
Differential expression of the actin-binding proteins, alpha-actinin-2 and -3, in different species:...
12
© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 13 1335–1346 ARTICLE Differential expression of the actin-binding proteins, α-actinin-2 and -3, in different species: implications for the evolution of functional redundancy Michelle A. Mills 1 , Nan Yang 1 , Ron P. Weinberger 3 , Douglas L. Vander Woude 4 , Alan H. Beggs 4 , Simon Easteal 5 and Kathryn N. North 1,2, * 1 Neurogenetics Research Unit, Children’s Hospital at Westmead, Sydney, NSW, Australia, 2 Department of Paediatrics and Child Health, University of Sydney, Sydney, NSW, Australia, 3 Oncology Research Unit, Children’s Hospital at Westmead, Sydney, NSW, Australia, 4 Genetics Division, Children’s Hospital, Harvard Medical School, Boston, MA, USA and 5 Centre for Bioinformation Science, John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia Received February 19, 2001; Revised and Accepted April 12, 2001 The α-actinins are a multigene family of four actin-binding proteins related to dystrophin. The two skeletal muscle isoforms of α-actinin (ACTN2 and ACTN3) are major structural components of the Z-line involved in anchoring the actin-containing thin filaments. In humans, ACTN2 is expressed in all muscle fibres, while ACTN3 expression is restricted to a subset of type 2 fibres. We have recently demonstrated that α-actinin-3 is absent in ∼18% of individuals in a range of human populations, and that homozygosity for a premature stop codon (577X) accounts for most cases of true α-actinin-3 deficiency. Absence of α-actinin-3 is not associated with an obvious disease phenotype, raising the possibility that ACTN3 is functionally redundant in humans, and that α-actinin-2 is able to compensate for α-actinin-3 deficiency. We now present data concerning the expression of ACTN3 in other species. Genotyping of non-human primates indicates that the 577X null mutation has likely arisen in humans. The mouse genome contains four orthologues which all map to evolutionarily conserved syntenic regions for the four human genes. Murine Actn2 and Actn3 are differentially expressed, spatially and temporally, during embryonic development and, in contrast to humans, α-actinin-2 expression does not completely overlap α-actinin-3 in postnatal skeletal muscle, suggesting independent function. Furthermore, sequence comparison of human, mouse and chicken α-actinin genes demonstrates that ACTN3 has been conserved over a long period of evolutionary time, implying a constraint on evolutionary rate imposed by continued function of the gene. These observations provide a real framework in which to test theoretical models of genetic redundancy as they apply to human populations. In addition we highlight the need for caution in making conclusions about gene function from the phenotypic consequences of loss-of-function mutations in animal knockout models. INTRODUCTION The α-actinins are a family of actin-binding proteins related to dystrophin and the spectrins (1). In non-muscle cells, the cytoskeletal isoforms, α-actinin-1 and -4, are found along microfilament bundles where they mediate membrane attach- ment at adherans-type junctions in a dynamic manner regulated by calcium binding (2,3). In skeletal muscle, α-actinin-2 and -3 are major structural components of sarcomeric Z-lines where they function to anchor actin-containing thin filaments in a constitutive manner (4). Recent studies suggest additional roles for the α-actinins in skeletal muscle. Sarcomeric α-actinins bind to other thin filament and Z-line proteins including nebulin, myotilin, CapZ and myozenin (5–7), the intermediate filament proteins, synemin and vinculin (8,9), and the sarcolemmal membrane proteins, dystrophin and β1 integrin (10,11). These binding studies suggest that the α-actinins play a role in thin filament organization and the interaction between the sarcom- eric cytoskeleton and the muscle membrane. In addition, sarcom- eric α-actinin binds phosphatidylinositol 4,5-bisphophate (12), phosphatidylinositol 3-kinase (13) and PDZ-LIM adaptor proteins *To whom correspondence should be addressed at: Clinical Sciences Building, Children’s Hospital at Westmead, Locked Bag 4001, Westmead, Sydney, NSW 2145, Australia; Tel: +61 2 9845 3011; Fax: +61 2 9845 3082; Email: [email protected] The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors
Transcript of Differential expression of the actin-binding proteins, alpha-actinin-2 and -3, in different species:...

© 2001 Oxford University Press Human Molecular Genetics, 2001, Vol. 10, No. 13 1335–1346
ARTICLE
Differential expression of the actin-binding proteins,α-actinin-2 and -3, in different species: implications forthe evolution of functional redundancyMichelle A. Mills1, Nan Yang1, Ron P. Weinberger3, Douglas L. Vander Woude4,Alan H. Beggs4, Simon Easteal5 and Kathryn N. North1,2,*
1Neurogenetics Research Unit, Children’s Hospital at Westmead, Sydney, NSW, Australia, 2Department ofPaediatrics and Child Health, University of Sydney, Sydney, NSW, Australia, 3Oncology Research Unit, Children’sHospital at Westmead, Sydney, NSW, Australia, 4Genetics Division, Children’s Hospital, Harvard Medical School,Boston, MA, USA and 5Centre for Bioinformation Science, John Curtin School of Medical Research, The AustralianNational University, Canberra, ACT, Australia
Received February 19, 2001; Revised and Accepted April 12, 2001
The α-actinins are a multigene family of four actin-binding proteins related to dystrophin. The two skeletalmuscle isoforms of α-actinin (ACTN2 and ACTN3) are major structural components of the Z-line involved inanchoring the actin-containing thin filaments. In humans, ACTN2 is expressed in all muscle fibres, while ACTN3expression is restricted to a subset of type 2 fibres. We have recently demonstrated that α-actinin-3 is absentin ∼18% of individuals in a range of human populations, and that homozygosity for a premature stop codon(577X) accounts for most cases of true α-actinin-3 deficiency. Absence of α-actinin-3 is not associated with anobvious disease phenotype, raising the possibility that ACTN3 is functionally redundant in humans, and thatα-actinin-2 is able to compensate for α-actinin-3 deficiency. We now present data concerning the expressionof ACTN3 in other species. Genotyping of non-human primates indicates that the 577X null mutation has likelyarisen in humans. The mouse genome contains four orthologues which all map to evolutionarily conservedsyntenic regions for the four human genes. Murine Actn2 and Actn3 are differentially expressed, spatially andtemporally, during embryonic development and, in contrast to humans, α-actinin-2 expression does notcompletely overlap α-actinin-3 in postnatal skeletal muscle, suggesting independent function. Furthermore,sequence comparison of human, mouse and chicken α-actinin genes demonstrates that ACTN3 has beenconserved over a long period of evolutionary time, implying a constraint on evolutionary rate imposed bycontinued function of the gene. These observations provide a real framework in which to test theoreticalmodels of genetic redundancy as they apply to human populations. In addition we highlight the need forcaution in making conclusions about gene function from the phenotypic consequences of loss-of-functionmutations in animal knockout models.
INTRODUCTION
The α-actinins are a family of actin-binding proteins related todystrophin and the spectrins (1). In non-muscle cells, thecytoskeletal isoforms, α-actinin-1 and -4, are found alongmicrofilament bundles where they mediate membrane attach-ment at adherans-type junctions in a dynamic manner regulatedby calcium binding (2,3). In skeletal muscle, α-actinin-2 and-3 are major structural components of sarcomeric Z-lineswhere they function to anchor actin-containing thin filamentsin a constitutive manner (4). Recent studies suggest additional
roles for the α-actinins in skeletal muscle. Sarcomeric α-actininsbind to other thin filament and Z-line proteins including nebulin,myotilin, CapZ and myozenin (5–7), the intermediate filamentproteins, synemin and vinculin (8,9), and the sarcolemmalmembrane proteins, dystrophin and β1 integrin (10,11). Thesebinding studies suggest that the α-actinins play a role in thinfilament organization and the interaction between the sarcom-eric cytoskeleton and the muscle membrane. In addition, sarcom-eric α-actinin binds phosphatidylinositol 4,5-bisphophate (12),phosphatidylinositol 3-kinase (13) and PDZ-LIM adaptor proteins
*To whom correspondence should be addressed at: Clinical Sciences Building, Children’s Hospital at Westmead, Locked Bag 4001, Westmead, Sydney, NSW2145, Australia; Tel: +61 2 9845 3011; Fax: +61 2 9845 3082; Email: [email protected] authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors

1336 Human Molecular Genetics, 2001, Vol. 10, No. 13
(14,15), suggesting a role in the regulation of myofibre differen-tiation and/or contraction.
In humans, the α-actinin-2 gene, ACTN2, is expressed in allskeletal muscle fibres, whereas expression of ACTN3, encodingα-actinin-3, is limited to a subset of type 2 (fast) fibres (16).We have recently demonstrated that α-actinin-3 is absent in∼18% of individuals in a range of human populations and thathomozygosity for a premature stop codon (577X) accounts forall cases of true α-actinin-3 deficiency identified to date. Anadditional polymorphism (523R) occurs in linkage disequilibriumwith 577X, but does not appear to exert a deleterious effect whenexpressed in the heterozygous state in coupling with 577R.Absence of α-actinin-3 is not associated with an obvious diseasephenotype, suggesting that ACTN3 is redundant in humans (17).
Functional redundancy occurs when two genes performoverlapping functions so that inactivation of one of the genes haslittle or no effect on the phenotype (reviewed in 18). α-actinin-2expression in human skeletal muscle completely overlapsα-actinin-3. ACTN2 and ACTN3 are 80% identical and 90%similar (4). In addition, α-actinin-2 and α-actinin-3 formheterodimers in vitro and in vivo, suggesting structural simi-larity and lack of significant functional differences between thetwo skeletal muscle α-actinin isoforms (19). On this basis wehypothesize that α-actinin-2 is able to compensate for theabsence of α-actinin-3 in type 2 (fast) fibres in humans.
Despite the apparent functional redundancy of ACTN3 inhumans, sequence comparison of human and chicken skeletalmuscle α-actinin genes suggest that human ACTN2 andACTN3 have both evolved very slowly since their divergencemore than 300 million years ago, implying strong functionalconservation (17). Here we demonstrate that ACTN2 andACTN3 are differentially expressed, spatially and temporally,during mouse embryonic development. Unlike humans andnon-human primates, ACTN2 expression does not completelyoverlap ACTN3 in postnatal mouse skeletal muscle, suggesting
that ACTN3 may not be functionally redundant in the mouse.We provide further evidence for the evolutionary conservationof the ACTN3 gene and demonstrate that the null mutation haslikely arisen in humans, with marked variation in frequencyamong different human populations.
RESULTS
There is marked population variation in the allelicfrequency of 577R and 577X
We have previously demonstrated that the stop codon poly-morphism (577X) is present in different ethnic populations(17). To determine the population frequency and distributionof each allele at positions 523 and 577, we genotyped 485DNA samples (970 chromosomes) derived from anonymousindividuals whose DNA was collected for other purposes,including the four major human groups (from Asia/Americas,Australasia, Africa and Europe; Table 1). The 577X allele ismost frequent in Eurasia (0.51) and least frequent in Africa (0.16).Frequency of the 577X allele in the African Bantu sample issignificantly lower than that of all other samples. The 577X allelefrequencies in African Americans and Aboriginal Australians arealso significantly lower than the Asian and European groups.
The null mutation (577X) likely arose in humans
To determine the origin of the 577X allele (and the 523R allele,which occurs in strong linkage disequilibrium with 577X) (17),we genotyped 36 unrelated baboons (diverged from humanlineage ∼25 × 106 years ago) and 33 unrelated chimpanzees(diverged from human lineage ∼5 × 106 years ago). All 69non-human primates were homozygous for the ‘wild-type’alleles in exons 15 (523Q) and 16 (577R), suggesting that thepolymorphisms originated after the separation of the human
Table 1. Genotype and allelic frequencies of ACTN3 577R/X alleles in human populations
Ethnic group No. of chromosomes No. of genotypes Relative allele frequency of 577X
RX XX
Asian 56 14 7 0.5 ± 0.07
Javanese 96 28 12 0.54 ± 0.05
Native American 14 2 2 0.43 ± 0.14
Asia/Americas 166 44 21 0.52 ± 0.04
Hispanic 64 16 5 0.41 ± 0.06
White 214 47 21 0.42 ± 0.03
Europe 278 63 26 0.41 ± 0.03
Aboriginal Australian 174 33 9 0.29 ± 0.03
PNG Highlander 78 16 6 0.36 ± 0.05
Australasia 252 49 15 0.31 ± 0.03
African American 90 12 6 0.27 ± 0.05
African Bantu 156 14 1 0.10 ± 0.05
Africa 246 26 7 0.16 ± 0.05
Unknown 152 50 11 0.47
Total 1094 232 80

Human Molecular Genetics, 2001, Vol. 10, No. 13 1337
and chimpanzee lineages, or that they have a very lowfrequency in non-human primates.
Identification of mouse Actn genes
To determine the number and identity of α-actinin genes in themurine genome, the chromosomal localizations for mouseorthologues of the four human ACTN genes were identifiedusing the Jackson Laboratory interspecific backcross mappingpanel C57BL/6JEi × SPRET/Ei)F1 × SPRET/Ei (Jackson BSS)(20). PCR primers were designed based on human exonsequences to amplify across small murine introns that wouldpresumably vary between Mus musculus and Mus spretus.Each PCR product was sequenced and shown to represent themurine orthologue of a human ACTN gene, confirming that themouse genome contains a single orthologous locus corres-ponding to each human gene. SSCP analysis of these PCRproducts on the backcross mapping panel revealed that Actn1is at 36.2 cM on mouse chromosome 12, Actn2 is at 2.1 cM onchromosome 13, Actn3 is in the most proximal (centromeric)group of loci on chromosome 19 and Actn4 is at 5.3 cM onchromosome 7 (Fig. 1). These localizations are supported bythe observation that there are genes with human orthologueslinked to the corresponding human ACTN genes at each of theselocations (http://www.ncbi.nlm.nih.gov/ Homology/index.html).
Cloning of mouse Actn2
We determined the full-length coding sequence of mouseActn2 by screening a mouse diaphragm cDNA library(GenBank accession no. AF248643). The mouse Actn3 codingsequence is available from GenBank (accession no.AF093775). The similarity between mouse Actn2 and Actn3 is
the same as between human ACTN2 and ACTN3, i.e. 88%similar and 79% identical. The mouse proteins are colinear andhave the same functional domains as the human proteins—anN-terminal actin-binding domain, four central repeat domainsand C-terminal EF-hands (4).
ACTN3 is conserved during evolution
We have previously shown strong sequence conservationbetween human and chicken skeletal muscle α-actinin genes(17). There is only one skeletal muscle ACTN gene in thechicken (4), whereas we have now demonstrated that the mousegenome contains four orthologues which all map to evolution-arily conserved syntenic regions for the four human genes.Sequence comparison between mouse and human ACTN2 andACTN3 suggests that the evolution of the α-actinins has beenslow relative to other genes (Table 2). The nucleotide substitu-tion rate at non-synonymous sites between the human andmouse ACTN3 genes (0.016 substitutions/site) is substantiallylower than the average rate for a sample of more than 1000 genes
Figure 1. Haplotypes from Jackson BSS backcross mapping of four murine α-actinin genes and neighboring loci. (A) Actn1; (B) Actn2; (C) Actn3; (D) Actn4.Loci are listed in order with the most proximal at the top. Black boxes represent the C57BL6/JEi allele and white boxes represent the SPRET/Ei allele. The numberof animals with each haplotype is given at the bottom of each column of boxes. The percentage recombination (R) between adjacent loci and standard error (SE)for each R is given at the right.
Table 2. Numbers of nucleotide substitutions per 100 synonymous (Ks) andnon-synonymous sites (Ka) between humans and rodents
aData from Duret and Mouchiroud (21).
Ks Ka
α-Actinin-3 45 ± 3.5 1.6 ± 0.3
1296 genesa 49 7.2
101 ubiquitous genesa 47 2.9
1195 tissue-specific genesa 49 7.6

1338 Human Molecular Genetics, 2001, Vol. 10, No. 13
(0.072 substitutions/site). It is also lower than the mean rate ofubiquitously expressed genes (0.029 substitutions/site), whichgenerally evolve more slowly than genes with tissue-specificpatterns of expression (21). The nucleotide substitution rate atsynonymous sites between the human and mouse ACTN3 genes(approximately 0.45 substitutions/site) is similar to the averagerate for the same sample of more than 1000 genes (approxi-mately 0.49 substitutions/site), implying that the low rate ofsubstitution in ACTN3 is not due to an intrinsically lowmutation rate in this gene.
Tissue-specific expression of the skeletal muscle α-actininisoforms
α-Actinin-2 is more widely expressed than α-actinin-3. Northernblot analysis was used to assess mRNA transcript expression inmouse tissues. For α-actinin-2, a 3.5 kb transcript was detectedin skeletal and cardiac muscle and in the brain but was notexpressed in smooth muscle, kidney, liver or lung. Themembrane was stripped and reprobed to detect a 3.4 kb tran-script representing α-actinin-3. A transcript for α-actinin-3was detected only in skeletal muscle (data not shown).
We further determined the tissue-specific expression ofthe sarcomeric α-actinins using western blot analysis. In themouse, antibodies directed against α-actinin-2 identified a96 kDa protein band in skeletal and cardiac muscles, thediaphragm and brain. α-Actinin-3 antibodies recognized aband of similar size in skeletal muscle and diaphragm, butnot in cardiac muscle or brain. In humans, α-actinin-2 waspresent in skeletal, cardiac and extraocular muscles and inthe brain, with greater amounts in grey matter than in whitematter, but not in liver or lung. α-Actinin-3 was detected inlimb skeletal muscle and at very low levels in the brain(grey matter) (Fig. 2). The results of western blot analysis ofbaboon tissue were identical to human, although brainsamples were not available for analysis.
To further define the expression of α-actinin isoforms in thebrain, we stained serial sections of mouse brain from postnatalday 3 (P3) mice with antibodies to α-actinin-2, α-actinin-3 anddesmin (as a negative control). α-Actinin-2 stained manystructures including pyramidal cells in the hippocampus,choroid plexus, meninges and cells of the olfactory bulb(Fig. 3). Staining was enriched in the pyramidal cell dendritesand puncta of the dentate gyrus. These puncta may representsynaptic interactions of the perforant pathway with thegranule cells. α-Actinin-3 also stained pyramidal cells ofthe hippocampus, meninges and cells of the olfactory bulb.Positive staining was present in the dendritic processes ofpyramidal and olfactory bulb cells. There was negativestaining with antibodies to desmin and with secondary anti-body alone.
Expression of α-actinin-2 does not completely overlapα-actinin-3 in the mouse skeletal muscle
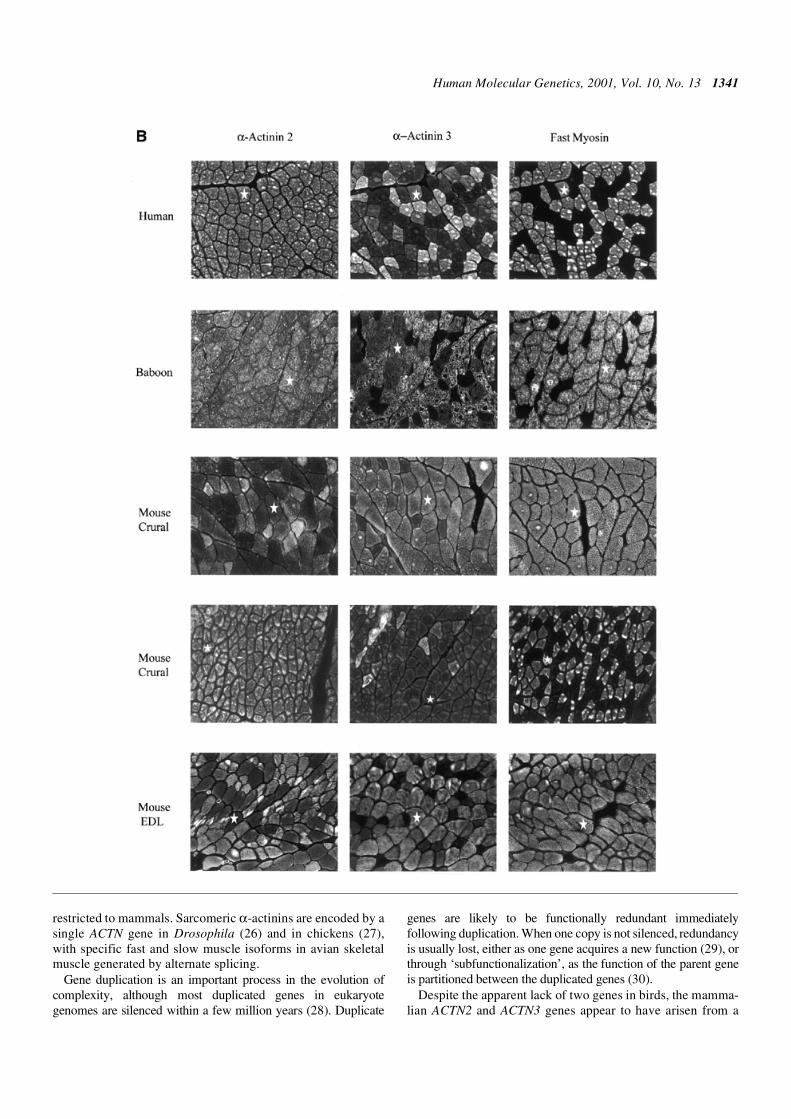
We performed immunocytochemistry on sections of skeletalmuscle from control human quadriceps muscle. As notedpreviously, α-actinin-2 is expressed in all muscle fibres andα-actinin-3 is expressed in type 2 fibres (16). Fibre typeexpression of the α-actinins in baboon skeletal muscle demon-strates the same staining patterns as for humans (Fig. 4B).
Type 2 (fast) fibres are the predominant fibre type in themouse (80% of fibres, compared with 40–50% in humans).Three muscles from the mouse were analysed; extensor digito-rium longus (EDL, predominantly fast fibres), soleus (predomi-nantly slow fibres) and crural (mixed). Quantitation of themouse skeletal muscle α-actinin isoforms demonstrated that α-actinin-2 is expressed in all type 1, 2A and 2X fibres (Fig. 4A).α-Actinin-2 was also expressed in a subset of type 2B fibres, butthe proportion varied between different muscles and betweendifferent areas of the same muscle. In general, ∼60% of type 2Bfibres positively stain with α-actinin-2. α-Actinin-3 expressionis restricted to type 2B fibres. Consequently, there is some
Figure 2. Western blot analysis of human α-actinin-2 and -3. In humans, α-actinin-2 is present in skeletal, cardiac and extraocular muscles, and in the brain, withgreater amounts in grey matter than in white matter. α-Actinin-3 was detected in limb skeletal muscle and at very low levels in the brain (grey matter). Notably,neither protein was seen in smooth muscle (uterine myometrium) demonstrating a lack of cross reactivity of the antibodies with the non-muscle isoforms (ACTN1and ACTN4), both of which are abundant in this tissue.

Human Molecular Genetics, 2001, Vol. 10, No. 13 1339
co-expression of α-actinin-2 and -3. For example, in soleus(80% type 1 and 20% type 2A fibres), all fibres express α-actinin-2 but not α-actinin-3. In the EDL (50% type 2A and 50%type 2B fibres), ∼60% of fibres stained positively for α-actinin-2 and >95% of fibres stained positively for α-actinin-3, withvariable expression of α-actinin-2 in different areas of themuscle. Three separate areas of the crural were analysed. Inareas predominantly composed of type 2B fibres, all fibresstained positive for α-actinin-3 and 60–70% expressed α-actinin-2, i.e. there was high co-expression of the two α-actininskeletal muscle isoforms (∼60% of fibres). However, in areas ofthe same specimen with high fibre type 1 expression, α-actinin-3 expression is low (17%) and subsequently, co-expression of α-actinin-2 and -3 is also low (∼15%) (Fig. 4B). Thus, althoughthere is some co-expression of α-actinin-2 and -3 in mouse skel-etal muscle, unlike humans and non-human primates, there aremany fibres in the mouse that express α-actinin-3 only.
Developmental expression of sarcomeric α-actinins inmouse and human
ACTN2 is expressed before ACTN3 with different tissue-specificpatterns of expression. To determine the developmental expres-sion of α-actinin-2 and -3 in mouse embryos, we performedimmunocytochemistry on sections of whole mouse embryos at
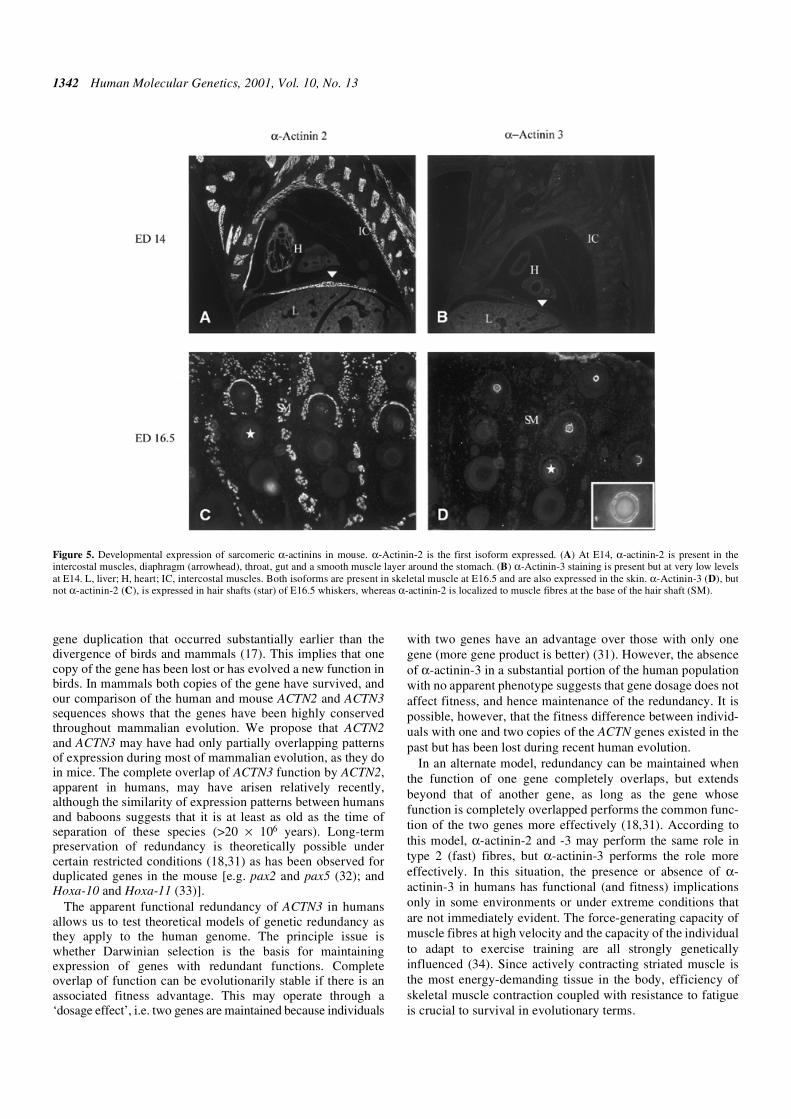
embryonic days (E) 11.5, 12.5 (period of organogenesis), 14.5and 16.5 (fetal growth and development). The first skeletalmuscle isoform expressed is α-actinin-2. At E11.0, α-actinin-2staining was localized to the mesenchyme next to the firstbrachial arch and cardiac tissue. α-Actinin-3 staining wasnegative. At E12.5, α-actinin-2 was expressed in cardiacmuscle, and also in intercostal muscles, in neuroepithelial andlaryngopharyngeal tissues and in the intestinal region, particu-larly the duodenum. α-Actinin-3 was not expressed at E12.5.
At E14.5, α-actinin-2 was the predominant isoformexpressed, with positive staining of the intercostal muscles,diaphragm, throat, gut and a smooth muscle layer around thestomach. At high power, α-actinin-2 stained in a striatedpattern consistent with localization at the Z-line. At thisstage, α-actinin-3 was present in skeletal muscle only, at avery low level compared with α-actinin-2 (Fig. 5). The patternof staining in skeletal muscle is similar at E16.5 and, in addition,both α-actinin-2 and -3 were detected in the skin. α-Actinin-3,but not α-actinin-2, was expressed in hair shafts of E16.5whiskers, whereas α-actinin-2 was localized to muscle fibresat the base of the hair shaft (Fig. 5). An actin complex whichcontains α-actinin has been identified in human epithelialhair follicles, and may function to maintain hair folliclestructure (22). By P1, α-actinin-2 and -3 have approximately
Figure 3. Sarcomeric α-actinin isoforms are present in the brain of P3 mice. Staining with α-actinin-2 [(A) low power, (C) high power] is enriched in the pyramidalcell dendrites (arrowhead) and puncta of the dentate gyrus. α-Actinin-3 (B and D) is also present in the dendritic processes of pyramidal cells of the hippocampus.SP, stratum pyramidale; SO, stratum oriens; SLM, stratum lacunosum moleculare; DG, dentate gyrus.

1340 Human Molecular Genetics, 2001, Vol. 10, No. 13
equal staining intensities in mouse skeletal muscle. α-Actinin-3is the predominant skeletal muscle isoform by P30 (Fig. 4B).
Western blot analysis with antibodies to α-actinin-2 and -3 wasperformed on fetal human muscle protein lysates. Thespecimen ages ranged from 16 weeks gestation to newbornsamples. Both α-actinin-2 and -3 were expressed from theearliest age studied in human muscle and were expressedbefore fast myosin isoforms (data not shown).
DISCUSSION
We have identified two α-actinin genes in the mouse (Actn2and Actn3) which correspond to human ACTN2 and ACTN3,allowing us to directly compare the expression of geneticallydistinct isoforms in different species. In humans, the potentialfor α-actinin-2/-3 functional redundancy is indicated by completeoverlapping expression of α-actinin-3 by α-actinin-2 at theprotein level, and confirmed by the presence of an ACTN3 nullvariant with no detectable phenotypic effect. The proteins arestructurally and functionally similar, and it is likely that α-actinin-2 can compensate for α-actinin-3 deficiency. We haveshown that the ACTN3 null allele (577X) probably results froma single mutation that occurred after the divergence of humans
and chimpanzees. The variant occurs with moderate frequencyin human populations from Africa, Europe, Asia and Australasia.It does not appear to contribute to major forms of musculardisease.
In contrast, we have shown that α-Actinin-3 may not befunctionally redundant in mouse skeletal muscle. α-Actinin-3is the predominant isoform in postnatal mouse muscle and,unlike human and baboon, murine α-actinin-2 expression doesnot completely overlap that of α actinin-3. α-Actinin-2 and -3are differentially expressed, spatially and temporally, duringembryonic development, suggesting independent transcrip-tional regulation and different roles during myogenesis. Thetissue distribution of the sarcomeric α-actinins is wider thanpreviously reported as both α-actinin-2 and -3 are expressed inmouse and human brain. Wyszynski et al. (23) have previouslydemonstrated expression of α-actinin-2, but not α-actinin-3, inrat cerebellum and suggested that α-actinin-2 may providestructural support or regulation of cytoskeletal remodelling ofthe NMDA receptors.
In other mammals, such as rabbits and pigs, there are alsofast- and slow-muscle-specific isoforms of α-actinin, althoughthe gene(s) responsible have not been isolated (24,25). Thepresence of two sarcomeric α-actinin genes may, however, be
Figure 4. (A) Fibre type-specific expression of sarcomeric α-actinins in mouse skeletal muscle. Double label indirect immunofluorescence analysis of fourconsecutive cryosections of mouse skeletal muscle for ACTN2 (top panels) or ACTN3 (bottom panels) and type 1 (slow) fibre-specific myosin heavy chain (MHC;left) or type 2A (fast) fibre-specific MHC (right). Rabbit anti-α-actinin antibodies were visualized with Cy3-anti rabbit IgG (red) and mouse anti-MHC antibodieswere stained with fluorescein-anti-mouse IgG (green). Fibres labelling with both antibodies appear yellow. Selected fibre types are indicated (1, 2A or 2B).α-Actinin-2 is expressed in all type 1, 2A and 2X fibres, as well as a subset of type 2B fibres. α-Actinin-3 expression is restricted to type 2B fibres, which is thepredominant fibre type in the mouse. (B; opposite) Comparison of α-actinin-2 and -3 expression in skeletal muscle of humans, baboon and mouse. In human andbaboon skeletal muscle, α-actinin-2 is expressed in all muscle fibres and α-actinin-3 is expressed in type 2 fibres only. Three muscles from the mouse wereanalysed, EDL (predominantly fast fibres), and two components of the crural muscles, soleus (predominantly slow fibres) and gastrocnemius (mixed). Unlikehuman and baboon, the expression of α-actinin-2 does not completely overlap α-actinin-3 in mouse skeletal muscle; there are many fibres in the mouse that expressα-actinin-3 only. The proportion of type 2B fibres expressing α-actinin-2 varies between different muscles and between different areas of the same muscle. Stain-ing for fast myosin demonstrates type 2 (2A and 2B) fibres in each section.
Human Molecular Genetics, 2001, Vol. 10, No. 13 1341
restricted to mammals. Sarcomeric α-actinins are encoded by asingle ACTN gene in Drosophila (26) and in chickens (27),with specific fast and slow muscle isoforms in avian skeletalmuscle generated by alternate splicing.
Gene duplication is an important process in the evolution ofcomplexity, although most duplicated genes in eukaryotegenomes are silenced within a few million years (28). Duplicate
genes are likely to be functionally redundant immediatelyfollowing duplication. When one copy is not silenced, redundancyis usually lost, either as one gene acquires a new function (29), orthrough ‘subfunctionalization’, as the function of the parent geneis partitioned between the duplicated genes (30).
Despite the apparent lack of two genes in birds, the mamma-lian ACTN2 and ACTN3 genes appear to have arisen from a
1342 Human Molecular Genetics, 2001, Vol. 10, No. 13
gene duplication that occurred substantially earlier than thedivergence of birds and mammals (17). This implies that onecopy of the gene has been lost or has evolved a new function inbirds. In mammals both copies of the gene have survived, andour comparison of the human and mouse ACTN2 and ACTN3sequences shows that the genes have been highly conservedthroughout mammalian evolution. We propose that ACTN2and ACTN3 may have had only partially overlapping patternsof expression during most of mammalian evolution, as they doin mice. The complete overlap of ACTN3 function by ACTN2,apparent in humans, may have arisen relatively recently,although the similarity of expression patterns between humansand baboons suggests that it is at least as old as the time ofseparation of these species (>20 × 106 years). Long-termpreservation of redundancy is theoretically possible undercertain restricted conditions (18,31) as has been observed forduplicated genes in the mouse [e.g. pax2 and pax5 (32); andHoxa-10 and Hoxa-11 (33)].
The apparent functional redundancy of ACTN3 in humansallows us to test theoretical models of genetic redundancy asthey apply to the human genome. The principle issue iswhether Darwinian selection is the basis for maintainingexpression of genes with redundant functions. Completeoverlap of function can be evolutionarily stable if there is anassociated fitness advantage. This may operate through a‘dosage effect’, i.e. two genes are maintained because individuals
with two genes have an advantage over those with only onegene (more gene product is better) (31). However, the absenceof α-actinin-3 in a substantial portion of the human populationwith no apparent phenotype suggests that gene dosage does notaffect fitness, and hence maintenance of the redundancy. It ispossible, however, that the fitness difference between individ-uals with one and two copies of the ACTN genes existed in thepast but has been lost during recent human evolution.
In an alternate model, redundancy can be maintained whenthe function of one gene completely overlaps, but extendsbeyond that of another gene, as long as the gene whosefunction is completely overlapped performs the common func-tion of the two genes more effectively (18,31). According tothis model, α-actinin-2 and -3 may perform the same role intype 2 (fast) fibres, but α-actinin-3 performs the role moreeffectively. In this situation, the presence or absence of α-actinin-3 in humans has functional (and fitness) implicationsonly in some environments or under extreme conditions thatare not immediately evident. The force-generating capacity ofmuscle fibres at high velocity and the capacity of the individualto adapt to exercise training are all strongly geneticallyinfluenced (34). Since actively contracting striated muscle isthe most energy-demanding tissue in the body, efficiency ofskeletal muscle contraction coupled with resistance to fatigueis crucial to survival in evolutionary terms.
Figure 5. Developmental expression of sarcomeric α-actinins in mouse. α-Actinin-2 is the first isoform expressed. (A) At E14, α-actinin-2 is present in theintercostal muscles, diaphragm (arrowhead), throat, gut and a smooth muscle layer around the stomach. (B) α-Actinin-3 staining is present but at very low levelsat E14. L, liver; H, heart; IC, intercostal muscles. Both isoforms are present in skeletal muscle at E16.5 and are also expressed in the skin. α-Actinin-3 (D), butnot α-actinin-2 (C), is expressed in hair shafts (star) of E16.5 whiskers, whereas α-actinin-2 is localized to muscle fibres at the base of the hair shaft (SM).

Human Molecular Genetics, 2001, Vol. 10, No. 13 1343
Under this model, α-actinin-3 would be conserved because itfunctions more effectively in type 2 fibres. α-Actinin-2 wouldbe conserved because of its more general function. Thefunctional overlap of the genes would eventually be lostthrough mutation, as individuals with genes of overlappingfunction have no advantage over those in which the genesfunctioned independently. It is difficult to explain the spread ofthe ACTN3 null allele under this model, since the modelimplies that individuals with ACTN3 have an advantage overthose that lack the gene. However, it is possible that the advan-tage conferred by ACTN3 has been lost during recent humanevolution. The occurrence of the allele at intermediate frequen-cies in different human populations suggests a relatively earlyorigin of the allele, probably >100 000 years ago. Any changein physiology, lifestyle or environment that might have causedthe loss of a fitness difference between individuals with andwithout the ACTN gene would have to have persisted for atleast this long.
The persistence of α-actinin-3 redundancy in humans maybe better explained by a model put forward by Gibson andSpring (35), who suggest that there is no need to invoke activeselection for redundant gene expression in vertebrate develop-ment. They propose that, following gene duplication in multi-domain proteins, selection operates to reject deleterious pointmutations in redundant genes because the majority of codingpoint mutations have a ‘dominant-negative’ effect on theprotein relative to its normal function in the cell. As a result theredundant gene is maintained with a high degree of evolu-tionary conservation. In contrast, mutations that result incomplete loss of gene expression may occur with little effecton phenotype or fitness because of the redundancy. The redun-dancy itself is a historical consequence of either polyploidy inthe vertebrate common ancestor (36) or subsequent gene dupli-cation. Redundancy is preserved because the rate of mutationsleading to complete loss of function is much lower than the rateof (usually deleterious) mutations leading to altered function.Redundancy cannot be maintained indefinitely in this way andeventually it is lost as rare loss-of-function mutations occur.Under this model, the intermediate frequency of the ACTN3577X null allele represents a transitory stage in the eventualloss, by genetic drift, of the highly conserved but redundantACTN3 gene. The complete loss of the gene is unlikely tooccur unless the human population is drastically reduced insize, since the mean time to fixation of a neutral mutation isfour times the effective population size (37). As long as thehuman population is extremely large, the null allele willremain at its current intermediate frequency.
ACTN3 577X is the human equivalent of a ‘gene knockout’or homozygous null mutation of single genes in mice and otherexperimental species that produce no phenotype or a subtlephenotype compared with double knockouts for more than onemember of a multigene family (38,39). In humans, as inexperimental species, there is a need for caution in drawinginferences about function from the phenotypic consequencesof loss-of-function mutations. No disease-associated ACTN3mutations have been reported to date, but we predict that,despite the absence of any detectable phenotypic effect of theloss-of-function 577X allele, ACTN3 missense mutations mayhave dominant-negative phenotypes (40).
MATERIALS AND METHODS
ACTN3 genotyping
Human genomic DNA was isolated from blood by phenol–chloroform extraction following cell lysis with Triton X-100 anddigestion with Proteinase K, or from muscle (∼7 mg of tissue)using QIAamp tissue kits (Qiagen). Exons 15 and 16 ofACTN3 were amplified from genomic DNA. The primersfor exon 15 were: forward, 5′-GGTGGGTAGGTGGGT-GAGGC-3′; reverse, 5′-GAGTGTACCAGCCACACGTCC-3′.The primers for exon 16 were: forward, 5′-CTGTTGCCTG-TGGTAAGTGGG-3′; reverse, 5′-TGGTCACAGTATGCAGGA-GGG-3′, corresponding to adjacent intronic sequences. ThePCR reaction cycle for both sets of primers was: 94°C for5 min, 94°C for 30 s and 70°C for 60 s, for 35 cycles, with afinal extension of 72°C for 10 min. The Q523R alleles(codons CAG and CGG, respectively) can be distinguishedby the presence (523R) or absence (523Q) of an MspI restrictionsite (C↓CGG). 523Q PCR products have 118 and 74 bpfragments. 523R PCR products have 21, 74 and 97 bp fragments.The R577X alleles (codons CGA and TGA, respectively) can bedistinguished by the presence (577X) or absence (577R) of aDdeI restriction site in exon 16. 577R PCR products have 205and 85 bp fragments; 577X PCR products have 108, 97 and86 bp fragments. Digested PCR fragments were separated by10% polyacrylamide gel electrophoresis and stained withethidium bromide.
Genomic DNA was also isolated from 36 unrelated baboonsand 33 unrelated chimpanzees. For chimpanzees, ACTN3exons 15 and 16 were amplified using primers designed for thehuman sequence (see above). To amplify baboon DNA, wedesigned primers complementary to human ACTN3 sequencewithin exons. The forward primer for exon 15 was 5′-CACCTC-TAGCGGATGGAGAA-3′ and the reverse primer was 5′-CGA-GTCCTCCACAGAGTG-3′. The forward primer for exon 16 was5′-CCAGAGCCTGCTGACAGC-3′ and the reverse primerwas 5′-TCCCACTTGGTGTTGATGTC-3′. PCR fragmentswere digested with MspI and DdeI as detailed above. PCRfragments of exons 15 and 16 from two baboons and onechimpanzee were sequenced to confirm amplification of theappropriate sequence and the results of restriction enzymeanalysis.
Chromosomal localization of mouse Actn genes
The chromosomal localizations for the four mouse Actn geneswere identified using the Jackson Laboratory interspecificbackcross panel (C57BL/6JEi × SPRET/Ei)F1 × SPRET/Ei(Jackson BSS) as described by Rowe et al. (20). PCR wasperformed using primers based on human cDNA sequences anddesigned to amplify across small introns in the four murineActn genes as follows: Actn1 forward, 5′-CAGATGAATG-AGTTCCGGGCCTCCTTCAAC-3′; Actn1 reverse, 5′-TAACC-CAAGCTGATGAGGCAGGCTT-3′; Actn2 forward, 5′-TTC-CAATCCTTCATCGACTTCATGAC-3′; Actn2 reverse, 5′-GGC-ATCCTCTTGATGCAGTACTGGGCCT-3′; Actn3 forward,5′-AACGAGTTCCGAGCATCCTTCAACC-3′; Actn3 reverse,5′-CTTCCCCCAGGTCATAGCCCATGGAGAT-3′; Actn4 for-ward, 5′-CTGCACCCTGGGCCCCAGCGGACC-3′; Actn4reverse, 5′-ATGAAGGCTTGGAAGGTCACAAAG-3′.

1344 Human Molecular Genetics, 2001, Vol. 10, No. 13
PCR reaction cycles for Actn1 and Actn4 primers were 30cycles of: 94°C for 5 min, 94°C for 30 s, 55°C for 30 s, 72°Cfor 60 s, with a final extension of 72°C for 10 min. Actn2 andActn3 were amplified under similar conditions, but theannealing temperature was 60°C. Several primer pairs ampli-fied multiple murine loci that segregated in the Jackson BSSpanel; however, the correct α-actinin loci were easily identi-fied by DNA sequencing of the PCR products as, in each case,the amplified coding regions of each murine locus were iden-tical to the orthologous human gene. Variation between thedifferent mice breeds, M.musculus and M.spretus, weredetected by separating radiolabelled products on MDE SSCPgels following the manufacturer’s protocols (FMCBioproducts). Segregation of the C57BL/6JEi alleles fourgenes was determined in the 94-backcross offspring and themap locations were determined by the Jackson LaboratoryBackcross Mapping Group. Missing typings were inferredfrom surrounding data where assignment was unambiguous.Map locations in cM are given relative to the most centromericmarker on the Jackson BSS map as of January 2001. Raw datafrom The Jackson Laboratory are available for these loci athttp://www.jax.org/resources/documents/cmdata.
Cloning and sequencing cDNA of mouse Actn2
PCR amplification of mouse Actn2 was performed usingprimers designed from human cDNA sequence. The primersused were: forward, 5′-GCCATGAACCAGATAGAGCC-3′(from –3 to 17 bp), covering the ATG start codon at position 1;reverse, 5′-TCCATCTTGTTCTGGATCCAG-3′ (from 1953to 1973 bp). The PCR product was sequenced and used toscreen a mouse diaphragm muscle cDNA library. Two cloneswere isolated from the library, sequenced and compared withhuman ACTN2 sequence.
Sequence and statistical analysis
Sequences were aligned using GDE (Genetic Data Environ-ment) 2.2 (41). Substitution rates at synonymous and non-synonymous sites were estimated by the method of Li (42),with correction for multiple substitution using the two-parameter method of Kimura (37). Allele frequency differ-ences among population groups were assessed by the methodof Raymond and Rousset (43) implemented in the computerpackage ‘Arlequin’ (http://anthropologie.unige.ch/arlequin).Haplotype frequencies were obtained using the Expectation-Maximization algorithm (44) implemented in Arlequin.Linkage disequilibrium was estimated by the method ofSlatkin and Excoffier (45) implemented in Arlequin.
Northern blot analysis
RNA was isolated from frozen mouse (C57Black) tissue viaguanidine thiocyanate extraction (46) with some modifications.Approximately 750 mg of tissue was crushed in a liquid-nitrogen-chilled mortar and pestle and prepared with denaturingsolution (4 M guanidium thiocyanate, 25 mM sodium citrate pH7.0, 0.5% sarcosyl and 0.1 M 2-mercaptoethanol), precipitatedwith isopropanol and finally dissolved in 0.1% diethyl pyro-carbonate water.
RNA was prepared for electrophoretic separation by themethod described by Lehrach et al. (47). Transfer of RNA to
Hybond N+ membrane was performed following the protocolrecommended by the manufacturer (Amrad PharmaciaBiotech). The probe for hybridization was prepared usingGigaprime DNA labelling kit (Bresatec). Hybridizations wereperformed in 50% formamide, 5× SSPE (3.6 M NaCl, 0.2 MNa2HPO4, 0.02 M Na2EDTA, pH 7.7), 1× Denhardt’s solution(2% BSA, 2% Ficoll 400 and 2% polyvinylpyrrolidine) and10 mg/ml denatured salmon sperm DNA. The membrane waswashed in 2× SSC at room temperature and 0.1× SSPE at65°C. The membrane was exposed to X-ray film (Kodak)overnight at –70°C.
Western blot analysis
Mouse and human protein samples were prepared as describedby Hoffman et al. (48), with some modifications. Approxi-mately 50 mg of tissue was cut from frozen samples main-tained at –70°C and crushed in a liquid-nitrogen-cooled mortarand pestle. Samples were transferred to a microcentrifuge tubeand homogenized and vortexed in sample buffer (62.5 µMTris–PO4, 0.1% glycerol, 1 mM EDTA, 1.5 mg/ml aprotonin,1.25 mg/ml leupeptin, 1.0 mg/ml pepstatin, 0.04 mg/ml PMSF,2.5% SDS and 0.05 M DTT). Before electrophoresis, sampleswere boiled for 5 min and centrifuged. Protein concentrationsof muscle lysates were estimated using the amido black proteinestimation protocol (49).
Supernatants containing ∼50 µg of protein sample wereloaded into wells of SDS–polyacrylamide gel (5% stacking geland 7.5% separating gel) and were electrophoresed for 1 h at200 V. Transfer onto prepared PVDF membrane (AmershamPharmacia Biotech) was performed overnight at 40 V usingMini Protean II gel electrophoresis and transfer unit (Bio-RadLaboratories). Membranes were blocked in 5% non-fat milkpowder in TBST [0.01 M Tris–HCl, 0.05% (v/v) Tween-20and 9% (w/v) NaCl]. Primary antibodies (see below) wereapplied for 2 h. The membrane was washed and alkaline phos-phatase-conjugated secondary antibody was applied to themembrane for 1 h. BCIP-NBT tablets (Sigma) (one tablet in 10ml of deionized water) were applied to the membrane for ∼10min or until bands were detected.
Immunocytochemistry
Frozen P1 mouse brains were stained according to theprocedure detailed by Weinberger et al. (50) with somemodifications. After dewaxing, sections were blocked in 10%goat serum before adding the primary antibodies, α-actinin-2(1/500) and α-actinin-3 (1/200) and the negative control,desmin (1/800). Images were taken with an Olympus micro-scope with digital SPOT camera and software.
Gastrocnemius (including soleus) and EDL muscle from1- and 6-month-old mice were removed, stretched toprevent contracture, frozen in tissue-freezing medium(ProSciTech) and stored at –70°C. Quadriceps muscle frombaboon and human samples were also analysed. Five micronsequential sections were mounted on gelatin-coated slides,fixed in 2% paraformaldehyde in phosphate-buffered saline(PBS) on ice for 2 min, rinsed in PBS, incubated inethanolamine for 10 min and rinsed again before blockingwith 10% fetal calf serum (FCS). Primary antibodies usedwere anti-α-actinin-2 and -3 (16,19), and fibre typing wasperformed using antibodies directed against fast myosin

Human Molecular Genetics, 2001, Vol. 10, No. 13 1345
isoforms 2A (SC-71) and 2B (BF-F3) (51), and slow myosin(Chemicon). All antibodies were diluted in 10% FCS andapplied to sections. In mice, those fibres that remainedunstained with the above-mentioned fibre typing antibodieswere classified as type 2X fibres. For baboon and humanmuscle, antibodies directed against fast myosin isoforms(MY32; Sigma) were also used to assess fibre typing. Afterrinsing with PBS, secondary antibodies conjugated tocyanine 3 were applied. Sections were mounted and viewedusing the Olympus fluorescent microscope and imagescaptured using SPOT digital imaging software.
Preparation and histochemistry of polyester-embeddedmouse embryos
Embryonic tissue was prepared according to the procedureoutlined by Sheppard et al. (52). Briefly, tissue was fixed in5% (v/v) glacial acetic acid in absolute ethanol for 6 h at 4°C,followed by dehydration and embedding in polyester wax(BDH). Sections were cut at 4 µm thickness. Slides werestored at 4°C and dewaxed in 50% xylene/50% histolene andrehydrated with decreasing grades of ethanol. Sections wereblocked using 10% FCS in PBS and stained according to theprocedure outlined above.
ACKNOWLEDGEMENTS
We are grateful to Dr Peter Jeffrey (Children’s MedicalResearch Institute) for providing the mouse embryonictissue, David Fribourg for α-actinin PCR primer design, andLucy Rowe and Mary Barter for outstanding assistance withmouse backcross mapping. We also thank Professor PeterGunning, Dr Sandra Cooper and Ms Biljana Ilkovski for theirthoughtful reviews of the manuscript. Baboon blood andtissue was obtained from the Baboon colony facility,Wallacia, NSW. This work was supported by an RACP GlaxoWellcome Australia Fellowship to K.N.N. and generoussupport from the Ramaciotti Foundation, the Australian BrainFoundation (K.N.N.) and the Joshua Frase Foundation, theMuscular Dystrophy Association of USA, and National Insti-tutes of Health (NIAMS) grants R01 AR44345 and K02AR02026 to A.H.B.
REFERENCES
1. Blanchard, A., Ohanian, V. and Critchley, D. (1989) The structure andfunction of α-actinin. J. Muscle Res. Cell Motil., 10, 280–289.
2. Honda, K., Yamada, T., Endo, R., Ino, Y., Gotoh, M., Tsuda, H.,Yamada, Y., Chiba, H. and Hirohashi, S. (1998) α-Actinin-4, a novelactin-bundling protein associated with cell motility and cancer invasion.J. Cell Biol., 140, 1383–1393.
3. Youssoufian, H., McAfee, M. and Kwiatkowski, D.J. (1990) Cloning andchromosomal localization of the human cytoskeletal α-Actinin genereveals linkage to the β-spectrin gene. Am. J. Hum. Genet., 47, 62–71.
4. Beggs, A.H., Byers, T.J., Knoll, J.H., Boyce, F.M., Bruns, G.A. andKunkel, L.M. (1992) Cloning and characterization of two human skeletalmuscle α-actinin genes located on chromosomes 1 and 11. J. Biol. Chem.,267, 9281–9288.
5. Nave, R., Furst, D.O. and Weber, K. (1990) Interaction of α-Actinin andnebulin in vitro. Support for the existence of a fourth filament system inskeletal muscle. FEBS Lett., 269, 163–166.
6. Papa, I., Astier, C., Kwiatek, O., Raynaud, F., Bonnal, C., Lebart, M.C.,Roustan, C. and Benyamin, Y. (1999) α-Actinin-CapZ, an anchoring com-plex for thin filaments in Z-line. J. Muscle Res. Cell Motil., 20, 187–197.
7. Salmikangas, P., Mykkanen, O.M., Gronholm, M., Heiska, L., Kere, J.and Carpen, O. (1999) Myotilin, a novel sarcomeric protein with two Ig-likedomains, is encoded by a candidate gene for limb-girdle musculardystrophy. Hum. Mol. Genet., 8, 1329–1336.
8. Bellin, R.M., Sernett, S.W., Becker, B., Ip, W., Huiatt, T.W. andRobson, R.M. (1999) Molecular characteristics and interactions of the inter-mediate filament protein synemin: interactions with α-actinin may anchorsynemin-containing heterofilaments. J. Biol. Chem., 274, 29493–29499.
9. McGregor, A., Blanchard, A.D., Rowe, A.J. and Critchley, D.R. (1994)Identification of the vinculin-binding site in the cytoskeletal protein α-actinin.Biochem. J., 301, 225–233.
10. Hance, J.E., Fu, S.Y., Watkins, S.C., Beggs, A.H. and Michalak, M.(1999) α-Actinin-2 is a new component of the dystrophin-glycoproteincomplex. Arch. Biochem. Biophys., 365, 216–222.
11. Otey, C.A., Vasquez, G.B., Burridge, K. and Erickson, B.W. (1993)Mapping of the α-actinin binding site within the β 1 integrin cytoplasmicdomain. J. Biol. Chem., 268, 21193–21197.
12. Fukami, K., Endo, T., Imamura, M. and Takenawa, T. (1994) α-Actininand vinculin are PIP2-binding proteins involved in signaling by tyrosinekinase. J. Biol. Chem., 269, 1518–1522.
13. Shibasaki, F., Fukami, K., Fukui, Y. and Takenawa, T. (1994) Phosphati-dylinositol 3-kinase binds to α-actinin through the p85 subunit. Biochem.J., 302, 551–557.
14. Pomies, P., Louis, H.A. and Beckerle, M.C. (1997) CRP1, a LIM domainprotein implicated in muscle differentiation, interacts with α-actinin.J. Cell Biol., 139, 157–168.
15. Pomies, P., Macalma, T. and Beckerle, M.C. (1999) Purification and char-acterization of an α-actinin-binding PDZ-LIM protein that is up-regulatedduring muscle differentiation. J. Biol. Chem., 274, 29242–29250.
16. North, K.N. and Beggs, A.H. (1996) Deficiency of a skeletal muscle iso-form of α-actinin (α-actinin-3) in merosin-positive congenital musculardystrophy. Neuromusc. Disord., 6, 229–235.
17. North, K.N., Yang, N., Wattanasirichaigoon, D., Mills, M., Easteal, S. andBeggs, A.H. (1999) A common nonsense mutation results in α-actinin-3deficiency in the general population. Nat. Genet., 21, 353–354.
18. Nowak, M.A., Boerlijst, M.C., Cooke, J. and Smith, J.M. (1997)Evolution of genetic redundancy. Nature, 388, 167–171.
19. Chan, Y., Tong, H.Q., Beggs, A.H. and Kunkel, L.M. (1998) Humanskeletal muscle-specific α-actinin-2 and -3 isoforms form homodimersand heterodimers in vitro and in vivo. Biochem. Biophys. Res. Commun.,248, 134–139.
20. Rowe, L.B., Nadeau, J.H., Turner, R., Frankel, W.N., Letts, V.A.,Eppig, J.T., Ko, M.S., Thurston, S.J. and Birkenmeier, E.H. (1994) Mapsfrom two interspecific backcross DNA panels available as a communitygenetic mapping resource. Mamm. Genome, 5, 253–274.
21. Duret, L. and Mouchiroud, D. (2000) Determinants of substitution rates inmammalian genes: expression pattern affects selection intensity but notmutation rate. Mol. Biol. Evol., 17, 68–74.
22. Furamura, M. and Ishikawa, H. (1996) Actin bundles in human hairfollicles as revealed by confocal laser microscopy. Cell Tissue Res., 283,425–434.
23. Wyszynski, M., Lin, J., Rao, A., Nigh, E., Beggs, A.H., Craig, A.M. andSheng, M. (1997) Competitive binding of α-actinin and calmodulin to theNMDA receptor. Nature, 385, 439–442.
24. Schachat, F.H., Canine, A.C., Briggs, M.M. and Reedy, M.C. (1985)The presence of two skeletal muscle α-actinins correlates with troponin-tropomyosin expression and Z-line width. J. Cell Biol., 101, 1001–1008.
25. Suzuki, H., Komiyama, M., Konno, A. and Shimada, Y. (1998) Exchange-ability of actin in cardiac myocytes and fibroblasts as determined byfluorescence photobleaching recovery. Tissue Cell, 30, 274–280.
26. Fyrberg, E., Kelly, M., Ball, E., Fyrberg, C. and Reedy, M.C. (1990)Molecular genetics of Drosophila α-actinin: mutant alleles disrupt Z discintegrity and muscle insertions. J. Cell Biol., 110, 1999–2011.
27. Kobayashi, R., Itoh, H. and Tashima, Y. (1989) α-Actinin expressionduring avian myogenesis in vivo. Evidence for the existence of an embryo-specific isoform of α-actinin. Eur. J. Biochem., 185, 297–302.
28. Lynch, M. and Conery, J.S. (2000) The evolutionary fate and consequencesof duplicate genes. Science, 290, 1151–1155.
29. Li, W.H. (1997) Molecular Evolution. Sinauer Press, Sunderland, MA,pp. 279–284.
30. Force, A., Lynch, M., Pickett, F.B., Amores, A., Yan, Y.L. andPostlethwait, J. (1999) Preservation of duplicate genes by complementary,degenerative mutations. Genetics, 151, 1531–1545.

1346 Human Molecular Genetics, 2001, Vol. 10, No. 13
31. Krakauer, D.C. and Nowak, M.A. (1999) Evolutionary preservation ofredundant duplicated genes. Semin. Cell Dev. Biol., 10, 555–559.
32. Bouchard, M., Pfeffer, P.L. and Busslinger, M. (2000) Functionalequivalence of the transcription factors pax2 and pax5 in mousedevelopment. Development, 127, 3703–3713.
33. Branford, W.W., Benson, G.V., Ma, L., Maas, R.L. and Potter, S.S. (2000)Characterization of Hoxa-10/Hoxa-11 transheterozygotes revealsfunctional redundancy and regulatory interactions. Dev. Biol., 224, 387.
34. Simoneau, J.A. and Bouchard, C. (1998) The effects of genetic variationon anaerobic perfomance. In Van Praagh (ed.), Paediatric AnaerobicPerformance. Human Kinetics Publishers Inc., Champaign, IL, pp. 5–21.
35. Gibson, T.J. and Spring, J. (1998) Genetic redundancy in vertebrates:polyploidy and persistence of genes encoding multidomain proteins.Trends Genet., 14, 46–49.
36. Meyer, A. and Schartl, M. (1999) Gene and genome duplications invertebrates: the one-to-four (-to-eight in fish) rule and the evolution ofnovel gene functions. Curr. Opin. Cell Biol., 11, 699–704.
37. Kimura, M. (1980) A simple method for estimating evolutionary rates ofbase substitutions through comparative studies of nucleotide sequences.J. Mol. Evol., 16, 111–120.
38. Thomas, J.H. (1993) Thinking about genetic redundancy. Trends Genet.,9, 395–399.
39. Cooke, J., Nowak, M.A., Boerlijst, M. and Maynard-Smith, J. (1997)Evolutionary origins and maintenance of redundant gene expressionduring metazoan development. Trends Genet., 13, 360–364.
40. Chan, Y., Tong, H.Q., Beggs, A.H. and Kunkel, L.M. (1998) Humanskeletal muscle-specific α-actinin-2 and -3 isoforms form homodimersand heterodimers in vitro and in vivo. Biochem. Biophys. Res. Commun.,248, 134–139.
41. Smith, S.W., Overbeek, R., Woese, C.R., Gilbert, W., Gillevet, P.M.,Slatkin, M., Excoffier, L., Krakauer, D.C., Nowak, M.A., Pfeffer, P.L.et al. (1994) Testing for linkage disequilibrium in genotypic data using theExpectation-Maximization algorithm. Comput. Appl. Biosci., 10, 671–675.
42. Li, W.H. (1993) Unbiased estimation of the rates of synonymous and non-synonymous substitution. J. Mol. Evol., 36, 96–99.
43. Raymond, M. and Rousset, F. (1995) An exact test for populationdifferentiation. Evolution, 49, 1280–1283.
44. Dempster, A., Laird, N. and Rubin, D. (1977) Maximum likelihoodestimation from incomplete data via the EM algorithm. J. R. Stat. Soc., 39,1–38.
45. Slatkin, M. and Excoffier, L. (1996) Testing for linkage disequilibrium ingenotypic data using the Expectation-Maximization algorithm. Heredity,76, 377–383.
46. Chomczynski, P. and Sacchi, N. (1987) Single-step method of RNAisolation by acid guanidium thiocyanate–phenol–chloroform extraction.Anal. Biochem., 162, 156–159.
47. Lehrach, H., Diamond, D., Wozney, J.M. and Boedtker, H. (1977) RNAmolecular weight determinations by gel electrophoresis under denaturingconditions, a critical reexamination. Biochemistry, 16, 4743.
48. Hoffman, E.P., Fischbeck, K.H., Brown, R.H., Johnson, M., Medori, R.,Loike, J.D., Harris, J.B., Waterston, R., Brooke, M., Specht, L. et al.(1988) Characterisation of dystrophin in muscle biopsy specimens frompatients with Duchenne or Becker muscular dystrophy. N. Engl. J. Med.,318, 1363–1368.
49. Nakamura, K., Tanaka, T., Kuwahara, A. and Takeo, K. (1985)Microassay for proteins on nitrocellulose filter using protein dye stainingprocedure. Anal. Biochem., 148, 311–319.
50. Weinberger, R., Schevzov, G., Jeffrey, P., Gordon, K., Hill, M. andGunning, P. (1996) The molecular composition of neuronal micro-filamants is spatially and temporally regulated. J. Neurosci., 16, 238–252.
51. Schiaffino, S., Gorza, L., Sartore, S., Saggin, L., Ausoni, S., Vianello, M.and Gundersen, K. (1989) Three myosin heavy chain isoforms in type 2skeletal muscle fibres. J. Muscle Res. Cell Motil., 10, 197–205.
52. Sheppard, A.M., Konopka, M. and Jeffrey, P.L. (1988) The developmentalappearance of Thy-1 in the avian cerecellum. Dev. Brain Res., 40, 181–192.
.




















