
GUÍAS FISICOQUÍMICA: Fisicoquímica I y II Programa de Química
174
1 GUÍAS FISICOQUÍMICA: Fisicoquímica I y II Programa de Química Compiladas por: Lis Manrique Losada y Vladimir Sánchez Tovar Docentes Programa de Química
Transcript of GUÍAS FISICOQUÍMICA: Fisicoquímica I y II Programa de Química

1
GUÍAS FISICOQUÍMICA: Fisicoquímica I y II Programa de Química
Compiladas por: Lis Manrique Losada y Vladimir Sánchez Tovar Docentes Programa de Química

2
TABLA DE CONTENIDO PÁG
A.PROPIEDADES DE LOS GASES ................................................................................. 4
PRÁCTICA DE LABORATORIO No. 1: DETERMINACIÓN DE PESO MOLECULAR DE
LÍQUIDOS VOLÁTILES. APLICACIÓN DE LA LEY DE LOS GASES IDEALES. ............ 5
PRÁCTICA DE LABORATORIO No. 2: DETERMINACIÓN DE LA MASA MOLAR Y
DENSIDAD DE UN GAS: CASO DEL BUTANO. ............................................................ 8
PRÁCTICA DE LABORATORIO No. 3. PROPIEDADES DE LOS GASES: LEY DE
BOYLE Y LEY DE CHARLES. (Propiedades De Los Gases Ideales) ........................... 11
B. LEYES TERMODINÁMICAS....................................................................................... 18
PRÁCTICA DE LABORATORIO No. 4: CONCEPTOS TERMODINÁMICOS ............... 19
C. TERMOQUÍMICA ........................................................................................................ 26
PRÁCTICA DE LABORATORIO No. 5. CALOR DE SOLUCIÓN Y DE REACCIÓN. .... 26
PRÁCTICA DE LABORATORIO No. 6. CALOR PERDIDO EN FUSIÓN TOTAL Y
PARCIAL DEL HIELO Y CALOR DE NEUTRALIZACIÓN. ........................................... 32
PRÁCTICA DE LABORATORIO No. 7: DETERMINACIÓN DE LA CAPACIDAD
CALORÍFICA DE UN SÓLIDO. .................................................................................... 37
D. EQUILIBRIO QUÍMICO ............................................................................................... 41
PRÁCTICA DE LABORATORIO No. 8: EQUILIBRIO QUÍMICO. ................................. 42
E. EQUILIBRIO DE FASES ............................................................................................ 46
PRÁCTICA DE LABORATORIO No. 9: EQUILIBRIO DE FASES. (Equilibrio Liquido
Vapor Y Equilibrio Sólido Líquido Para Mezclas Binarias) ............................................ 47
F. PROPIEDADES COLIGATIVAS: ................................................................................ 55
PRACTICA DE LABORATORIO No. 10: ASCENSO PUNTO DE EBULLICIÓN Y
DESCENSO CRIOSCÓPICO. ...................................................................................... 56
G. CINÉTICA ................................................................................................................... 63
PRACTICA DE LABORATORIO No.11: CINÉTICA DE LA VITAMINA C. (estudio de la
cinética de oxidación de la vitamina c con ferricianuro de potasio. determinación de la
ley experimental de rapidez) ........................................................................................ 64
PRÁCTICA DE LABORATORIO No. 12: RELOJ DE YODO ......................................... 67
PRÁCTICA DE LABORATORIO No. 13: LA BOTELLA AZUL ...................................... 72
PRÁCTICA DE LABORATORIO No. 14: REDUCCIÓN DEL ION PERMANGANATO. . 79

3
PRÁCTICA DE LABORATORIO No.15: REACCIONES OSCILANTES ........................ 82
PRÁCTICA DE LABORATORIO No.16: SOLVÓLISIS DEL CLORURO DE TERT-
BUTILO. ....................................................................................................................... 91
H. CATALISIS HOMOGENEA ......................................................................................... 97
PRÁCTICA DE LABORATORIO No. 17: DESCOMPOSICIÓN CATALIZADA DEL
PERÓXIDO DE HIDRÓGENO ..................................................................................... 97
PRÁCTICA DE LABORATORIO No. 18: CINÉTICA DE LA REACCIÓN ENTRE EL
YODO Y LA ACETONA CATALIZADA POR UN ÁCIDO. ........................................... 106
I. CATALISIS HETEROGENEA .................................................................................... 112
PRÁCTICA DE LABORATORIO No. 19: ADSORCIÓN SOBRE CARBÓN ACTIVADO.
................................................................................................................................... 112
PRACTICA DE LABORATORIO No. 20: REACCIONES FOTOQUÍMICAS ................ 116
J. FENOMENOS SUPERFICIALES .............................................................................. 122
PRÁCTICA DE LABORATORIO No. 21: MEDICION DE LA TENSION SUPERFICIAL:
METODO DE LA GOTA Y METODO DEL CAPILAR. ............................................... 123
PRÁCTICA DE LABORATORIO No. 22: PROPIEDADES DE LOS COLOIDES: DOSIS
DE COAGULANTE EN TRATAMIENTO DE AGUAS. ................................................ 134
K. FENÓMENOS DE TRANSPORTE ............................................................................ 143
PRÁCTICA DE LABORATORIO No. 23. LEY DE STOKES Y SEDIMENTACIÓN ..... 144
PRÁCTICA DE LABORATORIO No. 24: FLUJO DE POISEUILLE. ............................ 151
PRÁCTICA DE LABORATORIO No. 25: TRANSPORTE DE MASA POR DIFUSIÓN A
TRAVÉS DE UN GEL. ................................................................................................ 160
PRÁCTICA DE LABORATORIO No. 26: DISOLUCIÓN DE UN SÓLIDO. .................. 165
PRÁCTICA DE LABORATORIO No. 27: DESORCIÓN DEL AMONIACO:
TRANSPORTE DE MATERIA A TRAVÉS DE LA INTERFACE LIQUIDO-GAS. ........ 170
REFERENCIAS ............................................................................................................. 173

4
A.PROPIEDADES DE LOS GASES

5
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO NO. 1: DETERMINACIÓN DE PESO MOLECULAR DE
LÍQUIDOS VOLÁTILES. APLICACIÓN DE LA LEY DE LOS GASES IDEALES.
(Determinación De Peso Molecular De Líquidos Volátiles. Aplicación De La Ley De
Los Gases Ideales)
1 . 1FUNDAM ENTOS TEÓ RICO S: Una de las más importantes aplicaciones de la ley de los gases ideales,
𝑷𝑽 = 𝒏𝑹𝑻
(1.1)
es la determinación del peso molecular de un líquido que pueda transformarse en vapor. Si se conocen la temperatura, presión, volumen y peso del vapor, el peso molecular se calcula a partir de
𝐧 =𝐩𝐞𝐬𝐨 𝐝𝐞𝐥 𝐠𝐚𝐬 𝐞𝐧 𝐠𝐫𝐚𝐦𝐨𝐬
𝐩𝐞𝐬𝐨 𝐝𝐞 𝟏 𝐦𝐨𝐥 𝐝𝐞𝐥 𝐠𝐚𝐬 𝐨 𝐧 =
𝐩𝐞𝐬𝐨
𝐩𝐞𝐬𝐨 𝐦𝐨𝐥
( 1.2)
Por lo tanto, la Ec. 1 puede expresarse como
𝑷𝑽 = (𝒑𝒆𝒔𝒐
𝒑𝒆𝒔𝒐 𝒎𝒐𝒍) (𝑹𝑻) =
(𝒑𝒆𝒔𝒐)(𝑹)(𝑻)
𝒑𝒆𝒔𝒐 𝒎𝒐𝒍
(1.3)
De lo cual se obtiene la expresión del peso molecular:
𝒑𝒆𝒔𝒐 𝒎𝒐𝒍 = (𝒑𝒆𝒔𝒐)(𝑹)(𝑻)
(𝑷)(𝑽)
(1.4)
1.2 MATERIAL Y EQUIPO
beaker de 600 ml mechero banda de hule
soporte de anillo rejilla anillo de hierro perlas de ebullición

6
Trozo de papel de aluminio matraz Erlenmeyer de 125-250 ml alfiler pinzas
termómetro de mercurio balanza
1.3 PROCEDIMIENTO 1. Obténgase un matraz Erlenmeyer de 125-250 ml de capacidad, un trozo de papel de
aluminio para tapar la boca del matraz y una banda de hule o un alambre para sujetar el papel de aluminio.
2. Pese todo el conjunto. 3. Introdúzcanse 3-5 ml de un líquido desconocido (cuyo punto de ebullición sea inferior
al del agua) y sujétese el aluminio alrededor de la boca del matraz con el hule o el alambre.
4. Con un alfiler delgado hágase una perforación, tan pequeña como sea posible, en el centro del aluminio.
5. Colóquese el matraz en un beaker con agua hirviendo. 6. Inclínese y manténgase casi completamente sumergido, deteniéndolo con una pinza
como lo muestra la Fig. 1. Añádanse unas perlas de ebullición al beaker para evitar saltos. Bajo estas condiciones, el líquido del matraz se vaporizará y el exceso de vapor escapará a través del orificio de alfiler.
7. Después de que se haya consumido todo el líquido y ya no salgan vapores por el orificio (asegúrese de que no se condense el líquido en la boca del matraz), mídase la temperatura del agua, suspéndase el calentamiento y espérese hasta que el agua deje de hervir.
8. Retírese el matraz del beaker y colóquese sobre una plancha de asbesto para que se enfríe y se seque. El vapor encerrado en el matraz se condensará formando un líquido.
9. Después de enfriar hasta la temperatura ambiente, asegúrese de que todo el conjunto está completamente limpio y seco, y regístrese el peso exacto del matraz con el líquido y el aluminio. Determínese entonces el peso del líquido condensado. Este peso corresponde al peso del vapor encerrado en el matraz a la temperatura del agua hir-viendo.
10. Mídase el volumen total del matraz. 11. Léase la presión barométrica y calcúlese el peso molecular del líquido. 12. Repítase el mismo procedimiento con otra muestra del mismo líquido y compárense las
respuestas. Sí son muy diferentes, efectúese una tercera determinación.

7
Fig. 1. Dispositivo para la determinación de pesos moleculares 1.4 PREGUNTAS DE REVISIÓN
1. Supóngase que esta determinación de peso molecular se efectuó muy precipitadamente y que no se llegó a vaporizar todo el líquido del matraz. ¿Sería el peso molecular calculado muy pequeño o muy grande? ¿Qué tipo de error de procedimiento podría conducir a un error de resultado que fuera opuesto al interior? 2. ¿Por qué no puede determinarse el peso molecular del n-butanol (por ejemplo, 118°C) con este método?
Líquido problema: Cloroformo (CHCl3 )61.2 °C , Benceno (C6H6) 80.1 °C, Alcohol etílico, (CH3CH2OH), 78.4 °C, Alcohol metílico (CH3OH), 64.7 °C 1.5 BIBLIOGRAFÍA:
Hess, George G. y Kask, Uno. 1970. “Química General Experimental”. CECSA, México. Pp. 105 a 107.
Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-México.
G.M. Barrow. 1976. Química Física para las Ciencias de la Vida. Ed. Reverté, I.N. Levine. 1996. Fisicoquímica. Ed. Mc Graw Hill. Levine, Y.1992. Fisicoquímica, 3a. edición Mc Graw-Hill México.

8
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO No. 2: DETERMINACIÓN DE LA MASA MOLAR Y
DENSIDAD DE UN GAS: CASO DEL BUTANO. (Determinación de la masa molar y la
densidad de un gas, caso del Butano)
OBJETIVOS El objetivo principal de la práctica es la determinación del peso molecular y de la densidad del butano. Para Este fin se debe considerar el gas butano como un gas ideal. Así podemos relacionar variables macroscópicas tales como: presión P; temperatura T; masa del gas Mgas y volumen V, mediante la ecuación de estado de los gases ideales. 2.1 FUNDAMENTO TEÓRICO El butano, a presiones cercanas a la presión atmosférica y temperaturas alejadas de su punto de ebullición se comporta en forma similar a un gas ideal, por lo que podemos estudiarlo utilizando la ecuación de estado de los gases ideales:
𝑷. 𝑽 = 𝒏.𝑹. 𝑻 =𝒎
𝑴. 𝑹. 𝑻
Donde P es la presión absoluta del gas, V el volumen, n el número de moles, R la constante universal de los gases, T la temperatura absoluta, m la masa del gas y M la masa molecular del gas. Para determinar la densidad y masa molecular del butano, se pueden medir distintos volúmenes de gas de masa conocida a presión y temperaturas constantes. Si el butano se comporta como un gas ideal, graficando la masa del gas en función del volumen a P y T constantes, esperaríamos obtener una recta cuya pendiente sería:
𝑷𝒆𝒏𝒅𝒊𝒆𝒏𝒕𝒆 =𝑴.𝑷
𝑹.𝑻
2.2 PROCEDIMIENTO

9
Utilizar para las mediciones una probeta graduada cada 10 ml, una balanza digital con una sensibilidad de 0.01 g y un encendedor como fuente de butano. Medir temperatura en el laboratorio y consultar la presión atmosférica y la presión de vapor de agua a la misma temperatura de laboratorio. Llenar la probeta con agua y colocarla invertida dentro de un recipiente con agua teniendo cuidado de que no entre aire a la probeta al invertirla (ver figura 1).
Figura 1. Dispositivo utilizado para medir el volumen de agua desplazado por le gas.
Se pesa el encendedor antes de utilizarlo, una vez armado el dispositivo se coloca la manguerilla debajo de la probeta invertida dejando salir el gas dentro de la misma. Se mide el volumen de agua desplazada por el gas, teniendo cuidado de que el nivel de agua de la probeta coincida con el del recipiente exterior, para asegurarnos que la presión dentro de ella sea la misma que la atmosférica. Obtenemos así el dato de Vgas de gas
liberado dentro de la probeta (𝑚𝑔𝑎𝑠).
Luego de medir varios volúmenes de gas para varias masas del mismo, graficamos 𝑚𝑔𝑎𝑠
vs Vgas según:
𝒎𝒈𝒂𝒔 =𝑴𝒈𝒂𝒔.𝑷
𝑹.𝑻. 𝑽𝒈𝒂𝒔
Donde 𝑚𝑔𝑎𝑠 es la diferencia de peso del encendedor lleno y luego de liberar el gas, Vgas el
volumen de agua desplazada por el butano, T la temperatura ambiente en grados Kelvin, R la constante universal de los gases, mgas la masa molecular del gas que se desea calcular
y P es la presión absoluta del gas, la cual debe corregirse del siguiente modo:
𝑷 = 𝑷𝒂𝒕𝒎 − 𝑷𝑽𝒂𝒑.𝒂𝒈𝒖𝒂
Siendo Pvap agua la presión de vapor del agua a la temperatura de trabajo, dato que se
obtiene de tablas.

10
Tabla 1. Datos del experimento
Volumen desplazado
Peso mechero inicial
Peso mechero final
Masa de gas
NOTA: Llevar para la práctica un encendedor de gas. 2.3 ELABORAR Y CALCULAR 1.1 Realice la gráfica propuesta (graficamos mgas versus Vgas).
1.2 Determine la ecuación que rige los datos y su coeficiente de determinación, con el valor de la pendiente determinar la masa molar del gas butano y su densidad, y con los datos teóricos calcular el porcentaje de error. i. Bajo condiciones anaeróbicas una suspensión de la bacteria Micrococcusdenitrificans reduce cuantitativamente el nitrato a gas nitrógeno si también se suple con un exceso de sustrato oxidable, por ejemplo succinato.
𝟐 𝑵𝑶𝟑 − → 𝑵𝟐 + 𝟐 𝑶𝑯
− + 𝟒 𝑯𝟐𝑶
En una botella cerrada de 2 dm3 de capacidad con una atmósfera inicial de oxígeno libre de N2, y a 100 kPa, se incuban 200 cm3 de una suspensión lavada de M. denitrificans, suplementada con un exceso de succinato y 0.25 mol de nitrato, temperatura de incubación 303 K ¿Cuál será la presión final de gas en la botella después de la reducción completa del nitrato? (Considerar solubilidad despreciable de N2 en el medio de cultivo). ii. La presión total de una mezcla de oxígeno e hidrógeno es 1 atm. La mezcla se incendia y el agua formada se separa. El gas restante es hidrógeno puro y ejerce una presión de 0,4 atm. Cuando se mide en las mismas condiciones de T y V que la mezcla original. ¿Cuál era la composición original de la mezcla en porcentaje en mol? 2.4 BIBLIOGRAFÍA DE CONSULTA
Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-México.
G.M. Barrow. 1976. Química Física para las Ciencias de la Vida. Ed. Reverté, I.N. Levine. 1996. Fisicoquímica. Ed. Mc Graw Hill.

11
Levine, Y.1992. Fisicoquímica, 3a. edición Mc Graw-Hill México.
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO No. 3. PROPIEDADES DE LOS GASES: LEY DE
BOYLE Y LEY DE CHARLES. (Propiedades De Los Gases Ideales)
OBJETIVOS
1. Comprobar experimentalmente la ley de Boyle y de Charles con un gas real. 2. Determinar el peso molecular de un vapor condensable empleando varias muestras
problema. 3. Comparar la eficiencia de métodos en la determinación del peso molecular de una
sustancia. 4. Aplicar la ecuación del gas ideal. 5. Diferenciar entre un gas ideal y un gas real.
3.1 MARCO TEÓRICO De acuerdo con la teoría cinética, el gas perfecto está compuesto por partículas extremadamente pequeñas (sus moléculas) que poseen un movimiento continuo, al azar e independiente. Durante su movimiento al azar, las moléculas chocan incesantemente contra las paredes del recipiente y es este continuo bombardeo de las paredes lo que se conoce como, presión del gas. Las "partículas" componentes del gas perfecto son absolutamente elásticas y rebotan con una energía igual a la que tenían en el momento del choque. Además las moléculas de un gas perfecto no deben ocupar volumen (lo cual confirma que el gas perfecto es una ficción útil). En virtud del movimiento independiente y al azar de sus moléculas, cuando un gas de una determinada densidad se introduce en un volumen mayor que el que ocupaba anteriormente a la misma temperatura, las moléculas se redistribuyen de forma que cada una tiene una libertad máxima de movimiento. El gas ocupa totalmente el nuevo volumen con la disminución correspondiente de su densidad. Esta tendencia de las moléculas gaseosas a moverse de una zona de densidad mayor a otra de densidad menor y así conseguir una densidad media de equilibrio, se conoce como fuerza de difusión. De aquí se deduce que se debe comprimir un gas para aumentar su densidad-fuerza de compresión. El efecto de los cambios de la temperatura sobre un gas también se puede interpretar por medio de la teoría cinética. Un aporte de calor aumenta la energía cinética de las moléculas, favorece su tendencia a moverse incluso a más distancia unas de otras y por tanto provoca
12
una expansión del gas a presión constante. El descenso de temperatura disminuye la movilidad de las moléculas y la tendencia del gas a presión constante es a contraerse. Por tanto, en cierto sentido, el aumento de la presión y el descenso de la temperatura tienden al mismo fin, a la disminución del volumen del gas. De aquí se deduce que la condición de un gas perfecto está afectada por cuatro variables independientes: (i) volumen, (ii) presión, (iii) temperatura y (iv) moles. El análisis del efecto de los cambios de presión y/o temperatura sobre el volumen de una masa dada de gas ideal ha determinado el establecimiento de ciertas relaciones entre estos factores, las cuales se conocen como leyes de gas ideal. La Relación Presión-Volumen: Ley De Boyle. Un aumento isotérmico de la presión disminuirá proporcionalmente el volumen de una cierta cantidad de gas y viceversa.
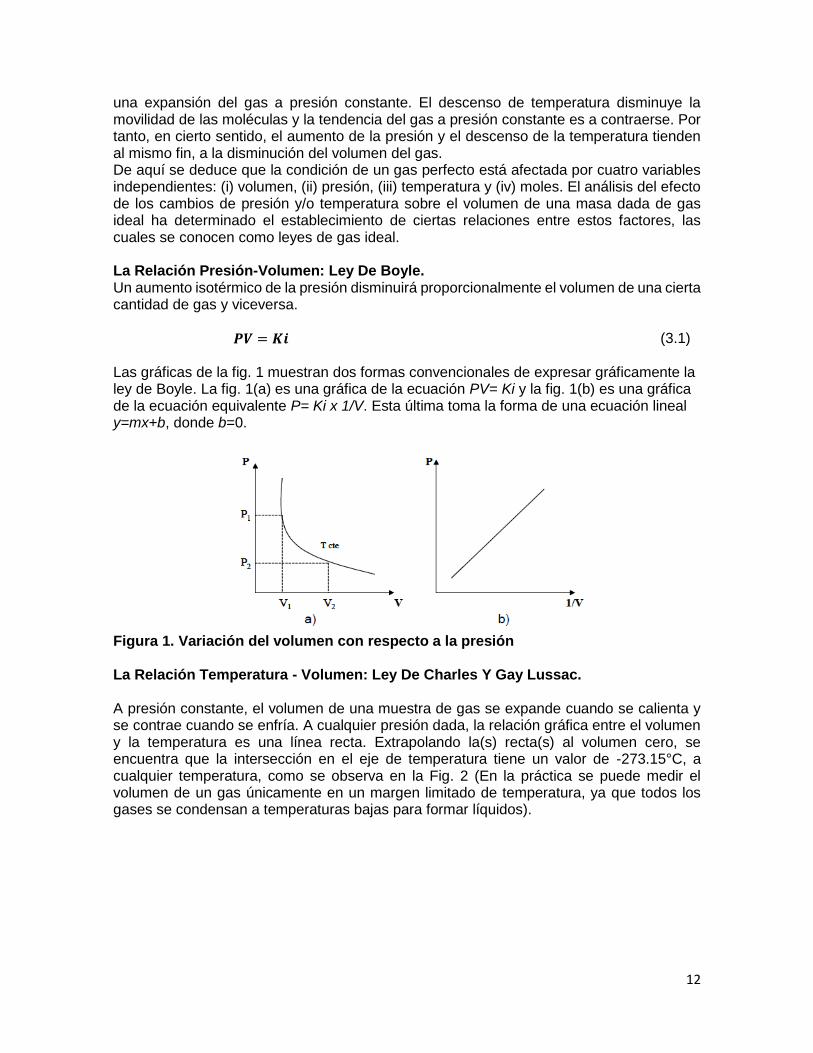
𝑷𝑽 = 𝑲𝒊 (3.1)
Las gráficas de la fig. 1 muestran dos formas convencionales de expresar gráficamente la ley de Boyle. La fig. 1(a) es una gráfica de la ecuación PV= Ki y la fig. 1(b) es una gráfica de la ecuación equivalente P= Ki x 1/V. Esta última toma la forma de una ecuación lineal y=mx+b, donde b=0.
Figura 1. Variación del volumen con respecto a la presión La Relación Temperatura - Volumen: Ley De Charles Y Gay Lussac. A presión constante, el volumen de una muestra de gas se expande cuando se calienta y se contrae cuando se enfría. A cualquier presión dada, la relación gráfica entre el volumen y la temperatura es una línea recta. Extrapolando la(s) recta(s) al volumen cero, se encuentra que la intersección en el eje de temperatura tiene un valor de -273.15°C, a cualquier temperatura, como se observa en la Fig. 2 (En la práctica se puede medir el volumen de un gas únicamente en un margen limitado de temperatura, ya que todos los gases se condensan a temperaturas bajas para formar líquidos).

13
Figura 2. Variación de la temperatura con respecto al volumen
𝑽
𝑻= 𝑲𝟐
(3.2)
La Relación Volumen - Cantidad: Ley De Avogadro. El volumen de cualquier gas debe ser proporcional al número de moles de moléculas presentes, es decir:
𝑽 = 𝑲𝟑𝒏 (3.3)
Donde n representa el número de moles y K3 es la constante de proporcionalidad. La última ecuación es la expresión matemática de Ley de Avogadro, la cual establece que a presión y temperatura constantes, el volumen de un gas es directamente proporcional al número de moles del gas presente. Ecuación Del Gas Ideal Resumiendo las leyes de los gases que se han analizado hasta el momento se pueden combinar las tres expresiones anteriores para obtener una sola ecuación que describa el comportamiento de los gases:
𝑷𝑽 = 𝒏𝑹𝑻 (3.4)
Donde R, es la constante de proporcionalidad y se denomina la constante universal de los gases y la ecuación se conoce como la ecuación del gas ideal y describe la relación entre las cuatro variables P, V, T y n. Ecuación de estado de gases reales

14
La ecuación de estado del gas ideal no es del todo correcta: los gases reales no se comportan exactamente así. En algunos casos, la desviación puede ser muy grande. Por ejemplo, un gas ideal nunca podría convertirse en líquido o sólido por mucho que se enfriara o comprimiera. Por eso se han propuesto modificaciones de la ley de los gases ideales
𝑷𝑽 = 𝒏𝑹𝑻 (3.5)
Una de ellas, muy conocida y particularmente útil, es la ecuación de estado de Van der Waals
(𝑷 +𝒂
𝒗𝟐) . (𝒗 − 𝒃) = 𝑹. 𝑻 (3.6)
Donde v = V/n, a y b son parámetros ajustables determinados a partir de medidas experimentales en gases reales. Son parámetros de la sustancia y no constantes universales, puesto que sus valores varían de un gas a otro. La ecuación de Van der Waals también tiene una interpretación microscópica. Las moléculas interaccionan entre sí. La interacción es muy repulsiva a corta distancia, se hace ligeramente atractiva a distancias intermedias y desaparece a distancias más grandes. La ley de los gases ideales debe corregirse para considerar las fuerzas atractivas y repulsivas. Por ejemplo, la repulsión mutua entre moléculas tiene el efecto de excluir a las moléculas vecinas de una cierta zona alrededor de cada molécula. Así, una parte del espacio total deja de estar disponible para las moléculas en su movimiento aleatorio. En la ecuación de estado, se hace necesario restar este volumen de exclusión (b) del volumen del recipiente (V); de ahí el término (V - b). Actualmente existen gran variedad de ecuaciones de estado que pretenden predecir cada vez mejor y acertadamente el comportamiento de un gas en todas las condiciones de presión y temperatura posibles. En ocasiones involucran una solución matemática compleja que requiere métodos numéricos y necesariamente un ordenador para ser solucionadas. 3.2 MATERIALES Y REACTIVOS
- Montaje de Mariotte: erlenmeyer de 500 mL con desprendimiento lateral, manguera de látex, aguja hipodérmica, balón fondo plano, tapones de caucho, estufa, probeta 100mL.
- Montaje desplazamiento de volumen: probeta 500 mL, cubeta de plástico, manguera de látex con diámetro pequeño.
- Montaje ley de Boyle: jeringa de plástico, masas regulares, tapón de caucho. 3.3 METODOLOGÍA Ley de Charles
Construya un montaje Mariotte de la siguiente manera: un balón fondo plano con tapón de caucho, el tapón debe contener una salida para el gas con una manguera que debe estar conectada a un erlenmeyer donde se llevará a cabo el desplazamiento de agua; este erlenmeyer se encuentra por lo menos a un metro abajo del balón para garantizar la entrada

15
del gas sin mezclas no deseadas. El erlenmeyer inicialmente está lleno de agua y el volumen de gas desplazará este líquido que es evacuado por una aguja hipodérmica. En este montaje se desarrollarán dos experiencias: A. Cambios en el volumen del aire por cambios en la temperatura: Prepare tres diferentes baños: agua a 40◦C, agua a 70◦C , agua a 80 ◦C y agua hirviendo. Someta el balón seco al baño de agua ambiente espere 2-3 min y registre el volumen desplazado de agua en el erlenmeyer. Repita el procedimiento con agua a las otras temperaturas. Registre volúmenes. Grafique V vs T, compare resultados con gráficas teóricas para gas ideal y real (use al menos Ecuación de Van der Waals) de V vs T. Genere al menos tres réplicas para cada caso. De la gráfica extrapole y prediga la temperatura a la cual el volumen es cero. B. Volumen desplazado por un vapor condensable: Caliente una muestra conocida (pese el balón con la muestra líquida) y evapore una cierta cantidad (pese el balón después de evaporar la muestra), mida el volumen desplazado de agua en el erlenmeyer. Repita este procedimiento con diferentes masas de muestra. Calcule la densidad del vapor y su peso molecular. Genere al menos 3 réplicas en cada caso y calcule porcentajes de error, aplique estadística descriptiva. Ley de Boyle y Avogadro
A. Cambio de volumen de una muestra de aire al aplicar diferentes presiones: Estas presiones serán diferentes según la masa utilizada, y deben convertirse a unidades
absolutas con la siguiente información: El área de la cabeza del émbolo es . La masa empleada se divide cada vez entre el área obtenida. La presión manométrica se calcula
considerando que la masa de 1 g sobre un área de 1 cm2 ejerce una presión de 1012.3 Pa. La presión absoluta se obtiene sumando la manométrica y la atmosférica. Introduzca una jeringa sin aguja sobre un tapón de hule y sujétela a un soporte sobre una mesa de tal manera que se mantenga vertical. Succione a un volumen fijo el aire. Agregue pesas una a una sobre una plataforma del émbolo iniciando con las de menor valor. Permita que el sistema se estabilice por unos minutos y mida el volumen de aire. Después de cada adición permita que el émbolo regrese a su volumen original. Haga diez mediciones. Repita el procedimiento agregando los pesos en la misma secuencia. Haga por lo menos tres réplicas con cada cambio de presión. Haga una gráfica de V vs 1/P. Grafique los datos de P vs V; trace la curva que mejor se aproxime a los datos. Compare la gráfica P vs V experimental con teóricas considerando gas ideal y gas real. B. Gas butano contenido en una mechera: El objetivo de este procedimiento es determinar la variación de la relación masa-volumen, en diferentes cantidades de gas butano medidas a temperatura y presión constantes. Además se trata de calcular la presión del gas contenido en la mechera. A una mechera conectada a un sistema de desplazamiento de agua (probeta llena de agua invertida sobre una cubeta con agua) realice el siguiente procedimiento: Pese el encendedor a gas antes de utilizarlo, registre su peso en la tabla. Inserte con cuidado al encendedor, la conexión flexible que lleva el gas al interior de la probeta. Descargue del encendedor el volumen de gas indicado en la tabla. Retire el encendedor y obtenga nuevamente su peso. Repita el procedimiento para cada uno de los volúmenes solicitados
16
en la tabla. Luego de medir los volúmenes de gas y determinar su masa, graficar mgas vs
𝐕𝐠𝐚𝐬 según la tabla de datos obtenida.
No olvide que la presión absoluta del gas debe corregirse del siguiente modo: P´ = Patm – Pvapor agua donde P’ =presión del gas dentro del recipiente Calcule la presión a la que se encuentra el gas butano en el mechero a partir de los datos experimentales, a partir de ecuación ideal de gases y a partir de al menos dos ecuaciones para gases reales. Compare la gráfica m vs V experimental con gráficas para el butano teóricas considerándolo gas ideal y real (utilice van der waals y RedlichKwong). Determine la densidad del gas y su peso molecular. La presión atmosférica en Florencia, la temperatura del agua y la temperatura del recinto deben consultarse. Calcule porcentajes de error y aplique conceptos de cifras significativas, rechazo de datos y estadísticos descriptivos en el informe. Tabla 1. Datos
Prueba No.
Volumen de gas Desprendido (mL)
Masa inicial del
Encendedor (g)
Masa Final del
Encendedor (g)
Masa del gas Desprendido
(g)
1 30
2 60
3 90
4 120
5 150
6 180
7 210
8 240
9 270
10 300
11 330
12 360
13 390
14 420
15 450
16 480
3.3 BIBLIOGRAFÍA
Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-México.
Garland, C., Nibler, J., Shoemaker, David. 2003. Experimental in Physical Chemistry. SéptimaEdición. Mc. Graw Hill.
Peña A.B. Fisicoquímica, Manual de laboratorio. Universidad de Medellin. 2007. Hernandez G., Suarez, M.F. 2002. Fisicoquímica experimental, procesos de
transporte y cinética química. Facultad de ciencias (notas de clase). Universidad Nacional de Colombia.

17

18
B. LEYES TERMODINÁMICAS
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química
19
Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO No. 4: CONCEPTOS TERMODINÁMICOS
OBJETIVOS 1. Determinar el coeficiente de expansión térmica a partir de mediciones de volumen
específico. 2. Determinar la razón de los gases por el método de Clement y Desormes.
3. Determinar el trabajo producido por el vapor en una máquina térmica. 4. Determinar la eficiencia teórica y real de una máquina térmica. 4.1 MARCO TEÓRICO La razón de las capacidades caloríficas a presión constante (Cp) y volumen constante (Cv),
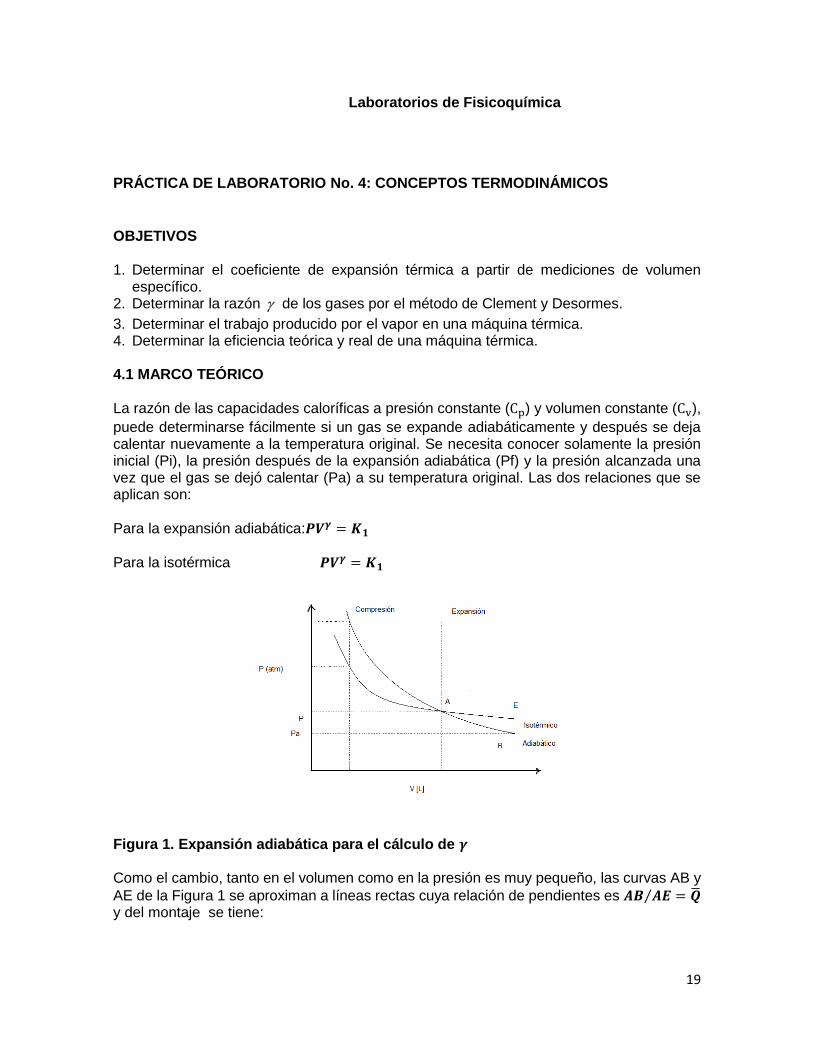
puede determinarse fácilmente si un gas se expande adiabáticamente y después se deja calentar nuevamente a la temperatura original. Se necesita conocer solamente la presión inicial (Pi), la presión después de la expansión adiabática (Pf) y la presión alcanzada una vez que el gas se dejó calentar (Pa) a su temperatura original. Las dos relaciones que se aplican son: Para la expansión adiabática:𝑷𝑽𝜸 = 𝑲𝟏
Para la isotérmica 𝑷𝑽𝜸 = 𝑲𝟏
Figura 1. Expansión adiabática para el cálculo de 𝜸 Como el cambio, tanto en el volumen como en la presión es muy pequeño, las curvas AB y
AE de la Figura 1 se aproximan a líneas rectas cuya relación de pendientes es 𝑨𝑩 𝑨𝑬⁄ = �̅� y del montaje se tiene:

20
𝜸 = 𝑨𝑩
𝑨𝑬=𝑷𝒐−𝑷𝒂
𝑷𝒐−𝑷=
𝒉𝒐
𝒉𝒐−𝒉
Dónde: La diferencia inicial en el manómetro es:
𝑷𝒐 −𝑷 = 𝒉𝒐 = 𝑫𝒐 − 𝑰𝒐 y la diferencia entre la presión final e inicial es:
𝑷𝒐 −𝑷 = 𝒉𝒐 − 𝒉, 𝒄𝒐𝒏 𝒉 = 𝑫 − 𝑰 Do = presión inicial en cm, Io = Presión inicial en cm D = Presión final en cm, I = Presión final en cm La segunda ley de la termodinámica define una función de estado que es la entropía (S) mediante la cual se puede definir cuándo un proceso es espontáneo o no, cuándo el proceso alcanza el equilibrio y cuál es la energía mínima requerida para que un proceso no espontáneo ocurra. Una máquina térmica es un dispositivo mecánico que actúa cíclicamente y tiene por objeto transformar la energía térmica en energía mecánica. Trabaja entre dos fuentes de temperatura o de calor. De la fuente caliente toma calor, parte de este lo convierte en trabajo y el calor no aprovechado lo envía a una fuente fría (Figura 3).
Figura 2. Máquina Térmica Para la máquina térmica de la Figura 3 se tiene que:
𝜟𝑺𝒄 + 𝜟𝑺𝒇 = 𝟎
𝑒𝑛𝑡𝑜𝑛𝑐𝑒𝑠 𝚀𝒄
𝑻𝒄=𝚀𝒇
𝑻𝒇
(4.1)
Se define como eficiencia () para una máquina térmica como la relación entre el trabajo
realizado y el calor recibido de la fuente caliente (𝜺 = 𝑾 𝚀⁄ ).
Eficiencia máxima (Carnot):
𝜺 =𝑾
𝚀𝒄=𝚀𝒄−𝚀𝒇
𝚀𝒄=𝑻𝒄−𝑻𝒇
𝑻𝒄
(4.2)

21
4.2 MATERIALES Y REACTIVOS
Para coeficiente Picnómetro, pipetas, beaker 250 mL, placa de calefacción.
Para Cp/Cv
Botella de 20 L sencilla o con salida lateral (frasco de decantación) Bomba de hule. Manómetro en U Pinzas tapones y mangueras de látex.
Para máquina térmica
Balón o erlenmeyer de 500 mL, que hace las veces de caldera para generar el vapor. Termómetro. Plancha eléctrica o mechero de gas para calentamiento. Erlenmeyer de 500 mL como depósito de auga a transferir. Beaker de 250 mL como depósito del agua transferida. Mangueras y tapones de conexión. Regla para medir alturas. 4.3 PROCEDIMIENTO 1. Determinación del coeficiente de expansión térmica para líquidos (Propiedad termodinámica). Para determinar el coeficiente de expansión térmica de un líquido en este laboratorio se deben tener mediad de la densidad del líquido y por tanto del volumen molar. La primera parte consiste entonces en determinar este volumen molar a diferentes temperaturas: 1.1 Determinación del volumen molar de un líquido: La densidad, ρ, es una variable de estado que se puede determinar pesando la masa de un volumen conocido de líquido. La densidad cambia con la temperatura y la presión. El inverso de la densidad es el volumen específico, V. Se puede determinar experimentalmente la dependencia del volumen específico con la temperatura a presión constante, por medición directa a varias temperaturas del sistema. Para observar el comportamiento VT del líquido, mida la densidad del mismo a diferentes temperaturas de la siguiente manera: A un beaker de 250 mL agregue entre 50 a 100 mL del líquido problema, al que antes, se le ha medido su densidad con un picnómetro. Mida la temperatura inicial (ambiente). Con ayuda de una placa de calefacción, mida la densidad del líquido cada vez que el líquido aumente su temperatura en 10ºC como mínimo, hasta antes de la temperatura de ebullición. Repita el experimento por lo menos dos veces para garantizar validez estadística de los resultados. Repita el procedimiento con dos líquidos más.

22
1.2 Determinación del coeficiente de expansión térmica: El coeficiente de expansión térmica, β, que se define como
𝜷 =𝟏
�̅�(𝝏�̅�
𝝏𝑻)𝑷= −
𝟏
𝝆(𝝏𝑷
𝝏𝑻)𝑷
El coeficiente de expansión térmica se puede estimar a partir de los datos [T, 𝜌] a presión constante de varias formas, cuya diferencia está en la manera de evaluar la derivada:
1. Se grafican los datos T vs. ρ y se ajustan a una línea recta. La pendiente de la 𝐴 =𝜋𝑟2recta esel valor de la derivada. 2. Si la gráfica no se ajusta a una recta, se prueba una cuadrática (o en general, una forma polinomial):
𝝆 = 𝒂𝑻𝟐 + 𝒃𝑻 + 𝒄 De donde
(𝝏𝑷
𝝏𝑻)𝑷= 𝟐𝒂𝑻 + 𝒃 𝑦 𝜷 =
𝟐 𝒂𝑻+𝒃
𝒂𝑻𝟐+𝒃𝑻+𝒄
3. Se estima la derivada localmente por diferencias finitas y se traza una gráfica de
𝑻 𝒗𝒔 (∆𝝆
∆𝑻) . De la forma de la gráfica se decide si la derivada es una constante o si se ajusta
a una línea recta o a un polinomio. Para los resultados y el análisis de resultados: A partir de las mediciones de densidad, determine el volumen específico como función de la temperatura. Elabore gráficas de T vs V para cada una de las sustancias. Haga ajustes lineales o polinomiales y presente la correlación𝑽 = 𝒇(𝑻) Se sugiere encontrar el coeficiente de expansión térmica para cada sustancia a partir de las correlaciones encontradas para T y ρ y a partir de la aproximación de la derivada como un cociente incremental de las diferencias entre dos puntos, para los datos T vs V (método de diferencias finitas). Aquí las diferencias se pueden tomar hacia delante, hacia atrás o como el promedio de ambas. ¿Hay diferencia en los resultados, para las diferencias consideradas (% error)? ¿Qué valores están más cercanos a los datos de la literatura? ¿A qué puede deberse esto?
2. Determinación de la razón𝜸 =𝑪𝒑
𝑪𝒗de los gases
Se dispone de un montaje que consiste de una botella preferiblemente de 20L que debe contener silica gel para asegurar que durante el experimento el aire no contenga humedad. Se introduce el aire en el aparato, bombeando el aire con la bomba de hule. Mantener la pinza abierta, de tal forma que la presión aumente ligeramente arriba de la presión

23
atmosférica (de 30 a 40 cm en la escala del manómetro). Cerrar la pinza y dejar que el aire en la botella alcance la temperatura ambiente, lo que se indica por la constancia en la lectura manométrica. Escribir longitudes de las columnas de agua a la izquierda y la derecha
(I0 y D0 respectivamente). Destapar la botella por medio de un orificio en el tapón por un instante y cerrar finalmente; un poco del aire escapa. La expansión es tan rápida que es adiabática; en la expansión el aire se enfría, por que debe dejarse calentar hasta la temperatura ambiente. Escribir longitudes de las columnas de agua a la izquierda y a la derecha (I y D respectivamente). Repetir las lecturas las veces que sea necesario. Para convertir las lecturas manométricas a cm de Hg, se necesita multiplicar aquellas por la razón de densidades
𝒉𝑯𝒈 ∗ 𝝆𝑯𝒈 = 𝒉𝒇𝒕𝒂𝒍𝒂𝒕𝒐 ∗ 𝝆𝒇𝒕𝒂𝒍𝒂𝒕𝒐
Llenar la siguiente tabla: Tabla 1.Datos del experimento
Prueba 𝐃𝟎 (cm) 𝐈𝟎 (cm) 𝐡 = 𝐃𝟎 − 𝐈𝟎 (cm)
D (cm) 𝐈 (cm) 𝐡 = 𝐃 − 𝐈 (cm)
1
2
3
4
5
Para el análisis de resultados: Determinar experimental.
Predecir los valores de Cv y Cp y calcular para el aire. Compara con el resultado anterior.
Suponer que el aire es un gas perfecto compuesto de 79% de N2 y y 21% O2 en volumen. Calcular el trabajo de expansión del aire expulsado. Preguntas: Explique por qué para gases ideales Cp >Cv.
Demuestre que Cp >Cv = R para gases ideales.
Establezca la diferencia entre el proceso adiabático y el isotérmico de la práctica. 3. Trabajo producido por el vapor en una máquina térmica Preparar el montaje de la Figura 2. Colocar 300 mL de agua en el recipiente generador de vapor. Colocar 300 mL de agua en el recipiente de transferencia, al cual se le coloca un tubo que esté aproximadamente en la mitad de la altura del líquido. Colocar calentamiento hasta la ebullición del agua (Tc) para que el vapor generado pase al recipiente de transferencia y empuje el agua hacia el recipiente receptor (Tf). Cuando pase el agua que llega al tope del tubo del recipiente de transferencia al recipiente receptor, quitar el calentamiento. Medir la altura máxima (∆ℎ𝑚𝑎𝑥), la altura neta ∆ℎ𝑛𝑒𝑡𝑜 el volumen final de agua en recipiente generador del vapor y el volumen en el recipiente receptor.

24
Figura 2. Montaje para máquina térmica
Para los cálculos y resultados: Calcular el trabajo máximo:
𝑊𝑚𝑎𝑥 = 𝑚𝑎𝑔𝑢𝑎,𝑡𝑟𝑎𝑛𝑠𝑓𝑒𝑟𝑖𝑑𝑎 ∗ 𝑔 ∗ ∆ℎ𝑚𝑎𝑥 (4.3)
Calcular el trabajo neto:
𝑊𝑚𝑎𝑥 = 𝑚𝑎𝑔𝑢𝑎,𝑡𝑟𝑎𝑛𝑠𝑓𝑒𝑟𝑖𝑑𝑎 ∗ 𝑔 ∗ ∆ℎ𝑚𝑒𝑡𝑎 (4.4)
donde g =gravedad Calcular Eficiencia máxima (Carnot) y ecuación: (4.2) Eficiencia Real:
휀𝑟𝑒𝑎𝑙 =𝑊𝑟𝑒𝑎𝑙
𝚀𝑟𝑒𝑎𝑙,𝑣𝑎𝑝𝑜𝑟
(4.5)
Donde:
𝑊𝑟𝑒𝑎𝑙 = 𝑃𝑒𝑥𝑡∆𝑉𝑣𝑎𝑝𝑜𝑟
(4.6)
𝑃𝑒𝑥𝑡 = 𝑃𝑎𝑡𝑚 ∆𝑉𝑣𝑎𝑝𝑜𝑟 = 𝑉𝑓 − 𝑉𝑖 =𝑛𝑣𝑎𝑝𝑜𝑟∗𝑅∗𝑇𝑣𝑎𝑝𝑜𝑟
𝑃𝑟𝑒𝑎𝑙 (4.7)
𝑇𝑣𝑎𝑝𝑜𝑟 = 𝑇𝑒𝑏𝑢𝑙𝑙𝑖𝑐𝑖ó𝑛 𝑛𝑣𝑎𝑝𝑜𝑟 =𝑚𝑣𝑎𝑝𝑜𝑟,𝐻2𝑂
𝑀𝑀𝐻2𝑂 (4.8)
𝑚𝑣𝑎𝑝𝑜𝑟 = 𝑚𝑎𝑠𝑎 𝑖𝑛𝑖𝑐𝑖𝑎𝑙 − 𝑚𝑎𝑠𝑎 𝑓𝑖𝑛𝑎𝑙 𝑒𝑛 𝑟𝑒𝑐𝑖𝑝𝑖𝑒𝑛𝑡𝑒 𝑔𝑒𝑛𝑒𝑟𝑎𝑑𝑜𝑟
𝑃𝑔𝑎𝑠 = 𝑃𝑎𝑡𝑚 +∆ℎ𝑚𝑎𝑥 + ∆ℎ𝑛𝑒𝑡𝑎
2 𝑒𝑛 𝑎𝑡𝑚
(4.9)

25
𝚀𝑟𝑒𝑎𝑙,𝑣𝑎𝑝𝑜𝑟 = 𝑚𝑣𝑎𝑝𝑜𝑟 ∗ ∆𝐻𝑣𝑎𝑝 (4.10)
𝑑𝑜𝑛𝑑𝑒 ∆𝐻𝑣𝑎𝑝 = 𝑐𝑎𝑙𝑜𝑟 𝑙𝑎𝑡𝑒𝑛𝑡𝑒 𝑑𝑒 𝑣𝑎𝑝𝑜𝑟𝑖𝑧𝑎𝑐𝑖ó𝑛 𝑑𝑒𝑙 𝑎𝑔𝑢𝑎
Calcular el error relativo (%error relativo)= %𝐸𝑟 =[ 𝑟𝑒𝑎𝑙− 𝑚á𝑥𝑖𝑚𝑎,𝑐𝑎𝑟𝑛𝑜𝑡]
𝑚á𝑥𝑖𝑚𝑎,𝑐𝑎𝑟𝑛𝑜𝑡∗ 100
(4.11) Datos requeridos: Masa agua transferida, masa agua generadora inicial, masa agua generadora final,
∆𝐻𝑚𝑎𝑥,∆ℎ𝑛𝑒𝑡𝑎, temperatura ambiente (inicial) y temperatura final (ebullición). Consultar: gravedad, Cp del agua y ∆𝐻𝑣𝑎𝑝
Preguntas para el análisis de resultados:
Por qué la eficiencia térmica de Carnot es mayor que la eficiencia térmica real de una máquina térmica?
El tipo de fuente de calentamiento del agua generadora de vapor, afectará la eficiencia térmica del sistema?
Si de dos sistemas térmicos uno es mejor que otro, ¿en qué emplea el “exceso” de energía que malgasta el más ineficiente?
Cómo afecta la temperatura de la fuente caliente y la temperatura de la fuente fría la eficiencia del sistema?
4.4 BIBLIOGRAFÍA Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-
México. Garland, C., Nibler, J., Shoemaker, David. 2003. Experimental in Physical Chemistry.
SéptimaEdición. Mc. Graw Hill. Peña A.B. Fisicoquímica, Manual de laboratorio. Universidad de Medellin. 2007. Hernandez G., Suarez, M.F. 2002. Fisicoquímica experimental, procesos de transporte
y cinética química. Facultad de ciencias (notas de clase). Universidad Nacional de Colombia.

26
C. TERMOQUÍMICA
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO No. 5. CALOR DE SOLUCIÓN Y DE REACCIÓN.

27
OBJETIVOS 1. Inferir el comportamiento de las mezclas de ácidos y bases fuertes en una experiencia
de neutralización. 2. Aplicar los principios de calorimetría a las experiencias a realizar. 5.1 Introducción Todas las reacciones químicas van acompañadas ya sea por una absorción o un desarrollo de energía, que en general se manifiesta como calor. La Termoquímica es la parte de la Termodinámica que estudia los cambios de energía en las reacciones químicas. El calor de una reacción química es el calor intercambiado en el curso de tal reacción, o, en un sentido más general, es igual al cambio de entalpía del sistema cuando la reacción ocurre a presión constante. En general, este calor de reacción depende no sólo de la naturaleza química de cada producto y cada reactivo, sino también de sus estados físicos. El calor absorbido o cedido por un sistema a presión constante (el cambio de entalpía), puede determinarse usando calorimetría adiabática, que como su nombre lo indica, utiliza un recipiente que aísla el sistema químico de los alrededores de modo que el calor desprendido o absorbido es igual, pero de signo contrario, al absorbido o desprendido por la parte interna del calorímetro. Como el sistema está aislado de los alrededores, es decir no interactúa térmicamente con ellos, se cumple: ∆𝑯𝒏𝒆𝒕𝒐 = 𝟎 = ∆𝑯𝒑𝒓𝒐𝒄𝒆𝒔𝒐 + ∆𝑯𝒄𝒂𝒍𝒐𝒓𝒊𝒎𝒆𝒕𝒓𝒐 (5.1)
O lo que es lo mismo:
∆𝑯𝒑𝒓𝒐𝒄𝒆𝒔𝒐 = ∆𝑯𝒄𝒂𝒍𝒐𝒓𝒊𝒎𝒆𝒕𝒓𝒐 (5.2)
El primer término indica el cambio térmico relacionado con el proceso que afecta el sistema estudiado y el segundo el calor absorbido o desprendido por el calorímetro (partes internas). Se tiene entonces:
∆𝑯𝒑𝒓𝒐𝒄𝒆𝒔𝒐 = − ∫ 𝑪𝒅𝑻
𝑻𝟐
𝑻𝟏
(𝟓. 𝟑)
Donde C representa la capacidad calorífica del sistema global incluyendo partes internas del calorímetro (especies involucradas, termómetro, agitador, resistencias, etc) que están en contacto con el sistema químico. Para cambios razonables de temperatura C es constante y la ecuación anterior puede resolverse obteniéndose:
∆𝐻𝑝𝑟𝑜𝑐𝑒𝑠𝑜 = −𝐶. ∆𝑇 (5.4)

28
Donde ΔT = T2 – T1 es el cambio de temperatura durante el proceso. La determinación de la capacidad calorífica del sistema se hace calibrando con un líquido cuya capacidad calorífica se conoce a varias temperaturas, o bien suministrando su trabajo eléctrico conocido el sistema y midiendo la diferencia de temperatura. Se utilizará la calorimetría adiabática para determinar los cambios entálpicos asociados al calor de solución y al calor de neutralización. 5.2 MATERIALES Calorímetro Dewar Termómetro -10 a 120 oC Fuente de poder Espátula Juego de caimanes Vidrio reloj Beaker 400 mL (2) Probeta 50 mL 30 g de NaOH 300 mL HCl 0.5 M Determinar el cambio térmico asociado a cada uno de los procesos y posteriormente medir la capacidad calorífica del sistema.
a) Disolución de NaOH.
El proceso de disolución de una sustancia suele ir acompañado de una absorción o desprendimiento de calor que, referido a un mol de sustancia, se conoce con el nombre de “calor molar de disolución”. Sin embargo, esta magnitud no es constante sino que depende de la cantidad de disolvente y, por lo tanto, de la concentración de la disolución. Al disolver gradualmente una masa de sustancia en una cantidad de disolvente dada, la composición de la disolución varía desde la del disolvente puro hasta la de la disolución final. La expresión:
(𝝏(∆𝑯)
𝝏𝒏)𝒑,𝒕
(5.4)
Sin embargo, es más interesante conocer la cantidad de calor absorbida o desprendida por mol de sustancia en el proceso completo, es decir, cuando se disuelve toda la sustancia en la cantidad de disolvente elegida. A la cantidad de calor generada en estas condiciones se le llama “calor integral de disolución”, y viene dado por:
∆𝐻 = {∫𝜕(∆𝐻)
𝜕𝑛
𝑛
0
}𝑝,𝑡
. 𝑑𝑛 (5.5)

29
Como el calor integral de disolución depende de la concentración es preciso especificar esta. En la práctica, se supondrá que 0.5 M es una concentración lo suficientemente baja como para suponer que se encuentra en el límite de dilución ∞. Por lo tanto, tanto para el calibrado como para la medida con la sustancia problemas se usarán los valores de la entalpía de disolución a dilución infinita (ΔH∞). Entalpía de disolución infinita: Definida como la variación de entalpía en la disolución de un mol de soluto en una cantidad infinita de agua. Como se trata de electrolitos, esta entalpía será un balance entre la entalpía de ruptura de la red y las de hidratación de aniones y cationes. 5.3 PROCEDIMENTO Colocar en el calorímetro 150 mL de agua destilada a temperatura ambiente y registrar las temperaturas cada 15 segundos durante tres o cuatro minutos. Adicionar la cantidad x gramos de NaOH para formar una solución 0,5 N, continuar agitando y leyendo la temperatura en función del tiempo hasta que ésta permanezca constante o presente un descenso regular durante 5 minutos. Una vez el sistema se encuentre a temperatura inicial adecuada y estable, conectar la resistencia de calentamiento a una fuente de poder y ajustar el voltaje para dar un valor cercano a 9 voltios, anotar los voltios y los amperios, determine el tiempo requerido para alcanzar el cambio inicial tomando datos de temperatura cada 30 segundos. Una vez desconectada la fuente, se continúan las lecturas de temperatura en función del tiempo hasta que permanezca constante o presente un descenso regular. b) Calor de neutralización. Calor de neutralización: El calor de neutralización es definido como el calor producido cuando un equivalente gramo de ácido es neutralizado por una base. El calor de neutralización tiene un valor aproximadamente constante, en la neutralización de un ácido fuerte con una base fuerte, ya que en esta reacción se obtiene como producto en todos los casos un mol de agua, que es formada por la reacción:
𝐇+ + 𝐎𝐇− → 𝐇𝟐𝐎 ó 𝐇𝟑𝐎+ + 𝐎𝐇− → 𝟐𝐇𝟐𝐎
En cada una de las reacciones anteriores se obtienen 13,7 kcal. Esta constancia en la entalpía de neutralización, se entiende fácilmente cuando se recuerda que los ácidos y bases fuertes y las sales, están completamente disociados en sus soluciones diluidas; y, en tal consecuencia el efecto químico común a todas estas neutralizaciones, que es sustancialmente el único cambio responsable para el efecto térmico observado, es la unión de los iones hidratados hidrogeno e hidroxilo para formar agua no ionizada. O sea, si la ecuación anterior de neutralización se escribe en forma iónica, se tiene que:
Na(aq)+ + OH(aq)
− +H(aq)+ + Cl(aq)
− → Na(aq)+ + Cl(aq)
− + H2O (5.6)
Y cancelando los iones comunes en ambos miembros de la igualdad:

30
OH(aq)− + H(aq)
+ → H2O (5.7)
Esta constancia en la entalpía de neutralización no se mantiene en la neutralización de soluciones de ácidos débiles por bases fuertes, bases débiles por ácidos fuertes o de ácidos débiles por bases débiles. En todos estos casos el valor de ΔH es menor y mucho menor en el siguiente caso: HCN(aq) + NaOH(aq) → NaCN(aq) +H2O
(5.8)
En donde se obtiene -2.9 kcal. En estos últimos casos el ΔH de neutralización difiere del valor constante citado, porque la formación del agua a partir de sus iones no es el único proceso químico que acompaña a la neutralización, ya que paralelamente a la combinación de los iones hidratados hidrogeno e hidroxilo, va ocurriendo la ionización de los solutos débiles, siendo el efecto térmico observado la suma de las entalpías de ionización y neutralización. 5.3.1 PROCEDIMIENTO Al calorímetro que contiene la solución de NaOH, agregar 150 mL de HCl 0.5 N a temperatura ambiente (revisar que tanto el termómetro como la resistencia de calentamiento estén sumergidos en la solución), continuar agitando y leyendo la temperatura en función del tiempo hasta que ésta permanezca constante o presente un descenso regular durante 5 minutos. 5.4 CÁLCULOS Debido que en la práctica resulta imposible alcanzar una adiabaticidad total y además los procesos no son instantáneos, la determinación de los cambios de temperatura debe hacerse considerando estas pérdidas. Construir las curvas temperatura-tiempo tanto para el proceso como para el trabajo eléctrico equivalente (Ver figura 1). Determine la capacidad calorífica del sistema calorimétrico suministrando un trabajo eléctrico, que produzca un cambio de temperatura similar al del proceso en estudio
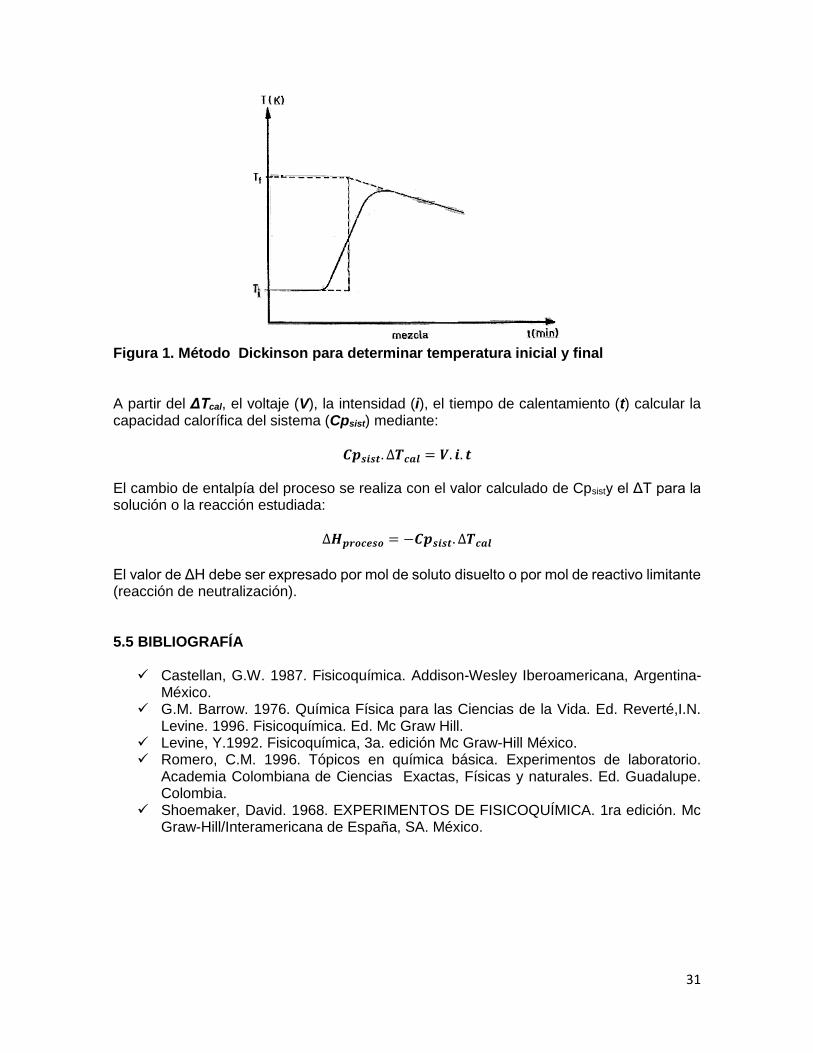
31
Figura 1. Método Dickinson para determinar temperatura inicial y final A partir del ΔTcal, el voltaje (V), la intensidad (i), el tiempo de calentamiento (t) calcular la capacidad calorífica del sistema (Cpsist) mediante:
𝑪𝒑𝒔𝒊𝒔𝒕. ∆𝑻𝒄𝒂𝒍 = 𝑽. 𝒊. 𝒕 El cambio de entalpía del proceso se realiza con el valor calculado de Cpsisty el ΔT para la solución o la reacción estudiada:
∆𝑯𝒑𝒓𝒐𝒄𝒆𝒔𝒐 = −𝑪𝒑𝒔𝒊𝒔𝒕. ∆𝑻𝒄𝒂𝒍
El valor de ΔH debe ser expresado por mol de soluto disuelto o por mol de reactivo limitante (reacción de neutralización). 5.5 BIBLIOGRAFÍA
Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-México.
G.M. Barrow. 1976. Química Física para las Ciencias de la Vida. Ed. Reverté,I.N. Levine. 1996. Fisicoquímica. Ed. Mc Graw Hill.
Levine, Y.1992. Fisicoquímica, 3a. edición Mc Graw-Hill México. Romero, C.M. 1996. Tópicos en química básica. Experimentos de laboratorio.
Academia Colombiana de Ciencias Exactas, Físicas y naturales. Ed. Guadalupe. Colombia.
Shoemaker, David. 1968. EXPERIMENTOS DE FISICOQUÍMICA. 1ra edición. Mc Graw-Hill/Interamericana de España, SA. México.

32
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO No. 6. CALOR PERDIDO EN FUSIÓN TOTAL Y
PARCIAL DEL HIELO Y CALOR DE NEUTRALIZACIÓN.
OBJETIVOS El presente experimento tiene por objetivo poner de manera explícita la variable tiempo, para partir de las leyes que gobiernan la transferencia de calor, poder evaluar directamente el proceso de fusión del hielo.
TRANSPORTE DE CALOR DURANTE LA FUSIÓN DEL HIELO Los procesos de transferencia de calor son procesos termodinámicos irreversibles que ocurren en situaciones de no-equilibrio y no se pueden describir en el marco de la termodinámica clásica. Recordemos que esta última tiene que ver con estados de equilibrio termodinámico. El presente experimento tiene por objetivo poner de manera explícita la variable tiempo, para partir de las leyes que gobiernan la transferencia de calor, poder evaluar directamente el proceso de fusión del hielo. Es decir que a partir del correcto planteamiento de los procesos de transferencia de calor podemos seguir la “cinética” de fusión del hielo. Para lograr el objetivo debemos proponer un modelo que nos ayude a entender el trasporte de calor, desde los alrededores hacia el hielo, como un proceso de conducción, convección o advección. 6.1 Fundamentos En condiciones normales el hielo funde de manera espontánea cuando se expone a un ambiente que se encuentra a una temperatura mayor a la del punto de fusión. La primera pregunta debe ser, por qué? La respuesta es dada por la segunda ley de la termodinámica. Ahora nos preguntamos, ¿cómo ocurre el proceso? Las evidencias empíricas muestran que la fusión se da por la continua transferencia de calor desde los alrededores hacia el hielo. Ahora pensemos en las siguientes preguntas: ¿cuánta energía se necesita para fundir una determinada masa de hielo?, ¿cuál es la composición hielo-agua después de haber suministrado cierta cantidad de energía? Las respuestas a este tipo de preguntas están bien establecidas a partir de la primera ley de la termodinámica. Finalmente, y es el objetivo de este experimento, hacernos preguntas como las siguientes: ¿en cuánto tiempo funde cierta masa de hielo, o a determinado tiempo cuánta masa de hielo se ha fundido?. La respuesta a estas preguntas no las encontramos en la termodinámica clásica, sino en las leyes que gobiernan los procesos de transporte. Para dar respuesta a este último tipo de preguntas es necesario asumir un modelo para el proceso de transporte de calor.

33
6.1.1 Transporte de calor por conducción Como una aproximación para comprender el proceso de transporte consideremos como válido el siguiente modelo: un cubo de hielo de lado X (puede ser cualquier forma geométrica regular o irregular) expuesto al aire en condiciones normales de temperatura y presión se funde por transferencia de calor. Entre el hielo y el aire se crea una superficie de contacto (una película de agua y una de aire), que asumimos de espesor constante y propiedades térmicas constantes, a través de la cual ocurre la transferencia de calor por conducción. La ecuación de balance de energía para el sistema se escribe de la siguiente manera: 𝒅𝑬𝒔𝒊𝒔𝒕𝒆𝒎𝒂 𝒅𝒕⁄ = 𝑬𝒆𝒏𝒕𝒓𝒂 +𝑬𝒈𝒆𝒏𝒆𝒓𝒂 − 𝑬𝒔𝒂𝒍𝒆 (6.1)
Ya que ningún proceso interno genera energía en el sistema y que este tampoco transfiere energía en ninguna dirección:
𝒅𝑬𝒔𝒊𝒔𝒕𝒆𝒎𝒂 𝒅𝒕⁄ = 𝑬𝒆𝒏𝒕𝒓𝒂 (6.2)
Para resolver la ecuación anterior es necesario especificar correctamente las condiciones iníciales, por simplicidad a sumados que el cubo de hielo funde sin pérdida de la forma geométrica. Como en nuestro modelo la transferencia de energía desde el aíre se da por conducción a través de la superficie de contacto, entonces tenemos
𝑬𝒆𝒏𝒕𝒓𝒂 = (𝑼)(𝟔𝑿𝟐)(𝑻𝒂𝒊𝒓𝒆 − 𝑻𝒉𝒊𝒆𝒍𝒐) (6.3)
Donde 𝑼 = 𝟏 (𝑳𝒂𝒊𝒓𝒆
𝑲𝒂𝒊𝒓𝒆+𝑳𝒂𝒈𝒖𝒂
𝑲𝒂𝒈𝒖𝒂)⁄ es el coeficiente global de transferencia, Laire es el espesor de
la película de aire y Lagua el de la película de agua, K son las respectivas conductividades térmicas y 6x2 es el área superficial del cubo (área de transferencia). A 25°C la
conductividad térmica del aire es 0.0267𝑊 ∗ 𝑚−1 ∗ 𝐾−1 y la capacidad calorífica es
de1005𝐽 ∗ 𝐾𝑔−1 ∗ 𝐾−1 y la conductividad térmica del agua es 𝑘 = 19,6𝑊 ∗𝑚−1 ∗ 𝐾−1. El cambio en la energía del sistema es un proceso isotérmico gobernado por la primera ley de la termodinámica, que debe ser consistente con la ecuación 3.2 Así, la ecuación 3.2 específica que el cambio en la energía del sistema debe ser una cantidad positiva, y esto es consistente con el aumento en la masa de agua proveniente de la fusión o con la disminución de la masa de hielo:
𝑑𝐸𝑠𝑖𝑠𝑡𝑒𝑚𝑎 𝑑𝑡⁄ =𝑑𝑚𝑎𝑔𝑢𝑎
𝑑𝑡∗ ∆𝐻𝑓𝑢𝑠𝑖ó𝑛
(6.4)
𝑑𝐸𝑠𝑖𝑠𝑡𝑒𝑚𝑎 𝑑𝑡⁄ =𝑑𝑚ℎ𝑖𝑒𝑙𝑜𝑑𝑡
∗ ∆𝐻𝑓𝑢𝑠𝑖ó𝑛 (6.5)

34
Finalmente a reemplazar 6.3 y 6.5 en 6.2 tenemos:
−𝒅𝒎𝒉𝒊𝒆𝒍𝒐
𝒅𝒕∗ ∆𝑯𝒇𝒖𝒔𝒊ó𝒏 = (𝑼)(𝟔𝑿
𝟐)(𝑻𝒂𝒊𝒓𝒆 − 𝑻𝒉𝒊𝒆𝒍𝒐) (6.6)
Como la masa de hielo varía en el tiempo, entonces 𝑥 cambia y resulta conveniente expresar el área de contacto en función del volumen (masa y densidad) del hielo:
𝑨𝒓𝒆𝒂 = 𝟔(𝒎 𝝆⁄ )𝟐 𝟑⁄ (6.7)
Entonces de las ecuaciones [3] y [5], la ecuación [2] se escribe:
𝒅𝒎𝒉𝒊𝒆𝒍𝒐
𝒅𝒕∗ ∆𝑯𝒇𝒖𝒔𝒊ó𝒏 = −(𝟔𝑼)(𝝆
𝟐 𝟑⁄ )(𝑻𝒂𝒊𝒓𝒆 − 𝑻𝒉𝒊𝒆𝒍𝒐)𝒎𝟐 𝟑⁄
(6.8)
La ecuación anterior se integra conociendo que para t = 0 la masa del hielo es mo y que en el tiempo t la mas es m(t):
𝑚1 3⁄ (𝑡) = −(2𝑈∆𝑇 𝜌2 3⁄ ∆𝐻𝑓𝑢𝑠𝑖ó𝑛)𝑡 +𝑚01 3⁄⁄ (6.9)
La anterior ecuación se puede escribir, sin pérdida de generalidad, de la siguiente manera:
𝑚1 3⁄ (𝑡) = −𝑎𝑡 + 𝑏 (6.10)
6.1.2 Transporte de calor por convección El planteamiento del problema es igual a como se realizó en la sección anterior. La única diferencia es que se asume para el proceso de transferencia de calor un modelo de convección, donde U es el coeficiente de transferencia de calor por convección:
Eentra = U(6X2)(Taire − Tagua) (6.11)
dmhielodt
∗ ∆Hfusión = −(6U)(ρ2 3⁄ )(Taire − Thielo)m
2 3⁄ (6.12)
m1 3⁄ (t) = −(2U∆T ρ2 3⁄ ∆Hfusión)t + m01 3⁄⁄
(6.13)
Datos necesarios:

35
𝒌(𝒉𝒊𝒆𝒍𝒐,𝑾 ∗𝒎−𝟏𝑲−𝟏) = 𝟏, 𝟖𝟖; 𝒌(𝒂𝒈𝒖𝒂,𝑾 ∗𝒎−𝟏𝑲−𝟏) = 𝟏𝟗, 𝟔
𝑪𝒑(𝒉𝒊𝒆𝒍𝒐, 𝑱 ∗ 𝑲𝒈−𝟏𝑲−𝟏) = 𝟐, 𝟎𝟒𝟎; 𝑪𝒑(𝒂𝒈𝒖𝒂, 𝑱 ∗ 𝑲𝒈−𝟏𝑲−𝟏) = 𝟒𝟏𝟕𝟗
∆𝑯𝒇𝒖𝒔𝒊ó𝒏 = 𝟔𝟎𝟎𝟐 𝑱 𝒎𝒐𝒍⁄
𝒅(𝒂𝒈𝒖𝒂, 𝟎°𝑪) = 𝟎. 𝟗𝟗𝟗𝟖𝟕 𝒈 𝒄𝒎𝟑⁄
𝒅(𝒉𝒊𝒆𝒍𝒐, 𝟎°𝑪) = 𝟎. 𝟗𝟏𝟔𝟕𝟒 𝒈 𝒄𝒎𝟑⁄
6.2 Trabajo experimental 1. Determine la masa inicial del bloque de hielo y su geometría.
2. A tiempo cero, suspenda un cubo de hielo de un soporte. Debajo del hielo debe
encontrase un recipiente que reposa sobre una balanza.
3. Cada 30 segundo registre la variación en la masa de agua recogida en el recipiente.
4. Lleve a cabo el experimento en ausencia y presencia de una corriente de aire (ventilador).
5. Realice los experimentos por duplicado.
Figura 3.1: Esquema de montaje experimental para el estudio “cinético” de la fusión de hielo. 6.3 Cuestionario
1. A partir de los datos experimentales de masa y tiempo, verifique la validez de las ecuaciones 3.9 y 3.10. Cuando la masa de hielo está expuesta a una masa fija de

36
aire la transferencia se puede asumir por conducción y convección, pero si la masa de aire es circulante (ventilador) entonces la transferencia se puede asumir por conducción y advección.
2. Determine el tiempo que le toma a la masa de hielo fundir completamente. Discuta este valor del predicho por las ecuaciones dadas.
3. Halle el coeficiente de transferencia de calor experimental según el modelo
adoptado. Analice el coeficiente𝑼 = 𝟏 (𝑳𝒂𝒊𝒓𝒆
𝑲𝒂𝒊𝒓𝒆+𝑳𝒂𝒈𝒖𝒂
𝑲𝒂𝒈𝒖𝒂)⁄ , e identifique el factor
determinante de U en las condiciones del presente experimento. De acuerdo a su análisis calcule el espesor de la película que limita la transferencia de calor.
4. Si la propiedad de transporte (coeficiente de conductividad térmica o coeficiente de convección) es dependiente del área de la superficie de contacto, entonces se debería hablar de un valor efectivo para el coeficiente de transporte. Así para el caso de convección:
𝑈𝑒𝑓𝑒𝑐 = (1 𝐴𝑠⁄ )∫ 𝑈𝑑𝐴𝑠
𝐴𝑠 (6.14)
Discuta la expresión anterior.
5. Deducir las ecuaciones relevantes para el caso en el que se tiente una masa esférica
de hielo.
6. ¿Será posible calcular la curva de distribución de temperatura en el hielo durante el proceso de fusión? Recuerde el experimento de conducción de calor en la barra metálica.
7. Discuta el planteamiento del proceso de fusión del hielo a partir de la ley de conservación de la masa:
𝒅𝒎𝒉𝒊𝒆𝒍𝒐 𝒅𝒕⁄ = 𝒎𝒆𝒏𝒕𝒓𝒂 −𝒎𝒔𝒂𝒍𝒆 (6.15)
6.4 Bibliografía
W. E. Boyce and R. C. DiPrima. Elementary differential equations and boundary value problems, John Wiley, 1992.
D. R. Pitts y L. E. Sissom. Transferencia de calor, Schaum McGraw Hill. 1977.
Universidad de la Amazonia

37
Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO No. 7: DETERMINACIÓN DE LA CAPACIDAD
CALORÍFICA DE UN SÓLIDO.
OBJETIVOS 1. Calcular la capacidad calorífica de algunos sólidos. 2. Armar un calorímetro, entrenarse en su uso. 3. Aplicar los principios de calorimetría a las experiencias a realizar.
7.1 Introducción
Concepto de temperatura. La temperatura es la sensación física que nos produce un cuerpo cuando entramos en contacto con él. Observamos cambios en los cuerpos cuando cambian su temperatura, por ejemplo, la dilatación que experimenta un cuerpo cuando incrementa su temperatura. Esta propiedad se usa para medir la temperatura de un sistema, como ocurre en los termómetros, los cuales consisten en un pequeño depósito de mercurio que asciende por un capilar a medida que se incrementa la temperatura. Concepto de calor. Cuando dos cuerpos A y B que tienen diferentes temperaturas se ponen en contacto térmico, después de un cierto tiempo, alcanzan la condición de equilibrio en la que ambos cuerpos están a la misma temperatura. Cuando un sistema de masa grande se pone en contacto con un sistema de masa pequeña que está a diferente temperatura, la temperatura de equilibrio resultante está próxima a la del sistema grande. Decimos que una cantidad de calor Q se transfiere desde el sistema de mayor temperatura al sistema de menor temperatura. La cantidad de calor transferida es proporcional al cambio de temperatura ΔT. La constante de proporcionalidad Cp se denomina capacidad calorífica del sistema:
𝑸 = 𝑪𝒑. ∆𝑻 (7.1)
Si los cuerpos A y B son los dos componentes de un sistema aislado, el cuerpo que está a mayor temperatura transfiere calor al cuerpo que está a menos temperatura hasta que ambas se igualan. Si TA>TB
El cuerpo A cede calor: 𝑸𝑨 = 𝑪𝒑𝑨. (𝑻 − 𝑻𝑨), entonces QA<0 El cuerpo B recibe calor: 𝑸𝑨 = 𝑪𝒑𝑨. (𝑻 − 𝑻𝑨), entonces QB<0

38
Como 𝑸𝑨 +𝑸𝑩 = 𝟎 La temperatura de equilibrio se obtiene mediante la media ponderada:
T =CpATA + CpBTB
CpA + CpB
(7.2)
La capacidad calorífica de la unidad de masa se denomina calor específico cp.
𝑪𝒑 = 𝒎𝑪𝒑 (7.3)
La fórmula para la transferencia de calor entre los cuerpos se expresa en términos de la masa m del calor específico cp y del cambio de temperatura.
𝑸 = 𝒎.𝑪𝒑. (𝑻𝒇 − 𝑻𝒊) (7.4)
Donde Tf es la temperatura final y Ti es la temperatura inicial. El calor específico es la cantidad de calor que hay que suministrar a un gramo de una sustancia para que eleve en un grado centígrado su temperatura. Joule demostró la equivalencia entre calor y trabajo 1cal = 4.186 J. El calor específico del agua es 𝑪𝒑 = 𝟏𝑪𝒂𝒍 (𝒈. °𝑪)⁄ . Hay que suministrar una caloría para que un gramo de agua
eleve su temperatura en un grado centígrado. Cuando varios cuerpos a diferentes temperaturas se encuentran en un recinto adiabático se producen intercambios caloríficos entre ellos alcanzándose la temperatura de equilibrio al cabo de cierto tiempo. Cuando se ha alcanzado este equilibrio se debe cumplir que la suma de las cantidades de calor intercambiadas es cero. Tabla 1. Calor especifico de algunas sustancias.
Sustancia Calor específico (J/kg·K)
Acero 460
Aluminio 880
Cobre 390
Estaño 230
Hierro 450
Mercurio 138
Oro 130
Plata 235
Plomo 130
Sodio 1300
Fuente: Manual de Física, Koshkin, Shirkévich. Editorial Mir, pág 74-75 7.2 MATERIALES Y REACTIVOS

39
Vasos Dewar (5) Termómetros de -10 a 120 grados Celsius (5) Malla de asbesto, Trípode Beaker 1000 mL , Beaker 500 mL Balanza Triple brazo Cobre (300 g) Hierro (300 g) Aluminio (300 g) Plomo (300 g) Guantes de asbesto 7.3 PROCEDIMIENTO
La experiencia se realiza en un calorímetro consistente en un vaso (Dewar) o en su defecto, convenientemente aislado. El vaso se cierra con una tapa hecha de material aislante, con dos orificios por los que salen un termómetro y el agitador. Supongamos que el calorímetro está a la temperatura inicial T0, y sea:
mv es la masa del vaso del calorímetro y cpv su calor específico. mt la masa de la parte sumergida del termómetro y cpt su calor específico ma la masa de la parte sumergida del agitador y cpa su calor específico M la masa de agua que contiene el vaso, su calor específico es la unidad
Por otra parte: Sean m y cp las masa y el calor específico del cuerpo problema a la temperatura inicial T. En el equilibrio a la temperatura Te se tendrá la siguiente relación:
(𝑴 +𝒎𝒗. 𝑪𝒑𝒗 +𝒎𝒕. 𝑪𝒑𝒕 +𝒎𝒂. 𝑪𝒑𝒂)(𝑻𝒆 − 𝑻𝟎) +𝒎.𝑪𝒑(𝑻𝒆 − 𝑻) = 𝟎
La capacidad calorífica del calorímetro es: 𝑲 = 𝒎𝒗. 𝑪𝒑𝒗 +𝒎𝒕. 𝑪𝒑𝒕 +𝒎𝒂. 𝑪𝒑𝒂se le
denomina equivalente en agua del calorímetro, y se expresa en gramos de agua. Por tanto, representa la cantidad de agua que tiene la misma capacidad calorífica que el vaso del calorímetro, parte sumergida del agitador y del termómetro, y es una constante para cada calorímetro. 1. Determinación del equivalente en agua del calorímetro Se ponen M gramos de agua en el calorímetro, se agita, y después de un poco de tiempo, se mide su temperatura T0. A continuación se vierten m gramos de agua a la temperatura T. Se agita la mezcla y después de un poco de tiempo, se mide la temperatura de equilibrio Te. Como el calorímetro es un sistema adiabáticamente aislado tendremos que (M + k). Cp. (Te − To) + mCa(Te − T) = 0 (7.5)
k =m(T − Te)
Te − To−M
(7.6)
2. Determinación del calor específico del sólido Calentar m gramos de agua en un Beaker con ayuda de un mechero hasta la temperatura T, colocarla en el calorímetro y cerrar la tapa, simultáneamente pesar M gramos del sólido,

40
introducirlo dentro del agua. Se agita la mezcla y después de un poco de tiempo, se mide la temperatura de equilibrio Te. La experiencia real se debe hacer con mucho cuidado, para que la medida del calor específico sea suficientemente precisa. Tenemos que tener en cuenta el intercambio de calor entre el calorímetro y la atmósfera que vienen expresadas por la denominada ley del enfriamiento de Newton. El calor específico desconocido del sólido será por tanto:
c =(M + k) ∗ (Te − T0)
m(T − Te)
(7.7)
1 cal/(g.ºC)
7.4 BIBLIOGRAFÍA
Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-México.
G.M. Barrow. 1976. Química Física para las Ciencias de la Vida. Ed. Reverté, I.N. Levine. 1996. Fisicoquímica. Ed. Mc Graw Hill.
Levine, Y.1992. Fisicoquímica, 3a. edición Mc Graw-Hill México. Shoemaker, David. 1968. EXPERIMENTOS DE FISICOQUÍMICA. 1ra edición. Mc
Graw-Hill/Interamericana de España, SA. México.

41
D. EQUILIBRIO QUÍMICO
Universidad de la Amazonia Facultad de Ciencias Básicas

42
Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA No. 8: EQUILIBRIO QUÍMICO.
OBJETIVO Determinar la constante de equilibrio de la reacción de formación del Ion complejo monotiocianato férrico. 8.1 FUNDAMENTO TEÓRICO La reacción de formación del ion complejo monotiocianato férrico, Fe(SCN)2+, a partir de ion tiocianato, SCN-, y férrico, Fe3+, se describe mediante la siguiente ecuación:
𝑺𝑪𝑵(𝒂𝒄)− + 𝑭𝒆(𝒂𝒄)
𝟑+ ↔ 𝑭𝒆(𝑺𝑪𝑵)(𝒂𝒄)𝟐+
Para determinar la constante de equilibrio de la misma se debe conocer la concentración de cada una de las especies presentes en el equilibrio. El ion Fe(SCN)2+ es la única especie coloreada que se forma en concentración apreciable en las condiciones de reacción de este trabajo práctico, y por lo tanto, es la única especie presente que presenta absorción en la región visible del espectro electromagnético. Por ello, su concentración se puede medir espectrofotométricamente. Las concentraciones de las otras especies pueden calcularse a partir de ésta mediante relaciones estequiométricas. Equipo a utilizar y procedimiento Se utilizará un espectrofotómetro de absorción ultravioleta-visible monohaz. 1) Seleccionar la longitud de onda a la cual se determinará la transmitancia de la muestra (para este caso λ = 450 nm). 2) Ajustar el cero de absorbancia con el solvente a utilizar, "blanco". 3) Colocar la cubeta con la muestra y medir la absorbancia. 8.2 MATERIALES Balón aforado 100 mL Pipeta aforada 25 mL 1 celda espectrofotométrica Pipeta aforada 10 mL 7 Tubos de ensayo Gradilla Pera pipeteadora Varilla de agitación Reactivos (5 grupos) 25 mL de Fe(NO3)3 0.2 M preparado en HCLO4 0.5 M. 100 mL de KSCN 0.001 M 250 mL HCLO4 2 M 8.3 PROCEDIMIENTO En un matraz aforado de 100,0 mL colocar 10,00 mL de KSCN 0,001 M medidos con pipeta aforada y 25,00 mL de HClO4 2M (también usando pipeta aforada) y llevar a volumen. Los pasos que se enumeran a continuación conviene realizarlos al lado del espectrofotómetro donde se realizarán las mediciones correspondientes.

43
Tomar una porción de 10,00 mL de dicha solución, agregar 0,1 mL de Fe(NO3)3 0,2 M en ácido perclórico 0,5 M, agitar con varilla para homogeneizar e inmediatamente transferir a la cubeta para la medición espectrofotométrica (usar pipeta graduada de 1 mL). Repetir agregando 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8 mL de Fe(NO3)3. Nota: es importante medir estos volúmenes con mucha exactitud, ya que pequeños errores conducen a serios problemas en los resultados del trabajo práctico. 8.4 ANÁLISIS DE LOS DATOS El ion tiocianato reacciona con el ion Fe3+ en solución ácida formando una serie de complejos:
𝑭𝒆𝟑+ + 𝑺𝑪𝑵− ↔ 𝑭𝒆(𝑺𝑪𝑵)𝟐+
𝑭𝒆(𝑺𝑪𝑵)𝟐+ + 𝑺𝑪𝑵− ↔ 𝑭𝒆(𝑺𝑪𝑵)𝟐
+
Si la concentración de ion tiocianato se mantiene baja, la concentración de los iones
complejos 𝑭𝒆(𝑺𝑪𝑵)𝒏(𝟑−𝒏)+
con n ≥ 2 es muy pequeña, y se puede suponer que el único ion
complejo presente en el equilibrio es el monotiocianato férrico (n = 1). El procedimiento consiste en agregar cantidades variables de una solución conteniendo Fe3+ a un volumen conocido de solución conteniendo SCN-. Al aumentar la cantidad de Fe3+ agregado, aumenta la concentración de monotiocianato férrico, y lasolución se torna cada vez más coloreada (es decir, aumenta su absorbancia). La determinación se realiza a una longitud de onda donde el ion Fe(SCN)2+ es la única especie que absorbe. Según la ley de Lambert-Beer:
𝑨 = 𝜺. 𝒃[𝑭𝒆(𝑺𝑪𝑵)𝟐+]𝒆𝒒
(8.1)
Donde ε es el coeficiente de absorción molar del compuesto y b el paso óptico de la cubeta. Las concentraciones de las otras especies en el equilibrio pueden obtenerse a partir de los siguientes balances de masa:
[𝑺𝑪𝑵−]𝟎 = [𝑺𝑪𝑵−]𝒆𝒒 + [𝑭𝒆(𝑺𝑪𝑵)
𝟐+]𝒆𝒒
(8.2)
[𝑭𝒆𝟑+]𝟎= [𝑭𝒆𝟑+]
𝒆𝒒+ [𝑭𝒆(𝑺𝑪𝑵)𝟐+]
𝒆𝒒 (8.3)
Donde el subíndice “0” indica concentración inicial y el subíndice “eq”, el valor en equilibrio. La constante de equilibrio correspondiente a la formación del ion Fe(SCN)2+ se puede escribir como:
𝑲 = [𝑭𝒆(𝑺𝑪𝑵)𝟐+]𝒆𝒒([𝑭𝒆𝟑+]
𝒆𝒒∗ [𝑺𝑪𝑵−]𝒆𝒒)⁄ (8.4)

44
Donde los corchetes representan la concentración molar de las respectivas especies en el equilibrio. NOTA: Los equilibrios en los que participan especies iónicas son afectados por la presencia de todos los iones en la solución. La fuerza iónica, una forma de expresar la concentración iónica total, se define como:
𝑰 = (𝟏
𝟐)∑𝑪𝒊𝒁𝒊
𝟐
DondeCi representa la concentración de cada especie y zisu carga. Estrictamente, la constante de equilibrio termodinámica es el cociente de las actividades de cada especia en el equilibrio y no el de sus concentraciones. Cuando la fuerza iónica es baja la actividad de cada especie es muy parecida a su concentración y el cociente de concentraciones tiende al valor de la verdadera constante termodinámica, que es el que se obtendría por extrapolación a fuerza iónica nula. Notar que en el trabajo práctico las mediciones se realizan a fuerza iónica constante y elevada. En estas condiciones, las correcciones necesarias exigen conocimientos que van más allá del alcance de la materia. La constante de equilibrio obtenida es válida, estrictamente, sólo en estas condiciones de fuerza iónica. Relacionando (2), (3) y (4) se obtiene:
𝑲 =[𝑭𝒆𝑺𝑪𝑵𝟐+]
([𝑭𝒆𝟑+]𝟎 − [𝑭𝒆𝑺𝑪𝑵𝟐+])([𝑺𝑪𝑵−]𝟎 − [𝑭𝒆𝑺𝑪𝑵
𝟐+])
(8.5)
La constante de equilibrio podría calcularse si se conociera la concentración del complejo, pero para ello habría que conocer su coeficiente de absorción molar, dado que lo que se mide es la absorbancia (ecuación 1). La determinación de ε requiere obtener una solución de complejo de concentración conocida. Una posible forma de lograrlo consiste en utilizar un gran exceso de alguno de los reactivos para desplazar completamente el equilibrio hacia la formación del complejo. Sin embargo, en este caso se generarían los siguientes problemas: a) si se agrega un gran exceso de ion SCN- se obtendrían complejos con n ≥ 2; b) en exceso de ion Fe3+, dado que hay que agregar cantidades grandes de este ión, la fuerza iónica de la solución sería diferente. Veremos sin embargo que K y ε pueden determinarse simultáneamente reordenando la ecuación (5) y realizando una aproximación cuya validez puede ser verificada a posteriori. Debido a que en las condiciones del trabajo práctico [SCN-]o es mucho menor que [Fe3+]o, se puede suponer que la concentración de Fe3+ no varía apreciablemente por la formación del ion complejo, es decir [Fe3+] ≈ [Fe3+]o. Matemáticamente esto equivale a suponer [Fe(SCN)2+] << [Fe3+]o y, por lo tanto, laecuación (5) se convierte en:
𝑲 =[𝑭𝒆𝑺𝑪𝑵𝟐+]
[𝑭𝒆𝟑+]𝟎([𝑺𝑪𝑵−]𝟎 − [𝑭𝒆𝑺𝑪𝑵
𝟐+])
(8.6)
y, combinando la ecuación anterior con la ecuación (1):

45
𝑲 =𝑨 𝜺. 𝒃⁄
[𝑭𝒆𝟑+]𝟎([𝑺𝑪𝑵−]𝟎 − 𝑨 𝜺. 𝒃⁄
(8.7)
Reordenando esta ecuación se obtiene:
𝑲 = 𝜺. 𝒃. [𝑺𝑪𝑵−]𝟎 −𝑨
𝑲. [𝑭𝒆𝟑+]𝟎
(8.8)
Si se grafica A vs A / [Fe3+]o se obtiene una recta de pendiente K-1 y ordenada al origen ε.b. Una vez obtenido el valor de K, se puede calcular la concentración de todas las especies para cada punto de la recta y verificar la suposición hecha para obtener la ecuación (6). CUESTIONARIO 1) ¿Se puede reemplazar el HClO4 por HCl o por HNO3? 2) Prediga cómo se modificaría la posición del equilibrio si: - se aumenta la temperatura. - se agrega KSCN. - se diluye la solución a la mitad con solución de HClO4. 3) La constante de equilibrio para la siguiente reacción:
𝑭𝒆𝟑+ + 𝑺𝑪𝑵− ↔ 𝑭𝒆(𝑺𝑪𝑵)𝟐+ vale 13 cuando la concentración se expresa en unidades molares. Calcule la concentración de Fe(SCN)2+ en una solución obtenida al agregar 5 mL de solución de Fe3+ de acuerdo a lo indicado en la parte experimental. 4) ¿Por qué las determinaciones se realizan a 450 nm? ¿Cómo lo demostraría experimentalmente? 8.5 BIBLIOGRAFÍA
Castellan, G.W. 1987. Fisicoquímica. Addison-Wesley Iberoamericana, Argentina-México.
G.M. Barrow. 1976. Química Física para las Ciencias de la Vida. Ed. Reverté,I.N.
Levine. 1996. Fisicoquímica. Ed. McGraw Hill.
Shoemaker, David. 1968. EXPERIMENTOS DE FISICOQUÍMICA. 1ra edición. Mc Graw-Hill/Interamericana de España, SA. México.
Journal of ChemicalEducation 40,71 (1963)

46
E. EQUILIBRIO DE FASES
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica

47
PRÁCTICA No. 9: EQUILIBRIO DE FASES. (Equilibrio Liquido Vapor Y Equilibrio
Sólido Líquido Para Mezclas Binarias)
OBJETIVOS
1. Determinar las curvas de equilibrio líquido-vapor para mezclas binarias de líquidos totalmente miscibles.
2. Determinar si las mezclas obedecen a la ley de Raoult o presentan desviación Positiva o negativa.
3. Construir el diagrama de fases sólido-líquido de un sistema binario que presenta punto eutéctico simple, mediante la aplicación del análisis térmico.
9.1 MARCO TEÓRICO Equilibrio de fases Una fase de acuerdo a la definición clásica, se define como materia uniforme en todas sus partes, tanto en composición química como física, ejemplos de ésta son: agua líquida, hielo, vapor de agua, solución de sacarosa en agua, la mezcla de gases en una muestra de aire etc. Dos o más fases se presentan cuando hay una superficie macroscópica de separación entre las fases homogéneas, ejemplos son: un sistema formado por tres fases (hielo-agua-aire), sistemas formados por dos fases (líquidos inmiscibles) y mezclas de sólidos o formas cristalinas diferentes del mismo sólido. El número de fases identificables depende de cómo se observe la muestra, por ejemplo, una solución de DNA es homogénea a simple vista, y a nivel microscópico se observa no homogénea. Existen diferentes tipos de equilibrios entre fases: sólido–líquido (S-L), líquido-líquido (L-L), líquido-vapor (L-V), etc., y también, pueden existir equilibrios de un solo componente, de dos componentes llamados binarios y de tres componentes denominados ternarios. El estado de una muestra de materia se define por dos tipos de variables: intensivas y extensivas. Las variables intensivas son aquellas que no dependen de la cantidad de materia presente como la temperatura, presión y concentración; y las variables extensivas son aquellas que si presentan dependencia de la cantidad de materia presente como por ejemplo la masa y volumen. En un sistema de una o más fases, generalmente se requiere conocer al menos una variable extensiva por fase, para determinar la masa de cada una de ellas, y el resto de las propiedades del sistema se pueden determinar a partir de propiedades intensivas. La regla de las fases de Gibbs define el número mínimo de variables intensivas necesarias para describir un sistema:
f = c − p + 2 (9.1)

48
Donde f son los grados de libertad del sistema, y se define como el número mínimo de variables intensivas necesarias para describir un sistema, c es el número de componentes químicos y p es el número de fases del sistema. El estado en equilibrio de una sustancia pura puede representarse en forma gráfica como se muestra en la Fig. 1, y se le conoce como diagrama de fase.
Figura 1. Diagrama de fase del agua. El diagrama de fase del agua (Fig. 1) muestra cómo pueden existir simultáneamente las fases sólida, líquida y vapor, en equilibrio. A lo largo de la línea de sublimación están en equilibrio la fase sólida y vapor; a lo largo de la línea de fusión lo están las fases sólida y líquida; y a lo largo de la línea de vaporización están la fase líquida y vapor. El único punto en el cual pueden estar las tres fases en equilibrio, es el punto triple. Equilibrio líquido-vapor en mezclas azeotrópicas El equilibrio liquido-vapor de sistemas ideales se puede describir de acuerdo a las leyes de Raoult y Dalton. La Ley de Raoult determina que:
PA = XAPoA (9. 2)
PB = XBPoB (9.3)
Donde PA y PB son las presiones parciales en la solución de los componentes A y B; P°A y P°B son las presiones de vapor de las sustancias puras, XA y XB son las fracciones mol. De acuerdo a la Ley de Dalton la presión total del sistema queda definida como:
PT = PA + PB (9.4)

49
En una mezcla homogénea de dos líquidos volátiles A y B, de acuerdo a la ley de Raoult se pueden presentar dos casos: 1. Si las fuerzas intermoleculares entre las moléculas A y B son más débiles que las que existen entre las moléculas de A y entre las de B, se dice que existe una mayor tendencia para considerarse una mezcla real que en el caso de una solución ideal. En consecuencia, la presión de vapor de la solución es mayor que la predicha por la ley de Raoult para la misma concentración. Esta conducta da origen a la desviación positiva. El mezclado es un proceso endotérmico. 2. Si las moléculas A y B se atraen con mayor fuerza que entre las del mismo tipo, la presión de vapor de la solución es menor que la suma de las presiones parciales, según la ley de Raoult. Dicha conducta se le conoce como desviación negativa. El mezclado es un proceso exotérmico. El comportamiento de cada uno de los casos descritos se puede apreciar en la Fig. 2
Figura 2. Diagrama de presión de vapor y punto de ebullición para sistemas que muestran: (a) Idealidad, (b) desviación positiva y (c) desviación negativa según la ley de Raoult.
Los diagramas de fases líquido-vapor, y en particular los de punto de ebullición, son de importancia para la destilación, la cual generalmente tiene como objetivo la separación parcial o completa de una solución líquida en sus componentes. Una destilación simple, "un plato" de un sistema binario, no tiene máximo o mínimo en su curva de punto de ebullición, como se observa en la figura 3. La fracción mol de B en la solución inicial se representa por χBL1. Cuando ebulle y una pequeña porción del vapor se condensa, se obtiene una gota de destilado con una fracción mol χBV1. Ya que éste es más rico en A que el residuo del matraz, el residuo es ligeramente más rico en B y se presenta por χBL2. La siguiente gota de destilado χBV2 es más rica en B que la primera. Si la destilación se continúa hasta que el residuo no ebulla más, la última gota del condensado será B puro. Para obtener una separación completa de la solución en A y B puros por destilaciones de esta clase es necesario separar el destilado cambiando el frasco receptor durante la destilación y posteriormente volver a destilar las fracciones separadas y repetir en varias ocasiones. El

50
mismo resultado se puede obtener en una sola destilación con el uso de una columna de fraccionamiento que contenga un gran número de "platos". Si hay un máximo en la curva de punto de ebullición (Fig. 2c) la composición de vapor y residuo no están cerca de A o B puros, sino a la composición correspondiente al máximo. Una mezcla con esta composición destilará sin cambio en composición y se conoce, como "mezcla de ebullición constante" o "azeótropo". Estos términos se aplican a una mezcla con punto de ebullición mínimo. En ocasiones son útiles (como el HCl-H2O de punto de ebullición constante usado como estándar analítico), y en otros casos un problema (como el azeótropo de 95% etanol y 5% de agua, que no permite la obtención de etanol absoluto por destilación directa de soluciones acuosas de etanol).
Figura 3. Variación dela composición de liquido y vapor durante la destilación. Equilibrio sólido líquido Considérese un sistema de dos componentes, a una de terminada presión, cuando el intervalo de temperaturas utilizado corresponde a la formación de una o más fases sólidas. El comportamiento más sencillo lo presentan aquellos sistemas para los que los líquidos son totalmente miscibles entre sí, y para los que las únicas fases sólidas que se pueden presentar, son las formas cristalinas puras de los componentes. Este comportamiento se presenta en la figura (1).
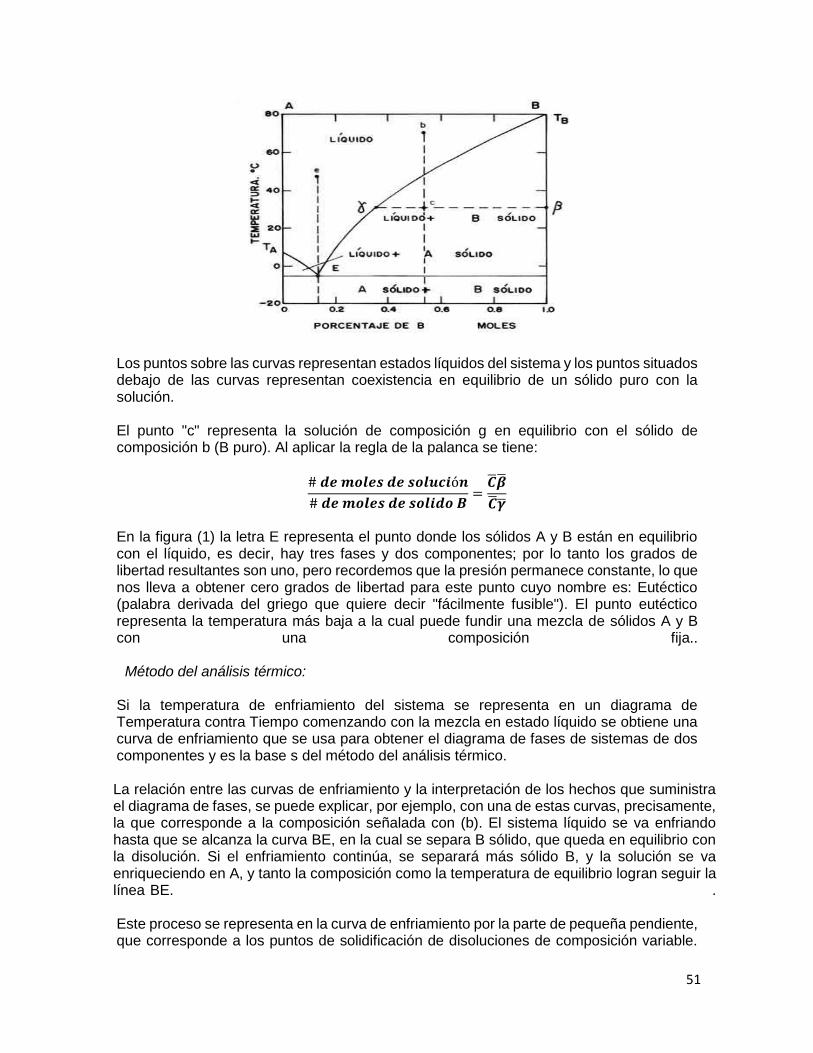
51
Los puntos sobre las curvas representan estados líquidos del sistema y los puntos situados debajo de las curvas representan coexistencia en equilibrio de un sólido puro con la solución. El punto "c" representa la solución de composición g en equilibrio con el sólido de composición b (B puro). Al aplicar la regla de la palanca se tiene:
# 𝒅𝒆 𝒎𝒐𝒍𝒆𝒔 𝒅𝒆 𝒔𝒐𝒍𝒖𝒄𝒊ó𝒏
# 𝒅𝒆 𝒎𝒐𝒍𝒆𝒔 𝒅𝒆 𝒔𝒐𝒍𝒊𝒅𝒐 𝑩=�̅��̅�
�̅��̅�
En la figura (1) la letra E representa el punto donde los sólidos A y B están en equilibrio con el líquido, es decir, hay tres fases y dos componentes; por lo tanto los grados de libertad resultantes son uno, pero recordemos que la presión permanece constante, lo que nos lleva a obtener cero grados de libertad para este punto cuyo nombre es: Eutéctico (palabra derivada del griego que quiere decir "fácilmente fusible"). El punto eutéctico representa la temperatura más baja a la cual puede fundir una mezcla de sólidos A y B con una composición fija.. Método del análisis térmico: Si la temperatura de enfriamiento del sistema se representa en un diagrama de Temperatura contra Tiempo comenzando con la mezcla en estado líquido se obtiene una curva de enfriamiento que se usa para obtener el diagrama de fases de sistemas de dos componentes y es la base s del método del análisis térmico. La relación entre las curvas de enfriamiento y la interpretación de los hechos que suministra el diagrama de fases, se puede explicar, por ejemplo, con una de estas curvas, precisamente, la que corresponde a la composición señalada con (b). El sistema líquido se va enfriando hasta que se alcanza la curva BE, en la cual se separa B sólido, que queda en equilibrio con la disolución. Si el enfriamiento continúa, se separará más sólido B, y la solución se va enriqueciendo en A, y tanto la composición como la temperatura de equilibrio logran seguir la línea BE. . Este proceso se representa en la curva de enfriamiento por la parte de pequeña pendiente, que corresponde a los puntos de solidificación de disoluciones de composición variable.

52
Debe señalarse que, aunque la temperatura y composición total coloca al sistema en el área por debajo de la curva BE, no puede existir ninguna fase con esa composición. Solo se pueden observar dos fases, una a la derecha y otra a la izquierda de la línea de puntos que representa la composición total. Como indica la figura (1) éstas se señalan con una línea horizontal de trazos, que pasa por la composición total en c, por ejemplo, para unir a ligar las dos fases que están presentes en el equilibrio. Pero en realidad, estas líneas no siempre es necesario dibujarlas.
9.2 MATERIALES Y REACTIVOS Montaje destilación simple Refractómetro, 11 tubos de ensayo, vaso de precipitado 250 mL, 1 tripode, 1 malla de asbesto, 1 pinza para tubo de ensayo, 1 mechero, 1 termómetro, 1 cronómetro, 1 soporte universal con pinza, difenilo y naftaleno. 9.3 PROCEDIMIENTO
1. Equilibrio líquido vapor El laboratorio consiste en la determinación de curvas de punto de ebullición o diagrama de fases para uno de los siguientes sistemas binarios: a) benceno-tolueno b) benceno-hexano c) cloroformo-acetona d) agua-ácido acético e) alcohol isopropílico-benceno f) cloroformo-metanol g) metanol-benceno 1. Preparar mezclas binarias que contengan 10, 20, 40, 60, 80 y 90% de uno de los componentes. 2. Medir los índices de refracción de cada mezcla a una temperatura determinada (25°C), empleando un refractómetro, así como los índices de refracción de los componentes puros.

53
3. Graficar la relación entre composición (𝑥) y el índice de refracción (𝑦). 4. Determinar teóricamente el punto de ebullición de los componentes puros a la presión atmosférica en Florencia. 5. Determinar los puntos de ebullición de cada una de las mezclas en equilibrio del vapor y del líquido: para esta etapa se debe realizar una destilación simple midiendo 20 ml. de mezcla. (nota: el bulbo del termómetro debe estar inmerso en el vapor generado). 6. Cuando la temperatura del sistema sea constante, anotar y colectar aproximadamente 5 ml en un matraz con tapón. 7. Determinar los índices de refracción del destilado (vapor) y del residuo que quedó en el matraz de destilación (líquido) con el refractómetro. El índice de refracción permite conocer la composición del líquido y del vapor en equilibrio de manera indirecta. 8. Proceder de la misma forma con cada mezcla binaria. PARA TENER CLARO ANTES DEL LABORATORIO 1. Definir los siguientes términos: Solución ideal y no ideal, Azeotrópo, Variable intensiva y extensiva (indicar 3 ejemplos de cada una), Grados de libertad, Tipos de equilibrio, Índice de refracción. 2. Reportar el diagrama de bloques de la metodología a desarrollar. 3. Determinar los grados de libertad para un sistema etanol-metanol-agua en equilibrio. PARA TENER EN CUENTA EN EL INFORME 1. Reportar los resultados en forma tabular y gráfica. 2. Realizar las siguientes gráficas: * Índice de refracción Vs Concentración * Diagrama de fase en equilibrio 3. De acuerdo a los resultados analizar si la mezcla presenta un punto azeotrópico yademás, determinar qué tipo de comportamiento presenta de acuerdo a la ley de Raoult. 4. Análisis de resultados y Conclusiones.
2. Equilibrio sólido líquido a. Se pesan las cantidades de reactivos indicados en la tabla y se colocan en los tubos
correspondientes.
Tubo 1 2 3 4 5 6 7 8 9 10 11
Dif (g) 5.0 4.6 4.0 3.75 3.35 3.0 2.0 1.65 1.25 1.0 0.0
Naf(g) 0.0 0.4 1.0 1.25 1.65 2.0 3.0 3.35 3.75 4.0 5.0
% Dif
b. Una vez preparados los tubos con las cantidades necesarias, cada una de las mezclas formadas se funde por separado en el baño maría y se continua el calentamiento hasta alcanzar la temperatura de 80ºC. Logrado esto, los tubos se sacan del baño y se colocan firmemente en un soporte, con la finalidad de evitar movimientos que perturben el enfriamiento de las mezclas.

54
NOTA: Ocurrida la fusión, durante el calentamiento posterior debe agitarse continuamente la mezcla, con el objeto de homogeneizarla.
c. A partir de este momento (80ºC) se toman lecturas del termómetro cada 30 segundos hasta llegar a 30ºC aproximadamente. Con los datos obtenidos se trazan las curvas de enfriamiento correspondientes a cada mezcla y a partir de éstas se traza el diagrama de fases.
9.4 CUESTIONARIO:
1. ¿Qué representan las temperaturas TA y TB en la figura (1)?. 2. Dibuje las curvas de enfriamiento que esperarían para los componentes puros?
Explique su forma. 3. ¿A qué se debe el cambio de pendiente de la curva de enfriamiento (b) de la figura
(2)?. 4. ¿Por qué en la zona de detención eutéctica la temperatura permanece constante?. 5. Dibuje la curva de enfriamiento que se esperaría para un tubo con la composición
eutéctica. Dibuje su forma.
Concluida la práctica, las sustancias se calientan hasta fusión y el residuo se deposita en el frasco de desechos. Repetir esta operación tantas veces como sea necesario. Cuando el tubo esté prácticamente limpio, se lava con un escobillón, detergente y agua hirviendo hasta que se logre la limpieza total de este.
. 9.5 BIBLIOGRAFÍA
D.P. Shoemaker, C.W. Garland, J.I. Steinfeld., EXPERIMENTS IN PHYSICALCHEMISTRY, Mc. Graw Hill, 1962.Pags.228-237.
Sime R. J., PHYSICAL CHEMISTRY: METHODS, TECHNIQUES
ANDEXPERIMENTS, Saunders College.Publishing, 1988, USA, Pag.449-451.
Barrow, G.M. Química Física Ed, Reverté, S.A.
Ferguson, F.D. La Regla de las Fases Ed. Alhambra, S.A.
Urquiza, M. Experimentos de Fisicoquímica Ed. LimusaWiley, S.A.

55
F. PROPIEDADES COLIGATIVAS:
Universidad de la Amazonia Facultad de Ciencias Básicas

56
Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 10: ASCENSO PUNTO DE EBULLICIÓN Y
DESCENSO CRIOSCÓPICO.
OBJETIVOS
1. Comprender las propiedades coligativas de soluciones con soluto no electrolito y electrolito.
2. Analizar el efecto que tiene la adición de cantidades diferentes de un soluto no electrolito y electrolito, sobre el abatimiento de la temperatura de fusión de un disolvente.
Objetivos específicos
Determinar las constantes ebulloscópica molal 𝐾𝑏 y crioscópica molal 𝐾𝑐 del agua a partir de soluciones de cloruro de sodio y de soluciones no electrolíticas. Determinar la temperatura de congelación de disoluciones acuosas de un no electrolito, a diferentes concentraciones, a partir de curvas de enfriamiento. Calcular la constante crioscópica del agua con base en el efecto de la concentración de un no electrolito sobre la temperatura de congelación del agua. 10.1 MARCO TEÓRICO El punto de congelación de una solución acuosa cualquiera es menor que el del agua pura. Esta propiedad se denomina «descenso crioscópico» y es una propiedad coligativa. Una de las prácticas fraudulentas en la producción e industria de la leche, es la adición de agua a la leche con el objeto de aumentar su volumen. Los métodos que pueden aplicarse a la detección de agua adicionada a la leche, están basados en la medición de una propiedad física que varia proporcionalmente a la cantidad de agua adicionada al producto, tal como ocurre con el punto de congelación. La leche por poseer numerosas sustancias en solución, tiene un punto de congelación inferior al del agua. El descenso crioscópico normal observado en la leche se debe principalmente a la lactosa y sales minerales que se encuentra en solución. Cuando se le agrega agua a la leche, se diluyen sus solutos y el punto de congelación aumenta, acercándose al del agua pura. El punto de congelación aumenta en proporción a la cantidad de agua adicionada. Las propiedades de una solución son una mezcla de las propiedades del soluto y solvente. En soluciones diluidas de cualquier solvente, existen algunas propiedades físicas que no depende de la naturaleza del soluto. Estas propiedades, llamadas propiedades coligativas, dependen únicamente de la concentración de soluto. Las Propiedades coligativas son la presión osmótica, el descenso de la presión de vapor del solvente, el ascenso del punto de ebullición con referencia al del solvente puro y el descenso del punto de congelación con referencia al del solvente puro. El ascenso del punto de ebullición de soluciones se describe mediante la relación

57
∆𝑇𝑏 = 𝑇𝑏 − 𝑇𝑏0 = 𝑘𝑏𝐶𝑚
10.1
Donde Tb es la temperatura de ebullición de la solución, 𝑇𝑏0 la temperatura de ebullición del
solvente puro, kb una constante de proporcionalidad llamada constante ebulloscópica molal que depende de la naturaleza del solvente, y Cm la concentración molal de la solución. De manera análoga, la relación para el descenso del punto de congelación es
∆𝑇𝑐 = 𝑇𝑐 − 𝑇𝐶0 = −𝑘𝑐𝐶𝑚
10.2
Donde Tc es la temperatura de congelación de la solución, 𝑇𝐶0 la temperatura de
congelación del solvente puro, kc una constante de proporcionalidad llamada constante crioscópica molal que depende de la naturaleza del solvente, y 𝐶𝑚 la concentración molal de la solución. 10.2 MATERIALES Y REACTIVOS: Para solución con electrolito
Materiales Reactivos
2 vasos de precipitado de 500 mL
Agua destilada
Tubo de ensayo Cloruro de sodio (NaCl) Termómetro Hielo machacado Plancha eléctrica
Para solución con no electrolito
Agua destilada 5 tubos de ensayo de 15 mL
Soluciones de Urea(0.25, 0.5, 0.75, 1.0 molal)
1 gradilla para tubos de ensayo
Soluciones de dextrosa (0.25, 0.5, 0.75, 1.0 molal)
1 vaso de unicel con tapa de un litro
Sal de grano (NaCl) 1 termómetro de décimas de grado de -10 a 32°C o digital
Hielo 1 cronometro
Benceno, Ácido benzoico, nafataleno y ciclohexano. 10.3 PROCEDIMIENTO EXPERIMENTAL
1. Electrolito
1.1 Ascenso del punto de ebullición:

58
Llenar 3/4 de un vaso de precipitado con agua destilada, y llevarlo a ebullición sobre la
hornilla. Registrar la temperatura de ebullición del agua 𝑇𝑏0con el termómetro. Al hacer
lecturas con el termómetro, el bulbo no debe hacer contacto con el fondo ni las paredes del vaso. Retirar el vaso de la hornilla, dejar enfriar por un lapso corto de tiempo, y trasvasar 200 g de agua caliente al otro vaso. Agregar 10 g de cloruro de sodio al segundo vaso, disolver completamente, y llevar la mezcla a ebullición. Registrar la temperatura de ebullición de la mezcla Tb. Retirar el vaso de la hornilla, dejar enfriar por un lapso corto de tiempo, añadir 15 g de cloruro de sodio a la mezcla anterior, disolver, y llevar la nueva mezcla a ebullición. Repetir los pasos 5 y 6 hasta completar 55 g de cloruro de sodio. 1.2 Descenso del punto de congelación:
Agua destilada 5 tubos de ensayo de 15 mL
Soluciones de Urea(0.25, 0.5, 0.75, 1.0 molal)
1 gradilla para tubos de ensayo
Soluciones de dextrosa (0.25, 0.5, 0.75, 1.0 molal)
1 vaso de unicel con tapa de un litro
Sal de grano (NaCl) 1 termómetro de décimas de grado de -10ª 32°C o digital
Hielo 1 cronometro
1. Preparar una mezcla frigorífica en un vaso con hielo machacado mediante la adición de sal sobre el hielo en un proporción de 1 a 3 en peso.
2. Agregar 10 g de agua destilada en el tubo de ensayo, e introducir el tubo en la mezcla frigorífica.
3. Introducir el termómetro en el tubo evitando el contacto con las paredes, y remover el tubo con precaución hasta congelamiento.
4. Registrar la temperatura de congelación del agua 𝑇𝑐0.
5. Retirar el tubo, descongelar el agua, agregar tantos g de cloruro de sodio como sea necesario para una solución 0,25 m, y disolver completamente.
6. Introducir el tubo en la mezcla frigorífica, introducir el termómetro en el tubo, y remover hasta congelamiento.
7. Registrar la temperatura de la mezcla cada 30 seg en la tabla 1 y finalmente la temperatura de congelación de la mezcla Tc.
8. Repetir los pasos 5 -7para soluciones 0,5, 0,75 y 1 m de cloruro de sodio. 9. Repetir los pasos 1-7 con soluciones de cloruro de calcio (Registrar en tabla anexa
2).
2. No electrolito
2.1 Soluciones de urea y dextrosa
Se desean obtener datos de temperaturas de congelamiento para soluciones a diferentes concentraciones y en diferentes tiempos. Las tablas4, 5 y 6almacenarán los resultados que se requieren (Ver tablas anexas).
2.2 Otros no electrolitos

59
En este caso dada la importancia de la precisión experimental, todas las soluciones se preparan por pesada y de manera independiente en vez de hacer adiciones sucesivas. Preparar soluciones por pesada cuyas concentraciones sean las siguientes: 2,5m; 2,0m; 1,5m; 1,0m; 0,7m; 0,5m; 0,3m. De naftaleno en benceno; naftaleno en ciclohexano y ácido benzoico en benceno. Determinar la temperatura de congelación. CÁLCULOS, RESULTADOS Y ANÁLISIS DE RESULTADOS 1.1 Para Procedimiento 1.1
1. Completar la siguiente tabla a partir de los datos experimentales.
Mezcla Masas H2O Masa de NaCl
𝐶𝑚 𝑇𝑏 ∆𝑇𝑏
1 200g 10 g 2 200g 25 g 3 200g 40 g 4 200g 55 g
2. Con los datos calculados de la tabla anterior, determinar la constante ebulloscópica
molal 𝑘𝑏 del agua.
3. Determinar el error relativo del valor experimental de 𝑘𝑏 con respecto al valor teórico de 0,52 °C kg/mol para el agua. 4. Calcular el coeficiente de actividad experimental del soluto y del solvente. Determine porcentajes de error con los esperados teóricamente. 1.2 Para procedimiento 1.2 1. Trazar las curvas de enfriamiento (Temperatura vs tiempo) para cada sistema,
utilizando los datos de las tablas 1 y 2. 2. Completar la siguiente Tabla 3 (Ver anexo) con los datos experimentales, 3. Construir el gráfico de la disminución de la temperatura de congelación en función de
la concentración de las disoluciones de cloruro de sodio y de cloruro de calcio. Utilizando los datos de la tabla 3.
4. Con los datos calculados de la tabla 3 anterior, determinar la constante crioscópica del agua kc.
5. Construir el gráfico de ∆𝑇𝑓(𝑁𝑎𝐶𝑙 𝑦 𝐶𝑎𝐶𝑙)𝑣 𝑠 ∆𝑇𝑓 (no electrolito, teórico y experimental).
6. Explicar cómo varia la temperatura de congelación de las disoluciones en función de la concentración de cloruro de sodio y del cloruro de calcio, de acuerdo a las datos incluidos en las tablas 1 y 2.
7. Explicar por qué la temperatura de los sistemas objeto de estudio permanece constante en cierto intervalo de tiempo.
8. Explicar el comportamiento del gráfico de la disminución de la temperatura de congelación en función de las concentraciones de cloruro de sodio y cloruro de calcio; proponer una ecuación que los describa.
9. Analizar el gráfico de ∆𝑇𝑓(𝑁𝑎𝐶𝑙 𝑦 𝐶𝑎𝐶𝑙)𝑣 𝑠 ∆𝑇𝑓 (no electrolito), proponer una ecuación
que lo describa. Explicar cuál es el significado de cada uno de los términos de la ecuación.
10. Comparar el valor del factor de van’tHoff teórico con el experimental.

60
11. Determinar el error relativo del valor experimental de kc con respecto al valor teórico
de 1,86 ℃ kg/mol para el agua. 12. Calcule el coeficiente de actividad experimental del solvente y del soluto. Compare con
valores teóricos. 1.3 Para procedimiento 2.1 Trazar las curvas de enfriamiento (temperatura vs. tiempo) para cada sistema, utilizando los datos de las tablas 4 y 5. 2. Construir el grafico de la disminución de la temperatura de congelación en función de la concentración de las disoluciones de urea y dextrosa. Utilizando los datos de la tabla 6. 3. Explicar cómo varia la temperatura de congelación de las disoluciones en función de la concentración de urea y de la dextrosa, de acuerdo a los datos incluidos en las tablas 4 y 5. 4. Explicar porque la temperatura de los sistemas objeto de estudio permanece constante en cierto intervalo de tiempo. 5. Explicar el comportamiento del grafico de la disminución de la temperatura de congelación en función de la concentración de urea y de dextrosa proponer una ecuación que lo describa. 6. Calcular el valor de las pendientes de los gráficos del punto (3), analizar sus unidades y explicar que representan estos datos. 7. Comparar el valor obtenido en el punto (4) con el reportado en la literatura y calcular el por ciento de error. 8. Determine los coeficientes de actividad para el soluto y el solvente experimentales en cada caso y compare con los esperados teóricamente. Determine las actividades. 9. Elabore análisis y conclusiones detallados. 1.4 Para procedimiento 2.2
1. Construir una gráfica de Θ (descenso crioscópico) vs concentración de soluto de peso molecular conocido. Calcular Kc
2. Analice las diferencias entre descensos crioscópicos para solventes con solutos no volátiles electrolito, no electrolito, iónicos y no polares.
3. Determine los coeficientes de actividad y las actividades experimentales para los
solutos y solventes en este punto.
4. Análisis y conclusiones 10.4 BIBLIOGRAFÍA
David W. Ball, (2004), Fisicoquímica, Editorial Thomson, Keith J. Laidler, (1997), Fisicoquímica, Editorial CECSA. Lange, N. (1998), Lange. Manual de Química. McGraw-Hill.
ANEXOS Tabla 1. Datos experimentales de tiempo y temperatura para el agua y las soluciones de NaCl
61
Temperatura (°C)
Sistema H2O NaCl/ H2O
Tiempo (min) 0.0 m 0.25m 0.50m 0.75 1.0m
0.0
Tabla 2. Datos experimentales de tiempo y temperatura para el agua y las soluciones de NaCl Tabla 3. Datos experimentales de tiempo y temperatura para el agua y las soluciones de NaCl
m/(moles Kg-1) t/(°C) T/(°K) ∆T//(°K)
NaCl/Agua
0.0
0.25
0.50
0.75
1.0
CaCl2/Agua
0.0
0.25
0.50
0.75
1.0
Tabla 4: Datos experimentales de tiempo y temperatura para el agua y las soluciones de dextrosa
Temperatura (°C)
Sistema H2O CaCl/ H2O
Tiempo (min)
0.0 m 0.25m 0.50m 0.75 1.0m
0.0
Temperatura (°C)
Sistema H2O H2O/dextrosa
62
Tabla 5. Datos experimentales de tiempo y temperatura para el agua y las soluciones de urea
Temperatura (°C)
Sistema H2O H2O/Urea
Tiempo (min) 0.0 m 0.25m 0.50m 0.75 1.0m
0.0
Tabla 6. Valores de la temperatura de congelación del agua y de las soluciones de urea y de dextrosa.
m/(moles Kg-1) t/(°C) T/(°K) ∆T//(°K)
Agua/Urea
0.0
0.25
0.50
0.75
1.0
Agua/dextrosa
0.0
0.25
0.50
0.75
1.0
Tiempo (min)
0.0 m 0.25m 0.50m 0.75 1.0m
0.0

63
G. CINÉTICA
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica

64
PRACTICA DE LABORATORIO NO.11: CINÉTICA DE LA VITAMINA C. (estudio de la
cinética de oxidación de la vitamina c con ferricianuro de potasio. determinación de
la ley experimental de rapidez)
OBJETIVO PRINCIPAL. Determinar la ley experimental de rapidez de la reacción de oxidación de vitamina C con ferricianuro de potasio Objetivos Secundarios.
Utilizar el método de aislamiento de Ostwald para el diseño del experimento.
Determinar los órdenes parciales, aplicando el método integral para el análisis de los datos cinéticos.
11.1 MATERIALES Y REACTIVOS. Cronómetro Matraz erlenmeyer 50 mL (3) Pipeta 10 mL, 5 mL Espectrofotómetro con su celda Beaker 50 mL (3) Pera Pipeteadora Gradilla Tubos de ensayo 10 mL, (5) Ferricianuro de potasio (K3Fe(CN)6) 0.0025 M , 50 mL Ácido nítrico (HNO3) 0.1 M, 25 mL Ácido ascórbico (C6O6H8) 0.004 M, 25 mL
11.2 PREVIAMENTE. 1. Investigue cuál es el fundamento de la espectrofotometría y de la ley de Lambert-Beer. 2. ¿Qué es una curva patrón y para qué se utiliza en esta práctica? 3. Defina los siguientes términos: a) Orden de Reacción b) Constante de Velocidad c)
Reactivo Limitante. 4. Balancee la ecuación redox (medio ácido) que se efectúa entre la vitamina C y el
ferricianuro de potasio. C6O6H8 + Fe(CN)6
3- → C6O6H6 + Fe(CN)64-
11.3 Técnica experimental.
Conectar los equipos: Calibrar el espectrofotómetro: = 418 nm, blanco: agua destilada.
Etiquete, para cada corrida, 3 vasos de precipitados, limpios y secos, de la siguiente manera: Ácido ascórbico H2A, Ferricianuro de potasio FP y Mezcla reactiva M.
Preparar la curva patrón de ferricianuro de potasio (Tabla 1). Calcular la concentración de cada solución. Leer absorbancia de la curva patrón, después programar el espectrofotómetro para medir la cinética de la reacción. Tabla 1. Curva Patrón de Ferricianuro de Potasio

65
K3Fe(CN)6 (0.0025 mol.dm-3) ml
HNO3 (0.1
mol.dm-3) ml
H2O destilada.
ml
Concentración molar final
FP
Absorbancia
4 1 5
3 1 6
2 1 7
1 1 8
0.25 1 8.75
Una vez que tenga sus vasos de ferricianuro (FP) y vitamina (H2A), preparados según la corrida que vaya a trabajar (ver tabla 2), vacíelos SIMULTÁNEAMENTE en el vaso M y al mismo tiempo dispare el cronómetro.
Tabla 2. Soluciones reactivas de acuerdo al cambio de concentración del reactivo limitante
Vaso FP Vaso H2A
Corrida Num.
ml K3Fe(CN)6 0.0025 M
ml HNO3 0.1M
ml H2O (destil)
ml Vit C 0.004M
ml H2O (destil)
Intervalo de Lectura (s)
1 8 2.0 0 5 5 30
2 6.4 2.0 1.6 4 6 60
3 4 2.0 4 2.5 7.5 90
4 3.2 2.0 4.8 2 8 120
Vacíe cuidadosamente en la celda del espectrofotómetro un volumen ligeramente arriba de la mitad, nunca completamente llena y tome 10 lecturas de concentración a 418 nm a los intervalos de tiempo dados en la tabla 2.
NOTA: El tiempo entre lecturas, puede variar según avanza la reacción. PARA EL INFORME. 1. Calcule las concentraciones iníciales de la vitamina C y del ferricianuro de potasio en las mezclas de reacción e indica cuál es el reactivo “aislado” y cuál el “desbordado” en este experimento. 2. Realice Curva Patrón y gráfica correspondiente. (ABS vs C), datos de regresión lineal 3. Método Diferencial: elaborar las gráficas C vs t, obtener la rapidez en cada punto, elaborar la gráfica Log r vs Log C. 4. Método Integral: Aplicar las ecuaciones lineales de orden “0”, “1” y “2” a los datos de cada corrida. Datos de la regresión lineal de cada gráfico. Gráficas de cada orden. Presentar los gráficos de cada “orden” para cada juego de corridas. 5. Determinar orden de reacción y constante de rapidez, por cada método. Comparar. 6. Calcular la rapidez para cada corrida. Cuestionario:

66
1. Qué proporcionalidad guarda la velocidad respecto a la concentración en una reacción de 0, 1º y 2º orden? 2. Mencione las ventajas y desventajas del método diferencial frente al integral.
11.4 BIBLIOGRAFÍA RECOMENDADA
Atkins, P.W., "Fisicoquímica", Addison Wesley Iberoamericana, 3ª edición, 1991. Castellan, G.W., "Fisicoquímica", Addison Wesley, 2ª edición, 1987. Levine, I.N., "Fisicoquímica", 2 vols, Mc. Graw Hill Interamericana de España, 3ª
edición, 1997.
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
67
PRÁCTICA DE LABORATORIO NO. 12: RELOJ DE YODO
OBJETIVOS En medio ácido el bromato oxida los iones yoduro hasta yodato, siguiendo un complejo mecanismo de reacción que involucran 8 intermediarios de reacción (HBrO2, HOBr, Br ̄, HIO2, HOI, HOI, IBr, I2, Br2). Está reacción es de especial interés en el área de la Dinámica Química No Lineal, ya que cuando se lleva a cabo en un CSTR exhibe biestabilidad, oscilaciones en el potencial redox y en las concentraciones de los intermediarios. Esta reacción presenta una marcada dependencia en las concentraciones iníciales de los reactivos de partida (bromato, yoduro y ácido). El objetivo de este laboratorio es determinar cinéticamente los ordenes de reacción en cada uno de ellos para una ley de velocidad
propuesta: −𝑑[𝐵𝑟𝑂3− ]/𝑑𝑡 = 𝐾´[𝐵𝑟𝑂3
−]𝑥[𝐼−]𝑦 [𝐻+]𝑧 12.1 FUNDAMENTOS La reacción de oxidación de los iones yoduro por bromato en medio ácido se puede seguir por varios métodos: a) espectrofotométrico, b) Potenciométrico, c) visual. Los intermediarios de reacción que permiten el estudio cinético de la reacción por espectrofotometría son los siguientes:
Especie λ (máx) (nm) ϵ (M-1 cm-1)
I2/I3−(ac) 460 770
Br2(ac) 390 161
IBr(ac) 390 346
Esta reacción se caracteriza por exhibir un comportamiento tipo “reloj” es decir que después de cierto tiempo (conocido como “tiempo de inducción”) la concentración de alguno de los intermediarios de la reacción sufre un repentino y fuerte cambio en la concentración. Aquí el tiempo de inducción se debe a la presencia de los iones yoduro, los cuales se consumen lentamente en el medio de reacción. Una vez que el yoduro ha desaparecido del medio de reacción el yodo experimenta una repentina y fuerte disminución (hasta desaparecer) en la concentración, como se indica en la siguiente figura:
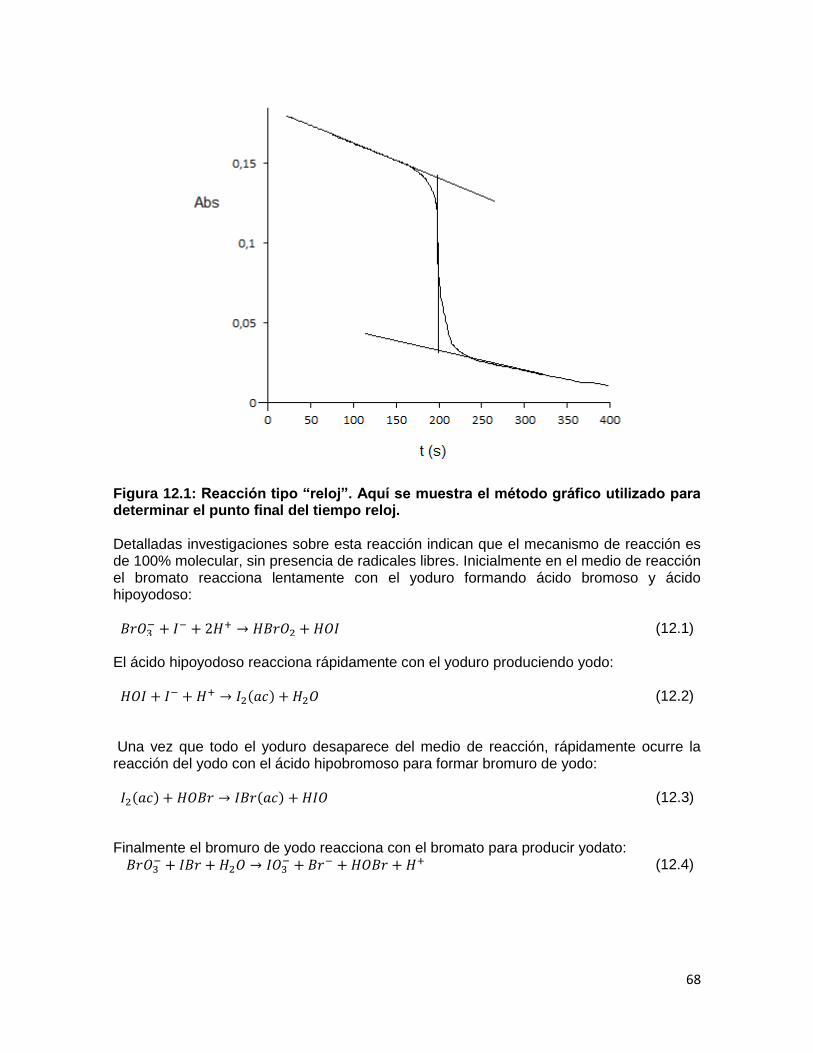
68
Figura 12.1: Reacción tipo “reloj”. Aquí se muestra el método gráfico utilizado para determinar el punto final del tiempo reloj. Detalladas investigaciones sobre esta reacción indican que el mecanismo de reacción es de 100% molecular, sin presencia de radicales libres. Inicialmente en el medio de reacción el bromato reacciona lentamente con el yoduro formando ácido bromoso y ácido hipoyodoso:
𝐵𝑟𝑂3− + 𝐼− + 2𝐻+ → 𝐻𝐵𝑟𝑂2 +𝐻𝑂𝐼 (12.1)
El ácido hipoyodoso reacciona rápidamente con el yoduro produciendo yodo: 𝐻𝑂𝐼 + 𝐼− +𝐻+ → 𝐼2(𝑎𝑐) + 𝐻2𝑂 (12.2)
Una vez que todo el yoduro desaparece del medio de reacción, rápidamente ocurre la reacción del yodo con el ácido hipobromoso para formar bromuro de yodo:
𝐼2(𝑎𝑐) + 𝐻𝑂𝐵𝑟 → 𝐼𝐵𝑟(𝑎𝑐) + 𝐻𝐼𝑂 (12.3)
Finalmente el bromuro de yodo reacciona con el bromato para producir yodato: 𝐵𝑟𝑂3
− + 𝐼𝐵𝑟 + 𝐻2𝑂 → 𝐼𝑂3− + 𝐵𝑟− +𝐻𝑂𝐵𝑟 + 𝐻+ (12.4)

69
El bromuro y el ácido hipobromoso reaccionan produciendo bromo molecular que se acumula en el medio de reacción.
𝐵𝑟− +𝐻𝑂𝐵𝑟 + 𝐻+ → 𝐵𝑟2(𝑎𝑐) + 𝐻2𝑂 (12.5)
Detallados estudios cinéticos revelan que la reacción sigue dos estequiometrias:
a) Cuando el bromato es un exceso:
𝐵𝑟𝑂3− + 6𝐼− + 6𝐻+ → 𝐵𝑟− + 3𝐼2(𝑎𝑐) +
3𝐻2𝑂
(12.6)
b) Cuando el yoduro es en exceso:
𝐵𝑟𝑂3− + 9𝐼− + 6𝐻+ → 𝐵𝑟− + 3𝐼3
−(𝑎𝑐) + 3𝐻2𝑂 (12.7)
A pesar de la complejidad de la reacción, la marcada dependencia de las concentraciones iníciales de los reactivos de partida: bromato, yoduro y ácido, permite proponer la siguiente ley de velocidad:
−𝑑[𝐵𝑟𝑂3− ]/𝑑𝑡 = 𝐾´[𝐵𝑟𝑂3
−]𝑥[𝐼−]𝑦 [𝐻+]𝑧 (12.8)
12.2 TRABAJO EXPERIMENTAL 1. Preparar las siguientes soluciones: 1000mL de H2SO4 0,7M (solución A), 300 mL de
𝐾𝐵𝑟𝑂3 0,01M en H2SO4 0,7M (solución B), 250 mL de KI 0,005M en H2SO4 0,7M (solución C). Las soluciones de KBrO3y KI se deben preparar con la solución A.
2. Estudio cinético: Realice las siguientes mezclas (todos los valores están en cm³) y tome tiempo cero cuando haya adicionado la mitad del volumen de KI (esta adición se debe hacer lo más rápido posible). Para cada una de las mezclas de reacción anote el tiempo en el cual la mezcla de reacción cambia repentinamente de color (de un tono café-rojizo, típico del yodo, aun color amarillo nítidamente definido). Mezclas:
Reactivo / Mezcla 1 2 3 4 5 6
Determinación del orden de reacción en bromato
10 15 20 25 30 35
30 25 20 15 10 5
Agua 0 0 0 0 0 0
KI 10 10 10 10 10 10
Determinación del orden de reacción en el ácido
10 10 10 10 10 10
5 10 15 20 25 30
Agua 25 20 15 10 5 0

70
KI 10 10 10 10 10 10
Determinación del orden de reacción en yoduro
10 10 10 10 10 10
36 32 28 24 20 16
Agua 0 0 0 0 0 0
KI 4 8 12 16 20 24
3. Recoja todos los residuos en una solución de NaOH.
4. Como una variante al experimento, algunos grupos pueden realizar el estudio cinético
utilizando ácido perclórico y realizando ajuste de fuerza iónica con perclorato de sodio.
12.3 TRATAMIENTO DE DATOS Y CUESTIONARIO 1. Construya las gráficas de concentración (bromato, yoduro, ácido) contra “tiempo de
inducción”. 2. Analice detenidamente las gráficas. Revise los experimentos de reducción del ion
permanganato y botella azul para decidir como procesar los datos para determinar los ordenas de reacción 𝑥, 𝑦, 𝑧.
3. Explique el significado cinético de los resultados obtenidos.
4. Cómo cree que se afectaría la cinética de la reacción si se colocara una concentración
inicial de yodo molecular?
5. Una forma de describir lo que ocurre en esta reacción es la siguiente: a) Lento consumo de yoduro b) Rápida formación y acumulación de yodo en solución c) Repentina y autocatalítica disminución del yodo d) Rápida y repentina formación y, rápida y repentina disminución del brumuro de yodo. e) Formación de bromo molecular y de yodato
Explique cada uno de los pasos de acuerdo al mecanismo anexo.
6. Del mecanismo de reacción analice la competencia cinética de:
a) Conjunto de reacciones que involucran bromato b) Reacciones que involucran yoduro c) Reacciones que involucran yodo
12.4 BIBLIOGRAFÍA
Clarkm R.H., J. Phys. Chem., 10, 679, 1958. King, D. E., Lister, W. M., Can. J. Chem., 46, 279, 1968. Simoyi, R. H., et. Al., J. Phys. Chem., 100, 1643, 1996. Simoyi, R. H., et. Al., J. Phys. Chem., 90, 4126, 1986. Simoyi, R. H., et. Al., J. Phys. Chem., 89, 3570, 1985.
12.5 ANEXO 1

71
La primera reacción con comportamiento tipo “reloj” se debe a H. Landolt (1985). Esta reacción es también un “reloj de yodo”, en la cual se estudia la reducción de los iones yodato a yoduro en presencia de bisulfito:
𝐼𝑂3− + 3𝐻𝑆𝑂3
− → 𝐼− + 3𝐻𝑆𝑂4− (12.9)
El yoduro formado reacciona rápidamente con el yodato para producir yodo molecular:
𝐼𝑂3− + 5𝐼− + 6𝐻+ → 3𝐼2 + 3𝐻2𝑂 (12.10)
Pero el yodo producido es rápidamente reducido a yoduro en presencia del bisulfito:
𝐼2 +𝐻𝑆𝑂3− +𝐻2𝑂 → 2𝐼
− +𝐻𝑆𝑂4− + 2𝐻+ (12.11)
Esta última reacción es tan rápida que el yodo no aparece en el medio de reacción sino hasta que todo el bisulfilto ha sido oxidado. En este punto, en presencia de una pequeña cantidad de almidón, la solución se oscurece rápidamente. El tiempo que toma al yodo molecular para aparecer es función de las concentraciones y la temperatura. Se ha estimado que a 23ºC el tiempo del “reloj” obedece la siguiente relación:
𝑡(𝑠) =3,7 𝑥10−3
[𝐼𝑂3−][𝐻𝑆𝑂3
−]
(12.12)
Nota usted cuál es la diferencia cinética entre el reloj de yodo de Landolt y el propuesto en este libro?. 12.6 Anexo 2: Mecanismo de reacción No. Reacción Constante cinética 1 𝐵𝑟𝑂𝟑
− + 𝐼− + 2𝐻+ → 𝐻𝐵𝑟𝑂2 +𝐻𝑂𝐼 44.3M-1S-1
2 𝐵𝑟𝑂𝟑− +𝐻𝑂𝐼 + 𝐻+ → 𝐻𝐵𝑟𝑂2 +𝐻𝑂𝐼2 0.26M-1S-1
3 𝐵𝑟𝑂𝟑− +𝐻𝐼𝑂2 → 𝐻𝐵𝑟𝑂2 + 𝐼𝑂3
− 9M-1S-1
4 𝐵𝑟𝑂𝟑− + 𝐵𝑟− + 2𝐻+ → 𝐻𝐵𝑟𝑂2 +𝐻𝑂𝐵𝑟 2.1M-1S-1, 1x104 M-1S-1
5 𝐵𝑟𝑂3− + 𝐼𝐵𝑟 + 𝐻2𝑂 → 𝐼𝑂3
− +𝐵𝑟− +𝐻𝐵𝑟𝑂 + 𝐻+ 8x10-4M-1S-1 6 𝐻𝐵𝑟𝑂2 +𝐻𝑂𝐼 → 𝐻𝑂𝐵𝑟 + 𝐻𝐼𝑂2 2.5x108M-1S-1 7 𝐻𝐵𝑟𝑂2 + 𝐵𝑟
− +𝐻+ → +2𝐻𝑂𝐵𝑟 2x106M-1S-1 8 2𝐻𝐵𝑟𝑂2 → 𝐻𝑂𝐵𝑟 + 𝐵𝑟𝑂3
− +𝐻+ 2.2x102M-1S-1 9 𝐻𝑂𝐵𝑟 + 𝐼2 → 𝐻𝑂𝐼 + 𝐼𝐵𝑟 8x108M-1S-1,1x102 M-1S-1 10 𝐵𝑟− +𝐻𝑂𝐵𝑟 + 𝐻+ → 𝐵𝑟2(𝑎𝑐) + 𝐻2𝑂 4.1x109M-1S-1,1.1x10 M-1S-1 11 𝐻𝑂𝐵𝑟 + 𝐻𝐼𝑂2 → 𝐼𝑂3
− + 𝐵𝑟− + 2𝐻+ 1x106M-1S-1 12 𝐻𝑂𝐵𝑟 + 𝐻𝐼𝑂 → 𝐻𝐼𝑂2 + 𝐵𝑟
− + 2𝐻+ 1x106M-1S-1 13 𝐻𝐼𝑂2 +𝐻𝑂𝐼 → 𝐼𝑂3
− + 𝐼− + 2𝐻+ 6x102M-1S-1 14 𝐼− +𝐻𝑂𝐼 + 𝐻+ → 𝐼2 +𝐻2𝑂 3.1x1012M-1S-1,2.2 S-1 15 𝐼𝐵𝑟 + 𝐻2𝑂 → 𝐻𝑂𝐼 + 𝐵𝑟
− +𝐻+ 0.8 S-1,1x108 M-1S-1 16 𝐼𝐵𝑟 + 𝐼− → 𝐼2 +𝐵𝑟− 2x1010M-1S-1,4.7x102 M-1S-1 17 𝐵𝑟2 + 𝐼2 → 2𝐼𝐵𝑟 1.3x105M-1S-1,1.0 M-1S-1

72
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO NO. 13: LA BOTELLA AZUL
OBJETIVOS La siguiente práctica es una adaptación de la presentada por Campbell (1,2). La botella azul es una famosa demostración de química (3,4), que consiste en un recipiente que contiene un líquido acuoso incoloro que al agitarse se torna azul, y después de un tiempo en reposo vuelve a la apariencia original. El ciclo coloreado – reposo. En este experimento se realiza el estudio cinético de la reacción de oxidación de glucosa en medio básico y en presencia de oxígeno y azul de metileno. 13.1 FUNDAMENTOS Cualquier especulación sobre lo que suceda en la botella azul debe basarse en la composición y los cambios químicos que ocurren en el interior del reactor. La solución es inicialmente una mezcla de 2% glucosa (un agente reductor), 2% NaOH y 0.005% de azul de metileno (AM), este último es el causante del cambio de color en el reactor. El AM es un indicador redox, que es azul en su forma oxidada, per que vira a incoloro cuando el potencial redox baja de cierto valor, según la reacción: Se puede suponer razonablemente que el azúcar se oxida en el medio y agota el oxidante disponible (el oxígeno), el medio se vuelve reductor y hace virar el AM a la forma incolora. La agitación transporta rápidamente oxígeno, que permanece como reserva en la fase gaseosa, a la fase acuosa, cambia el potencia redox y el AM se regresa a su forma coloreada. La saturación de oxígeno se alcanza para unas pocas ppm, así que una reacción de velocidad moderada puede agotar el oxígeno.
CH3
CH3
N
SN
CH3
N+
CH3
H
CH3
CH3
N
SN
CH3
N CH3
+ 2e-+H+
Figura 13. 1 Transferencia electrónica sobre una molécula de azul de metileno. En unos pocos minuto, y volver el medio reductor. El viraje de coloración del AM es reversible en el sentido cinético. Algunos hechos apoyan el mecanismo general que se acaba de postular:
Aún en el estado incoloro la solución muestra una delgada banda en la interfase gas – líquido que tiene color azul: esto indica que la difusión molecular de oxígeno mantiene oxidante la interfase.

73
La duración del color es más corta si no se renueva el aire del espacio gaseoso de la botella. Si se elimina completamente el aire por arrastre con un gas inerte el sistema no puede alcanzar el estado azul.
Unas gotas de peróxido de hidrógeno, que se descompone con generación de oxígeno, impiden que la solución cambie de color
Entre los productos de oxidación de la glucosa se encuentran los ácidos carboxílicos arabinoico, fórmico, oxálico y eritrónico. Un observador cuidadoso puede apreciar la turbidez creciente de la solución y la intensificación de olores característicos. Proponer un esquema más detallado de la reacción requiere numerosos experimentos y mentes entrenadas en la metodología de la cinética. Un razonable mecanismo de reacción es(1):
𝑂2(𝑔) ↔ 𝑂2(𝑖𝑛𝑡𝑒𝑟𝑓) ↔ 𝑂2(𝑎𝑐)
(13.1)
𝑂2(𝑎𝑐) + 𝐴𝑀(𝑖𝑛𝑐𝑜𝑙𝑜𝑟𝑜) →𝐴𝑀+(𝑎𝑧𝑢𝑙); 𝑟á𝑝𝑖𝑑𝑎
(13.2)
𝐺𝑙𝑢𝑂𝐻 + 𝑂𝐻 ↔ 𝐺𝑙𝑢𝑂− +𝐻2𝑂; 𝑟á𝑝𝑖𝑑𝑎 (13.3)
𝐺𝑙𝑢𝑂− + 𝐴𝑀+ → 𝐴𝑀 + 𝑂𝐻− + 𝑆𝑎𝑙𝑒𝑠 Á𝑐𝑖𝑑𝑜𝑠 𝐶𝑎𝑟𝑏𝑜𝑥𝑖𝑙í𝑐𝑜𝑠, 𝑙𝑒𝑛𝑡𝑎 (13.4)
La reacción neta es:
𝑂2(𝑎𝑐) + 𝐺𝑙𝑢𝑂𝐻 → 𝑆𝑎𝑙𝑒𝑠 (13.5)
en la cual no aparecen el hidróxido de sodio ni el AM. El papel de estas sustancias es el de producir intermediarios reactivos y de esta manera facilitar la reacción. En otras palabras, el NaOH y el AM son catalizadores. A partir del mecanismo de reacción se puede deducir una ley de velocidad. Esta consiste en una ecuación diferencial que relaciona la tasa de variación de la concentración de productos o reactantes con una función de las concentraciones de los reactantes. Para deducir la ley de velocidad se pueden usar varias aproximaciones, entre ellas las más comunes son: (a) aproximación del estado estacionario y (b) aproximación de la etapa que controla la velocidad de reacción (etapa lenta) (5,6). En este caso, la ley se puede deducir de la molecularidad de la etapa más lenta, que es la reacción 4:
𝑟 =𝑑[𝑃]
𝑑𝑡= 𝑘[𝐴𝑀+][𝐺𝑙𝑢𝑂−]
(13.6)
Donde [P] es la concentración de los productos. La ecuación 4 muestra que el papel del hidróxido es proporcionar un medio muy básico que facilite la disociación del protón de la glucosa. De allí se sigue que:

74
𝐾𝑒𝑝 =[𝐺𝑙𝑢𝑂−][𝐻2𝑂]
[𝑂𝐻−][𝐺𝑙𝑢𝑂𝐻]
(13.7)
[𝐺𝑙𝑢𝑂−] =𝐾𝑒𝑝
[𝐻2𝑂][𝑂𝐻−][𝐺𝑙𝑢𝑂𝐻]~𝐾´[𝑂𝐻−][𝐺𝑙𝑢𝑂𝐻]
(13.8)
𝑟 =𝑑[𝑃]
𝑑𝑡= 𝑘𝐾´[𝐴𝑀+][𝐻𝑂−][𝐺𝑙𝑢𝑂𝐻]
(13.9)
Según el mecanismo, la velocidad de reacción es de orden uno respecto a la glucosa, AM y HO−. La velocidad no depende de la concentración de oxígeno, porque el consumo de oxígeno sigue un proceso de orden cero en el tiempo. Si estas afirmaciones con respecto a los reactantes pudieran confirmarse experimentalmente el mecanismo propuesto sería una hipótesis aceptable. Una manera de poner a prueba la ecuación 10 es por variación de las concentraciones iníciales de los reactivos. Las concentraciones iníciales de azul de metileno y NaOH no cambian significativamente durante el curso de la reacción, ya que son catalizadores. La concentración de glucosa, comparada con la de oxígeno, es muy alta por lo tanto se puede sumir que durante el curso de la reacción permanece constante. Es obvio que el tiempo de la desaparición de color azul es una medida de la velocidad con que se consume el oxígeno, o se forman los productos. Un problema asociado con las medidas anteriores que las comparaciones son válidas para iguales concentraciones de AM+, que se forma por la reacción 3. Si la concentración del colorante fuera mayor tardaría más en desaparecer el color, aunque aumente “r”. Esto indica que a partir de medidas de tiempo de decoloración no se pueden hallar los órdenes de reacción con respecto al oxígeno o el AM. Los experimentos que se describen más adelante se limitan a encontrar
los órdenes de la glucosa y del HO−. 13.2 Trabajo experimental Se tiene listas las siguientes soluciones: A. 4% NaOH acuoso. B. 4% Glucosa acuosa C. 1% Azul de metileno en etanol.
Se ensambla el reactor de la figura. E el reactor se prepara alguna de las mezclas indicadas más adelante y en la jeringa superior se colocan 60ml de agua. Colocar el burbujeador en posición y hacer pasar aire durante 5 minutos a los 60 ml de agua.
Por el reactor se pasa nitrógeno o butano gaseoso hasta decoloración de la solución. Registre la temperatura.
Abra la válvula que comunica la jeringa con el reactor y drene exactamente 50 ml de agua. Cuando se haya desocupado la mitad ponga en marcha el cronómetro. Registre el tiempo que tarda la solución en tornarse incolora.
Antes de iniciar la práctica, en una caja de Petri mezcle 5 mL de NaOH 1M,1.6 mL de glucosa 0.5M, 0.5mL de AM+ 10-3M y 2.9 mL de agua. De manera opcional coloque al
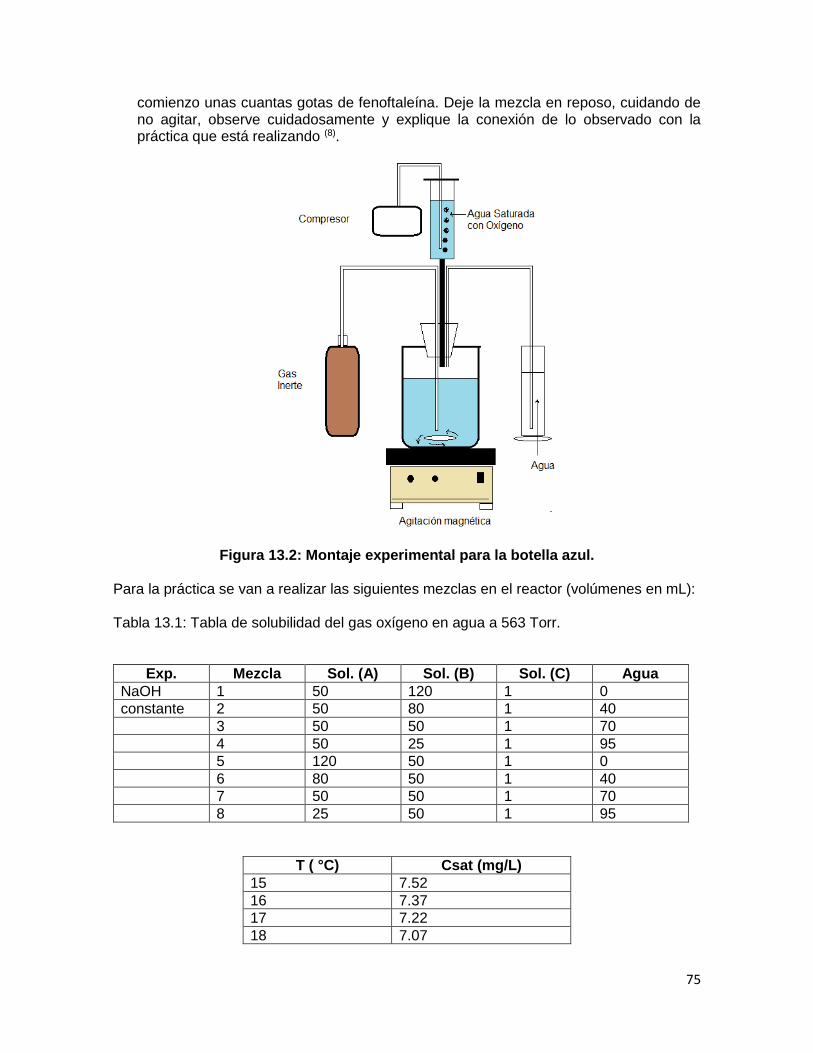
75
comienzo unas cuantas gotas de fenoftaleína. Deje la mezcla en reposo, cuidando de no agitar, observe cuidadosamente y explique la conexión de lo observado con la práctica que está realizando (8).
Figura 13.2: Montaje experimental para la botella azul.
Para la práctica se van a realizar las siguientes mezclas en el reactor (volúmenes en mL): Tabla 13.1: Tabla de solubilidad del gas oxígeno en agua a 563 Torr.
Exp. Mezcla Sol. (A) Sol. (B) Sol. (C) Agua
NaOH 1 50 120 1 0
constante 2 50 80 1 40
3 50 50 1 70
4 50 25 1 95
5 120 50 1 0
6 80 50 1 40
7 50 50 1 70
8 25 50 1 95
T ( °C) Csat (mg/L)
15 7.52
16 7.37
17 7.22
18 7.07

76
19 6.93
20 6.79
13.3 Tratamiento de datos 1. La velocidad de reacción es:
𝑟 =∆[𝑂2]
𝑡𝑖𝑒𝑚𝑝𝑜 𝑑𝑒 𝑑𝑒𝑐𝑜𝑙𝑜𝑟𝑎𝑐𝑖ó𝑛
(13.10)
La concentración inicial de oxígeno puede hallarse con la tabla de solubilidades: convierta el valor a mol/L y multiplíquelo por el factor de dilución 50/221.
2. Como las concentraciones de los catalizadores no varían y la concentración de glucosa
es prácticamente constante, entonces 𝑟 = 𝑎[𝐺𝑙𝑢𝑂𝐻]0𝑛 para la serie de [NaOH] constante
y 𝑟 = 𝑏[𝑁𝑎𝑂𝐻]0𝑛 para la serie de glucosa constante ”n” y ”m” son ordenes de la reacción
para la glucosa y el NaOH respectivamente. Haga gráficas de 𝑙𝑛 r vs 𝑙𝑛 [GluOH]0 y 𝑙𝑛 r vs ln [NaOH]0, según sea el caso. La pendiente de estas gráficas será “n” o “m” y el intercepto 𝑙𝑛 a o 𝑙𝑛 b según el caso. Verifique si los órdenes experimentales se ajustan al modelo teórico.
3. Calcule en todos los casos Kap, que es igual a kK´, por medio de la ecuación 10, y verifique su reproducibilidad. Haga el cálculo a partir de “a” y “b” hallados en el punto anterior. Compare los resultados.
13.4 Cuestionario La “botella azul” se puede estudiar con muchas variantes: (a) a diferentes temperaturas, (b) en un CSTR, (c) en medio no agitado, (d) en medio gelificado, (e) en presencia de reactivos adicionales (p.ej: peróxido de hidrógeno), (f) en ausencia de oxígeno, y (g) todas las que la mente de un químico creativo considere de interés. Sevcik y colaboradores (7) estudiaron cinéticamente la “botella azul” en ausencia de oxígeno. Entre sus evidencias experimentales se encuentran: (a) la reacción no exhibe el comportamiento reloj (después de cierto tiempo repentina desaparición del color azul de la solución), (b) la concentración del AM+ disminuye exponencialmente en el tiempo, (c) la
disminución de la concentración de AM+, GluOH y HO−, (d) evidencia de “saturación “ al variar las concentraciones de los reactivos. Estos resultados permiten proponer la siguiente ley de velocidad:
𝑟 =𝑑[𝐴𝑀+]0𝑑𝑡
= 𝑘𝑜𝑏𝑠[𝐴𝑀+]0
(13.11)
Donde los valores de kobs dependen de las concentraciones iniciales de GluOH y OH¯ y de
la temperatura, p. ej: kobs = 0.04 𝑠−1 para [𝐺𝑙𝑢𝑂𝐻]0 = 0,08 𝑀, [HO−]0 =0.1M 20° C. Al estudiar la dependencia de kobs (ó las velocidades iniciales) de las concentraciones de

77
GluOH y HO− se obtiene evidencia experimental de suturación cinética (a altas
concentraciones de GluOH y HO−, Kobs se hace independiente de los reactivos), lo cual sugiere la siguiente expresión cinética:
𝐾𝑜𝑏𝑠 =𝑎[𝐺𝑙𝑢𝑂𝐻][𝑂𝐻−]
1 + 𝑏[𝐺𝑙𝑢𝑂𝐻][𝑂𝐻−]
(13.12)
1. Explique cómo se determina experimentalmente el valor de kobs.
2. Explique cómo se verifica a partir de datos experimentales la ecuación 13 y cómo se
obtienen los valores de las constantes a y b.
3. Explique el significado o con qué se relacionan las constantes a y b de la ecuación 13.
4. Para la “botella azul” en ausencia de oxígeno considere como “factible” el siguiente
mecanismo de reacción: (𝑎) 𝐺𝑙𝑢𝑂𝐻 + 𝑂𝐻− ↔ 𝐺𝑙𝑢𝑂− +𝐻2𝑂, (𝑏) 𝐺𝑙𝑢𝑂− + 𝐴𝑀+ ↔𝐺𝑙𝑢𝑂𝐴𝑀, 𝐺𝑙𝑢𝑂𝐴𝑀 → 𝑃𝑟𝑜𝑑𝑢𝑐𝑡𝑜𝑠.Use la aproximación de estado estacionario para los
intermediarios𝐺𝑙𝑢𝑂− y 𝐺𝑙𝑢𝑂𝐴𝑀 y obtenga una expresión de velocidad para el consumo de AM+ y una para la producción de los productos. Discuta con validez de la aproximación, lo obtenido y si las expresiones son consistentes con el mecanismo propuesto (recomendación: use la referencia 6).
5. Si del mecanismo anterior, la etapa (c) es la limitante de la velocidad de reacción, ¿qué aproximación utilizar para obtener un ley de velocidad consistente con el mecanismo?
6. Considere que los resultados de Sevcik y colaboradores(8) son extrapolables a la “botella
azul” en presencia de oxígeno, y que el consumo de oxígeno sigue la siguiente expresión cinética:
𝑟 = −𝑑[𝑂2]
𝑑𝑡=
𝑐[𝐺𝑙𝑢𝑂𝐻][𝑂𝐻−]
1 + 𝑑[𝐺𝑙𝑢𝑂𝐻][𝑂𝐻−]
(13.13)
Verifique con sus datos experimentales la validez de la ecuación anterior, obtenga los valores de las constantes c y de 7. Compare los resultados obtenidos a partir de la ecuación 9 y 13 (constantes cinéticas
de índices de reacción) y analice cuál de las ecuaciones es más válida.
13.5 Bibliografía
Campbell, J. A., J. Chem. Educ. , 40, 578-583,196. Campbell, J.A. Why do chemical reactions occur? Prentice-Hall: Englewood Cliffs,
NJ, 1965, Chapter 2.
Cook, A.G., Tolliver, R.M., Williams, J.E., J. Chem. Edu., 71, 160-160, 1994.
Engerer, S. C., Cook, A.G., J. Chem. Educ., 76, 1519-1520, 1999.

78
Para decidir que aproximación es la más adecuada el investigador debe hacer uso de toda la evidencia experimental disponible, de las constantes cinéticas de cada una de las etapas del mecanismo propuesto, de sentido común y de un poco de álgebra.
James H. Espenson. Chemical Kinetics and Reaction Mechanisms. Mc-Graw
HILL. 1995
Bees, M.A., Pons, a.j., Sorensen, PG., Sagués, F. J. Chem. Phys., 114, 1932-1943, 2001
Adamcikova, L., Pavlikova, K., Sevcik, P. Int. J. Chm. Kinet., 31, 463-468,
1999.
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica

79
Práctica de Laboratorio No. 14: REDUCCIÓN DEL ION PERMANGANATO.
Objetivos
Los Agentes reductores, en medio ácido, reducen el ion 𝑀𝑛𝑂4− hasta ion 𝑀𝑛2+. Se sabe
que este último acelera la reacción, es decir actúa como catalizador de la reducción del ion 𝑀𝑛𝑂4
−. Por ésta razón se le da a esta reacción el calificativo de “autocatalítica” o “catalizada por productos”. En experimento se estudia la cinética de la reducción del ion 𝑀𝑛𝑂4
− en medio ácido a diferentes temperaturas. 14.1Fundamentos La naturaleza del agente reductor es uno de los factores que mas influye en la velocidad de reacción. Así, en los cuatro casos siguientes hay diferencias significativas substanciales en la velocidad de reacción n(2).
2𝑀𝑛𝑂4− + 5𝐻2 + 6𝐻
+ → 8𝐻2𝑂 + 2𝑀𝑛2+ (14.1)
𝑀𝑛𝑂4− + 5𝐹𝑒2+ + 8𝐻+ → 4𝐻2𝑂 +𝑀𝑛
2+ + 5𝐹𝑒3+ (14.2)
2𝑀𝑛𝑂4− + 5𝐶2𝐻2𝑂4 + 6𝐻
+ → 8𝐻2𝑂 + 2𝑀𝑛2+ + 10𝐶𝑂2 (14.3)
2𝑀𝑛𝑂4− + 5𝑆𝑂4(𝑎) + 2𝐻2𝑂 → 8𝑆𝑂4
2− + 2𝑀𝑛2+ + 4𝐻+ (14.4)
De otra parte, la concentración de las especies que reaccionan también influye en la velocidad de reacción, lo mismo que la temperatura a la cual se realiza el proceso y la adición de sustancias distintas de los reactivos, como el cloruro MnCl2 que tiene un efecto catalítico en la reacción. Todas la reacciones anteriores están lejos de ser sencillas u los mecanismos de tallados de las mismas aun no se han se conocen. En este experimento el permanganato será
reducido según la reacción 𝑥, la cual fue estudiada por R. M. Noyes y coloaboradores (3). Una solución correctamente preparada de permanganato (libre de impurezas) no reacciona apreciablemente con el oxalato de calcio puro, sino que la reacción ocurre con complejos de oxalato-manganeso (II). Durante la reacción el permanganato es reducido y el manganeso (II) es oxidado en una secuencia de reacciones que forman el complejo
𝑀𝑛(𝐶2𝑂4)𝑛3−2𝑛. Los valores de n dependen de la concentración del oxalato:
𝑀𝑛𝑂4− + 4𝑀𝑛2+ + 5𝑛𝐶2𝑂4
2− + 8𝐻+ → 4𝐻2𝑂 + 5𝑀𝑛(𝐶2𝑂4)𝑛3−2𝑛 (14.5)
2𝑀𝑛(𝐶2𝑂4)𝑛3−2𝑛 → 2𝑀𝑛2+ + (2𝑛 − 1)𝐶2𝑂4
2− + 2𝐶𝑂2 (14.6)

80
2𝑀𝑛𝑂4− + 8𝑀𝑛2+ + 5𝐶2𝑂4
2− + 16𝐻+ → 8𝐻2𝑂 + 10𝐶𝑂2 + 10𝑀𝑛2+ (14.7)
La descomposición de los complejos de oxalato –manganeso (III), paso x, constituye la etapa determinante de la velocidad global de la reacción. La reacción global x es autocatalítica en la producción de Mn2+. El experimento que se describe a continuación estudia el efecto que produce cada uno de los siguientes factores: concentración, temperatura y adición de catalizador como el MnCl2. 14.2 Trabajo Experimental
14.2.1 Efecto de la concentración del ion 𝑴𝒏𝑶𝟒−.
1. Aliste 5 tubos de ensayo de capacidad de 15 mL. Coloque en ellos 1, 2,3, 4 y 5 mL
respectivamente de la solución acuosa 0.01M de KMnO4 y adicione la cantidad de agua destilada necesaria para completar un volumen de 5 mL en cada uno de ellos.
2. Coloque los tubos de ensayo en un baño termostato a una temperatura entre 0 y 40°C dejándolos en reposo por 10 minutos. En el mismo baño se mantiene un balón con 100 Ml de solución 0.5 M de ácido oxálico preparada en acido sulfúrico 1.0 M.
3. Con una pipeta transfiera 5 mL de la última solución al tubo de ensayo que contiene la solución diluida de KMnO4, y mida el tiempo transcurrido desde que se adicionó la mitad del volumen hasta observar la aparición de un color amarillo pálido. Durante todo el proceso agite permanentemente la solución con un agitador de vidrio
4. Repita el último paso con las soluciones restantes de KMnO4
Se aconseja realizar el experimento anterior a temperaturas de 0, 20, 25, 30, 35 y 45°C.
14.2.2 Efecto del ion Mn2+
Proceda de manera similar a la propuesta anterior, pero agregando 0.5 ml de una solución acuosa de MnCl2, a cada una de las mezclas contenidas en los tubos de ensayo
14.3 tratamiento de los datos
1. Calcule la concentración inicial de KMnO4 en cada tubo después de la adición de la solución de acido oxálico y coloque estos datos en una tabal junto con los tiempos de decoloración. Calcule la velocidad de reacción para cada caso dividiendo la concentración inicial de KMnO4 por el tiempo de decoloración en cada caso.
2. Construya la grafica de velocidad de decoloración en función de la concentración de KMnO4 para cada temperatura de trabajo y determine el orden de reacción de la reacción respecto al ion permanganato, calcule la constante de velocidad para la reacción.
3. haga una grafica de logaritmo natural de la constante de velocidad en función del inverso de la temperatura absoluta, y determine la energía de activación para el proceso.
4. Explique los cambios de color que produjeron en el curso de la reacción.

81
5. discuta el efecto de ion Mn2+ en al velocidad de reacción y explique por qué asumimos que la velocidad de reacción permanece constante durante el tiempo de reacción para cada experimento a diferentes concentraciones de 𝑀𝑛𝑂4
−.
6. Considere como “aceptable” el siguiente modelo para describir la reacción estudiada:
[1]Permangato(X ) + Oxalato + (Y) − k1 → Manganeso(II)(Z) + Otros
[2]Manganeso(II)(Z ) + Oxalato + (Y) − k2 → Complejo
[3] Complejo + Permangato(X ) − k3 → 2Manganeso(II)(Z) + Otros
De acuerdo con lo descrito en este libro, el paso 1 ocurre lentamente, el paso 3 es una
reacción rápida y el paso 2ocurre a una velocidad intermedia,𝑘1 < 𝑘2, 𝑘3 ≫ 𝑘2
Entonces la ley de velocidad para la disminución de la concentración de permanganato (X) en el tiempo es la siguiente:−𝑑[𝑋]/𝑑𝑡 = 𝐾1[𝑋][𝑌] + 𝑘2[𝑌][𝑍]. También podemos considerar que [𝑍]0 = 0 y que [𝑌]0 ≫ [𝑋]0, entonces[𝑍]𝑡 = [𝑋]0 [𝑋]𝑡. A partir de las consideraciones anteriores obtenga la expresión integrada que describa la variación de la concentración de 𝑥 en el tiempo. En una grafica de 𝑥 vs 𝑡, muestre la validez del modelo propuesto al comparar con lo observado experimentalmente (para esto se deben encontrar los valores adecuados, usando el sentido común, para 𝑘1, 𝑘2, [𝑋]0, [𝑌]0).
14.4 Anexos
A: la estandarización de soluciones de KMnO4 es una de las prácticas volumétricas más comunes de la química analítica. La valoración sigue obviamente la cinética estudiada es esta practica y tradicionalmente se realiza en caliente (60°-70°C), adicionando el permanganato a la solución ácida de acido oxálico. Una variante para la estandarización de la solución es realizarla a temperatura ambiente pero en presencia de acido ascórbico (p. ej: para valorar acido oxálico 0.05 M con permanganato 0.05M, adicionar a alícuota 0.6 mL de ascórbico 1.0x10-4 M y 6 mL de H2SO4 5N). Recuerde los conceptos fundamentales del análisis volumétrico y el manejo del equivalente-gramo.
B: Para apreciar experimentalmente la transición del manganeso por los estados de oxidación +7(violeta), +6(verde obscuro),+5 (azul cielo),+4 (precipitado café9, realice el siguiente experimento. A, 400 ml de NaOH 6M que se encuentre a una temperatura de aproximada de 0°C y en continua agitación, adiciones lentamente (de gota ) una solución compuesta de 1 mL de agua y 0.03 g de KMnO4, luego inicie adición , gota a gota de H2O2 al 0.1 %.
14.5 Bibliografías
James H. Espeson. Chemikal Kinectics an Reaction Mecahanisms, McGraw-Hill, 1995. pp 36-37.
Gran numero de ejemplos de agentes reductores que reaccionan con el permanganato en medio básico o ácido se encuentran en los textos de análisis Cualitativo/Cuantitativo.
Malcon, J., Noyes, R.M. J. Am Chem. Soc., 742769-2775,1952. (b) Alder, S.J., Noyes, R. M. J. Am Chem. Soc., 77, 2036-2042,1955.

82
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
Práctica de Laboratorio No.15: REACCIONES OSCILANTES
OBJETIVOS

83
Los fenómenos periódicos u oscilantes se presentan en física, astronomía, biología y química. Van desde el movimiento de los péndulos hasta las órbitas de los planteas y los complejos relojes biológicos que gobiernan el comportamiento diario y estacional de los organismos. El objetivo de esta práctica es el de analizar de manera general las condiciones dinámicas mínimas necesarias para que se presenten reacciones químicas oscilantes. 15.1 Fundamentos 15.1.1 Reacción de Briggs-Rahscher (BR) La mayoría de las reacciones químicas oscilantes conocidas tienen en común tres características que se han propuesto como necesarias para que se presente comportamientos oscilatorios en sistemas químicos. La primera es que los sistemas químicos oscilan sólo si están “lejos del equilibrio”. La expresión “lejos del equilibrio” puede generar confusión respecto a lo que significa. Erróneamente los autores la relacionan con la termodinámica de los procesos irreversibles, indicando que los sistemas que exhiben comportamiento oscilatorio se encuentran “lejos del equilibrio termodinámico”. Para nosotros, las reacciones químicas oscilantes son sistemas dinámicos gobernados por no-linealidades (esto es lo que quiere decir “lejos de equilibrio”). Cuando el sistema dinámico se aleja de un punto fijo punto de reposo o punto de equilibrio su dinámico se aleja de un punto fijo, punto de reposo o punto de equilibrio su dinámica puede ser gobernada por linealidades (cerca del equilibrio en el sentido dinámico) o por no-linealidades (lejos del equilibrio dinámico). La segunda característica es que en algún paso del mecanismo de reacción se debe presentar una reacción simple autocatalítica, de retroalimentación o alguna clase de “feedback”, esto implica que un producto de un paso de la secuencia de reacciones debe influir en su propia velocidad de formación. Esta última característica se conoce como INESTABILIDAD. En sistemas químicos se han identificado tres tipos de inestabilidades: 1) autocatálisis directa, 2) autocatálisis cruzada y 3) inhibidores de producto final. La tercera condición es que el sistema químico debe presentar biestabilidad. Lo anterior quiere decir que bajo el mismo conjunto de condiciones externas, a las que nos referimos como factores limitantes (ó parámetros de control), el sistema ha de poder existir en dos estados estables diferentes. De un estado decimos que es estable si es capaz de regresar a su estado inicial después de realizar una pequeña perturbación en él. Por ejemplo una canica en el fondo de un recipiente cónico esta en un estado estable, pero apoyada en la punta de una pirámide se encuentra en un estado inestable [1]. La época moderna de las reacciones químicas oscilantes comienza con el descubrimiento, obtenido al azar en 1985, por el químico ruso B.P. Belousov. Él observó que al disolver en agua ácido cítrico, ácido sulfúrico, bromato de potasio y una En poco tiempo, aparecieron muchas variaciones de ésta reacción donde se sustituyeron uno o más componentes por especies químicas análogas. Así surgieron osciladores más rápidos o lentos, o con cambios de distintos colores. La variante más espectacular, desde el punto de vista cromático, es un híbrido de la reacción de Bray (reacción de descomposición del H2O2 catalizada por IO3
−) y la reacción de Belousov[2] que fue desarrollada por los profesores de bachillerato del Galileo High School de San Francisco USA en 1973, Thomas S. Briggs y Warren C. Rauscher[3]. Su reacción, que utiliza peróxido de hidrógeno, ácido malónico, sulfato de manganeso y almidón, cambia de incoloro a dorado y pasa a azul, para volver a empezar.

84
El mecanismo de reacción propuesto para esta reacción es tomado del libro de Shakhashiri[2]. En la reacción BR la velocidad de producción de oxígeno y de dióxido de carbono, al igual que las concentraciones de yodo y yoduro oscilan. Un mecanismo simplificado de la reacción global está representado por los siguientes pasos fundamentales:
𝐼𝑂3− + 2𝐻2𝑂2 + 𝐶𝐻2(𝐶𝑂𝑂𝐻)2 +𝐻
+ → 𝐼𝐶𝐻(𝐶𝑂𝑂𝐻)2 + 2𝑂2 + 3𝐻2𝑂 (15.1)
esta transformación está acompañada por dos pasos fundamentales:
𝐼𝑂3− + 2𝐻2𝑂2 +𝐻
+ → 𝐼𝑂𝐻 + 2𝑂2 + 2𝐻2𝑂 (15.2)
𝐻𝐼𝑂 + 𝐶𝐻2(𝐶𝑂𝑂𝐻)2 → 𝐼𝐶𝐻(𝐶𝑂𝑂𝐻)2 +𝐻2𝑂 (15.3)
La reacción 2 puede ocurrir por dos vías diferentes, una donde intervienen y otra donde no participan radicales libres. La vía que tome la reacción está determinada por la concentración de ion yoduro en la solución. Cuando la concentración de yoduro es baja la vía donde intervienen radicales es la más importante, pero cuando la concentración de yoduro es alta la reacción se da sin la intervención de radicales libres. Cuando interviene radicales en la reacción la producción de HIO se da más lentamente que cuando no
intervienen. EL HIO que no reaccione en la ecuación 3 es reducido a I− por la acción del H2O2 como uno de los pasos complementarios de la vía donde no interviene radicales. La explicación detallada requiere analizar los pasos individuales de ambos procesos. Si el ion yoduro está presente en alta concentración, la reacción 2 se da sin la intervención de radicales y el yoduro reacciona lentamente con el ion yodato, 𝐼𝑂3− + 𝐼− + 2𝐻+ → 𝐻𝐼𝑂2 +𝐻𝐼𝑂 (15.4)
El ácido yodoso (HIO2) es reducido a ácido hipoyodoso (HIO),
𝐻𝐼𝑂2 + 𝐼− +𝐻+ → 2𝐻𝐼𝑂 (15.5)
El ácido hipoyodoso es entonces reducido por el H2O2,
𝐻𝐼𝑂 + 𝐻2𝑂2 → 𝐼− + 𝑂2 +𝐻2𝑂 (15.6)
La reacción neta presentada por la ecuación 2 es obtenida por la adición estequiométrica de las reacciones 4, 5 y 6. Ya que la reacción 2 es más lenta que la reacción 3 el consumo de HIO por esta última es grande, impidiendo la recuperación de los iones I− consumidos en las reacciones 4 y 5. Así en este punto de la reacción la concentración de I− disminuye constantemente. Una vez la
concentración de I− se hace suficientemente baja, la reacción 2 se da por la vía donde intervienen radicales.

85
En este punto de la reacción interviene posiblemente la siguiente secuencia de reacciones [4]:
𝐼𝑂3− +𝐻𝐼𝑂2 +𝐻
+ → 2𝐼𝑂2. +𝐻2𝑂 (15.7)
𝐼𝑂2. +𝑀𝑛2+ +𝐻2𝑂 → 𝐻𝐼𝑂2 +𝑀𝑛(𝑂𝐻)2
+ (15.8)
𝑀𝑛2+ +𝐻2𝑂2 → 𝑀𝑛
2+ +𝐻2𝑂 + 𝐻𝑂𝑂. (15.9)
2𝐻𝑂𝑂. → 𝐼𝑂3
− +𝐻𝐼𝑂 +𝐻+ (15.10)
2𝐻𝐼𝑂2 → 𝐼𝑂3
− +𝐻𝐼𝑂 + 𝐻+ (15.11)
Estos pasos combinados estequiométricamente dan como resultado la reacción 2,
2(𝐸𝑞. 7) + 4(𝐸𝑞. 8) + 2(𝐸𝑞. 9) + (𝐸𝑞. 10) + (𝐸𝑞. 11) = (𝐸𝑞. 2) Una característica importante de este proceso es que, tomando conjuntamente las reacciones 7 y 8, representa un proceso autocatalítico donde se producen dos moléculas de HIO2 por una molécula consumida. De éste modo la velocidad de reacción de los pasos 7 y 8 aumenta a medida que estas reacciones proceden. Éste proceso autocatalítico hace que la concentración de HIO aumente drásticamente (Eq. 11) hasta que las reacciones 7 a la 11 llegan a un estado estacionario. Las ecuaciones 8 y 9 muestran la función catalítica del Mn2+. El manganeso es oxidado en la reacción 8 y es reducido en la reacción 9. Su efecto catalítico en la reacción global esta dado por su capacidad para reducir el radical 𝐼𝑂2
. a HIO2, de este modo completando el ciclo autocatalítico de las ecuaciones 7 y 8. El ácido hipoyodoso, producido en la reacción 11, reacciona con el ácido malónico mediante la reacción 3. Sin embargo la producción del HIO es mucho más rápida que su consumo dado en la reacción3, lo que hace que el exceso de HIO reaccione con el H2O2 mediante la reacción 6. Como resultado se da un incremento en
la concentración de I−, que a la vez ocasiona el cambio de vía por el cual se da la reacción 2, de la vía mediada por radicales a la vía, más lenta, donde no intervienen radicales iniciada por la Eq. 4. El cambio dramático de color se da porque la reacción 3 no se da en un paso simple sino que se da por la secuencia de reacciones dada por las expresiones 12 y 13.
𝐼− +𝐻𝐼𝑂 + 𝐻+ → 𝐼2 +𝐻2𝑂 (15.12)
𝐼2 + 𝐶𝐻2(𝐶𝑂𝑂𝐻)2 → 𝐼𝐶𝐻(𝐶𝑂𝑂𝐻)2 +𝐻
+ + 𝐼− (15.13)
La producción de I2 a partir de la ecuación 12 le da un color dorado a la solución. Este color
permanece mientras se mantenga la concentración de HIO mayor que la de 𝐼−, y la reacción 2 se dé por la vía mediada por radicales. El exceso de HIO es convertido a 𝐼− a través de
la Eq. 6. La solución entonces se torna azul oscura cuando la concentración de 𝐼− es mayor que la de HIO y el 𝐼− puede combinarse con I2 y el almidón para formar un complejo de

86
color azul. La alta concentración 𝐼− ocasiona el cambio de mecanismo hacia el proceso lento donde no intervienen radicales en la Eq. 2. El color azul entonces empieza a desaparecer lentamente a medida que la reacción 3 consume el yodo más rápidamente de lo que es producido. Cuando el sistema cambia nuevamente al mecanismo donde intervienen radicales, comienza un nuevo ciclo de la reacción. Las oscilaciones continúan
hasta que ácido malónico o el 𝐼𝑂3− se consumen totalmente. Las reacciones anteriores
constituyen el esqueleto básico del mecanismo de la reacción oscilante BR. Concentraciones de iones cloruro mayores a 0.07 M inhiben las oscilaciones [5]. Por ésta razón, los recipientes usados para esta reacción deben estar libres de cloruros y debe usar agua destilada para la preparación de todas las soluciones. 15.2 Trabajo experimental 15.2.1 Soluciones
Vierta cuidadosamente 410 ml de peróxido de hidrógeno al 30% en un matraz de un litro y complete a volumen con agua destilada. Utilice guantes mientras esté manipulando el peróxido de hidrógeno (solución A).
En un vaso de precipitados de dos litros coloque aproximadamente 800 ml de agua destilada y 43 g de KIO3. Adicione 4.3 ml de H2SO4 concentrado y caliente la mezcla con un mechero. Agite la mezcla.
Figura 16.1: Registro potenciométrico de una reacción oscilante Continuamente hasta que toda la sal se halla disuelto. Transfiera la mezcla a un balón de un litro y lleve a volumen la solución (solución B).
En un vaso de precipitados de dos litros disuelva 16 g de ácido malónico y 3.4 g de sulfato de manganeso hidratado en aproximadamente 500ml de agua destilada. En otro vaso de precipitados de 100 ml colocar 50 ml de agua destilada y calentar hasta ebullición. En un vaso separado de 100 ml coloque 10 ml de agua y 3 gramos de almidón soluble. Agitar la mezcla anterior hasta formar una suspensión homogénea y agréguesela a los 50 ml de agua caliente. Continué la agitación y el calentamiento hasta

87
que el almidón se halla disuelto. Finalmente mezcle la solución de almidón con la solución de ácido malónico y sulfato de manganeso. Transfiera finalmente esta última mezcla a un balón de un litro y complete a volumen con agua destilada (solución C).
15.2.2 Procedimiento En un vaso de precipitados de 250 ml mezcle 50 ml de la solución B. Introduzca una barra de agitación y coloque el vaso en una plancha de agitación. Empiece a agitar vigorosamente, hasta que se vea un vortex bien definido. Adiciones 50 mL de la solución C a la mezcla anterior. La solución se debe tornar de color dorado casi inmediatamente. Después de unos segundos debe tornarse rápidamente de color azul. Después la solución azul debe cambiar a incolora para volver a comenzar nuevamente el ciclo. El periodo de oscilación inicialmente es de unos 15 segundos pero gradualmente se va incrementando. Después de unos minutos la solución no cambia más de color, permaneciendo azul oscura. Repita el experimento anterior sin agitación y observe cuidadosamente la zona de formación de color. Otra variante al experimento es colocar 50 mL de la mezcla de reacción en un recipiente plástico de aproximadamente 100 mL, y seguir el transcurso de la reacción y las oscilaciones con medidas de presión. Se recomienda utilizar un sistema de medida tipo LabPro. 15.2.3 Reacción química oscilante no catalizada por iones metálicos En la reacción oscilante clásica de Belousov-Zhabotinisky (BZ) se requiere de una pareja redox como catalizador que sea capaz de reducirse y oxidarse reversiblemente como los son Ce(IV)/Ce(III) o Mn(III)/Mn(II). Sin embargo Körös y Orban[6] reportaron que es posible encontrar oscilaciones en el potencial redox, concentración de bromuros y color durante la reacción química entre el ion bromato y el ácido gálico en ácido sulfúrico, sin la necesidad de adicionar un ion metálico como catalizador. Los mismos autores [7] reportaron que las oscilaciones se podían seguir presentando si se remplazaba el ácido gálico por uno de 23 compuestos orgánicos derivados del fenol y la anilina con al menos una posición orto o para libre.
𝐵𝑟𝑂3− + 𝐵𝑟− + 2𝐻+ → 𝐻𝐵𝑟𝑂2 +𝐻𝑂𝐵𝑟 (15.14)
𝐻𝐵𝑟𝑂2 + 𝐵𝑟− +𝐻+ → 2𝐻𝑂𝐵𝑟 (15.15)
𝐵𝑟𝑂3− +𝐻𝐵𝑟𝑂2 + 2𝐻
+ → 2𝐵𝑟𝑂2. +𝐻2𝑂 (15.16)
𝐵𝑟𝑂2. +Ѱ− 𝑂𝐻 → 𝐻𝐵𝑟𝑂2 +Ѱ
−𝑂. (15.17)
2𝐻𝐵𝑟𝑂2 → 𝐵𝑟𝑂3− +𝐻𝑂𝐵𝑟 + 𝐻+ (15.18)
O
OHH
O
OHH
+ Br.
HOBr +
(15.19)
𝐵𝑟𝑂2. +Ѱ− 𝑂𝐻 → 𝐻𝐵𝑟𝑂2 +Ѱ
−𝑂.
(15.20)

88
O
OH
C+
+ BrO2
.+H2OBrO3
-+
H+
O
OHH
(15.22)
𝐻𝑂𝐵𝑟 + 𝐵𝑟− → 𝐻2𝑂 + 𝐵𝑟2 (15.23)
𝐵𝑟2 +Ѱ− 𝑂𝐻 → 𝑜 − 𝑏𝑟𝑜𝑚𝑜𝑓𝑒𝑛𝑜𝑙 + 𝐻+ + 𝐵𝑟− (15.24)
𝐻𝑂𝐵𝑟 + Ѱ− 𝑂𝐻 → 𝑜 − 𝑏𝑟𝑜𝑚𝑜𝑓𝑒𝑛𝑜𝑙 + 𝐻2𝑂 (15.25)
O.O
.+
O.
2
(15.26)
Al parecer el mecanismo de esta reacción no catalizada por iones metálicos está relacionado con el mecanismo FKN para la reacción clásica BZ[8,9]. Herbine y Field[10]
propusieron el siguiente mecanismo de reacción para la reacción entre fenol y el ion bromato en medio ácido: 15.2.4 Soluciones A. Ácido sulfúrico 4 M, 250 ml. B. KBrO3 0.12 M, 250ml. C. Fenol 0.028 M, 250 ml D. KI
0.1 M, 50 ml.
Temperatura (°C) Periodo de Tiempo de registro (segundos)
Sin I− Con I−
17 5000 3000
30 1200 1200
40 800 800
15.2.5 Procedimiento 1. Para esta práctica se necesita un sistema para capturar señales analógicas, con el
objeto de poder registrar el potencial entre un electrodo de trabajo de platino con respecto a un electro de referencia de calomel cada 10 segundos durante 2 horas. De otro modo los datos tiene que ser registrados manualmente lo que hace la experiencia muy dispendiosa.
2. En un vaso de precipitados de 150 ml mezcle 20 ml de la solución A con 20 ml de la solución B. Adicione 20 ml de agua destilada a la mezcla anterior y coloque el vaso de un sistema de agitación magnética. Introduzca los electrodos de trabajo y de referencia en un vaso de precipitados. Verifique que la barra de agitación no golpea los electrodos e inicie la agitación vigorosa de la mezcla. Programe el dispositivo para la captura de datos de potencial en función de tiempo y conecte los electrodos a la interfase. Adicione rápidamente 20 ml de la solución C y ponga en marcha el sistema de captura de datos. Registre el potencial por el periodo de tiempo que se indica en el numeral 4.

89
3. Para ver el efecto de la adición de iones 𝐼− en la cinética de la reacción proceda de la misma manera que en el punto anterior mezclando 20 ml de A, 20ml de B, 19 ml de agua destilada y 1 ml de D. después adicione 20 ml de la solución C y registre el potencial entre los electrodos durante el periodo de tiempo que indica el numeral 4.
4. Para ver el efecto de la temperatura en el número y la frecuencia de las oscilaciones el
experimento se puede realizar a las siguientes temperaturas de trabajo: Si después de los tiempos señalados no se observan oscilaciones suspenda el experimento y repítalo de nuevo.
15.3 Tratamiento de datos y cuestionario Analice los siguientes mecanismos de reacción hipotéticos:
[h]
Mecanismo A Mecanismo B
∎𝑘1→ 𝐶
𝑍 + 𝐶2→ 2𝐷
𝑍𝑘3→ 𝑃
∎𝑘1→ 𝐶
𝐴 + 𝐶𝑘2→ 2𝐶
𝑍 + 𝐶𝑘3→ 2𝑍
𝑍𝑘4→ 𝑃
1. ¿Cree usted que a partir de los mecanismos anteriores se puede obtener un
comportamiento oscilatorio?. Para dar respuesta a esta pregunta escriba las ecuaciones diferenciales aplicando ley de acción de masas (asuma pasos elementales de reacción, así las velocidades de reacción son proporcionales a las concentraciones de los reactivos), y analice el sistema de ecuaciones para cada uno de modelos. Si es posible integre el sistema de ecuaciones utilizando algún programa de aplicaciones tipo Matlab, Mathematica, etc.
2. Considere un gran “espacio” n-dimensional de todos los posibles “parámetros de control” para los mecanismos anteriores. ¿En qué región de ese espacio considera usted que se encuentra el comportamiento oscilatorio? Ayuda: tenga muy presente su trabajo en el laboratorio.
3. ¿Cuáles especies de ambos mecanismos pueden cambiar de manera oscilante sus respectivas concentraciones en función del tiempo?
4. Describa dos fenómenos distintos a los anteriores que estén regido por reacciones químicas oscilantes.
5. ¿En la reacción oscilante BR por qué cree usted que el periodo de oscilación va
aumentando a medida que la reacción avanza?
6. ¿Cuál es el efecto de la adición del ion yoduro en la reacción de oxidación del fenol por el ion yodato en medio ácido?

90
7. Analice detenida y detalladamente los resultados experimentales obtenidos en el
laboratorio: duración de la reacción, tiempo de inducción, número de oscilaciones, amplitud, período, etc.
Justifique detalladamente todas sus repuestas. 15.4 Instrucciones de seguridad . El peróxido de hidrógeno al 30% es un agente altamente corrosivo y un agente oxidante fuerte. Se debe evitar cualquier contacto de éste reactivo con la piel o los ojos. . El ácido sulfúrico concentrado puede ocasionar quemaduras severas al entrar en contacto con la piel. Además éste reactivo es un agente deshidratante fuerte y libera bastante calor cuando es diluido con agua. En caso de accidente remueva la mayor cantidad del reactivo con una toalla desechable seca y lave rápidamente la zona afectada con agua, fría si es posible. Finalmente lave la zona afectada con bicarbonato de sodio al 10% aproximadamente. Si el ácido es derramado en el piso neutralice el ácido con bicarbonato de sodio sólido y lave con abundante agua. 15.5 Bibliografía
Epstein, I.R., Kustin K., De Kepper P., Orbán M. Orden y Caos, Libros de investigación y ciencia, Scientific American. Barcelona, España, pag. 56-65,1990.
B.Z. Shakhashiri, Chemical Demonstrations: A Handbook for Teachers of
Chemistry, Volume 2, Wisconsin, US, The University of Wisconsin Press, 248-256, 1985.
T.S. Briggs & W.C. Rauscher, J. Chem. Educ., 50, 496.1973. b) www.chem.leeds.ac.uk/delights P. De Kepper & I.R. Epstein, J. Am. Chem. Soc., 104, 49,1982. D.O. Cooke, React. Kinet. Catal. Lett., 3, 377, 1975.
E. Körös & M. Orban, Nature (London), 273, 371, 1978.
M. Orban & E. Körös, J. Phys. Chem., 82, 1672, 1978.
R. M. Noyes, J. Am. Chem. Soc. 102, 4644, 1980.
Cadena A.O. “Efecto de la adición de fenol en la reacción de Belousov –
Zhabotinsky”, Universidad Nacional de Colombia, 2001. Bogotá, Colombia. (Este es un trabajo de grado que se encuentra en la biblioteca del Departamento de Química de la Universidad Nacional).
P. Herbine & R. J. Field, J. Phys. Chem., 84, 1330, 1980.

91
Berenstein, I., Ágreda, J & Barragán, D. Effect of Methyl Ketones in the Belousov – Zhabotinskii Reaction. J. Phys. Chem. A., 103(48), 9780-9782, 1999.
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO NO.16: SOLVÓLISIS DEL CLORURO DE TERT-
BUTILO.
OBJETIVOS
En este experimento se estudia la cinética de la reacción de solvólisis del cloruro de terbutilo mediante un método conductimétrico
La sustitución nucleofílica alifática es una de las áreas de la química orgánica mas ampliamente estudiada, En este experimento se estudia la cinética de la reacción de solvólisis del cloruro de terbutilo mediante un método conductimétrico. Además se busca constractar los datos experimentales contra un mecanismo propuesto por la reacción de solvólisis y así determinar la constante de la velocidad para el proceso
16.1 Fundamentos
La reacción a estudiar es:
(𝐶𝐻3)3𝐶𝐶𝑙+𝐻2𝑂 → (𝐶𝐻3)3𝐶𝑂𝐻 + 𝐻++𝐶𝑙− (16.1)
El mecanismo de la reacción anterior comprende, al menos, los siguientes pasos:
(𝐶𝐻3)3𝐶𝐶𝑙𝑙𝑒𝑛𝑡𝑎→ (𝐶𝐻3)3𝐶
++𝐶𝑙− (16.2)
(𝐶𝐻3)3𝐶++𝐻2𝑂
𝑟á𝑝𝑖𝑑𝑎→ (𝐶𝐻3)3𝐶𝑂𝐻 + 𝐻
+ (16.3)

92
El medio de reacción es una mezcla de etanol /agua, cura proporción puede variarse con el fin de modificar la constante dieléctrica del medio (a 25°C ϵ=78.41 para el agua, ϵ=24,75 para el etanol, ϵ=32,63 para el metanol). Una disminución de este parámetro incrementa la interacción entre cargas y dificulta la formación del carbocatión dada la ecuación 2. Este hecho hace que la constante cinética de la reacción dependa de la constante dieléctrica del medio y por ende de la proporción de etanol en le medio de reacción. Por ultimo, como la molecularidad de la etapa lenta es uno, reacción 2, la reacción debe ser de uno.
16.2 Método conductimétrico
La solución inicial prácticamente no contiene iones, a no ser impurezas, pero en el curso de la reacción se forman los iones cloruro e hidronio, que tienden aumentar la conductividad Λ (el inverso de la resistividad eléctrica) del medio de acuerdo de la ecuación:
Λ𝑡 = Λ𝑚[𝐻𝐶𝑙] + Λ𝑖 (16.4)
Donde Λ𝑡 es la conductividad en un tiempo dado, Λ𝑚 la conductividad molar de HCl, y Λ𝑖 la conductividad inicial. La velocidad reacción se puede expresar en función de la variación de [HCl] con el tiempo, o bien, de la variación Λ𝑡 en función del tiempo:
𝑟 =𝑑[𝐻𝐶𝑙]
𝑑𝑡=1
Λ𝑚
𝑑Λ𝑡𝑑𝑡
(16.5)
La velocidad de reacción expresada en función de la concentración del cloruro de ter-butilo (CTB) es igual a:
𝑟 = −𝑑[𝐶𝑇𝐵]
𝑑𝑡= 𝑘[𝐶𝑇𝐵]
(16.6)
Como [𝐶𝑇𝐵] = [𝐶𝑇𝐵]𝑖 − [𝐻𝐶𝑙](donde el subíndice i representa la propiedad inicial), podemos escribir la ecuación:
𝑟 = −𝑑[𝐶𝑇𝐵]𝑖 − [𝐻𝐶𝑙]
𝑑𝑡= 𝑘[𝐶𝑇𝐵]𝑖 − [𝐻𝐶𝑙]
(16.7)
Integrando la ecuación anterior obtenemos que:
[𝐻𝐶𝑙] = [𝐶𝑇𝐵]𝑖(1 − 𝑒−𝑘𝑡)
(16.8
Expresando la ecuación anterior en función de la conductividad obtenemos la siguiente ecuación:
[𝐶𝑇𝐵]𝑖 = [𝐻𝐶𝑙]∞ =Λ∞ − Λ𝑖Λ𝑚
(16.9)
Donde los subíndices ∞ representan la propiedad en un tiempo infinito. Reemplazando el valor de [𝐶𝑇𝐵]𝑖 dado en la ecuación 9 y el valor de [HCl] dado por la ecuación 4en la ecuación 8 obtenemos:

93
Λt − Λ𝑖Λ𝑚
=Λ∞ − Λ𝑖Λ𝑚
(1 − 𝑒−𝑘𝑡)
ó
Λt = Λ∞ + (Λ𝑖 − Λ∞)𝑒−𝑘𝑡
(16.10)
Si no se conocen los valores de Λ𝑖 ni de Λ∞, un procedimiento debido a Guggenheim(4) permite determinar k sin necesidad de conocer estos valores. Suponga que en un
tiempo 𝑡 + 𝛿 la conductancia es Λ𝑡+𝛿 . Aplicando a la ecuación 10 obtenemos:
Λ𝑡+𝛿 = Λ∞ + (Λ𝑖 − Λ∞)𝑒−𝑘(𝑡+𝛿)
(16.11)
Restando a la ecuación 20.10 la ecuación 20.11 obtenemos:
Λ𝑡+𝛿−Λt = [(Λ∞ − Λ𝑖)(1 − 𝑒−𝑘𝛿)]𝑒−𝑘𝑡
(16.12)
Si el intervalo de tiempo 𝛿 es constante en una serie de medidas, el producto de las cantidades encerradas en el paréntesis cuadrado de la ecuación anterior es constante y se llega a la ecuación de trabajo (ecuación de Guggenheim):
Λ𝑡+𝛿−Λt = 𝑲𝑒−𝑘𝑡
ln(Λ𝑡+𝛿−Λt) = −𝑘𝑡 + 𝑙𝑛𝑲
(16.13)
Si se hace una grafica de ln(Λ𝑡+𝛿−Λt) vs 𝑡 se puede determinar la constante de velocidad del proceso a partir de la pendiente de esta gráfica. Nota: Un tratamiento alternativo se basa en el metodo de kezdy swibourne donde se obtiene que
Λ𝑡 = Λ∞(1 − 𝑒𝑘𝛿) + Λ𝑡+𝛿𝑒𝑘𝛿. De una grafica de Λ𝑡 contra Λ𝑡+𝛿, la cual se espera sea una
línea recta para una cinética de primer orden, se obtiene que:𝑘 = ln(𝑝𝑒𝑛𝑑𝑖𝑒𝑛𝑡𝑒) /𝛿.
También se puede obtener Λ∞ del intercepto de la recta obtenida con una línea de 45°C.
20.3 trabajo experimental
20.3.1 Soluciones
A. Solución de CTB: 1 mL del compuesto puro se lleva a 50 mL con etanolal 95%. La velocidad de descomposición de CTB en esta solución es muy lenta
B.Solucion acuosa de etanol al 42%.

94
Figura 16.1: Reactor para estudiar la cinética de solvólisis mediante un método conductimétrico.
1. Al reactor termostato provisto de agitador u una celda de conductividad (ver diagrama), agregue 40 mL de la solución B. Deje que se alcance el equilibrio térmico y agregue 2 mL de la solución A. Ponga en marcha el cronometro cuando haya descargado la mitad del CTB.
2. Haga medidas cada 30 s de la conductancia durante 30 minutos o hasta que la variación de la conductancia sea muy pequeña.
3. Efectúe un duplicado del experimento anterior. Al final de cada experimento lava la celda y el reactor con agua y purgue con la solución B.
4. Puede observar le efecto del medio en la velocidad de reacción agregando 5 mL de agua pura o etanol al 95% a 35 mL de la solución B y con esta mezcla repita el experimento anterior.
5. Realice el experimento a 15, 20, 25, 30, 35, y 40°C.
20.4 tratamiento de los datos
1. Para cada experimento haga una grafica de conductancia vs tiempo.
2. Seleccione los datos de conductancia que están a intervalos iguales de tiempo (𝛿) de 2, 4 y 6 minutos y realice las gráficas de Guggenheim y kezdy swibourne.
Verifique que efectivamente se obtienen graficas de rectas, y a partir de las pendientes de la grafica anterior determine la contante de velocidad para cada caso. Discuta los resultados.
3. El efecto del solvente sobre la constante las constantes se pueden observar tratando los datos de la misma forma. La concentración final de etanol se calcula con la formula:

95
(%𝐸𝑡𝑂𝐻)𝑓𝑖𝑛𝑎𝑙 =∑𝑉𝑖((%𝐸𝑡𝑂𝐻)𝑖
𝑉𝑡𝑜𝑡𝑎𝑙
𝑘
𝑖
(16.14)
Donde el subíndice 𝑖 se refiere a todas las soluciones empleadas para hacer la mezcla final.
4. Discuta el efecto de la constante dieléctrica en la velocidad de reacción. La constante dieléctrica de la mezcla final se puede calcular aproximadamente a partir de la ecuación:
ε = 80 − 0.57 (%EtOH)final
(16.15)
5. La energía de activación para el proceso se puede determinar a partir de la ecuación de Arrhenius. Haga una gráfica de ln 𝑘 𝑣𝑠. 1/𝑇 (°𝐾). De la pendiente y el intercepto se puede calcular la energía de activación y el factor exponencial.
Una alternativa para obtener estos mismos datos es haciendo la gráfica de conductancia vs tiempo y ajustando los datos a una ecuación cuadrática de la forma:
Λt = 𝑎 + 𝑏𝑡 + 𝑐𝑡2
(16.16)
6. La velocidad inicial es proporcional a la derivada a 𝑡 = 0(es decir 𝑟0 𝛼 𝑏). Se construye una gráfica de Arrhenius con ln 𝑟0 𝑣𝑠 1/𝑇 (°𝐾), cuya pendiente es 𝐸𝑎/𝑅. Demuestre que las relaciones son validas.
16.5 cuestionario
1. Discuta el significado de los siguientes términos comparándolos entre si: hidratación, solvatación y solvólisis.
2. Discuta brevemente la conexión existente entre la naturaleza del solvente y la cinética de reacción.
3. En este experimento se estudia la reacción de solvolisiside un sustrato alquílico terciario, ¿Discuta este experimento para la reacción de solvolisis de sustrato alquílico secundario?
4. Una de las preguntas relevante a la cinetica estudiada es , ¿depende o no la velocidad de reacción de la concentración del nucleófilo?.¿Qué opina usted?
16.6 Bibliografía
Chesick. J.P., Patterson Ajr., J. Chem. Educ., 37, 242, 1960 Olsen, DE, J. Chem. Educ., 47, 542, 1970. (Influencia del solvente). Grunwald, E., Winstein, SJ., J. Amer. Chem. Soc. 70, 846, 1948. (Valores de k) Espenson, James. CHEMICAL KINETICS AND REACTION MECHANISMS.
Second Edition , McGraw-Hill, 1995. p 25 y siguientes.

96

97
H. CATALISIS HOMOGENEA
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 17: DESCOMPOSICIÓN CATALIZADA DEL
PERÓXIDO DE HIDRÓGENO
OBJETIVOS
Determinar la constante de velocidad para la reacción de descomposición del peróxido de hidrogeno en presencia de iones yoduro
El presente experimento ilustra el efecto de un catalizador en la velocidad de una reacción
química. Si bien la reacción de descomposición del HIO2 es lenta a temperatura ambiente con pequeños incrementos de temperatura la reacción ocurre vigorosamente. Esta reacción de descomposición pude ser acelerada dramáticamente, inclusive a temperatura ambiente, mediante el uso de un catalizador. Una gran variedad de sustancias puede utilizarse como catalizadores en esta reacción como son: dióxido de manganeso, sangre, partículas de polvo e inclusive superficies rugosas. Este experimento busca determinar la constante de velocidad para la reacción de descomposición del peróxido de hidrogeno en presencia de iones yoduro, y analizar la validez del mecanismo de reacción propuesto para este proceso.

98
17.1 Fundamentos
El peróxido de hidrogeno en solución es inestable y se descompone lentamente para formar oxígeno y agua según la reacción:
2H2O2(ac) → 2H2O + O2(g) (17.1)
El catalizador que se va utilizar en esta la practica es el ion yoduro. El mecanismo de reacción que se propone es
H2O2 + I−K1→ H2O + IO
− (17.2)
H2O2 + IO− K2→ H2O + I
− +O2 (g)
(17.3)
La ley cinética se puede derivar fácilmente para el caso en que 𝐾1 ≪ 𝐾2. Si esta condición se da, el ´´cuello de la botella´´ para la reacción será la etapa dada en la reacción 3, que es una reacción bimolecular donde la ley de velocidad esta dada por la ecuación.
r =d[IO−]
dt= K1[H2O2][I
−]
(17.4)
La relación de constantes cinéticas indica que el intermediario, una vez formado, se descompone rápidamente según la reacción 3. Teniendo en cuenta las anteriores consideraciones cinéticas la velocidad de formación del oxigeno será prácticamente igual a la velocidad de formación del intermediario:
r =d[O2]
dt=d[IO−]
dt= K1[H2O2][I
−]
(17.5)
En otras palabras, con los compuestos hechos, la descomposición del peróxido debe seguir una cinética de segundo orden. Uno con respecto al peróxido y uno con respecto al ion yoduro. Por definición la concentración de un catalizador no cambia durante la reacción química, por tanto la ley cinética es de orden aparente uno con respecto al peróxido:
r = Kap[H2O2]; Kap = K1[I−]0
(17.6)
Un par de hechos experimentales observados al realizar el experimento ponen en entre dicho el mecanismo propuesto de reacción.
El flujo de oxigeno se tarda unos pocos minutos en establecerse, lo que indica que hay un periodo de inducción antes de que la reacción principal tome el control.
Realmente, la descomposición del peróxido de hidrógeno es una reacción extremadamente compleja, que fluye por muchas vías y pasa por numerosos estados intermedios, incluyendo radicales libres. En esta práctica solo se intenta investigar las reacciones más importantes del mecanismo completo de reacción y en un rango limitado de concentraciones, pH y temperatura.

99
Una de las reacciones mas ampliamente estudiada de descomposición catalizada del peróxido de hidrogeno, en fase homogénea, se conoce con el nombre de “Reacción de Bray-Liebhsfsky” (4-7) (BL). La reacción BL es la descomposición del peróxido de hidrógeno por iones yodato en medio acido:
𝟓𝑯𝟐𝑶+ 𝟐𝑰𝑶𝟑− + 𝟐𝑯+ → 𝑰𝟐 + 𝟓𝑶𝟐 + 𝟔𝑯𝟐𝑶 . . . . . [𝑬𝒕𝒂𝒑𝒂 𝑨] (17.7)
𝟓𝑯𝟐𝑶+ 𝑰𝟐 → 𝟐𝑰𝑶𝟑− + 𝟐𝑯+ + 𝟒𝑯𝟐𝑶 . . . . . [𝑬𝒕𝒂𝒑𝒂 𝑩] (17.8)
𝟐𝑯𝟐𝑶 → 𝑰𝟐 +𝑶𝟐 + 𝟐𝑯𝟐𝑶 . . . . . [𝑬𝒕𝒂𝒑𝒂 𝑮𝒍𝒐𝒃𝒂𝒍]
(17.9)
El mecanismo de la reacción BL es bastante complejo e involucra un gran número de rutas e intermediarios. A continuación presentamos algunas de las etapas que pueden tener conexión con la reacción que se propone en esta guía.
H2O2 + I
− +H+ → HOI + H2O . . . . . [R1] (17.10)
HOI + I− + H+ → I2 + H2O . . . . . [R2]
(17.11)
H2O2 + HOI → I− + O2 + H2O+ H
+ . . . . . [R3]
(17.12)
Donde la reacción R1 parece que se da muy lentamente a temperatura ambiente y en las condiciones de la reacción BL; la reacción R2 directa es muy rápida 𝑘𝑅+2 =4.3𝑥1012 𝑀−2𝑆−1; 𝑘𝑅−2 = 18 𝑆
−1 ) y la reacción R3 tiene una constante cinética de 𝑘𝑅3 =110 𝑀−1𝑆−1. La etapa B es muy compleja y no hay evidencia de reacción directa en el peróxido y el yodo molecular.
El estudiante debe discutir y analizar las reacciones anteriormente presentadas.
187.2 Trabajo experimental
17.2.1 Procedimiento 1: método de flujo volumétrico
El volumen de oxígeno que fluye por unidad de tiempo, jt es una medida de la velocidad de reacción, como se indica en la siguiente demostración:
[H2O2] = [H2O2]0e−Kapt
(17.13)
Derivando respecto al tiempo se obtiene:
d[H2O2]
dt= −Kap[[H2O2]0e
−Kapt
(17.14)
Como:

100
dnH2O2dt
= Vrd[H2O2]
dt= −2
dnO2dt
(17.15)
Donde V es el volumen de la solución
Para un gas ideal se cumplen las siguientes ecuaciones
𝑉𝑔 = (𝑅𝑇
𝑝)𝑛
(17.16)
y
Jt=dVg
dt= (RT
p)dnO2dt
(17.17)
Haciendo las sustituciones adecuadas podemos llegar a la ecuación:
𝐽𝑡 = (𝑉𝑟𝐾𝑎𝑝[𝐻2𝑂2]0 𝑅𝑇
2𝑝)𝑒−𝐾𝑎𝑝𝑡
(17.18)
con,
Jt = ae−Kapt
(17.19)
Donde a es una constante.
Tomando logaritmo natural a ambos lados de la ecuación anterior obtenemos
𝑙𝑛(𝐽𝑡) = 𝑙𝑛𝑎 − 𝐾𝑎𝑝𝑡
(17.20)
Esta ultima ecuación constituye la ecuación de trabajo para el experimento, ya que la gráfica del logaritmo del flujo medido en función del tiempo debe dar una línea recta con una pendiente igual a −𝑲𝒂𝒑𝒕.
Desarrollo experimental
Aparato

101
Figura 17.1. Método de flujo volumétrico para seguir la cinética de reacción
El dispositivo consiste en un vaso de reacción, con agitación y una salida para gases que se conecta a un tubo graduado, el medidor de flujo.
El reactor se carga con una solución de KI determinada. La jeringa de la parte superior se carga con un determinado volumen de 𝐻2𝑂2 que se descarga en el reactor al tiempo cero.
La jeringa en la parte inferior del medidor de flujo sirve para inyectar una pequeña cantidad de solución jabonosa en el sitio donde entra la corriente de oxígeno, producido por la reacción. Como resultado se produce una burbuja que fluye por el tubo a una velocidad medible.
Soluciones
A) Peróxido de hidrogeno al 6% en peso nominal: 20 cm3 de reactivo analítico se diluyen a 100 cm3 con agua.
B) Yoduro de potasio a 0.10 M.
Ensayos
Tenga listos dos cronómetros: uno para medir el tiempo que ha trascurrido de la reacción y el otro par a medir el flujo en un tiempo dado.
A (cm3) B (cm3) H2O
1 10 8 12
2 10 12 8
3 10 16 4
4 10 20 0

102
1. De acuerdo con el ensayo, agregue la solución de KI y el agua y ponga el reactor en agitación
2. Cargue la jeringa con 10 cm3 de la solución A y de un solo empujo transfiera la solución al reactor. Ponga en marcha el primer cronometro.
3. Cuando se establezca un flujo apreciable mida con el segundo cronometro el tiempo que se requiere para que la burbuja barra 1-3 cm3
4. Para cada ensayo haga medidas de flujo cada 3 minutos durante media hora. 5. Otros posibles ensayos incluyen la variación de temperatura (para hallar la energía
de activación y el empleo de diferentes catalizadores. 17.2.2 Tratamiento de datos 1. Suponga que se refiere a la cuarta medida de flujo en cierto ensayo, y que esta
se barrieron 3 cm3 en 39.3 s. obviamente el flujo es 3 cm3 /39.3 = 0.0752 cm3/s. pero este valor no es el de un flujo instantáneo, sino el que se da entre los tiempos de 12 min y 12 nub 39.3 s(o 12.665min). el tiempo mas representativo será el tiempo (t1/2) de 12.3.3 min. Proceda de esta manera con todos los datos y elabora una tabla de Jt VS tiempo promedio (t1/2).
2. Construya, el mismo plano, una grafica de Jt vs t1/2 para distintos ensayo. Las
pendientes iniciales a 𝑡 = 0 son una medida de la velocidad inicial. 3. En el mismo plano haga una grafica de 𝑙𝑛 Jt vs t1/2. Una buena correlación lineal
indica que se trata de un proceso de primer orden. Las pendientes de las líneas semilogaritmicas son iguales a −𝐾𝑎𝑝. si obtuvo estas contantes min-1
conviértalas a s -1. 4. Calcule la concentración final KI en mol/L para cada ensayo y grafique 𝐾𝑎𝑝 vs
[𝐾𝐼]. Una línea recta que pase por el punto (0,0) indica que la reacción es de primer orden con respecto al ion yoduro y la pendiente es igual a k1, la constante verdadera de segundo orden en unidades de M-1 S-1.
5. Un procedimiento mas general es el de hacer una grafica de ln𝐾𝑎𝑝 vs 𝑙𝑛 [𝐾𝐼],
done se obtiene una recta cuya pendiente es el orden con respecto al catalizador
y el intercepto ln𝐾1. 6. Discuta los resultados de manera coherente y con criterio científico
17.2.3 Procedimiento 2: método de medida del volumen de O2 desprendido
Aparato

103
Figura 17.2. Medida del volumen de oxígeno desprendido
El equipo que se va a emplear para la determinación del oxígeno es el que aparece en la figura 2. El dispositivo consiste en un vaso de reacción con agitación y una salida para gases que se conecta a un tubo graduado el medidor del volumen de gas desprendido. A medida que se desprende O2 se debe mover el bulbo para que el nivel de agua en el y en la bureta sea el mismo. De esta manera se asegura que la presión dentro el reactor es siempre la misma e igual a la presión atmosférica.
Ensayos
A (cm3) B (cm3) H2O
1 5 5 20
2 5 10 15
3 5 15 10
4 5 20 15
5 10 10 10
6 15 10 5
Soluciones
A) Peróxido de hidrógeno 3% en peso nominal: 10 cm3 de reactivo analítico se diluyen a 100 cm3 de agua.
B) Yoduro de potasio 0.10M 1. Tenga listo un cronometro para medir el tiempo que se desocupa un
volumen determinado. De acuerdo con el ensayo, agregue la solución de KI y el agua, y ponga el reactor en agitación.

104
2. Agregar agua a la bureta hasta que esta quede llena y el bulbo nivelador contenga una quinta parte de su volumen en agua. Los niveles de agua en el bulbo y en la bureta deben ser iguales.
3. Mida la cantidad de peróxido de hidrogeno necesaria para el peróxido necesaria para el experimento con una pipeta graduada y transfiera la solución lo mas rápido posible.
4. Conecte lo más rápidamente posible el sistema para medir el volumen de gas desprendido. Ponga en marcha el cronometro. ajuste permanentemente la altura del bulbo nivelador para que el nivelador para que el nivel de agua en la bureta y el bulbo sean el mismo. Esto último se hace para asegurar que la presión dentro del reactor sea siempre la misma e igual a la presión atmosférica.
5. Registrar el tiempo en función del volumen de gas producido, tomando lecturas el tiempo en función del volumen de gas producido, tomando lecturas a intervalos de 2 ml hasta que el volumen total del oxígeno sea 14 mL.
6. Repita el mismo procedimiento para todos los ensayos. 7. Otros posibles ensayos incluyen la variación de temperatura (para hallar la
energía de activación y el empleo de diferentes catalizadores.
Tratamiento de datos
1. Se debe determinar para cada mezcla las cantidades iniciales, en moles de H2O2 y de 𝐾𝐼 y sus respectivas concentraciones.
2. Con los volúmenes de gas producido y de acuerdo con la estequiometria de la reacción determinar, a la temperatura de trabajo, las moles de H2O2 consumida y la concentración de H2O2 remanente expresado en moles por litro. Se debe hacer una tabla de resultados en función del tiempo.
3. Construya una gráfica de 𝑙𝑛 [H2O2] en función del tiempo para cada uno de los ensayos. Si se obtiene, rectas, mediante la pendiente determinar la constante aparente de velocidad, 𝐾𝑎𝑝.
4. Con los resultados obtenidos para los ensayos 2,5 y 6 verificar si la reacción es
de primer orden respecto al H2O2. 5. Calcule la concentración final del KI en mol/L para cada ensayo y haga la gráfica de
𝐾𝑎𝑝 vs [𝐾𝐼], una línea recta que pases por (0,0) indica que la reacción es de primer
orden con respecto al ion yoduro y la pendiente es igual a K1, la constante verdadera de segundo orden en unidades M-1 s-1.
6. Un procedimiento mas general es el de hacer la grafica de lln 𝐾𝑎𝑝vs ln[𝐾𝐼]donde se
obtiene una recta cuya pendiente es el orden con respecto al catalizador y el
intercepto es ln 𝐾1. 7. Discuta los resultados de manera coherente y con criterio científico
17.2.4 Procedimiento 3: método de medida de la presion del O2 desprendido.
Aparato: el equipo que se va a emplear para la determinación de la cantidad de oxigeno producto en función del tiempo consiste en una botella plásticas de 2 litros de alguna bebida gaseosa comercial, con la cantidad de H2O2 y catalizador indica, a la que le ha adaptado

105
un sesión de presión en la boca. El sensor de presión esta conectado a una interface análoga–digital, la cual tiene la función de recopilar los datos de presión en función del tiempo. A medida que se desprende O2 la presión interna de la botella va aumentado por lo tanto hay que resisar meticulosamente todas la uniones para verificar que no excitan fugas de gas durante el experimento. La botella va aumentado por lo tanto hay que revisar meticulosamente todas las uniones para verificar que no existan fugas de gas durante el experimento. Una mala agitación va a hacer que la reacción ocurra lentamente y que los datos sean reproducibles.
Las soluciones y los ensayos a realizar son los mismos que en el procedimiento 1.
1. De acuerdo con el ensayo, agregue la solución de 𝐾𝐼 y el agua. Agregue la cantidad de peróxido indicada por la pared de la botella rápidamente pero sin agitar la botella.
2. Conecte rápidamente el sensor depresión, verificando que no existan fugas de gas.
3. Al mismo tiempo ponga en marcha el sistema de captura de datos, previamente programado para captura los datos depresión durante 30 minutos cada 30 segundos, e inicie la agitación vigorosa del reactor.
4. Repita el procedimiento anterior para cada ensayo. 5. El tratamiento de datos es similar al expuesto ene procedimiento 2. Lo único que
cambia es que con los datos de presión parcial de gas producido, y de acuerdo con la estequiometria de la reacción se debe calcular las moles de H2O2 consumida y la concentración de H2O2 remante expresada en moles por litro.
Instrucciones de seguridad: el reactivo comercial de H2O2 viene a una concentración de 30% la cual es muy corrosiva y un agente oxidante fuerte, por este motivo se de evitar cualquier contacto de este reactivo con la piel o los ojos.
Bibliografías.
Harned, H.S., J. Am Chem. Soc., 40, 1416, 1918 Martin A. et al., Physical Pharmacy, 1983, tercera edición , Lea y Febiger,
Philadelpihia. www.chemleeds.ac.uk/delights. Sevick, P.,Kissimonová, K., Adamaciková, L. J.Phys. Chem.,a.,104, 3958-3963,
2000. Genigová, J.,Melichercik, M., Olexová, A., Treindl, L., J.Phys. Chem., A. 103, 4960-
4962, 1999. Schmitz, G, Phys. Chem, Phys. 1, 4605-4608, 1999. Stanisvljev, D., Begovic, N., Vukojevic, V.J. Phys. Chem A., 102, 6887-6891, 1998.

106
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 18: CINÉTICA DE LA REACCIÓN ENTRE EL YODO
Y LA ACETONA CATALIZADA POR UN ÁCIDO.
OBJETIVOS
1. Determinar secuencialmente parámetros cinéticos: órdenes parciales, constantes aparentes y constantes absolutas de velocidad.

107
2. Aplicar el método de aislamiento de Ostwald. 3. Comprobar la coherencia entre la ley de velocidad y el mecanismo de reacción.
18.1 Fundamentos teóricos
En disolución acuosa la reacción de iodación de la acetona es lenta pero se puede acelerar si es catalizada por ácido:
𝐶𝐻3 − 𝐶𝑂 − 𝐶𝐻3 + 𝐼2 𝐻+
→ 𝐶𝐻3 − 𝐶𝑂 − 𝐶𝐻2𝐼 + 𝐻𝐼 Esta reacción procede según un mecanismo en tres etapas: las dos primeras corresponden al equilibrio cetoenólico en medio ácido y la tercera consiste en la reacción entre el enol y el yodo. Cinéticamente, los pasos (1) y (3) son rápidos mientras que (2) es lento, por lo que es la etapa determinante del mecanismo
𝐶𝐻3 − 𝐶𝑂 − 𝐶𝐻3 + 𝐻+
(1) → 𝐶𝐻3 − 𝐶𝑂𝐻
+ − 𝐶𝐻3 (2) → 𝐶𝐻3 − 𝐶𝑂𝐻 = 𝐶𝐻2 + 𝐻
+
𝐶𝐻3 − 𝐶𝑂𝐻 = 𝐶𝐻2 + 𝐼2 (3) → 𝐶𝐻3 − 𝐶𝑂 − 𝐶𝐻2𝐼 + 𝐻𝐼
La velocidad de la reacción puede expresarse:
𝑉 = −𝑑[𝐼2]
𝑑𝑡= 𝐾[𝐴𝑐𝑒𝑡]𝑎[𝐻+]𝑏[𝐼2]
𝐶
(18.1)
en la que k es la constante absoluta de velocidad y a, b y c son los órdenes parciales respecto a la acetona (Acet), los protones y el yodo, respectivamente. En las condiciones de la práctica, las concentraciones de acetona y ácido permanecen constantes a lo largo de la reacción, de forma que se puede seguir la cinética respecto del iodo (reactivo test o indicador). Estamos, pues, utilizando el método de aislamiento de Ostwald. En este caso, se consigue partiendo de una concentración inicial de acetona muy grande y teniendo en cuenta que el HCl es el catalizador (los moles que quedan en cada instante son prácticamente igual a los iniciales y, por tanto, las concentraciones constantes a lo largo de la reacción: [𝐻+]0 , [𝐴𝑐𝑒𝑡]0 ≅ [𝐻
+], [𝐴𝑐𝑒𝑡] ≅ 𝑐𝑡𝑒𝑠). Así pues, la reacción será de pseudo-orden c y su velocidad se simplificará a:
V = Kap[I2]c
(18.2)
donde la constante aparente de velocidad es:

108
𝐾𝑎𝑝 = 𝐾[𝐴𝑐𝑒𝑡]0𝑎[𝐻+]0
𝑏
(18.3)
puesto que el iodo no interviene en la etapa determinante de la velocidad, ésta no dependerá de su concentración, es decir será de orden cero respecto al yodo (c=0), y:
V = Kap
(18.4)
Para seguir la evolución de la concentración de iodo, se toman muestras alícuotas de reacción y, tras detenerla, se valoran con tiosulfato de sodio, según:
𝐼2 + 2𝑁𝑎2𝑆2𝑂3 → 𝑁𝑎2𝑆4𝑂6 + 2𝑁𝑎𝐼 Dado que la reacción es catalizada por ácido, la reacción se detiene y se elimina el catalizador por adición de una base o una sal básica. Efectuando diversas experiencias, y variando en cada una de ellas únicamente la concentración de uno de los reactivos (la acetona o el ácido) se puede medir la influencia de éstos sobre la velocidad de reacción y hallar sus órdenes parciales de reacción. Disoluciones 1. Disolución 0.06 M de yodo (I2) común a toda la mesa (ya está preparada). 2. Disolución de NaOH 1 M estandarizada común a toda la mesa (ya está preparada). 3. 500 mL de disolución 0.005 M de tiosulfato sódico (𝑁𝑎2𝑆2𝑂3 pentahidratado). Trasvasar a un frasco de color topacio ya que debe mantenerse en la medida de lo posible en ausencia de luz. 4. 250 mL de HCl 1.2 M (común a toda la mesa), a partir del comercial (en vitrina). 5. 250 mL de acetato de sodio (AcNa) al 2.5 % en peso. (OJO: el acetato de sodio es trihidrato). 18.2 Procedimiento Experimental 1. Conectar el baño termostático a 25 ºC. 2. Preparar las disoluciones 2, 3 (compartida), 4 y 5. 3. Valorar la disolución de ácido clorhídrico (tomar una muestra de 10 mL) con la disolución de NaOH 1M. Cada pareja realiza una valoración. 4. Se realizan tres series de experiencias donde, comparadas dos a dos, se varía la concentración de uno de los reactivos, ácido o acetona, y se mantiene constante la del otro. Serie 1: 5 mL de acetona y 5 mL de ácido clorhídrico 1.2 M 5. En un matraz aforado de 100 mL preparar la mezcla de reacción del siguiente modo y orden: 50 mL de agua + 5 mL de HCl + 5 mL de acetona y aforar con agua. 6. Trasvasar esta disolución a un erlenmeyer con un imán y colocarla en el baño termostático sobre el agitador. 7. Disponer 7 erlenmeyers con unos 10 mL de acetato de sodio al 2.5 %. 8. Llenar la bureta con tiosulfato sódico y enrasarla.

109
9. Cuando se alcance el equilibrio térmico (unos 5 minutos) añadir 10 mL de disolución de I2 al erlenmeyer. Poner el cronómetro en marcha (t = 0), cuando la pipeta se encuentre a mitad de vaciar. 10. A los 2-3 minutos, tomar una muestra de reacción de 10 mL, Valíc, y vaciarla sobre un erlenmeyer con acetato de sodio al 2.5 %. Tomar el tiempo cuando la pipeta se encuentre a mitad de vaciar (t = tiempo al que se detiene la reacción). 11. Valorar la muestra con Na2S2O3 0.005 M hasta que se decolore. ¡OJO! Cuando se está a punto de llegar al final, añadirle el indicador de almidón. 12. Tomar muestras de 10 mL cada ocho minutos y valorar como se ha indicado en el apartado anterior, hasta completar un total de siete muestras. Serie 2: 15 mL de acetona y 3 mL de ácido clorhídrico Repetir el experimento, según lo indicado para la serie 1, extrayendo las muestras cada 6 minutos. Serie 3: 15 mL de acetona y 5 mL de ácido clorhídrico Repetir el experimento, según lo indicado para la serie 1, extrayendo las muestras cada 4 minutos. Nota: Recordad que hay recipientes para desechar los residuos al acabar la experiencia. 18.3 Resultados experimentales: presentación de los datos 1. Presentar de forma clara los cálculos realizados para preparar las diferentes disoluciones indicando: las masas o volúmenes teóricos, los reales utilizados y recalcular todas las concentraciones (Tabla 1). 2. Presentar los datos de la valoración de la disolución de ácido clorhídrico con sosa y calcular la concentración del ácido clorhídrico con su error aleatorio (Tabla 2). 3. Tabular los resultados de cada serie: tiempos de reacción y volúmenes de tiosulfato utilizados (Tabla 3). 18.4 Tratamiento y Discusión de Resultados
1. Comprobar que la reacción es de orden cero respecto al yodo (c=0). Para ello hay que representar la ecuación integrada de velocidad de orden cero donde la concentración de iodo varía linealmente con el tiempo, según:
[I2]𝑡 = [I2]0 −𝐾𝑎𝑝𝑡
(18.5)
En lugar de esta ecuación, representamos una ecuación equivalente en función del volumen
de tiosulfatoconsumido en la valoración de la muestra 𝑉𝑡𝑖𝑜𝑠, ya que la relación estequiométrica entre el número de moles de iodo y el de tiosulfato es 𝑛𝐼2 − 1/2𝑛𝑡𝑖𝑜𝑠 , por
lo que la concentración de yodo en un momento dado, [I2]𝑡 , es directamente proporcional al volumen de tiosulfato, según

110
[I2]𝑡 =1
2
[𝑆2𝑂32−]𝑉𝑡𝑖𝑜𝑠(𝑡)
𝑉𝐼2= 𝐴. 𝑉𝑡𝑖𝑜𝑠(𝑡)
(18.6)
siendo A una constante cuya expresión es 𝐴 =1
2[𝑆2𝑂3
2−]/𝑉𝐼2.Sustituyendo la ecuación (6)
en la (5) obtenemos:
𝑉𝑡𝑖𝑜𝑠(𝑡) = 𝑉𝑡𝑖𝑜𝑠(0) − 𝐾𝑎𝑝, 𝑡 (18.7)
donde
𝐾𝑎𝑝, =
𝐾𝑎𝑝𝐴
(18.8)
y 𝑉𝑡𝑖𝑜𝑠(0) es el volumen de tiosulfato que se utilizaría para valorar una alícuota cuando
comienza la reacción, t = 0. Si para cada serie, la representación del volumen de tiosulfato en función del tiempo es una recta, habremos comprobado que la reacción es de orden cero respecto al iodo. Hacer la representación de las tres series en la misma gráfica. 2. Calcular las constantes de velocidad aparentes. Del ajuste de las rectas anteriores (ecuación 7), se obtendrá k’ap,i de la pendiente y con el valor de la constante A obtendremos las constantes aparentes de cada serie: kap,i. 3. Calcular las concentraciones iniciales de acetona y de ácido de cada serie. 4. Calcular los órdenes parciales respecto a la acetona (a) y el ácido (protones) (b). Para ello vamos a aplicar la ecuación (3) a las series tomadas de dos en dos, de forma que la concentración de un reactivo sea la misma. 4.1. [HCl] igual en las series 1 y 3:
𝑠𝑒𝑟𝑖𝑒 1
𝑠𝑒𝑟𝑖𝑒 3 →𝐾𝑎𝑝.1
𝐾𝑎𝑝.3= ([𝐴𝑐𝑒𝑡]0.1[𝐴𝑐𝑒𝑡]0.3
)𝑎
𝑡𝑜𝑚𝑎𝑛𝑜𝑠 𝑙𝑜𝑔𝑎𝑟𝑖𝑡𝑚𝑜𝑠: 𝑙𝑛 (𝐾𝑎𝑝.1
𝐾𝑎𝑝.3)
= 𝑎 𝑙𝑛 ([𝐴𝑐𝑒𝑡]0.1[𝐴𝑐𝑒𝑡]0.3
) 𝑦 𝑑𝑒𝑠𝑝𝑒𝑗𝑎𝑚𝑜𝑠:
𝑎 =𝑙𝑛𝐾𝑎𝑝.1 −𝐾𝑎𝑝.3
ln [𝐴𝑐𝑒𝑡]0.1 − 𝑙𝑛[𝐴𝑐𝑒𝑡]0.3
(18.9)
4.2. [Acet] igual en las series 2 y 3:
𝑠𝑒𝑟𝑖𝑒 2
𝑠𝑒𝑟𝑖𝑒 3 →𝐾𝑎𝑝. 2𝐾𝑎𝑝.3
= ([𝐻𝐶𝑙]0.2[𝐻𝐶𝑙]0.3
)𝑏
𝑡𝑜𝑚𝑎𝑛𝑜𝑠 𝑙𝑜𝑔𝑎𝑟𝑖𝑡𝑚𝑜𝑠: 𝑙𝑛 (𝐾𝑎𝑝.2𝐾𝑎𝑝.3
)

111
= 𝑎 𝑙𝑛 ([𝐻𝐶𝑙]0.2[𝐻𝐶𝑙]0.3
) 𝑦 𝑑𝑒𝑠𝑝𝑒𝑗𝑎𝑚𝑜𝑠:
𝑎 =𝑙𝑛𝐾𝑎𝑝.2 − 𝐾𝑎𝑝.3
ln [𝐻𝐶𝑙]0.2 − 𝑙𝑛[𝐻𝐶𝑙]0.3
(18.10)
5. Calcular la constante de velocidad absoluta con su error aleatorio.
Con los valores obtenidos de a y b (a=b=1), aplicamos de nuevo la ecuación (3) para cada serie, y tendremos:
𝐾𝑖 =𝐾𝑎𝑝, 𝑖
[𝐴𝑐𝑒𝑡]𝑜,𝑖 [𝐻+]𝑜,𝑖
(18.11)
Calcular el valor medio, k , de los tres y su error aleatorio. 6. Deducir y discutir, a partir de las gráficas volumen-tiempo y de las constantes de velocidad aparentes obtenidas en cada serie, cómo influyen las concentraciones de acetona y de ácido clorhídrico en la velocidad de la reacción. 7. Comprobar que el mecanismo propuesto es coherente con la ley de velocidad experimental obtenida.

112
I. CATALISIS HETEROGENEA
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 19: ADSORCIÓN SOBRE CARBÓN ACTIVADO.
OBJETIVOS
1. Determinar la constante de adsorción del acido Oxálico. 2. Determinar el área superficial de una muestra de carbón activado

113
Adsorción
La adsorción es la acumulación de una sustancia en una interface. Hay dos clases de adsorción física y la quimiadsorcion. Irving lagmuir fue quien estableció en 1996 que aparte de los procesos de absorción física o adsorción de van der Waals, existían los procesos de quimioadsorción al medir los calor es liberado durante los proceso de adsorción: 100 a 500 kj/mol para la quimioadsorcion (valores del orden de los del enlace químico) y<20kj/mol para la adsorción física. La adsorción física no es específica y es un proceso relativamente rápido y reversible. Por el contrario en la quimioadsorción de la formación de enlace químicos definidos y asean iónicos y covalentes. La interfaces donde se puede presentar el fenómeno son generalmente gas-solido, sólido-líquido y gas –líquido entre el carbón activado y una solución acuosa de un ácido carboxílico, con el propósito de determinar la constante de adsorción y e área superficial de una muestra de carbón activado.
19.1 Fundamentos
La presencia de una interface implica que la adsorción suceda. Sin embargo, para que sea notorio el fenómeno se requiere que la interface tenga un tamaño considerable. En el caso de un sólido esto se logra cuando la relación área/masa es grande, como por ejemplo en solidos finamente divididos o muy porosos. La cantidad de substancia adsorbida disminuye al aumentar la temperatura ya que todos los proceso de adsorción son exotérmicos.
Para determinar la energía de adsorción (física) se pueden realizar medidas calorimétricas directas, para obtener la “energía molar diferencial de adsorción”. Una forma alternativa y muy llamativa de determinar esta energía consiste en realizar las isotermas de adsorción a diferentes temperaturas. La energía asociada al proceso de adoración física (calor liberado) se conoce en la literatura especializada como “entalpia isostérica de adsorción”. Recordemos que el proceso termodinámico de adsorción ocurre en sistema cerrado y a temperatura y volumen constantes.
A temperatura constante la cantidad adsorbida aumenta con la concentración de equilibrio del adsorbato en la fase liquida o con la presión parcial del adsorbato en el caso de una fase gaseosa. A la relación de la cantidad de adsorbida 𝑋𝑎𝑑 y la concentración de adsorbato, o presión parcial en el caso de los gases, en el equilibrio a una temperatura determinada se le llama isoterma de adsorción. Empíricamente se ha encontrado que muchas isotermas de adsorción siguen ecuación de la forma:
𝑋𝑎𝑑 = 𝐾𝐶𝑛 (19.1)
Donde 𝐾y 𝑛 son cosntantes, y 𝐶 es la concentración molar del adsorbato en el equilibrio. Esta relación se conoce como isoterma de adsorción de frendlich
Langmuir desarrolló el primer modelo teórico de la adsorción y encontró la ecuación mas sencilla para describir el proceso. Las consideración principales del modelo son: la superficie del adsorbente contiene un número fijo de lugares donde se puede l levar a cabo

114
la adsorción y cada uno de ellos pude acomodar solo una molécula, no hay interacción entre las moléculas adsorbida, y el calor de adsorción es el mismo para todos lo lugres y no depende de la superficie cubierta.
Para llegar a la ecuación de Langmuir se consideran dos tendencias opuestas: la condensación de moléculas de adsorbato en la superficie del adsorbente y la desorción de
las mismas hacia la solución. Si 𝛿 es la fracción de superficie cubierta, se puede considerar que:
𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑑𝑒 𝑐𝑜𝑛𝑑𝑒𝑛𝑠𝑎𝑐𝑖ó𝑛 = 𝐾𝑎(1 − 𝛿)𝐶 (19.2)
𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑 𝑑𝑒 𝑎𝑑𝑠𝑜𝑟𝑐𝑖ó𝑛 = 𝐾𝑑𝛿
Donde C es la concentración del absorvato en solución. Cuando el sistema esta en equilibrio tenemos la igualdad:
𝐾𝑎(1 − 𝛿)𝐶 = 𝐾𝑑𝛿 (19.3)
𝛿 =𝑏𝐶
1 + 𝑏𝐶
(19.4)
𝑏 =𝐾𝑎𝐾𝑏
la cantidad "𝑏" es la constante conocida como el coeficiente de adsorción ( o constante de
Langmuir). Si se define a 𝑋𝑎 como el número de moles de adsorbato por gramo de adsorbente y Г como el número de moles necesarias para cubrir completamente la superficie disponible de un gramo de adsorbente, se puede reescribir la ecuación (4) como:
𝐶
𝑋𝑎=1
Г𝑏+𝐶
Г
(19.5)
1
𝑋𝑎=
1
𝐶Г𝑏+1
Г
(19.6)
Si se conoce el área que ocupa una molécula de asorbato sobre la superficie del adsorbente (𝜎), y se supone que las moléculas de adsorbato forman una monocapa, se puede calcular el área especifica del adsorbente (A) con la ecuación:
𝐴 = 𝜎Г𝑁𝑎 (19.7)
Donde Na es el número de Avogadro.
19.2 Trabajo experimental.

115
19.1 Adsorción de ácido acético
1. Tenga en cuenta que el presente experimento las medidas cuantitativas se deben hacer con mucho cuidado, porque los efectos que se van a medir son muy pequeños y cualquier error en manipulación ocasiona un error grande en los resultados.
2. Trepare 2501 ml de una solución 1M de acido oxálico o acético y 500 ml de solución 0.1 M de NaOH. La solución de NaOH debe ser valorada con la solución de NaOH para determinar las verdaderas concentraciones de los reactivos. Finalmente prepare por dilución 250 ml de solución 0.005M y 0.002 de NaOH a partir de solución patrón.
3. Prepare frascos volumétricos soluciones del acido seleccionado tomando los volúmenes de 100, 50, 30, 20, 10, 7, 5, 2, y 1 ml de la solución patrón 1M, y llevándolos a un volumen total de 100 ml.
4. A ocho frascos con tapa coloque 1 g de carbón activado pesado a la centésima de gramo.
5. Agregue al primer frasco 100 ml de agua destilada (mezcla blanco) y a los demás frascos agregue los 100 ml de una de las siete soluciones del ácido preparadas anteriormente. Agite las mezclas por 60 minutos.
6. Filtre aproximadamente 30 ml de la mezcla de cada frasco y valore por duplicado una alícuota razonable de sobrenadante con la solución de NaOH mas apropiada, esto significa que los volúmenes de titulación deben estar entre 10 y 20 ml, y utilizando como indicado fenolftaleína.
7. Una variante al procedimiento anterior es utilizar la gravimetría para preparar y valorara las soluciones. Rediseñe antes de la sesión de laboratorio, el experimento anterior para trabajar gravimétricamente (p.e: concentraciones en moles/Kg de solución y alícuotas en gramos).
19.2.2 (B) Adsorción de azul de metileno (2)
1. Preparar una solución de azul de metileno en agua destilada, de concentración 25 mg/L.
2. En 10 frascos distribuya las siguientes cantidades de carbón activado (mg) 1, 5, 10, 13, 25, 30, 35, 40, 70, 100. A cada frasco, debidamente rotulado, adicione 100 ml de la solución de azul de metileno. Deje los frascos en fuerte agitación durante 3 días.
3. Luego de agitación deje los frascos en reposo hasta sedimentación del carbón activado, tome alícuotas (si es necesario use una centrifuga) y determine la absorbancia a 630 𝑛𝑚.
4. El tratamiento de los datos es el mismo descrito para la adsorción del ácido acético (área de una molécula de azul de metileno, 120 Å2)
19.3 Tratamiento de datos
1. Calcule la concentración final del ácido de cada uno de los frascos y réstele a cada una de ellas la concentración del blanco. Este valor es la concentración real C del acido en equilibrio.
2. Calcule el número de moles de acido adsorbido pro gramo de adsorbente 𝑋𝑎. Este valor se determina a partir del número de moles de ácido iniciales en los 100 ml de solución y el número de moles de ácido que quedan en la solución cuando se alcanza el equilibrio.

116
3. Con los datos obtenidos construya las graficas de ln 𝑋𝑎 𝑣𝑠 𝑙𝑛 𝐶, 𝐶/𝑋𝑎 𝑣𝑠 𝐶 𝑦 1/𝑋𝑎 𝑣𝑠 1/𝐶 correspondiente a ls isotermas de Freundlich y Langmuir. Si el sistema se ajusta a una o ambas isotermas determine las constantes respectivas.
4. Algunos valores estimados para e área ocupada por molécula son: 21 Å para una molécula de ácido acético y 26 A para una de acido oxálico. Use estos datos para calcular el área específica del adsorbente si el sistema sigue el comportamiento descrito por la ecuación de langmuir.
5. Analice la propagación de error en las dos diferentes formas matemáticas de escribir la ecuación de Langmuir. Para esto genere numéricamente una isoterma y propague a través de los datos un 20% de error aleatorio
19.4 Bibliografía.
Castellam, G.W. FISICOQUíMICA, segunda edición, Addison-Wesley Iberoamericana, 1987.p 452.
Potgieter, J. H., J. Chem. Educ., 68, 349-350, 1991. Silva, G., Pinzón, J.A. “Aplicación de la isoterma de lagmuir a la adsorción
competitiva de cadmio, níquel y zinc sobre una bentonita natural.”. Rev. Col. Quím.,28(2), 25-36,1999.
Silva, G., Pinzón, J.A. “Aplicación de la isoterma de lagmuir a la adsorción competitiva de cadmio, níquel y zinc sobre una bentonita natural.”. Rev. Col. Quím., 28(2), 37-42 ,1999.
Pinzón, J.A. “superficie específica de una bentonita mediante adsorción de azul de metileno”. Rev. Col. Quím., 26 (1), 1-14, 1997.
Lozano de yunda, A., Guzmán, G., ferrucho, A. “Isotermas de adsorción de potasio en oxisol de los llanos orientales de Colombia”. Rev. Col. Quím., 26 (1), 23-30, 1997.
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 20: REACCIONES FOTOQUÍMICAS
OBJETIVOS
Analizar el uso de las reacciones fotoquímicas en la degradación de colorantes.
La fotoquímica es la investigación del efecto de la luz sobre las reacciones químicas. Solo la luz absorbida por el sistema puede tener efectos químicos. El primer paso de un proceso

117
fotoquímico es la absorción de un cuanto de energía lumínica sobre un determinado átomo, molécula, ion o partícula, que por el acto pasa a un estado excitado mucho mas reactivo químicamente puesto que tiene una energía de exceso igual a ℎ𝑣. En la presente práctica, la luz no actúa directamente sobre el reactante, sino que produce un estado excitado en las partículas del semiconductor anatasa (una forma meta estable de TiO2) que conlleva a la formación de electrones con una energía suficiente que le permite ser transferidos a sustancias aceptoras de electrones (agentes oxidantes) y “huecos” positivos capaces de recibir electrones de sustancias donoras de electrones (agentes reductores) presentes en la interface partícula anatasa–solución. Este experimento ilustra las técnicas básicas utilizadas para el estudio de las reacciones fotoquímicas y la catálisis heterogénea.
20.1 Fundamentos
Como se indica en la Fig. 1. A, para muchos compuestos, a medida que el número de unidades monomiarias (N) en una partícula incrementa, la energía necesaria para promover
un electrón de la partícula desde la banda de valencia (BV) a la banda de conducción (BC) disminuye. En el límite cuando N ≫ 2000 es posible encontrar partículas que exhiben una estructura electrónica de bandas características de un semiconductor, como se ilustra en la figura 1.A, donde la diferencia de energía entre la banda de valencia y la banda de conducción es igual a ∆E
La Figura 20.1.B muestra que la activación del semiconductor se inicia mediante la absorción de un fotón cuya energía es mayor o igual a AE. Como resultado de la promoción de un electrón a la banda de conducción se genera un “huevo” positivo (h+) en la banda de valencia. La doble capa eléctrica, que se forma espontáneamente en la interface semiconductor-solución cuando los semiconductores de tipo (usualmente óxidos metálicos) son sumergidos en una solución, favorece la separación en el espacio de los huecos

118
positivos (h+) y los electrones excitados e inhibe la recombinación de estos. Como
consecuencia, en los semiconductores de tipo 𝑛 (que se caracterizan porque la conducción eléctrica se hace mediante el transporte de carga negativa), el electrón fotón foto generado se mueve al interior de la partícula donde puede ser trasferido ya sea a un alambre conductor o a otro electrodo no fotoactivo, o a otro sitio de la superficie donde puede ser transferido a un aceptor de electrones A según la reacción:
A + e− → A− ( 20.1)
Los h+ pueden migrar bajo la influencia del campo eléctrico hacia la superficie del semiconductor donde puede oxidar posibles sustancias capaces de donar electrones (D) según la reacción:
D + h+ → D− (20.2)
Dependiendo de las condiciones de pH de la solución los electrones y h+ generados pueden oxidar o reducir respectivamente, el agua (1,2).
Una de las aplicaciones más comunes de los semiconductores es su aplicación como foto catalizadores en reacciones que involucran la degradación de compuestos orgánicos o biomasa en general (1,3,4). La degradación completa de biomasa se puede dar después de mucho tiempo de irradiación en un medio libre de oxigeno según la reacción:
Biomasa + H2O →TiO2 Pt⁄
hv > ∆E→ CO2 + H2
(20.3)
En presencia de oxígeno la reacción más probable es:
𝐵𝑖𝑜𝑚𝑎𝑠𝑎 + 𝑂2 →𝑇𝑖𝑂2 𝑃𝑡⁄
ℎ𝑣 > ∆𝐸→ 𝐶𝑂2 +𝐻2𝑂 + 𝐴𝑐𝑖𝑑𝑜𝑠 𝑚𝑖𝑛𝑒𝑟𝑎𝑙𝑒𝑠
(20.4)
Otra aplicación importante de la anatasa es como catalizador de la reacción de oxidación de SO2 hasta acido sulfúrico en medio acuoso según el siguiente mecanismo de reacción (6): El estudio de esta reacción es importante puesto que está directamente relacionado con el problema ambiental bien conocido de la formación de lluvia ácida , y con el estudio de la remoción de SO2 los gases de desecho de industrias relacionadas con la producción de H2 SO4 o plantas termoeléctricas que usan combustibles fósiles para la producción de electricidad (5).
21.2 Trabajo experimental
El montaje experimental consiste en una lámpara ultravioleta de 6 W o 4W (de la misma clase que usan los probadores de billetes) protegida por un tubo de vidrio que se introduce en un recipiente que contiene los reactivos y la anatasa. Un termómetro esta acondicionado al recipiente para medir la temperatura durante el curso de la reacción y un orificio lateral

119
permite, ya sea sacar muestras del recipiente el curso de la reacción para ser analizadas, o la introducción de un sensor que nos permita seguir el curso de la reacción.
Reacciones del SO2 SO2(g) → SO2(aq)
SO2(aq) + H2O → 𝐻2SO3(ac) HSO3(ac) → HSO3
−(ac) + H+
𝐻2SO3−(ac) → HSO3
−(ad)
Reacciones del oxígeno molecular
O2(g) → O2(aq) O2(ac) → O2(ad)
TiO2 hv → e− + h+
H2O(ad) + h+HO. + H+
1. HSO3(ad) + 2HO. → HSO4
−(ad) + H2O
2. HSO3−(ad) + 2HO2 → HSO4
−(ad) + H2O + O2
O2(ad) → O2−(aq)
O2−(ad) + H+ → 2HO2
HSO3−(ad) + 1/2O2(ad)
TiO2 hv → HSO4
−(ad)
HSO4−(ad) → HSO4
−(ac)
Figura 20.2: Mecanismo de reacción
20.3 Foto degradación de colorantes orgánicos
Figura 20.3: Reactor Fotoquímico.

120
20.3.1 Foto degradación de rojo congo(4)
1. Para la práctica hay que disolver 20 mg de rojo congo en 850 ml de agua, directamente en el recipiente de trabajo.
2. Una vez disuelto el colorante medir la absorbancia de la solución 𝜆 = 598 𝑛𝑚.
3. Adicionar una cantidad exactamente pesada de anatasa entre 200 y 500 mg, e iniciar la continua agitación de la suspensión.
4. Después de 10 minutos tomar dos muestras de 10 mL cada una. Una muestra debe ser centrifugada durante 10 minutos, después el sobrenadante debe ser transferido a otro tubo de ensayo para ser centrifugado nuevamente durante 5 minutos más y finalmente se le mide el porcentaje de transmitancia (%T) a la solución sobrenadante. La segunda muestra debe ser guardada en la oscuridad y ser agitada periódicamente hasta el final de la práctica, cuando deber ser procesada de manera similar a la primera muestra en el montaje para determinar el %T sobrenadante.
5. Colocar el reactor tal como se muestra en el montaje experimental y encender la lámpara UV. Este momento se toma como tiempo cero.
6. Tomar muestras de la suspensión cada 15 minutos y medirles el %T al sobrenadante siguiendo el mismo procedimiento indicado anteriormente. Se deben tomar muestras durante un tiempo no menor a 3 horas de reacción o hasta que la absorbancia sea menor al 25% de la absorbancia inicial.
20.4 Tratamiento de datos
1. Discuta el posible mecanismo de reacción del proceso de degradación del Rojo Congo. Analice si la reacción es controlada por el proceso de transporte (difusión, convección o migración) de los reactivos desde el interior de la solución hacia la superficie del catalizador, o por procesos cinéticos sobre la superficie. Analice en que circunstancia la reacción puede ser de orden cero o uno. Si no entiende la pregunta consulte este punto con el instructor.
2. Haga la grafica de absorbancia (A) vs tiempo y ln A vs tiempo. Una línea recta de la ultima gráfica indica un orden uno para la reacción y a partir de la pendiente se puede hallar la constante de velocidad para la reacción.
3. Compare la absorbancia de la muestra que se dejó en la oscuridad con la absorbancia de la muestra al tiempo cero y la del final del experimento bajo irradiación UV. Explique la diferencia.
20.4.1 Reducción fotoquímica del Naranja de Metilo
Los compuestos aromáticos son de importancia comercial, siendo el naranja de metilo uno de los más usados. Este compuesto es estable bajo la acción de la radiación UV-VS, pero en soluciones acuosas puede ser reducido por varios agentes reductores, dando como producto un derivado de la hidracina incoloro:
(𝐶𝐻3)𝑁𝐶6𝐻4𝑁 = 𝑁𝐶6𝐻4𝑆𝑂3− + 2𝐻+ + 2𝑒− → (𝐶𝐻3)𝑁𝐶6𝐻4𝑁𝐶6𝐻4𝑆𝑂3
− (20.5)
121
Sin embargo, la velocidad de esta reacción es usualmente muy baja, lo que le confiere cierta estabilidad a los soluciones acuosa de naranja de metilo, aun en presencia de agentes reductores como el ácido ascórbico. La velocidad de reducción del naranja de metilo en presencia de un agente reductor puede ser incrementada si se adiciona TiO2
como catalizador y se irradia la solución con una lámpara capaz de emitir luz de una longitud de onda igual o menor a 388 nm (cercano ultravioleta).
Para el experimento vamos a seguir el siguiente protocolo:
1. Disolver una cantidad entre 0.018 y 0.010 g de Naranja de Metilo y entre 0.025 g y 1 g de ácido ascórbico en 850 mL de agua directamente en el recipiente de trabajo.
2. Una vez disuelto el colorante medir la absorbancia de la solución 𝜆 = 463 𝑛𝑚
3. Adicionar una cantidad exactamente pesada de anatasa entre 0.050 y 0.400g, e iniciar la agitación continua de la suspensión.
4. A partir de este momento se debe seguir el mismo procedimiento y el tratamiento de los datos del experimento anterior, pero aplicados para el caso del naranja de metilo.
Para un estudio sistemático del efecto del ácido ascórbico y la anatasa en la velocidad de reacción se recomienda estudiar las siguientes mezclas:
Mezcla 1 2 3 4 5 6 7 8
Ácido ascórbico
0,150 0,150 0,150 0,150 0,025 0,050 0,200 1,00
Naranja de metilo
0,015 0,200 0,200 0,200 0,012 0,012 0,012 0,012
Anatasa 0,050 0,200 0,200 0,200 0,200 0,200 0,200 0,200
20.3 Bibliografía Mills. A., Le Hunte, S., “An overevew of semiconductor photocatalysis”, Photchem
Photobiol A Chem 108,35,1997. Rajeshward, k., Ibañez, J.G. “Electrochemical aspectos of photocatalysis: aplication
to detoxificaión an desinfection scenarios” J.Chem. Educ. 72, 1044-49, 1995. Peral, J., Trillas, M., Domonech, X., “Heterogeneous Photochemistry”, J. Chem
Educ., 72, 565-566,1995.

122
J. FENOMENOS SUPERFICIALES

123
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 21: MEDICION DE LA TENSION SUPERFICIAL:
METODO DE LA GOTA Y METODO DEL CAPILAR.
OBJETIVOS Se pretende determinar las tensiones superficiales del etanol y de agua con una elevada concentración de un electrolito fuerte (NaCl), así como realizar una medida de la disminución en la tensión superficial del agua cuando se le añade como soluto un detergente comercial (mezcla de tensioactivos aniónicos y no iónicos) y un tensioactivo aniónico como el dodecilbencenosulfonato sódico, de interés comercial.
21.1 FUNDAMENTO TEÓRICO
¿Quién no ha invertido algunos minutos de su vida en observar cómo el agua se acumula en elextremo de un grifo, formando gotas que caen sucesivamente? Inicialmente, puede observarse unapequeña superficie ovalada. Después, a medida que el agua se acumula, esta superficie va tomandoforma esférica y finalmente cae. Se observa que las gotas siempre caen cuando alcanzan un determinado volumen. Es decir, no caen a veces gotas

124
pequeñas y luego gotas grandes. Si el flujo de agua se mantiene constante, es un hecho que las gotas desprendidas tienen siempre el mismo tamaño. Si en vez de agua, el grifo diera alcohol, las gotas se desprenderían antes y su volumen sería notablemente más pequeño. La explicación que justifica el hecho de que líquidos diferentes generen gotas de distinto tamaño, reside en la misma explicación que justifica que algunos insectos puedan “caminar” sobre la superficie del agua. La misma que argumenta el uso de servilletas de papel como absorbente, y la que igualmente explica por qué la sabia accede desde las raíces hasta las hojas y porqué el detergente sirve para lavar. La explicación de todos estos fenómenos reside en una propiedad que tienen todas las sustancias que presentan un límite en su extensión, una frontera que la separe de otra fase diferente. Esta propiedad se denomina tensión superficial. Analicemos la estructura microscópica de la gota de agua. En ella podemos distinguir entre el volumen que constituye su interior y la superficie que delimita su forma. En el agua, al igual que ocurre en todos los líquidos, las moléculas establecen interacciones atractivas que las mantienen cohesionadas. De hecho si no existiesen estas fuerzas, nuestro sistema no sería líquido sino gaseoso. Estaríamos hablando de un gas ideal. En el interior, una molécula de agua está rodeada de otras de su misma especie. Como se ilustra en la figura 1, las interacciones se distribuyen en todas las direcciones sin existir ninguna privilegiada. Sin embargo en la interfase que limita la gota y la separa del aire, la situación es diferente. Una molécula de agua que ocupe cualquier posición de esta superficie, no tiene a otras sobre ella, lo que significa que no está sometida a interacciones con otras moléculas de agua, más allá de la interfase. En consecuencia se da una asimetría en la distribución de interacciones y la aparición de una fuerza resultante neta que apunta hacia el interior de la gota.
Figura 21.1 interacción entre las moléculas de agua.
En términos de estabilidad, una molécula de agua se encuentra en una situación mucho más favorable cuando se encuentra en el interior que cuando se encuentra en la interfase. Esto es así porque debido a la fuerza resultante que apunta hacia el interior, el acceso de moléculas internas hacia la superficie corresponde con un proceso energéticamente costoso. Y es que para vencer esta fuerza, el sistema debe consumir parte de su energía interna en realizar el trabajo necesario para llevarla hasta allí. Evidentemente si acceden un número determinado de moléculas hasta la superficie, ésta se vería incrementada. El trabajo necesario dW, para incrementar en dA la superficie de la gota se calcula como,
𝑑𝑤 = 𝛾𝛼𝛽𝑑𝐴, donde,

125
𝛾𝛼𝛽, representa la fuerza neta que apunta al interior de la gota y se denomina tensión superficial ab, representa a las interfases limítrofes (en el ejemplo agua y aire)
Las unidades de tensión superficial serían 𝑈𝑛𝑖𝑑𝑎𝑑𝑒𝑠 𝑑𝑒 𝑒𝑛𝑒𝑟𝑔í𝑎
𝑈𝑛𝑖𝑑𝑎𝑑𝑒𝑠 𝑑𝑒 𝑆𝑢𝑝𝑜𝑒𝑟𝑓𝑐𝑖𝑒 . Empleando las unidades
recomendadas por el sistema internacional se expresarían como 𝑚2
𝐽. Si desarrollamos este
cociente, se llega a:𝐽
𝑚2=𝑁.𝑚
𝑚2=𝑁
𝑚;
Es decir, 1𝐽
𝑚2 equivale a 1
𝑁
𝑚. En estas unidades, las tensiones superficiales de la mayor
parte de los líquidos son del orden de las centésimas o las milésimas. Por ello, en vez de
emplear 𝑁
𝑚, se utiliza el submúltiplo
𝑚𝑁
𝑚 , que se lee como mili newton por metro.
Según estas unidades, la tensión superficial se puede expresar como fuerza por unidad de longitud. Veamos porqué. Imagine que disponemos de dos globos exactamente iguales y que lo inflamos hasta alcanzar volúmenes diferentes. Evidentemente el globo de mayor volumen soporta una tensión superior a la del otro. Ahora imagine que nos disponemos a efectuar un corte de igual longitud en sus superficies. ¿Cómo serán la fuerzas de cohesión que deberíamos aplicar en ambos globos a lo largo de la longitud del corte, para que las superficies no se separen?. Lógicamente será mayor en el globo de mayor volumen, puesto que la tensión a la que está sometida su superficie, es superior a del globo más pequeño. En este ejemplo queda reflejado cómo la tensión superficial puede medirse en unidades de fuerza por longitud. Volviendo a la gotas, ¿cómo se justifica en términos de tensión superficial, que las gotas de alcohol que se desprenden del grifo, sean más pequeñas que las de agua?. Hemos afirmado que en todos los líquidos existen interacciones moleculares o iónicas que los mantienen cohesionados. Estas fuerzas son de magnitud diferente para cada líquido y en el caso del agua, son sustancialmente elevadas en relación con las de otros líquidos, ya que en su seno se establecen unas interacciones relativamente intensas denominadas puentes de hidrógeno. En el alcohol, las interacciones moleculares son bastante menos intensas y como consecuencia la tensión superficial también es relativamente pequeña en relación con la del agua. A medida que la gota acumula líquido, sobre ella se aprecian dos fuerzas verticales de sentido opuesto. Por un lado el peso (𝑚 ∗ 𝑔) que apuntando al centro de la tierra trata que la gota caiga. Por otra parte la tensión superficial, que al igual que la superficie del globo, mantiene cohesionado el contenido de la gota. Esta fuerza que apunta en sentido contrario al peso, está aplicada a lo largo de la curva cerrada de contacto entre la gota y el grifo, y corresponde a la tensión superficial (véase figura 2). Como el agua tiene una tensión superficial relativamente elevada, la masa de agua necesaria para que el peso de la gota supere en una cantidad infinitesimal a la tensión superficial, es relativamente grande. Sin embargo, la cantidad de masa de alcohol requerida para que el peso de la gota supere su tensión superficial es menor. Por ello, en el momento en que se desprenden, las gotas de agua son de mayor volumen que las de alcohol.

126
Un sistema para medir la tensión superficial: el método del peso de la gota Como ya hemos visto, el proceso de formación de una gota en el extremo de una superficie sólida, es un fenómeno regido por la tensión superficial. Para que una gota de líquido se desprenda y caiga, es preciso que su peso supere en una cantidad infinitesimal al trabajo ejercido por la tensión superficial para la ampliación de su superficie. En el caso de que consideremos la situación de un líquido que gotea en el extremo de un tubo capilar de radio r, la condición de equilibrio en el instante anterior al desprendimiento de la gota es,
𝛾 =𝐹
𝐿=𝑚𝑔
2𝜋𝑟=𝑚´𝑔
2𝜋𝑟ф =
𝑉 ,𝜌𝑔
2𝜋𝑟ф
(21.1)
donde m es la masa de la “gota ideal”, m’ es la masa de la gota desprendida, medida experimentalmente, V’ es el volumen de esa gota, g es la aceleración de la gravedad, r es la densidad del líquido, r es el radio exterior del capilar (mejor dicho el radio de la circunferencia de contacto líquido - vidrio) y ф es una función correctora que tiene en cuenta los restos de masa del líquido que no se desprenden del extremo del capilar y que distingue a la “gota ideal” de la desprendida. En efecto, la masa de la gota obtenida por este método (m’), es menor que el valor ideal (m). La razón de ello es fácil de comprender tras observar detenidamente el proceso de formación de una gota por desprendimiento desde un capilar de vidrio (figura 2).
Figura 21. 2. Formación de la gota en la bureta La parte superior de la gota en formación, corresponde con un cuello cilíndrico, mecánicamente poco estable. Debido a la existencia de este cuello, se observa que sólo se desprende de la gota en formación una pequeña porción, pudiendo quedar hasta un 40% del líquido adherido al capilar para entrar a formar parte de la siguiente gota. Para compensar este efecto se incorpora la función ф. Su valor depende de la relación entre el
radio externo del capilar y la raíz cúbica del volumen real de la gota desprendida (𝑟/𝑉1/3) y sus valores pueden calcularse por la expresión empírica de Harkins y Brown (apéndice 1). Tensioactivos

127
Se dice que una sustancia es un agente tensioactivo o surfactante cuando da lugar a un descenso significativo en la tensión superficial de un líquido. Los tensioactivos más efectivos (jabones, detergentes y colorantes), presentan en su estructura molecular, una parte hidrofílica y otra hidrófoba Generalmente, la parte hidrofílica es un grupo iónico, ya que los iones suelen presentar una fuerte afinidad por el agua, motivada por su atracción electrostática hacia sus dipolos. La parte hidrófoba suele consistir en una larga cadena hidrocarbonada, una estructura muy parecida a la de las grasas y parafinas, hacia las que experimenta atracción. Se clasifican en aniónicos, catiónicos y no iónicos según la carga de la parte hidrofílica. En la siguiente tabla se pueden ver algunos ejemplos de las tres clases.
Anionicos Estreato sódico CH3(CH2)16COO Oleato sódico CH3(CH2)7CH=CH(CH2)7COO- Dodecilsilfato sódico CH3(CH2)11SO4
- Dodecilbencenosulfonato sódico CH3(CH2)11C6H4·SO4
-
Cationicos Clorhidrato de laurilamina CH3(CH2)11NH3
+ No ionicos Óxidos de politileno, como CH3(CH2)7C6H4(OCH2CH2)8OH
La disminución de la tensión superficial de un líquido implica que la fuerza con la que cada molécula de la superficie es atraída hacia el interior, también disminuye. Por tanto el trabajo necesario para incrementar la superficie de la gota también lo hace, reduciendo así la capacidad de formar gotas esféricas (volumen que presenta superficie mínima) y aumentando la capacidad de extensión, es decir, de mojado. 21.2 MATERIAL Y REACTIVOS MATERIAL 2 Vasos de precipitados de 100 ml. 2 Vasos de precipitados de 250 ml. 3 Matraces Erlenmeyer de 100 ml. 1 Matraz aforado de 100 ml. 1 Picnómetro. 2 Buretas de 25 ml. 1 Probeta de 100 ml 1Varilla de vidrio. 1 Jeringuilla. 1 Aguja de jeringuilla. REACTIVOS

128
Detergente comercial. Etanol. NaCl. Agua purificada. Dodecilbencenosulfonato sódico 21.3 PROCEDIMIENTO EXPERIMENTAL Proceso experimental Una simple bureta, con una punta lo más fina posible, es un instrumento adecuado para una primera aproximación a la determinación de tensiones superficiales. La punta debe estar completamente limpia. Para la obtención de una adecuada precisión, el sistema debe estar exento de vibraciones. La primera gota debe formarse lentamente y despreciarse ésta y las siguientes, hasta obtener un régimen estacionario de caída. Metodología del proceso Calibración de las buretas con agua purificada. Cálculo del radio estimado de cada bureta. Las operaciones que aquí se describen deben realizarse con ambas buretas.
Enrase la bureta con agua destilada. Abra la llave de manera que caiga su contenido, gota a gota, a razón de unas cuatro por minuto y deseche las 10 primeras que caerán sobre uno de los vasos de precipitado.
Utilice un matraz erlenmeyer, previamente tarado, para contener las siguientes 50 gotas.
Vuelva a pesar y calcule el peso de las 50 gotas de agua por diferencia con el peso del matraz vacío. Anótelo en la fila correspondiente a la muestra 1 de la tabla 5.1, (si se trata de la bureta 1), o de la tabla 5.2, (si se trata de la bureta 2).
Seque perfectamente el matraz y repita la experiencia. Anote la masa de las gotas en la fila correspondiente a la muestra 2 de la tabla 5.1, (si se trata de la bureta 1), o de la tabla 5.2, (si se trata de la bureta 2).
Calcule la densidad del agua con la siguiente expresión,
𝑑𝐴𝑔𝑢𝑎 = (30.0658 − 7.48𝑥10−3 ∗ 𝑇)/30 (21.2)
𝑑𝐴𝑔𝑢𝑎, es la densidad del agua expresada en 𝑔/𝑐𝑚3
T, es la temperatura en grados centígrados

129
Calcule el volumen medio de la gota en cada bureta y anótelo en las tablas 5.1 y 5.2. (Estos volúmenes no tienen porqué coincidir). Utilizando la expresión (1) y el valor de tensión superficial que se proporciona en la siguiente tabla, calcule el valor del radio de la circunferencia de contacto líquido-vidrio (r) que se empleará en las próximas medidas. Tome ф =1 como factor corrector.
tensiones superficiales del agua a distintas temperaturas
𝛾(𝑚𝑁/𝑚) Temperatura (°C) 𝛾(𝑚𝑁/𝑚) Temperatura (°C)
73.05 18 72.28 23
72.90 19 72.13 24
72.75 20 71.97 25
72.59 21 71.82 26
72.44 22 71.66 27
Anote los resultados de r Bureta (1) y r Bureta (2) en la tablas 5.1 y 5.2, respectivamente Determinación de la tensión superficial del etanol
Calcule la densidad del etanol utilizando el picnómetro como se explica en el apéndice 2. Anote el resultado en la tabla 5.3.
Opere del mismo modo que en el proceso de calibración pero, en este caso, con la bureta enrasada con etanol. Anote las masas de las 50 gotas en la tabla 5.3.
Realice la determinación dos veces (muestra 1 y muestra 2) con la misma bureta y lleve a cabo los cálculos necesarios para rellenar la tabla 5.3, excepto los valores
de 𝑟𝐵𝑢𝑟𝑒𝑡𝑎( )/�̅�)1/3, ф 𝑦 𝛾 .
Anote en esta tabla la bureta empleada, incluyendo el número correspondiente en el paréntesis de la variable, 𝑟𝐵𝑢𝑟𝑒𝑡𝑎( ) .
Determinación de la tensión superficial de una disolución de 100 ml de NaCl 1M.
Calcule la densidad de la disolución utilizando el picnómetro como se explica en el apéndice 2. Anote el resultado en la tabla 5.4.
Prepare 100 ml de una disolución 1M de NaCl y opere de la misma manera que en el caso anterior. Anote las masas de las 50 gotas en la tabla 5.4.
Realice la determinación dos veces (muestra 1 y muestra 2) con la misma bureta y lleve a cabo los cálculos necesarios para rellenar la tabla 5.4 excepto los valores de
𝑟𝐵𝑢𝑟𝑒𝑡𝑎( )/�̅�)1/3, ф 𝑦 𝛾.
Anote en esta tabla la bureta empleada, incluyendo el número correspondiente en el paréntesis de la variable, 𝑟𝐵𝑢𝑟𝑒𝑡𝑎( ).

130
Determinación de la tensión superficial del agua con detergente comercial.
Calcule la densidad de la disolución utilizando el picnómetro como se explica en el apéndice 2.
Pese 1 g de detergente en un vaso de 250 ml. Añada 100 ml de agua medidos en la probeta y homogenice por agitación con una varilla de vidrio.
Opere de la misma manera que en el caso anterior y anote las masas de las 50 gotas en la tabla 5.5.
Realice la determinación dos veces (muestra 1 y muestra 2) con la misma bureta y lleve a cabo los cálculos necesarios para rellenar la tabla 5.5 excepto los valores de
𝑟𝐵𝑢𝑟𝑒𝑡𝑎( )/�̅�)1/3, ф 𝑦 𝛾.
Anote en esta tabla la bureta empleada, incluyendo el número correspondiente en el paréntesis de la variable, 𝑟𝐵𝑢𝑟𝑒𝑡𝑎( ).
Calcule la densidad de la disolución utilizando el picnómetro como se explica en el apéndice 2.
Pese 1 g de disolución de DSS. Añada 100 ml de agua medidos en la probeta y homogenice por agitación con una varilla de vidrio.
Opere de la misma manera que en el caso anterior y anote las masas de las 50 gotas en la tabla5.6.
Realice la determinación dos veces (muestra 1 y muestra 2) con la misma bureta y lleve a cabo los cálculos necesarios para rellenar la tabla 5.6 excepto los valores
de𝑟𝐵𝑢𝑟𝑒𝑡𝑎( )/�̅�)1/3, ф 𝑦 𝛾
Anote en esta tabla la bureta empleada, incluyendo el número correspondiente en el paréntesis de la variable, 𝑟𝐵𝑢𝑟𝑒𝑡𝑎( ).
21.3 Cálculos.
Realice las operaciones necesarias para calcular para cada una de las 𝑟𝐵𝑢𝑟𝑒𝑡𝑎( )/�̅�)1/3, ф 𝑦 𝛾
determinaciones y anótelas en las tablas correspondientes, expresando los datos de
tensión superficial en 𝑚𝑁
𝑚.
APENDICE 1 FUNCION DE AJUSTE DE LA ECUACIÓN DE HARHINS Y BROWN
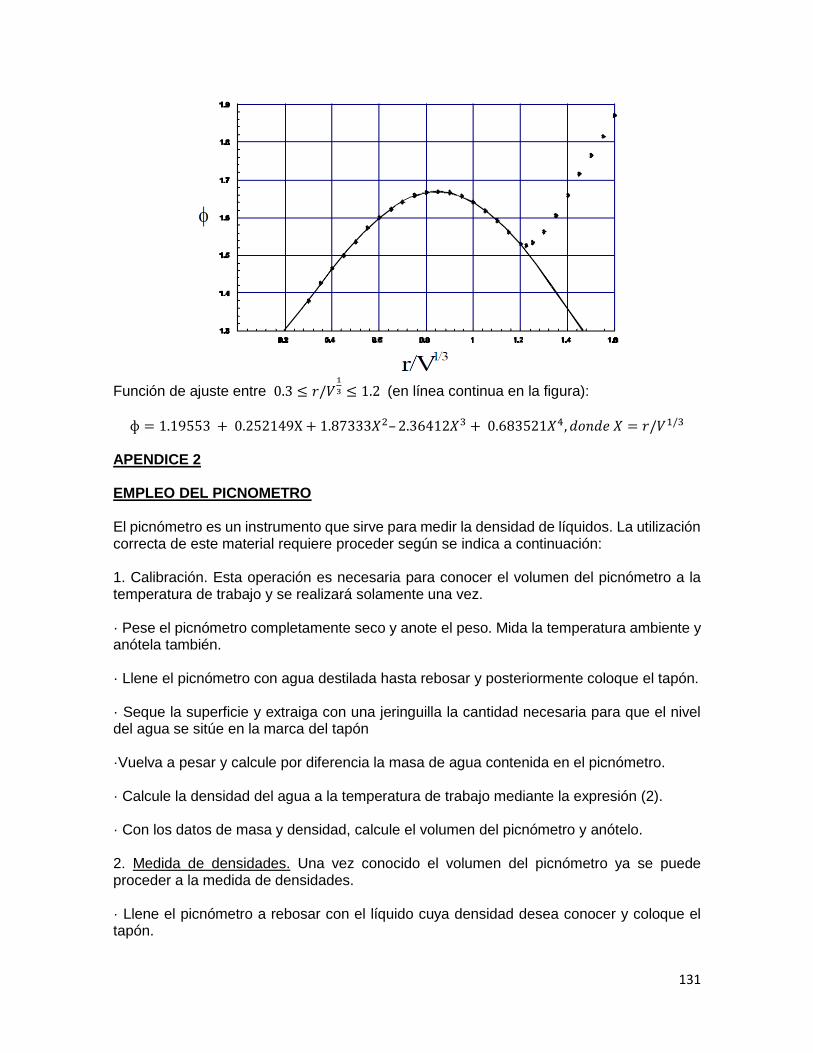
131
Función de ajuste entre 0.3 ≤ 𝑟/𝑉1
3 ≤ 1.2 (en línea continua en la figura):
ф = 1.19553 + 0.252149X + 1.87333𝑋2–2.36412𝑋3 + 0.683521𝑋4, 𝑑𝑜𝑛𝑑𝑒 𝑋 = 𝑟/𝑉1/3 APENDICE 2 EMPLEO DEL PICNOMETRO El picnómetro es un instrumento que sirve para medir la densidad de líquidos. La utilización correcta de este material requiere proceder según se indica a continuación: 1. Calibración. Esta operación es necesaria para conocer el volumen del picnómetro a la temperatura de trabajo y se realizará solamente una vez. · Pese el picnómetro completamente seco y anote el peso. Mida la temperatura ambiente y anótela también. · Llene el picnómetro con agua destilada hasta rebosar y posteriormente coloque el tapón. · Seque la superficie y extraiga con una jeringuilla la cantidad necesaria para que el nivel del agua se sitúe en la marca del tapón ·Vuelva a pesar y calcule por diferencia la masa de agua contenida en el picnómetro. · Calcule la densidad del agua a la temperatura de trabajo mediante la expresión (2). · Con los datos de masa y densidad, calcule el volumen del picnómetro y anótelo. 2. Medida de densidades. Una vez conocido el volumen del picnómetro ya se puede proceder a la medida de densidades. · Llene el picnómetro a rebosar con el líquido cuya densidad desea conocer y coloque el tapón.

132
· Seque la superficie y extraiga con una jeringuilla la cantidad necesaria para que el nivel se sitúen la marca del tapón. · Pese y calcule por diferencia la masa de líquido contenido en el picnómetro. · Finalmente calcule la densidad dividiendo la masa de líquido entre el volumen del picnómetro. 21.4 RESULTADOS EXPERIMENTALES
5.1.- Calibración de la bureta 1
Numero de gotas
Masa de las gotas
Masa de una gota
Masa media de una gota
Densidad del agua
Volumen medio de la gota
Muestra 1
Muestra 2
𝑟𝐵𝑢𝑟𝑒𝑡𝑎(1 ) =
5.2.- Calibración de la bureta 2
Numero de gotas
Masa de las gotas
Masa de una gota
Masa media de una gota
Densidad del agua
Volumen medio de la gota
Muestra 1
Muestra 2
𝑟𝐵𝑢𝑟𝑒𝑡𝑎(2 ) =
5.3.-Tension superficial del etanol
Numero de gotas
Masa de las gotas
Masa de una gota
Masa media de una gota
Densidad del agua
Volumen medio de la gota
Muestra 1
Muestra 2
𝑟𝐵𝑢𝑟𝑒𝑡𝑎(2 )/�̅�)1/3 = ф 𝐸𝑡𝑎𝑛𝑜𝑙 =
𝛄𝐃𝐢𝐬𝐨𝐥𝐮𝐜𝐢ó𝐧
Tensión superficial de una disolución con detergente comercial
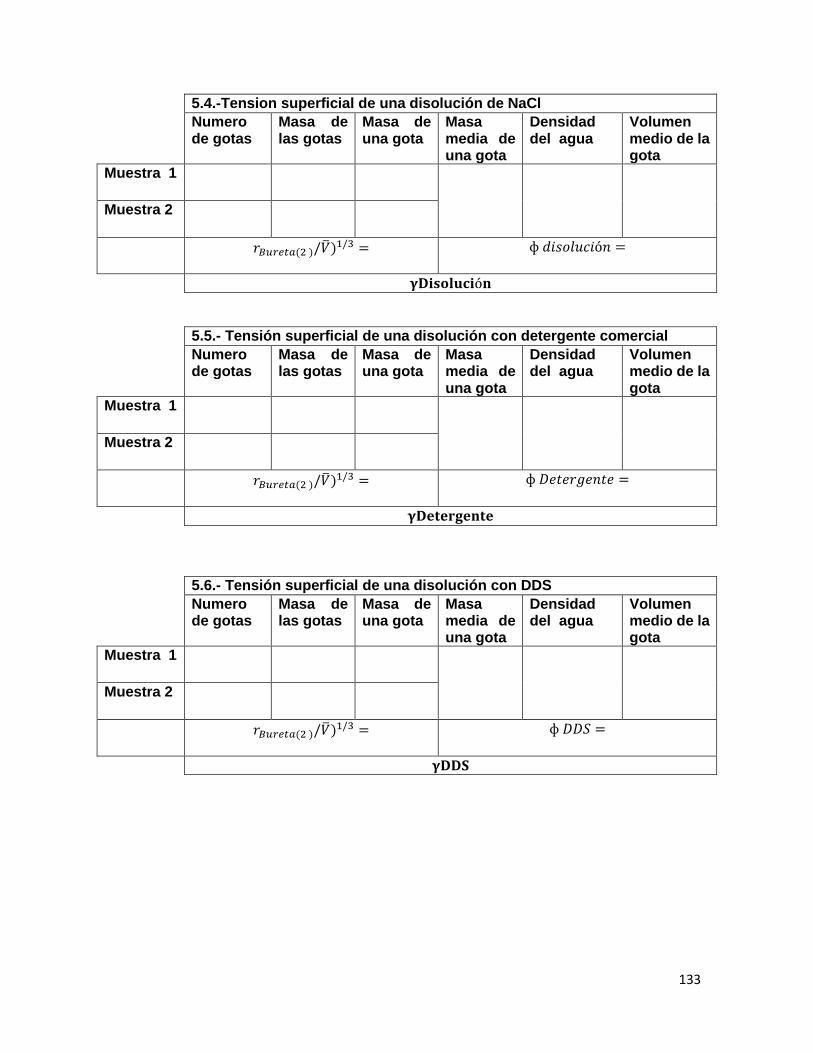
133
5.4.-Tension superficial de una disolución de NaCl
Numero de gotas
Masa de las gotas
Masa de una gota
Masa media de una gota
Densidad del agua
Volumen medio de la gota
Muestra 1
Muestra 2
𝑟𝐵𝑢𝑟𝑒𝑡𝑎(2 )/�̅�)1/3 = ф 𝑑𝑖𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 =
𝛄𝐃𝐢𝐬𝐨𝐥𝐮𝐜𝐢ó𝐧
5.5.- Tensión superficial de una disolución con detergente comercial
Numero de gotas
Masa de las gotas
Masa de una gota
Masa media de una gota
Densidad del agua
Volumen medio de la gota
Muestra 1
Muestra 2
𝑟𝐵𝑢𝑟𝑒𝑡𝑎(2 )/�̅�)1/3 = ф 𝐷𝑒𝑡𝑒𝑟𝑔𝑒𝑛𝑡𝑒 =
𝛄𝐃𝐞𝐭𝐞𝐫𝐠𝐞𝐧𝐭𝐞
5.6.- Tensión superficial de una disolución con DDS
Numero de gotas
Masa de las gotas
Masa de una gota
Masa media de una gota
Densidad del agua
Volumen medio de la gota
Muestra 1
Muestra 2
𝑟𝐵𝑢𝑟𝑒𝑡𝑎(2 )/�̅�)1/3 = ф 𝐷𝐷𝑆 =
𝛄𝐃𝐃𝐒

134
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 22: PROPIEDADES DE LOS COLOIDES: DOSIS
DE COAGULANTE EN TRATAMIENTO DE AGUAS.
Objetivos.
1. Determinar qué tipo de coagulantes son más eficaces para el agua utilizada. 2. Establecer criterios adecuados para los procesos de coagulación, floculación y
sedimentación de un agua problema. 3. Realizar ensayos a escala de laboratorio con coagulantes como Sulfato de Aluminio
tipo B, Ultrión 8157, Polisulfato de Aluminio, Policloruro de Aluminio, Hidroxicloruro de Aluminio, para determinar cuál de ellos presenta mejores resultados en el proceso de Coagulación-Floculación.
4. Analizar los fenómenos superficiales que conducen a resultados óptimos de coagulación, floculación y sedimentación.
Introducción

135
Coagulación Definición La coagulación puede entenderse como la desestabilización eléctrica de algunas partículas media te la adición de sustancia químicas que son los coagulantes. Esta operación se efectúa en unidades y tanques de mezcla rápida, en los cuales el agua se somete a agitación muy intensa para formar una solución homogénea de los coagulantes con el agua en el menor tiempo posible. Este proceso se usa para:
Remoción de turbiedad orgánica o inorgánica que no se puede sedimentar rápidamente.
Remoción de color verdadero y aparente. Eliminación de bacteria, virus y organismos patógenos susceptibles de ser
separados por coagulación.
Destrucción de algas y plancton en general. Eliminación de sustancias productoras de sabor y olor, en algunos casos de
precipitados químicos suspendidos en otros.
El uso de cualquier otro proceso para la remoción de partículas muy finas, como la sedimentación simple, resulta muy poco económico y en ocasiones imposible, debido al alto tiempo requerido. Para la evaluación de este proceso es necesario tener en cuenta las características físicas y químicas del agua, la dosis del coagulante, la concentración del coagulante, el punto de aplicación del coagulante, la intensidad y el tiempo de mezcla y el tipo de dispositivo de mezcla. Teoría de la Coagulación
Las partículas que forman la turbiedad y el color de las aguas naturales, poseen cargas eléctricas que normalmente son negativas, pero como también existen cargas eléctricas positivas, se puede afirmar que el agua y las soluciones son eléctricamente neutras. Las cargas eléctricas de las partículas generan fuerzas de repulsión entre ellas, por lo cual se mantienen suspendidas y separadas en el agua. Es por esto que dichas partículas no se sedimentan. El conjunto formado por estas partículas constituye un sistema coloidal, formado por una doble capa de iones1, el cual es sometido a un potencial en la superficie inferior del doble lecho, denominado potencial Z. Este potencial tiene un valor crítico, por encima del cual los coloides son estables, y por debajo de él, la repulsión en las partículas se reduce a un grado tal que chocando con cierta velocidad pueden unirse y flocular. El problema en la coagulación consiste en disminuir el potencial Z por uno de los siguientes métodos:
Coagulación por neutralización de la carga: Esta se realiza cuando coloides de diferente signo se mezclan en el agua. Esto es lo que sucede cuando se agrega alumbre o sales de hierro al agua.
Coagulación por disminución del espesor de la doble capa (distancia d). Al incrementarse la concentración de iones en el agua la “distancia d” disminuye, hasta hacer el valor del potencial Z inferior al punto crítico.

136
El fenómeno de la desestabilización se efectúa mediante una serie de reacciones químicas bastante complejas, de las cuales algunas no se han podido entender lo suficiente. Dentro de esas reacciones se encuentran las que se efectúan con las diversas formas de alcalinidad, por lo cual su contenido disminuye. Además, algunas de estas reacciones producen CO2, cuyo efecto consiste fundamentalmente en el incremento de la acidez del agua y por consiguiente la disminución del pH. Factores que influyen en la Coagulación
Valencia: Entre mayor sea la valencia del ion, más efectivo resulta como coagulante.
Capacidad de cambio: Es una medida de la tendencia a remplazar cationes de baja valencia por otros de mayor valencia, provocando la desestabilización y aglomeración de partículas en forma muy rápida. 1 Modelo de Stern, 1924
Tamaño de las partículas: Las partículas deben poseer el diámetro inferior a una
micra. Las partículas con diámetro entre una y cinco micras, sirven como núcleos de floc, en cambio de diámetro superior a cinco micras, son demasiado grandes para ser incorporadas en el floc.
Temperatura: La temperatura cambia el tiempo de formación del floc, entre más fría el agua, la reacción es más lenta y el tiempo de formación del floc es mayor.
Concentración de iones H+ o pH: Para cada coagulante hay por lo menos una zona de pH óptima, en la cual una buena floculación ocurre en el tiempo más corto y con la misma dosis de coagulante.
Relación cantidad-tiempo: La cantidad de coagulante es inversamente
proporcional al tiempo de formación del floc.
Alcalinidad: La alcalinidad guarda la relación con el pH y por lo tanto el contenido de alcalinidad del agua es uno de los factores por considerar en la coagulación.
Clases de Coagulantes Los coagulantes que se utilizan en la práctica para agua potable son los siguientes:
Sales de Aluminio: Forman un floc ligeramente pesado. Las más conocidas son: El Sulfato de Aluminio, Al2(SO3) *14H2O, que en la práctica se le denomina como Alumbre; el Sulfato de Aluminio Amoniacal y el Aluminato Sódico. El primero es el que se usa con mayor frecuencia dado su bajo costo y manejo relativamente sencillo.
Sales de Hierro: Se utiliza el Cloruro Férrico, FeCl3, y los Sulfatos de Hierro Férrico y Ferroso, Fe(SO4)3 y FeSO4. Forman un floc más pesado y de mayor velocidad de asentamiento que las sales de aluminio.
Polímeros o polielectrolitos: Son compuestos complejos de alto peso molecular que se utilizan no propiamente como coagulantes sino como ayudantes de coagulación. La dosificación de estas sustancias se lleva a cabo en concentraciones muy bajas, lo cual es una gran ventaja y compensa el costo del polímero. Están

137
siendo ampliamente empleados en el tratamiento de aguas potables ya que se produce una menor cantidad de lodos, adicionalmente el lodo producido es más fácilmente tratable.
Floculación La floculación consiste en la aglomeración, mediante la agitación moderada del agua, de las partículas que se desestabilizaron durante la coagulación, formando otras de mayor tamaño y peso específico –flóculos. Los objetivos básicos de la floculación son reunir microflóculos para formar partículas con peso específico superior al del agua y compactar el flóculo disminuyendo su grado de hidratación para producir baja concentración volumétrica, lo cual produce una alta eficiencia en los procesos posteriores como sedimentación y filtración. Cinética de la Floculación Tan pronto como se agregan coagulantes a una suspensión coloidal, se inician una serie de reacciones hidrolíticas que adhieren iones a la superficie de las partículas presentes en la suspensión, las cuales tienen así oportunidad de unirse por sucesivas colisiones hasta formar flóculos que crecen con el tiempo. La rapidez con que esto ocurre depende del tamaño de las partículas con relación al estado de agitación del líquido, de la concentración de las mismas y de su “grado de desestabilización”, que es el que permite que las colisiones sean efectivas para producir adherencia. Los contactos pueden realizarse por dos modos distintos:
Floculación Pericinética: Contactos por bombardeo de las partículas producidos por el movimiento de las moléculas del líquido (movimiento browniano) que sólo influye en partículas de tamaños menores a un micrón. Sólo actúa al comienzo del proceso, en los primeros 6 a 10 s y es independiente del tamaño de la partícula.
Floculación Ortocinética: Contactos por turbulencia del líquido, esta turbulencia causa el movimiento de las partículas a diferentes velocidades y direcciones, lo cual aumenta notablemente la probabilidad de colisión. Efectivo sólo con partículas mayores a un micrón. Actúa durante el resto del proceso, de 20 a 30 min.
Factores que influyen en la Floculación
Concentración y naturaleza de las partículas: La velocidad de formación del floc es proporcional a la concentración de partículas en el agua y del tamaño inicial de estas.
Tiempo de detención: La velocidad de aglomeración de las partículas es proporcional al tiempo de detención. Debe estar lo más cerca posible al óptimo determinado por medio de ensayos de jarras, esto se puede lograr dividiendo la unidad de floculación en cámaras. Se puede decir que una eficiencia dada, se obtiene en tiempos cada vez menores a medida que se aumenta el número de cámaras de floculación en serie. Por razones de orden práctico el número de cámaras no puede ser muy grande, estableciéndose un mínimo de tres (3) unidades.

138
Gradiente de velocidad: Este es un factor proporcional a la velocidad de aglomeración de las partículas. Existe un límite máximo de gradiente que no puede ser sobrepasado, para evitar el rompimiento del floc. El gradiente a través de las cámaras debe ser decreciente y no se deben tener cámaras intermedias con gradientes elevados.
Prueba de Jarras
La coagulación química y la dosificación apropiada de reactivos deben ser seleccionadas por la simulación del paso de clarificación en un laboratorio a escala. La Prueba de Jarras es la que mejor simula la química de la clarificación y la operación llevada a cabo. Un arreglo simple de vasos de precipitado y paletas permite comparar varias combinaciones químicas, las cuales todas están sujetas a condiciones hidráulicas similares. Esta prueba se realiza con el fin de determinar la concentración óptima de coagulante necesaria para obtener un floc de las mejores características. Metodología Dosis óptima
Procedimiento:
1. Determinar la temperatura del agua cruda, la turbiedad, el pH, y la alcalinidad. 2. Medir las cantidades de coagulantes (Sulfato de aluminio 1% para dosis de 5, 10,
15, 20, 30 y 40 ppm. 3. Hacer girar las paletas del equipo a 100 rpm e inyectar el coagulante utilizando
jeringas hipodérmicas. 4. Mantener a esta velocidad por 1 minuto (Mezcla rápida). 5. Disminuir la velocidad de 40 rpm y mantenerlo por 20 minutos (mezcla lenta). 6. Luego suspender la agitación, retirar las jarras, colocar los sifones para la toma de
muestra y dejar sedimentar el agua por 10 minutos. 7. Tomar las muestras descartando los primeros 10 ml y proceder a determinar la
turbiedad.
RESULTADOS
Turbiedad inicial : Alcalinidad total : Temperatura : pH : Volumen de jarras : MEZCLA RÁPIDA Tiempo : Velocidad : Gradiente : MEZCLA LENTA : Tiempo :

139
Velocidad : Gradiente : SEDIMENTACIÓN Tiempo :
pH óptimo
Procedimiento:
1. Ajustar el pH del agua cruda para los valores de: 4, 5, 6, 7, 8 y 9. 2. Efectuar la prueba de jarras en forma convencional. Mezcla rápida, floculación y
decantación. 3. Determinar la turbiedad residual vs pH. El pH óptimo será aquel con el que se obtiene
la máxima remoción de la turbiedad.
Concentración óptima de coagulante
Procedimiento:
1. Con la dosis óptima y el pH óptimo previamente determinados, realizar la prueba de jarras en forma convencional.
2. En esta prueba la dosificación se realiza a diferentes concentraciones del coagulante (sulfato de aluminio): 0.5, 1, 2, 3, 4, y 5%.
3. Se grafican las turbiedades residuales vs concentración de coagulante y se determina la concentración optima como aquella que produce la menor turbiedad residual.
Parámetros de floculación Procedimiento:
1. Con las variables previamente determinadas efectuar la mezcla rápida. 2. Al finalizar la mezcla rápida se inicia el proceso de floculación, para lo cual se
modifica la velocidad de acuerdo a los gradientes: 20, 40, 60, y 80. 3. Luego de floculada el agua por 5 minutos, se retira el primer vaso, se coloca el
tomador de muestra se deja sedimentar por 10 minutos. 4. Los siguientes vasos (2, 3, 4, 5, 6,) se retiran a los tiempos 10´, 15´, 20´, 25´ y 30´)
minutos respectivamente. 5. Se determina la turbiedad residual para cada muestra. 6. Se repite en ensayo incrementando el gradiente de acuerdo al intervalo
seleccionado.
Parámetros de decantación.
Procedimiento:
1. Efectuar la prueba de jarras convencional. Mezcla rápida por 1 minuto a 100 rpm, floculación por 20 minutos y 40 rpm. Trabajar solo con tres vasos.
2. Terminada la floculación detener el equipo y colocar los flotadores para tomar las muestras.

140
3. Tomar muestras a 6 cm de profundidad, y a los tiempos de 30¨, 1´, 2´, 3´, 4´, 5´, y 10´.
4. Determinar la turbiedad residual (Tf) a cada una de las muestras. 5. Calcular los valores de Vs=h/T. 6. Graficar los valores de 𝐶0 = 𝑇𝑓/𝑇0 vs 𝑣 para obtener la curva de sedimentación
para el agua estudiada. 7. Para diferentes cargas superficiales determinar: el porcentaje total de remoción
(𝑅𝑡), la turbiedad removida 𝑇𝑟 y la turbiedad final o remanente 𝑇𝑓.
𝑅𝑡 = [1 − (𝐶0 − 𝐶𝑓)] + ((𝑎 + 𝑉𝑠2𝑉𝑠
) (𝐶0 − 𝐶𝑓)
𝑇𝑟 = 𝑅𝑡 ∗ 𝑇0 , 𝑇𝑓 = 𝑇0 − 𝑇𝑟
8. La carga superficial a de decantador será aquella que corresponda al valor de
turbiedad final deseado en el afluente de la unidad.
Parámetros de filtración directa
Procedimiento: Primera prueba. Dosis de pH optimo
1. Efectuar la prueba de jarras variando el pH para los valores de: 6.0, 6.5, 7.0 y 7.33. 2. Seleccionar las dosis óptimas de sulfato de aluminio para cada valor de pH. 3. En cada ensayo proceder del siguiente modo:
-Graduar la muestra al pH seleccionado para la prueba. -preparar los embudos con el papel Whatman N° 40. -Llenar las jarras con las muestras de agua y colocar los estatores. -encender el equipo y graduar a 100 rpm. -agregar simultáneamente las dosis al tiempo. “0”. -mezclar durante 1 minuto y apagar el equipo. -tomar las muestras simultáneamente, trasegando suavemente de las jarras a los embudos. -obtener una muestra de 50 ml y determinar la turbiedad remanente y el pH.
RESULTADOS
Turbiedad inicial : Alcalinidad inicial : Temperatura : pH : : MEZCLA RÁPIDA Tiempo : Velocidad : Gradiente :
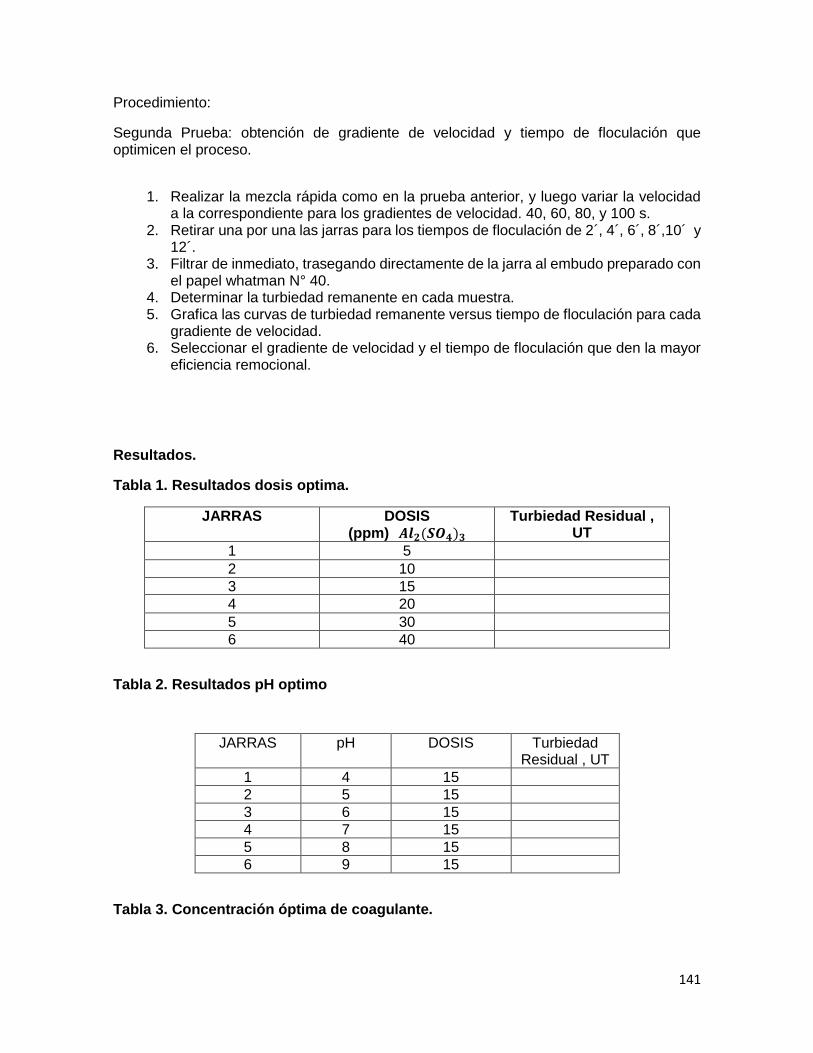
141
Procedimiento:
Segunda Prueba: obtención de gradiente de velocidad y tiempo de floculación que optimicen el proceso.
1. Realizar la mezcla rápida como en la prueba anterior, y luego variar la velocidad
a la correspondiente para los gradientes de velocidad. 40, 60, 80, y 100 s. 2. Retirar una por una las jarras para los tiempos de floculación de 2´, 4´, 6´, 8´,10´ y
12´. 3. Filtrar de inmediato, trasegando directamente de la jarra al embudo preparado con
el papel whatman N° 40. 4. Determinar la turbiedad remanente en cada muestra. 5. Grafica las curvas de turbiedad remanente versus tiempo de floculación para cada
gradiente de velocidad. 6. Seleccionar el gradiente de velocidad y el tiempo de floculación que den la mayor
eficiencia remocional.
Resultados.
Tabla 1. Resultados dosis optima.
JARRAS DOSIS (ppm) 𝑨𝒍𝟐(𝑺𝑶𝟒)𝟑
Turbiedad Residual , UT
1 5
2 10
3 15
4 20
5 30
6 40
Tabla 2. Resultados pH optimo
JARRAS pH DOSIS Turbiedad Residual , UT
1 4 15
2 5 15
3 6 15
4 7 15
5 8 15
6 9 15
Tabla 3. Concentración óptima de coagulante.
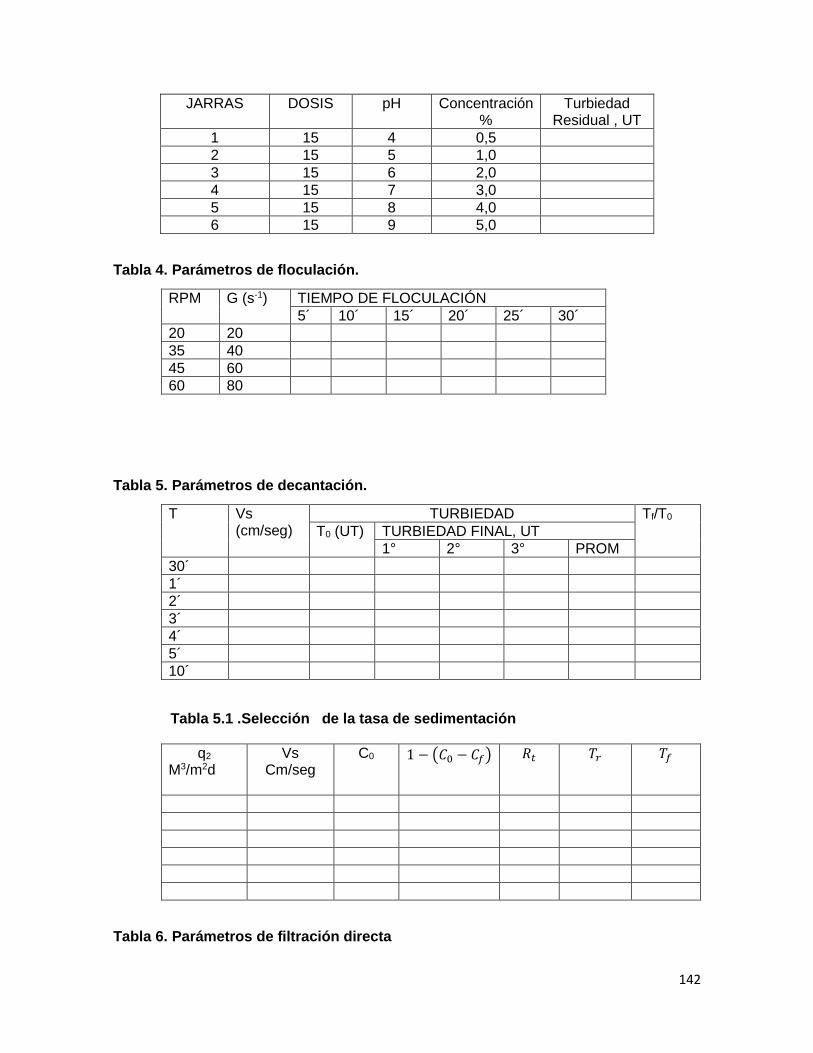
142
JARRAS DOSIS pH Concentración %
Turbiedad Residual , UT
1 15 4 0,5
2 15 5 1,0
3 15 6 2,0
4 15 7 3,0
5 15 8 4,0
6 15 9 5,0
Tabla 4. Parámetros de floculación.
RPM G (s-1) TIEMPO DE FLOCULACIÓN
5´ 10´ 15´ 20´ 25´ 30´
20 20
35 40
45 60
60 80
Tabla 5. Parámetros de decantación.
T Vs (cm/seg)
TURBIEDAD Tf/T0
T0 (UT) TURBIEDAD FINAL, UT
1° 2° 3° PROM
30´
1´
2´
3´
4´
5´
10´
Tabla 5.1 .Selección de la tasa de sedimentación
q2 M3/m2d
Vs Cm/seg
C0 1 − (𝐶0 − 𝐶𝑓) 𝑅𝑡 𝑇𝑟 𝑇𝑓
Tabla 6. Parámetros de filtración directa
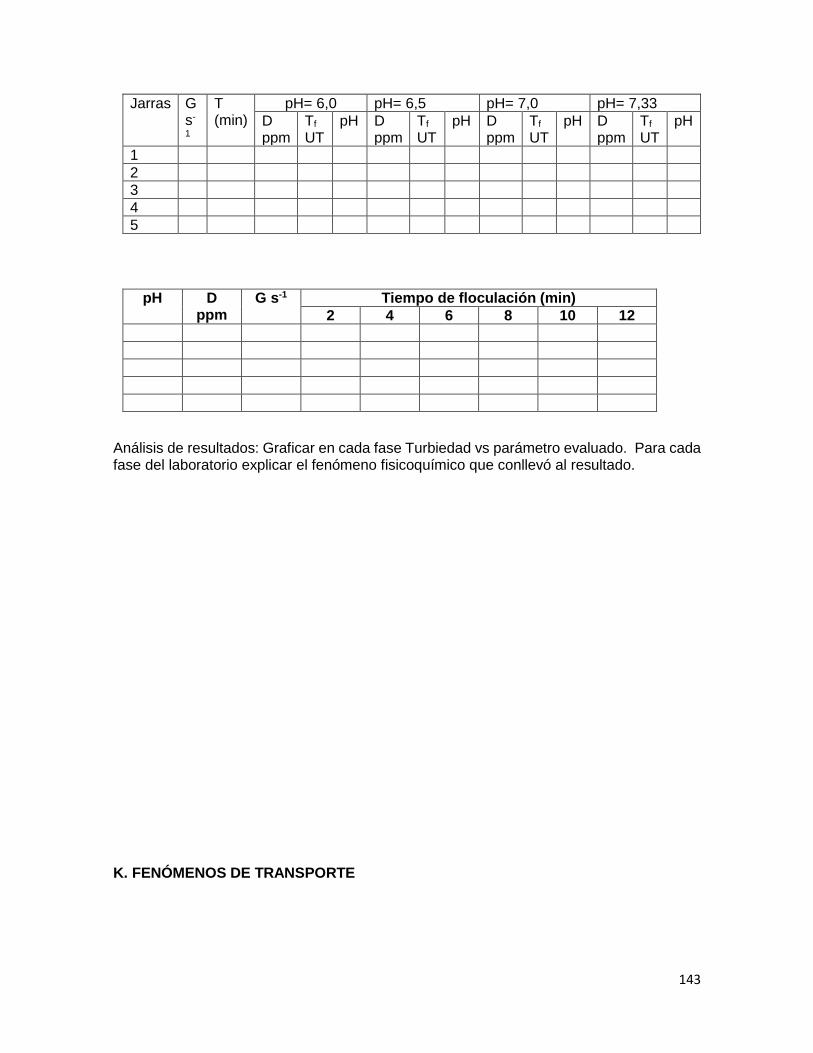
143
Jarras G s-
1
T (min)
pH= 6,0 pH= 6,5 pH= 7,0 pH= 7,33
D ppm
Tf UT
pH D ppm
Tf UT
pH D ppm
Tf UT
pH D ppm
Tf UT
pH
1
2
3
4
5
pH D ppm
G s-1 Tiempo de floculación (min)
2 4 6 8 10 12
Análisis de resultados: Graficar en cada fase Turbiedad vs parámetro evaluado. Para cada fase del laboratorio explicar el fenómeno fisicoquímico que conllevó al resultado.
K. FENÓMENOS DE TRANSPORTE

144
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO NO. 23. LEY DE STOKES Y SEDIMENTACIÓN
OBJETIVOS
Una muestra real de un sólido finamente dividido está compuesta de diferentes radios y geometrías. El siguiente desarrollo experimental permite comprender como se combina la ley de Stokes con métodos para hallar la distribución de partículas según el radio.
23.1 Fundamentos
George Gabriel Stokes fue un matemático irlandés que paso gran parte de su vida trabajando con las propiedades de los fluidos. En 1856 los resultados de su extensivo estudio sobre la resistencia al movimiento de una partícula esférica en u medio viscoso debido a fuerzas de fricción de partícula esférica redirija de radio r no hidratada, que se mueve en un fluido de viscosidad n a una velocidad de 𝑑𝑥/𝑑𝑡, la fuerza de resistencia
friccional (𝐹𝑓) esta dad por
𝐹𝑗 = 6𝜋ɳ𝑟𝑑𝑥
𝑑𝑡
(23.1)

145
Esta relación es conocida como la ley de Stokes y se aplica cuando las partículas son mucho mas grandes que las moléculas que componen el fluido y su concentración es lo suficiente baja como para afectar la viscosidad del fluido.
La ecuación de Stokes fue refinada posteriormente por Gibson y Jacobs en 1920 para tener en cuenta el llamado ̈ efecto paredes¨ del recipiente donde está contenido el fluido y “efecto final” cuando el movimiento termina, generalmente en el fondo del recipiente. Estos efectos san como resultado una velocidad de movimiento menor de la esperada. La corrección por efectos de las paredes tiene encuentra la comprensión del fluido contra los lados del recipiente, debido al movimiento de las partículas esféricas a través de este. Esta corrección es mayor en la medida que la relación entre el radio de las partículas y el radio interno del recipiente sea mayor.
La corrección por el efecto final tiene en cuenta el hecho que las partículas al sedimentarse no se mueven indefinidamente. Este efecto es mayor en la medida que la relación entre el radio de la partícula esférica y la altura total del recipiente sea mayor. La corrección por este efecto generalmente es mucho menos que la correlación por el efecto de las paredes.
23.1.1 Fuerza boyante
El matemático griego Arquímedes (287-212 AC) mostró que el peso de un objeto suspendido en un líquido disminuye en la misma proporción que el peso del líquido desplazado por el objeto sólido. Esta ley se conoce como el principio de Arquímedes. Cuando un objeto es sumergido en un líquido se dice que sobre el actúa una fuerza boyante:
𝐹𝑏 = 𝑉𝑙𝜌𝑙𝑔 (23.2)
Donde 𝑉𝑙 es el volumen y d la densidad de la partícula. Si la partícula esta completamente
sumergirse el sólido y d es la densidad del líquido. La fuerza total (𝐹𝑡) que actúa sobre una partícula esférica sumergida en un fluido será igual a la fuerza de la gravedad (𝐹𝑔) menos
la fuerza boyante:
𝐹𝑡 = 𝐹𝑔 − 𝐹𝑏 = 𝑉𝑠𝜌𝑠𝑔 − 𝑉𝑙𝜌𝑔 (23.3)
Donde 𝑉𝑠 es le volumen y 𝜌𝑠 la densidad de la partícula. Si la partícula está completamente sumergida en le liquido 𝑉𝑙 = 𝑉𝑠 y su volumen es el doble de una esfera la ecuación 3 se transforma en:
𝐹𝑡 = 𝑉𝑔(𝜌𝑠 − 𝜌𝑙) =4𝜋𝑟2
3 (𝜌𝑠 − 𝜌𝑙)𝑔
(23.4)
Durante el proceso de sedimentación las partículas se aceleran debido a Ft hasta que se llega a una velocidad límite donde esta fuerza es contrarrestada por la fuerza de fricción
𝐹𝑡 = 𝐹𝑓 , 𝑜 4𝜋𝑟2
3 (𝜌𝑠 − 𝜌𝑙)𝑔 = 6𝜋ɳ𝑟
𝑑𝑥
𝑑𝑡
(23.5)
Si despejamos r de la ecuación anterior obtenemos

146
𝑟 = √9ɳ 𝑑𝑥
2𝑔(𝜌𝑠 − 𝜌𝑙) 𝑑𝑡
(23.6)
Si la velocidad de decantación se asume constante a lo largo de su caída esta se puede aproximar a L/t donde L es la longitud total que recorren las partículas al caer y T el tiempo de caída libre.
𝑟 = √9ɳ
2𝑔(𝜌𝑠 − 𝜌𝑙)(𝐿
𝑡) ; 𝑜 𝑡 =
9ɳ
2𝑔(𝜌𝑠 − 𝜌𝑙)(𝐿
𝑟2)
(23.7)
23.1.2 Distribución de tamaño de partículas
Consideramos, en primer lugar, que las partículas que componen la muestra tienen igual radio y están suspendidas homogéneamente en un líquido. Las partículas cercanas al fondo del recipiente decantan primero y los granos que inicialmente se hallaban cerca de la superficie del líquido son los últimos en depositarse. La masa sedimentada (𝑀) aumenta
constantemente con el tiempo, hasta que un tiempo 𝑡1 se ha depositado todo el sólido (figura. 1) y la masa no varía. La expresión matemática que describe esta situación es:
𝑀 = 𝑚1𝑡 𝑝𝑎𝑟𝑎 𝑡 < 𝑡1 𝑦 𝑀 = 𝑀1 𝑝𝑎𝑟𝑎 𝑡 > 𝑡1 (23.8)
𝑀 es la masa depositada en el tiempo 𝑡, 𝑀1 es la masa total de partículas radio uniforme 𝑟1 y 𝑚1 es la pendiente (constante) de la Fig. 1 de la izquierda.
Un tratamiento similar es válido para cualquier población de partículas radio uniforme. Si los radios son cada vez más pequeños los tiempos de caída serán cada vez más grandes y mi ira disminuyendo. Suponga ahora que se prepara una mezcla bien dispersa de 𝑀1 (𝑔) de partículas de radio 𝑟1 𝑀2 de partículas de radio 𝑟2, etc., donde el mayor subíndice indica menor radio. La curva total de sedimentación será la suma de las contribuciones individuales de cada grupo de partículas de radios homogéneos. Por ejemplo en la parte de la derecha de la Fig. 1, para el segundo donde se halla en punto 𝐴(𝑡1 < 𝑡2 < 𝑡3) obtenemos que
𝑀 = (𝑚3 +𝑚4 +𝑚5) 𝑡 + 𝑀1 +𝑀2) (23.9)
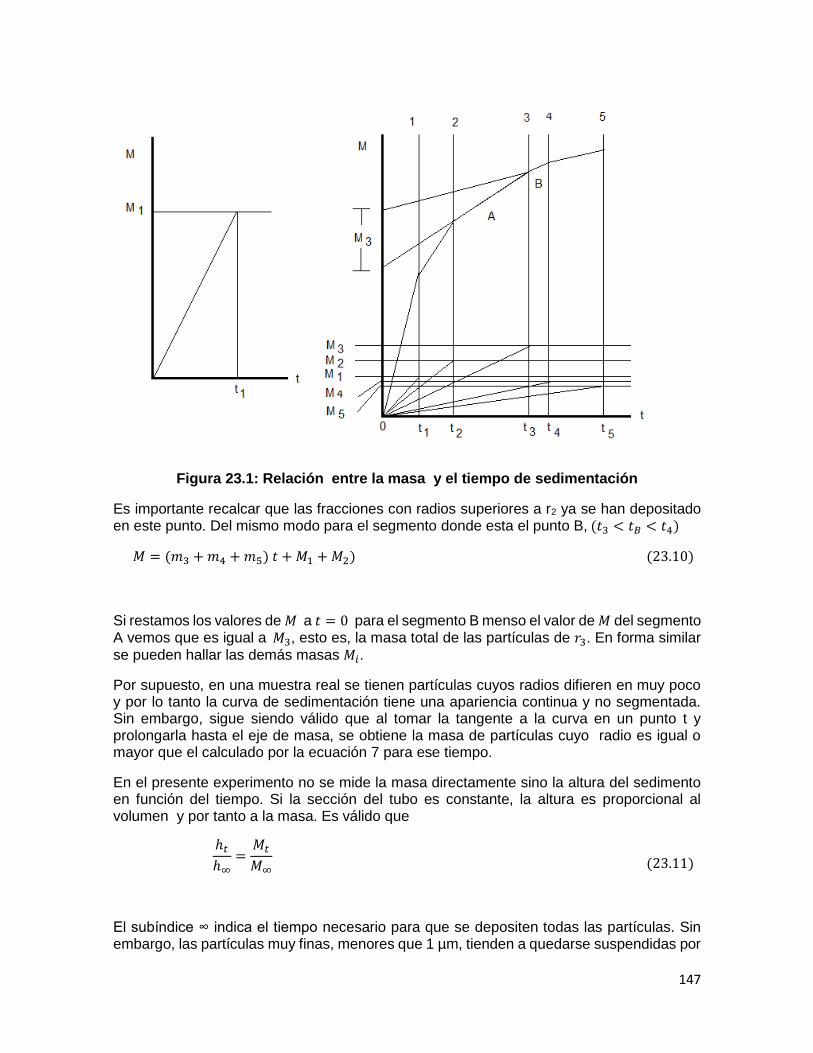
147
Figura 23.1: Relación entre la masa y el tiempo de sedimentación
Es importante recalcar que las fracciones con radios superiores a r2 ya se han depositado en este punto. Del mismo modo para el segmento donde esta el punto B, (𝑡3 < 𝑡𝐵 < 𝑡4)
𝑀 = (𝑚3 +𝑚4 +𝑚5) 𝑡 + 𝑀1 +𝑀2) (23.10)
Si restamos los valores de 𝑀 a 𝑡 = 0 para el segmento B menso el valor de 𝑀 del segmento A vemos que es igual a 𝑀3, esto es, la masa total de las partículas de 𝑟3. En forma similar
se pueden hallar las demás masas 𝑀𝑖.
Por supuesto, en una muestra real se tienen partículas cuyos radios difieren en muy poco y por lo tanto la curva de sedimentación tiene una apariencia continua y no segmentada. Sin embargo, sigue siendo válido que al tomar la tangente a la curva en un punto t y prolongarla hasta el eje de masa, se obtiene la masa de partículas cuyo radio es igual o mayor que el calculado por la ecuación 7 para ese tiempo.
En el presente experimento no se mide la masa directamente sino la altura del sedimento en función del tiempo. Si la sección del tubo es constante, la altura es proporcional al volumen y por tanto a la masa. Es válido que
ℎ𝑡ℎ∞=𝑀𝑡𝑀∞
(23.11)
El subíndice ∞ indica el tiempo necesario para que se depositen todas las partículas. Sin embargo, las partículas muy finas, menores que 1 µm, tienden a quedarse suspendidas por

148
efecto del movimiento Browniano, o por perturbación mecánicas o térmicas. Si la fracción de las partículas que se queda en suspensión es menos que 5% el error introducido no es muy significativo.
23.2 Trabajo experimental
Figura 23.2: Tubo de sedimentación
1. la cantidad de azufre necesaria para el experimento depende de las dimensiones de la columna donde la altura de la mezcla azufre-agua sea de 60 cm y el tubo de recolección tenga un diámetro de 0.5 cm por 10 cm de largo.
2. Pese aproximadamente 2 g de azufre y tritúrelos en un mortero por 5 minutos. 3. Transfiera el azufre a un recipiente de 250 Ml y agregue 10 de jabón líquido para
manos, o vajilla y 100 mL de agua. Agite vigorosamente la mezcla con agitador magnético hasta que no se observen agregados o grumos
4. Manteniendo una agitación constante, transfiera rápidamente la suspensión al tubo de sedimentación. Cuando haya llenado la mitad ponga en marcha el cronometro.
5. Golpee suavemente la parte inferior del tubo con el fin de producir un nivel horizontal y una buena compactación de las partículas. Con ayuda de una lupa y un calibrador mida las alturas del sedimento cada 30 segundos por 5 minutos después tome las medidas cada minuto hasta los 20 min y a partir de este tiempo puede hacer las medidas cada 5 min.
6. Siga tomando las medidas hasta que no observe variación significativa en la altura y por ultimo tome la última medida después de 2 horas.
7. Mida L, la trayectoria de sedimentación, entre el nivel del líquido y el fondo del tubo recolector.
8. Si es posible mida la viscosidad y la densidad de la mezcla remanente en el tubo de decantación.
9. Repita el experimento pero utilizando una suspensión de azufre en agua y sin jabón.

149
23.3 Tratamiento de los datos 1. Calcule el porcentaje de masa sedimentada (%Mt) como
% 𝑀𝑡 = 100ℎ𝑡ℎ∞
(23.12)
2. Haga una gráfica suavizada de % 𝑚 𝑣𝑠 𝑡 en una hoja de papel milimetrado (no
por computador). En una hoja aparte haga una ampliación de la gráfica anterior de la zona de tiempo comprendida entre 0 s y 100 s.
3. Calcule el tiempo necesario para que las partículas de radios 5, 10, 15, 20, 25,
30, 35, 40, 45 y 50 µm se sedimenten utilizando la ecuación 7. La densidad del azufre es 1.96 g cm-3 y como aproximación puede suponer que la densidad del líquido es 1 g cm-3 y su viscosidad es 0.0134 poises. La trayectoria de
sedimentación puede asumir que es 𝐿∗,
𝐿∗ = 𝐿 −1
2ℎ∞
(23.13)
4. Indique sobre la gráfica los tiempos necesarios para que sedimenten las
partículas que tengan como radio los indicados en el punto anterior (𝑡𝑟). halle esos tiempos indique los porcentajes de masa (%M). en ese punto trace una recta tangente prolongada hasta el eje de %M, como se indica en la fig.3. el tratamiento grafico anterior se puede realizar de una forma analítica utilizando las herramientas del análisis numérico (ajuste de funciones) para determinar derivadas en un punto, de un conjunto de datos discretos, y el corte de esta recta tangente con el eje de las ordenadas. Para este punto se recomienda ajustar los datos %M vs t a una función del tipo:
%𝑀 = 100(1 − 𝑒−𝑘𝑡) (23.14)
donde 𝑘 es la constante a determinar mediante un análisis de mínimos cuadrados.
5. El porcentaje de masa que corresponde a las partículas cuyos radios están entre
𝑟𝑛 y 𝑟𝑚 (%𝑀𝑟𝑛−𝑟𝑚) va a ser igual a la diferencia entre los respectivos valores de
los cortes con el eje de las ordenadas (%M) de las rectas tangentes que pasa por 𝑟𝑛 y 𝑟𝑚, donde asdfadfa (Fig. 3).
6. De la misma manera como se indicó anteriormente calcule (%𝑀𝑟𝑛−𝑟𝑚) para las
partículas cuyos radios están en los intervalos (𝑟𝑛−𝑟𝑚) de 0-5, 5-10, 10-15…y
45-50 µm. 7. Dibuje un diagrama de barras (histograma) donde se presente %𝑀𝑟𝑛−𝑟𝑚 en el
eje de las ordenadas y los intervalos (𝑟𝑛−𝑟𝑚) en el eje de las abscisas.
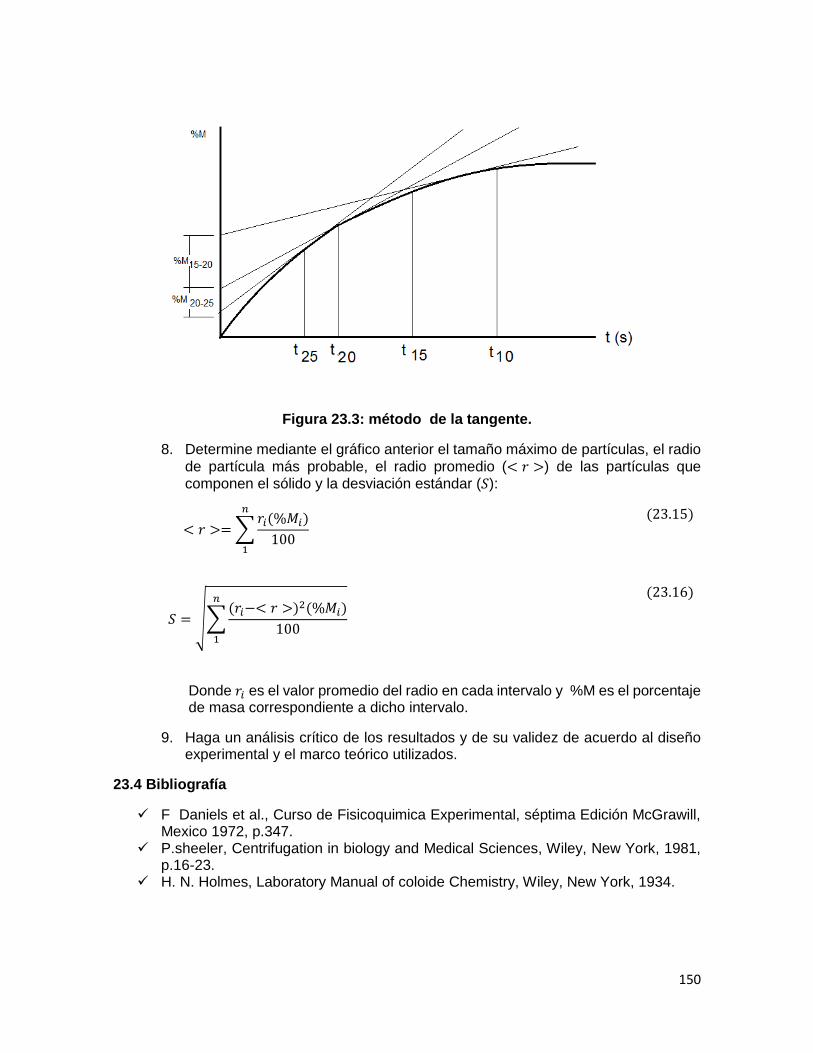
150
Figura 23.3: método de la tangente.
8. Determine mediante el gráfico anterior el tamaño máximo de partículas, el radio
de partícula más probable, el radio promedio (< 𝑟 >) de las partículas que componen el sólido y la desviación estándar (𝑆):
< 𝑟 >=∑𝑟𝑖(%𝑀𝑖)
100
𝑛
1
(23.15)
𝑆 = √∑(𝑟𝑖−< 𝑟 >)
2(%𝑀𝑖)
100
𝑛
1
(23.16)
Donde 𝑟𝑖 es el valor promedio del radio en cada intervalo y %M es el porcentaje de masa correspondiente a dicho intervalo.
9. Haga un análisis crítico de los resultados y de su validez de acuerdo al diseño experimental y el marco teórico utilizados.
23.4 Bibliografía
F Daniels et al., Curso de Fisicoquimica Experimental, séptima Edición McGrawill, Mexico 1972, p.347.
P.sheeler, Centrifugation in biology and Medical Sciences, Wiley, New York, 1981, p.16-23.
H. N. Holmes, Laboratory Manual of coloide Chemistry, Wiley, New York, 1934.

151
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO NO. 24: FLUJO DE POISEUILLE.
OBJETIVOS
Éste experimento tiene por objetivo estudiar los parámetros de la ecuación de Poiseuille.
24.1 Introducción
Jean-Luis-Marie (1799-1869) fue un físico y fisiólogo francés que desarrollo una expresión matemática para fluidos que pasa por un tubo circular, con las dimensiones del tubo y las diferencias de presión a la entrada y salida este. Esta relación fue descubierta independiente por el ingeniero hidráulico alemán Gotthilf Hagen, por esta razón esta relación es también conocida como la ecuación de Hagen-Poiseuille. Poiseuille obtuvo el grado de medico en 1828 y ejerció su práctica en parís. El desarrollo un método para medir la presión sanguínea y se cree que fue el primer científico en usar un manómetro de mercurio para medir la presión sanguínea.
Un modelo simple que describe el flujo de un fluido a través de un tubo está dado por la ecuación de Bernoulli, que es simplemente una aplicación masa de la ley de la conservación de la energía mecánica de un fluido en movimiento:
𝑃 + 𝜌𝑔ℎ +𝜌𝑣2
2= 𝑐𝑜𝑛𝑠𝑡𝑎𝑛𝑡𝑒
(24.1)
Donde 𝑃 es la presión a la que está sometido el fluido, 𝜌 es la densidad del fluido, ℎ es la altura de la cabeza de presión con respecto a la salida del fluido y 𝑣 es la velocidad lineal del fluido. Este modelo asume que el fluido es incompresible y que no esta sujeto a ninguna fuerza de fricción durante su movimiento a través del tubo. El uso de un valor único de 𝑣 en la ecuación de Bernoulli indica que esta ecuación asume que todas las opciones del fluido que esta en directo contacto con las paredes del tubo se mueve mas rápidamente. La fricción interna que causa este gradiente de velocidades es la llamada viscosidad del fluido.
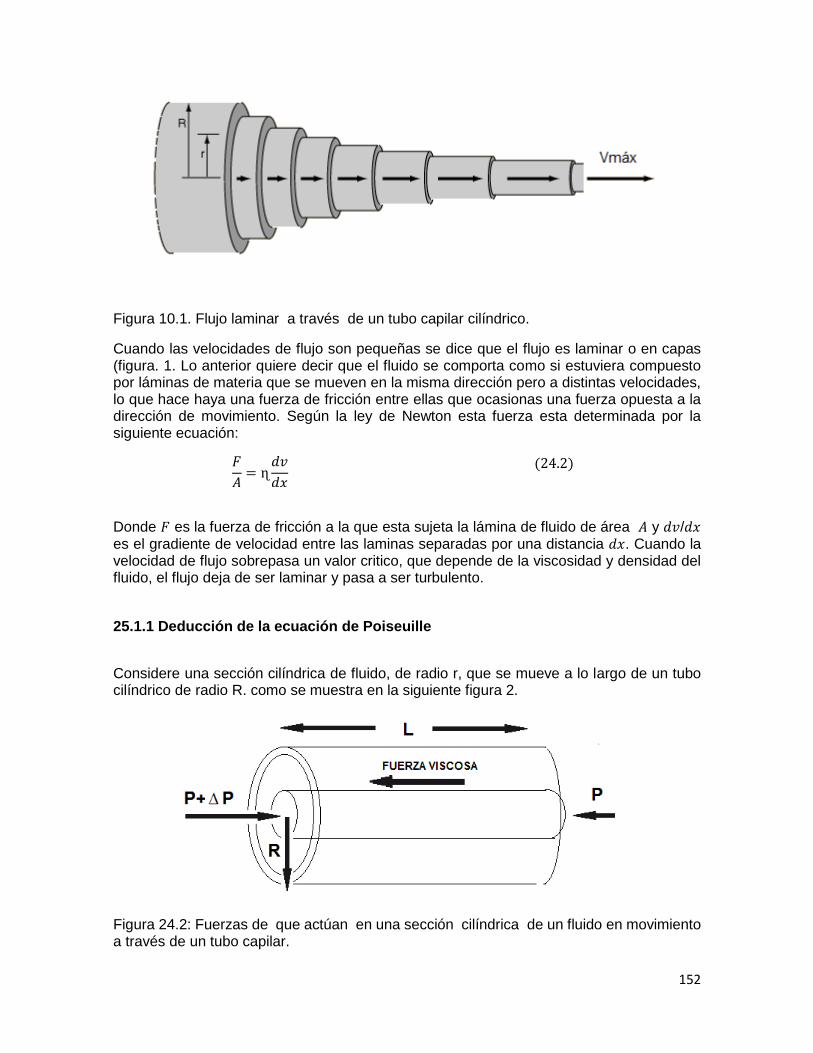
152
Figura 10.1. Flujo laminar a través de un tubo capilar cilíndrico.
Cuando las velocidades de flujo son pequeñas se dice que el flujo es laminar o en capas (figura. 1. Lo anterior quiere decir que el fluido se comporta como si estuviera compuesto por láminas de materia que se mueven en la misma dirección pero a distintas velocidades, lo que hace haya una fuerza de fricción entre ellas que ocasionas una fuerza opuesta a la dirección de movimiento. Según la ley de Newton esta fuerza esta determinada por la siguiente ecuación:
𝐹
𝐴= ɳ
𝑑𝑣
𝑑𝑥
(24.2)
Donde 𝐹 es la fuerza de fricción a la que esta sujeta la lámina de fluido de área 𝐴 y 𝑑𝑣/𝑑𝑥 es el gradiente de velocidad entre las laminas separadas por una distancia 𝑑𝑥. Cuando la velocidad de flujo sobrepasa un valor critico, que depende de la viscosidad y densidad del fluido, el flujo deja de ser laminar y pasa a ser turbulento.
25.1.1 Deducción de la ecuación de Poiseuille
Considere una sección cilíndrica de fluido, de radio r, que se mueve a lo largo de un tubo cilíndrico de radio R. como se muestra en la siguiente figura 2.
Figura 24.2: Fuerzas de que actúan en una sección cilíndrica de un fluido en movimiento a través de un tubo capilar.

153
La fuerza que impulsa a la sesión cilíndrica a moverse (Fp) y la fuerza viscosa (Fv) que
tiende a contrarrestar esta fuerza son respectivamente:
𝐹𝑝 = ∆𝑃(𝜋𝑟2) (24.3)
𝐹𝑣 = −ɳ(2𝜋𝑟𝐿)𝑑𝑣
𝑑𝑟
(24.4)
En condiciones estacionarias estas dos fuerzas son iguales y el fluido en su totalidad se mueve a velocidad constante. En términos matemáticos tenemos:
𝐹𝑝 = 𝐹𝑣; 𝑜 ∆𝑃(𝜋𝑟2) = −ɳ(2𝜋𝑟𝐿)
𝑑𝑣
𝑑𝑟
(24.5)
Despejando el gradiente de velocidad obtenemos
𝑑𝑣
𝑑𝑟= −(
∆𝑃
2ɳ𝐿) 𝑟
(24.6)
De lo anterior ecuación se puede obtener una expresión matemática que relaciones la velocidad en función del radio mediante integración:
∫ 𝑑𝑣 =0
𝑣
(∆𝑃
2ɳ𝐿)∫ 𝑟𝑑𝑟
𝑅
𝑟
(24.7)
𝑣(𝑟) = (∆𝑃
2ɳ𝐿) (𝑅2 − 𝑟2)
(24.8)
En la anterior ecuación se observa que el perfil de velocidades desarrollado es parabólico como se observa en la figura 3, y que la velocidad del fluido es máxima en el centro del tubo y cero en la superficie interna del tubo

154
Figura 24.3 Perfil de velocidades de un fluido en movimiento por un tubo capilar.
El flujo volumétrico total a través del tubo (𝑗) se puede relacionar con la velocidad lineal de cada lámina de flujo según la ecuación:
𝑗 =𝑑𝑉
𝑑𝑡= ∫𝑣𝑑𝐴
(24.9)
Donde A representa el área perpendicular a la dirección de flujo. Si hacemos las
correspondientes de 𝑣 y A en la ecuación 24.9, utilizando los resultados de la ecuación 24.8, obtenemos:
𝑗 = ∫𝑣𝑑𝐴 ∫ (∆𝑃
2ɳ𝐿) [𝑅2 − 𝑟2](2𝜋𝑟𝑑𝑟)
𝑅
0
(24.10)
𝑗 = (𝜋∆𝑃
2ɳ𝐿)∫ [𝑅2 − 𝑟3](𝑑𝑟)
𝑅
0
(24.11)
𝑗 = (𝜋∆𝑃
2ɳ𝐿) ⌊𝑅4
2−𝑅4
4⌋ = (
𝜋∆𝑃𝑅4
8ɳ𝐿)
(24.12)
La ecuación anterior es la denominada ecuación Poiseuille. Hay que hacer notar la fuerte dependencia del flujo con respecto al radio del tubo (a la cuarta potencia), lo que hace que inclusive pequeñas variaciones del radio van a causar grandes variaciones en el flujo volumétrico a través del tubo.
Como el flujo (𝑗) y el ∆𝑃 son proporcionales (ecuación 25.12), como lo son corriente y el voltaje en la ley de Ohm, se puede escribir la ecuación 25.12 en una forma análoga a la ley de Ohm introduciendo un nuevo concepto, la resistencia del flujo (Ω):
Ω =∆𝑃
𝑗=8ɳ𝐿
𝜋𝑟4
(24.13)
La ecuación de Poiseuille se cumple tanto para gases como para líquidos a una temperatura constante. Pero la variación de la viscosidad con la temperatura es diferente para gases y líquidos. Al aumentar la temperatura se incrementa la viscosidad de un gas, mientras por el contrario, la viscosidad de líquido disminuye. Esta diferencia en comportamiento se puede explicar si se examinan detalladamente las causas de la viscosidad. La resistencia que un fluido ofrece al corte depende de las fuerzas de cohesión y la rapidez de la trasferencia de la cantidad de movimiento entre moléculas. En un líquido, las fuerzas de cohesión son mucho mayores que las presentes en un gas, debido a que las moléculas se encuentran más próximas entre si. La cohesión parece ser la causa predominante de la viscosidad en un líquido, y como la cohesión disminuye al incrementarse la temperatura lo mismo le sucede a la viscosidad.

155
Por otro lado en un gas las fuerzas de cohesión son muy débiles y la mayor parte de su resistencia al esfuerzo cortante resulta de la transferencia de cantidad de movimiento molecular. Dentro de un fluido siempre existe el paso en uno y otro sentido de moléculas a través de cualquier superficie ficticia que se considere. Cuando una capa de fluido se mueve relativamente a otra capa adyacente, la transferencia molecular de cantidad de movimiento acarrea una transferencia neta de movimiento de una capa a otra, de tal modo que se origina un esfuerzo cortante aparentemente que tiende a neutralizar el movimiento relativo y tiende a igualar las velocidades de las capas. La actividad molecular en los gases ocasiona un esfuerzo cortante aparente, el cual es mas importante que las fuerzas de cohesión y, dado que la actividad molecular crece con la temperatura, la viscosidad del gas también aumenta con esta.
24.1.2 Numero de Reynolds
El número de Reynolds (Re) es un número experimental usado en mecánica de fluidos para predecir si el flujo en determinadas condiciones es turbulento o laminar. Este número es definido como:
𝑅𝑒=𝐿𝜌𝑣𝑚𝑎𝑥ɳ
(24.14)
Donde vmax es la velocidad máxima para el flujo laminar en un tubo. Si el flujo es laminar está dada por la ecuación:
𝑣𝑚𝑎𝑥 =∆𝑃𝑅2
4ɳ𝐿=2𝑗
𝜋𝑅2
(24.15)
Así el número de Reynolds se puede expresar, para el caso de tubos cilíndricos, en función del flujo volumétrico como:
𝑅𝑒 =2𝐿𝑗𝜌
𝜋ɳ𝑅2
(24.16)
Para tubo es cilíndricos Reynolds encontró que el flujo pasaba de laminar a turbulento cuando este numero excedía el valor de 2000.
24.2. Trabajo experimental
24.2.1 Procedimiento 1
Utilice el montaje que se muestra en la figu.4. La columna de líquido ejerce una presión sobre el fluido del capilar, que es proporcional a la diferencia de alturas entre el nivel del
líquido y la horizontal definida por el eje del tubo capilar (ℎ):

156
∆𝑃 = 𝜌𝑔ℎ (24.17)
Donde g es la aceleración debida a la gravedad (981 cm s.2) y d es la densidad del líquido. Si todos los parámetros de la ecuación 10.12 se mantienen constantes el flujo va a ser proporcional a (ℎ):
𝑗 =∆𝑉
∆𝑡=𝜋∆𝑃𝑅4
8ɳ𝐿=𝜋𝑅4𝜌𝑔ℎ
8ɳ𝐿= 𝐾ℎ
(24.18)
Una medida aceptable del flujo consiste en tomar el tiempo necesario para desocupar un volumen pequeño de la bureta, digamos 2 cm3. Para obtener buenos resultados se deben utilizar capilares largos y de radios pequeños.
10.3 Desarrollo experimental.
Figura 24.4: Montaje experimental para el procedimiento 1.
1. Deje descargar la bureta y mida la altura del nivel del liquido (ℎ) en función del tiempo cada 2 cm3 descargados.
2. Repita el experimento hasta obtener 10 medidas y mida la temperatura del líquido al final de cada experimento.
3. La altura de la cabeza de presión (ℎ) se determina en una escala de milimetros y la longitud del capilar con un calibrador.
4. El radio del capilar es muy pequeño de modo que se debe determinar de la siguiente manera:
Seque muy bien el capilar y péselo en una balanza analítica.

157
Llene con mucho cuidado el capilar con mercurio y mida, con un calibrador, la longitud de la columna de mercurio.
Pese nuevamente el capilar con el mercurio. Busque la densidad del mercurio a la temperatura de trabajo y determine el radio del capilar utilizando los datos anteriores.
5. Para observar el efecto de la longitud y radio del capilar en el flujo, escoja tres capilares, dos de los cuales tengan igual radio pero diferente longitud y otros dos que igual longitud pero diferente radio. Para cada capilar realice el mismo procedimiento del numeral 1 al 4.
6. Para observar el efecto de la viscosidad en el flujo realice el mismo procedimiento del numeral 1 al 3 utilizando un líquido diferente (solución de sacarosa al 40% por ejemplo).
24.3.1 Tratamiento de los datos y cuestionario
Si tenemos en cuenta la ∆𝑉 = 𝐴∆ℎ la ecuación 25.18 se transforma en
𝑗 =∆𝑉
∆𝑡=𝐴∆ℎ
∆𝑡=𝜋𝑅4𝜌𝑔ℎ
8ɳ𝐿
(24.19)
Si integramos la ecuación anterior obtenemos la ecuación:
ℎ(𝑡) = ℎ0𝑒𝑥𝑝(𝜋𝑅4𝜌𝑔ℎ
8ɳ𝐿)
(24.20)
Donde ℎ0 es la altura inicial del líquido en la probeta.
1. Haga una tabla de flujo y el número de Reynolds en función de altura h. tenga en cuenta que para calcular el flujo se divide 2 cm3 en el tiempo que tardaron en fluir h es la altura promedio a la que estuvieron los 2 cm3 de liquido. Con los datos experimentales realice los siguientes gráficos:ℎ(𝑡)𝑣𝑠 𝑡, ln(ℎ) 𝑣𝑠 𝑡 𝑦 𝑗 𝑣𝑠 ℎ.
2. Calcule la viscosidad de cada líquido trabajado utilizando las graficas anteriores para cada tubo capilar trabajado.
3. ¿Cuánto varia el flujo volumétrico si el radio aumenta un 5% de su valor?
4. ¿En sus propias palabras de una definición para el flujo turbulento?
5. Compare las viscosidades halladas experimentales con las reportadas en literatura y haga un análisis crítico, y en lo posible cuantitativo, de las causas de error experimental más importantes.
24.3.2 Procedimiento 2

158
Figura 25.5: dispositivo experimental para el procedimiento 2.
1. Prepare el montaje experimental que se muestra en la figura 5. Cuando introduzca el líquido asegúrese muy bien que no hayan burbujas desde el embudo de decantación hasta la salida del líquido por el capilar.
2. Determine la altura h con una precisión de milímetro. El montaje debe estar dispuesto de tal manera que h se puede variar de 80 cm a 20 cm.
3. Una vez fijada h abra la llave del embudo y cuente el tiempo que tardan en fluir 5 o 10 cm3 de líquido. Mida por lo menos por triplicado este tiempo de flujo.
4. Repita los pasos 2 y 3 para las alturas de 20, 40, 60 y 80 cm.
24.3.3 Tratamiento de los datos y cuestionario.
1. Como la altura no varía significativamente durante el tiempo que fluyen los 5 o 10 cm3 se puede asumir como constante. Para que esto sea aproximadamente cierto el embudo debe ser por lo menos de 250 cm3 y el nivel de líquido debe estar alrededor de la parte media del embudo, donde hay mayor área superficial.
2. Haga las gráficas de flujo promedio vs ℎ para cada capilar, de radio conocido. Determine la viscosidad de cada líquido y haga el mismo tratamiento de datos del procedimiento 1.

159
Bibliografías.
R. Lemilich, Two Pennyv experiments in chemical engineering. J. Chem. Educ., 34, 489. 1957.
V. L. Streeter, E. B. Wylie, Mecánica de fluidos , Mc Graw-Hill, Cali, Colombia, 1979, p. 21-23.
http://www.sc.ehu.es/sbweb/física/fluidos/dinámica/viscosidad/viscosidad.htm http://hyperphysics.phy-astr.gsu.edu/hbase/pturb.html#reym http//landau1.physi.virginia.eduIclasses/241L/poise/poise.htm

160
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRÁCTICA DE LABORATORIO NO. 25: TRANSPORTE DE MASA POR DIFUSIÓN A
TRAVÉS DE UN GEL.
Objetivos
El presente experimento es un ejemplo sencillo de un proceso termodinámico irreversible, y tiene por objetivo la verificación y cuantificación experimental del transporte de materia por difusión
25.1 Fundamentos
El constante movimiento aleatorio de las moléculas, a temperatura y presión constantes, es la propiedad responsable del transporte de masa en los líquidos cuando hay gradientes de concentración. En un fluido no sujeto a fuerzas externas las moléculas se encuentran en continuo movimiento, pero con un desplazamiento neto igual a cero. Si el fluido se expone a un gradiente constante de concentración, las moléculas se desplazan espontáneamente de las regiones de alta concentración a las de baja concentración. En esta situación decimos que la materia se transporta por DIFUSIÓN espontanea y que la razón de transporte de materia por unidad de área es proporcional al gradiente normal de concentración: (𝐽𝑐/𝐴 = −𝐷𝜕𝑐/𝜕𝑥). Esta relación experimental se conoce como primera ley de Fick, y es una de las leyes fenomenológicas lineales de la termodinámica lineal de los procesos irreversibles. Si el gradiente de concentración no es constante, es decir que el flujo de materia es dependiente del espacio y del tiempo, el proceso de transporte es
descrito por la segunda ley de Fick (𝜕/𝜕𝑡 = 𝐷𝜕2𝑐/𝜕𝑥2)..
La termodinámica lineal de los procesos irreversibles (1) postula una relación lineal entre las fuerzas termodinámicas que actúan sobre un sistema y los lujos termodinámicos inducidos por estas.
𝐽 = 𝐿𝑋 (25.1)
Cuando la fuerza termodinámica que actúa sobre el sistema es un gradiente de concentración, a temperatura y presión constantes, el flujo inducido por esta es un transporte de materia, y la ecuación 26.1 toma la siguiente forma.
𝐽 = −𝐿∇(𝜇/𝑇) (25.2)
1 Ley empírica establecida por le Fisiólogo alemán Adolf Eugen Fick (1829-1901).

161
Donde L es el coeficiente fenomelogico de Onsager, 𝜇 es el potencial químico, y T es la temperatura termodinámica. Las ecuaciones termodinámicas 26.1 y 26.2 no se deben confundir con las leyes empíricas para los procesos de transporte (Fourier, de Fick, de Ohm, de Poiseuille).
Como la fuerza termodinámica responsable del transporte de materia es el gradiente de potencial químico (3,4).
𝐹𝑢𝑒𝑟𝑧𝑎 = −(𝜕𝜇/𝜕𝑥) (25.3)
y asumiendo un soluto termodinámicamente ideal.
𝜇 = 𝜇0 + 𝑅𝑇𝑙𝑛𝐶 (25.4)
La fuerza termodinámica se puede escribir como
𝐹𝑢𝑒𝑟𝑧𝑎 = −(𝑅𝑇
𝑐) (𝜕𝑐
𝜕𝑥)
(25.5)
Lo que finalmente lleva a la primera ley de Fick
𝐽 = −𝐷∇(𝑐(𝑥)) (25.6)
La ecuación anterior es consistente con la primera y segunda ley de la termodinámica, y define positivo el coeficiente de difusión “D” del soluto transportado. Si el transporte de materia se lleva a cabo en una dirección, la ecuación anterior se escribe.
𝐽𝑥 = −𝐷𝜕𝑐/𝜕𝑥 (25.7)
Donde 𝐷 es dado en unidades de cm2/s.
La ecuación anterior supone un gradiente constante de concentración para poderla aplicar directamente en la cuantificación del coeficiente de difusión. Esto es posible en la práctica utilizando un reactor de flujo continuo sin agitación.
Si el gradiente de concentración no es constante, como el experimento a realizar, el flujo de materia se hace dependiente del espacio y del tiempo, y el proceso de transporte de describe por la llamada segunda ley de Fick.
(𝜕𝑐(𝑥, 𝑡)/𝜕𝑡)𝑥 = 𝐷(𝜕2𝑐(𝑥, 𝑡)/𝜕𝑥2)𝑡 (25.8)

162
Para resolver esta ecuación diferencial parcial de orden 2 es necesario especificar las condiciones de frontera (2):
𝑡 = 0, 𝐶 = 𝐶0 𝑝𝑎𝑟𝑎 𝑥 > 0
𝑡 > 0, 𝐶 → 𝐶0 𝑐𝑢𝑎𝑛𝑑𝑜 𝑥 → ∞ (25.9)
𝑡 → ∞, 𝑒𝑛𝑡𝑜𝑛𝑐𝑒𝑠 𝐶 → 𝑐𝑜𝑛𝑠𝑡𝑎𝑛𝑡𝑒
Las condiciones anteriores son consistentes con el trabajo experimental a realizar y la solución que satisface las ecuaciones anteriores son consistentes con el trabajo experimental a realizar y la solución que satisface las ecuaciones anteriores tiene la siguiente forma:
D = (L2(C/C0)0π)t (25.10)
25.2 Trabajo experimental
Realice el montaje experimental para estudiar la difusión de un electrolito o de un colorante (dependiendo del sensor de concentración que vaya a utilizar). El soluto se encuentra disperso en un gel que se pone en contacto con agua y buena agitación
25.2.1 Difusión de Azul de Bromofenol
Lleve aprox. 30 mg de azul de bromofenol y 0.5 g de acetato de sodio (este último del pH para un adecuado desarrollo del color) a cantidad exacta de 30 cm3 completada con agua (Solución I). Luego 5 cm3 de la solución I se diluyen a 250 cm3 con agua destilada, y 10 cm3 de esta última solución se lleva a 100 cm3 (solución II). Mida la absorbancia de la
solución II a 590 𝑛𝑚, usando como blanco agua destilada (esta es la lectura de referencia). El volumen restante de la solución I (25 cm3), se mezclan en un vaso con aprox. 0.5 de agar-agar, se calientan hasta el primer hervor y se vierte en un tubo de 10 cm de longitud y unos 2 cm de diámetro (el tubo esta previamente sellado por unos de sus extremos con un plástico o papel adecuado). Luego de que la solución de agar se ha enfriado y gelificado en el tubo, determine la longitud L del gel y libere el extremo sellado.

163
Figura 26.1: Dispositivo experimental para el estudio de la difusión de materia a través de un gel.
Luego en un vaso que contiene 150 cm3 de agua en continua agitación, sumerja el tubo con el gel (aprox. 2 cm del fondo del vaso) y tome tiempo cero. Cada 5 minutos, y durante hora y media, tome una alícuota de la solución y mida su absorbancia a 590 nm, luego retorne la alícuota al vaso del experimento. Mida continuamente la temperatura de la solución utilizando un termómetro de ±0.05°C.
25.2.2 Difusión del cloruro de sodio
Disuelva aprox. 0.3 g de la sal en un volumen de 30 cm3 (solución I). Luego 5 cm3 de la solución I se diluyen a un volumen de 250 cm3 (solución II). Determine el valor de la
conductividad eléctrica de la solución II. Este es el valor de la diferencia.
Con el volumen restante de la solución I (25 cm3 ) prepare el gel como se describió en el procedimiento anterior, y siga el mismo procedimiento2.
25.3 Cuestionario 1. Basándose en la ecuación 26.10 represente correctamente en una grafica los datos
experimentales para obtener el valor del coeficiente de difusión del soluto. Para esto debe determinar C/C0 a partir de los datos tomados y manejando correctamente los factores de dilución realizados. Por ejemplo: para las lecturas de absorbancia:
C/C0 = (𝐴 (𝑡)
𝐴𝑟𝑒𝑓) /10/100)(5/250)(150/𝑉. Donde 𝑉 es el volumen del gel.
2. Analice los procedimientos seguidos y los resultados obtenidos. Discuta las propiedades de transporte del soluto y del medio usados en el experimento.
3. ¿Cómo se afectan sus resultados si la temperatura del sistema durante el proceso no es constante?
4. ¿Qué otras propiedades del soluto o del medio podrían obtenerse a partir del trabajo experimental realizado?
5. ¿es posible tener procesos de transporte de materia que no obedezcan las leyes de Fick?
6. Todo proceso macrascópico consistente con las leyes de la termodinámica debe satisfacer las leyes de conservación de la energía, de la masa y de la cantidad del
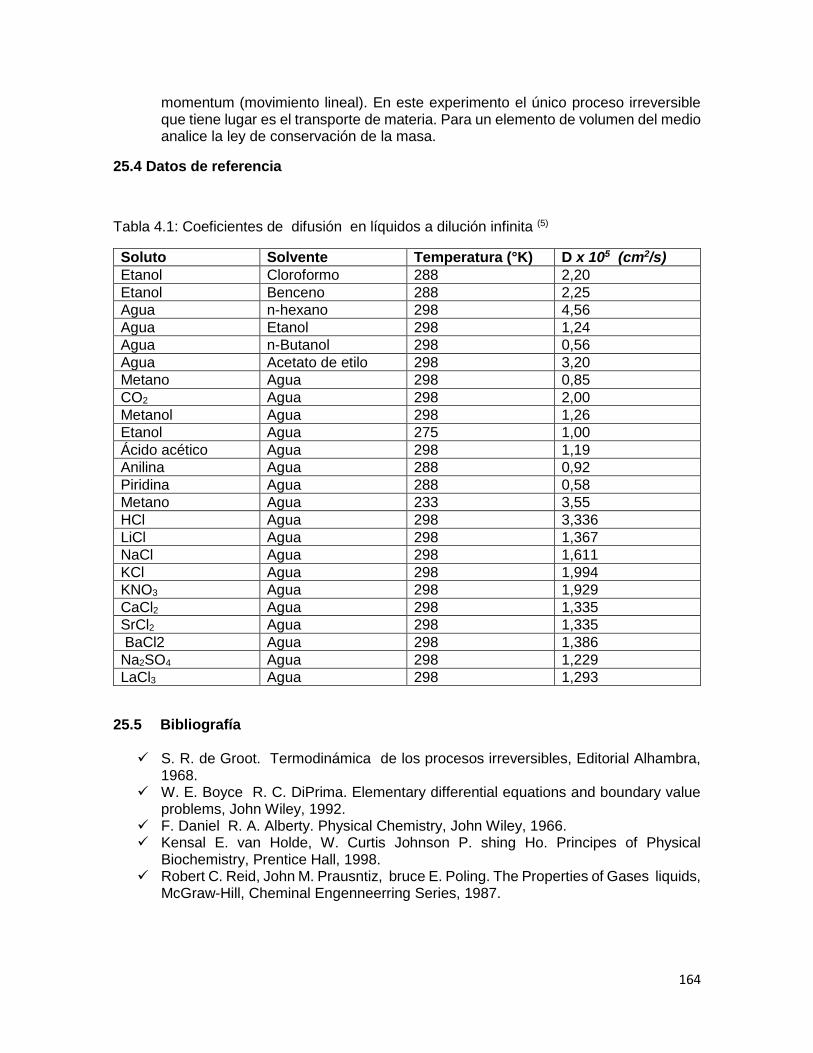
164
momentum (movimiento lineal). En este experimento el único proceso irreversible que tiene lugar es el transporte de materia. Para un elemento de volumen del medio analice la ley de conservación de la masa.
25.4 Datos de referencia
Tabla 4.1: Coeficientes de difusión en líquidos a dilución infinita (5)
Soluto Solvente Temperatura (°K) D x 105 (cm2/s)
Etanol Cloroformo 288 2,20
Etanol Benceno 288 2,25
Agua n-hexano 298 4,56
Agua Etanol 298 1,24
Agua n-Butanol 298 0,56
Agua Acetato de etilo 298 3,20
Metano Agua 298 0,85
CO2 Agua 298 2,00
Metanol Agua 298 1,26
Etanol Agua 275 1,00
Ácido acético Agua 298 1,19
Anilina Agua 288 0,92
Piridina Agua 288 0,58
Metano Agua 233 3,55
HCl Agua 298 3,336
LiCl Agua 298 1,367
NaCl Agua 298 1,611
KCl Agua 298 1,994
KNO3 Agua 298 1,929
CaCl2 Agua 298 1,335
SrCl2 Agua 298 1,335
BaCl2 Agua 298 1,386
Na2SO4 Agua 298 1,229
LaCl3 Agua 298 1,293
25.5 Bibliografía S. R. de Groot. Termodinámica de los procesos irreversibles, Editorial Alhambra,
1968. W. E. Boyce R. C. DiPrima. Elementary differential equations and boundary value
problems, John Wiley, 1992. F. Daniel R. A. Alberty. Physical Chemistry, John Wiley, 1966. Kensal E. van Holde, W. Curtis Johnson P. shing Ho. Principes of Physical
Biochemistry, Prentice Hall, 1998. Robert C. Reid, John M. Prausntiz, bruce E. Poling. The Properties of Gases liquids,
McGraw-Hill, Cheminal Engenneerring Series, 1987.

165
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica
PRACTICA DE LABORATORIO NO. 26: DISOLUCIÓN DE UN SÓLIDO.
OBJETIVOS
1. En el presente experimento se pretende estudiar los parámetros físicos que afectan el proceso de disolución de un sólido (un dulce) en un líquido (agua).
2. Mediante esta práctica el estudiante debe aprender a diferenciar cuando una reacción está controlada por los procesos de transporte de materia o por proceso científicos químicos en la interface sólido-líquido.
26.1 Fundamentos
Desde las primeras etapas de la era científica moderna, la ciencia química se ha centrado en los todos de reacciones que tienen lugar en fases homogéneas, especialmente en gases o líquidos. Los métodos de síntesis, las técnicas analíticas, las ecuaciones cinéticas y termodinámicas fueron diseñados para el estudio de sistemas homogéneos debido a la alta complejidad de los sistemas homogéneos debido a la alta complejidad de los sistemas heterogéneos. Sin embargo siempre se ha reconocido que la mayoría de los procesos naturales no se dan en fases homogéneas.
Muchos factores se pueden identificar como la causa del reciente interés en las reacciones químicas en interfaces. En primer lugar, la química de los sistemas homogéneos ya ha llegado a un grado de madurez en su conocimiento, lo que hace más fácil sobreponer al estudio la complejidad adicional de los sistemas heterogéneos. En segundo lugar, en las últimas décadas se han dado grandes avances en la sensibilidad y el diseño de las técnicas analíticas necesarias para el estudio de interfaces y de los medios computacionales necesarios para contrastar los experimentos con teorías que tienen en cuenta un gran número de variables. Estos avances han abierto la posibilidad de estudiar con precisión y a nivel molecular el efecto de un gran número de variables físicas en los procesos químicos en interfaces. En tercer lugar, y posiblemente es el factor más importante, la complejidad impuesta por la heterogeneidad, que siempre rica para el diseño de nuevas estructuras moleculares que presentan propiedades físicas interesantes, y útiles, que no se presentan en sistemas homogéneos.
Un gran número de procesos naturales e industriales involucran reacciones en interfaces sólido-líquido tales como el crecimiento y disolución de cristales o sólidos amorfos a partir de soluciones. Los cristales son usados como componentes electrónicos, gemas, reactivos, etc., y suspensiones de micro cristales o sólidos amorfos de distintas sustancias se usan como medicamentos, catalizadores a o reactivos químicos. En la naturaleza, cristales y son disueltos y atacados por otros, y la formación del cristal depende de toda una gama de reacciones elementales que determinaran finalmente si el cristal crece o se disuelve.

166
El proceso de disolución de sólidos se da mediante un mecanismo bastante complejo donde la reacción total involucra los siguientes pasos:
1. Ruptura de los enlaces que unen los iones o moléculas a la superficie 2. Difusión superficial de los iones o moléculas liberadas. 3. Procesos de desorción y adsorción. 4. Procesos de transferencia de masa desde o hacia la superficie del sólido.
El último paso involucra usualmente el transporte de materia debido a procesos de difusión, convección, migración en la interface sólido-líquido y posiblemente difusión, convección, migración en la interface sólido-líquido. Los pasos 1, 2 y 3 son procesos superficiales, y cuando uno de estos pasos es muy lento comparado con los procesos de transporte se dice que la reacción de disolución está controlada cinéticamente por procesos superficiales. Si el paso lento de la reacción global es el cuarto se dice que la reacciono está controlada por los procesos de transporte (1,2).
En la presente práctica de laboratorio se a estudiar el proceso de disolución de un sólido (dulce compuesto principalmente de sacarosa) esférico en agua. Si el proceso de disolución esta controlado por los procesos difusivos de transporte de materia la ecuación diferencial que describiría el fenómeno seria.
𝑑𝑚
𝑑𝑡= 𝐽 = −𝐷𝐴
𝑑𝐶
𝑑𝑥
(26.1)
Donde M representa la masa del sólido, J el flujo difusivo de materia del sólido hacia el interior del líquido, D y C son el coeficiente de difusión y la concentración en el líquido de la
sustancia que compone el sólido, A es el área superficial del sólido y 𝑥 es la coordenada que representa la distancia desde la superficie del solido hacia el interior del líquido.
Para poder resolver la anterior ecuación se hace necesario expresarla únicamente en función de la masa, el tiempo y constantes. Con este fin debemos expresar el área en función de la masa, para ello utilizamos las ecuaciones para el área (A) y el volumen (V) de una esfera:
𝑉 =4
3𝜋𝑟3; 𝐴 = 4𝜋𝑟2
(26.2)
De las ecuaciones anteriores se puede expresar el área en función del volumen del siguiente modo:
𝐴 = 22/3𝜋1/332/3𝑉2/3 ≈ 4.836 𝑉2
3 (26.3)
Como el volumen es igual a la masa sobre la masa sobre la densidad del solido (p), la ecuación anterior se transforma en
𝐴 ≈4.836
𝜌2/3𝑚2/3
(26.4)

167
Por otro lado si suponemos que se forma una capa de difusión estacionaria el gradiente de concentración lo podemos expresar como
𝑑𝐶
𝑑𝑥=𝐶𝑠𝑎𝑡−𝐶𝑙𝛿
≈𝐶𝑠𝑎𝑡
𝛿
(26.5)
Donde 𝐶𝑠𝑎𝑡 y 𝐶𝑙son la concentración de saturación o solubilidad (expresada en gramos por centímetro cúbico de solución) y la concentración en el interior del líquido
respectivamente de la sustancia que compone el sólido en el líquido, y 𝑥es la capa de difusión de Nernst. En nuestras condiciones experimentales 𝐶𝑠𝑎𝑡 ≫ 𝐶𝑙 por lo tanto 𝐶𝑙 no se tiene en cuenta para nuestro desarrollo teórico.
Si remplazamos las ecuaciones 4 y5 en la ecuación 1 la ecuación,
𝑑𝑚
𝑑𝑡= −
4.836𝐷𝐶𝑠𝑎𝑡
𝛿𝜌𝑠2/3
𝑚2/3 = −𝐾1𝑚2/3
(26.6)
Donde 𝐾1 es la constante cinética del proceso. Finalmente resolviendo la anterior ecuación diferencial llegamos a la ecuación de trabajo:
∫ 𝑚−2/3𝑑𝑚 = ∫ 𝐾1𝑑𝑡0
𝑡
𝑚
𝑚0
(26.7)
𝑚1/3 = 𝑚01/3−𝐾13𝑡
(26.8)
Si la reacción esta controlada cinéticamente por la superficie la velocidad de disolución se puede expresar como,
𝑑𝑚
𝑑𝑡= −𝐾2𝐴 = −
4.836𝐾2
𝜌𝑠2/3
𝑚2/3
= −𝐾3𝑚2/3
(26.9)
Si resolvemos la anterior ecuación diferencial, obtenemos una ecuación similar a la
ecuación 8 pero el significado de las constantes cinéticas 𝐾1 y 𝐾2es totalmente distinto.
26.2 Trabajo experimental
1. El reactor es un vaso de precipitado de 500 mL, por lo menos, el cual se ha sumergido en un termostato y al que se le ha acondicionado un agitador de aspas, o magnético, para poder agitar la solución dentro del vaso.

168
2. Para comenzar la práctica se introducen 400 mL de agua al vaso de precipitados y se inicia la agitación vigorosa y constante. Se deben esperar unos 10 minutos a que se equilibre la temperatura.
3. Pesar un dulce esférico (se consiguen en el comercio con el nombre de bola rayada), preferiblemente en una balanza digital que se pueda tarar. Introduzca el dulce en el vaso de precipitados.
4. El dulce se debe sacar rápidamente, con la ayudad de una cuchara de brazo largo, después de un minuto exacto. Seque el dulce suavemente con una servilleta y péselo.
5. Repita el procedimiento de introducir el dulce por un minuto en el vaso y pesar el dulce hasta que el peso del dulce sea menor a 30% del peso original.
6. Los datos se deben reportar en una tabla donde este anotado el peso del dulce en función del tiempo acumulado de disolución.
7. Para ver el efecto de la temperatura en la velocidad de disolución se aconseja hacer el experimento a las temperaturas de 20, 25, 30 y 35 centígrados utilizando el mismo volumen de agua.
26.2.1 Variantes experimentales
1. Efectos de la agitación: el mismo experimento se lleva a cabo a una sola temperatura y sin agitación.
2. Efecto del volumen utilizado de líquido: el mismo experimento se lleva a cabo a una sola temperatura con agitación, pero se deben utilizar los siguientes volúmenes de líquido: 100, 200, 300 400 mL.
3. Efecto de la concentración de sacarosa. El experimento se lleva a cabo a una sola temperatura, con agitación y a un volumen constante de líquido pero donde el líquido utilizado es una solución de sacarosa de las siguientes concentraciones. 10, 20, y 30% en peso a peso.
26.3 Tratamiento de los datos
1. Realice un análisis crítico de las gráficas de masa remanente del dulce (m) en función del tiempo acumulado de disolución (𝑡) y m1/3 en función de 𝑡.
2. ¿Usted cree que el proceso de disolución está controlado por procesos de transporte o por procesos en la superficie del sólido?
3. Analice si las condiciones de trabajo en el laboratorio representan adecuadamente las condiciones planteadas durante el desarrollo teórico de las ecuaciones 8 y 9.
4. Haga discusión crítica de como esperaría que varíe la constante 𝐾1 y 𝐾3 con la temperatura, con las condiciones hidrodinámicas (velocidad de movimiento de la solución, forma del sólido, etc.) y con la forma cristalina del sólido.
Si asumimos que las constantes físicas de la sacarosa son:
Densidad de la sacarosa sólida a 25ºC (Merk Index): 1.587 g/cm3.
Coeficiente de difusión a 12ºC (Handbook of chemistry and physics 14th, pg 1743): 0.254 cm2dia-1 (3x10-6cm2s-1).
Como aproximación suponga que este coeficiente de difusión no varía con la temperatura.
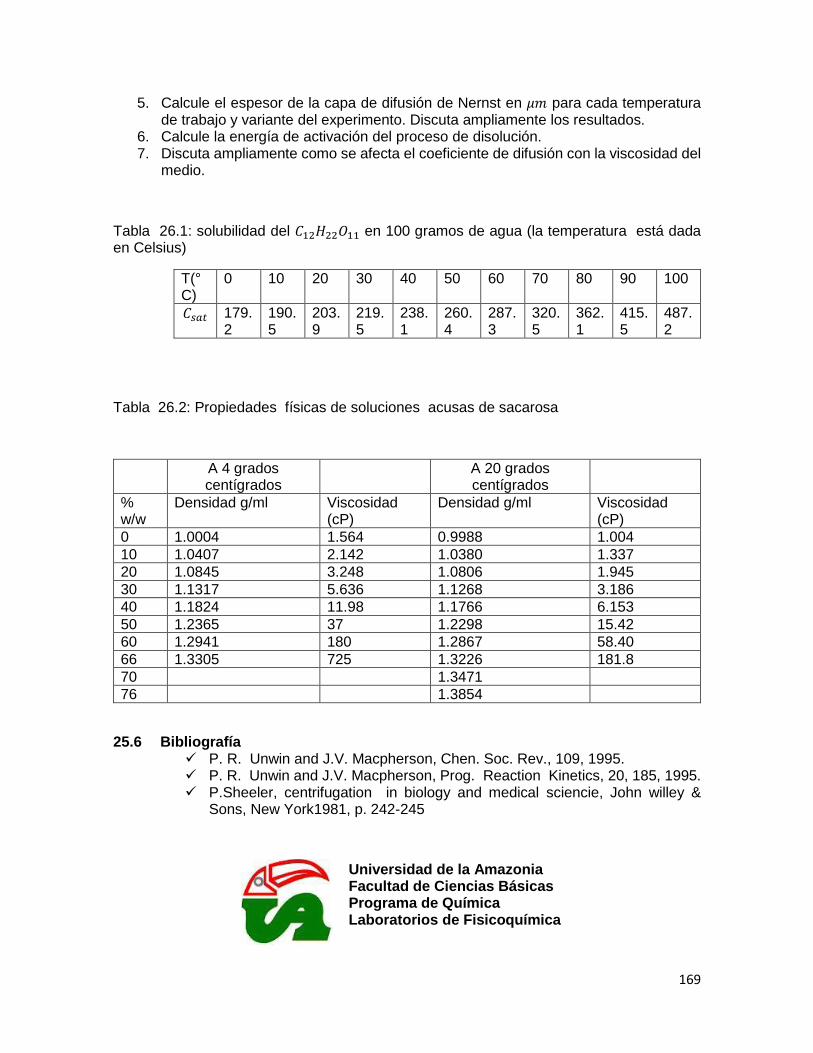
169
5. Calcule el espesor de la capa de difusión de Nernst en 𝜇𝑚 para cada temperatura de trabajo y variante del experimento. Discuta ampliamente los resultados.
6. Calcule la energía de activación del proceso de disolución. 7. Discuta ampliamente como se afecta el coeficiente de difusión con la viscosidad del
medio.
Tabla 26.1: solubilidad del 𝐶12𝐻22𝑂11 en 100 gramos de agua (la temperatura está dada en Celsius)
T(°C)
0 10 20 30 40 50 60 70 80 90 100
𝐶𝑠𝑎𝑡 179.2
190.5
203.9
219.5
238.1
260.4
287.3
320.5
362.1
415.5
487.2
Tabla 26.2: Propiedades físicas de soluciones acusas de sacarosa
A 4 grados centígrados
A 20 grados centígrados
% w/w
Densidad g/ml Viscosidad (cP)
Densidad g/ml Viscosidad (cP)
0 1.0004 1.564 0.9988 1.004
10 1.0407 2.142 1.0380 1.337
20 1.0845 3.248 1.0806 1.945
30 1.1317 5.636 1.1268 3.186
40 1.1824 11.98 1.1766 6.153
50 1.2365 37 1.2298 15.42
60 1.2941 180 1.2867 58.40
66 1.3305 725 1.3226 181.8
70 1.3471
76 1.3854
25.6 Bibliografía P. R. Unwin and J.V. Macpherson, Chen. Soc. Rev., 109, 1995. P. R. Unwin and J.V. Macpherson, Prog. Reaction Kinetics, 20, 185, 1995. P.Sheeler, centrifugation in biology and medical sciencie, John willey &
Sons, New York1981, p. 242-245
Universidad de la Amazonia Facultad de Ciencias Básicas Programa de Química Laboratorios de Fisicoquímica

170
PRACTICA DE LABORATORIO NO. 27: DESORCIÓN DEL AMONIACO:
TRANSPORTE DE MATERIA A TRAVÉS DE LA INTERFACE LIQUIDO-GAS.
OBJETIVOS.
El objetivo de este experimento es estudiar el proceso de desorción del amoniaco desde el agua hasta el aire, pasando por la interfase líquido-vapor, en condiciones controladas de temperatura, presión y en presencia de convección forzada (advenccion) en el aire.
27.1 Fundamentos
El transporte de materia a través de interface liquido-vapor es de gran importancia en proceso industriales y ambientales algunos ejemplos son la producción industrial NH3(ac) por flujo en concentraciones entre NH3(g) y agua, o los proceso de absorción/desorción de gases entre ríos o lagos y el medio ambiente. Estos procesos de trasporte dependen de varios factores externos, entre ellos de la presión, de la temperatura, o de contaminantes.
La fuerza termodinámica que conduce los procesos de transporte a través de interfaces es el ingrediente de potencial químico de las sustancia presente en las diferentes fases. Entonces para que ocurra transporte de amoníaco desde el agua el aire:
𝜇𝑁𝐻3(𝑙) > 𝜇𝑁𝐻3(𝑔) (27.1)
El trasporte termina cuando se igualan los potenciales químicos, y esto solo se puede dar en un sistema cerrado, es decir cuando el vapor se satura con amoníaco y el sistema ha alcanzado un estado de equilibrio dinámico en el cual la velocidad de transporte de amoniaco desde el agua hacia el aire es la misma que desde el aire hacia el agua:
𝜇𝑁𝐻3(𝑙) = 𝜇𝑁𝐻3(𝑔) (27.2)
El equilibrio establecido por la ecuación 2 depende de factores como la temperatura, la presión y la composición, es decir que a diferentes temperaturas presiones o composiciones siempre se tendrán diferentes equilibrios liquido-vapor pero todos cumpliendo la ecuación 2.
El estudiante debe tener presente que el experimento se realizara siempre bajo condiciones de la ecuación 1, es decir que se trata de un proceso termodinámico alejado del equilibrio termodinámico, en el marco de la teoría termodinámica de los procesos irreversibles, el presente experimento, es un claro ejemplo de un proceso que a través de la continua disipación de materia y energía (transporte) conduce irreversiblemente el sistema hacia el estado de equilibrio (estado de máxima entropía).
El modelo propuesto (1) para explicar el proceso de desorción del amoniaco desde el agua hacia el aire involucra las siguientes etapas:
𝑁𝐻3(𝑠𝑒𝑛𝑜 𝑑𝑒 𝑙𝑎 𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 ) → 𝑁𝐻3(𝑖𝑛𝑡𝑒𝑟𝑓𝑎𝑠𝑒 𝑙𝑖𝑞. )

171
𝑁𝐻3( 𝑖𝑛𝑡𝑒𝑟𝑓𝑎𝑠𝑒 𝑙𝑖𝑞. ) → 𝑁𝐻3(𝑖𝑛𝑡𝑒𝑟𝑓𝑎𝑠𝑒 𝑔𝑎𝑠. )
𝑁𝐻3( 𝑖𝑛𝑡𝑒𝑟𝑓𝑎𝑠𝑒 𝑔𝑎𝑠) → 𝑁𝐻3(𝑎𝑖𝑟𝑒)
Lo cual lo resumimos en el siguiente esquema:
𝑁𝐻3(𝑠𝑜𝑙𝑢𝑐𝑖ó𝑛 )//𝑁𝐻3(𝑖𝑛𝑡𝑒𝑟𝑙𝑖𝑞 )//𝑁𝐻3(𝑖𝑛𝑡𝑒𝑟𝑔𝑎𝑠 )//𝑁𝐻3(𝑎𝑖𝑟𝑒) (27.3)
Para tratar de poner sobre bases formales el anterior esquema se han producido varias teorías, una de ellas es la conocida “teoría de resistencias” por analogía con los circuitos eléctricos. En pocas palabras esta teoría postula que la resistencia al transporte de masa a través de la interface esta dada únicamente por las mismas fases, liquido y vapor, mientas la interfase compuesta por capas estacionarias de líquido y gas permanece en un continuo estado de equilibrio que obedece la ley de Henry (el transporte a través de la interface se da únicamente por difusión y gracias a los gradientes de concentración que se generan a lado y lado de la interface):
1
𝐾𝑓=
1
𝐾𝐻𝑘𝑔+1
𝑘𝑙
(27.4)
Donde 𝑘𝑙 es una constante global de trasferencia de masa desde la fase liquida hacia el aire, 𝐾𝐻es la constante de Henry, 𝑘𝑔 es la constante local de trasferencia de masa entre la
interface y el aire y 𝑘𝑙 es la constante local de trasferencia de masa entre el líquido y la
interface. La ecuación 4 se lee, con sentido físico, como que 1
𝑘𝑙 es una resistencia que se
opone al transporte y que es igual a la suma de dos resistencias 1
𝐾𝐻𝑘𝑔 y1
𝑘𝑙 . ¿Cómo disminuir
esa resistencia al transporte de materia manejando adecuadamente los parámetros de control (temperatura, presión, agitación, etc) de modo que se logre un aumento en la magnitud de las constantes de transporte?
Para el esquema 3 escribanos las concentraciones, en fracciones molares de la fase, de la
consiguiente manera: 𝑋(fracción molar del amoníaco en el seno de la solución) 𝑋𝑖 (Fracción molar del amoníaco en la interface líquido), 𝑌𝑖(fracción molar del amoniaco en la interface
gas/vapor) y 𝑌 (fracción molar del amoníaco en el aire), entonces durante:
𝑋 > 𝑋𝑖; 𝑌𝑖 ≫ 𝑌 (27.5)
Donde 𝑌𝑖 = 𝐾𝐻𝑋𝑖 (Ley de Henry para soluciones diluidas).
Al realizar un balance de masa en el plano de la interfase, entre las dos capas estacionarias de fluido, obtenemos (aplicando la primera ley de Fick: 𝑑𝑚/𝑑𝑡 = 𝐽 = −𝐴𝐷𝜕𝐶/𝜕𝑥)
𝑑𝑚𝑖𝑛𝑡𝑑𝑡
= 𝐽𝑙𝑖𝑞−𝑖𝑛𝑡 − 𝐽𝑖𝑛𝑡−𝑎𝑖𝑟𝑒 =𝐴𝐷𝑙𝛿𝑙(𝑋𝑖 − 𝑋) +
𝐴𝐷𝑔
𝛿𝑔(𝑌 − 𝑌𝑖)
(27.6)

172
Donde 𝛿 es la capa de difusión de nerst, 𝑘𝑙 =𝐴𝐷𝑙
𝛿𝑙 y 𝑘𝑔 =
𝐴𝐷𝑔
𝛿𝑔
El experimento que se va a realizar en el laboratorio tiene que ver con medidas directas de 𝑋𝑖 durante el proceso de transporte, pero no con𝑋𝑖 , 𝑌𝑖, 𝑌. ¿Por qué? Es decir que se estudiara directamente el proceso global de trasferencia de materia (amoniaco) desde el líquido hacia el aire y no el proceso de transporte a través del plano de la interfase (ecuación 6):
𝐽 = −𝑉𝑑𝑋
𝑑𝑡= −𝐴𝐾𝑖(𝑋
ɵ − 𝑋)
27.7
La ecuación 7 es la ecuación de trabajo para el presente experimento, donde la K es la constante global de trasferencia en la fase liquida para el amoniaco localizado en la interfase gas y 𝑋 es una variable fantasma que representa, la concentración de una fase líquida fantasma en equilibrio con el aire. La utilidad “practica” de 𝑋, se relaciona con la
forma como se realiza el experimento (medidas directas de 𝑋), además como el proceso de desorción del amoniaco se realiza en sistema abiertos entonces 𝑋ɵ = 0, es decir que el trasnporte de amoniaco a través de la interfase liquido-vapor depende solo de la concentración de este en el liquido.
𝑑𝑥
𝑑𝑡= −𝐹𝑋; 𝑋 = 𝑋0𝑒
−𝐹𝑡
(27.8)
27.2 Trabajo experimental
1. En recipiente plástico (cubeta) que permita dejar expuesta un gran área superficial, coloque agua de modo que la profundidad sea de aprox. 5 cm. Agite la superficie del líquido con un agitador de paletas. Para el caso de provocar convección forzada del aire (advención) coloque un ventilador en la dirección de la superficie del líquido. Registre periódicamente la temperatura del sistema.
2. Adicione 20 cm3 de solución concentrada de amoniaco, después de un par de minutos tome alícuotas de 5 cm3 (por duplicado y en un recipiente de boca angosta) a la vez que pone en marcha el cronometro. Titule con HCl 0.01 N estandarizado utilizando como indicador azul de bromofenol. Esta es la concentración inicial a tiempo cero.
3. Luego tome alícuotas cada 15 minutos, por duplicado, y titule con el acido estandarizado. Después de una hora el tiempo de toma de las alícuotas se puede extender a 20 y luego a 30 minutos. El experimento se realiza hasta que la concentración en la cubeta sea de aprox. ¼ de la inicial.
4. Las variatne4s a realizar en este experimento son: i) diferentes cubetas (relación área/volumen), ii) Diferente temperatura: ambiente, 25°C, 30°C, iii) diferente agitación, mayor revolución o mayor número de agitadores a lo largo de la superficie del líquido, iv) líquidos con diferente viscosidad, v) transporte libre o forzado a través de la interfase.

173
Figura 27.1: desorción de amoníaco.
27.3 tratamiento de los datos
1. Para un mismo tiempo de desorción, exprese el promedio estadístico de las titulaciones en 𝑝𝑝𝑚.
2. En una grafica muestre la variación de la concentración en el tiempo. 3. Verifique se los datos experimentales cumple la siguiente ecuación 8: [𝑁𝐻3]𝑡 =
[𝑁𝐻3]0𝑒−𝐹𝑡
4. En el caso de haber realizado el experimento con diferentes restricciones externas, discuta el efecto de las restricciones sobre el valor de la constante 𝐹.
27.4 Bibliografía.
Treybal, R.B. Mass Transfer operations. Second Edition, McGrawHill (1968), 94-98.
REFERENCIAS
GUIAS TOMADAS DE:

174
1. Fisicoquímica Manual de laboratorio, Ana Beatriz peña Santamaría, Juan Martin céspedes Galeano, Universidad de Medellín.
2. Fisicoquímica Experimental, Daniel Barragán, Marco Fidel Suarez, Guillermo Hernández, Departamento de química–Facultad de ciencias, Universidad Nacional de Colombia.
3. Ensayos de laboratorio, Ingeniero Víctor Maldonado Yactayo, Profesor. Universidad Nacional de Ingeniería Perú, agosto de 1993.
4. Evaluación del proceso de coagulación – floculación de una planta de tratamiento de agua potable, Hernán Alonso Restrepo Osorno, Universidad Nacional de Colombia, sede Medellín, Facultad de Minas 2009.




















