
A methodology to reduce thermal gradients due to the exothermic reactions in composites processing
Upload
independentCategory
view
5download
0
Physica D 146 (2000) 261–274
Exothermic and endothermic chemical reactions involvingvery many particles modeled with molecular dynamics
Witold Aldaa, David A. Yuenb,∗, Hans-Peter Lüthic, James R. Rustadda Institute of Computer Science, University of Mining and Metallurgy AGH, 30-059 Krakow, Poland
b Department of Geology and Geophysics and Minnesota Supercomputing Institute,University of Minnesota, 1200 Washington Ave. South, Minneapolis, MN 55415-1227, USA
c Swiss Center for Scientific Computing, ETH, Zurich CH-8092, Switzerlandd The Pacific Northwest National Lab, W.R. Wiley EMSL, Richland, WA 99352, USA
Received 5 August 1999; accepted 3 July 2000Communicated by R.P. Behringer
Abstract
The traditional continuum approach of modeling chemical reactions with specified kinetic rates suffers from numericaldifficulties in reactive flows and other highly non-equilibrium situations due to the stiffness of the differential equations inboth space and time. These drawbacks can be eliminated within the framework of the discrete-particle approach in whichthe chemical reactions are modeled by means of two-body interparticle potentials with classical molecular dynamics. Herewe present a simple model for prescribing the binary endothermic and exothermic reactions of the type A+ B → C, in thepresence of many (greater than 104) reacting particles. Two-body Lennard–Jones based potentials,Vij , have been utilized, inwhich the reaction takes place via an attractive potentialVAB with a deep enough potential well. The other potentialsVAC
andVBC have deeper wells thanVAB for creating a suitable situation for inducing exothermic reactions, while a high-energyplateau forVAC andVBC is used for simulating endothermic reactions. We have deployed 20 000 to 100 000 particles in acomputational domain with an area of around 1 million Å2. These ensembles have all been integrated out to around the order ofnanoseconds. We have examined the bimolecular reactions between two layers initially consisting of A particles lying atop Bparticles, equally divided in the total population. Reactions take place first along this boundary. There is a nonlinear thresholdphenomenon in the exothermic reactions, which are greatly accelerated by the heat liberated from the neighboring reactions.In contrast, the endothermic reactions are more subdued, as there is no positive feedback. Exothermic reactions occur in aspatially heterogeneous manner, especially at the lower temperatures, in which no single reaction rate can be assigned to theentire reaction front. More coherent spatial structures are found with decreasing temperature. These spatial structures caninfluence the overall flow field associated with the motions of the reacting particles. Timescales of the reactions are greatlylengthened by a reduction in the temperature in a threshold manner. © 2000 Published by Elsevier Science B.V.
Keywords:Exothermic and endothermic reactions; Molecular dynamics; Lennard–Jones potential
∗ Corresponding author. Tel.:+1-612-625-1818;fax: +1-612-624-8861E-mail address:[email protected] (D.A. Yuen).
1. Introduction
Today chemical reacting flows form indeed impor-tant and fundamental foci for many scientific and engi-neering endeavors. Numerous examples can be drawnfrom fields as diverse as metamorphic reacting fronts
0167-2789/00/$ – see front matter © 2000 Published by Elsevier Science B.V.PII: S0167-2789(00)00150-0

262 W. Alda et al. / Physica D 146 (2000) 261–274
imprinted in Alpine rocks and the ionization processesaccompanying the reentry of hypersonic vehicles de-scending back into the earth’s atmosphere. Most of theemphasis on the study of chemically reacting flows hasbeen directed toward the continuum approach basedon solving nonlinear partial differential equations withthe chemical reactions added in as a set of nonlinearordinary differential equations in time. These chem-ical conservation relations are generally prescribedby given kinetic-rate laws, which are thermally acti-vated, and hence strongly temperature-dependent inthe Arrhenius sense. However, solving these coupledspecies-energy-momentum equations is beset by themultiple-scale nature of the coupled equations, whichbaffle even the best of numerical methods, especiallyat the chemically reacting fronts with sharp gradients.
The stiffness of the partial differential equationsarising from the use of both the spatial and temporaloperators in the continuum approach can be amelio-rated by using the discrete methods, such as cellularautomata, lattice-gas, lattice-Boltzmann and molecu-lar dynamics (MD), as they operate over much shortertimescales, many orders in magnitude smaller thanthe time steps taken in the continuum approach. Theabove methods are based on very simplified modelsand often they do not give accurate or even realis-tic results, but according to Binder et al. [1], “. . .
the goal of such simulations is not to predict num-bers but to provide better understanding”. In our case,this means a better understanding of the dynamics ofchemical reaction processes with many particles inhighly non-equilibrium situations.
Cellular-automaton methods have been used tostudy diffusion and reaction processes by Mai andvon Niessen [2], Chopard and Drozd [3] as well asWeimar and Boon [4]. There have been some previousmodeling of chemical reactions at mineral surfacesby lattice gas automata (LGA) model by Boon et al.[5], Wells et al. [6] as well as Rothman and Zaléski[7]. They found that LGA calculations appeared to bewell suited for problems in the mesoscopic scale asin the range of pore-fluid domain, of the order of tensof microns.
Classical MD along with simple two-body po-tentials has been used to study phase transitions in
two-dimensional Lennard–Jones (LJ) binary systemsby Li et al. [8]. The efficacy of this simplified ap-proach for long-time integration, involving 1000 to10 000 particles for over 107 time steps, was em-phasized because of the need to monitor the onsetof melting, a relatively long timescale phenomenon.Chemical reaction simulations on the atomic levelhave been performed by Wilson and coworkers us-ing classical MD (e.g. [9]) or transition state theory[10]. Stillinger and Weber [11] have used two-bodyand three-body potentials in MD simulations to ob-serve spontaneous forming of diatomic molecules.Ortoleva and Yip [12] used a hard disks system forbinary reactions, where particles react by collisions(forward reactions) and by spontaneous transitions(backward reactions), giving different steady statesdepending on the reaction rates. In contrast to ourapproach, all these simulations were performed inequilibrium systems with exchange reactions onlyand constant number of molecules. On the other hand,direct integration of the coupled quantum-mechanicalself-consistent molecular dynamics (SCMD) problem[13–15] for several hundred atoms and for timescales,up to the nanosecond range, remains somewhat out ofreach with the present generation of computers. Thus,the time is now for some middle-ground solution forintermediate timescales by means of MD, which canaddress this problem of bimolecular reactions witha large swarm of heterogeneous atoms which candisplay their collective fluid-like nature [16]. Somemacroscopic fluid-dynamical phenomenon can bemanifested, as in the MD study of the fluid-dynamicalinstabilities by Moscinski et al. [17], Alda et al. [18]and Dzwinel et al. [19], in which as many as 107
particles were integrated on massively parallel com-puters for around 106 time steps, in order to simulatemesoscale fluid-dynamical instabilities, such as theRayleigh–Taylor instability.
In this same spirit, we present in this paper a simplerconceptual and computational model for investigatingbinary chemical reactions, based on classical MD andtwo-body interparticle potentials. With the MD simu-lations, we can systematically investigate the essentialcharacteristics of chemically reacting hydrodynam-ics with far more participating atoms, exceeding by

W. Alda et al. / Physica D 146 (2000) 261–274 263
several orders in magnitude what would be feasible bythe SCMD technique, and for much longer timescales.The physical scale in which we will be operating stillfalls within the microscale range of under 1mm butthe spatial range can be extended to the mesoscale do-main of Wells et al. [6] by combining the dissipativeparticle dynamics (DPD) method [20,21], where parti-cles represent clusters of atoms, with classical atomicMD particles. In the above approach, one can thinkof a system built up of large DPD particles repre-senting non-reacting solvent with dissolved reactingMD atoms. The method described here will poten-tially constitute a powerful new tool for the computa-tional treatment of chemical reacting hydrodynamicsin the microscale and also eventually be connected tothe mesoscale of the order of microns by DPD meth-ods [22]. Our approach stands in sharp contrast to MDstudies in which the chemical kinetics are assumed apriori for the reacting particles [23].
2. MD model
Our simulations have been carried out in two di-mensions. We have employed a two-body LJ-basedinterparticle potential, for both exothermic and en-dothermic reactions, modified for certain interactions:
V LJαβ (rij ) = 4εαβ
[(σαβ
rij
)12
−(
σαβ
rij
)6]
, (1)
whereα and β denote the particle species,εαβ thewell depth of the potential,σαβ a parameter determin-ing the nearest neighbor distance in the interaction,and rij the distance between the particlesi and j .All of the potentials are truncated at certainrcut andshifted up, so as to reach zero potential at cut-off andachieve potential continuity at the cut-off radiusrcut.
In this model, three different types of particles, A, Band C, which may differ in mass and interaction poten-tial parameters, are employed. All interactions are as-sumed to have identicalσαβ = σ parameters; thus allparticles have the same size. The total number of par-ticles in the system changes, as the reactions develop.
The VAB potential for both types of reactions hasbeen modified in this study by including a hump
(Figs. 1a and 2a) in the overall potential, which isgiven by
VAB(rij ) = V LJAB + Vhump. (2)
The hump is modeled by piecing together two cubicsplines, which start atV LJ
AB(rsh), wherersh = (267 )1/6σ ,
go through the top of the hump atrh and end atV LJ
AB(reh), wherereh = 2rh − rsh. VAB is truncated atrcut = 2.5σ ; thus the hump lies within the interac-tion range. The idea of using this artificial feature isto enable the reactions to take place only for particleswhich have a sufficiently high kinetic energy.
The VAA , VBB andVCC potentials, which are alsoidentical for both the exothermic and endothermic re-actions (Figs. 1c and 2c), are truncated atrcut =1.12σαβ , a minimum ofV LJ
αβ , and shifted up byεαβ ,thus making the potential purely repulsive in this study.
The VBC and VAC potentials differ for both typesof reactions. For exothermic reactions, the potentialwell depth,ε, in VBC andVAC is deeper than inVAB
(Fig. 1b). This results in smaller average potentialenergy of A–C and B–C interactions than A–B thatoccurred before the reaction. Assuming that the totalenergy of the system is kept constant, the averagekinetic energy increases.
Changing the shape of theVAC andVBC functions bymoving them up, increases the average A–C and B–Cpotential energy, eventually allowing the endothermicreactions to occur. For this purpose, we have chosena very simple criterion, by introducing the repulsivepotential with a flat plateau (Fig. 2b), where particlesin A–C and B–C interactions can lose their kineticenergy.
In this way it is possible to find the A–C and B–Cpotential for which there will be zero heat generatedby the reaction. With A–B and A–A, B–B, C–C po-tentials used, this can be achieved with a very shal-low potential well depth,ε = 20 J/K, in the A–Cand B–C interactions. In addition to the exothermicand endothermic runs, we have carried out a seriesof zero heat runs at different temperatures in order todemonstrate the temperature dependence of the reac-tion speed. These results are shown at the end of thispaper. The parameters of the exothermic and endother-mic potentials are listed in Table 1.
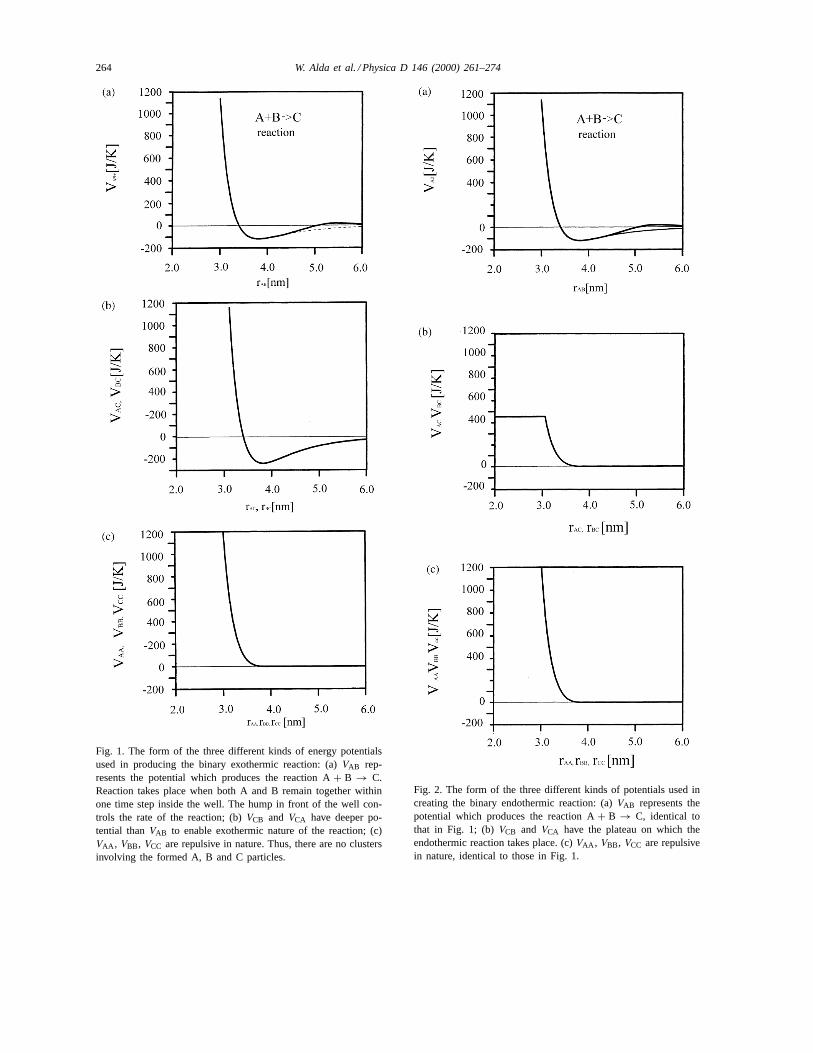
264 W. Alda et al. / Physica D 146 (2000) 261–274
Fig. 1. The form of the three different kinds of energy potentialsused in producing the binary exothermic reaction: (a)VAB rep-resents the potential which produces the reaction A+ B → C.Reaction takes place when both A and B remain together withinone time step inside the well. The hump in front of the well con-trols the rate of the reaction; (b)VCB and VCA have deeper po-tential thanVAB to enable exothermic nature of the reaction; (c)VAA , VBB, VCC are repulsive in nature. Thus, there are no clustersinvolving the formed A, B and C particles.
Fig. 2. The form of the three different kinds of potentials used increating the binary endothermic reaction: (a)VAB represents thepotential which produces the reaction A+ B → C, identical tothat in Fig. 1; (b)VCB and VCA have the plateau on which theendothermic reaction takes place. (c)VAA , VBB, VCC are repulsivein nature, identical to those in Fig. 1.

W. Alda et al. / Physica D 146 (2000) 261–274 265
Table 1Potential parameters used in the three-component system with exothermic and endothermic reactions
Interaction εαβ (J/kB) σαβ (Å) rcut (σαβ/Å) Plateau cut-off (σαβ/Å) Hump height (J/kB) Hump distance (σαβ/Å)
Potentials equal for exothermic and endothermic reactionsA–B 119.8 3.4 2.5 – 20.0 1.6A–A 119.8 3.4 1.12 – – –B–B 119.8 3.4 1.12 – – –C–C 119.8 3.4 1.12 – – –Exothermic onlyC–B 239.6 3.4 2.5 – – –C–A 239.6 3.4 2.5 – – –Endothermic onlyC–B 119.6 3.4 1.12 0.9 – –C–A 119.6 3.4 1.12 0.9 – –
The mass of A and B particles has been set to40 amu, while the mass of C particle, a product ofA + B reaction, to 80 amu. The particles are confinedwithin a rectangular box, and initially are placed on agrid forming a mesh built up from 2D regular squares.With the value ofσ assumed, the size of such a cellis about 3.53 Å × 3.53 Å, which yields a total area ofthe computational box greater than 106 Å2 (Fig. 3) fora simulation with 105 particles. The initial configura-tion is not close packed, so the particles can diffusemore easily. This involves the initial stage of the
Fig. 3. Schematic diagram of the computational domain, where the exothermic reactions take place (see colliding particles depicted). Theinitial grid layout with the regular square configuration of particles A and B is displayed.
simulation, as the particles can start reacting earlier. Inthe course of simulation the structure is not kept andthe reacting particles can form a collective fluid-likeparticle flow. The size of the box is kept constant. Pe-riodic boundary conditions have been applied alongthe vertical edges, while for the horizontal boundarywe have introduced reflecting walls.
The time step,1t , has been set to 10−14 s, whichputs an upper-bound on the relative velocity of theinteracting particles. With this model, we can achievea speed of around two time steps per second for 105

266 W. Alda et al. / Physica D 146 (2000) 261–274
particles on a 600 MHz workstation. We can then com-plete 106 time step run (or 10 nanoseconds with thegiven1t) within 1 day.
3. Algorithms for inducing binary endothermicand exothermic reactions
For simulating chemical reactions with both en-dothermic and exothermic nature in MD, we have im-plemented simple algorithms to a model of A+ B →C chemical reactions to a classical MD scheme basedon LJ particles [24]. By a reaction, we mean here an“instantaneous” or an extremely short time-instant in-volving the transformation of particles A and B intoa single particle C, when certain conditions concern-ing the duration of time spent inside the well ofVAB
and velocity magnitudes of particles are fulfilled. Theoccurrence and reaction speed depend on the fourcriteria:1. the potential depths and widths applied,2. the average energies or speed of the ensemble of
particles,3. the global thermodynamic conditions, such as the
density and the temperature, and4. the initial boundary conditions.
In order for a reaction to occur between particlesA and B, theVAB potential must have a potentialwell deep enough. The “hump” added inVAB can beespecially important in low-density systems, wheremeanrAB goes beyond the hump. In such a case, thehump acts as a threshold. The non-reacting A–A, B–Band C–C pairs, interact by repulsive potential only(Fig. 1b). Repulsion prevents the forming of clustersof same-kind particles and enables better mixing. Par-ticles A–C and B–C attract each other; however, thepotential well depth is deeper than in the A–B inter-action. Moreover, we do not allow C to react with Aand B in this study, i.e. no back reaction.
In this model, the principal criterion for the occur-rence of a chemical reaction, is the interparticle dis-tance. We assume that a reaction takes place for thecondition rAB < rcrit, wherercrit is a certain criticaldistance. We also assume that therAB < rcrit stateshould be kept for a certain number of time steps, con-
tinuously. The reaction rate can be slowed down then,by decreasingrcrit or increasing the time interval. Inthe simulations performed, we have assumed that theradius for reactive occurrence,rAB < rcrit, in a singletime step, O(10−14) s, is sufficient for the reaction. Ingeneral,rcrit is chosen to lie within 0.8 . . . 0.9 of σ inA–B interaction. To avoid very long runs with veryfew reactions, we have chosen a relatively large valueof rcrit = 0.87 to speed up the reactions.
The model reactions do fulfill the conservation laws.Conservation of mass and momentum is straightfor-ward, asmC = mA + mB and EpC = EpA + EpB. Thecenter of mass of C is equal to the center of mass ofA +B. The total energy, however, is not automaticallyconserved because each time the reaction occurs, twoparticles A and B are replaced with one particle C,which interacts with its nearest neighbors by a differ-ent potential. The particle C has no internal dynamicalstructure and cannot keep any hidden potential energy.To keep the total energy constant, we can only changethe kinetic energy of some particles whenever a reac-tion takes place. We accomplish this task at each timestep by adjusting the kinetic energy of all particleswithin 2.5σαβ radius around C, so as to conserve thetotal energy of the neighboring area at each reaction.
4. Results
We have used a constant energy scaling in conduct-ing the simulations of a system involving 20 000 par-ticles for both exothermic and endothermic reactionsfor a range of temperatures in order to see some no-ticeable effects. The changes of potential, kinetic andtotal energies with time are displayed in Figs. 4 and 5.
Exothermic runs have been performed in three ini-tial dimensionless temperatures,T = 0.1, 0.3 and 0.7in reduced LJ units. As shown in Fig. 4, forT = 0.1and 0.7 runs, the total energy is conserved, while thepotential and kinetic energies vary with time. Thekinetic energy increases because of the exothermicnature of the reaction. Due to the increase in the num-ber of particles C and mixing at the interface area,the potential energy now has a tendency to decrease,while the temperature of the entire system increases.A similar, but reversed situation is shown in Fig. 5,

W. Alda et al. / Physica D 146 (2000) 261–274 267
Fig. 4. Conservation of the total energy with time for exothermicreactions. Dimensionless initial temperature is 0.1–0.5 by the endof the run. 20 000 particles consisting of A and B initially wereemployed. The total number of particles changes in time due tothe reactions.
in which with endothermic reactions the potentialenergy increases, while the temperature decreasesmonotonically.
Figs. 6–8 show the snapshots of the particle loca-tions for the exothermic reactions in three differentinitial temperatures,T = 0.7, 0.3 and 0.1, and in threedifferent instants of time. Gray areas represent atomsA and B in the lower and upper parts of the box, while
Fig. 5. Conservation of the total energy with time for endothermicreactions. Dimensionless initial temperature is 0.2 decreasing to0.08 by the end of the run. 20 000 particles consisting of A andB initially were employed. The total number of particles varieswith time due to the chemical reactions.
the atoms C, the product of the reaction, are shownas darker dots. The latter are displayed in larger for-mat for better visualization. Initially the system con-sists only of atoms A and B occupying the upper andlower halves of the box, separated by a straight line ofcontact. It can be observed from the snapshots, that inhigher temperature the reaction starts much faster andquickly spreads over the entire interface. At the high-est temperature (T = 0.7) the particle motion is morechaotic and the reactions take place along the entirefront. On the other hand, reactions atT = 0.1 are spo-radic and induce a heterogeneous character to the in-terface. This is due to the exothermic character of thereactions and the fact that, once the reaction occurs,the next reaction becomes more probable to occur inthe neighborhood. Timescales are faster at the highertemperatures. One can also see that the reaction frontforms a curve line, which is rugged but its shape re-mains periodic with a dominant wavelength. The cur-vature of the reaction interface can be explained by theexothermic character of the reactions and the tendencyof the heat liberated to stretch the interface boundary,which is energetically adjusted. On the other hand, thereactions rate is slowed down with time due to thegrowing thickness of C particles layer which separatesthe reacting A and B species. In these three runs, start-ing from T = 0.7, 0.3 and 0.1, the temperatures riseasymptotically toT = 0.98, 0.60 and 0.42, respec-tively.
Similar snapshots showing the endothermic reac-tions with the initial temperatures,T = 0.6, 0.4 and0.2, are shown in Figs. 9–11. We observe that the im-age of the initial phase of the reactions in Figs. 9 and10 is similar to that in the exothermic model. Fur-thermore, the interface line remains rather straight,with only short-wavelength fluctuations. Particles Care more scattered (especially in Fig. 10) due to thesmaller probability of the next reaction in the vicinityof an already reacted site.
A larger domain has been established by elongat-ing the computational domain by fivefold. Two sim-ulations for both exothermic and endothermic reac-tions have been performed for the larger box in theinitial temperatureT = 0.3. We observe in Fig. 12,that the spatial heterogeneity of the reactions is much
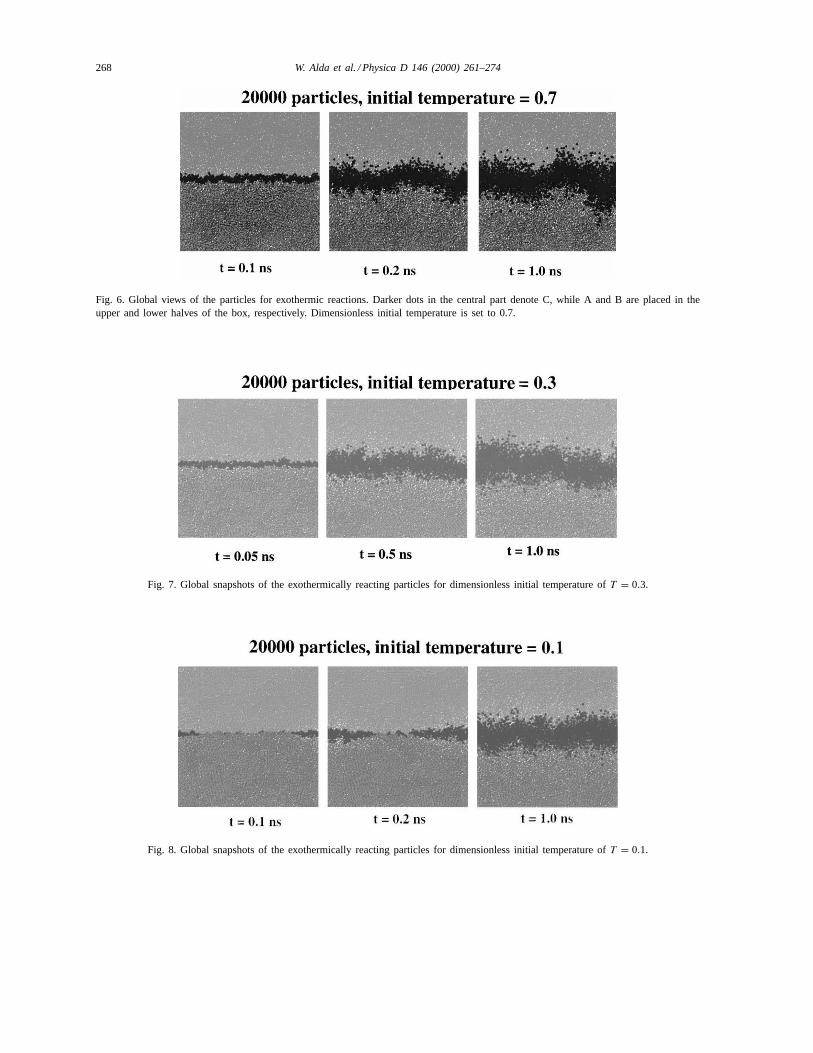
268 W. Alda et al. / Physica D 146 (2000) 261–274
Fig. 6. Global views of the particles for exothermic reactions. Darker dots in the central part denote C, while A and B are placed in theupper and lower halves of the box, respectively. Dimensionless initial temperature is set to 0.7.
Fig. 7. Global snapshots of the exothermically reacting particles for dimensionless initial temperature ofT = 0.3.
Fig. 8. Global snapshots of the exothermically reacting particles for dimensionless initial temperature ofT = 0.1.

W. Alda et al. / Physica D 146 (2000) 261–274 269
Fig. 9. Global views of the particle distribution for endothermic reactions. Darker dots in the central part denote C, while A and B areplaced in the upper and lower halves of the box, respectively. The dimensionless initial temperature is set to 0.6.
Fig. 10. Global snapshots of the endothermically reacting particles for dimensionless initial temperature ofT = 0.4.
Fig. 11. Global snapshots of the endothermically reacting particles for a dimensionless initial temperature ofT = 0.2.

270 W. Alda et al. / Physica D 146 (2000) 261–274
Fig. 12. Global views of run with 100 000 initial particles for exothermic reactions. Dimensionless temperature is set to 0.3. The configurationhas an aspect ratio of 5 and an area of 500 Å× 2500 Å.
stronger than in the smaller boxes. This tendency isprobably due to greater length of the reaction frontand smaller influence of periodic boundary conditions.The occurrence of the reaction generates heat, whichis then distributed over the neighboring particles. Theenergy distribution along the interface is slower, whennot constrained by the end conditions. This also showsthat the speed and the range of the kinetic energy be-ing distributed about, can greatly influence the overalldynamics of the reaction, thus indicating some posi-tive feedback tendency. The shape of the growing Cparticle cluster is shown in Fig. 13. In contrast, for
the endothermic reaction shown in Fig. 14, in spite ofthe much lower reaction rate, the C particles remainin separated spots, uniformly distributed along theinterfacial line.
Fig. 15 shows the increasing number of C atomswith the development of the exothermic reactions. Theresults come from three 20 000-atom runs in tempera-turesT = 0.1, 0.3 and 0.7. As expected, the reactionproceeds much faster at the higher temperature. InT = 0.7 and 0.3 the speed of reaction monotonicallydecreases with time, as the contact line becomes filledwith C atoms which separate A and B atoms. On the

W. Alda et al. / Physica D 146 (2000) 261–274 271
Fig. 13. Zoomed-in shot for the aspect ratio 5 run shown in Fig. 12. A and B particles are placed in the upper and lower halves of thebox, respectively, while C particles are shown as darker dots.
Fig. 14. Global views of run with 100 000 initial particles for endothermic reactions. Dimensionless temperature is set to 0.3. Theconfiguration has an aspect ratio of 5 and an area of 500 Å× 2500 Å.

272 W. Alda et al. / Physica D 146 (2000) 261–274
Fig. 15. Number of exothermic reactions taken globally as afunction of time for various temperatures. Three temperatures havebeen considered,T = 0.1, 0.3 and 0.7.
other hand, at the lowest temperature,T = 0.1, thereaction speed is slow initially, then increases, andafter roughly 0.4 ns decreases. This phenomenon iscaused by the fact, that at low temperature only a fewparticles can react, only after a relatively long incuba-tion time. Once a reaction takes place, a local increaseof the temperature enhances the rate of the reactionsand acts as a source of positive feedback. We do notobserve this effect with endothermic reactions. Thenumber of reactions with time (Fig. 16) shows thatthe speed of reaction decreases monotonically for alltemperatures.
Fig. 17 displays the temperature dependence of
Fig. 16. Number of endothermic reactions taken globally as afunction of time for several temperatures. Five temperatures havebeen considered,T = 0.2, 0.4, 0.6, 0.8 and 1.0.
Fig. 17. Number of reactions as a function of temperature forapproximately a zero heat of reaction. Curves indicate subsequentinstants.
number of reactions obtained in 10 runs with zeroheat of the reaction. Runs have been performed withthe same constant energy, as in the previous cases.Due to the potential chosen, the temperature remainedroughly constant during the entire run, fluctuatingonly, with no drift. Two regions consisting of fasterand slower growth of number of reactions varyingwith the temperature can be observed. There existsa threshold temperature dividing the two regions. Itis roughly equal to 0.25 for short simulation timeand moves down toT = 0.2 for t = 1.0 ns. Thisphenomenon can be explained by the scarcity of reac-tions at lower temperature due to the absence of anyheat liberated by the reactions. Thus, the interfacialgrowth involving the C particles does not deceleratethe global reaction rates.
5. Concluding remarks
In this work, we have introduced for the first time asimple model for simulating chemical reactions, basedon the formalism of classical MD. This techniquehas great potential of being utilized for investigat-ing the collective nonlinear interaction involving mil-lions to billions of reacting species being coupled toa larger-scale flow, thus allowing the possibilities forupscaling of chemical kinetics. Some of our prelim-

W. Alda et al. / Physica D 146 (2000) 261–274 273
inary results already offer some interesting insights,especially those concerning the enhancing effects ofincreasing the temperature, the marked heterogeneitiesproduced at the reacting interface, the threshold natureof increasing the temperature and the fundamental is-sue regarding the concepts of local versus global ratesof reaction.
Our approach differs fundamentally from previousstudies using discrete particles, by means of cellu-lar automata or lattice-gas techniques, because wedo not employ rules for updating the lattice dynam-ics but only two-body, LJ-like potentials with somechemical–physical meaning. This method is also dif-ferent from other MD approaches, employing alreadyassumed kinetic-rate equations [23]. Previous worksin this area of MD with reactions have been concernedwith equilibrium systems of rare gas solutions con-sisting of hard-spheres [25] and with a single LJ po-tential but relatively few particles, O(100), [26]. Weare aware of the simplifications and drawbacks in thismodel, but our primary interest lies in building a com-putationally efficient scheme to study the collective in-teraction of millions to billions of reacting particles inthe hydrodynamic mode and to understand some basicchemical–physical principles in many-body reactions.First, we do not account for quantum-mechanical ef-fects, which would slow down considerably the com-putations and limit the number of participating atomsto at most several hundred. Second, this model lacks athree-body interaction term [11] for ensuring the for-mation of chemical bonds between the correct numberof atoms. Third, we have not allowed for the possibil-ities of back reactions, involving C+ A and C+ B.Back reactions would certainly have a substantial non-linear feedback effects and studies are currently underway to address this important issue. Lastly, there isthe issue of longer timescales, necessary for achievingboth hydrodynamic effects and more realistic reactiontime rates.
Extending these calculations to the temporal do-main of microseconds would definitely require theuse of massively parallel processors with large sharedmemory architecture of 10 GB or more. By elongat-ing the simulation time, it is possible to observe thereactions along with their interactions with mixing
processes induced by larger-scale external flow fields,pressure or velocities applied to particles, or by thereaction itself as well. In this case, we can expect col-lective hydrodynamic-type motion of particles with awide-range of length-scales. In the very near future,we will complete simulations of up to 107 reactingparticles. The initial conditions we have employedis only an end-member. Other initial conditions in-volving, e.g., a prepared state of well-mixed reactantswould also be of considerable interest in studies ofchemical combustion. This method may be extendedto DPD for studying at the mesoscale the interactionbetween mixing and chemical reactions in the novelcross-scale sense [24,27]. Many of the analytical toolsdeveloped for mixing in the macroscale (e.g. [28]),can be brought to bear on these newer cross-scaleissues in the micro- to mesoscale. By sacrificingsome of the details of fine-scale structures found inquantum-chemical reactions, we may now be able toconnect, with this approach, chemical reacting flowsacross considerable spatial–temporal scales, whichwill be of direct relevance in the environmental andengineering disciplines [29,30], as well as in thechaos control of chemical reaction systems [31].
Acknowledgements
We thank Witold Dzwinel, Rashid Zia and JulioOttino for stimulating discussions. This research hasbeen supported by the Geosciences Program of theDepartment of Energy (DOE).
References
[1] K. Binder, M. Müller, F. Schmid, Comput. Sci. Eng. 3 (1999)10.
[2] J. Mai, W. von Niessen, J. Chem. Phys. 98 (1993) 2032.[3] B. Chopard, M. Drozd, Europhys. Lett. (Switzerland) 15
(1991) 459.[4] J.R. Weimar, J.P. Boon, Phys. Rev. E 49 (1994) 1749.[5] J.P. Boon, D. Dab, R. Kapral, A. Lawniczak, Phys. Rep. 273
(1996) 556.[6] J.T. Wells, D.R. Janecky, B.J. Travis, Physica D 47 (1991)
115.[7] D. Rothman, S. Zaléski, Rev. Mod. Phys. 66 (1994) 1417.[8] M. Li, W.L. Johnson, W.A. Goddard III, Phys. Rev. B 54
(1996) 12067.

274 W. Alda et al. / Physica D 146 (2000) 261–274
[9] L.L. Lee, Y.S. Li, K.R. Wilson, J. Chem. Phys. 95 (1991)2458.
[10] J.P. Bergsma, J.T. Hynes, J.R. Reimers, K.R. Wilson, J. Chem.Phys. 85 (1986) 5625.
[11] F.H. Stillinger, Th.A. Weber, J. Chem. Phys. 88 (1988)5123.
[12] P. Ortoleva, S. Yip, J. Chem. Phys. 65 (1976) 2045.[13] I. Stich, R. Car, M. Parinello, Phys. Rev. B 44 (1991) 4262.[14] J.R. Chelikowsky, N. Troullier, Y. Saad, Phys. Rev. Lett. 72
(1994) 1240.[15] K.C. Hass, W.F. Schneider, A. Curioni, W. Andreoni, Science
282 (1998) 265.[16] J.P. Boon, S. Yip, Molecular Hydrodynamics, McGraw-Hill,
New York, 1980.[17] J. Moscinski, W. Alda, M. Bubak, W. Dzwinel, J. Kitowski,
M. Pogoda, D.A. Yuen, in: D. Stauffer (Ed.), Annual Reviewof Computational Physics, Vol. V, World Scientific, Singapore,1997, p. 97.
[18] W. Alda, W. Dzwinel, J. Kitowski, J. Moscinski, M. Pogoda,D.A. Yuen, Comput. Phys. 12 (1998) 595.
[19] W. Dzwinel, W. Alda, M. Pogoda, D.A. Yuen, Physica D 137(2000) 157.
[20] P.J. Hoogerbrugge, J.M.V.A. Koelman, Europhys. Lett. 19(1992) 155.
[21] P. Español, Phys. Rev. E 57 (1998) 2930.[22] W. Dzwinel, D.A. Yuen, Mol. Sim. 22 (1999) 369.[23] K. Geisshirt, E. Praestgaard, S. Toxvaerd, J. Chem. Phys. 107
(1997) 9406.[24] W. Dzwinel, W. Alda, D.A. Yuen, Mol. Sim. 22 (1999) 397.[25] J. Gorecki, J. Gryko, J. Stat. Phys. 48 (1987) 329.[26] J. Gorecki, J. Gryko, Comput. Phys. Commun. 54 (1989) 245.[27] W. Dzwinel, D.A. Yuen, J. Colloid Interf. Sci. 225 (2000)
179.[28] J.M. Ottino, Scient. Am. 260 (1989) 40.[29] A.A. Ten, Y.Y. Podladchikov, D.A. Yuen, T.B. Larsen, A.V.
Malevsky, Geophys. Res. Lett. 25 (1998) 3205.[30] J.M. Ottino, A. Souvaliotis, G. Metcalfe, Chaos, Solitons and
Fractals 6 (1995) 425.[31] A.A. Hoff, H.H. Diebner, G. Baier, Z. Naturforsch 50a (1995)
1141.
Copyright © 2022 FDOKUMEN




















