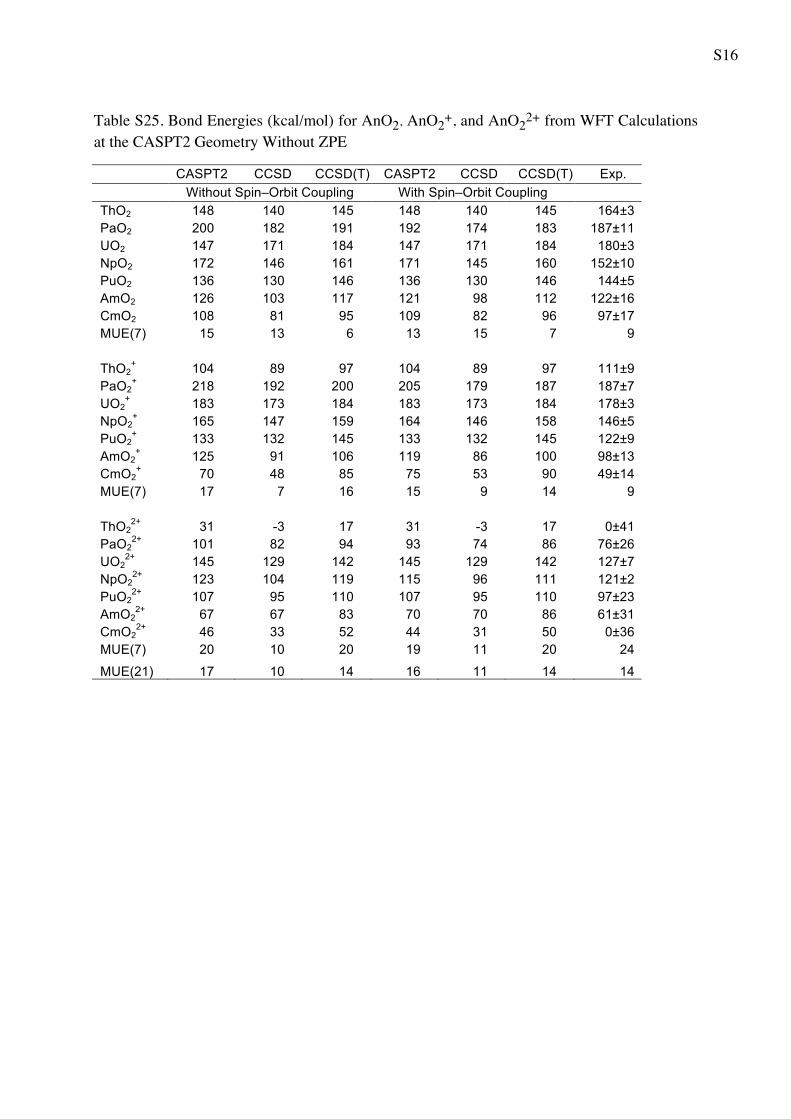
Table S15. First and Second Ionization Potentials (eV) of AnO2 from WFT Calculations at the CASPT2 Geometry Without ZPE
CASPT2 CCSD CCSD(T) CASPT2 CCSD CCSD(T) Exp. Without Spin–Orbit Coupling With Spin–Orbit Coupling
ThO2 8.50 8.59 8.59 8.50 8.59 8.59 8.9 ± 0.4 PaO2 5.70 5.92 6.03 5.70 5.92 6.03 5.9 ± 0.2 UO2 6.20 6.01 6.03 6.21 6.02 6.04 6.128 ± 0.003 NpO2 6.33 5.94 6.04 6.27 5.88 5.98 6.33 ± 0.18 PuO2 6.43 5.82 5.93 6.20 5.59 5.70 7.03 ± 0.12 AmO2 6.71 6.57 6.46 6.76 6.62 6.51 7.23 ± 0.15 CmO2 8.39 7.85 7.68 8.27 7.73 7.56 8.5 ± 1.0 MUE(7) 0.27 0.48 0.50 0.32 0.53 0.55 0.29 ThO2
+ 15.10 15.87 15.40 15.10 15.87 15.40 16.6 ± 1.0 PaO2
+ 16.99 16.94 16.88 16.99 16.94 16.88 16.6 ± 0.4 UO2
+ 14.51 14.26 14.14 14.36 14.11 13.99 14.6 ± 0.4 NpO2
+ 15.37 15.22 15.04 15.58 15.43 15.25 15.1 ± 0.4 PuO2
+ 15.48 15.94 15.72 15.37 15.83 15.61 15.1 ± 0.4 AmO2
+ 16.75 16.91 16.79 16.28 16.44 16.32 15.7 ± 0.6 CmO2
+ 15.97 15.90 15.75 16.15 16.08 15.93 17.9 ± 1.0 MUE(7) 0.80 0.80 0.84 0.74 0.74 0.76 0.60 MUE(14) 0.54 0.64 0.67 0.53 0.63 0.66 0.45
Table S16. First and Second Ionization Potentials (eV) of AnO2 from WFT Calculations at the CASPT2 Geometry With ZPE
CASPT2 CCSD CCSD(T) CASPT2 CCSD CCSD(T) Exp. Without Spin–Orbit Coupling With Spin–Orbit Coupling
ThO2 8.49 8.58 8.58 8.49 8.58 8.58 8.9 ± 0.4 PaO2 5.71 5.93 6.04 5.71 5.93 6.04 5.9 ± 0.2 UO2 6.23 6.04 6.06 6.24 6.05 6.07 6.128 ± 0.003 NpO2 6.36 5.97 6.07 6.30 5.91 6.01 6.33 ± 0.18 PuO2 6.46 5.85 5.96 6.23 5.62 5.73 7.03 ± 0.12 AmO2 6.75 6.61 6.50 6.80 6.66 6.55 7.23 ± 0.15 CmO2 8.43 7.89 7.72 8.31 7.77 7.60 8.5 ± 1.0 MUE(7) 0.26 0.46 0.48 0.31 0.51 0.53 0.29 ThO2
+ 15.09 15.86 15.39 15.09 15.86 15.39 16.6 ± 1.0 PaO2
+ 16.98 16.93 16.87 16.98 16.93 16.87 16.6 ± 0.4 UO2
+ 14.53 14.28 14.16 14.38 14.13 14.01 14.6 ± 0.4 NpO2
+ 15.38 15.23 15.05 15.59 15.44 15.26 15.1 ± 0.4 PuO2
+ 15.49 15.95 15.73 15.38 15.84 15.62 15.1 ± 0.4 AmO2
+ 16.76 16.92 16.80 16.29 16.45 16.33 15.7 ± 0.6 CmO2
+ 15.99 15.92 15.77 16.17 16.10 15.95 17.9 ± 1.0 MUE(7) 0.80 0.80 0.83 0.74 0.74 0.76 0.60 MUE(14) 0.53 0.63 0.66 0.53 0.62 0.65 0.45

S12
Table S17. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the CASPT2 Geometry Without ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.17 8.40 7.94 8.57 8.07 8.61 8.40 8.9 ± 0.4 PaO2 5.77 6.10 6.57 6.33 7.19 6.35 6.28 5.9 ± 0.2 UO2 6.06 6.03 5.78 6.18 5.91 6.22 6.27 6.128 ± 0.003 NpO2 6.32 6.20 5.71 6.28 6.08 6.32 6.42 6.33 ± 0.18 PuO2 6.62 6.73 6.22 6.62 6.41 6.66 6.73 7.03 ± 0.12 AmO2 6.95 6.92 6.48 7.07 6.43 7.11 6.77 7.23 ± 0.15 CmO2 8.34 8.32 7.84 8.37 7.59 8.41 7.95 8.5 ± 1.0 MUE(7) 0.26 0.24 0.69 0.22 6.86 7.15 7.02 0.29 ThO2
+ 16.49 16.46 16.11 16.25 15.90 16.28 16.38 16.6 ± 1.0 PaO2
+ 16.68 17.02 16.49 17.07 17.63 17.10 16.86 16.6 ± 0.4 UO2
+ 15.58 15.35 15.02 15.23 14.60 15.25 15.09 14.6 ± 0.4 NpO2
+ 16.17 16.11 15.82 16.00 15.21 16.03 15.71 15.1 ± 0.4 PuO2
+ 16.59 16.53 16.04 16.42 15.83 16.44 16.23 15.1 ± 0.4 AmO2
+ 17.00 17.07 16.65 16.91 16.37 16.93 16.72 15.7 ± 0.6 CmO2
+ 16.19 16.27 15.81 16.33 15.47 16.36 15.90 17.9 ± 1.0 MUE(7) 0.96 0.97 0.82 0.92 15.91 16.39 16.18 0.60 MUE(14) 0.61 0.61 0.75 0.57 11.38 11.77 11.60 0.45
Table S18. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the DFT Geometry Without ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.31 8.53 8.01 8.57 8.02 8.62 8.39 8.9 ± 0.4 PaO2 5.81 6.12 6.57 6.33 7.15 6.35 6.28 5.9 ± 0.2 UO2 6.04 6.03 5.78 6.23 5.88 6.28 6.26 6.128 ± 0.003 NpO2 6.34 6.24 5.80 6.39 6.14 6.42 6.46 6.33 ± 0.18 PuO2 6.66 6.80 6.37 6.81 6.52 6.84 6.81 7.03 ± 0.12 AmO2 6.89 6.87 6.49 7.08 6.17 7.12 6.79 7.23 ± 0.15 CmO2 8.30 8.30 7.84 8.37 7.58 8.41 7.94 8.5 ± 1.0 MUE(7) 0.24 0.22 0.64 0.20 0.72 0.20 0.34 0.29 ThO2
+ 16.31 16.44 16.07 16.29 15.94 16.32 16.38 16.6 ± 1.0 PaO2
+ 16.70 17.04 16.45 16.96 17.47 17.00 16.70 16.6 ± 0.4 UO2
+ 15.48 15.27 14.99 15.21 14.64 15.24 15.11 14.6 ± 0.4 NpO2
+ 16.08 16.05 15.81 15.99 15.24 16.02 15.73 15.1 ± 0.4 PuO2
+ 16.55 16.50 16.04 16.42 15.84 16.45 16.24 15.1 ± 0.4 AmO2
+ 16.96 17.04 16.64 16.90 16.37 16.92 16.73 15.7 ± 0.6 CmO2
+ 16.21 16.29 15.81 16.32 15.42 16.35 15.86 17.9 ± 1.0 MUE(7) 0.95 0.94 0.82 0.90 0.80 0.91 0.81 0.60 MUE(14) 0.60 0.58 0.73 0.55 0.76 0.55 0.57 0.45

S13
Table S19. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the CASPT2 Geometry With ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.17 8.39 7.93 8.56 8.07 8.61 8.40 8.9 ± 0.4 PaO2 5.78 6.11 6.59 6.34 7.19 6.34 6.28 5.9 ± 0.2 UO2 6.09 6.06 5.80 6.19 5.91 6.22 6.27 6.128 ± 0.003 NpO2 6.35 6.23 5.74 6.31 6.08 6.32 6.42 6.33 ± 0.18 PuO2 6.65 6.76 6.26 6.65 6.41 6.66 6.72 7.03 ± 0.12 AmO2 6.99 6.96 6.52 7.11 6.43 7.11 6.77 7.23 ± 0.15 CmO2 8.37 8.36 7.86 8.39 7.59 8.41 7.95 8.5 ± 1.0 MUE(7) 0.24 0.22 0.67 0.21 0.70 0.20 0.35 0.29 ThO2
+ 16.52 16.45 16.11 16.25 15.90 16.28 16.38 16.6 ± 1.0 PaO2
+ 16.67 17.01 16.48 17.04 17.63 17.10 16.86 16.6 ± 0.4 UO2
+ 15.60 15.37 15.04 15.24 14.60 15.25 15.09 14.6 ± 0.4 NpO2
+ 16.17 16.12 15.83 16.01 15.21 16.03 15.71 15.1 ± 0.4 PuO2
+ 16.60 16.54 16.05 16.43 15.83 16.44 16.23 15.1 ± 0.4 AmO2
+ 17.01 17.09 16.66 16.92 16.37 16.93 16.72 15.7 ± 0.6 CmO2
+ 16.20 16.29 15.83 16.35 15.47 16.36 15.90 17.9 ± 1.0 MUE(7) 0.96 0.97 0.82 0.92 0.81 0.93 0.82 0.60 MUE(14) 0.60 0.60 0.75 0.56 0.76 0.57 0.58 0.45
Table S20. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the DFT Geometry With ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.31 8.52 8.00 8.56 8.02 8.62 8.39 8.9 ± 0.4 PaO2 5.82 6.13 6.59 6.34 7.15 6.35 6.27 5.9 ± 0.2 UO2 6.07 6.06 5.80 6.24 5.88 6.28 6.26 6.128 ± 0.003 NpO2 6.37 6.27 5.83 6.42 6.14 6.42 6.46 6.33 ± 0.18 PuO2 6.69 6.83 6.40 6.84 6.52 6.84 6.81 7.03 ± 0.12 AmO2 6.93 6.91 6.53 7.12 6.17 7.12 6.79 7.23 ± 0.15 CmO2 8.33 8.34 7.86 8.39 7.58 8.41 7.94 8.5 ± 1.0 MUE(7) 0.23 0.20 0.62 0.20 0.72 0.20 0.34 0.29 ThO2
+ 16.34 16.43 16.07 16.30 15.94 16.32 16.38 16.6 ± 1.0 PaO2
+ 16.70 17.03 16.44 16.93 17.47 17.00 16.70 16.6 ± 0.4 UO2
+ 15.50 15.29 15.01 15.23 14.64 15.24 15.11 14.6 ± 0.4 NpO2
+ 16.09 16.06 15.81 16.00 15.24 16.02 15.73 15.1 ± 0.4 PuO2
+ 16.56 16.51 16.05 16.43 15.83 16.45 16.24 15.1 ± 0.4 AmO2
+ 16.97 17.06 16.65 16.91 16.37 16.92 16.72 15.7 ± 0.6 CmO2
+ 16.23 16.30 15.82 16.34 15.42 16.35 15.86 17.9 ± 1.0 MUE(7) 0.95 0.94 0.83 0.90 0.80 0.91 0.81 0.60 MUE(14) 0.59 0.57 0.73 0.55 0.76 0.55 0.57 0.45

S14
Table S21. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the CASPT2 Geometry Without ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.17 8.40 7.94 8.57 8.07 8.61 8.40 8.9 ± 0.4 PaO2 5.77 6.10 6.57 6.33 7.19 6.35 6.28 5.9 ± 0.2 UO2 6.07 6.04 5.79 6.19 5.92 6.23 6.28 6.128 ± 0.003 NpO2 6.26 6.14 5.65 6.22 6.02 6.26 6.36 6.33 ± 0.18 PuO2 6.39 6.50 5.99 6.39 6.18 6.43 6.50 7.03 ± 0.12 AmO2 7.00 6.97 6.53 7.12 6.48 7.16 6.82 7.23 ± 0.15 CmO2 8.22 8.20 7.72 8.25 7.47 8.29 7.83 8.5 ± 1.0 MUE(7) 0.31 0.29 0.74 0.27 0.75 0.25 0.38 0.29 ThO2
+ 16.49 16.46 16.11 16.25 15.90 16.28 16.38 16.6 ± 1.0 PaO2
+ 16.68 17.02 16.49 17.07 17.63 17.10 16.86 16.6 ± 0.4 UO2
+ 15.43 15.20 14.87 15.08 14.45 15.10 14.94 14.6 ± 0.4 NpO2
+ 16.38 16.32 16.03 16.21 15.42 16.24 15.92 15.1 ± 0.4 PuO2
+ 16.48 16.42 15.93 16.31 15.72 16.33 16.12 15.1 ± 0.4 AmO2
+ 16.53 16.60 16.18 16.44 15.90 16.46 16.25 15.7 ± 0.6 CmO2
+ 16.37 16.45 15.99 16.51 15.65 16.54 16.08 17.9 ± 1.0 MUE(7) 0.86 0.87 0.72 0.82 0.75 0.83 0.72 0.60 MUE(14) 0.58 0.58 0.73 0.55 0.75 0.54 0.55 0.45
Table S22. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the DFT Geometry Without ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.31 8.53 8.01 8.57 8.02 8.62 8.39 8.9 ± 0.4 PaO2 5.81 6.12 6.57 6.33 7.15 6.35 6.28 5.9 ± 0.2 UO2 6.05 6.04 5.79 6.24 5.89 6.29 6.27 6.128 ± 0.003 NpO2 6.28 6.18 5.74 6.33 6.08 6.36 6.40 6.33 ± 0.18 PuO2 6.43 6.57 6.14 6.58 6.29 6.61 6.58 7.03 ± 0.12 AmO2 6.94 6.92 6.54 7.13 6.22 7.17 6.84 7.23 ± 0.15 CmO2 8.18 8.18 7.72 8.25 7.46 8.29 7.82 8.5 ± 1.0 MUE(7) 0.29 0.27 0.69 0.24 0.77 0.23 0.38 0.29 ThO2
+ 16.31 16.44 16.07 16.29 15.94 16.32 16.38 16.6 ± 1.0 PaO2
+ 16.70 17.04 16.45 16.96 17.47 17.00 16.70 16.6 ± 0.4 UO2
+ 15.33 15.12 14.84 15.06 14.49 15.09 14.96 14.6 ± 0.4 NpO2
+ 16.29 16.26 16.02 16.20 15.45 16.23 15.94 15.1 ± 0.4 PuO2
+ 16.44 16.39 15.93 16.31 15.73 16.34 16.13 15.1 ± 0.4 AmO2
+ 16.49 16.57 16.17 16.43 15.90 16.45 16.26 15.7 ± 0.6 CmO2
+ 16.39 16.47 15.99 16.50 15.60 16.53 16.04 17.9 ± 1.0 MUE(7) 0.85 0.84 0.72 0.80 0.73 0.81 0.71 0.60 MUE(14) 0.57 0.56 0.71 0.52 0.75 0.52 0.54 0.45

S15
Table S23. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the CASPT2 Geometry With ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.17 8.39 7.93 8.56 8.07 8.61 8.40 8.9 ± 0.4 PaO2 5.78 6.11 6.59 6.34 7.19 6.34 6.28 5.9 ± 0.2 UO2 6.10 6.07 5.81 6.20 5.92 6.23 6.28 6.128 ± 0.003 NpO2 6.29 6.17 5.68 6.25 6.02 6.26 6.36 6.33 ± 0.18 PuO2 6.42 6.53 6.03 6.42 6.18 6.43 6.49 7.03 ± 0.12 AmO2 7.04 7.01 6.57 7.16 6.48 7.16 6.82 7.23 ± 0.15 CmO2 8.25 8.24 7.74 8.27 7.47 8.29 7.83 8.5 ± 1.0 MUE(7) 0.28 0.27 0.72 0.26 0.75 0.25 0.38 0.29 ThO2
+ 16.52 16.45 16.11 16.25 15.90 16.28 16.38 16.6 ± 1.0 PaO2
+ 16.67 17.01 16.48 17.04 17.63 17.10 16.86 16.6 ± 0.4 UO2
+ 15.45 15.22 14.89 15.09 14.45 15.10 14.94 14.6 ± 0.4 NpO2
+ 16.38 16.33 16.04 16.22 15.42 16.24 15.92 15.1 ± 0.4 PuO2
+ 16.49 16.43 15.94 16.32 15.72 16.33 16.12 15.1 ± 0.4 AmO2
+ 16.54 16.62 16.19 16.45 15.90 16.46 16.25 15.7 ± 0.6 CmO2
+ 16.38 16.47 16.01 16.53 15.65 16.54 16.08 17.9 ± 1.0 MUE(7) 0.86 0.87 0.72 0.82 0.75 0.83 0.72 0.60 MUE(14) 0.57 0.57 0.72 0.54 0.75 0.54 0.55 0.45
Table S24. First and Second Ionization Potentials (eV) of AnO2 from DFT Calculations at the DFT Geometry With ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 8.31 8.52 8.00 8.56 8.02 8.62 8.39 8.9 ± 0.4 PaO2 5.82 6.13 6.59 6.34 7.15 6.35 6.27 5.9 ± 0.2 UO2 6.08 6.07 5.81 6.25 5.89 6.29 6.27 6.128 ± 0.003 NpO2 6.31 6.21 5.77 6.36 6.08 6.36 6.40 6.33 ± 0.18 PuO2 6.46 6.60 6.17 6.61 6.29 6.61 6.58 7.03 ± 0.12 AmO2 6.98 6.96 6.58 7.17 6.22 7.17 6.84 7.23 ± 0.15 CmO2 8.21 8.22 7.74 8.27 7.46 8.29 7.82 8.5 ± 1.0 MUE(7) 0.27 0.25 0.67 0.24 0.77 0.23 0.38 0.29 ThO2
+ 16.34 16.43 16.07 16.30 15.94 16.32 16.38 16.6 ± 1.0 PaO2
+ 16.70 17.03 16.44 16.93 17.47 17.00 16.70 16.6 ± 0.4 UO2
+ 15.35 15.14 14.86 15.08 14.49 15.09 14.96 14.6 ± 0.4 NpO2
+ 16.30 16.27 16.02 16.21 15.45 16.23 15.94 15.1 ± 0.4 PuO2
+ 16.45 16.40 15.94 16.32 15.72 16.34 16.13 15.1 ± 0.4 AmO2
+ 16.50 16.59 16.18 16.44 15.90 16.45 16.25 15.7 ± 0.6 CmO2
+ 16.41 16.48 16.00 16.52 15.60 16.53 16.04 17.9 ± 1.0 MUE(7) 0.85 0.84 0.73 0.80 0.73 0.81 0.71 0.60 MUE(14) 0.56 0.55 0.70 0.52 0.75 0.52 0.54 0.45
S16
Table S25. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from WFT Calculations at the CASPT2 Geometry Without ZPE
CASPT2 CCSD CCSD(T) CASPT2 CCSD CCSD(T) Exp. Without Spin–Orbit Coupling With Spin–Orbit Coupling
ThO2 148 140 145 148 140 145 164±3 PaO2 200 182 191 192 174 183 187±11 UO2 147 171 184 147 171 184 180±3 NpO2 172 146 161 171 145 160 152±10 PuO2 136 130 146 136 130 146 144±5 AmO2 126 103 117 121 98 112 122±16 CmO2 108 81 95 109 82 96 97±17 MUE(7) 15 13 6 13 15 7 9 ThO2
+ 104 89 97 104 89 97 111±9 PaO2
+ 218 192 200 205 179 187 187±7 UO2
+ 183 173 184 183 173 184 178±3 NpO2
+ 165 147 159 164 146 158 146±5 PuO2
+ 133 132 145 133 132 145 122±9 AmO2
+ 125 91 106 119 86 100 98±13 CmO2
+ 70 48 85 75 53 90 49±14 MUE(7) 17 7 16 15 9 14 9 ThO2
2+ 31 -3 17 31 -3 17 0±41 PaO2
2+ 101 82 94 93 74 86 76±26 UO2
2+ 145 129 142 145 129 142 127±7 NpO2
2+ 123 104 119 115 96 111 121±2 PuO2
2+ 107 95 110 107 95 110 97±23 AmO2
2+ 67 67 83 70 70 86 61±31 CmO2
2+ 46 33 52 44 31 50 0±36 MUE(7) 20 10 20 19 11 20 24
MUE(21) 17 10 14 16 11 14 14

S17
Table S26. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from WFT Calculations at the CASPT2 Geometry With ZPE.
CASPT2 CCSD CCSD(T) CASPT2 CCSD CCSD(T) Exp. Without Spin–Orbit Coupling With Spin–Orbit Coupling
ThO2 147 139 144 147 139 144 164±3 PaO2 198 180 189 190 172 181 187±11 UO2 146 170 183 146 170 183 180±3 NpO2 170 144 159 169 143 158 152±10 PuO2 134 128 144 134 128 144 144±5 AmO2 124 101 115 119 96 110 122±16 CmO2 107 80 94 108 81 95 97±17 MUE(7) 15 15 6 14 17 7 9 ThO2
+ 103 88 96 103 88 96 111±9 PaO2
+ 216 190 198 203 177 185 187±7 UO2
+ 181 171 182 181 171 182 178±3 NpO2
+ 163 145 157 162 144 156 146±5 PuO2
+ 131 130 143 131 130 143 122±9 AmO2
+ 123 89 104 117 84 98 98±13 CmO2
+ 68 46 83 73 51 88 49±14 MUE(7) 16 8 14 13 9 13 9 ThO2
2+ 30 -4 16 30 -4 16 0±41 PaO2
2+ 100 81 93 92 73 85 76±26 UO2
2+ 143 127 140 143 127 140 127±7 NpO2
2+ 121 102 117 113 94 109 121±2 PuO2
2+ 104 92 107 104 92 107 97±23 AmO2
2+ 64 64 80 67 67 83 61±31 CmO2
2+ 43 30 49 41 28 47 0±36 MUE(7) 18 9 18 18 10 19 24
MUE(21) 16 11 13 15 12 13 14
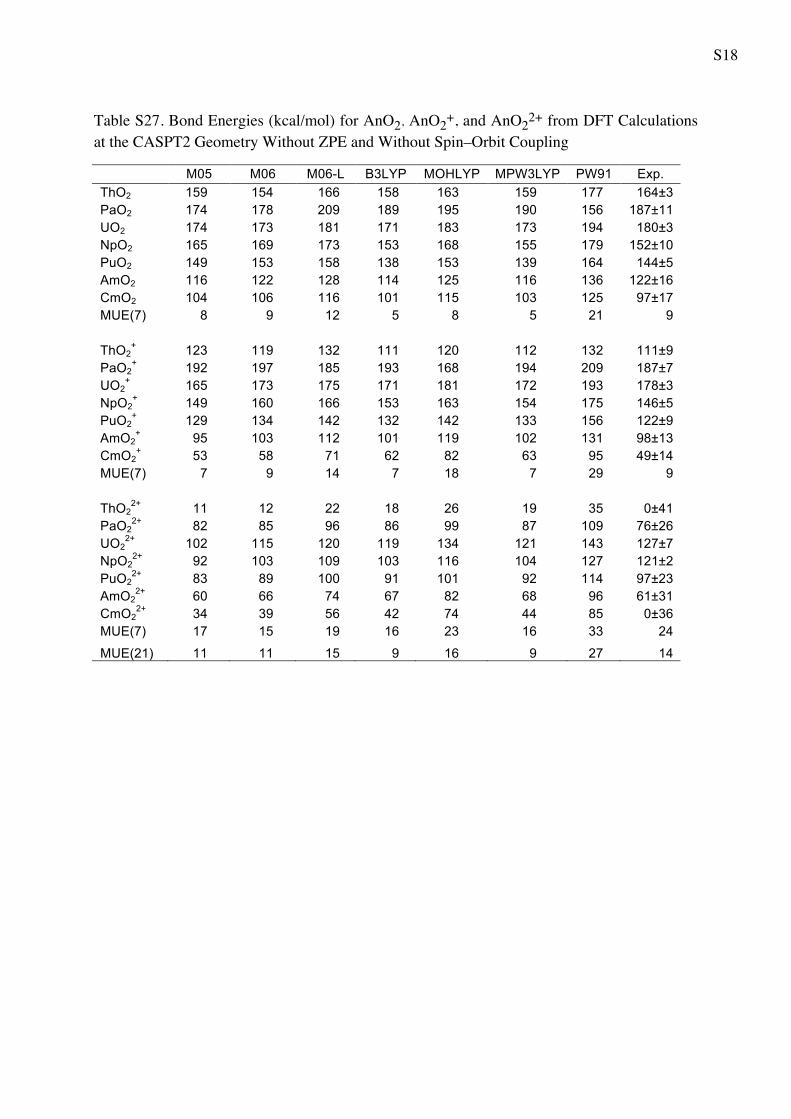
S18
Table S27. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the CASPT2 Geometry Without ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 159 154 166 158 163 159 177 164±3 PaO2 174 178 209 189 195 190 156 187±11 UO2 174 173 181 171 183 173 194 180±3 NpO2 165 169 173 153 168 155 179 152±10 PuO2 149 153 158 138 153 139 164 144±5 AmO2 116 122 128 114 125 116 136 122±16 CmO2 104 106 116 101 115 103 125 97±17 MUE(7) 8 9 12 5 8 5 21 9 ThO2
+ 123 119 132 111 120 112 132 111±9 PaO2
+ 192 197 185 193 168 194 209 187±7 UO2
+ 165 173 175 171 181 172 193 178±3 NpO2
+ 149 160 166 153 163 154 175 146±5 PuO2
+ 129 134 142 132 142 133 156 122±9 AmO2
+ 95 103 112 101 119 102 131 98±13 CmO2
+ 53 58 71 62 82 63 95 49±14 MUE(7) 7 9 14 7 18 7 29 9 ThO2
2+ 11 12 22 18 26 19 35 0±41 PaO2
2+ 82 85 96 86 99 87 109 76±26 UO2
2+ 102 115 120 119 134 121 143 127±7 NpO2
2+ 92 103 109 103 116 104 127 121±2 PuO2
2+ 83 89 100 91 101 92 114 97±23 AmO2
2+ 60 66 74 67 82 68 96 61±31 CmO2
2+ 34 39 56 42 74 44 85 0±36 MUE(7) 17 15 19 16 23 16 33 24
MUE(21) 11 11 15 9 16 9 27 14
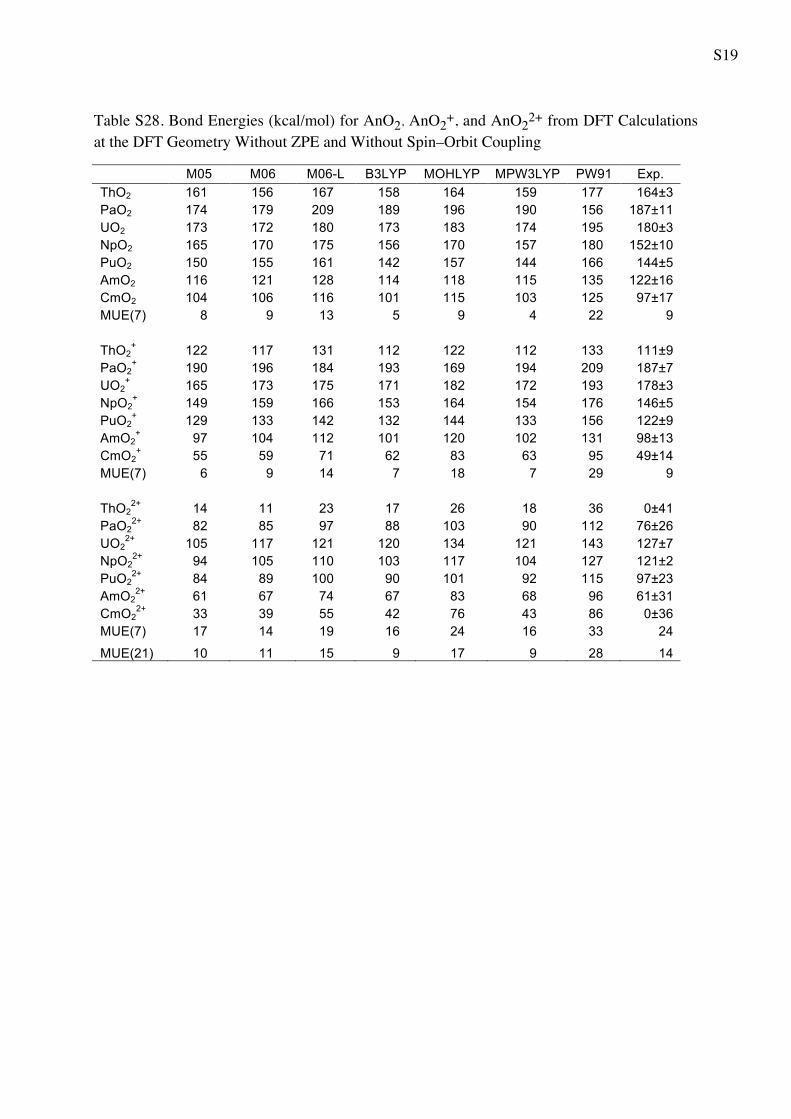
S19
Table S28. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the DFT Geometry Without ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 161 156 167 158 164 159 177 164±3 PaO2 174 179 209 189 196 190 156 187±11 UO2 173 172 180 173 183 174 195 180±3 NpO2 165 170 175 156 170 157 180 152±10 PuO2 150 155 161 142 157 144 166 144±5 AmO2 116 121 128 114 118 115 135 122±16 CmO2 104 106 116 101 115 103 125 97±17 MUE(7) 8 9 13 5 9 4 22 9 ThO2
+ 122 117 131 112 122 112 133 111±9 PaO2
+ 190 196 184 193 169 194 209 187±7 UO2
+ 165 173 175 171 182 172 193 178±3 NpO2
+ 149 159 166 153 164 154 176 146±5 PuO2
+ 129 133 142 132 144 133 156 122±9 AmO2
+ 97 104 112 101 120 102 131 98±13 CmO2
+ 55 59 71 62 83 63 95 49±14 MUE(7) 6 9 14 7 18 7 29 9 ThO2
2+ 14 11 23 17 26 18 36 0±41 PaO2
2+ 82 85 97 88 103 90 112 76±26 UO2
2+ 105 117 121 120 134 121 143 127±7 NpO2
2+ 94 105 110 103 117 104 127 121±2 PuO2
2+ 84 89 100 90 101 92 115 97±23 AmO2
2+ 61 67 74 67 83 68 96 61±31 CmO2
2+ 33 39 55 42 76 43 86 0±36 MUE(7) 17 14 19 16 24 16 33 24
MUE(21) 10 11 15 9 17 9 28 14
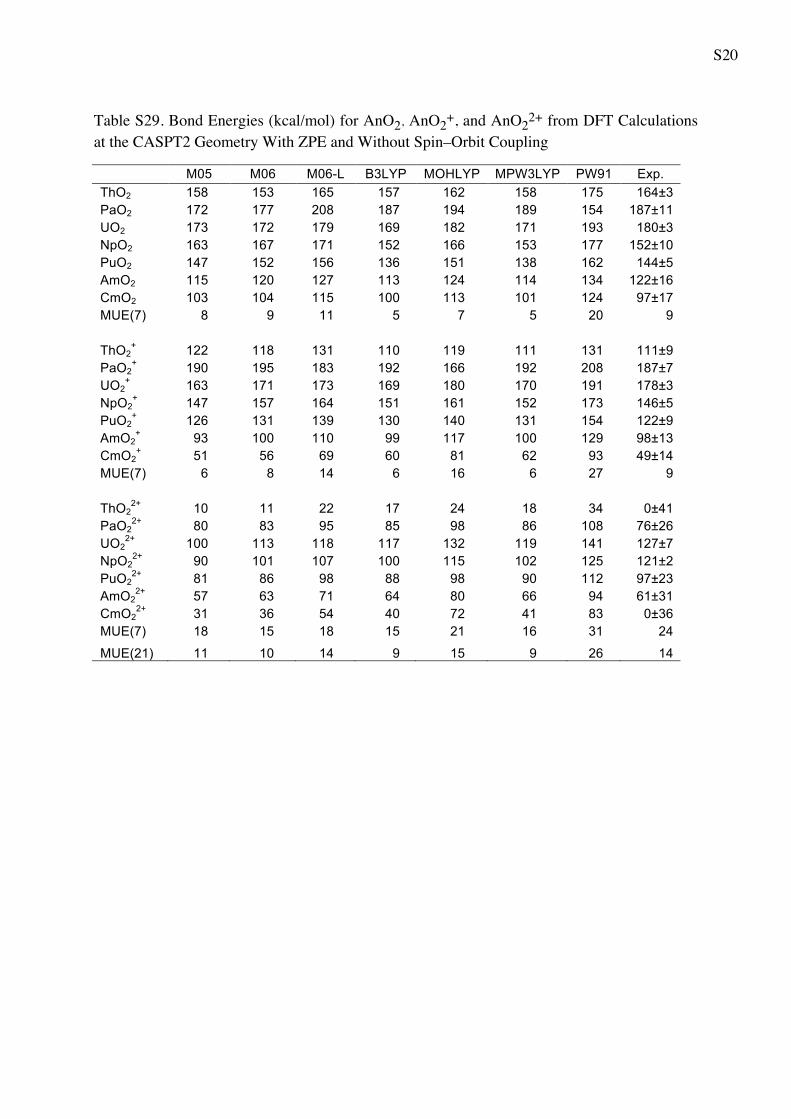
S20
Table S29. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the CASPT2 Geometry With ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 158 153 165 157 162 158 175 164±3 PaO2 172 177 208 187 194 189 154 187±11 UO2 173 172 179 169 182 171 193 180±3 NpO2 163 167 171 152 166 153 177 152±10 PuO2 147 152 156 136 151 138 162 144±5 AmO2 115 120 127 113 124 114 134 122±16 CmO2 103 104 115 100 113 101 124 97±17 MUE(7) 8 9 11 5 7 5 20 9 ThO2
+ 122 118 131 110 119 111 131 111±9 PaO2
+ 190 195 183 192 166 192 208 187±7 UO2
+ 163 171 173 169 180 170 191 178±3 NpO2
+ 147 157 164 151 161 152 173 146±5 PuO2
+ 126 131 139 130 140 131 154 122±9 AmO2
+ 93 100 110 99 117 100 129 98±13 CmO2
+ 51 56 69 60 81 62 93 49±14 MUE(7) 6 8 14 6 16 6 27 9 ThO2
2+ 10 11 22 17 24 18 34 0±41 PaO2
2+ 80 83 95 85 98 86 108 76±26 UO2
2+ 100 113 118 117 132 119 141 127±7 NpO2
2+ 90 101 107 100 115 102 125 121±2 PuO2
2+ 81 86 98 88 98 90 112 97±23 AmO2
2+ 57 63 71 64 80 66 94 61±31 CmO2
2+ 31 36 54 40 72 41 83 0±36 MUE(7) 18 15 18 15 21 16 31 24
MUE(21) 11 10 14 9 15 9 26 14

S21
Table S30. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the DFT Geometry With ZPE and Without Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 159 155 166 157 163 158 176 164±3 PaO2 173 177 207 187 194 189 154 187±11 UO2 172 170 179 171 182 172 193 180±3 NpO2 164 168 173 155 169 156 179 152±10 PuO2 148 153 159 141 155 142 165 144±5 AmO2 114 120 127 113 117 114 134 122±16 CmO2 103 105 115 100 113 101 124 97±17 MUE(7) 8 9 12 5 9 5 21 9 ThO2
+ 121 116 130 111 121 112 132 111±9 PaO2
+ 189 194 182 191 167 192 208 187±7 UO2
+ 163 171 173 169 180 170 191 178±3 NpO2
+ 147 157 164 151 162 152 174 146±5 PuO2
+ 126 131 139 130 141 131 154 122±9 AmO2
+ 94 101 110 99 118 100 129 98±13 CmO2
+ 53 57 69 60 81 62 93 49±14 MUE(7) 6 7 14 5 17 6 27 9 ThO2
2+ 12 11 22 17 25 18 35 0±41 PaO2
2+ 80 84 96 87 102 89 111 76±26 UO2
2+ 102 115 119 118 132 119 141 127±7 NpO2
2+ 92 102 108 101 115 102 125 121±2 PuO2
2+ 81 87 98 88 99 90 112 97±23 AmO2
2+ 58 64 71 64 80 66 94 61±31 CmO2
2+ 30 36 53 39 73 41 84 0±36 MUE(7) 17 14 18 16 22 16 32 24
MUE(21) 10 10 15 9 16 9 26 14
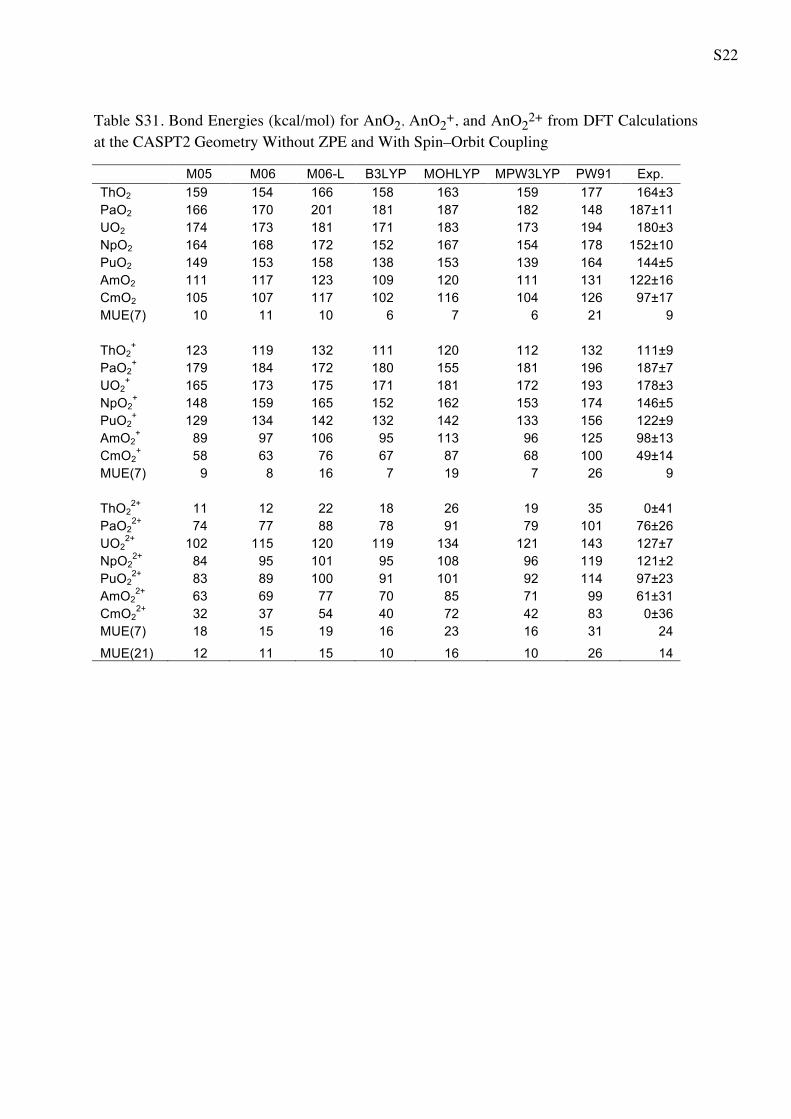
S22
Table S31. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the CASPT2 Geometry Without ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 159 154 166 158 163 159 177 164±3 PaO2 166 170 201 181 187 182 148 187±11 UO2 174 173 181 171 183 173 194 180±3 NpO2 164 168 172 152 167 154 178 152±10 PuO2 149 153 158 138 153 139 164 144±5 AmO2 111 117 123 109 120 111 131 122±16 CmO2 105 107 117 102 116 104 126 97±17 MUE(7) 10 11 10 6 7 6 21 9 ThO2
+ 123 119 132 111 120 112 132 111±9 PaO2
+ 179 184 172 180 155 181 196 187±7 UO2
+ 165 173 175 171 181 172 193 178±3 NpO2
+ 148 159 165 152 162 153 174 146±5 PuO2
+ 129 134 142 132 142 133 156 122±9 AmO2
+ 89 97 106 95 113 96 125 98±13 CmO2
+ 58 63 76 67 87 68 100 49±14 MUE(7) 9 8 16 7 19 7 26 9 ThO2
2+ 11 12 22 18 26 19 35 0±41 PaO2
2+ 74 77 88 78 91 79 101 76±26 UO2
2+ 102 115 120 119 134 121 143 127±7 NpO2
2+ 84 95 101 95 108 96 119 121±2 PuO2
2+ 83 89 100 91 101 92 114 97±23 AmO2
2+ 63 69 77 70 85 71 99 61±31 CmO2
2+ 32 37 54 40 72 42 83 0±36 MUE(7) 18 15 19 16 23 16 31 24
MUE(21) 12 11 15 10 16 10 26 14
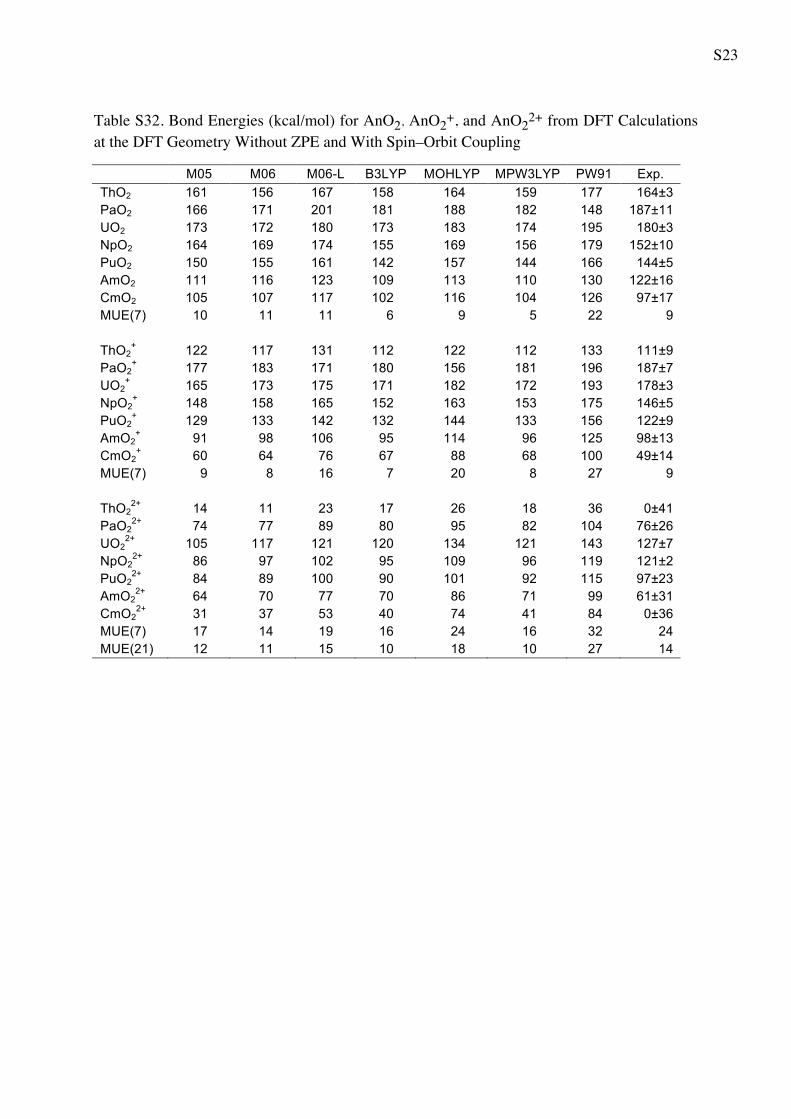
S23
Table S32. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the DFT Geometry Without ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 161 156 167 158 164 159 177 164±3 PaO2 166 171 201 181 188 182 148 187±11 UO2 173 172 180 173 183 174 195 180±3 NpO2 164 169 174 155 169 156 179 152±10 PuO2 150 155 161 142 157 144 166 144±5 AmO2 111 116 123 109 113 110 130 122±16 CmO2 105 107 117 102 116 104 126 97±17 MUE(7) 10 11 11 6 9 5 22 9 ThO2
+ 122 117 131 112 122 112 133 111±9 PaO2
+ 177 183 171 180 156 181 196 187±7 UO2
+ 165 173 175 171 182 172 193 178±3 NpO2
+ 148 158 165 152 163 153 175 146±5 PuO2
+ 129 133 142 132 144 133 156 122±9 AmO2
+ 91 98 106 95 114 96 125 98±13 CmO2
+ 60 64 76 67 88 68 100 49±14 MUE(7) 9 8 16 7 20 8 27 9 ThO2
2+ 14 11 23 17 26 18 36 0±41 PaO2
2+ 74 77 89 80 95 82 104 76±26 UO2
2+ 105 117 121 120 134 121 143 127±7 NpO2
2+ 86 97 102 95 109 96 119 121±2 PuO2
2+ 84 89 100 90 101 92 115 97±23 AmO2
2+ 64 70 77 70 86 71 99 61±31 CmO2
2+ 31 37 53 40 74 41 84 0±36 MUE(7) 17 14 19 16 24 16 32 24 MUE(21) 12 11 15 10 18 10 27 14

S24
Table S33. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the CASPT2 Geometry With ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 158 153 165 157 162 158 175 164±3 PaO2 164 169 200 179 186 181 146 187±11 UO2 173 172 179 169 182 171 193 180±3 NpO2 162 166 170 151 165 152 176 152±10 PuO2 147 152 156 136 151 138 162 144±5 AmO2 110 115 122 108 119 109 129 122±16 CmO2 104 105 116 101 114 102 125 97±17 MUE(7) 10 11 9 7 7 7 20 9 ThO2
+ 122 118 131 110 119 111 131 111±9 PaO2
+ 177 182 170 179 153 179 195 187±7 UO2
+ 163 171 173 169 180 170 191 178±3 NpO2
+ 146 156 163 150 160 151 172 146±5 PuO2
+ 126 131 139 130 140 131 154 122±9 AmO2
+ 87 94 104 93 111 94 123 98±13 CmO2
+ 56 61 74 65 86 67 98 49±14 MUE(7) 8 8 15 7 18 7 25 9 ThO2
2+ 10 11 22 17 24 18 34 0±41 PaO2
2+ 72 75 87 77 90 78 100 76±26 UO2
2+ 100 113 118 117 132 119 141 127±7 NpO2
2+ 82 93 99 92 107 94 117 121±2 PuO2
2+ 81 86 98 88 98 90 112 97±23 AmO2
2+ 60 66 74 67 83 69 97 61±31 CmO2
2+ 29 34 52 38 70 39 81 0±36 MUE(7) 18 15 18 16 22 16 30 24 MUE(21) 12 11 14 10 15 10 25 14
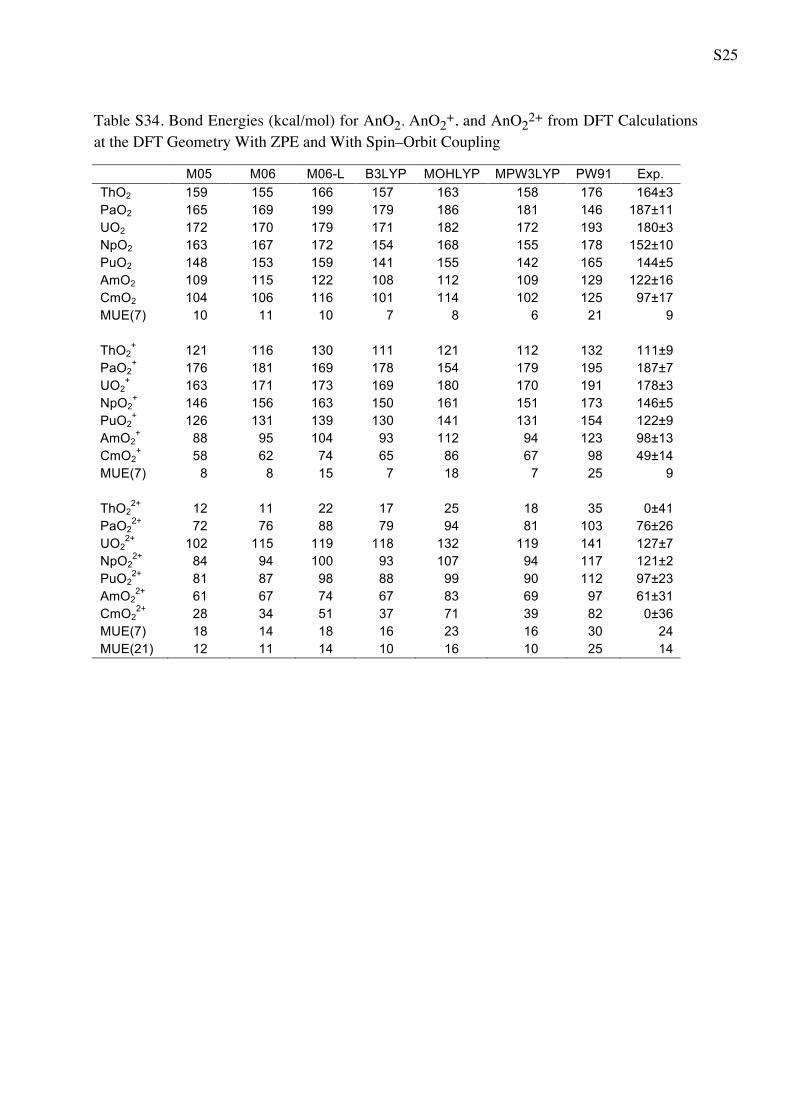
S25
Table S34. Bond Energies (kcal/mol) for AnO2, AnO2+, and AnO22+ from DFT Calculations at the DFT Geometry With ZPE and With Spin–Orbit Coupling
M05 M06 M06-L B3LYP MOHLYP MPW3LYP PW91 Exp. ThO2 159 155 166 157 163 158 176 164±3 PaO2 165 169 199 179 186 181 146 187±11 UO2 172 170 179 171 182 172 193 180±3 NpO2 163 167 172 154 168 155 178 152±10 PuO2 148 153 159 141 155 142 165 144±5 AmO2 109 115 122 108 112 109 129 122±16 CmO2 104 106 116 101 114 102 125 97±17 MUE(7) 10 11 10 7 8 6 21 9 ThO2
+ 121 116 130 111 121 112 132 111±9 PaO2
+ 176 181 169 178 154 179 195 187±7 UO2
+ 163 171 173 169 180 170 191 178±3 NpO2
+ 146 156 163 150 161 151 173 146±5 PuO2
+ 126 131 139 130 141 131 154 122±9 AmO2
+ 88 95 104 93 112 94 123 98±13 CmO2
+ 58 62 74 65 86 67 98 49±14 MUE(7) 8 8 15 7 18 7 25 9 ThO2
2+ 12 11 22 17 25 18 35 0±41 PaO2
2+ 72 76 88 79 94 81 103 76±26 UO2
2+ 102 115 119 118 132 119 141 127±7 NpO2
2+ 84 94 100 93 107 94 117 121±2 PuO2
2+ 81 87 98 88 99 90 112 97±23 AmO2
2+ 61 67 74 67 83 69 97 61±31 CmO2
2+ 28 34 51 37 71 39 82 0±36 MUE(7) 18 14 18 16 23 16 30 24 MUE(21) 12 11 14 10 16 10 25 14
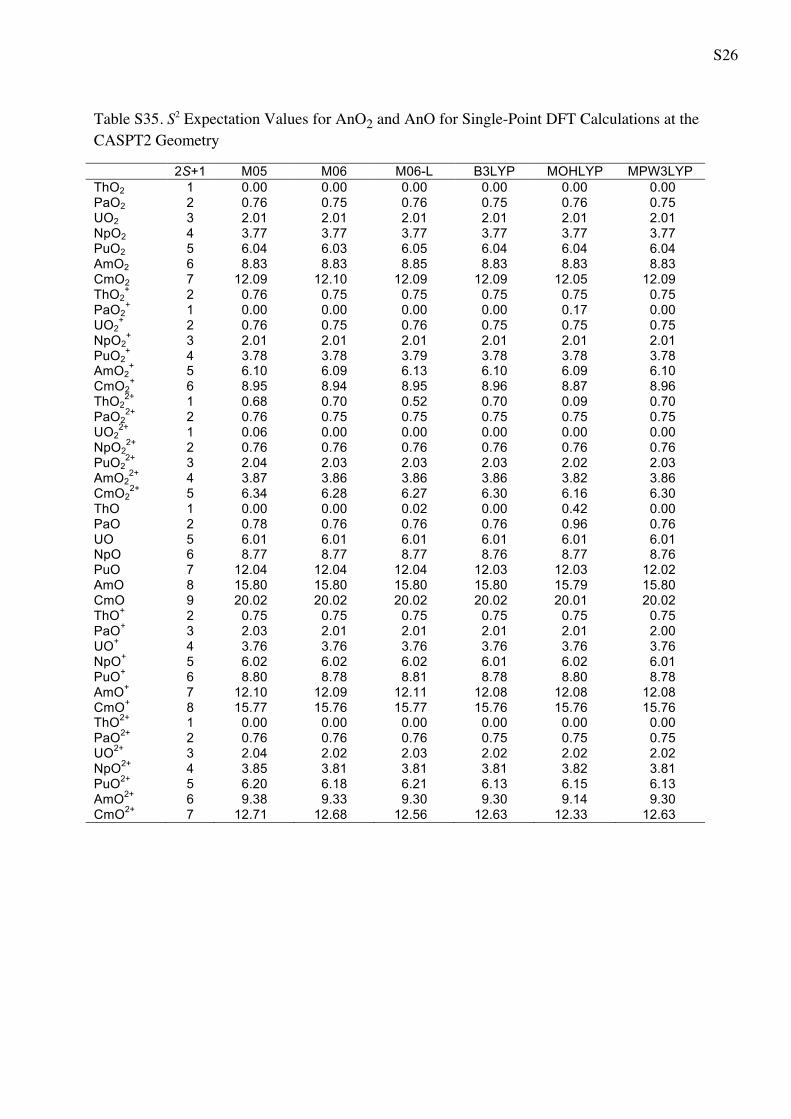
S26
Table S35. S2 Expectation Values for AnO2 and AnO for Single-Point DFT Calculations at the CASPT2 Geometry
2S+1 M05 M06 M06-L B3LYP MOHLYP MPW3LYP ThO2 1 0.00 0.00 0.00 0.00 0.00 0.00 PaO2 2 0.76 0.75 0.76 0.75 0.76 0.75 UO2 3 2.01 2.01 2.01 2.01 2.01 2.01 NpO2 4 3.77 3.77 3.77 3.77 3.77 3.77 PuO2 5 6.04 6.03 6.05 6.04 6.04 6.04 AmO2 6 8.83 8.83 8.85 8.83 8.83 8.83 CmO2 7 12.09 12.10 12.09 12.09 12.05 12.09 ThO2
+ 2 0.76 0.75 0.75 0.75 0.75 0.75 PaO2
+ 1 0.00 0.00 0.00 0.00 0.17 0.00 UO2
+ 2 0.76 0.75 0.76 0.75 0.75 0.75 NpO2
+ 3 2.01 2.01 2.01 2.01 2.01 2.01 PuO2
+ 4 3.78 3.78 3.79 3.78 3.78 3.78 AmO2
+ 5 6.10 6.09 6.13 6.10 6.09 6.10 CmO2
+ 6 8.95 8.94 8.95 8.96 8.87 8.96 ThO2
2+ 1 0.68 0.70 0.52 0.70 0.09 0.70 PaO2
2+ 2 0.76 0.75 0.75 0.75 0.75 0.75 UO2
2+ 1 0.06 0.00 0.00 0.00 0.00 0.00 NpO2
2+ 2 0.76 0.76 0.76 0.76 0.76 0.76 PuO2
2+ 3 2.04 2.03 2.03 2.03 2.02 2.03 AmO2
2+ 4 3.87 3.86 3.86 3.86 3.82 3.86 CmO2
2+ 5 6.34 6.28 6.27 6.30 6.16 6.30 ThO 1 0.00 0.00 0.02 0.00 0.42 0.00 PaO 2 0.78 0.76 0.76 0.76 0.96 0.76 UO 5 6.01 6.01 6.01 6.01 6.01 6.01 NpO 6 8.77 8.77 8.77 8.76 8.77 8.76 PuO 7 12.04 12.04 12.04 12.03 12.03 12.02 AmO 8 15.80 15.80 15.80 15.80 15.79 15.80 CmO 9 20.02 20.02 20.02 20.02 20.01 20.02 ThO+ 2 0.75 0.75 0.75 0.75 0.75 0.75 PaO+ 3 2.03 2.01 2.01 2.01 2.01 2.00 UO+ 4 3.76 3.76 3.76 3.76 3.76 3.76 NpO+ 5 6.02 6.02 6.02 6.01 6.02 6.01 PuO+ 6 8.80 8.78 8.81 8.78 8.80 8.78 AmO+ 7 12.10 12.09 12.11 12.08 12.08 12.08 CmO+ 8 15.77 15.76 15.77 15.76 15.76 15.76 ThO2+ 1 0.00 0.00 0.00 0.00 0.00 0.00 PaO2+ 2 0.76 0.76 0.76 0.75 0.75 0.75 UO2+ 3 2.04 2.02 2.03 2.02 2.02 2.02 NpO2+ 4 3.85 3.81 3.81 3.81 3.82 3.81 PuO2+ 5 6.20 6.18 6.21 6.13 6.15 6.13 AmO2+ 6 9.38 9.33 9.30 9.30 9.14 9.30 CmO2+ 7 12.71 12.68 12.56 12.63 12.33 12.63
S27
Table S36. Bond Distances (Ǻ) for AnO2 Obtained by Using a Different Basis Set for Oxygen , in Particular, the Correlation-Consistent aug-cc-pVTZ Basis
B3LYP B3P86 MPW1-PW91 PBE0 PBE TPSS HCTH BLYP BP86
ThO2a 1.898 1.884 1.882 1.881 1.899 1.900 1.879 1.917 1.902
PaO2 1.806 1.793 1.789 1.787 1.828 1.813 1.802 1.847 1.833 UO2 1.789 1.814 1.812 1.810 - b - b - b - b - b NpO2 1.821 1.804 1.801 1.800 - b - b - b - b - b PuO2 1.814 1.797 1.794 1.792 - b - b - b - b - b AmO2 1.826 1.809 1.807 1.805 - b 1.835 - b - b 1.835 CmO2 1.839 1.820 1.819 1.817 1.839 1.844 1.826 1.863 1.844 ThO2
+ 1.868 1.851 1.847 1.846 1.870 1.872 1.845 1.868 1.874 PaO2
+ 1.767 1.756 1.752 1.751 1.777 1.776 1.757 1.793 1.780 UO2
+ 1.753 1.741 1.737 1.736 1.764 1.763 1.743 1.781 1.768 NpO2
+ 1.732 1.720 1.715 1.714 1.745 1.744 1.724 1.763 1.749 PuO2
+ 1.714 1.700 1.695 1.694 1.728 1.726 1.708 1.747 1.731 AmO2
+ 1.716 1.701 1.695 1.694 1.733 1.732 1.716 1.754 1.736 CmO2
+ 1.748 1.729 1.726 1.724 1.759 1.762 1.746 1.783 1.762 ThO2
2+ 1.850 1.832 1.827 1.825 1.855 1.856 1.829 1.880 1.861 PaO2
2+ 1.772 1.759 1.753 1.752 1.784 1.784 - b 1.804 1.788 UO2
2+ 1.693 1.683 1.678 1.677 1.710 1.707 1.690 1.725 1.713 NpO2
2+ 1.688 1.677 1.671 1.670 1.707 1.705 1.687 1.724 1.711 PuO2
2+ 1.673 1.661 1.655 1.654 1.694 1.691 1.675 1.713 1.698 AmO2
2+ 1.666 1.652 1.646 1.644 1.687 1.684 1.670 1.707 1.691 CmO2
2+ 1.688 1.668 1.664 1.663 1.706 1.706 1.696 1.732 1.710 aMolecule has bent geometry (C2v symmetry) With angle 111.9 degrees. b SCF did not converge. The test calculations in Table S36 show that changing the basis set for oxygen makes only negligible differences (for example, only in the fourth digit after the decimal point for B3LYP calculations) in the bond distances. These results have been obtained with the Gaussian 03 program.




















