
BIRS-CMO 2020 Annual Report - Banff International Research ...
Upload
khangminh22Category
view
2download
0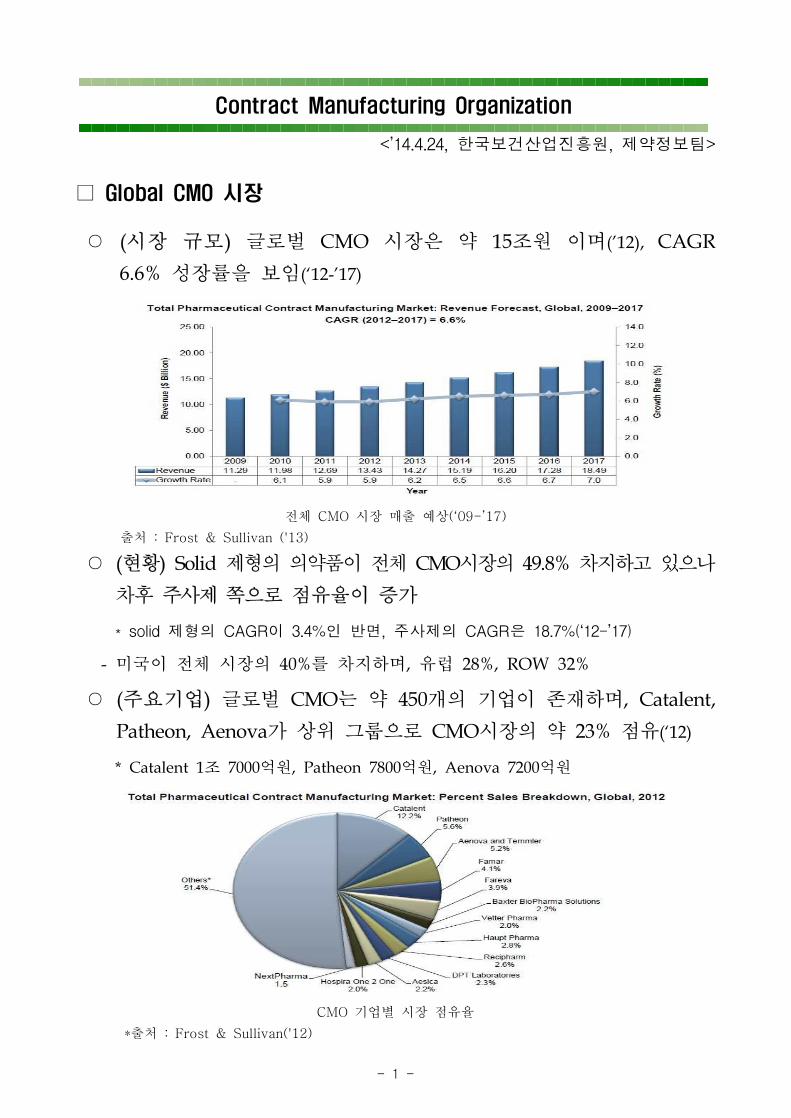
- 1 -
Contract Manufacturing Organization
<’14.4.24, 한국보건산업진흥원, 제약정보팀>
□ Global CMO 시장
○ (시장 규모) 글로벌 CMO 시장은 약 15조원 이며('12), CAGR
6.6% 성장률을 보임(‘12-’17)
전체 CMO 시장 매출 예상(‘09-’17)
출처 : Frost & Sullivan ('13)
○ (현황) Solid 제형의 의약품이 전체 CMO시장의 49.8% 차지하고 있으나
차후 주사제 쪽으로 점유율이 증가
* solid 제형의 CAGR이 3.4%인 반면, 주사제의 CAGR은 18.7%(‘12-’17)
- 미국이 전체 시장의 40%를 차지하며, 유럽 28%, ROW 32%
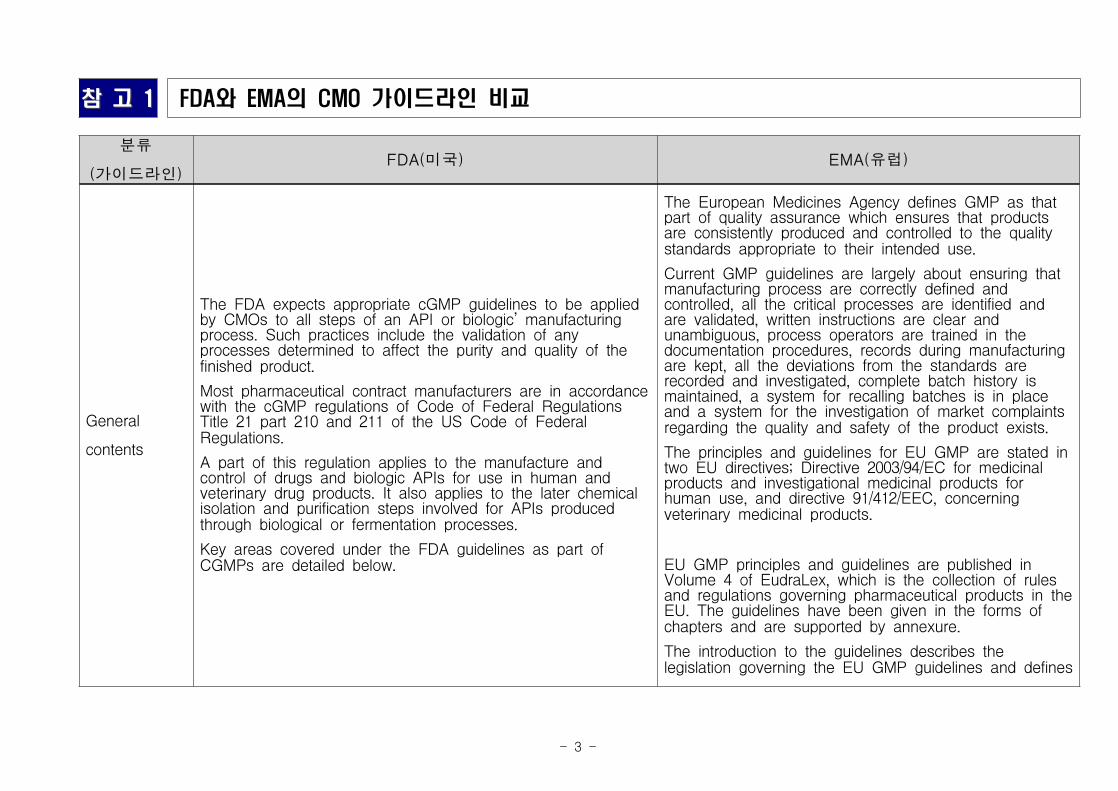
○ (주요기업) 글로벌 CMO는 약 450개의 기업이 존재하며, Catalent,
Patheon, Aenova가 상위 그룹으로 CMO시장의 약 23% 점유(‘12)
* Catalent 1조 7000억원, Patheon 7800억원, Aenova 7200억원
CMO 기업별 시장 점유율
*출처 : Frost & Sullivan('12)

- 2 -
Molecule별 CMO 현황
※ CMO시장은 Solid Dose Formulations, Liquid and Semi-solid Dose
Formulations, Injectable Dose Formulations으로 3개의 주요 부분으로 나뉨
○ Solid Dose Formulations
- (시장규모) 6조 9500억원 규모로 전체 CMO 시장에서 가장 많은 비율을
차지하고 있으며(49.8%), CAGR 3.4%(‘12)
* SDF는 Advanced Oral Solids와 Basic Oral Solids로 나뉘며, 각각 65%와
35% 점유(‘12)
- (장·단점) Sterile 제형과는 달리, 제조가 용이하며, 적은 자본과 운영비용으로
수익 창출이 가능하나, 차후 IDF성장으로 시장 점유 축소가 예상됨
○ Liquid and Semi-solid Dose Formulations
- (시장규모) 2조 6500억원 규모로, CAGR은 2.5%이며, 전체 CMO 시장의 19%
차지(‘12)
* Sterile과 Non-sterile로 분류되며, 각각 29%, 71%를 차지함
- (장·단점) 낮은우영비용과자본지출에도불구하고, 운송과저장에대한어려움으로
점차적으로 감소하는 추세
○ Injectable Dose Formulations
- (시장규모) 4조 3500억원, CAGR 18.7%로 강한 시장성장세를 보임.이며, 전체
CMO 시장의 31.2% 차지(‘12)
* 저용량과 고용량으로 분류되며, 각각 89.9%, 10.1%의 점유를 보임
- (장·단점) 빠른 발현 및 치료 효과와 항암제 시장의 강한 수요현상으로 CMO
시장의 지속적인 성장이 예상되나, 기술적인 어려움과 제조 및 운영비용 高
☞ 글로벌 제약기업들이 제약생산의 아웃소싱으로서 CMO의 역할 및 Asian
국가들의 몫이 점차 커지고 있으며, 비용절감 효과와 높은 퀄리티로 인하여
향후 지속적인 성장이 기대됨. 또한, Solid 제형에서 Injectable 제형으로의 시장
점유율 변화가 예상됨
- 3 -
분류
(가이드라인)FDA(미국) EMA(유럽)
General
contents
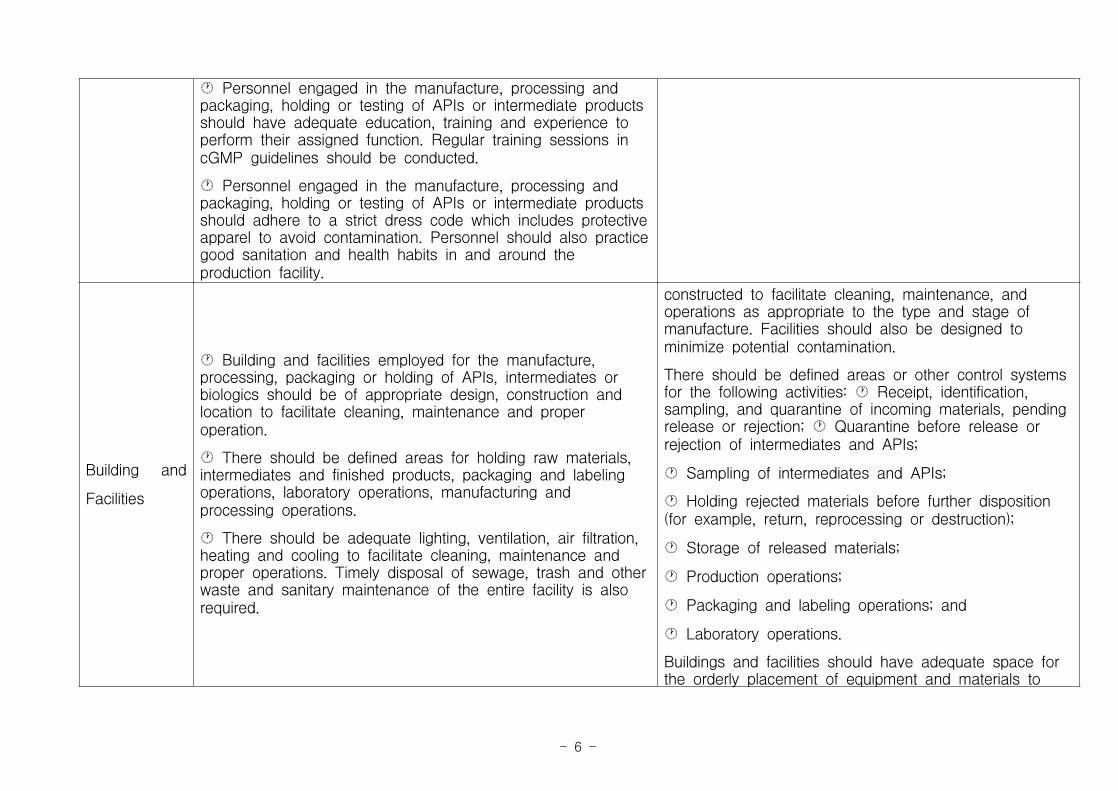
The FDA expects appropriate cGMP guidelines to be appliedby CMOs to all steps of an API or biologic’ manufacturingprocess. Such practices include the validation of anyprocesses determined to affect the purity and quality of thefinished product.
Most pharmaceutical contract manufacturers are in accordancewith the cGMP regulations of Code of Federal RegulationsTitle 21 part 210 and 211 of the US Code of FederalRegulations.
A part of this regulation applies to the manufacture andcontrol of drugs and biologic APIs for use in human andveterinary drug products. It also applies to the later chemicalisolation and purification steps involved for APIs producedthrough biological or fermentation processes.
Key areas covered under the FDA guidelines as part ofCGMPs are detailed below.
The European Medicines Agency defines GMP as thatpart of quality assurance which ensures that productsare consistently produced and controlled to the qualitystandards appropriate to their intended use.
Current GMP guidelines are largely about ensuring thatmanufacturing process are correctly defined andcontrolled, all the critical processes are identified andare validated, written instructions are clear andunambiguous, process operators are trained in thedocumentation procedures, records during manufacturingare kept, all the deviations from the standards arerecorded and investigated, complete batch history ismaintained, a system for recalling batches is in placeand a system for the investigation of market complaintsregarding the quality and safety of the product exists.
The principles and guidelines for EU GMP are stated intwo EU directives; Directive 2003/94/EC for medicinalproducts and investigational medicinal products forhuman use, and directive 91/412/EEC, concerningveterinary medicinal products.
EU GMP principles and guidelines are published inVolume 4 of EudraLex, which is the collection of rulesand regulations governing pharmaceutical products in theEU. The guidelines have been given in the forms ofchapters and are supported by annexure.
The introduction to the guidelines describes thelegislation governing the EU GMP guidelines and defines
참 고 1참 고 1 FDA와 EMA의 CMO 가이드라인 비교

- 4 -
quality management in brief.
Part one of the guidelines covers basic requirements formanufacturing medicinal products. Chapters in theguidelines cover quality management, personnelrequirements, premise and equipment requirements,documentation guidelines, production guidelines, qualitycontrol guidelines, contract manufacture andanalysis-related guidelines, complaints and productrecall-related guidelines and self inspection-relatedguidelines.
Part two deals with the basic requirements for activesubstances used as starting materials for themanufacturing of finished dosages. Manufacturingguidelines for sterile products are covered in Annex 1,and guidelines for biologics manufacturing are coveredin Annex 2. Annex 7 deals with packaging materials andAnnex 8 deals with the manufacturing of creams, liquidsand ointments.
Chapter 7 of the guidelines defines the contract giver,contract acceptor, the contract and principles of contractmanufacturing. It also provides guidelines for the partiesto the manufacturing contract.
Figure 21 shows the applicability of EU GMP guidelinesin various steps of API manufacturing. Therepresentation also shows increasing GMP requirementsin the manufacturing process.

- 5 -
Key areas covered under the EU GMP guidelinesinclude the following sections: Quality Management,Production Activities, Buildings and Facilities, ProcessEquipment, Materials Management, and Packaging andLabeling.
Organization
and
Personnel
· According to the FDA, CMOs should have a QualityControl (QC) unit which has the responsibility and authority toapprove or reject all raw materials, packaging materials, labelsand finished products (APIs).
The QC unit should maintain proper production records andhave robust validation protocols. It should also have adequatelaboratory facilities for testing and approval and shouldperform periodic assessments of procedures, policies andmanufacturing.
- 6 -
· Personnel engaged in the manufacture, processing andpackaging, holding or testing of APIs or intermediate productsshould have adequate education, training and experience toperform their assigned function. Regular training sessions incGMP guidelines should be conducted.
· Personnel engaged in the manufacture, processing andpackaging, holding or testing of APIs or intermediate productsshould adhere to a strict dress code which includes protectiveapparel to avoid contamination. Personnel should also practicegood sanitation and health habits in and around theproduction facility.
Building and
Facilities
· Building and facilities employed for the manufacture,processing, packaging or holding of APIs, intermediates orbiologics should be of appropriate design, construction andlocation to facilitate cleaning, maintenance and properoperation.
· There should be defined areas for holding raw materials,intermediates and finished products, packaging and labelingoperations, laboratory operations, manufacturing andprocessing operations.
· There should be adequate lighting, ventilation, air filtration,heating and cooling to facilitate cleaning, maintenance andproper operations. Timely disposal of sewage, trash and otherwaste and sanitary maintenance of the entire facility is alsorequired.
constructed to facilitate cleaning, maintenance, andoperations as appropriate to the type and stage ofmanufacture. Facilities should also be designed tominimize potential contamination.
There should be defined areas or other control systemsfor the following activities: · Receipt, identification,sampling, and quarantine of incoming materials, pendingrelease or rejection; · Quarantine before release orrejection of intermediates and APIs;
· Sampling of intermediates and APIs;
· Holding rejected materials before further disposition(for example, return, reprocessing or destruction);
· Storage of released materials;
· Production operations;
· Packaging and labeling operations; and
· Laboratory operations.
Buildings and facilities should have adequate space forthe orderly placement of equipment and materials to

- 7 -
prevent mix-ups and contamination. The flow ofmaterials and personnel through the building or facilitiesshould be designed to prevent mix-ups orcontamination.
Laboratory areas/operations should normally beseparated from production areas. Some laboratory areas,in particular those used for in-process controls, can belocated in production areas, provided the operations ofthe production process do not adversely affect theaccuracy of the laboratory measurements, and thelaboratory and its operations do not adversely affect theproduction process or intermediate or API.
Process
Equipment
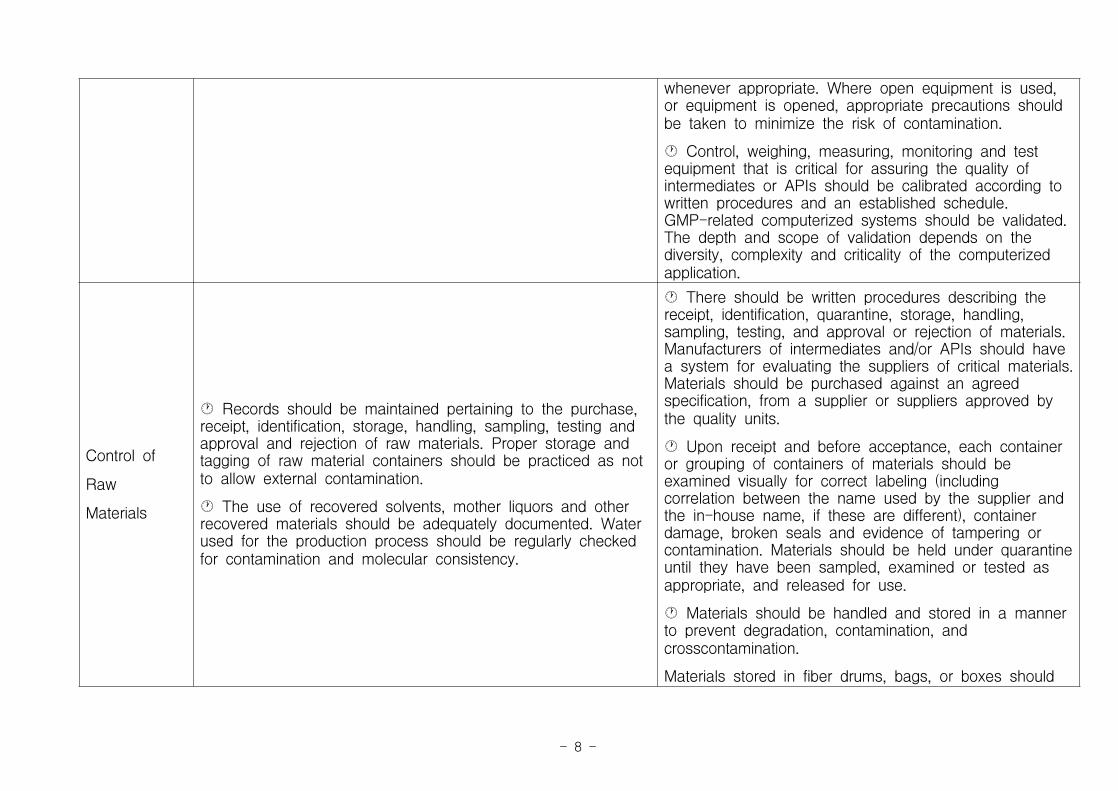
· In-house protocols should be established and followed formaintaining and cleaning equipment involved in manufacturing,storing and packaging the API. Regular equipment inspectionsshould be conducted for contamination, damage or anyperformance inconsistencies.
· Routine calibration of automatic, mechanical or electronicprocessing equipment, which include computers, should beperformed. Written records of these checks should be madeand maintained.
· Equipment used in the manufacture of intermediatesand APIs should be of appropriate design and adequatesize, and suitably located for its intended use, cleaning,sanitization (where appropriate), and maintenance.
· Equipment should be constructed so that surfacesthat contact raw materials, intermediates, or APIs do notalter the quality of the intermediates and APIs beyondthe official or other established specifications. Productionequipment should only be used within its qualifiedoperating range.
· Any substances associated with the operation ofequipment, such as lubricants, heating fluids or coolants,should not contact intermediates or APIs so as to altertheir quality beyond the official or other establishedspecifications. Any deviations from this should beevaluated to ensure that there are no detrimental effectsupon the fitness for purpose of the material. Whereverpossible, food grade lubricants and oils should be used.Closed or contained equipment should be used
- 8 -
whenever appropriate. Where open equipment is used,or equipment is opened, appropriate precautions shouldbe taken to minimize the risk of contamination.
· Control, weighing, measuring, monitoring and testequipment that is critical for assuring the quality ofintermediates or APIs should be calibrated according towritten procedures and an established schedule.GMP-related computerized systems should be validated.The depth and scope of validation depends on thediversity, complexity and criticality of the computerizedapplication.
Control of
Raw
Materials
· Records should be maintained pertaining to the purchase,receipt, identification, storage, handling, sampling, testing andapproval and rejection of raw materials. Proper storage andtagging of raw material containers should be practiced as notto allow external contamination.
· The use of recovered solvents, mother liquors and otherrecovered materials should be adequately documented. Waterused for the production process should be regularly checkedfor contamination and molecular consistency.
· There should be written procedures describing thereceipt, identification, quarantine, storage, handling,sampling, testing, and approval or rejection of materials.Manufacturers of intermediates and/or APIs should havea system for evaluating the suppliers of critical materials.Materials should be purchased against an agreedspecification, from a supplier or suppliers approved bythe quality units.
· Upon receipt and before acceptance, each containeror grouping of containers of materials should beexamined visually for correct labeling (includingcorrelation between the name used by the supplier andthe in-house name, if these are different), containerdamage, broken seals and evidence of tampering orcontamination. Materials should be held under quarantineuntil they have been sampled, examined or tested asappropriate, and released for use.
· Materials should be handled and stored in a mannerto prevent degradation, contamination, andcrosscontamination.
Materials stored in fiber drums, bags, or boxes should
- 9 -
be stored off the floor and, when
appropriate, suitably spaced to permit cleaning andinspection. Materials should be stored under conditionsand for a period that have no adverse affect on theirquality, and should normally be controlled so that theoldest stock is used first. Certain materials in suitablecontainers can be stored outdoors, provided identifyinglabels remain legible and containers are appropriatelycleaned before opening and use.
Production
and Process
Controls
· Maintaining production and process control protocolsmaking regular updates to ensure the purity and quality ofAPIs and intermediates. Appropriate weighing of raw materialsto ensure proper usage and to avoid wastage.
· Yields should be identified and documented regularly toanalyze inconsistencies in production and identifycontamination. Deviations from expected yields should beinvestigated and documented.
· Appropriate time limits should be established and adheredto for various production processes to ensure quality andpurity of the end product. In-process blending of intermediatesor products should
be adequately controlled and documented.
P a c k a g i n g
and Labeling
· Strict controls should be exercised over labels issued foruse in labeling operations. The number of labels issued orreturned should be noted and any discrepancies should beinvestigated. Protocols should be established for the correctlabeling and packaging of finished APIs.
· Spatial separation from other finished products should beprovided in order to avoid mix-ups and contamination. TheAPI packaging material should be non-reactive andnon-absorptive and should provide adequate protection
· Labeling operations should be designed to preventmix-ups. There should be physical or spatial separationfrom operations involving other intermediates or APIs.Labels used on containers of intermediates or APIsshould indicate the name or identifying code, the batchnumber of the product, and storage conditions, whensuch information is critical to assure the quality of theintermediate or API.
· Packaged and labeled intermediates or APIs should
- 10 -
against product deterioration or contamination.
· Wherever necessary, APIs (antibiotic and biologics) shouldbe labeled with the correct expiration dates.
For others, the container label or Certificate of Analysis (COA)should specify an appropriate expiration or re-test date.
be examined to ensure that containers and packages inthe batch have the correct label. This examinationshould be part of the packaging operation. The resultsof these examinations should be recorded in the batchproduction or control records.
· Facilities should be available for the storage of allmaterials under appropriate conditions (for example,controlled temperature and humidity when necessary).Records of these conditions should be maintained ifthey are critical for the maintenance of materialcharacteristics. Unless there is an alternative system toprevent the unintentional or unauthorized use ofquarantined, rejected, returned, or recalled materials,separate storage areas should be assigned for theirtemporary storage until the decision as to their futureuse has been taken.
Holding and
Distribution
· Protocols should be established for the storage andquarantine of finished products under specified temperature,humidity and light to ensure the quality and purity of the API.
· The maintenance of records on the dispatch of finishedproducts is mandatory to ensure no wastage.
Any discrepancies in records should be dealt withimmediately.
Key Changes
in Guidelines
In August 2002, the FDA announced the PharmaceuticalcGMPs for the 21st Century initiative. The primary objective ofthis was to integrate quality and risk management approachesinto the FDA’ existing protocols and programs. The previousrevision on the cGMP regulations was carried out in 1978 andsince then there have been many advances in themanufacture and production of pharmaceutical products. Since2002 there have been many changes and revisions to theseguidelines.
In 2007, the EMA made changes to its CPMP/3097/02guidelines with respect the comparability of biotechnologyproducts.
Under the new draft, the need, extent and nature ofnon-clinical and clinical comparability studies will beconsidered on a case-by-case basis, based on variousfactors, including:
· Process complexity, the nature of the change, the

- 11 -
potential impact on the molecule structure and on thefinal product profile.
· The nature and extent of differences demonstratedby the physicochemical and quality-related biologicalcharacterization, including product-related substances,impurity profile, stability and excipients. Thus,well-characterized differences may provide a backgroundfor a rational and focused approach with respect to theneed for non-clinical and clinical studies.
· Product complexity, including heterogeneity and higherorder structure, and the availability, capabilities andlimitations of analytical tests. If the analytical proceduresused are not sufficient to discern relevant differencesthat can impact the safety and efficacy of the product,additional nonclinical and/or confirmatory clinical testingmay be necessary.
* 출처 : Frost & Sullivan ('12)
Copyright © 2022 FDOKUMEN
















