
Down-regulation of tyrosine aminotransferase at a frequently deleted region 16q22 contributes to the...
11
Down-Regulation of Tyrosine Aminotransferase at a Frequently Deleted Region 16q22 Contributes to the Pathogenesis of Hepatocellular Carcinoma Li Fu, 1,2* Sui-Sui Dong, 2* Yi-Wu Xie, 2 Lai-Shan Tai, 2 Leilei Chen, 2 Kar Lok Kong, 2 Kwan Man, 3 Dan Xie, 1 Yan Li, 1 Yingduan Cheng, 4 Qian Tao, 4 and Xin-Yuan Guan 1,2 Loss of 16q is one of the most frequent alterations in many malignancies including hepato- cellular carcinomas (HCC), suggesting the existence of a tumor suppressor gene (TSG) within the frequently deleted region. In this report we describe the identification and char- acterization of one candidate TSG, tyrosine aminotransferase gene (TAT), at 16q22.1. Loss of one TAT allele was detected in 27/50 (54%) of primary HCCs by quantitative real-time polymerase chain reaction. In addition, homo-deletion of TAT alleles was detected in two cases. Down-regulation of TAT was detected in 28/50 (56%) of HCCs, which was signifi- cantly associated with the loss of TAT allele and hypermethylation of TAT 5 0 CpG island (CGI) region (P < 0.001). Functional studies found that TAT has a strong tumor suppres- sive ability. Introduction of the TAT gene into HCC cell lines could effectively inhibit col- ony formation in soft agar, foci formation, and tumor formation in nude mice. Further study found that the tumor suppressive mechanism of TAT was associated with its proa- poptotic role in a mitochondrial-dependent manner by promoting cytochrome-c release and activating caspase-9 and PARP. Conclusion: Taken together, our findings suggest that TAT plays an important suppressive role in the development and progression of HCC. (HEPATOLOGY 2010;51:1624-1634) H epatocellular carcinoma (HCC) is one of the most common cancers in the world, espe- cially in Asia and Africa, with a very poor prognosis. 1 It is believed that the pathogenesis of HCC is a long-term process that involves multiple genetic alterations. Deletion of 16q is one of the most frequent chromosomal alterations in primary HCC, as observed in studies using loss of heterozygosity (LOH) 2 and comparative genomic hybridization (CGH). 3 In our previous CGH study the loss of 16q was observed at a strikingly high rate of 70% in 50 primary HCC cases and this deletion may be an early event in the pathogenesis of HCC. 3 Loss of tumor suppressor gene (TSG), E-cadherin at 16q22, has been reported in hepatitis B virus-associated HCC. 4 Using a fine mapping strategy, several distinct minimal deleted regions on 16q were found, 2 suggesting the existence of other TSGs on 16q associated with HCC pathogenesis. In order to isolate down-regulated tran- scripts at 16q, complementary DNAs (cDNAs) gener- ated from a primary HCC tumor with the loss of 16q have been applied to subtract cDNAs generated from its matched nontumor liver tissue. Most of the subtracted genes are localized at commonly deleted chromosomal regions in HCC including 1p, 4q, 8p, 16q, and 17p (unpublished data). One of the isolated genes is the tyro- sine aminotransferase (TAT) gene located at 16q22.1. Abbreviations: CGH, comparative genomic hybridization; HCC, hepatocellular carcinoma; LOH, loss of heterozygosity; MSP, methylation-specific PCR; qPCR, quantitative real-time PCR; TAT, tyrosine aminotransferase; TMA, tissue microarray; TSG, tumor suppressor gene. From the 1 State Key Laboratory of Oncology in Southern China, Cancer Center, Sun Yat-sen University, Guangzhou, China; 2 Department of Clinical Oncology and Center for Cancer Research; 3 Department of Surgery, University of Hong Kong, Pokfulam, Hong Kong; and 4 Department of Clinical Oncology, Chinese University of Hong Kong, Hong Kong. Additional supporting information may be found in the online version of this article. Received July 27, 2009; accepted December 17, 2009. Supported by Research Grant Council Grant (HKU 7393/04M), Hong Kong Research Grant Council Central Allocation (HKU 1/06C & HKU 5/ CRF/08), Sun Yat-Sen University ‘‘Hundred Talents Program’’ (85000-3171311), and the Major State Basic Research Program of China (2006CB910104). *These authors contributed equally to this work. Address reprint requests to: Xin-Yuan Guan, State Key Laboratory of Oncology in Southern China, Cancer Center, Sun Yat-Sen University, Room 605, 651 Dongfeng Road East, Guangzhou 510060, China. E-mail: xyguan@ hkucc.hku.hk; . Copyright V C 2010 by the American Association for the Study of Liver Diseases. Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hep.23540 Potential conflict of interest: Nothing to report. 1624
Transcript of Down-regulation of tyrosine aminotransferase at a frequently deleted region 16q22 contributes to the...

Down-Regulation of Tyrosine Aminotransferase at aFrequently Deleted Region 16q22 Contributes to the
Pathogenesis of Hepatocellular CarcinomaLi Fu,1,2* Sui-Sui Dong,2* Yi-Wu Xie,2 Lai-Shan Tai,2 Leilei Chen,2 Kar Lok Kong,2 Kwan Man,3 Dan Xie,1
Yan Li,1 Yingduan Cheng,4 Qian Tao,4 and Xin-Yuan Guan1,2
Loss of 16q is one of the most frequent alterations in many malignancies including hepato-cellular carcinomas (HCC), suggesting the existence of a tumor suppressor gene (TSG)within the frequently deleted region. In this report we describe the identification and char-acterization of one candidate TSG, tyrosine aminotransferase gene (TAT), at 16q22.1. Lossof one TAT allele was detected in 27/50 (54%) of primary HCCs by quantitative real-timepolymerase chain reaction. In addition, homo-deletion of TAT alleles was detected in twocases. Down-regulation of TAT was detected in 28/50 (56%) of HCCs, which was signifi-cantly associated with the loss of TAT allele and hypermethylation of TAT 50 CpG island(CGI) region (P < 0.001). Functional studies found that TAT has a strong tumor suppres-sive ability. Introduction of the TAT gene into HCC cell lines could effectively inhibit col-ony formation in soft agar, foci formation, and tumor formation in nude mice. Furtherstudy found that the tumor suppressive mechanism of TAT was associated with its proa-poptotic role in a mitochondrial-dependent manner by promoting cytochrome-c releaseand activating caspase-9 and PARP. Conclusion: Taken together, our findings suggest thatTAT plays an important suppressive role in the development and progression of HCC.(HEPATOLOGY 2010;51:1624-1634)
Hepatocellular carcinoma (HCC) is one of themost common cancers in the world, espe-cially in Asia and Africa, with a very poor
prognosis.1 It is believed that the pathogenesis ofHCC is a long-term process that involves multiplegenetic alterations. Deletion of 16q is one of the mostfrequent chromosomal alterations in primary HCC, asobserved in studies using loss of heterozygosity(LOH)2 and comparative genomic hybridization(CGH).3 In our previous CGH study the loss of 16qwas observed at a strikingly high rate of 70% in 50primary HCC cases and this deletion may be an earlyevent in the pathogenesis of HCC.3
Loss of tumor suppressor gene (TSG), E-cadherin at16q22, has been reported in hepatitis B virus-associatedHCC.4 Using a fine mapping strategy, several distinctminimal deleted regions on 16q were found,2 suggestingthe existence of other TSGs on 16q associated with HCCpathogenesis. In order to isolate down-regulated tran-scripts at 16q, complementary DNAs (cDNAs) gener-ated from a primary HCC tumor with the loss of 16qhave been applied to subtract cDNAs generated from itsmatched nontumor liver tissue. Most of the subtractedgenes are localized at commonly deleted chromosomalregions in HCC including 1p, 4q, 8p, 16q, and 17p(unpublished data). One of the isolated genes is the tyro-sine aminotransferase (TAT) gene located at 16q22.1.
Abbreviations: CGH, comparative genomic hybridization; HCC,hepatocellular carcinoma; LOH, loss of heterozygosity; MSP, methylation-specificPCR; qPCR, quantitative real-time PCR; TAT, tyrosine aminotransferase;TMA, tissue microarray; TSG, tumor suppressor gene.From the 1State Key Laboratory of Oncology in Southern China, Cancer
Center, Sun Yat-sen University, Guangzhou, China; 2Department of ClinicalOncology and Center for Cancer Research; 3Department of Surgery, University ofHong Kong, Pokfulam, Hong Kong; and 4Department of Clinical Oncology,Chinese University of Hong Kong, Hong Kong.
Additional supporting information may be found in the online version ofthis article.Received July 27, 2009; accepted December 17, 2009.Supported by Research Grant Council Grant (HKU 7393/04M), Hong
Kong Research Grant Council Central Allocation (HKU 1/06C & HKU 5/CRF/08), Sun Yat-Sen University ‘‘Hundred Talents Program’’(85000-3171311), and the Major State Basic Research Program of China(2006CB910104).
*These authors contributed equally to this work.Address reprint requests to: Xin-Yuan Guan, State Key Laboratory of
Oncology in Southern China, Cancer Center, Sun Yat-Sen University, Room605, 651 Dongfeng Road East, Guangzhou 510060, China. E-mail: [email protected]; .Copyright VC 2010 by the American Association for the Study of Liver Diseases.Published online in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/hep.23540Potential conflict of interest: Nothing to report.
1624

The TAT gene encodes a mitochondrial protein ty-rosine aminotransferase which is present in the liverand breaks down tyrosine in a five-step process intoharmless molecules that are either excreted by the kid-neys or used in reactions that produce energy. Theliver is the principle site of tyrosine formation as wellas degradation. Under normal conditions, intracellulartyrosine levels are tightly controlled; transported tyro-sine and tyrosine synthesized from phenylalanine arein different metabolic pools.5 Deficiency of hepatic ty-rosine aminotransferase results in tyrosinemia type II(Richner-Hanhart syndrome, RHS). Tyrosinemia is ahereditary disease characterized by elevated blood levelsof tyrosine, a building block of most proteins. Muta-tions in the TAT gene cause a shortage of the enzyme,leading to a toxic accumulation of tyrosine and itsbyproducts, which can damage the liver, kidneys, nerv-ous system, and other organs and tissues.6 Tyrosinemiahas long been considered an important risk factor forHCC.7,8 Although this correlation was primarily basedon data from tyrosinemia type I, it is fairly reasonableto speculate that loss of TAT may also be a high riskfor HCC, because loss of TAT results in similar eleva-tions of tyrosine and its catabolites, as in tyrosinemiatype I. In the present study, down-regulation of TATwas frequently detected in primary HCCs. Introduc-tion of TAT gene into HCC cells could effectively in-hibit their tumorigenicity, strongly suggesting thatTAT plays a tumor suppressive role in the pathogenesisof HCC.
Materials and MethodsHCC Cell Lines and Tissue DNA/RNA Samples.
Fifty primary HCC samples and their surroundingnontumor liver tissues were collected at the time ofsurgical resections from the Cancer Center, Sun Yat-Sen University (Guangzhou, China). Samples used inthis study were approved by the Committees for Ethi-cal Review of Research Involving Human Subjects atCancer Center, Sun Yat-Sen University. HCC cell linesQGY-7703, BEL7402, and PLC-8024 were obtainedfrom the Institute of Virology, Chinese Academy ofMedical Sciences (Beijing, China). Genomic DNA andtotal RNA from these samples were extracted asdescribed.9
Semiquantitative and Quantitative PolymeraseChain Reaction (PCR) (qPCR). Details are describedin the Supporting Materials and Methods.Southern and Northern Blot Hybridization. A
1401-bp probe of the TAT cDNA covering its whole
coding region was synthesized by PCR (the primersare listed in Supporting Table 1). This probe was thenlabeled with 32P and hybridized to the membranetransferred with HCC genomic DNA or total RNA bysouthern and northern blot analyses as described.10
Detection of Homo-Deletion by PCR. PCR wascarried out for 28 cycles with genomic DNA from 50HCC cases as a template. Five sets of primers used inthe PCR are listed in Supporting Table 1.Tissue Microarrays (TMA) and Immunohisto-
chemistry (IHC). Details are described in the Sup-porting Materials and Methods.11
Immunofluorescence. Details are described in theSupporting Materials and Methods.5-Aza-20-Deoxycytidine (5-Aza) Treatment. To
study whether demethylation could restore TAT expres-sion in QGY-7703 cells, 2 � 105 cells were treatedwith the DNA demethylating agent 5-Aza (Sigma-Aldrich, St. Louis, MO) at the indicated concentrationfor 72 hours. Drugs and culture medium wererefreshed every day during treatment.Bisulfite Treatment and Methylation-Specific PCR
(MSP). Genomic DNA was chemically modified with2.4 mol/L of sodium metabisulfite for 4 hours asdescribed.9 The bisulfite-modified DNA was amplifiedusing primers for either methylated or unmethylatedsequences of the 50 CpG island of TAT. The primers formethylated and unmethylated TAT are listed in Support-ing Table 1. MSP was performed with 36 cycles.Tumor-Suppressive Function of TAT. To test the
tumor-suppressive function of TAT, the full-lengthcDNA and mutant (deletion of 77 amino acids at C-terminal) of the gene was PCR amplified, cloned intopcDNA3.1/V5-His TOPO TA vector (Invitrogen,Carlsbad, CA), and transfected into HCC cell lineQGY-7703 and BEL7402 cells. Stable TAT-expressingclones were selected for further study. Empty vectortransfected QGY-7703 and BEL7402 cells (Vec-7703/Vec-7402) were used as control. MTT assay, foci for-mation assay, and colony formation in soft agar wascarried out as described.9
Tumor Formation in Nude Mice. The in vivo tu-mor-suppressive ability of TAT was investigated by tu-mor xenograft experiment. About 4 � 106 TAT-expressing QGY-7703 cells or control Vec-7703 cellswere injected subcutaneously into the right and lefthind legs of 4-week-old nude mice (10 mice for TAT-c2 and 10 mice for TAT-c3 cells), respectively. Tumorformation in nude mice was monitored over a 4-weekperiod.Cell Cycle Analysis. Details are described in the
Supporting Materials and Methods.
HEPATOLOGY, Vol. 51, No. 5, 2010 FU ET AL. 1625
Detection of Apoptosis by Transferase-MediateddUTP Nick-End Labeling (TUNEL) Assay. TAT-transfected and vector-transfected QGY-7703 cellswere treated with straurosporine (STS; 1 lM) for 4
hours. Morphological changes in the nuclear chro-matin undergoing apoptosis were detected byterminal deoxynucleotidyl TUNEL assay accordingto the manufacturer’s protocol (Roche, Mannheim,
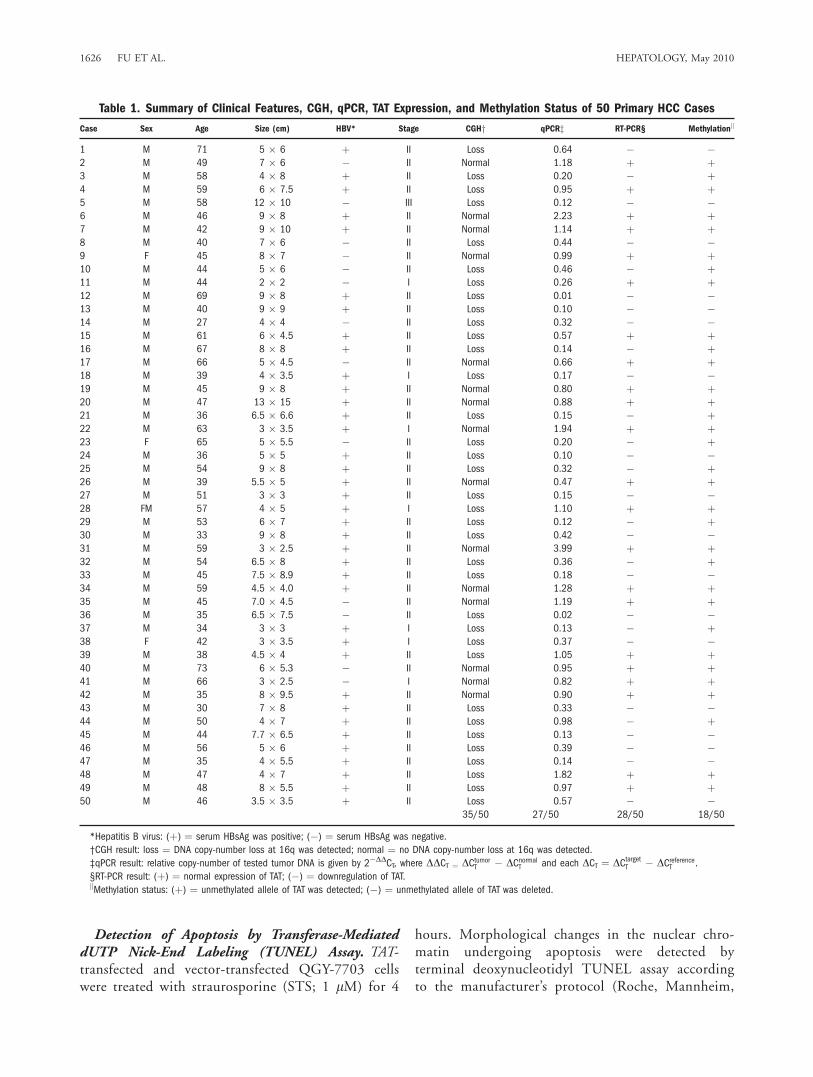
Table 1. Summary of Clinical Features, CGH, qPCR, TAT Expression, and Methylation Status of 50 Primary HCC Cases
Case Sex Age Size (cm) HBV* Stage CGHy qPCRz RT-PCR§ Methylationk
1 M 71 5 � 6 þ II Loss 0.64 � �2 M 49 7 � 6 � II Normal 1.18 þ þ3 M 58 4 � 8 þ II Loss 0.20 � þ4 M 59 6 � 7.5 þ II Loss 0.95 þ þ5 M 58 12 � 10 � III Loss 0.12 � �6 M 46 9 � 8 þ II Normal 2.23 þ þ7 M 42 9 � 10 þ II Normal 1.14 þ þ8 M 40 7 � 6 � II Loss 0.44 � �9 F 45 8 � 7 � II Normal 0.99 þ þ10 M 44 5 � 6 � II Loss 0.46 � þ11 M 44 2 � 2 � I Loss 0.26 þ þ12 M 69 9 � 8 þ II Loss 0.01 � �13 M 40 9 � 9 þ II Loss 0.10 � �14 M 27 4 � 4 � II Loss 0.32 � �15 M 61 6 � 4.5 þ II Loss 0.57 þ þ16 M 67 8 � 8 þ II Loss 0.14 � þ17 M 66 5 � 4.5 � II Normal 0.66 þ þ18 M 39 4 � 3.5 þ I Loss 0.17 � �19 M 45 9 � 8 þ II Normal 0.80 þ þ20 M 47 13 � 15 þ II Normal 0.88 þ þ21 M 36 6.5 � 6.6 þ II Loss 0.15 � þ22 M 63 3 � 3.5 þ I Normal 1.94 þ þ23 F 65 5 � 5.5 � II Loss 0.20 � þ24 M 36 5 � 5 þ II Loss 0.10 � �25 M 54 9 � 8 þ II Loss 0.32 � þ26 M 39 5.5 � 5 þ II Normal 0.47 þ þ27 M 51 3 � 3 þ II Loss 0.15 � �28 FM 57 4 � 5 þ I Loss 1.10 þ þ29 M 53 6 � 7 þ II Loss 0.12 � þ30 M 33 9 � 8 þ II Loss 0.42 � �31 M 59 3 � 2.5 þ II Normal 3.99 þ þ32 M 54 6.5 � 8 þ II Loss 0.36 � þ33 M 45 7.5 � 8.9 þ II Loss 0.18 � �34 M 59 4.5 � 4.0 þ II Normal 1.28 þ þ35 M 45 7.0 � 4.5 � II Normal 1.19 þ þ36 M 35 6.5 � 7.5 � II Loss 0.02 � �37 M 34 3 � 3 þ I Loss 0.13 � þ38 F 42 3 � 3.5 þ I Loss 0.37 � �39 M 38 4.5 � 4 þ II Loss 1.05 þ þ40 M 73 6 � 5.3 � II Normal 0.95 þ þ41 M 66 3 � 2.5 � I Normal 0.82 þ þ42 M 35 8 � 9.5 þ II Normal 0.90 þ þ43 M 30 7 � 8 þ II Loss 0.33 � �44 M 50 4 � 7 þ II Loss 0.98 � þ45 M 44 7.7 � 6.5 þ II Loss 0.13 � �46 M 56 5 � 6 þ II Loss 0.39 � �47 M 35 4 � 5.5 þ II Loss 0.14 � �48 M 47 4 � 7 þ II Loss 1.82 þ þ49 M 48 8 � 5.5 þ II Loss 0.97 þ þ50 M 46 3.5 � 3.5 þ II Loss 0.57 � �
35/50 27/50 28/50 18/50
*Hepatitis B virus: (þ) ¼ serum HBsAg was positive; (�) ¼ serum HBsAg was negative.
†CGH result: loss ¼ DNA copy-number loss at 16q was detected; normal ¼ no DNA copy-number loss at 16q was detected.
‡qPCR result: relative copy-number of tested tumor DNA is given by 2�DDCT, where DDCT ¼ DCtumorT � DCnormalT and each DCT ¼ DCtargetT � DCreferenceT .
§RT-PCR result: (þ) ¼ normal expression of TAT; (�) ¼ downregulation of TAT.kMethylation status: (þ) ¼ unmethylated allele of TAT was detected; (�) ¼ unmethylated allele of TAT was deleted.
1626 FU ET AL. HEPATOLOGY, May 2010
Germany). Triplicate independent experiments wereperformed.Mitochondrial Membrane Potential Assay. Loss of
mitochondrial membrane potential (DWm), indicativeof apoptosis, was detected using the MitoPT JC-1detection kit (Immunochemistry Technologies, Bloo-mington, MN) according to the manufacturer’s proto-col. Briefly, cells were cultured on the coverslips to80% confluence in a 6-well plate. After STS treatment,cells were washed twice with phosphate-buffered saline(PBS) and then incubated with the DWm-sensitive dyesJC-1 at 37�C for 15 minutes. Images were capturedusing a Leica DMRA fluorescence microscope (Rueil-Malmaison, France).RNA Interference. Details are described in the Sup-
porting Materials and Methods.Western Blot Analysis. Western blot analyses were
performed with the standard method with antibodiesto TAT (Uscnlife Science & Technology, Wuhan,China), PAPR (Boster Biotechnology, Wuhan, China),cytochrome-c (Cyt-c), caspase-9, caspase-8, and b-actin(Cell Signaling Technology, Danvers, MA).Statistical Analysis. Statistical analysis was per-
formed with the SPSS standard version 13.0 (Chicago,IL). The statistical significance of the correlations
between TAT expression and loss of TAT allele, pro-moter methylation, as well as consistency betweenCGH and qPCR were assessed by a chi-square test.Results expressed as mean 6 SD were analyzed usingStudent’s t test. Differences were considered significantwhen P was less than 0.05.
ResultsDeletion of TAT Allele in HCC. In our previous
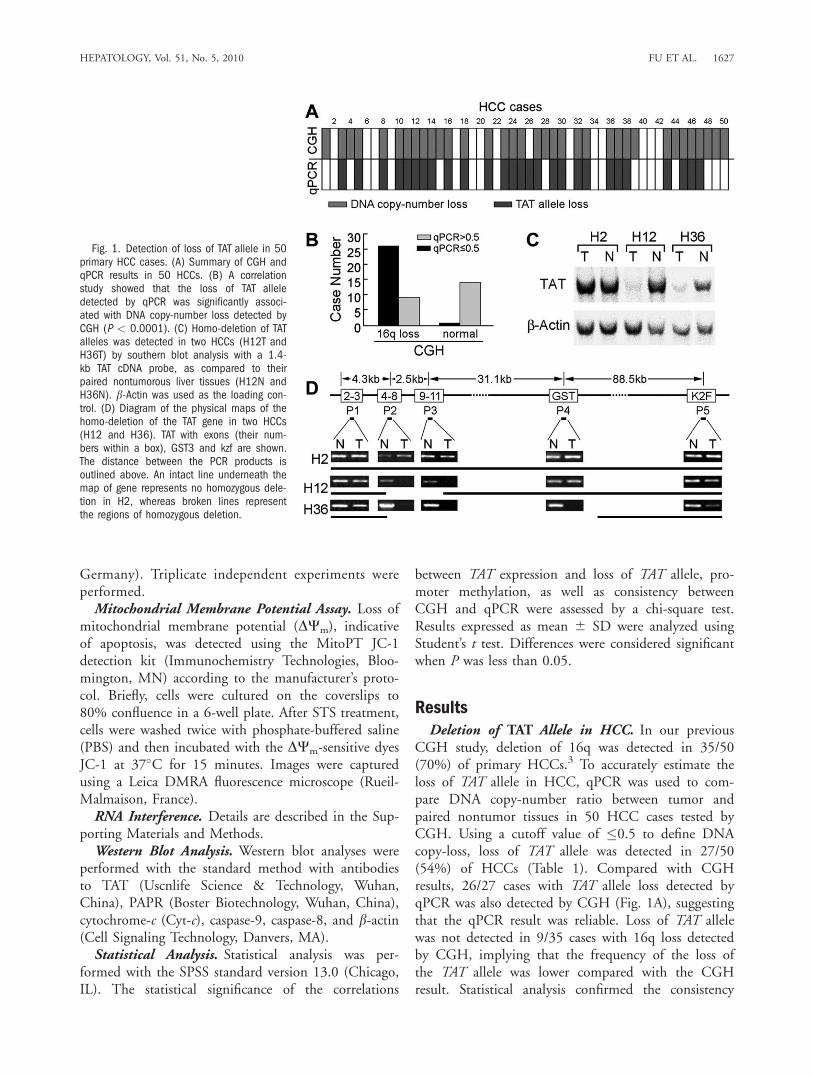
CGH study, deletion of 16q was detected in 35/50(70%) of primary HCCs.3 To accurately estimate theloss of TAT allele in HCC, qPCR was used to com-pare DNA copy-number ratio between tumor andpaired nontumor tissues in 50 HCC cases tested byCGH. Using a cutoff value of �0.5 to define DNAcopy-loss, loss of TAT allele was detected in 27/50(54%) of HCCs (Table 1). Compared with CGHresults, 26/27 cases with TAT allele loss detected byqPCR was also detected by CGH (Fig. 1A), suggestingthat the qPCR result was reliable. Loss of TAT allelewas not detected in 9/35 cases with 16q loss detectedby CGH, implying that the frequency of the loss ofthe TAT allele was lower compared with the CGHresult. Statistical analysis confirmed the consistency
Fig. 1. Detection of loss of TAT allele in 50primary HCC cases. (A) Summary of CGH andqPCR results in 50 HCCs. (B) A correlationstudy showed that the loss of TAT alleledetected by qPCR was significantly associ-ated with DNA copy-number loss detected byCGH (P < 0.0001). (C) Homo-deletion of TATalleles was detected in two HCCs (H12T andH36T) by southern blot analysis with a 1.4-kb TAT cDNA probe, as compared to theirpaired nontumorous liver tissues (H12N andH36N). b-Actin was used as the loading con-trol. (D) Diagram of the physical maps of thehomo-deletion of the TAT gene in two HCCs(H12 and H36). TAT with exons (their num-bers within a box), GST3 and kzf are shown.The distance between the PCR products isoutlined above. An intact line underneath themap of gene represents no homozygous dele-tion in H2, whereas broken lines representthe regions of homozygous deletion.
HEPATOLOGY, Vol. 51, No. 5, 2010 FU ET AL. 1627
between the CGH and qPCR results (Fig. 1B, R ¼0.528; P < 0.0001).
In the qPCR test we noted that the ratio betweentumor and nontumor tissue was extremely low in H12and H36, implying the possibility of homo-deletion ofthe TAT allele in these cases. The homo-deletion ofTAT gene was further confirmed by southern blotanalysis (Fig. 1C) using a full-length TAT probe.Compared with the TAT band detected in paired non-tumor liver tissue, a very weak band was detected inHCC tissues in H12 and H36. To identify the homo-deleted region, five sets of primers were designed toamplify TAT, GST3 (�30 kb from the 30 of TAT) andK2F gene (�90 kb from the 30 of TAT, Fig. 1D).PCR results indicated that the homo-deletion regionin H12 was from exon 4 to 11 of TAT, whereas thedeleted region in H36 was from exon 4 of TAT toGST3 (Fig. 1D).Down-Regulation of TAT in HCC. Semiquantita-
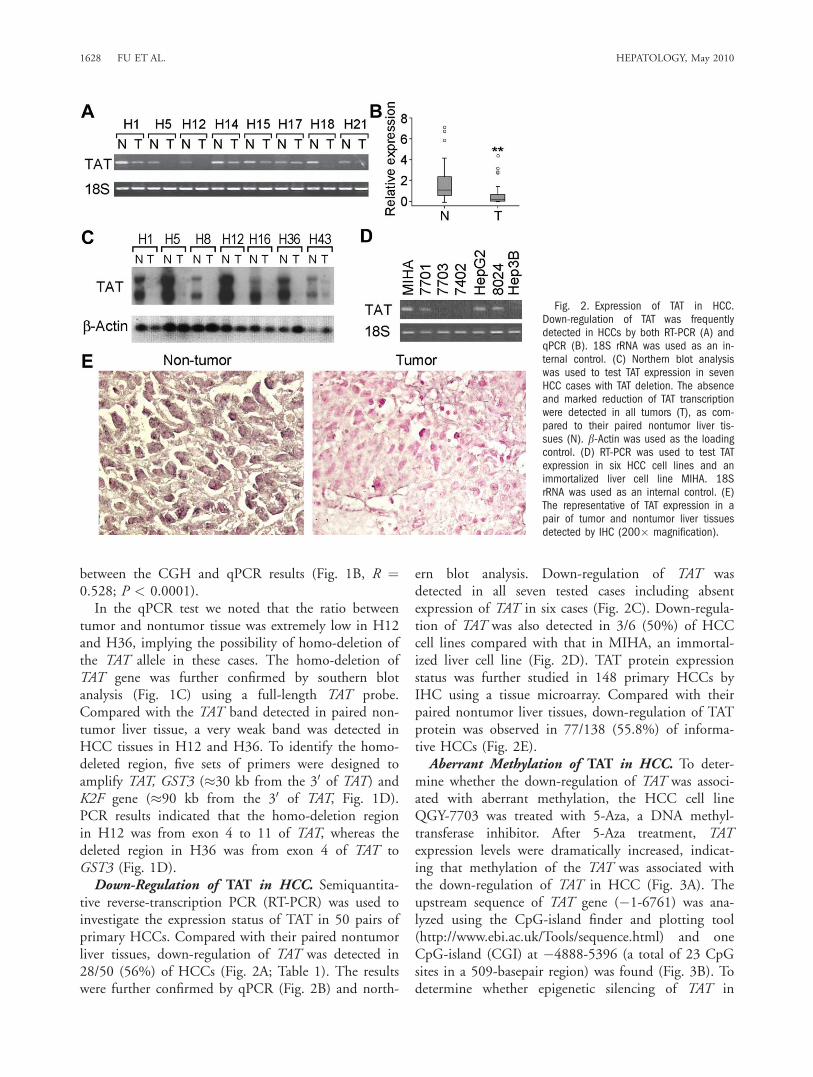
tive reverse-transcription PCR (RT-PCR) was used toinvestigate the expression status of TAT in 50 pairs ofprimary HCCs. Compared with their paired nontumorliver tissues, down-regulation of TAT was detected in28/50 (56%) of HCCs (Fig. 2A; Table 1). The resultswere further confirmed by qPCR (Fig. 2B) and north-
ern blot analysis. Down-regulation of TAT wasdetected in all seven tested cases including absentexpression of TAT in six cases (Fig. 2C). Down-regula-tion of TAT was also detected in 3/6 (50%) of HCCcell lines compared with that in MIHA, an immortal-ized liver cell line (Fig. 2D). TAT protein expressionstatus was further studied in 148 primary HCCs byIHC using a tissue microarray. Compared with theirpaired nontumor liver tissues, down-regulation of TATprotein was observed in 77/138 (55.8%) of informa-tive HCCs (Fig. 2E).Aberrant Methylation of TAT in HCC. To deter-
mine whether the down-regulation of TAT was associ-ated with aberrant methylation, the HCC cell lineQGY-7703 was treated with 5-Aza, a DNA methyl-transferase inhibitor. After 5-Aza treatment, TATexpression levels were dramatically increased, indicat-ing that methylation of the TAT was associated withthe down-regulation of TAT in HCC (Fig. 3A). Theupstream sequence of TAT gene (�1-6761) was ana-lyzed using the CpG-island finder and plotting tool(http://www.ebi.ac.uk/Tools/sequence.html) and oneCpG-island (CGI) at �4888-5396 (a total of 23 CpGsites in a 509-basepair region) was found (Fig. 3B). Todetermine whether epigenetic silencing of TAT in
Fig. 2. Expression of TAT in HCC.Down-regulation of TAT was frequentlydetected in HCCs by both RT-PCR (A) andqPCR (B). 18S rRNA was used as an in-ternal control. (C) Northern blot analysiswas used to test TAT expression in sevenHCC cases with TAT deletion. The absenceand marked reduction of TAT transcriptionwere detected in all tumors (T), as com-pared to their paired nontumor liver tis-sues (N). b-Actin was used as the loadingcontrol. (D) RT-PCR was used to test TATexpression in six HCC cell lines and animmortalized liver cell line MIHA. 18SrRNA was used as an internal control. (E)The representative of TAT expression in apair of tumor and nontumor liver tissuesdetected by IHC (200� magnification).
1628 FU ET AL. HEPATOLOGY, May 2010
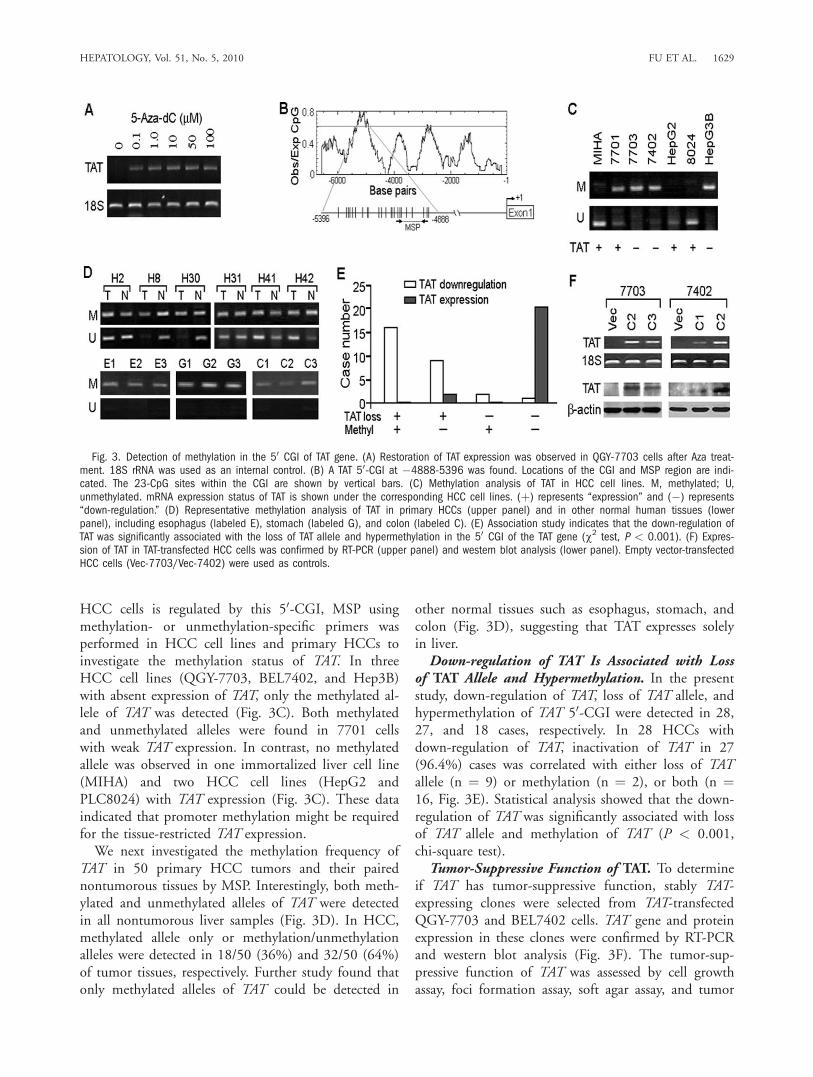
HCC cells is regulated by this 50-CGI, MSP usingmethylation- or unmethylation-specific primers wasperformed in HCC cell lines and primary HCCs toinvestigate the methylation status of TAT. In threeHCC cell lines (QGY-7703, BEL7402, and Hep3B)with absent expression of TAT, only the methylated al-lele of TAT was detected (Fig. 3C). Both methylatedand unmethylated alleles were found in 7701 cellswith weak TAT expression. In contrast, no methylatedallele was observed in one immortalized liver cell line(MIHA) and two HCC cell lines (HepG2 andPLC8024) with TAT expression (Fig. 3C). These dataindicated that promoter methylation might be requiredfor the tissue-restricted TAT expression.We next investigated the methylation frequency of
TAT in 50 primary HCC tumors and their pairednontumorous tissues by MSP. Interestingly, both meth-ylated and unmethylated alleles of TAT were detectedin all nontumorous liver samples (Fig. 3D). In HCC,methylated allele only or methylation/unmethylationalleles were detected in 18/50 (36%) and 32/50 (64%)of tumor tissues, respectively. Further study found thatonly methylated alleles of TAT could be detected in
other normal tissues such as esophagus, stomach, andcolon (Fig. 3D), suggesting that TAT expresses solelyin liver.Down-regulation of TAT Is Associated with Loss
of TAT Allele and Hypermethylation. In the presentstudy, down-regulation of TAT, loss of TAT allele, andhypermethylation of TAT 50-CGI were detected in 28,27, and 18 cases, respectively. In 28 HCCs withdown-regulation of TAT, inactivation of TAT in 27(96.4%) cases was correlated with either loss of TATallele (n ¼ 9) or methylation (n ¼ 2), or both (n ¼16, Fig. 3E). Statistical analysis showed that the down-regulation of TAT was significantly associated with lossof TAT allele and methylation of TAT (P < 0.001,chi-square test).Tumor-Suppressive Function of TAT. To determine
if TAT has tumor-suppressive function, stably TAT-expressing clones were selected from TAT-transfectedQGY-7703 and BEL7402 cells. TAT gene and proteinexpression in these clones were confirmed by RT-PCRand western blot analysis (Fig. 3F). The tumor-sup-pressive function of TAT was assessed by cell growthassay, foci formation assay, soft agar assay, and tumor
Fig. 3. Detection of methylation in the 50 CGI of TAT gene. (A) Restoration of TAT expression was observed in QGY-7703 cells after Aza treat-ment. 18S rRNA was used as an internal control. (B) A TAT 50-CGI at �4888-5396 was found. Locations of the CGI and MSP region are indi-cated. The 23-CpG sites within the CGI are shown by vertical bars. (C) Methylation analysis of TAT in HCC cell lines. M, methylated; U,unmethylated. mRNA expression status of TAT is shown under the corresponding HCC cell lines. (þ) represents ‘‘expression’’ and (�) represents‘‘down-regulation.’’ (D) Representative methylation analysis of TAT in primary HCCs (upper panel) and in other normal human tissues (lowerpanel), including esophagus (labeled E), stomach (labeled G), and colon (labeled C). (E) Association study indicates that the down-regulation ofTAT was significantly associated with the loss of TAT allele and hypermethylation in the 50 CGI of the TAT gene (w2 test, P < 0.001). (F) Expres-sion of TAT in TAT-transfected HCC cells was confirmed by RT-PCR (upper panel) and western blot analysis (lower panel). Empty vector-transfectedHCC cells (Vec-7703/Vec-7402) were used as controls.
HEPATOLOGY, Vol. 51, No. 5, 2010 FU ET AL. 1629
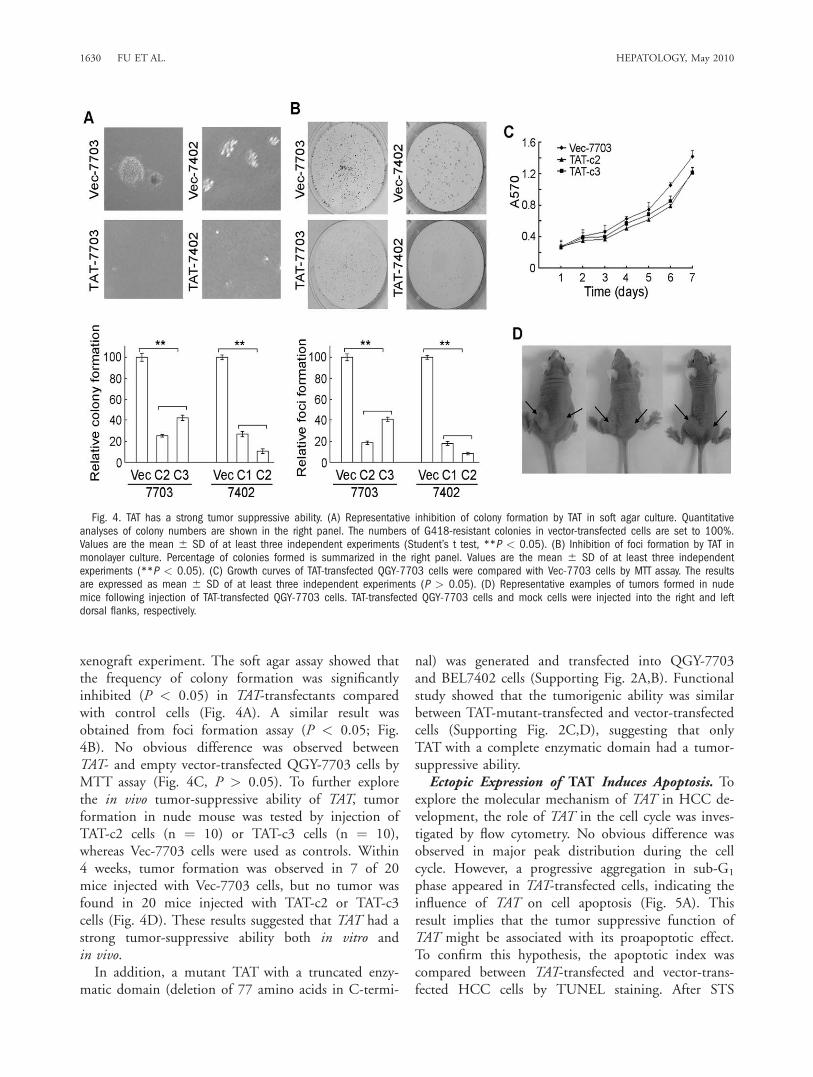
xenograft experiment. The soft agar assay showed thatthe frequency of colony formation was significantlyinhibited (P < 0.05) in TAT-transfectants comparedwith control cells (Fig. 4A). A similar result wasobtained from foci formation assay (P < 0.05; Fig.4B). No obvious difference was observed betweenTAT- and empty vector-transfected QGY-7703 cells byMTT assay (Fig. 4C, P > 0.05). To further explorethe in vivo tumor-suppressive ability of TAT, tumorformation in nude mouse was tested by injection ofTAT-c2 cells (n ¼ 10) or TAT-c3 cells (n ¼ 10),whereas Vec-7703 cells were used as controls. Within4 weeks, tumor formation was observed in 7 of 20mice injected with Vec-7703 cells, but no tumor wasfound in 20 mice injected with TAT-c2 or TAT-c3cells (Fig. 4D). These results suggested that TAT had astrong tumor-suppressive ability both in vitro andin vivo.In addition, a mutant TAT with a truncated enzy-
matic domain (deletion of 77 amino acids in C-termi-
nal) was generated and transfected into QGY-7703and BEL7402 cells (Supporting Fig. 2A,B). Functionalstudy showed that the tumorigenic ability was similarbetween TAT-mutant-transfected and vector-transfectedcells (Supporting Fig. 2C,D), suggesting that onlyTAT with a complete enzymatic domain had a tumor-suppressive ability.Ectopic Expression of TAT Induces Apoptosis. To
explore the molecular mechanism of TAT in HCC de-velopment, the role of TAT in the cell cycle was inves-tigated by flow cytometry. No obvious difference wasobserved in major peak distribution during the cellcycle. However, a progressive aggregation in sub-G1
phase appeared in TAT-transfected cells, indicating theinfluence of TAT on cell apoptosis (Fig. 5A). Thisresult implies that the tumor suppressive function ofTAT might be associated with its proapoptotic effect.To confirm this hypothesis, the apoptotic index wascompared between TAT-transfected and vector-trans-fected HCC cells by TUNEL staining. After STS
Fig. 4. TAT has a strong tumor suppressive ability. (A) Representative inhibition of colony formation by TAT in soft agar culture. Quantitativeanalyses of colony numbers are shown in the right panel. The numbers of G418-resistant colonies in vector-transfected cells are set to 100%.Values are the mean 6 SD of at least three independent experiments (Student’s t test, **P < 0.05). (B) Inhibition of foci formation by TAT inmonolayer culture. Percentage of colonies formed is summarized in the right panel. Values are the mean 6 SD of at least three independentexperiments (**P < 0.05). (C) Growth curves of TAT-transfected QGY-7703 cells were compared with Vec-7703 cells by MTT assay. The resultsare expressed as mean 6 SD of at least three independent experiments (P > 0.05). (D) Representative examples of tumors formed in nudemice following injection of TAT-transfected QGY-7703 cells. TAT-transfected QGY-7703 cells and mock cells were injected into the right and leftdorsal flanks, respectively.
1630 FU ET AL. HEPATOLOGY, May 2010
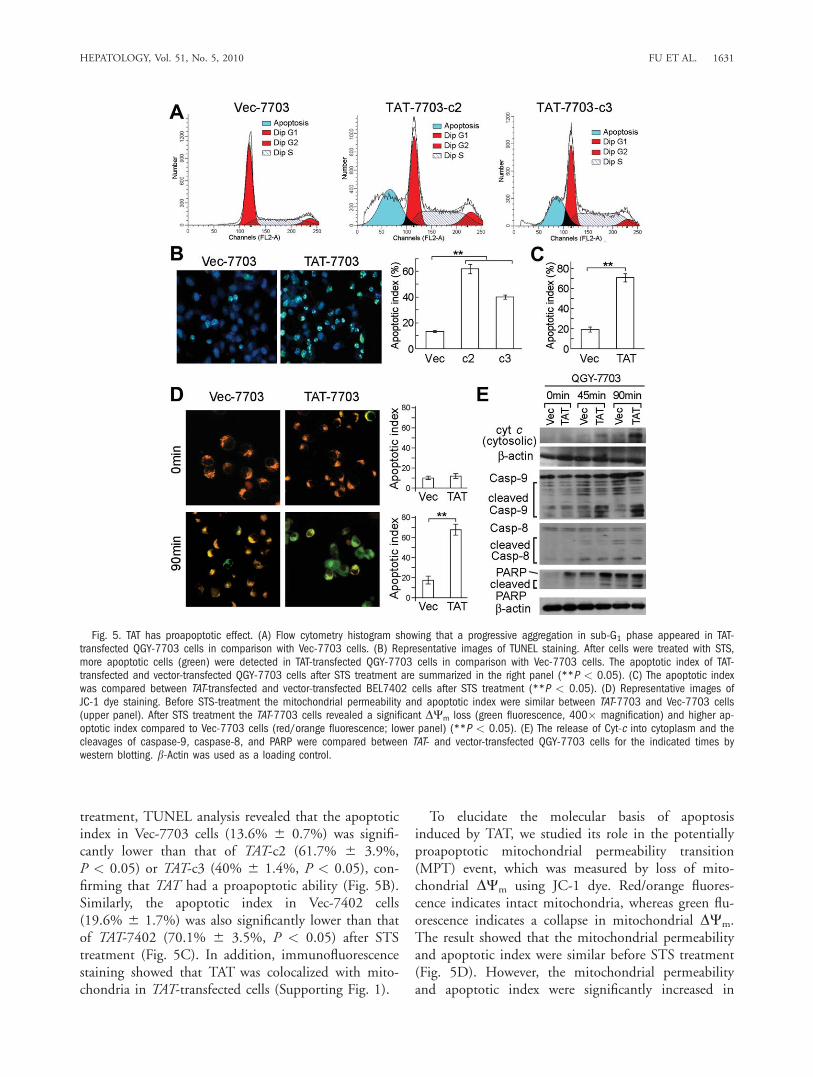
treatment, TUNEL analysis revealed that the apoptoticindex in Vec-7703 cells (13.6% 6 0.7%) was signifi-cantly lower than that of TAT-c2 (61.7% 6 3.9%,P < 0.05) or TAT-c3 (40% 6 1.4%, P < 0.05), con-firming that TAT had a proapoptotic ability (Fig. 5B).Similarly, the apoptotic index in Vec-7402 cells(19.6% 6 1.7%) was also significantly lower than thatof TAT-7402 (70.1% 6 3.5%, P < 0.05) after STStreatment (Fig. 5C). In addition, immunofluorescencestaining showed that TAT was colocalized with mito-chondria in TAT-transfected cells (Supporting Fig. 1).
To elucidate the molecular basis of apoptosisinduced by TAT, we studied its role in the potentiallyproapoptotic mitochondrial permeability transition(MPT) event, which was measured by loss of mito-chondrial DWm using JC-1 dye. Red/orange fluores-cence indicates intact mitochondria, whereas green flu-orescence indicates a collapse in mitochondrial DWm.The result showed that the mitochondrial permeabilityand apoptotic index were similar before STS treatment(Fig. 5D). However, the mitochondrial permeabilityand apoptotic index were significantly increased in
Fig. 5. TAT has proapoptotic effect. (A) Flow cytometry histogram showing that a progressive aggregation in sub-G1 phase appeared in TAT-transfected QGY-7703 cells in comparison with Vec-7703 cells. (B) Representative images of TUNEL staining. After cells were treated with STS,more apoptotic cells (green) were detected in TAT-transfected QGY-7703 cells in comparison with Vec-7703 cells. The apoptotic index of TAT-transfected and vector-transfected QGY-7703 cells after STS treatment are summarized in the right panel (**P < 0.05). (C) The apoptotic indexwas compared between TAT-transfected and vector-transfected BEL7402 cells after STS treatment (**P < 0.05). (D) Representative images ofJC-1 dye staining. Before STS-treatment the mitochondrial permeability and apoptotic index were similar between TAT-7703 and Vec-7703 cells(upper panel). After STS treatment the TAT-7703 cells revealed a significant DWm loss (green fluorescence, 400� magnification) and higher ap-optotic index compared to Vec-7703 cells (red/orange fluorescence; lower panel) (**P < 0.05). (E) The release of Cyt-c into cytoplasm and thecleavages of caspase-9, caspase-8, and PARP were compared between TAT- and vector-transfected QGY-7703 cells for the indicated times bywestern blotting. b-Actin was used as a loading control.
HEPATOLOGY, Vol. 51, No. 5, 2010 FU ET AL. 1631
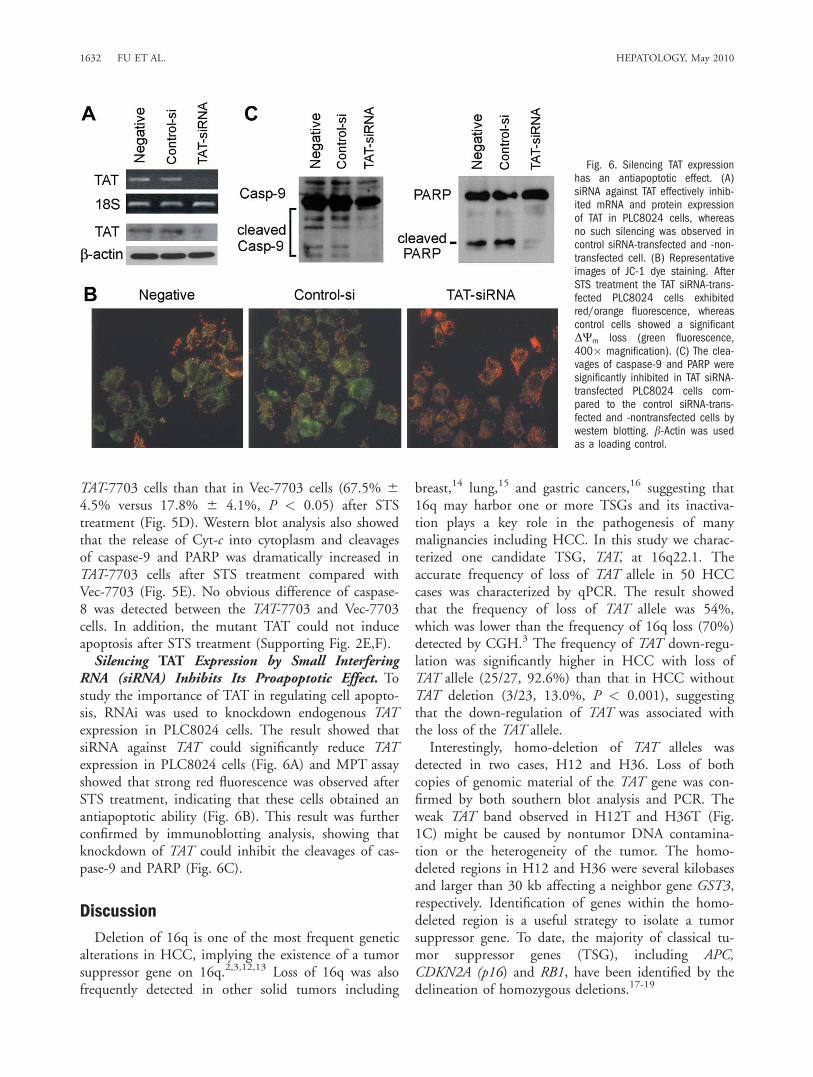
TAT-7703 cells than that in Vec-7703 cells (67.5% 64.5% versus 17.8% 6 4.1%, P < 0.05) after STStreatment (Fig. 5D). Western blot analysis also showedthat the release of Cyt-c into cytoplasm and cleavagesof caspase-9 and PARP was dramatically increased inTAT-7703 cells after STS treatment compared withVec-7703 (Fig. 5E). No obvious difference of caspase-8 was detected between the TAT-7703 and Vec-7703cells. In addition, the mutant TAT could not induceapoptosis after STS treatment (Supporting Fig. 2E,F).Silencing TAT Expression by Small Interfering
RNA (siRNA) Inhibits Its Proapoptotic Effect. Tostudy the importance of TAT in regulating cell apopto-sis, RNAi was used to knockdown endogenous TATexpression in PLC8024 cells. The result showed thatsiRNA against TAT could significantly reduce TATexpression in PLC8024 cells (Fig. 6A) and MPT assayshowed that strong red fluorescence was observed afterSTS treatment, indicating that these cells obtained anantiapoptotic ability (Fig. 6B). This result was furtherconfirmed by immunoblotting analysis, showing thatknockdown of TAT could inhibit the cleavages of cas-pase-9 and PARP (Fig. 6C).
Discussion
Deletion of 16q is one of the most frequent geneticalterations in HCC, implying the existence of a tumorsuppressor gene on 16q.2,3,12,13 Loss of 16q was alsofrequently detected in other solid tumors including
breast,14 lung,15 and gastric cancers,16 suggesting that16q may harbor one or more TSGs and its inactiva-tion plays a key role in the pathogenesis of manymalignancies including HCC. In this study we charac-terized one candidate TSG, TAT, at 16q22.1. Theaccurate frequency of loss of TAT allele in 50 HCCcases was characterized by qPCR. The result showedthat the frequency of loss of TAT allele was 54%,which was lower than the frequency of 16q loss (70%)detected by CGH.3 The frequency of TAT down-regu-lation was significantly higher in HCC with loss ofTAT allele (25/27, 92.6%) than that in HCC withoutTAT deletion (3/23, 13.0%, P < 0.001), suggestingthat the down-regulation of TAT was associated withthe loss of the TAT allele.Interestingly, homo-deletion of TAT alleles was
detected in two cases, H12 and H36. Loss of bothcopies of genomic material of the TAT gene was con-firmed by both southern blot analysis and PCR. Theweak TAT band observed in H12T and H36T (Fig.1C) might be caused by nontumor DNA contamina-tion or the heterogeneity of the tumor. The homo-deleted regions in H12 and H36 were several kilobasesand larger than 30 kb affecting a neighbor gene GST3,respectively. Identification of genes within the homo-deleted region is a useful strategy to isolate a tumorsuppressor gene. To date, the majority of classical tu-mor suppressor genes (TSG), including APC,CDKN2A (p16) and RB1, have been identified by thedelineation of homozygous deletions.17-19
Fig. 6. Silencing TAT expressionhas an antiapoptotic effect. (A)siRNA against TAT effectively inhib-ited mRNA and protein expressionof TAT in PLC8024 cells, whereasno such silencing was observed incontrol siRNA-transfected and -non-transfected cell. (B) Representativeimages of JC-1 dye staining. AfterSTS treatment the TAT siRNA-trans-fected PLC8024 cells exhibitedred/orange fluorescence, whereascontrol cells showed a significantDWm loss (green fluorescence,400� magnification). (C) The clea-vages of caspase-9 and PARP weresignificantly inhibited in TAT siRNA-transfected PLC8024 cells com-pared to the control siRNA-trans-fected and -nontransfected cells bywestern blotting. b-Actin was usedas a loading control.
1632 FU ET AL. HEPATOLOGY, May 2010

Down-regulation of TAT was detected in 28/50(56%) of 50 primary HCCs by RT-PCR. A similarfrequency of TAT down-regulation (77/138, 55.8%)was detected in 148 primary HCCs by IHC, com-pared with their paired nontumor liver tissues. Thedown-regulation of TAT was not only associated withthe loss of TAT allele, but also DNA hypermethylationin the 50-CGI of TAT. It has been well documentedthat DNA methylation of CpG islands located neargene promoters affects the transcription of specificgenes.20,21 Aberrant DNA methylation of two geneslocated at 16q22.1, E-cadherin and TAT, have beenreported in pretumorous conditions and HCCs.22,23
Methylation of one TAT allele could be detected in all50 adjacent nontumor liver samples, indicating thatone TAT allele was inactivated in early development.Loss of the active (unmethylated) allele of TAT, whichwas detected in 18/50 (36%) of HCCs, might be oneof the major mechanisms of TAT inactivation. Statisti-cal analysis further confirmed that the down-regulationof TAT was significantly associated with both TAT de-letion and methylation (P < 0.001).The functional study provided the first evidence
that TAT has strong tumor-suppressive ability, includ-ing the inhibition of foci formation, colony formationin soft agar, and tumor formation in nude mice. Amechanism study found that the tumor-suppressiveeffect of TAT was associated with its proapoptoticeffect. The apoptosis induced by TAT was mediatedthrough the intrinsic mitochondrial pathway becausecaspase-9, but not caspase-8, was activated.A molecular study found that the proapoptotic
effect of TAT was triggered by the stimulation of apo-ptotic agents such as STS. Before STS treatment, theapoptotic index between TAT-7703 and Vec-7703 wassimilar. However, the apoptotic index was significantlyincreased in TAT-7703 compared to Vec-7703 cells (P< 0.05) after STS treatment. Because TAT is a mito-chondrial protein, it is reasonable to speculate that theproapoptotic effect of TAT may be associated with mi-tochondrial membrane potential (DWm) change. TheMPT assay found that the mitochondrial permeabilitywas dramatically increased in TAT-7703 cells after STStreatment, which subsequently increased the release ofCyt-c into the cytoplasm by way of permeability tran-sition mechanisms. Once released, Cyt-c is able tochange the conformation of Apaf-1, initiating the apo-ptosis by the activation of caspase-9 and the subse-quent cleavages of caspase-3 and PARP.24 Western blotanalysis confirmed that the STS stimulation could dra-matically increase Cyt-c release, cleavages of caspase-9and PARP in TAT-transfected cells compared to empty
vector-transfected cells. Furthermore, silencing TATexpression by RNAi could effectively inhibit its proa-poptotic ability upon apoptotic stimulation.In summary, our findings demonstrate that TAT is a
novel TSG and its inactivation caused by gene deletionand hypermethylation contributes to the pathogenesisof HCC. A better understanding of the molecularmechanism of TAT in promoting tumor cell apoptosiswould provide novel therapeutic strategies to HCCcancer patients.
References1. Harris CC. Hepatocellular carcinogenesis: recent advances and specula-
tions. Cancer Cells 1990;2:146-148.2. Piao Z, Park C, Kim JJ, Kim H. Deletion mapping of chromosome
16q in hepatocellular carcinoma. Br J Cancer 1999;80:850-854.3. Guan XY, Fang Y, Sham JST, Kwong DLW, Zhang Y, Liang Q, et al.
Recurrent chromosome alterations in hepatocellular carcinoma detectedby comparative genomic hybridization. Genes Chromosomes Cancer2000;29:110-116.
4. Slagle BL, Zhou YZ, Birchmeier W, Scorsone KA. Deletion of the E-cadherin gene in hepatitis B virus-positive Chinese hepatocellular carci-nomas. HEPATOLOGY 1993;18:757-762.
5. Shiman R, Gray DW. Formation and fate of tyrosine: intracellular par-titioning of newly synthesized tyrosine in mammalian liver. J BiolChem 1998;273:34760-34769.
6. Huhn R, Stoermer H, Klingele B, Bausch E, Fois A, Farnetani M,et al. Novel and recurrent tyrosine aminotransferase gene mutations intyrosinemia type II. Hum Genet 1998;102:305-313.
7. Paraskevopoulos JA. Management options for primary hepatocellularcarcinoma, an overview. Acta Oncol 1994;33:895-900.
8. Paradis K. Tyrosinemia: the Quebec experience. Clin Invest Med 1996;19:311-316.
9. Fu L, Qin YR, Xie D, Hu L, Kwong DL, Srivastava G, et al. Charac-terization of a novel tumor-suppressor gene PLC delta 1 at 3p22 inesophageal squamous cell carcinoma. Cancer Res 2007;67:10720-10726.
10. Guan XY, Sham JST, Tang TCM, Fang Y, Huo KK, Yang JM. Isola-tion of a novel candidate oncogene within a frequently amplified regionat 3q26 in ovarian cancer. Cancer Res 2001;61:3806-3809.
11. Xie D, Sham JS, Zeng WF, Lin HL, Che LH, Wu HX, et al. Hetero-geneous expression and association of beta-catenin, p16 and c-myc inmultistage colorectal tumorigenesis and progression detected by tissuemicroarray. Int J Cancer 2003;107:896-902.
12. Okabe H, Ikai I, Matsuo K, Satoh S, Momoi H, Kamikawa T, et al. Com-prehensive allelotype study of hepatocellular carcinoma: potential differen-ces in pathways to hepatocellular carcinoma between hepatitis B virus-positive and -negative tumors. HEPATOLOGY 2000;31:1073-1079.
13. Nidoridawa Y, Yamamoto S, Ishikawa S, Kaminura N, Igarashi H,Sugimura H, et al. Molecular karyotyping of human hepatocellular car-cinoma using single-nucleotide polymorphism arrays. Oncogene 2006;25:5581-5590.
14. Poylance R, Gorman P, Papior T, Wan YL, Ives M, Watson JE, et al. Acomprehensive study of chromosome 16q in invasive ductal and lobularcarcinoma using array CGH. Oncogene 2006;25:6544-6553.
15. Stanton SE, Shin SW, Johnson BE, Meyerson M. Recurrent allelic dele-tions of chromosome arms 15q and 16q in human small cell lung car-cinomas. Genes Chromosomes Cancer 2000;27:323-331.
16. Suzuki S, Egami K, Sasajima K, Ghazizadeh M, Shimizu H, WatanabeH, et al. Comparative study between DNA copy number aberrationsdetermined by quantitative microsatellite analysis and clinical outcomein patients with stomach cancer. Clin Cancer Res 2004;10:3013-3019.
HEPATOLOGY, Vol. 51, No. 5, 2010 FU ET AL. 1633

17. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, AlbertDM, et al. A human DNA segment with properties of the gene thatpredisposes to retinoblastoma and osteosarcoma. Nature 1986;323:643-646.
18. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, AlbertsenH, et al. Identification and characterization ofthe familial adenomatouspolyposis coli gene. Cell 1991;66:589-600.
19. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavti-gian SV, et al. A cell cycle regulator potentially involved in genesis ofmany tumour types. Science 1994;264:436-440.
20. Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP.Allele-specific hypermethylation of the retinoblastoma tumor suppressorgene. Am J Hum Genet 1991;48:880-888.
21. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Bay-lin SB, et al. 50 CpG island methylation is associated with transcrip-tional silencing of the tumour suppressor p16/CDKN2/MTS1 inhuman cancers. Nat Med 1995;1:686-692.
22. Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, HirohashiS. Silencing of the E-cadherin invasion-suppressor gene by CpG meth-ylation in human carcinomas. Proc Natl Acad Sci U S A 1995;92:7416-7419.
23. Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Sugimura T, HirohashiS. Aberrant DNA methylation on chromosome 16 is an early event inhepatocarcinogenesis. Jpn J Cancer Res 1996;87:1210-1217.
24. Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holo-enzyme. Genes Dev 1999;13:3179-3184.
1634 FU ET AL. HEPATOLOGY, May 2010




















