
Cortical Fluoro-Jade staining and blunted adrenomedullary response to hypoglycemia after noncoma...
11
Cortical Fluoro-Jade staining and blunted adrenomedullary response to hypoglycemia after noncoma hypoglycemia in rats Nancy C Tkacs 1 , Yanhua Pan 1 , Ramesh Raghupathi 2 , Ambrose A Dunn-Meynell 3 and Barry E Levin 3,4 1 School of Nursing, University of Pennsylvania, Philadelphia, Pennsylvania, USA; 2 Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, Pennsylvania, USA; 3 Neurology Service, VA Medical Center, East Orange, New Jersey, USA; 4 Department of Neurology and Neurosciences, NJ Medical School, Newark, New Jersey, USA Intensive insulin therapy in patients with type 1 diabetes mellitus reduces long-term complications; however, intensive therapy is also associated with a three-fold increase in hypoglycemic episodes. The present study in conscious rats characterizes the physiologic and neuropathologic consequences of a single episode of moderate hypoglycemia. In this model, intravenous insulin is used to reduce plasma glucose to 30 to 35 mg/dL for 75 mins. This single hypoglycemic insult acutely induces hypoglycemia-associated autonomic failure (HAAF), with epinephrine responses to hypoglycemia reduced more than 36% from control. Neuropathology after this insult includes the appearance of dying cells, assessed with the marker Fluoro-jade B (FJ). After hypoglycemic insult, FJ þ cells were consistently seen in subdivisions of the medial prefrontal cortex, the orbital cortex, and the piriform cortex. There was a significant correlation between depth of hypoglycemia and number of FJ þ cells, suggesting that there is a critical threshold below which vulnerable cells begin to die. These data suggest that there is a population of cells that are vulnerable to moderate levels of hypoglycemia commonly experienced by patients with insulin-treated diabetes. These cells, which may be neurons, are primarily found in cortical regions implicated in visceral perception and autonomic control, raising the possibility that their loss contributes to clinically reported deficits in autonomic and perceptual responses to hypoglycemia. Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655. doi:10.1038/sj.jcbfm.9600152; published online 18 May 2005 Keywords: central autonomic regulation; hypoglycemia; insulin; neuron death; visceral cortex Introduction Hypoglycemia is a common occurrence in indivi- duals treated with insulin for type 1 diabetes mellitus, and the incidence of hypoglycemia in- creases in patients striving to maintain optimal glycemic control to reduce long-term diabetic com- plications (DCCT, 1993, 1997). When hypoglycemia occurs because of exogenous insulin administration for diabetes, recovery from hypoglycemia is largely integrated by the central nervous system (CNS). In response to insulin-induced hypoglycemia, the brain initiates behavioral, autonomic, and endocrine responses including food intake, increased gastric motility, secretion of epinephrine, glucocorticoids, and growth hormone, and increased sympathetic nervous system activity. Recurrent episodes of hypo- glycemia are associated with decreased conscious perception of the hypoglycemic state and blunted or absent secretion of counterregulatory hormones. These sequelae of recurrent hypoglycemia are termed ‘hypoglycemia unawareness’ and ‘hypoglycemia- associated autonomic failure’ (HAAF), respectively (Gerich et al, 1991; Cryer, 2001). One approach to elucidating the nature of these hypoglycemia- induced deficits is to study animal models of HAAF. When rats sustain 2 to 7 episodes of mild hypoglycemia (40 to 60 mg/dL), they have dimin- ished epinephrine responses to hypoglycemic challenge administered 1 day later. Variable effects Received 28 August 2004; revised 12 April 2005; accepted 13 April 2005; published online 18 May 2005 Correspondence: Dr NC Tkacs, School of Nursing, University of Pennsylvania, 420 Guardian Drive, Philadelphia, PA 19104-6096, USA. E-mail: [email protected] Funding for this project was provided by NIH DK02899; NIH/JDRF Research Award DK59754, and the University of Pennsylvania Research Foundation (NCT) and the VA Research Service and NIH DK53181 (BEL). Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655 & 2005 ISCBFM All rights reserved 0271-678X/05 $30.00 www.jcbfm.com
Transcript of Cortical Fluoro-Jade staining and blunted adrenomedullary response to hypoglycemia after noncoma...

Cortical Fluoro-Jade staining and bluntedadrenomedullary response to hypoglycemiaafter noncoma hypoglycemia in rats
Nancy C Tkacs1, Yanhua Pan1, Ramesh Raghupathi2, Ambrose A Dunn-Meynell3
and Barry E Levin3,4
1School of Nursing, University of Pennsylvania, Philadelphia, Pennsylvania, USA; 2Department ofNeurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, Pennsylvania, USA;3Neurology Service, VA Medical Center, East Orange, New Jersey, USA; 4Department of Neurology andNeurosciences, NJ Medical School, Newark, New Jersey, USA
Intensive insulin therapy in patients with type 1 diabetes mellitus reduces long-term complications;however, intensive therapy is also associated with a three-fold increase in hypoglycemic episodes.The present study in conscious rats characterizes the physiologic and neuropathologicconsequences of a single episode of moderate hypoglycemia. In this model, intravenous insulinis used to reduce plasma glucose to 30 to 35 mg/dL for 75 mins. This single hypoglycemic insultacutely induces hypoglycemia-associated autonomic failure (HAAF), with epinephrine responses tohypoglycemia reduced more than 36% from control. Neuropathology after this insult includes theappearance of dying cells, assessed with the marker Fluoro-jade B (FJ). After hypoglycemic insult,FJþ cells were consistently seen in subdivisions of the medial prefrontal cortex, the orbital cortex,and the piriform cortex. There was a significant correlation between depth of hypoglycemia andnumber of FJþ cells, suggesting that there is a critical threshold below which vulnerable cells beginto die. These data suggest that there is a population of cells that are vulnerable to moderate levels ofhypoglycemia commonly experienced by patients with insulin-treated diabetes. These cells, whichmay be neurons, are primarily found in cortical regions implicated in visceral perception andautonomic control, raising the possibility that their loss contributes to clinically reported deficits inautonomic and perceptual responses to hypoglycemia.Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655. doi:10.1038/sj.jcbfm.9600152; published online18 May 2005
Keywords: central autonomic regulation; hypoglycemia; insulin; neuron death; visceral cortex
Introduction
Hypoglycemia is a common occurrence in indivi-duals treated with insulin for type 1 diabetesmellitus, and the incidence of hypoglycemia in-creases in patients striving to maintain optimalglycemic control to reduce long-term diabetic com-plications (DCCT, 1993, 1997). When hypoglycemiaoccurs because of exogenous insulin administrationfor diabetes, recovery from hypoglycemia is largely
integrated by the central nervous system (CNS).In response to insulin-induced hypoglycemia, thebrain initiates behavioral, autonomic, and endocrineresponses including food intake, increased gastricmotility, secretion of epinephrine, glucocorticoids,and growth hormone, and increased sympatheticnervous system activity. Recurrent episodes of hypo-glycemia are associated with decreased consciousperception of the hypoglycemic state and blunted orabsent secretion of counterregulatory hormones.These sequelae of recurrent hypoglycemia are termed‘hypoglycemia unawareness’ and ‘hypoglycemia-associated autonomic failure’ (HAAF), respectively(Gerich et al, 1991; Cryer, 2001). One approach toelucidating the nature of these hypoglycemia-induced deficits is to study animal models of HAAF.
When rats sustain 2 to 7 episodes of mildhypoglycemia (40 to 60 mg/dL), they have dimin-ished epinephrine responses to hypoglycemicchallenge administered 1 day later. Variable effects
Received 28 August 2004; revised 12 April 2005; accepted 13April 2005; published online 18 May 2005
Correspondence: Dr NC Tkacs, School of Nursing, University ofPennsylvania, 420 Guardian Drive, Philadelphia, PA 19104-6096,USA. E-mail: [email protected]
Funding for this project was provided by NIH DK02899; NIH/JDRF
Research Award DK59754, and the University of Pennsylvania
Research Foundation (NCT) and the VA Research Service and NIH
DK53181 (BEL).
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655& 2005 ISCBFM All rights reserved 0271-678X/05 $30.00
www.jcbfm.com

on norepinephrine, glucagon, and corticosteroneresponses to hypoglycemia have been reported(Evans et al, 2001; Shum et al, 2001; Flanaganet al, 2003; De Vries et al, 2004). A recent reportindicates that a single prolonged episode of moder-ate hypoglycemia does not significantly reduce theepinephrine response to subsequent hypoglycemicchallenge (Paranjape and Briski, 2004). We pre-viously reported that three episodes of moderatehypoglycemia (30 to 35 mg/dL) delivered at 48 hintervals resulted in decreased epinephrine res-ponses. Rats killed after one to three hypoglycemicepisodes also had positive TUNEL staining in cellsof the arcuate nucleus, indicating DNA damage,and reduced mRNA for arcuate neuropeptide Y(NPY) and proopiomelanocortin (POMC) (Tkacset al, 2000).
Pathologic effects of hypoglycemia on the brainhave also been studied using a model of severehypoglycemia induced in anesthetized rats whilecontinuously monitoring EEG. Neuroglycopenia ismaintained at a level to produce cerebral isoelec-tricity for 30 to 60 mins duration, with blood glucosevalues less than 15 mg/dL. After this insult, exten-sive neuron death occurs in cortical layers 2 and 3,in the striatum, and in hippocampal neurons,particularly in the dentate gyrus (Auer et al, 1984;Ouyang et al, 2000). The effect of severe hypogly-cemic coma on counterregulatory responses tohypoglycemia has not been reported.
The first experiment we report here aimed to con-firm and extend our previous finding that a singleepisode of moderate hypoglycemia is sufficient toreduce counterregulatory hormone responses tosubsequent hypoglycemia (Tkacs et al, 2000). Thetime points and experimental manipulations werebased on our prior study with the following modi-fications: epinephrine, norepinephrine, glucagon,and corticosterone responses to equivalent mildhypoglycemia were compared between rats studied48 h after a single episode of moderate hypoglycemiaand control, pair-fasted rats. Immediately afterblood sampling for hormone measurements wascompleted, these rats were euthanized and perfusedfor subsequent neuropathologic assessment usingFluoro-Jade B (FJ), a recently developed putativemarker of neuron death (Schmued and Hopkins,2000; Sato et al, 2001; Munoz et al, 2001). Thesecond experiment characterized the time courseof appearance of FJ and other injury markers aftermoderate hypoglycemia in rats that were not sub-jected to blood sampling for hormone measure-ments.
Materials and methods
All protocols used in these experiments were reviewedand approved by the University of Pennsylvania Institu-tional Animal Care and Use Committee. At all times,the work performed was in accord with the Guide for the
Care and Use of Laboratory Animals of the Instituteof Laboratory Animal Resources, Commission on LifeSciences, National Research Council, 1996.
Animal Preparation and Handling
The experiments were performed on adult male Sprague–Dawley rats. After shipping, the rats were group-housedand held for 1 week before surgery to allow recovery fromstress. Rats were maintained in a Research Animal Facilityof the University of Pennsylvania in a temperature andhumidity controlled room with 12:12 h light:dark cycle(lights on at 0700) and with free access to rodent chow(Lab Diets) and water. Surgery was performed under acombined anesthetic/analgesic mixture of ketamine/xyla-zine/buprenorphine (50, 10, and 0.2 mg/kg, respectively,delivered by intraperitoneal injection). The right jugularvein was isolated and a saline-filled silastic catheter wasintroduced and advanced to the approximate level of thesuperior vena cava/right atrium junction. The catheter wastunneled subcutaneously to exit between the scapulae andwas closed with a stainless steel plug. Daily catheter careinvolved aspiration of any clots and refilling with asuspension of polyvinylpyrrolidone (20% PVP-40) insterile heparinized saline (50 U/mL heparin). Rats weremaintained until they recovered preoperative weightbefore initiating the experimental protocol 5 to 7 daysafter surgery.
Experiment 1: The Effect of a Single Episode ofHypoglycemia on Counterregulatory HormoneResponses to Subsequent Hypoglycemia andBrain Fluoro-Jade B Staining
Male Sprague–Dawley rats were obtained from CharlesRiver Laboratories, Kingston, NY, USA. Initial weightsranged from 320 to 360 g. Rats were surgically preparedwith jugular venous catheters as described above. Ratswere maintained postoperatively until they regainedpreoperative weight. During this time, the rats were givena 2-h period of training to the experimental conditions inthe procedure room. For the first hypoglycemic episode,rats were fasted overnight before the treatment day. In themorning, rats were brought to the procedure room andwere administered insulin 4 U/kg (intravenous) (insulin–insulin n¼ 10) or saline (saline–insulin n¼ 8). Bloodsamples (75 mL) for glucose and lactate were drawn every15 mins after insulin and every 30 mins after saline andwere analyzed using an YSI 2700 glucose/lactate analyzer(Yellow Springs Instruments, Yellow Springs, OH, USA).Additional doses of insulin 0.5 to 1 U/kg (intravenous)were administered to maintain hypoglycemia at a levelbetween 30 and 35 mg/dL for 75 mins. After the 90 minsblood sample was drawn, insulin-treated rats wereadministered dextrose 1 g/kg intravenously to restoreeuglycemia, and food was returned to all rats.
For the second hypoglycemic episode, 48 h after theinitial hypoglycemic episode, all rats were fasted over-night before hypoglycemic challenge. An initial bloodsample was drawn to measure baseline levels of glucose,
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1646
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655

lactate, epinephrine, norepinephrine, glucagon, and corti-costerone. Insulin was administered at a dose of 0.75 U/kg(intravenous). This dose was repeated for a total of 3 timesat 5 mins intervals, and a small sample for blood glucosewas drawn 5 mins after the third dose. This insulin-dosingschedule was developed in pilot studies with the aim ofmaintaining plasma glucose at 40 to 45 mg/dL for 2 h.Additional doses of 0.25 to 0.75 U/kg were administeredas necessary to maintain the desired level of hypoglyce-mia. Blood was drawn for glucose, lactate, and hormonemeasurements at 20, 40, 60, 80, and 120 mins after insulin.In each case, the blood sample volume was 0.8 mL. Theblood was centrifuged, plasma was removed and red bloodcells were resuspended in saline and returned to the ratvia the intravenous catheter. After the final blood samplethe rats were deeply anesthetized with sodium pentobar-bital and were perfused via the aorta with heparinizedsaline followed by 4% paraformaldehyde in phosphatebuffer. Brains were removed and were postfixed in 4%paraformaldehyde for several hours. Forebrain and brain-stem blocks were then cryoprotected in 30% sucroseovernight, frozen on powdered dry ice, and kept at �801Cuntil sectioning. Forebrain and brainstem sections werecut at 40 mm thickness on a Leica cryostat and were savedserially in six wells with an ethylene glycol-basedcryoprotectant (Watson et al, 1986). Sections were heldat �201C until staining.
Histology
For the FJ staining procedure, one set of sections(comprising a one-in-six series through the forebrain orbrainstem) was removed from the cryoprotectant, rinsedin PBS, after which sections were mounted on gelatin-coated slides and were air-dried for at least 24 h. Fluoro-Jade B (Histo-Chem, Jefferson, AR, USA) staining wasperformed following the protocol described by Hopkins etal (2000), except that the incubation time in potassiumpermanganate was reduced to 5 mins. Sections wereexamined using an Olympus fluorescence microscopeand Chroma filter set 41001 (FITC/Bodipy/Fluo3/DiO). Allfluorescent cells were counted for each set of brainsections by an observer who was masked to the treatmentgroups. Forebrain sections from the first six rats repre-sented levels from 1 mm anterior to bregma through theposterior hypothalamus, based on atlas plates (Paxinosand Watson, 1998). After this processing was completed, itwas apparent that FJ staining was maximal in the mostrostral sections, so forebrain blocks for the remaining 12rats included the entire forebrain from the olfactory bulbsthrough the posterior hypothalamus.
Statistical analysis was performed using two-wayrepeated measures analysis of variance and area underthe curve measurements were compared between treat-ment groups using unpaired t-tests.
Hormone Assays
Plasma for catecholamine assay was vortexed with 5 Nperchloric acid and then immediately frozen on dry ice
and held at �701C for further processing. After all ratscompleted the protocol, the samples were briefly thawedand centrifuged, and 100mL of supernatant was mixedwith the internal standard dihydroxybenzylamine.Catecholamine assay using HPLC with electrochemicaldetection was performed at the VAMC in East Orange, NJ,USA. Plasma for glucagon assay was mixed with proteaseinhibitor consisting of leupeptin, trasylol, and EDTA,following the protocol of the University of PennsylvaniaDiabetes Center radioimmunoassay core facility. Glucagonradioimmunoassay was performed using the Linco anti-body and corticosterone assay was performed using anenzyme immunoassay kit from ALPCO.
Experiment 2: Time Course of Fluoro-Jade BAppearance
Experiment 2A. The results of experiment 1 indicated thatFJ was present in brains collected 2 h into a secondepisode of hypoglycemia, and we hypothesized that thisstaining was the result of the initial episode of moderatehypoglycemia 48 h before killing. We thus elected toassess the time course of FJ staining after a single episodeof moderate hypoglycemia. The time course of FJ stainingwas evaluated in 33 rats in which brains were collected 4,8, 12, 24, 48, 72 h or 5 or 10 days after one episode ofhypoglycemia.
Experiment 2B. Previous work in this lab and othersstudied hypoglycemia repeated at least two times, whichmight be expected to exacerbate neuronal damage anddeath. Therefore, the neuropathologic consequences ofrepeated hypoglycemia were studied in an additional fourrats. These rats were subjected to 2 episodes of hypo-glycemia, 2 days apart, and were perfused 24 h (n¼ 2) or48 h (n¼ 2) after the second episode of hypoglycemia.
Tissue Processing for Experiments 2A and 2B
Brain sections from all rats subjected to hypoglycemicinsult were processed for FJ. FJþ cells were counted onsections representing 1 mm intervals through the forebrainfrom 4.7 mm anterior to bregma through 4.3 mm posteriorto bregma. Selected additional sections from rats at 12, 24,or 48 h after hypoglycemic insult were processed usingimmunohistochemistry for the following markers ofneuron injury or death: 72 kDa heat shock protein(HSP70), activated caspase-3, a calpain-generated spectrincleavage product or a caspase-generated spectrin cleavageproduct. Antibody detection was performed using a Vector‘Elite’ ABC kit and diaminobenzidine as the chromogen.Reactive gliosis was assessed using a mouse monoclonalantibody to glial fibrillary acidic protein (GFAP). Addi-tional selected sections were used to assess double-labelfluorescent immunohistochemistry with FJ staining usingantibodies for neurons (NeuN, MAP2), astrocytes (GFAP),and glutamate receptor subunits 2/3. A list of antibodiesused in these experiments and their sources is shown inTable 1.
Experiment 3. The cardiovascular effects of hypoglyce-mic insult were assessed in two rats. In these rats, carotid
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1647
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655
artery catheters were inserted in addition to jugularvenous catheters. After recovery, rats were subjected to90 mins of insulin-induced hypoglycemia after an over-night fast, with continuous recording of blood pressure andheart rate using the Micro-Med BPA system (Louisville,KY, USA).
Data Analysis
Data were entered into SPSS 11.0 (SPSS, Chicago, IL,USA) for statistical analysis. Data are expressed as
mean7standard deviation throughout the report. Statis-tical comparison for hormone measurements used two-way repeated measures analysis of variance. Area underthe curve measurements were compared with unpairedt-test. Counts of FJþ cells over time after hypoglycemiawere compared with one-way analysis of variance.Significance was set at Po0.05 for all statistical tests.
Results
Experiment 1
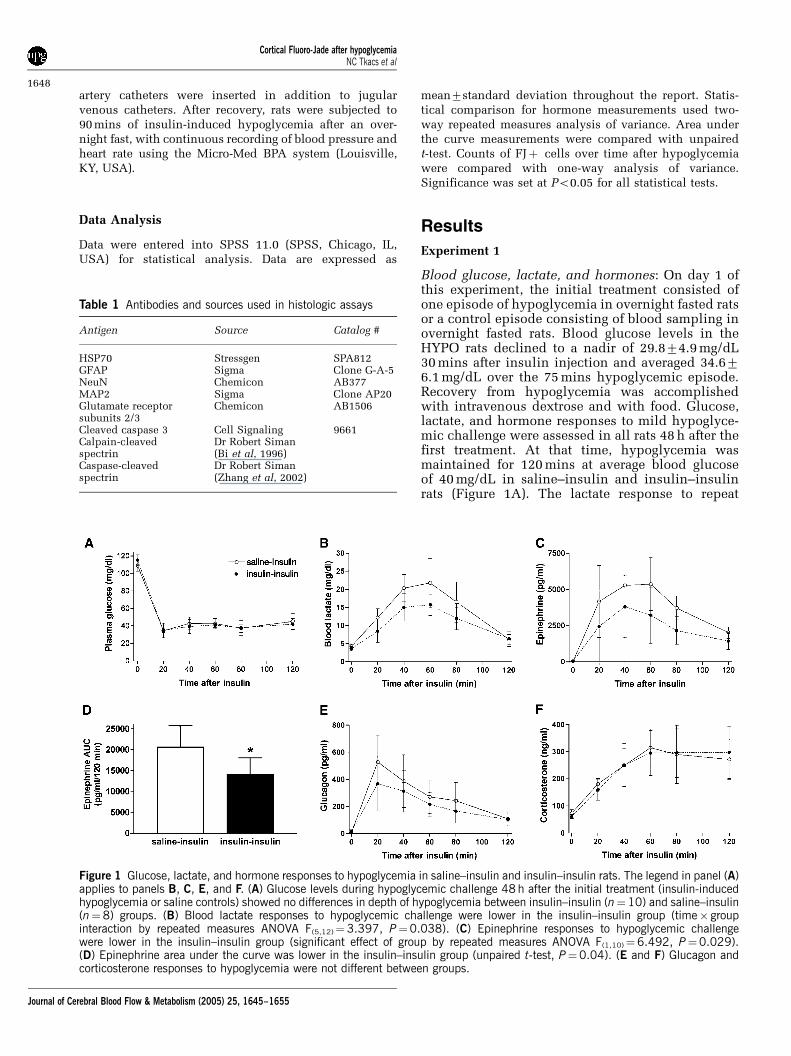
Blood glucose, lactate, and hormones: On day 1 ofthis experiment, the initial treatment consisted ofone episode of hypoglycemia in overnight fasted ratsor a control episode consisting of blood sampling inovernight fasted rats. Blood glucose levels in theHYPO rats declined to a nadir of 29.874.9 mg/dL30 mins after insulin injection and averaged 34.676.1 mg/dL over the 75 mins hypoglycemic episode.Recovery from hypoglycemia was accomplishedwith intravenous dextrose and with food. Glucose,lactate, and hormone responses to mild hypoglyce-mic challenge were assessed in all rats 48 h after thefirst treatment. At that time, hypoglycemia wasmaintained for 120 mins at average blood glucoseof 40 mg/dL in saline–insulin and insulin–insulinrats (Figure 1A). The lactate response to repeat
Table 1 Antibodies and sources used in histologic assays
Antigen Source Catalog #
HSP70 Stressgen SPA812GFAP Sigma Clone G-A-5NeuN Chemicon AB377MAP2 Sigma Clone AP20Glutamate receptorsubunits 2/3
Chemicon AB1506
Cleaved caspase 3 Cell Signaling 9661Calpain-cleavedspectrin
Dr Robert Siman(Bi et al, 1996)
Caspase-cleavedspectrin
Dr Robert Siman(Zhang et al, 2002)
Figure 1 Glucose, lactate, and hormone responses to hypoglycemia in saline–insulin and insulin–insulin rats. The legend in panel (A)applies to panels B, C, E, and F. (A) Glucose levels during hypoglycemic challenge 48 h after the initial treatment (insulin-inducedhypoglycemia or saline controls) showed no differences in depth of hypoglycemia between insulin–insulin (n¼10) and saline–insulin(n¼8) groups. (B) Blood lactate responses to hypoglycemic challenge were lower in the insulin–insulin group (time� groupinteraction by repeated measures ANOVA F(5,12)¼3.397, P¼0.038). (C) Epinephrine responses to hypoglycemic challengewere lower in the insulin–insulin group (significant effect of group by repeated measures ANOVA F(1,10)¼6.492, P¼0.029).(D) Epinephrine area under the curve was lower in the insulin–insulin group (unpaired t-test, P¼0.04). (E and F) Glucagon andcorticosterone responses to hypoglycemia were not different between groups.
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1648
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655
hypoglycemia was significantly lower in insulin–insulin rats than in saline–insulin rats, whichexperienced hypoglycemia for the first time (Figure1B, interaction of time and group F(5,12) ¼ 3.397,P¼ 0.038, repeated measures ANOVA). Epinephrineresults from the first 6 rats were lost because oftechnical difficulties with the assay, so the epi-nephrine data are based on measurements from thefinal 12 rats (7 insulin–insulin, 5 saline–insulin).The epinephrine response to hypoglycemia wassignificantly blunted in insulin–insulin rats, relativeto saline–insulin rats (Figures 1C and 1D, nosignificant group� time interaction; effect of groupF(1,10)¼ 6.492, P¼ 0.029; mean AUC is different by
unpaired t-test, P¼ 0.04). Glucagon (Figure 1E),corticosterone (Figure 1F), and norepinephrine(not shown) responses to hypoglycemia were notsignificantly different between treatment groups.
Histology
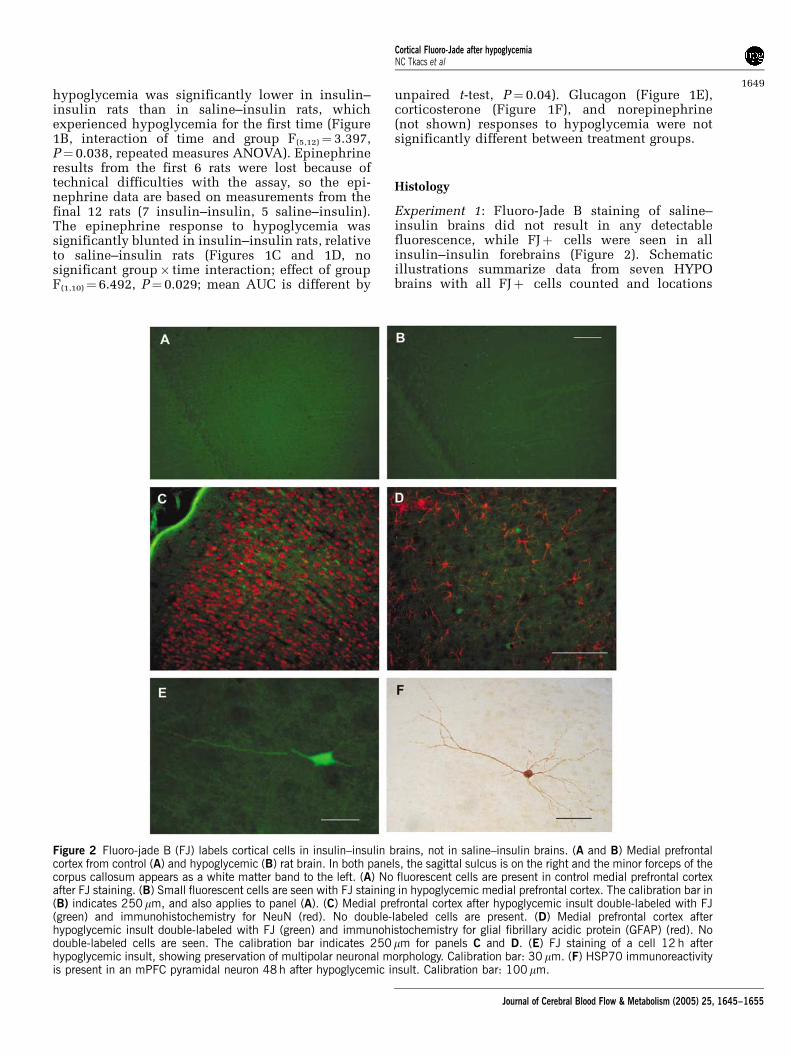
Experiment 1: Fluoro-Jade B staining of saline–insulin brains did not result in any detectablefluorescence, while FJþ cells were seen in allinsulin–insulin forebrains (Figure 2). Schematicillustrations summarize data from seven HYPObrains with all FJþ cells counted and locations
Figure 2 Fluoro-jade B (FJ) labels cortical cells in insulin–insulin brains, not in saline–insulin brains. (A and B) Medial prefrontalcortex from control (A) and hypoglycemic (B) rat brain. In both panels, the sagittal sulcus is on the right and the minor forceps of thecorpus callosum appears as a white matter band to the left. (A) No fluorescent cells are present in control medial prefrontal cortexafter FJ staining. (B) Small fluorescent cells are seen with FJ staining in hypoglycemic medial prefrontal cortex. The calibration bar in(B) indicates 250 mm, and also applies to panel (A). (C) Medial prefrontal cortex after hypoglycemic insult double-labeled with FJ(green) and immunohistochemistry for NeuN (red). No double-labeled cells are present. (D) Medial prefrontal cortex afterhypoglycemic insult double-labeled with FJ (green) and immunohistochemistry for glial fibrillary acidic protein (GFAP) (red). Nodouble-labeled cells are seen. The calibration bar indicates 250 mm for panels C and D. (E) FJ staining of a cell 12 h afterhypoglycemic insult, showing preservation of multipolar neuronal morphology. Calibration bar: 30 mm. (F) HSP70 immunoreactivityis present in an mPFC pyramidal neuron 48 h after hypoglycemic insult. Calibration bar: 100 mm.
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1649
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655
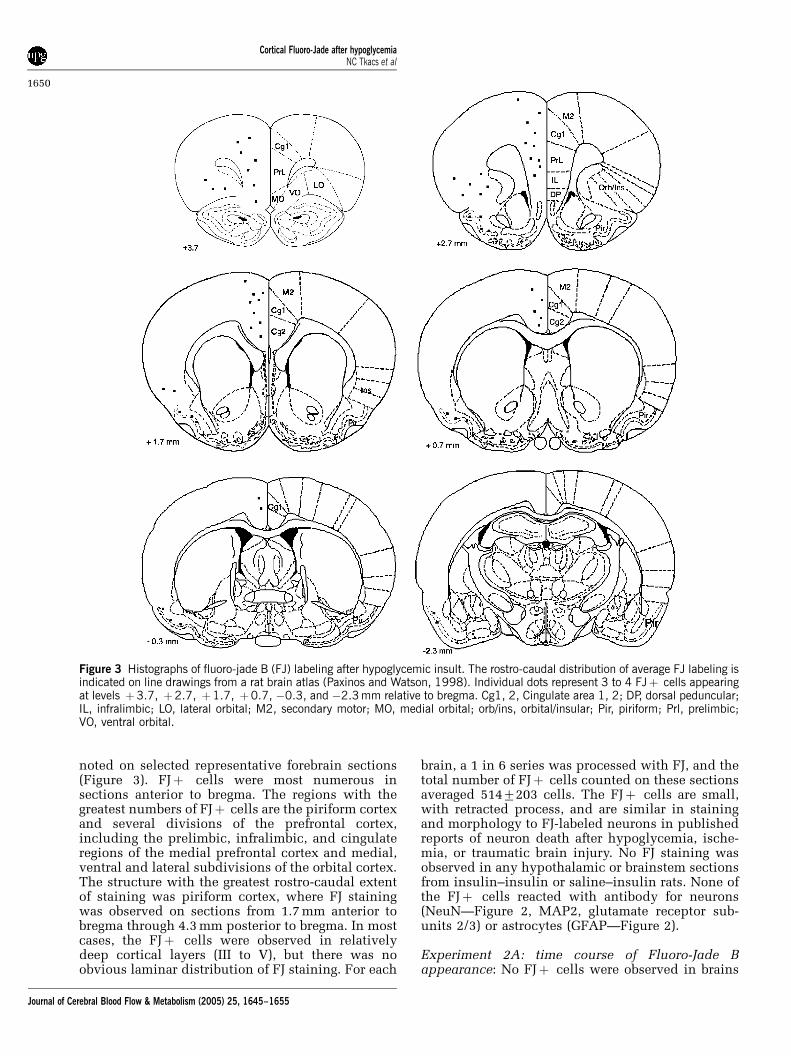
noted on selected representative forebrain sections(Figure 3). FJþ cells were most numerous insections anterior to bregma. The regions with thegreatest numbers of FJþ cells are the piriform cortexand several divisions of the prefrontal cortex,including the prelimbic, infralimbic, and cingulateregions of the medial prefrontal cortex and medial,ventral and lateral subdivisions of the orbital cortex.The structure with the greatest rostro-caudal extentof staining was piriform cortex, where FJ stainingwas observed on sections from 1.7 mm anterior tobregma through 4.3 mm posterior to bregma. In mostcases, the FJþ cells were observed in relativelydeep cortical layers (III to V), but there was noobvious laminar distribution of FJ staining. For each
brain, a 1 in 6 series was processed with FJ, and thetotal number of FJþ cells counted on these sectionsaveraged 5147203 cells. The FJþ cells are small,with retracted process, and are similar in stainingand morphology to FJ-labeled neurons in publishedreports of neuron death after hypoglycemia, ische-mia, or traumatic brain injury. No FJ staining wasobserved in any hypothalamic or brainstem sectionsfrom insulin–insulin or saline–insulin rats. None ofthe FJþ cells reacted with antibody for neurons(NeuN—Figure 2, MAP2, glutamate receptor sub-units 2/3) or astrocytes (GFAP—Figure 2).
Experiment 2A: time course of Fluoro-Jade Bappearance: No FJþ cells were observed in brains
Figure 3 Histographs of fluoro-jade B (FJ) labeling after hypoglycemic insult. The rostro-caudal distribution of average FJ labeling isindicated on line drawings from a rat brain atlas (Paxinos and Watson, 1998). Individual dots represent 3 to 4 FJþ cells appearingat levels þ3.7, þ2.7, þ1.7, þ0.7, �0.3, and �2.3 mm relative to bregma. Cg1, 2, Cingulate area 1, 2; DP, dorsal peduncular;IL, infralimbic; LO, lateral orbital; M2, secondary motor; MO, medial orbital; orb/ins, orbital/insular; Pir, piriform; Prl, prelimbic;VO, ventral orbital.
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1650
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655
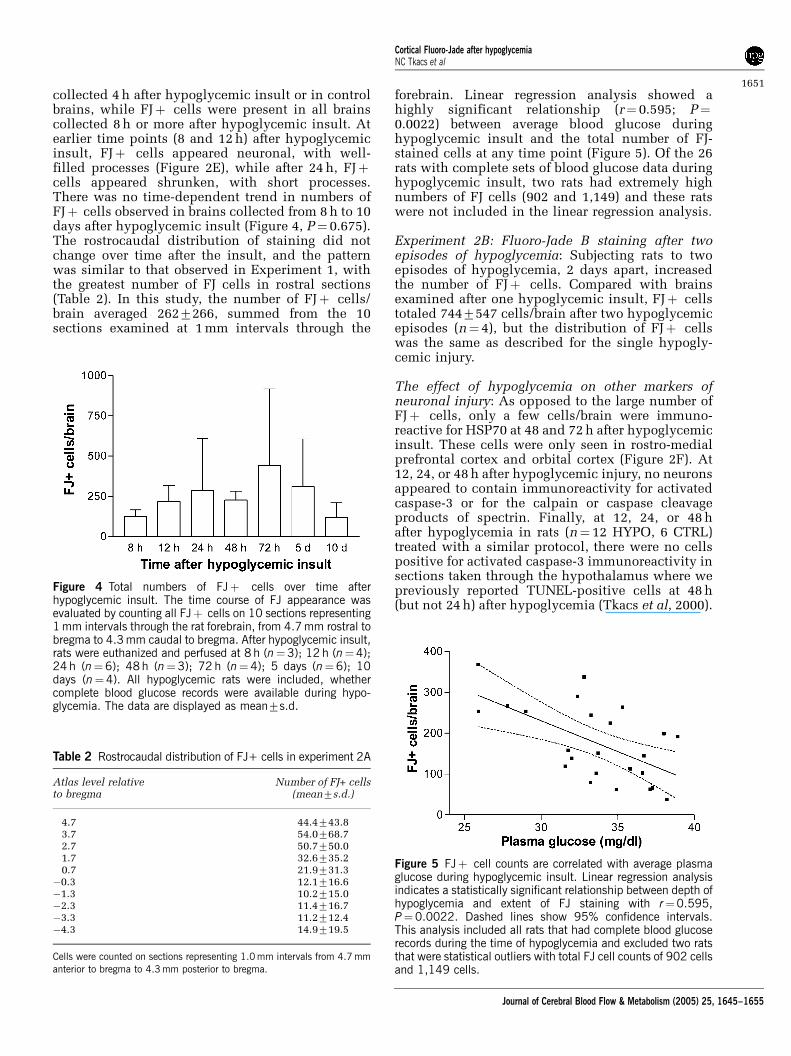
collected 4 h after hypoglycemic insult or in controlbrains, while FJþ cells were present in all brainscollected 8 h or more after hypoglycemic insult. Atearlier time points (8 and 12 h) after hypoglycemicinsult, FJþ cells appeared neuronal, with well-filled processes (Figure 2E), while after 24 h, FJþcells appeared shrunken, with short processes.There was no time-dependent trend in numbers ofFJþ cells observed in brains collected from 8 h to 10days after hypoglycemic insult (Figure 4, P¼ 0.675).The rostrocaudal distribution of staining did notchange over time after the insult, and the patternwas similar to that observed in Experiment 1, withthe greatest number of FJ cells in rostral sections(Table 2). In this study, the number of FJþ cells/brain averaged 2627266, summed from the 10sections examined at 1 mm intervals through the
forebrain. Linear regression analysis showed ahighly significant relationship (r¼ 0.595; P¼0.0022) between average blood glucose duringhypoglycemic insult and the total number of FJ-stained cells at any time point (Figure 5). Of the 26rats with complete sets of blood glucose data duringhypoglycemic insult, two rats had extremely highnumbers of FJ cells (902 and 1,149) and these ratswere not included in the linear regression analysis.
Experiment 2B: Fluoro-Jade B staining after twoepisodes of hypoglycemia: Subjecting rats to twoepisodes of hypoglycemia, 2 days apart, increasedthe number of FJþ cells. Compared with brainsexamined after one hypoglycemic insult, FJþ cellstotaled 7447547 cells/brain after two hypoglycemicepisodes (n¼ 4), but the distribution of FJþ cellswas the same as described for the single hypogly-cemic injury.
The effect of hypoglycemia on other markers ofneuronal injury: As opposed to the large number ofFJþ cells, only a few cells/brain were immuno-reactive for HSP70 at 48 and 72 h after hypoglycemicinsult. These cells were only seen in rostro-medialprefrontal cortex and orbital cortex (Figure 2F). At12, 24, or 48 h after hypoglycemic injury, no neuronsappeared to contain immunoreactivity for activatedcaspase-3 or for the calpain or caspase cleavageproducts of spectrin. Finally, at 12, 24, or 48 hafter hypoglycemia in rats (n¼ 12 HYPO, 6 CTRL)treated with a similar protocol, there were no cellspositive for activated caspase-3 immunoreactivity insections taken through the hypothalamus where wepreviously reported TUNEL-positive cells at 48 h(but not 24 h) after hypoglycemia (Tkacs et al, 2000).
Figure 4 Total numbers of FJþ cells over time afterhypoglycemic insult. The time course of FJ appearance wasevaluated by counting all FJþ cells on 10 sections representing1 mm intervals through the rat forebrain, from 4.7 mm rostral tobregma to 4.3 mm caudal to bregma. After hypoglycemic insult,rats were euthanized and perfused at 8 h (n¼3); 12 h (n¼4);24 h (n¼6); 48 h (n¼3); 72 h (n¼4); 5 days (n¼6); 10days (n¼4). All hypoglycemic rats were included, whethercomplete blood glucose records were available during hypo-glycemia. The data are displayed as mean7s.d.
Table 2 Rostrocaudal distribution of FJ+ cells in experiment 2A
Atlas level relativeto bregma
Number of FJ+ cells(mean7s.d.)
4.7 44.4743.83.7 54.0768.72.7 50.7750.01.7 32.6735.20.7 21.9731.3
�0.3 12.1716.6�1.3 10.2715.0�2.3 11.4716.7�3.3 11.2712.4�4.3 14.9719.5
Cells were counted on sections representing 1.0 mm intervals from 4.7 mmanterior to bregma to 4.3 mm posterior to bregma.
Figure 5 FJþ cell counts are correlated with average plasmaglucose during hypoglycemic insult. Linear regression analysisindicates a statistically significant relationship between depth ofhypoglycemia and extent of FJ staining with r¼0.595,P¼0.0022. Dashed lines show 95% confidence intervals.This analysis included all rats that had complete blood glucoserecords during the time of hypoglycemia and excluded two ratsthat were statistical outliers with total FJ cell counts of 902 cellsand 1,149 cells.
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1651
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655

As we found in brains collected 48 h after insult, FJstaining did not colocalize with NeuN or GFAP inbrains collected at earlier time points (8 or 12 h).
Cardiovascular measurements during hypoglycemia:To evaluate the extent of cardiovascular perturba-tions during hypoglycemic insult, blood pressureand heart rate were measured in two rats followingour usual protocol. Overnight-fasted rats wereadministered regular human insulin, 4 U/kg (intra-venous). Hypoglycemia was maintained between30 and 35 mg/dL for 90 mins, and the hypoglycemicepisode was terminated with 50% dextrose andfood. In both rats, hypoglycemia was accompaniedby bradycardia with heart rate decreasing from thecontrol level of 318 beats/min to 240 and 260 beats/min during hypoglycemia. Mean arterial pressurewas approximately 110 mm Hg in each rat andremained constant in both rats for the duration ofthe hypoglycemic episode.
Discussion
Reports of rodent HAAF models generally showblunted hormone responses to hypoglycemia occur-ring within 24 h after recurrent hypoglycemia (2 to 7episodes of prior hypoglycemia) but not after asingle hypoglycemic episode. In our previous study,we observed apparent blunting of hormone res-ponses to repeated hypoglycemia delivered at 48 hintervals; however, our study did not include acontrol group of pair-fasted rats. In the presentstudy, we compared hormone responses to hypo-glycemia between rats with a single episode ofmoderate hypoglycemia and pair-fasted controlsexposed to identical experimental conditions asthe hypoglycemic rats. We assessed hormone res-ponses 48 h after the initial insult to allow 1 day forrecovery from the initial insult followed by a nightof fasting. Despite the fact that we only exposed therats to one episode of hypoglycemia and we waited48 h for recovery, we still found significant bluntingof epinephrine responses to hypoglycemia after asingle episode of moderate hypoglycemia. Thisrodent model of HAAF is unique in that suppressedepinephrine responses are seen after a singleepisode of moderate hypoglycemia, rather thanmultiple episodes used in other models (Evanset al, 2001; Sivitz et al, 2001; Inouye et al, 2002;Flanagan et al, 2003).
In this model, we also aimed to study neuro-pathologic consequences of a single episode ofhypoglycemia using the histologic marker FJ. Thedata presented here show positive staining with FJafter a single 75 mins episode of moderate, hypo-glycemia that did not cause loss of consciousness.Fluoro-Jade B has been used as a marker of neurondeath in a variety of brain injury models includingischemia, traumatic brain injury, and excitotoxicity(Sato et al, 2001; Butler et al, 2002). A previous
study using FJ to assess brain injury after hypo-glycemic coma indicated that FJ labels neurons inthe cortex, hippocampus, and subiculum after30 mins of cerebral isoelectricity (Suh et al, 2003).In agreement with our model, in young rats,recurrent hypoglycemia without loss of conscious-ness (3 episodes averaging 40 mg/dL) was reportedto induce FJ staining in a few neurons in the sagittaland temporal cortex (averaging 17 and 6 neurons/section, respectively) at a mid-hypothalamic level(3 mm posterior to bregma) (Yamada et al, 2004).
The present data are consistent with these find-ings, but indicate that cells in far rostral corticalregions may be more vulnerable to hypoglycemiathan previous reports indicated. Specifically, wefound a highly reproducible pattern of FJ staining inthe medial prefrontal cortex, including the pre-limbic, infralimbic, anterior cingulate regions; theorbital cortex, including the medial, ventral, andlateral orbital regions; and the piriform cortex. TheFJ-stained cells appear to be neurons, although themorphology changes significantly from 12 to 48 hafter hypoglycemic insult. Early time points show FJextending well into cell processes, while at latertime points the FJþ cells appear smaller thansurrounding neurons, with retracted or absentprocesses. This is consistent with other descriptionsof FJ staining after traumatic brain injury orexcitotoxicity (Sato et al, 2001; Gilliams-Franciset al, 2003; Singleton and Povlishok, 2004). Sincethe initial description of FJ as a marker of neurondeath, there have been only two reports of FJstaining nonneuronal cells in rat CNS injury models.In one report, glial staining was restricted to thespinal cord, and in the other report, glial FJ stainingin the CNS was observed weeks after the injury(Butler et al, 2002; Anderson et al, 2003) Thus,under conditions similar to ours, FJ staining hasbeen used as a valid and reliable marker of neurondeath.
The fact that the FJþ cells do not react withantibodies to neuron markers such as NeuN orglutamate receptor subunits may indicate that theFJ-stained cells in this study are not neurons.However, it is possible that NeuN is rapidly clearedduring a cell death process, because proteolysis is anearly event after injury in neurons (Lee et al, 2003).Although cells undergoing apoptosis may retainsome neuronal markers until late stages of the deathprocess, the same has not been shown for neuronsdying by necrosis or autophagy. Further work isneeded to confirm the neuronal nature of the FJ-stained cells after this moderate hypoglycemic insult.
The mechanism of cell death is not indicated bythese results, because FJ, like silver staining, markscells dying from either apoptosis or necrosis (Satoet al, 2001). We did not detect labeled cells afterhypoglycemic insult by using immunohistochem-ical probes of caspase and calpain pathways.However, these do not represent the only modes ofneuron death as cathepsins are also implicated in
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1652
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655

neuron death, for example, after cerebral ischemia(Seyfried et al, 1997; Hill et al, 1997; Unal-Ceviket al, 2004). Because the number of dying cells issmall in this model, it is not likely that methods forquantifying neuron loss (such as Nissl staining) willdetect significant depletion of neurons. The appear-ance of HSP70 immunoreactivity in the orbital andmedial prefrontal cortex neurons after hypoglycemicinsult supports the possibility that the FJ stainingindicates dying neurons, and points to the relativevulnerability of these brain regions to moderatehypoglycemic insult. However, the lack of FJstaining in the hippocampus at any time point afterhypoglycemia indicates that our insult was sub-threshold, relative to the effects of hypoglycemiccoma (Suh et al, 2003). The extent of cell deathsignificantly correlated with the depth of hypogly-cemia during the insult. Based on the regressionequation developed, we would predict that therewould be little to no cell death if hypoglycemia iskept higher than 40 mg/dL. In fact, we have not seenFJ staining in rats subjected to hypoglycemia at45 mg/dL for 2 h (data not shown).
The depth of hypoglycemia during the insult isassociated with cardiovascular alterations indicatedby moderate bradycardia. This is consistent withother reports showing bradycardia during eitheracute or chronic severe hypoglycemia in rats (Bryanand Pelligrino, 1988; Bryan et al, 1994). At the levelof hypoglycemia (o2.0 mmol/L) achieved in ourmodel, it is also assumed that there would be anabrupt increase in cerebral blood flow, as shown byother investigators (Bryan et al, 1994; Horinaka et al,1997; Choi et al, 2001). Using nuclear magneticresonance spectroscopy to measure brain glucoselevels, the cerebral blood flow increase duringhypoglycemia occurs at a plasma glucose thresholdof 2.0 mmol/L, at which point the brain glucoseconcentration approaches zero (Choi et al, 2001).However, even at very low brain glucose levels,astrocyte-derived lactate would contribute to main-tenance of cerebral function for at least 30 mins(Ransom and Fern, 1997; Pellerin and Magistretti,2004). Because our rats were responsive to stimu-lation during the entire hypoglycemic episode, itis likely that some substrate (glucose and/orlactate) was available throughout the 75 mins ofhypoglycemia.
In our experiments, FJþ cells are seen reprodu-cibly when moderate/severe hypoglycemia is main-tained below 40 mg/dL (2.2 mmol/L) for at least60 mins. However, the global reduction in energysubstrate does not explain the specific vulnerabilityof neurons in the medial prefrontal, orbital, andpiriform cortices observed in this study. Onepossible explanation for this vulnerability is thatthese neurons may be activated during hypoglyce-mia, accelerating their rate of energy depletion. Arecent positron emission tomography (PET) studyindicates that, in humans studied during hypogly-cemia, portions of the medial prefrontal and orbital
cortex were activated, while the remaining cere-brum, brainstem, and cerebellum showed decreasedactivity (Teves et al, 2004). Hypoglycemic activationof homologous cortical regions in rats could predis-pose neurons to rapid energy depletion and neurondeath.
Surprisingly, we did not observe FJ staining in thearcuate nucleus of the hypothalamus, where wepreviously identified TUNEL staining and alteredgene expression after hypoglycemic insult (Tkacset al, 2000). The reversibility of hypoglycemicDNA damage in neurons has been shown in vitro,as striatal neurons exposed to aglycemia for 24,48, or 72 h show increased TUNEL staining thatreverses when the neurons are returned to glucose-containing culture medium (McDermott et al, 2003).In fact, even though we previously showed evidencefor hypoglycemia-induced TUNEL staining in thehypothalamic arcuate nucleus cells (Tkacs et al,2000), neither FJþ nor activated caspase-3-positivecells were found in the arcuate. This suggests thatsome minor DNA fragmentation may occur butthis does not necessarily lead to cell death in thehypothalamus.
Future neuropathologic studies of noncoma hy-poglycemia are needed to identify the phenotype ofthe cells positive for FJ and other injury markers.In addition, the location of these vulnerable cellsmay be significant to the well-described deficitsin hypoglycemia responses of diabetic patients.Patients with recurrent hypoglycemia have bluntedor absent warning signs of hypoglycemia, that is,they suffer from hypoglycemia unawareness (Gerichet al, 1991). Given the human PET data showingmedial prefrontal and orbital cortex activationduring hypoglycemia, it is possible that these brainregions participate in hypoglycemia awareness, theconscious perception of the symptoms engenderedby autonomic activation (including palpitations,hunger, sweating, tingling) (Teves et al, 2004).Alternatively, functional imaging in humans andneuroanatomic tracing studies in rats have consis-tently suggested a role of medial prefrontal cortex invisceral sensation and regulation (Terreberry andNeafsey, 1983; Jansen et al, 1997; Ter Horst andPostema, 1997; King et al, 1999; Buijs et al, 2001;Westerhaus and Loewy, 2001; Owens and Verberne,2001; Floyd et al, 2001; Cameron and Minoshima,2002). Thus, the loss of neurons in the medialprefrontal and orbital cortex may be one elementcontributing to reduced compensatory adreno-medullary responses to hypoglycemia in our model.
Many, if not most, children and adults with type 1diabetes mellitus sustain single or multiple episodesof hypoglycemia at the level we use to inducehypoglycemic insult in this rodent study. Thepreponderance of the evidence from studies ofdiabetic patients who experience repeated moderatehypoglycemia shows sustained preservation ofcognitive function and gross brain integrity. Thisimplies that neuroprotective mechanisms, such as
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1653
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655

the presence of the ATP-sensitive potassium chan-nel, prevent large-scale neuron death during hypo-glycemia (Mobbs et al, 2001). However, recurrenthypoglycemia can result in long-term reductions inhypoglycemia awareness and compensatory auto-nomic responses that are not always fully reversiblewith avoidance of hypoglycemia (Mitrakou et al,1993). Our results using a sensitive marker of celldeath show that there are vulnerable cells, whichmay be neurons, in the medial prefrontal, orbital,and piriform cortex, that die after a single episode ofmoderate hypoglycemia. It remains to be determinedwhether persistent deficits such as hypoglycemiaawareness and counterregulatory responses result,in part, from a selective vulnerability of these brainregions to hypoglycemic injury.
Acknowledgements
The authors gratefully acknowledge the experttechnical assistance of Antoinette Moralishvili. Theauthors thank Jason Davis for advice on Fluoro-JadeB staining, Dr Hilaire Thompson for dedicatedassistance with photomicrographs, and Dr TracyMcIntosh for helpful discussions related to modelsof neuron death. The authors thank Dr Robert Simanfor the generous gift of the antibodies to calpain- andcaspase-cleaved spectrin. The radioimmunoassayswere performed in the core facility of the Universityof Pennsylvania Diabetes Endocrinology ResearchCenter (DK19525).
References
Anderson KJ, Fugaccia I, Scheff SW (2003) Fluoro-jade Bstains quiescent and reactive astrocytes in the rodentspinal cord. J Neurotrauma 20:1223–31
Auer RN, Wieloch T, Olsson Y, Siesjo B (1984) Thedistribution of hypoglycemic brain damage. ActaNeuropathol 64:177–91
Bi X, Chang V, Siman R, Tocco G, Baudry M (1996)Regional distribution and time-course of calpainactivation following kainate-induced seizure activityin adult rat brain. Brain Res 726:98–108
Bryan RM, Eichler MY, Johnson TD, Woodward WT,Williams JL (1994) Cerebral blood flow, plasma cate-cholamines, and electroencephalogram during hypo-glycemia and recovery after glucose infusion.J Neurosurg Anesthesiol 6:24–34
Bryan Jr RM, Pelligrino DA (1988) Cerebral blood flowduring chronic hypoglycemia in the rat. Brain Res475:397–400
Buijs RM, Chun SJ, Niijima A, Romijn HJ, Nagai K (2001)Parasympathetic and sympathetic control of pancreas:a role for the suprachiasmatic nucleus and otherhypothalamic centers that are involved in the regula-tion of food intake. J Comp Neurol 431:405–23
Butler TL, Kassed CA, Sanberg PR, Willing AE, Penny-packer KR (2002) Neurodegeneration in the rat hippo-campus and striatum after middle cerebral arteryocclusion. Brain Res 929:252–60
Cameron OG, Minoshima S (2002) Regional brain activa-tion due to pharmacologically induced adrenergicinteroceptive stimulation in humans. Psychosom Med64:851–61
Choi I-Y, Lee S-P, Kim S-G, Gruetter R (2001) In vivomeasurements of brain glucose transport using thereversible Michaelis–Menten model and simultaneousmeasurements of cerebral blood flow changes duringhypoglycemia. J Cereb Blood Flow Metab 21:653–63
Cryer PE (2001) Hypoglycemia-associated autonomicfailure in diabetes. Am J Physiol 281:E1115–21
DCCT (1993) The effect of intensive treatment of diabeteson the development and progression of long-termcomplications in insulin-dependent diabetes mellitus.N Engl J Med 329:977–86
DCCT (1997) Hypoglycemia in the diabetes control andcomplications trial. Diabetes 46:271–86
De Vries MG, Lawson MA, Beverly JL (2004) Dissociationof hypothalamic noradrenergic activity and sympatho-adrenal responses to recurrent hypoglycemia. Am JPhysiol Regul Integrative Comp Physiol 286:R910–5
Evans SB, Wilkinson CW, Bentson K, Gronbeck P, ZavoshA, Figlewicz DP (2001) PVN activation is suppressed byrepeated hypoglycemia but not antecedent corticoster-one in the rat. Am J Physiol 281:R1426–36
Flanagan DE, Keshavarz T, Evans ML, Flanagan S, Fan X,Jacob RJ, Sherwin RS (2003) Role of corticotrophin-releasing hormone in the impairment of counterregu-latory responses to hypoglycemia. Diabetes 52:605–13
Floyd NS, Price JL, Ferry AT, Keay KA, Bandler R (2001)Orbitomedial prefrontal cortical projections to hy-pothalamus in the rat. J Comp Neurol 432:307–28
Gerich JE, Mokan M, Veneman T, Korytkowski M,Mitrakou A (1991) Hypoglycemia unawareness. EndocrRev 12:356–71
Gilliams-Francis KL, Quaye AA, Naegele JR (2003) PARPcleavage, DNA fragmentation, and pyknosis duringexcitotoxin-induced neuronal death. Exp Neurol184:359–72
Hill IE, Preston E, Monette R, MacManus JP (1997) Acomparison of cathepsin B processing and distributionduring neuronal death in rats following global ischemiaor decapitation necrosis. Brain Res 751:206–16
Hopkins KJ, Wang G-J, Schmued LC (2000) Temporalprogression of kainic acid induced neuronal andmyelin degeneration in the rat forebrain. Brain Res864:69–80
Horinaka N, Artz N, Jehle J, Takahashi S, Kennedy C,Sokoloff L (1997) Examination of potential mechanismsin the enhancement of cerebral blood flow by hypogly-cemia and pharmacological doses of deoxyglucose.J Cereb Blood Flow Metab 17:54–63
Inouye K, Shum K, Chan O, Mathoo JMR, Matthews SG,Vranic M (2002) Effects of recurrent hyperinsulinemiawith and without hypoglycemia on counterregulationin diabetic rats. Am J Physiol 282:E1369–79
Jansen ASP, Hoffman JL, Loewy AD (1997) CNS sitesinvolved in sympathetic and parasympatheticcontrol of the pancreas: a viral tracing study. BrainRes 766:29–38
King AB, Menon RS, Hachinski V, Cechetto DF (1999)Human forebrain activation by visceral stimuli. J CompNeurol 413:572–82
Lee DR, Helps SC, Gibbins IL, Nilsson M, Sims NR (2003)Losses of NG2 and NeuN immunoreactivity but notastrocytic markers during early reperfusion followingsevere focal cerebral ischemia. Brain Res 989:221–30
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1654
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655

McDermott CJ, Bradley KN, McCarron JG, Palmer AM,Morris BJ (2003) Striatal neurones show sustainedrecovery from severe hypoglycaemic insult. J Neuro-chem 86:383–93
Mitrakou A, Fanelli C, Veneman T, Perriello G, CalderoneS, Platanisiotis D, Rambotti A, Raptis S, Brunetti P,Cryer P, Gerich J, Bolli G (1993) Reversibility ofunawareness of hypoglycemia in patients with insuli-nomas. N Engl J Med 329:834–9
Mobbs CV, Kow L-M, Yang X (2001) Brain glucose-sensingmechanisms: ubiquitous silencing by aglycemia versushypothalamic neuroendocrine responses. Am J PhysiolEndo Metab 281:E649–54
Munoz A, Lopez A, Caruncho HJ, Guerra MJ, Labandeira-Garcıa JL (2001) Long-term cortical atrophy afterexcitotoxic striatal lesion: Effects of intrastriatal fetal-striatum grafts and implications for Huntington Disease.J Neuropathol Exp Neurol 60:786–97
Ouyang Y-B, He Q, Li P-A, Janelidze S, Wang G-X, Siesj B(2000) Is neuronal injury caused by hypoglycemiccoma of the necrotic or apoptotic type? NeurochemRes 25:661–7
Owens NC, Verberne AJM (2001) Regional haemodynamicresponses to activation of the medial prefrontal cortexdepressor region. Brain Res 919:221–31
Paranjape SA, Briski KP (2004) Recurrent insulin-inducedhypoglycemia causes site-specific patterns of habitua-tion or amplification of CNS neuronal genomic activa-tion. Neurosci 130:957–70
Paxinos G, Watson C (1998) The rat brain in stereotaxiccoordinates. San Diego: Academic Press
Pellerin L, Magistretti PJ (2004) Neuroenergetics: callingupon astrocytes to satisfy hungry neurons. Neuroscien-tist 10:53–62
Ransom BR, Fern R (1997) Does astrocytic glycogenbenefit axon function and survival in CNS white matterduring glucose deprivation. Glia 21:134–41
Sato M, Chang E, Igarashi T, Noble LJ (2001) Neuronalinjury and loss after traumatic brain injury: time courseand regional variability. Brain Res 917:45–54
Schmued LC, Hopkins KJ (2000) Fluoro-jade B: a highaffinity fluorescent marker for the localization ofneuronal degeneration. Brain Res 874:123–30
Seyfried D, Han Y, Zheng Z, Day N, Moin K, Rempel S,Sloane B, Chopp M (1997) Cathepsin B andmiddle cerebral artery occlusion in the rat. J Neurosurg87:716–23
Shum K, Inouye K, Chan O, Mathoo JMR, Bilinski D,Matthews SG, Vranic M (2001) Effects of ante-cedent hypoglycemia, hyperinsulinemia, and excess
corticosterone on hypoglycemic counterregulation. AmJ Physiol 281:E455–65
Singleton RH, Povlishok JT (2004) Identification andcharacterization of heterogeneous neuronal injury anddeath in regions of diffuse brain injury: evidence formultiple independent injury phenotypes. J Neurosci24:3543–53
Sivitz WI, Herlein JA, Morgan DA, Fink BD, Phillips BG,Haynes WG (2001) Effect of acute and antecedenthypoglycemia on sympathetic neural activity andcatecholamine responsiveness in normal rats. Diabetes50:1119–25
Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, GumE, Liu J, Swanson RA (2003) Hypoglycemic neuronaldeath and cognitive impairment are prevented bypoly(ADP-ribose) polymerase inhibitors administeredafter hypoglycemia. J Neurosci 23:10681–90
Ter Horst GJ, Postema F (1997) Forebrain parasympatheticcontrol of heart activity: retrograde transneuronal virallabeling in rats. Am J Physiol 273:H2926–30
Terreberry RR, Neafsey EJ (1983) Rat medial frontal cortex:a visceral motor region with a direct projection to thesolitary nucleus. Brain Res 278:245–9
Teves D, Videen TO, Cryer PE, Powers WJ (2004)Activation of human medial prefrontal cortex duringautonomic responses to hypoglycemia. Proc Natl AcadSci USA 101:6217–21
Tkacs NC, Dunn-Meynell AA, Levin BE (2000) Presumedapoptosis and reduced arcuate nucleus neuropeptide Yand pro-opiomelanocortin mRNA in non-coma hypo-glycemia. Diabetes 49:820–6
Unal-Cevik I, Kilinc M, Can A, Gursoy-Ozdemir Y, DalkaraT (2004) Apoptotic and necrotic death mechanisms areconcomitantly activated in the same cell after cerebralischemia. Stroke 35:2189–94
Watson RE, Wiegand SJ, Clough RW, Hoffman GE (1986)Use of cryoprotectant to maintain long-term peptideimmunoreactivity and tissue morphology. Peptides7:155–9
Westerhaus MJ, Loewy AD (2001) Central representationof the sympathetic nervous system in the cerebralcortex. Brain Res 903:117–27
Yamada KA, Rensing N, Izumi Y, De Erausquin GA, GazitV, Dorsey DA, Herrera DG (2004) Repetitive hypogly-cemia in young rats impairs hippocampal long-termpotentiation. Pediatric Res 55:372–9
Zhang C, Siman R, Xu YA, Mills AM, Frederick JR,Neumar RW (2002) Comparison of calpain and caspaseactivities in the adult rat brain after transient forebrainischemia. Neurobiol Dis 10:289–305
Cortical Fluoro-Jade after hypoglycemiaNC Tkacs et al
1655
Journal of Cerebral Blood Flow & Metabolism (2005) 25, 1645–1655









![Molecular Imaging of Murine Intestinal Inflammation With 2-Deoxy-2-[18F]Fluoro-d-Glucose and Positron Emission Tomography](https://static.fdokumen.com/doc/165x107/6344fffc596bdb97a908b96f/molecular-imaging-of-murine-intestinal-inflammation-with-2-deoxy-2-18ffluoro-d-glucose.jpg)




![Molecular Imaging of Murine Intestinal Inflammation With 2-Deoxy-2-[ 18F]Fluoro- d-Glucose and Positron Emission Tomography](https://static.fdokumen.com/doc/165x107/6344fff26cfb3d4064097a1a/molecular-imaging-of-murine-intestinal-inflammation-with-2-deoxy-2-18ffluoro-.jpg)





