
complete dissertation.pdf
147
Pathogenesis of neurotoxicity and role of signaling in the efficacy of anticancer agents Abolfazl Avan A flower can grow even in a hostile desert! With the optimal treatment, the cancer patient can also live a happy life!
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of complete dissertation.pdf

Pathogenesisofneurotoxicityandroleofsignalingintheefficacyofanticanceragents
AbolfazlAvan
A flower can grow even in a hostile desert! With the optimal treatment,
the cancer patient can also live a happy life!

ThestudiesdescribedinthepresentthesiswereperformedattheLaboratoryofMedicalOncology, Department ofMedical Oncology, VU UniversityMedical Center Amsterdam,Amsterdam,theNetherlands.
AbolfazlAvan’sPhDThesisVUUniversityMedicalCenter,Amsterdam,TheNetherlandsDigitalcopy:http://dare.ubvu.vu.nl/handle/1871/53558
ThefrontandbackcoverswerephotographedanddesignedbyAbolfazlAvan.ThelayoutwaspreparedbyAbolfazlAvanwiththePagesv.5.6,AppleInc©.186pages,about38,000wordsbodytext,inadditionto18tables,32figures,and732citations.TextFont:Candara,size9,spacing1line.ThecostofprintingwassponsoredbyVrijeUniversiteit,GreinerBio-One®,andABN-AMRObank.PrintedbyOffPage,Amsterdam,TheNetherlands
Copyright © 2015 by Abolfazl Avan, Amsterdam, the Netherlands. All rights reserved. Publishedarticleswerereproducedandreprintedwithpermission.Thereproduction,storage,transmission,orusageofthisbookinwholeorinpartinanymannerisprohibitedwithoutthewrittenpermissionoftheauthor,orwhenappropriatethepublisherofthearticles.
ISBN978-94-6182-628-2

VRIJEUNIVERSITEIT
Pathogenesisofneurotoxicityandroleofsignalingintheefficacyofanticanceragents
ACADEMISCHPROEFSCHRIFT
terverkrijgingvandegraadDoctoraandeVrijeUniversiteitAmsterdam,opgezagvanderectormagnificus
prof.dr.V.Subramaniam,inhetopenbaarteverdedigen
tenoverstaanvandepromotiecommissievandeFaculteitderGeneeskunde
opmaandag16november2015om11.45uurindeaulavandeuniversiteit,
DeBoelelaan1105
door
AbolfazlAvangeborenteMashhad,Iran

promotor: prof.dr.G.J.Peterscopromotor: dr.E.Giovannetti

Membersofthereadingcommittee:
dr.J.Buterdr.T.U.Hoogenraaddr.J.R.Kroepdr.T.J.Postmaprof.dr.E.F.Smitprof.dr.T.Würdinger

Human beings are members of a whole, In creation of one essence and soul. If one member is afflicted with pain, Other members uneasy will remain. If you've no sympathy for human pain, The name of human you cannot retain!
Iranian poet Sa'adi
بنی آدم اعضای یکدیگرند که در آفرينش ز یک گوهرند
چو عضوى به درد آورد روزگار دگر عضو ها را نماند قرار
تو کز محنت دیگران بی غمینشاید که نامت نهند آدمی
This poem from the 13th century became a motto and decorates the gate of the United Nations building entrance
Rhyming translation by M. Aryanpoor
… whoever saves a soul, it is as if he had saved entire mankind … Quran, (chapter 5: verse 32)

Contents (Avan’sdissertation)
ContentsCHAPTER
1Introductionandoutl ine Pages
1-10
Part 1: Drug-induced Neurotoxicity: Pathogenesis and TreatmentCHAPTER
2Platinum-inducedneurotoxicityandpreventivestrategies: past, present, andfuture
Pages13-44
CHAPTER3
Oxal iplatin, bortezomibandepothi lone-B inducedneurotoxicity: characterizationandprotection
Pages45-54
CHAPTER4
Calcium/magnesiuminfusionforoxal iplatin- inducedneuropathy: protectiveornot?
Pages55-58
Part 2: Signaling and the Efficacy of Cancer TreatmentCHAPTER
5RoleofAkts ignal ing inresistancetoDNA-targetedtherapy Pages
61-84
CHAPTER6
Phospho-Aktoverexpression is prognosticandcanbeusedtotai lorthesynergist ic interactionofthenovel Akt inhibitorperifosinewithgemcitabine inpancreaticcancer
Pages85-102
CHAPTER7
Predictiveroleofrepair enzymes intheeff icacyofcisplatincombinations inpancreaticandlungcancer
Pages103-114
CHAPTER8
Modulationofs ignal ingenhancestheeff icacyofthecombinationofsatraplatinanderlotinib
Pages115-130
CHAPTER9
AKT1andSELPpolymorphismspredictr iskofdevelopingcachexia inpancreaticcancerpatients
Pages131-146
CHAPTER10
SNPs inPI3K–PTEN–mTORandBrainMetastases inNSCLC Pages147-150
Discussion and SummaryCHAPTER
11Discussion Pages
153-162
CHAPTER12
SummaryinEnglish,Dutch,andPersian Pages163-169
CURRICULUMVITAE Pages170-175
ACKNOWLEDGEMENTS Pages176-177
�viiivii

Contents (Avan’sdissertation)
�ixviii

CHAPTER1IntroductionandOutline
CharacteristicsofneurotoxicAgentsandSignaling
1

Chapter1(Avan’sdissertation) Introduction
Neuropathy:anintricateconsequenceofplatinumagents
1.Generaloverview
Peripheral neuropathy due to chemotherapy is one of the major dose-limiting side effects of manyanticancermedicationsandtheircharacteristicsareoftenrelatedtoboththechoiceofanticancerdrugsandthecumulativedoses[1].Sinceperipheralnervoussystemisnotprotectedasefficientlyasthecentralnervoussystemfromexogenousagents,itismoresensitivetothetoxicaction.Theoverallincidenceofchemotherapy-inducedperipheralneuropathyisestimatedtoberoughly38%,whichmayvarydependingontypeanddurationoftreatmentaswellastheassessmenttool[2;3].Higherriskandincidenceofthissideeffect isreportedwithplatinumcompounds,vincaalkaloids,bortezomib,and/ortaxanesamongstothers[2;4].
Newinsightsintheunderstandingofthepathophysiology,pathogenesis,andindividualsusceptibilityfactorsmaypave theway for thedevelopmentof newprophylactic and therapeuticmeasures. Thepathophysiologyandpathogenesisofplatinum-inducedperipheralneurotoxicityhasbeenreviewedaswell as thepotencyofdifferentneurotropicandneuroprotectivedrugs (chapter2). Thesedatamayhelp to design novel models to develop alternative options in the treatment of platinum-inducedneuropathy.
Among more than 3000 platinum compounds, cisplatin, carboplatin, and oxaliplatin are the mostcommonly used in the clinic, while other analogs of platinum, including nedaplatin, lobaplatin, andheptaplatinareonlyapprovedinJapan,China,andSouthKorea,respectively[5].Theantitumoreffectofplatinumcompounds ismediatedvia the formationofDNA-platinumadducts,whichmayactivatesignalingpathwaysandapoptosis.Thisprocessmayinduceneurotoxicity.
1.2.History
In1965,Rosenbergserendipitouslydiscoveredcisplatinwhileevaluatingtheeffectofelectricfieldsonthecelldivisionofbacteria[6].Later,in1969,itsantineoplasticcharacteristicswereidentified[7],andintheearly70s,itwasappliedinclinicaltreatment.Platinumcompoundshavebeenincreasinglyusedin routineoncological clinical practice, andare currently themainstayof cancer treatment. TheU.S.FoodandDrugAdministrationapprovedcisplatinforuseintesticularandovariancancersin1978andin the UK (and in several other European countries) in 1979. Insight intomechanisms of antitumoraction for cisplatin led to thedesignofnovelplatinumanalogswithbetterpharmacologicalprofilesandsafety,suchascarboplatinandoxaliplatin[5].OxaliplatinreceivedEuropeanapprovalin1996andapproval by theU.S. FoodandDrugAdministration in 2002.While the toxicityprofilediffers amongplatinumagents,peripheralneuropathyisacommonfeature,particularlywithcisplatinandoxaliplatin.
1.3.Pathophysiology
Peripheralneurotoxicityofplatinumcompoundsisrelatedtoseveralmolecularmechanisms,includingdorsalrootgangliacytotoxic inflammatorychanges,mitotoxicity,enhancedoxidativestress,voltage-gated ion(sodium/potassium/calcium)channeldysfunction, functional impairmentof ionchannelsofthe transient receptor potential family, induction of neuronal apoptosis in dorsal root ganglia, anddemyelination [8]. Accumulation of platinum products mainly in the dorsal root ganglia, as well asperipheral neuronsmay provoke chronic neuropathy. Since these neurons are post-mitotic and notdividing, theformationofDNAadducts isnot lethal,butresults inDNA-strandbreaks.TheextentofDNA crosslinks in dorsal root ganglia at specific cumulative dose corresponds to the degree ofneurotoxicity[9].Cisplatinproducesmoreadductsinthedorsalrootgangliacomparedtooxaliplatin,whichmayexplainitshigherneurotoxicity.Platinumadductsmayalsoaffectneurons,whilebrainandspinalcordareprotectedbythebloodbrainbarrier[9].
�2
1

Chapter1(Avan’sdissertation) Introduction
1.4.NeuroprotectionandTreatment
Several model systems have been used to study the nature of overall neurotoxicity and the effectof potential neuroprotective drugs. There are heterogeneous results on the efficacy ofneuroprotective agents (i.e. detoxicants, nerve growth factor stimulants, antioxidants, electrolytes,chelators,ionchannelmodulators,etc.),butdoseadjustmentand/ordrugwithdrawalseemtobethemosteffectiveandcommonlyusedtherapeuticagainstplatinum-inducedperipheralneurotoxicity. Inaddition, different antidepressants, anticonvulsants, and some other compounds have been testedagainst platinum-induced peripheral neurotoxicity (chapter 2, 3, and 4); however, only duloxetine isrecommendedfortherapeuticpurposes.
2.CharacteristicsofPlatinumanalogs
Cisplatin,carboplatin,andoxaliplatinarenowadaysinroutineclinicaluse.Thecharacteristicsofthesethreeplatinumcompoundsandanoveloralanalogarediscussedinthenextsections.
2.1.Cisplatin
Cisplatin,cis-diamminedichloroplatinum(Figure1.1)isaheavymetalcomplexcontainingacentralatomofplatinumsurroundedbytwochlorideatomsandtwoammoniamoleculesinthecisposition.Asfirstlinechoiceoftreatment,cisplatinisapprovedagainstmetastatictesticulartumors,ovariancarcinoma,advancedbladdercancer,andfrequentlyused,oftenincombination,againstheadandnecksquamouscellcancer,cancersofesophagus,malignantpleuralmesothelioma,non-smallcelllungcancer,cervix,endometrium,andosteogenicsarcomas.
Cisplatin ishighlymutagenicandcarcinogenic inboth in vitro and invivomodels[10].Theantitumorpropertiesofcisplatinareascribedtothe kinetics of its chloride ligand displacement reactions leading toDNAcrosslinks.ThiscausesDNAbending,whichleadstoimpedingofDNA replication, transcription and other nuclear functions, whichfinally interferes with cancer cell proliferation and tumor growth[5;11].Thenucleotideexcisionrepair,e.g.ERCC-1,istheprincipalrepairpathwayforremovalofcisplatin-adducts[12-14],butmayalsoinvolverecognitionofthedamagebyHighMobilityGroup(HMG),nonhistoneproteins, andmismatch repairproteins [15].Accordingly,DNA repairproduces resistance tocisplatin through the failure to recognize thecisplatin-DNA adduct and propagates a signal to the apoptoticmachinery.
A number of additional properties of cisplatin are now emerging,which confer drug resistance, including activation of several signaltransduction pathways, such as Akt, ABL, p53, and MAPK/JNK/ERKleading to apoptosis [16]. Cisplatin may activate or inhibit thesepathwaysthroughalterationofreceptororlipidmoleculesinthecellmembrane, regulation of protein kinases, or activation of the DNArepair pathways after DNA damage (reviewed in [5]). Cisplatinresistancecouldalsoarisefromdecreasedtumorbloodflow,reducedplatinumuptake, increasedefflux, intracellulardetoxification, e.g.by
glutathione,decreasedbinding(duetohighintracellularpH),DNArepair,decreasedmismatchrepair,defectiveapoptosis,antiapoptoticfactors,modulationofsignalingpathways,orpresenceofquiescentnon-cyclingcells [17].Extracellularenvironmentanddietmayalso influencecisplatinefficacy,ashighextracellular PH [18;19], presence of extracellular matrix proteins fibronectin, type IV collagen andlaminin[20],extracellulargamma-glutamyltransferase[21],andbicarbonate intake[22]maydecreasetheintakecisplatinbytumorcells.
�3
NH3
Pt
Cl
NH3Cl
Figure 1.1. Cisplatin, is a yellowto orange crystalline powderwith the molecular formulaPtCl2H6N2, and a molecularweight of 300.1. Cisplatin is aheavymetalcomplexcontaininga central atom of platinumsurrounded by two chlorideatoms and two ammoniamoleculesinthecisposition.Itissolubleinwaterorsalineat1mg/mLandindimethylformamideat24mg/mL.Ithasameltingpointof207°C.
1

Chapter1(Avan’sdissertation) Introduction
Some therapeutic interventions at different molecular levels have been identified to sensitize thecancercellstocisplatin(chapter3).Thediscoveryofnovelplatinummoleculeswithhigherspecificityfor the targets and better safety profile could also lead to a breakthrough in bypassing cisplatinresistance[23].
2.2.Carboplatin
Carboplatin(Figure1.2)wasintroducedinthe1980s,followingajointdrug development and screening process between industry andacademia. It has markedly less toxicity to the kidneys and nervoussystem at conventional doses than cisplatin and causes lessgastrointestinalproblems,while retainingcomparableantineoplasticactivity, particularly in ovarian cancers [2]. Carboplatin resistancecouldarisebysimilarmechanismsascisplatin[17].Nevertheless, themost recent Cochrane review comparing the toxicity of carboplatinversus cisplatin in combination with third-generation drugs foradvanced non-small cell lung cancer reported an almost two-timeshigherrateofneurotoxicityinthecarboplatingroup[24].
2.3.Oxaliplatin
Oxaliplatin, (trans-R,R-1,2-diaminocyclohexane) oxalate platinum (II)(Figure 1.3), is a third generation platinum drug with adiaminocyclohexane (DACH) entity, which is approved for the clinical treatment of colon cancer.Oxaliplatinproduces the same typeof inter- and 1,2-GG intrastrandcross-linksas cisplatin,buthasaspectrum of activity andmechanisms of action and resistance different from those of cisplatin andcarboplatin[5],suggestingadifferentmechanismofaction.Thecellularandmolecularaspectsofthemechanismofactionofoxaliplatinarenotyetcompletelyelucidated.
The pattern of oxaliplatin side effects, include neurotoxicity,hematologicaltoxicity,andgastrointestinaltracttoxicitycorrelatedwiththe cumulative-doseof oxaliplatin [25]. Theneurologicalmanifestationsof oxaliplatin-induced peripheral neuropathy may vary between asubacute transientperipheral sensoryneuropathy (i.e. paresthesias anddysesthesia in the extremities sometimes accompanied by muscularcramps)anda late-onsetneuropathy(i.e.deepsensory loss,ataxia,andfunctional impairment). The peripheral neuropathy of oxaliplatinmightbecausedby itsability todecreasebothNa+andK+currentsand thusmodifying the voltage-dependent ionic channels mainly by altering theexternalsurfacemembranepotential[26].
Compared to cisplatin and carboplatin, oxaliplatin is approved for anarrowerspectrumofcancers,includingnon-smallcelllungcancerandcolorectal cancers, and it is usually administered in combination withother anticancer agents, such as 5-fluorouracil, gemcitabine,topoisomeraseinhibitors,andtaxanes[27;28).
2.4.Satraplatin
Satraplatin (JM216; Figure 1.4) is the first oral platinum analog [29] having greater lipophilicitycomparedtocarboplatinandcisplatin[30],favorablebioavailability[29;31],andeffectivepenetrationthroughbloodbrain barrier comparable to that of carboplatin and cisplatin [30] plus a good safetyprofile (lackingnephrotoxicity andneurotoxicity) compared to cisplatin in vivo [32;33]. Someclinicalstudieshaveshowntheefficacyofsatraplatininchemonaivesmall-celllungcancerandadvancednon-
�4
OOPt
NH2
H2N O O
Figure 1.3. Oxaliplatin is a thirdgeneration platinum analogwith molecu lar formulaC8H14N2O4Pt, andamolecularmass of 397.2858 g/mol. Itfeatures the bidentate ligand1,2-diaminocyclohexaneinplaceof the two monodentateammineligands.
O
OPt
NH3
NH3
O
OFigure 1.2. Carboplatin is asecond generation platinumanalogwithmolecular formulaC6H12N2O4Pt,andamolecularmassof371.294g/mol. Ithasabidentate dicarboxylate ligandin place of the two chlorideligand.
1

Chapter1(Avan’sdissertation) Introduction
small cell lung cancer [34;35], as well as cervical cancer, hormonerefractoryprostatecancer,andbreastcancer[36-38].
3.Etiologyofplatinum-inducedneurotoxicityandresistance
3.1.Polymorphismsandmodulationofsignaling
Association of polymorphisms in some genes with platinum-inducedperipheral neuropathy is a matter of debate. There are controversialreportsaboutsomegenesthatmaycontributetotheincidenceand/orseverity of neuropathy in patients being treated with platinumanalogues (chapter 2). The genes, include ATP-binding cassette sub-familyBmember1(ABCB1),ATP-bindingcassettesub-familyBmember1or 2 (ABCC1, C2 or CG2), alanine-glyoxylate aminotransferase (AGXT),cyclin H (CCNH), catechol O-methyltransferase (COMT), excision repaircross-complementation group 1 (ERCC1) and ERCC2 (alias XPD,Xeroderma-Pigmentosumgroup-D), integrinbeta3 (ITGβ3),glutathioneS-transferases (e.g. GSTM1, GSTM3 and GSTT1), voltage-gated sodiumchannel genes (SCNAs), thiopurine S-methyltransferase (TPMT), and X-ray repair cross-complementing protein 1 (XRCC1). Although some data support the role of thementioned genetic variations in the presentation and severity of platinum-induced peripheralneurotoxicity,theheterogeneityoftheresultsdiverse(chapter2).
3.2.Roleofsignalingintreatmentefficacy
The Phosphatidylinositol 3/protein kinase B (PI3K/Akt) signal transduction pathway controls mosthallmarks of cancer, including metabolism, cell survival, cell cycle progression or regulation ofapoptosis, protein synthesis, motility and genomic instability by phosphorylation of the substrates(chapters 5-10). Aberrant activation of this pathway has been associated with the development ofcancer.TheAktpathwayservesasasurvivalpathway,meaningthatitsactivationinhibitsmajorstepsin cell death regulation and directly stimulates other survival pathways. Moreover, consecutiveevidence affirms overactivation of PI3K/Akt pathway in nearly one third of carcinomas. Given thepromotionofthemalignantphenotypeandimplicationofdrugresistanceduetoexcessiveactivationofAktcascade,modulationofthissurvivalpathwaymayenhancetheefficacyofchemoradiotherapy.Accordingly,differentAkt inhibitors,e.g.perifosineandMK-2206,orPI3Kmodulators,e.g.LY294002andwortmannin,havebeenusedinnumerouspreclinicalandclinicalstudiestoenhancethecytotoxiceffectsoftheDNA-targetedtreatmentstosensitizethetumortotheanticanceragent(chapters5,6).
3.3.Repairenzymesandplatinumefficacy
Antineoplasticactivityofplatinumcompoundsismainlymediatedviatheformationoftoxicplatinum-DNA adducts. Removal of these adducts, as the normal protective mechanism, causeschemoresistance. Nucleotide excision repair and mismatch repair system are the principal repairpathwaysforremovalofplatinum-DNAadducts.EnzymesinvolvedintherecognitionofDNAdamage,unwinding, subsequent excisionof thedamagednucleotides, and insertionof newdeoxynucleosidetriphosphates into theDNAareTranscriptionFactor IIH,XerodermaPigmentosumC,D, Fenzymes,excision-repaircross-complementinggroup1,DNApolymerasesδ andε,andligase1.Singlenucleotidepolymorphisms in any of these genes may affect the repair capacity and contribute to individualvariationsinchemotherapyresponse(chapters7-10).
�5
NH2
Pt
Cl
Cl NH3
OC
O
CH3
OCO
CH3
Figure1.4.Satraplatinisanorallyactiveplatinum-basedantineoplasticagent,withmolecularformulaC10H22N2O4Pt,andamolecularmassof500.277g/mol.
1

Chapter1(Avan’sdissertation) Introduction
5.Outlineofthethesis
Part1.Drug-inducedNeurotoxicity:PathogenesisandTreatment
5.1.Chapter2
Neurotoxicity is an undesirable consequence of platinum-based chemotherapy, which preventsadministration of the full efficacious dosage. It often leads to treatment withdrawal, affects thepatients’ quality of life, and sometimes is irreversible. Chapter 2 discusses available preclinical andclinical evidence of the pathogenesis and pathophysiology of platinum-induced peripheralneurotoxicityaswellasavailableneuroprotectiveandtherapeuticstrategiestoavertthissideeffect.Thesedatamayhelpto improveordevelopalternativeoptions inthetreatmentofplatinum-inducedneuropathy, along with in vitro models, and appropriate trials planning to find the best patient-orientedsolution.
5.2.Chapter3
Different models have been introduced to study chemotherapy-induced neurotoxicity. Chapter 3describes a method for evaluation of the neurotoxicity using the neurite outgrowth in PC12 ratpheochromocytoma cells. With this method, we investigated the neurotoxicity of oxaliplatin,bortezomib, and epothilone-B, and tested the potential neuroprotection of amifostine. We alsoinvestigatedthesuitabilityoftwomarkersofneuronaldifferentiation,cyclin-B2andBIRC5.
5.3.Chapter4
Calcium/magnesium infusion is one of the popular preventive strategies against oxaliplatin-inducedneurotoxicity.Chapter4discusseswhetherornotcurrentevidencesupportsitsefficacy.
Part2.SignalingandtheEfficacyofCancerTreatment
5.4.Chapter5
ActivationoftheAkt-survivalpathwayisamechanismofresistancetoDNA-targeteddrugs.Chapter5reviews the effect of common anticancer drugs, i.e. platinum agents, taxanes, antimetabolites, andtumorantibioticsonAktpathway,andwhetherAktinhibitorsmayenhancethecytotoxiceffectsoftheDNA-targeted treatments by antagonizing the Akt survival pathway and sensitizing tumors toanticanceragent.
5.5.Chapter6
ThereisincreasingevidenceofaconstitutiveactivationofAktinpancreaticcancer.Chapter6discussesthe therapeutic potential of the novel Akt inhibitor perifosine in combination with gemcitabine inpancreaticductal adenocarcinomacells, showing thatperifosinecan interferewithcellproliferation,induceapoptosis,reducemigration/invasion,andsynergisticallyinteractwithgemcitabineincellswithphospho-Aktoverexpression.
5.6.Chapter7
Increasing the formation and retention of platinum-DNA adducts by decreasing the DNA-repairenzymesmayboosttheantineoplasticeffectsofthetreatmentandoverallsurvival.Chapter7explorestheimportanceofprotein/mRNAexpression-analysis,aswellastheroleofpolymorphismintheDNArepairenzymesinnon-smallcelllungcancerandpancreaticductaladenocarcinoma.
5.7.Chapter8
Satraplatinistheonlyoralanalogamongplatinumagents.Chapter8discussesthepotentialsynergismbetween erlotinib, an epidermal growth factor receptor (EGFR) inhibitor and JM118, the activemetaboliteofsatraplatin.Forthispurpose,sevencancercelllineswereexaminedfortheexpressionof
�6
1

Chapter1(Avan’sdissertation) Introduction
Akt, Erk and p38, and cell cycle proteins, as well as cell cycle distribution, cell death, and adductformation.
5.8.Chapter9
Singlenucleotidepolymorphismsmaybeindicativeofashortersurvival.Chapter9explorestheroleofnovel predictive biomarkers for cachexia, as a direct cause of reduced quality of life and shortersurvival,inpancreaticductaladenocarcinoma.SELPandAKT1polymorphismsmaybepredictiveoftheriskofcachexiaanddeathinthistypeofcancer.
5.9.Chapter10
Singlenucleotidepolymorphismsmaypredictbrainmetastases innon-small-cell lungcancer.Chapter10discussestheroleofsinglenucleotidepolymorphisms inpredictingbrainmetastases innon-small-celllungcancer.
5.10.Chapter11
Chapter11discussestheresultspresentedinthisdissertation.
References
1. Balayssac D, Ferrier J, Descoeur J, Ling B, Pezet D, Eschalier A, Authier N: Chemotherapy-inducedperipheralneuropathies:fromclinicalrelevancetopreclinicalevidence.ExpertOpinDrugSaf2011;10:407-417.
2. Argyriou AA, Bruna J, Marmiroli P, Cavaletti G: Chemotherapy-induced peripheralneurotoxicity(CIPN):anupdate.CritRevOncolHematol2012;82:51-77.
3. CavalettiG,ZannaC:Currentstatusandfutureprospectsforthetreatmentofchemotherapy-inducedperipheralneurotoxicity.EurJCancer2002;38:1832-1837.
4. HershmanDL,LacchettiC,DworkinRH,LavoieSmithEM,BleekerJ,CavalettiG,ChauhanC,GavinP,LavinoA,LustbergMB,PaiceJ,SchneiderB,SmithML,SmithT,TerstriepS,Wagner-Johnston N, Bak K, Loprinzi CL: Prevention and management of chemotherapy-inducedperipheral neuropathy in survivors of adult cancers: American Society of Clinical Oncologyclinicalpracticeguideline.JClinOncol2014;32:1941-1967.
5. BoulikasT,PantosA,BellisE,ChristofiP:Designingplatinumcompoundsincancer:structuresandmechanisms.CancerTherapy2007;5:537-583.
6. RosenbergB,Vancampl,KrigasT:InhibitionofcelldivisioninEscherichiacolibyelectrolysisproductsfromaplatinumelectrode.Nature1965;205:698-699.
7. ROSENBERGB, VANCAMP L, Trosko JE,Mansour VH: Platinum compounds: a new class ofpotentantitumouragents.Nature1969;222:385-386.
8. Diezi M, Buclin T, Kuntzer T: Toxic and drug-induced peripheral neuropathies: updates oncauses,mechanismsandmanagement.CurrOpinNeurol2013;26:481-488.
9. GreggRW,MolepoJM,MonpetitVJ,MikaelNZ,RedmondD,GadiaM,StewartDJ:Cisplatinneurotoxicity: the relationship between dosage, time, and platinum concentration inneurologictissues,andmorphologicevidenceoftoxicity.JClinOncol1992;10:795-803.
10. SandersonBJ,FergusonLR,DennyWA:Mutagenicandcarcinogenicpropertiesofplatinum-basedanticancerdrugs.MutatRes1996;355:59-70.
11. PetersGJ,AvanA,RuizMG,OrsiniV,AvanA,GiovannettiE,SmitEF:Predictiveroleofrepairenzymes in theefficacyofCisplatincombinations inpancreaticand lungcancer.AnticancerRes2014;34:435-442.
12. Postel-Vinay S, Vanhecke E, Olaussen KA, Lord CJ, Ashworth A, Soria JC: The potential ofexploiting DNA-repair defects for optimizing lung cancer treatment. Nat Rev Clin Oncol2012;9:144-155.
13. SancarA:DNArepairinhumans.AnnuRevGenet1995;29:69-105.�7
1

Chapter1(Avan’sdissertation) Introduction
14. Kamileri I, Karakasilioti I, Garinis GA:Nucleotide excision repair: new trickswith old bricks.TrendsGenet2012;28:566-573.
15. Siddik SH: Mechanisms of action of cancer chemotherapeutic agents: DNA-interactivealkylating agents and antitumour platinum-based drugs; in Alison MR, (ed): The cancerhandbook.London,UK,NaturePublishingGroup,2002,pp1299-1313.
16. WangD,LippardSJ:Cellularprocessingofplatinumanticancerdrugs.NatRevDrugDiscov2005;4:307-320.
17. StewartDJ:Mechanismsof resistance to cisplatin and carboplatin. CritRevOncolHematol2007;63:12-31.
18. RaghunandN,GilliesRJ:pHanddrugresistanceintumors.DrugResistUpdat2000;3:39-47. 19. PrescottDM, CharlesHC, Poulson JM, PageRL, Thrall DE, Vujaskovic Z,DewhirstMW: The
relationship between intracellular and extracellular pH in spontaneous canine tumors. ClinCancerRes2000;6:2501-2505.
20. BerubeM,TalbotM,CollinC,Paquet-BouchardC,GermainL,GuerinSL,PetitclercE:Roleofthe extracellular matrix proteins in the resistance of SP6.5 uveal melanoma cells towardcisplatin.IntJOncol2005;26:405-413.
21. Pompella A, De T, V, Paolicchi A, Zunino F: Expression of gamma-glutamyltransferase incancercellsanditssignificanceindrugresistance.BiochemPharmacol2006;71:231-238.
22. Raghunand N, He X, van SR,Mahoney B, Baggett B, Taylor CW, Paine-Murrieta G, Roe D,BhujwallaZM,GilliesRJ:EnhancementofchemotherapybymanipulationoftumourpH.BrJCancer1999;80:1005-1011.
23. McKeageMJ:New-generationplatinumdrugsinthetreatmentofcisplatin-resistantcancers.ExpertOpinInvestigDrugs2005;14:1033-1046.
24. deCastriaTB,daSilvaEM,GoisAF,RieraR:Cisplatinversuscarboplatinincombinationwiththird-generationdrugsforadvancednon-smallcelllungcancer.CochraneDatabaseSystRev2013;8:CD009256.
25. PasettoLM,D'AndreaMR,RossiE,MonfardiniS:Oxaliplatin-relatedneurotoxicity:howandwhy?CritRevOncolHematol2006;59:159-168.
26. BenoitE,BrienzaS,DuboisJM:Oxaliplatin,ananticanceragentthataffectsbothNa+andK+channelsinfrogperipheralmyelinatedaxons.GenPhysiolBiophys2006;25:263-276.
27. RansonM, Thatcher N: Paclitaxel: a hope for advanced non-small cell lung cancer? ExpertOpinInvestigDrugs1999;8:837-848.
28. Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E: Cellular and molecularpharmacologyofoxaliplatin.MolCancerTher2002;1:227-235.
29. McKeageMJ,Mistry P,Ward J, Boxall FE, Loh S, O'Neill C, Ellis P, Kelland LR,Morgan SE,Murrer B, .: A phase I and pharmacology study of an oral platinum complex, JM216: dose-dependentpharmacokineticswithsingle-doseadministration.CancerChemotherPharmacol1995;36:451-458.
30. Marcus L, Murphy R, Fox E, McCully C, Cruz R, Warren KE, Meyer T, McNiff E, Balis FM,WidemannBC:Theplasmaandcerebrospinalfluidpharmacokineticsoftheplatinumanalogsatraplatin after intravenous administration in non-human primates. Cancer ChemotherPharmacol2012;69:247-252.
31. RicartAD,SarantopoulosJ,CalvoE,ChuQS,GreeneD,NathanFE,PetroneME,TolcherAW,PapadopoulosKP: Satraplatin, anoralplatinum,administeredonafive-dayevery-five-weekschedule:apharmacokineticandfoodeffectstudy.ClinCancerRes2009;15:3866-3871.
32. McKeageMJ,MorganSE,BoxallFE,MurrerBA,HardGC,HarrapKR:Lackofnephrotoxicityoforal ammine/amine platinum (IV) dicarboxylate complexes in rodents. Br J Cancer1993;67:996-1000.
33. McKeage MJ, Boxall FE, Jones M, Harrap KR: Lack of neurotoxicity of oralbisacetatoamminedichlorocyclohexylamine-platinum(IV) in comparison to cisplatin andtetraplatinintherat.CancerRes1994;54:629-631.
�8
1

Chapter1(Avan’sdissertation) Introduction
34. FokkemaE,GroenHJ,BauerJ,UgesDR,WeilC,SmithIE:PhaseIIstudyoforalplatinumdrugJM216 as first-line treatment in patients with small-cell lung cancer. J Clin Oncol1999;17:3822-3827.
35. Judson I, Cerny T, Epelbaum R, Dunlop D, Smyth J, Schaefer B, Roelvink M, Kaplan S,HanauskeA:PhaseIItrialoftheoralplatinumcomplexJM216innon-small-celllungcancer:anEORTCearlyclinicalstudiesgroupinvestigation.AnnOncol1997;8:604-606.
36. TrudeauM,StuartG,HirteH,DrouinP,PlanteM,BessetteP,DuludeH,LebwohlD,FisherB,Seymour L: A phase II trial of JM-216 in cervical cancer: anNCICCTG study.GynecolOncol2002;84:327-331.
37. LatifT,WoodL,ConnellC,SmithDC,VaughnD,LebwohlD,PeereboomD:PhaseIIstudyoforalbis (aceto)amminedichloro (cyclohexamine)platinum (IV) (JM-216,BMS-182751)givendailyx5inhormonerefractoryprostatecancer(HRPC).InvestNewDrugs2005;23:79-84.
38. Smith JW, McIntyre KJ, Acevedo PV, Encarnacion CA, Tedesco KL, Wang Y, Asmar L,O'ShaughnessyJA:ResultsofaphaseIIopen-label,nonrandomizedtrialoforalsatraplatininpatientswithmetastaticbreastcancer.BreastCancerResTreat2009;118:361-367.
�9
1

Chapter1(Avan’sdissertation) Introduction
�10
1

Chapter1(Avan’sdissertation) Introduction
�11
PART 1

Chapter1(Avan’sdissertation) Introduction
�12

CHAPTER2
Platinum-InducedNeurotoxicityandPreventiveStrategies:Past,Present,andFuture
AbolfazlAvan,1TjeerdJ.Postma,2CeciliaCeresa,3AmirAvan,1,4GuidoCavaletti,3ElisaGiovannetti,1GodefridusJ.Peters1*
Departmentsof1MedicalOncologyand2Neurology,VUUniversityMedicalCenter,Amsterdam,TheNetherlands;3DepartmentofSurgeryandInterdisciplinaryMedicine,
UniversityofMilano-Bicocca,Monza,Italy;4DepartmentofNewSciencesandTechnology,SchoolofMedicine,MashhadUniversityofMedicalSciences,Mashhad,Iran
TheOncologist2015;20:411-432
CME

Chapter2 TheOncologist2015;20:411-432
Platinum-inducedneurotoxicityandpreventivestrategies:past,present,andfuture
ABSTRACT:Neurotoxicityisaburdensomesideeffectofplatinum-basedchemotherapythatpreventsadministration of the full efficacious dosage and often leads to treatment withdrawal. Peripheralsensory neurotoxicity varies from paresthesia in fingers to ataxic gait, whichmight be transient orirreversible. Because the number of patients being treated with these neurotoxic agents is stillincreasing,theneedforunderstandingthepathogenesisofthisdramaticsideeffectiscritical.Platinumderivatives, suchas cisplatin and carboplatin, harmmainlyperipheral nervesanddorsal rootganglianeurons, possibly because of progressive DNA-adduct accumulation and inhibition of DNA repairpathways (e.g., extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase/stress-activatedproteinkinase,andp38mitogen-activatedproteinkinass),whichfinallymediateapoptosis.Oxaliplatin,with a completely different pharmacokinetic profile, may also alter calcium-sensitive voltage-gatedsodiumchannelkineticsthroughacalciumionimmobilizationbyoxalateresidueasacalciumchelatorandcauseacuteneurotoxicity.Polymorphismsinseveralgenes,suchasvoltage-gatedsodiumchannelgenesorgenesaffecting theactivityofpivotalmetal transporters (e.g.,organic cation transporters,organiccation/carnitine transporters,andsomemetal transporters, suchas thecopper transporters,and multidrug resistance-associated proteins), can also influence drug neurotoxicity and treatmentresponse.However,mostpharmacogeneticsstudiesneedtobeelucidatedbyrobustevidence.Thereare supportive reports about the effectiveness of several neuroprotective agents (e.g., vitamin E,glutathione, amifostine, xaliproden, and venlafaxine), but dose adjustment and/or drug withdrawalseem to be themost frequently usedmethods in themanagement of platinum-induced peripheralneurotoxicity.Todevelopalternativeoptionsinthetreatmentofplatinum-inducedneuropathy,studieson in vitro models and appropriate trials planning should be integrated into the future design ofneuroprotectivestrategiestofindthebestpatient-orientedsolution.TheOncologist2015;20:411–432
ImplicationsforPractice:Neurotoxicityisaburdensomesideeffectofplatinum-basedchemotherapythatpreventsadministrationofthefullefficaciousdosageandoftenleadstotreatmentwithdrawal.This review summarizes preclinical and clinical evidence of pathogenesis and pathophysiology ofplatinum-induced peripheral neurotoxicity, as well as available evidence of neuroprotective andtherapeutic strategies. These data may help to develop alternative options in the treatment ofplatinum-inducedneuropathy,studieson invitromodels,andappropriatetrialsplanningtofindthebestpatient-orientedsolution.
Introduction
Since the discovery of cisplatin in the mid-1960s, many platinum compounds (more than 3,000compounds) have been developed. Thirty-five of these compounds have exhibited adequatepharmacologicaladvantages(e.g.,reachingsufficientlyhighplasmalevelsnotassociatedwithcommontoxicities,suchasrenaltoxicityandthrombocytopenia)[1].Someofthemhavebeenregisteredorarebeingconsideredforregistrationfortreatmentofdifferentcancers,suchasthesecond(carboplatin,nedaplatin, tetraplatin, and iproplatin) and third (oxaliplatin, lobaplatin, heptaplatin, satraplatin, andLA-12)generation,usuallywithbettersafetyprofiles[2–4].
Despitetheefficacyofplatinumanalogsincancertreatment,serioussideeffects,especiallyperipheralsensory neurotoxicity, often prevent their administration at their full efficacious doses or mayconsiderablyaffectthequalityof lifeofcancerpatientsbeingtreatedwiththem[5,6].Cisplatinwasthefirstheavymetalusedinseveralkindsofsolidtumors,includinglung,ovary,testis,bladder,headandneck,andendometrium[7,8];mostpatientsdevelopasymptomaticneuropathy[9].Secondandthirdgenerationsofplatinumcompoundshaveemergedinattemptstoreducethetoxicityofcisplatin.Carboplatin,asecondgenerationofplatinumsusedtotreatovarian,non-smallcelllung,andrefractorytesticular cancers, was thought to be associated with a lower risk of developing neurotoxicity [9].However, themost recentCochrane reviewcomparing the toxicityof carboplatin versus cisplatin in
�14
CME

Chapter2 TheOncologist2015;20:411-432
combinationwiththird-generationdrugsforadvancednon-smallcell lungcancerreportedanalmosttwotimeshigherrateofneurotoxicityinthecarboplatingroup[10].Oxaliplatin,asawidelyusedthird-generationplatinumanalogapprovedforuseinthetreatmentofmetastaticcoloncancer,isreportedby the Food and Drug Administration to be responsible for more than 70% rate of symptomaticneurotoxicity with any severity [11] and often leads to treatment discontinuation [12–14]. In otherstudies, approximately 80% of colorectal cancer patients treated with oxaliplatin alone or incombinationwithotherchemotherapeuticsexperiencedneurotoxicity[15–17],andimpairmentmaybepermanent.Becausethenumberofpatientsbeingtreatedwithaneurotoxicagent is increasing, it isessential to understand the nature of such a problematic side effect. Furthermore, testing andvalidatingavailableprotectivestrategiesinpreclinicalandclinicalsettingsshouldbethenextstepsinovercomingplatinum-inducedperipheralneurotoxicity.
Clinicalfeaturesofneurotoxicity
Platinum drugs are almost always given in combination with other chemotherapy drugs and/orradiationthatmaybeneurotoxicintheirownright.Earlypresentationofperipheralneurotoxicitycanbe with numbness, tingling, or paresthesia in fingers and/or toes, a decreased distal vibratorysensitivity, and/or loss of ankle jerks [5]. Moreover, prolonged treatment may also affectproprioception,whichmayresultinataxicgait.
Oxaliplatin and cisplatin are the two most commonly used neurotoxic platinum agents. Platinum-inducedperipheralneurotoxicitycanpresentastwoclinicallydistinctsyndromes.Theacutetransientparesthesia in the distal extremities, which is only commonly seen with oxaliplatin, usually occurswithin the early phase of drug administration, whereas the chronic cumulative sensory neuropathycausesmore persistent clinical impairments [5]. The latter deteriorates with cumulative doses [18],followed by “coasting,” wherein symptoms worsen even months after treatment withdrawal.Furthermore,patientscandevelopLhermitte’ssyndrome,whichisashocklikesensationofparesthesiaradiating from the neck to the feet triggered by neck flexion. This phenomenon indicates theinvolvementofthecentripetalbranchofthesensorypathwaywithinthespinalcord[19].Neuropathycan also become irreversible. In a prospectivemulticenter study, Argyriou et al. [20] reported thatoxaliplatincanresult inanacuteandchronicrateofneuropathyin85%(169patientsof200)and73%(145patientsof200)ofpatients,respectively.
Hearing loss or ototoxicity is another progressive and irreversible adverse effect of platinumchemotherapy[21]withahighfrequencyofalmost88%[22],whichusuallypresentsbilaterallyandcanoccur during or years after treatment [23]. Nevertheless, the risk of ototoxicity may vary betweencisplatin,carboplatin,andoxaliplatintreatments,andcisplatinisbelievedtobethemostototoxicandoxaliplatin is believed to be the least [24]. In one study, 19%–77% of patients treated with cisplatindeveloped bilateral sensorineural hearing loss, and 19%–42% developed permanent tinnitus [25].Cisplatin accumulates in the cochlear tissue, forms DNA adducts, and causes inefficient anddysfunctionalproteinandenzymesynthesisleadingtoapoptosisofauditorysensorycells[26].
DiagnosisandEvaluation
The clinical diagnosis is generally not very difficult [27]. Nerve biopsies and neurophysiologicassessments are helpful for the examination of pathological and functional nerve damage (e.g.,demyelinating versus axonal pathology; abnormalities in nerve conduction studies, somatosensoryevoked potentials, magnetic resonance imaging, threshold tracking techniques, and quantitativesensorytesting)[27].Objectiveelectromyographyassessmentofmotornerveexcitabilityisasensitiveand specific endpoint of acute oxaliplatin-induced motor nerve hyperexcitability, which has theadvantageofbeingwidelyavailable[28,29].Additionally,thethresholdtrackingtechniqueisusedto
�15
2

Chapter2 TheOncologist2015;20:411-432
assess axonal excitability [30]. This technique allows the detection of sensory axonal dysfunctionbeforeclinicalsymptoms[18]andcanbeusedasapredictivemarkerfornervedysfunction.
Chemotherapy-inducedperipheralneurotoxicityistypicallyamultidisciplinarymedicalissue,leadingtodifferent terminology, measurement, clinical evaluation, and grading, precluding the reliability ofneurologicalassessment.However,standardizationisimproving.TheFunctionalAssessmentofCancerTherapy/Gynecologic Oncology Group-Neurotoxicity Scale, the FACT-Taxane scales, the PatientNeurotoxicityQuestionnaire, EuropeanOrganization forResearchandTreatmentofCancer (EORTC)quality of life questionnaire [QLQ] to assess chemotherapy-induced peripheral neuropathy, and theEORTCQLQC30questionnairearescoringsystemsthathavebeenusedforneurotoxicityassessmenttoquantifytheimpactofchemotherapy-inducedneurotoxicityonpatients’qualityoflife[31].Amongthe questionnaires, the EORTC questionnaires are widely used nowadays [32]. Among differentcommon toxicity criteria scales that are used for peripheral neurotoxicity assessment, the onedevelopedbytheEasternCooperativeOncologyGroupandNationalCancerInstitute(NCI-CTC)ismostwidelyused [19,33–35].Although the reliabilityofdifferentassessmentmethodshasbeen tested indifferent settings, there are vast discrepancies between patient perception and objective tools,particularly in intermediate grades [32, 36], which increase the need for a more effective andstandardizedmethod[19].
NatureofNeurotoxicity
Thepathophysiologyofplatinum-inducedperipheralneurotoxicityisnotcompletelyelucidated.Basedonavailabledata,platinumcompoundsmayactivelyenterthetumorandnormalcellsthroughorganiccationtransporters[37],organiccation/carnitinetransporters[38],andsomemetaltransporters,suchas the copper transporters [39, 40]. Platinum compounds can be excreted via platinum effluxtransporters (e.g., ATP7A, ATP7B, andMRP2) [41–45] (Figure 2.1). The platinum adducts are formed
�16
and/or DNA-protein cross-links with platinum, affecting DNAsynthesis in cancer cells [47] and mediating apoptosis [48].Extensive DNA repair is considered as a major mechanism ofchemotherapy resistance (Fig. 1), but efficient DNA repair canpossibly preventdevelopmentof neurotoxicity. In dividing tumorcells, the formation of DNA adducts is supposed to cause growthinhibition and cell kills, hence eliminating the tumor cells.
Platinum products accumulate in the dorsal root ganglia(DRG), which is the main target, and in peripheral neurons(Fig. 2). Because these cells are postmitotic and not dividing,the formationofDNAadducts isnot lethal, although theextentofDNAcross-links inDRGneuronsata specific cumulativedosestrongly correlates with the degree of neurotoxicity [49].Cisplatin produces approximately three timesmore adducts inthe DRG compared with oxaliplatin [50], which is consistentwith its higher neurotoxicity [12]. Platinum adducts probablycause axonal changes secondary to the neuronal damage [51],whereas brain and spinal cord are to some extent protected bythe blood-brain barrier (BBB) [52]. However, there are datashowingthatcisplatin crosses theBBBandcanaccumulatewhenrepeated dosages are given [53]. This can cause demyelinationand vacuolar changes in thewhitematter [54]. It is believed thatimpaired axonal voltage-gated sodium channels kinetics caninterferewithchannelkineticsbyoxalate (ametabolicproductofoxaliplatin) [55] and can also reduce sodium ions current [56].Sittl et al. [57] also showed that cooling in the presence ofoxaliplatin induced bursts of action potentials in myelinatedA but not unmyelinated C-fibers from human and mouseperipheral axons. Consequently, these alterations led to en-hanced resurgent and persistent current amplitudes in large,
but not small, diameter DRG neurons. Potassium channel block-ade and calcium chelation are also two other etiologic possi-bilities [56, 58–60]. Besides, oxidative stress and mitochondrialdysfunction are regarded as another probable etiology of theapoptosis [61]. Podratz et al. [62] showed that cisplatin mightinhibit mitochondrial DNA replication and cause mitochondrialvacuolizationanddegradation inDRGneurons invitroand invivo.These events can, to some extent, explain themechanism of theneurotoxic effect of platinum compounds.
PHARMACOGENETICSSingle-nucleotide polymorphisms (SNPs) may play a key rolein determining the induction of neurotoxicity, as well asapoptosis, because they may impair DNA repair pathways,including genes in base excision repair, nucleotide excisionrepair, mismatch repair, and double-strand break repairpathways [63] (Fig. 2). Moreover, SNPs can alter the drugmetabolism, cell cycle control, detoxification, or excretionpathways, which finally may lead to drug toxicities, e.g.,neurotoxicity. Several studies evaluated the pharmacogeneticassociation of SNPs with potential functional changes in theencodedproteinthatplayarole indrugdisposition,metabolism,and detoxification, DNA repair, and cancer-cell resistance andthat may lead to platinum peripheral neurotoxicity [19]. How-ever, the results are scattered and diverse with several meth-odological flaws, including small sample size, retrospectivestudy design, and the implementation of a post hoc analysis ofoncology-based databases of different, not preplanned sizes aswell as lackingaprestudyhypothesisbasedontheknownroleofthe investigated targets in the peripheral nervous system and
Figure 1. Effects of platinated compounds (Pt) and potentialmechanisms of action. Ptmay enter tumor cells (Pt influx) via copper transporter,organic cation transporters, and organic cation/carnitine transporters or by passive diffusion. DNA-platinum adducts block DNA replication,transcription,andothernuclear functionsandalsoactivate signal transductionpathways,which result in apoptosis andnecrosis in tumorcells. Individing tumor cells, the formation of DNA adducts is supposed to cause growth inhibition and cell kill, hence eliminating the tumor cells. DNAdamage is recognizedviahighmobilitygroupnonhistoneproteins (HMG1andHMG2)and/or variousDNArepair pathways, dependingon thePtanalog.GSHandMTNcanneutralizePt (e.g., byacomplex thatcanbeeffluxed).MRPs (multidrugresistance-associatedproteins,e.g.,MRP2,alsoknownasABCC2)andsomeothereffluxtransporters (ATP7AandATP7B)canexcretePt fromcells (Ptefflux).The increasedrepairofDNAdamageand protectionwith GSH, aswell as dysregulation in apoptosis pathways and reduced Pt influx and increased Pt efflux, can induce Pt resistance.
Abbreviations: ERK, extracellular signal-regulated kinase; GSH, reduced glutathione; HMG, high mobility group nonhistone protein;JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MTN, metallothionein protein; PKB (Akt), protein kinase B; Pt,platinated compounds; Sapk, stress-activated protein kinase.
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 413
CME
Figure 2.1. Effects of Pt and potential mechanisms of action. Pt may enter tumor cells (Pt influx) via copper transporter, organic cationtransporters,andorganiccation/carnitinetransportersorbypassivediffusion.DNA-platinumadductsblockDNAreplication,transcription,andothernuclearfunctionsandalsoactivatesignaltransductionpathways,whichresultinapoptosisandnecrosisintumorcells.Individingtumorcells,theformationofDNAadductsissupposedtocausegrowthinhibitionandcellkill,henceeliminatingthetumorcells.DNAdamageisrecognizedviahighmobilitygroupnonhistoneproteins(HMG1andHMG2)and/orvariousDNArepairpathways,dependingonthePtanalog.GSHandMTNcanneutralizePt(e.g.,byacomplexthatcanbeeffluxed).MRPs(multidrugresistance-associatedproteins,e.g.,MRP2,alsoknownasABCC2)andsomeothereffluxtransporters(ATP7AandATP7B)canexcretePtfromcells(Ptefflux).TheincreasedrepairofDNAdamageandprotectionwithGSH,aswellasdysregulationinapoptosispathwaysandreducedPtinfluxandincreasedPtefflux,caninducePtresistance.
Abbreviations:ERK,extracellularsignal-regulatedkinase;GSH,reducedglutathione;HMG,highmobilitygroupnonhistoneprotein;JNK,c-JunN-terminalkinase;MAPK,mitogen-activatedproteinkinase;MTN,metallothioneinprotein;PKB(Akt),proteinkinaseB;Pt,platinatedcompounds;Sapk,stress-activatedproteinkinase.
CME

Chapter2 TheOncologist2015;20:411-432
intracellularly because of a hydrolysis process [46], resulting in interstrand cross-links, intrastrandcross-links and/orDNA-protein cross-linkswithplatinum, affectingDNA synthesis in cancer cells [47]and mediating apoptosis [48]. Extensive DNA repair is considered as a major mechanism ofchemotherapy resistance (Figure 2.1), but efficientDNA repair can possibly prevent development ofneurotoxicity. In dividing tumor cells, the formation of DNA adducts is supposed to cause growthinhibitionandcellkills,henceeliminatingthetumorcells.
Platinum products accumulate in the dorsal root ganglia (DRG), which is the main target, and inperipheralneurons(Figure2.2).Becausethesecellsarepostmitoticandnotdividing,theformationofDNA adducts is not lethal, although the extent of DNA cross-links in DRG neurons at a specificcumulative dose strongly correlates with the degree of neurotoxicity [49]. Cisplatin producesapproximately three times more adducts in the DRG compared with oxaliplatin [50], which isconsistent with its higher neurotoxicity [12]. Platinum adducts probably cause axonal changessecondarytotheneuronaldamage[51],whereasbrainandspinalcordaretosomeextentprotectedbytheblood-brainbarrier(BBB)[52].However,therearedatashowingthatcisplatincrossestheBBBandcan accumulate when repeated dosages are given [53]. This can cause demyelination and vacuolarchanges in thewhitematter [54]. It isbelieved that impairedaxonalvoltage-gatedsodiumchannelskineticscaninterferewithchannelkineticsbyoxalate(ametabolicproductofoxaliplatin)[55]andcanalso reduce sodium ions current [56]. Sittl et al. [57] also showed that cooling in the presence ofoxaliplatin induced bursts of action potentials in myelinated A but not unmyelinated C-fibers fromhumanandmouseperipheral axons.Consequently, thesealterations led toenhanced resurgentandpersistent current amplitudes in large, but not small, diameter DRG neurons. Potassium channelblockadeandcalciumchelationarealsotwootheretiologicpossibilities[56,58–60].Besides,oxidativestressandmitochondrialdysfunctionareregardedasanotherprobableetiologyoftheapoptosis[61].Podratz et al. [62] showed that cisplatin might inhibit mitochondrial DNA replication and causemitochondrialvacuolizationanddegradationinDRGneuronsinvitroandinvivo.Theseeventscan,tosomeextent,explainthemechanismoftheneurotoxiceffectofplatinumcompounds.
Pharmacogenetics
Single-nucleotide polymorphisms (SNPs) may play a key role in determining the induction ofneurotoxicity,aswellasapoptosis,becausetheymayimpairDNArepairpathways,includinggenesinbase excision repair, nucleotide excision repair, mismatch repair, and double-strand break repairpathways [63] (Figure 2.2). Moreover, SNPs can alter the drug metabolism, cell cycle control,detoxification, or excretion pathways, which finally may lead to drug toxicities, e.g., neurotoxicity.SeveralstudiesevaluatedthepharmacogeneticassociationofSNPswithpotentialfunctionalchanges
�17
the inappropriate outcome measures for neurological impair-ment [64] (Table 1).
There are controversial reports on the association ofpolymorphisms in some genes with platinum-induced neuro-toxicity. These genes include ATP-binding cassette subfamily Bmember 1 (ABCB1) [65–68], ATP-binding cassette subfamily Cmember 1 or 2 (ABCC1, C2, or CG2) [69, 70], alanine-glyoxylateaminotransferase (AGXT) [69, 72, 73, 77, 94], cyclin H (CCNH)[70], catechol O-methyltransferase (COMT) [76], cytochromeP450s(CYPs;e.g.,CYP2C8,CYP3A5exons3and5)[65–68],excisionrepair cross-complementation group 1 (ERCC1) and ERCC2 (aliasXPD, xerodermapigmentosumgroupD) [67,68,71,74,75,77–87,88], integrin b3 (ITGB3) [92], glutathione S-transferases (e.g.,GSTM1 [69, 77, 78, 85, 86, 88, 89–91],GSTM3 [84, 75], andGSTT1[88,91]),voltage-gatedsodiumchannelgenes(SCNAs)[20,71,98],thiopurine S-methyltransferase (TPMT) [76], and x-ray repaircross-complementing protein 1 (XRCC1) [71, 73]. Although somedata support the role of the mentioned genetic variations in thepresentation and severity of platinum-induced peripheral neuro-toxicity, the results are scattered and diverse (Tables 1, 2), whichmay form leads for future research.
ABCB1,ABCC1,ABCC2,ABCG2, and probably several othersubfamily members mediate the cellular trafficking of drugs,theirmetabolites,and theirendogenous factors, e.g., platinumefflux [99, 100]. CCNH plays an important role in the cell cycleprogression, the transcriptionalactivityof theRNApolymeraseII, and theDNA repairing process [101].Thus, itmayderegulatethe repair after platinum damage to the dorsal root ganglianeurons [102]. COMT and TPMT, which encode enzymes thatmetabolize catecholamine-containing chemical and thiopur-ine drugs via methylation [103], respectively, might be as-sociated with cisplatin-related hearing loss [104, 105].
Glutathione S-transferases (GSTs), a familyofenzymes thathavean important role indetoxification, havebeenextensivelystudied for the relation of SNPs with neurotoxicity induced byplatinated compounds. GSTs are involved in detoxificationthrough glutathione conjugation of electrophilic compounds(e.g., GSTM1 and GSTM3). A GSTP1 SNP (rs16953), for exam-ple, has been investigated in relation to peripheral neuro-toxicity of platinum compounds in 24 studies (Table 1).
Among these, 9 studies reported an association of this SNPwith the course and severity of peripheral neurotoxicity [68,74, 77, 80, 85, 88–90, 93], whereas other researchers reportedcontradicting results in 15 studies with regard to the as-sociation of GSTP1 gene variants with neurotoxicity [67, 71,72, 75, 78, 81, 83, 84, 86, 87, 91, 94–97]. Moreover, recentmeta-analysis showed no significant associations betweenGSTP1 Ile105Val polymorphism and oxaliplatin-induced neu-ropathy in a dominant model (odds ratio [OR] 5 1.08, 95%confidence interval [CI]0.67–1.74,p5 .754),a recessivemodel(OR 5 1.67, 95% CI 0.56–4.93, p 5 .357), and allelic analysis(OR51.22,95%CI0.67–2.24,p5 .513) [106].This inconsistencybetween the findingsmightbeexplainedby thedifference in thecancer type, ethnicity of the population studied, and/or numberof the patients enrolled in each study [19].
Other studies evaluated the association of platinum-induced peripheral neurotoxicity with different SNPs in ERCC1[67, 68, 77, 71, 74, 78–87], ERCC2 [71, 88], and XRCC1 [71, 73],which are parts of the nucleotide excision repair (ERCC1 andERCC2) and base excision repair (XRCC1) pathways and arerequired for repairofDNA lesions [107].AlthoughLeeetal. [73]reported the polymorphism Arg399Gln (rs25487) in XRCC1associated with less grade 2–4 sensory neuropathy in Koreanpatients treated with oxaliplatin-based treatment, the recentmeta-analysis found it to be generally associated with poorclinical outcomes [108]. AGXT prevents accumulation ofglyoxylate in the cytosol by converting it into glycolate, whichis subsequently metabolized by lactate dehydrogenase intooxalate, the metabolite of oxaliplatin [109]. Pharmacogeneticanalyses evaluated also cytochrome P450s [65–68], which aremajor enzymes of drug metabolism and bioactivation (e.g.,CYP2C8 and CYP3A5), and ITGB3 [92], which belongs to thelarge family of integrins, known to participate in cell adhesionand cell surface-mediated signaling.
A recent study has provided evidence that SNPs in voltage-gated sodium channel genes (SCNAs; e.g., SCN4A-rs2302237and SCN10A-rs1263292) can play a causal role in oxaliplatin-based peripheral neurotoxicity [20, 57] (Table 1). A poly-morphism in SCN1A (rs3812718) was also reported to beassociated with decreased neurotoxicity [85]. However, these
Figure 2. Mechanism of acute andchronic platinum-induced neurotoxicity.Oxaliplatin may impair normal calcium-sensitive voltage-gated sodium channels,which cause acute neurotoxicity. Platinatedcompound (Pt) adducts can accumulatein dorsal root ganglia and lead to chronicneurotoxicity. Because these cells are post-mitotic and not dividing, the formation ofDNA adducts is not lethal to neurons.Increased Pt influx by organic cationtransporters (OCTs) and organic cation/carnitine transporters (OCTNs), as well aspolymorphisms and/or overexpression ofsomegenesthatplayarole inPtmetabolism(e.g., inOCT,OCTN,andGSH),cancontributeto Pt-induced neurotoxicity.
Abbreviations: GSH, reduced gluta-thione; Pt, platinated compounds; VG,voltage-gated.
©AlphaMed Press 2015TheOncologist®
414 Platinum-Induced Neurotoxicity
CME
Figure 2.2.Mechanismof acute and chronicplatinum-induced neurotoxicity. Oxaliplatinmayimpairnormalcalcium-sensitivevoltage-gated sodium channels, which cause acuteneurotoxicity. Pt adducts can accumulate indorsal root ganglia and lead to chronicneurotoxicity. Because these cells arepostmitotic and not dividing, the formationof DNA adducts is not lethal to neurons.Increased Pt influx by organic cationtransporters (OCTs) and organic cation/carnitine transporters (OCTNs), as well aspolymorphisms and/or overexpression ofsomegenesthatplayaroleinPtmetabolism(e.g.,inOCT,OCTN,andGSH),cancontributetoPt-inducedneurotoxicity. Abbreviations:GSH,reducedglutathione;Pt,platinatedcompounds;VG,voltage-gated.
2

Chapter2 TheOncologist2015;20:411-432
intheencodedproteinthatplayaroleindrugdisposition,metabolism,anddetoxification,DNArepair,andcancer-cell resistanceandthatmay leadtoplatinumperipheralneurotoxicity [19].However, theresults are scattered and diverse with several methodological flaws, including small sample size,retrospective study design, and the implementation of a post hoc analysis of oncology-baseddatabases of different, not preplanned sizes aswell as lacking a prestudy hypothesis based on theknown role of the investigated targets in the peripheral nervous system and the inappropriateoutcomemeasuresforneurologicalimpairment[64](Table2.1).
There are controversial reports on the association of polymorphisms in some geneswith platinum-inducedneurotoxicity.Thesegenes includeATP-bindingcassettesubfamilyBmember1(ABCB1)[65–68],ATP-binding cassette subfamily Cmember 1 or 2 (ABCC1,C2, orCG2) [69, 70], alanine-glyoxylateaminotransferase (AGXT) [69, 72, 73, 77, 94], cyclin H (CCNH) [70], catechol O-methyltransferase(COMT) [76], cytochromeP450s (CYPs; e.g.,CYP2C8,CYP3A5 exons 3 and 5) [65–68], excision repaircross-complementationgroup1(ERCC1)andERCC2(aliasXPD,xerodermapigmentosumgroupD)[67,68,71,74,75,77–87,88], integrinβ3(ITGB3)[92],glutathioneS-transferases(e.g.,GSTM1 [69,77,78,85,86,88,89–91],GSTM3 [84,75],andGSTT1 [88,91]),voltage-gatedsodiumchannelgenes(SCNAs)[20,71,98],thiopurineS-methyltransferase(TPMT)[76],andx-rayrepaircross-complementingprotein1 (XRCC1) [71, 73]. Although some data support the role of thementioned genetic variations in thepresentationandseverityofplatinum-inducedperipheralneurotoxicity, theresultsarescatteredanddiverse(Tables2.1,2.2),whichmayformleadsforfutureresearch.
ABCB1, ABCC1, ABCC2, ABCG2, and probably several other subfamily members mediate the cellulartraffickingof drugs, theirmetabolites, and their endogenous factors, e.g., platinumefflux [99, 100].CCNH plays an important role in the cell cycle progression, the transcriptional activity of the RNApolymeraseII,andtheDNArepairingprocess[101].Thus, itmayderegulatetherepairafterplatinumdamage to the dorsal root ganglia neurons [102]. COMT and TPMT, which encode enzymes thatmetabolize catecholamine-containing chemical and thiopurine drugs via methylation [103],respectively,mightbeassociatedwithcisplatin-relatedhearingloss[104,105].
GlutathioneS-transferases (GSTs),a familyofenzymes thathavean important role indetoxification,have been extensively studied for the relation of SNPs with neurotoxicity induced by platinatedcompounds. GSTs are involved in detoxification through glutathione conjugation of electrophiliccompounds (e.g.,GSTM1 andGSTM3). AGSTP1 SNP (rs16953), for example, has been investigated inrelationtoperipheralneurotoxicityofplatinumcompoundsin24studies(Table2.1).
Amongthese,9studiesreportedanassociationofthisSNPwiththecourseandseverityofperipheralneurotoxicity[68,74,77,80,85,88–90,93],whereasotherresearchersreportedcontradictingresultsin15studieswithregardtotheassociationofGSTP1genevariantswithneurotoxicity[67,71,72,75,78,81, 83, 84, 86, 87, 91, 94–97]. Moreover, recent meta-analysis showed no significant associationsbetweenGSTP1Ile105Valpolymorphismandoxaliplatin-inducedneuropathyinadominantmodel(oddsratio[OR]=1.08,95%confidenceinterval[CI]0.67–1.74,p=.754),arecessivemodel(OR=1.67,95%CI0.56–4.93,p= .357),andallelicanalysis(OR=1.22,95%CI0.67–2.24,p= .513)[106].This inconsistencybetween the findings might be explained by the difference in the cancer type, ethnicity of thepopulationstudied,and/ornumberofthepatientsenrolledineachstudy[19].
Other studies evaluated the association of platinum-induced peripheral neurotoxicity with differentSNPs in ERCC1 [67, 68, 77, 71, 74, 78–87], ERCC2 [71, 88], andXRCC1 [71, 73], which are parts of thenucleotide excision repair (ERCC1 and ERCC2) and base excision repair (XRCC1) pathways and arerequiredforrepairofDNAlesions[107].AlthoughLeeetal.[73]reportedthepolymorphismArg399Gln(rs25487)inXRCC1associatedwithlessgrade2–4sensoryneuropathyinKoreanpatientstreatedwithoxaliplatin-based treatment, the recentmeta-analysis found it to be generally associatedwith poorclinicaloutcomes[108].AGXTpreventsaccumulationofglyoxylateinthecytosolbyconvertingitintoglycolate,whichissubsequentlymetabolizedbylactatedehydrogenaseintooxalate,themetaboliteof
�18
CME

Chapter2 TheOncologist2015;20:411-432
�19
Table 1. Studies on the genes with or without significant correlations with incidence and/or severity of platinum-inducedperipheral neurotoxicity
Target geneRelevance to platinumagents SNP/deletion
With association Without association
Studies Patients Studies Patients
ABC ABCB1 Drug transporters(membrane effluxproteins)
rs2032582 4 [65–68] 1,591
ABCC1 rs2074087 1 [69] 144
rs35587 1 [69] 144
ABCC2 rs1885301 1 [69] 144
rs2273697 1 [69] 144
rs3740066 1 [69] 144
rs4148396 1 [69] 144
rs717620 1 [69] 144
ABCG2 rs2622604 1 [69] 144
rs3114018 1 [70] 181 1 [70] 206
ACYP2 rs843748 2 [71]a 343
AGXT Detoxification enzyme rs34116584 1 [72] 135 3 [69, 73, 74] 518
rs4426527 1 [72] 135 3 [69, 73, 77] 570
N/A del74bp 1 [69] 144
BTG4 rs4936453 2 [71] 343
CAMK2N1 rs12023000 2 [71] 343
CCNH Cell cycle progression rs2230641 2 [70] 206 1 [70] 181
COMT Detoxification enzyme rs4646316 1 [76] 66
DLEU7 rs797519 2 [71] 343
ERCC ERCC1 DNA repair mechanisms rs11615 2 [74, 75] 169 15 [67, 68,71, 77–88]
3,242
rs3212986 1 [71] 247
ERCC2 rs13181 1 [71] 247
rs179993 1 [71] 247
rs1052559 1 [88] 63
FARS2 rs17140129 2 [71] 343
rs6924717 2 [71] 343
FOXC1 rs2338 2 [71] 343
GST GSTM1 Detoxification enzymes N/A deletion 1 [88] 63 9 [69, 75, 78,85, 86, 88–91]
1,472
GSTM3 N/A deletion 1 [84] 1 [92] 107
GSTP1 rs1695 9 [68, 74,77, 80, 85,88–90, 93]
1,408 15 [67, 71, 72, 75,78, 81, 83, 84, 86,87, 91, 94–97]
2,747
rs947894 1 [69] 144
rs1138272 1 [69] 144
rs6591256 1 [76] 66
GSTT1 Deletion 2 [88, 92] 170
ITG ITGA1 Cell adhesion and cellsurface-mediatedsignaling
rs830884 2 [71] 343
ITGB3 rs5918 1 [92] 55
SCNA SCN1A Voltage-gated sodiumchannels
rs2298771 1 [71] 247
SCN2A rs17183814 1 [98] 62
SCN4A rs2302237 1 [20] 200
SCN9A rs6746030 1 [20] 200
SCN10A rs1263292 1 [20] 200
SCN10A rs6800541 1 [20] 200
TAC1 rs10486003 2 [71] 343
TPMT Detoxification enzyme rs4380755 1 [76] 66
rs5008499 1 [76] 66
XRCC1 DNA repair mechanism rs25487 1 [73] 292 1 [71] 247
aWon et al. conducted the study in two settings, with 96 discovery and 247 validation samples.Abbreviations: ABCB1 or C1/C2/G2, ATP-binding cassette subfamily B, member 1 or C1/C2/G2; ACYP2, acylphosphatase 2, muscle type; BTG4, B-celltranslocation gene 4; AGXT, alanine-glyoxylate aminotransferase; CCNH, cyclin H; COMT, Catechol-O-methyltransferase; CAMK2N1, calcium/calmodulin-dependent protein kinase II inhibitor 1; DLEU7, deleted in lymphocytic leukemia, 7; ERCC1, excision repair cross-complementation group 1;ERCC2, alias XPD, Xeroderma-Pigmentosum group-D; FARS2, phenylalanyl-tRNA synthetase 2, mitochondrial; FOXC1, forkhead box C1;GSTM,m class ofglutathione S-transferases; GSTP1/TT1, glutathione S-transferases P1/TT1; ITGA1, integrin, a1; ITGB3, integrin b3; N/A, not applicable; SCNA,voltage-gated sodium channel a-subunit; SNP, single nucleotide polymorphism; TAC1, tachykinin, precursor 1; TPMT, thiopurine S-methyltransferase;XRCC1, x-ray repair cross-complementing protein 1.
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 415
CME
Table2.1.Studiesonthegeneswithorwithoutsignificantcorrelationswithincidenceand/orseverityofplatinum-induced
peripheralneurotoxicity
2

Chapter2 TheOncologist2015;20:411-432
�20
Table 2. Polymorphisms associated with platinum-induced peripheral neuropathy
Gene SNPCancer(patients) Neurotoxic agent Assessment Association Reference
ABCC1 rs2074087 Colorectal(144)
Oxaliplatin (FOLFOX4) Oxaliplatinspecific scale
More severe PIPN forC/C or G/C vs. G/Ggenotypes (p5 .0170)
Cecchin et al.(2013) [69]
ABCC2 rs717620 Colorectal(144)
Oxaliplatin (FOLFOX4) Oxaliplatinspecific scale
More severe PIPN forTTor C/T vs. C/Cgenotypes (p5 .0164)
Cecchin et al.(2013) [69]
rs1885301 More severe PIPN forA/A or G/A vs. G/Ggenotypes (p5 .0072)
rs3740066 More severe PIPNfor T/Tor C/T vs. C/Cgenotypes (p5 .0231)
rs4148396 More severe PIPNfor T/Tor C/T vs. C/Cgenotypes (p5 .0048)
ABCG2 rs3114018 Colon(181)
Oxaliplatin(FOLFOX/CAPOX)
NCI-CTC v2or v3
Increased rate of severePIPN for A/A vs. A/C vs.C/C genotypes (p5 .016)
Custodio et al.(2014) [70]
ACYP2 rs843748 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with G allele(p5 1.013 1025)
Wo et al.(2012) [71]
AGXT rs34116584 Colorectal(135)
Oxaliplatin (FOLFOX4) NCI-CTC v1 |andOxaliplatinspecific scale
More severe PIPN forC/T and T/T vs. C/Cgenotypes (p, .001)
Gamelin et al.(2007) [72]
rs4426527 More severe PIPN forA/G and G/G vs. A/Agenotypes (p, .001)
BTG4 rs4936453 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated withT allele (p5 9.863 1025)
Wo et al.(2012) [71]
CAMK2N1 rs12023000 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with A allele(p5 8.813 1025)
Wo et al.(2012) [71]
CCNH rs2230641 Colon(206)
Oxaliplatin (FOLFOXor CAPOX)
NCI-CTC v3 Increased rate of severePIPN for C/C vs. C/T vs.T/T (p5 .042)
Custodio et al.(2014) [70]
DLEU7 rs797519 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with G allele(p5 8.213 1025)
Wo et al.(2012) [71]
ERCC1 rs11615 Colorectal(51)
Oxaliplatin (FOLFOX6) NCI-CTC v3 Higher incidence of PIPN(grade 1) for C/T and T/T vs.C/C genotypes (p5 .016)
Inada et al.(2010) [74]
rs3212986 Ovarian(118)
Cisplatin/carboplatin(1 paclitaxel/docetaxel)
NCI-CTC v2 Higher incidence of PIPN(grades 3–4) for C/C vs.C/A or A/A genotypes(p5 .019)
Kim et al.(2009) [75]
FARS2 rs17140129 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with A allele(p5 3.233 1025)
Wo et al.(2012) [71]
rs6924717 Oxaliplatin (FOLFOXor XELOX)
More severe neuropathyassociated with C allele(p5 3.233 1025)
Wo et al.(2012) [71]
FOXC1 rs2338 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with G allele(p5 4.633 1026)
Wo et al.(2012) [71]
GSTM1 Deletion Colorectal(63)
Oxaliplatin (mFOLFOX-6) NCI-CTC v3 More severe PIPN(grades 2–3) for A/G orG/G vs. A/A genotypes(p5 .03)
Kumamoto et al.(2013) [88]
GSTM3 rs1799735 Ovarian(104)
Cisplatin NCI-CTCb Lower incidence of PIPNfor AGG/AGG genotype(p5 .055)c
Khrunin et al.(2010) [84]
(continued)
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 417
CME
Table2.2.Polymorphismsassociatedwithplatinum-inducedperipheralneuropathy
CME

Chapter2 TheOncologist2015;20:411-432
�21
Table 2. (continued)
Gene SNPCancer(patients) Neurotoxic agent Assessment Association Reference
GSTP1 rs16953 Colorectal(52)
Oxaliplatin NCI-CTC v3 Higher incidence of PIPN(grades 2–3) for A/G or G/G vs.A/A genotypes (p5 .03)
Hong et al.(2011) [77]
Colorectal(166)
Oxaliplatin(FOLFOX4)
NCI-CTCb Higher incidence of PIPN:for G/G and A/G vs. A/Agenotypes (p, .01)
Chen et al.(2010) [80]
Colorectal(51)
Oxaliplatin(FOLFOX6)
NCI-CTC v3 Higher incidence of PIPN(grade 1) for A/A vs. A/Gand G/G genotypes(p5 .032)
Inada et al.(2010) [74]
Colorectal(520)
Oxaliplatin (FOLFOX4or IROX)
NCI-CTC v2 More severe PIPN(grades 3–4) for T/Tgenotype (p5 .003)
McLeod et al.(2010) [68]
Colorectal(166)
Oxaliplatin (FOLFOX4) Oxaliplatinspecific scale
More severe PIPN forG/G. A/G. A/Agenotypes (p, .001)
Ruzzo et al.(2007) [85]
Colorectal(59)
Oxaliplatin Oxaliplatinspecific scale
Higher PIPN incidence(grade 3) for A/A vs. A/Gand G/G genotypes (p5 .02)
Lecomte et al.(2006) [89]
Colorectal(66)
Oxaliplatin (mFOLFOX-6) NCI-CTC v3 More severe PIPN(grades 2–3) for A/G or G/Gvs. A/A genotypes (p5 .05)c
Kumamoto et al.(2013) [88]
Gastric(85)
Oxaliplatin (FOLFOX6) NCI-CTC v2 More severe PIPN for A/A vs.A/G and G/G genotypes(p5 .005)
Li et al.(2010) [93]
Testicular(238)
Cisplatin (1 bleomycin,etoposide or vinblastine)
SCIN More severe PIPN forA/A genotype (p5 .012)or A/G vs. G/G genotypes(p5 .003)
Oldenburg et al.(2007) [90]
ITGA1 rs830884 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with T allele(p5 1.743 1026)
Wo et al.(2012) [71]
ITGB3 rs5918 Colorectal(55)
Oxaliplatin TNS More severe PIPN forT/T genotype vs. C/T andC/C genotypes (p5 .044)
Antonacopoulouet al. (2010) [92]
SCNA rs2302237 Colorectal(200)
Oxaliplatin (FOLFOX4) NCI-CTC v3and TNS
More severe (p5 .0029)and higher incidence ofacute (p5 .019) and chronic(p5 .037) PIPN for C/T vs.C/C and T/T genotypes
Argyriou et al.(2013) [20]
rs1263292 Higher incidence of PIPNfor C/T vs. C/C and T/Tgenotypes (p5 .023)
TAC1 rs10486003 Colorectal(343)a
Oxaliplatin (FOLFOXor XELOX)
NCI-CTC v3 More severe neuropathyassociated with C allele(p5 4.843 1027)
Wo et al.(2012) [71]
XRCC1 rs23885 Colon(292)
Oxaliplatin NCI-CTC v3 Less severe (p5 .050)c
and latter-onset (p5 .041)PIPN for A/G and A/A vs.G/G genotype
Lee et al.(2013) [73]
aThis studyhasbeenconducted in twosettings,with96discoveryand247validationsamples.Thepvaluesmentionedare for thecombinedgroups,whichwere also significant for each SNP in each group.bVersion of the scoring scale was not reported.cTrends toward significance.Abbreviations:ABCC1/C2/G2, ATP-binding cassette subfamilyC,member1orC2/G2;ACYP2, acylphosphatase2,muscle type;AGXT, alanine-glyoxylateaminotransferase;BTG4, B-cell translocationgene4;CAMK2N1, calcium/calmodulin-dependentprotein kinase II inhibitor 1;CAPOX, capecitabine andoxaliplatin; CCNH, cyclin H; DLEU7, deleted in lymphocytic leukemia, 7; ERCC1, excision repair cross-complementing group 1; FARS2, phenylalanyl-tRNAsynthetase 2,mitochondrial; FOLFOX, folinic acid (leucovorin), fluorouracil andoxaliplatin; FOXC1, forkheadboxC1;GSTM,m class ofglutathioneS-transferases; GSTP1, glutathione S-transferases P1; IROX, irinotecan plus oxaliplatin; ITGA1, integrin a1; ITGB3, integrin b3; mFOLFOX, modifiedFOLFOX; NCI-CTC, National Cancer Institute–Common Toxicity Criteria; PIPN, platinum-induced peripheral neurotoxicity; SCIN, scale for chemotherapy-induced long-term neurotoxicity; SCNA, voltage-gated sodium channel a-subunit; SNP, single-nucleotide polymorphism; TAC1, tachykinin, precursor 1;TNS, total neuropathy score; v, version; XELOX, oxaliplatin, capecitabine; XRCC1, x-ray repair cross-complementing protein 1.
©AlphaMed Press 2015TheOncologist®
418 Platinum-Induced Neurotoxicity
CME
Table2.2.Continued
2

Chapter2 TheOncologist2015;20:411-432
oxaliplatin [109]. Pharmacogenetic analyses evaluated also cytochrome P450s [65–68], which aremajorenzymesofdrugmetabolismandbioactivation(e.g.,CYP2C8andCYP3A5),andITGB3[92],whichbelongstothelargefamilyofintegrins,knowntoparticipateincelladhesionandcellsurface-mediatedsignaling.
ArecentstudyhasprovidedevidencethatSNPs involtage-gatedsodiumchannelgenes(SCNAs;e.g.,SCN4A-rs2302237 and SCN10A-rs1263292) can play a causal role in oxaliplatin-based peripheralneurotoxicity [20, 57] (Table 2.1). A polymorphism in SCN1A (rs3812718) was also reported to beassociated with decreased neurotoxicity [85]. However, these results still need to be validated byappropriate larger and prospective studies. Won et al. [71], in a genome-wide pharmacogenomicapproach, identified nine novel polymorphisms associatedwith and predictors of severe oxaliplatin-induced peripheral neurotoxicity, including rs10486003 (tachykinin, precursor 1 [TAC1]), rs2338(forkheadboxC1 [FOXC1]), rs830884(integrinα1 [ITGA1]), rs843748(acylphosphatase2,muscle type[ACYP2]),rs4936453(B-celltranslocationgene4[BTG4]),rs17140129andrs6924717(phenylalanyl-tRNAsynthetase 2 [FARS2]), rs12023000 (calcium/calmodulin-dependent protein kinase II inhibitor 1[CAMK2N1]),andrs797519(deletedinlymphocyticleukemia,7[DLEU7])[71].Thesegenesmayaccountforthemechanismofneurotoxicitypreventionbycalcium-magnesiuminfusionsormaybeassociatedwiththe importantoxalateandglyoxylateoutcomepathway[72].However,noneoftheSNPs inthediscovery samples (96 patients with colon cancer) surpassed genome-wide significance, and theseSNPswerenotsignificantintheirvalidationset(247patientswithcolorectalcancer;p=.05–.19)[71].However,theauthorsnotedthatthislimitationmightbeovercomebyincreasingthesamplesizeinaprospectiveanalysis.
Someevidencedemonstratedthatmitogen-activatedproteinkinasepathways,includingextracellularsignal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK)/ stress-activated protein kinase(Sapk), and p38,might also have a causal effect on chemotherapy-induced peripheral neuropathies[110]. Normally, there is a balance between ERK1/2 and p38 activation, which regulates neuronalapoptosis, and JNK/Sapk, which preserves neuronal degeneration. This balance is also altered byplatinumderivatives[109].
Neuroprotection
NeuroprotectiveAgents,MechanismsandControversies
Severalmodelsystemshavebeenusedtostudythenatureofoverallneurotoxicityandtheeffectofpotential neuroprotective drugs. These include overall neurotoxicity signs in the animal, specificmodels includingtheDRGofrats[111–113], thestructureofthecerebralgangliaofsnails[114–116], invitromodelssuchasneuriteextension[117–121],andevaluationofbiomarkersofneurotoxicitysuchascyclinB[122].Thesemodelshavebeenveryusefultoselectproperpotentialneuroprotectivedrugstobe evaluated in the clinic. Unfortunately, preventive and therapeutic treatment options are notsufficientsofartobypassneurotoxicity[9,19,123].However,afewdrugscan,tosomeextent,protectagainst platinum-induced peripheral neurotoxicity (Table 2.3). Neuroprotective drugs include thefollowing.
Detoxicants
Sodium thiosulfate (STS) is a reactive thiol agent used clinically as an antidote to cyanide ornitroprussidepoisoning,andathighmolarexcess,itbindstoandinactivatestheelectrophilicplatinumcompound. Its use includes otoprotection [169–173] (discussed separately under “Neuro- VersusChemoprotection”).
�22
CME
Chapter2 TheOncologist2015;20:411-432
�23
Chapter$2 The$Oncologist$2015;20:411Y432
�20
Table3.
Rand
omized
controlledtrialson
neurop
rotectiveagen
tsforthepreven
tion
ofplatinum
-indu
cedpe
riph
eralne
urotoxicity
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aRe
ference
NAC
Anu
tritionalsup
plem
ent
thou
ghttoincrease
who
lebloo
dconcen
trations
ofglutathion
e;italso
activatesER
Ksandp3
8MAPK
,tomed
iate
the
neurop
rotectiveeffectof
NAC
14(5:9)/colorectalcancer
Oxaliplatin
Decreaseinincide
nceand
severityof
grades
2–4
neurotoxicityassessed
byNCI-CTC
(p,
.05);no
significant
changesin
incide
nceandmeanSN
AP
amplitud
e
Prospective,rand
omized,
placebo-controlled/
interm
ediate(dissimilarity
ofgroups,small-size
dtrial,
random
ization,blinding,
concealmentallocatio
n,outcom
edatanot
describ
ed,etc.)
Linet
al.
2006
[124]
a-Lipoicacid
Aph
ysiologicantioxidant
withne
urop
rotective
activity
243(122:121);29%
completed
thetrial(34:
36)/gastrointestinal
cancers(176),lung
cancer
(27),gen
itou
rinary
cancers(14),other
(15)
Cisplatinor
oxaliplatin
Nosignificantdecrease
inincidenceandseverityof
neurotoxicity
assessed
byNCI-CTC,FAC
T/GOG-Ntx,
andBPIscore
Prospective,rand
omized,
doub
le-blind,placebo-
controlledtrialb
Guo
etal.
2014
[125]
Amifo
stine
Detoxicant:an
organic
thioph
osph
atecystam
ine
analog
with
cytoprotective
activity
92(46:46)/advanced
orrelapsed
colorectalor
gastriccancer
with
variablemetastatic
diseaseto
liver,lun
g,and
othe
rsites
Oxaliplatin(FOLFOX4
)Decreaseinincide
nceof
grades
1–4ne
urotoxicity
assessed
byNCI-CTC
(p,
.007);changesinseverity
andne
urop
hysiologic
assessmen
tNR
Prospective,rand
omized,
placebocontrolledtrial/
low(blinding
and
concealmentallocatio
nno
tdescribed)
Luet
al.
2008
[126]
90(45:45)/advanced
ovariancarcinom
aCarbop
latin,paclitaxel
Decreaseinincide
nceof
neurotoxicitygrades
2–3
assessed
byNCI-CTC
(p,
.05);changes
inseverity
andne
urop
hysiologic
assessmen
tNR
Rand
omized
phaseII
stud
y/high
(non
placeb
otrial,ob
serversun
blinded
unclearparticipant
blinding,and
incomplete
datarepo
rting,
rand
omizationand
concealmentallocatio
nno
tdescribed)
DeVo
set
al.
2005
[127]
72(37:34)/ovariancancer
Carbop
latin,paclitaxel
Asignificantp
rotective
effectobserved
for2-PD,
TRA,VPT,VD
T,andNCI-CTC
neuropathy
scores
Rand
omized
doub
le-blind
placeb
o-controlledtrial/
low(unableto
separate
theeffectsof
carbop
latin
from
thoseof
paclitaxel,
similarityof
grou
psun
clear,etc.)
Hilperte
tal.
2005
[128]
38(19:19)/NSCLC
Carbop
latin,paclitaxel
Decreaseinincide
nceof
grades
1–2ne
urotoxicity
assessed
byNCI-CTC
(p5
.018);no
significant
declineinmeanSN
AP
amplitud
es
Prospective,rand
omized/
interm
ediate(small
samplesize,unb
linded
participants,noplacebo,
incompleteou
tcom
edata,
etc.)
Kanate
tal.
2003
[129]
60(30:30)/un
resectable
stageIIINSCLC
Carbop
latin,paclitaxel
Nosignificant
changesin
incide
nceor
neurop
hysiologic
Rand
omized
doub
le-blind
trial/low(onlytw
odo
ses
ofcarbop
latin,sm
all
samplesize,etc.)
Leon
get
al.
2003
[130]
(con
tinu
ed)
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 419
CME
Table!2.3.$Ran
domized
$con
trolled$trials$on$ne
urop
rotective$ag
ents$fo
r$the
$preve
ntion$of$platin
umYin
duce
d$pe
riphe
ral$n
euro
toxicity
2

Chapter2 TheOncologist2015;20:411-432
�24
Table3.
(con
tinu
ed)
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aRe
ference
assessmen
t;changesin
severityNR
187(93:94)/advanced
ovariancancer
Carbop
latin,paclitaxel
Decreaseinseverityof
grades
3–4neurotoxicity;
assessed
byNCI-CTC
(p5
.021);changesinincidence
andseverityNR
PhaseIIIprospe
ctive
multicenter
rand
omized
trial/low(not
placeb
ocontrolled,un
clear
blinding,incom
plete
outcom
edata
etc.)
Lorussoet
al.
2003
[131]
20(10:10)/cervicalcancer
Cisplatin
Nosignificant
changesin
incide
nceandseverityof
neurop
athy
Anop
en,single-blinde
dpilotstud
y/high
(ope
n,single-blinde
d,no
allocation
concealm
ent,
smalltrial,etc.)
Gallardoet
al.
1999
[132]
73(36:37)/advanced
head
andne
ckcancer
Cisplatin
Decreaseinincide
nceof
subclinicalne
urotoxicity
assessed
byVPT
(p5
.03);
changesrepo
rted
inseverityNR
Prospective,rand
omized,
placebo-controlled/
interm
ediate
(unclear
blinding,rando
mization
andallocatio
nconcealm
ent,etc.)
Planting
etal.
1999
[133]
242(122:120)/ovarian
cancer
Cisplatin
Decreaseinseverityof
grades
1–3ne
urotoxicity
(p5
.029);assessed
byNCI-CTC
;inciden
ceand
neurop
hysiologic
assessmen
tNR
Prospective,rand
omized/
low(not
placebo-
controlled,etc.)
Kempet
al.
1996
[134]
BNP7
787
Detoxicant:
neurop
rotective
151(76:75)/advanced
NSCLC
Cisplatin,do
cetaxel
Unableto
separate
the
effectsof
cisplatinfrom
thoseof
docetaxel
PhaseIIrand
omized
stud
yb(unb
linde
dandno
tplaceb
o-controlled)
Miller
etal.
2008
[135]
Calcium-m
agne
sium
Chelatewithoxaliplatin
metaboliteandprotect
thevoltage-gatedsodium
channe
lsfrom
alteration
20(10:10)/colorectal
cancer
Oxaliplatin(XELOXor
mFO
LFOX6
)Nosignificantdecreasein
severityofneurotoxicity
basedon
motornerve
excitabilityassessed
byEM
G
Rand
omized
,dou
ble-
blinde
d,placeb
o-controlled,crossover
stud
yb
Han
etal.
2013
[136]
353(118:116:119)divided
into
threegrou
psof
CaMg/CaMg;CaMg/
placeb
o;placeb
o/placeb
obe
fore
andafter
chem
othe
rapy/colon
cancer
Oxaliplatin(FOLFOX4
or6)
Nosignificant
changesin
severityof
grades
2–4
assessed
byNCI-CTC
;no
significant
changesin
EORT
CCIPN
-20sensory,
motor,orautono
mic
scales;changes
inincide
nceand
neurop
hysiologic
assessmen
tNR
PhaseIIIrand
omized
,placeb
o-controlled,
doub
le-blindstud
y/low
Loprinziet
al.
2014
[137]
102(50:52)/colorectal
cancer
Oxaliplatin
Lower
incide
nceofgrades
2–4ne
urotoxicity;
assessed
byNCI-CTC
(p5
.018);changesinseverity
Prospective,rand
omized,
placebo-controlled,
doub
le-blind/low
(insufficient
samplesize
Grothey
etal.
2011
[138]
(con
tinu
ed)
©AlphaMed Press 2015TheOncologist®
420 Platinum-Induced Neurotoxicity
CME
Table2.3.(Co
ntinue
d)
CME

Chapter2 TheOncologist2015;20:411-432
�25Table3.
(con
tinu
ed)
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aReferen
ce
andne
urop
hysiologic
assessmen
tNR
andfollow-up,un
clear
allocatio
nconcealment,
etc.)
732(551
:181
)/colorectal
cancer
Oxaliplatin(plus
cape
citabine
,be
vacizumab
withor
witho
utcetuximab
Onlyredu
cedgrade1
neurotoxicityassessed
byNCI-CTC
;changes
inseverityand
neurop
hysiologic
assessmen
tNR
Retrospe
ctiveanalyses
witho
utrand
omization/
high
(neither
rand
omized
orblinde
d,andno
allocation
concealm
ent)
Knijn
etal.
2011
[139
]
33(17:16
)/metastatic
colorectalcancer
Oxaliplatin(FOLFOX6
)Nosignificant
changesin
incide
nceandseverityof
neurotoxicityassessed
byDEB
-NTS
andNCI-CTC
Prospective,rand
omized,
placebo-controlled/low
(smallsam
plesize)
Ishibashieta
l.20
10[140
]
19(9:10)/colorectal
cancer
Oxaliplatin(FOLFOX4
orXELO
X)Nosignificant
changesin
incide
nceandseverityof
neurotoxicitygrades
1–3
assessed
byNCI-CTC
,oxaliplatin-specificscale
andNCS
Prospective,rand
omized,
placebo-controlled/low
(smalltrial)
Chay
etal.
2010
[141
]
Carbam
azep
inAsodium
channe
lblocker
that
exertsitseffectby
inhibitin
ghyperexcitability
ofthechannelon
oxaliplatin
36(19:17
)/advanced
colorectalcancer
Oxaliplatin(FOLFOX)
Nosignificant
changesin
incide
nceandseverityof
neurotoxicityassessed
byLevi’sscale
Rand
omized
,con
trolled
multicenter
phaseII
stud
y/low(smallsam
ple
size)
vonDeliuset
al.
2007
[142
]
Diethyldithio-carbamate
Chelatingagen
tand
antioxidant:preven
tsthe
degradationofextracellular
matrix,asan
initialstep
incancermetastasis
and
angiogenesis
195(96:99
)/ovarian,
SCLC,and
NSCLC
Cisplatin
Adverse
effectswere
repo
rted
inallstudy
participantsandlower
levelsof
cisplatin
administration;
changes
inseverityand
neurop
hysiologic
assessmen
tNR
Prospective,rand
omized,
placebocontrolled
multicentertrial/low
(insufficient
samplesize
consideringthevarietyof
center
types,etc.)
Gandara
etal.
1995
[143
]
Glutamine
Neu
rotrop
hic:indu
ces
NGFsynthe
sis
86(42:44
)/metastatic
colorectalcancer
Oxaliplatin
Lower
incide
nceofgrades
1–2ne
urotoxicity;
assessed
byNCI-CTC
(p5
.04);severityassessmen
tNR;
nosignificant
changes
inneurophysio
logic
assessment
Rand
omized
pilotstud
y/high
(neither
blinde
dno
rplaceb
o-controlled)
Wanget
al.
2007
[144
]
Glutathione
Antioxidant:a
physiologic
free
radicalscavenger,
chelator
forhe
avymetals
that
decreasestheinitial
accumulationof
platinum
addu
ctsinthene
uron
s,
185(94:91
)/ovarian,
fallopian
tube
,and
prim
arype
ritone
alcancers(86),lun
gcancer
(53),other
type
s(22)
Carbop
latin,paclitaxel
Nosignificant
changesin
incide
nceandseverityof
neurop
athy
grades
1–4
assessed
byEO
RTCQLQ
-CIPN
20,and
NCI-CTC
repo
rted
;changes
in
PhaseIIIrand
omized
,do
uble-blindplaceb
o-controlledstud
y/low
Lealet
al.
2014
[145
]
(con
tinu
ed)
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 421
CME
Table2.3.(Co
ntinue
d)2
Chapter2 TheOncologist2015;20:411-432
�26
Chapter$2 The$Oncologist$2015;20:411Y432
�23
Table3.
(con
tinu
ed)
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aReferen
ce
andmod
ulator
ofMAPK
s,includ
ingJNK/Sapk
ERKs
andp3
8pathwayswith
which
apop
tosisis
inhibited
neurop
hysiologic
assessmen
tNR
27(14:13
)/colorectal
cancer
Oxaliplatin(FOLFOX4
)Lower
incide
nceofgrades
1and3ne
urotoxicity;
assessed
byNCI-CTC
(p5
.003
7);changesinseverity
andne
urop
hysiologic
assessmen
tNR
Prospective,rand
omized,
placebo-controlled/
interm
ediate
(small
samplesize,unclear
rand
omizationand
blinding,etc.)
Millaet
al.
2009
[146
]
52(26:26
)/colorectal
cancer
Oxaliplatin
Decreaseinincidenceand
severityofgrades
1–4
neurotoxicity;assessedby
NCI-CTC,and
sensory
nervecond
uctio
nstud
ies
(sural,m
edian,or
ulnar);
severityassessmentN
R;no
significant
changesin
neurop
hysiologic
assessment
Prospe
ctive,rand
omized
placeb
o-controlled/low
(incompleteou
tcom
edata,etc.)
Cascinuet
al.
2002
[147
]
20(11:9)/N
SCLC
andhe
adandne
ckcancer
Cisplatin
Nosignificant
decrease
inincidenceof
neurotoxicity
assessed
byWHO
neurotoxicity
measureand
motor
nervecond
uctio
nstud
ies
Prospectivelyrand
omized
placebo-
controlledpilot
trial/interm
ediate
(insufficient
samplesize,
unclearrando
mization,
andblinding)
Schm
idingeret
al.
2000
[148
]
151(74:77
)/ovarian
cancer
Cisplatin
Significantlylesstingling
assessed
byNCI/W
HO;
HADandRo
tterdamscales
(fun
ctionalscales);
changesinseverityand
neurop
hysiologic
assessmen
tNR
Prospective,rand
omized,
placebo-controlled/low
(blinding
andou
tcom
edatano
tclearlydescribed,
etc.)
Smythet
al.
1997
[149
]
50(25:25)/ovariancancer
Cisplatin
Decreaseinincide
nceof
neurotoxicityassessed
byNCI/W
HOcriteriaand
sensorynervecond
uctio
nstud
ies(sural,m
edian,or
ulnar;p,
.01);changes
inseverityNR
Prospective,rand
omized,
placebo-controlled/low
(relativelysm
alltrial,lon
g-term
follow-upun
clear,
incompleteou
tcom
edata,
etc.)
Cascinuet
al.
1995
[150
]
33(16:17
)/relapsing
ovariancancer
Cisplatin
Nosignificant
changesin
incide
nceandseverityof
neurop
athy
assessed
bySN
APandNCI/W
HO
Prospective,rand
omized,
placebo-controlledstud
y/low(lack
ofinform
ation
abou
trando
mizationand
blinding
andsm
allsam
ple
Colombo
etal.
1995
[151
]
(con
tinu
ed)
©AlphaMed Press 2015TheOncologist®
422 Platinum-Induced Neurotoxicity
CME
Table!2.3.!Con
tinue
dTa
ble2.3.(Co
ntinue
d)CME

Chapter2 TheOncologist2015;20:411-432
�27
Table3.
(con
tinu
ed)
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aRe
ference
size,incom
pleteou
tcom
edata,etc.)
54(27:27)/ovariancancer
Cisplatin
DecreaseinSN
AP
amplitud
e(sural,m
edian,
orulnar;p5
.043)and
VPT;changes
inseverityof
neurotoxicityno
tsignificant;changes
inincidenceNR
Prospective,rand
omized,
placebo-controlled/
interm
ediate
(unb
linded,
relativelysm
alltrial,
unclearrando
mizationand
allocatio
nconcealment,
repo
rtingbias,etc.)
Bogliunet
al.
1992
[152]
Goshajinkigan(Kampo
med
icine;compo
sedof10
naturalingredien
ts)
Actsviareleaseof
dyno
rphin,nitric-oxide
prod
uction
,and
redu
cing
transm
itterproteins
and
sensoryreceptors
associated
withC-fib
eractivation
89(44:45)/colorectal
cancer
Oxaliplatin(FOLFOX4
ormFO
LFOX6
)Suggestive
ofade
crease
inincide
nceof
grade2–
3ne
urotoxicityassessed
byNCI-CTC
andFA
CT/G
OG-
Ntx,but
insignificant;
changesinseverityand
neurop
hysiologic
assessmen
tNR
PhaseII,multicenter,
rand
omized
,dou
ble-
blind,placeb
o-controlled
trialb
Kono
etal.
2013
[153]
45(22:23)/no
nresectable
orrecurren
tcolorectal
cancer
Oxaliplatin(FOLFOX6
)Lower
incide
nceofgrades
3ne
urotoxicity;assessed
byDEB
-NTC
(p,
.01;un
blinde
dcontrol);
Severityand
neurop
hysiologic
assessmen
tNR
Prospe
ctiverand
omized
controlledstud
y/low
(relativelysm
allsam
ple
size
withun
blinde
dcontrolgroup
,etc.)
Nishiokaet
al.
2011
[154]
Nim
odipine
Calcium
channe
lantagonistthat
might
attenu
atechelatingeffect
byincreasing
calcium
serum
levels
50(24:26)/ovariancancer
Cisplatin
Thetrialw
asterm
inated
becauseof
side
effects;
theavailabledata
suggestedthatnimodipine
exacerbated(not
prevented)neurotoxicity;
incidence,severityand
neurophysiologic
assessmentN
R
Rand
omized
placeb
o-controlledstud
y/high
(smalltrialwith
unreliable
measuresandinadequate
follow-upperio
d)
Cassidyet
al.
1998
[155]
Org
2766
Neu
rotrop
hic:an
adreno
corticotropic
horm
oneanalog
with
neurotroph
iceffects
196(129:67)/epithelial
ovariancancer
Cisplatin
Nosignificant
changesin
incide
nceandseverityof
neurotoxicity;VPT
increasedinbo
thgrou
psdu
ring
thestud
y(w
orse
outcom
e)
Prospective,rand
omized,
placebo-controlled/low
(unclearrand
omization
andallocatio
nconcealment,etc.)
Robe
rtset
al.
1997
[156]
42(19:23)/testicular
and
aden
ocarcino
maof
unknow
nprim
ary
Cisplatin(and
differen
tcombination
sof
etop
oside,bleo
micin,and
ifosphamide)
Neu
roph
ysiologic
assessmen
tno
tsuggestive
ofne
urop
rotection;changes
Prospective,rand
omized,
placebo-controlled/low
(insufficient
samplesize,
unclearrando
mizationand
vanGervenet
al.
1994
[157]
(con
tinu
ed)
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 423
CME
Table2.3.(Co
ntinue
d)2

Chapter2 TheOncologist2015;20:411-432
�28
Table3.
(con
tinu
ed)
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aReferen
ce
inincide
nceandseverity
NR
blinding,incom
plete
outcom
edata,etc.)
20(7:11)/epithelial
ovariancancer
Cisplatin
VPT
increased
significantlylessinactive
arm
than
placeb
o;changesinincide
nceand
severityNR
Prospective,rand
omized,
placebo-controlled/
interm
ediate
(insufficient
samplesize,unclear
rand
omization,substantial
drop
out,incomplete
outcom
edata,etc.)
Hovestadt
etal.
1992
[158]
55(33:22)/ep
ithe
lial
ovariancancer
Cisplatin
VPT
increased
approxim
atelytw
ofold,
significantlylessthan
the
nearlyeightfoldincrease
intheplaceb
ogrou
p;changesinincide
nceand
severityNR
Prospective,rand
omized,
placebo-controlled/low
(relativelysm
alltria
l,etc.)
vande
rHoo
pet
al.
1990
[159]
Oxcarbazepine
Sodium
channe
linh
ibitor:
protectsthealteration
ofvoltage-gatedsodium
channe
lsby
oxalate
40(20:20)/coloncancer
Cisplatin
Significant
decrease
inincide
nceandseverityof
neurotoxicityassessed
byNSS,TNS,SN
AP,and
perone
almotorrespon
ses
Prospective,random
ized,
open
labelw
ithblind
assessment/interm
ediate
(relativelysm
alltrialw
ithan
unblindedcontrolarm
,etc.)
Argyriouet
al.
2006
[160]
Retino
icacid(all-trans-
retino
icacid)
Stim
ulator
ofNGFandthe
expression
ofitsreceptor,
activatorof
retino
idacid
receptorswith
neurop
rotectiveprofile;it
isalso
aprod
ifferen
tiative
agen
tableto
activate
both
JNK/Sapk
andER
K1/
2
92(45:47)/advanced
NSCLC
Cisplatin,paclitaxel
Decreaseinincide
nceand
severityof
grade2–
4ne
urop
athy
assessed
bymotor
andsensory
respon
seam
plitud
esand
NCI-CTC
Prospective,rand
omized,
placebo-controlled/low
(sho
rtperiod
ofevaluatio
n,etc.)
Arrieta
etal.
2005
[161]
rhuLIF
Neu
rotrop
hic:aIL-6
class
cytokine
thatup
regulates
NGFsynthesis,which
affectsnerveregeneratio
nby
inhibitingdifferentiation
117(75:42)/differen
tsolid
tumors
Carbop
latin,paclitaxel
Worse
neurop
athy
endp
ointsintheactive
therapyarmforchangesin
velocityinthemed
ian
nerve;no
significant
changesinCP
NEscoreand
VPT;changesinincidence
ofneurop
athy
NR
Rand
omized
,dou
ble-
blinde
d,placeb
o-controlledph
aseIItrial/
low
Daviset
al.
2005
[162]
Ven
lafaxine
Serotonin-
norepine
phrine
reup
take
inhibitor:
mod
ulates
theoxidative
stressinthene
rvou
ssystem
andmay
block
sodium
channe
ls
48(24:24)/differen
tsolid
tumors
Carbop
latin/paclitaxel
DecreaseinNPSI(pins
and
need
les;p,
.001);no
significant
decrease
inNPSIpaintriggeredby
cold;changes
inincide
nce
andne
urop
hysiologic
assessmen
tNR
Rand
omized
,dou
ble-
blind,placeb
o-controlled
phaseIIItrial/
interm
ediate
(small
samplesize,unclear
allocation
concealm
ent,etc.)
Durandet
al.
2012
[163]
(con
tinu
ed)
©AlphaMed Press 2015TheOncologist®
424 Platinum-Induced Neurotoxicity
CME
Table2.3.(Co
ntinue
d)CME

Chapter2 TheOncologist2015;20:411-432
�29Table3.
(con
tinu
ed)
Agent
Mecha
nism
ofaction
Totalpatients(treated
vs.
control)/tumor
type
Neu
rotoxicagen
t(s)
Neu
roprotection
Stud
yde
sign/overall
risk
ofbias
aRe
ference
Vitam
inE
Antioxidant:protects
biologicmem
branes
byinhibiting
peroxidation
ofpo
lysaturatedfattyacids,
andaprotectoragainst
platinum
accumulationin
theDRG
189(96:93
)/colorectal,
breast,lun
g,andothe
rcancers
Cisplatin(8),carbop
latin
(2),oxaliplatin(50),
taxane
s(109
),or
combination
sof
each
Inclusionof
alarge
numbe
rof
participants
receivingtaxane
swas
confou
nding;changesin
neurop
hysiologic
assessmen
tNR
Rand
omized
,dou
ble-
blind,placebo-
controlled
phaseIIItrial/low
(differenttypes
ofcancers
with
nocomparable
distrib
utionand
insufficient
samplesize
for
platinum
grou
ps,etc.)
Kottschade
etal.
2011
[164
]
108(54:54
)after
drop
out
(17:24
)/differen
tsolid
tumors
Cisplatin
Significant
decrease
inincide
nceandseverityof
neurotoxicitygrades
3–4
assessed
byTN
SandSN
AP
(suralandmed
ian)
Rand
omized
,placebo
-controlledtrial/high
(sub
stantialdrop
outrate,
exclud
ingintention-to-
treata
nalyses,etc.)
Pace
etal.
2010
[165
]
30(14:16
)/solid
tumors
Cisplatin,paclitaxel
Significant
decrease
inincide
nceandseverityof
neurotoxicityassessed
byNSS/N
DS;redu
ced
med
ianSN
APam
plitud
esincontrolgroup
Prospective,rand
omized,
open
labelw
ithblind
assessment/high
(relativelysm
alltrialand
lackofaplacebogrou
pand
reliablemeasures;
exclud
ingintention-to-
treatanalyses,etc.)
Argyriouet
al.
2006
[166
]
27(13:14
)/lung,ovarian,
rhino-ph
aryngeal,gastric,
testicular,esoph
agus,
ethm
oid,andtongue
cancers
Cisplatin
Significant
decrease
inincide
nceandseverityof
neurotoxicity;redu
ced
med
ianSN
APam
plitud
esincontrolgroup
Prospective,rand
omized,
placebo-controlled/high
(smallsizeandexclud
ing
intention-to-treat
analyses;con
trol
participantswere
untreated)
Pace
etal.
2003
[167
]
Xaliprode
nAsynthe
tic
neurom
odulantthat
activatesM
APK
pathways,
which
minim
izes
neuritic
damage
649(325
:324
)/metastatic
colorectalcancer
Oxaliplatin(FOLFOX4
)Efficient
inredu
cing
grade
3–4ne
urotoxicity
evaluatedby
sensory
action
potential(17%vs.
11%withplaceb
o);but
nosignificant
differen
cein
overallinciden
ce(73%
ofbo
tharms)
Rand
omized
,dou
ble-
blind,placeb
o-controlled
phaseIIItrialb
(unp
ublishe
ddata)
Cassidyet
al.
2006
[168
]
a Stren
gthof
eviden
ceisaccordingto
the2014
American
Societyof
ClinicalOncologyClinicalPractice
Guide
line[123]and
2014
Cochrane
system
aticreview
[9].
bNot
assessed
byAmerican
Societyof
ClinicalOncologyor
Cochrane
stud
ies.
Abb
reviations:2-PD,two-po
intd
iscrim
ination;CP
NE,compo
sitepe
riph
eralne
rveelectrop
hysiology;DEB
-NTS,D
ebioph
armNeu
rotoxicityScale;DRG
,dorsalroo
tganglion;EM
G,electromyography;EORT
CQLQ
-CIPN20,
Europe
anOrganizationforR
esearchandTreatm
ento
fCancerq
ualityof
lifequ
estion
naireto
assesschem
othe
rapy-in
ducedpe
riph
eralne
urop
athy;ERK
,extracellularsignal-regulated
kinase;FACT/G
OG-Ntx,Fun
ctional
Assessm
entofCancerThe
rapy/G
ynecologicOncologyGroup
-Neu
rotoxicityScale;FO
LFOX,folinicacid(le
ucovorin),flu
orou
racil,andoxaliplatin;HAD,hospitalanxietyandde
pression
;IL,interleu
kin;JNK,c-JunN-terminal
kinase;M
APK
,mitogen
-activated
proteinkinase;m
FOLFOX,mod
ified
FOLFOX;NAC,N-acetylcysteine;NCI-CTC
,NationalCancerInstitute–
Common
ToxicityCriteria;N
CS,nerve
condu
ctionstud
ies;NDS,ne
urolog
ical
disabilityscore;NGF,ne
rvegrow
thfactor;N
IS,neu
ropathy
impairm
entscale;N
PSI,Neu
ropa
thicPainSymptomInventory;NR,notrep
orted;NSS,neu
rologicalsym
ptom
score;NSC
LC,non
-smallcelllun
gcancer;OSS,
oxaliplatin
-specific
scale;rhuLIF,recom
binanthu
man
leukem
iainhibitoryfactor;Sapk,stress-activated
proteinkinase;SCLC,sm
allcelllun
gcancer;SNAP,sural,superficialperon
eal,andulnarsensorynerveactio
npo
tential;TN
S,totalneuropathyscore;TRA,tendo
nreflexactivity;V
DT,vibrationdisapp
earancethreshold;VPT,vibratio
nperceptio
nthreshold;WHO,W
orldHealth
Organization;XELO
X,oxaliplatin
andcapecitabine.
www.TheOncologist.com ©AlphaMed Press 2015
Avan, Postma, Ceresa et al. 425
CME
Table2.3.(Co
ntinue
d)2

Chapter2 TheOncologist2015;20:411-432
Amifostine isanorganicthiophosphatealsoregardedasacytoprotectiveanddetoxicantagent[117].Therearesuccessful invitro results supportingamifostineneuroprotectionagainstcisplatin [120],aswellasagainstoxaliplatin[122].
Someclinicaldata,withdifferentlevelsofreliability(Table2.3),indicatedthatamifostineexertssomeprotection against peripheral neurotoxicity of carboplatin plus paclitaxel combination therapy [127–129, 131], oxaliplatin [126], and cisplatin [133, 134], whereas two other studies have failed to showsignificantneuroprotectionagainstcarboplatinpluspaclitaxelcombinationtherapy[130]andcisplatin[132](Table2.3).BNP7787(Dimesna,Tavocept,2,29-dithio-bis-ethanesulfonate)hasalsoshownsomecytoprotectiveactivitiesinvitro[174].Thiseffectwasnotconfirmedintheclinicalsetting,althoughthestudywasunblindedwithnoplacebo-controlledgroupandhighriskofbias[135].
NGFStimulants
Circulatingnervegrowthfactor(NGF)levelsarereducedincancerpatientswithneuropathycausedbyneurotoxicagents[175].Inaddition,Schmidtetal.[176]showedinamousemodelthatNGFexertedamajor effect on themetabolism of transmitters associatedwith nociception, pain, and sensation incervical dorsal root ganglia in various models of neurotoxicity, including the cisplatin-inducedneuropathy.Thus,NGF inhighdosesmayprotectDRGneuronsexposedtocisplatin[176],aswellasagainstoxaliplatin-inducedperipheralneurotoxicity[177].TheyhypothesizedthatthiseffectcouldbeduetoNGF’sabilitytopreservethecorrectneuronaldifferentiationstatusbyblockingthecellcycleintheG0phase.
ThesynthesisofNGFcanbestimulatedbyOrg2766[34,156,159],leukemiainhibitoryfactor(rhuLIF)[162],retinoicacid[161,178],glutamine[144],andacetyl-L-carnitine[179–181].Thetwolattermayalsoincrease glutathione production [182, 183]. Two clinical trials [158, 159] with relatively small samplesizes showed some degree of Org 2766 neuroprotection in patients with peripheral neurotoxicityinducedbycisplatin,whereastwootherstudiescouldnotfindasignificantdecreaseintheincidenceandseverityofneuropathy[156,157](Table2.3).Furthermore,derivativesoferythropoietin,aproteinsignalingcytokine,(e.g.,carbamylatederythropoietinandasialo-erythropoietin)havebeensuccessfulinvitroandinanimalmodels[184,185].
Retinoicacid(all-trans-retinoicacid)isastimulatorofNGFandtheexpressionofitsreceptor,activatorof retinoidacid receptorswithneuroprotectiveprofile [177]. It isalsoaprodifferentiatingagent thatcounteractsplatinum-inducedneuronalapoptosisthroughactivatingbothJNK/SapkandERK1/2[177].Additionally,Arrietaetal.[186]reportedadecreaseinincidenceandseverityofneuropathyinducedbycisplatin-paclitaxelcombinationwhenretinoicacidwasadministered.
AntioxidantsorAntioxidant-RelatedAgents
α-Lipoic acid, a physiologic antioxidant with some neuroprotective activity [187], has recently beentestedinawell-designedclinicaltrial[125]inwhichnosignificantdecreaseinincidenceandseverityofperipheral neurotoxicity induced by cisplatin and oxaliplatin has been reported (Table 2.3). α-Tocopherol(vitaminE),asanotherantioxidant,actsagainstfreeradicals.Fourtrialshaveevaluatedtheeffect of vitamin E in preventing platinum, mainly cisplatin-induced peripheral neurotoxicity, andshowedsignificantlylowerincidenceandseverityofneuropathyinthevitaminEgroupcomparedwiththe control group [164–167], although all werewith high risk of bias and low strength of evidence(Table2.3).
Reduced glutathione is a natural neuroprotectant antioxidant derived from the γ-glutamyltranspeptidasewithahighaffinityforheavymetals,whichmaypreventtheaccumulationofplatinumintheDRG[146,147,149,150].Additionally,itisanaturalfree-radicalscavengerandcanalsostimulateNGFreceptors[188].Fivedifferentclinicaltrialsdemonstratedthepotentialofglutathioneinreducingthe incidenceandseverityofneuropathy inducedbyoxaliplatin [146, 147]orcisplatin [149, 150, 152],
�30
CME

Chapter2 TheOncologist2015;20:411-432
whereasthreeotherscouldnotfindsignificantneuroprotectiveeffectagainstcarboplatin(carboplatin-paclitaxel combination) [145] or cisplatin-induced peripheral neurotoxicity [148, 151] (Table 2.3).Similarly, oral glutamine, another derivative of the γ-glutamyl transpeptidase, may reduce theincidence and severity of oxaliplatin-induced peripheral neuropathy [144], although based on arandomized,butneitherblindednorplacebo-controlledtrial.
N-Acetylcysteine is a glutathione precursor that is believed to increase the blood concentration ofglutathione [124]. The only available clinical trial onN-acetylcysteine in a small population revealedsome potential neuroprotective effects against oxaliplatin-induced peripheral neurotoxicity [124](Table 2.3). D-Methionine, a sulfur-containing nucleophilic antioxidant, has also shown successfulneuroprotectionagainstcisplatin-inducedneurotoxicityincorticalnetworkinvitro[189].
Electrolytes,chelators,andionchannelmodulators
Theelectrolytes calciumandmagnesiummayact as chelators againstoxalateaccumulationandwillprobably protect the voltage-gated sodium channels from alteration [140]. Among six availablerandomized clinical trials conducted to evaluate the efficacy of calcium/magnesium infusion againstoxaliplatin-inducedperipheralneurotoxicity (Table2.3), twopreliminary studieswereunsuccessful inshowing any protection by intravenous calcium/magnesium [140, 141]. Knijn et al. [139], in aretrospectiveanalysis studyonpatientswithoxaliplatin-inducedperipheral neurotoxicity, couldonlyfind reduced rate of grade 1 peripheral neurotoxicity, considering the high risk of bias. Later, twoclinical studies have shown some levels of neuroprotection against the development of oxaliplatin-inducedneuropathy [138].However, the twomost recent trialsdidnot confirmtheneuroprotectiverole of calcium/ magnesium against oxaliplatin-induced neuropathy [137, 136]. Moreover, Han et al.[136]haveshownthatcalciumandmagnesium infusionsdonotalter thepharmacokineticsofeitherintactoxaliplatinor freeplatinum,whereas therewasnoevidenceof apharmacokinetic interactionbetweencalcium/magnesiumandoxaliplatin,meaningthattheseinfusionsmayprovidenobenefitinreducingacuteoxaliplatin-inducedperipheralneurotoxicity.
Carbamazepine and oxcarbazepine are known as antiseizure drugs. They block voltage-sensitivesodiumchannelsandsomecalciumchannels,whichmightprotectthevoltage-gatedsodiumchannelsfrom alteration by oxalate [55, 190]. These two agents have also been tested in clinical trials withrelativelysmallsamplesizes,withoneshowingneutraleffect[142]andtheothersuggestingbenefit,althoughwithanunblindedcontrolarm[160](Table2.3).
Nimodipineisacalciumchannelblockerthatdidnotshowsignificantneuroprotectionagainstcisplatinin the only clinical trial ever done [155]. Although the trial had to be terminated because of severegastrointestinaltoxicity,theresultsbythattimedidnotsupportneuroprotection.Amulticentertrialon diethyldithiocarbamate, a chelating agent and antioxidant that prevents the degradation ofextracellularmatrix as an initial step in cancermetastasis and angiogenesis, did not demonstrate asignificantchemoprotectiveeffectagainstcisplatin-inducedneurotoxicity[143](Table2.3).
Othercompounds
Therearealsosomedatasupporting thepreventiveeffectofotheragentsagainstplatinum-inducedperipheral neurotoxicity. Acetyl-L-carnitine is a natural compound that plays a role in intermediarymetabolismandhasanantioxidantactivity [191]. Invitrodatasupport itseffectiveness forplatinum-induced neurotoxicity [179], but a recent randomized double-blinded placebo-controlled trialdiscouraged its administration for a non-platinum agent [192]. Xaliproden is a 5-hydroxytryptamine(HT)1Aagonist that alsoacts as aneuromodulatorwithneurotrophic andneuroprotectiveeffects invitro[193]andhadpositiveresultsinaclinicalsettingaswell[168],althoughtheresultshaveyettobepublished. Venlafaxine is a serotonin-norepinephrine reuptake inhibitor that also modulates theoxidative stress in the nervous system and may block sodium channels, which showed someneuroprotectiveeffectinasmallclinicaltrial[163].
�31
2

Chapter2 TheOncologist2015;20:411-432
Goshajinkigan(Kampomedicine),composedof10naturalingredients,isfrequentlyusedforalleviatingsymptoms of diabetic peripheral neuropathy in Japan; it is shown to have some neuroprotectivepotentials [194, 195].Moreover, its safety and efficacy for preventing oxaliplatin-induced peripheralneurotoxicityhavebeentestedintwoclinicaltrials[153,154](Table2.3).Nishiokaetal.[154]reportedasignificantlylowerincidenceofgrade3peripheralneurotoxicity,althoughbasedonasmallsamplesizewith unblinded control group. The findings of a phase II, multicenter, randomized, double-blind,placebo-controlledtrialbyKonoetal. [153]werealsosuggestiveof reducedbut insignificant rateofperipheral neurotoxicity grade 2 and 3 in patients treated with oxaliplatin compared with placebo(incidenceofgrade2neuropathyuntiltheeighthcycle:39%and51%intheKampoandplacebogroups,respectively[relativerisk,0.76;95%CI0.47–1.21];andgrade3:7%versus13%[0.51,0.14–1.92]).
NondrugApproach
Thereareothermodalities thatmightenhance theeffectivenessof the treatmentwhilediminishingside effects or prevent peripheral neurotoxicity. It might be helpful to identify risk factors forneurotoxicity, such as pre-existing neuropathy, inherited neuropathies, age-related axonal loss,diabetes mellitus, alcohol abuse, and poor nutritional status, that may predispose to more severesymptomsfromplatinum-inducedperipheralneurotoxicity[196].
Timing in drug administration to account for biological rhythms (chronotherapy) seems also veryimportant [197], because there aredrugs anddisease conditions, including cancers suggestiveof anoptimal circadian timeofdrugadministration [198].However, inameta-analysisonfive randomizedcontrolled trialswith958patients, therewasno significantdifference in the incidenceofperipheralsensoryneuropathyafterchronomodulation[199].
Finally, regarding the paucity of evidence about preventive and therapeutic strategies, treatmentmodificationanddrugwithdrawal remain themosteffectivemodalities formajorityofpatients [31],whichindeednecessitatesmoreadequatelypoweredpreclinicalandclinicalresearchestofindbetteralternativemodalities[200,201].
Neuro-VersusChemoprotection
It isessentialtodemonstratewhethertheapplicationofaneuroprotectiveagentmightdiminishtheefficacyofthetherapeuticagent.However,thispotentialadverseeffecthasbeentestedwithdifferentagents. As an example, cisplatin and carboplatin are used in induction and myeloablativechemotherapy for high-risk neuroblastoma, but because of significant ototoxicity in children, theiradministration may be compromised. Harned et al. [169] showed that the exposure of sixneuroblastomacelllinestoSTS,at6hoursaftercisplatin,didnotbindtoandeliminatethecirculatingcytotoxiccompoundandthusdidnotaffect theantitumoreffectof theplatinumagent,evenunderhypoxicconditions.However,asignificantundesiredprotectionagainstcisplatincytotoxicitywasseenwhentheneuroblastomacellsweresimultaneouslyexposedtobothcisplatinandSTScombinations.Moreover,Harnedetal.[169]demonstratedthatinasubcutaneousneuroblastomaxenograftmodelinnu/numice,mice receiving cisplatin aloneor cisplatinplus STS after6hourshad significantlybetterprogression-free survival rates (p < .03) compared with controls or mice treated with concurrentcisplatin and STS administration. Likewise, Muldoon et al. [170] reported that delaying theadministrationofSTSfor6–8hoursaftercarboplatindidnotreduceitsantitumoractivityinahumansmallcelllungcancerxenograftmodelintherat,butstillprotectedagainstototoxicityinguineapigs.Moreover, Dickey et al. [171] found that adding STS simultaneously or up to 2 hours postcisplatinprotected against the antitumor effect of cisplatin in glioblastoma, SKOV3 ovarian carcinoma,medulloblastoma,andsmallcelllungcancercelllines,butthatdelayedSTSadministrationfor6hoursdidnotshowasignificantchemoprotectioninanyofthecelltypes.
�32
CME

Chapter2 TheOncologist2015;20:411-432
TheuseofSTStopreventhearinglossinchildrenwithavarietyofmalignancieshasbeentestedintwophase III randomizedtrialsSIOPEL6(NCT00652132) [172]andCOGACCL0431 (NCT00716976) [173]. Inthe preliminary report of COG, presented at the 2014 annual American Society of Clinical Oncology(ASCO)meeting[173],aprotectiveeffectofSTSwasfoundinreducingtheproportionofhearinglosscompared with observation (29% versus 55%; p = .006). In this trial, 126 cancer patients wererandomizedtoeithercisplatin infusionsaloneor,topreventcisplatin-inducedhearingloss,combinedwithSTSat16g/m2IVover15minutesbeginning6hoursafterthecompletionofeachcisplatindose.Themedianpostdiagnosisfollow-upwas2.1years.However,thepotentiallylowersurvivalseeninthepatientswithdisseminateddisease receivingSTS (event-freesurvival60%versus70%,p = .53;overallsurvival75%versus89%,p=.50)raisessomeconcernofatumor-protectiveeffectofSTS.
Treatment
Theefficacyofsomeantidepressants,anticonvulsants,andatopicalgelhasbeentestedinsixtrialsfortreatment of platinum-induced peripheral neurotoxicity. Smith et al. [202] studied the effect ofduloxetineinarandomized,placebo-controlled,crossovertrialof231patientswitheitherplatinumortaxane neurotoxicity. Patients received 30mg of duloxetine for the first week and 60mg of dailyduloxetine for 4 more weeks. Duloxetine significantly reduced pain and paresthesia, especially inoxaliplatingroup.Incontrast,50mgofdailyamitriptylineor100mgofnortriptyline, intwoseparatetrials, failed todemonstrate any significant improvements inpatient-reported sensory symptoms, aswell asobjective scorings [203]. Similarly, trials testinggabapentinat a targetdoseof 2,700mg/day[204] or lamotrigine at a target dose of 300 mg/day [205] failed to demonstrate any benefit fortreatmentofperipheralneurotoxicity.Finally,onetrialevaluatedacompoundedtopicalgelcontainingbaclofen(10mg),amitriptylineHCl(40mg),andketamine(20mg)on208randomlyallocatedpatients[206],andasignificantimprovementinmotorsubscalescoreswasobserved.
Future
Thereare somepromising results favoring theabilityof someneuroprotectiveagents to reduce therateof subsequentneurotoxicity inducedbyplatinumanalogs.However, themost recentupdateoftheCochranereviewonchemo-neuroprotectiveagentsfoundinsufficientdatatoconcludethatanyoftheavailablechemoprotectiveagentsissufficientlyeffectiveinpreventingorlimitingtheneurotoxicityofplatinumdrugs.Albersetal. [9] reviewed29randomizedcontrolledtrials (RCTs)orquasi-RCTs, inwhich2,906participants receivedchemotherapywithcisplatinor relatedcompounds.Patientswerealsoevaluatedforquantitativesensorytesting(primaryoutcome)orothermeasures includingnerveconductionorneurologicalimpairmentratingusingvalidatedscales(secondaryoutcomes)beforeand6monthsaftercompletingchemotherapy(Table2.3).Likewise,themostrecentASCOClinicalPracticeGuideline [123] based on a systematic review on 48 RCTs, including 35 RCTs on platinum-inducedperipheral sensory neurotoxicity, did not recommend any established agent for the prevention ofplatinum-inducedperipheralneurotoxicity.Only for thetreatmentofexistingoxaliplatinneuropathy,they advised duloxetine, for which intermediate strength of evidence is present, considering thebalancebetweenbenefitandharm[123].
Altogether, no neuroprotective strategy can yet be recommended for prevention and treatment ofplatinum- induced neurotoxicity, with a possible exception for duloxetine for oxaliplatin. There is ageneticdiversitybetweenpatients, leadingtodifferencesindrugresponseincludingthesideeffects;henceaneuropathypreventivestrategyshouldbeindividualizedforeachpatient.
Apharmacogeneticapproachmightbeuseful inunderstandingthecauseofperipheralneurotoxicityand tailoring the most suitable chemotherapy for each patient. A genome-wide pharmacogenomicapproachmayalsobeusefulinidentifyingnovelpolymorphismpredictorsofsevereplatinum-inducedperipheral neurotoxicity thatmay be used in personalized chemotherapy [71]. However, it is highly
�33
2

Chapter2 TheOncologist2015;20:411-432
recommendedthatthepositiveandnegativeeffectsoftheantineoplasticagentsbestudiedindetailinpreclinicalsettingsbeforeimplementationinclinicalpractice.Inparticular,thecentralnervoussystemofanimalscanbeusedtoquantifytheeffectsofplatinatedcompoundsonneuronsthatcorroborateclinical data and suggest them as suitable models for studying possible neurotoxicity of platinumagents [111–121]. Some in vitro models can also be used to investigate morphological parametersaffected by platinum compounds [122]. Thesemodels enablemeasuring the effect of the drugs onneuronsalongwithtestingtheneurogenicpotentialofneuroprotectivecompounds.
Conclusion
Our knowledge about the pathophysiology of platinum-induced peripheral neurotoxicity andsuggestedneuroprotectivestrategies isdiverseandnotadequatelypowered.Therefore,a thoroughinvestigation of available evidence is important to design new, solid studies to tailor appropriatetreatmenttoindividualpatients.Thiswillminimizetheburdenofperipheralneurotoxicity,optimizingthepotentiallypositiveimpactofthechemotherapeuticmedication.
Correspondenceto:GodefridusJ.Peters,Ph.D.,DepartmentofMedicalOncology,CancerCenterAmsterdam,VUUniversityMedicalCenter,DeBoelelaan 1117, 1081HVAmsterdam,TheNetherlands.Telephone:31-20-4442633;E-Mail:[email protected]
Acknowledgement This work was partially supported by a grant from Cancer Center AmsterdamFoundation2012(toElisaGiovannetti,AmirAvan,andGodefridusJ.Peters).
AuthorContributions
Conception/Design:AbolfazlAvan,TjeerdJ.Postma,GodefridusJ.PetersProvisionofstudymaterialorpatients:AbolfazlAvan,GodefridusJ.PetersCollectionand/orassemblyofdata:AbolfazlAvanDataanalysisandinterpretation:AbolfazlAvan,TjeerdJ.Postma,GuidoCavaletti,ElisaGiovannettiManuscriptwriting:AbolfazlAvan,Tjeerd J.Postma,CeciliaCeresa,AmirAvan,GuidoCavaletti, ElisaGiovannetti,GodefridusJ.PetersFinal approval of manuscript: Abolfazl Avan, Tjeerd J. Postma, Guido Cavaletti, Elisa Giovannetti,GodefridusJ.Peters
DisclosuresTheauthorsindicatednofinancialrelationships.
References
1. FuertesMA,CastillaJ,AlonsoCetal.Novelconceptsinthedevelopmentofplatinumantitumordrugs.CurrMedChemAnticancerAgents2002;2:539–551.
2. Boulikas T, Pantos A, Bellis E et al. Designing platinum compounds in cancer: Structures andmechanisms.CancerTher2007;5:537–583.
3. PichlerV,MayrJ,HeffeterPetal.Maleimide-functionalisedplatinum(IV)complexesasasyntheticplatformfortargeteddrugdelivery.ChemCommun(Camb)2013;49:2249–2251.
4. Ali I,WaniWA,SaleemKet al. Platinumcompounds:Ahope for future cancer chemotherapy.AnticancerAgentsMedChem2013;13:296–306.
5. ArgyriouAA,BrunaJ,MarmiroliPetal.Chemotherapy-inducedperipheralneurotoxicity(CIPN):Anupdate.CritRevOncolHematol2012;82:51–77.
6. CavalettiG.Chemotherapy-inducedperipheralneurotoxicity(CIPN):Whatweneedandwhatweknow.JPeripherNervSyst2014;19:66–76.
7. MollmanJE.Cisplatinneurotoxicity.NEnglJMed1990;322:126–127.
�34
CME

Chapter2 TheOncologist2015;20:411-432
8. PrestaykoAW,D’AoustJC,IssellBFetal.Cisplatin(cis-diamminedichloroplatinumII).CancerTreatRev1979;6:17–39.
9. Albers JW, Chaudhry V, Cavaletti G et al. Interventions for preventing neuropathy caused bycisplatinandrelatedcompounds.CochraneData-baseSystRev2014;3:CD005228.
10. deCastriaTB,daSilvaEM,GoisAFetal.Cisplatinversuscarboplatin incombinationwiththird-generationdrugs for advancednon-small cell lungcancer.CochraneDatabaseSystRev2013;8:CD009256.
11. Ibrahim A, Hirschfeld S, Cohen MH et al. FDA drug approval summaries: Oxaliplatin. TheOncologist2004;9:8–12.
12. McWhinneySR,GoldbergRM,McLeodHL.Platinumneurotoxicitypharmacogenetics.MolCancerTher2009;8:10–16.
13. GrotheyA.Oxaliplatin-safetyprofile:Neuro-toxicity.SeminOncol2003;30(suppl15):5–13.14. GrotheyA,GoldbergRM.Areviewofoxaliplatinand itsclinicaluse incolorectalcancer.Expert
OpinPharmacother2004;5:2159–2170.15. ArgyriouAA,BrianiC,CavalettiGetal.Advancedageandliabilitytooxaliplatin-inducedperipheral
neuropathy:Posthocanalysisofapro-spectivestudy.EurJNeurol2013;20:788–794.16. Argyriou AA, Cavaletti G, Briani C et al. Clinical pattern and associations of oxaliplatin acute
neurotoxicity:Aprospectivestudyin170patientswithcolorectalcancer.Cancer2013;119:438–444.17. ArgyriouAA,VelascoR,BrianiCetal.Peripheralneurotoxicityofoxaliplatinincombinationwith5-
fluorouracil(FOLFOX)orcapecitabine(XELOX):Aprospectiveevaluationof150colorectalcancerpatients.AnnOncol2012;23:3116–3122.
18. ParkSB,LinCS,KrishnanAVetal.Oxaliplatin-inducedneurotoxicity:Changesinaxonalexcitabilityprecededevelopmentofneuropathy.Brain2009;132:2712–2723.
19. CavalettiG,AlbertiP,MarmiroliP.Chemotherapy-inducedperipheralneurotoxicity intheeraofpharmacogenomics.LancetOncol2011;12:1151–1161.
20. ArgyriouAA,CavalettiG,AntonacopoulouAetal.Voltage-gatedsodiumchannelpolymorphismsplay a pivotal role in the development of oxaliplatin- induced peripheral neurotoxicity: Resultsfromaprospectivemulticenterstudy.Cancer2013;119:3570–3577.
21. vanAsJW,vandenBergH,vanDalenEC.Medicalinterventionsforthepreventionofplatinum-inducedhearinglossinchildrenwithcancer.CochraneDatabaseSystRev2014;7:CD009219.
22. McHaney VA, Thibadoux G, Hayes FA et al. Hearing loss in children receiving cisplatinchemotherapy.JPediatr1983;102:314–317.
23. BertoliniP,LassalleM,MercierGetal.Platinumcompound-relatedototoxicityinchildren:Long-termfollow-uprevealscontinuousworseningofhearing loss.JPediatrHematolOncol2004;26:649–655.
24. McKeageMJ.Comparativeprofilesofplatinumdrugs.DrugSaf1995;13:228–244.25. BrydøyM,OldenburgJ,KleppOetal.Observationalstudyofprevalenceoflong-termRaynaud-
like phenomena and neurological side effects in testicular cancer survivors. J Natl Cancer Inst2009;101:1682–1695.
26. ThomasJP,LautermannJ,LiedertBetal.Highaccumulationofplatinum-DNAadducts in strialmarginal cells of the cochlea is an early event in cisplatin but not carboplatin ototoxicity.MolPharmacol2006;70:23–29.
27. Verstappen CC, Heimans JJ, Hoekman K et al. Neurotoxic complications of chemotherapy inpatientswithcancer:Clinicalsignsandoptimalmanagement.Drugs2003;63:1549–1563.
28. HillA,BerginP,HanningFetal.Detectingacuteneurotoxicityduringplatinumchemotherapybyneurophysiologicalassessmentofmotornervehyperexcitability.BMCCancer2010;10:451.
29. WilsonRH,LehkyT,ThomasRRetal.Acuteoxaliplatin-inducedperipheralnervehyperexcitability.JClinOncol2002;20:1767–1774.
30. KrishnanAV,GoldsteinD,FriedlanderMetal.OxaliplatinandaxonalNa+channelfunctioninvivo.ClinCancerRes2006;12:4481–4484.
31. Cavaletti G, Alberti P, Frigeni B et al. Chemotherapy-induced neuropathy. Curr Treat OptionsNeurol2011;13:180–190.
�35
2

Chapter2 TheOncologist2015;20:411-432
32. AlbertiP,RossiE,CornblathDRetal.Physician-assessedandpatient-reportedoutcomemeasuresinchemotherapy-inducedsensoryperipheralneurotoxicity:Twosidesofthesamecoin.AnnOncol2014;25:257–264.
33. BrundageMD, Pater JL, Zee B. Assessing the reliability of two toxicity scales: Implications forinterpretingtoxicitydata.JNatlCancerInst1993;85:1138–1148.
34. KoeppenS,VerstappenCC,KorteRetal.LackofneuroprotectionbyanACTH(4-9)analogue.ArandomizedtrialinpatientstreatedwithvincristineforHodgkin’sornon-Hodgkin’slymphoma.JCancerResClinOncol2004;130:153–160.
35. Postma TJ, Heimans JJ, Muller MJ et al. Pitfalls in grading severity of chemotherapy-inducedperipheralneuropathy.AnnOncol1998;9:739–744.
36. InoueN,IshidaH,SanoMetal.DiscrepancybetweentheNCI-CTCAEandDEB-NTCscalesintheevaluationofoxaliplatin-relatedneurotoxicityinpatientswithmetastaticcolorectalcancer.IntJClinOncol2012;17:341–347.
37. SprowlJA,CiarimboliG,LancasterCSetal.Oxaliplatin-inducedneurotoxicityisdependentontheorganiccationtransporterOCT2.ProcNatlAcadSciUSA2013;110:11199–11204.
38. Jong NN, Nakanishi T, Liu JJ et al. Oxaliplatin transport mediated by organic cation/carnitinetransporters OCTN1 and OCTN2 in overexpressing human embryonic kidney 293 cells and ratdorsalrootganglionneurons.JPharmacolExpTher2011;338:537–547.
39. Ip V, Liu JJ, McKeage MJ. Evaluation of effects of copper histidine on copper transporter 1-mediatedaccumulationofplatinumandoxaliplatin-inducedneurotoxicityinvitroandinvivo.ClinExpPharmacolPhysiol2013;40:371–378.
40. LiuJJ,KimY,YanFetal.ContributionsofratCtr1totheuptakeandtoxicityofcopperandplatinumanticancerdrugsindorsalrootganglionneurons.BiochemPharmacol2013;85:207–215.
41. IshidaS,LeeJ,ThieleDJetal.Uptakeof theanticancerdrugcisplatinmediatedbythecoppertransporterCtr1inyeastandmammals.ProcNatlAcadSciUSA2002;99:14298–14302.
42. Safaei R. Role of copper transporters in the uptake and efflux of platinum containing drugs.CancerLett2006;234:34–39.
43. GuminskiAD,BalleineRL,ChiewYEetal.MRP2(ABCC2)andcisplatinsensitivity inhepatocytesandhumanovariancarcinoma.GynecolOncol2006;100:239–246.
44. IpV,LiuJJ,MercerJFetal.DifferentialexpressionofATP7A,ATP7BandCTR1inadultratdorsalrootgangliontissue.MolPain2010;6:53.
45. YamasakiM,MakinoT,MasuzawaTetal.Roleofmultidrugresistanceprotein2(MRP2)inchemo-resistanceand clinical outcome inoesophageal squamous cell carcinoma.Br JCancer 2011;104:707–713.
46. Zhu C, Raber J, Eriksson LA. Hydrolysis process of the second generation platinum-basedanticancerdrugcis-amminedichlorocyclohexylamineplatinum(II).JPhysChemB2005;109:12195–12205.
47. ChválováK,BrabecV,KaspárkováJ.MechanismoftheformationofDNA-proteincross-linksbyantitumorcisplatin.NucleicAcidsRes2007;35:1812–1821.
48. McDonald ES, Randon KR, Knight A et al. Cisplatin preferentially binds to DNA in dorsal rootganglionneuronsinvitroandinvivo:Apotentialmechanismforneurotoxicity.NeurobiolDis2005;18:305–313.
49. DzagnidzeA, Katsarava Z,Makhalova J et al. Repair capacity for platinum-DNA adducts deter-minestheseverityofcisplatin-inducedperipheralneuropathy.JNeurosci2007;27:9451–9457.
50. TaLE,EspesetL,PodratzJetal.Neurotoxicityofoxaliplatinandcisplatinfordorsalrootganglionneuronscorrelateswithplatinum-DNAbinding.Neurotoxicology2006;27:992–1002.
51. Gregg RW, Molepo JM, Monpetit VJ et al. Cisplatin neurotoxicity: The relationship betweendosage, time, and platinum concentration in neurologic tissues, andmorphologic evidence oftoxicity.JClinOncol1992;10:795–803.
52. McKeageMJ,HsuT,ScrenciDetal.Nucleolardamagecorrelateswithneurotoxicityinducedbydifferentplatinumdrugs.BrJCancer2001;85:1219–1225.
53. NamikawaK,AsakuraM,MinamiTetal.Toxicityofcisplatintothecentralnervoussystemofmalerabbits.BiolTraceElemRes2000;74:223–235.
�36
CME

Chapter2 TheOncologist2015;20:411-432
54. OliviA,GilbertM,DuncanKLetal.Directdeliveryofplatinum-basedantineoplasticstothecentralnervoussystem:Atoxicityandultrastructuralstudy.CancerChemotherPharmacol1993;31:449–454.
55. AdelsbergerH,QuasthoffS,GrosskreutzJetal.Thechemotherapeuticoxaliplatinaltersvoltage-gatedNa+channelkineticsonratsensoryneurons.EurJPharmacol2000;406:25–32.
56. GrolleauF,StankiewiczM,Birinyi-StrachanLetal.Electrophysiologicalanalysisoftheneurotoxicactionofafunnel-webspidertoxin,delta-atracotoxin-HV1a,oninsectvoltage-gatedNa+channels.JExpBiol2001;204:711–721.
57. Sittl R, Lampert A, Huth T et al. Anticancer drug oxaliplatin induces acute cooling-aggravatedneuropathyviasodiumchannelsubtypeNaV1.6-resurgentandpersistentcurrent.ProcNatlAcadSciUSA2012;109:6704–6709.
58. DimitrovVG,ArabadzhievTI,DimitrovaNAetal.Thespectralchanges inEMGduringasecondbouteccentriccontractioncouldbeduetoadaptationinmusclefibresthemselves:Asimulationstudy.EurJApplPhysiol2012;112:1399–1409.
59. KagiavaA,TsingotjidouA,EmmanouilidesCetal.Theeffectsofoxaliplatin,ananticancerdrug,onpotassiumchannelsof theperipheralmyelinatednervefibresof theadult rat.Neurotoxicology2008;29:1100–1106.
60. MogyorosI,LinCS,KuwabaraSetal.Strength-durationpropertiesandtheirvoltagedependenceasmeasuresofa thresholdconductanceat thenodeofRanvierofsinglemotoraxons.MuscleNerve2000;23:1719–1726.
61. RaymondE,ChaneySG,TaammaAetal.Oxaliplatin:Areviewofpreclinicalandclinicalstudies.AnnOncol1998;9:1053–1071.
62. PodratzJL,KnightAM,TaLEetal.Cisplatin inducedmitochondrialDNAdamage indorsal rootganglionneurons.NeurobiolDis2011;41:661–668.
63. SukR,GurubhagavatulaS,ParkSetal.Poly-morphismsinERCC1andgrade3or4toxicityinnon-smallcelllungcancerpatients.ClinCancerRes2005;11:1534–1538.
64. Argyriou AA, Kyritsis AP, Makatsoris T et al. Chemotherapy-induced peripheral neuropathy inadults:Acomprehensiveupdateoftheliterature.CancerManagRes2014;6:135–147.
65. Bergmann TK, Gréen H, Brasch-Andersen C et al. Retrospective study of the impact ofpharmacogeneticvariantsonpaclitaxeltoxicityandsurvivalinpatientswithovariancancer.EurJClinPharmacol2011;67:693–700.
66. GréenH,SöderkvistP,RosenbergPetal.PharmacogeneticstudiesofPaclitaxelinthetreatmentofovariancancer.BasicClinPharmacolToxicol2009;104:130–137.
67. Marsh S, Paul J, King CR et al. Pharmacogenetic assessment of toxicity and outcome afterplatinumplustaxanechemotherapyinovariancancer:TheScottishRandomisedTrial inOvarianCancer.JClinOncol2007;25:4528–4535.
68. McLeod HL, Sargent DJ, Marsh S et al. Pharmacogenetic predictors of adverse events andresponse to chemotherapy in metastatic colorectal cancer: Results from North AmericanGastrointestinalIntergroupTrialN9741.JClinOncol2010;28:3227–3233.
69. CecchinE,D’AndreaM,LonardiSetal.Aprospectivevalidationpharmacogenomicstudyintheadjuvantsettingofcolorectalcancerpatientstreatedwiththe5-fluorouracil/leucovorin/oxaliplatin(FOLFOX4)regimen.PharmacogenomicsJ2013;13:403–409.
70. CustodioA,Moreno-Rubio J,Aparicio J et al. Pharmacogenetic predictors of severeperipheralneuropathy in colon cancer patients treated with oxaliplatin-based adjuvant chemotherapy: AGEM-CADgroupstudy.AnnOncol2014;25:398–403.
71. WonHH,Lee J,Park JOetal.Polymorphicmarkersassociatedwith severeoxaliplatin-induced,chronicperipheralneuropathyincoloncancerpatients.Cancer2012;118:2828–2836.
72. Gamelin L, Capitain O, Morel A et al. Predictive factors of oxaliplatin neurotoxicity: Theinvolvementoftheoxalateoutcomepathway.ClinCancerRes2007;13:6359–6368.
73. Lee KH, Chang HJ, Han SW et al. Pharmacogenetic analysis of adjuvant FOLFOX for Koreanpatientswithcoloncancer.CancerChemotherPharmacol2013;71:843–851.
�37
2

Chapter2 TheOncologist2015;20:411-432
74. InadaM,SatoM,MoritaSetal.Associationsbetweenoxaliplatin-inducedperipheralneuropathyandpolymorphismsoftheERCC1andGSTP1genes.IntJClinPharmacolTher2010;48:729–734.
75. KimHS,KimMK,ChungHHetal.Geneticpolymorphismsaffectingclinicaloutcomesinepithelialovarian cancer patients treatedwith taxanes and platinum compounds: A Korean population-basedstudy.GynecolOncol2009;113:264–269.
76. FungC,VaughnDJ,MitraNetal.Chemotherapyrefractorytesticulargermcell tumor isassoci-atedwithavariant inArmadilloRepeatgenedeleted inVelco-Cardio-Facial syndrome (ARVCF).FrontEndocrinol(Lausanne)2012;3:163.
77. Hong J, Han SW, Ham HS et al. Phase II study of biweekly S-1 and oxaliplatin combinationchemotherapyinmetastaticcolorectalcancerandpharmacogeneticanalysis.CancerChemotherPharmacol2011;67:1323–1331.
78. BoigeV,MendiboureJ,PignonJPetal.Pharmacogeneticassessmentoftoxicityandout-comeinpatients with metastatic colorectal cancer treated with LV5FU2, FOLFOX, and FOLFIRI: FFCD2000-05.JClinOncol2010;28:2556–2564.
79. CaponigroF,LacombeD,TwelvesCetal.AnEORTCphaseIstudyofBortezomibincombinationwithoxaliplatin, leucovorinand5-fluorouracil inpatientswithadvancedcolorectalcancer.EurJCancer2009;45:48–55.
80. Chen YC, Tzeng CH, Chen PM et al. Influence of GSTP1 I105V polymorphism on cumulativeneuropathy and outcome of FOLFOX-4 treatment in Asian patientswith colorectal carcinoma.CancerSci2010;101:530–535.
81. Goekkurt E, Al-Batran SE, Hartmann JT et al. Pharmacogenetic analyses of a phase III trial inmetastatic gastroesophageal adenocarcinoma with fluorouracil and leucovorin plus eitheroxaliplatinorcisplatin:AstudyoftheArbeitsgemeinschaftInternistischeOnkologie.JClinOncol2009;27:2863–2873.
82. Isla D, Sarries C, Rosell R et al. Single nucleotide polymorphisms and outcome in docetaxel-cisplatin-treatedadvancednon-small-celllungcancer.AnnOncol2004;15:1194–1203.
83. KeamB, Im SA,Han SWet al.Modified FOLFOX- 6 chemotherapy in advanced gastric cancer:Results of phase II study and comprehensive analysis of polymorphisms as a predictive andprognosticmarker.BMCCancer2008;8:148.
84. KhruninAV,MoisseevA,GorbunovaVetal.Geneticpolymorphismsandtheefficacyandtoxicityofcisplatin-basedchemotherapyinovariancancerpatients.PharmacogenomicsJ2010;10:54–61.
85. Ruzzo A, Graziano F, Loupakis F et al. Pharmacogenetic profiling in patients with advancedcolorectalcancertreatedwithfirst-lineFOLFOX-4chemotherapy.JClinOncol2007;25:1247–1254.
86. SeoBG,KwonHC,OhSYetal.Comprehensiveanalysisofexcisionrepaircomplementationgroup1, glutathione S-transferase, thymidylate synthase and uridine diphosphate glucuronosyltransferase 1A1 polymorphisms predictive for treatment out- come in patients with advancedgastriccancertreatedwithFOLFOXorFOLFIRI.OncolRep2009;22:127–136.
87. ZarateR,RodríguezJ,BandresEetal.Oxaliplatin,irinotecanandcapecitabineasfirst-linetherapyinmetastaticcolorectalcancer(mCRC):Adose-findingstudyandpharmacogenomicanalysis.BrJCancer2010;102:987–994.
88. Kumamoto K, Ishibashi K, Okada N et al. Polymorphisms of GSTP1, ERCC2 and TS-39UTR areassociated with the clinical outcome of mFOLFOX6 in colorectal cancer patients. Oncol Lett2013;6:648–654.
89. LecomteT,LandiB,BeaunePetal.GlutathioneS-transferaseP1polymorphism(Ile105Val)predictscumulative neuropathy in patients receiving oxaliplatin-based chemotherapy. Clin Cancer Res2006;12:3050–3056.
90. OldenburgJ,KraggerudSM,BrydøyMetal.Associationbetween long-termneuro-toxicities intesticular cancer survivors and polymorphisms in glutathione-S-transferase-P1 and -M1, aretrospectivecrosssectionalstudy.JTranslMed2007;5:70.
91. StoehlmacherJ,ParkDJ,ZhangWetal.AssociationbetweenglutathioneS-transferaseP1,T1,andM1geneticpolymorphismandsurvivalofpatientswithmetastaticcolorectalcancer.JNatlCancerInst2002;94:936–942.
�38
CME

Chapter2 TheOncologist2015;20:411-432
92. AntonacopoulouAG,ArgyriouAA,ScopaCDetal. Integrinbeta-3L33P:Anew insight into thepathogenesisofchronicoxaliplatin-inducedperipheralneuropathy?EurJNeurol2010;17:963–968.
93. LiQF,YaoRY,LiuKWetal.Geneticpoly-morphismofGSTP1:Predictionofclinicaloutcometooxaliplatin/5-FU-basedchemotherapyinadvancedgastriccancer.JKoreanMedSci2010;25:846–852.
94. KanaiM,YoshiokaA,TanakaSetal.AssociationsbetweenglutathioneS-transferasepiIle105Valand glyoxylate aminotransferase Pro11- Leu and Ile340Met polymorphisms and early-onsetoxaliplatin-inducedneuropathy.CancerEpidemiol2010;34:189–193.
95. Booton R, Ward T, Heighway J et al. Glutathione-S-transferase P1 isoenzyme polymorphisms,platinum-basedchemotherapy,andnon-smallcelllungcancer.JThoracOncol2006;1:679–683.
96. Kweekel DM, GelderblomH, Antonini NF et al. Glutathione-S-transferase pi (GSTP1) codon 105polymorphismisnotassociatedwithoxaliplatinefficacyortoxicityinadvancedcolorectalcancerpatients.EurJCancer2009;45:572–578.
97. ParéL,MarcuelloE,AltésAet al.Pharmacogeneticpredictionof clinicaloutcome inadvancedcolorectal cancer patients receiving oxaliplatin/5- fluorouracil as first-line chemotherapy. Br JCancer2008;99:1050–1055.
98. ArgyriouAA,AntonacopoulouAG,ScopaCDetal.Liabilityofthevoltage-gatedsodiumchannelgeneSCN2AR19Kpolymorphismtooxaliplatin-inducedperipheralneuropathy.Oncology2009;77:254–256.
99. AshrafT,KisO,BanerjeeNetal.Drugtransportersatbrainbarriers:Expressionandregulationbyneurologicaldisorders.AdvExpMedBiol2012;763:20–69.
100.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: Role of ATP-dependenttransporters.NatRevCancer2002;2:48–58.
101. LolliG,JohnsonLN.CAK-cyclin-dependentactivatingkinase:Akeykinaseincellcyclecontrolandatargetfordrugs?CellCycle2005;4:572–577.
102.WuG,CaoJ,PengCetal.TemporalandspatialexpressionofcyclinH in rat spinalcord injury.NeuromolecularMed2011;13:187–196.
103.Weinshilboum RM. Pharmacogenomics: Catechol O-methyltransferase to thiopurine S-methyl-transferase.CellMolNeurobiol2006;26:539–561.
104.RossCJ,Katzov-EckertH,DubéMPetal.GeneticvariantsinTPMTandCOMTareassociatedwithhearinglossinchildrenreceivingcisplatinchemotherapy.NatGenet2009;41:1345–1349.
105.PussegodaK,RossCJ,VisscherHetal.ReplicationofTPMTandABCC3geneticvariantshighlyassociatedwithcisplatin-inducedhearinglossinchildren.ClinPharmacolTher2013;94:243–251.
106.PengZ,WangQ,GaoJetal.AssociationbetweenGSTP1Ile105Valpolymorphismandoxaliplatin-induced neuropathy: A systematic re- view and meta-analysis. Cancer Chemother Pharmacol2013;72:305–314.
107.Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use ofpharmacogenomicstoindividualizetherapy.CancerTreatRev2005;31:90–105.
108.ZhangX, JiangLP,YinYetal.XRCC1andXPDgeneticpolymorphismsandclinicaloutcomesofgastric cancer patients treatedwith oxaliplatin- based chemotherapy: Ameta-analysis. TumourBiol2014;35:5637–5645.
109.SégurelL,LafosseS,HeyerEetal.FrequencyoftheAGTPro11Leupolymorphisminhumans:Doesdietmatter?AnnHumGenet2010;74:57–64.
110. Cavaletti G, Miloso M, Nicolini G et al. Emerging role of mitogen-activated protein kinases inperipheralneuropathies.JPeripherNervSyst2007;12:175–194.
111. Bianchi R, Gilardini A, Rodriguez-Menendez V et al. Cisplatin-induced peripheral neuropathy:Neuroprotectionbyerythropoietinwithoutaffect-ingtumourgrowth.EurJCancer2007;43:710–717.
112. Cavaletti G, Gilardini A, Canta A et al. Bortezomib-induced peripheral neurotoxicity: Aneurophysiologicalandpathologicalstudyintherat.ExpNeurol2007;204:317–325.
113. Meregalli C, Canta A, Carozzi VA et al. Bortezomib-induced painful neuropathy in rats: Abehavioral,neurophysiologicalandpathologicalstudyinrats.EurJPain2010;14:343–350.
�39
2

Chapter2 TheOncologist2015;20:411-432
114. Müller LJ, Moorer-van Delft CM, Roubos EW. Snail neurons as a possible model for testingneurotoxicsideeffectsofantitumoragents:Para-crystalformationbyVincaalkaloids.CancerRes1988;48:7184–7188.
115. MüllerLJ,Moorer-vanDelftCM,RoubosEWetal.Quantitativeultrastructuraleffectsofcisplatin(Platinol), carboplatin (JM8), and iproplatin (JM9) on neurons of freshwater snail Lymnaeastagnalis.CancerRes1992;52:963–973.
116.Müller LJ, Moorer-van Delft CM, Zijl R et al. Use of snail neurons in developing quantitativeultrastructural parameters for neurotoxic side effects of Vinca antitumor agents. Cancer Res1990;50:1924–1928.
117. DiPaolaRS,SchuchterL.Neurologicprotectionbyamifostine.SeminOncol1999;26(suppl7):82–88.
118. Geldof AA. Nerve-growth-factor-dependent neurite outgrowth assay; a research model forchemotherapy-inducedneuropathy.JCancerResClinOncol1995;121:657–660.
119. Geldof AA, Minneboo A, Heimans JJ. Vinca- alkaloid neurotoxicity measured using an in vitromodel.JNeurooncol1998;37:109–113.
120.VerstappenCC,GeldofAA,PostmaTJetal.Invitroprotectionfromcisplatin-inducedneurotoxicitybyamifostineanditsmetaboliteWR1065.JNeurooncol1999;44:1–5.
121. VerstappenCC,PostmaTJ,GeldofAAetal.Amifostineprotectsagainstchemotherapy-inducedneurotoxicity:Aninvitroinvestigation.AnticancerRes2004;24:2337–2341.
122. Ceresa C, Avan A, Giovannetti E et al. Characterization of and protection from neurotoxicityinducedbyoxaliplatin,bortezomibandepothilone-B.AnticancerRes2014;34:517–523.
123.Hershman DL, Lacchetti C, Dworkin RH et al. Prevention andmanagement of chemotherapy-inducedperipheralneuropathyinsurvivorsofadultcancers:AmericanSocietyofClinicalOncologyclinicalpracticeguideline.JClinOncol2014;32:1941–1967.
124.LinCH,KuoSC,HuangLJetal.Neuro-protectiveeffectofN-acetylcysteineonneuronalapoptosisinducedbyasyntheticgingerdionecompound:InvolvementofERKandp38phosphorylation.JNeurosciRes2006;84:1485–1494.
125.GuoY,JonesD,PalmerJLetal.Oralalpha-lipoicacidtopreventchemotherapy-inducedperipheralneuropathy: A randomized, double-blind, placebo-controlled trial. Support Care Cancer 2014;22:1223–1231.
126.LuP,FanQX,WangLXetal.[Prophylacticeffectofamifostineonoxaliplatin-relatedneurotoxicityinpatientswithdigestivetracttumors].AiZheng2008;27:1117–1120.
127.DeVosFY,BosAM,SchaapveldMetal.ArandomizedphaseIIstudyofpaclitaxelwithcarboplatin1/2amifostineasfirstlinetreatmentinadvancedovariancarcinoma.GynecolOncol2005;97:60–67.
128.HilpertF,StähleA,Tome Oetal.Neuro-protectionwithamifostineinthefirst-linetreatmentofadvanced ovarian cancer with carboplatin/paclitaxel-based chemotherapy: A double-blind,placebo-controlled, randomized phase II study from the Arbeitsgemeinschaft GynäkologischeOnkologoie(AGO)OvarianCancerStudyGroup.SupportCareCancer2005;13:797–805.
129.KanatO,EvrenselT,BaranIetal.Protectiveeffectofamifostineagainsttoxicityofpaclitaxelandcarboplatininnon-smallcelllungcancer:Asinglecenterrandomizedstudy.MedOncol2003;20:237–245.
130.LeongSS,TanEH,FongKWetal.Randomizeddouble-blindtrialofcombinedmodalitytreatmentwithorwithoutamifostineinunresectablestageIIInon-smallcelllungcancer.JClinOncol2003;21:1767–1774.
131. LorussoD, FerrandinaG,GreggiSet al.Phase IIImulticenter randomized trialof amifostineascytoprotectant in first-line chemotherapy in ovarian cancer patients. Ann Oncol 2003;14:1086–1093.
132. GallardoD,MoharA, CalderilloG et al. Cisplatin, radiation, and amifostine in carcinomaof theuterinecervix.IntJGynecolCancer1999;9:225–230.
133.PlantingAS,CatimelG,deMulderPHetal.Randomizedstudyofashortcourseofweeklycisplatinwithorwithoutamifostineinadvancedheadandneckcancer.AnnOncol1999;10:693–700.
�40
CME

Chapter2 TheOncologist2015;20:411-432
134.KempG,RoseP,LurainJetal.Amifostinepretreatmentforprotectionagainstcyclophosphamide-induced and cisplatin-induced toxicities: Results of a randomized control trial in patients withadvancedovariancancer.JClinOncol1996;14:2101–2112.
135.MillerAA,WangXF,GuLetal.PhaseIIrandomizedstudyofdose-densedocetaxelandcisplatinevery 2weekswith pegfilgrastim and darbepoetin alfawith andwithout the chemoprotectorBNP7787 in patientswith advanced non-small cell lung cancer (CALGB 30303). J ThoracOncol2008;3:1159–1165.
136.HanCH,KhwaounjooP,KilfoyleDHetal.PhaseIdrug-interactionstudyofeffectsofcalciumandmagnesiuminfusionsonoxaliplatinpharmacokineticsandacuteneurotoxicityincolorectalcancerpatients.BMCCancer2013;13:495.
137.LoprinziCL,QinR,DakhilSRetal.PhaseIIIrandomized,placebo-controlled,double-blindstudyofintravenouscalciumandmagnesiumtopreventoxaliplatin-inducedsensoryneurotoxicity(N08CB/Alliance).JClinOncol2014;32:997–1005.
138.Grothey A, Nikcevich DA, Sloan JA et al. Intravenous calcium and magnesium for oxaliplatin-inducedsensoryneurotoxicityinadjuvantcoloncancer:NCCTGN04C7.JClinOncol2011;29:421–427.
139.KnijnN,TolJ,KoopmanMetal.Theeffectofprophylacticcalciumandmagnesiuminfusionsonthe incidence of neurotoxicity and clinical outcome of oxaliplatin-based systemic treatment inadvancedcolorectalcancerpatients.EurJCancer2011;47:369–374.
140.Ishibashi K,OkadaN,Miyazaki T et al. Effect of calcium andmagnesiumon neurotoxicity andblood platinum concentrations in patients receiving mFOLFOX6 therapy: A prospectiverandomizedstudy.IntJClinOncol2010;15:82–87.
141. ChayWY,TanSH,LoYLetal.Useofcalciumandmagnesiuminfusionsinpreventionofoxaliplatininducedsensoryneuropathy.AsiaPacJClinOncol2010;6:270–277.
142.von Delius S, Eckel F,Wagenpfeil S et al. Carbamazepine for prevention of oxaliplatin-relatedneurotoxicity in patients with advanced colorectal cancer: Final results of a randomised,controlled,multicenterphaseIIstudy.InvestNewDrugs2007;25:173–180.
143.Gandara DR, Nahhas WA, Adelson MD et al. Randomized placebo-controlled multicenterevaluationof diethyldithiocarbamate for chemoprotection against cisplatin-induced toxicities. JClinOncol1995;13:490–496.
144.Wang WS, Lin JK, Lin TC et al. Oral glutamine is effective for preventing oxaliplatin-inducedneuropathyincolorectalcancerpatients.TheOncologist2007;12:312–319.
145.LealAD,QinR,AthertonPJetal.NorthCentralCancerTreatmentGroup/AlliancetrialN08CA:Theuse of glutathione for prevention of paclitaxel/carboplatin-induced peripheral neuropathy: Aphase3randomized,double-blind,placebo-controlledstudy.Cancer2014;120:1890–1897.
146.Milla P, AiroldiM,Weber G et al. Administration of reduced glutathione in FOLFOX4 adjuvanttreatmentforcolorectalcancer:Effectonoxaliplatinpharmacokinetics,Pt-DNAadductformation,andneurotoxicity.AnticancerDrugs2009;20:396–402.
147.Cascinu S, Catalano V, Cordella L et al. Neuroprotective effect of reduced glutathione onoxaliplatin-based chemotherapy in advanced colorectal cancer: A randomized, double-blind,placebo-controlledtrial.JClinOncol2002;20:3478–3483.
148.SchmidingerM,BudinskyAC,WenzelCetal.Glutathione inthepreventionofcisplatin inducedtoxicities.Aprospectivelyrandomizedpilottrial inpatientswithheadandneckcancerandnonsmallcelllungcancer.WienKlinWochenschr2000;112:617–623.
149.SmythJF,BowmanA,PerrenTetal.Glutathionereducesthetoxicityandimprovesqualityoflifeof women diagnosed with ovarian cancer treated with cisplatin: Results of a double-blind,randomisedtrial.AnnOncol1997;8:569–573.
150.CascinuS,CordellaL,DelFerroEetal.Neuroprotectiveeffectofreducedglutathioneoncisplatin-basedchemotherapyinadvancedgastriccancer:Arandomizeddouble-blindplacebo-controlledtrial.JClinOncol1995;13:26–32.
151. ColomboN,BiniS,MiceliDetal.Weeklycisplatin1/2glutathioneinrelapsedovariancarcinoma.IntJGynecolCancer1995;5:81–86.
�41
2

Chapter2 TheOncologist2015;20:411-432
152.BogliunG,MarzoratiL,CavalettiGetal.Evaluationbysomatosensoryevokedpotentialsoftheneurotoxicityofcisplatinaloneorincombinationwithglutathione.ItalJNeurolSci1992;13:643–647.
153.KonoT,HataT,MoritaSetal.Goshajinkiganoxaliplatinneurotoxicityevaluation(GONE):Aphase2, multicenter, randomized, double-blind, placebo-controlled trial of goshajinkigan to preventoxaliplatin-inducedneuropathy.CancerChemotherPharmacol2013;72:1283–1290.
154.NishiokaM,ShimadaM,KuritaNetal.TheKampomedicine,Goshajinkigan,preventsneuropathyinpatientstreatedbyFOLFOXregimen.IntJClinOncol2011;16:322–327.
155.Cassidy J, Paul J, SoukopM et al. Clinical trials of nimodipine as a potential neuroprotector inovariancancerpatientstreatedwithcisplatin.CancerChemotherPharmacol1998;41:161–166.
156. RobertsJA,JenisonEL,KimKetal.Arandomized,multicenter,double-blind,placebo-controlled,dose-findingstudyofORG2766 in thepreventionordelayof cisplatin-inducedneuropathies inwomenwithovariancancer.GynecolOncol1997;67:172–177.
157.van Gerven JM, Hovestadt A, Moll JW et al. The effects of an ACTH (4-9) analogue ondevelopmentofcisplatinneuropathy intesticularcancer:Arandomizedtrial.JNeurol 1994;241:432–435.
158.HovestadtA, vanderBurgME,VerbiestHBet al. The courseofneuropathyafter cessationofcisplatintreatment,combinedwithOrg2766orplacebo.JNeurol1992;239:143–146.
159.vanderHoopRG,VechtCJ,vanderBurgMEetal.PreventionofcisplatinneurotoxicitywithanACTH(4-9)analogueinpatientswithovariancancer.NEnglJMed1990;322:89–94.
160.ArgyriouAA,ChroniE,PolychronopoulosPetal.Efficacyofoxcarbazepineforprophylaxisagainstcumulativeoxaliplatin-inducedneuropathy.Neurology2006;67:2253–2255.
161. ArrietaO,García-NavarreteR,ZúñigaSetal.Retinoicacidincreasestissueandplasmacontentsofnervegrowthfactorandpreventsneuropathyindiabeticmice.EurJClinInvest2005;35:201–207.
162.Davis ID,KiersL,MacGregorLetal.A randomized,double-blinded,placebo-controlledphase IItrial of recombinant human leukemia inhibitory factor (rhuLIF, emfilermin, AM424) to preventchemotherapy-inducedperipheralneurop-athy.ClinCancerRes2005;11:1890–1898.
163.DurandJP,DeplanqueG,MontheilVetal.Efficacyofvenlafaxineforthepreventionandreliefofoxaliplatin-inducedacuteneurotoxicity:ResultsofEFFOX,a randomized,double-blind,placebo-controlledphaseIIItrial.AnnOncol2012;23:200–205.
164.Kottschade LA, Sloan JA, Mazurczak MA et al. The use of vitamin E for the prevention ofchemotherapy-induced peripheral neuropathy: Results of a randomized phase III clinical trial.SupportCareCancer2011;19:1769–1777.
165.Pace A, Giannarelli D, Galiè E et al. Vitamin E neuroprotection for cisplatin neuropathy: A ran-domized,placebo-controlledtrial.Neurology2010;74:762–766.
166.ArgyriouAA,ChroniE,KoutrasAetal.Arandomizedcontrolledtrialevaluatingtheefficacyandsafety of vitamin E supplementation for protection against cisplatin-induced peripheralneuropathy:Finalresults.SupportCareCancer2006;14:1134–1140.
167.Pace A, Savarese A, PicardoM et al. Neuroprotective effect of vitamin E supplementation inpatientstreatedwithcisplatinchemotherapy.JClinOncol2003;21:927–931.
168.CassidyJ,BjarnasonGA,HickishTetal.Randomizeddoubleblind(DB)placebo(Plcb)controlledphase III study assessing the efficacy of xaliproden (X) in reducing the cumulative peripheralsensoryneuropathy(PSN)inducedbytheoxaliplatin(Ox)and5-FU/LVcombination(FOLFOX4)infirst- line treatment of patients (pts) with metastatic colorectal cancer (MCRC). J Clin Oncol2006;24(suppl18):3507a.
169.HarnedTM,KalousO,NeuweltAetal.Sodiumthiosulfateadministeredsixhoursaftercisplatindoesnotcompromiseantineuroblastomaactivity.ClinCancerRes2008;14:533–540.
170.Muldoon LL, PagelMA, Kroll RA et al. Delayed administration of sodium thiosulfate in animalmodels reduces platinum ototoxicity without reduction of antitumor activity. Clin Cancer Res2000;6:309–315.
171. Dickey DT, Wu YJ, Muldoon LL et al. Protection against cisplatin-induced toxicities by N-acetylcysteineandsodiumthiosulfateasassessedatthemolecular,cellular,andinvivolevels.JPharmacolExpTher2005;314:1052–1058.
�42
CME

Chapter2 TheOncologist2015;20:411-432
172.MaibachR,ChildsM,RajputKetal.SIOPEL6:Amulticenteropen-labelrandomizedphaseIIItrialoftheefficacyofsodiumthiosulphate(STS)inreducingototoxicityinpatientsreceivingcisplatin(Cis) monotherapy for standard-risk hepatoblastoma (SR-HB). J Clin Oncol 2014;32(suppl 5):TPS10094a.
173.FreyerDR.Theeffectsofsodiumthiosulfate(STS)oncisplatin-inducedhearingloss:AreportfromtheChildren’sOncologyGroup.JClinOncol2014;32(suppl5):10017a.
174.ParkerAR,PetluruPN,WuMetal.BNP7787-mediatedmodulationofpaclitaxel- andcisplatin-inducedaberrantmicrotubuleproteinpolymerizationinvitro.MolCancerTher2010;9:2558–2567.
175.DeSantisS,PaceA,BoveLetal.Patientstreatedwithantitumordrugsdisplayingneurologicaldeficitsarecharacterizedbyalowcirculatinglevelofnervegrowthfactor.ClinCancerRes2000;6:90–95.
176.SchmidtY,UngerJW,BartkeIetal.Effectofnervegrowthfactoronpeptideneuronsindorsalroot ganglia after taxol or cisplatin treatment and in diabetic (db/db) mice. Exp Neurol1995;132:16–23.
177. ScuteriA,GalimbertiA,RavasiMetal.NGFprotectsdorsalrootganglionneuronsfromoxaliplatinbymodulatingJNK/SapkandERK1/2.Neuro-sciLett2010;486:141–145.
178.CorcoranJ,MadenM.Nervegrowth factoractsvia retinoicacid synthesis to stimulateneuriteoutgrowth.NatNeurosci1999;2:307–308.
179.BianchiG,VitaliG,CaraceniAetal.Symptomaticandneurophysiologicalresponsesofpaclitaxel-orcisplatin-inducedneuropathytooralacetyl-L-carnitine.EurJCancer2005;41:1746–1750.
180.De Grandis D. Acetyl-L-carnitine for the treatment of chemotherapy-induced peripheralneuropathy:Ashortreview.CNSDrugs2007;21(suppl1):39–46.
181. Ghirardi O, Lo Giudice P, Pisano C et al. Acetyl- L-carnitine prevents and reverts experimentalchronicneurotoxicityinducedbyoxaliplatin,with-outalteringitsantitumorproperties.AnticancerRes2005;25:2681–2687.
182.GwagBJ,SesslerFM,RobineVetal.EndogenousglutamatelevelsregulatenervegrowthfactormRNAexpressionintheratdentategyrus.MolCells1997;7:425–430.
183.Taglialatela G, Navarra D, Cruciani R et al. Acetyl-L-carnitine treatment increases nerve growthfactorlevelsandcholineacetyltransferaseactivityinthecentralnervoussystemofagedrats.ExpGerontol1994;29:55–66.
184.Bianchi R, Brines M, Lauria G et al. Protective effect of erythropoietin and its carbamylatedderivativeinexperimentalcisplatinperipheralneurotoxicity.ClinCancerRes2006;12:2607–2612.
185.LeistM,GhezziP,GrassoGetal.Derivativesoferythropoietinthataretissueprotectivebutnoterythropoietic.Science2004;305:239–242.
186.Arrieta Ó, Hernández-Pedro N, Fernández- González-Aragón MC et al. Retinoic acid reduceschemotherapy-inducedneuropathyinananimalmodelandpatientswithlungcancer.Neurology2011;77:987–995.
187.IbrahimpasicK.Alphalipoicacidandglycaemiccontrolindiabeticneuropathiesattype2diabetestreatment.MedArh2013;67:7–9.
188.Barhwal K, Hota SK, Prasad D et al. Hypoxia- induced deactivation of NGF-mediated ERK1/2signaling in hippocampal cells: Neuroprotection by acetyl-L-carnitine. J Neurosci Res 2008;86:2705–2721.
189.GopalKV,WuC,ShresthaBetal.D-Methionineprotectsagainstcisplatin-inducedneurotoxicityincorticalnetworks.NeurotoxicolTeratol2012;34:495–504.
190.Schmutz M, Brugger F, Gentsch C et al. Oxcarbazepine: Preclinical anticonvulsant profile andputativemechanismsofaction.Epilepsia1994;35(suppl5):S47–S50.
191. BarhwalK,HotaSK,JainVetal.Acetyl-L-carnitine(ALCAR)preventshypobarichypoxia-inducedspatial memory impairment through extra- cellular related kinase-mediated nuclear factorerythroid2-relatedfactor2phosphorylation.Neu-roscience2009;161:501–514.
192.Hershman DL, Unger JM, Crew KD et al. Randomized double-blind placebo-controlled trial ofacetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoingadjuvantbreastcancertherapy.JClinOncol2013;31:2627–2633.
193.SusmanE.Xaliprodenlessensoxaliplatin-mediatedneuropathy.LancetOncol2006;7:288.�43
2

Chapter2 TheOncologist2015;20:411-432
194.NagakiY,HayasakaS,HayasakaYetal.Effectsofgoshajinkiganoncornealsensitivity,superficialpunctatekeratopathyandtearsecretioninpatientswithinsulin-dependentdiabetesmellitus.AmJChinMed2003;31:103–109.
195.Kono T, Mishima H, Shimada M et al. Preventive effect of goshajinkigan on peripheralneurotoxicityofFOLFOXtherapy:Aplacebo-controlleddouble-blindrandomizedphaseIIstudy(theGONEStudy).JpnJClinOncol2009;39:847–849.
196.Grisold W, Cavaletti G, Windebank AJ. Peripheral neuropathies from chemotherapeutics andtargetedagents:Diagnosis,treatment,andprevention.Neuro-oncol2012;14(suppl4):iv45–iv54.
197.KaurG,PhillipsC,WongKetal.Timingisimportantinmedicationadministration:Atimelyreviewofchronotherapyresearch.IntJClinPharmacol2013;35:344–358.
198.MormontMC,LéviF.Circadian-systemalterationsduringcancerprocesses:Areview.IntJCancer1997;70:241–247.
199.LiaoC,LiJ,BinQetal.Chronomodulatedchemotherapyversusconventionalchemotherapyforadvancedcolorectalcancer:Ameta-analysisoffiverandomizedcontrolledtrials.IntJColorectalDis2010;25:343–350.
200.CavalettiG.Calciumandmagnesiumprophylaxisforoxaliplatin-relatedneurotoxicity:Isitatrade-offbetweendrugefficacyandtoxicity?TheOncologist2011;16:1667–1668.
201.KhattakMA.Calciumandmagnesiumprophylaxisforoxaliplatin-relatedneurotoxicity:Isitatrade-offbetweendrugefficacyandtoxicity?TheOncologist2011;16:1780–1783.
202.SmithEM,PangH,CirrincioneCetal.Effectofduloxetineonpain, function,andqualityof lifeamongpatientswithchemotherapy-inducedpainfulperipheralneuropathy:Arandomizedclinicaltrial.JAMA2013;309:1359–1367.
203.HammackJE,MichalakJC,LoprinziCLetal.PhaseIIIevaluationofnortriptylineforalleviationofsymptomsofcis-platinum-inducedperipheralneuropathy.Pain2002;98:195–203.
204.RaoRD,MichalakJC,SloanJAetal.Efficacyofgabapentininthemanagementofchemotherapy-induced peripheral neuropathy: A phase 3 randomized, double-blind, placebo-controlled,crossovertrial(N00C3).Cancer2007;110:2110–2118.
205.RaoRD,FlynnPJ,Sloan JAetal. Efficacyof lamotrigine in themanagementof chemotherapy-induced peripheral neuropathy: A phase 3 randomized, double-blind, placebo-controlled trial,N01C3.Cancer2008;112:2802–2808.
206.BartonDL,WosEJ,QinRetal.Adouble-blind,placebo-controlledtrialofatopicaltreatmentforchemotherapy-inducedperipheralneuropathy:NCCTGtrialN06CA.SupportCareCancer2011;19:833–841.
ReceivedFebruary5, 2014; accepted forpublicationDecember 11, 2014;publishedOnlineFirstonMarch 12, 2015.©AlphaMedPress1083-7159/2015/$20.00/0http://dx.doi.org/10.1634/theoncologist.2014-0044
�44
of the efficacy of sodium thiosulphate (STS) inreducing ototoxicity in patients receiving cisplatin(Cis) monotherapy for standard-risk hepatoblas-toma (SR-HB). J Clin Oncol 2014;32(suppl 5):TPS10094a.
173. Freyer DR. The effects of sodium thiosulfate(STS) on cisplatin-induced hearing loss: A reportfrom the Children’s Oncology Group. J Clin Oncol2014;32(suppl 5):10017a.
174. Parker AR, Petluru PN,WuM et al. BNP7787-mediated modulation of paclitaxel- and cisplatin-induced aberrant microtubule protein polymeriza-tion in vitro. Mol Cancer Ther 2010;9:2558–2567.
175. De Santis S, Pace A, Bove L et al. Patientstreated with antitumor drugs displaying neurolog-ical deficits are characterized by a low circulatinglevel of nerve growth factor. Clin Cancer Res 2000;6:90–95.
176. Schmidt Y, Unger JW, Bartke I et al. Effect ofnerve growth factor on peptide neurons in dorsalroot ganglia after taxol or cisplatin treatment and indiabetic (db/db) mice. Exp Neurol 1995;132:16–23.
177. Scuteri A, Galimberti A, Ravasi M et al. NGFprotects dorsal root ganglion neurons from oxali-platin by modulating JNK/Sapk and ERK1/2. Neuro-sci Lett 2010;486:141–145.
178. Corcoran J, Maden M. Nerve growth factoracts via retinoic acid synthesis to stimulate neuriteoutgrowth. Nat Neurosci 1999;2:307–308.
179. Bianchi G, Vitali G, Caraceni A et al. Symp-tomatic and neurophysiological responses of pacli-taxel-orcisplatin-inducedneuropathy tooralacetyl-L-carnitine. Eur J Cancer 2005;41:1746–1750.
180. De Grandis D. Acetyl-L-carnitine for thetreatment of chemotherapy-induced peripheralneuropathy: A short review. CNS Drugs 2007;21(suppl 1):39–46.
181. GhirardiO, LoGiudiceP, PisanoCetal. Acetyl-L-carnitine prevents and reverts experimentalchronic neurotoxicity induced by oxaliplatin, with-outaltering its antitumorproperties.AnticancerRes2005;25:2681–2687.
182. Gwag BJ, Sessler FM, Robine V et al. Endog-enousglutamate levels regulatenervegrowthfactormRNA expression in the rat dentate gyrus.Mol Cells1997;7:425–430.
183. Taglialatela G, Navarra D, Cruciani R et al.Acetyl-L-carnitine treatment increases nervegrowth factor levels and choline acetyltransferaseactivity in the central nervous system of aged rats.Exp Gerontol 1994;29:55–66.
184. Bianchi R, Brines M, Lauria G et al. Protectiveeffect of erythropoietin and its carbamylated de-rivative in experimental cisplatin peripheral neuro-toxicity. Clin Cancer Res 2006;12:2607–2612.
185. LeistM,Ghezzi P,GrassoGetal.Derivativesoferythropoietin that are tissue protective but noterythropoietic. Science 2004;305:239–242.
186. Arrieta O, Hernandez-Pedro N, Fernandez-Gonzalez-Aragon MC et al. Retinoic acid reduceschemotherapy-induced neuropathy in an animalmodel and patients with lung cancer. Neurology2011;77:987–995.
187. Ibrahimpasic K. Alpha lipoic acid and glycae-mic control in diabetic neuropathies at type 2diabetes treatment. Med Arh 2013;67:7–9.
188. Barhwal K, Hota SK, Prasad D et al. Hypoxia-induced deactivation of NGF-mediated ERK1/2signaling in hippocampal cells: Neuroprotection byacetyl-L-carnitine. J Neurosci Res 2008;86:2705–2721.
189. Gopal KV, Wu C, Shrestha B et al. D-Methionine protects against cisplatin-induced neu-rotoxicity in cortical networks. Neurotoxicol Teratol2012;34:495–504.
190. Schmutz M, Brugger F, Gentsch C et al.Oxcarbazepine: Preclinical anticonvulsant profileand putative mechanisms of action. Epilepsia 1994;35(suppl 5):S47–S50.
191. Barhwal K, Hota SK, Jain V et al. Acetyl-L-carnitine (ALCAR) prevents hypobaric hypoxia-induced spatial memory impairment through extra-cellular related kinase-mediated nuclear factorerythroid 2-related factor 2 phosphorylation. Neu-roscience 2009;161:501–514.
192. Hershman DL, Unger JM, Crew KD et al.Randomized double-blind placebo-controlled trialof acetyl-L-carnitine for the prevention of taxane-inducedneuropathy inwomenundergoingadjuvantbreast cancer therapy. J Clin Oncol 2013;31:2627–2633.
193. Susman E. Xaliproden lessens oxaliplatin-mediated neuropathy. Lancet Oncol 2006;7:288.
194. Nagaki Y, Hayasaka S, Hayasaka Y et al. Effectsof goshajinkigan on corneal sensitivity, superficialpunctate keratopathy and tear secretion in patientswith insulin-dependentdiabetesmellitus. Am J ChinMed 2003;31:103–109.
195. Kono T, Mishima H, Shimada M et al. Pre-ventive effect of goshajinkigan on peripheralneurotoxicity of FOLFOX therapy: A placebo-controlled double-blind randomized phase II study
(the GONE Study). Jpn J Clin Oncol 2009;39:847–849.
196. Grisold W, Cavaletti G, Windebank AJ. Pe-ripheral neuropathies from chemotherapeutics andtargeted agents: Diagnosis, treatment, and pre-vention. Neuro-oncol 2012;14(suppl 4):iv45–iv54.
197. Kaur G, Phillips C, Wong K et al. Timing isimportant in medication administration: A timelyreview of chronotherapy research. Int J ClinPharmacol 2013;35:344–358.
198.Mormont MC, Levi F. Circadian-system alter-ations during cancer processes: A review. Int JCancer 1997;70:241–247.
199. Liao C, Li J, Bin Q et al. Chronomodulatedchemotherapy versus conventional chemotherapyfor advanced colorectal cancer: A meta-analysis offive randomized controlled trials. Int J Colorectal Dis2010;25:343–350.
200. Cavaletti G. Calcium and magnesium pro-phylaxis for oxaliplatin-related neurotoxicity: Is ita trade-off between drug efficacy and toxicity? TheOncologist 2011;16:1667–1668.
201. Khattak MA. Calcium and magnesium pro-phylaxis for oxaliplatin-related neurotoxicity: Is ita trade-off between drug efficacy and toxicity? TheOncologist 2011;16:1780–1783.
202. Smith EM,PangH, CirrincioneCet al. Effect ofduloxetine on pain, function, and quality of lifeamong patients with chemotherapy-induced pain-ful peripheral neuropathy: A randomized clinicaltrial. JAMA 2013;309:1359–1367.
203. Hammack JE, Michalak JC, Loprinzi CL et al.Phase III evaluation of nortriptyline for alleviation ofsymptoms of cis-platinum-induced peripheral neu-ropathy. Pain 2002;98:195–203.
204. RaoRD,Michalak JC, Sloan JA et al. Efficacy ofgabapentin in the management of chemotherapy-induced peripheral neuropathy: A phase 3 random-ized, double-blind, placebo-controlled, crossovertrial (N00C3). Cancer 2007;110:2110–2118.
205. Rao RD, Flynn PJ, Sloan JA et al. Efficacy oflamotrigine in the management of chemotherapy-induced peripheral neuropathy: A phase 3 random-ized, double-blind, placebo-controlled trial, N01C3.Cancer 2008;112:2802–2808.
206. BartonDL,Wos EJ, Qin R et al. A double-blind,placebo-controlled trial of a topical treatment forchemotherapy-induced peripheral neuropathy:NCCTG trial N06CA. Support Care Cancer 2011;19:833–841.
CME This article is available for continuing medical education credit at CME.TheOncologist.com.
©AlphaMed Press 2015TheOncologist®
432 Platinum-Induced Neurotoxicity
CME
CME

CHAPTER3
CharacterizationofandProtectionfromNeurotoxicityInducedbyOxaliplatin,BortezomibandEpothilone-B
CeciliaCeresa1,*,AbolfazlAvan2,3,*,ElisaGiovannetti3,AlbertA.Geldof4,AmirAvan3,GuidoCavaletti1,GodefridusJPeters3#
1DepartmentofSurgeryandInterdisciplinaryMedicine,UniversityofMilano-Bicocca,Monza,Italy;2DepartmentofNeurology,SchoolofMedicine,MashhadUniversityofMedical
Sciences,Mashhad,Iran;3DepartmentofMedicalOncology,and4DepartmentofUrology,VUUniversityMedicalCenter,Amsterdam,TheNetherlands
(*Theseauthorscontributedequallytothisstudy)
AnticancerRes2014;34(1):517-523
3

Chapter3 AnticancerRes2014;34(1):517-523
Oxaliplatin,bortezomibandepothilone-Binducedneurotoxicity:characterizationandprotection
ABSTRACT:Aim:Tocharacterizeneurotoxicityinducedbyoxaliplatin,bortezomib,andepothilone-Baswell as protection against their neurotoxicity using an in vitro model. Materials and Methods:Neurotoxicitywas evaluated using the neurite outgrowthmethod in PC12 rat pheochromo- cytomacells differentiated towards a mature neuronal phenotype, while neuroprotection was explored bysimultaneousexposureto0.5mMamifostine.Thepotentialmarkersofneuronaldifferentiation,cyclin-B2 (Ccnb2) and baculoviral inhibitor of apoptosis repeat-containing 5 (Birc5), were evaluated byquantitative reverse transcription polymerase chain reaction (RT-PCR). Results: Bortezomib,epothilone-B,andoxaliplatin reducedneurite length to68%,78%and66%, respectively (p<0.05).Thepercentageofneurite-forming-cells(discriminatingneurotoxicityfromgeneralcytotoxicity)decreasedfrom 70% (control) to 55% (bortezomib), 46% (epothilone-B), and 51% (oxaliplatin). Amifostine wasneuroprotectiveagainstoxaliplatin-inducedneurotoxicity, increasingbothneuritelengthandneurite-forming-cells. Quantitative-RT-PCR showed a 2.7-fold decrease in Ccnb2 expression in differentiatedPC12vs.undifferentiatedcells.Conclusion:Oxaliplatin,bortezomib,andepothilone-BareneurotoxicinthePC12model.Amifostinehasaneuroprotectiveeffectonlyagainstoxaliplatin-inducedneurotoxicity,suggesting that these compounds have different mechanisms of neurotoxicity. Anticancer Res2014;34(1):517-523
Keywords:Neurotoxicity,oxaliplatin,amifostine,neuroprotection,PC12model,Cyclin-B2
Introduction
Chemotherapy-induced peripheral neurotoxicity (CIPN) represents a clinically relevant problem inanticancer therapy. CIPNmainly presentswith sensory symptoms, and is usually dose-dependent. Ittypicallydevelopsafteracumulativedose,evenifacutetoxicneuropathyismorecommonwithsomedrugs (e.g. oxaliplatin and taxanes) (1). This side-effect might be dose-limiting or even lead totreatmentwithdrawal.However,evenwhenneurotoxicityisnotdose-limiting,itisanimportantside-effectthatcanirreversibly impairthepatients’qualityof lifebycausingchronicdiscomfort(2-5).Notonly established anticancer drugs, such as platinum compounds and taxanes, but also novelcompounds,suchasepothilonesandbortezomib,havebeenreportedtobeneurotoxic,althoughthemechanismsoftoxicityarelikelytobedifferent(6-8).
Bortezomibisaproteasomeinhibitorregisteredforthetreatmentofmultiplemyelomaandmantlecellmyeloma (9, 10), and is also tested against other diseases, usually in combination with platinatedcompounds(11).Peripheralneuropathyisamongthemostfrequently-observedtoxicitiesnecessitatingdosereductionsortreatmentdiscontinuation(11).Atleastone-thirdofpatientsundertreatmenthaveevidence of clinical sensory peripheral neuropathy (11-13). Although, it is not clear how and wherebortezomib affects the peripheral nervous system, treatment withdrawal can result in clinicalimprovementoftheinducedneurotoxicity.Asaresult,furtherstudiesareessentialinordertodefinethemechanismofthistoxiceffect,bothaloneandincombinationwithplatinatedcompounds.
Epothilone-B is another new chemotherapeutic agent causing peripheral neurotoxicity due to itsinteraction with microtubule polymerisation inducing arrest in the G2/M transition (14), similarly totaxanes. Nevertheless, epothilones and taxanes differ in terms of resistance conferred by specificpoint-mutationsinthegeneencodingβ-tubulin(15-17).Epothilonesalsoinducedose-limitingperipheralneurotoxicity (18).Despite the fact that sensoryperipheralneurotoxicity is regardedasamain toxiceffectonthenervoussystem,motoror(rarely)autonomicneuropathyhasoccasionallybeenreported.Althoughnopathologicaldataareavailableonepothilone-treatedpatients,dose-dependentaxonaldamagewasdemonstratedinanimalmodels,inwhichrecoverywasobtainedwithinfewweeksaftertreatmentwithdrawal(19).
�46
3

Chapter3 AnticancerRes2014;34(1):517-523
Oxaliplatinisaplatinum-basedcompoundwithhighefficacyincolorectalcancer,whenadministeredincombination with 5-fluorouracil and folinic acid. Oxaliplatin exerts its cytotoxic effects through theformation of DNA adducts, with subsequent impairment of DNA replication and transcription, andcausessevereanddisablingsensoryperipheralneurotoxicitydue toaccumulationof thedrug in thedorsalrootganglia(DRG),wheresensoryneuronsarelocated(20,21).
Theuseofneuroprotectiveagentsmayhelpreduceneurotoxicitycausedbychemotherapeuticagents,thusallowingforintensificationofchemotherapy.Onepotentialstrategyinneuroprotectionistheuseofdetoxicantssuchasamifostine.Amifostineisaphosphorylatedamino-thiolpro-drug,whichneedsto be de-phosphorylated in order to exert its protective effect, which is selective in normal cells.Amifostinehasbeenpostulatedtobeselectivelyprotectiveagainstnormaltissuewithoutreducingitsantitumoractivity(22-24).Amifostineisabletoprovideprotectionfromthetoxiceffectofcisplatininperipheralnerves(25-27).
Few pre-clinicalmodels have been used to study the neurotoxic effects of novel chemotherapeuticdrugsandneuroprotectiveagents(6,28).Hence,theaimsofthepresentstudywere:i)tocharacterizethe extent of neurotoxicity induced by bortezomib, epothilone-B and oxaliplatin using the rat PC12pheochromocytoma cell line to investigate the neurotoxic effects of different drugs; ii) to test thepotential roleofamifostine in thepreventionofchemotherapy-inducedneurotoxicity. In thismodel,neurotoxicitywasassessedbystandardquantitativemorphologicalmethods,includingthecountingofcells exhibiting neurites and the measurement of neurite length (28). Moreover, we evaluated thepotentialroleofcyclin-B2(Ccnb2)andbaculoviralinhibitorofapoptosisrepeat-containing5(Birc5)assurrogatebiomarkersofneuronaldifferentiationusingquantitativeRT-PCR.
MaterialsandMethods
Drugsandchemicals.BortezomibwasagiftfromMilleniumPharmaceuticals,Inc.(Johnson&Johnson,Cambridge,MA,USA),whileoxaliplatinwasobtained fromSigmaChemicalCo. (St.Louis,MO,USA)and epothilone-B from Calbiochem (Calbiochem, Darmstadt, Germany). Amifostine (S-2-(3-aminopropylamino)- ethylphosphorothioic acid, WR2721, Ethyol1) was obtained from USB Pharma(Nijmegen, the Netherlands). Epothilone-B and bortezomib were dissolved in dimethyl sulfoxide(DMSO),whereasoxaliplatinandamifostineweredissolved in sterilewaterandphosphatebufferedsaline (PBS), respectively. The drugs were diluted in culture medium before use. RPMI-1640 andDulbecco’smodified Eagle’smedium (DMEM), fetal bovine serum (FBS), penicillin and streptomycinwerefromGibco(Gaithersburg,MD,USA).AllotherchemicalswerefromSigma.
Cell culture. PC12 rat pheocromocytoma cells (29), were cultured in RPMI (containing 2 mM L-glutamine) supplemented with 10% heat-inactivated FBS, 1 mM sodium pyruvate, insulin-transferrin-sodium selenite medium supplement, penicillin (50 IU/ml) and streptomycin (50 μg/ml). Cells weremaintainedasmonolayerculturesin75cm2cultureflasks(Costar,Cambridge,MA,USA),at37Cin5%CO2and95%air,andharvestedwithtrypsin-EDTAwhentheywereinexponentialgrowth.
Cytotoxicitystudies.Thegrowthinhibitioncausedbybortezomib,epothilone-BandoxaliplatinonPC12cellswasevaluatedwiththesulforhodamine-B(SRB)assay(30,31).Forthispurpose,cellswereplatedatadensityof20,000cells/wellin96-wellflat-bottomplates(Costar).Twenty-fourhourslater(day-0)cellswere treated using different concentration ranges (0.0001-0.5 μM bortezomib, 0.0001- 0.5 μMepothilone-B,and0.05-100μMoxaliplatin).Thedrugexposuretimewas72h(day3).TheIC50andtheIC80were thedrug concentrations atwhich cell-growthwas inhibited to 50% and80%, respectively,basedonthedifferenceofabsorbancevaluesonday0andday3ofdrugexposure,ascalculatedbythesigmoidinhibitionmodel(GraphPadPRISMversion4.0;IntuitiveSoftwareforScience,SanDiego,CA,USA).
�47
3

Chapter3 AnticancerRes2014;34(1):517-523
NeurotoxicityandPC12-neuriteoutgrowthassay.Theneurotoxiceffectsofbortezomib,epothiolone-Band oxaliplatin were investigated using the nerve growth factor (NGF)-induced neurite outgrowthassay,aspreviouslydescribed(29,32).Briefly,undifferentiatedPC12cellswerepre-treatedforfivedaysbyaddingNGF2.5S(NGFfrommousesource;Promega,Madison,WI,USA)toafinalconcentrationof50ng/ml, trypsinized,washedusingPBS,andplated in60-mmculturedishes(Costar)atadensityof25,000cells/dish.Optimaladherenceandneuriteformationrequiredprecoatingtheplasticculturewellusing0.5mg/mlwatersolutionofpolylysine-hydrobromide(Sigma) followedbywashingwithsterilewater.After48hoursofculturewith10ng/mlNGF,differentiatedcellswereexposedtobortezomib,epothilone-Bor oxaliplatin at IC50 concentrations (0.95nMbortezomib, 0.75nMepothilone-B, 0.07μMoxaliplatin).Nodrugwasaddedtocontroldishes.
After72hofculture,theinterferenceofcytostaticcompoundsonneuriteoutgrowthwasevaluatedbymeasuringtheneuritelengthwithanautomaticimageanalyser(Quantimet,Leica,Cambridge,UK)onat least 200 randomly selected cells, and the percentage of differentiated cells per culture wascalculated(33).Forthispurpose,cellswereconsidereddifferentiatedwhenthelengthsoftheirneuritewere longer thanone-fold thecellbody length (34).Themorphometricdeterminationswerealwaysperformed by the same examiner who was blinded to the treatment of the cell culture underobservation. To discriminate specific neurotoxicity from general cytotoxicity, the analysis wasperformedonthebasisofthefractionofdifferentiatedcellsinsteadoftheabsolutenumberofneurite-formingcells.Theresults,therefore,aregivenasthepercentageofdifferentiatedcells.
Inordertoevaluatetheneuroprotectiveeffectofamifostine,co-incubationexperimentswerecarriedout as follows. Differentiated PC12 cells were incubated with 0.5 mM amifostine and 2.7 nMbortezomib,1.8nMepothilone-Bor0.2μMoxaliplatin.After72hoursofculturecellswerescoredforneurites.
Quantitative-RT-PCR.TotalRNAwasextractedusingTRIzol (Invitrogen,Carlsbad,CA,USA).RNAwasdissolvedinRNasefree-water,andmeasuredat260nm.RNA(1μg)wasreverse-transcribedat37Cforonehourin100-μlreactionvolumecontaining0.8mMdNTPs,200UofMoloneymurineleukemiavirusreverse transcriptase (MMLV-RT), 40 U of RNase inhibitor, and 0.05 μg/ml of random primers. ThecDNA was amplified by quantitative-PCR with the Applied Biosystems 7500HT sequence detectionsystem(AppliedBiosystems,FosterCity,CA,USA).PCRreactionswereperformedintriplicateusing5μl of cDNA, 12.5μl of TaqManUniversal PCRMasterMix, 2.5μl of probe and 2.5μl of forward andreverseprimersinafinalvolumeof25μl.Sampleswereamplifiedusingthefollowingthermalprofile:an initial incubation at 50 C for 5min, to prevent the re-amplification of carryover-PCRproducts byAmpEraseuracil-N-glycosylase(UNG),followedbyincubationat95Cfor10min,tosuppressAmpEraseUNGactivityanddenaturetheDNA,40cyclesofdenaturationat95Cfor15secfollowedbyannealingandextensionat60for1min.ForwardandreverseprimersandprobesforratCcnb2(Rn01530826_g1)and rat glyceraldehyde 3-phosphate dehydrogenase (Gapdh, Rn99999916_s1) were obtained fromTaqMan®GeneExpressionAssays(AppliedBiosystems),whileforwardandreverseprimersandprobesforratBirc5wereobtainedascustomTaqMan®GeneExpressionAssays(AppliedBiosystems),usingtheFileBuilderversion2.0software,onthebasisofGenBankdatabase.
Amplification data were normalized to the housekeeping gene Gapdh. Quantification of geneexpressionwasperformedusing standardcurvesobtainedwithdilutionsof rat cDNA fromamixofPC-12,BCLO,Bara-CandCC531ratcelllines,andwithdilutionsofcDNAobtainedfromquantitative-PCRHumanReferenceTotalRNA(Stratagene,LaJolla,CA,USA).PCRefficiencieswerebetween96.4and100%.
Statisticalevaluation.Allexperimentswereperformedintriplicateandrepeatedatleastthreetimes.Dataareexpressedasmeanvalues±SDandwereanalysedbyANOVAfollowedbytheTukey’smultiplecomparison.Differenceswereconsideredsignificantwhenp<0.05.
�48
3

Chapter3 AnticancerRes2014;34(1):517-523
Results
Cytotoxicityexperiments.Aconcentration-dependent inhibitionofcellgrowthwasobservedforPC12cells.PC12Cellswereequallysensitivetobortezomibandepothilone-B(IC50valuesof0.9and0.7nM,respectively). Oxaliplatin had a higher IC50 (70 nM). The IC50 of the single drugs was chosen toevaluate the neurotoxic effects, while the IC80 was used to evaluate the neuroprotective role ofamifostine.
In vitro neurotoxicity and protective role of amifostine. The standard neurotoxicity assaymeasuringNGF-dependent neurite outgrowth from the PC12 pheocromocytoma cell line was used to predictneurotoxicityafter72hincubationwithbortezomib,epothilone-B,andoxaliplatin.DrugtreatmentofdifferentiatedPC12cellsresultedinastatisticallysignificantdecreaseofneuritelengthwithrespecttocontrol cells. Bortezomib, epothilone-B, and oxaliplatin significantly reduced the neurite length to68±5, 78±3, and 66±4%, respectively, compared to controls (Figure 3.1A). The percentages of cellsexpressing neurites, which were used to discriminate neurotoxicity from general cytotoxicity, alsodecreasedsignificantlyfrom±3% inthecontrolto55±2% inbortezomib–,49±3% inepothilone-B–,and51±3%inoxaliplatin-treatedcells(Figure3.1B).
To investigate whether amifostine was able to prevent neurotoxicity induced by bortezomib,epothilone-B,andoxaliplatininPC12cells,theneuritelengthwasalsomeasuredinPC12cellsexposedto amifostine for 72 h together with the single drugs, at their IC80. Amifostine partially reversedoxaliplatin-dependent neurotoxicity by reducing the shortening of neurite length from 59±3%(oxaliplatin alone) to 74±3% (oxaliplatin with amifostine). The percentage of neurite-forming cellsincreased from 42±5% to 59±3% for the combination. On the contrary, amifostine did not protectagainst the decrease of neurite length nor the percentage of neurite-forming cells induced bybortezomibandepothilone-Btreatments(Figures3.2AandB).
Modulationof Ccnb2 andBirc5mRNAbybortezomib, epothilone-B, andoxaliplatin.Quantificationofneuriteoutgrowthisthestandardmethodtoinvestigateneurotoxicityinvitro,butitisimpracticalonalargescale.Forthisreason,weexaminedchangesinCcnb2andBirc5mRNAexpressioninPC12cellsaspotentialbiomarkersofneuronaldifferentiation.Afterdifferentiationtowardsaneuronalphenotype,
�49
A" B"
Figure"1"
Figure 3.1. Effect of bortezomib, epothilone-B and oxaliplatin on neurite length and celldifferentiation in PC12 cells. Nerve growth factor (NGF)- differentiated PC12 cells wereexposed to half-maximal inhibitory concentrations (IC50s) of bortezomib, epothilone-Bandoxaliplatinfor72h.Theneuritelengthvalues(a)andthepercentageofdifferentiatedcellsperculture(b)werecalculatedasthepercentageofvaluesobtainedincontrolcells.Columns, mean values obtained from three independent experiments; bars, SD.*Significantlydifferentfromcontrolcells(p<0.001).
3

Chapter3 AnticancerRes2014;34(1):517-523
PC12 cells showeda 1.6-folddecrease inCcnb2expression compared to theundifferentiated controlcells(Figure3.3).Birc5mRNAexpressiondidnotchange(datanotshown).Themostneurotoxicdrugin theneuriteoutgrowthassay,oxaliplatin, ledtoa2.0-folddecrease inCcnb2expressionat its IC50(Figure3.4),whilebortezomibandepothilone-BinducedaslightermodulationofCcnb2.
�50
induced neuropathy with no adverse effect on treatmentefficacy, but not against bortezomib and epothilone-B,suggesting a different mechanism of neurotoxicity.
Bortezomib-induced neurotoxicity was mainly characterizedin rat models by axonopathy of unmyelinated fibres in nervesand pathological alteration in DRG satellite cells, while DRG
ANTICANCER RESEARCH 34: 517-524 (2014)
520
Figure 2. Effect of amifostine (WR2721) on bortezomib, epothilone-B and oxaliplatin PC12 neurotoxicity in in vitro. Nerve growth factor (NGF)-differentiated PC12 cells were exposed to 80% maximal-inhibitory concentrations (IC80s) of bortezomib, epothilone-B and oxaliplatin alone or incombination with amifostine (0.5 mM) for 72 h. The neurite length values (a) and the percentage of differentiated cells (b) were calculated as thepercentage of values obtained in control cells. Columns, mean values obtained from three independent experiments; bars, SD. Significantly differentfrom *control cells (p<0.001), #oxaliplatin-treated cells (p<0.01).
Figure 1. Effect of bortezomib, epothilone-B and oxaliplatin on neurite length and cell differentiation in PC12 cells. Nerve growth factor (NGF)-differentiated PC12 cells were exposed to half-maximal inhibitory concentrations (IC50s) of bortezomib, epothilone-B and oxaliplatin for 72 h. Theneurite length values (a) and the percentage of differentiated cells per culture (b) were calculated as the percentage of values obtained in controlcells. Columns, mean values obtained from three independent experiments; bars, SD. *Significantly different from control cells (p<0.001).
Figure3.2.Effectofamifostine(WR2721)onbortezomib,epothilone-BandoxaliplatinPC12neurotoxicity in in vitro. Nerve growth factor (NGF)- differentiated PC12 cells wereexposed to 80% maximal-inhibitory concentrations (IC80s) of bortezomib, epothilone-Band oxaliplatin alone or in combinationwith amifostine (0.5mM) for 72 h. The neuritelength values (a) and the percentage of differentiated cells (b) were calculated as thepercentageofvaluesobtainedincontrolcells.Columns,meanvaluesobtainedfromthreeindependent experiments; bars, SD. Significantly different from*control cells (p<0.001),#oxaliplatin-treatedcells(p<0.01).
neuron degeneration was observed only in mice (28, 35).Epothilone-B effects have not been studied extensively in pre-clinical settings. It is assumed that epothilone-B reduces thenerve conduction velocity (NCV), although no correlation hasbeen found between the concentrations of the drug in tissueswith NCV changes (35). Based on two studies, sensoryneuropathy induced by ixabepilone (epothilone-B analog)varied from 3 to 59% (37, 38). Oxaliplatin-inducedneurotoxicity is characterized by cumulative sensoryneurotoxicity due to increased neuronal excitability byalteration of the voltage-gated sodium channels throughchelation of calcium by the oxaliplatin metabolite (39).Further characterization of this induced neurotoxicity is crucialin order to find the most effective neuroprotective treatmentand minimize the incidence and consequences of such aserious side-effect. Some agents such as vitamin E,carbamazepine, calcium and magnesium infusion, reducedglutathione, N-acetylcysteine and amifostine are considered asneuroprotective treatments, but they have not yet proven to becompletely effective (1).
In the present work, we evaluated the potentialneuroprotective effect of amifostine on bortezomib, epothilone-B, and oxaliplatin. Oxaliplatin-induced neurotoxicity waspartially protected, by restoring neurite length and neurite-forming cells between 35-50% of the neurotoxicity induced by
oxaliplatin alone. Importantly, in a clinical trial performed byLu and colleagues, amifostine reduced the peripheralneurotoxicity due to oxaliplatin by about 60%, resulting inabout 20% increase in the number of patients who receivedmultiple chemotherapy cycles (40). The results obtained in ourexperimental setting thus confirm the reliability of themorphological method used in this study to test the potentialneuroprotective activity of different drugs.
In order to evaluate potential biomarkers for neuriteoutgrowth, we also measured the mRNA expression ofCcnb2 and Birc5. Decreased levels of Ccnb2 and Birc5have been correlated with neurite outgrowth induced byNGF in SH-SY5Y HN cells (41). Accordingly, our studiesshowed a significant reduction in Ccnb2 expression inNGF differentiated PC12 cells with respect to the control.Conversely, Birc5 mRNA level was not down-regulatedfollowing differentiation. Ccnb2 is an important regulatorof the cell cycle and it associates with cyclin-dependentkinase 1 as its positive regulatory subunit. The decrease inCcnb2 expression observed in PC12 cells following NGFdifferentiation is in accordance with previous data obtained
Ceresa et al: Protection from Oxiliplatin Neurotoxicity
521
Figure 3. Effect of differentiation on cyclin B2 (CB2) level in PC12cells. Modulation of CB2 expression in PC12 cells, as determined byquantitative real-time reverse transcription polymerase chain reaction(RT-PCR). Columns, mean values obtained from three independentexperiments calculated in comparison with standard curves and withrespect to the respective expression values of the housekeeping geneGapdh; bars, SE.
Figure 4. Effects of bortezomib, epothilone-B and oxaliplatin on cyclin-B2 levels in PC12 cells. Modulation of cyclin-B2 (CB2) expression asdetermined by real-time reverse transcription polymerase chain reaction(RT-PCR). CB2 expression was studied in nerve growth factor (NGF)-differentiated PC12 cells treated for 72 h with bortezomib, epothilone-B and oxaliplatin at their half-maximal inhibitory concentrations (IC50s)with respect to undifferentiated and NGF-differentiated PC12 cells.Columns, mean values obtained from three independent experimentscalculated in comparison with standard curves and with respect to therespective expression values of the housekeeping gene Gadph; bars, SE.Significantly different from *undifferentiated control cells (p<0.05);
Figure 3.3. Effect of differentiation oncyclin B2 (CB2) level in PC12 cells.Modulation of CB2 expression in PC12cells, as determined by quantitative real-time reverse transcription polymerasechain reaction (RT-PCR). Columns, meanvalues obtained from three independentexperiments calculated in comparisonwith standard curves andwith respect tothe respective expression values of thehousekeepinggeneGapdh;bars,SE.
Figure 3.4. Effects of bortezomib, epothilone-B and oxaliplatin oncyclin-B2 levels in PC12 cells. Modulation of cyclin-B2 (CB2)expression as determined by real-time reverse transcriptionpolymerasechainreaction(RT-PCR).CB2expressionwasstudiedinnervegrowthfactor(NGF)-differentiatedPC12cellstreatedfor72hwithbortezomib,epothilone-Bandoxaliplatinattheirhalf-maximalinhibitory concentrations (IC50s) with respect to undifferentiatedandNGF-differentiated PC12 cells. Columns,mean values obtainedfromthreeindependentexperimentscalculatedincomparisonwithstandard curves and with respect to the respective expressionvalues of the housekeeping gene Gadph; bars, SE. Significantlydifferentfrom*undifferentiatedcontrolcells(p<0.05).
3

Chapter3 AnticancerRes2014;34(1):517-523
Discussion
In the present study, the neurotoxic effects of bortezomib, epothilone-B and oxaliplatin weresuccessfullyevaluatedinPC12ratpheocromocytomacellsafterneuronaldifferentiation.Inagreementwithclinicaldata,neurotoxicity inducedbybortezomib,epothilone-Bandoxaliplatinhasbeenshownin ourmodel, by reducingbothneurite length and thepercentageof neurite-forming cells.We alsodemonstrated the potential role of the expression of Ccnb2 mRNA as biomarkers of neuronaldifferentiationinPC12ratcells.Moreover,amifostineprotectedagainstoxaliplatin-inducedneuropathywithnoadverseeffectontreatmentefficacy,butnotagainstbortezomibandepothilone-B,suggestingadifferentmechanismofneurotoxicity.
Bortezomib-induced neurotoxicity was mainly characterized in rat models by axonopathy ofunmyelinated fibres in nerves and pathological alteration in DRG satellite cells, while DRG neurondegeneration was observed only in mice (28, 35). Epothilone-B effects have not been studiedextensively in pre- clinical settings. It is assumed that epothilone-B reduces the nerve conductionvelocity (NCV), although no correlation has been found between the concentrations of the drug intissues with NCV changes (35). Based on two studies, sensory neuropathy induced by ixabepilone(epothilone-Banalog)variedfrom3to59%(37,38).Oxaliplatin-inducedneurotoxicityischaracterizedbycumulativesensoryneurotoxicityduetoincreasedneuronalexcitabilitybyalterationofthevoltage-gated sodium channels through chelation of calcium by the oxaliplatin metabolite (39). Furthercharacterization of this induced neurotoxicity is crucial in order to find the most effectiveneuroprotectivetreatmentandminimizetheincidenceandconsequencesofsuchaseriousside-effect.SomeagentssuchasvitaminE,carbamazepine,calciumandmagnesiuminfusion,reducedglutathione,N-acetylcysteineandamifostineareconsideredasneuroprotectivetreatments,buttheyhavenotyetproventobecompletelyeffective(1).
Inthepresentwork,weevaluatedthepotentialneuroprotectiveeffectofamifostineonbortezomib,epothilone-B, and oxaliplatin. Oxaliplatin-induced neurotoxicitywas partially protected, by restoringneurite length and neurite-forming cells between 35-50% of the neurotoxicity induced by oxaliplatinalone.Importantly,inaclinicaltrialperformedbyLuandcolleagues,amifostinereducedtheperipheralneurotoxicityduetooxaliplatinbyabout60%,resultinginabout20%increaseinthenumberofpatientswhoreceivedmultiplechemotherapycycles(40).Theresultsobtainedinourexperimentalsettingthusconfirm the reliability of the morphological method used in this study to test the potentialneuroprotectiveactivityofdifferentdrugs.
In order to evaluate potential biomarkers for neurite outgrowth, we also measured the mRNAexpressionofCcnb2andBirc5.DecreasedlevelsofCcnb2andBirc5havebeencorrelatedwithneuriteoutgrowth induced by NGF in SH-SY5Y HN cells (41). Accordingly, our studies showed a significantreductioninCcnb2expressioninNGFdifferentiatedPC12cellswithrespecttothecontrol.Conversely,Birc5mRNAlevelwasnotdown-regulatedfollowingdifferentiation.Ccnb2isanimportantregulatorofthe cell cycle and it associateswith cyclin-dependent kinase 1 as its positive regulatory subunit. ThedecreaseinCcnb2expressionobservedinPC12cellsfollowingNGFdifferentiationisinaccordancewithpreviousdataobtained inthesamecellularmodel.Thesedatasuggestedthatthedecrease inCcnb2expression after treatment with agents inducing neuronal differentiation could lead to cyclin-dependent kinase 1 inactivation with a consequent increase in neurite outgrowth (42, 43). Theobserved effect of oxaliplatin is probably due to reduction of cyclin- dependent kinase 1 activationinducedby theantineoplasticdrugwitha consequentdecrease inCcnb2expression, asobserved inHCT116colorectalcancercellstreatedwithoxaliplatin(44).
Inconclusion,theexperimentalmethodsusedinthepresentinvitrostudy,includingaPCRanalysisforCcnb2 as a surrogate biomarker of neuronal differentiation, showed similar neurotoxic effects,especially for oxaliplatin. Moreover, we demonstrated that amifostine partially protects againstoxaliplatin-inducedneurotoxicity.Theseresultspromptfuturepre-clinicalandtranslationalstudiestotest neurotoxicity of novel platinated compounds, as well as new neuroprotective agents, beforestartingthetreatment.
�51
3

Chapter3 AnticancerRes2014;34(1):517-523
Correspondenceto:Dr.E.GiovannettiandProfessorDr.G.J.Peters,DepartmentofMedicalOncology,VUUniversityMedical Center, De Boelelaan 1117, 1081 HV Amsterdam, the Netherlands. E-mail: [email protected],[email protected]
References
1. Cavaletti G, Alberti P, Frigeni B, PiattiM and Susani E: Chemotherapy-induced neuropathy.CurrTreatOptionsNeurol13(2):180-190,2011.
2. Argyriou AA, Bruna J, Marmiroli P and Cavaletti G: Chemotherapy-induced peripheralneurotoxicity(CIPN):Anupdate.CritRevOncolHematol82(1):51-77,2012.
3. StillmanMandCataJP:Managementofchemotherapy-inducedperipheralneuropathy.CurrPainHeadacheRep10(4):279-287,2006.
4. ArmstrongT,AlmadronesLandGilbertMR:Chemotherapy- inducedperipheralneuropathy.OncolNursForum32(2):305-311,2005.
5. Cavaletti G and Marmiroli P: Chemotherapy-induced peripheral neurotoxicity. Expert OpinDrugSaf3(6):535-546,2004.
6. CavalettiG,GilardiniA,CantaA,RigamontiL,Rodriguez-MenendezV,CeresaC,MarmiroliP,Bossi M, Oggioni N, D’Incalci M and De Coster R: Bortezomib-induced peripheralneurotoxicity: a neurophysiological and pathological study in the rat. Exp Neurol 204(1):317-325,2007.
7. CersosimoRJ:Oxaliplatin-associatedneuropathy:Areview.AnnPharmacother39(1):128-135,2005.
8. LeeJJandSwainSM:Peripheralneuropathyinducedbymicrotubule-stabilizingagents.JClinOncol24(10):1633-1642,2006.
9. Adams J: The proteasome: A suitable antineoplastic target. Nat Rev Cancer 4(5): 349-360,2004.
10. AltunM,GalardyPJ, ShringarpureR,HideshimaT, LeBlancR,AndersonKC,PloeghHLandKessler BM: Effects of PS-341 on the activity and composition of proteasomes in multiplemyelomacells.CancerRes65(17):7896-7901,2005.
11. VoortmanJ,SmitEF,HoneywellR,KuenenBC,PetersGJ,vandeVeldeHandGiacconeG:Aparallel dose-escalation study ofweekly and twice-weekly bortezomib in combinationwithgemcitabineandcisplatin inthefirst-linetreatmentofpatientswithadvancedsolidtumors.ClinCancerRes13(12):3642-3651,2007.
12. Aghajanian C, Dizon DS, Sabbatini P, Raizer JJ, Dupont J and Spriggs DR: Phase I trial ofbortezomib and carboplatin in recurrent ovarian or primary peritoneal cancer. J ClinOncol23(25):5943-5949,2005.
13. Orlowski RZ, Voorhees PM, Garcia RA, Hall MD, Kudrik FJ, Allred T, Johri AR, Jones PE,IvanovaA,VanDeventerHW,GabrielDA,SheaTC,MitchellBS,AdamsJ,EsseltineDL,TrehuEG,GreenM,LehmanMJ,NatoliS,CollinsJM,LindleyCMandDeesEC:Phase1 trialof theproteasome inhibitor bortezomib and pegylated liposomal doxorubicin in patients withadvancedhematologicmalignancies.Blood105(8):3058-3065,2005.
14. GoodinS,KaneMPandRubinEH:Epothilones:Mechanismofactionandbiologicactivity.JClinOncol22(10):2015-2025,2004.
15. AltmannKH,WartmannMandO’ReillyT:Epothilonesandrelatedstructures−anewclassofmicrotubule inhibitorswithpotent invivoantitumoractivity.BiochimBiophysActa 1470(3):M79-M91,2000.
16. GiannakakouP,GussioR,NogalesE,DowningKH,ZaharevitzD,BollbuckB,PoyG,SackettD,NicolaouKCandFojoT:A commonpharmacophore forepothiloneand taxanes:Molecularbasis for drug resistance conferred by tubulin mutations in human cancer cells. Proc NatlAcadSciUSA97(6):2904-2909,2000.
17. GoodinS:Ixabepilone:Anovelmicrotubule-stabilizingagentforthetreatmentofmetastaticbreastcancer.AmJHealthSystPharm65(21):2017-2026,2008.
�52
3

Chapter3 AnticancerRes2014;34(1):517-523
18. Lee JJ and Swain SM: Development of novel chemotherapeutic agents to evade themechanismsofmultidrugresistance(MDR).SeminOncol32(6Suppl7):S22-S26,2005.
19. ChiorazziA,NicoliniG,CantaA,OggioniN,RigolioR,CossaG,LombardiR,RoglioI,CervelliniI, Lauria G, Melcangi RC, Bianchi R, Crippa D and Cavaletti G: Experimental epothilone Bneurotoxicity:Resultsofinvitroandinvivostudies.NeurobiolDis35(2):270-277,2009.
20. Authier N, Gillet JP, Fialip J, Eschalier A and Coudore F: An animal model of nociceptiveperipheralneuropathyfollowingrepeatedcisplatininjections.ExpNeurol182(1):12-20,2003.
21. McDonaldESandWindebankAJ:Cisplatin-inducedapoptosisofDRGneurons involvesBAXredistributionandcytochromec releasebutnotFAS receptor signaling.NeurobiolDis9(2):220-233,2002.
22. Capizzi RL: Protection of normal tissues from the cytotoxic effects of chemotherapy byamifostine(Ethyol):Clinicalexperiences.SeminOncol21(5Suppl11):8-15,1994.
23. NgTY,NganHY,ChengDKandWongLC:Theeffectofamifostineontheinvitrocytotoxicityof chemotherapeutic agents in three epithelial ovarian carcinoma cell lines. Gynecol Oncol75(2):194-197,1999.
24. PetersGJ and vanderVijghWJ:Protectionofnormal tissues from the cytotoxic effectsofchemotherapy and radiation by amifostine (WR-2721): preclinical aspects. Eur J Cancer31A(Suppl1):S1-S7,1995.
25. Mollman JE, Glover DJ, Hogan WM and Furman RE: Cisplatin neuropathy. Risk factors,prognosis,andprotectionbyWR-2721.Cancer61(11):2192-2195,1988.
26. TreskesMandvanderVijghWJ:WR2721asamodulatorofcisplatin-andcarboplatin-inducedside-effectsincomparisonwithotherchemoprotectiveagents:Amolecularapproach.CancerChemotherPharmacol33(2):93-106,1993.
27. Verstappen CC, Geldof AA, Postma TJ and Heimans JJ: In vitro protection from cisplatin-inducedneurotoxicitybyamifostineanditsmetaboliteWR1065.JNeurooncol44(1):1-5,1999.
28. MeregalliC,CantaA,CarozziVA,ChiorazziA,OggioniN,GilardiniA,CeresaC,AvezzaF,CrippaL,MarmiroliP,andCavalettiG:.Bortezomib-inducedpainfulneuropathyinrats:abehavioral,neurophysiologicalandpathologicalstudyinrats.EurJPain14(4):343-350,2010.
29. Geldof AA:Nerve-growth-factor-dependent neurite outgrowth assay; A researchmodel forchemotherapy-inducedneuropathy.JCancerResClinOncol121(11):657-660,1995.
30.Skehan P, Storeng R, Scudiero D,Monks A,McMahon J, Vistica D,Warren JT, Bokesch H,KenneySandBoydMR:Newcolorimetriccytotoxicityassayforanticancer-drugscreening.JNatlCancerInst82(13):1107-1112,1990.
31. KeepersYP,PizaoPE,PetersGJ,vanArk-OtteJ,WinogradBandPinedoHM:Comparisonofthe sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivitytesting.EurJCancer27(7):897-900,1991.
32. GeldofAA,MastbergenSC,HenrarREandFairclothGT:Cytotoxicityandneurocytotoxicityofnewmarineanticanceragentsevaluatedusinginvitroassays.CancerChemotherPharmacol44(4):312-318,1999.
33. Nicolini G, Miloso M, Zoia C, Di Silvestro A, Cavaletti G and Tredici G: Retinoic aciddifferentiated SH-SY5Y human neuroblastoma cells: An in vitro model to assess drugneurotoxicity.AnticancerRes18(4A):2477-2481,1998.
34. CasafontI,BercianoMTandLafargaM:Bortezomibinducestheformationofnuclearpoly(A)RNAgranulesenrichedinSAM68andPABPN1insensoryganglianeurons.NeurotoxRes17(2):167-178,2010.
35. Bruna J, Udina E, Ale A, Vilches JJ, Vynckier A, Monbaliu J, Silverman L and Navarro X:Neurophysiological, histological and immunohistochemical characterization of bortezomib-inducedneuropathyinmice.ExpNeurol223(2):599-608,2010.
36. ChiorazziA,HochelJ,StockigtD,CantaA,CarozziVA,MeregalliC,AvezzaF,CrippaL,SalaB,Ceresa C, Oggioni N and Cavaletti G: Exposure-response relationship of the syntheticepothilone sagopilone in a peripheral neurotoxicity rat model. Neurotox Res 22(2): 91-101,2012.
�53
3

Chapter3 AnticancerRes2014;34(1):517-523
37. HuangH,MenefeeM,EdgerlyM,ZhuangS,KotzH,PoruchynskyM,HuffLM,BatesS,andFojo T: A phase II clinical trial of ixabepilone (Ixempra; BMS-247550; NSC 710428), anepothiloneBanalog, inpatientswithmetastaticrenalcellcarcinoma.ClinCancerRes16(5):1634-1641,2010.
38. LowJA,WedamSB,LeeJJ,BermanAW,BrufskyA,YangSX,PoruchynskyMS,SteinbergSM,Mannan N, Fojo T and Swain SM: Phase II clinical trial of ixabepilone (BMS-247550), anepothilone B analog, inmetastatic and locally advanced breast cancer. J ClinOncol 23(12):2726-2734,2005.
39. TanMH,ChayWY,NgJH,TehBTandChewL:Transientbilateralabducensneuropathywithpost-tetanic facilitationandacutehypokalemiaassociatedwithoxaliplatin:A case report. JMedCaseRep4:36,2010.
40. LuP,FanQX,WangLX,WangX,ZongHandWangRL:Prophylacticeffectofamifostineonoxaliplatin-related neurotoxicity in patients with digestive tract tumors. Ai Zheng 27(10):1117-1120,2008
41. OeT,NagashimaT,MuramotoM,Yamazaki T,MorikawaN,OkitsuO,NishimuraS,Aoki T,Katayama Y and Kita Y: Cyclin B2 and BIRC5 genes as surrogate biomarkers for neuriteoutgrowthinSH-SY5Ysubclonalcells.Neuropharmacology50(8):1041-1047,2006.
42. ErhardtJAandPittmanRN:Ectopicp21(WAF1)expressioninducesdifferentiation-specificcellcycle changes in PC12 cells characteristic of nerve growth factor treatment. J Biol Chem273(36):23517-23523,1998.
43. DobashiY,ShojiM,KitagawaM,NoguchiTandKameyaT:SimultaneoussuppressionofCdc2and Cdk2 activities induces neuronal differentiation of PC12 cells. J Biol Chem 275(17):12572-12580,2000.
44. Chiu SJ, Lee YJ, Hsu TS and ChenWS:Oxaliplatin-induced γ- H2AX activation via both p53-dependentand-independentpathwaysbutisnotassociatedwithcellcyclearrest inhumancolorectalcancercells.ChemBiolInteract182(2-3):173-182,2009.
ReceivedSeptember18,2013RevisedNovember27,2013AcceptedNovember28,2013
�54
3

CHAPTER4
Calcium/magnesiuminfusionforoxaliplatin-inducedneuropathy:protectiveornot?
AbolfazlAvan1*,AmirAvan1,ElisaGiovannetti1,GodefridusJPeters1
1VUUniversityMedicalCenter,Amsterdam,theNetherlands
JClinOncol2014;10;32(29):3341
4

Chapter4 JClinOncol2014;10;32(29):3341
Calcium/magnesiuminfusionforoxaliplatin-inducedneuropathy:protectiveornot?
TOTHEEDITOR:Loprinzietal [1] recentlypublishedresultsofaphase III trialshowingthatcalcium/magnesiuminfusiondoesnotdecreaseoxaliplatin-relatedneurotoxicity.Thistrialenrolled353patientswithcoloncancerwhowereundergoingadjuvant therapycontainingoxaliplatinandwere randomlyassigned into three groups, including two groups who received calcium/magnesium according todifferentschedules.TheauthorsusedthesensoryscaleoftheEuropeanOrganisationforResearchandTreatmentofCancerQualityofLifeQuestionnaire–Chemotherapy-InducedPeripheralNeuropathy20tool (QLQ-CIPN20). This trial is of utmost importance because it might lead to a change in clinicalpractice.However,inouropinion,somekeypointsshouldbediscussedinmoredetail.
First, theauthors [1]mentioned thesubjective (patient reported)outcomemeasuresasbetter toolsformeasuringsymptoms.However,onlyalowcorrelationbetweenallsubjectiveandobjectivesensorygradingscoreswasfoundinthecitedstudybyLavoieSmithetal[2].Furthermore,theauthorsusedtwo different versions of the Common Toxicity Criteria for validation (ie, versions 3 and 4).Nevertheless,inarecentstudy,theEuropeanOrganisationforResearchandTreatmentofCancerQLQ-CIPN20sensoryscorewashighlyrelatedtosensoryitemsinCommonToxicityCriteriaversion2.0[3].For now, no evidence demonstrates the superiority of a given method, whereas there are vastdiscrepancies between patients’ perception and objective tools, particularly in intermediate grades[3,4].Inaddition,onthebasisofdatainTable1[1],theauthorsmeasuredtotalQLQ-CIPN20scoresforsensoryaswellasautonomicandmotorneuropathies.However,thecorrelationsbetweenautonomicandmotorneuropathiesandQLQ-CIPN20scoresarequitelow[2,4],andadditionalinformationshouldbeprovidedonhowpatientswereeducatedtoreportthesesymptoms.
Second,theinterpretationofpriorbeliefswithevidenceisbestachievedbyBayesianmethods,notbyBonferroniadjustments,whicharehelpfulindecisionmakingbutnotinassessingevidenceindata[5].Inparticular, in theBonferronimethod, auniversal null hypothesismustbepostulated inwhich thetwogroupsareidenticalwithrespecttoallvariables.Thismethodwillprovideacorrectanswerifthevariantsarecompletelyindependent,butthatisnotthecaseinthisstudy.Moreover,thelikelihoodoftype II errors is inflated by this statistical analysis,5 so that truly important differences are deemednonsignificant. Similarly, regarding the data in Table 1 [1], the Kruskal-Wallis test for nonordinalvariablessuchasscoringsandage,Fisher’sexacttestforcomparingfourrace/ethnicitysubgroups,andthe 2 test for staging as a nominal variable should be replaced with more appropriate statisticalmethods[6].
Finally, according to the CONSORT statement [7], the authors should report a more detaileddescriptionofthemethodsandtypesofrandomizationandblindingtheyusedinthetrial,andshouldindicatewhetherthosewhoassessedtheoutcomeswerealsoblinded.
Overall, incorporation of the proper methodology in the conducting of the trial as well as in dataanalysis and reporting of results is essential to obtain solid evidence that can be used in clinicalpractice.Therefore,despitetheimportanceofthetrialconductedbyLoprinzietal[1],webelievethatthe question of whether calcium/magnesium infusion can protect against oxaliplatin-inducedneuropathyisstillopen,andtheresultsofthisstudyneedappropriateconfirmationandreplication.
AbolfazlAvan
VUUniversityMedicalCenter,Amsterdam,theNetherlandsAmirAvan
VU University Medical Center, Amsterdam, the Netherlands; and Mashhad University of MedicalSciences,Mashhad,IranElisaGiovannettiandGodefridusJ.PetersVUUniversityMedicalCenter,Amsterdam,theNetherlands
�56
4

Chapter4 JClinOncol2014;10;32(29):3341
AUTHORS’DISCLOSURESOFPOTENTIALCONFLICTSOFINTEREST
Theauthor(s)indicatednopotentialconflictsofinterest.
Correspondence to: Abolfazl Avan, MD; Department of Medical Oncology, VU University Medical Center, DeBoelelaan1117,1081HVAmsterdam,theNetherlands.E-mail:[email protected]
REFERENCES
1. Loprinzi CL,QinR,Dakhil SR, et al: Phase III randomized,placebo- controlled, double-blindstudy of intravenous calcium and magnesium to prevent oxaliplatin-induced sensoryneurotoxicity(N08CB/Alliance).JClinOncol32:997-1005,2014
2. LavoieSmithEM,BartonDL,QinR,etal:Assessingpatient-reportedperipheralneuropathy:The reliability and validity of the European Organization for Research and Treatment ofCancerQLQ-CIPN20Questionnaire.QualLifeRes22:2787-2799,2013
3. Alberti P, Rossi E, Cornblath DR, et al: Physician-assessed and patient- reported outcomemeasuresinchemotherapy-inducedsensoryperipheralneu-rotoxicity:Twosidesofthesamecoin.AnnOncol25:257-264,2014
4. InoueN,IshidaH,SanoM,etal:DiscrepancybetweentheNCI-CTCAEandDEB-NTCscalesinthe evaluation of oxaliplatin-related neurotoxicity in patients with metastatic colorectalcancer.IntJClinOncol17:341-347,2012
5. PernegerTV:What’swrongwithBonferroniadjustments.BMJ316:1236-1238,19986. GreenhalghT:Howtoreadapaper:Statisticsforthenon-statistician.I:Differenttypesofdata
needdifferentstatisticaltests.BMJ315:364-366,19977. SchulzKF,AltmanDG,MoherD:CONSORT2010statement:Updatedguidelinesforreporting
parallelgrouprandomisedtrials.BMJ340:c332,2010
DOI:10.1200/JCO.2014.55.1846;publishedonlineaheadofprintatwww.jco.orgonAugust18,2014
�57
4

Chapter4 JClinOncol2014;10;32(29):3341
�58
4

PART 2


CHAPTER5RoleofAktsignalinginresistancetoDNA-
targetedtherapy
AbolfazlAvan1,RaviNarayan2,ElisaGiovannetti1,GodefridusJ.Peters1*
1DepartmentofMedicalOncologyand2DepartmentofRadiationOncology,VUUniversityMedicalCenter,Amsterdam,TheNetherlands.
Avanetal.AktandDNA-targetedagents(submitted)
5

Chapter5 Avanetal.AktandDNA-targetedagents(submitted)
RoleofAktsignalinginresistancetoDNA-targetedtherapy
ABSTRACT:TheAktsignaltransductionpathwaycontrolsmosthallmarksofcancer.ActivationoftheAkt cascade promotes a malignant phenotype and is also widely implicated in drug resistance.Therefore,themodulationofAktactivityisregardedasanattractivestrategytoenhancetheefficacyof cancer therapy and irradiation. This pathway consists of phosphatidylinositol 3 kinase (PI3K),mammaliantargetofrapamycin(mTOR),andtheserine-threoninekinaseAkt,alsoknownasproteinkinaseB(PKB).DNA-targetedagents,suchasplatinumagents,taxanes,andantimetabolites,aswellasradiation have a significant impact on cancer treatment by affecting DNA replication, which isaberrantly activated in malignancies. However, they may also trigger the activation of repairmechanisms,suchasupstreamanddownstreamcascadeofAktsurvivalpathway.Thus, inhibitionofthis pathway can possibly improve the efficacy without compromising the effect of conventionaltreatment. Akt inhibitors, e.g. MK-2206 and perifosine, or PI3K modulators, e.g. LY294002 andwortmannin,haveshownpromisingresults in favorofsensitizingcancercells tothetherapy invitroand in vivo, which have provided the rationale for incorporation of some of these inhibitors intomultimodality treatment of differentmalignancies. Despite the acceptable safety profile of someofthese agents in clinical studies the results are still too preliminary. Ongoing trials and translationalstudies will be critical to exploit these targeted compounds as well as to design more effectivecombination strategies with chemotherapeutic agents. Therefore, this review examines boththeeffectsofdifferentchemotherapeuticdrugsonAktsignaling,andtheeffectsofofAkt-inhibitionontheefficacyofdifferentanticanceragents.
Keywords:PI3K/Akt,platinum,taxane,antimetabolite,radiation.
1.Aktpathwaysignalingoverview
The Akt signal transduction pathway controls most hallmarks of cancer, including metabolism, cellsurvival, cell cycle progression, regulation of apoptosis, protein synthesis, motility, and genomicinstabilitybyphosphorylationofthesubstrates(reviewedin[1]).AberrantlossorgainofAktactivationhasbeenassociatedwith thedevelopmentof variousdiseases, e.g. diabetes, autoimmunediseases,andcancer[2-5].
The Akt pathway consists of phosphatidylinositol 3 kinase (PI3K), mammalian target of rapamycin(mTOR), and the transforming serine-threonine kinase Akt protein isoforms (further referred to asAkt),alsoknownasproteinkinaseB(PKB)andphosphateandtensinhomologue(PTEN)asacriticaltumor suppressor. PI3K enzymes phosphorylate phosphatidylinositol-4,5-biphosphate (PIP2) togeneratephosphatidylinositol-3,4,5-triphosphate(PIP3)at thecellmembranethat is requiredfor therecruitmentandactivationofAkt.[6,7](Figure5.1).Thesephospholipidsareconstitutivelyelevatedinmostcancercells.DockingofAkttothecellmembranecausesaconformationalchange,whichinturnleads to phosphorylationof the two critical amino acid residues, threonine 308 and serine 473, andfinally leads to the activation of Akt [8]. After the activation, Akt is translocated to intracellularcompartmentswhereitphosphorylatesseveralsubstrateproteins.ThedownstreamtargetsofAktarenumerous due to the multiple interactions with its consensus sequence [1].In summary, the mostimportanteffectsofAktactivationare:(a)cellsurvivalthroughinhibitionofBAD,caspase-9,andFOX[9-11];(b)cellproliferationandgluconeogenesisthroughinhibitionofGSK3,P21,P27,etc.[12];and(c)proteinsynthesisandcellgrowththroughactivationofmTOR[13](Figure5.1).
Todate,threeAktfamilymembershavebeen identified inmammals, i.e.Akt1(alsoknownasPKBα),Akt2(PKBβ)andAkt3(PKBγ).Havingshownhighlyconservedproperties,thesehomologuesmaybeactivatedbythesamemechanism[14].However,beingencodedbythreedifferentregionsat 14q32,19q13, and 1q44, respectively, these three isoformsaredistinct substrateswithdistinctphysiologicaloutcomes,andalsoopposingtoeachother.AccumulatingevidenceputsAkt1andAkt2functionalmost
�62
5
SubmittedPaper(Confidentialdata)
Pages62-84

CHAPTER6Phospho-Aktoverexpressionisprognosticandcanbe
usedtotailorthesynergisticinteractionofthenovelAktinhibitorperifosinewithgemcitabineinpancreaticcancer
AmirAvan1,3,*MinaMaftouh1,*,AbolfazlAvan1,*,NicolevanGrieken2,AnnevanKrieken1,RajivRaktoe1,NiccolaFunel4,KaamarAzijli1,SaraCaponi5,UgoBoggi6,
BabetteAicher8,LeticiaL.Leon9,GodefridusJ.Peters1,ElisaGiovannetti1,7
Departmentsof1MedicalOncologyand2Pathology,VUUniversityMedicalCenter,Amsterdam,DeBoelelaan1117,1081HVAmsterdam,TheNetherlands,3DepartmentofNewSciencesandTechnology,FacultyofMedicine,MashhadUniversityofMedicalSciences,Mashhad,Iran;Departmentsof4PathologicalSurgery,5Oncology,6GeneralSurgeryand7Start-UpUnitUniversityofPisa,viaRoma55,56100Pisa,Italy;8ÆternaZentarisGmbH,
Weismuellerstrasse50,60314FrankfurtamMain,Germany;9CenterforBiomedicalResearchoftheCanaryIslands,InstitutodeTecnologiasBiomedicas,UniversityofLaLaguna,La
Laguna,Spain.*Theseauthorsequallycontributedtothiswork
Avanetal.Perifosine-gemcitabinecombinationinpancreaticcancer(submitted)
6

Chapter6 Avanetal.Perifosine-gemcitabinecombinationinpancreascancer
Phospho-Akt overexpression is prognostic and can be used to tailor the synergisticinteractionofthenovelAktinhibitorperifosinewithgemcitabineinpancreaticcancer
ABSTRACT: There is increasing evidence of a constitutive activation of Akt in pancreatic ductaladenocarcinoma (PDAC), associated with poor prognosis and chemoresistance. Therefore, weevaluatedtheexpressionofphospho-Akt inPDACtissuesandcells,and investigatedthetherapeuticpotentialofthenovelAktinhibitorperifosineincombinationwithgemcitabineinPDACcells.Phospho-Akt was overexpressed in 60% of PDACs as determined by immunohistochemistry, which wascorrelatedwith shorter survival in 50 resected PDAC patients.mRNA and protein expression levelsvaried considerably in 14 PDAC cells, as assessed by RT-PCR, immunocytochemistry and ELISA.Perifosineinhibitedcellgrowthinmonolayercellculturesandspheroids,andsynergisticallyenhancedthe antiproliferative activity of gemcitabine, with combination index values of 0.4 and 0.8 in cellscharacterized by high phospho-Akt expression (LPC028 and CFPAC-1), while this combination wasantagonistic inLPC006cells,with lowphospho-Aktexpression.Thesynergisticeffectwasassociatedwith reduction of the expression of the ribonucleotide reductase subunit-M2, potentially facilitatinggemcitabine cytotoxicity. Moreover, perifosine decreased cell migration and invasion, which wasadditionally reduced by the perifosine/gemcitabine combination. The combination enhanced thepercentages of cells in S-phase, and significantly increased apoptosis, associated with induction ofcaspase-3/6/8/9, PARP and BAD, and inhibition of Bcl-2 and NF-κB. In summary, these data providenovelinsightsintotheabilityofperifosinetointerferewithcell-proliferation,induceapoptosis,reducemigration/invasion and synergistically interact with gemcitabine in cells with phospho-Aktoverexpression. Furthermore these results support the analysis of phospho-Akt expression as abiomarkerfortherationaldevelopmentofthisinnovativetherapeuticapproach.
Keywords:Pancreaticductaladenocarcinoma,Akt,perifosine,gemcitabine
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is among themost lethal solid tumors.Despite extensivepreclinicalandclinicalresearch,theprognosisofthisdiseasehasnotimproved,witha5-yearsurvivalrate below 5% [1]. This dismal outcome can partially be explained by the lack of biomarkers forscreeninganddiagnosis at earlier stages, andby the resistance to currently available chemotherapyregimens[2].
ActivatingKRASmutationsoccurearly,followedbylossofp16,andthenlater,inactivationofTP53andSMAD4 [3]; however, targeting these events has proven to be very difficult. Studies on new drugstargeting additional oncogenic pathways and/or downstream effectors are warranted. Thephosphatidylinositol-3 kinase (PI3K)/Akt pathway emerged as one of the core signaling pathways inPDAC [4]. In particular, the serine/threonine kinaseAkt,which is coded in three highly homologousisoforms(Akt1,Akt2,andAkt3),isoverexpressedinmorethan40%ofPDACpatients[5,6].MechanismsunderlyingaberrantAktactivationincancerincludedirectalterationssuchasmutations,amplification,oroverexpression,but alsoactivationofupstreamsignalingevents, suchas activationofHER-2/neusignalingorPTENmutation/loss[7].
ThePI3K/Aktpathwayplays a key role in cell proliferation, survival andmotility [8].Deregulationofcomponentsinvolvedinthispathwaycouldconferresistancetochemotherapy[9,10],whileblockageofAktsignalingresultinprogrammedcelldeathandinhibitionoftumorgrowth[11].
ActivationofAktisafrequenteventinPDACandhasbeencorrelatedtoitspoorprognosis[12,13].FahyandcollaboratorsshowedthatinhibitionofthePI3K/Aktpathwaysensitizespancreaticcancercellsbyincreasingapoptosisbothinvitroandinvivo[14].
�86
6
SubmittedPaper(Confidentialdata)
Pages86-102

CHAPTER7
Predictiveroleofrepairenzymesintheefficacyofcisplatincombinationsinpancreaticandlungcancer
GodefridusJPeters1,#,AbolfazlAvan1,MarielleGallegosRuiz,VanessaOrsini1,AmirAvan1,2,ElisaGiovannetti1,EgbertF.Smit3
Departmentsof1MedicalOncologyand3Pulmonology,VUUniversityMedicalCenter,Amsterdam,theNetherlands;2CardiovascularResearchCenterandDepartmentofNewSciencesandTechnology,SchoolofMedicine,MashhadUniversityofMedicalScience,
Mashhad,Iran
AnticancerRes2014;34(1):435-442
7

Chapter7 AnticancerRes2014;34(1):435-442
Predictiveroleofrepairenzymesintheefficacyofcisplatincombinationsinpancreaticandlungcancers
ABSTRACT: Platinum combinations are the mainstay of treatment for non-small cell lung cancer(NSCLC), while for pancreatic cancer platinum combinations are being given to good- performancestatuspatients.Theseplatinumcombinationsconsistofcis-orcarboplatinwithgemcitabine,while,fornon- squamous NSCLC and mesothelioma, of pemetrexed. The combination of gemcitabine andcisplatin is based on gemcitabine-induced increased formation and retention of DNA–platinumadducts, which can be explained by a decrease of excision repair cross-complementing group-1(ERCC1)-mediatedDNArepair.Inthesepatients,survivalandresponseisprolongedwhenERCC1hasalowprotein ormRNAexpression. A low expression of ribonucleotide reductase (RR) is related to abetter treatment outcome after both gemcitabine and gemcitabine- platinum combinations. Forpemetrexed combinations, ERCC1 expression was not related to survival. For both NSCLC andpancreatic cancer, polymorphisms in ERCC1 (C118T) and Xeroderma pigmentosum group D (XPD)(A751C)wererelatedtosurvival.Incurrentlyongoingandfutureprospectivestudies,patientsshouldbeselectedbasedontheirDNArepairstatus,butitstillhastobedeterminedwhetherthisshouldbebyimmunohistochemistry,mRNAexpression,orapolymorphism.AnticancerRes2014;34(1):435-442
KeyWords:Non-small cell lung cancer,pancreatic cancer,gemcitabine, cisplatin,DNA repair, ERCC1,XPD,ribonucleotidereductase,review.
Introduction
Non-small cell lung cancer (NSCLC) and pancreatic ductal adenocarcinoma (PDAC) account forapproximately85%oflungandpancreaticcancer,respectively,andhavea5-yearsurvivaloflessthan5% (1). Platinum combinations are standard therapy for both NSCLC and malignant pleuralmesothelioma(MPM)butarestillexperimentalinPDAC(2).Pemetrexed-platinumtherapyisstandardforMPMandnon- squamousNSCLC,while for squamousNSCLC,gemcitabine-cisplatin isoftenused(3).ThecombinationofraltitrexedandcisplatinwasassimilarlyactiveinMPMaspemetrexed−cisplatinorpemetrexed−carboplatin(4).
Allcombinationsarebasedontheassumptionthatinthecombination,DNAdamageismoreextensiveand repair is inhibited.Among the variousDNA repair systems, thenucleotideexcision repair (NER)systemseemsthemajorsystem involved in therepairofDNA−platinumadducts (5,6),althoughthemismatch repair system also plays a role for cisplatin and carboplatin, but not for oxaliplatin (7).Severalproteinsare involved in the recognitionofDNAdamage,unwinding, subsequentexcisionofthe damaged nucleotides (AG and GG Pt−adducts), and insertion of new deoxynucleosidetriphosphates (dNTP) in the DNA. These include transcription factor II H (TFIIH), the xerodermapigmentosumgroupenzymesXPD,XPC,ERCC1/XPF,DNApolymerasesδandε,andligase1(Figure7.1).Single nucleotide polymorphisms (SNP) in any of these genes may affect the repair capacity andcontributeto individualvariations inchemotherapyresponse.Hencetheserepairsystemshavebeenextensivelybeeninvestigatedasanexplanationfortheinteractionofgemcitabinewithcisplatin(8,9).Suchpharmacogeneticstudiesmayallowthevariationinhowindividualpatientsrespondtomedicinestobereducedbytailoringtherapiestotheirgeneticprofile(10).
Pre-clinicalBasisforTheInteractionofGemcitabineandCisplatin
Themolecularbasisfortheinteractionbetweencisplatinandgemcitabinewasinitiallyinvestigatedincelllinesofovarian,headandneck,andNSCLC(11-13).Inthesemodelsystems,wedemonstratedoneof themostpronouncedsynergismsbetween twodrugs,withcombination indices (CI)of0.001andless(11).Thecombinationwasalsomorethanadditiveininvivotumorsgeneratedfromthesecelllines(12-14);themostactivescheduleconsistedofevery3-daygemcitabineschedule(fourtimes),combined
�104
7

Chapter7 AnticancerRes2014;34(1):435-442
with cisplatin only on the first day. Both gemcitabine-preceding-cisplatin and cisplatin precedinggemcitabinewereveryactive,althoughthegemcitabineprecedingcisplatinbecamethemostwidelyusedschedule.Detailedanalysisofthemolecularinteractiondemonstratedthatgemcitabineincreasedtheformationofcisplatin−DNAadducts(13-16),bothinvitroandinvivo.AdetailedanalysisinmurineLewis lungwithNSCLCshowedthatPt−DNAadduct levels(especiallyPt−GG),evaluatedastheareasunder the concentration−time curves of Pt−DNA in the tumors, were associated with reduceddoubling-time of tumors treatedwith the combination and thiswas related to the antitumor effect(Figure7.2).InvitroitwasalsodemonstratedthatcisplatinincreasedtheincorporationofgemcitabineintoDNA(13).Most likelythis increased incorporationwasresponsibleforastructuralchange intheDNA,allowingmoreDNAadductstobeformed;thischangemightalsoaffectinhibitionofDNArepair.TheextentofERCC-mediatedDNArepairinthegemcitabine−cisplatincombinationwascorrelatedwiththe extent of synergism (17). Selvakumaran et al. demonstrated in an in vivo system with ERCC1antisensetransfectantsthatERCC1isessentialforinvivorepairofDNAplatinumadducts(18).
For the repair of DNA damage, DNA polymerases require a sufficient supply of dNTP which areprovided by the action of ribonucleotide reductase (RR),which has two subunits RRM1 and RRM2.
�105
we demonstrated one of the most pronounced synergismsbetween two drugs, with combination indices (CI) of 0.001and less (11). The combination was also more than additivein in vivo tumors generated from these cell lines (12-14); themost active schedule consisted of every 3-day gemcitabine
schedule (four times), combined with cisplatin only on thefirst day. Both gemcitabine-preceding-cisplatin and cisplatinpreceding gemcitabine were very active, although thegemcitabine preceding cisplatin became the most widelyused schedule. Detailed analysis of the molecular interactiondemonstrated that gemcitabine increased the formation ofcisplatin−DNA adducts (13-16), both in vitro and in vivo. Adetailed analysis in murine Lewis lung with NSCLC showedthat Pt−DNA adduct levels (especially Pt−GG), evaluated asthe areas under the concentration−time curves of Pt−DNA inthe tumors, were associated with reduced doubling-time oftumors treated with the combination and this was related tothe antitumor effect (Figure 2). In vitro it was alsodemonstrated that cisplatin increased the incorporation ofgemcitabine into DNA (13). Most likely this increasedincorporation was responsible for a structural change in theDNA, allowing more DNA adducts to be formed; this changemight also affect inhibition of DNA repair. The extent ofERCC-mediated DNA repair in the gemcitabine−cisplatincombination was correlated with the extent of synergism(17). Selvakumaran et al. demonstrated in an in vivo systemwith ERCC1 antisense transfectants that ERCC1 is essentialfor in vivo repair of DNA platinum adducts (18).
For the repair of DNA damage, DNA polymerases requirea sufficient supply of dNTP which are provided by the actionof ribonucleotide reductase (RR), which has two subunitsRRM1 and RRM2. Inhibition of RR by gemcitabine reducesthe concentration of dATP, dGTP and dCTP (19), thisdecrease hampers the repair of both AG and GG adducts.Hence RR expression is likely to play a role in the repair ofPt−DNA adducts, as outlined below.
ANTICANCER RESEARCH 34: 435-442 (2014)
436
Figure 1. Schematic action of the nucleotide excision repair (NER)pathway. In NER, after DNA damage is recognized, the DNA helix isunwound by transcription factor II H (TFIIH), which is stabilized byXeroderma pigmentosum group D (XPD). The TFIIH subunits of XPDand XPB/C act as a helicase and ATPase, respectively. Next incisionsare made both up- and downstream of the lesion by XPG and XPF-excision repair cross-complementing group 1 (ERCC1). XPG acts as anendonuclease which cuts DNA damage on the 3’ side while the XPF-ERCC1 heterodimeric protein cuts on the 5’ side. The dual incisionleads to the removal of a ssDNA with a single-strand gap of 25-30nucleotides. DNA polymerase then uses the undamaged single-strandedDNA as a template to synthesize a short complementary sequence,followed by ligation via DNA ligase to complete NER and form adouble-stranded DNA. Partially modified from (6).
Figure 2. Correlation between Pt−DNA adduct formation and retention(expressed as area under the curve of adducts) and sensitivity of Lewislung tumors to various gemcitabine−cisplatin treatments. The treatmentwith the highest extent of adducts was also the most effective treatment(Van Moorsel et al. (15), with permission).
Figure7.1.Schematicactionofthenucleotideexcisionrepair(NER)pathway.InNER,afterDNAdamage is recognized, theDNAhelix is unwound by transcription factor II H (TFIIH),which isstabilizedbyXerodermapigmentosumgroupD(XPD).TheTFIIHsubunitsofXPDandXPB/CactasahelicaseandATPase,respectively.Nextincisionsaremadebothup-anddownstreamofthelesion by XPG and XPF- excision repair cross-complementing group 1 (ERCC1). XPG acts as anendonucleasewhichcutsDNAdamageonthe3’sidewhiletheXPF-ERCC1heterodimericproteincutsonthe5’side.ThedualincisionleadstotheremovalofassDNAwithasingle-strandgapof25-30 nucleotides. DNA polymerase then uses the undamaged single-stranded DNA as atemplatetosynthesizeashortcomplementarysequence,followedbyligationviaDNAligasetocompleteNERandformadouble-strandedDNA.Partiallymodifiedfrom(6).
we demonstrated one of the most pronounced synergismsbetween two drugs, with combination indices (CI) of 0.001and less (11). The combination was also more than additivein in vivo tumors generated from these cell lines (12-14); themost active schedule consisted of every 3-day gemcitabine
schedule (four times), combined with cisplatin only on thefirst day. Both gemcitabine-preceding-cisplatin and cisplatinpreceding gemcitabine were very active, although thegemcitabine preceding cisplatin became the most widelyused schedule. Detailed analysis of the molecular interactiondemonstrated that gemcitabine increased the formation ofcisplatin−DNA adducts (13-16), both in vitro and in vivo. Adetailed analysis in murine Lewis lung with NSCLC showedthat Pt−DNA adduct levels (especially Pt−GG), evaluated asthe areas under the concentration−time curves of Pt−DNA inthe tumors, were associated with reduced doubling-time oftumors treated with the combination and this was related tothe antitumor effect (Figure 2). In vitro it was alsodemonstrated that cisplatin increased the incorporation ofgemcitabine into DNA (13). Most likely this increasedincorporation was responsible for a structural change in theDNA, allowing more DNA adducts to be formed; this changemight also affect inhibition of DNA repair. The extent ofERCC-mediated DNA repair in the gemcitabine−cisplatincombination was correlated with the extent of synergism(17). Selvakumaran et al. demonstrated in an in vivo systemwith ERCC1 antisense transfectants that ERCC1 is essentialfor in vivo repair of DNA platinum adducts (18).
For the repair of DNA damage, DNA polymerases requirea sufficient supply of dNTP which are provided by the actionof ribonucleotide reductase (RR), which has two subunitsRRM1 and RRM2. Inhibition of RR by gemcitabine reducesthe concentration of dATP, dGTP and dCTP (19), thisdecrease hampers the repair of both AG and GG adducts.Hence RR expression is likely to play a role in the repair ofPt−DNA adducts, as outlined below.
ANTICANCER RESEARCH 34: 435-442 (2014)
436
Figure 1. Schematic action of the nucleotide excision repair (NER)pathway. In NER, after DNA damage is recognized, the DNA helix isunwound by transcription factor II H (TFIIH), which is stabilized byXeroderma pigmentosum group D (XPD). The TFIIH subunits of XPDand XPB/C act as a helicase and ATPase, respectively. Next incisionsare made both up- and downstream of the lesion by XPG and XPF-excision repair cross-complementing group 1 (ERCC1). XPG acts as anendonuclease which cuts DNA damage on the 3’ side while the XPF-ERCC1 heterodimeric protein cuts on the 5’ side. The dual incisionleads to the removal of a ssDNA with a single-strand gap of 25-30nucleotides. DNA polymerase then uses the undamaged single-strandedDNA as a template to synthesize a short complementary sequence,followed by ligation via DNA ligase to complete NER and form adouble-stranded DNA. Partially modified from (6).
Figure 2. Correlation between Pt−DNA adduct formation and retention(expressed as area under the curve of adducts) and sensitivity of Lewislung tumors to various gemcitabine−cisplatin treatments. The treatmentwith the highest extent of adducts was also the most effective treatment(Van Moorsel et al. (15), with permission).
Figure 7.2. Correlation between Pt−DNA adductformationandretention(expressedasareaunderthecurveofadducts)andsensitivityofLewislungtumors to various gemcitabine−cisplatintreatments.Thetreatmentwiththehighestextentofadductswasalso themosteffective treatment(VanMoorseletal.(15),withpermission).
7

Chapter7 AnticancerRes2014;34(1):435-442
InhibitionofRRbygemcitabinereducestheconcentrationofdATP,dGTPanddCTP(19),thisdecreasehamperstherepairofbothAGandGGadducts.HenceRRexpressionislikelytoplayaroleintherepairofPt−DNAadducts,asoutlinedbelow.
Genetic and epigenetic alterations, such as gene mutations, amplification, deletions, polymorphicstatus, or altered gene/protein expression have been shown to be correlatedwith drug responses,includingofgemcitabineandplatinumanalogs(6,8,20).Amongthepossiblepredictiveorprognosticfactorsofsurvivalbenefittoaspecifictreatment,germlinepolymorphismshavebeenidentifiedasanattractive target, specifically for advanced cancer, since their analysis canbemoreeasilyperformedcompared to tumormutationalanalysisandgeneexpressionarrays (21).Therefore, in the following,wemainlyfocusedongeneexpressionalterationsorpolymorphicstatusofthegenesinvolvedinDNArepairsystemsanddrugmetabolism.
ERCC1ExpressionandRepairofPt−DNAAdductsinLungCancer
Since its introduction into the clinic, the cisplatin−gemcitabine combination is still considered astandardregimenforthetreatmentofadvancedNSCLC(22).Inarandomizedfour-armphaseIIIstudy,nodifferencewasobservedintime-to-progression(TTP),andoverallsurvival(OS)betweenpaclitaxel−cisplatin,docetaxel−cisplatin,paclitaxel−carboplatin,andgemcitabine−cisplatincombinations(2)andinathree-armstudybetweenpaclitaxel-cisplatin,gemcitabine-cisplatinorpaclitaxel-gemcitabine(23).Becauseofbetter tolerance, thegemcitabine−cisplatin (orcarboplatin) regimen is still consideredasthestandardregimen,althoughpemetrexedhasreplacedgemcitabinefornon-squamousNSCLCbasedonaphaseIIIstudy(3).
InaninitialstudybyLordetal.(24), itwasdemonstratedthatpatientswithNSCLCwithalowERCC1mRNAexpression(asdeterminedbyPCR)hadincreasedsurvivalcomparedtothosewithhighERCC1expression. In subsequentanalysesbothmRNAexpressionbyPCRand immunohistochemistrywereused to determine the expression of ERCC1. Although some antibodies against ERCC1 recognizedanotherprotein(25-27),ameta-analysisfrom12studiesand836patientsclearlydemonstratedthatlowlevels of ERCC1 mRNA or protein expression were associated with a longer survival [odds ratio(OR)=0.77, 95% confidence interval (CI)=0.47-1.07, p<0.00001] and a superior major response rate(OR=0.48,95%CI=0.35-0.64,p<0.0001)(28).Anevenstrongercorrelationwasobservedwhendifferentparameterswerecombined.Ceppietal. (29)demonstratedthata lowexpressionofERCC1orRRM1each were associated with a longer survival for patients treated with gemcitabine−cisplatin (ERCC1p=0.0032;RRM1p=0.039)butwhenthetwowerecombined(bothlow),thesurvivalbenefitincreasedand was more significant (p=0.0023); similar results were observed by Bepler et al. (30). Thisinformationwas recently applied inorder to selectpatientson the treatment armmost likely tobesensitive to these combinations (31); from 275 eligible patients, those with a low RRM1/ERCC1expressionwere randomized for the gemcitabine−carboplatin combination, thosewith a highRRM1and low ERCC1 for docetaxel−cisplatin, and those with a high RRM1 and high ERCC1 for docetaxel−vinorelbine. No statistically significant differences were observed between the experimental andcontrolarmsregardingprogression-freesurvival(6.1vs.6.9months)andoverallsurvival(11.0vs.11.3months).However,allpatientswhoreceivedthesametreatmentandhadalowexpressionofERCC1/RRM1 had better progression-free survival (8.1 months) in the control group, compared to theexperimental arm (5.0 months). Another major conclusion of this study was that measurement ofproteinexpressionwasfeasibleandveryreproducible.
BothimmunohistochemistryandPCRanalysismayhaveproblemsregardingtheirusein largegroupsofpatients,sinceantibodieshavetobevalidated(whichisnotalwaysdoneproperly)andmaychangein time (25-27), while sufficient RNA cannot always be isolated from tumor samples, albeit thetechnology to isolate sufficient and high quality RNA from paraffin-embedded tissues has improvedconsiderablyinthelastdecade.Wecomparedimmunohistochemistry,PCRandgeneticpolymorphismsfor several biomarkers for their potential in patientswithMPM (32). Indeed immunohistochemistry
�106
7
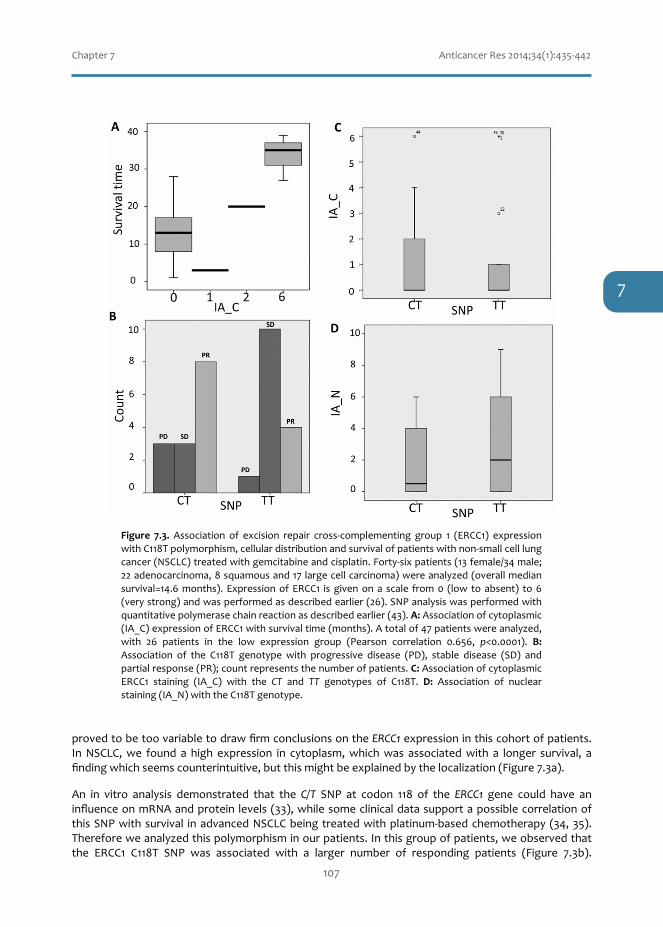
Chapter7 AnticancerRes2014;34(1):435-442
provedtobetoovariabletodrawfirmconclusionsontheERCC1expressioninthiscohortofpatients.InNSCLC,we found a high expression in cytoplasm,whichwas associatedwith a longer survival, afindingwhichseemscounterintuitive,butthismightbeexplainedbythelocalization(Figure7.3a).
An in vitro analysis demonstrated that the C/T SNP at codon 118 of the ERCC1 gene could have aninfluenceonmRNAandprotein levels(33),whilesomeclinicaldatasupportapossiblecorrelationofthisSNPwithsurvival inadvancedNSCLCbeingtreatedwithplatinum-basedchemotherapy(34,35).Thereforeweanalyzedthispolymorphisminourpatients.Inthisgroupofpatients,weobservedthatthe ERCC1 C118T SNP was associated with a larger number of responding patients (Figure 7.3b).
�107
combination (37), as well as NSCLC (38). It can beconcluded that in NSCLC, ERCC1 and RRM1 expressionand ERCC1 and XPD polymorphisms may be associatedwith response to cisplatin therapy. However, a recent meta-analysis concluded that the predictive value of ERCC1 andXPD polymorphisms in patients with advanced NSCLCreceiving platinum-based chemotherapy may both be
important (39). The discrepancies observed among thestudies may be due to differential methods, treatmentheterogeneity, and relatively small sample size.
Triggered by the analysis of various DNA repair systemsin the process of DNA platinum adduct repair, we analyzedthese parameters in MPM, also treated with a platinumcombination. The standard treatment for MPM is a
ANTICANCER RESEARCH 34: 435-442 (2014)
438
Figure 3. Association of excision repair cross-complementing group 1 (ERCC1) expression with C118T polymorphism, cellular distribution andsurvival of patients with non-small cell lung cancer (NSCLC) treated with gemcitabine and cisplatin. Forty-six patients (13 female/34 male; 22adenocarcinoma, 8 squamous and 17 large cell carcinoma) were analyzed (overall median survival=14.6 months). Expression of ERCC1 is given ona scale from 0 (low to absent) to 6 (very strong) and was performed as described earlier (26). SNP analysis was performed with quantitativepolymerase chain reaction as described earlier (43). A: Association of cytoplasmic (IA_C) expression of ERCC1 with survival time (months). A totalof 47 patients were analyzed, with 26 patients in the low expression group (Pearson correlation 0.656, p<0.0001). B: Association of the C118Tgenotype with progressive disease (PD), stable disease (SD) and partial response (PR); count represents the number of patients. C: Association ofcytoplasmic ERCC1 staining (IA_C) with the CT and TT genotypes of C118T. D: Association of nuclear staining (IA_N) with the C118T genotype.
Figure 7.3.Association of excision repair cross-complementing group 1 (ERCC1) expressionwithC118Tpolymorphism,cellulardistributionandsurvivalofpatientswithnon-smallcelllungcancer(NSCLC)treatedwithgemcitabineandcisplatin.Forty-sixpatients(13female/34male;22adenocarcinoma,8squamousand17largecellcarcinoma)wereanalyzed(overallmediansurvival=14.6months). ExpressionofERCC1 is givenona scale from0 (low toabsent) to6(verystrong)andwasperformedasdescribedearlier(26).SNPanalysiswasperformedwithquantitativepolymerasechainreactionasdescribedearlier(43).A:Associationofcytoplasmic(IA_C)expressionofERCC1withsurvivaltime(months).Atotalof47patientswereanalyzed,with 26 patients in the low expression group (Pearson correlation 0.656, p<0.0001). B:Association of the C118T genotype with progressive disease (PD), stable disease (SD) andpartialresponse(PR);countrepresentsthenumberofpatients.C:AssociationofcytoplasmicERCC1 staining (IA_C) with the CT and TT genotypes of C118T. D: Association of nuclearstaining(IA_N)withtheC118Tgenotype.
7

Chapter7 AnticancerRes2014;34(1):435-442
Expression of ERCC1 was also related to theC118T SNP; in cytoplasm of tumor cells ofpatientswithaTTgenotype, therewasa lowintensity (0-1), in thosewith aCT genotype atwo-foldhigherintensity(0-2).Inthenucleioftumor cells of patients with a TT genotype,
therewasahighintensity(0-6),butinthosewithaCTgenotypealowerintensity(0-4)(Figure7.3c-d).Another intriguing finding in this cohort of patients was the association of an XPD polymorphism(A751C lys-gln) with longer survival of these patients (Figure 7.4). For the pemetrexed−carboplatincombinationA751gln-glnwasassociatedwithashortersurvival(36).AnotherpolymorphismsofXPD(Asp312asn) was associated with longer survival in patients treated with a carboplatin−taxanecombination (37), as well as NSCLC (38). It can be concluded that in NSCLC, ERCC1 and RRM1expressionandERCC1andXPDpolymorphismsmaybeassociatedwithresponsetocisplatintherapy.However,arecentmeta-analysisconcludedthatthepredictivevalueofERCC1andXPDpolymorphismsinpatientswithadvancedNSCLCreceivingplatinum-basedchemotherapymaybothbeimportant(39).The discrepancies observed among the studies may be due to differential methods, treatmentheterogeneity,andrelativelysmallsamplesize.
TriggeredbytheanalysisofvariousDNArepairsystemsintheprocessofDNAplatinumadductrepair,we analyzed these parameters in MPM, also treated with a platinum combination. The standardtreatment for MPM is a combination of the antifolate pemetrexed and cisplatin or carboplatin.Pemetrexed is a multi-targeting antifolate, which not only inhibits its major target thymidylatesynthase,butalsodihydrofolatereductase,aswelldenovopurinenucleotidesynthesis. Inhibitionofthymidylate synthase will lead to a decrease in dTTP, which will affect DNA repair, but differentlycomparedto inhibitionofRRM1.Inthisstudy,a lowthymidylatesynthaseexpressionwasassociatedwithalongersurvival(18.0months)comparedtothosepatientswhohasahighexpression(9months,p=0.022) (32). In the same cohort of patients, ERCC1 expression by immunohistochemistry failed toshowanysignificantassociation,withanoverallsurvivalof12monthsforthelowexpressionand18forthehigherexpressiongroups.
TheRoleofDNARepairinPDAC
StandardtreatmentinPDACisgemcitabine,whichreplaced5-fluorouracil(5FU)inthelate1990s.Fromallcombinationswhichhavebeeninvestigated,theplatinumcombinationsseemedmostpromising,inasettingeitherwithcisplatinoroxaliplatin(40,41).ThecombinationtreatmentofFOLFIRINOX(5FUwith leucovorin, irinotecan and oxaliplatin) produced the best results in patients with a goodperformancestatuswhowereabletotoleratethispotentiallytoxicregimen.Analternative,theoral5FUformulationS-1withcisplatinproducedalongersurvivalinpatientswiththeERCC1C118Tgenotype(CT and TT; p=0.030) (42). In an analysis of 122 patients, treatment with gemcitabine-basedpolychemotherapy, the TT genotype was associated with a 13.3-month median survival (95%CI=9.7-17.0) compared toCC+TT (11.8months,95%CI=10.4-13.4;p=0.44) (43). ForXPD, anassociationbetween polymorphisms was found for both the XPD Asp312Asn and XPD lys-751gln. The Asn-Asngenotype was associated with a shorter overall survival (11.2 months, 95% CI=10.9-15.7 months)
�108
combination of the antifolate pemetrexed and cisplatin orcarboplatin. Pemetrexed is a multi-targeting antifolate, whichnot only inhibits its major target thymidylate synthase, butalso dihydrofolate reductase, as well de novo purinenucleotide synthesis. Inhibition of thymidylate synthase willlead to a decrease in dTTP, which will affect DNA repair, butdifferently compared to inhibition of RRM1. In this study, alow thymidylate synthase expression was associated with alonger survival (18.0 months) compared to those patientswho has a high expression (9 months, p=0.022) (32). In thesame cohort of patients, ERCC1 expression byimmunohistochemistry failed to show any significantassociation, with an overall survival of 12 months for the lowexpression and 18 for the higher expression groups.
The Role of DNA Repair in PDAC
Standard treatment in PDAC is gemcitabine, which replaced5-fluorouracil (5FU) in the late 1990s. From allcombinations which have been investigated, the platinumcombinations seemed most promising, in a setting either withcisplatin or oxaliplatin (40, 41). The combination treatmentof FOLFIRINOX (5FU with leucovorin, irinotecan andoxaliplatin) produced the best results in patients with a goodperformance status who were able to tolerate this potentiallytoxic regimen. An alternative, the oral 5FU formulation S-1with cisplatin produced a longer survival in patients with theERCC1 C118T genotype (CT and TT; p=0.030) (42). In an
analysis of 122 patients, treatment with gemcitabine-basedpolychemotherapy, the TT genotype was associated with a13.3-month median survival (95% CI=9.7-17.0) compared toCC+TT (11.8 months, 95% CI=10.4-13.4; p=0.44) (43). ForXPD, an association between polymorphisms was found forboth the XPD asp312Asn and XPD lys-751gln. The Asn-Asngenotype was associated with a shorter overall survival (11.2months, 95% CI=10.9-15.7 months) compared to the Asp-Asp phenotype (15.1 months, 95% CI=10.4-19.4 months;p=0.010). Similarly the XPD GLn751Gln had a shortersurvival of 10.3 (95% CI=4.0-16.5) months compared to theLysLys + Lys+Gln cohort (13.3 months, 95% CI=10.9-15.7;p=0.003). Survival of those with XPD Gln751Gln wasshorter compared to those with other genotypes. In a largercohort, 247 patients were treated with multiple drugcombinations (PEXG: gemcitabine-cisplatin + epirubicin-xeloda; PDXG: gemcitabine-cisplatin + docetaxel-xeloda;EC-Gem-Cap: gemcitabine-cisplatin (i.a.) + Epirubicin (i.a.)-xeloda (i.a.) or gemcitabine alone (90 patients). In thispolychemotherapy schedule, the XPD Gln751Gln conferred apoor survival (Figure 5a) (44). In this study, the geneticpolymorphisms were also investigated for a functionalassociation assuming that a decreased repair ability is relatedwith the formation of more Pt–DNA adducts; indeed, whiteblood cells with the specific genotype (Lys751Lys) have alower repair capacity compared with cells with Gln751Gln);this formation of Pt−DNA adducts was enhanced bygemcitabine in the Lys751Lys cohort (Figure 5b). Hence, theanalysis of the polymorphism by a simple blood test offers aninnovative tool for optimizing palliative chemotherapy inpatients with advanced PDAC.
Conclusion
The efficacy of platinum combinations is dependent on theformation of platinum adducts. The extent of their formationand retention are controlled by DNA repair enzymes, but thecontribution of each enzyme may be different for variousdiseases and combinations. For example, in a recent study in19 head and neck cancer cell lines, the sensitivity to cisplatinwas associated with DNA adduct formation and retention butnot with the expression of any repair enzyme or otherpotential markers of resistance, such as drug transporters,which were shown to play a role in cisplatin resistance (45).It seems that in NSCLC treated with gemcitabine−cisplatincombinations, ERCC1 expression is related to efficacy, butthis does not hold true for pemetrexed combinations in MPM.
Another major problem is the method used to determinethe expression of the mRNA the protein. Althoughimmunohistochemistry is a sensitive and versatile method fordetermination of protein expression, it is largely empirical,and the outcome mainly depends on which antibody is beingused and also on the pathologist’s expertise. A major
Peters et al: DNA Repair and Platinum Combination Therapy (Review)
439
Figure 4. Association of the Xeroderma pigmentosum group D (XPD)A751C (Lys/Gln) genotype (67% AA and 33 AT genotype; Hardy-Weinberg equilibrium 0.147) with survival of patients with non-smallcell lung cancer (NSCLC) treated with gemcitabine and cisplatin.
Figure 7.4.Associationof theXerodermapigmentosum group D (XPD) A751C (Lys/Gln) genotype (67% AA and 33 ATgenotype; Hardy-Weinberg equilibrium0.147)with survival of patientswith non-small cell lung cancer (NSCLC) treatedwithgemcitabineandcisplatin.
7

Chapter7 AnticancerRes2014;34(1):435-442
comparedtotheAsp-Aspphenotype(15.1months,95%CI=10.4-19.4months;p=0.010).SimilarlytheXPDGLn751Glnhada shorter survivalof 10.3 (95%CI=4.0-16.5)months compared to theLysLys+Lys+Glncohort (13.3 months, 95% CI=10.9-15.7; p=0.003). Survival of those with XPD Gln751Gln was shortercomparedtothosewithothergenotypes.Ina largercohort,247patientsweretreatedwithmultipledrug combinations (PEXG: gemcitabine-cisplatin + epirubicin- xeloda; PDXG: gemcitabine-cisplatin +docetaxel-xeloda; EC-Gem-Cap: gemcitabine-cisplatin (i.a.) + Epirubicin (i.a.)- xeloda (i.a.) orgemcitabinealone (90patients). In thispolychemotherapy schedule, theXPDGln751Gln conferredapoorsurvival(Figure7.5a)(44).Inthisstudy,thegeneticpolymorphismswerealsoinvestigatedforafunctionalassociationassumingthatadecreasedrepairability is relatedwith the formationofmorePt–DNAadducts;indeed,whitebloodcellswiththespecificgenotype(Lys751Lys)havealowerrepaircapacity compared with cells with Gln751Gln); this formation of Pt−DNA adducts was enhanced bygemcitabineintheLys751Lyscohort(Figure7.5b).Hence,theanalysisofthepolymorphismbyasimplebloodtestoffersaninnovativetoolforoptimizingpalliativechemotherapyinpatientswithadvancedPDAC.
Conclusion
Theefficacyofplatinumcombinationsisdependentontheformationofplatinumadducts.TheextentoftheirformationandretentionarecontrolledbyDNArepairenzymes,butthecontributionofeachenzymemaybedifferentforvariousdiseasesandcombinations.Forexample, inarecentstudy in19headandneckcancercelllines,thesensitivitytocisplatinwasassociatedwithDNAadductformationand retention but not with the expression of any repair enzyme or other potential markers ofresistance,suchasdrugtransporters,whichwereshowntoplaya role incisplatin resistance(45). It
�109
disadvantage of protein expression is the potential lack ofspecificity of antibodies (26, 27), potentially leading toincorrect expression data. Another problem is the cellulardistribution of the protein, as shown for the cytoplasmic andnuclear staining (Figure 3). Hence new antibodies should becharacterized thoroughly, preferably in model systemslacking or having high expression. Several studies have usedthe quantitative RT-PCR technique, but mRNA expressioncan differ from protein expression. mRNA isolation wasconsidered a problem, but nowadays mRNA can be isolatedin reliable quantities, even from paraffin-embedded tissues.The disadvantage is the lack of pathological confirmation ofthe sample, although RNA can be isolated from tumor-enriched parts, or one can use laser microdissection.Therefore, optimization and standardization of these twomodalities with appropriate controls, which can be used forinter-laboratory validation, are essential before largerprospective investigations in homogeneously treated patients,which can address the same pharmacogenetic question.
From the same sample, DNA can be isolated for SNPanalysis. Although SNP analysis using DNA is much morereliable, most data are not strong enough (small groups) to usein prospective studies. These studies are essential to select thosepatients who are likely to respond to the standard treatment andto select patients who are eligible for therapy with tyrosinekinase inhibitors such as erlotinib and gefitinib for patients with
activating mutations for EGFR, or with crizotinib for patientswith ALK expression (46). Since many of these tyrosine kinaseinhibitors are synergistic with DNA-targeted therapy, such asgemcitabine−cisplatin or pemetrexed−cisplatin, pre-treatmentanalysis of gene expression or SNP will help select thetreatment most likely to be effective.
Further investigations are needed to evaluate theseemerging biomarkers, as well as to identify and select theoptimal patient populations that will benefit from specifictreatments. Together with the standardized techniques forsample collection and processing, larger and uniformlytreated populations, and integration with functional data arenecessary in order to validate the best markers forpersonalized treatment of patients.
References
1 Siegel R, Naishadham D and Jemal A:Cancer statistics, 2012.CA Cancer J Clin 62: 10-29, 2012.
2 Schiller JH, Harrington D, Belani CP, Langer C, Sandler A,Krook J, Zhu J, Johnson DH and Eastern Cooperative OncologyGroup: Comparison of four chemotherapy regimens for advancednon-small cell lung cancer. N Engl J Med 346: 92-98, 2002.
3 Scagliotti GV, Parikh P, von Pawel J, Biesma B, VansteenkisteJ, Manegold C, Serwatowski P, Gatzemeier U, Digumarti R,Zukin M, Lee JS, Mellemgaard A, Park K, Patil S, Rolski J,Goksel T, de Marinis F, Simms L, Sugarman KP and Gandara
ANTICANCER RESEARCH 34: 435-442 (2014)
440
Figure 5. A: Association of the Xeroderma pigmentosum group D (XPD) A751C genotype with overall survival of patients with pancreatic ductaladenocarcinoma (PDAC) (213 patients with XPD Lys751Lys, or XPD Lys751Gln and 33 with XPD Gln751Gln, median overall survival 13.0 (11.4-14.6) and 7.0 (4.0-10.0) months) treated with gemcitabine- and cisplatin-containing poly-chemotherapy regimen (44). From (44) with permission.B: White blood samples from five volunteers per group A751A, A751C and C751C were tested for their DNA repair ability after exposure to 1 μMgemcitabine, 200 μM cisplatin or the combination for 24 h, after which a target sequence of β-globin was amplified with extra-long PCR (XL-PCR)as described earlier (44). The extent of DNA repair ability was based on the reduction of PCR amplification of the target sequence. Data are means±SD. Significantly different from *XPD Gln751Gln, **XPD Gln751Gln, or XPD Lys751Gln.
Figure7.5.A:AssociationoftheXerodermapigmentosumgroupD(XPD)A751Cgenotypewithoverallsurvivalofpatientswithpancreaticductaladenocarcinoma(PDAC)(213patientswithXPDLys751Lys,orXPDLys751Glnand33withXPDGln751Gln,medianoverallsurvival13.0(11.4-14.6) and 7.0 (4.0-10.0) months) treated with gemcitabine- and cisplatin-containing poly-chemotherapy regimen (44). From (44)with permission.B:White blood samples from fivevolunteersper groupA751A,A751C andC751Cwere tested for theirDNA repair ability afterexposure to 1μMgemcitabine, 200μMcisplatinor thecombination for 24h, afterwhichatargetsequenceofβ-globinwasamplifiedwithextra-longPCR(XL-PCR)asdescribedearlier(44).TheextentofDNArepairabilitywasbasedonthereductionofPCRamplificationofthetarget sequence. Data are means±SD. Significantly different from *XPD Gln751Gln, **XPDGln751Gln,orXPDLys751Gln.
7

Chapter7 AnticancerRes2014;34(1):435-442
seemsthatinNSCLCtreatedwithgemcitabine−cisplatincombinations,ERCC1expressionisrelatedtoefficacy,butthisdoesnotholdtrueforpemetrexedcombinationsinMPM.
Anothermajor problem is themethod used to determine the expression of themRNA the protein.Although immunohistochemistry is a sensitive and versatile method for determination of proteinexpression, it is largelyempirical,andtheoutcomemainlydependsonwhichantibody isbeingusedandalsoon thepathologist’s expertise.Amajordisadvantageofproteinexpression is thepotentiallack of specificity of antibodies (26, 27), potentially leading to incorrect expression data. Anotherproblem is thecellulardistributionof theprotein,asshownfor thecytoplasmicandnuclearstaining(Figure7.3).Hencenewantibodiesshouldbecharacterized thoroughly,preferably inmodel systemslackingorhavinghighexpression.Several studieshaveused thequantitativeRT-PCR technique,butmRNAexpressioncandifferfromproteinexpression.mRNAisolationwasconsideredaproblem,butnowadays mRNA can be isolated in reliable quantities, even from paraffin-embedded tissues. Thedisadvantageisthelackofpathologicalconfirmationofthesample,althoughRNAcanbeisolatedfromtumor- enriched parts, or one can use laser microdissection. Therefore, optimization andstandardization of these two modalities with appropriate controls, which can be used for inter-laboratoryvalidation,areessentialbeforelargerprospectiveinvestigationsinhomogeneouslytreatedpatients,whichcanaddressthesamepharmacogeneticquestion.
Fromthesamesample,DNAcanbeisolatedforSNPanalysis.AlthoughSNPanalysisusingDNAismuchmore reliable,most data are not strongenough (small groups) to use in prospective studies. Thesestudiesareessentialtoselectthosepatientswhoarelikelytorespondtothestandardtreatmentandto select patients who are eligible for therapy with tyrosine kinase inhibitors such as erlotinib andgefitinib for patients with activating mutations for EGFR, or with crizotinib for patients with ALKexpression (46). Since many of these tyrosine kinase inhibitors are synergistic with DNA-targetedtherapy, such as gemcitabine−cisplatin or pemetrexed−cisplatin, pre-treatment analysis of geneexpressionorSNPwillhelpselectthetreatmentmostlikelytobeeffective.
Further investigationsareneeded toevaluate theseemergingbiomarkers, aswell as to identifyandselect the optimal patient populations thatwill benefit from specific treatments. Togetherwith thestandardized techniques for sample collection and processing, larger and uniformly treatedpopulations,and integrationwithfunctionaldataarenecessary inordertovalidatethebestmarkersforpersonalizedtreatmentofpatients.
Correspondenceto:GodefridusJ.Peters,PhD,DepartmentofMedicalOncology,VUUniversityMedicalCenter,POBox7057,1007MBAmsterdam,theNetherlands.Tel:+31204442633,e-mail:[email protected]
References
1. SiegelR,NaishadhamDandJemalA:Cancerstatistics,2012.CACancerJClin62:10-29,2012.2. Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, JohnsonDH and
Eastern Cooperative Oncology Group: Comparison of four chemotherapy regimens foradvancednon-smallcelllungcancer.NEnglJMed346:92-98,2002.
3. ScagliottiGV,ParikhP,vonPawelJ,BiesmaB,VansteenkisteJ,ManegoldC,SerwatowskiP,GatzemeierU,DigumartiR,ZukinM,LeeJS,MellemgaardA,ParkK,PatilS,RolskiJ,GokselT,deMarinisF,SimmsL,SugarmanKPandGandaraD:PhaseIIIstudycomparingcisplatinplusgemcitabinewithcisplatinpluspemetrexed inchemotherapy-naivepatientswithadvanced-stagenon-small-celllungcancer.JClinOncol26:3543-3551,2008.
4. vanMeerbeeckJP,GaafarR,ManegoldC,VanKlaverenRJ,VanMarckEA,VincentM,LegrandC,BottomleyA,DebruyneC,GiacconeG;EuropeanOrganisationforResearchandTreatmentofCancerLungCancerGroupandNationalCancerInstituteofCanada:RandomizedphaseIIIstudyofcisplatinwithorwithoutraltitrexedinpatientswithmalignantpleuralmesothelioma:
�110
7

Chapter7 AnticancerRes2014;34(1):435-442
an intergroup study of the European Organisation for Research and Treatment of CancerLungCancerGroupandtheNationalCancerInstituteofCanada.JClinOncol23:6881-6889,2005.
5. SancarA:DNArepairinhumans.AnnuRevGenet29:69-105,1995.6. Postel-VinayS,VanheckeE,OlaussenKA,LordCJ,AshworthAandSoriaJC:Thepotentialof
exploitingDNA-repairdefectsforoptimizinglungcancertreatment.NatRevClinOncol9:144-155,2012.
7. Siddik ZH: Mechanisms of action of cancer chemotherapeutic agents: DNA-interactivealkylatingagentsandantitumourplatinum-baseddrugs.In:TheCancerHandbook.AlisonMR(ed.).NaturePublishingGroup,London,UK,pp.1295-1313,2002.
8. Giovannetti E, Toffalorio F, De Pas T and Peters GJ: Pharmacogenetics of conventionalchemotherapy innon-small- cell lungcancer:A changing landscape?Pharmacogenomics 13:1073-1086,2012.
9. MathiauxJ,LeMorvanV,PulidoM,JougonJ,BégueretHandRobertJ:RoleofDNArepairgene polymorphisms in the efficiency of platinum-based adjuvant chemotherapy for non-smallcelllungcancer.MolDiagnTher15(3):159-166,2011.
10. GoldsteinDB,TateSKandSisodiyaSM:Pharmacogeneticsgoesgenomic.NatRevGenet4:937-947,2003.
11. Bergman AM, Ruiz van Haperen VWT, Veerman G, Kuiper CM and Peters GJ: Synergisticinteractionbetweengemcitabineandcisplatininvitro.ClinCancerRes2:521-530,1996.
12. Braakhuis BJM, Ruiz van Haperen VWT, Welters MJP, Peters GJ. Schedule-dependenttherapeuticefficacyofthecombinationofgemcitabineandcisplatininheadandneckcancerxenografts.EurJCancer31A:2335-2340,1995
13. VanMoorselCJA,PinedoHM,VeermanG,BergmanAM,KuiperCM,VermorkenJB,VanderVijghWJF and Peters GJ: Mechanisms of synergism between cisplatin and gemcitabine inovarianandnon-smallcelllungcancercelllines.BrJCancer35:808-814,1999.
14. Van Moorsel CJA, Pinedo HM, Veerman G, Vermorken JB, Postmus PE and Peters GJ:SchedulingofgemcitabineandcisplatininLewislungtumourbearingmice.EurJCancer35:808-814,1999.
15. VanMoorselCJA,PinedoHM,SmidK,ComijnEM,VoornDA,VeermanG,LakerveldB,VanderVijghWJF,GiacconeG,PostmusPE,PetersGJ.ScheduledependentpharmacodynamiceffectsofgemcitabineandcisplatininmicebearingLewisLungmurinenon-smallcell lungtumors.EurJCancer36:2420-2429,2000.
16. PetersGJ,VanMoorselCJ,LakerveldB,SmidK,NoordhuisP,ComijnEC,WeaverD,WilleyJC,VoornD,VanderVijghWJandPinedoHM:Effectsofgemcitabineoncis-platinum-DNAadductformation and repair in a panel of gemcitabine and cisplatin-sensitive or -resistant humanovariancancercelllines.IntJOncol28(1):237-244,2006.
17. YangLY,LiL,JiangH,ShenYandPlunkettW:ExpressionofERCC1antisenseRNAabrogatesgemcitabine-mediated cytotoxic synergism with cisplatin in human colon tumor cellsdefective inmismatch repairbutproficient innucleotideexcision repair. ClinCancerRes6:773-781,2000.
18. Selvakumaran M, Pisarcik DA, Bao R, Yeung AT and Hamilton TC. Enhanced cisplatincytotoxicitybydisturbingthenucleotideexcisionrepairpathwayinovariancancercelllines.CancerRes63:1311-1316,2003.
19. Van Moorsel CJA, Smid K, Voorn DA, Bergman AM, Pinedo HM and Peters GJ: Effects ofgemcitabineandcis-platinumcombinationsonribonucleotideanddeoxyribonucleotidepoolsinovariancancercelllines.IntJOncol22:201-207,2003.
20.ElnaggarM,GiovannettiEandPetersGJ:Moleculartargetsofgemcitabineaction:Rationalefor development of novel drugs and drug combinations. Curr Pharm.Design 18: 2811-2829,2012.
21. Danesi R, Pasqualetti G, Giovannetti E, Crea F, Altavilla G, Del Tacca M and Rosell R:Pharmacogenomics in non-small cell lung cancer chemotherapy. Adv Drug Deliv Rev 61:408-417,2009.
�111
7

Chapter7 AnticancerRes2014;34(1):435-442
22. PfisterDG,JohnsonDH,AzzoliCG,SauseW,SmithTJ,BakerS,Jr.,OlakJ,StoverD,StrawnJR,TurrisiAT,SomerfieldMRandAmericanSocietyofClinicalOncology:Americansocietyofclinical oncology treatment of unresectable non-small-cell lung cancer guideline: Update2003.JClinOncol22:330-353,2004.
23. SmitEF,vanMeerbeeckJP,LianesP,DebruyneC,LegrandC,SchramelF,SmitH,GaafarR,Biesma B, Manegold C, Neymark N, Giaccone G; Europea Organization for Research andTreatmentofCancerLungCancerGroup.Three-armrandomizedstudyoftwocisplatin-basedregimensandpaclitaxelplusgemcitabineinadvancednon-small-cell lungcancer:aphaseIIItrialoftheEuropeanOrganizationforResearchandTreatmentofCancerLungCancerGroup-EORTC08975.JClinOncol21:3909-3917,2003.
24. LordRV,BrabenderJ,GandaraD,AlberolaV,CampsC,DomineM,CardenalF,SánchezJM,GumerlockPH,TarónM,SánchezJJ,DanenbergKD,DanenbergPVandRosellR:LowERCC1expressioncorrelateswithprolongedsurvivalaftercisplatinplusgemcitabinechemotherapyinnon-smallcelllungcancer.ClinCancerRes8:2286-2291,2002.
25. OlaussenKA,DunantA, Fouret P, Brambilla E, André F, Haddad V, Taranchon E, FilipitsM,PirkerR,PopperHH,StahelR,SabatierL,PignonJP,TurszT,LeChevalierT,SoriaJCandIALTBio Investigators: DNA repair by ERCC1 in non-small cell lung cancer and cisplatin-basedadjuvantchemotherapy.NEnglJMed355:983-991,2006.
26. Bhagwat NR, Roginskaya VY, Acquafondata MB, Dhir R, Wood RD and Niedernhofer LJ:Immunodetection of DNA repair endonuclease ERCC1-XPF in human tissue. Cancer Res 69:6831-6838,2009.
27. FribouletL,OlaussenKA,PignonJP,ShepherdFA,TsaoMS,GrazianoS,KratzkeR,DouillardJY, Seymour L, Pirker R, FilipitsM, André F, Solary E, Ponsonnailles F, Robin A, Stoclin A,DorvaultN,CommoF,AdamJ,VanheckeE,SaulnierP,ThomaleJ,LeChevalierT,DunantA,RousseauV,LeTeuffG,BrambillaEandSoriaJC:ERCC1isoformexpressionandDNArepairinnon-smallcelllungcancer.NEnglJMed368:1101-1110,2013.
28. ChenS,ZhangJ,WangR,LuoXandChenH:Theplatinum-basedtreatmentsforadvancednon-smallcelllungcancer,islow/negativeERCC1expressionbetterthanhigh/positiveERCC1expression?Ameta-analysis.LungCancer70:63-70,2010.
29. CeppiP,VolanteM,NovelloS,RapaI,DanenbergKD,DanenbergPV,CambieriA,SelvaggiG,SaviozziS,CalogeroR,PapottiMandScagliottiGV:RRM1geneexpressionsbutnotEGFRarepredictive of shorter survival in advanced non-small- cell lung cancer treatedwith cisplatinandgemcitabineAnn.Oncol17:1818-1825,2006.
30.Bepler G, Kusmartseva I, Sharma S, Gautam A, Cantor A, Sharma A and Simon G: RRM1modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small cell lungcancer.JClinOncol24:4731-4737,2006.
31. BeplerG,WilliamsC,SchellMJ,ChenW,ZhengZ,SimonG,GadgeelS,ZhaoX,SchreiberF,Brahmer J, Chiappori A, Tanvetyanon T, Pinder-SchenckM, Gray J, Haura E, Antonia S andFischer JR: Randomized international phase III trial of ERCC1 and RRM1 expression-basedchemotherapyversusgemcitabine/carboplatin inadvancednon-smallcell lungcancer.JClinOncol31(19):2404-2412,2013.
32. Zucali PA, Giovannetti E, Destro A,MencoboniM, Ceresoli GL, Gianoncelli L, Lorenzi E, DeVincenzoF,SimonelliM,PerrinoM,BruzzoneA,ThunnissenE,TunesiG,GiordanoL,RoncalliM,PetersGJandSantoroA:Thymidylatesynthaseandexcisionrepair-cross-complementinggroup-1aspredictorsofresponsivenessinmesotheliomapatientstreatedwithpemetrexed-carboplatin.Clin.CancerRes17:2581-2590,2011.
33. Yu JJ, LeeKB,MuC, LiQ,AbernathyTV,Bostick-BrutonF andReedE.Comparisonof twohuman ovarian carcinoma cell lines (a2780/cp70 and mcas) that are equally resistant toplatinum,butdifferatcodon118oftheercc1gene.IntJOncol16:555-560,2000.
34. IslaD,SarriesC,RosellR,AlonsoG,DomineM,TaronM,Lopez-VivancoG,CampsC,BotiaM,NunezL,Sanchez-RoncoM,SanchezJJ,Lopez-BreaM,BarnetoI,ParedesA,MedinaB,ArtalAandLianesP:Singlenucleotidepolymorphismsandoutcomeindocetaxel-cisplatin-treatedadvancednon-smallcelllungcancer.AnnOncol15:1194-1203,2004.
�112
7

Chapter7 AnticancerRes2014;34(1):435-442
35. RyuJS,HongYC,HanHS,LeeJE,KimS,ParkYM,KimYCandHwangTS:Associationbetweenpolymorphismsofercc1 andxpdand survival innon-small cell lung cancerpatients treatedwithcisplatincombinationchemotherapy.LungCancer44:311-316,2004.
36. TiseoM,GiovannettiE,TibaldiC,CameriniA,DiCostanzoF,BarbieriF,BurgersJA,VincentA,PetersGJ,SmitEFandArdizzoniA:Pharmacogeneticstudyofpatientswithadvancednon-smallcelllungcancertreatedwithsecond-linepemetrexedorpemetrexed-carboplatin.LungCancer78:92-99,2012.
37. GurubhagavatulaS,LiuG,ParkS,ZhouW,SuL,WainJC,LynchTJ,NeubergDSandChristianiDC:XPDandXRCC1geneticpolymorphismsareprognosticfactorsinadvancednon-smallcelllung cancer patients treated with platinum chemotherapy. J Clin Oncol 22(13): 2594-2601,2004.
38. Tibaldi C, Giovannetti E, Vasile E,Mey V, Laan AC, Nannizzi S, DiMarsico R, Antonuzzo A,OrlandiniC,RicciardiS,DelTaccaM,PetersGJ,FalconeAandDanesiR:CorrelationofCDA,ERCC1, andXPDpolymorphismswith responseand survival ingemcitabine/cisplatin-treatedadvancednon-smallcelllungcancerpatients.ClinCancerRes14:1797-1803,2008.
39. Wei SZ, Zhan P, ShiMQ, ShiY, QianQ,Yu LK and SongY: Predictive value of ercc1 and xpdpolymorphisminpatientswithadvancednon-smallcelllungcancerreceivingplatinum-basedchemotherapy:Asystematicreviewandmeta-analysis.MedOncol28:315-321,2011.
40.Conroy T, Desseigne F, YchouM, BouchéO, GuimbaudR, Bécouarn Y, Adenis A, Raoul JL,Gourgou-BourgadeS,delaFouchardièreC,BennounaJ,BachetJB,Khemissa-AkouzF,Péré-VergéD,DelbaldoC,AssenatE,ChauffertB,MichelP,Montoto-GrillotC,DucreuxM;GroupeTumeursDigestivesofUnicancerandPRODIGEIntergroup:FOLFIRINOXversusgemcitabineformetastaticpancreaticcancer.NEnglJMed364:1817-1825,2011.
41. Reni M, Cordio S, Milandri C, Passoni P, Bonetto E, Oliani C, Luppi G, Nicoletti R, Galli L,BordonaroR,PassardiA,ZerbiA,BalzanoG,AldrighettiL,StaudacherCandVillaE,DiCarloV:Gemcitabineversuscisplatin,epirubicin,fluorouracil,andgemcitabineinadvancedpancreaticcancer:ArandomisedcontrolledmulticentrephaseIIItrial.LancetOncol6:369-376,2005.
42. Kamikozuru H, Kuramochi H, Hayashi K, Nakajima G and Yamamoto M: ERCC1 codon 118polymorphismisausefulprognosticmarker inpatientswithpancreaticcancertreatedwithplatinum-basedchemotherapy.IntJOncol32:1091-1096,2008.
43. GiovannettiE,PacettiP,ReniM,LeonLG,MambriniA,VasileE,GhidiniM,FunelN,LucchesiM,CeredaS,PetersGJandCantoreM:AssociationbetweenDNArepairpolymorphismsandsurvival in pancreatic cancer patients treated with combination chemotherapy.Pharmacogenomics12:1641-1652,2011.
44. Avan A, Pacetti P, Reni M, Mambrini A, Cereda S, Peters GJ, Cantore M and Giovannetti:Prognosticfactorsingemcitabine/cisplatinpolychemotherapyregimensinpancreaticcancer:XPD-Lys751Glnpolymorphismstrikesback.Int.JCancer133:1016-1022,2013.
45. Martens-de Kemp SR, Dalm SU,Wijnolts FMJ, Brink A, Honeywell RJ, Peters GJ, BraakhuisBJMandBrakenhoffRH:DNA-boundplatinumisthemajordeterminantofcisplatinsensitivityinheadandnecksquamouscarcinomacells.PLoSOne8(4):e61555,2013.
46. GalvaniE,PetersGJandGiovannettiE:EGFreceptor-targetedtherapy innon-smallcell lungcancer:Roleofgermlinepolymorphismsinoutcomeandtoxicity.FutureOncol8:1015-1029,2012.
ReceivedSeptember24,2013RevisedNovember27,2013AcceptedNovember29,2013;
�113
7

Chapter7 AnticancerRes2014;34(1):435-442
�114
7

CHAPTER8ModulationofSignalingEnhancestheEfficacyof
theCombinationofSatraplatinandErlotinib
AbolfazlAvan,1AukeD.Adema,1EvelineK.Hoebe,1CharlotteM.Huijts,1AmirAvan,1,2GarethJ.Veal,3R.Ruitenbeek,4K.Wosikowski,5GodefridusJ.Peters1*
1DepartmentofMedicalOncology,VUUniversityMedicalCenter,Amsterdam,TheNetherlands;2DepartmentofNewSciencesandTechnology,FacultyofMedicine,MashhadUniversityofMedicalSciences,Mashhad,Iran;3NorthernInstituteforCancerResearch,
NewcastleUniversity,NewcastleuponTyne,UnitedKingdom;4Pamgene,‘sHertogenbosch,theNetherlands;5PreviouslyatGPCBiotech,Munich,Germany;currentaddress:Isarna
Therapeutics,Munich,Germany
CurrDrugTargets2014;15(14):1312-1321
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
Modulationofsignalingenhancestheefficacyofthecombinationofsatraplatinanderlotinib
ABSTRACT: The active metabolite (JM118) of the oral platinum analog satraplatin (JM216) wasinvestigated for potential synergism with erlotinib, an epidermal growth factor receptor (EGFR)inhibitor.JM118sensitivityof7cancercelllines(ovarian:2008,A2780;colon:Lovo92,WiDr;lung:A549,SW1573; epidermoid: A431),was enhancedmost pronouncedwhen JM118 preceded erlotinib,whichwasassociatedwith increasedformationofDNA-platinumadducts.Thecombination increasedG2/Mphase accumulation and enhanced apoptosis. JM118 increased the phosphorylation of the cell cycleproteinsCDK2andCHK1after24hrexposure.JM118/erlotinibenhancedErkandAktphosphorylationafter 2hr. JM118 significantlydecreased thephosphorylationofPTEN,VEGFR,EPHA1,ERBB4,FGF-R,andSTAT3by20(PTEN)to>90%(STAT3).Conclusion:ErlotinibenhancedtheeffectsofJM118,evenincellswithmutationsinRas.Themechanismofsynergyinvolvedacombinationofeffectsonplatinum-DNAadductformation,cellcycledistributionandsignaling.CurrDrugTargets2014;15(14):1312-1321
Keywords:Satraplatin,erlotinib,signaling,Akt,Erk,coloncancer,lungcancer
Introduction
Platinumcompoundsarethemainstayoftreatmentofvariousmalignancies.Cisplatin isusedforthetreatment of cancers such as lung, cervical, ovarian, testicular and squamous cell carcinoma of theheadandneck[1],whileoxaliplatinismainlyusedforthetreatmentofcolorectalcancers[2].Platinumcompounds are always given parenterally, usually in combination with other drugs such asgemcitabine, taxanes,or5-fluorouracil (5-FU) [3-5].Platinumderivatives formadductswithDNA [6],which issensedasdamagebyrepairpathwaysandwilleitherberepaired,or thecellswillentertheapoptosispathway[7].Cisplatin-DNAadductscanbe recognizedby themismatch repairpathwayorthe nucleotide excision repair pathway, while oxaliplatin-DNA adducts are only recognized by thenucleotideexcisionrepairpathway[3,8].
Satraplatin (JM216) is the first oral platinum analog [9], exhibiting greater lipophilicity compared tocarboplatin and cisplatin [10], favorable bioavailability [9, 11, 12], effective penetration through thebloodbrainbarrier[10]andagoodsafetyprofilelackingnephrotoxicityandneurotoxicitycomparedtocisplatininvivo[13,14].
Adductformationbysatraplatinismostlycomparabletocisplatin,sincewhenJM118losesitsacetategroupitisstructurallysimilartocisplatin[15].Nevertheless,satraplatincompoundshavemuchhigherpotencycomparedtocisplatin[16],possiblyduetoDNAreplicationandtranscription, leadingtocellcycle arrest in theG2phase andfinally apoptosis [17].Moreover, adducts formedby satraplatin arebulkier, more efficient in inhibiting DNA synthesis and less recognizable by DNA mismatch repairsystemcomparedtocisplatinandcarboplatin[8,18].
Satraplatin has shown some efficacy in Phase II studies with chemo naive small-cell lung cancerpatientsandadvancednon-smallcelllungcancer(NSCLC)[19,20]aswellascervicalcancer,hormonerefractory prostate cancer and breast cancer [21-23]. More promising efficacy was found in acombinationofsatraplatinplusprednisoneversusprednisonealoneinpatientswithcastrate-resistantprostatecancer[24],withanincreasedmedianprogression-freesurvival(PFS)(5.2vs2.5months;p=.023) compared to the prednisone-alone arm. However, in a larger Phase III trial (Satraplatin andPrednisoneAgainstRefractoryProstateCancer[SPARC])with950pretreatedprostatecancerpatients,despite the mild toxicity along with improved PFS and the palliative effects, satraplatin plusprednisolonefailedtodemonstrateasignificantimprovementinoverallsurvival[25].
Satraplatin has also been tested in combination with bevacizumab, a monoclonal antibody againstvascularendothelialgrowthfactor,[26]withsomepromisingactivityobserved[27,28].Bevacizumab-satraplatin plus a low dose prednisolone in docetaxel-pretreated patients with metastatic castrate-
�116
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
resistantprostatecancerledtoa30%declineinprostate-specificantigen,amediantimetoprogressionof7.0monthsandmedianoverallsurvivalof11.2months[29].
Thereisevidencethatcisplatinmaymodulateseveralsignaltransductionpathways,includingAKT,ABL,p53,MAPK/JNK/ERK[30].Akt,alsoknownasserine/threoninekinaseorproteinkinaseB,pathway,hasacritical regulatory role in diverse cellular processes, including cell cycle progression, protein synthesis,glucosemetabolismanddown-regulationofcelldeath[31].Epidermalgrowthfactorreceptor(EGFR)isoneofthefourkeymoleculesofthispathway,whichisalsoimportantinK-RasgeneactivityandinAktkinase induction[32].Thus,overexpressionofEGFRmay leadtodrugresistanceandpotentiallytumorgrowth[33,34].Additionally, there isadirect interactionbetweenphosphatidylinositol3kinase(PI3K)andRasprotein for activationofPI3K/Akt cascade [35], andRasactivity is important in stimulationofPI3K/Aktsignaling[36].K-Ras-mutatedtumorcellsstimulatetheexpressionofEGFRligands,whichleadstodownstreamsignalingthroughphosphorylatedErk1/2andAkt[37-39].Moreover,NSCLCtumorcellswithoverexpressingAkt1seemresistanttocisplatin[40].MutationsinTP53andPTENwerefoundinhigh-gradeovarian carcinomaand resistant colorectal carcinoma [41-43].Moreover, anti EGFR therapeuticssuchasthemonoclonalantibodycetuximabandthesmallmoleculeserlotinib,gefitinibandafatinibareregistered for treatment of NSCLC and colon cancer [44-47]. Earlier, we demonstrated that posttreatmentwitherlotinib (Tarceva®)enhanced theeffectof themultitargetedantifolatepemetrexed inNSCLCcellsbyaffectingcellcycleregulatingproteins[48].Sinceerlotinibisoftencombinedwithcisplatinor carboplatin, and since platinum compounds affect signaling pathways [13], we postulated thaterlotinibmightincreasetheefficacyofsatraplatin.Aclinicaladvantagemightbethecombinationoftwooraldrugs.Thereforewetestedwhethersatraplatinanderlotinibinteractedsynergisticallyinlung,colonand ovarian cancer cells, and investigated the role of DNA adduct formation, cell cycle and signalingpathwaysintheinteraction.
MATERIALSANDMETHODS
DrugsandChemicals
Satraplatin (JM216) and JM118 were provided by GPC Biotech (Munich, Germany), and erlotinib,oxaliplatin and cisplatin were obtained from Roche, Sanofi-Aventis and Sigma (Zwijndrecht, theNetherlands), respectively. The drugswere dissolved in ethanol or dimethyl sulfoxide (DMSO), anddilutedinculturemediumbeforeuse.RPMI-1640orDMEMmedium,fetalcalfserum(FCS),penicillin(50IU/ml)andstreptomycin(50g/ml)werefromGibco(Gaithersburg,MD).AllotherchemicalswerepurchasedfromSigma-Aldrich(Zwijndrecht,theNetherlands).
CellCulture
Seven different cell lines were included in this study: ovarian cancer (A2780, 2008), NSCLC (A549,SW1573),andcoloncancer(Lovo92,WiDr),whileA431(epidermoidcarcinoma)wasincludedbecauseofitshighEGFRexpression.CellsweregrowninmonolayersandmaintainedinexponentialgrowthineitherRPMI1640(WiDr,Lovo92,2008)orDMEM(SW1573,A549,A2780,A431)supplementedwith10%heat-inactivated FCS and 20 mM HEPES at 37°C under an atmosphere of 5% carbon dioxide. Celldoublingtimesvariedbetween22-28hrundertheseconditions.
DrugSensitivityandCombinationStudies
The cell growth inhibitory effects of the drugs were determined after a 72 hr exposure by thesulforhodamine B (SRB) assay in all cell lines as previously described [49]. The combinations weredesignedeitherbysimultaneoustreatmentwithJM118/erlotinibfor72hr,or24hrofpreincubationwitheitherJM118orerlotinib followedby48hrexposure to thecombinationoferlotinib/JM118 (Table8.1).Drugrangesvariedfrom0.005to20μMforJM118orfrom0.01-30μMforerlotinib.Combinationsweredesignedbasedonafixedratioofthehalfmaximalinhibitoryconcentration(IC50)valuesofeachdrug.
�117
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
Thecombinationswereevaluatedusingthemultiple-drugeffectanalysisaccordingtoChouandTalalay[50].CalcuSynsoftwareversion2.0wasusedtoanalysethedata(ElsevierBiosoft,Oxford,UK).Resultswere expressed asmutually non-exclusive combination index (CI) values for every fraction affected
�118
Satraplatin and Erlotinib Synergism Current Drug Targets, 2014, Vol. 15, No. 14 3
Table 1. Characterization of EGFR, PIK3CA, k-Ras, PTEN, TP53 and p16 gene status in seven cancer cells lines. Drug sensitivity to and cell death induced by erlotinib, JM118, and their combination.
Epidermoid Carcinoma�
Non Small Cell Lung Cancer� Ovarian Cancer� Colon Cancer�
� A431� SW1573� A549� 2008� A2780� Lovo92� WiDr�
Mutationsa� � � � � � � �
EGFR� Wt� Wt� Wt� Wt� Wt� Wt� Wt�
PIK3CA� Wt� Mut (K111E)� Wt� Wt� Wt� Wt� Wt�
k-Ras� Wt� Mut (G12C)� Mut (G12S)� Wt� Wt� Mut (G13D)� Wt�
PTEN� Wt� Wt� Wt� Wt�Mut (K128-
R130del)�Wt� Wt�
TP53�Mut
(R273H)�Wt� Wt� TP53 (Y126C)� Wt� Wt� Mut (R273H)�
p16� Wt�Mut (M1_ *157del)�
Wt� Wt� Wt� Wt� Wt�
IC50 (�M)� � � � � � � �
Cisplatin� -� 2.8 ± 0.6� 3.9 ± 0.6� 1.4 ± 0.1� 0.5 ± 0.1� 1.5 ± 0.4� 6.9 ± 1.0�
Oxaliplatin� -� 0.7 ± 0.1� 2.2 ± 0.9� 0.4 ± 0.0� 0.3 ± 0.0� 0.2 ± 0.0� 0.9 ± 0.1�
JM216� -� 1.1 ± 0.1� 3.0 ± 0.6� 2.1 ± 0.6� 0.9 ± 0.1� 2.3 ± 0.3� 3.5 ± 0.8�
JM118� 1.6 ± 0.2� 1.5 ± 0.4� 2.2 ± 0.6� 0.9 ± 0.4� 0.2 ± 0.0� 0.3 ± 0.1� 1.0 ± 0.4�
Erlotinib� 0.9 ± 0.1� 4.9 ± 0.4� 4.6 ± 0.6� 6.0 ± 0.7� 5.2 ± 0.7� 6.4 ± 1.5� 5.4 ± 0.6�
CIb value (�M)� � � � � � � �
JM118+erlotinib (72h)� 0.55 ± 0.07� 1.11 ± 0.13� 0.60 ± 0.08� 1.04 ± 0.21� 0.96 ± 0.18� 0.78 ± 0.05� 1.20 ± 0.14�
Erlotinib (24h)=> JM118+erlotinib (48h)�
0.68 ± 0.08� 1.36 ± 0.08� 0.63 ± 0.13� 0.56 ± 0.16� 0.91 ± 0.17� 0.85 ± 0.08� 0.88 ± 0.01�
JM118 (24h)=> JM118+erlotinib (48h)�
0.52 ± 0.10� 0.78 ± 0.10� 0.69 ± 0.09� 0.68 ± 0.07� 0.90 ± 0.04� 0.76 ± 0.12� 0.74 ± 0.16�
Sub-G1 cellsc� � � � � � � �
Control� � 3.4� 1.4� 1.3� 1� 1.7� 2.7�
JM118 (48h)� � 4.6 ± 2.4� 1.8 ± 1.0� 1.5 ± 0.9� 14.0 ± 0.7**� 6.1 ± 1.2� 5.3 ± 1.9�
JM118 (72h)� � 6.2 ± 2.4� 2.5 ± 1.0� 3.8 ± 0.9� 28.4 ± 0.7**� 14.5 ± 1.2**� 13.7 ± 1.9**�
Erlotinib (72h)� � 1.7 ± 2.4� 2.0 ± 1.0� 2.7 ± 0.9� 6.2 ± 0.7#� 1.3 ± 1.2� 2.8 ± 1.9�
JM118+erlotinib (72h)� � 7.8 ± 2.4� 4.5 ± 1.0� 4.6 ± 0.9� 20.2 ± 0.7**� 11.7 ± 1.2**� 7.9 ± 1.9#�
Erlotinib => JM118+erlotinib (72h)� � 2.3 ± 2.4� 0.9 ± 1.0� 1.3 ± 0.9� 4.0± 0.7� 2.3 ± 1.2� 1.8 ± 1.9�
JM118 => JM118+erlotinib (72h)� � 10.9 ± 2.4*� 3.9 ± 1.0� 4.8 ± 0.9� 15.4 ± 0.7**� 8.5 ± 1.2#� 5.7 ± 1.9�
a. mutation analysis data were as reported in earlier papers (48, 53, 54) and from www.sanger.ac.uk/genetics/CGP/. b. Cells were exposed to a concentration range of drug in a fixed ratio based on IC50s of, JM118 and erlotinib. Mean combination index (CI) of the erlotinib/JM118 combinations at fraction affected (FA) values of 0.5, 0.75 and 0.9 were averaged for each experiment, and this value was used to calculate the mean between experiments, as described in the Materials and Methods section. c. Cells were exposed to IC50s of JM216, JM118 and erlotinib. The apoptotic index was calculated as the percentage cells in the sub-G1 phase compared to all counted cells after being exposed for 48-72 hours to IC50 concentrations of the drugs and analyzed by FACS simultaneously with the cell cycle. #, P<0.05; *, P<0.01; **, P<0.0001 compared to controls
Table8.1.CharacterizationofEGFR,PIK3CA,K-Ras,PTEN,TP53andp16genestatusinsevencancercellslines.Drugsensitivityto
andcelldeathinducedbyerlotinib,JM118,andtheircombination.
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
(FA),whileforthefinalevaluationweusedtheaveragedCIatFAsof0.50,0.75and0.90,representingrelevant growth inhibition values. The classification for the effect is as follows: CI <0.70 (highlysynergistic/synergistic); 0.70-0.90 (moderately synergistic); 0.90-1.10 (additive); >1.10-1.45(antagonistic).
MeasurementofPlatinum-DNAAdductFormation
Platinum adduct formationwasmeasured in the A2780, A549 and SW1573 cell lines. The cellswereselectedbasedontheirsensitivitytothedifferentsingleorcombinationtreatmentschedules.Initiallyadductformationwasdeter-minedafterexposureto10Moxaliplatin,or5Mcisplatin,JM216,JM118.
Forthispurpose5x105cellswereseededin6wellsplatesanddrugswereaddedafter24hr.Cellswereharvestedafter72hr.Forthecombinations,differentexposuretimeswereused:24hrto5MJM118aloneorincombinationorfollowedby5Merlotinibfor24hr.Alternatively,cellswerepretreatedwitherlotiniborJM118followedbyJM118/erlotinibfor24hr.DNAwasisolatedfromthecellpelletsusingaQIAamp DNA isolation kit (Qiagen, Venlo, the Netherlands). DNA yield and purity were checked at260-280 nm with NanoDrop 1000 Detector (NanoDrop Technologies, Wilmington, DE, USA) andsamples were analyzed on an inductively coupled plasma mass spectrometer (ICP-MS) followingovernightacidhydrolyzationaspreviouslydescribed[51].
MeasurementofCellDeathbyAnalysisofSub-G1Region
Toevaluatetheeffectofthedrugsonmodulationofcellcycle,thecellsweretreatedfor24,48and/or72hrwithJM118,erlotinibortheircombinationattheirIC50concentrations.Thecellswerestainedbyhighlyfluorescentdyepropidiumiodide(PI)solution,containing50g/mlPI,0.1%(Tri-)sodiumcitrate,0.1% TritonX-100 and0.1mg/mlRNAaseA. TotalDNA contentweremeasuredon a FlowCytometer(Fluorescence-ActivatedCellSorter[FACS],BDBenelux,Erembodegem-Aalst,Belgium)andtheresultswereanalyzedwiththeCELLQuestsoftware,asdescribedearlier[48,49,53].Moreover,theabilityofthedrugstoinducecelldeathwasevaluatedbymeasuringsub-G1regionsduringcellcycleanalysis.
WesternBlotAnalysis
Inordertoevaluateexpressionofrepairenzymes(themismatchrepairenzymesMSH2andMLH1,andtheexcisionrepairenzymeERCC1),EGFR,themodulationofsignalingpathwayproteins(Akt,Erkandp38) and the expression of cell cycle regulatory proteins (CDC25c, CDK2, CHK1, CHK2 and E2F-1),westernblotanalysiswasdone,asdescribedearlier(53,54).Twentyμgofeachproteinwasseparatedon a 10% sodium dodecyl sulfate (SDS)- polyacrylamide gel and transferred to the polyvinylidenedifluoride (PVDF) membrane (Immobilion®-FL, Millipore, Billerica, MA, USA). The membrane wasincubatedovernightwiththeanti-MSH2,MLH1,ERCC1,Akt,Erk,p38,CDC25c,CDK2,CHK1,CHK2andE2F-1(1:1000,dilutedintheblockingsolution;CellSignalingTechnology,Danvers,MA,USA)andmouseanti-β-actin(1:10000;Sigma-AldrichChemicals,Zwijndrecht,theNetherlands).ForEGFRweused1:1000Rabbit-anti-EGFR (Cell Signaling Technology,Danvers,MA,USA) andDonkey-anti- Rabbit at a 1:2000dilution(DAKOCytomation,Glostrup,Denmark).Themembranewas incubatedwiththeHorseradishPeroxidaseConjugatedsecondaryantibody(DAKOCytomation,Glostrup,Denmark).ThebandswerevisualizedusingECLPlusAgent(AmershamBiosciences,Piscataway,NJ,USA).
AnalysisofKinaseActivityinPeptideArray
ThePamchiparraycontains136phosphorylationsitesof144peptidesconsistingof15aminoacids.The13amino-carboxylicacid(R-COOH)ofeachpeptidecorrespondstoknownorputativephosphorylationsitesinavarietyofhumanproteins.
TheA549cellswereexposedtoerlotiniband/orJM118for2hrattheirIC50values,harvested,rapidlyfrozeninliquidnitrogenandstoredat-80°Cuntilanalysis.Beforeanalysiscellswerecompletelylysedonicefor20minusingMammalianProteinExtractionReagent(M-PER),whichcontainsphosphatase
�119
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
andproteaseinhibitors(ThermoScientific,Rockford,IL,USA).Lysateswerecentrifugedfor15minat10000rpm;thesupernatantwascollectedandstoredat-80°C.40µlcontrolsamplemixforthekinaseactivityarraywassubsequentlypreparedbyusing reactionbuffer, containingABLbuffer (Westburg,Leusden,theNetherlands),100μMadenosinetriphosphate(ATP;Sigma-Aldrich),fluorescein-labeledantibodyPY20(Exalpha,Maynard,MA,USA),7.5glysateproteinandDMSO.Afterblockingthearrayswith 2% bovine serum albumin (BSA) and subsequent loading of the samplemix onto the arrays intriplicate, incubation (at 30 °C) was started for 60 cycles utilizing a PamStation®12 instrument.Repeatedfluorescentimagingofeacharraywasperformedwitha12-bitcharge-coupleddevice(CCD)camera, monitoring fluorescence in- tensities in real time. Spot intensity at each time point wasquantified(beingcorrectedforthelocalbackground)bythemeansofBionavigatorsoftwareversion5.1(PamGeneInternational,'s-Hertogenbosch,theNetherlands).Basedontheresultingtime-resolvedcurves, the initial phosphorylation rate (V-ini) was calculated using specific kinetic algorithms andappropriatestatisticalmethods.
StatisticalAnalysis
All experiments were performed in triplicate and repeated at least twice. Data were expressed asmeanvalues±standarderrorofmean(SEM).Theresultswereanalyzedbyregular two-wayANOVAfollowed by Dunnett's multiple comparison tests. Statistical significance was set at 95% confidenceintervalofdifferenceofmeansandatwo-tailedmultiplicityadjustedPvalueof<0.05.
RESULTS
CharacteristicsoftheCellLines
Sevencelllinesdifferingintheexpressionofrepairenzymesandsignalingpathways(Table8.1;Figure8.1)wereused.Lovo92ismismatchrepairdeficientbecauseofthelackofexpressionofDNAmismatchrepairproteinMutLhomolog1(MLH1);theovariancancercelllines2008andA2780haveasomewhathigherexpressionofMLH1;A431,A549andWiDrshowedahigherexpressionofMLH1.TheotherDNAmismatchrepairproteinMSH2wasrelativelyhighinLovo92andWiDr.A549,WiDRandLovo92haveahigher expressionof excision repair cross-complementation group 1 (ERCC1) compared to theothercell lines.Epidermalgrowthfactorreceptor(EGFR)isexpressedinallcell lines,atalowlevelinA549andSW1573cells,butnotdetectablebywesternblottinginA2780andWiDrcells.However,withFACSanalysisalowbutdetectableEGFRexpressioncouldbedeterminedinthesecelllines;intheothercelllinesEGFRexpressionbyFACSwashigher(datanotshown).AhighexpressionwasfoundinA431cells,apositivecontrolforEGFRoverexpression.
�120
A
MSH2
tubulin
SW1573
A549WiDR
Lovo92
A2008
A2780
tubulin
ERCC1
MSH2
Tubulin
ERCC1
Tubulin
MLH1
Tubulin
A431
EGFR
Β-actin
A
B
Figure 8.1. Western blot of selectedrepairproteinsusingtubulinas loadingcontrol (A) and EGFR using β-actin asloading control (B). For the analysiscellsinexponentialgrowthphasewereharvested and analysed on WesternBlot as described in the Materials andMethods. Analysis of the repairenzymes inA431cellswasnotdoneonthe same blot as the other cell lines.Hence an example with a similarprotein loading (20 μg) and a similarexpressionofβ-actinwaschosentobeadded to this figure. The blots consistof a representative example of 3separateblots.
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
Theovariancellline2008hadaTP53(Y126C)mutationandA2780aPTEN(K128-R130del)mutation.TheNSCLCcell linesA549hadaK-Ras(G12S)mutation,andSW1573PIK3CA (K111E),K-Ras(G12C),andp16(M1_*157del)mutations.ThecoloncancercelllinesLovo92hadaK-Ras(G13D)mutation,andtheWiDRaTP53(R273H)mutation(Table8.1).A431cellswereincludedbecauseoftheoverexpressionofEGFR(Table8.1,Figure8.1)
PreincubationandSimultaneousJM118and/orErlotinibIncreasedtheSensitivity
JM118wasthemostactiveplatinumanalog inallcell lines investigated,exceptforSW1573and2008cells, which were more sensitive to oxaliplatin (Table 8.1). Despite being considered as the activemetabolite,SW1573cellswerelesssensitivetoJM118.A2780andLovo92weremostsensitivetoJM118,whiletheywerealsomostsensitivetooxaliplatin.A549andSW1573cellswerelesssensitivetoJM118,butA549wasleastsensitive.
Theinteractionbetweenthedrugswasstudiedatdifferentschedules,sinceearlierevidenceindicatedthat post treatmentwith erlotinibmight prevent repair of DNAdam- age or delay cell cycling [48].SimultaneoustreatmentwitherlotinibandJM118onlyshowedsynergisminA431andA549cells,andadditivityintheothercelllines.Pretreatmentwitherlotinibwasgenerallysimilarorlesseffective,andevenantagonistic inSW1573cells.Only in the2008cellswas this schedulemoreeffective (Table8.1,Figure 8.2). Pretreatment with JM118 followed by a combination of the two drugs was the mosteffectiveschedule,showingsynergisminallcelllines,whichwasmostpronouncedinA431,A549and2008cells.
JM118-ErlotinibCombinationIncreasedPlatinum-DNAAdductFormation
Theeffectofplatinumanalogs ismediatedby the formationofPlatinum-DNAadducts. Therewas adifference in adduct formation between cisplatin, oxaliplatin, JM118 and JM216, with the highestaccumulation in cells treatedwith JM118 (Figure 8.3a; 2-6 fold higher than that of JM216). Platinumadduct levels in cisplatin andoxaliplatin -treated cellswere approximately 50%of thoseobserved inJM216 treated cells (Figure 8.3a). The highest accumulation of platinumDNA adductswas found inA2780cells,whichwere themost sensitive cells, and the lowest inA549cells,whichwere the leastsensitiveofthispanel(Figure8.3a).Platinumadductformationafter24hrdecreasedwhencellswere
�121
A
B
0.2 0.4 0.6 0.8 1.0
JM118/erlotinib (72h)
Com
bina
tion
Inde
x
+/- 1
.96
SD
0
0.5
1.0
0 0.2 0.4 0.6 0.8 1.00
0.5
1.0
1.5
F rac tional2E ffect
CI2+
/:21.96
2s.d.
Fractional Effect
0 0.2 0.4 0.6 0.8 1.00
0.2
0.4
0.6
0.8
1.0
F rac tional1E ffect
CI1+
/911.96
1s.d.
Fractional Effect 0.2 0.4 0.6 0.8 1.0
JM118 (24h) => JM118/erlotinib (48h)
0
0.5
1.0
SW1573 cells
0.01 0.1 1 10 1000
20
40
60
80
100
Drug (µM)
% C
ell g
row
th
JM118 (72h)Erlotinib (72h)JM118 (24h) =>JM118/erlotinib (48h)
Figure8.2.Inhibitionofcellproliferationandpharmacologicalinter-actionoferlotinib,JM118andthecombinationinSW1573non-smallcelllungcancer(NSCLC)cells.A.Representativecurvesofgrowthinhibitoryeffectsoferlotinib,JM118andtheerlotinib/JM118combinationinSW1573cellswithfixedratiosaroundtheirhalfmaximalinhibitoryconcentrations(IC50s):a24-hrpreincubationwithJM118wasfollowedbya48hrexposuretoerlotinib/JM118,showingasynergismwiththecombinationtherapy.B.CI/FAgraphsofthe72hrerlotinib/JM118combinationinSW1573andofthe24hrJM118followedby48hrerlotinib/JM118combinationinSW1573cells. 8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
postincubatedwitherlotinib.EvensimultaneousexposuretoJM118anderlotinibfor48hrdecreasedtheamountof adducts com-pared to 24hrexposure to JM118alone.However, 24hrpretreatmentwitherlotinib increasedtheplatinumadduct formationofasequential24hrexposuretoJM118.Thehighestaccumulationwasfound incellspretreatedwithJM118alone,followedbya24hr incubationwith the drug combination. This was observed in all cell lines, with the relative effect being mostpronouncedinSW1573cells.
JM118-ErlotinibCombinationModulatedCellCycle
Sincewedemonstratedthatposttreatmentwitherlotinibenhanceddrugsensitivitybyaffectingthecell cycle, we investigated the effect of different schedules of JM118 and erlotinib on cell cycledistribution. Ingeneral, theeffectson thevariouscell lineswerecomparable,with theexceptionofA2780cells.A24hrexposuretoJM118ledtoanaccumulationintheSphase,whichturnedintoaG2/Maccumulationafter48and72hrexposureinallcelllinesapartfromA2780cells(Figure8.4).Incontrasterlotinibdidnotshowanyeffectatany incubationtime,exceptfor intheA2780cell linewherecellsaccumulated in theG1phase. In2008cellssimultaneousexposuretoJM118anderlotinibshowedanintermediate effectwith fewer cells in S-phase as compared tomonotherapy regimes. However, atpreincubation with JM118 the G2/M phase accumulation was retained. On the other hand,preincubationwitherlotinibdecreasedtheG2/Mphaseac-cumulationinboth2008andA549cancercellscomparedtotheJM118pre-incubatedgroup.InA549cellstheeffectofJM118wasmoredominantanderlotinibinfluencedtheeffectofJM118toalesserextent.
Theinductionofcelldeath(measuredassub-G1accumulation)byJM118wasmostpronouncedinthemostsensitiveA2780cellsandleastpronouncedinthelesssensitiveA549cells.Erlotinibhadaminoreffectinallcelllines.SimultaneousexposuretoJM118anderlotinibwaseitheradditive,orresultedinadecreasedlevelofcellkillinthemostsensitivecelllines(A2780,Lovo92andWiDr).Pretreatmentwitherlotinibalsoreducedtheeffectinmostcelllines.However,pretreatmentwithJM118followedbythe
�122
JM118 (24h)
JM118+erlotinib (48h)
JM118 (24h) ⇒ erlotinib (24h)
Erlotinib (24h) ⇒ JM118 (24h)
Erlotinib (24h) ⇒ combination (24h)
JM118 (24h) ⇒ combination (24h)
0
50
100
150
Pla
tinum
-DN
A a
dduc
ts
(nm
ol/g
DN
A)
A549A2780SW1573**
**
*
****
** ****
***
*
**
**
Oxalip
latin
Cisplat
in
JM21
6
JM11
80
100
200
300
400 A549A2780SW1573
Plat
inum
-DN
A ad
duct
s (n
mol
/g D
NA
)
Figure8.3.(a)ComparisonofPlatinum-DNAadductformationafterexposurefor72hrto10Moxaliplatin,or5Mcisplatin,JM216,JM118.(b)Platinum-DNAadductformationatdifferentschedulesof5Merlotiniband5MJM118inA549,A2780andSW1573cells.JM118(24h):adductsformedafter24hrexposuretoJM118;JM118+erlotinib(48h),:adductsformedafter48hrsimultaneousexposuretoJM118anderlotinib;JM118(24h)=>erlotinib (24h): 24hrexposure to JM118,wash, followedby24hrexposure toerlotinib;Erlotinib (24h)=>JM118 (24h): 24 hr exposure to erlotinib, wash, followed by 24 hr exposure to JM118; erlotinib (24h) =>combination(24h):24hrexposuretoerlotinib,followedbya24hrexposuretoerlotinibandJM118;JM118(24h) => combination (24h): 24 hr exposure to JM118, followedby 24 hr exposure to the combination oferlotinibandJM118.Valuesmeans±SEoftriplicates.*,P=0.015;**,P<0.001,comparedtomonotherapy.
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
drugcombinationincreasedtheinductionofcelldeathinthecelllinesleastsensitivetoJM118,butnotincelllineswhichwerealreadysensitivetothesingledrug(Table8.1).
JM118-ErlotinibCombinationIncreasedPhosphorylationofCDK2andCHK1
Inordertoelucidatethemolecularmechanismbehindthecellcyclemodulation,weinvestigatedtheeffect of the drugs on the expression and phosphorylation of several cell cycle proteins. ErlotinibslightlyincreasedthephosphorylationoftheG2/MregulatingproteinCDC25candofCDK2inWiDRandLovo92 cells (Figure 8.5). JM118 increased the phosphorylation of CDK2 and CHK1 in all cell lines,including2008,A549,WiDRandLovo92,whichwasalso seen in thecombination.Regulationof theother cell cycle proteins was variable between the cell lines studied. After 48 hr incubation, thechanges in phosphorylationwere less prominent,with the possible exception of the A549 cell line,where JM118 decreased the phosphorylation of all targets. Preincubation with erlotinib increasedexpressionofE2F-1inallcelllinestested.
�123
1 2 3 4 5 6 7 8 9 10 11 12 1 2 3 4 5 6 7 8 9 10 11 12
1 2 3 4 5 6 7 8 9 10 11 12 1 2 3 4 5 6 7 8 9 10 11 12
A549 2008
A2780 Lovo92
0
20
40
60
80
100
120A
ccum
ulat
ion
of c
ells
(%)
A549 cells
******
****
0
20
40
60
80
100
120
Acc
umul
atio
n of
cel
ls (%
)
2008 cells**** **
****
0
20
40
60
80
100
120
Acc
umul
atio
n of
cel
ls (%
)
A2780 cells
* ******* **** ****
0
20
40
60
80
100
120
Acc
umul
atio
n of
cel
ls (%
)
Lovo92 cells
G0/G1 phaseS phaseG2/M phase
****
Figure 8.4.Effect on cell cycle distribution in A549NSCLC cells, 2008 andA2780 ovarian cancer cells andLovo92coloncancercells.Cellcycle(G0/G1,S,G2/Mphases)distributionwasevaluatedbyFACSafter24,48and72hr.CellswereexposedtotheirrespectiveIC50valuesof72hrexposure.24,48and72hrdenotescellcycle distribution after these time points. Combinationswere evaluated only after 72 hr, at simultaneousexposuretothedrugs,at24hrpretreatmentwithJM118followedby48hrexposuretothecombination(24hrJM118=>48hrerlotinib),orat24hrpretreatmentwitherlotinibfollowedby48hrexposuretoerlotinib+JM118(24hJM118=>48hJM118).). 1:Control,2:Erlotinib(24hr),3:Erlotinib(48hr),4:Erlotinib(72hr),5:JM118 (24 hr), 6: JM118 (48 hr), 7: JM118 (72 hr), 8: JM118/erlotinib (24 hr), 9: JM118/erlotinib (48 hr), 10:JM118/erlotinib(72hr),11:Erlotinib(24hr)=>JM118/erlotinib(48hr),12:JM118(24hr)=>JM118/erlotinib(48hr). Values are means ± SE of three separate experiments. Effects in the other two cell lines werecomparable.*,p=0.0189;**,p=0.0280;***,p=0.0014;****,p<0.0001comparedtocontrol.
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
IncreasedPhosphorylationofAktandErkinJM118-ErlotinibCombination
Since the Akt pathway often acts as a survival pathway, we investigated key kinases for theirexpressionandphosphorylation.IntheA2780,2008andWiDRcelllines,2hrincubationwitherlotinibincreasedthephosphorylationofbothAktandErk,butonlyofErkinA549cells(Figure8.6;Table8.S1).JM118showed increasedphosphorylationofAkt inA549,2008andLovo92,andofErk inA2780andLovo92.ThecombinationofJM118anderlotinibshowedanadditionaleffectonphosphorylationofErkinLovo92,comparedtothatoferlotinibalone.
In SW1573 cells 24hr incubationwitherlotinib increased thephosphorylationofAkt andErk, and inA549ofAkt(Figure8.6;Table8.S1). InA549andSW1573cells24hr incubationwithJM118 increasedthephosphorylationofbothAktandErk, inA2780cellsofAktalone,and inWiDRcellsofErkalone.PhosphorylationofErkwasfurtheraffectedinA549andWiDRafter24hr incubationwithJM118anderlotinibcomparedtoerlotinibalone.
�124
CDC25c
p-CDC25c
CDK2
p-CDK2
CHK1
p-CHK1
CHK2
p-CHK2
E2F1
β-actin
1 2 3 4 5 6 7 8 9 10
CDC25c p-CDC25c
CDK2 p-CDK2
CHK1 p-CHK1
CHK2 p-CHK2
E2F1 β-actin
Lovo 92 WiDR
A549 2008
CDC25c
p-CDC25c
CDK2
p-CDK2
CHK1
p-CHK1
CHK2
p-CHK2
E2F1
β-actin
1 2 3 4 5 6 7 8 9 10
CDC25c p-CDC25c
CDK2 p-CDK2
CHK1 p-CHK1
CHK2 p-CHK2
E2F1 β-actin
CDC25c
p-CDC25c
CDK2
p-CDK2
CHK1
p-CHK1
CHK2
p-CHK2
E2F1
β-actin
1 2 3 4 5 6 7 8 9 10
CDC25c p-CDC25c
CDK2 p-CDK2
CHK1 p-CHK1
CHK2 p-CHK2
E2F1 β-actin
CDC25c
p-CDC25c
CDK2
p-CDK2
CHK1
p-CHK1
CHK2
p-CHK2
E2F1
β-actin
1 2 3 4 5 6 7 8 9 10
CDC25c p-CDC25c
CDK2 p-CDK2
CHK1 p-CHK1
CHK2 p-CHK2
E2F1 β-actin
Figure8.5.Westernblotresultsofthecellcycleregulatoryproteinsin2008,A549,Lovo92andWiDrcellsafterexposuretothedrugsattheirIC50.1.Control(24h);2.JM118(24h);3.Erlotinib(24h);4.Combination(24h);5.Control(48h);6.Preincubationwitherlotinib(24h)followedbyJM118(24h);7.PreincubationwithJM118(24h)followedbyerlotinib(24h);8.Combinationtreatment(48h);9.Erlotinib(48h);10.JM118(48h).β-actinwasincludedasacontrolforloading.Representativeblotoutofthreeseparateexperiments.
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
IncreasedKinasePhosphorylationonPeptideArray
Inordertoinvestigatewhethertheproteinswerenotonlyphosphorylated,butalsoaffectedintheirfunction, we used a functional phosphorylation array for A549 cells. In the PAMgene array, kinaseactivitycanbeevaluatedby real-timemeasurementofproteinphosphorylationusingshortpeptidesspecific forphosphorylationsitesof 136tyrosinekinases.Phosphorylationofmostof thesepeptideswas onlymarginally (less than 30%) affected after treatmentwith IC50s of erlotinib, JM118 and thecombination.Hencewefocusedonthosephosphorylationsiteswhereoneofthedrugshadaneffect>30%. Erlotinib decreased the phosphorylation of PTEN, STAT3, FGF-R, ERBB4, VEGFR and EPHA1 by40-60%(Figure8.7),whileJM118decreasedthatofSTAT3bymorethan99%andofERBB5,FGFRandEPHA1between60-80%.Surprisinglythecombinationeitherdidnotfurtheraffectthephosphorylationorcompletelyreversedtheinhibition,suchasforSTAT3,PTEN,FGFRandEPHA1.
�125
VEGFR
ERBB4
STAT3PTEN
FGFR
EPHA10
20
40
60
80
100
120
ErlotinibJM118Combination
Kin
ase
phos
phor
ylat
ion
(%)
Figure 8.7. Kinase phosphorylation on apeptide array, including phosphatase andtensin homolog (PTEN), signal transducerand activator of transcription 3 (STAT3),fibroblastgrowthfactor receptor (FGF-R),v-erb-b2avianerythroblasticleukemiaviraloncogene homolog 4 (ERBB4), vascularendothelial growth factor (VEGF), ephrintype-Areceptor1(EPHA1).A549cellswereexposedfor2hrtoIC50concentrationsoferlotinib and/or JM118. Phosphorylationactivity in untreated cellswas set at 100%foreachseparatekinase.Valuesaremeans±SEofexperimentsintriplicate.
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +- - - - + + + +- - - - + + + +
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
Akt
Erk
p38
EGF
p-Akt
β-actin
p-Erk
p-p38
ErlCon
JMComErl
ConJMCom Erl
ConJMComErl
ConJMComErl
ConJMComErl
ConJMCom Erl
ConJMComErl
ConJMCom
- - - - + + + +- - - - + + + +
Lovo 92 WiDR SW1573 A549 A2780 2008
Akt Erk
P38 p-Akt p-Erk
p-P38 β-actin
Figure8.6.WesternblotresultsofphosphorylationofAkt,Erk,andP38,withβ-actinasacontrolforloading.Cellswereexposedfor2hrtoIC50concentrationsoferlotiniborJM118,ortheircombination.CellswerefirststimulatedbyapulseofEGF[54].Representativeblotsoutofthreeseparateexperiments.
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
DISCUSSION
The present study demonstrated that the combination of JM118 and the EGFR inhibitor erlotinib,showedascheduledependentsynergisminapanelofNSCLC,ovarianandcoloncancercelllineswithdifferentmolecularproperties.However,simultaneousexposurewasleasteffectiveinmostcelllines.ThemostsynergisticschedulewasJM118followedbythecombinationoferlotinibandJM118,evenincells with a Ras mutation. Interestingly the most pronounced synergism was found in cells leastsensitive to JM118, suchasA549cells, and inA431 cells,whichweremost sensitive toerlotinib. Thesynergistic effect was associated with increased platinum-adduct formation, increased cell kill andincreasedAktandErkphosphorylation.
Erlotinib is an EGFR inhibitor [45, 46] and is an effective treatment for tumorswith activatingEGFRmutationsandnomutationsintheRASpathway[47].Inearlierstudiesitwasfoundthaterlotinibandgefitinib showed synergism with DNA damaging drugs when given after the drug being used incombination,suchastheantifolatepemetrexed[48,52].However,inourexperiments,efficacyofthecombination was also found in cells with an intrinsically low sensitivity to erlotinib. Apparently theeffectoferlotinibissufficienttoinhibitDNArepairpathwaysascanbeconcludedfromtheincreasedplatinum-DNAadductformationandcellkill.Thiscanpossiblybeexplainedbytwodifferenteffectsofthecombination, theeffectonthecellcycleproteinsandonthesurvivalpathwaysaswasobservedearlierforoxaliplatin[53].TheaccumulationofcellsintheG2/Mphasewasassociatedwithchangesinphosphorylationofcellcycleproteinsthatregulatethetransitionfromonephasetoanother,suchasCDC25candCHK1andCHK2.Moreover,JM118inducedphosphorylationofAkt,whichwasneutralizedby erlotinib treatment. Similar changes were observed for combination of EGFR inhibitors withantifolatesandcisplatin[30,48].
To the best of our knowledge, this is the first study evaluating the synergistic effect of JM118 anderlotinib.Theeffectoferlotiniboncell signalingproteinswasmoderateandvariedbetweenthecelllines studied, possibly due to their different properties. In wild-type cells, especially with activatingmutations,erlotinibwilldecreasephosphorylationofEGFRanddownstreamproteins[54,55],but incellswithmutantEGFRandRasgenes,theseeffectsarenotexpected.Onoetal.[56]hypothesizedthatincellswithmutantEGFR, thecellsmaydependontheEGFRpathwayfortheirsurvival,thusmakingthem sensitive toEGFR inhibitorydrugs. Conversely, cellswithwild typeEGFRgenemaydependonseveral receptors [46, 57], making the EGFR pathway redundant; hence, the cells become moreresistanttoEGFRinhibitorydrugs.[55].
Inthepresentstudy,havingshownanincreasedphosphorylationofErkafter2hrincubationwithbothdrugs,thecelllinesweresynergisticinthecombinationstudy.EventhecelllineswitheitherincreasedphosphorylationofbothAktandErkornomodulationwereadditivetosynergisticinthecombinationstudy.ThisindicatesthatthephosphorylationofErkseemsimportantinresponsetothecombinationandAktcancounteractthiseffect,whichneedsfurtherinvestigation.However,withalongerexposuretime,theeffectofthedrugsonthecellsignalingproteinswasmorecelllinede-pendent.
Consequently, it seems thatpreincubationwithJM118 is thebest schedule for thecombinationwitherlotinib.Thesynergisticeffectofthecombinationregimenresulted inan increasedaccumulationofplatinum adducts in the DNA, and thereby enhanced the disruption of the cell signaling pathways,which led to growth inhibition. Finally, the results suggest the ability of JM118 in targeting keypathwaysanditssynergismwitherlotinib.
CONFLICTOFINTERESTTheauthorsconfirmthatthisarticlecontenthasnoconflictofinterest.ACKNOWLEDGEMENTSThestudywassupportedbyagrantfromGPCBiotech.SUPPLEMENTARYMATERIALSSupplementarymaterialisavailableonthepublisherswebsitealongwiththepublishedarticle.
�126
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
*Correspondenceto:GodefridusJ.Peters,PhD,DepartmentofMedicalOncology,VUUniversityMedicalCenter,DeBoelelaan 1117, 1081 HV Amsterdam, The Netherlands; Tel: (31) - 20 - 4442633; Fax: (31) - 20 - 4443844; E-mail:[email protected]
REFERENCES
1. SiddikZH.Mechanismsofactionofcancerchemotherapeuticagents:DNA-interactivealkylatingagents and antitumour platinum-based drugs. In: The Cancer Handbook. Alison MR (Ed.).NaturePublishingGroup,London,UK,1295-1313(2002).
2. CunninghamD,AtkinW,LenzHJ,etal.Colorectalcancer.Lancet2010;375:1030-47.3. PetersGJ,AvanA,RuizMG,etal.PredictiveroleofrepairenzymesintheefficacyofCisplatin
combinationsinpancreaticandlungcancer.AnticancerRes2014;34:435-42.4. GrivicichI,MansDR,PetersGJ,SchwartsmannG.Irinotecanandoxaliplatin:anoverviewofthe
novelchemotherapeuticoptionsforthetreatmentofadvancedcolorectalcancer.BrazJMedBiolRes2001;34:1087-103.
5. WisinskiKB,WahlAO,SmallW,Jr.,BensonAB,3rd.Inoperablepancreaticcancer:standardofcare.Oncology(WillistonPark).2007;21:1558-64;discussion65,70-2.
6. Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecularpharmacologyofoxaliplatin.MolCancerTher2002;1:227-35.
7. Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene2003;22:7265-79.
8. FinkD,Nebel S, Aebi S, et al. The role of DNAmismatch repair in platinumdrug resistance.CancerRes1996;56:4881-6.
9. McKeageMJ,MistryP,Ward J, et al.Aphase I andpharmacology studyof anoralplatinumcomplex, JM216: dose-dependent pharmacokinetics with single-dose administration. CancerChemotherPharmacol1995;36:451-8.
10. MarcusL,MurphyR,FoxE,etal.Theplasmaandcerebrospinalfluidpharmacokineticsoftheplatinum analog satraplatin after intravenous administration in non-human primates. CancerChemotherPharmacol2012;69:247-52.
11. RicartAD,SarantopoulosJ,CalvoE,etal.Satraplatin,anoralplatinum,administeredonafive-dayevery-five-weekschedule:apharmacokineticandfoodeffectstudy.ClinCancerRes2009;15:3866-71.
12. Rose WC, Crosswell AR, Schurig JE, Casazza AM. Preclinical antitumor activity of orallyadministeredplatinum(IV)complexes.CancerChemotherPharmacol1993;32:197-203.
13. McKeage MJ, Morgan SE, Boxall FE, et al. Lack of nephrotoxicity of oral ammine/amineplatinum(IV)dicarboxylatecomplexesinrodents.BrJCancer1993;67:996-1000.
14. McKeage MJ, Boxall FE, Jones M, Harrap KR. Lack of neurotoxicity of oralbisacetatoamminedichlorocyclohexylamine- platinum(IV) in comparison to cisplatin andtetraplatinintherat.CancerRes1994;54:629-31.
15. Poon GK, Mistry P, Raynaud FI, et al. Determination of metabolites of a novel platinumanticancerdrugJM216inhumanplasmaultrafiltrates.JPharmBiomedAnal1995;13:1493-8.
16. Twentyman PR, Wright KA, Mistry P, Kelland LR, Murrer BA. Sensitivity to novel platinumcompoundsofpanelsofhumanlungcancercelllineswithacquiredandinherentresistancetocisplatin.CancerRes1992;52:5674-80.
17. O'Neill CF, Koberle B, Masters JR, Kelland LR. Gene-specific repair of Pt/DNA lesions andinductionofapoptosisbytheoralplatinumdrugJM216inthreehumanovariancarcinomacelllinessensitiveandresistanttocisplatin.BrJCancer1999;81:1294-303.
18. McKeage MJ. Satraplatin in hormone-refractory prostate cancer and other tumour types:pharmacologicalpropertiesandclinicalevaluation.Drugs2007;67:859-69.
19. Fokkema E, GroenHJ, Bauer J, et al. Phase II study of oral platinumdrug JM216 as first-linetreatmentinpatientswithsmall-celllungcancer.JClinOncol1999;17:3822-7.
�127
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
20. JudsonI,CernyT,EpelbaumR,etal.PhaseIItrialoftheoralplatinumcomplexJM216innon-small-cell lungcancer:anEORTCearlyclinical studiesgroup investigation.AnnOncol 1997;8:604-6.
21. TrudeauM,StuartG,HirteH, et al.Aphase II trialof JM-216 in cervical cancer: anNCICCTGstudy.GynecolOncol2002;84:327-31.
22. Latif T, Wood L, Connell C, et al. Phase II study of oral bis (aceto) ammine dichloro(cyclohexamine) platinum (IV) (JM-216, BMS- 182751) given daily x 5 in hormone refractoryprostatecancer(HRPC).InvestNewDrugs2005;23:79-84.
23. SmithJW,2nd,McIntyreKJ,AcevedoPV,etal.ResultsofaphaseIIopen-label,nonrandomizedtrialoforalsatraplatininpatientswithmetastaticbreastcancer.BreastCancerResTreat2009;118:361-7.
24. SternbergCN,WhelanP,HetheringtonJ,etal.PhaseIIItrialofsatraplatin,anoralplatinumplusprednisonevs.prednisonealoneinpatientswithhormone-refractoryprostatecancer.Oncology2005;68:2-9.
25. SternbergCN,PetrylakDP,SartorO,etal.Multinational,double-blind,phaseIIIstudyofprednisoneandeithersatraplatinorplaceboinpatientswithcastrate-refractoryprostatecancerprogressingafterpriorchemotherapy:theSPARCtrial.JClinOncol2009;27:5431-8.
26. Doshi G, Sonpavde G, Sternberg CN. Clinical and pharmacokinetic evaluation of satraplatin.ExpertOpinDrugMetabToxicol2012;8:103-11.
27. SandlerA,GrayR,PerryMC,et al.Paclitaxel-carboplatinaloneorwithbevacizumab fornon-small-celllungcancer.NEnglJMed2006;355:2542-50.
28. Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin,fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer:results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol 2007; 25:1539-44.
29. VaishampayanUN,FontanaJ,HeilbrunLK,etal.PhaseIItrialofbevacizumabandsatraplatinindocetaxel-pretreated metastatic castrate-resistant prostate cancer. Urol Oncol 2014; 32: 31e25-33.
30.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov2005;4:307-20.
31. VivancoI,SawyersCL.Thephosphatidylinositol3-KinaseAKTpathwayinhumancancer.NatRevCancer2002;2:489-501.
32. MukoharaT,KudohS,MatsuuraK,et al.ActivatedAktexpressionhas significant correlationwithEGFRandTGF-alphaexpressionsinstageINSCLC.AnticancerRes2004;24:11-7.
33. KimHS,KimJW,GangJ,etal.Thefarnesyltransferaseinhibitor,LB42708,inhibitsgrowthandinduces apoptosis irreversibly in H- ras and K-ras-transformed rat intestinal epithelial cells.ToxicolApplPharmacol2006;215:317-29.
34. Giralt J,de lasHerasM,CerezoL, et al. Theexpressionofepidermalgrowth factor receptorresults in a worse prognosis for patients with rectal cancer treated with preoperativeradiotherapy:amulticenter,retrospectiveanalysis.RadiotherOncol2005;74:101-8.
35. KarasaridesM,Anand-ApteB,WolfmanA.AdirectinteractionbetweenoncogenicHa-Rasandphosphatidylinositol3-kinaseisnotrequiredforHa-Ras-dependenttransformationofepithelialcells.JBiolChem2001;276:39755-64.
36. CaronRW,YacoubA, LiM, et al.Activated formsofH-RASandK-RASdifferentially regulatemembraneassociationofPI3K,PDK-1,andAKTandtheeffectoftherapeutickinase inhibitorsoncellsurvival.MolCancerTher2005;4:257-70.
37. Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP. Radioresistance of K-Rasmutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKTpathway.RadiotherOncol2005;76:143-50.
38. ToulanyM,BaumannM,RodemannHP.StimulatedPI3K-AKTsignalingmediatedthroughligandor radiation-induced EGFRdepends indirectly, but not directly, on constitutiveK-Ras activity.MolCancerRes2007;5:863-72.
�128
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
39. ToulanyM,MinjgeeM,SakiM,etal.ERK2-dependentreactivationofAktmediatesthelimitedresponseoftumorcellswithconstitutiveK-RASactivitytoPI3Kinhibition.CancerBiolTher2014;15(3):317-28.
40.HovelmannS,BeckersTL,SchmidtM.Molecularalterations inapoptoticpathwaysafterPKB/Akt-mediatedchemoresistanceinNCIH460cells.BrJCancer2004;90:2370-7.
41. PeretsR,WyantGA,MutoKW,etal.Transformationofthefallopiantubesecretoryepitheliumleadstohigh-gradeserousovariancancerinBrca;Tp53;Ptenmodels.CancerCell2013;24:751-65.
42. WangQ,WeiF,LvG,etal.TheassociationofTP53mutationswiththeresistanceofcolorectalcarcinoma to the insulin-likegrowth factor-1 receptor inhibitorpicropodophyllin.BMCCancer2013;13:521.
43. WarrenRS,AtreyaCE,NiedzwieckiD,etal.AssociationofTP53mutationalstatusandgenderwithsurvivalafteradjuvanttreatmentforstageIIIcoloncancer:resultsofCALGB89803.ClinCancerRes2013;19:5777-87.
44. KlamerusJF,BrahmerJR.Optimizingthedoseandscheduleofanti-vascularendothelialgrowthfactorantibodiesinnon-small-celllungcancer.ClinLungCancer2008;9Suppl2:S51-6.
45. PirkerR.Noveldrugsagainstnon-small-celllungcancer.CurrOpinOncol2014;26:145-51.46. RolfoC,GiovannettiE,HongDS,etal.NoveltherapeuticstrategiesforpatientswithNSCLCthat
donotrespondtotreatmentwithEGFRinhibitors.CancerTreatRev2014;40:990-1004.47. Kishiki T, Ohnishi H,Masaki T, et al. Impact of genetic profiles on the efficacy of anti-EGFR
antibodiesinmetastaticcolorectalcancerwithKRASmutation.OncolRep2014;32:57-64.48. Giovannetti E, Lemos C, Tekle C, et al. Molecular mechanisms underlying the synergistic
interactionoferlotinib,anepidermalgrowthfactorreceptortyrosinekinaseinhibitor,withthemultitargetedantifolatepemetrexed innon-small-cell lungcancer cells.MolPharmacol 2008;73:1290-300.
49. AvanA,CreaF,PaolicchiE,etal.MolecularmechanismsinvolvedinthesynergisticinteractionoftheEZH2inhibitor3-deazaneplanocinAwithgemcitabineinpancreaticcancercells.MolCancerTher2012;11:1735-46.
50. ChouTC,TalalayP.Quantitativeanalysisofdose-effectrelationships:thecombinedeffectsofmultipledrugsorenzymeinhibitors.AdvEnzymeRegul1984;22:27-55.
51. Cooper BW, Veal GJ, Radivoyevitch T, et al. A phase I and pharmacodynamic study offludarabine,carboplatin,andtopotecaninpatientswithrelapsed,refractory,orhigh-riskacuteleukemia.ClinCancerRes2004;10:6830-9.
52. Li T, Ling YH, Goldman ID, Perez-Soler R. Schedule-dependent cytotoxic synergism ofpemetrexedanderlotinib inhumannon-smallcell lungcancercells.ClinCancerRes2007;13:3413-22.
53. Noordhuis P, Laan AC, van de Born K, et al. Oxaliplatin activity in selected and unselectedhumanovarianandcolorectalcancercelllines.BiochemPharmacol2008;76:53-61.
54. Janmaat ML, Giaccone G. Small-molecule epidermal growth factor receptor tyrosine kinaseinhibitors.Oncologist2003;8:576-86.
55. WangCY,ChaoTT,ChangFY,etal.CIP2Amediateserlotinib-inducedapoptosisinnon-smallcelllungcancercellswithoutEGFRmutation.LungCancer2014;85:152-60.
56. OnoM,HirataA,KometaniT,etal.Sensitivitytogefitinib(Iressa,ZD1839)innon-smallcelllungcancer cell lines correlateswithdependenceon theepidermalgrowth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation.MolCancerTher2004;3:465-72.
57. Giovannetti E, Labots M, Dekker H, et al. Molecular mechanisms and modulation of keypathwaysunderlyingthesynergisticinteractionofsorafenibwitherlotinibinnon-small-cell-lungcancer(NSCLC)cells.CurrPharmDes2013;19:927-39.
Received: August 25, 2014 Revised: September 15, 2014 Accepted: October 21, 2014; DOI:10.2174/1389450115666141107110321
�129
8

Chapter8 CurrDrugTargets2014;15(14):1312-1321
SupplementaryTable
�130
Supplementary Materials Current Drug Targets, 2014, Vol. 15, No. 14 i
Supplementary Materials
Modulation of Signaling Enhances the Efficacy of the Combination of Satraplatin and Erlotinib
Abolfazl Avan1, Auke D. Adema1, Eveline K. Hoebe1, Charlotte M. Huijts1, Amir Avan1,2, Gareth J. Veal3, R. Ruijtenbeek4, K. Wosikowski5 and Godefridus J. Peters1
1Department of Medical Oncology, VU University Medical Center, Amsterdam, The Netherlands; 2Department of New Sciences and Technology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran; 3Northern Institute for Cancer Research, Newcastle University, Newcastle upon Tyne, United Kingdom; 4Pamgene, ‘s Hertogenbosch, the Netherlands; 5Previously at GPC Biotech, Munich, Germany; current address: Isarna Therapeutics, Munich, Germany
Table S1. The effects of single and combination treatment on phosphorylation of Akt and Erk signaling pathways in different cell lines after 2 and 24 hours incubation.
Erlotinib JM118 Erlotinib + JM118
Cell line Gene 2 hours 24 hours 2 hours 24 hours 2 hours 24 hours
AKT + – + + + + 2008
ERK +++ + 0 – + +
AKT + 0 0 + + + A2780
ERK + 0 + 0 + +
AKT 0 + + + 0 0 A549
ERK + – 0 + + +
AKT 0 + 0 + 0 0 SW1573
ERK 0 + 0 + 0 0
AKT 0 0 + 0 0 0 Lovo92
ERK 0 0 + 0 + +
AKT + 0 0 – 0 0 WiDR
ERK + 0 0 + 0 0
Note: (+): increased phosphorylation; (–): decreased phosphorylation; (0): no obvious change in phosphorylation compared to the control group.
Table8.S1.
8

CHAPTER9AKT1andSELPpolymorphismspredictriskof
developingcachexiainpancreaticcancerpatients
AbolfazlAvan1*,AmirAvan1,2*,TessaY.S.LeLarge1,AndreaMambrini3,
NiccolaFunel4,MinaMaftouh1,MajidGhayour-Mobarhan2,MaurizioCantore3,UgoBoggi4,GodefridusJ.Peters1,PaolaPacetti3*,ElisaGiovannetti1,4*§
1DepartmentofMedicalOncology,VUUniversityMedicalCenter,Amsterdam,TheNetherlands;2BiochemistryofNutritionResearchCenter,andDepartmentofNewSciencesandTechnology,SchoolofMedicine,MashhadUniversityofMedicalSciences,Mashhad,
Iran;3DepartmentofMedicalOncology,CarraraCivicHospital,Carrara,Italy;4Start-UpUnit,UniversityofPisa,Pisa,Italy
*Theseauthorsequallycontributedtothestudy
PLoSOne2014;19;9(9):e108057
9

Chapter9 PLoSOne2014;19;9(9):e108057
AKT1andSELPpolymorphismspredicttheriskofdevelopingcachexiainpancreaticcancerpatients
ABSTRACT: Pancreatic ductal adenocarcinoma (PDAC) patients have the highest risk of developingcachexia,which isadirect causeof reducedqualityof lifeandshorter survival.Novelbiomarkers toidentify patients at risk of cachexia are needed and might have a substantial impact on clinicalmanagement.HereweinvestigatedtheprognosticvalueandassociationofSELP-rs6136,IL6-rs1800796and AKT1-rs1130233 polymorphismswith cachexia in PDAC. Genotypingwas performed in DNA frombloodsamplesofatestandvalidationcohortsof151and152chemo-naivelocally-advanced/metastaticPDAC patients, respectively. The association of SELP-rs6136, IL6-rs1800796 and AKT1-rs1130233polymorphisms with cachexia as well as the correlation between cachexia and the candidatepolymorphisms and overall survival were analyzed. Akt expression and phosphorylation in musclebiopsieswereevaluatedbyspecificELISAassays.SELP-rs6136-AAandAKT1-rs1130233-AA/GAgenotypeswere associatedwith increased riskofdeveloping cachexia inboth cohorts (SELP:p = 0.011 andp =0.045;AKT1:p = 0.004 andp = 0.019 for the first and second cohorts, respectively), while patientscarrying AKT1-rs1130233-GG survived significantly longer (p = 0.002 and p = 0.004 for the first andsecond cohorts, respectively). In the multivariate analysis AKT1-rs1130233-AA/GA genotypes weresignificantpredictorsforshortersurvival,withanincreasedriskofdeathof1.7(p=0.002)and1.6(p=0.004), in the first and second cohorts, respectively. This might be explained by the reducedphosphorylationofAkt1 inmusclebiopsiesfrompatientsharboringAKT1-rs1130233-AA/GA(p=0.003),favoringapoptosisinduction.Inconclusion,SELPandAKT1polymorphismsmayplayaroleintheriskofcachexia anddeath inPDACpatients, and shouldbe furtherevaluated in largerprospective studies.PLoSOne2014;19;9(9):e108057
Keywords:cachexia;pancreaticcancer;polymorphisms,SELP,AKT1
Introduction
Cachexia is a multi-factorial, systemic syndrome characterized by pathological wasting of skeletalmuscleandadipose tissuemass that leads topronouncedweight loss. It canoccur in the courseofseveral chronic illnesses, but it ismost frequently observed concomitantlywithmalignancies [1]. Inparticular, patients with pancreatic ductal adenocarcinoma (PDAC) have the highest prevalence ofcachexia,andoftenexperiencethemostseveresymptomsofthisdisease[2].Recentstudiesshowedthat existing preoperative cachexia reduces the post-operative outcome of PDAC patients [3,4]. Inadvanced PDAC the presence of cachexia is also associatedwith aworse prognosis, and stabilizingweightcanbecrucialtoprolongsurvival[5].Moreover,cachexiadecreasesthetolerancetosystemictreatmentsanddramaticallyaffectsthequalityoflife[6].
Extensive preclinical and clinical studies have evaluated the underlying mechanisms of cachexia inPDAC, revealing a complex of multiple interdependent patient- and cancer-specific components.Several hormones and tumor-derived factors can contribute to tissue catabolism and appetiteregulation, leading to anorexia and fatigue, whereas symptoms such as chronic pain, nausea andpancreatic insufficiency, as well as adverse effects of anticancer therapies, might further reduceappetiteandnutritionalintake[7].
However,incontrasttocachecticpatientswithchronicpancreatitis,PDACpatientswithcachexiahavesignificantlyreducedhemoglobinandalbumin levels,associatedtothesystemicreactiontoboththetumorandthe inflammatoryprocesses[4].Specificpro-inflammatorycytokinessuchas interleukin1-beta(IL-1β),interleukin6(IL-6),tumornecrosisfactoralpha(TNFα),andinterferongammahavebeenshowntobeassociatedwithprogressiveweightloss,throughtheacutephaseproteinresponseintheliver [8–10]. This systemic inflammation can also convey its action via local inflammatory signaling,activatedbythenucleartranscriptionfactorkB(NF-κB),andinbothmousemodelsandmusclebiopsyspecimensNF-κB–mediatedPax7dysregulationemergedamongthecausesofmusclewastinginPDAC[11]. Furthermore, in cachexia thebalancebetween the anabolismand the catabolismof proteins is
�132
9

Chapter9 PLoSOne2014;19;9(9):e108057
tippedtowardacatabolicstateresultingfromactivatedubiquitinproteasomeandautophagysystemsthat promote protein breakdown, as well as from reduced Akt activity, that decreases proteinsynthesis[1,12].Theregulationofmuscledifferentiationisindeeddependentontheactivationofsignaltransduction cascades with the complex involvement of key kinases, such as the serine/threoninekinaseAkt,andpreviousstudiesdemonstratedthatAktisessentialtopromoteproteinsynthesisandcellsurvivalandtoblockproteindegradation[13],Allthisevidencesupportsitsbiologicalrelevanceinthecontextofcachexia.
Although the variable production of the pro-inflammatory cytokines, NF-κB activation and Aktphosphorylation status depends on a variety of clinical and pathological factors, there is growingevidencethatgeneticvariationsmightalsoaffectthesekeydeterminantsofcachexia.Inparticular,theG-allele of the IL6-rs1800796 polymorphism was associated with decreased survival and increasedsusceptibility to cachexia in ChinesePDACpatients [14].Moreover, in a systematic studyongeneticdeterminants of cachexia, out of 92 potential candidate genes with 184 polymorphisms relating tocancer cachexia, 42 polymorphisms across 33 genes have been identified for having a functional orclinicalrelevanceinmorethanonestudy[15].Ofthese42polymorphisms,13hadmorethanoneeffecton clinical features associatedwith cancer cachexia (i.e. inflammation, loss of fatmass and/or leanmass,andreducedsurvival).Furtherpathwayanalysisofthesecandidatesrevealed4genes(ADIPOQ,IL6,NFKB1andTLR4)interlinkingtwoputativemajornetworksinvolvedinthedevelopmentofcancercachexia.However,inarecentstudyusingtheseselectedpolymorphismsonlytheC-alleleofthesinglenucleotidepolymorphism(SNP)rs6136-SELPwasfoundtobeassociatedwithweightlossof>10%,bothinthefirststudycohortof775,including114PDACpatients,andinthevalidationcohortof101cancerpatients,including6PDACpatients[16].TheSELPgeneencodesa140kDaproteinstoredinthealpha-granulesofplateletsandWeibel-Paladebodiesofendothelialcells,whichredistributestotheplasmamembrane during platelet activation and degranulation and functions as a cell adhesion molecule(CAM)mediatingtheinteractionofactivatedendothelialcellsorplateletswithleukocytesduringtheinitialstepsininflammation[17].
However,thelargeststudyoncancercachecticpatients(N=1797),including35PDACpatients,failedtovalidate thepredictive roleof9candidateSNPs [18].Oneof thepossibleexplanations fornotbeingable toconfirmgeneticassociationsmaybe thegene–environment relationsspecific for thestudieddisease. Previous analysis supported the role of the IL6-rs1800796 and SELP-rs6136 SNPs assusceptibilitybiomarkersforPDACcachexia[13,15],whileourrecentstudiesrevealedakeyfunctionalrole of theAKT1-rs1130233 SNP [19]. Therefore in the present studywe evaluated the association ofthesecandidatepolymorphismsintheAKT1,IL6,andSELPgeneswithcachexiainatotalpopulationof303PDACpatients,intwoindependentcohorts.
MaterialsandMethods
Cachexiaphenotypedefinition
To ensure that patients with or without cachexia were clearly distinguished, we used the ‘‘2011InternationalConsensus,’’accordingtowhichcachexia isdefinedbyunintendedweight lossofmorethan5%ofbodyweightorweightlossofmorethan2%inalreadydepletedindividualswithabodymassindex(BMI)lessthan20kg/m2over6months[20].
Patients
Inthecurrentstudyweselectedafirstcohortof151patients,enrolledbetween31stMarch2004and10thJanuary2009,andasecond/validationcohortof152patients,enrolledbetween15thJanuary2009and 15th January 2013. All the eligible patients were chemo-naive patients with diagnosis ofhistologically confirmed locally advanced or metastatic PDAC, treated at the Carrara Civic-Hospital(Carrara,Italy).
�133
9

Chapter9 PLoSOne2014;19;9(9):e108057
Ethics
All the patients gave theirwritten informed consent to the sample collection and analysis, and thestudy has received approval from the Ethics Committeeof Carrara Civic-Hospital (Carrara, Italy) andEthicsCommitteeofPisaUniversityHospital(Pisa,Italy)asafollow-upstudyoftheresearchprotocolentitled‘‘Pharmacogeneticsofgemcitabine-relatedgenesinpancreascancer:correlationwithclinicaloutcome and tolerability’’. The responsible investigators ensure that this study was conductedaccording to the Declaration of Helsinki, the European Guidelines on Good Clinical Practice, andrelevantnationalandregionalauthorityrequirements.
Genotyping
Genomic DNA was extracted from blood samples at the Laboratory Medical Oncology (VUmc,Amsterdam, The Nether- lands) using the QIAamp DNA Mini-Kit according to the manufacturerprotocol (Qiagen, San Diego, CA). The concentration and purity of DNAs was determined with theNanoDrop-1000-Detector(NanoDrop-Technologies,Wilmington,USA).Genotypeanalysisofrs1800796,rs6136,andrs1130233polymorphismswasperformedusingTaqman-basedPCRreactionscarriedoutin12.5mltotalvolume,using20ngofDNAdilutedinTaqManUniversalMasterMixwithspecificprimersandprobes (SNPGenotypingAssaysproductsC__11326893_10,C__11975277_20,andC__7489835_10,respectively).TheABIPRISM-7500instrument(AppliedBiosystems,LifeTechnologies,FosterCity,CA)equipped with the SDS version-2.0 software was employed to evaluate the allelic content of eachsampleintheplatebyreadingthegeneratedfluorescence.
AnalysisofAktexpressionandphosphorylationinhomogenizedmusclebiopsies
Previous studies evaluated the relationships of AKT1 polymorphisms with AKT1 mRNA and proteinexpressioninlymphoblastoidsandlungcancercells[19,21],butnodataareavailableonthecorrelationbetweenAKT1-rs1130233 and Akt1 protein expression and phosphorylation status in skeletal muscle.There-fore,wecollectedrectusabdominismusclebiopsiesobtainedduringresectionofprimaryPDAC.Smalltissuepieces(about100mg)wereimmediatelyfrozeninliquidnitrogen,usingisopentane,aftersurgeryat theUniversityHospitalofPisa (Pisa, Italy), according toaprotocolapprovedby the localHospitalEthicCommittee.Atotalof18samplesfromchemonaivepatients(ninefortheAKT1-rs1130233-AA/GAandninefortheAKT1-rs1130233-GGgenotype,asdeterminedinpreliminarygenotypinganalysisfrombloodsamples,respectively)wereevaluated.
Thefrozentissueswerehomogenizedusingthemicro-dismembrator,asdescribedpreviously[22].Thefrozenpowderwasextractedwith2.5%w/vsulfosalicylacidandcentrifuged(10min,8000RPM)andtheproteinconcentrationintheextractswasdeterminedusingtheBioradproteinassay(LifeScience,Bio-Rad Laboratories B.V., Veenendaal, The Netherlands). Total Akt1 and Akt1 phosphorylation atserine-473(Akt[pS473])wereevaluatedwithspecificELISAassays(Invitrogen,LifeTechnologies),andnormalized to standard curvesofHuman-total-Akt1 andHuman- phospho-Akt1, aswell as toproteincontent,asdescribedpreviously[23].
Statistics
DemographicandclinicalinformationwerecomparedacrossgenotypeusingPearson’sx2andlogisticregression. In agreement with previous studies, the correlation with candidate genotypes wasperformedcombiningthelesscommonhomozygousandheterozygousgenotypes[24].
Toevaluatewhethercachexiaandtheotherclinicalvariablesaswellasthecandidatepolymorphismsaffectedclinicaloutcome,overallsurvival(OS)curveswereanalyzedfromthedayoftreatmentstarttotheendpoint(deathorcensoring)accordingtoKaplan–Meiermethod,andcomparedbylog-rank.Thesignificantprognosticvariables in theunivariateanalysiswere included inmultivariateanalysis,usingCox’s proportional hazards model. This analysis included a step down procedure based on the
�134
9
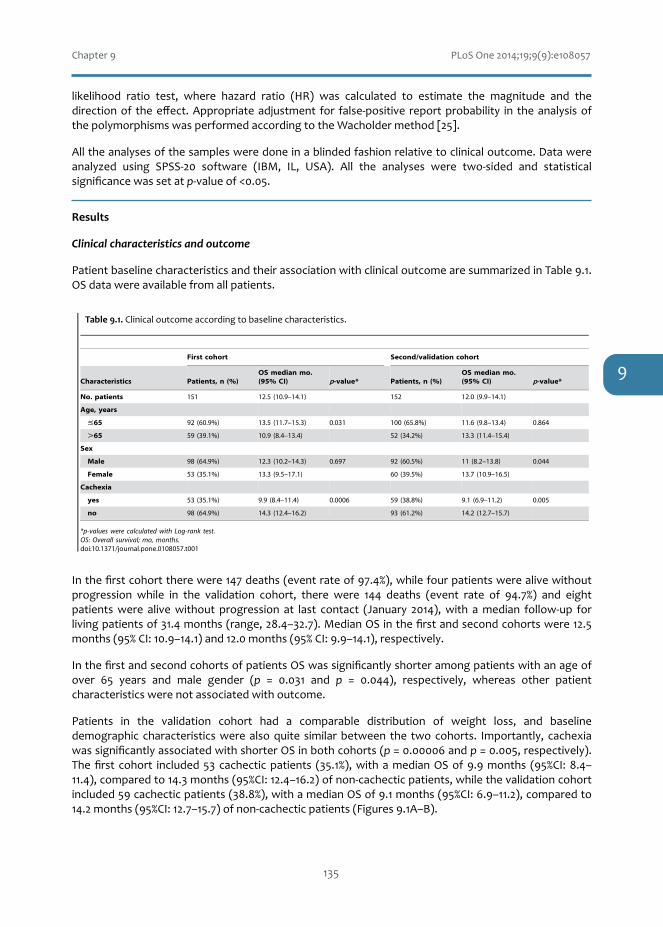
Chapter9 PLoSOne2014;19;9(9):e108057
likelihood ratio test, where hazard ratio (HR) was calculated to estimate the magnitude and thedirectionof theeffect.Appropriateadjustment for false-positive reportprobability in theanalysisofthepolymorphismswasperformedaccordingtotheWacholdermethod[25].
Alltheanalysesofthesamplesweredoneinablindedfashionrelativetoclinicaloutcome.Datawereanalyzed using SPSS-20 software (IBM, IL, USA). All the analyses were two-sided and statisticalsignificancewassetatp-valueof<0.05.
Results
Clinicalcharacteristicsandoutcome
PatientbaselinecharacteristicsandtheirassociationwithclinicaloutcomearesummarizedinTable9.1.OSdatawereavailablefromallpatients.
Inthefirstcohorttherewere147deaths(eventrateof97.4%),whilefourpatientswerealivewithoutprogression while in the validation cohort, there were 144 deaths (event rate of 94.7%) and eightpatientswere alivewithout progression at last contact (January 2014),with amedian follow-up forlivingpatientsof31.4months(range,28.4–32.7).MedianOSinthefirstandsecondcohortswere12.5months(95%CI:10.9–14.1)and12.0months(95%CI:9.9–14.1),respectively.
InthefirstandsecondcohortsofpatientsOSwassignificantlyshorteramongpatientswithanageofover 65 years and male gender (p = 0.031 and p = 0.044), respectively, whereas other patientcharacteristicswerenotassociatedwithoutcome.
Patients in the validation cohort had a comparable distribution of weight loss, and baselinedemographic characteristicswerealsoquite similarbetween the twocohorts. Importantly, cachexiawassignificantlyassociatedwithshorterOSinbothcohorts(p=0.00006andp=0.005,respectively).Thefirst cohort included53 cachecticpatients (35.1%),withamedianOSof9.9months (95%CI:8.4–11.4),comparedto14.3months(95%CI:12.4–16.2)ofnon-cachecticpatients,whilethevalidationcohortincluded59cachecticpatients(38.8%),withamedianOSof9.1months(95%CI:6.9–11.2),comparedto14.2months(95%CI:12.7–15.7)ofnon-cachecticpatients(Figures9.1A–B).
�135
Table 1. Clinical outcome according to baseline characteristics.
First cohort Second/validation cohort
Characteristics Patients, n (%)OS median mo.(95% CI) p-value* Patients, n (%)
OS median mo.(95% CI) p-value*
No. patients 151 12.5 (10.9–14.1) 152 12.0 (9.9–14.1)
Age, years
#65 92 (60.9%) 13.5 (11.7–15.3) 0.031 100 (65.8%) 11.6 (9.8–13.4) 0.864
.65 59 (39.1%) 10.9 (8.4–13.4) 52 (34.2%) 13.3 (11.4–15.4)
Sex
Male 98 (64.9%) 12.3 (10.2–14.3) 0.697 92 (60.5%) 11 (8.2–13.8) 0.044
Female 53 (35.1%) 13.3 (9.5–17.1) 60 (39.5%) 13.7 (10.9–16.5)
Cachexia
yes 53 (35.1%) 9.9 (8.4–11.4) 0.0006 59 (38.8%) 9.1 (6.9–11.2) 0.005
no 98 (64.9%) 14.3 (12.4–16.2) 93 (61.2%) 14.2 (12.7–15.7)
*p-values were calculated with Log-rank test.OS: Overall survival; mo, months.doi:10.1371/journal.pone.0108057.t001
Figure 1. Clinical outcome according to cachexia and AKT1-rs1130233 polymorphism A and B) Kaplan–Meier survival curves according tocachexia in the first and second cohort of patients. (C and D) Kaplan–Meier survival curves according to the AKT1-rs1130233 polymorphism in the firstand second cohort of patients. p-values were calculated with the log-rank test. OS: Overall survival.doi:10.1371/journal.pone.0108057.g001
Association of AKT1 and SELP Polymorphisms with Cachexia
PLOS ONE | www.plosone.org 4 September 2014 | Volume 9 | Issue 9 | e108057
. (
Table9.1.Clinicaloutcomeaccordingtobaselinecharacteristics.
9

Chapter9 PLoSOne2014;19;9(9):e108057
Polymorphismsandcachexia
ToinvestigatewhetherthereisanassociationbetweencachexiaandIL6-rs1800796,AKT1-rs1130233andSELP-rs6136polymorphisms,weperformedgenotypingusinggenomicDNAextractedfromperipheralbloodsamples.
Previous studies showed no differences in polymorphisms analyzed in tumors and normal tissues[26,27]. However, since high level gene amplification in tumor cells might result in homozygousgenotypes in individualswhoareheterozygous inthegermline,weperformedpreliminarystudiesofAKT1 polymorphisms in 45 paired samples of germ-line and cancer DNA, showing identicalinterindividual genotypes between normal and malignant tissues [19]. Therefore, for the patientsenrolled in this study the genotyping was performed in DNA extracted from the available bloodsamples.Genotypingwassuccessfullycarriedout inall theDNAsamples,andnodiscrepancieswerefoundinthesamplesanalyzedinduplicate(approximately10%).
In the first cohort of patients the wild-type SELP-rs6136 genotype (AA) had a frequency of 46.4%,whereastheACandCCgenotypeswerefoundin43.7%and9.9%ofthepatients,respectively.FortheIL6-rs1800796 polymorphism, the frequenciesof theGG,GC andCC genotypeswere59.3%, 37.4%and3.3%, respectively. Regarding the AKT1-rs1130233 polymorphism, the GG, GA and AA variants wereobserved in 52.8%, 39.3% and 7.9% of the cases, respectively. Similar results were observed in the
�136
OS
prob
abili
ty (%
)
OS
prob
abili
ty (%
)
OS
prob
abili
ty (%
)
OS
prob
abili
ty (%
)
Time (months) Time (months)
Time (months) Time (months)
A B
C D
Figure9.1.ClinicaloutcomeaccordingtocachexiaandAKT1-rs1130233polymorphism.(AandB)Kaplan–Meiersurvivalcurvesaccordingtocachexiainthefirstandsecondcohortofpatients.(CandD)Kaplan–MeiersurvivalcurvesaccordingtotheAKT1-rs1130233polymorphisminthefirstandsecondcohortofpatients.p-valueswerecalculatedwiththelog-ranktest.OS:Overallsurvival.
9

Chapter9 PLoSOne2014;19;9(9):e108057
second cohort of patients, as reported in the Table 9.2. Indeed, no significant differences weremeasured both the baseline demographic characteristics and in the frequencies of the candidatepolymorphicgenotypesinthetwocohortsofPDACpatients.
All the polymorphisms followed Hardy–Weinberg equilibrium, as calculated with the SNP analyzersoftware (http://snp.istech21. com/snpanalyzer/2.0/, Table 9.S1) and their allelic frequencies werecomparablewiththosereportedinCaucasianpopulations,andinNCBIandNCI-SNP500databases.Nosignificant correlations were detected between genotype and baseline demographic characteristics(i.e.ageandsex,datanotshown).
Bygroupingpatientswith IL6-rs1800796-GGversusthosewithGCorCCgenotypeswedidnotfindanassociation with cachexia (Table 9.2). Conversely, a correlation was observed between the AKT1-rs1130233genotypeandcachexia,withsignificantlyhigherproportionofpatientsharboringatleastoneA-allelehavingcachexia(i.e.37outof81,versus16outof70GGpatients,p=0.004).AsshowninTable9.2, the SELP-rs6136 polymorphism was also significantly (p = 0.011) associated with cachexia. Inparticular,44%ofthepatientsharboringtheSELP-rs6136-AAvariantsexperiencedcachexia,comparedto24%ofthepatientsharboringtheSELP-rs6136-AC/CCgenotype.
Remarkably,weobservedsimilarresults inthevalidationcohortofpatients,showingthatindividualswho carry theAKT1-rs1130233-GG genotype or the C-allele of the SELP-rs6136 polymorphismwere atreducedriskofdevelopingcachexia(Table9.2).Theseresultswereconfirmedbyunconditionallogisticregression, which calculated ORs and their 95%CI for association with cachexia phenotype of eachindividualSNP,asreportedinTable9.S2.
Polymorphismsandoutcome
Considering that cachexia is a key determinant of cancer-related death, we hypothesized that thecandidatepolymorphismsassociatedwithcachexiamightalsoaffecttheclinicaloutcome.
At univariate analysis, theAKT1-rs1130233 polymorphismwas associatedwith significantly differentialOS.Inparticular,significantlylongersurvivalwasobservedinpatientsharboringtheGGgenotype(14.3months;95%CI:12.1–16.5),incomparisonwithpatientscarryingtheGA/AAgenotype,whohadamedianOS of 10.3 months (95% CI: 8.1–12.4; p = 0.002; Figure 9.1C). Similar results were observed in thevalidation cohort: patients with the GA and AA genotypes had a worse prognosis (median OS 9.9
�137
Polymorphisms and outcomeConsidering that cachexia is a key determinant of cancer-
related death, we hypothesized that the candidate polymorphismsassociated with cachexia might also affect the clinical outcome.
At univariate analysis, the AKT1-rs1130233 polymorphismwas associated with significantly differential OS. In particular,significantly longer survival was observed in patients harboring theGG genotype (14.3 months; 95% CI: 12.1–16.5), in comparisonwith patients carrying the GA/AA genotype, who had a medianOS of 10.3 months (95% CI: 8.1–12.4; p = 0.002; Figure 1C).Similar results were observed in the validation cohort: patientswith the GA and AA genotypes had a worse prognosis (median OS9.9 months, 95% CI: 8.6–11.1), compared to the patientsharbouring the GG genotype (14.3 months, 95% CI: 11.7–16.8;p = 0.004; Table 3; Figure 1D). To keep at minimum theprobability to find a statistically significant difference purely bychance, the usual nominal level (p = 0.05) has been lowered to0.017 by Bonferroni adjustment for multiple comparisons. Afterthe Bonferroni adjustment, the AKT1-rs1130233 SNP was stillsignificantly correlated to OS. In contrast, SELP-rs6136 and IL6-rs1800796 polymorphisms were not associated with the outcome,as reported in Table 3.
In the multivariate analysis, cachexia and age .65 weresignificantly associated with increased risk of death (Table 4). TheCox proportional hazard regression model also showed theprognostic significance of the AKT1-rs1130233 polymorphism.In particular, the GA/AA genotype emerged as a significantpredictor for shorter survival, with an increased risk of death of 1.7(95% CI: 1.2–2.4; p = 0.002) and 1.6 (95% CI: 1.2–2.3; p = 0.004),in the first and second cohorts, respectively.
AKT1-rs1130233 and expression of Akt1 andphospho-Akt1 in skeletal muscle
The significant association of the AKT1-rs1130233 polymor-phism with cachexia and clinical outcome prompted us to performan exploratory study on its role on the level and activity of Akt1 ina panel of muscle biopsies. As illustrated in Figure 2, the sampleswith the GA or AA genotypes had a trend toward a significantlylower protein expression (approximately 230%) of Akt1 incomparison to the samples with AKT1-rs1130233-GG genotype(p = 0.050). These results were similar to the reduction in phospho-Akt1 levels, resulting in a significant lower average level ofphosphorylation at Ser437 of Akt1 in the GA/AA samples
Table 2. Correlation between cachexia and candidate SELP, AKT1 and IL-6 SNPs.
First cohort Second cohort
SNP Cachexia Genotype Patients n (%) p-value* Patients n (%) p-value*
SELP rs6136 Yes AA 36 (23.8%) 0.011 36 (23.8%) 0.045
AC/CC 17 (11.2%) 23 (15.1%)
No AA 45 (29.8%) 40 (26.3%)
AC/CC 53 (35.2%) 53 (34.8%)
AKT1 rs1130233 Yes GG 16 (10.1%) 0.004 21 (13.8%) 0.019
GA/AA 37 (24.5%) 36 (23.7%)
No GG 54 (35.7%) 53 (34.9%)
GA/AA 44 (29.7%) 40 (27.6%)
IL-6 rs1800796 Yes GG 17 (11.2%) 0.169 18 (11.8%) 0.486
GC/CC 36 (23.4%) 41 (27%)
No GG 43 (28.5%) 34 (22.4%)
GC/CC 55 (36.9%) 59 (38.8%)
*p-values were calculated with Fisher’s exact test.SNPs, single nucleotide polymorphisms.Note: SELP and IL6 SNPs was detectable in all the samples, while the AKT1 genotype could not be determined in 2 of the patients of the second cohort.doi:10.1371/journal.pone.0108057.t002
Table 3. Clinical outcome according to candidate SELP, AKT1 and IL-6 SNPs.
First cohort Second/validation cohort
SNP Genotype OS median mo. (95% CI) p-value OS median mo. (95% CI) p-value
SELP rs6136 AA 11.4 (8.4–14.4) 0.665 11.0 (9.1–12.9) 0.519
AC/CC 12.7 (10.1–15.2) 13.7 (10.2–17.1)
AKT1 rs1130233 GG 14.3 (12.1–16.5) 0.002 14.3 (11.7–16.8) 0.004
GA/AA 10.3 (8.1–12.4) 9.9 (8.6–11.1)
IL-6 rs1800796 GG 11.5 (7.1–16.0) 0.259 11.6 (9.1–14.2) 0.651
GC/CC 12.5 (10.8–14.3) 12.5 (9.7–15.3)
OS: overall survival; mo: months.doi:10.1371/journal.pone.0108057.t003
Association of AKT1 and SELP Polymorphisms with Cachexia
PLOS ONE | www.plosone.org 5 September 2014 | Volume 9 | Issue 9 | e108057
Table9.2.CorrelationbetweencachexiaandcandidateSELP,AKT1andIL-6SNPs.
9

Chapter9 PLoSOne2014;19;9(9):e108057
months,95%CI:8.6–11.1),comparedtothepatientsharbouringtheGGgenotype(14.3months,95%CI:11.7–16.8;p=0.004;Table9.3;Figure9.1D).Tokeepatminimumtheprobabilitytofindastatisticallysignificantdifferencepurelybychance,theusualnominallevel(p=0.05)hasbeenloweredto0.017byBonferroniadjustmentformultiplecomparisons.AftertheBonferroniadjustment,theAKT1-rs1130233SNPwasstillsignificantlycorrelatedtoOS. Incontrast,SELP-rs6136and IL6-rs1800796polymorphismswerenotassociatedwiththeoutcome,asreportedinTable9.3.
Inthemultivariateanalysis,cachexiaandage>65weresignificantlyassociatedwith increasedriskofdeath (Table 9.4). The Cox proportional hazard regression model also showed the prognosticsignificance of the AKT1-rs1130233 polymorphism. In particular, the GA/AA genotype emerged as asignificant predictor for shorter survival, with an increased risk of death of 1.7 (95% CI: 1.2–2.4; p =0.002)and1.6(95%CI:1.2–2.3;p=0.004),inthefirstandsecondcohorts,respectively.
AKT1-rs1130233andexpressionofAkt1andphospho-Akt1inskeletalmuscle
The significant association of the AKT1-rs1130233 polymorphism with cachexia and clinical outcomepromptedustoperformanexploratorystudyonitsroleonthelevelandactivityofAkt1inapanelofmusclebiopsies.AsillustratedinFigure2,thesampleswiththeGAorAAgenotypeshadatrendtowarda significantly lower protein expression (approximately 230%) of Akt1 in comparison to the sampleswithAKT1-rs1130233-GGgenotype(p=0.050).Theseresultsweresimilartothereduction inphospho-Akt1levels,resultinginasignificantloweraveragelevelofphosphorylationatSer437ofAkt1intheGA/AAsamplescomparedtotheGGsamples(p=0.006).However,whenwenormalizedthephospho-Akt1levelstothetotal-Akt1levelsineachsampleweobservedthatinallexceptonecase,thesampleswithGA/AAgenotypehada lowerratiobetweenphospho-Akt1andtotalAkt1vs.thesampleswiththeGGgenotype(Figure9.S1,p=0.003).ThesedatasuggestthattheAKT1-rs1130233-GA/AAgenotypesmightconferareducedactivitytoAkt1,andthusreducetheantiapoptoticactivityofthispivotalregulatorofapoptoticsignaling.
�138
Polymorphisms and outcomeConsidering that cachexia is a key determinant of cancer-
related death, we hypothesized that the candidate polymorphismsassociated with cachexia might also affect the clinical outcome.
At univariate analysis, the AKT1-rs1130233 polymorphismwas associated with significantly differential OS. In particular,significantly longer survival was observed in patients harboring theGG genotype (14.3 months; 95% CI: 12.1–16.5), in comparisonwith patients carrying the GA/AA genotype, who had a medianOS of 10.3 months (95% CI: 8.1–12.4; p = 0.002; Figure 1C).Similar results were observed in the validation cohort: patientswith the GA and AA genotypes had a worse prognosis (median OS9.9 months, 95% CI: 8.6–11.1), compared to the patientsharbouring the GG genotype (14.3 months, 95% CI: 11.7–16.8;p = 0.004; Table 3; Figure 1D). To keep at minimum theprobability to find a statistically significant difference purely bychance, the usual nominal level (p = 0.05) has been lowered to0.017 by Bonferroni adjustment for multiple comparisons. Afterthe Bonferroni adjustment, the AKT1-rs1130233 SNP was stillsignificantly correlated to OS. In contrast, SELP-rs6136 and IL6-rs1800796 polymorphisms were not associated with the outcome,as reported in Table 3.
In the multivariate analysis, cachexia and age .65 weresignificantly associated with increased risk of death (Table 4). TheCox proportional hazard regression model also showed theprognostic significance of the AKT1-rs1130233 polymorphism.In particular, the GA/AA genotype emerged as a significantpredictor for shorter survival, with an increased risk of death of 1.7(95% CI: 1.2–2.4; p = 0.002) and 1.6 (95% CI: 1.2–2.3; p = 0.004),in the first and second cohorts, respectively.
AKT1-rs1130233 and expression of Akt1 andphospho-Akt1 in skeletal muscle
The significant association of the AKT1-rs1130233 polymor-phism with cachexia and clinical outcome prompted us to performan exploratory study on its role on the level and activity of Akt1 ina panel of muscle biopsies. As illustrated in Figure 2, the sampleswith the GA or AA genotypes had a trend toward a significantlylower protein expression (approximately 230%) of Akt1 incomparison to the samples with AKT1-rs1130233-GG genotype(p = 0.050). These results were similar to the reduction in phospho-Akt1 levels, resulting in a significant lower average level ofphosphorylation at Ser437 of Akt1 in the GA/AA samples
Table 2. Correlation between cachexia and candidate SELP, AKT1 and IL-6 SNPs.
First cohort Second cohort
SNP Cachexia Genotype Patients n (%) p-value* Patients n (%) p-value*
SELP rs6136 Yes AA 36 (23.8%) 0.011 36 (23.8%) 0.045
AC/CC 17 (11.2%) 23 (15.1%)
No AA 45 (29.8%) 40 (26.3%)
AC/CC 53 (35.2%) 53 (34.8%)
AKT1 rs1130233 Yes GG 16 (10.1%) 0.004 21 (13.8%) 0.019
GA/AA 37 (24.5%) 36 (23.7%)
No GG 54 (35.7%) 53 (34.9%)
GA/AA 44 (29.7%) 40 (27.6%)
IL-6 rs1800796 Yes GG 17 (11.2%) 0.169 18 (11.8%) 0.486
GC/CC 36 (23.4%) 41 (27%)
No GG 43 (28.5%) 34 (22.4%)
GC/CC 55 (36.9%) 59 (38.8%)
*p-values were calculated with Fisher’s exact test.SNPs, single nucleotide polymorphisms.Note: SELP and IL6 SNPs was detectable in all the samples, while the AKT1 genotype could not be determined in 2 of the patients of the second cohort.doi:10.1371/journal.pone.0108057.t002
Table 3. Clinical outcome according to candidate SELP, AKT1 and IL-6 SNPs.
First cohort Second/validation cohort
SNP Genotype OS median mo. (95% CI) p-value OS median mo. (95% CI) p-value
SELP rs6136 AA 11.4 (8.4–14.4) 0.665 11.0 (9.1–12.9) 0.519
AC/CC 12.7 (10.1–15.2) 13.7 (10.2–17.1)
AKT1 rs1130233 GG 14.3 (12.1–16.5) 0.002 14.3 (11.7–16.8) 0.004
GA/AA 10.3 (8.1–12.4) 9.9 (8.6–11.1)
IL-6 rs1800796 GG 11.5 (7.1–16.0) 0.259 11.6 (9.1–14.2) 0.651
GC/CC 12.5 (10.8–14.3) 12.5 (9.7–15.3)
OS: overall survival; mo: months.doi:10.1371/journal.pone.0108057.t003
Association of AKT1 and SELP Polymorphisms with Cachexia
PLOS ONE | www.plosone.org 5 September 2014 | Volume 9 | Issue 9 | e108057
Table9.3.ClinicaloutcomeaccordingtocandidateSELP,AKT1andIL-6SNPs.
com
pared
toth
eG
Gsam
ples
(p=
0.0
06).
How
ever,w
hen
we
norm
alizedth
ep
hosp
ho-A
kt1levels
toth
eto
tal-Akt1
levelsin
eachsam
ple
we
ob
servedth
atin
allexcep
ton
ecase,
the
samp
lesw
ithG
A/A
Agen
otyp
eh
ada
low
erratio
betw
eenp
hosp
ho-A
kt1an
dto
talA
kt1vs.
the
samp
lesw
ithth
eG
Ggen
otyp
e(F
ig.S
1,
p=
0.0
03).
Th
esed
atasu
ggestth
atth
eA
KT
1-rs1130233-G
A/A
Agen
otyp
esm
ight
con
fera
redu
cedactivity
toA
kt1,an
dth
us
redu
ceth
ean
tiapop
totic
activityof
this
pivo
talregu
lator
of
apop
totic
signalin
g.
Discu
ssion
Inth
ep
resent
stud
yw
ed
emon
stratedth
eim
portan
ceofA
KT
1-rs1130233
polym
orp
hism
asa
pred
ictivem
arkerof
riskof
cachexia
and
death
inp
atients
with
locally-ad
vanced
or
metastatic
PD
AC
.O
ur
ph
armaco
genetic
analysis
alsoid
entified
anasso
cia-tio
nb
etween
cachexia
and
the
SEL
P-rs6136-A
Agen
otyp
e,in
agreemen
tw
itha
previo
us
stud
yin
120
PD
AC
patien
ts[1
6].
Con
versely,n
ostatistically
significan
tco
rrelation
was
detected
for
the
G-allele
of
the
rs1800796p
olym
orp
hism
inth
eIL
6gen
e,w
hich
was
associated
with
increased
suscep
tibility
tocach
exiaan
dd
ecreasedsu
rvivaltim
eof
stageII
and
IIIC
hin
eseP
DA
Cp
atients
[14].
Th
esed
iscrepan
ciesco
uld
suggest
that
ph
armaco
genetic
associatio
ns
aren
ot
always
repro
du
cible
instu
dies
inp
op
ulatio
ns
with
differen
teth
nic
backgro
un
ds,
asw
ellas
ind
ifferent
clinical
stages.H
ow
ever,th
em
inor
allelefreq
uen
cies,of
app
roxim
ately4%
,in
ou
rco
horts,
greatlylim
itedth
estatistical
pow
erfo
rth
ean
alysisof
this
polym
orp
hism
.L
argerstu
dies
with
hom
ogen
eou
ssettin
gsof
patien
tsare
essential
toin
vestigateth
ero
leof
emergin
gb
iom
arkersb
efore
plan
nin
gof
pro
spective
trials.P
DA
Cp
atients
have
the
high
estrisk
of
develo
pin
gcach
exiaam
on
gth
egastro
intestin
altu
mors,
and
ith
asb
eensh
ow
nth
atcach
exiais
correlated
with
poor
pro
gnosis,
redu
cedtreatm
ent
toleran
cean
da
significan
tred
uctio
nin
the
qu
alityof
lifeof
these
patien
ts[2
8,2
9].
Cach
exiacan
be
caused
by
aco
mp
lexin
terplay
of
mech
anism
sin
clud
ing
med
icalp
rob
lems
such
asd
iabetes,
tum
or
stage,an
dd
uod
enal
or
com
mon
bile
du
ctob
structio
n,
wh
ichcau
sep
ain,
nau
sea,d
ysph
agia,gastro
paresis,
pan
creaticin
sufficien
cyan
dm
alabso
rptio
n[3
0].
Recen
tstu
dies
have
show
nth
atalso
neu
ralin
vasion
,w
hich
com
mon
lyoccu
rsin
PD
AC
,is
relatedto
cachexia.
Nerve
dam
agefro
mP
DA
Ccan
ind
eedactivate
astrocytes,
wh
ichsu
bseq
uen
tlyin
du
celip
olysis
and
mu
scleatro
ph
y[3
1].
Fu
rther-
more,
the
increase
inth
esym
path
eticn
ervou
ssystem
activitym
ight
cause
lipolysis
inad
ipose
tissue
and
mu
scleatro
ph
y[3
2].
Inad
ditio
nto
these
mech
anical
factors,
severaloth
erm
echan
isms
have
been
pro
posed
tod
riveth
ep
athop
hysio
logy
of
PD
AC
cachexia
and
there
isevid
ence
that
anorexia
and
hyp
ercatabolism
canb
etriggered
by
cytokin
es,circu
lating
horm
on
es,n
euro
pep
-tid
es,n
euro
transm
itters,an
dtu
mor-d
erivedfacto
rs.S
everalstud
iessh
ow
edth
atin
creasedlevels
of
cytokin
es,su
chas
IL-6
,w
ereasso
ciatedw
ithw
eight
loss
and
poor
pro
gnosis
inP
DA
Cp
atients
[8–10].
How
ever,th
evariab
lep
redisp
ositio
nto
cachexia
may
be
alsod
ue
toth
ep
atient’s
genotyp
e,an
da
com
preh
ensive
ph
armaco
genetic
stud
yd
emon
stratedth
easso
cia-tio
nof
cachexia
with
the
rs6136p
olym
orp
hism
of
the
gene
SEL
P.
Th
isgen
een
cod
esth
ecell
adh
esion
pro
teinP
-selectin,
wh
ichw
asfo
un
dto
be
up
regulated
inm
urin
ean
drats
mod
elsof
cachexia
caused
by
both
acute
and
chro
nic
inflam
mato
ryin
sults
[16].T
hese
data
revealedth
atP
-selectinh
asa
relevant
role
inb
oth
anim
alm
od
elsan
din
cachectic
cancer
patien
ts.H
ow
ever,n
od
ataare
yetavailab
leon
itsro
leas
arisk
factor
or
asa
poten
tialmed
iator
of
the
cachectic
pro
cess.
Table 4. Factors associated with overall survival in the multivariate analysis.
First cohort Second/validation cohort
Covariates for OS HR (95%CI) df p-value HR (95%CI) df p-value
Age, years #65 1 (ref) 1 0.032 1 (ref) 1 0.864
.65 1.4 (1.0–2.0) 1.0 (0.7–1.4)
Sex Male 1.1 (0.8–1.5) 1 0.698 1.4 (1.0–1.9) 1 0.046
Female 1 (ref) 1 (ref)
Cachexia Yes 2.2 (1.6–3.2) 1 0.0001 1.6 (1.1–2.3) 1 0.006
No 1 (ref) 1 (ref)
AKT1 rs1130233 GG 1 (ref) 1 0.002 1 (ref) 1 0.004
GA/AA 1.7 (1.2–2.4) 1.6 (1.2–2.3)
df: degrees of freedom; HR: hazard ratio; OS: overall survival.doi:10.1371/journal.pone.0108057.t004
Asso
ciation
of
AKT1
and
SELPP
olym
orp
hism
sw
ithC
achexia
PLO
SO
NE
|w
ww
.plo
son
e.org
6Sep
temb
er2014
|V
olu
me
9|
Issue
9|
e108057
Table9.4.Factorsassociatedwithoverallsurvivalinthemultivariateanalysis.
9

Chapter9 PLoSOne2014;19;9(9):e108057
Discussion
InthepresentstudywedemonstratedtheimportanceofAKT1-rs1130233polymorphismasapredictivemarker of risk of cachexia and death in patients with locally-advanced or metastatic PDAC. Ourpharmacogenetic analysis also identified an association between cachexia and the SELP-rs6136-AAgenotype,inagreementwithapreviousstudyin120PDACpatients[16].
Conversely, no statistically significant correlation was detected for the G-allele of the rs1800796polymorphism in the IL6 gene, which was associated with increased susceptibility to cachexia anddecreased survival time of stage II and III Chinese PDAC patients [14]. These discrepancies couldsuggestthatpharmacogeneticassociationsarenotalwaysreproducibleinstudiesinpopulationswithdifferent ethnic backgrounds, as well as in different clinical stages. However, the minor allelefrequencies,ofapproximately4%,inourcohorts,greatlylimitedthestatisticalpowerfortheanalysisofthispolymorphism.Largerstudieswithhomogeneoussettingsofpatientsareessentialtoinvestigatetheroleofemergingbiomarkersbeforeplanningofprospectivetrials.
PDACpatientshavethehighestriskofdevelopingcachexiaamongthegastrointestinaltumors,andithasbeenshownthatcachexia iscorrelatedwithpoorprognosis, reducedtreatmenttoleranceandasignificantreductioninthequalityoflifeofthesepatients[28,29].
Cachexia can be caused by a complex interplay ofmechanisms includingmedical problems such asdiabetes, tumor stage, and duodenal or common bile duct obstruction, which cause pain, nausea,dysphagia,gastroparesis,pancreaticinsufficiencyandmalabsorption[30].Recentstudieshaveshownthatalsoneuralinvasion,whichcommonlyoccursinPDAC,isrelatedtocachexia.NervedamagefromPDAC can indeed activate astrocytes, which subsequently induce lipolysis and muscle atrophy [31].Furthermore,theincreaseinthesympatheticnervoussystemactivitymightcauselipolysisinadiposetissueandmuscleatrophy[32].Inadditiontothesemechanicalfactors,severalothermechanismshavebeenproposedtodrivethepathophysiologyofPDACcachexiaandthereisevidencethatanorexiaandhypercatabolism can be triggered by cytokines, circulating hormones, neuropeptides,neurotransmitters,andtumor-derivedfactors.
Several studiesshowedthat increased levelsofcytokines,suchas IL-6,wereassociatedwithweightlossandpoorprognosisinPDACpatients[8–10].However,thevariablepredispositiontocachexiamaybealsoduetothepatient’sgenotype,andacomprehensivepharmacogeneticstudydemonstratedtheassociation of cachexiawith the rs6136 polymorphismof the geneSELP. This gene encodes the celladhesionproteinP-selectin,whichwasfoundtobeupregulatedinmurineandratsmodelsofcachexiacausedbybothacuteandchronicinflammatoryinsults[16].ThesedatarevealedthatP-selectinhasa
�139
Figure 2
GG GA-AA0.0
0.3
0.6
0.9
1.2
*p=0.01
AKT1-rs1130233 polymorphismGG GA-AA
0.0
0.5
1.0
1.5
2.0
2.5
p=0.05
AKT1-rs1130233 polymorphism
A B
Pho
spho
-Akt
1 (U
/ml)
Tota
l Akt
1 (m
g/m
l)
Figure 9.2. Akt1 expression inmuscle samples according to theAKT1-rs1130233 polymorphism. Bargraphsillustratingthemean±SDexpressionoftotalAkt1(A)andphospho-Akt1(B)inmusclesamplesfrompatientswithdifferentialAKT1-rs1130233genotypes(N=9samplesineachgroup)*p<0.05.doi:10.1371/journal.pone.0108057.g002
9

Chapter9 PLoSOne2014;19;9(9):e108057
relevant role in both animal models and in cachectic cancer patients. However, no data are yetavailableonitsroleasariskfactororasapotentialmediatorofthecachecticprocess.
Sinceapoptosisisreportedtotakeplaceinwastingmuscleincachexia,severalotherstudiesevaluatedthekeyroleofAkt1indevelopingcancercachexia[33–35].Akt1isaserine/threoninekinaseactingasacritical mediator of growth factor-induced survival. Survival factors can suppress apoptosis in atranscription- independent manner by activating Akt1, which then phosphorylates and inactivatescomponentsoftheapoptoticmachinery[32,36].Moreover,inskeletalmuscle,Akt1playsaverycentralrole in the control of both muscle protein synthesis, via mTOR, and protein degradation, via thetranscriptionfactorsoftheFoxOfamily.Thissuggestsapivotalrole inexcessivelossofmusclemassassociatedwithseveraldiseases,includingmyopathiesandmusculardystrophies,aswellasincachexiaassociatedwithsystemicdisorderssuchascancer,diabetes,sepsisandheartfailure[13].Schmittandcolleagues demonstrated a cachexia-associated loss of Akt-dependent signaling in human skeletalmuscleofcachecticpatientscomparedtonon-cachecticpatients,usingmusclebiopsiesfrom16PDACpatientsundergoingpancreatectomy[33].Notably,AKT1isahighlypolymorphicgene,andfunctionalSNPsmightaffectAkt1levelsandinfluenceapoptosisinduction[21].
Therefore,inthecurrentstudyweinvestigatedtheassociationwithcachexiaandtheprognosticvalueof the candidate functional polymorphisms IL6-rs1800796, AKT1-rs1130233 and SELP-rs6136 in a firstcohort of 151 patients with locally-advanced or metastatic PDAC. Then we validated the results byreplicatingtheassociationstudyinanindependentlyrecruitedgroupof152patients.
TothebestofourknowledgethisisthefirststudydemonstratingthatindividualswhocarriedtheA-alleleforAKT1-rs1130233polymorphismwereatincreasedriskofdevelopingcachexia.Moreover,thesepatients survived significantly shorter, compared to patients carrying theGG genotype.Remarkably,the Cox proportional hazards regression model used for the multivariate analysis illustrated theindependentprognosticvalueofAKT1-rs1130233.
Ofnote,theAKT1-rs1130233 isasynonymouspolymorphism,i.e.apolymorphismwherethechangeinthebase in theDNAsequencedoesnotalter theaminoacidencodedduetotheredundancyof thegeneticcode.BecausesynonymousSNPsdonotchangethecompositionoftheproteinproduct,theyhave largelybeenassumed toexert nodiscernible effectongene functionorphenotype.However,studieson artificial site-directed silentmutagenesisof synonymous codons in several genes supportthe hypothesis that altered translation kinetics ofmRNA, caused by altered translation kinetics andfolding, might affect final protein conformation [37]. Moreover, Kimchi-Sarfaty and collaboratorsdemonstratedthattheP-glycoproteininhibitorscyclosporinandverapamilwerelesseffectiveagainstproteins that were produced from polymorphic haplotypes that did not change the amino acidsequence but slow down the ribosome traffic at the corresponding mRNA regions [38]. Thesealterations may thus affect the cotranslational folding pathway, resulting in a different finalconformation and function. Although we could not test this hypothesis because of the lack ofconformation-sensitive mono- clonal antibodies for Akt1, previous studies in different tissue typesshowedthattheAKT1-rs1130233-AAvariantcorrelatedwithlowerAKT1mRNAexpression[19],andwithlowerproteinlevels,contributingtolowerapoptoticresponse[21].
To gain further insight into the mechanisms behind our findings, we performed additional studiesshowing a significant association of Akt1 phosphorylation status in muscle biopsies and the AKT1-rs1130233 polymorphism. In particularweobserved a significant reductionof thephosphorylation atSer437ofAkt1intheGA/AAsamplescomparedtotheGGsamples.TheseresultssuggestthattheAKT1-rs1130233-GA/AA genotypesmight reduce the activity to Akt1 and favour the induction of apoptosis,whichinturncausesmuscleatrophyandincreasescachexia,inagreementwiththeclinicalresults.
Remarkably,arecentstudybyHeandcolleaguesshowedthattumor-secretedmicrovesiclescontainanelevatedexpressionofmicroRNA-21 (miR-21)and inducemyoblastapoptosis incancercachexiaviaaToll-like receptor 7-c-Jun N-terminal kinase- dependent pathway [39].We have recently shown that
�140
9

Chapter9 PLoSOne2014;19;9(9):e108057
miR-21isup-regulatedandactsasanoncogeneinpancreaticintraductalpapillarymucinousneoplasmsand PDAC [40,41]. Moreover, we demonstrated that modulation of Akt1 phosphorylation andapoptosis induction may contribute to the prognostic role of miR-21, as well as in gemcitabinechemoresistance[42].Indeed,amongthemultipletargetsofmiR-21inPDACweshowedthekeyroleofPTEN,leadingtoAKT1regulation.Therefore,wemighthypothesizethatthedifferentAKT1genotypesmightaffectitsinhibitionbyPTENafterstimulationbymiR-21,favouringtumor-inducedmusclewastingthroughapoptosisinduction.
However, previous studies reported controversial relationshipbetweenmiR-21 expression andPTENregulation,bothinthepreclinicalandclinicalsetting[43],aswellasonthefunctionalroleofcandidateAKT1 SNPs [44–46].More genotype-phenotype correlation studies and functional analyses of othercriticalgenes involved intheAkt1pathwayarewarranted.Furtherresearchtoelucidatethe intricatemechanisms involved in the induction and maintenance of PDAC cachexia, should aid in thedevelopmentoffuturetherapeutictargets.Inparticular,itremainstobedeterminedifmodulationofphospho-Akt by specific drugs might alter the development of cachexia. Akt1 might indeed be acandidatetherapeutictarget incancercachexiaandevensurvivalofPDAC,aftertheselectionofthepatientsaccordingtotheirgenotype.
Amajorstrengthofthepresentstudyisthatitwascarriedoutinahomogeneoussettingofpatientswith pancreatic cancer. The results of multivariate analysis indicate the noteworthiness of theprognostic role of AKT1-rs1130233. Moreover, the minor allele frequency of this polymorphism in arandomCaucasianpopulationisfrequent(i.e.,28%accordingtotheSNP-NCBIcancerdatabase).Thus,thesefindingsmightberelevanttoalargenumberofpatients.Conversely,themainlimitationsofthisstudy include the retrospective explorative study design and the lack of prospective randomizedstudiesonthepotentialpredictiveroleofAKT1-rs1130233forchemotherapyactivity.
ConclusionandFuturePerspective
AKT1-rs1130233 and SELP-rs6136 polymorphisms emerged as a predictive risk factor of developingcachexia in locally-advanced and metastatic PDAC. Moreover AKT1 polymorphisms may play aprognosticrole.Sincepancreaticcancerissuchalethaldisease,anybiomarkerthatcanhelptobetterstratifypatientsfordevelopingcachexiamighthavecrucialclinicalapplications.Ultimately,validationofthevalueoftheemergingcandidatepolymorphismsinfutureprospectivetrialswilloffernewtoolstoimprovetheclinicalmanagementofadvancedPDACpatients.
SupportingInformation
Figure 9.S1 Phospho/Total Akt1 expression in muscle samples according to the AKT1-rs1130233polymorphism.Bargraphsillustratingthemean±SDexpressionoftheratiooftotalAkt1andphospho-Akt1inmusclesamplesfrompatientswithdifferentialAKT1-rs1130233genotypes.*p<0.05.(PPT)
Table9.S1GenotypingofPDACpatientsforthecandidateSELP,AKT1andIL-6SNPs.(DOC)
Table9.S2LogisticregressionanalysisofcachexiaandSELP,AKT1andIL-6SNPs.(DOC)
AuthorContributionsConceivedanddesignedtheexperiments:EGPPMGMGJP.Performedtheexperiments:AbolfazlAvanAmirAvanMMNFTYSL.Analyzedthedata:AbolfazlAvanAmirAvanMMNFTYSLEGPPMGMGJP.Contributed reagents/materials/analysis tools: PP NF AMMC UB. Contributed to the writing of themanuscript:AbolfazlAvanAmirAvanPPTYSLEG.Citation:AvanA,AvanA,LeLargeTYS,MambriniA,FunelN,etal.(2014)AKT1andSELPPolymorphismsPredict theRiskofDevelopingCachexia inPancreaticCancerPatients.PLoSONE9(9):e108057.doi:10.1371/journal.pone.0108057
�141
9

Chapter9 PLoSOne2014;19;9(9):e108057
Editor:FranciscoX.Real,CentroNacionaldeInvestigacionesOncológicas(CNIO),SpainReceivedMay24,2014;AcceptedAugust19,2014;PublishedSeptember19,2014Copyright:ß2014Avanetal.Thisisanopen-accessarticledistributedunderthetermsoftheCreativeCommonsAttribution License,which permits unrestricted use, distribution, and reproduction in anymedium,providedtheoriginalauthorandsourcearecredited.DataAvailability:Theauthorsconfirmthatalldataunderlyingthefindingsarefullyavailablewithoutrestriction.AllrelevantdataarewithinthepaperanditsSupportingInformationfiles.Funding: This work was partially supported by grants from Netherlands Organization for ScientificResearch,VENIgrant(projectnumber91611046(ElisaGiovannetti))andStimuleringfondsOpenAccess(ElisaGiovannetti),CCAFoundation2012(AmirAvan,ElisaGiovannetti,GodefridusJ.Peters),ResearchCouncil at the Mashhad University of Medical Sciences, Mashhad (Amir Avan, Majid Ghayour-Mobarhan), Istituto ToscanoTumori (ElisaGiovannetti,Niccola Funel,UgoBoggi) andAIRCStart-Upgrant(ElisaGiovannetti).Thefundershadnoroleinstudydesign,datacollectionandanalysis,decisiontopublish,orpreparationofthemanuscript.CompetingInterests:Theauthorshavedeclaredthatnocompetinginterestsexist.
Correspondenceto:ElisaGiovannetti,MD,PhD,DepartmentofMedicalOncology,VUUniversityMedicalCenter,DeBoelelaan1117,1081HVAmsterdam,theNetherlands.E-mail:[email protected]
References
1. TisdaleMJ(2002)Cachexiaincancerpatients.NatRevCancer2(11):862–871.2. BachmannJ,KettererK,MarschC,FechtnerK,Krakowski-RoosenH,etal.(2009)Pancreatic
cancer related cachexia: influence on metabolism and correlation to weight loss andpulmonaryfunction.BMCCancer28(9):255.
3. Pausch T, HartwigW,HinzU, Swolana T, BundyBD, et al. (2012) Cachexia but not obesityworsens the postoperative outcome after pancreatoduodenectomy in pancreatic cancer.Surgery152(3Suppl1):S81–S88.
4. BachmannJ,BuchlerMW,FriessH,MartignoniME(2013)Cachexia inpatientswithchronicpancreatitisandpancreaticcancer:impactonsurvivalandoutcome.NutrCancer65(6):827–833.
5. DiSebastianoKM,YangL,ZbukK,WongRK,ChowT,etal. (2013)Acceleratedmuscleandadipose tissue lossmaypredict survival inpancreatic cancerpatients: the relationshipwithdiabetesandanaemia.BrJNutr109(2):302–312.
6. Uomo G, Gallucci F, Rabitti PG (2006) Anorexia-cachexia syndrome in pancreatic cancer:recentdevelopmentinresearchandmanagement.JOP7(2):157–162.
7. Blum D, Omlin A, Baracos VE, Solheim TS, Tan BH, et al. (2011) European Palliative CareResearchCollaborative.Cancercachexia:asystematicliteraturereviewofitemsanddomainsassociatedwithinvoluntaryweightlossincancer.CritRevOncolHematol80(1):114–144.
8. Argiles JM, Busquets S, Lopez-Soriano FJ (2003) Cytokines in the pathogenesis of cancercachexia.CurrOpinClinNutrMetabCare6(4):401–406.
9. Ebrahimi B, Tucker SL, Li D, Abbruzzese JL, Kurzrock R (2004) Cytokines in pancreaticcarcinoma: correlationwithphenotypic characteristics andprognosis. Cancer 101(12): 2727–2736.
10. Martignoni ME, Kunze P, Hildebrandt W, Ku nzli B, Berberat P, et al. (2005) Role ofmononuclear cells and inflammatory cytokines in pancreatic cancer- related cachexia. ClinCancerRes11(16):5802–5808.
11. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, et al. (2013) NF-κB–mediated Pax7dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest123(11):4821–4835.
12. Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, et al. (2013)Nuclear transcription factor k B activation and protein turnover adaptations in skeletal
�142
9

Chapter9 PLoSOne2014;19;9(9):e108057
muscle of patientswith progressive stages of lung cancer cachexia. Am J Clin Nutr 98(3):738–748.
13. BonaldoP,SandriM(2013)Cellularandmolecularmechanismsofmuscleatrophy.DisModelMech6(1):25–39.
14. Zhang D, Zhou Y, Wu L, Wang S, Zheng H, et al. (2008) Association of IL-6 genepolymorphisms with cachexia susceptibility and survival time of patients with pancreaticcancer.AnnClinLabSci38(2):113–119.
15. Tan BH, Ross JA, Kaasa S, Skorpen F, Fearon KC (2011) European Palliative Care ResearchCollaborative.Identificationofpossiblegeneticpolymorphismsinvolvedincancercachexia:asystematicreview.JGenet90(1):165–177.
16. Tan BH, Fladvad T, Braun TP, Vigano A, Strasser F, et al. (2012) European Palliative CareResearch Collaborative. P-selectin genotype is associated with the development of cancercachexia.EMBOMolMed4(6):462–471.
17. StenbergPE,McEverRP,ShumanMA,JacquesYV,BaintonDF(1985)Aplateletalpha-granulemembraneprotein(GMP-140) isexpressedontheplasmamembraneafteractivation.JCellBiol101(3):880–886.
18. SolheimTS,FayersPM,FladvadT,TanB,SkorpenF,etal.(2011)Isthereageneticcauseforcancercachexia?-aclinicalvalidationstudyin1797patients.BrJCancer105(8):1244–1251.
19. Giovannetti E, Zucali PA, Peters GJ, Cortesi F, D’Incecco A, et al. (2010) Association ofpolymorphisms in AKT1 and EGFRwith clinical outcome and toxicity in non-small cell lungcancerpatientstreatedwithgefitinib.MolCancerTher9(3):581–593.
20. FearonK,StrasserF,AnkerSD,BosaeusI,BrueraE,etal.(2011)Definitionandclassificationofcancercachexia:aninternationalconsensus.LancetOncol12(5):489–495.
21. Harris SL, Gil G, Robins H, HuW, Hirshfield K, et al. (2005) Detection of functional singlenucleotide polymorphisms that affect apoptosis. Proc Natl Acad Sci USA 102(45): 16297–16302.
22. AvanA, Caretti V, FunelN,Galvani E,MaftouhM, et al. (2013) Crizotinib inhibitsmetabolicinactivationofgemcitabine in c-Met-drivenpancreatic carcinoma.CancerRes 73(22): 6745–6756.
23. GiovannettiE,LabotsM,DekkerH,GalvaniE,LindJS,etal.(2013)Molecularmechanismsandmodulationofkeypathwaysunderlyingthesynergisticinteractionofsorafenibwitherlotinibinnon-small-cell-lungcancer(NSCLC)cells.CurrPharmDes19(5):927–939.
24. GiovannettiE,PacettiP,ReniM,LeonLG,MambriniA,etal.(2011)AssociationbetweenDNA-repair polymorphisms and survival in pancreatic cancer patients treated with combinationchemotherapy.Pharmacogenomics12(12):1641–1652.
25. Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N (2004) Assessing theprobabilitythatapositivereport isfalse:anapproachformolecularepidemiologystudies.JNatlCancerInst96(6):434–442.
26. Marsh S, Mallon MA, Goodfellow P, McLeod HL (2005) Concordance of pharmacogeneticmarkersingermlineandcolorectaltumourDNA.Pharmacogenomics6(8):873–877.
27. vanHuis-TanjaL,KweekelD,GelderblomH,KoopmanM,PuntK,etal.(2013)Concordanceofgenotype for polymorphisms in DNA isolated from peripheral blood and colorectal cancertumorsamples.Pharmacogenomics14(16):2005–2012.
28. Fearon KC (2008) Cancer cachexia. developingmultimodal therapy for a multidimensionalproblem.EurJCancer44(8):1124–1132.
29. TucaA,Jimenez-FonsecaP,GasconP(2013)Clinicalevaluationandoptimalmanagementofcancercachexia.CritRevOncolHematol88(3):625–636.
30. Tan CR, Yaffee PM, Jamil LH, Lo SK, Nissen N, et al. (2014) Pancreatic cancer cachexia: areviewofmechanismsandtherapeutics.FrontPhysiol5:88.
31. ImotoA1,MitsunagaS, InagakiM,AoyagiK, SasakiH, et al. (2012)Neural invasion inducescachexia via astrocytic activation of neural route in pancreatic cancer. Int J Cancer 131(12):2795–2807.
�143
9

Chapter9 PLoSOne2014;19;9(9):e108057
32. LiG,KleinRL,MathenyM,KingMA,MeyerEM,etal.(2002)Inductionofuncouplingprotein1by central interleukin-6 gene delivery is dependent on sympathetic innervation of brownadiposetissueandunderliesonemechanismofbodyweightreductioninrats.Neuroscience115(3):879–889.
33. SchmittTL,MartignoniME,BachmannJ,FechtnerK,FriessH,etal.(2007)ActivityoftheAkt-dependent anabolic and catabolic pathways in muscle and liver samples in cancer-relatedcachexia.JMolMed(Berl)85(6):647–54.
34. BodineSC,StittTN,GonzalezM,KlineWO,StoverGL,etal. (2001)Akt/mTORpathway isacrucialregulatorofskeletalmusclehypertrophyandcanpreventmuscleatrophyinvivo.NatCellBiol3(11):1014–1119.
35. SugitaH,KanekiM,SugitaM,YasukawaT,YasuharaS,etal.(2005)Burninjuryimpairsinsulin-stimulatedAkt/PKBactivationinskeletalmuscle.AmJPhysiolEndocrinolMetab288(3):585–591.
36. KimAH,KhursigaraG,SunX,FrankeTF,ChaoMV(2001)Aktphosphorylatesandnegativelyregulatesapoptosissignal-regulatingkinase1.MolCellBiol21(3):893–901.
37. KomarAA,LesnikT,ReissC(1999)Synonymouscodonsubstitutionsaffectribosometrafficandproteinfoldingduringinvitrotranslation.FEBSLett462(3):387–391.
38. Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, et al. (2007) A ‘‘silent’’polymorphismintheMDR1genechangessubstratespecificity.Science315(5811):525–528.
39. HeWA, Calore F, Londhe P, Canella A, GuttridgeDC, et al. (2014)Microvesicles containingmiRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci USA111(12):4525–4529.
40. CaponiS,FunelN,FramptonAE,MoscaF,SantarpiaL,etal.(2013)Thegood,thebadandtheugly: a tale of miR-101, miR-21 and miR-155 in pancreatic intraductal papillary mucinousneoplasms.AnnOncol24(3):734–741.
41. Frampton AE, Castellano L, Colombo T, Giovannetti E, Krell J, et al. (2014) MicroRNAscooperatively inhibit a network of tumor suppressor genes to promote pancreatic tumorgrowthandprogression.Gastroenterology146(1):268–277.
42. Giovannetti E, Funel N, Peters GJ, Del Chiaro M, Erozenci LA, et al. (2010) MicroRNA-21(miR-21) in pancreatic adenocarcinoma (PDAC): correlation with clinical outcome andpharmacologicalaspectsunderlyingitsroleinthemodulationofgemcitabineactivity.CancerRes70(11):4528–4538.
43. KrichevskyAM,GabrielyG(2009)miR-21:asmallmulti-facetedRNA.JCellMolMed13(1):39–53.
44. GalvaniE,PetersGJ,GiovannettiE(2012)EGFreceptor-targetedtherapyinnon-small-celllungcancer: role of germline polymorphisms in outcome and toxicity. Future Oncol 8(8): 1015–1029.
45. Li Q, Yang J, Yu Q, Wu H, Liu B, et al. (2013) Associations between single- nucleotidepolymorphisms inthePI3K-PTEN-AKT-mTORpathwayand increasedriskofbrainmetastasisinpatientswithnon-smallcelllungcancer.ClinCancerRes19(22):6252–6260.
46. AvanA,MaftouhM,AvanA,TibaldiC,ZucaliPA,etal. (2014)SNPs inPI3K-PTEN-mTORandBrainMetastasesinNSCLC-Letter.ClinCancerRes20(13):3623–3624.
ReceivedMay24,2014;AcceptedAugust19,2014;PublishedSeptember19,2014;DOI:10.1371/journal.pone.0108057
�144
9

Chapter9 PLoSOne2014;19;9(9):e108057
SupplementaryTables
�145
Supplemental Tables
Table 9.S1. Genotyping of PDAC patients for the candidate SELP, AKT1 and IL-6 SNPs
First cohort Second/validation cohort
SNP Patients n (%)
HWE p-value
Patients n (%)
HWE p-value
SELP-rs6136
AA 81 (53.6) 76 (50.0)
AC 60 (39.7) 66 (43.4)
CC 10 (6.6) 0.911 10 (6.6) 0.859
AKT1-rs1130233
GG 70 (46.4) 74 (49.3)
GA 65 (43.1) 64 (42.7)
AA 16 (10.6) 0.980 12 (8.0) 0.972
IL6-rs1800796
GG 91 (60.3) 100 (65.8)
GC 51 (33.8) 44 (29.0)
CC 9 (6.0) 0.923 8 (5.3) 0.527
HWE: Hardy–Weinberg equilibrium
Table 9.S2. !Logistic regression analysis of cachexia and SELP, AKT1 and IL-6 SNPs
First cohort Second cohort
SNP Risk genotype df OR (95%CI) p-value
OR (95%CI)
p-value
SELP-rs6136
AA 1 2.4 (1.1-5.0) 0.017 2.0 (1.0-4.0) 0.039
AKT1-rs1130233
GA-AA 1 3.0 (1.4-6.3) 0.003 2.1 (1.1-4.1) 0.035
IL6-rs1800796
GG 1 0.6 (0.3-1.2) 0.139 0.7 (0.4-1.5) 0.416 df: degree od freedom; OR: odds ratio; SNPs: single nucleotide polymorphisms
9

Chapter9 PLoSOne2014;19;9(9):e108057
�146
9

CHAPTER10SNPsinPI3K-PTEN-mTORandBrainMetastasesin
NSCLC—Letter
AbolfazlAvan*,1MinaMaftouh*,1AmirAvan,1,2CarmeloTibaldi,3PaoloA.Zucali,4ElisaGiovannetti1#
1DepartmentofMedicalOncology,VUUniversityMedicalCenter,Amsterdam,theNetherlands;2DepartmentofNewSciencesandTechnology,FacultyofMedicine,MashhadUniversityofMedicalSciences,Mashhad,Iran;3DepartmentofOncology,AziendaUSL-6ofLivorno,Livorno,Italy;4DepartmentofMedicalOncologyandHematology,IstitutoClinico
Humanitas,Rozzano,Milan,Italy.*Theseauthorsequallycontributedtothestudy
ClinCancerRes2014;20(13):3623-4
10

Chapter10 ClinCancerRes2014;20(13):3623-4
SNPsinPI3K/PTEN/mTORandbrainmetastasesinNSCLC–Letter
We have read with interest the study by Li and colleagues (1) on single-nucleotide polymorphisms(SNP) predicting brain metastases in non–small cell lung cancer (NSCLC). Patients with the GT/GGgenotypeofAKT1-rs2498804,CT/TTofAKT1-rs2494732,andAG/AAofPIK3CA-rs2699887hadhigherriskof brainmetastasis. These data support the feasibility of studying germinal polymorphisms for riskstratification.However,severalpointsshouldbediscussedinmoredetail.
First, thechoiceto focusonNSCLC isvaluable,as treatmentwithprophylacticcranial irradiation isamatter of debate in this tumor. However, according to the "rule of tens," in amultivariate analysisthereshouldbeaminimumof10eventsperpredictorvariable.As16SNPswereanalyzed,atotalof160patients would have to have brain metastases to prevent overfitting, because the results from anoverfittedmodelarenotgeneralizabletootherpopulations.Anothercriticalobstacletothesuccessfuldevelopmentofagenotype-basedtestisthehighnumberofspuriousassociations(2).Toavoidfalse-positiveassociations,aBonferronicorrectionconsideringallthestudiedSNPswouldrequireaPvalueof<0.05/16=0.003forstatisticalsignificance.
EmergingSNPsneedtobevalidatedinindependentcohorts,andourdatasetfromstageIIIB/IVNSCLC(3)confirmedtheassociationofAKT1-rs2498804,butnotofPIK3CA:rs2699887,withbrainmetastases(Table10.1).Theseconflictingdatamightbeexplainedbyseveralfactors,suchasethnicity,samplesize,different clinical settings, histotypes, stage, and treatment. However, Li and colleagues did notdescribetreatmentdetailsoftheircohort,whileallourpatientsreceivedgefitinib.Similarly,although50%oftheirpatientswerenever-smoker,noinformationwasprovidedonEGFRmutationsorALKtranslocations.Because almost half of ALK+ patients treated withcrizotinibhavecentralnervoussystemrelapse(4),wewonder whether the same results would have beenobtained after adjustment for EGFR/ALK geneticaberrations and targeted treatments. Ultimately, theroleofAKT1-rs2498804inclinicaldecision-makingshouldbe validatedwithin prospective trials in homogeneouspatientcohorts.
Inadditiontopharmacogenetictrials,furtherresearchisneeded to unravel the functional significance of thesepoly- morphisms. Specifically, AKT1-rs2498804, locatedat 30 untranslated region,may affect gene expressionthrough changes in transcription factor–binding sites,microRNA target sequences, and/or splicing variants.AlthoughthisSNPhasnotbeentested inexperimentalmodels (5), it couldbe evaluated at least in silico, usingpublicly available software such asPROMO(http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB1⁄4TF_8.3)todetecttranscriptionfactor–bindingsites,MicroSNiper(http://cbdb.nimh.nih.gov/microsniper)forpredictionofSNPeffectson microRNA targets, and Human Splicing Finder (http://www.umd.be/HSF/) to study pre-mRNAsplicing. In particular, additional studies should investigatewhether differential expression levels ofcandidate microRNAs might explain the fact that this polymorphism was not associated withmetastasisat sitesother than thebrain.Finally,directevidenceof theeffectsonproteinexpressioncouldbeobtainedbyimmunohistochemistryofNSCLCprimaryandmetastastictissues.
Inconclusion,wethankLiandcolleaguesfortheirstudy,butwebelievethatadditionalparametersareessential to validate candidate SNPs beyond already available clinical factors for predicting brainmetastasesinNSCLC.
�148
Letter to the Editor
SNPs in PI3K-PTEN-mTOR and Brain Metastases in NSCLC—Letter
Abolfazl Avan1, Mina Maftouh1, Amir Avan1,2, Carmelo Tibaldi3, Paolo A. Zucali4, and Elisa Giovannetti1
We have read with interest the study by Li and colleagues(1) on single-nucleotide polymorphisms (SNP) predictingbrain metastases in non–small cell lung cancer (NSCLC).Patients with the GT/GG genotype of AKT1-rs2498804, CT/TT of AKT1-rs2494732, and AG/AA of PIK3CA-rs2699887had higher risk of brain metastasis. These data support thefeasibility of studying germinal polymorphisms for riskstratification. However, several points should be discussedin more detail.First, the choice to focus on NSCLC is valuable, as
treatment with prophylactic cranial irradiation is a matterof debate in this tumor. However, according to the "rule oftens," in amultivariate analysis there should be aminimumof 10 events per predictor variable. As 16 SNPs were ana-lyzed, a total of 160 patients would have to have brainmetastases to prevent overfitting, because the results froman overfitted model are not generalizable to other popula-tions. Another critical obstacle to the successful develop-ment of a genotype-based test is the high number ofspurious associations (2). To avoid false-positive associa-tions, a Bonferroni correction considering all the studiedSNPs would require a P value of <0.05/16 ¼ 0.003 forstatistical significance.Emerging SNPs need to be validated in independent
cohorts, and our dataset from stage IIIB/IV NSCLC (3)confirmed the association of AKT1-rs2498804, but not ofPIK3CA:rs2699887, with brain metastases (Table 1). Theseconflicting data might be explained by several factors, suchas ethnicity, sample size, different clinical settings, histo-types, stage, and treatment. However, Li and colleagues didnot describe treatment details of their cohort, while all ourpatients received gefitinib. Similarly, although 50% of theirpatients were never-smoker, no information was providedon EGFR mutations or ALK translocations. Because almosthalf of ALKþ patients treated with crizotinib have centralnervous system relapse (4), we wonder whether the sameresults would have been obtained after adjustment forEGFR/ALK genetic aberrations and targeted treatments.Ultimately, the role of AKT1-rs2498804 in clinical deci-
sion-making should be validated within prospective trialsin homogeneous patient cohorts.
In addition to pharmacogenetic trials, further research isneeded to unravel the functional significance of these poly-morphisms. Specifically, AKT1-rs2498804, located at 30
untranslated region, may affect gene expression throughchanges in transcription factor–binding sites, microRNAtarget sequences, and/or splicing variants. Although thisSNP has not been tested in experimental models (5), itcould be evaluated at least in silico, using publicly availablesoftware such as PROMO (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB¼TF_8.3) to detecttranscription factor–binding sites, MicroSNiper (http://cbdb.nimh.nih.gov/microsniper) for prediction of SNPeffects on microRNA targets, and Human Splicing Finder(http://www.umd.be/HSF/) to study pre-mRNA splicing.In particular, additional studies should investigate whetherdifferential expression levels of candidatemicroRNAsmightexplain the fact that this polymorphism was not associatedwith metastasis at sites other than the brain. Finally, directevidence of the effects on protein expression could beobtained by immunohistochemistry ofNSCLCprimary andmetastastic tissues.
In conclusion, we thank Li and colleagues for their study,but we believe that additional parameters are essential tovalidate candidate SNPs beyond already available clinicalfactors for predicting brain metastases in NSCLC.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Table 1. Polymorphisms and brain metastasesin patients with advanced NSCLC treated withgefitinib
Polymorphism Patients, N Events, N (%) Pa
AKT1:rs2498804TT 36 3 (8)GTþGG 54 17 (31) 0.010
PIK3CA:rs2699887GG 79 15 (19)AGþAA 11 5 (45) 0.062
NOTE:Polymorphic loci inAKT1 andPIK3CAwere assessedin DNA isolated from blood samples and/or paraffin-embed-ded tumors from 90 patients with stage IIIB/IV NSCLC,treated with gefitinib (ClinicalTrials.gov ID-NCT00831454),as described previously (3).aP values were calculated by the Fisher exact test.
Authors' Affiliations: 1Department of Medical Oncology, VU UniversityMedical Center, Amsterdam, the Netherlands; 2Department of NewSciences and Technology, Faculty of Medicine, Mashhad University ofMedical Sciences,Mashhad, Iran; 3Department ofOncology, AziendaUSL-6 of Livorno, Livorno, Italy; and 4Department of Medical Oncology andHematology, Istituto Clinico Humanitas, Rozzano, Milan, Italy
Corresponding Author: Elisa Giovannetti, Department of Medical Oncol-ogy, VU University Medical Center, CCA Room 1.42, De Boelelaan 1117,1081HVAmsterdam, theNetherlands.Phone: 31-20-4442267; Fax: 31-20-4443844; E-mail: [email protected]
doi: 10.1158/1078-0432.CCR-13-3256
!2014 American Association for Cancer Research.
ClinicalCancer
Research
www.aacrjournals.org 3623
on December 1, 2014. © 2014 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst May 6, 2014; DOI: 10.1158/1078-0432.CCR-13-3256
Table10.1.PolymorphismsandbrainmetastasesinpatientswithadvancedNSCLCtreatedwithgefitinib
10

Chapter10 ClinCancerRes2014;20(13):3623-4
DisclosureofPotentialConflictsofInterestNopotentialconflictsofinterestweredisclosed.
Grant Support: This work was supported by grants from CCA foundation (to A. Avan and E.Giovannetti),FondazioneHumanitas(toE.GiovannettiandP.A.Zucali),theNetherlandsOrganizationforScientificResearch"NWO-Venigrant"andAIRC/Start-Up(toE.Giovannetti).
Correspondence to: ElisaGiovannetti,MD,PhD,DepartmentofMedicalOncology,VUUniversityMedicalCenter,CCARoom1.42,DeBoelelaan1117,1081HVAmsterdam,theNetherlands.Phone:31-20-4442267;Fax:31-20-4443844;E-mail:[email protected]
References
1. Li Q, Yang J, Yu Q, Wu H, Liu B, Xiong H, et al. Associations between Single- NucleotidePolymorphismsinthePI3K-PTEN-AKT-mTORPathwayandIncreasedRiskofBrainMetastasisinPatientswithNon-SmallCellLungCancer.ClinCancerRes.2013;19:6252-60.
2. MaitlandML,RatainMJ,CoxNJ.InterpretingPvaluesinpharmacogeneticstudies:acallforprocessandperspective.JClinOncol.2007;25:4513-5.
3. Giovannetti E, Zucali PA, Peters GJ, Cortesi F, D'Incecco A, Smit EF, et al. Association ofpolymorphisms in AKT1 and EGFRwith clinical outcome and toxicity in non-small cell lungcancerpatientstreatedwithgefitinib.MolCancerTher.2010;9:581-93.
4. OttersonGA,RielyGJ,ShawAT,CrinòL,KimDW,MartinsR,etal.ClinicalcharacteristicsofALK+NSCLCpatients(pts)treatedwithcrizotinibbeyonddiseaseprogression(PD):potentialimplicationsformanagement.JClinOncol.2012;30(suppl):abstr.7600.
5. YangCH,ChengYH,ChuangLY,ChangHW.Drug-SNPing:anintegrateddrug-based,proteininteraction-based tagSNP-based pharmacogenomics platform for SNP genotyping.Bioinformatics.2013;29:758-64.
Received December 5, 2013; revised April 13, 2014; accepted January 5, 2014; published online July 1, 2014. DOI:10.1158/1078-0432.CCR-13-3256
�149
10

Chapter10 ClinCancerRes2014;20(13):3623-4
�150
10

DISCUSSION AND
SUMMARY


Chapter11Discussion
Chemotherapy-inducedneurotoxicity(submitted)androleofsignalingintheefficacyofcancertreatment
11

Chapter11(Avan’sdissertation) Discussion
Discussion
Part1:Chemotherapy-inducedNeurotoxicity
Neurologiccomplicationscanbeseenthroughoutthecourseofcancerdiagnosisandtreatment.Sometoxicities may develop during or years after completion of treatment, which makes the diagnosischallenging.Neurologicdisordersaffectapproximately 15%ofpatientswithcancerandmaydiminishthe patient’s quality of life and directly shorten survival. They may be associated with metastaticcomplicationsofthecancerinthebrain,cranialnerves,spine,leptomeninges,andperipheralnerves,ornonmetastatic ones, including infection, toxic metabolites, vascular, paraneoplastic, or related tochemo-radiotherapy.Peripheralneurotoxicity isamajordose-limitingtoxicityofchemotherapyandafrequentsourceofpainfulanddisablingsymptomsthatdiminishqualityoflifeinpatientswithcancer.Earlyrecognitionmayresultintreatmentandimprovementofadverseneurologicsymptomsaswellasavoid permanent neurologic damage. It is also important to recognize neurologic complications ofcancer early and to appropriately distinguish symptoms directly related to the cancer, such asmetastatic involvement, from those associated with other neurologic disorders or with cancermedication(1).Moreover,certaincancers,suchassmallcelllungcancer,multiplemyeloma,thymoma,andWaldenstrommacroglobulinemiaareassociatedwithneuropathy,whichneedtobeconsidered.
Clinical manifestations of neurological side effects may vary depending upon the type, dose, andduration of treatment. Moreover, the type of the affected nerve fibers, i.e. sensory, motor, orautonomic, determines the signs and symptoms. Sensorynerve cell bodies reside in thedorsal rootganglia,unlikemotorneuroncellbodies,whichresideinthespinalcordandarenotprotectedbytheblood brain barrier. Hence, sensory nerve cell bodies have a much greater exposure to circulatingdrugs. Common sensory symptoms include symmetric painful paresthesia in the hands and feet,allodynia, lossof deep tendon reflexes, anddiminishedproprioception andmoreproximal vibratorysensitivityimpairment.
The chemotherapy agents most commonly associated with peripheral neuropathy (Table 11.1) aretaxanes, vinca alkaloids, platinum compounds, proteasome inhibitors (bortezomib), andantiangiogenic compounds (thalidomide). Given the clinical burden of chemotherapy-inducedperipheral neuropathy, there is a growing interest in better understanding its pathophysiology,standardizingevaluation,anddevelopingeffectivetreatments.
Platinum compounds are widely used in various anticancer regimens. Thus, in the first part of thethesis, a systematic review of the literature is provided for pathophysiology, pathogenesis, andcharacteristics of platinum-induced neurotoxicity along with the best available evidence on thepreventiveandtherapeuticstrategies(2).Platinumneurotoxicitymaypresentnotonlywithanacuteor chronic peripheral neuropathy, but also as central, e.g. chemo brain (short-term memory andconcentration problems and an inability to resume high-level tasks as a result of high dosechemotherapy)andototoxicity(sensorineuralhearingloss).Theincidenceofperipheralneurotoxicityis variable and ranges from30% to40%ofpatientswith cancer treatedwith chemotherapy (1),withsomestudiesreportinganincidenceashighas90%(3).
Peripheral neurotoxicity of platinum compounds is associated with several molecular alterations,including dorsal root ganglia cytotoxic inflammatory changes, mitotoxicity and enhanced oxidativestress, voltage-gated ion (sodium/potassium/calcium) channel dysfunction, functional impairment ofionchannelsofthetransientreceptorpotentialfamily,inductionofneuronalapoptosisindorsalrootganglia,anddemyelination(4).Accumulationofplatinumproducts,mainly inthedorsalrootganglia,aswellasperipheralneuronsmayprovokechronicneuropathy.Sincetheseneuronsarepost-mitoticand not dividing, the formation ofDNA adducts is not lethal, but results inDNA-strand breaks. TheextentofDNAcrosslinksindorsalrootgangliaandthedegreeofneurotoxicityisrelatedtothetotalcumulative dose of platinum agent (5). Cisplatin producesmore adducts in the dorsal root ganglia
�154
11

Chapter11(Avan’sdissertation) Discussion
comparedtooxaliplatin,whichmayexplainitshigherneurotoxicity.Platinumadductsmayalsoaffectneurons,whilebrainandspinalcordareprotectedbythebloodbrainbarrier(5).
Among the potentially neurotoxic agents with antineoplastic profiles, oxaliplatin, bortezomib, andepothilone-B are commonly used in daily practice, for which neurotoxicity may not only be dose-limiting, but also influence the patients’ quality of life. Hence, utilizing a representative model topredicttheneurotoxicityofdrugswouldbeideal,whichwouldenableustoadministertherightdrugtotherightpersonbytakingtheriskofneurotoxicityintoaccount.
Differentmodelshavebeenemployedtostudytheneurotoxicityofanti-canceragents,includingdorsalrootgangliaof rats, cerebralgangliaof snails, andmany invitromodels suchasneuriteoutgrowth.Giventhehighburdenofthisadverseevent,developmentofappropriateassessmenttoolsisessentialfor early characterizationof thedrug-inducedneuropathy and thus to improve thequality of life ofcancerpatients.Althoughstillfarfromanidealmodel,wecouldsuccessfullyevaluatetheneurotoxiceffectsofoxaliplatin,bortezomib,andepothilone-BinPC12ratpheocromocytomacellsafterneuronaldifferentiation (6). Having evaluated the potential efficacy of amifostine in reducing the risk ofneuropathyduetothesethreedrugs,wefounditupto50%protectiveonlyintheoxaliplatin-treatedcell lines.Thismodelhaspreviouslybeenestablishedtostudythepotentialofamifostine toprotectagainstcisplatin-,paclitaxel-,andvincristine-inducedneurotoxicity(7).Theneurotoxicityofthethree
�155
Table&11.1.&Neurotoxicity+associated+with+chemotherapy+drugs+
Drug Approved&application Target(s)/Nerve&Type Clinical&Presentations
Bortezomib Mantle+cell+lymphoma+
and+multiple+myeloma
Mitochondria,+
endoplasmic+reticulum/+
sensory+nerves
Distal+painful+paresthesia
Platinum+agents+
(Cisplatin)
Advanced+bladder+
cancer,+ovarian+cancer,+
lung+cancer,+and+
testicular+cancer+
Dorsal+root+ganglion/+
sensory+large+fibers
Numbness,+diminished+of+
proprioception,+loss+of+deep+tendon+
reflexes,+loss+of+vibratory+sense,+
Lhermitte+symptom,+ototoxicity,+
coasting+effect
+Platinum+agents+
(Oxaliplatin)
Advanced+colorectal+
cancer
Dorsal+root+ganglion,+
sodium+ion+channels/+
sensory+large+fibers
Acute+neuropathy,+chronic+
neuropathy,+oropharyngeal+
allodynia,+cold+hypersensitivity,+
Vinca+alkaloids+
(vincristine,+
vinblastine,+
vindensine)
Acute+leukemia,+
rhabdomyosarcoma,+
neuroblastoma,+Wilm’s+
tumor,+Hodgkin’s+
disease,+and+other+
lymphomas
Dorsal+root+ganglion,+
microtubules,+nerve+
terminals/+sensorimotor+
small+fibers,+autonomic+
and+cranial+nerves
Dizziness,+ocular+palsy,+facial+
weakness,+vocal+cord+paralysis,+
numbness,+pain,+extensor+weakness,+
constipation,+orthostatic+
hypotension+and+syncope
Taxanes+(docetaxel,+
paclitaxel)
Breast+cancer,+head+and+
neck+cancer,+gastric+
cancer,+hormoneO
refractory+prostate+
cancer,+and+non+smallO
cell+lung+cancer
Dorsal+root+ganglion,+
microtubules,+nerve+
terminals+or+channels/+
sensorimotor+large+and+
small+fibers
Painful+paresthesia,+myalgia
Thalidomide Multiple+myeloma Dorsal+root+ganglion,+
nerve+blood+supply,+
dysregulation+of+
neurotrophin/+
sensorimotor+nerves
Painful+paresthesia,+myalgia
Note:+Almost+all+signs+and+symptoms+are+usually+reversible+with+dose+reduction+or+cessation+of+the+responsible+drug
11

Chapter11(Avan’sdissertation) Discussion
drugs was shown by a significant reduction in neurite outgrowth without diminishing the drugcytotoxicity, amongwhich coincubation and 1-hour, not 24-hour postincubation of amifostine couldprevent paclitaxel and cisplatin neurotoxicity, respectively. Thus, amifostine hadnoneuroprotectivepropertyagainstvincristine-inducedneurotoxicitybeingcausedbybreakdownofneurotubulesintheaxon.Verstappenetal (7)postulatedthatvincristinehas,presumably,noeffectontheneuronalcellitself.Amifostine is thought toscavenge free radicals (8),bindtoplatinum(9)andalkylatingagents(10) and lead to secondary removal of preformedDNA-adducts (10) in cisplatin-induced toxicity andpresumablyalsoincisplatin-andpaclitaxel-inducedneurotoxicity.
Cyclins B1 and B2 are two known activators of Cyclin-dependent kinase 1 (CDK1) operating duringmitosis in human cells, which prevents DNA re-replication (11). Cyclins B2 has been found to beupregulatedinmanyhumancancerswithasignificantlyhigherrelativeexpressionofcirculatingCyclinsB2mRNAincancerpatientscomparedtonormalcontrolsandbenigndiseasesgroup(12).Moreover,circulatingCyclinsB2mRNAlevelwassignificantlyassociatedwithcancerstageandmetastasisstatusand thus can have potential clinical applications in screening and monitoring of metastasis andtherapeutictreatments(12;13).Thisoverexpressionmaybeindependentofthegeneamplificationandleadtothechromosomalinstabilityofcancerouscells(14).CyclinsB1andB2aswellasp53havebeenalsoinvolvedinapoptosisanddifferentiationofsymptomaticneurons,oligodendrocytesandPC12cells(15-17).Likewise,BaculoviralInhibitorofapoptosisRepeatContaining5(Birc5)isamultitaskingproteinthathasdual roles inpromotingcellproliferationandpreventingapoptosis,which isoverexpressedduringfetaldevelopmentandinmosttumors.
Neurite outgrowth plays a key role in neuronal development and regeneration, and is the hallmarkassay for the effects of neurotrophic factors such as nerve growth factor (NGF). NGF has beensuggestedtotriggerneuronaldifferentiation,followedbyneuriteoutgrowth(18).However,measuringneuriteoutgrowthisaslowandresource-intensiveprocess.Oeetal(19)suggestedCyclinsB2andBirc5as surrogate biomarkers formeasuringNGF-related neurite outgrowth. Nevertheless, association ofneurotoxicity of anticancer agents with dysregulation of these critical factors has never beenevaluated. Our results showed that downregulation of Cyclins B2 mRNA may predict oxaliplatinneuronalneurotoxicity(6),anditcanbeusedasapredictormarker.Birc5mRNAexpressionwasnotsignificantly different than the controls, thus we did not have enough evidence for or against itspredictivevalue(6).Ouroutcomespromptfuturestudiestotestneurotoxicityofnovelagentsbeforestarting the treatment. Accordingly, before coming to a decisive conclusion whether or not thesefindingscanbeutilizedintopractice,ourresultsneedtobereplicatedinanindependentstudy.
Manytreatmentshavebeentestedinclinictopreventchemotherapy-inducedperipheralneuropathy,none,however,havedemonstratedconsistentefficacywithrespecttosideeffects inclinicaltrialstobe recommended for routine administration (20;21). Calcium and magnesium infusion, selectiveserotonin and serotonin-norepinephrine reuptake inhibitors, vitaminE, etc. are regardedeffective intermsoftargetingproposedmechanismsofchemotherapy-inducedperipheralneuropathy.Theyare,despite lackingrobustevidence,used inclinicalpracticeasoff-labeloptions inordertodecreasetheriskand/orseverityofplatinum-inducedperipheralneuropathy.Somehypothesesareproposed,whichmayexplaintheinconsistencyofdrugefficacy.Theenvironmentisoneofthemajorfactorsthatimpactthecellularpumps,channels,transporters,andisoenzymestosomedegree,andtheyareallinvolvedincontrollingcellularpHandviceversa(22).
Therefore, the pH variability in the nervous system may influence the drug effects in the nervoussystem.Boththe2014CochranereviewandtheguidelineofAmericanSocietyofClinicalOncologydidnot recommendanyof the agents available for thepreventionof chemotherapy-inducedperipheralneuropathy(Table11.2)(20;21).
Hence, a critical appraisal of the fundamental data is needed to reassess and analyze currentknowledgeofthemechanismofdrug-inducedneurotoxicity.Inaddition,furtherbasicresearchisalso
�156
11
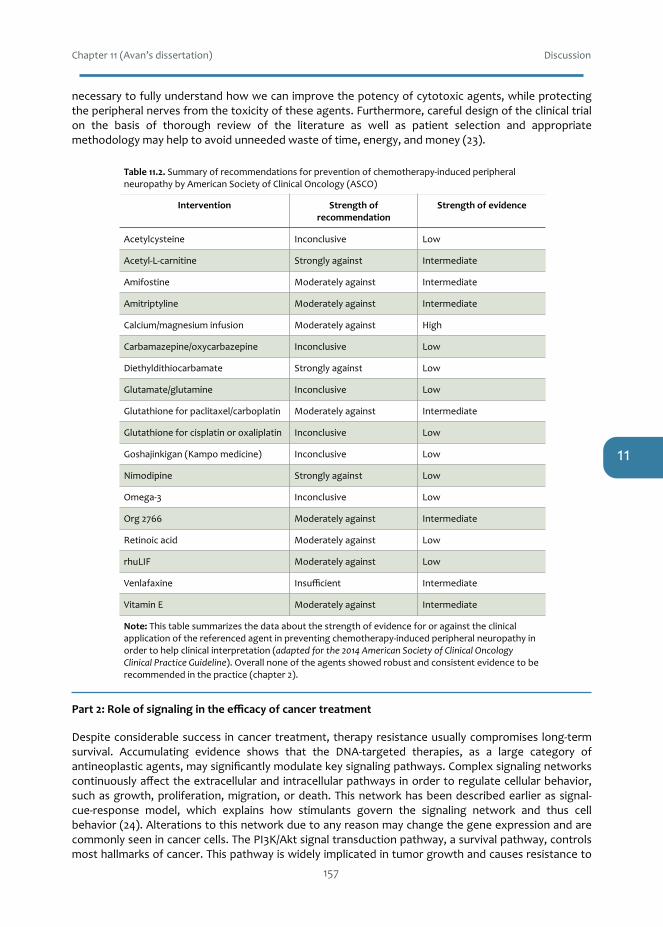
Chapter11(Avan’sdissertation) Discussion
necessarytofullyunderstandhowwecanimprovethepotencyofcytotoxicagents,whileprotectingtheperipheralnervesfromthetoxicityoftheseagents.Furthermore,carefuldesignoftheclinicaltrialon the basis of thorough review of the literature as well as patient selection and appropriatemethodologymayhelptoavoidunneededwasteoftime,energy,andmoney(23).
Part2:Roleofsignalingintheefficacyofcancertreatment
Despiteconsiderablesuccess incancertreatment, therapyresistanceusuallycompromises long-termsurvival. Accumulating evidence shows that the DNA-targeted therapies, as a large category ofantineoplasticagents,maysignificantlymodulatekeysignalingpathways.Complexsignalingnetworkscontinuouslyaffecttheextracellularand intracellularpathways inordertoregulatecellularbehavior,suchasgrowth,proliferation,migration,ordeath.Thisnetworkhasbeendescribedearlierassignal-cue-response model, which explains how stimulants govern the signaling network and thus cellbehavior(24).Alterationstothisnetworkduetoanyreasonmaychangethegeneexpressionandarecommonlyseenincancercells.ThePI3K/Aktsignaltransductionpathway,asurvivalpathway,controlsmosthallmarksofcancer.Thispathwayiswidelyimplicatedintumorgrowthandcausesresistanceto
�157
Table&11.2.&Summary+of+recommendations+for+prevention+of+chemotherapyOinduced+peripheral+neuropathy+by+American+Society+of+Clinical+Oncology+(ASCO)
Intervention Strength&of&recommendation
Strength&of&evidence
Acetylcysteine Inconclusive Low
AcetylOLOcarnitine Strongly+against Intermediate
Amifostine Moderately+against Intermediate
Amitriptyline Moderately+against Intermediate
Calcium/magnesium+infusion Moderately+against High
Carbamazepine/oxycarbazepine Inconclusive Low
Diethyldithiocarbamate Strongly+against Low
Glutamate/glutamine Inconclusive Low
Glutathione+for+paclitaxel/carboplatin Moderately+against Intermediate
Glutathione+for+cisplatin+or+oxaliplatin Inconclusive Low
Goshajinkigan+(Kampo+medicine) Inconclusive Low
Nimodipine Strongly+against Low
OmegaO3 Inconclusive Low
Org+2766 Moderately+against Intermediate
Retinoic+acid Moderately+against Low
rhuLIF Moderately+against Low
Venlafaxine Insufficient Intermediate
Vitamin+E Moderately+against Intermediate
Note:&This+table+summarizes+the+data+about+the+strength+of+evidence+for+or+against+the+clinical+application+of+the+referenced+agent+in+preventing+chemotherapyOinduced+peripheral+neuropathy+in+order+to+help+clinical+interpretation+(adapted&for&the&2014&American&Society&of&Clinical&Oncology&Clinical&Practice&Guideline).+Overall+none+of+the+agents+showed+robust+and+consistent+evidence+to+be+recommended+in+the+practice+(chapter+2).
11

Chapter11(Avan’sdissertation) Discussion
treatment.ActivationoftheAktcascadepromotesmalignantphenotypeandisalsowidelyimplicatedin drug resistance. Therefore,modulation of Akt activity, as one critical pathway, is regarded as anattractivestrategytoenhancetheefficacyofcancertherapyandirradiation.Wereviewed(chapter5)the current evidence on the efficacy of adding a PI3K/Akt inhibitor to the traditionally used DNA-targeted agent for specific cancer types in vitro, in vivo, and in the clinic. However, overall despitesome suggestive evidence in favor of the improving the cytotoxicity of cancer treatment, theheterogeneityofthedatadeteragainstadvisinganycombinationwhichwouldsignificantlybenefittheconventionalanticancerregimens.
Because of the treatment resistance that patients develop against standard platinum drugs, weevaluated the efficacy of satraplatin. Moreover, this compound can be given orally. Our results(Chapter 8) demonstrate that the combination of JM118 (active metabolite of satraplatin) and theepidermalgrowthfactorreceptor(EGFR)inhibitorerlotinib,showedascheduledependentsynergismin a panel of non-small cell lung cancer (NSCLC), ovarian, and colon cancer cell lineswith differentmolecular properties. Simultaneous exposure was least effective in most cell lines, while JM118followedby thecombinationoferlotinibandJM118was themost synergistic schedule,even in cellswithaRasmutation,bywhichtheexpressionofEGFRligandsisstimulatedandleadstodownstreamsignaling through phosphorylated Erk1/2 andAkt (25-27).Moreover, the amount of platinum-adductformation, cell kill, andAkt/Erkphosphorylationhadpositive correlationswith the synergisticeffect.Besides,theefficacyoferlotinibwascell linedependent,possiblyduetotheirdifferentproperties.Inwild type cells, especially with activating mutations, erlotinib has an inhibitory effect onphosphorylationofEGFRanddownstreamproteins[54,55],whilewithEGFR/Rasmutatedgenes,theseeffectsarenotexpected.ItispostulatedthatactivatingEGFRmutationsresultinthedominanceoftheEGFR, rather than other receptors, pathway in survival and thusmore sensitivity to EGFR inhibitors(28).Conversely,cellswithintactEGFRmaydependmainlyonseveralotherreceptorsthatcontrolcellsurvivalthroughactivationofAktandERk1/2pathways(29;30);hence,thecellsbecomemoreresistanttoEGFR inhibitorydrugs.Basedonthesedataandourfindings, theeffectoferlotinib issufficienttoinhibitDNArepairpathwaysascanbeconcludedfromtheincreasedplatinum-DNAadductformationandcellkill.Thiswasbecausethecombinationcould,inparallel,inhibitcellcycleproteinsandsurvivalpathways, which was also reported for oxaliplatin (31). The synergistic effect of the combinationregimenresultedinanincreasedaccumulationofplatinumadductsintheDNA,andtherebyenhancedthe disruption of the cell signaling pathways, which led to growth inhibition. Consequently,preincubationwithJM118followedbytheJM118/erlotinibseemsthemostcytotoxicscheduleforthecombinationoftwooralagents,whichisworthfurtherinvestigations.
IdentificationofgenesthatharborhighlycorrelatedSNPsiscritical,becausetheyusuallyresultinlargechanges ingeneexpression intumorversusnormaltissue.Genome-wideassociationstudies(GWAS)havereformedhumangeneticsandcanpotentiallyimpacthumanhealth(32).BythemeansofGWAS,amyriadoflocihavebeenidentifiedthatfosterriskstratificationtodiseases(33;34),andledtobetterunderstanding of the genetic architecture of complex traits in the hopes ofmoving closer to a fullmolecularcharacterizationofthegeneticcontributionstocancers,asoneareaofinterestforitsusage.However,tominimizethefalsepositiverates,ideallytheresultsareurgedtoaccompanyindependentreplicationandverification(35).Furthermore,additionalanalysis isalwaysneededtounderstandthefunctionalsignificanceoftheidentifiedSNPsinthepost-GWASera(36).
DifferenttagSNPshavebeenfoundtodirectlyorindirectlyassociatewithhigherriskand/orseverityofvaryingcancers(37-41)andthuscompromisethefinaloutcome.Comorbidities,suchascachexiamayalsofurthernegativelyinfluencetheclinicaloutcomeandqualityoflifeofcancerpatients(42-44)andthus correlate with the risk of associated SNPs. Pancreatic ductal adenocarcinoma (PDAC) patientshave the highest risk of developing cachexia, which is a direct cause of reduced quality of life andshortersurvival.Increasedlevelsofcytokines,suchasIL-6,wereassociatedwithweightlossandpoorprognosis in PDAC patients (45-47). A comprehensive pharmacogenetic study demonstrated anassociationofcachexiawiththeSELP-rs6136polymorphisminroughly1800cancerpatients(48).Since
�158
11

Chapter11(Avan’sdissertation) Discussion
apoptosis is reported to take place in wasting muscle in cancer cachexia, several other studiesevaluated the key role of Akt1 in developing cachexia (49-51). Akt1 plays a pivotal role in proteinsynthesis and protein degradation, which contribute to excessive muscular loss in many systemicdiseases (52). Thus, we studied the association of cachexia with the three of the most suggestivefactorsaccording to the literature inahomogeneous settingofpatientswithchemonaïveadvancedpancreaticcancerintwoindependentpopulations.
The resultspresented in chapter9 suggestanassociationbetweencachexiaand theSELP-rs6136-AAandAKT1-rs1130233-AA/GA genotypes in two independent cohorts.We postulate thatAKT1-rs1130233polymorphism may predict the risk of cachexia and death in patients with locally advanced ormetastaticPDAC.Inasystematicstudyongeneticdeterminantsofcachexia(53),SELP-rs6136-AAwastheonly significantly correlatedwith the riskof cachexia among 129polymorphisms in 80genes. Incontrast,despitetheincreasedsusceptibilityoftheIL6-rs1800796-GGgenotypepatientstodevelopingcachexiawithdecreased survival time inpatientswith stage II and III PDAC in aChinesepopulation(54),we could not find a statistically significant correlation. These discrepancies could suggest thatpharmacogeneticassociationsarenotalways reproducible in studieswithpopulationswithdifferentethnic backgrounds and/or different clinical stages. Accordingly, larger studies with homogeneoussettings of patients are essential to investigate the role of emergingbiomarkers beforeplanningofprospectivetrials.
Conclusion
Different tools have been developed to evaluate chemotherapy-induced peripheral neurotoxicity, tomeasureitsseverityandtoinvestigatetheneurotoxiceffectsofdrugs.Biomarkersmayhelptopredicttheriskandseverityofdrug-inducedneurotoxicity.Moreover,thevalidityandreliabilityofthesemarkersneed to be replicated and also carefully studied in pilot clinical setting. Platinum derivatives harmperipheralnervesanddorsalrootganglia,possiblybecauseofprogressiveDNA-adductaccumulationandinhibitionofDNArepairpathways.Therefore,carefulindividualselectionofappropriateneuroprotectiveagents may help to reduce neurotoxicity caused by chemotherapeutic agents and thus improve theprofileofthetreatmentincancerpatients.Inaddition,employingthebestavailableevidenceintheclinicalongwithextensivecollaborationbetweenscientistsandhealthprofessionalsmayhelptoelucidateabreakthroughincancertreatmentthatwouldalsoimprovethepatients’qualityoflife.Besides,preclinicalmodels, surrogatebiomarkers, andpotentially relatedgene studies canbe valuable to investigate thepathogenesisofdrug-related(e.g.neurotoxicity)anddisease-related(e.g.cachexia)sideeffects,and/orpossibly predict the associated risks. Furthermore, searching for the most effective schedule forcombiningnewertargeteddrugswithconventionalregimensiscritical incancermanagement.Inotherwords, simultaneous administration of two potentially effective drugs will not necessarily showsynergism, while preincubation with one of the two can improve the cytotoxicity. Current evidenceprovidesmanyleadsforfurtherinvestigationstooptimizethecytotoxicityofconventionalantineoplasticagents, but still many questions have remained unanswered with regard to pathogenesis ofchemotherapy-inducedperipheralneurotoxicityandroleofsignalinginimprovingtheefficacyofcancertreatment.
References
1. NolanCP,DeAngelisLM.Neurologiccomplicationsofchemotherapyand radiation therapy.Continuum(MinneapMinn)2015April;21(2Neuro-oncology):429-51.
2. Avan A, Postma TJ, Ceresa C, Avan A, Cavaletti G, Giovannetti E et al. Platinum-InducedNeurotoxicity and Preventive Strategies: Past, Present, and Future. Oncologist 2015 April;20(4):411-32.
�159
11

Chapter11(Avan’sdissertation) Discussion
3. ArgyriouAA,CavalettiG,BrianiC,VelascoR,BrunaJ,CampagnoloMetal.Clinicalpatternandassociations of oxaliplatin acute neurotoxicity: a prospective study in 170 patients withcolorectalcancer.Cancer2013January15;119(2):438-44.
4. Diezi M, Buclin T, Kuntzer T. Toxic and drug-induced peripheral neuropathies: updates oncauses,mechanismsandmanagement.CurrOpinNeurol2013October;26(5):481-8.
5. Gregg RW, Molepo JM, Monpetit VJ, Mikael NZ, Redmond D, Gadia M et al. Cisplatinneurotoxicity: the relationship between dosage, time, and platinum concentration inneurologictissues,andmorphologicevidenceoftoxicity.JClinOncol1992May;10(5):795-803.
6. CeresaC,AvanA,GiovannettiE,GeldofAA,AvanA,CavalettiGetal.Characterizationofandprotection from neurotoxicity induced by oxaliplatin, bortezomib and epothilone-B.AnticancerRes2014January;34(1):517-23.
7. Verstappen CC, Postma TJ, Geldof AA, Heimans JJ. Amifostine protects againstchemotherapy-induced neurotoxicity: an in vitro investigation. Anticancer Res 2004 July;24(4):2337-41.
8. MyersCE,McGuireWP,LissRH,IfrimI,GrotzingerK,YoungRC.Adriamycin:theroleoflipidperoxidationincardiactoxicityandtumorresponse.Science1977July8;197(4299):165-7.
9. TreskesM,NijtmansLG,Fichtinger-SchepmanAM,vanderVijghWJ.Effectsofthemodulatingagent WR2721 and its main metabolites on the formation and stability of cisplatin-DNAadducts in vitro in comparison to the effects of thiosulphate and diethyldithiocarbamate.BiochemPharmacol1992March3;43(5):1013-9.
10. DeNeveWJ,EverettCK,SuminskiJE,ValerioteFA.InfluenceofWR2721onDNAcross-linkingbynitrogenmustard innormalmousebonemarrowand leukemia cells in vivo.CancerRes1988November1;48(21):6002-5.
11. Bellanger S, de GA, Sobczak-Thepot J. Cyclin B2 suppresses mitotic failure and DNA re-replicationinhumansomaticcellsknockeddownforbothcyclinsB1andB2.Oncogene2007November8;26(51):7175-84.
12. MoML,ChenZ,LiJ,LiHL,ShengQ,MaHYetal.UseofserumcirculatingCCNB2 incancersurveillance.IntJBiolMarkers2010November23;25(4).
13. TakashimaS,SaitoH,TakahashiN,ImaiK,KudoS,AtariMetal.StrongexpressionofcyclinB2mRNAcorrelateswithapoorprognosis inpatientswithnon-smallcell lungcancer.TumourBiol2014May;35(5):4257-65.
14. Sarafan-VasseurN,LamyA,BourguignonJ,LePF,HieterP,SesboueRetal.OverexpressionofB-typecyclinsalterschromosomalsegregation.Oncogene2002March27;21(13):2051-7.
15. PoratS,SimantovR.Bcl-2andp53: role indopamine-inducedapoptosisanddifferentiation.AnnNYAcadSci1999;893:372-5.
16. ShirvanA,Ziv I,MachlinT,Zilkha-FalbR,MelamedE,BarzilaiA.Twowavesof cyclinBandproliferatingcellnuclearantigenexpressionduringdopamine-triggeredneuronalapoptosis.JNeurochem1997August;69(2):539-49.
17. ShirvanA,ZivI,BarzilaiA,DjaldetiR,Zilkh-FalbR,MichlinTetal.Inductionofmitosis-relatedgenesduringdopamine-triggeredapoptosis insympatheticneurons. JNeuralTransmSuppl1997;50:67-78.
18. EggertA,IkegakiN,LiuX,ChouTT,LeeVM,TrojanowskiJQetal.MoleculardissectionofTrkAsignal transduction pathways mediating differentiation in human neuroblastoma cells.Oncogene2000April13;19(16):2043-51.
19. OeT,NagashimaT,MuramotoM,YamazakiT,MorikawaN,OkitsuOetal.CyclinB2andBIRC5genes as surrogate biomarkers for neurite outgrowth in SH-SY5Y subclonal cells.Neuropharmacology2006June;50(8):1041-7.
20. AlbersJW,ChaudhryV,CavalettiG,DonehowerRC.Interventionsforpreventingneuropathycausedbycisplatinandrelatedcompounds.CochraneDatabaseSystRev2014;3:CD005228.
21. Hershman DL, Lacchetti C, Dworkin RH, Lavoie Smith EM, Bleeker J, Cavaletti G et al.Preventionandmanagementofchemotherapy-inducedperipheralneuropathyinsurvivorsofadult cancers:AmericanSocietyofClinicalOncology clinicalpracticeguideline. JClinOncol2014June20;32(18):1941-67.
�160
11

Chapter11(Avan’sdissertation) Discussion
22. CarlinK.ThenervoussystemandpH.OpenJournalofInternalMedicine2013;3:126-8. 23. AvanA,AvanA,GiovannettiE,PetersGJ.Calcium/magnesiuminfusionforoxaliplatin-induced
neuropathy:protectiveornot?JClinOncol2014October10;32(29):3341. 24. PawsonT,LindingR.Networkmedicine.FEBSLett2008April9;582(8):1266-70. 25. Toulany M, Minjgee M, Saki M, Holler M, Meier F, Eicheler W et al. ERK2-dependent
reactivation of Akt mediates the limited response of tumor cells with constitutive K-RASactivitytoPI3Kinhibition.CancerBiolTher2014March1;15(3):317-28.
26. Toulany M, Baumann M, Rodemann HP. Stimulated PI3K-AKT signaling mediated throughligandor radiation-inducedEGFRdepends indirectly, butnotdirectly, on constitutiveK-Rasactivity.MolCancerRes2007August;5(8):863-72.
27. Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP. Radioresistance of K-Rasmutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKTpathway.RadiotherOncol2005August;76(2):143-50.
28. OnoM,HirataA,KometaniT,MiyagawaM,UedaS,KinoshitaHetal.Sensitivitytogefitinib(Iressa, ZD1839) in non-small cell lung cancer cell lines correlateswith dependence on theepidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGFreceptor/Aktpathwayforproliferation.MolCancerTher2004April;3(4):465-72.
29. Rolfo C, Giovannetti E, Hong DS, Bivona T, Raez LE, Bronte G et al. Novel therapeuticstrategies forpatientswithNSCLC thatdonot respond to treatmentwithEGFR inhibitors.CancerTreatRev2014September;40(8):990-1004.
30. GiovannettiE,LabotsM,DekkerH,GalvaniE,LindJS,SciarrilloRetal.Molecularmechanismsand modulation of key pathways underlying the synergistic interaction of sorafenib witherlotinibinnon-small-cell-lungcancer(NSCLC)cells.CurrPharmDes2013;19(5):927-39.
31. NoordhuisP,LaanAC,vandeBornK,LosekootN,KathmannI,PetersGJ.Oxaliplatinactivityin selected and unselected human ovarian and colorectal cancer cell lines. BiochemPharmacol2008July1;76(1):53-61.
32. Barsh GS, Copenhaver GP, Gibson G,Williams SM. Guidelines for genome-wide associationstudies.PLoSGenet2012July;8(7):e1002812.
33. Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS et al. Potentialetiologicandfunctionalimplicationsofgenome-wideassociationlociforhumandiseasesandtraits.ProcNatlAcadSciUSA2009June9;106(23):9362-7.
34. ManolioTA.Genomewideassociationstudiesandassessmentoftheriskofdisease.NEnglJMed2010July8;363(2):166-76.
35. ThompsonJR,AttiaJ,MinelliC.Themeta-analysisofgenome-wideassociationstudies.BriefBioinform2011May;12(3):259-69.
36. SchaubMA,BoyleAP,KundajeA,BatzoglouS,SnyderM.Linkingdiseaseassociationswithregulatoryinformationinthehumangenome.GenomeRes2012September;22(9):1748-59.
37. YaoL,TakYG,BermanBP,FarnhamPJ.FunctionalannotationofcoloncancerriskSNPs.NatCommun2014;5:5114.
38. Garcia-ClosasM,ChanockS.Geneticsusceptibilitylociforbreastcancerbyestrogenreceptorstatus.ClinCancerRes2008December15;14(24):8000-9.
39. Roberts NJ, Klein AP. Genome-wide sequencing to identify the cause of hereditary cancersyndromes: with examples from familial pancreatic cancer. Cancer Lett 2013 November1;340(2):227-33.
40. Brennan P, Hainaut P, Boffetta P. Genetics of lung-cancer susceptibility. Lancet Oncol 2011April;12(4):399-408.
41. Li Q, Yang J, Yu Q, Wu H, Liu B, Xiong H et al. Associations between single-nucleotidepolymorphisms inthePI3K-PTEN-AKT-mTORpathwayand increasedriskofbrainmetastasisinpatientswithnon-smallcelllungcancer.ClinCancerRes2013November15;19(22):6252-60.
42. LasheenW,WalshD.Thecanceranorexia-cachexiasyndrome:mythorreality?SupportCareCancer2010February;18(2):265-72.
�161
11

Chapter11(Avan’sdissertation) Discussion
43. YangR,CheungMC,PedrosoFE,ByrneMM,KoniarisLG,ZimmersTA.Obesityandweightlossat presentationof lung cancer are associatedwithopposite effectson survival. J SurgRes2011September;170(1):e75-e83.
44. PauschT,HartwigW,HinzU,SwolanaT,BundyBD,HackertTetal.Cachexiabutnotobesityworsens the postoperative outcome after pancreatoduodenectomy in pancreatic cancer.Surgery2012September;152(3Suppl1):S81-S88.
45. ArgilesJM,BusquetsS,Lopez-SorianoFJ.Cytokinesinthepathogenesisofcancercachexia.CurrOpinClinNutrMetabCare2003July;6(4):401-6.
46. EbrahimiB, Tucker SL, LiD,Abbruzzese JL,KurzrockR. Cytokines inpancreatic carcinoma:correlationwithphenotypiccharacteristicsandprognosis.Cancer2004December15;101(12):2727-36.
47. Martignoni ME, Kunze P, Hildebrandt W, Kunzli B, Berberat P, Giese T et al. Role ofmononuclear cells and inflammatory cytokines in pancreatic cancer-related cachexia. ClinCancerRes2005August15;11(16):5802-8.
48. Tan BH, Fladvad T, Braun TP, Vigano A, Strasser F, Deans DA et al. P-selectin genotype isassociatedwiththedevelopmentofcancercachexia.EMBOMolMed2012June;4(6):462-71.
49. SchmittTL,MartignoniME,BachmannJ,FechtnerK,FriessH,KinscherfRetal.ActivityoftheAkt-dependentanabolicandcatabolicpathwaysinmuscleandliversamplesincancer-relatedcachexia.JMolMed(Berl)2007June;85(6):647-54.
50. BodineSC,StittTN,GonzalezM,KlineWO,StoverGL,BauerleinRetal.Akt/mTORpathwayisa crucial regulatorof skeletalmusclehypertrophy and canpreventmuscle atrophy in vivo.NatCellBiol2001November;3(11):1014-9.
51. SugitaH,KanekiM,SugitaM,YasukawaT,YasuharaS,MartynJA.Burninjuryimpairsinsulin-stimulatedAkt/PKBactivationinskeletalmuscle.AmJPhysiolEndocrinolMetab2005March;288(3):E585-E591.
52. BonaldoP,SandriM.Cellularandmolecularmechanismsofmuscleatrophy.DisModelMech2013January;6(1):25-39.
53. Tan BH, Ross JA, Kaasa S, Skorpen F, Fearon KC. Identification of possible geneticpolymorphisms involved in cancer cachexia: a systematic review. J Genet 2011 April;90(1):165-77.
54. ZhangD,ZhouY,WuL,WangS,ZhengH,YuBetal.AssociationofIL-6genepolymorphismswithcachexiasusceptibilityandsurvivaltimeofpatientswithpancreaticcancer.AnnClinLabSci2008;38(2):113-9.
�162
11

CHAPTER12
Summary,CurriculumVitae,
Acknowledgements
English,Dutch,Persian
12

Chapter12(Avan’sdissertation) Summary
Summary
Neuropathy and resistance to the treatment are two main burdensome consequences ofchemotherapy. Neurotoxicity is the most common dose-limiting side effect of platinumcompounds,whichoften leads to treatmentwithdrawal, affects thepatients’ qualityof life, andsometimesis irreversible.Chapter1shortlyreviewsthehistoryoftheconventionalandemergingplatinum-baseddrugs,andthecharacteristicsofthecommonplatinumagentsincancertreatment.Moreindepth,chapter2discussesavailableevidenceonthepathogenesisandpathophysiologyofplatinum-inducedperipheralneurotoxicityanddiscussesthecharacteristicsandtoolsforitstimelydiagnosis. This chapter also summarizes available evidence on neuroprotective and therapeuticstrategies, which indicates the heterogeneity and inconsistency of the data. Accordingly, theevidence does not clearly support the advantage of any of the tested agents to be utilized foraffectedpatientswithrespecttotheirsafetyandefficacy.
Identificationandquantificationofadverseoutcomesassociatedwithexposurestochemotherapyagents, however, has remained widely undetermined, and is still under investigation, andnumerouschallengesstillexistintranslatingbiomarkerresearchintotheclinicalpractice.Differentmodelshavebeenintroducedtostudychemotherapy-inducedneurotoxicity.TheresultsinChapter3 supports the importance of the neurite outgrowth in PC12 rat pheochromocytoma cells forevaluation of the neurotoxicity. With this method, we demonstrated the neurotoxicity ofoxaliplatin, bortezomib, and epothilone-B, and investigated the neuroprotective effect ofamifostine.Besides,wefoundthatupregulationofcyclin-B2mRNAwasassociatedwithneuronaldifferentiation and thus oxaliplatin-induced neurotoxicity. Hence, cyclin-B2 can be used as apredictivemarker.
Calciumandmagnesiuminfusionisoneofthecommonlyusedoff-labelstrategiesbyoncologistsinthe hope of preventing or at least, to some extent reducing the severity of oxaliplatin-inducedperipheralneurotoxicity.However,consistentevidenceislackingforthismethod,andrecentdatadidnotsupporttheefficacyofcalcium/magnesiuminfusioninpreventingthissideeffect.Chapter4shortlydiscussesthismethodandsometoolstomeasureitsefficacy,aswellastheimportanceofemployingsuitablemethodologyinanalysistheresults.
Activation of the Akt-survival pathway is a mechanism of resistance to DNA-targeted drugs.Chapter5 reviews theeffectof commonDNA-targetedanticancerdrugsonAktpathway, to seewhether antagonizing the Akt survival pathway acts synergistically with the conventionaltreatments. Available data suggest that the mechanism of drug resistance is complex andcomprises difference pathways. There is evidence in favor to improve the cytotoxicity of cancertreatment.However, the available preclinical data is so heterogeneous (varyingwith drugs) andclinicalevidenceislimited.
ThereisevidenceofaconstitutiveactivationofAkt inpancreaticcancer.Chapter6describesthetherapeutic potential of the novel Akt inhibitor perifosine in combination with gemcitabine inpancreatic ductal adenocarcinoma (PDAC) cells. Perifosine could effectively interfere with cellproliferation, induce apoptosis, reduce migration/invasion, and synergistically interact withgemcitabineincellswithphospho-Aktoverexpression.
Increasing the formation and retention of platinum-DNA adducts by decreasing the DNA-repairenzymes may boost the antineoplastic effects of the treatment and overall survival. Chapter 7explorestheimportanceofprotein/mRNAexpression-analysis,aswellastheroleofpolymorphismintheDNArepairenzymesinnon-smallcelllungcancerandPDAC.
Satraplatin is the only oral analog among platinum agents, which in case of success, maysignificantlydecreasetheburdenofparenteralcancertherapy.Chapter8discussesourresultswith
�164
12

Chapter12(Avan’sdissertation) Summary
regardtothepotentialsynergismbetweenerlotinib,anepidermalgrowthfactorreceptorinhibitor,andJM118,theactivemetaboliteofsatraplatin.ThesynergisticeffectofthecombinationregimenresultedinanincreasedaccumulationofplatinumadductsintheDNA,andtherebyenhancedthedisruptionofthecellsignalingpathways,whichledtogrowthinhibition.Consequently,wefoundthatpreincubationwithJM118followedbytheJM118/erlotinibcouldimprovetheprofileofthetwooralagents.
Singlenucleotidepolymorphismsmaybe indicativeof a shorter survival.Chapter 9 explores theroleof novel predictivebiomarkers for cachexia, as a direct causeof reducedquality of life andshortersurvival,inPDAC.InlocallyadvancedandmetastaticPDAC,AKT1-rs1130233andSELP-rs6136polymorphismswereassociatedwiththeriskofdevelopingcachexia.Moreover,AKT1-rs1130233-AA/GAgenotypesweresignificantpredictorsforshortersurvival,withanincreasedriskofdeathintwoindependentcohorts.WealsofoundacorrelationwithreducedphosphorylationofAkt1inmusclebiopsies frompatients harboring these particular Single nucleotide polymorphisms. Thus, itmayplayaprognosticrole,whichneedstobereplicatedinanotherindependentstudybeforeutilizingitintotheclinic.
Singlenucleotidepolymorphismsmaypredictbrainmetastasesinnon-small-celllungcancer.Chapter10discusses some controversies between our findings and some recent data in the role of singlenucleotidepolymorphismsofAktinpredictingbrainmetastasesinnon-small-celllungcancer.
Chapter11discussestheresultspresentedinthepresentthesiscomparedwithothersintheliterature.Currentevidenceprovidesmanyleadsforfurtherinvestigationstooptimizethecytotoxicityofconventionalantineoplasticagents,butstillmanyquestionsremainunansweredwithregardtothepathogenesisofchemotherapy-inducedperipheralneurotoxicityandroleofsignalinginimprovingtheefficacyofcancertreatment,whichwarrantsmorestudies.
�165
12

Chapter12(Avan’sdissertation) Summary
Samenvatting
Neurotoxiciteit en resistentie tegen een behandeling zijn twee mogelijke complicaties bij debehandeling van patiënten met kanker. Neurotoxiciteit is een van de meest voorkomendebijwerkingen van platinaverbindingen. Dit leidt vaak tot het stoppen van de behandeling en eenafname in de kwaliteit van leven.De toxiciteit is somsonomkeerbaar.Hoofdstuk 1geeft een kortoverzicht van platinaverbindingen, waaronder zowel het klassieke cisplatin als nieuweplatinaverbindingen,enhunrolindebehandelingvankanker.Hoofdstuk2geefteenoverzichtvandebeschikbare informatie over hoe platinaverbindingen neurotoxiciteit veroorzaken en wat dekarakteristieken van deze toxiciteit zijn. Dit is van belang bij het stellen van de diagnose. In dithoofdstuk worden naast de behandelingsmogelijkheden ook de verschillende strategieën omneurotoxiciteittegentegaansamengevat.Eriseengroteheterogeniteitinneurotoxiciteit,terwijldebeschrijving van deze toxiciteit vaak inconsistent is. Dat maakt het ook moeilijk om beschikbaremiddelenbetreffendebijwerkingeneneffectiviteitgoedtekunnenbeoordelen.
Het identificeren en kwantificeren van neurotoxiciteit van chemotherapie is nog steeds niet goedgevalideerd, zodat er nog veel uitdagingen zijn om biomarkers te vertalen naar de kliniek. Er zijnverschillendemodelsystemenbeschreven. InHoofdstuk 3wordende voordelen van de neurieten-uitgroeimethodebeschreven.Metditmodelsysteemisdeneurotoxiciteitvanoxaliplatin,bortezomibenepothiloneBonderzocht.Ookhetomkeren/beschermentegenneurotoxiciteitdooramifostineisnaderbekeken.Alseenalternatievemarkervoorneuronaledifferentiatie isdeverhogingvancyclinB2mRNAexpressiebestudeerd.
Calcium en magnesium infusen worden vaak off-label (oneigenlijk) gebruikt om perifereneurotoxiciteittevoorkomenofteverminderen.Hetprobleemhierbijisdatderesultatentotnutoenietovertuigendzijn.Dezemethodewordt inHoofdstuk4beschreven,waarbijookhetbelangvangoedeassaysbenadruktwordt.
Activering van de Akt overlevingsroute is een resistentie mechanisme tegen DNA gerichtegeneesmiddelen.InHoofdstuk5wordendeeffectenvaneenaantalvandegeneesmiddelen(platinaanaloga, gemcitabine, taxanen, antifolaten) op deze route beschreven. De hypothese is dat deeffectiviteitvanDNA-gerichtemiddelenversterktkanwordendoordeAKTrouteteremmen.
Hoofdstuk6beschrijftderolvanAKTactivatieinresistentievanalvleesklierkankertegenstandaardbehandeling met gemcitabine. Verder is de potentie van perifosine, een AKT antagonist, incombinatie met gemcitabine onderzocht. Perifosine remde de cellulaire proliferatie, induceerdeapoptoseen remdedemigratie/invasieenvan tumorcellen. Incellenmeteenhogeexpressievanfosfor-AKTwasperifosinesynergistischmetgemcitabine.
De vorming van platina-DNA adducten is cruciaal voor de antitumor activiteit van het antikankermiddel cisplatin. Deze adducten kunnen hersteldworden doorDNA repair enzymen,waardoor deeffectiviteit van cisplatin verminderdwordt. Een lage activiteit van deze enzymen zal dan ook deaanwezigheidvandezeadductenindetumorcellenverlengenendaarbijdeeffectiviteitvancisplatinverhogen.InHoofdstuk7beschrijvenwehetbelangvandeeiwitenRNAexpressievanDNA-repairenzymen.Ookwerdenpolymorfismes inDNArepairenzymen in longkankerenalvleesklierkankeronderzocht.
Satraplatin is een nieuw platina analoog wat oraal gegeven kan worden. Hoofdstuk 8 beschrijftsynergie van JM118, de actieve metaboliet van satraplatin, met erlotinib, een remmer van deepidermalegroei factor receptor. IndecombinatiewerdeenhogereconcentratievanDNAplatinaadducten gevonden en een verstoorde regulatie van signaaltransductieroutes. Dit leidde tot een
�166
12

Chapter12(Avan’sdissertation) Summary
verhoogdegroeiremming.VerdervondenwedateenvoorbehandelingmetJM118gevolgddoordecombinatieJM118/erlotinibhetbestegroeiremmendeeffecthad.
Singlenucleotidepolymorfismes(SNP’s)zijnveranderingenineenbepaaldebaseinhetDNA.SNP’skunnen het effect van een behandeling voorspellen. In Hoofstuk 9 beschrijven we de rol vanbepaaldeSNP’sindeaanwezigheidvancachexie,eenverstervingvannormaalweefsel,waardooreenpatiëntsnelgewichtverliest,dienskwaliteitvanlevenachteruitgaatensnelleroverlijdt.Cachexieiseenvandekenmerkenvanalvleesklierkanker;deSNP’sinhetAKT1gen(rs1130233)enhetSELPgen(rs6136)blekengerelateerdtezijnaanhetontwikkelenvancachexie.HetAKT1gen(rs1130233)AA-CAgenotypehadintweeapartegroepenpatiënteneensignificanterelatiemeteenverkorteoverlevingenverhoogdrisicoopoverlijden.WevondenookeencorrelatietussendezeSNP’seneenverlaagdefosforylering van AKT1 in spierbiopten van patiënten. SNP’s hebben wellicht een voorspellendewaarde,maarditdienteerstineengroterestudieaangetoondteworden.
SNP’s kunnen ook voorspellen of patiënten met longkanker hersenmetastases ontwikkelen. InHoofdstuk10wordtderolvanbepaaldeSNP’sinhetAKTgenindevormingvanhersenmetastasesvanlongkankerbesproken.
InHoofdstuk11wordenderesultatenuitditproefschriftvergelekenmetdie indeliteratuur.Erzijnvelemogelijkhedenomdeeffectiviteitvanconventionelecytotoxischemiddelenteverbeteren.Erisechter nog steeds veel onduidelijkheid over het ontstaan van perifere neuropathie en bij hoeveelpatiëntendittotcomplicatiesleidt.Ookdevraaghoehetbeïnvloedenvansignaaltransductieroutesdeeffectiviteitvantherapiekanverbeteren,moetverderonderzochtworden.
�167
12

Chapter12(Avan’sdissertation) Summary
PersianSummary:
عللسميتبرخىازداروهاىضدسرطانبرروىعصبهاىمحيطىونقشمسيرهاىعملكردىسلولىدر
اثربخشىداروهاىضدسرطان
(بKيمارىيKااخKتالليKكيKاچKندعKصبمKحيطىكKهمنجKربKهبKىحKسىيKامKورمKورعKضودرگKيرمKىشKود)ومKقاومKتبKه ١خHالصHه:نKوروپKاتKى
(آسKيبعKصببKهدلKيليKكيKاچKندعKامKلسKمى)،شKايKعتKريKن ٢درمKاندوعKارضKهمKهمدرشKيمىدرمKانKىسKرطKانهKاسKت.نKوروتKوكKسيسيتى
اسKKتكKKهنKKيازبKKهتKKعديKKلدوزيKKاقKKطعدرمKKانداردوبKKهشKKدتكKKيفيتزنKKدگKKىبKKيمارانراتKKحتتKKأثKKيرقKKرار ٣عKKارضKKهداروهKKاىبKKاتKKركKKيبپKKالتKKني
مىدهدكهدربرخىمواردممكناستاينعارضهقابلبرگشتنباشد.
فHصلاولايKنپKايKاننKامKهبKطورخKالصKهتKاريKخچهوويKژگKىهKاىداروهKاىقKديKمىوجKديKدتKرداروهKاىبKاتKركKيبپKالتKنيرامKرورمKىكKند.
فHصلدومبKهطKورجKامKعشKواهKدعKلمىمKوجKودرادرزمKينهعKلتومKكانKيسمآسKيبعKصبهKاىمKحيطىبKهدلKيلپKالتKنيرامKوردبKحثقKرار
دادهوويKKژگKKىهKKاىاخKKتصاصKKىبKKالKKينىوروشهKKاىمKKورداسKKتفادهبKKراىتKKشخيصبKKهمKKوقKKعآنرانKKيزمKKرورمKKىكKKند.ايKKنفKKصلهKKمچننيشKKواهKKدبKKالKKينىمKKوجKKوددرمKKوردروشهKKاىپKKيشگيرىودرمKKانKKىايKKنعKKارضKKهشKKايKKعرابKKطورخKKالصKKهبKKررسKKىمKKىكKKندكKKهگKKويKKاىتKKعددواخKتالفنKتايKجدرمKطالKعاتمKختلفاسKت.درنKتيجه،درحKالحKاضKرشKواهKدعKلمىمسKتندوكKافKىبKراىتKايKيدهKيچكKدامازايKناقKدامKات
پيشگيرىكنندهموجودنيستتابتواناستفادهازآنهارابطوررايجدرموردايناختاللتوصيهكرد.
هKKرچKKنداسKKتفادهبKKالKKينىازيKKافKKتههKKاىپKKاراكKKلينيكىوبKKيومKKاركKKرهKKاىنKKویKKنمKKرتKKبطبKKااخKKتالليKKابKKيمارىمKKوردبKKررسKKىبKKسيارمKKشكلاسKKت؛تKKشخيصوانKKدازهگKKيرىعKKوارضمKKرتKKبطبKKاداروهKKاىشKKيمىدرمKKانKKىيKKكىازمKKواردمKKهمدرحKKالتKKحقيقوپKKژوهKKشاسKKت.مKKدلهKKاىزيKKادى
بKراىمKطالKعهسKمیتداروهKاىشKيمىدرمKانKىبKررویعKصبهKامKعرفKىشKدهاسKت.درفHصلسHومكKتابنKتايKجمKوفKقيتآمKيزاسKتفادهاز
،بKورتKزومKاب درارزيKابKىايKنعKارضKهارائKهشKدهكKهدرآنبKخوبKىتKأثKيرداروهKاىاگKزالKيپالتKني ٦سKلولهKاىبKافKتفKئوكKرومKوسKايKتومKاىمKوش ٥ ٤
،بKKKهعKKنوانيKKكانKKتخاب بKKرروىسKKلولهKKایعKKصبینKKشاندادهشKKدهاسKKتوهKKمينطوردرايKKنمKKدل،داروىآمKKيفوسKKتني ٨واپKKوتKKايKKلون-بKKى ٧
پKيشگيرى-درمKانKىبKاعKثكKاهKشايKنسKميتاگKزالKيپالتKنيشKد.عKالوهبKرايKن،درايKنتKحقيقنKشاندادهشKدكKهافKزايKشبKيانژنآراناى
بKKابKKلوغسKKلولعKKصبىارتKKباطداشKKتهودرنKKتيجهاحKKتماالمKKىتKKوانKKدشKKانKKسسKKميتاگKKزالKKيپالتKKنيبKKرروىسKKلول ٩پKKيغامKKبرسKKایKKکلني-بKKى-٢
عصبیراپيشبينىكند.
تKKزريKKقتKKركKKيبكKKلسيم-مKKنيزيKKميKKكىازروشهKKاىمKKوردتKKوجKKهمKKتخصصنيخKKونوسKKرطKKانشKKناسKKىبKKودهكKKهبKKهجهKKتپKKيشگيرىويKKاكKKاهKKش
شKدتاثKرسKوءداروىاگKزالKيپالتKنيبKرروىعKصبمKورداسKتفادهقKرارمKىگKرفKتهاسKت؛هKرچKندنKتايKجتKحقيقهKاهKمراسKتابKاهKمنKبودهانKدو
مKKطالKKعهاخKKيرنKKيزاثKKربKKخشىآنرازيKKرسKKوالبKKردهاسKKت.فHصلچHهارمكKKتاببKKطورخKKالصKKهايKKنروشدرمKKانKKى،ارزيKKابKKىاثKKربKKخشىآنو
اهميتاستفادهازروشمناسبدرآناليزيافتههاراموردبحثقرارمىدهد.
كKKهدرحKKياتسKKلولهKKانKKقشدارد،بKKهعKKنوانيKKكىازمKKكانKKيسمهKKاىمKKقاومKKتبKKهداروهKKاىضKKدسKKرطKKانبKKا ١٠افKKزايKKشفKKعالKKيتمKKسيراكKKت
مKKكانKKيسممKKهارتKKكثيرژنKKىشKKناخKKتهشKKدهاسKKت.فHصلپنجHمكKKاركKKردايKKندسKKتهازداروهKKاىپKKرمKKصرفضKKدسKKرطKKانرامKKرورمKKىكKKند،و
اينكهبراساسشواهدموجودآيامهاراينمسيرمىتوانداثردرمانرابيشتركندياخيرموردبحثقرارگرفتهاست.هرچنداينمسيرتKنهامKسيركKنترلكKنندهمKقاومKتدارويKىنيسKتوبKاوجKودايKنكهمKمكناسKتاثKربKخشىبKرخKىازداروهKاراافKزايKشدهKد،دربKيمارانمKوردمKطالKعهنKتايKجمKختلفىبKهدسKتآمKدهودرنKتيجهنKمىتKوانبKهيKكنKتيجهگKيرىوتKوصKيهواحKدبKراىاسKتفادهازايKنمKهاركKنندههKادركKنار
داروهاىقديمىرسيد.
neuropathy١
neurotoxicity٢
platinum٣
PC12ratpheochromocytomacells٤
oxaliplatin٥
bortezomib٦
epothylone-B٧
amiphostine٨
mRNAcyclin-B2٩
Akt-signalingpathway١٠
�168
12

Chapter12(Avan’sdissertation) Summary
بKKهعKKنKوانمKKهKاركKKنKنKده ١١بKKاتKKوجKKهبKKهشKKواهKKدافKKزايKKشفKKعKالKKيKتمKKسKيKراكKKتدرسKKرطKKانپKKانKKكKراس،درفHHصHلشHHشHم،پKKتKانKKسKيKلدرمKKانKKىپKKريKKفKوزيKKن
مKKوردبKKررسKKىوآزمKKايKKشقKKرارگKKرفKKتKهاسKKت.نKKتKايKKجحKKاصKKلازايKKنتKKحKقKيKقنKKشKاندادپKKريKKفKوزيKKن ١٢مKKسKيKراكKKتدركKKنKارداروىجKKمKسKايKKتKوبKKني
راتKحKKریKKکوتKKهKاجKKمسKKلKولKKىراكKKاهKKشدهKKد؛وهKKمKنيطKKوربKKاعKKث ١٣مKKىتKKوانKKدجKKلKوىتKKكKثKيKرسKKلKولKKىراگKKرفKKتKه،مKKرگبKKرنKKامKKهريKKزىشKKدهسKKلKول
گKKردد.عKKالوهبKKرمKKكKانKKيKسKمفKKوق،سKKركKKوبمKKسKيKرتKKرمKKيKم ١٤افKKزايKKشاثKKرضKKدسKKرطKKانKKيجKKمKسKايKKتKوبKKنيدرسKKلKولهKKاىبKKاافKKزايKKشفKKسKفKو-اكKKت
نKKيKزمKKىتKKوانKKداثKKربKKخKشKىدرمKKانهKKاىضKKدسKKرطKKانوطKKولعKKمKربKKيKمKارانراافKKزايKKش ١٥ژنKKىبKKاافKKزايKKشتKKشKكKيKلكKKمKپKلKكKسپKKالتKKني-دىاناى
دهKKدکKKهدرفHHصHلهHHفHتHممKKوردمKKطKالKKعKهقKKرارگKKرفKKت؛واهKKمKيKتبKKيKانپKKروتKKئKني،بKKرخKKىژنهKKاواخKKتKالالتژنKKىدرآنKKزيKKمهKKاىتKKرمKKيKمكKKنKنKدهژن
درسKرطKانريKهوپKانKكراسبKررسKىگKردیKد.
تKKنهاداروىخKKوراكKKىازدسKKتهداروهKKاىبKKاسKKاخKKتارپKKالتKKنياسKKتكKKهدرصKKورتتKKايKKيداثKKربKKخشىمKKناسKKبوعKKوارضكKKم، ١٦سKKاتKKراپKKالتKKني
مKىتKوانKددرمKقايKسهبKاداروهKاىتKزريKقىمKتداولجKايKگزيKنمKناسKبىدردرمKانبKسياريازسKرطKانهKابKاشKد.درتKحقيقاتارايKهشKدهدر
،دركKنارسKاتKراپKالتKنياثKرضKدسKرطKانKيبهKترى ،مKهاركKنندهگKيرنKدهغKشايKىفKاكKتوررشKد ١٩فHصلهشHتمنKشاندادهشKدكKهارلKوتKينيب ١٨ ١٧
داردكهمكانيسمآننيزموردمطالعهقرارگرفت.
وجKودجKهشهKاىنKقطهاىخKاصدربKرخKىژنهKامKمكناسKتبKاطKولعKمركKمتربKيمارانسKرطKانKىدرارتKباطبKاشKد.درفHصلنHهم،نKقش
(الغKرىهKمراهبKاتحKليلعKضالنKى)كKهبKاطKولعKمركKمتربKيمارانسKرطKانپKانKكراسدر ٢٠بKرخKىازفKاكKتورهKاىپKيشبKينىكKنندهكKاشKكسى
ارتKباطهسKتدردومKطالKعهمسKتقلمKوردمKطالKعهقKرارگKرفKت.بKهطKورمKشابKه،وجKودجKهشهKاىنKقطهاىخKاصدرسKرطKانريKهمKمكناسKت
شانسانتشاربهمغزراپيشبينىكندكهبطورخالصهدرفصلدهمموردبحثقرارگرفتهاند.
درفKKصليKKازدهKKم،مجKKموعنKKتايKKجايKKنتKKحقيقاتمKKوردبKKحثقKKرارگKKرفKKتهاسKKت.بKKاوجKKودپيشKKرفKKتهKKاىمKKختلفدردرمKKانانKKواعسKKرطKKانهKKاونتايجحاصلازطرحهاىتحقيقاتىفوق،همچناننادانستههاىماازدانستههاىمادرزمینهسرطانبيشتراستوسؤاالتبسياريدرمKوردعKوارضعKصبىومKقاومKتدارويKىدرمKانهKاىضKدسKرطKانبKىپKاسKخمKانKدهاسKتكKهنKيازمKندتKحقيقاتبيشKترودقKيقتKردرايKن
زمينهاست.
perifosine١١
gemcitobine١٢
apoptosis١٣
phospho-Akt١٤
platinum-DNAadducts١٥
satraplatin١٦
erlotinib١٧
epidermalgrowthfactorreceptorinhibitor١٨
synergisticeffect١٩
cachexia٢٠
�169
12

Chapter12(Avan’sdissertation) CV
CURRICULUMVITAE AbolfazlAvan
Abolfazl Avan was born on the eighteenth of February 1982 inMashhad,Iran.HestudiedatAzarmaneshPrimarySchoolfrom1988to1993 (5 years), Din-o-Danesh Secondary School from 1993 to 1996 (3 years), Allameh
High School from 1996 to 1999 (3 years), and Sama Colleague from 1999 to 2000 (1year). He has been the top first or second student during his 12-year preuniversityeducationperiodwithgradepointaverageofhigherthan19outof20.Hewasrankedamongthetop0.1%candidates intheNationalUniversityEntranceExamandgrantedthe 7-year medical scholarship. He studied medicine at Tabriz University of Medical
Sciencesfrom2000to2006anddidhisoneandahalfyearsclinicalinternshipaswellashis medical doctorate thesis on malignant mesothelioma at Mashhad University ofMedicalSciences from2007 to2009.Abolfazlearnedhismedicaldoctorate fromthesixth top national university in November 2009. Later, heworked at the EmergencyDepartmentforabouttwoyearsthereafter.Besides,hehasbeenactivelycooperating
with neurologists and gastroenterologists, particularly in treating Wilson’s diseasepatientssince2009.Healsohaddifferentpresentationsatmajornationaluniversitiesand an international congress. Abolfazl has made an international network withworldwide seniors,which led to receiving the FreeCopperPrize in 2012, and agrantfromtheMovementDisorderSocietyin2013.Afterabout4yearsofclinicalpracticeand
research,hemovedtotheNetherlandstodoaPhDonpathogenesisofneurotoxicityandroleofsignalingintheefficacyofanticanceragentsatVUUniversityMedicalCenterinAmsterdamunderthesupervisionofProf.Dr.GodefridusJ.Peters,PhDandDr.ElisaGiovannetti,MD, PhD. In July 2015, hewas invited to collaboratewithDr.MahmoudRezaAzarpazhoohandaCanadianteamonasystematicreviewonneuropathologyof
dementia under the supervision of Prof. Vladimir Hachinski, MD at the WesternUniversityinLondon,Ontario,Canada.Furthermore,hejoinedaclinicalprojectoneyemovementdisordersinParkinson’sdiseaseandessentialtremorunderthesupervisionof Dr. Anne-Fleur van Rootselaar, MD, PhD at the Department of Neurology in theAcademicMedicalCenteroftheUniversityofAmsterdam.Heisalsoajuniormemberof
the American Academy of Neurology and the International Society of Parkinson’sdiseaseandMovementDisorders.
• DepartmentofNeurologyandNeurophysiology,AcademicMedicalCenter,UniversityofAmsterdam,Amsterdam,theNetherlands.July2015-Now
EyemovementsdisordersinParkinson’sdiseaseandessentialtremor(Supervisor:Dr.A.F.vanRootselaar,MD,PhD)
• CancerCenterAmsterdam,DepartmentofMedicalOncology,VUUniversity
MedicalCenter,Amsterdam,theNetherlands.2013-Now• CollaborationwiththeDepartmentsofNeurology,Gastroenterology,andPediatric
Gastroenterology,GhaemHospital,MashhadUniversityofMedicalSciences,Mashhad,Iran.(Clinician,treatingWilson’sdiseasepatients)2009-2013
�170
Experiences
Profile
12

Chapter12(Avan’sdissertation) CV
• EmergencyDepartment,Dr.ChamranHospital,Ferdows,BirjandUniversityofMedicalSciences.(EmergencyRoomClinician)2010-2011
• Generalmedicineandclinicaltraineeships2001-2006 TabrizUniversityofMedicalSciences(TUMS),Tabriz,Iran
(#6NationalMedicalUniversity)• ClinicalInternships(GPA:A–17.16outof20)2007-2009
-a)GhaemHospital,b)ImamRezaHospital,c)Dr.SheikHospital,
d)Ebn-eSinaHospital,MashhadUniversityofMedicalSciences(MUMS),Mashhad,Iran.
(#3NationalMedicalUniversity)-a)ImamKhomeiniHospital,b)SinaHospital,c)ImamRezaHospital,d)TelaghaniHospital,e)AzzahraHospital,andf)ShohadaHospital
TabrizUniversityofMedicalSciences(TUMS),Tabriz,Iran.(#6)
• AwardedtheMDS$2,000.00USDGrantby“TheMovementDisorderSociety'sAward
Committee,”17thInternationalCongressofParkinson'sDiseaseandMovementDisorders,June16-20,2013inSydney,Australia.2013
• AwardedHoogenraadPrize(FreeCopperPrize;TheBronzeOwl)byDr.TjaardU.Hoogenraad,promotorofzinctherapysince1978,formyworkonWilson’s
disease.2012• InvitedforthemanagementofpatientswithWilson'sdisease.MashhadUniversity
ofMedicalSciences,Mashhad,Iran.2011
• AwardedthetheGovernorGeneral’shonourasanactiveandcompassionatedoctorinDr.Chamranhospital,Ferdows,SouthKhorasan,Iran.2010
• Awarded€1,000.00grantfromtheStichtingDeKistvanBeresteyn(foundedin1606intheNetherlands)formyworksonWilson´sdisease;23April2009
• InvitedforthemanagementofpatientswithWilson'sdisease.TabrizUniversityofMedicalSciences,Tabriz,Iran.Asclinicianandconsultant.2009
• Grantedthe7-yearmedicaleducationNationalScholarshipfromtheMinistryofHealth
andMedicalEducation,Iran;(ranked617among0.6millioncandidates)2001
• AvanA,HoogenraadTU.ZincandCopper inAlzheimer’sdisease.JAlzheimerDis2015;46(1):inpress[IF:4.15]
• AvanA,PostmaTJ,CeresaC,AvanA,CavalettiG,GiovannettiE,PetersGJ.Platinum-inducedneurotoxicityandpreventivestrategies:past,present,andfuture.TheOncologist2015;20(4):411-432[IF:4.87]
• Nikkhah K,* Avan A,* Shoeibi A, Azarpazhooh A,* Ghandehary K, FoerchC, Saposnik G, Sasannejad P, Vakili V, Layegh P, Farzadfard MT, BavarsadShahripour R, Hoseini MR, Azarpazhooh MR. Gaps and hurdles deter againstfollowing stroke guidelines for thrombolytic therapy in Iran: exploring theproblem.JStrokeCerebrovascDis2015;24(2):408-415[IF:1.67]
�171
Neuro-Oncology
Platinum-Induced Neurotoxicity and Preventive Strategies: Past,Present, and FutureABOLFAZL AVAN,a TJEERD J. POSTMA,b CECILIA CERESA,c AMIR AVAN,a,d GUIDO CAVALETTI,c ELISA GIOVANNETTI,a GODEFRIDUS J. PETERSa
Departments of aMedical Oncology and bNeurology,VUUniversityMedical Center, Amsterdam,TheNetherlands; cDepartment of SurgeryandTranslationalMedicine,UniversityofMilano-Bicocca,Monza, Italy; dDepartmentofNewSciencesandTechnology, SchoolofMedicine,Mashhad University of Medical Sciences, Mashhad, IranDisclosures of potential conflicts of interest may be found at the end of this article.
Key Words. Neurotoxicity x Platinum x Pathogenesis x Polymorphism x Prevention x Models
ABSTRACT
Neurotoxicity is a burdensome side effect of platinum-basedchemotherapy that prevents administration of the fullefficacious dosage and often leads to treatment withdrawal.Peripheral sensory neurotoxicity varies from paresthesia infingers to ataxic gait, which might be transient or irreversible.Because the number of patients being treated with theseneurotoxic agents is still increasing, the need for understand-ing the pathogenesis of this dramatic side effect is critical.Platinum derivatives, such as cisplatin and carboplatin, harmmainly peripheral nerves and dorsal root ganglia neurons,possibly because of progressive DNA-adduct accumulationand inhibition of DNA repair pathways (e.g., extracellularsignal-regulated kinase 1/2, c-Jun N-terminal kinase/stress-activated protein kinase, and p38 mitogen-activated proteinkinass), which finally mediate apoptosis. Oxaliplatin, witha completely different pharmacokinetic profile, may also altercalcium-sensitive voltage-gated sodium channel kineticsthrough a calcium ion immobilization by oxalate residue as
a calcium chelator and cause acute neurotoxicity. Polymor-phisms in several genes, such as voltage-gated sodiumchannelgenes or genes affecting the activity of pivotal metal trans-porters (e.g., organic cation transporters, organic cation/carnitinetransporters, and some metal transporters, such as the coppertransporters, and multidrug resistance-associated proteins), canalso influence drug neurotoxicity and treatment response.However, most pharmacogenetics studies need to be elucidatedby robust evidence. There are supportive reports about theeffectiveness of several neuroprotective agents (e.g., vitamin E,glutathione, amifostine, xaliproden, and venlafaxine), but doseadjustment and/or drug withdrawal seem to be the mostfrequently used methods in the management of platinum-inducedperipheralneurotoxicity.Todevelopalternativeoptionsinthetreatmentofplatinum-inducedneuropathy, studieson invitromodels and appropriate trials planning should be integrated intothe future design of neuroprotective strategies to find the bestpatient-oriented solution.The Oncologist 2015;20:411–432
Implications for Practice: Neurotoxicity is a burdensome side effect of platinum-based chemotherapy that preventsadministration of the full efficacious dosage and often leads to treatment withdrawal. This review summarizes preclinical andclinical evidence of pathogenesis and pathophysiology of platinum-induced peripheral neurotoxicity, aswell as available evidenceof neuroprotective and therapeutic strategies.These data may help to develop alternative options in the treatment of platinum-induced neuropathy, studies on in vitro models, and appropriate trials planning to find the best patient-oriented solution.
INTRODUCTION
Since the discovery of cisplatin in the mid-1960s, manyplatinum compounds (more than 3,000 compounds) havebeen developed. Thirty-five of these compounds have exhi-bited adequate pharmacological advantages (e.g., reachingsufficiently high plasma levels not associated with commontoxicities, such as renal toxicity and thrombocytopenia) [1].Someof themhavebeen registeredorarebeing considered forregistration for treatment of different cancers, such as thesecond (carboplatin, nedaplatin, tetraplatin, and iproplatin)
and third (oxaliplatin, lobaplatin, heptaplatin, satraplatin, andLA-12) generation, usually with better safety profiles [2–4].
Despite the efficacy of platinum analogs in cancertreatment, serious side effects, especially peripheral sensoryneurotoxicity, often prevent their administration at their fullefficacious doses or may considerably affect the quality of lifeof cancer patients being treatedwith them [5, 6]. Cisplatinwasthe first heavy metal used in several kinds of solid tumors,including lung, ovary, testis, bladder, head and neck, and
Correspondence: Godefridus J. Peters, Ph.D., Department of Medical Oncology, Cancer Center Amsterdam, VU University Medical Center, DeBoelelaan 1117, 1081 HV Amsterdam, The Netherlands. Telephone: 31-20-4442633; E-Mail: [email protected] Received February 5, 2014;accepted for publication December 11, 2014; published Online First onMarch 12, 2015.©AlphaMed Press 1083-7159/2015/$20.00/0 http://dx.doi.org/10.1634/theoncologist.2014-0044
TheOncologist 2015;20:411–432 www.TheOncologist.com ©AlphaMed Press 2015
CME
MedicalEducation
AcademicHonors
Peer-ReviewedPublications
12

Chapter12(Avan’sdissertation) CV
• Avan A, Avan A, Maftouh M, Ghayour-Mobarhan M, Gholamin S. BiomarkerAnalysis in CLEOPATRA: Searching for a sensitiveprognostic factor. J ClinOncol2015;33.DOI:10.1200/JCO.2014.60.4702.Inpress[IF:18.43]
• Avan A, Avan A, Maftouh M, Ghayour-Mobarhan M, Gholamin S.PharmacogenomicModelingofCirculatingTumorandInvasiveCellsforPrediction
ofChemotherapyResponseandResistanceinPancreaticCancer-letter.ClinCancerRes2015;21(6):1497[IF:8.72]
• Avan A, Avan A, Giovannetti E, Peters GJ. Calcium/magnesium infusion foroxaliplatin-induced neuropathy: protective or not? J ClinOncol 2014; 32(29):3341[IF:18.43]
• AvanA,*MaftouhM,*AvanA,TibaldiC,ZucaliPA,Giovannetti E. SNPs inPI3K–PTEN–mTORandBrainMetastasesinNSCLC—Letter.ClinCancerRes2014;20(13):3623-3624[IF:8.72]
• Avan A, Adema AD, Hoebe EK, Huijts CM, Avan A, Peters GJ. Modulation ofsignaling enhances the efficacy of the combination of satraplatin and erlotinib.
CurrDrugTargets2014;15(14):1312-1321[IF:3.02]• Avan A,* Avan A,* Le Large TYS, Mambrini A, Funel N, Maftouh M, Ghayour-
MobarhanM,CantoreM,BoggiU,PetersGJ,PacettiP,*GiovannettiE*.AKT1andSELPpolymorphismspredicttheriskofdevelopingcachexia inpancreaticcancerpatients.PLOSONE2014.9(9):e108057.[IF:3.23]
• Ceresa C,* Avan A,* Giovannetti E, Geldof AA, Avan A, Cavaletti G, Peters GJ.Characterization of and Protection from Neurotoxicity Induced by Oxaliplatin,BortezomibandEpothilone-B.AntiCancerRes2014;34:517-523[IF:1.83]
• Peters GJ, Avan A, Gallegos Ruiz M, Orsini V, Avan A, Giovannetti E, Smit EF.Predictive Role of Repair Enzymes in the Efficacy of Cisplatin Combinations in
PancreaticandLungCancer.AntiCancerRes2014;34:435-442[IF:1.83]
• Avan A, Digaleh H, Behrouz R, Azarpazhooh MR, et al. Exploring the LinkbetweenSocioeconomicDeprivationandStroke.2015
• AvanA,NarayanR,GiovannettiE,PetersGJ.RoleofAktsignallinginresistancetoDNA-targetedtherapy.2015
• AvanA,*MaftouhM,*AvanA,*vanKriekenA,vanGriekenN,RaktoeR,FunelN,
CaponiS,BoggiU,LeonLL,PetersGJ,GiovannettiE.Phospho-AktoverexpressionisprognosticandcanbeusedtotailorthesynergisticinteractionofthenovelAktinhibitorperifosinewithgemcitabineinpancreaticcancer.2015
• Avan A, AzarpazhoohMR, Hoogenraad TU. Free copper intoxication instead ofcopperaccumulationinWilson'sdisease:acriticalappraisal.2015
• AvanA,KianifarHR,HoogenraadTU.Nomorelivertransplantation:successfulzinctherapyinacuteliverfailureduetoWilson’sdisease.2015
• AbolfazlAvan,PLOSONEReviewer.PLOSONE2014ReviewerThankYou.PLoSONE
2015;10(2):e0121093.doi:10.1371/journal.pone.0121093
�172
Manuscripts
Post-hocReview
Peer-ReviewedPublications(cont’d)
12

Chapter12(Avan’sdissertation) CV
• Avan, A., Azarpazhooh, M.R., Hoogenraad, T.U.; Zinc therapy reverses neuro-degeneration in Wilson’s disease patients with parkinsonism [abstract]. AD/PD2015:385(Nice,France)
• Avan,A.,Kianifar,H.R.,Hoogenraad,T.U.;SuccessfulinitialzinctherapyinWilson’sdisease with acute liver failure and copper intoxication: Wonder drug exists
[abstract].AD/PD2015:384(Nice,France)• Avan, A., Azarpazhooh, M.R., Hoogenraad, T.U.; Effective shift from traditional
chelation therapy to evidence based zinc monotherapy in four patients withWilson'sdiseaseandparkinsonism[abstract].MovementDisorders2013;28Suppl1:989(Sydney,Australia)
• Avan,A.,Kianifar,H.R.,Hoogenraad,T.U.;InitialzinctherapyinaWilson’sdiseasepatient with acute liver failure and copper intoxication: a clinical observation[abstract].MovementDisorders2013;28Suppl1:990(Sydney,Australia)
• Avan,A.,Azarpazhooh,M.,Kianifar,H.,Hoogenraad,T.;Patientswithfreecoppertoxicosis in Wilson's disease should be treated with zinc from the beginning
[abstract].MovementDisorders2012;27Suppl1:232(Dublin,Ireland)
• Movement disorders. CME 2015 – 8 AMA PRA Category 1 Credits; ContinuumAudio: (1) Parkinsonian Syndromes and Tremor Syndromes; (2) Chorea and GaitDisorders;(3)MyoclonusandCerebellarandAfferentAtaxias;(4)TicDisordersandPsychogenicMovementDisorders.
• Alzheimer’s disease & Dementia: CME 2015 – 8 AMA PRA Category 1 Credits:
Continuum Audio: (1) The Diagnostic Evaluation of a Patient with Dementia; (2)Biomarkers of Alzheimer Disease; (3) Nonpharmacologic Treatment andPrevention Strategies for Dementia; (4) Alzheimer Disease PharmacologicTreatment and TreatmentResearch; (5)NewDiagnostic Schemas,Genetics, andProteinopathy intheEvaluationofFrontotemporalDegeneration;(6)TheClinical
Problem ofNeuropsychiatric Signs and Symptoms in Dementia; (7) Dementia intheOldestOld;(8)UnderstandingandTreatingVascularCognitiveImpairment;(9)Ethics:WhenCognitivelyNormalPatientsAsktoBeTestedforAlzheimerDisease;(10)TestYourKnowledge:Dementia.
• MultipleSclerosis.CME2015–9AMAPRACategory1Credits;ContinuumAudio:(1)
Diagnosis and Differential Diagnosis of Multiple Sclerosis; (2) Current and NewDirectionsinMRIinMultipleSclerosis;(3)PathologyofMultipleSclerosis:WhereDo We Stand?; (4) Present and Emerging Therapies for Multiple Sclerosis; (5)GeneralHealthIssuesinMultipleSclerosis;(6)GaitDisordersinMultipleSclerosis;(7)OpticNeuritisandtheEvaluationofVisualImpairmentinMultipleSclerosis;(8)
Pediatric Demyelinating Diseases; (9) Disclosing a Misdiagnosis of MultipleSclerosis; (10) Acute Disseminated Encephalomyelitis, Transverse Myelitis, andNeuromyelitisOptica;(11)UnusualSymptomsandSyndromesinMultipleSclerosis.NeuroLearn: (12) Sick& Tired:Recognizing andTreating Fatigue in PersonsWithMultipleSclerosis.
• MouseMorphology&Genetics Course. OOACourse, AcademicMedical Center,
�173
MajorTrainings
Conferenceproceeding/PosterPresentation(cont’d)
12

Chapter12(Avan’sdissertation) CV
Amsterdam,theNetherlands,March23–April2,2015(72hours;3ECTSpoints).
• Alzheimer’s disease and Parkinson’s disease. CME/CPD 2015 – 30 AMA PRACategory 1Credits. 12th InternationalConferenceonAlzheimer'sandParkinson'sDiseases.Nice,France.March18-22,2015
• Alzheimer’sdisease&Dementia.CME2014–5.5AMAPRACategory1Credits:(1)
Alzheimer’sDiseaseDiagnosisandTreatmentUpdate;(2)PracticalApproachestoManaging Patients With Moderate to Severe Alzheimer Disease; (3) CaseChallengesinEarlyAlzheimer'sDisease;(4)EmergingImagingandTherapeuticsinAlzheimer’sDisease:EarlyDetectionandIntervention.
• MultipleSclerosisCME2014–2.15AMAPRACategory1Credits:(1)HotTopics in
MultipleSclerosisManagement:AnExpertDiscussion;(2)BrainAtrophy,MRI,andEvaluatingOralTherapyinPatientsWithRRMS;(3)EarlyTreatmentStrategiesandImproved Outcomes in RRMS; (4) Nurses and RRMSManagement: Real WorldExperience;(6)TheReal-WorldEvidenceforOralTherapiesinRRMS.
• Bioinformatics Sequence Analysis. The PhD course program 2013-2014, AMC
GraduateschoolforMedicalSciences,Amsterdam,theNetherlands
• Histopathologyof tumours,OOACourse,Amsterdam,theNetherlands,February10-11,2014.
• Basic Medical Statistics (SPSS), OOA Course, Amsterdam, the Netherlands,November25-29,2013.
• Movementdisorders:Awarded35AMAPRACategory1CreditsTM.TheMovementDisorder Society's 17th International Congress of Parkinson’s Disease andMovement Disorders at Sydney Convention and Exhibition Centre, Sydney,Australia,June16-20,2013.
• Movementdisorders:Awarded35AMAPRACategory1CreditsTM.TheMovement
Disorder Society's 16th International Congress of Parkinson’s Disease andMovementDisordersatTheConventionCentreDublin,Dublin,Ireland,June17-21,2012.
• Gastroenterology and Hepatology. Awarded 14 CME. 12th Iranian Congress ofGastroenterologyandHepatology,Tehran,Iran,onNovember28-30,2012
• Epilepsy:FirstInternationalCongressofEpilepsy,RazaviHospital,Mashhad,Iran.Dec2010(7scoreCME)
• Internship:ClinicalNeurology,InternalMedicine,PaediatricsMedicine,Cardiology,Infectious diseases and Clinical General Surgery at Ghaem Hospital, Imam RezaHospital, and Dr. Sheikh Hospital. Mashhad University of Medical Sciences.
Mashhad,Iran.2007-2009
• Internship:EmergencyMedicine,OrthopaedicSurgery,ENTSurgery,UrologyandRadiologyatImamKhomeiniHospitalandImamRezaHospital.TabrizUniversityofMedicalSciences.Tabriz,Iran.2007
• ResearchMethodologyWorkshop, Tabriz University ofMedical Sciences, Tabriz,
Iran.2004
�174
MajorTrainings(cont’d)
12

Chapter12(Avan’sdissertation) CV
• IELTSCertificateofEnglish,Score7.0.2012(speaking7.5)
• EnglishLearningCourses,CertificateofAdvancedEnglish (CAE),AllamehEnglishInstitute,Mashhad,Iran.2012
• Violin,ClassicalMusic(InIranandHolland),2010-now
• Back to life:miracle? (Wilson’s disease) In:Main NeurologyMeeting at AcademicMedicalCenter,UniversityofAmsterdam,Amsterdam, theNetherlands.October2015
• Wilson’sdiseaseTreatment:Firstdonoharm.In:Gastroenterologists’meetingatImamRezaHospital,Mashhad,Iran.August,2013.
• Reversal of fulminantWilson’s disease following twoweeks zinc therapy. IranianInternational Congress of Gastroenterology and Hepatology. Tehran, Iran. 28November,2012
• Evidence Based Decision Making; Effective, Safe and Cheaper Approach inTreatment of a Fatal Disease. Department ofNeurology,MashhadUniversity ofMedicalSciences(forneurologists,residentsandpeers),2010
• Wilson’s Disease; Prognosis and Differential Diagnoses. Mashhad University ofMedical Sciences. Department of Infectious Diseases (for professors, residentsandpeers),2009
• LiverTransplantationinWilson’sDisease;WhentoDecide.DepartmentofInternalMedicine, Mashhad University of Medical Sciences (For gastroenterologists,residentsandpeers),2009
• Decision Making; New Aspect in Treatment of Wilson’s Disease. Department ofInternalMedicine,TabrizUniversityofMedicalSciences(forgastroenterologists,residentsandpeers),2008
• The Problem of Wrong Decision. Department of Neurology, Tabriz University ofMedicalSciences(forneurologists,residentsandpeers),2008
• The Problem of Delayed Diagnosis; Appropriate Approach to Wilson’s Disease.Department of InternalMedicine,MashhadUniversity ofMedical Sciences (forgastroenterologists,residentsandpeers),2007
• GeneraloverviewofWilson’sdisease.DepartmentofNeurology,TabrizUniversityofMedicalSciences(forneurologists,residentsandpeers),2006
Evaluationofsigns,symptoms,treatmentandprognosisofpatientswithmalignantmesotheliomawhomwere admitted to Ghaem and Omid hospitals from 1982 to2008 Thesisscore:19.11outof20,Grade:“Aplus”November,2009 Supervisors:Dr.RezaBagheri,MD;Prof.SeyyedZiaallahHaghi,MD.
Twelveyearsofpreuniversityeducationperiod 1988-2000GPA:Aplus;Scores:higherthan19outof20;1stor2ndtopstudent
�175
MedicalDoctorate(MD)Thesis
Pre-graduationEducation
LecturesandPresentations
12

Chapter12(Avan’sdissertation) Acknowledgements
ACKNOWLEDGEMENTS
With love, I thank my father, Rahim Avan, and mother, Homa Khamseh, for their never-endingencouragement.Theykeptsupportingmeforallmyambitionsduringmylife,and inparticular,tostudymedicineinordertohelppatientshaveabetterlife.TheyalsoencouragedmetocometotheNetherlandstostrengthenmyknowledgeandexperiencetohopefullyhaveapositiveimpactonthefieldofmedicine.Theirlove,constantinspiration,encouragementandsupporthavealwaysbeenmystrength,Therefore,Iwouldliketodedicatethisbooktothemandwishthemhealth,comfort,andfortune.
Iwouldalsoliketoexpressmydeepestgratitudetoallmycolleaguesandfriendswithouthelpofwhom theworkspresented in thisdissertation couldhavenotbeendone. I am indebted toFritsPeters (my PhD promoter) and Elisa Giovannetti (my co-promoter) for giving the opportunity towork with them, for which I indeed feel privileged. I thoroughly appreciate their generosity,patience,andeffortintrainingme,andalsotheirfriendlyattitudetowardsmetoprovidemewithawell-roundedexperienceinthescientificresearch.Evenmoreso,theircarefulandcriticalcommentson my writings improved my scientific writing, so that my message was conveyed in the bestpossibleway.Mylearningfromthemdoesnotlimittotheaforementionedelements,butIhavealsolearnedalotfromthemintermsofmannerintreatingstudents,valueofclassifiedandprioritizedworking,andcollaboration.
Kindappreciationisexpressedtotheanonymousreviewersofthearticlespublishedinthisthesisfortheir precise and critical review, which helped me improve the quality and clarity of my works.Likewise, I amgrateful to theofficial reading committeemembers for their careful reviewofmydissertationandtheirinvaluablefeedbacks;dr.JanButer(Oncologist,VUUniversityMedicalCenter),dr. Tjaard U. Hoogenraad (Neurologist, University Medical Center Utrecht), dr. Judith R. Kroep(Oncologist, Leiden University Medical Center), dr. Tjeerd J. Postma (Neurologist, VU UniversityMedical Center Amsterdam), prof.dr. Egbert F. Smit (Pulmonary oncologist, Netherlands CancerInstituteandVUUniversityMedicalCenterAmsterdam),andprof.dr.ThomasWürdinger(Biologist,VUUniversityMedicalCenterAmsterdam).IhopeIcanextendmycollaborationswithallinfuture.Tjaard has been stimulating me in treating Wilson’s disease patients since 2007 when I saw hismonographonthediseaseinthelibraryofTabrizUniversityMedicalSciences,whereIwasstudyingmedicine.Hehasalwaysadvisedmetotreatpatientswiththebestavailableevidence-basedpatient-orientedscientificapproach.IowehimalotandIamhappytohavebeenworkingunderhisprecioussupervision,astheworld-knownexpertinWilson’sdisease.
KeesSmidhelpedmenotonlywiththeprojectfromtheverystartofthisproject,butalsowithmanyother problems that I encountered during my stay in the Netherlands, for which I am eternallythankful. Likewise, I received a considerable amount of help from Ietje Kathmann, RichardHoneywell,SaskiaBroekman,SimoneSpan,andHenkDekker,mycolleaguesandfriendsatCancerCenterAmsterdam.TheirfriendlyandopenattitudestowardsmeallowedlivingthestrenuousPhDproject as a totally joyful experience. I also met many good friends in CCA, including FrançoisRustenburg, Ingrid Garajová, Kaamar Azijli, Tessa Le Large, Laura Meijer, Ahmed Hassan, MayurYevgeri,ErikMeijer,DanieldeKlerk,andmanyothersthathopefullyforgivemefornotnamingthemhere,butIwillnotforgetthejoyfulmomentstheymadeformeinAmsterdam,whichismorethan6000kmfarfrommyhome.
I also had great moments with many Iranian friends in Amsterdam, including Mohammad Ali,Saeideh, Jafar, Leila,Mahdi, Samira,Mohammad,Maryam, Esmail, Elham,Hossein, Zahra,Hodjat,Marzieh, Shayan,Narges,MohammadAmin, Fatemeh,HamidReza,Marzieh,Naser,Aylar,Mahdi,Mahdieh,Masoud,Hoda, Amin, Parisa, Yasser,Hossein,Hadi,Mojtaba, and all those that Imighthave forgotten to mention. They all made me feel at home. I hope we will keep this fantasticfriendshipforever.
�176
12

Chapter12(Avan’sdissertation) Acknowledgements
I have also received a great amount of help and support from Mahmoud Reza Azarpazhooh(Neurologist, Mashhad University Medical Center) and Anne-Fleur van Rootselaar, JohannesKoelman,VincentOdekerken,andRobdeBie(Neurologists,AcademicMedicalCenter,UniversityofAmsterdam)whoacknowledgedmymotivationandpassioninneurologyandstudyingneurologicalproblemsbyinvitingmetotheirdepartmentsforinvaluablecollaborationswiththem,whichIhopewillcontinueduringmyfuturecareer.IamalsogratefultoFemkeVisser(NeurologyAssistant)andLoJBour(ClinicalPhysicist)forguidingmethroughtheeyemovementsprojectandtheirrespectfulattitudes.IshouldalsothankallgreatfriendsImetattheDepartmentofNeurologyatAMC,someofwhomwereRense,Joost,Sarvi,Arthur,Hanneke,José,Thijs,Erik,Stephanie,Janny,Edwin,Hanna,Shirly,Rosanna,Titia,Claudia,andmanyothersthatImighthaveforgottentomention.
Lastbutnotleast,Iwouldliketothankmyyoungerbrother,Amirandhiswife,MinaMaftouhwhohavehelpedmea lot,especiallyduring the firstyearofmyPhD.Seeingmy littlenephewSobhanwhowasbornduringthisperiodwasalsoagreatsourceofenergyforme!Ishouldalsothankmyelder brother, Mahdi and his wife, Samaneh Davoudi, and my niece, Zahra who always keptencouragingme to seekmy ambitions. I also had the support of allmy relativeswho frequentlycalledmetobesurethateverythingwasfinewithmehereandIdidnotfeellonely!
Aboveall,IoweitalltoGodforgrantingmewisdom,passion,curiosity,perseverance,motivation,health,andstrengthtofacedifficultiesinordertopavethewaytowardsabetterlifeformeandforothers. Ihopemyworkwillhelpscientists togo furtherwith findingbetter treatmentsandbringpatientshealthy,joyful,andproductivelives.
AbolfazlAvanOctober2015,Amsterdam
�177
12




















