
I ilMiiiliilg^B^M - You're automatically being redirected to ...
Upload
independentCategory
view
0download
0
IMMUNOBIOLOGY
Enhanced antilymphoma efficacy of CD19-redirected influenza MP1–specificCTLs by cotransfer of T cells modified to present influenza MP1Laurence J. N. Cooper, Zaid Al-Kadhimi, Lisa Marie Serrano, Timothy Pfeiffer, Simon Olivares, Adrian Castro, Wen-Chung Chang,Sergio Gonzalez, David Smith, Stephen J. Forman, and Michael C. Jensen
To enhance the in vivo antitumor activityof adoptively transferred, CD19-specificchimeric antigen receptor (CAR)–redirectedcytotoxic T lymphocytes (CTLs), we studiedthe effect of restimulating CAR� CTLsthrough their endogenous virus-specific T-cell antigen receptor (TcR) by the cotransferofengineeredT-cell antigen–presentingcells(T-APCs). Using influenza A matrix protein 1(MP1) as a model antigen, we show that exvivo–expanded CD4� and CD8� T-APCs ex-pressingahygromycinphosphotransferase-MP1 fusion protein (HyMP1) process andpresent MP1 to autologous human leuko-
cyte antigen (HLA)–restricted, MP1-specificCD4� and CD8� CTL precursors. The MP1-specific CTLs are amenable to subsequentgenetic modification to express a CD19-specific CAR, designated CD19R, and ac-quire HLA-unrestricted reactivity towardCD19� leukemia and lymphoma tumor tar-gets while maintaining HLA-restricted MP1specificity. The restimulation of MP1�CD19dual-specific CTLs in vivo by the adoptivetransfer of irradiated HyMP1� T-APCs re-sulted in the enhanced antilymphoma po-tency of bispecific effector cells, as mea-sured by elimination of the biophotonic
signal of established firefly luciferase–ex-pressing Burkitt lymphoma xenografts innonobese diabetic/severe combined immu-nodeficiency (NOD/scid) animals comparedwith control groups restimulated byHy�MP1neg T-APCs. Engineered T-APCs area novel and versatile antigen-delivery sys-tem for generating antigen-specific T cellsin vitro and enhancing the in vivo effectorfunctioning of CAR-redirected antitumor ef-fector cells. (Blood. 2005;105:1622-1631)
© 2005 by The American Society of Hematology
Introduction
Adoptive transfer of ex vivo–expanded T cells specific for immunodom-inant viral epitopes into immunocompromised hosts can reconstituteprotective antiviral immunity and can result in the long-term persistenceof transferred cells.1-5 In contrast, the application of adoptive T-celltransfer to the successful cellular immunotherapy of malignancy hasproved to be significantly more challenging, in part because of thedifficulty of isolating high-affinity, tumor-specific T cells that canmediate effective antitumor in vivo effector functions and the potentialfor tumors to evade immunologic clearance through a variety of escapemechanisms, including the down-regulation of restricting HLA mol-ecules.6-8 Several groups, including ours, are developing alternativestrategies for targeting tumors using genetically modified T cells that areendowed with redirected antigen specificity through the expression ofchimeric antigen receptors (CARs), such as a CD19-specific chimericimmunoreceptor. These chimeras typically use HLA-independent, high-affinity antigen recognition domains consisting of extracellular single-chain immunoglobulin variable fragments (scFvs) linked to cytoplasmicT-cell activation domain(s), such as CD3-�.9-19
Strategies to enhance the antitumor activity of adoptivelytransferred CAR� cytotoxic T lymphocytes (CTLs) and to over-come the potentially deleterious impact of in vivo recycling of
these cells solely through CAR-redirected engagement of tumorcells will likely be critical for achieving therapeutic efficacy.Because CAR-redirected T cells retain the specificity and functionof their endogenous T-cell antigen receptor (TcR), expressingCARs on virus-specific T cells, such as commonly acquired latentviruses (Epstein-Barr virus [EBV] and cytomegalovirus [CMV]), isa potential approach to maintain persistence in vivo throughre-encounter of these bispecific T cells with viral antigen presentedby professional antigen-presenting cells (APCs).9,20,21 Although thetiming and magnitude of latent virus reactivation makes the in vivorestimulation of bispecific T cells difficult to control, we hypoth-esize that the grafting of antitumor CARs to T cells specific forcommon nonlatent viruses and the delivery of a viral antigenvaccine boost(s) after adoptive transfer (transfer-boost strategy) isan approach amenable to iatrogenic regulation.
Here we describe the usefulness of ex vivo–expanded CD8�
and CD4� T cells to function as APCs by their genetic modificationto express a model viral antigen (influenza A MP1) for eliciting thein vitro expansion of MP1-specific CTLs and for augmenting theclearance of CD19� Daudi lymphoma in vivo, by CD19�MP1–bispecific CTLs by post-transfer boosting. Our finding that human
From the Divisions of Pediatric Hematology-Oncology, Molecular Medicine,Hematology and Hematopoietic Cell Transplantation, and Bioinformatics,Beckman Research Institute and City of Hope National Medical Center, Duarte,CA.
Submitted April 2, 2004; accepted October 13, 2004. Prepublished online asBlood First Edition Paper, October 26, 2004; DOI 10.1182/blood-2004-03-1208.
Supported by the National Institutes of Health (grants CA30206-23,CA033572-21), the Abe and Estelle Sanders Foundations, the Alliance forCancer Gene Therapy, the Altschul Foundation, the Amy PhillipsFoundation, the Leukemia and Lymphoma Society, the LymphomaResearch Foundation, the National Foundation for Cancer Research, the
Pediatric Cancer Research Foundation, and the Sidney Kimmel Foundationfor Cancer Research
L.J.N.C. and Z.A.-K. contributed equally to this work.
Reprints: Laurence J. N. Cooper, Division of Molecular Medicine, BeckmanResearch Institute, Division of Pediatric Hematology/Oncology, City of HopeNational Medical Center, 1500 E Duarte Rd, Duarte, CA 91010-3000; e-mail:[email protected].
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 U.S.C. section 1734.
© 2005 by The American Society of Hematology
1622 BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

T cells are amenable to genetic modification for expressing andpresenting viral antigens makes this transfer boost system wellsuited to augment the antilymphoma/leukemia effect of adoptivelytransferred CD19-specific CTLs in a variety of clinical settings.22
Materials and methods
Plasmid expression vectors
The pMG expression vector (InvivoGen, San Diego, CA) was modified bysite-directed mutagenesis to remove a PacI restriction enzyme (RE) site atposition 307 to generate pMGPac (Figure 1A). The plasmid vectorCD19R/HyTK-pMG was derived from pMGPac and has been describedpreviously.23 The HyMP1 fusion gene was assembled by polymerase chainreaction (PCR) splice overlap extension and consists of the 5� 972 basepairs (bps) of hygromycin phosphotransferase (Hy) gene and the 3� 759 bpsof the influenza virus A/WSN/33 MP1 gene (GenBank accession numberM19374), kindly provided by Dr Adolfo Garcia-Sastre (Mount Sinai Schoolof Medicine, NY). This construct incorporates unique 5� NheI and 3�BamHI RE sites for subcloning into plasmid ELF1� promoter andKanamycin-resistance gene (pEK) to generate the plasmid HyMP1-pEK(Figure 1A). The pEK plasmid was modified from pcDNA3.1� byreplacing the CMV promoter with human elongation factor 1� (hEF1�)promoter and replacing neomycin- and ampicillin-resistance genes with thekanamycin-resistance (KanR) gene. The bifunctional ffLucZeo fusion genethat coexpresses the North American firefly (Photinus pyralis) luciferase(ffLuc) and zeomycin-resistance genes (Zeo) were cloned from the plasmidpMOD-LucSh (InvivoGen) into pcDNA3.1� (Invitrogen, Carlsbad, CA),to create the plasmid ffLucZeo-pcDNA. Truncated CD19 (tCD19), lackingthe cytoplasmic domain, was expressed from the plasmid pCI-rCD19,
kindly provided by Dr Michio Kawano (Yamaguchi University School ofMedicine, Japan).24
Cell lines and primary human T-cell propagation
Lymphoblastoid cells (LCLs) and Daudi,25 T2,26 and K56227 cells wereobtained from ATCC (Manassas, VA) and were maintained in mediaconsisting of RPMI 1640 (Irvine Scientific, Santa Ana, CA) supplementedwith 2 mM L-glutamine (Irvine Scientific), 25 mM (N-2-hydroxyethylpipera-zine-N�-2-ethanesulfonic acid) HEPES (Irvine Scientific), 100 U/mLpenicillin, 0.1 mg/mL streptomycin (Irvine Scientific), and 10% heat-inactivated defined fetal calf serum (FCS) (Hyclone, Logan, UT), hereafterreferred to as culture media (CM). The U251T human glioblastoma cell linewas kindly provided by Dr Waldemar Debinski (Wake Forest University,NC), and were cultured in Dulbecco modified Eagle medium (IrvineScientific) supplemented with 10% heat-inactivated FCS and antibiotics, asdescribed. All cells were maintained at 37 °C in a humid atmosphere of 5%CO2 in air.
Human T-cell lines were derived from peripheral blood mononuclearcells (PBMCs) of healthy volunteers, who gave their consent, and werecultured using previously described methods.23 Briefly, stimulation/expansion cultures were established using 106 T cells, 30 ng/mL anti-CD3�(OKT3; Ortho Biotech, Raritan, NJ), 50 � 106 �-irradiated PBMCs (3500cGy), and 107 �-irradiated LCLs (8000 cGy) in 50 mL CM. Recombinanthuman interleukin-2 (rhIL-2) (Chiron, Emeryville, CA) was added toculture at 25 U/mL every 48 hours, beginning on day 1 of each 14-dayexpansion cycle. To obtain and expand CD4� T-APC, the cultures weresorted for binding of anti-CD4 28 days after electroporation. Antigen-specific expansion of MP1-specific T cells used �-irradiated autologousT-APCs (3500 cGy) expressing HyMP1 gene at a 5:1 responder/stimulator
Figure 1. Genetic modification of T-APCs. (A) Schematic of HyMP1 cDNA and plasmid expression vectors. The HyMP1 cDNA consisting of a 5� hygromycinphosphotransferase segment and a 3� influenza A matrix protein-1 (HyMP1) is shown with its flanking NheI and BamHI restriction enzyme (RE) sites. This transgene wascloned into the multiple cloning site (MCS) under control of the hEF1� hybrid promoter in the plasmid HyMP1-pEK. Plasmid pMGPac contains the hygromycinphosphotransferase (Hy) cDNA, under control of the human CMV immediate early (IE) promoter. The bovine growth hormone (bGhpA), late SV40 poly A sites (SV40pA),synthetic poly A and pause site (SpAn), Escherichia coli origin of replication (ori ColE1), and unique RE sites are shown. The PacI RE site was used to linearize the plasmidsbefore electroporation. (B) Chemiluminescence Western immunoblot of recombinant HyMP1. Whole-cell protein lysates from T-APCs genetically modified with HyMP1-pEK(lane 1) or pMGPac (lane 2) plasmids, along with molecular weight controls (not shown), were resolved by PAGE under reducing conditions. Western blotting with MP1-specificantibody was used to detect the approximately 176-kDa HyMP1 fusion protein. (C) Phenotype of T-APCs by flow cytometry. Histograms of binding of fluorescence-labeledcell-surface marker–specific mAbs (bold line) relative to isotype control or unstained cells (dotted line) to CD8� T-APCs genetically modified with HyMP1-pEK are shown.T-APCs were stained and analyzed by flow cytometry between days 10 and 14 of a 2-week in vitro OKT3 stimulation cycle. The flow cytometry histograms are almost identicalfor CD8� Hy�MP1neg T-APCs genetically modified with pMGPac (data not shown). The relative percentage of cells in each gate is indicated.
T-APCs ACTIVATE BISPECIFIC T CELLS 1623BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

ratio. Every 48 hours, 5 U/mL rhIL-2 was added to cultures beginning onday 1 of each 7-day culture cycle.
Flow cytometry
The following fluorescein isothiocyanate (FITC)–, phycoerythrin (PE)–,and CyChrome-conjugated reagents were obtained from BD Biosciences(San Jose, CA) and used according to the manufacturer’s instructions:anti–T-cell receptor�� (anti-TCR��), anti-CD2, anti-CD3�, anti-CD4,anti-CD8, anti-CD10, anti-CD11a, anti-CD18, anti-CD19, anti-CD27,anti-CD28, anti-CD50, anti-CD54, anti-CD58, anti-CD70, anti-CD80,anti-CD86, anti-HLA ABC, anti-HLA DR, and anti-NKG2D. A F(ab’)2
fragment of FITC-conjugated goat anti–human Fc�, (Jackson ImmunoRe-search, West Grove, PA) was used at a 1:20 dilution to detect cell-surfaceexpression of CD19R. Tetramer studies used the PE-conjugatedMP1(GILGFVFTL)–HLA-A2*0201 tetramer from Beckman Coulter (BCImmunomics Operations, San Diego, CA).28-30 The biotin-conjugatedmonoclonal antibody (mAb) specific for TCR V�17 (Immunotech; Beck-man Coulter, Fullerton, CA) was used with CyChrome-conjugated strepta-vidin (BD Biosciences). In some experiments, CyChrome-conjugatedmAbs were replaced with 1 g/mL propidium iodide (PI) to excludenonviable cells from analyses. Data acquisition was performed on aFACScalibur (BD Biosciences), and the percentage of cells in a region ofanalysis was calculated using CellQuest version 3.3 (BD Biosciences).Fluorescence-activated cell sorting using a MoFlo MLS (Dako-Cytoma-tion, Fort Collins, CO) was used to isolate CD4� T-APCs and MP1-HLAA2 tetramer� T cells.
Electrotransfer of plasmid vectors
To obtain T-APCs, OKT3-activated human PBMCs were geneticallymodified by electroporation using the Eppendorf Multiporator device(Eppendorf AG, Hamburg, Germany) based on the method previouslydescribed.31 Briefly, PBMCs (harvested from cultures on day 3 after OKT3stimulation) were resuspended in hypo-osmolar buffer (Eppendorf) at8 � 106/mL, and aliquots were apportioned into 0.2-cm cuvettes containing10 g linearized pMGPac or HyMP1-pEK DNA plasmids in a total volumeof 400 L. Each cuvette received a single 40-microsecond pulse of 250 V,followed by 5-minute room-temperature incubation, after which cells wereplaced back into rhIL-2–supplemented CM at 5 � 105 cells/mL. On day 2after electroporation and on day 5 for subsequent stimulation cycles,hygromycin B (Stratagene, Cedar Creek, TX) was added to cultures at acytocidal concentration of 0.2 mg/mL active drug.
To obtain bispecific T cells, 8 � 106 MP1-tetramer� in vitro–expandedT cells were electroporated per cuvette with linearized (at the PacI RE site)CD19R/HyTK-pMG plasmid DNA, as described in the preceding para-graph. The T cells were then stimulated on the same day with OKT3,50 � 106 �-irradiated PBMCs, and 107 �-irradiated LCLs. A cytocidalconcentration of hygromycin B was added beginning 5 days after theaddition of OKT3. Beginning the day after the addition of OKT3, 25 U/mLrhIL-2 was added to all T-cell cultures every other day. After 2 weeks ofculture, the T cells were restimulated for numerical expansion every 14days, as described, in the presence of cytocidal concentrations of hygro-mycin B.
Daudi lymphoma cells in log-phase growth were electroporated withlinearized ffLucZeo-pcDNA using the same conditions to express theffLucZeo gene. Two days after electroporation, zeocin (Invivogen) wasadded to the culture at a concentration of 0.2 mg/mL.
U251T (HLA A2�) was transfected in log-phase growth using 2 gpMGPac, pCI-rCD19-neo, or HyMP1-pEK linearized DNA in 2 Llipofectamine (Invitrogen) and/or was expanded in cytocidal concentra-tions of hygromycin B (0.2 g/mL) to obtain Hy� and HyMP1� U251Tand/or G418 (0.25 g/mL) to obtain tCD19�HyMP1� U251T ortCD19� U251T, respectively.
Western blots
T cells (2 � 107) were lysed on ice in 1 mL RIPA buffer (phosphate-buffered saline [PBS], 1% nonidet P40 (NP40), 0.5% sodium deoxycholate,
0.1% sodium dodecyl sulfate [SDS]) containing 1 tablet/10 mL Completeprotease inhibitor cocktail (Boehringer Mannheim, Penzberg, Germany).After 60 minutes, aliquots of centrifuged supernatant were boiled in anequal volume of loading buffer under reducing conditions and subjected toSDS–polyacrylamide gel electrophoresis (SDS-PAGE) on precast 12%acrylamide gels (Bio-Rad Laboratories, Hercules, CA). After transfer tonitrocellulose, membranes were blocked for 2 hours in Blotto solutioncontaining 0.07 g/mL nonfat dry milk. Membranes were washed in T-TBS(0.05% Tween 20 in Tris-buffered saline, pH 8.0) and were incubated for 2hours with 5 to 10 g goat antihuman influenza A MP1 (Immune SystemsLtd, Paignton, United Kingdom). After washing in T-TBS, the membraneswere incubated for 1 hour with a 1:500 dilution of alkaline phosphatase–conjugated mouse antibody specific for goat immunoglobulin G (IgG).After rinsing in T-TBS, the membranes were developed with 30 mL AKPsolution (Promega, Madison, WI) according to the manufacturer’s instruc-tions. Chemiluminescence was measured over a 2-hour period using anEpiChemi II Darkroom (UVP Inc, Upland, CA), and the images wereprocessed using LabWorks version 4.0.0.8 (UVP Inc).
Chromium-release assay
The cytolytic activity of T cells was determined by 4-hour chromium-release assay (CRA) using triplicate V-bottom wells in a 96-well plate(Costar, Cambridge, MA) containing Na51CrO4 (MP Biomedicals, Orange-burg, NY)–labeled Daudi, T2, T-APC, primary acute lymphoblastic leuke-mia (ALL) blast, or K562 target cells. Effector cells were harvested 10 to 14days after stimulation with OKT3, washed, and incubated with 5 � 103
target cells in triplicate, and the percentage of specific cytolysis wascalculated from the release of 51Cr, as described earlier, using a Cobra IIAutoGAMMA (Canberra Packard Ltd, Pangbourne, Berks, United King-dom) or TopCount NXT (PerkinElmer Life and Analytical Sciences, Inc,Boston, MA).23 Data are reported as mean SD.
Proliferation assay
T-cell responder cells were cocultured for 72 hours at a 1:1 ratio with�-irradiated autologous T-APCs (3500 cGy) or mitomycin C–treated (10%for 1 hour) HLA-restricted U251T stimulators in CM without rhIL-2.During the last 18 hours, cultures were pulsed with 1 Ci (0.037 MBq)/well3H-thymidine. Cells were then harvested with a Packard Cell Harvesteronto unifilter plates, and cell-associated radioactivity was measured byscintillation counting (Topcount NXT). Data are reported as mean SD.
Analysis of cytokine production
T-cell responder cells (106 cells)were cocultured at a 1:1 ratio in 12-welltissue culture plates with �-irradiated Daudi (8000 cGy), T-APC (3500cGy), or mitomycin C–treated HLA-restricted U251T stimulators in 2 mLCM. After a 48-hour incubation at 37°C, the conditioned media wereassayed by cytometric bead array (CBA) using the human TH1/TH2cytokine kit and array software according to the manufacturer’s instructions(BD Biosciences) on a FACScalibur.
Xenograft tumor model
On day 0, 6- to 10-week-old female NOD/scid (NOD/LtSz-Prkdcscid/J)mice (Jackson Laboratory, Bar Harbor, ME) were injected in the perito-neum with 5 � 106 ffLuc� Daudi cells. Beginning on day 2, tumorengraftment was evaluated by biophotonic imaging (see “Biophotonictumor imaging”) and was defined as increasing tumor ffLuc-mediated fluxover at least 2 imaging sessions. Mice with progressively growing tumorswere segregated into 4 treatment groups (5 mice/group) receiving combina-tions of intraperitoneal rhIL-2 (25 000 U/mouse on a Monday-Wednesday-Friday schedule), 20 � 106 effector T cells, and 5 � 106 �-irradiated (3500cGy) Hy�MP1� or HyMP1� T-APCs by additional separate intraperitonealinjections through 28-gauge hypodermic needles.
Biophotonic tumor imaging
Anesthetized mice were imaged using a Xenogen IVIS 100 series systembeginning approximately 15 minutes after intraperitoneal injection of
1624 COOPER et al BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

150 L (4.29 mg/mouse) of a freshly thawed aqueous solution ofD-luciferin potassium salt (Xenogen, Alameda, CA). Each animal wasserially imaged in an anterior-posterior orientation at the same relative timepoint after D-luciferin injection. Previous experiments established that thephoton flux from these ventral views of the abdomen was constant within6.32% 8.11% (mean SD) for mice bearing ffLuc� intraperitonealDaudi xenografts imaged over a 15-minute period (data not shown).Photons emitted from ffLuc� Daudi xenografts were quantified using thesoftware program Living Image (Xenogen), and the bioluminescence signalwas measured as total photon flux normalized for exposure time and surfacearea and expressed in units of photons (p) per second per cm2 per steradian(sr). For anatomic localization, a pseudocolor image representing lightintensity (blue, least intense; red, most intense) was superimposed over adigital grayscale body-surface reference image.
Statistical methods to analyze biophotonic data
To measure the differences between mouse treatment groups, we considereda primary end point evaluating tumor biophotonic signal over time. Bycalculating a cumulative area under the curve (AUC) for each mouse, anend point was generated that rewarded treatments that not only shranktumors but also kept them small over the course of the study. Mean AUCsbetween treatment groups were compared using an exact permutation testunder the Hothorn and Hornik exactRankTests package for the R lan-guage.32-35 Details for deriving the permutation P values in general arediscussed in Streitberg and Rohmel.36 Given the mouse data time points andthe photon flux, we plotted the connected points using time as the x-axis andthe end point as the y-axis. For any sequential time points, (xi, xj), and theircorresponding end points, (yi, yj), the area under this curve was calculatedby using the area of a trapezoid: 0.5 � (xj � xj) � (yi � yj). The cumula-tive AUC for the duration of the experiment was the sum of trapezoids. Weconsidered cumulative AUCs as an outcome for purposes of comparisonsamong groups. Groups with small y-values (ie, tumor flux) have small meanAUCs. When a mouse was killed for excessive tumor burden, we carried thelast measured tumor size through the end of the study. We chose a thresholdof 3.4 � 106 p/s per cm2/sr as the threshold for detectable tumor. This wasthe mean of the maximum flux of mice with no evidence of tumor after day31 and the minimum flux of mice with tumor after day 31. We defined thetime from initial treatment until the bioluminescence fell below the lowerthreshold as a time to remission end point. Similarly, we defined aprogression-free survival end point as the time from day 0 until tumorgrowth increased so that the mouse was killed for excessive tumor burden.Mice that went into remission were censored at the time of last evaluation.Based on these end points, we estimated time until remission andprogression-free survival.
Results
Generation of MP1� T-APCs
The influenza A cDNA encoding MP1 was fused to the 3� end of thehygromycin phosphotransferase cDNA by PCR splice overlapextension yielding an approximately 1746-bp transgene, desig-nated HyMP1 (Figure 1A). The HyMP1 cDNA was subcloned intothe mammalian expression vector pEK under the transcriptionalcontrol of a modified human EF-1� promoter.37 HyMP1� and Hy�
T-APCs were derived from OKT3-activated PBMCs by electrotrans-fer of the HyMP1-pEK and pMGPac plasmids, respectively,followed by selection and expansion in the presence of cytocidalconcentrations of hygromycin B, as previously described.23 Re-peated stimulation using this method typically results in preferen-tial in vitro expansion of CD8� T cells, but genetically modifiedCD4� T cells can be grown if isolated by flow cytometry sortingearly in the ex vivo–expansion process. Western blot analysis usingan MP1-specific antibody probe identified the expressed HyMP1fusion protein with an electrophoretic mobility consistent with the
expected molecular weight of approximately 176 kDa (Figure 1B,lane 1). A control T-APC line genetically modified with the plasmidpMGPac to express the Hy gene failed to exhibit a band at thismolecular weight (Figure 1B, lane 2).
T-APCs were subjected to flow cytometric analyses of their cell-surface phenotype toward the end of 14-day stimulation cycles (days12-14). Expanded CD4�CD8�HyMP1�, CD8�CD4�HyMP1�,CD4�CD8�Hy�, and CD8�CD4�Hy� T-APC lines coexpressed HLA-ABC, HLA-DR, CD2, CD11a, CD18, CD50, CD54, CD58, CD70, and,to a variable extent, CD80 and CD86 (Figure 1C; data not shown).Analysis of the CD8� T-APCs for expression of 4-1BBL, OX40L, orMICAat the end of an ex vivo expansion cycle, when they were used asstimulators, failed to detect expression over isotype and unstainedcontrols (data not shown).
HyMP1� T-APCs stimulate the in vitro expansion of MP1tetramer� CD8� T cells from PBMCs
The ability of autologous CD4� and CD8� HyMP1�–irradiatedT-APCs to stimulate MP1-specific precursors in PBMCs wasinvestigated by following the numbers of tetramer-positiveresponding cells emerging from the coculture. By day 7 ofcoculture with CD4� and CD8� T-APCs, the percentage of HLAA2� MP1-tetramer� T cells had increased to 2%, whichcompares favorably with the expansion of MP1-tetramer� Tcells cultured on mature dendritic cells (DCs) infected with liveinfluenza virus (Figure 2A-B).29 The percentage of MP1-tetramer� CD8� T cells continued to rapidly increase toapproximately 50% after 21 days of continuous coculture withCD8�HyMP1� T-APCs (Figure 2A). The outgrowth of MP1-tetramer� T cells could be explained by preferential expansionof these cells in response to stimulation with MP1 antigen—studies demonstrated that the T-APCs were capable of support-ing MP1-specific proliferation of the MP1-tetramer� T cells(Figure 2C). Furthermore, enumeration data demonstrated thatviable MP1-tetramer� CD8� T cells expanded up to 630-fold ina 3-week interval (Figure 2D). HLA-A2� PBMC responderswere cocultured under identical conditions without T-APC cellsor, with Hy�MP1� T-APCs, failed to expand MP1-tetramer�
CD8� CTLs.
HyMP1� T-APCs elicit functional CD8� and CD4�
MP1-specific CTLs
To demonstrate that MP1-tetramer� CD8� CTLs elicited by invitro stimulation with HyMP1-expressing T-APCs are function-ally intact, we assessed their lytic activity against MP1 peptide(GILGFVFTL)–loaded T2 target cells (Figure 3A). MP1-tetramer� effector CTLs lysed peptide-loaded T2 targets in aneffector/target (E/T) dose-dependent manner with negligiblebackground killing of T2 targets without peptide. In addition,these effector cells specifically lysed HyMP1� CD8� and CD4�
T-APCs, confirming that these APCs process and present MP1antigen through classical HLA class 1 (data not shown). Next,we evaluated the capacity of HyMP1-elicited MP1-tetramer�CD8� and CD4� effector T cells to be activated forcytokine secretion. Culture supernatants of these responders,incubated for 48 hours with autologous irradiated CD8� orCD4� HyMP1� T-APCs, were harvested and assayed forcytokine content by CBA. The interferon-gamma (IFN-�) andtumor necrosis factor-alpha (TNF-�) cytokines were 11-fold and7-fold, respectively, increased over MP1 tetramer�CD8� re-sponders cultured in media alone or with autologous irradiated
T-APCs ACTIVATE BISPECIFIC T CELLS 1625BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4
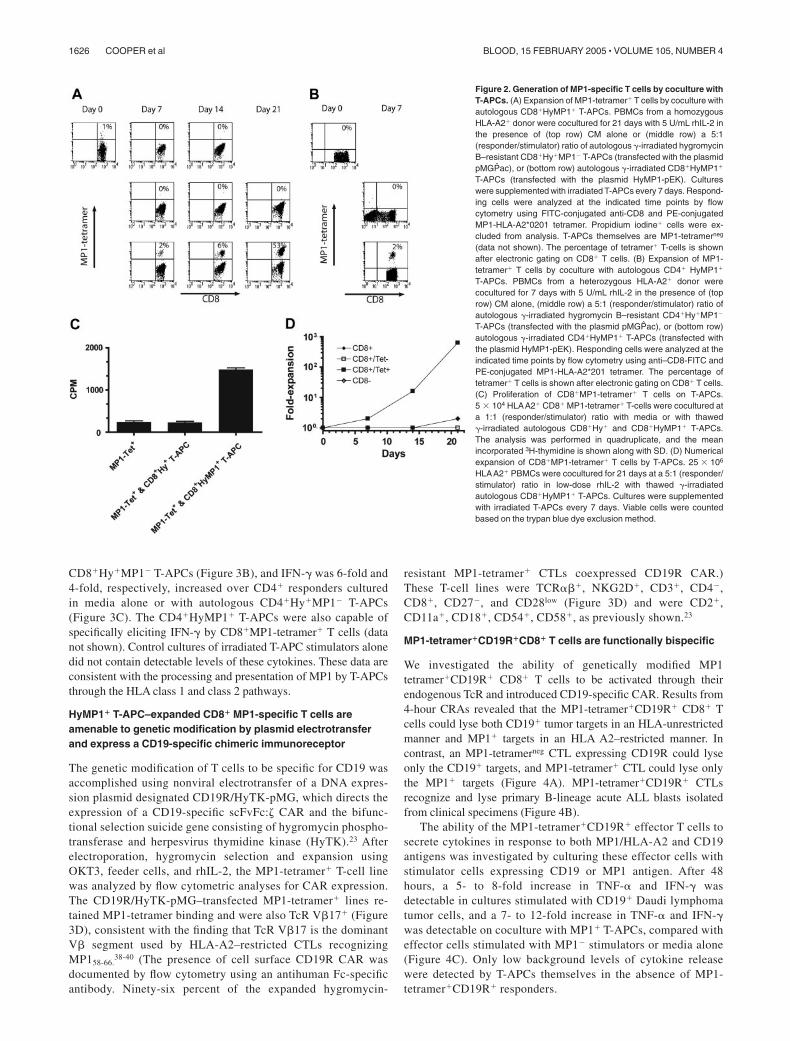
CD8�Hy�MP1� T-APCs (Figure 3B), and IFN-� was 6-fold and4-fold, respectively, increased over CD4� responders culturedin media alone or with autologous CD4�Hy�MP1� T-APCs(Figure 3C). The CD4�HyMP1� T-APCs were also capable ofspecifically eliciting IFN-� by CD8�MP1-tetramer� T cells (datanot shown). Control cultures of irradiated T-APC stimulators alonedid not contain detectable levels of these cytokines. These data areconsistent with the processing and presentation of MP1 by T-APCsthrough the HLA class 1 and class 2 pathways.
HyMP1� T-APC–expanded CD8� MP1-specific T cells areamenable to genetic modification by plasmid electrotransferand express a CD19-specific chimeric immunoreceptor
The genetic modification of T cells to be specific for CD19 wasaccomplished using nonviral electrotransfer of a DNA expres-sion plasmid designated CD19R/HyTK-pMG, which directs theexpression of a CD19-specific scFvFc:� CAR and the bifunc-tional selection suicide gene consisting of hygromycin phospho-transferase and herpesvirus thymidine kinase (HyTK).23 Afterelectroporation, hygromycin selection and expansion usingOKT3, feeder cells, and rhIL-2, the MP1-tetramer� T-cell linewas analyzed by flow cytometric analyses for CAR expression.The CD19R/HyTK-pMG–transfected MP1-tetramer� lines re-tained MP1-tetramer binding and were also TcR V�17� (Figure3D), consistent with the finding that TcR V�17 is the dominantV� segment used by HLA-A2–restricted CTLs recognizingMP158-66.
38-40 (The presence of cell surface CD19R CAR wasdocumented by flow cytometry using an antihuman Fc-specificantibody. Ninety-six percent of the expanded hygromycin-
resistant MP1-tetramer� CTLs coexpressed CD19R CAR.)These T-cell lines were TCR���, NKG2D�, CD3�, CD4�,CD8�, CD27�, and CD28low (Figure 3D) and were CD2�,CD11a�, CD18�, CD54�, CD58�, as previously shown.23
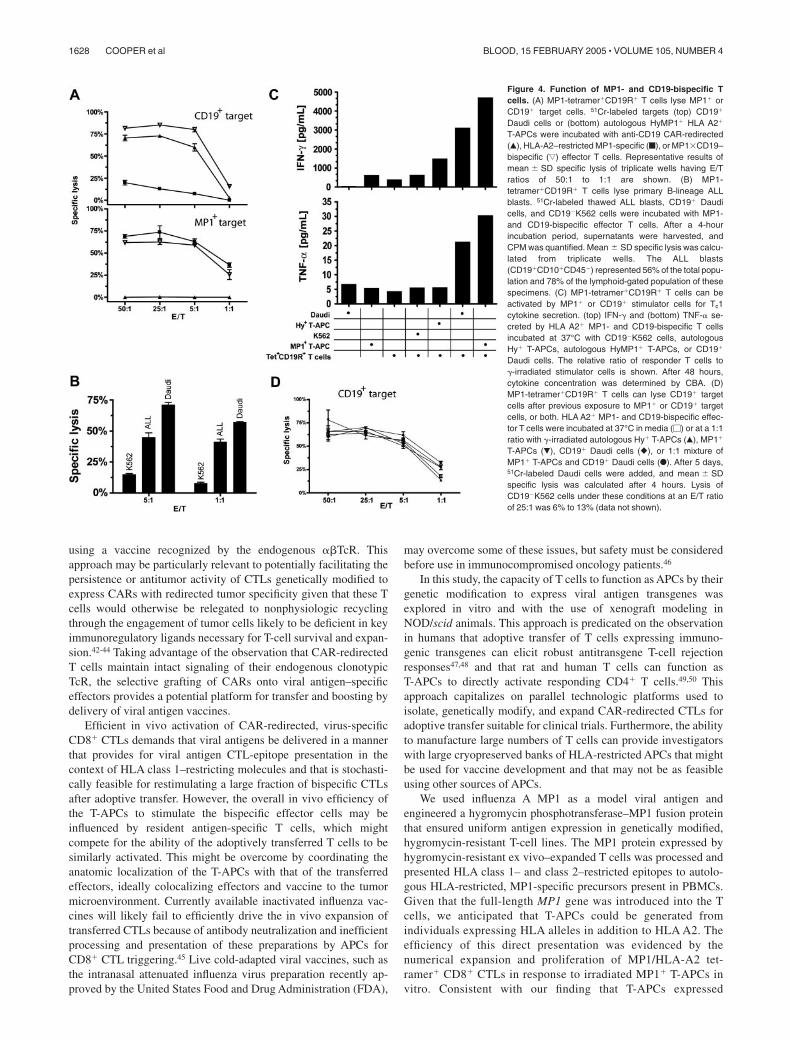
MP1-tetramer�CD19R�CD8� T cells are functionally bispecific
We investigated the ability of genetically modified MP1tetramer�CD19R� CD8� T cells to be activated through theirendogenous TcR and introduced CD19-specific CAR. Results from4-hour CRAs revealed that the MP1-tetramer�CD19R� CD8� Tcells could lyse both CD19� tumor targets in an HLA-unrestrictedmanner and MP1� targets in an HLA A2–restricted manner. Incontrast, an MP1-tetramerneg CTL expressing CD19R could lyseonly the CD19� targets, and MP1-tetramer� CTL could lyse onlythe MP1� targets (Figure 4A). MP1-tetramer�CD19R� CTLsrecognize and lyse primary B-lineage acute ALL blasts isolatedfrom clinical specimens (Figure 4B).
The ability of the MP1-tetramer�CD19R� effector T cells tosecrete cytokines in response to both MP1/HLA-A2 and CD19antigens was investigated by culturing these effector cells withstimulator cells expressing CD19 or MP1 antigen. After 48hours, a 5- to 8-fold increase in TNF-� and IFN-� wasdetectable in cultures stimulated with CD19� Daudi lymphomatumor cells, and a 7- to 12-fold increase in TNF-� and IFN-�was detectable on coculture with MP1� T-APCs, compared witheffector cells stimulated with MP1� stimulators or media alone(Figure 4C). Only low background levels of cytokine releasewere detected by T-APCs themselves in the absence of MP1-tetramer�CD19R� responders.
Figure 2. Generation of MP1-specific T cells by coculture withT-APCs. (A) Expansion of MP1-tetramer� T cells by coculture withautologous CD8�HyMP1� T-APCs. PBMCs from a homozygousHLA-A2� donor were cocultured for 21 days with 5 U/mL rhIL-2 inthe presence of (top row) CM alone or (middle row) a 5:1(responder/stimulator) ratio of autologous �-irradiated hygromycinB–resistant CD8�Hy�MP1� T-APCs (transfected with the plasmidpMGPac), or (bottom row) autologous �-irradiated CD8�HyMP1�
T-APCs (transfected with the plasmid HyMP1-pEK). Cultureswere supplemented with irradiated T-APCs every 7 days. Respond-ing cells were analyzed at the indicated time points by flowcytometry using FITC-conjugated anti-CD8 and PE-conjugatedMP1-HLA-A2*0201 tetramer. Propidium iodine� cells were ex-cluded from analysis. T-APCs themselves are MP1-tetramerneg
(data not shown). The percentage of tetramer� T-cells is shownafter electronic gating on CD8� T cells. (B) Expansion of MP1-tetramer� T cells by coculture with autologous CD4� HyMP1�
T-APCs. PBMCs from a heterozygous HLA-A2� donor werecocultured for 7 days with 5 U/mL rhIL-2 in the presence of (toprow) CM alone, (middle row) a 5:1 (responder/stimulator) ratio ofautologous �-irradiated hygromycin B–resistant CD4�Hy�MP1�
T-APCs (transfected with the plasmid pMGPac), or (bottom row)autologous �-irradiated CD4�HyMP1� T-APCs (transfected withthe plasmid HyMP1-pEK). Responding cells were analyzed at theindicated time points by flow cytometry using anti–CD8-FITC andPE-conjugated MP1-HLA-A2*201 tetramer. The percentage oftetramer� T cells is shown after electronic gating on CD8� T cells.(C) Proliferation of CD8�MP1-tetramer� T cells on T-APCs.5 � 104 HLAA2� CD8� MP1-tetramer� T-cells were cocultured ata 1:1 (responder/stimulator) ratio with media or with thawed�-irradiated autologous CD8�Hy� and CD8�HyMP1� T-APCs.The analysis was performed in quadruplicate, and the meanincorporated 3H-thymidine is shown along with SD. (D) Numericalexpansion of CD8�MP1-tetramer� T cells by T-APCs. 25 � 106
HLAA2� PBMCs were cocultured for 21 days at a 5:1 (responder/stimulator) ratio in low-dose rhIL-2 with thawed �-irradiatedautologous CD8�HyMP1� T-APCs. Cultures were supplementedwith irradiated T-APCs every 7 days. Viable cells were countedbased on the trypan blue dye exclusion method.
1626 COOPER et al BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

MP1-tetramer�CD19R�CD8� T cells retain cytolytic activitytoward CD19� tumor after coculture with MP1� T-APCs orCD19� tumor stimulators
To demonstrate that MP1-tetramer�CD19R� T cells can recycletheir CAR-regulated lytic effector function after activation throughthe endogenous TcR, CAR, or both, MP1�CD19 bispecific CTLswere cocultured with HyMP1� T-APCs, CD19� tumor cells, or a
mixture of T-APCs and tumor cells. After 5-day coculture, theseeffectors were harvested and subjected to 4-hour CRA againstCD19� tumor cells. The specific lysis of Daudi targets wasequivalent, regardless of the prior antigenic stimulation (endoge-nous TcR, CAR, or combined TcR/CAR) (Figure 4D).
Adoptive transfer of irradiated HyMP1� T-APCs augments theantilymphoma activity of MP1-tetramer�CD19R�CD8� CTLeffectors in vivo
The usefulness of adoptively transferred HyMP1� T-APCs forenhancing the in vivo antitumor potency of bispecific MP1�CD19CTLs was investigated. Using a noninvasive, biophotonic opticalimaging system, we tracked the regression of xenografted CD19�
ffLuc� Daudi lymphoma tumors.41 The in vitro ffLuc activity ofDaudi cells was approximately 2700-fold greater than that ofparental Daudi cells (data not shown). In this model system, ffLuc�
Daudi cells are seeded into the peritoneum of nonobese diabetic/severe combined immunodeficiency (NOD/scid) mice. Beforeadoptive therapy, tumor engraftment was verified by documenting2 successive biophotonic measurements with increasing ffLuc-mediated biophotonic tumor signal. Compared with tumor-bearingcontrol mice given rhIL-2 alone (group A), there was significant(P � .05) reduction of tumor ffLuc signal in mice givenMP1�CD19 bispecific CTLs in conjunction with 3 doses ofHyMP1� T-APCs and rhIL-2 (group B) (Figures 5 and 6A).Animals receiving bispecific effectors without MP1� T-APCs(group C, Hy�MP1�T-APCs/rhIL-2; group D, bispecific CTLs/rhIL-2) had intermediate responses (Figures 5 and 6A). Thereduction of biophotonic signal seen in mouse group B correlatedwith a lower cumulative biophotonic tumor flux for this group(Figure 6A), higher rates of biophotonic complete response rates(Figure 6B), and enhanced long-term progression-free survival(Figure 6C) compared with mouse groups A, C, and D.
To help understand the mechanism for the enhanced in vivoantitumor effect of combining T-APC with bispecific MP1�CD19CTLs, we generated a panel of HLA A2� artificial antigen-presenting cells (aAPCs) from U251T, which were geneticallymodified to express the genes Hy, HyMP1, or tCD19 or both tCD19and HyMP1. These cells served as a platform to test the ability ofthe bispecific MP1�CD19 T cells to respond to CD19, HyMP1, orboth. We demonstrate that MP1-tetramer�CD19R�CD8� T cellscan be activated for augmented proliferation and cytokine secretionafter the endogenous and chimeric immunoreceptors contact theirrespective antigens compared with when the TcR or CD19Rcontacts MP1 or CD19 antigen, respectively (Figure 7). We alsoobserved an augmentation of proliferation and cytokine releasewhen CD19� Daudi and autologous MP1� T-APCs were mixedwith bispecific MP1�CD19 CTLs compared with when thesestimulator cells were incubated separately with the responding Tcells (data not shown). This demonstrates that increased activationof bispecific T cells can occur when 2 different cells present thetargets. These data support the hypothesis that exposure of thebispecific T cells to MP1 and CD19 antigens results in augmentedT-cell activation, which, if it occurs in vivo, may lead to enhancedtumor control.
Discussion
Augmentation of the in vivo antitumor activity of adoptivelytransferred T cells is predicted to result in enhanced disease control.One strategy to achieve enhanced potency is to activate T cells
Figure 3. Function and phenotype of MP1-specific T cells. (A) T cellsexpanded on HyMP1� T-APCs exhibited HLA-restricted MP1-specific cytolyticresponse. 4-hour CRA using T-APC–elicited MP1-tetramer� effector T cells and51Cr-labeled HLA A2� T2 targets were performed to compare the lytic activityagainst T2 targets loaded in serum-free media with 10 M of the MP1-derivedpeptide GILGFVFTL (�) compared with mock-loaded targets (f). Representativeresults of mean SD specific lysis of triplicate wells having E/T ratios of 50:1 to1:1 are shown. (B) MP1-specific T cells elicited by HyMP1� T-APCs recognizeendogenously processed and presented HLA class 1 MP-1 epitopes and areactivated for cytokine secretion. CD8�MP1-tetramer� responder T cells cocul-tured with autologous CD8�HyMP1� T-APCs, followed by OKT3-based rapidexpansion, were incubated at a 1:1 responder/stimulator ratio with �-irradiatedautologous CD8�Hy� or CD8�HyMP1� T-APCs. Controls included T-APCswithout addition to culture of responders and responders incubated in CM withoutstimulators. After 48 hours, cell-free supernatants from these cultures wereharvested and subjected to CBA analysis for quantifying IFN-� and TNF-�content. (C) MP1-specific T cells elicited by HyMP1� T-APCs recognize endog-enously processed and presented HLA class 2 MP-1 epitopes and are activatedfor cytokine secretion. CD4� responder T cells cocultured for 21 days onautologous CD4�HyMP1� T-APCs, followed by OKT3-based rapid expansion,were incubated at a 1:1 responder/stimulator ratio with �-irradiated autologousCD4�Hy� or CD4�HyMP1� T-APCs. The CD4�HyMP1� T-APCs can alsostimulate MP1-specific secretion of IFN-� by CD8�MP1-tetramer� responder Tcells (data not shown). Controls included T-APCs without addition to culture ofresponders and responders incubated in CM without stimulators. After 48 hours,cell-free supernatants from these cultures were harvested and subjected to CBAanalysis for quantifying IFN-� content. (D) Phenotype of hygromycin-resistant,CD19R/HyTK-pMG–transfected MP1-specific CTLs. Histograms showing bindingof mAbs specific for T-cell cell-surface markers (bold lines), relative to isotypecontrol or unstained cells (dotted lines), to HyMP1� T-APC–elicited CD8�
MP1-tetramer� T cells, genetically modified with CD19R/HyTK-pMG and ex-panded by repeated OKT3-based 14-day stimulation cycles in the presence ofcytocidal concentrations of hygromycin B. The relative percentage of cells in eachgate is indicated.
T-APCs ACTIVATE BISPECIFIC T CELLS 1627BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4
using a vaccine recognized by the endogenous ��TcR. Thisapproach may be particularly relevant to potentially facilitating thepersistence or antitumor activity of CTLs genetically modified toexpress CARs with redirected tumor specificity given that these Tcells would otherwise be relegated to nonphysiologic recyclingthrough the engagement of tumor cells likely to be deficient in keyimmunoregulatory ligands necessary for T-cell survival and expan-sion.42-44 Taking advantage of the observation that CAR-redirectedT cells maintain intact signaling of their endogenous clonotypicTcR, the selective grafting of CARs onto viral antigen–specificeffectors provides a potential platform for transfer and boosting bydelivery of viral antigen vaccines.
Efficient in vivo activation of CAR-redirected, virus-specificCD8� CTLs demands that viral antigens be delivered in a mannerthat provides for viral antigen CTL-epitope presentation in thecontext of HLA class 1–restricting molecules and that is stochasti-cally feasible for restimulating a large fraction of bispecific CTLsafter adoptive transfer. However, the overall in vivo efficiency ofthe T-APCs to stimulate the bispecific effector cells may beinfluenced by resident antigen-specific T cells, which mightcompete for the ability of the adoptively transferred T cells to besimilarly activated. This might be overcome by coordinating theanatomic localization of the T-APCs with that of the transferredeffectors, ideally colocalizing effectors and vaccine to the tumormicroenvironment. Currently available inactivated influenza vac-cines will likely fail to efficiently drive the in vivo expansion oftransferred CTLs because of antibody neutralization and inefficientprocessing and presentation of these preparations by APCs forCD8� CTL triggering.45 Live cold-adapted viral vaccines, such asthe intranasal attenuated influenza virus preparation recently ap-proved by the United States Food and Drug Administration (FDA),
may overcome some of these issues, but safety must be consideredbefore use in immunocompromised oncology patients.46
In this study, the capacity of T cells to function as APCs by theirgenetic modification to express viral antigen transgenes wasexplored in vitro and with the use of xenograft modeling inNOD/scid animals. This approach is predicated on the observationin humans that adoptive transfer of T cells expressing immuno-genic transgenes can elicit robust antitransgene T-cell rejectionresponses47,48 and that rat and human T cells can function asT-APCs to directly activate responding CD4� T cells.49,50 Thisapproach capitalizes on parallel technologic platforms used toisolate, genetically modify, and expand CAR-redirected CTLs foradoptive transfer suitable for clinical trials. Furthermore, the abilityto manufacture large numbers of T cells can provide investigatorswith large cryopreserved banks of HLA-restricted APCs that mightbe used for vaccine development and that may not be as feasibleusing other sources of APCs.
We used influenza A MP1 as a model viral antigen andengineered a hygromycin phosphotransferase–MP1 fusion proteinthat ensured uniform antigen expression in genetically modified,hygromycin-resistant T-cell lines. The MP1 protein expressed byhygromycin-resistant ex vivo–expanded T cells was processed andpresented HLA class 1– and class 2–restricted epitopes to autolo-gous HLA-restricted, MP1-specific precursors present in PBMCs.Given that the full-length MP1 gene was introduced into the Tcells, we anticipated that T-APCs could be generated fromindividuals expressing HLA alleles in addition to HLA A2. Theefficiency of this direct presentation was evidenced by thenumerical expansion and proliferation of MP1/HLA-A2 tet-ramer� CD8� CTLs in response to irradiated MP1� T-APCs invitro. Consistent with our finding that T-APCs expressed
Figure 4. Function of MP1- and CD19-bispecific Tcells. (A) MP1-tetramer�CD19R� T cells lyse MP1� orCD19� target cells. 51Cr-labeled targets (top) CD19�
Daudi cells or (bottom) autologous HyMP1� HLA A2�
T-APCs were incubated with anti-CD19 CAR-redirected(Œ), HLA-A2–restricted MP1-specific (f), or MP1�CD19–bispecific (ƒ) effector T cells. Representative results ofmean SD specific lysis of triplicate wells having E/Tratios of 50:1 to 1:1 are shown. (B) MP1-tetramer�CD19R� T cells lyse primary B-lineage ALLblasts. 51Cr-labeled thawed ALL blasts, CD19� Daudicells, and CD19�K562 cells were incubated with MP1-and CD19-bispecific effector T cells. After a 4-hourincubation period, supernatants were harvested, andCPM was quantified. Mean SD specific lysis was calcu-lated from triplicate wells. The ALL blasts(CD19�CD10�CD45�) represented 56% of the total popu-lation and 78% of the lymphoid-gated population of thesespecimens. (C) MP1-tetramer�CD19R� T cells can beactivated by MP1� or CD19� stimulator cells for Tc1cytokine secretion. (top) IFN-� and (bottom) TNF-� se-creted by HLA A2� MP1- and CD19-bispecific T cellsincubated at 37°C with CD19�K562 cells, autologousHy� T-APCs, autologous HyMP1� T-APCs, or CD19�
Daudi cells. The relative ratio of responder T cells to�-irradiated stimulator cells is shown. After 48 hours,cytokine concentration was determined by CBA. (D)MP1-tetramer�CD19R� T cells can lyse CD19� targetcells after previous exposure to MP1� or CD19� targetcells, or both. HLA A2� MP1- and CD19-bispecific effec-tor T cells were incubated at 37°C in media (�) or at a 1:1ratio with �-irradiated autologous Hy� T-APCs (Œ), MP1�
T-APCs (�), CD19� Daudi cells (�), or 1:1 mixture ofMP1� T-APCs and CD19� Daudi cells (F). After 5 days,51Cr-labeled Daudi cells were added, and mean SDspecific lysis was calculated after 4 hours. Lysis ofCD19�K562 cells under these conditions at an E/T ratioof 25:1 was 6% to 13% (data not shown).
1628 COOPER et al BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

multiple costimulatory ligands used by T cells for full activa-tion, these T-APC–elicited MP1-specific responders were func-tional with respect to cytolytic killing, cytokine production, andproliferation. The ability to use CD4� T cells as APCs isexpected to be useful when the presence of antigen-specificCD8� CTLs present in the culture target and prevent theoutgrowth of autologous T-APCs. The MP1-specific lines couldbe modified to express a CD19-specific CAR. The derivedmodified CTLs display HLA-restricted MP1 and HLA-unrestricted CD19 bispecificity. In vitro, CD19-specific redi-rected lymphoma lysis was retained after activation by HyMP1�
T-APCs or CD19� tumor stimulators. In vivo, transfer ofMP1�CD19 bispecific effector/responder T cells, followed byirradiated HyMP1� autologous T-APCs, resulted in the aug-mented clearance of established Burkitt lymphoma xenograftscompared with control groups receiving Hy�MP1neg T-APCs.Although the mechanism responsible for this observation re-
mains to be established, we demonstrated that bispecificMP1�CD19 CTLs can be activated for augmented proliferationand cytokine production when the endogenous and introducedchimeric immunoreceptors are engaged. Presumably, it is thisactivation that leads to enhanced tumor clearance when bispe-cific effector T cells and T-APCs accumulate in the tumormicroenvironment. These data imply that combination therapyinfusing T-APCs and bispecific T cells could be improved if bothcell types could traffic to sites of tumor.
Whereas transgene-expressing T-APCs likely evoke antitrans-gene T-cell responses in immunocompetent syngeneic hosts bycross-priming through the uptake of antigen from apoptotic T-APCs by professional APCs, such as DCs and macrophages, oursystem clearly establishes that T-APCs themselves can directlystimulate antitransgene responses by processing and presentingtransgene-derived epitopes to CD4� and CD8� T cells. Thecapacity of T-APCs to present epitopes to HLA class 2–restricted
Figure 5. Biophotonic imaging of ffLuc� Daudi cells before and after adoptive T-cell therapy. Scatter graphs of tumor flux versus time and pseudocolor images ofselected mice (red lines) representing light intensity from ffLuc� Daudi cells in the peritoneum of NOD/scid mice serially imaged in ventral position. On day 0, NOD/scid micewere given 5 � 106 ffLuc� Daudi cells by intraperitoneal injection. (ffLuc� Daudi in vitro ffLuc activity was 141 8 CPM/cell (mean SD), compared with 0.05 CPM/cellbackground ffLuc activity in parental Daudi cells.) The mice with progressive disease, documented by 2 concurrent measurements demonstrating increase in tumor flux(measured on days 2 and 6), were among 4 treatment groups. The 5 mice from group A received no further cellular therapy. On day 7, the 5 mice in groups B, C, and D received20 � 106 MP1-tetramer�CD19R�CD8� T cells by intraperitoneal injection. Mice from group D received additional injections of 20 � 106 MP1-tetramer�CD19R�CD8� effectorT cells on days 9 and 12. On days 7, 9, 12, 21, 23, and 25, the mice in groups B and C received separate intraperitoneal injections of 5 � 106 thawed and �-irradiatedautologous hygromycin B-resistant T-APC that had been genetically modified with HyMP1-pMG (group B) or pMGPac (group C) coding for HyMP1 and Hy genes, respectively.All mice received rhIL-2 (25 000 U/mouse) by separate intraperitoneal injection on days 7, 9, 12, 21, 23, and 25. Each mouse was imaged at the same relative time point afterD-luciferin administration, which was within 19 minutes of injection. Data are presented as photon flux for a region of interest (ROI) encompassing the whole mouse. Similardata were obtained in repeated experiments (data not shown).
T-APCs ACTIVATE BISPECIFIC T CELLS 1629BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

CD4� TH from endogenously expressed transgenes is likelyimportant for coordinating the reactivation of CAR� CTL effectorswith the stimulation of endogenous CD4 antiviral help necessary tosupport these bispecific effectors. The capacity of T cells tostimulate an in vitro primary response is under investigation.However, the T-APC approach may be uniquely suited for selectivelyboosting the magnitude of adoptively transferred T cells when adminis-tered systemically, in part because of the expectation that theseautologous stimulators will not be rapidly neutralized by antibodyresponses, as might be predicted by the use of viral vectors or, aspreviously reported, the use of allogeneic PBMCs to drive allospecific,CAR-redirected effector cells.51 Preadoptive transfer lymphodepletionmay also be an important variable for facilitating the selective boostingof transferred bispecific effectors over a numerically larger endogenouspool of viral-specific responders.
Although the simplicity of generating T-APCs by nonviralgene transfer, as demonstrated in this work, offers feasibilityand safety advantages for boosting bispecific CTLs in vivo, wehypothesize that viral antigen transgene expression represents afirst-generation system amenable to significant additional en-
hancements by genetic engineering. Therefore, T-APCs mightbe further engineered to express transgenes to augment theirAPC activity, such as by enforced expression of CD19, desiredcostimulator molecules, or secretion of desired cytokines forloco-regional uptake. T cells used as APCs also offer thepotential for systemically targeting the APCs to defined ana-tomic locations beyond the peritoneal cavity described in thecurrent mouse model. These strategies might use the expressionof chemokine receptor and selectin transgenes that recapitulatethose used in the metastatic homing of tumors and, conceivably,may augment the antitumor activity of transferred bispecificeffectors through their restimulation in the tumor microenviron-ment. Our group is particularly interested in augmenting thelymph node and bone marrow homing of T-APCs to be used inconjunction with CD19-specific, CAR-expressing effector cellsfor immunotherapy of B-cell lymphomas and leukemias.
In conclusion, our studies demonstrate the usefulness of T cellsto function as APCs through their genetic modification to expressviral antigens. T-APCs have considerable potential to enhance theactivation of adoptively transferred, tumor-redirected effector Tcells with resultant improvements in antitumor responses. Applica-tions of T-APCs in transfer and boost clinical experimentation areanticipated to follow our currently active phase 1 trials for B-celllymphoma and leukemias using adoptive transfer of CD20– andCD19–CAR-redirected CTLs.
Acknowledgments
We thank Dr Michael Kalos (City of Hope) for critical reading ofthe manuscript, Dr David Senitzer (City of Hope) for assistancewith HLA typing, Lucy Brown for assistance with flow cytometrysorting, the Animal Resource Center at City of Hope, under thedirection of Dr Richard Ermel, and the Children’s Oncology GroupALL cell bank for providing primary ALL samples.
Figure 6. Adoptive immunotherapy of CD19� tumor using MP1- and CD19-bispecific T cells and T-APCs. (A) Serial measurement of flux from tumor. Trendlines for each group were derived by smoothing the tumor flux over all mice within a group.Background flux measurements, simultaneously measured from mice without ffLuc� tumorthat received D-luciferin, was between 106 and 107 p/s per cm2/sr, as discussed in“Statistical methods to analyze biophotonic data.” Tumor flux was measured periodically,as discussed in “Biophotonic tumor imaging.” Mouse treatment groups are as described inthe legend to Figure 5. Group A is represented by the solid blue line; group B, red dashedline; group C, orange dotted line; group D, green dashed and dotted line. (B) Percentage oftumor-bearing mice that achieved complete remission. Time to complete remission wascalculated from the beginning of the experiment until the first date when a mouse’s tumorflux measurement fell below the detection threshold. Complete remission was defined asmeasurable flux beneath the minimum threshold of tumor detection, estimated as3.4 � 106 p/s per cm2/sr, as described in “Statistical methods to analyze biophotonic data.”Mouse treatment groups are as described in the legend to Figure 5. (C) Progression-freesurvival. The tumor was defined as progressive when tumor flux was 10 times the valuerecorded on day 2. Mouse treatment groups are as described in the legend to Figure 5.
Figure 7. Stimulation of CD19- and MP1-bispecific T cells through introducedand endogenous immunoreceptor results in augmented activation. (A) Tomeasure proliferation, 106 HLA A2� MP1-tetramer�CD19R�CD8� T cells werecultured for 96 hours at a 1:1 ratio with irradiated HLA A2� aAPCs. For the last 18hours, the cells were pulsed with 3H-thymidine, and the incorporated thymidine wasdetermined using a scintillation counter. Data are presented as mean SD. (B) Tomeasure IFN-� cytokine production, bispecific MP1�CD19 CTLs were coculturedwith the aAPCs, and the conditioned tissue-culture supernatant was collected after48 hours and analyzed using a CBA. The U251T aAPCs were genetically modified todurably express tCD19, HyMP1, tCD19 and HyMP1, or Hy genes.
1630 COOPER et al BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4

References
1. Yee C, Thompson JA, Byrd D, et al. Adoptive Tcell therapy using antigen-specific CD8� T cellclones for the treatment of patients with meta-static melanoma: in vivo persistence, migration,and antitumor effect of transferred T-cells. ProcNatl Acad Sci U S A. 2002;99:16168-16173.
2. Walter EA, Greenberg PD, Gilbert MJ, et al. Re-constitution of cellular immunity against cytomeg-alovirus in recipients of allogeneic bone marrowby transfer of T-cell clones from the donor. N EnglJ Med. 1995;333:1038-1044.
3. Dudley ME, Wunderlich JR, Robbins PF, et al.Cancer regression and autoimmunity in patientsafter clonal repopulation with antitumor lympho-cytes. Science. 2002;298:850-854.
4. Brodie SJ, Patterson BK, Lewinsohn DA, et al.HIV-specific cytotoxic T lymphocytes traffic tolymph nodes and localize at sites of HIV replica-tion and cell death. J Clin Invest. 2000;105:1407-1417.
5. Heslop HE, Ng CY, Li C, et al. Long-term restora-tion of immunity against Epstein-Barr virus infec-tion by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551-555.
6. Wang RF, Rosenberg SA. Human tumor antigensfor cancer vaccine development. Immunol Rev.1999;170:85-100.
7. Pardoll D. Does the immune system see tumorsas foreign or self? Annu Rev Immunol. 2003;21:807-839.
8. Garrido F, Algarra I. MHC antigens and tumor es-cape from immune surveillance. Adv Cancer Res.2001;83:117-158.
9. Roessig C, Scherer SP, Baer A, et al. TargetingCD19 with genetically modified EBV-specific hu-man T lymphocytes. Ann Hematol. 2002;(suppl2):S42-S43.
10. Jensen MC, Cooper LJ, Wu AM, Forman SJ,Raubitschek A. Engineered CD20-specific pri-mary human cytotoxic T lymphocytes for targetingB-cell malignancy. Cytotherapy. 2003;5:131-138.
11. Brentjens RJ, Latouche JB, Santos E, et al.Eradication of systemic B-cell tumors by geneti-cally targeted human T lymphocytes co-stimu-lated by CD80 and interleukin-15. Nat Med. 2003;9:279-286.
12. Imai C, Mihara K, Andreansky M, et al. Chimericreceptors with 4-1BB signaling capacity provokepotent cytotoxicity against acute lymphoblasticleukemia. Leukemia. 2004;18:676-684.
13. Gross G, Eshhar Z. Endowing T-cells with anti-body specificity using chimeric T cell receptors.FASEB J. 1992;6:3370-3378.
14. Fitzer-Attas CJ, Schindler DG, Waks T, Eshhar Z.Harnessing Syk family tyrosine kinases as signal-ing domains for chimeric single chain of the vari-able domain receptors: optimal design for T cellactivation. J Immunol. 1998;160:145-154.
15. Finney HM, Lawson AD, Bebbington CR, WeirAN. Chimeric receptors providing both primaryand costimulatory signaling in T-cells from asingle gene product. J Immunol. 1998;161:2791-2797.
16. Geiger TL, Nguyen P, Leitenberg D, Flavell RA.Integrated src kinase and costimulatory activityenhances signal transduction through single-chain chimeric receptors in T lymphocytes. Blood.2001;98:2364-2371.
17. Pule M, Finney H, Lawson A. Artificial T-cell re-ceptors. Cytotherapy. 2003;5:211-226.
18. Sadelain M, Riviere I, Brentjens R. Targeting tu-
mours with genetically enhanced T lymphocytes.Nat Rev Cancer. 2003;3:35-45.
19. Finney HM, Akbar AN, Lawson AD. Activation ofresting human primary T-cells with chimeric re-ceptors: costimulation from CD28, inducible co-stimulator, CD134, and CD137 in series with sig-nals from the TCR� chain. J Immunol. 2004;172:104-113.
20. Jensen M, Tan G, Forman S, Wu AM, RaubitschekA. CD20 is a molecular target for scFvFc:zetareceptor redirected T-cells: implications for cellu-lar immunotherapy of CD20� malignancy. BiolBlood Marrow Transplant. 1998;4:75-83.
21. Rossig C, Bollard CM, Nuchtern JG, Rooney CM,Brenner MK. Epstein-Barr virus-specific human Tlymphocytes expressing antitumor chimeric T-cellreceptors: potential for improved immunotherapy.Blood. 2002;99:2009-2016.
22. Cooper LJ, Al-Kadhimi Z, Serrano LM, et al. Adop-tively transferred bispecific T cells expanded invivo by restimulation with a T-cell vaccine cantreat established B-lineage malignancy [abstract].Blood. 2003;102:2292.
23. Cooper LJ, Topp MS, Serrano LM, et al. T-cellclones can be rendered specific for CD19: towardthe selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637-1644.
24. Mahmoud MS, Fujii R, Ishikawa H, Kawano MM.Enforced CD19 expression leads to growth inhibi-tion and reduced tumorigenicity. Blood. 1999;94:3551-3558.
25. Klein E, Klein G, Nadkarni JS, Nadkarni JJ, Wig-zell H, Clifford P. Surface IgM-kappa specificity ona Burkitt lymphoma cell in vivo and in derived cul-ture lines. Cancer Res. 1968;28:1300-1310.
26. Salter RD, Howell DN, Cresswell P. Genes regu-lating HLA class I antigen expression in T-B lym-phoblast hybrids. Immunogenetics. 1985;2:235-246.
27. Koeffler HP, Golde DW. Human myeloid leukemiacell lines: a review. Blood. 1980;56:344-350.
28. Gotch F, Rothbard J, Howland K, Townsend A,McMichael A. Cytotoxic T lymphocytes recognizea fragment of influenza virus matrix protein in as-sociation with HLA-A2. Nature. 1987;326:881-882.
29. Larsson M, Messmer D, Somersan S, et al. Re-quirement of mature dendritic cells for efficientactivation of influenza A-specific memory CD8�
T-cells. J Immunol. 2000;165:1182-1190.
30. Morrison J, Elvin J, Latron F, et al. Identification ofthe nonamer peptide from influenza A matrix pro-tein and the role of pockets of HLA-A2 in its rec-ognition by cytotoxic T lymphocytes. Eur J Immu-nol. 1992;22:903-907.
31. Jensen MC, Clarke P, Tan G, et al. Human T lym-phocyte genetic modification with naked DNA.Mol Ther. 2000;1:49-55.
32. David HA. Robust estimation in the presence ofoutliers. Robustness in Statistics. 1979;1:61-74.
33. Guttman I, Smith DE. Investigation of rules fordealing with outliers in small samples from thenormal distribution I: estimation of the mean.Technometrics. 1969;11:527-550.
34. R Development Core Team. R: A Language andEnvironment for Statistical Computing. Vienna,Austria: R Foundation for Statistical Computing;2003.
35. Hothorn T, Hornik K. exactRankTests package forR. Available at: http://cran.r-project.org. AccessedNovember 23, 2004.
36. Streitberg B, Rohmel J. Exact distributions forpermutations and rank tests: an introduction tosome recently published algorithms. StatisticalSoftware Newsletter. 1986;12:10-17.
37. Uetsuki T, Naito A, Nagata S, Kaziro Y. Isolationand characterization of the human chromosomalgene for polypeptide chain elongation factor-1alpha. J Biol Chem. 1989;264:5791-5798.
38. Lehner PJ, Wang EC, Moss PA, et al. HumanHLA-A0201-restricted cytotoxic T lymphocyte rec-ognition of influenza A is dominated by T-cellsbearing the V beta 17 gene segment. J Exp Med.1995;181:79-91.
39. Lawson TM, Man S, Williams S, Boon AC, Zam-bon M, Borysiewicz LK. Influenza A antigen expo-sure selects dominant Vbeta17� TCR in humanCD8� cytotoxic T cell responses. Int Immunol.2001;13:1373-1381.
40. Moss PA, Moots RJ, Rosenberg WM, Rowland-Jones SJ, Bodmer HC, McMichael AJ. Extensiveconservation of alpha and beta chains of the hu-man T-cell antigen receptor recognizing HLA-A2and influenza A matrix peptide. Proc Natl AcadSci U S A. 1991;88:8987-8990.
41. Jenkins DE, Oei Y, Hornig YS, et al. Biolumines-cent imaging (BLI) to improve and refine tradi-tional murine models of tumor growth and metas-tasis. Clin Exp Metastasis. 2003;20:733-744.
42. Townsend SE, Allison JP. Tumor rejection afterdirect costimulation of CD8� T cells by B7-trans-fected melanoma cells. Science. 1993;259:368-370.
43. Cardoso AA, Veiga JP, Ghia P, et al. Adoptive T-cell therapy for B-cell acute lymphoblastic leuke-mia: preclinical studies. Blood. 1999;94:3531-3540.
44. D’Amico G, Vulcano M, Bugarin C, et al. Activa-tion of BCP-ALL cells through CD40 generatesIL-10 producing, IL-12 defective APC inducingallogeneic T-cell anergy. Blood. 2004;104:744-751.
45. Deng Y, Jing Y, Campbell AE, Gravenstein S.Age-related impaired type 1 T cell responses toinfluenza: reduced activation ex vivo, decreasedexpansion in CTL culture in vitro, and blunted re-sponse to influenza vaccination in vivo in the el-derly. J Immunol. 2004;172:3437-3446.
46. Nichol KL. Live attenuated influenza virus vac-cines: new options for the prevention of influenza.Vaccine. 2001;19:4373-4377.
47. Berger C, Huang ML, Gough M, Greenberg PD,Riddell SR, Kiem HP. Nonmyeloablative immu-nosuppressive regimen prolongs in vivo persis-tence of gene-modified autologous T cells in anonhuman primate model. J Virol. 2001;75:799-808.
48. Riddell SR, Elliott M, Lewinsohn DA, et al. T-cellmediated rejection of gene-modified HIV-specificcytotoxic T lymphocytes in HIV-infected patients.Nat Med. 1996;2:216-223.
49. Mannie MD, Walker MR. Feedback activation ofT-cell antigen-presenting cells during interactionswith T-cell responders. J Leukoc Biol. 2001;70:252-260.
50. Atanackovic D, Matsuo M, Ritter E, et al. Monitor-ing CD4� T cell responses against viral and tu-mor antigens using T cells as novel target APC.J Immunol Methods. 2003;278:57-66.
51. Kershaw MH, Westwood JA, Hwu P. Dual-specificT-cells combine proliferation and antitumor activ-ity. Nat Biotechnol. 2002;20:1221-1227.
T-APCs ACTIVATE BISPECIFIC T CELLS 1631BLOOD, 15 FEBRUARY 2005 � VOLUME 105, NUMBER 4
Copyright © 2022 FDOKUMEN




















