
Fabrication of biodendrimeric β-cyclodextrin via click reaction with potency of anticancer drug...
11
International Journal of Biological Macromolecules 79 (2015) 883–893 Contents lists available at ScienceDirect International Journal of Biological Macromolecules j ourna l h o mepa ge: www.elsevier.com/locate/ijbiomac Fabrication of biodendrimeric -cyclodextrin via click reaction with potency of anticancer drug delivery agent Yousef Toomari, Hassan Namazi ∗ , Ali Akbar Entezami Laboratory of Dendrimers and Nano-Biopolymers, Faculty of Chemistry, University of Tabriz, Tabriz, IranResearch Center for Pharmaceutical Nanotechnology (RCPN), Tabriz University of Medical Science, Tabriz, Iran a r t i c l e i n f o Article history: Received 22 February 2015 Received in revised form 19 May 2015 Accepted 4 June 2015 Available online 6 June 2015 Keywords: -Cyclodextrin Dendrimer Supramolecular inclusion complex a b s t r a c t The aim of this work was the synthesis of biodendrimeric -cyclodextrin (-CD) on the secondary face with encapsulation efficacy, with -CDs moiety to preserve the biocompatibility properties, also par- ticularly growth their loading capacity for drugs with certain size. The new dendrimer, having 14 -CD residues attached to the core -CD in secondary face (11), was prepared through click reaction. The encapsulation property of the prepared compound was evaluated by methotrexate (MTX) drug molecule. Characterization of compound 11 was performed with 1 H NMR, 13 C NMR and FTIR and its supramolec- ular inclusion complex structure was determined using FTIR, DLS, DSC and SEM techniques. In vitro cytotoxicity test results showed that compound 11 has very low or no cytotoxic effect on T47D cancer cells. In vitro drug release study at pHs 3, 5 and 7.4 showed that the release process was noticeably pH dependent and the dendrimer could be used as an appropriate controlled drug delivery system (DDS) for cancer treatment. © 2015 Elsevier B.V. All rights reserved. 1. Introduction Dendrimers are new artificial macromolecules having three- dimensional highly branched structures, uniform and monodis- persed, defined globular structure, densely packed structures, nanoscopic objects at the single molecular level and host–guest entrapment properties [1–4]. In this study we focused on den- drimers. These materials were first reported by Vögtle et al. as cascade molecules [5] and followed by Tomalia et al. [1] called starburst dendrimers, and Newkome et al. [6] as compact ‘arborol’ structures. The “dendrimer” term is now used almost generally to define highly branched with well-defined structure compounds. Moreover, these compounds possess empty internal cavities among their branches which create their potential in supramolecular chemistry or host–guest chemistry to encapsulate guest molecules [7]. Because of unique structures and properties, dendrimers have fascinated great attention in discovering their potential in biomed- ical applications such as gene transport [8,9], imaging and DDS [10–14]. ∗ Corresponding author at: Laboratory of Dendrimers and Nano-Biopolymers, Faculty of Chemistry, University of Tabriz, Tabriz, Iran. Tel.: +98 41 339 3121; fax: +98 41 334 0191. E-mail address: [email protected] (H. Namazi). -Cyclodextrin (-CD) is a highly biocompatible compound and one of the good candidates for the synthesis of star polymers [15]. Star polymers using core-first technique have been prepared [16]. The -CD is a cyclic oligosaccharide that is composed of seven glucose units joined by (1→4) glycosidic linkages to form a cylin- drical structure [17–20]. The outside of the CDs is hydrophilic while its interior cavity is hydrophobic. Because of this specific structure, -CDs can form host–guest inclusion complexes with hydrophobic molecules having the appropriate size by hydrophobic interac- tions and van der Waals forces [11,21–24]. In the pharmacological field, CDs have mainly been used in DDS via providing solubility of hydrophobic drugs thus increasing their bioavailability and/or eliminates adverse types of side effects of the drug [25–27]. So, inclusion complexes of CDs with hydrophobic compounds are of importance [28]. Dendrimers containing carbohydrates in their constructions are named ‘Glycodendrimer’ [29–31]. Glycodendrimers having CD part in their structures are named CD-dendrimers. CD dendrimers, dependent on the central and periphery groups can be divided into three main types. Firstly, CDs are coated on the outside of dendritic scaffold (CD-based dendrimers) [32–34], Secondly, a CD unit as the core where all outlets (in primary face) are joined (CD- scaffolded dendrimers) [35], and thirdly, CD moiety linked with dendrimers (CD-dendrimer conjugates) [36–38]. It is imaginable that CD dendrimers could show more suitable properties for DDS due to biocompatibility and water solubility. Also, it is commonly http://dx.doi.org/10.1016/j.ijbiomac.2015.06.010 0141-8130/© 2015 Elsevier B.V. All rights reserved.
Transcript of Fabrication of biodendrimeric β-cyclodextrin via click reaction with potency of anticancer drug...

Fp
YLN
a
ARRAA
K�DS
1
dpnedcssdMtc[fi[
Ff
h0
International Journal of Biological Macromolecules 79 (2015) 883–893
Contents lists available at ScienceDirect
International Journal of Biological Macromolecules
j ourna l h o mepa ge: www.elsev ier .com/ locate / i jb iomac
abrication of biodendrimeric �-cyclodextrin via click reaction withotency of anticancer drug delivery agent
ousef Toomari, Hassan Namazi ∗, Ali Akbar Entezamiaboratory of Dendrimers and Nano-Biopolymers, Faculty of Chemistry, University of Tabriz, Tabriz, IranResearch Center for Pharmaceuticalanotechnology (RCPN), Tabriz University of Medical Science, Tabriz, Iran
r t i c l e i n f o
rticle history:eceived 22 February 2015eceived in revised form 19 May 2015ccepted 4 June 2015vailable online 6 June 2015
eywords:
a b s t r a c t
The aim of this work was the synthesis of biodendrimeric �-cyclodextrin (�-CD) on the secondary facewith encapsulation efficacy, with �-CDs moiety to preserve the biocompatibility properties, also par-ticularly growth their loading capacity for drugs with certain size. The new dendrimer, having 14 �-CDresidues attached to the core �-CD in secondary face (11), was prepared through click reaction. Theencapsulation property of the prepared compound was evaluated by methotrexate (MTX) drug molecule.Characterization of compound 11 was performed with 1H NMR, 13C NMR and FTIR and its supramolec-
-Cyclodextrinendrimerupramolecular inclusion complex
ular inclusion complex structure was determined using FTIR, DLS, DSC and SEM techniques. In vitrocytotoxicity test results showed that compound 11 has very low or no cytotoxic effect on T47D cancercells. In vitro drug release study at pHs 3, 5 and 7.4 showed that the release process was noticeably pHdependent and the dendrimer could be used as an appropriate controlled drug delivery system (DDS) forcancer treatment.
© 2015 Elsevier B.V. All rights reserved.
. Introduction
Dendrimers are new artificial macromolecules having three-imensional highly branched structures, uniform and monodis-ersed, defined globular structure, densely packed structures,anoscopic objects at the single molecular level and host–guestntrapment properties [1–4]. In this study we focused on den-rimers. These materials were first reported by Vögtle et al. asascade molecules [5] and followed by Tomalia et al. [1] calledtarburst dendrimers, and Newkome et al. [6] as compact ‘arborol’tructures. The “dendrimer” term is now used almost generally toefine highly branched with well-defined structure compounds.oreover, these compounds possess empty internal cavities among
heir branches which create their potential in supramolecularhemistry or host–guest chemistry to encapsulate guest molecules7]. Because of unique structures and properties, dendrimers haveascinated great attention in discovering their potential in biomed-
cal applications such as gene transport [8,9], imaging and DDS10–14].∗ Corresponding author at: Laboratory of Dendrimers and Nano-Biopolymers,aculty of Chemistry, University of Tabriz, Tabriz, Iran. Tel.: +98 41 339 3121;ax: +98 41 334 0191.
E-mail address: [email protected] (H. Namazi).
ttp://dx.doi.org/10.1016/j.ijbiomac.2015.06.010141-8130/© 2015 Elsevier B.V. All rights reserved.
�-Cyclodextrin (�-CD) is a highly biocompatible compound andone of the good candidates for the synthesis of star polymers [15].Star polymers using core-first technique have been prepared [16].The �-CD is a cyclic oligosaccharide that is composed of sevenglucose units joined by � (1→4) glycosidic linkages to form a cylin-drical structure [17–20]. The outside of the CDs is hydrophilic whileits interior cavity is hydrophobic. Because of this specific structure,�-CDs can form host–guest inclusion complexes with hydrophobicmolecules having the appropriate size by hydrophobic interac-tions and van der Waals forces [11,21–24]. In the pharmacologicalfield, CDs have mainly been used in DDS via providing solubilityof hydrophobic drugs thus increasing their bioavailability and/oreliminates adverse types of side effects of the drug [25–27]. So,inclusion complexes of CDs with hydrophobic compounds are ofimportance [28].
Dendrimers containing carbohydrates in their constructionsare named ‘Glycodendrimer’ [29–31]. Glycodendrimers having CDpart in their structures are named CD-dendrimers. CD dendrimers,dependent on the central and periphery groups can be dividedinto three main types. Firstly, CDs are coated on the outside ofdendritic scaffold (CD-based dendrimers) [32–34], Secondly, a CDunit as the core where all outlets (in primary face) are joined (CD-
scaffolded dendrimers) [35], and thirdly, CD moiety linked withdendrimers (CD-dendrimer conjugates) [36–38]. It is imaginablethat CD dendrimers could show more suitable properties for DDSdue to biocompatibility and water solubility. Also, it is commonly
884 Y. Toomari et al. / International Journal of Biological Macromolecules 79 (2015) 883–893
of (T
ab
dfases
2
2
w(emat9d
Scheme 1. Synthetic routes employed for the synthesis
ccepted that the secondary face of the CD is chief mode for guestinding [34].
This study explains the synthesis of the new family of bioden-rimeric compound having on �-CD platforms on the secondaryace in their core and outside by using spacer arms, which waschieved by click reaction (Scheme 1). Also, the formation ofupramolecular inclusion complex and in vitro drug release prop-rty of obtained CD-dendrimers using MTX as the drug model wastudied.
. Materials and methods
.1. Materials
�-CD (98%, Merck) was purified by recrystallization fromater and dried under vacuum. p-Toluenesulfonyl chloride
p-TsCl, Merck) was purified with chloroform and petroleumther. Propargyl alcohol (99%), 3-bromopropionic acid (97%),ethotrexate (MTX, 99%), trimethylsilyl chloride (99%), �-
lanine (99%), MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyletrazolium bromide), tert-butyldimethylchlorosilane (TBDMSCl,7%), CuSO4·5H2O (99%), CuBr (99%), 2,2′-bipyridine (bpy, 99%),imethyl sulfoxide (DMSO; 99.9%) and acetonitrile (99.8%) were
BDMS)7-�-CD-(spacer-�-CD)14 (11) via click reactions.
purchased from Sigma–Aldrich and used as received. N,N′-Dimethylformamide (DMF; 99.8%, Merck) was dried over CaH2and purified under reduced pressure. Pyridine (C5H5N, 99.8%,Sigma–Aldrich) was dried over CaH2 and purified. Triethylamine(Et3N; Merck) was dried over CaH2 and then distilled. Thionylchloride (SOCl2, Merck) was purified from boiled linseed oilformer to use. N-Methyl-2-pyrrolidone (NMP), 4-dimethylamino-pyridine (DMAP), sodium azide (NaN3, 99%) and acetone (99.8%)were obtained from Merck and used as received. Dialysis tub-ing (benzoylated, molecular cut-off 2000 Da) was purchased fromSigma–Aldrich. T47D, a human breast cancer cells was achievedfrom Pasteur Institute Cell Bank of Iran (Tehran, Iran). All the sol-vents were purified before use. Deionized water was used throughthe experiments.
2.2. Synthesis of per-6-(tert-butyldimethylsilyl)-ˇ-cyclodextrin(per-6-TBDMS-ˇ-CD) (2)
The preparation of this compound (2) (Scheme 1) was similar
to previous literature [39]. Dry �-cyclodextrin (1) (1 g, 0.88 mmol)in dry pyridine (10 mL) was added to a solution of TBDMSCl (1.6 g,10.6 mmol) in 15 mL of dry pyridine dropwise at 0 ◦C over 3.5 h.The temperature of the solution was increased from 0 ◦C to the
Biolog
rv(trTFca
4732(
2
irt(adepca(
2
CC(2
2
CC(2
22(
03d6wtwbps(cwp
(
Y. Toomari et al. / International Journal of
oom temperature and stirred for 18 h. It was then removed byacuum to give a white powder, which was taken up in CH2Cl230 mL). The organic phase was washed with KHSO4 (20 mL, 1 M)o eliminate any residual C5H5N, followed by brine solution. Byemoving the CH2Cl2 with rotary evaporator, the product, per-6-BDMS-�-CD (2) (1.55 g, 91%) was recovered as a white powder.or further purification the crude product was purified by silica gelhromatogram using EtOAc/n-hexane as eluent to afford pure (2)s a white solid.
1H NMR (400 MHz, CDCl3; ı, ppm): 6.71 (s, 7H), 5.26 (s, 7H),.94–4.89 (d, J = 3 Hz, 7H), 4.01 (dd, J = 9.5 Hz, 7H), 3.88 (dd, J = 11 Hz,H), 3.72 (bd, J = 11 Hz, 7H), 3.65 (dd, J = 9.5 Hz, 7H), 3.62 (bs, 7H),.57–3.55 (dd, J = 9.5 Hz, 7H), 0.06 (s, 21H) 0.86 (s, 63H), 0.03 (s,1H). FTIR (KBr, thin film; cm−1): 3396 (str. OH), 2950 and 2927str. C H), 1470 (ben. C H), 1253 (str. Si C), 1028 (str. C O).
.3. Synthesis of 3-azidopropionyl chloride (4)
To a solution of 3-bromopropionic acid (3) (8 g, 52.3 mmol)n CH3CN (80 mL) was added NaN3 (6.8 g, 104.6 mmol) and theeaction mixture was stirred at reflux for 5 h. After evapora-ion of the CH3CN, the residue was suspended in ethyl acetate100 mL) and extracted with HCl (0.1 N, 3 × 80 mL), H2O (3 × 80 mL)nd brine (1 × 60 mL). The organic phase was dried over anhy-rous MgSO4. By filtering the MgSO4 and evaporating by rotaryvaporation, 3-azidopropionic acid (82% yield) was obtained asowder after dried in vacuum [40]. 3-Azidopropionic acid washlorinated with refluxing in SOCl2 for 6 h at 70 ◦C and 3-zidopropionyl chloride obtained as the brown solid, yield 96%Scheme 1).
.3.1. 3-Azidopropionic acid1H NMR (400 MHz, CDCl3; ı, ppm): 3.16–3.12 (t, J = 6.4 Hz,
H2 C O), 2.66–2.63 (t, J = 6.4 Hz, CH2 N3). 13C NMR (100 MHz,DCl3; ı, ppm); 178 (C O), 45 (HO2C CH2 CH2 N2), 38HO2C CH2 CH2 N2). FTIR (KBr, thin film; cm−1): 3360 (str. OH),926 (str. CH), 2100 (str. N3), 1712 (str. C O), 1169 (C O).
.3.2. 3-Azidopropionyl chloride1H NMR (400 MHz, CDCl3; ı, ppm): 3.50–3.47 (t, J = 6.4 Hz,
H2 C O), 2.55–2.5 (t, J = 6.4 Hz, CH2 N3). 13C NMR (100 MHz,DCl3; ı, ppm); 175 (C O), 45 (ClOC CH2 CH2 N2), 32ClOC CH2 CH2 N2). FTIR (KBr, thin film; cm−1): 2930 (str. CH),102 (str. N3), 1817 (str. C O), 1172 (C O).
.4. Synthesis of per-6-(tert-butyldimethylsilyl)per-,3-di-O-(3-azido propionyl)-ˇ-cyclodextrinper-6-TBDMS-ˇ-CD-(N3)14) (5)
In a dry round-bottom flask, per-6-TBDMS-�-CD (2) (0.7 g,.36 mmol) was dissolved in 5 mL dry NMP and was cooled to 0 ◦C.-Azido propionyl chloride (4) (1.35 g, 0.72 mmol) was dissolved inry NMP (3 mL). Then this solution was added dropwise to the per--TBDMS-�-CD (2) solution. Afterward DMAP (0.044 g, 0.36 mmol)as added to the resulted solution at 0 ◦C. The temperature of
he solution was kept at 0 ◦C for 2.5 h and then the temperatureas gradually reached to room temperature in 22 h. The solvent of
rown solution was removed under reduced pressure. Then for theurifying, the syrup was concentrated, the powder was then dis-olved in CH2Cl2 (50 mL) and washed successfully with 1 M NaHCO32 × 15 mL) solution and water (2 × 15 mL). The organic phase wasombined and dried over anhydrous Na2SO4 overnight, the solvent
as removed and then crystallized in n-hexane to prepare a brownowder (Scheme 1) (0.8 g, yield 68%).1H NMR (400 MHz, DMSO-d6; ı, ppm): 5.33 (m, H-1, 7H), 4.87m, H3 and H5, 14H), 4.2 (m, H2, 7H), 3.86 (m, H6, 14H), 3.5 (m, H4,
ical Macromolecules 79 (2015) 883–893 885
7H), 2.27 (t, N3 CH2 CH2 C O, 28H), 1.9 (t, N3 CH2 CH2 C O,28H), 1.21 (s, tert-butyl groups, 63H), 0.07 (s, methyl groups, 42H).13C NMR (100 MHz, CDCl3; ı, ppm): 172.4 (C O), 107.96 (C1 �-CD), 83 (C4 �-CD), 80 (C2 �-CD), 72 (C3 and C5 �-CD), 64 (C6�-CD), 45 (O C CH2 CH2 N3 ), 37 (O C CH2 CH2 N3 ), 30.6( Si C(CH3)3), and 25.9 ( Si C(CH3)3). FTIR (KBr, thin film; cm−1):2927 and 2845 (str. CH), 2100 (str. N3), 1731 (str. C O), 1248 (str.Si O), 1156–1024 (str. C O).
2.5. Synthesis of 6-mono (p-toluenesulfonyl)-ˇ-cyclodextrin(ˇ-CD-OTs) (6)
6-Mono (p-toluenesulfonyl)-�-cyclodextrin derivative(Scheme 1) was prepared in line with a previously describedmethod [36]. The ultimate pure compound 6 was achieved as awhite powder. The yield was 30%.
�-CD-OTs: m.p.: 182–183 ◦C (dec.). 1H NMR (400 MHz, DMSO-d6; ı, ppm): 7.75–7.73 (d, 2H, Ar), 7.43–7.41 (d, 2H, Ar), 5.72 (m,OH-2, OH-3), 4.83 (7H, H-1), 4.76 (H-1′), 4.51 (m, 14H, OH-6),4.33–4.3 (m, H-6-OTs), 3.64–3.2 (m, 28H, H-3, H-4, H-5, H-2), 2.4 (s,3H, Me-OTs) ppm. FTIR (KBr, thin film; cm−1): 3382(str. OH), 2927(str. C H), 1645 (str. C C), 1601 (str. C C aromatic), 1414 (str. SO2,asym.), 1158 (str. SO2, sym.), 1027 (str. C O).
2.6. Synthesis of ˇ-alanine propargyl ester hydrochloride (9)
In order to synthesize �-alanine propargyl ester hydrochlo-ride (9) (Scheme 1), propargyl alcohol (8) (10 mL) was added toa mixture of �-alanine (7) (1 g, 11.25 mmol) and freshly distilledtrimethylsilyl chloride (2.45 mL, 22.5 mmol) with stirring. After15 h stirring under an atmosphere of argon at room temperature,the reaction solution was removed by rotary evaporator and theresidual dissolved in a smallest volume of methanol and the mix-ture was precipitated by adding ether. The precipitate was collectedby filtration and washed by ether twice (20 mL) and dried undervacuum. The yield was 95%.
1H NMR (400 MHz, CDCl3; ı, ppm): 4.7–4.65 (d, CH2 O),3.23–3.2 (H-alkyne), 2.86–2.84 (t, CH2 C O), 2.79–2.76(CH2 NH2). 13C NMR (100 MHz, CDCl3; ı, ppm): 173 (C O),78 (HC C), 77 (HC C), 54 (CH2 O), 36 (CH2 C O), 32 (CH2 NH2).FTIR (KBr, thin film; cm−1): 3245 (str. CH-alkyne), 2980 and 2826(str. CH), 2128 (str. C C), 1742 (str. C O), 1012 (C- O).
2.7. Synthesis of mono alkyne-terminated ˇ-CD on the primaryface (10)
The synthesis of alkyne-terminated �-CD (10) (Scheme 1) isdescribed as follows. To a solution of �-alanine propargyl esterhydrochloride (9) (0.21 g, 1.3 mmol) and triethylamine (0.12 g,0.43 mmol) in dry DMF (6 mL) was slowly added mono [6-O-(p-toluenesulfonyl)]-�-cyclodextrin (6) (0.56 g, 0.43 mmol) at 0 ◦Cwith stirring for 2 h under an atmosphere of argon. The solutionwas increased in temperature from 0 ◦C to room temperature andkept for 24 h, and then the reaction temperature was increased to35 ◦C and kept for 12 h. The solvent was removed under reducedpressure, and the crude product was precipitated by addition ofacetone as a brown solid. The brown precipitate was filtered off,and washed with water. Recrystallization from ethanol provided�-alanine propargyl ester modified �-CD (10) (65%) as brownpowder.
1H NMR (400 MHz, DMSO-d6; ı, ppm): 5.76–5.66 (m, OH-2,
OH-3), 4.88–4.82 (d, H-1 and H-1′), 4.68 (O CH2 C C), 4.56–4.49(m, 14H, OH-6), 3.61–3.2 (m, 29H, H-3, H-4, H-5, H-2, C CH),3.09–3.04 (t, CH2 NH), 2.96–2.93 (m, NH CH2 CH2 C O), 2.5(NH CH2 CH2 C O), 2.21–2.2 (m, NH). 13C NMR (100 MHz,
8 Biolog
CC�5(O1
2r
oiatC
(1dwbmracfctG(v
(arH(0(o((((
3t
2d
dB0tttpmw
C
86 Y. Toomari et al. / International Journal of
DCl3; ı, ppm): 170.4 (C O), 101.96 (C1 �-CD), 82 (C4 �-D), 78 (O CH2 C C), 73.3–73 (C3 and C5 �-CD), 72 (C2-CD and O CH2 C C), 60.5 (C6 �-CD), 53 (O CH2 C C),1 (NH CH2 CH2 C O), 45 (�-CD CH2 NH ) and 35NH CH2 CH2 C O). FTIR (KBr, thin film; cm−1): 3423 (str.H), 3245 (str. H C C), 2924 and 2856 (str. CH), 2126 (str. C C),740 (str. C O), 1080–1052 (str. C O).
.8. Synthesis of 14-arm star ˇ-CD dendrimer by click couplingeaction (TBDMS)7-ˇ-CD-(spacer-ˇ-CD)14 (11)
Biodendrimeric �-CD with 14 �-CD group at the peripheryf secondary face, assigned as (TBDMS)7-�-CD-(spacer-�-CD)14n this research, was prepared by click reaction between perzido-�-CD and mono alkyne-terminated �-CD (Scheme 1). Typicalechniques applied for the synthesis of (TBDMS)7-�-CD-(spacer-�-D)14 were as follows:
A mixture of mono alkyne-terminated �-CD precursor (10)3.08 g, 2.47 mmol) and per-6-TBDMS-�-CD-(N3)14 (5) (0.3 g,.65 mmol azide groups) were dissolved in dry DMF (3 mL) in ary flask with a magnetic stir bar. Then the mixture was purgedith argon for three times and CuBr (49.5 mg, 0.34 mmol) and 2,2′-
ipyridine (150 mg, 0.96 mmol) were then added. Immediately, theixture was purged with argon and the solution was stirred at
oom temperature for 1 d and then the reaction was continuedt 70 ◦C for 48 h. The crude product was obtained through pre-ipitation into acetone as a brown powder. Then, the residue wasrequently washed with NH4OH (10%) for elimination of remainingopper and finally washed with acetone and desiccated. The resul-ant crude product was purified by chromatography on a sephadex-50 column by deionized water as eluent. A fine brown solid
TBDMS)7-�-CD-(spacer-�-CD)14 (0.8 g, 45%) was obtained underacuum.
1H NMR (400 MHz, DMSO-d6; ı, ppm): 7.93 (s, triazole), 5.72m, OH-2, 3), 5.4 (s, triazole ring CH2 O C O), 4.94–4.89 (m, H-1nd H-1′ of �-CD), 4.5 (m, OH-6), 4.12 (t, O C CH2 CH2 triazoleing), 3.92–3.86 (m, �-CD CH2 NH ), 3.72–3.3 (m, H-3, H-4,-5, H-2, H-6 and H-6′), 2.87 (t, O C CH2 CH2 NH ), 2.72
t, O C CH2 CH2 triazole ring), 2.5 (t, O C CH2 CH2 NH ),.90 (s, tert-butyl groups), 0.07 (s, methyl groups). 13C NMR100 MHz, CDCl3; ı, ppm): 170.61 (C O), 143 (CH of triaz-le ring), 129.31 (C of triazole ring), 102 (C1 �-CD), 81.6C4 �-CD), 73.1 (C3 �-CD), 72.5–72.1 (C2 and C5 �-CD), 70C6 �-CD), 60.01 (triazole CH2 O ), 51 (�-CD CH2 NH ), 41O C CH2 CH2 NH ), 39 (O C CH2 CH2 triazole ring), 35.5O C CH2 CH2 NH ), 30 (O C CH2 CH2 triazole ring andSi C(CH3)3) and 25.08 ( Si C(CH3)3). FTIR (KBr, thin film; cm−1):350 (str. OH), 2921 (str. C H), 1724 (str. C O ester), 1658 (str.riazole ring), 1252 (str. Si O), 1148 and 1028 (str. C O).
.9. Determination of ˇ-CD in (TBDMS)7-ˇ-CD-(spacer-ˇ-CD)14endrimer
The �-CD content of the (TBDMS)7-�-CD-(spacer-�-CD)14 wasetermined according to the previously described method [41,42].riefly, the solution of dried compound 11 (25 mg) in H2SO4 (15 mL,.5 M) was refluxed at 100 ◦C for 10 h. The solution was then dilutedo 50 mL with H2SO4 (0.5 M). Then, the mixture of phenol solu-ion (5 wt%, 0.75 mL) with portion (1.25 mL) of diluted solution wasreated with concentrated H2SO4 (4.5 mL). The absorbance of the
repared solution was estimated by UV–vis spectrophotometricethod at 490 nm (A490 nm). The concentration of glucose (Cglucose)as estimated by a standard curve as follows:glucose = 0.301 A490 nm + 0.014
ical Macromolecules 79 (2015) 883–893
The amount of �-CD in compound 11 was calculated by thefollowing formula:
�-CD (%) = Cglucose (mg mL−1) × 50 mL × M�-CD/7
× Mglucose × Mcompound 11
Here, Mglucose, Mcompound 11 and M�-CD denotes the molecularweight of the glucose, (TBDMS)7-�-CD-(spacer-�-CD)14 and �-CD,respectively. The number of 7 is the content of glucose units in the�-CD part.
2.10. Solid samples preparation
An inclusion complexes of MTX with (TBDMS)7-�-CD-(spacer-�-CD)14 (11) and �-CD were prepared by the common co-precipitation method. Amounts of compound 11 (100 mg), �-CD(25 mg) and MTX (10 mg) were exactly weighed. To a solution ofcompound 11 and �-CD in distilled water with stirring at 50 ◦C, asolution of MTX in water was slowly added at the same temper-ature and rate. The mixtures were magnetically stirred for 24 h inthe dark at 37 ◦C to form drug inclusion complexes. After reach-ing equilibrium, the temperature of solutions were decreased to4 ◦C and maintained overnight at 4 ◦C. Finally, the cold precipi-tated MTX/compound 11 and MTX/�-CD inclusion complexes werefiltered and dried under vacuum. The obtained complexes werewashed with acetone to eliminate MTX which were absorbed onthe exterior of compound 11 and �-CD and then dried in desicca-tor at room temperature. In order to describe inclusion complex,the physical mixture of MTX and compound 11 was obtained bymixing MTX and compound 11 at the same proportion in a ceramicmortar till a uniform mixture.
2.11. Drug loading and in vitro release
The encapsulation efficiency (EE) of �-CD and nanocarrier com-pound 11 were calculated by the isolation of complexes from theaqueous phase having free MTX by centrifugation at 13,000 rpmfor 25 min under dark conditions. The content of MTX loaded intothe �-CD and nanocarrier compound 11 were evaluated as thedifference between the total content used to obtain the inclusioncomplex of �-CD, nanocarrier compound 11 and free MTX contentin the solution. The content of free MTX in the solutions were ana-lyzed by spectrophotometer at � = 303 nm in appropriate dilutionwith HCl (0.1 N, 5 mL). The EE is defined as following:
EE (%) = (Mass of drug in �-CD or compound 11 (mg))
/(Mass of feed drug (mg)) × 100
In vitro release of MTX from �-CD and nanocarrier compound11 formulation were evaluated using a dialysis technique in phos-phate buffer saline (PBS) pH 7.4, sodium acetate buffer solutionpH 5 and sodium acetate buffer solution pH 3 as release media.The powdered inclusion complexes (30 mg) were dissolved in 3 mLof desired buffer solutions. The solution was then taken in a dial-ysis bag (molecular cut-off 2000) and placed in 30 mL of releasemedium with stirring at 100 rpm at 37 ± 0.5 ◦C. After each with-drawal, medium was replaced with equal volume of fresh releasemedium for analysis. The release study was performed for 72 h.The content of drug release was studied by UV spectrophotometerat � = 303 nm (pH 7.4 and 5) and � = 307 nm (pH 3). Each samplewas tested in triplicate.
2.12. Characterization
2.12.1. Nuclear magnetic resonance spectroscopy (NMR)All NMR spectra were recorded on a Bruker Avance spectrom-
eter (400 MHz for 1H and 100 MHz for 13C) used in the Fourier

Biolog
tu
2
tt
2
flspf
2
Tipue
2
sSgcBo
2
tbf
2
fcctdsft23otcv5t
3
sspss
the appearance of new absorbance alkyne peak at 3245 and
Y. Toomari et al. / International Journal of
ransform mode at room temperature. DMSO-d6 and CDCl3 weresed as the solvent. The data are defined as chemical shift (ı, ppm).
.12.2. Fourier transform infrared spectroscopy (FTIR)The FTIR spectra were recorded on a Bruker Tensor 27 spec-
rophotometer by KBr disk method at frequencies ranging from 400o 4000 cm−1.
.12.3. Dynamic light scattering (DLS)DLS experiments were carried out at 25 ◦C using Nanotrac Wave
rom Microtrac instruments at scattering angle of 170◦ and a He–Neaser (10 mW, 780 nm). The experiments were analyzed in CONTINtyle. All samples for DLS were filtered through a 0.45 �m filterrior to measurements. Triplicate measurements were performedor all samples.
.12.4. Differential scanning calorimetry (DSC)DSC curves were recorded on a Linseis-L 63/45 DSC instrument.
hermal behaviors of the samples (5 ± 0.05 mg) were examinedn sealed aluminum pans. The heating of powder samples wereerformed between 20 and 400 ◦C at scanning rate of 10 ◦C min−1
nder argon flow of 40 mL/min. An empty pan was used as a refer-nce.
.12.5. Scanning electron microscopy (SEM)Images of the MTX, compound 11, their supramolecular inclu-
ion complexes and physical mixtures were recorded using aEM (MIRA3 FEG-SEM), Tescan on aluminum stumps attached toraphite support parts using carbon adhesive tape. Samples wereoated with gold–palladium (plasma deposition method) with aIO-RAD AC500. Images were obtained at an accelerating voltagef 15 kV.
.12.6. UV–vis spectral analysis (UV-vis)The formation of complex in solution state was analyzed
hrough UV–vis spectrophotometer. UV–vis spectra were recordedy 1700 Shimadzu spectrophotometer in the wavelength rangerom 200 to 400 nm. Each test was performed in triplicate.
.12.7. MTT assay and cell treatmentThe in vitro cytotoxic effect of �-CD dendrimer on the secondary
ace, MTX, and their inclusion complex on T47D (human breast can-er cells) was evaluated by MTT tests. Briefly, 5000 cell/well wereultured in a 96-well culture plate and incubated at 37 ◦C for 24 ho allow cell attachment. The moderate was replaced every otheray. The cells were incubated with the �-CD dendrimer, MTX andupramolecular inclusion complex at concentrations of 2–64 �Mor 72 h in the quadruplicate manner. After incubation, the cul-ure medium was completely removed, and MTT solution (in 50-�L,
mg/mL) was added to each wells and further incubated for 4.5 h at7 ◦C, monitored by addition of 150 �L DMSO. Then, the absorbancef the solution was measured in a plate reader spectrophotome-er at 570 nm. Cell viability (%) was calculated through the livingells (%) comparative to controls. An inhibitory concentration (IC50)alue was distinct as the drug concentration of compound at which0% cell growing reserved. Then the IC50 value was considered byhe Prism 6.0 software (Graphpad, San Diego, USA).
. Results and discussion
The detailed synthetic route for biodendrimeric �-CD (11) washown in Scheme 1. As shown, biodendrimeric �-CD (11) on the
econdary face containing �-CD in both core and surface was pre-ared via click reaction. First, per-6-TBDMS-�-CD-(N3)14 (5) wasynthesized as the core molecule with terminal azide groups on theecondary face. Second, a novel mono alkyne-terminated �-CD (10)ical Macromolecules 79 (2015) 883–893 887
precursor was synthesized. Finally, azido groups were grafted to thejunction points of mono alkyne-terminated �-CD group via clickreaction to prepare �-CD dendrimer (11) on the secondary face.
3.1. Preparation of per azide-functionalized ˇ-CD precursor onthe secondary face (5)
The compound per azide-functionalized �-CD precursor on thesecondary face (5) was synthesized from �-CD in three steps(Scheme 1). The synthetic process for the preparation of 5 is basedon an ester bond between the 3-azidopropionyl chloride (4) andper-6-(tert-butyldimethylsilyl)-�-cyclodextrin (2) moiety. For thispurpose, the primary hydroxyl groups at O-6 position of �-CD wereprotected with TBDMSCl in dry pyridine [39]. Then, the use of 28equivalent of 3-azidopropionyl chloride and 28 equivalent of DMAPled to the pure fully acylated product 5 with 14 azido terminatedspacer at secondary face of the �-CD. The compound 5 is recrys-tallized from n-hexane (yield 68%). The structures of compoundswere confirmed by FTIR and 1H and 13C NMR spectroscopy. The 1HNMR spectrum of per-6-(tert-butyldimethylsilyl)-�-cyclodextrin(2) revealed the complete vanishing of OH-6 of �-CD signal at4.5 ppm. The study of per azide-functionalized �-CD precursor onthe secondary face by 1H NMR in DMSO-d6 showed the thoroughvanishing of hydroxyl proton signals OH-2 and OH-3 at 5.26 ppm,which was existent in �-CD. According to the corresponding 1HNMR spectrum with integration values, the successful acylationreaction was proved between compound 2 and 4 (Scheme 1).These experiments demonstrated that 3-azidopropionyl chloride(4) spacer can be efficiently attached to secondary hydroxylgroups of per-6-(tert-butyldimethylsilyl)-�-cyclodextrin (2). 13CNMR analysis of (5) showed the appearance of signals at 172.4 ppm,which could be ascribed to carbonyl of ester. FTIR analysis (Fig. 1)of per azide-functionalized �-CD (5) showed the appearance ofnew absorbance azide peak at 2100 cm−1, ester peak at 1731 cm−1
and disappearance of hydroxyl absorbance peaks at 3396 cm−1. Theachieved results established that the secondary hydroxyl groups of�-CD were selectively substituted by spacer containing terminatedazide groups. This substitution by azido-terminated spacer on thesecondary face of �-CD was confirmed by 1H and 13C NMR and FTIR.
3.2. Preparation of mono-functionalized alkyne-terminated ˇ-CDprecursor (10)
The multistep synthetic method to fabricate of mono-functionalized alkyne-terminated �-CD (10) is as follows(Scheme 1): compound 9 was achieved by the treatment of�-alanine and propargyl alcohol in the presence of trimethylsilylchloride. Then �-CD-OTs (6) was prepared and converted to(10) by substitution of the tosyl group with �-alanine propargylester via nucleophilic substitution (Scheme 1) in the presenceof triethylamine. FTIR, 1H and 13C NMR analysis were used toapprove the building and purity of compound 10. In the 1H NMRspectrum of �-CD-OTs (6) analytic peaks characteristic of tosylgroup were shown, especially the peaks of aromatic hydrogens oftosyl group at 7.75–7.73 and 7.43–7.41 ppm, methylene adjacentto the sulfonyl group at 4.33–4.3 ppm, and methyl group of thetosyl moiety at 2.4 ppm. In the spectrum of compound 10, allsignals relative to the tosyl group vanished after reaction of with�-alanine propargyl ester. 13C NMR analysis of (10) showed theappearance of signals at 170.4 ppm, which could be attributed tocarbonyl of ester. FTIR analysis (Fig. 1) of compound 10 showed
2126 cm−1, ester peak at 1740 cm−1 and disappearance of peaksat 1601, 1414 and 1158 cm−1 (consistent to the aromatic sectionand O S O) and confirmed the substitution of tosyl moiety withmono alkyne terminated spacer.

888 Y. Toomari et al. / International Journal of Biolog
Fig. 1. FTIR spectra of (a) 3-azidopropionic acid (3), (b) 3-azidopropionyl chlo-ride (4), (c) per-6-TBDMS-�-CD (2), (d) per-6-TBDMS-�-CD-(N3)14 (5), (e) monoalkyne-terminated �-CD (10), (f) (TBDMS)7-�-CD-(spacer-�-CD)14 (11), (g) inclu-sion complex, (h) respective physical mixture and (i) pure MTX.
ical Macromolecules 79 (2015) 883–893
3.3. Synthesis of (TBDMS)7-ˇ-CD-(spacer-ˇ-CD)14 dendrimer byclick reaction (11)
Compound (TBDMS)7-�-CD-(spacer-�-CD)14 (11) was preparedby Cu(I)-catalyzed click reaction between per-6-TBDMS-�-CD-(N3)14 (5) and alkyne-terminated �-CD (10). The structure ofcompound 11 was characterized and confirmed by 1H, 13C NMRand FTIR. The 1H NMR spectrum of this compound and the relatedsignal assignments are illustrated in Fig. 2. The 1H NMR spectrumof compound 11 (Fig. 2) shows not only the typical resonancepeaks at ı = 4.5 (OH-6 of �-CD), 4.94–4.89 (H-1 of �-CD) and 5.72(OH-2, OH-3 of �-CD) for the �-CD units, but also the proton sig-nals at ı = 5.4, 4.12, 2.87, 2.72, 2.5 (protons of spacer) and ı = 7.93(protons of triazole ring), corresponding to the protons in spacergroups. By click reaction 1H NMR study showed the presence of atriazole proton at 7.93 ppm and vanishing of the terminal alkyneproton, which indicated the formation of the compound 11 com-pletely (Scheme 1). The integral ratio of triazole proton (placed at7.93 ppm) to anomeric proton (placed at 4.94–4.89 ppm) is about1:7.23, which shows the successful “click” reaction between com-pound 5 and 10. 13C NMR spectrum and the assignments shown inFig. 3 further confirmed the formation of the desired compound 11.The appearance of signals at 143, 129.2 ppm for carbon of triazolering was confirmed by 13C NMR analyses. In addition, the structureof target molecule can be also checked by FTIR spectra, as shownin Fig. 1. Appearance of triazole ring peaks at 1658 cm−1 and disap-pearance of azide peak at 2100 cm−1 and alkyne peak at 2126 cm−1
in the FTIR spectra of compound 11 confirmed a successful clickreaction.
3.4. Characterization of MTX:(TBDMS)7-ˇ-CD-(spacer-ˇ-CD)14inclusion complex
3.4.1. FTIR study of complexThe FTIR analysis is a valuable technique in studying the
supramolecular inclusion complex. The FTIR spectra of (TBDMS)7-�-CD-(spacer-�-CD)14 (11), MTX, the physical mixture andrespective inclusion complex are shown in Fig. 1. Compound 11showed stretching vibration band at 3350 cm−1 (O H), 2922 cm−1
(C H), 1724 cm−1 (C O), 1658 cm−1 (triazole ring), 1252 cm−1
(Si O), and 1148 and 1032 cm−1 (O C O). The FTIR spectrum ofMTX (Fig. 1) showed characteristic stretching vibration bands at1706 cm−1 (C O of carboxylic acid groups), 1657 cm−1 (C O ofamide groups), 1595 and 1450 cm−1 (C C), 1203 and 1097 cm−1
(C O). The FTIR spectra of the physical mixtures MTX/compound11 displayed approximate mixed spectra of MTX and compound 11with slight shift. In the spectrum of inclusion complex, the bandsplaced at 3402, 2915, 1705, 1634, 1540 and 1396 cm−1 shifted tolower or higher frequencies with decreases in intensity, and theother characteristic peaks of MTX were almost totally covered bythe bands of compound 11. The differences between the physicalmixture and inclusion complex indicate that MTX were includedinto the dendrimer cavity to form the supramolecular inclusioncomplex.
3.4.2. Determination of ˇ-CD in (TBDMS)7-ˇ-CD-(spacer-ˇ-CD)14 dendrimer
The content of �-CD in the compound 11 was measured by theKoh and Tucker method [41,42] and the result showed that com-pound 11 had 81.04% �-CD in its structure, which is analogous tothe actual amount (79.65%).
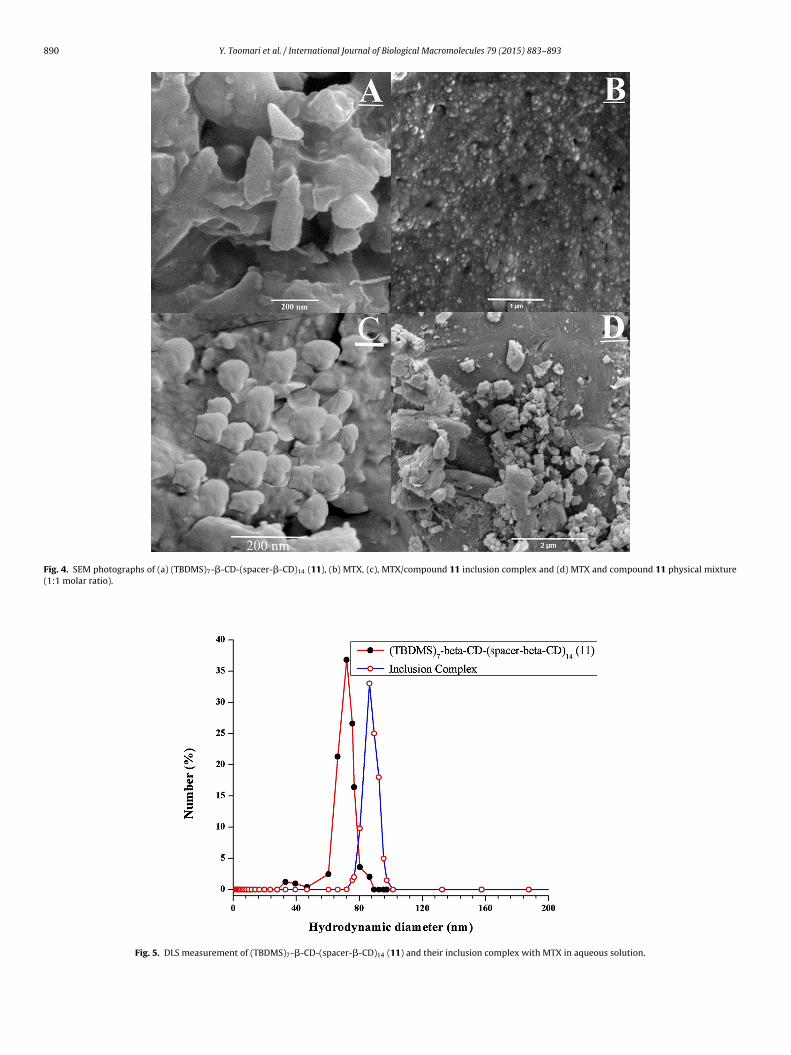
3.4.3. SEM analysisThe surface morphology of pure MTX, (TBDMS)7-�-CD-(spacer-
�-CD)14 dendrimer (11), their inclusion complex and physicalmixture were examined by SEM analysis (Fig. 4). As displayed

Y. Toomari et al. / International Journal of Biological Macromolecules 79 (2015) 883–893 889
D-(sp
ispipstc
Fig. 2. 1H NMR spectrum of (TBDMS)7-�-C
n SEM images (i) (TBDMS)7-�-CD-(spacer-�-CD)14 dendrimerhowed a crystalline cone-like structure, (ii) MTX showed amor-hous and crystalline shapes, (iii) the inclusion complex showed
rregular spherical crystals and (iv) their physical mixture showed
arts of MTX surrounded with compound 11. In this complex,izes and shapes of MTX and compound 11 were changed, andhe initial morphologies of both constituent vanished. These out-omes in morphology, confirmed the formation of a solid inclusionFig. 3. 13C NMR spectrum of (TBDMS)7-�-CD-(sp
acer-�-CD)14 dendrimer (11) in DMSO-d6.
complex with dendritic network of (TBDMS)7-�-CD-(spacer-�-CD)14 dendrimer (11) or nano cavity of �-CD.
3.4.4. DLS analysis
To calculate the number-average diameter of compounds andformation of supramolecular complex, DLS studies were performedin water (Fig. 5). According to DLS results, a number-average diame-ter of 72 ± 2.8 nm for (TBDMS)7-�-CD-(spacer-�-CD)14 dendrimer
acer-�-CD)14 dendrimer (11) in DMSO-d6.
890 Y. Toomari et al. / International Journal of Biological Macromolecules 79 (2015) 883–893
Fig. 4. SEM photographs of (a) (TBDMS)7-�-CD-(spacer-�-CD)14 (11), (b) MTX, (c), MTX/compound 11 inclusion complex and (d) MTX and compound 11 physical mixture(1:1 molar ratio).
Fig. 5. DLS measurement of (TBDMS)7-�-CD-(spacer-�-CD)14 (11) and their inclusion complex with MTX in aqueous solution.

Y. Toomari et al. / International Journal of Biolog
F1
(Tsc
3
odoam1p�2
F
ig. 6. DSC curves of (a) (TBDMS)7-�-CD-(spacer-�-CD)14 (11); (b); MTX/compound1 inclusion complex (c) physical mixture and (d) MTX.
11) and 86 ± 2.5 nm for its supramolecular complex was found.he exchange in dimension is because of the formation of inclu-ion complex at the network of dendrimer and or cyclodextrinavity.
.4.5. DSC analysisThe DSC studies were performed to characterize the formation
f inclusion complex between (TBDMS)7-�-CD-(spacer-�-CD)14endrimer (11) and MTX. The effect of compound 11 on the curvebtained by DSC was detected for disappearance, shifting andppearance of novel peaks. Results are shown in Fig. 6. The ther-ogram of MTX exhibited two endothermic peaks at 122.9 and
◦
80.9 C corresponding to their relevant dehydration and meltingoints, respectively. The thermogram of (TBDMS)7-�-CD-(spacer--CD)14 dendrimer (11) exhibited three endothermic peak at 137,36 and 320 ◦C matching to dehydration, phase transition (whereig. 7. Cumulative drug release (%) of MTX from �-CD (pH 3 and 7.4) and MTX:�-CD dend
ical Macromolecules 79 (2015) 883–893 891
the breakage of the ester bond may also occur) and melting points,respectively. The DSC pattern of the physical mixture exhibitedboth characteristic peaks of MTX and (TBDMS)7-�-CD-(spacer-�-CD)14 dendrimer (11) with a slight shift. The DSC thermogram of theinclusion complex obtained by co-precipitation method indicatedthat the dehydration process at 141 ◦C (where the decomplexa-tion drug may also occur) and melting/decomposition of MTX at207 ◦C with shifting value. Also, it appears that the complexationof the MTX into the compound 11 increases the thermal stabil-ity. This obviously recommended the successful inclusion complexformation of the MTX within the (TBDMS)7-�-CD-(spacer-�-CD)14dendrimer (11) cavity.
3.5. Drug loading and in vitro drug release study
In this work, UV spectrophotometric technique was estab-lished and confirmed for calculation of MTX amount encapsulatedinto �-CD and dendrimer (11) and released from these materi-als. Drug EE of (TBDMS)7-�-CD-(spacer-�-CD)14 dendrimer (11)and �-CD were found to be 84.7% and 52%, respectively. From theresults achieved by drug EE of MTX with compound (11) and �-CD(comparison of �-CD dendrimer (11) with �-CD), it seemed thatapproximately 52% MTX may be encapsulated in the cavity of �-CD and 32.7% MTX was entrapped in the dendritic networks. TheEE of �-CD dendrimer (11) significantly improved compare with�-CD. The high drug loading in compound 11 could be ascribedto the entrapment of drug into dendritic network and �-CD cav-ity, in which the inclusion of �-CD cavity to MTX made a greaterinvolvement for MTX loading. The high MTX loading in dendriticnanocarrier may be ascribed to the following reasons: (a) inclusionof �-CD cavity to MTX, (b) the hydrogen bonding might be existingbetween the COO− groups and amino groups in MTX molecules (thenet charge of MTX was negative at the drug loading medium (pH 7)(MTX pKa 3.8, 4.8 and 5.6) [43]) with the hydroxyl groups of �-CDand amine groups of spacer in compound 11 at pH 7. High encap-sulation efficiency values indicate the advantage of compound 11over the single �-CD. The in vitro cumulative release pattern of
MTX from the MTX/compound 11 and MTX/�-CD were evaluatedat pH 3, 5 (lysosomal pH) and 7.4 (physiological pH) and the tem-perature of 37 ± 0.5 ◦C for 72 h (Fig. 7). As shown in Fig. 7 the initialburst release (about 20.4% of the drug was released within the firstrimer nanoparticles in buffer solution (pH 3, 5 and 7.4) at 37 ◦C (Mean ± SD; n = 3).

892 Y. Toomari et al. / International Journal of Biolog
Fr
3SmbtooTapapt�trrcsTa
3
tbtb(e3ai3eccwM
4
t(
[[[[[[[[
[[[
[[[[
[
[
[[
[
[[[[
[
[35] G.R. Newkome, L.A. Godinez, C.N. Moorefield, Chem. Commun. (1998)
ig. 8. In vitro cytotoxicity of nanocarrier, MTX and inclusion complex of nanocar-ier:MTX measured by the MTT test against T47D cells (Mean ± SD; n = 3).
.6 h) followed by the sustained release up to 72 h for compound 11.ustained release was related to drug diffusion from the dendriticatrix, CD cavity and the dendrimer degradation and the primary
urst release perhaps initiated by the surface absorbent drug, sincehe MTX might form hydrogen bonds with the hydroxyl groupsf dendrimer. In this case, approximately 85.3%, 81.7% and 77.5%f loaded MTX released at pH 3, 5 and 7 after 72 h, respectively.he cumulative release patterns after primary burst displayed thatlmost 64.9%, 61.3% and 57.1% of MTX was released at pH 3, 5 andH 7.4, respectively. Moreover, compound 11 was showed to have
more sustained release behavior for the loaded MTX when com-ared �-CD. These results can be due to the lower solubility ofhe MTX/�-CD in pH 7.4 than in pH 3. It has been described that-CD could increase the release of hydrophobic drugs or dissolu-
ion profiles in acidic conditions [44]. Also, may be the electrostaticepulsion between largely protonated hydroxyl groups of nanocar-ier and amino groups of MTX which were generated in acidic pHan increase the drug release from the nanocarrier. These outcomeshowed that the release process was noticeably pH dependent.hese results exhibited that the synthesized nanocarrier could be
useful controlled DDS for cancer treatment.
.6. MTT assay
The cytotoxicity is a significant subject for both the carrier andhe drug. The aim of this biological experiment is to estimate theiocompatibility of the prepared dendrimer. A main difference inhe in vitro cytotoxic effect profiles of the nanocarrier and MTX cane detected from Fig. 8. As shown in Fig. 8, the (TBDMS)7-�-CD-spacer-�-CD)14 dendrimer (11) practically had very low cytotoxicffect on T47D cells in the concentrations extended as high as2 �M through the incubation period. Up to 90% cell viability waschieved. Data analysis of MTT test exhibited that IC50 of MTX andnclusion complex of compound 11:MTX on T47D cells was 7.4 and.2 �M for 72 h MTT tests, respectively. Commonly, drugs typicallymploy a somewhat upper toxicity in living cells. Then the lowytotoxic effect showed that the MTX-loaded dendrimer structuresould continue a low cytotoxicity even MTX was loaded in the net-ork and cavity of dendrimer. Also, controlled release property ofTX from this nanocarrier can be endorsed.
. Conclusions
In conclusion, we have demonstrated a facile strategy forhe fabrication of (TBDMS)7-�-CD-(spacer-�-CD)14 dendrimer11) from per-6-TBDMS-�-CD-(N3)14 (5) core moiety and mono
[
[
ical Macromolecules 79 (2015) 883–893
alkyne-terminated �-CD (10) by azide-alkyne click coupling reac-tion and its chemical structure was confirmed by NMR, FTIR andDLS analysis. The obtained dendrimer was employed as a hostcompound to fabricate supramolecular inclusion complex havingMTX as drug model. The formation of inclusion complex of theobtained dendrimer with MTX was confirmed by SEM, DLS, DSCand FTIR techniques. The obtained compound presented higherencapsulation efficiency (due to presences of dendritic networkand cavity of �-CD) and pH-sensing controlled release profile ofMTX. The in vitro toxicity results confirmed that compound 11has very low toxicity on T47D cells. Therefore, this compoundcould be utilized as controlled release nanocarrier in DDS. Thein vitro drug release results in buffer solution exhibited that thecompound 11 could be an appropriate controlled DDS for cancertreatment.
Acknowledgements
Authors thankfully acknowledge the financial funding from theUniversity of Tabriz and RCPN of Tabriz University of MedicalScience.
References
[1] D.A. Tomalia, H. Baker, J.R. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J.Ryder, P. Smith, Polym. J. 17 (1985) 117–132.
[2] H. Namazi, M. Adeli, Biomaterials 26 (2005) 1175–1183.[3] H. Namazi, S. Jafariradl, J. Appl. Polym. Sci. 117 (2010) 1085–1094.[4] K. Milowska, T. Gabryelak, M. Bryszewska, A.-M. Caminade, J.-P. Majoral, Int. J.
Biol. Macromol. 50 (2012) 1138–1143.[5] E. Buhleier, W. Wehner, F. Vögtle, Synthesis (1978) 155–158.[6] G.R. Newkome, Z. Yao, G.R. Baker, V.K. Gupta, J. Org. Chem. 50 (1985)
2003–2004.[7] J.F. Jansen, E.M. de Brabander-van den Berg, E.W. Meijer, Science 266 (1994)
1226–1229.[8] C.M. Paleos, D. Tsiourvas, Z. Sideratou, Mol. Pharm. 4 (2007) 169–188.[9] H. Arima, Y. Chihara, M. Arizono, S. Yamashita, K. Wada, F. Hirayama, K.
Uekama, J. Control. Release 116 (2006) 64–74.10] H. Namazi, A. Heydari, Polym. Int. 63 (2014) 1447–1455.11] H. Namazi, Y. Toomari, Adv. Pharm. Bull. 1 (2011) 40–47.12] H. Namazi, S.H. Motamedi, M. Namvari, BioImpacts 1 (2011) 63–69.13] H. Namazi, S. Jafarirad, Int. J. Pharm. 407 (2011) 167–173.14] M. Sun, A. Fan, Z. Wang, Y. Zhao, Soft Matter. 8 (2012) 4301–4305.15] H. Ritter, M. Tabatabai, Prog. Polym. Sci. 27 (2002) 1713–1720.16] C.J. Hawker, A.W. Bosman, E. Harth, Chem. Rev. 101 (2001) 3661–3688.17] W.R. Saenger, J. Jacob, K. Gessler, T. Steiner, D. Hoffmann, H. Sanbe, K.
Koizumi, S.M. Smith, T. Takaha, Chem. Rev. 98 (1998) 1787–1802.18] H. Namazi, A. Kanani, J. Bioact. Compat. Polym. 22 (2007) 77–88.19] H. Namazi, A. Kanani, Carbohydr. Polym. 76 (2009) 46–50.20] W. Buranaboripan, W. Lang, E. Motomura, N. Sakairi, Int. J. Biol. Macromol. 69
(2014) 27–34.21] K. Uekama, F. Hirayama, T. Irie, Chem. Rev. 98 (1998) 2045–2076.22] M.V. Rekharsky, Y. Inoue, Chem. Rev. 98 (1998) 1875–1918.23] T. Loftsson, M.E. Brewster, J. Pharm. Sci. 85 (1996) 1017–1025.24] L. Haidong, Y. Fang, T. Zhihong, R. Changle, Int. J. Biol. Macromol. 49 (2011)
561–566.25] T. Loftsson, H. Frikdriksdóttir, A.M. Sigurkdardóttir, H. Ueda, Int. J. Pharm. 110
(1994) 169–177.26] G. Dollo, P. Le Corre, F. Chevanne, R. Le Verge, Int. J. Pharm. 131 (1996)
219–228.27] H. Kono, T. Teshirogi, Int. J. Biol. Macromol. 72 (2015) 299–308.28] J. Chao, H. Wang, W. Zhao, M. Zhang, L. Zhang, Int. J. Biol. Macromol. 50 (2012)
277–282.29] Y.M. Chabre, C. Contino-Pepin, V. Placide, T.C. Shiao, R. Roy, J. Org. Chem. 73
(2008) 5602–5605.30] Y.M. Chabre, R. Roy, Adv. Carbohydr. Chem. Biochem. 63 (2010) 165–393.31] M.J. Cloninger, Curr. Opin. Chem. Biol. 6 (2002) 742–748.32] M. Adeli, M. Kalantari, Z. Zarnega, R. Kabiri, RSC Adv. 2 (2012) 2756–2758.33] S. Menuel, R.E. Duval, D. Cuc, P. Mutzenhardt, A. Marsura, New J. Chem. 31
(2007) 995–1000.34] S. Menuel, S. Fontanay, I. Clarot, R.E. Duval, L. Diez, A. Marsura, Bioconjug.
Chem. 19 (2008) 2357–2362.
1821–1822.36] I. Baussanne, J.M. Benito, C.O. Mellet, J.M. Garcia Fernandez, H. Law, J. Defaye,
Chem. Commun. (2000) 1489–1490.37] J.M. Benito, M. Gomez-Garcia, C. Ortiz Mellet, I. Baussanne, J. Defaye, J.M.
Garcia Fernandez, J. Am. Chem. Soc. 126 (2004) 10355–10363.

Biolog
[[
[
Y. Toomari et al. / International Journal of
38] H. Wang, N. Shao, S. Qiao, Y. Cheng, J. Phys. Chem. B 116 (2012) 11217–11224.39] P.R. Ashton, R. Königer, J.F. Stoddart, D. Alker, V.D. Harding, J. Org. Chem. 61
(1996) 903–908.40] R. Srinivasan, L.P. Tan, H. Wu, P.Y. Yang, K.A. Kalesh, S.Q. Yao, Org. Biomol.
Chem. 7 (2009) 1821–1828.
[[[
[
ical Macromolecules 79 (2015) 883–893 893
41] J. Xu, X. Li, F. Sun, Acta Biomater. 6 (2010) 486–493.42] K. Geok-Lay, I.G. Tucker, Int. J. Pharm. 34 (1986) 183–184.43] H.K. Kimelberg, T.F. Tracy Jr., S.M. Biddlecome, R.S. Bourke, Cancer Res. 36
(1976) 2949–2957.44] M. Stojanov, K.L. Larsen, Drug. Dev. Ind. Pharm. 38 (2012) 1061–1067.















![Chiral separation by a monofunctionalized cyclodextrin derivative: From selector to permethyl-[beta]-cyclodextrin bonded stationary phase](https://static.fdokumen.com/doc/165x107/63327b24576b626f850d70ad/chiral-separation-by-a-monofunctionalized-cyclodextrin-derivative-from-selector.jpg)




