
Artificial symmetry breaking in radicals is avoided by the use of the ensemble-referenced...
11
Artificial symmetry breaking in radicals is avoided by the use of the Ensemble-Referenced Kohn–Sham (REKS) method Michael Filatov * , Sason Shaik Department of Organic Chemistry, Lise Meitner-Minerva Centre for Computational Quantum Chemistry, The Hebrew University, 91904 Jerusalem, Israel Received 25 August 2000; in final form 10 October 2000 Abstract The computational scheme, termed spin-restricted ensemble-referenced Kohn–Sham (REKS) method (M. Filatov, S. Shaik, Chem. Phys. Lett. 304 (1999) 429; M. Filatov, S. Shaik, J. Phys. Chem. A 104 (2000) 6628), developed earlier to treat the strong non-dynamic correlation in singlet diradicals is extended for doublet states of triradicals. The energy and the density in the REKS method are represented as weighted sums of energies and densities of several KS de- terminants. The new method, dubbed REKS(3,3), is applied to calculation of several model problems where the conventional spin-unrestricted density functional calculations break the spatial symmetry. The results of the REKS(3,3) calculations show that the method avoids an artificial symmetry breaking in open-shell systems. Ó 2000 Elsevier Science B.V. All rights reserved. 1. Introduction A while ago it has been recognized [1] that since the total non-relativistic many-electron molecular Hamiltonian commutes with all respective spin and spatial symmetry operators, its exact eigen- function should also be an eigenfunction of all these symmetry operators. However, an applica- tion of the variational principle to an approximate wavefunction does not guarantee that this wave- function will possess all necessary symmetry properties [1]. Thus, an approximate wavefunction which breaks the molecular symmetry may fre- quently have a lower energy. This phenomenon, known as the ‘symmetry dilemma’ [1], appears in many situations in quantum-chemical calculations. The well-known examples are the diradicaloid situations in formally closed-shell molecules with stretched single bonds, twisted double bonds, etc., where the symmetry-broken spin-unrestricted so- lutions possess lower energy than the symmetry- adapted solutions at the Hartree–Fock (HF) and frequently at the post-HF (MPn, CC, etc.) levels of theory [2,3]. In spin-unrestricted calculations on radicals, the spatial symmetry breaking may occur in addition to the spin symmetry breaking [4]. This leads to total densities and molecular geometries that are distorted away from the correct molecular symmetry [4–7]. The now-classical examples are the allyl radical [5], formyloxyl radical [6], NO 2 [7], etc., where even the post-HF treatments based on a spin-unrestricted reference yield incorrect mo- lecular structures, dipole moments and vibrational frequencies [6,8]. 22 December 2000 Chemical Physics Letters 332 (2000) 409–419 www.elsevier.nl/locate/cplett * Corresponding author. Present address: Department of Theoretical Chemistry, Goteborg University, Reutersgatan 2, S-41320 Goteborg, Sweden. Fax: +31-773-5590. E-mail address: fi[email protected] (M. Filatov). 0009-2614/00/$ - see front matter Ó 2000 Elsevier Science B.V. All rights reserved. PII: S 0 0 0 9 - 2 6 1 4 ( 0 0 ) 0 1 2 5 7 - 4
Transcript of Artificial symmetry breaking in radicals is avoided by the use of the ensemble-referenced...

Arti®cial symmetry breaking in radicals is avoided by the useof the Ensemble-Referenced Kohn±Sham (REKS) method
Michael Filatov *, Sason Shaik
Department of Organic Chemistry, Lise Meitner-Minerva Centre for Computational Quantum Chemistry, The Hebrew University,
91904 Jerusalem, Israel
Received 25 August 2000; in ®nal form 10 October 2000
Abstract
The computational scheme, termed spin-restricted ensemble-referenced Kohn±Sham (REKS) method (M. Filatov,
S. Shaik, Chem. Phys. Lett. 304 (1999) 429; M. Filatov, S. Shaik, J. Phys. Chem. A 104 (2000) 6628), developed earlier
to treat the strong non-dynamic correlation in singlet diradicals is extended for doublet states of triradicals. The energy
and the density in the REKS method are represented as weighted sums of energies and densities of several KS de-
terminants. The new method, dubbed REKS(3,3), is applied to calculation of several model problems where the
conventional spin-unrestricted density functional calculations break the spatial symmetry. The results of the REKS(3,3)
calculations show that the method avoids an arti®cial symmetry breaking in open-shell systems. Ó 2000 Elsevier
Science B.V. All rights reserved.
1. Introduction
A while ago it has been recognized [1] that sincethe total non-relativistic many-electron molecularHamiltonian commutes with all respective spinand spatial symmetry operators, its exact eigen-function should also be an eigenfunction of allthese symmetry operators. However, an applica-tion of the variational principle to an approximatewavefunction does not guarantee that this wave-function will possess all necessary symmetryproperties [1]. Thus, an approximate wavefunctionwhich breaks the molecular symmetry may fre-quently have a lower energy. This phenomenon,
known as the `symmetry dilemma' [1], appears inmany situations in quantum-chemical calculations.The well-known examples are the diradicaloidsituations in formally closed-shell molecules withstretched single bonds, twisted double bonds, etc.,where the symmetry-broken spin-unrestricted so-lutions possess lower energy than the symmetry-adapted solutions at the Hartree±Fock (HF) andfrequently at the post-HF (MPn, CC, etc.) levels oftheory [2,3]. In spin-unrestricted calculations onradicals, the spatial symmetry breaking may occurin addition to the spin symmetry breaking [4]. Thisleads to total densities and molecular geometriesthat are distorted away from the correct molecularsymmetry [4±7]. The now-classical examples arethe allyl radical [5], formyloxyl radical [6], NO2 [7],etc., where even the post-HF treatments based ona spin-unrestricted reference yield incorrect mo-lecular structures, dipole moments and vibrationalfrequencies [6,8].
22 December 2000
Chemical Physics Letters 332 (2000) 409±419
www.elsevier.nl/locate/cplett
* Corresponding author. Present address: Department of
Theoretical Chemistry, Goteborg University, Reutersgatan 2,
S-41320 Goteborg, Sweden. Fax: +31-773-5590.
E-mail address: ®[email protected] (M. Filatov).
0009-2614/00/$ - see front matter Ó 2000 Elsevier Science B.V. All rights reserved.
PII: S 0 0 0 9 - 2 6 1 4 ( 0 0 ) 0 1 2 5 7 - 4

Within the conventional approach, CASSCFand MR-(S)DCI calculations with very largeactive space are necessary to eliminate the spatialsymmetry breaking [4]. Such calculations de-scribe properly the mixing of near-degeneratecon®gurations (non-dynamic correlation). How-ever, the high computational cost of such cal-culations limits their applicability to relativelysmall molecules. Furthermore, for the most reli-able conventional multireference methods, theCASPT2, CASPT3 and MR-(S)DCI, the analyticenergy derivatives with respect to the atomiccoordinates are not as yet implemented. Thisrenders the routine use of these methods ex-tremely costly.
Density functional theory (DFT) [9] representsan e�ective alternative to the conventional quan-tum-chemical calculations. Since density func-tionals include electron correlation e�ects byconstruction, this gives in principle some hope thatthe optimization of the Kohn±Sham orbitals willresist the spatial symmetry breaking in radicals.The performance of DFT for symmetry-breakingcases has been studied by Head-Gordon et al. [10].DFT methods indeed tend to avoid arti®cialsymmetry breaking in situations where the con-ventional spin-unrestricted approaches fail [10].The pure density functionals, like BLYP [11,12],BP86 [13,14], BPW91 [15±17], etc., appear to bemore stable with respect to symmetry breakingthan the hybrid density functionals, like B3LYP[18], B3PW91 [19], etc.
However, incorporation of the correlation ef-fects into the one-electron orbitals, like in theKohn±Sham (KS) method [9] in DFT, cannot byconstruction be a full proof against spatialsymmetry breaking. Thus, the spin-unrestrictedB3LYP calculations on the cation-radical of¯uoranthene result in breaking of the expectedC2v symmetry and in vibrational spectrum that isinconsistent with the experiment [20]. The BP86calculations on the same cation-radical do notbreak the expected spatial symmetry and yieldvibrational spectrum in good accord with theexperiment [20]. The spin symmetry breaking inradicals may also lead to unrealistic results ofDFT calculations. Thus, for the ClOO andBrOO radicals the hybrid B3LYP calculations
predict a much too long halogen±oxygen bondlength and unrealistic vibrational frequencies[21]. This arises from a severe spin contamina-tion �S2 > 1:5� in these calculations, but the Cs
spatial symmetry of the total density is notbroken [21].
Although, the pure density functionals are morestable with respect to both spin and spatial sym-metry breaking [10,22], there exist numerous ex-amples where even these functionals fail to resistthe symmetry breaking. A few such examples willbe presented here. All these situations share thecommon feature of the strong non-dynamic cor-relation in doublet state radicals associated withthe presence of a low lying quartet states. In suchcases, the erroneous spin polarization due to anodd electron [23] results in a considerable distor-tion of the total density within the spin-unre-stricted formalism [2±8].
The spin-restricted method with integer occu-pations of the KS orbitals may occasionally fail todescribe the strong non-dynamic correlationproperly, even if the exact (and yet unknown)density functional could have been applied [24,25].Examples of such formally closed-shell systems areC2 [24] and H2 �H2 [25], where it has been dem-onstrated that it is impossible to ®t the density of ahigh-level MR-(S)DCI calculation to a KS for-mulation unless certain orbitals are taken withfractional occupations. Since the phenomenon ofthe non-dynamic correlation is common forclosed- and open-shell systems [1], an idea to usethe fractional occupation numbers of the KSorbitals to describe the strong non-dynamic cor-relation in radicals is deemed both appealing andnecessary. In order to conform with quantumstatistics, these fractional occupation numbers(FONs) must follow from ensemble averaging [26],i.e., the total density should be represented as aweighted sum of densities of several KS determi-nants. Since both the total electronic energy andtotal density are linear in the N-particle densitymatrix, the ensemble representation for the densityimplies the same ensemble representation for thetotal electronic energy [27±29].
In previous Letters [30±32], we relied on a rep-resentation of the energy and density of thestrongly correlated system as weighted sums of
410 M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419

energies and densities of several KS determinants,and thereby have developed a spin-restricted en-semble-referenced Kohn±Sham (REKS) methodapplicable to singlet state diradicals. The testsshowed [31,32] that the REKS method yields re-sults of essentially the same quality as the CASPT2and MR-(S)DCI approaches. Encouraged by thesuccess of the REKS method for diradicals (whichby analogy with CASSCF terminology is denotedhereafter as REKS(2,2)) we extend here this for-malism to doublet states of the triradicaloid sys-tems, i.e., systems where doublet and quartet statesare nearly degenerate.
The present Letter reports the derivation of theworking equations of the new method (denotedfurther as REKS(3,3)) and the results of bench-mark calculations for several triradicaloid systems(alternatively, radicals with strong non-dynamiccorrelation). In Section 2 the equations for theenergy and density in REKS(3,3) are derived fromconsideration of the strong non-dynamic correla-tion in radicals. Section 3 describes details of thedensity functional and conventional multireferencecalculations undertaken in the present study. Sec-tion 4 compares the results of the REKS(3,3) andUKS calculations with the MR-(S)DCI results fora number of triradicaloid situations, such astwisting in the r-cation-radical of ethylene, dis-sociation of C±H bond in CH3 and dissociation ofO±H bond in hydroperoxo radicals, and the elec-tronic structure of the cycloheptatriene-2,5,7-triyltriradical.
2. REKS(3,3) method
To derive the equations for the REKS(3,3) (i.e.,REKS method with 3 `active' electrons in 3 `active'orbitals) we utilize the following strategy. First,the orbitally degenerate states of a certain highlysymmetric system are considered. For this systemthe density functional energy, given as a weightedsum of the single determinant energies, may bederived from symmetry considerations [33]. Sec-ond, a weak perturbation that lifts the orbital de-generacy is applied and the energy expression isderived from consideration of the secular problemin the basis of the states of the highly symmetric
system. Then, the resulting energy expression isrescaled to reproduce the results of the conven-tional spin-restricted single-reference KS calcula-tion for the `normal' systems without strong non-dynamic correlation. Although a number of ap-proximations is introduced during the derivation,the resulting working equations of the REKSmethod provide reasonably good description ofthe non-dynamic correlation. This has been es-tablished for the singlet states of diradicaloids[31,32] and will be demonstrated in the subsequentsections for the doublet states of triradicals.
Let us consider a r-cation-radical of ethylenewhich is 90°-twisted as an example of a triradica-loid system. The molecular symmetry is D2d andthe lowest doublet state is 2B1. The other doubletstates in the same electronic con®guration are the2A2, 2B2, and 2A1 states. If we apply a small twiston the CH2 groups, the symmetry will change to D2
and the 2B1 and 2A1 states become both 2A. Theother doublet states will belong to the B irreduciblerepresentation. Thus, to derive the energy anddensity expressions for the lowest doublet state ofthe r-cation-radical of ethylene at arbitrary twist-ing angle it is su�cient to consider a secularproblem in the basis of the two states, 2B1 and 2A1.
The energies of the 2B1 and 2A1 states may bederived from symmetry considerations and repre-sented as sums of energies of single KS determi-nants. In the 90°-twisted r-cation-radical ofethylene there are three singly occupied orbitals,one r-orbital /r and two p-type orbitals /r and /s
which are subspecies of the two-dimensional irre-ducible representation. Applying the formalismdeveloped by von Barth [33] to this orbitally de-generate situation, it is possible to obtain [30] forthe energies of the 2B1 and 2A1 states Eqs. (1) and(2), respectively.
E�2B1� � 12E�/r/r/r� � 1
2E�/s/s/r�
ÿ 12E�/r/s/r� ÿ 1
2E�/s/r/r�
� 12E�/r/s/r� � 1
2E�/r/s/r�; �1�
E�2A1� � 12E�/r/r/r� � 1
2E�/s/s/r�
� 12E�/r/s/r� � 1
2E�/s/r/r�
ÿ 12E�/r/s/r� ÿ 1
2E�/r/s/r�: �2�
M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419 411

In these equations, E�/r/r/r� denotes the densityfunctional energy of the (single-determinant) con-®guration �� � �/2
r /0s /
1r�, etc. All single KS deter-
minants whose energies enter into Eqs. (1) and (2)are constructed from the same set of orbitals, withthe doubly occupied `core' orbitals /k (not writtenin Eqs. (1) and (2) for brevity) and the `active'orbitals /r, /s, and /r. The orbitals are restrictedto be the same for both, up and down, spin vari-eties.
It should be noted, that despite the use of neg-ative weighting factors the restrictions imposed onthe KS determinants in Eqs. (1) and (2) guaranteethat the energy remains ®nite during variationaloptimization of the orbitals. Furthermore, the useof the negative weighting factors enables one toobtain the energy and the density of a speci®cspectroscopic state from a single variational cal-culation [33]. Had strictly positive weighting fac-tors been employed, then only a mixture of thedoublet states 2B1 and 2A1 would have resulted byuse of rigorous ensemble approach [27±29]. Insuch an event, it would have been necessary tosolve several variational problems for various en-sembles of di�erent spectroscopic states in order toobtain energy of a speci®c state. Thus, we stick tothe state-speci®c approach as proposed and justi-®ed in Ref. [33].
Let us now twist the CH2 groups slightly,thereby lifting the degeneracy of /r and /s orbi-tals. In the D2 symmetric group, both states 2B1
and 2A1 belong to the same irreducible repre-sentation A, and should therefore mix. Theenergy of the lowest 2A state may be obtainedfrom the two-dimensional secular problem inthe basis of the 2B1 and 2A1 states whose non-interacting reference wave functions are given inEqs. (3) and (4)
W 2B1
ÿ � � 1���2p /r/r/r
�� ��ÿ ÿ /s/s/r
�� ���; �3�
W 2A1
ÿ � � 1���2p /r/r/r
�� ��ÿ � /s/s/r
�� ���: �4�
The coupling matrix element between these statesis given by Eq. (5)
D � W 2B1
ÿ �H��� ���W 2A1
ÿ �D E� 1
2E /r/r/r
ÿ �ÿ ÿ E /s/s/r
ÿ ��; �5�
where H is an e�ective Hamiltonian, which uponacting on the reference wave function yields therespective density functional energy.
Resolving the aforementioned secular problemfor the lowest energy state and imposing a re-quirement that the resulting energy expressionshould reproduce the conventional spin-restrictedopen-shell KS calculation for a normal state(therefore, no double counting of correlation)[31,32], we arrive at the expression for the energyof the 2A state, Eq. (6)
E 2Aÿ � � nr
2E /r/r/r
ÿ �� ns
2E /s/s/r
ÿ �� nr; ns� �3=4 1
2E /r/s/r� ��
� 12E /r/s/r
ÿ �ÿ 12E /r/s/r
ÿ �ÿ 1
2E /s/r/r
ÿ ��: �6�
Here nr and ns are the fractional occupationnumbers of orbitals /r and /s which are obtainedvariationally together with orbitals. The same ex-pression holds also for the density, with the singledeterminant energies in Eq. (6) replaced by thedensities of single KS determinants. It is easy tosee that with the quali®cations adopted in REKS,such a density reduces to density with FONs,Eq. (7)
qREKS�3;3� r� � �X
k 2 core
2 /k�r�j j2 � nr /r�r�j j2
� ns /s r� �j j2 � /r r� �j j2: �7�With this energy expression, Eq. (6), it is possibleto describe the twisting curve of the ethylener-cation-radical all the way from the normal 2Ag
state of planar species to the strongly correlated2B1 state of 90°-twisted species. However, the sameformula, Eq. (6), when applied to another situa-tion with strong non-dynamic correlation in radi-cals, e.g. the dissociation of the C±H bond in CH3,yields an incorrect answer for the completely dis-sociated bond. Instead of yielding the sum of en-ergies of the 3B1 state of methylene and the 1S stateof hydrogen at the dissociation limit, this formula
412 M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419

yields higher energy. This implies that such a for-mula is not size-consistent for problems of bonddissociation in radicals.
An analysis of the reasons for such a behaviorof Eq. (6) reveals (see Appendix A) that in the caseof bond dissociation in CH3, it is not su�cient toconsider the secular problem in the basis of onlytwo states given in Eqs. (3) and (4). Now, it isnecessary to consider a 4� 4 secular problem inthe basis of the four states given in Appendix A.The solution of the 2� 2 secular problem, repre-sented by Eq. (6), contains contributions fromhigh-lying doublet states. It does not seem possibleto derive a compact energy expression of the typeof Eq. (6) from the full 4� 4 secular problem.However, it is possible to mimic the 4� 4 solution(see Appendix A) and to impose the correct dis-sociation limit in the case of the dissociating rad-ical by adding a simple correcting term,12E�/r/s/r� ÿ 1
2E�/r/s/r�, to the bracketed term
in Eq. (6). This term restores the correct energy ofthe dissociated CH3 radical and eliminates theadmixture of the high-lying doublet state. Thus,Eq. (6) turns to Eq. (8)
EREKS�3;3� � nr
2E /r/r/r
ÿ �� ns
2E�/s/s/r�
� �nrns�3=4 E�/r/s/r��
ÿ 12E�/r/s/r� ÿ
1
2E�/s/r/r�
�: �8�
Eq. (8) is adopted in the REKS(3,3) methodfor the general case of a doublet state triradical.Although, this expression does not converge toEq. (1) for 90°-twisted r-cation radical of ethylene,the discrepancy is small, of the order of a few kcal/mol. With that, Eq. (8) is perfectly size-consistentand avoids the arti®cial symmetry breaking instrongly correlated states of radicals. In contrast toEq. (6), Eq. (8) is derived from consideration of astrongly correlated system where two fractionallyoccupied orbitals, /r and /s, belong to the sameirreducible representation. Thus, Eq. (8) may alsobe applied to the calculation of non-symmetric(C1) systems. As will be demonstrated, REKS(3,3)compares well with MR-(S)DCI, and the use ofEq. (8) does not result in any distortion of thepotential energy surfaces of the studied species.
The one-electron orbitals and fractional occu-pation numbers are obtained in the REKS(3,3)method self-consistently by minimizing the energy,Eq. (8), with respect to the density, Eq. (7). Sincethe REKS ground-state energy, Eq. (8), is not anexplicit functional of the density, Eq. (7), itsfunctional derivative is not easily evaluated. Thus,instead of constructing a multiplicative KS po-tential from the orbitals by the optimized e�ectivepotential method, we stick to an alternative ap-proach [34] adopted in DFT when dealing withorbital-dependent and hybrid density functionals.Namely, the energy is minimized with respect tothe orbitals (under constraint of orthonormality)and construction of the multiplicative KS potentialis abandoned [34]. The further details of theREKS(3,3) implementation are the same as thoseof REKS(2,2) and may be found in our previousworks [30,31].
Finally, a comment on the calculation of thehigh-spin (quartet) states of triradicaloids is deemednecessary. In the REKS method, the orbitals forthe spin-up and spin-down electrons are restrictedto be the same. Thus, the same spin-symmetry re-striction should be used in the calculation of thehigh-spin states. This suggests that the naturalcounterpart of REKS(3,3) for the calculation ofquartet triradicaloid species is the spin-restrictedopen-shell KS method described, e.g. in Ref. [30].
3. Details of calculations
The REKS(3,3) method has been implementedin the CADPAC5 quantum-chemical package [35].The self-consistent calculations as well as the an-alytical gradients are available. All REKS andmost of UKS calculations reported have beendone with the CADPAC5 program. These calcu-lations employ the BLYP density functional[11,12]. Conventional UKS density functionalcalculations which employ the B3LYP [18] andB3PW91 [19] functionals have been done with theGAUSSIANAUSSIAN98 package [36]. Geometries of allspecies are optimized with REKS and UKSmethods.
The reference wave functions for MR-(S)DCI(corrected to full CI [37]) have been obtained from
M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419 413

the CASSCF calculations, and both methods uti-lized the MOLPRO2000 package 1. CASSCF op-timized geometries are used and the active space inthe CASSCF and MR-(S)DCI calculations in-volved all valence electrons and valence orbitals(unless noted otherwise).
The basis sets cc-pVTZ [38] and 6-31G* [39]were used and involve Cartesian d- and f-func-tions.
4. Results and discussion
4.1. Bond dissociation curves
The utility of the REKS(3,3) method can bedemonstrated by studying the dissociation of sin-gle bonds in radicals. Let us compare C±H and O±Hbond dissociation for CH3 and OOH as a test ofREKS(3,3) and UKS against the MR-(S)DCIbenchmark method.
For CH3 we studied the C±H dissociation underC2v symmetry with the principal symmetry axisalong the dissociating C±H bond. The groundstate of CH3 is 2B1 in C2v and it dissociates into amethylene in the ground 3B1 state and a hydrogenatom. The calculations employ the cc-pVTZ basisset. In the REKS(3,3) calculation the singly occu-pied orbital is the 1b1 orbital and the fractionallyoccupied orbitals are the 2a1 and 3a1 orbitalsshown in Fig. 1. All bond lengths and valenceangles except for the dissociating C±H bond lengthwere optimized. The dissociation curve is pre-sented in Fig. 1. The zero line corresponds to thesum of energies of the triplet methylene and thehydrogen atom calculated separately. The curvesin Fig. 1 show that both density functional meth-ods, REKS(3,3) and UKS, are size-consistent andare capable to describe the dissociation of singlebond in CH3 radical as well as MR-(S)DCI.
The hydroperoxo radical OOH has two lowlying doublet states, the ground 2A00 state and ex-cited 2A00 state. The O±H bond dissociation in the
ground state results in oxygen molecule in its3Rÿg �3A00 in Cs) ground state and hydrogen atom.The excited state OOH dissociates into singletstate oxygen (1A00 in Cs) and hydrogen atom. Forboth states, the calculations have been done withthe cc-pVTZ basis set. The O±O bond length andOOH valence angle were optimized.
Fig. 2 shows the dissociation curve for the 2A00
OOH radical. Basically, the curve looks very sim-ilar to the previous case (CH3 in Fig. 1) and bothREKS(3,3) and UKS describe this dissociationreasonably well. However, according to MR-(S)DCI the 2A0 state of OOH features a smallpotential barrier on the dissociation curve. Thisbarrier is reproduced by REKS(3,3) but not byUKS (Fig. 3).
Let us assume that the OOH radical lies in thexz-plane with both oxygen atoms lying along the z-axis. Then, in the 2A00 state, the 2a00 molecularorbital which originates from the dioxygen p�y or-bital is singly occupied. The doubly occupied 8a0
orbital is a bonding combination of the dioxygenp�x orbital and the hydrogen s-orbital while the 9a0
is the antibonding counterpart. These orbitals areincluded into the active space of REKS(3,3) wherethe 2a00 orbital is singly occupied and the 8a0 and
1 MOLPROOLPRO is a package of ab initio prorgams written by H.-J.
Werner, P.J. Knowles, with contributions from R.D. Amos
et al.
Fig. 1. C±H bond dissociation in CH3 radical. The MR-(S)DCI
curve is drawn with a solid line, the REKS(3,3)/BLYP curve
with a dashed±dotted line and the UKS curve with a dotted
line. The horizontal line corresponds to sum of energies of
dissociation products, triplet (3B1) methylene and hydrogen
atom. The inset in the plot shows the frontier orbitals of CH3
used in the REKS(3,3) calculation. The 1b1 orbital is singly
occupied and the 2a1 and 3a1 orbitals are fractionally occupied
in the REKS(3,3) calculation.
414 M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419
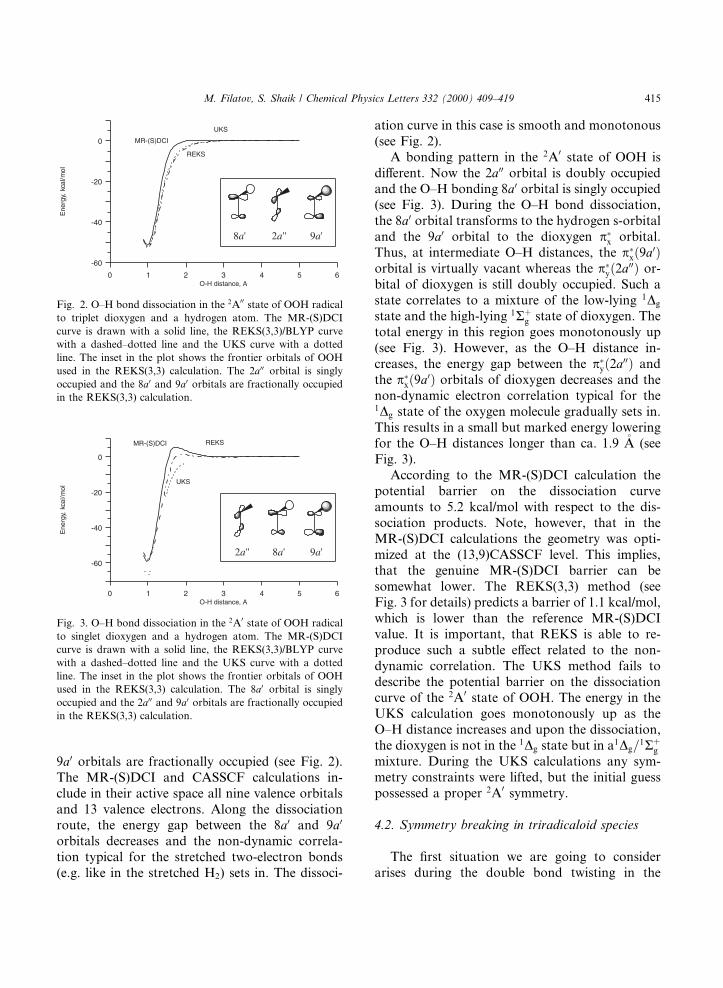
9a0 orbitals are fractionally occupied (see Fig. 2).The MR-(S)DCI and CASSCF calculations in-clude in their active space all nine valence orbitalsand 13 valence electrons. Along the dissociationroute, the energy gap between the 8a0 and 9a0
orbitals decreases and the non-dynamic correla-tion typical for the stretched two-electron bonds(e.g. like in the stretched H2) sets in. The dissoci-
ation curve in this case is smooth and monotonous(see Fig. 2).
A bonding pattern in the 2A0 state of OOH isdi�erent. Now the 2a00 orbital is doubly occupiedand the O±H bonding 8a0 orbital is singly occupied(see Fig. 3). During the O±H bond dissociation,the 8a0 orbital transforms to the hydrogen s-orbitaland the 9a0 orbital to the dioxygen p�x orbital.Thus, at intermediate O±H distances, the p�x�9a0�orbital is virtually vacant whereas the p�y�2a00� or-bital of dioxygen is still doubly occupied. Such astate correlates to a mixture of the low-lying 1Dg
state and the high-lying 1R�g state of dioxygen. Thetotal energy in this region goes monotonously up(see Fig. 3). However, as the O±H distance in-creases, the energy gap between the p�y�2a00� andthe p�x�9a0� orbitals of dioxygen decreases and thenon-dynamic electron correlation typical for the1Dg state of the oxygen molecule gradually sets in.This results in a small but marked energy loweringfor the O±H distances longer than ca. 1.9 �A (seeFig. 3).
According to the MR-(S)DCI calculation thepotential barrier on the dissociation curveamounts to 5.2 kcal/mol with respect to the dis-sociation products. Note, however, that in theMR-(S)DCI calculations the geometry was opti-mized at the (13,9)CASSCF level. This implies,that the genuine MR-(S)DCI barrier can besomewhat lower. The REKS(3,3) method (seeFig. 3 for details) predicts a barrier of 1.1 kcal/mol,which is lower than the reference MR-(S)DCIvalue. It is important, that REKS is able to re-produce such a subtle e�ect related to the non-dynamic correlation. The UKS method fails todescribe the potential barrier on the dissociationcurve of the 2A0 state of OOH. The energy in theUKS calculation goes monotonously up as theO±H distance increases and upon the dissociation,the dioxygen is not in the 1Dg state but in a1Dg=
1R�gmixture. During the UKS calculations any sym-metry constraints were lifted, but the initial guesspossessed a proper 2A0 symmetry.
4.2. Symmetry breaking in triradicaloid species
The ®rst situation we are going to considerarises during the double bond twisting in the
Fig. 3. O±H bond dissociation in the 2A0 state of OOH radical
to singlet dioxygen and a hydrogen atom. The MR-(S)DCI
curve is drawn with a solid line, the REKS(3,3)/BLYP curve
with a dashed±dotted line and the UKS curve with a dotted
line. The inset in the plot shows the frontier orbitals of OOH
used in the REKS(3,3) calculation. The 8a0 orbital is singly
occupied and the 2a00 and 9a0 orbitals are fractionally occupied
in the REKS(3,3) calculation.
Fig. 2. O±H bond dissociation in the 2A00 state of OOH radical
to triplet dioxygen and a hydrogen atom. The MR-(S)DCI
curve is drawn with a solid line, the REKS(3,3)/BLYP curve
with a dashed±dotted line and the UKS curve with a dotted
line. The inset in the plot shows the frontier orbitals of OOH
used in the REKS(3,3) calculation. The 2a00 orbital is singly
occupied and the 8a0 and 9a0 orbitals are fractionally occupied
in the REKS(3,3) calculation.
M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419 415
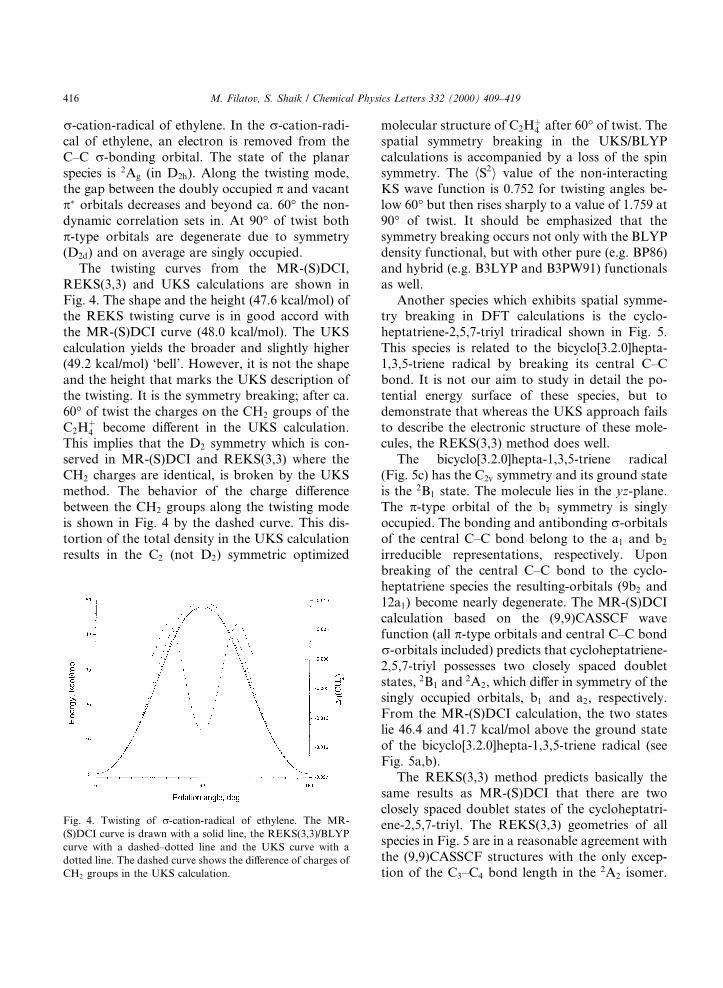
r-cation-radical of ethylene. In the r-cation-radi-cal of ethylene, an electron is removed from theC±C r-bonding orbital. The state of the planarspecies is 2Ag (in D2h). Along the twisting mode,the gap between the doubly occupied p and vacantp� orbitals decreases and beyond ca. 60° the non-dynamic correlation sets in. At 90° of twist bothp-type orbitals are degenerate due to symmetry(D2d) and on average are singly occupied.
The twisting curves from the MR-(S)DCI,REKS(3,3) and UKS calculations are shown inFig. 4. The shape and the height (47.6 kcal/mol) ofthe REKS twisting curve is in good accord withthe MR-(S)DCI curve (48.0 kcal/mol). The UKScalculation yields the broader and slightly higher(49.2 kcal/mol) `bell'. However, it is not the shapeand the height that marks the UKS description ofthe twisting. It is the symmetry breaking; after ca.60° of twist the charges on the CH2 groups of theC2H�4 become di�erent in the UKS calculation.This implies that the D2 symmetry which is con-served in MR-(S)DCI and REKS(3,3) where theCH2 charges are identical, is broken by the UKSmethod. The behavior of the charge di�erencebetween the CH2 groups along the twisting modeis shown in Fig. 4 by the dashed curve. This dis-tortion of the total density in the UKS calculationresults in the C2 (not D2) symmetric optimized
molecular structure of C2H�4 after 60° of twist. Thespatial symmetry breaking in the UKS/BLYPcalculations is accompanied by a loss of the spinsymmetry. The hS2i value of the non-interactingKS wave function is 0.752 for twisting angles be-low 60° but then rises sharply to a value of 1.759 at90° of twist. It should be emphasized that thesymmetry breaking occurs not only with the BLYPdensity functional, but with other pure (e.g. BP86)and hybrid (e.g. B3LYP and B3PW91) functionalsas well.
Another species which exhibits spatial symme-try breaking in DFT calculations is the cyclo-heptatriene-2,5,7-triyl triradical shown in Fig. 5.This species is related to the bicyclo[3.2.0]hepta-1,3,5-triene radical by breaking its central C±Cbond. It is not our aim to study in detail the po-tential energy surface of these species, but todemonstrate that whereas the UKS approach failsto describe the electronic structure of these mole-cules, the REKS(3,3) method does well.
The bicyclo[3.2.0]hepta-1,3,5-triene radical(Fig. 5c) has the C2v symmetry and its ground stateis the 2B1 state. The molecule lies in the yz-plane.The p-type orbital of the b1 symmetry is singlyoccupied. The bonding and antibonding r-orbitalsof the central C±C bond belong to the a1 and b2
irreducible representations, respectively. Uponbreaking of the central C±C bond to the cyclo-heptatriene species the resulting-orbitals (9b2 and12a1) become nearly degenerate. The MR-(S)DCIcalculation based on the (9,9)CASSCF wavefunction (all p-type orbitals and central C±C bondr-orbitals included) predicts that cycloheptatriene-2,5,7-triyl possesses two closely spaced doubletstates, 2B1 and 2A2, which di�er in symmetry of thesingly occupied orbitals, b1 and a2, respectively.From the MR-(S)DCI calculation, the two stateslie 46.4 and 41.7 kcal/mol above the ground stateof the bicyclo[3.2.0]hepta-1,3,5-triene radical (seeFig. 5a,b).
The REKS(3,3) method predicts basically thesame results as MR-(S)DCI that there are twoclosely spaced doublet states of the cycloheptatri-ene-2,5,7-triyl. The REKS(3,3) geometries of allspecies in Fig. 5 are in a reasonable agreement withthe (9,9)CASSCF structures with the only excep-tion of the C3±C4 bond length in the 2A2 isomer.
Fig. 4. Twisting of r-cation-radical of ethylene. The MR-
(S)DCI curve is drawn with a solid line, the REKS(3,3)/BLYP
curve with a dashed±dotted line and the UKS curve with a
dotted line. The dashed curve shows the di�erence of charges of
CH2 groups in the UKS calculation.
416 M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419

Note, however, that the ab initio molecularstructures are obtained at the CASSCF level whichdoes not treat the dynamic electron correlationsu�ciently. The energy di�erences obtained at theREKS(3,3) level are in good agreement with theMR-(S)DCI values. In the REKS(3,3) calculationsthe singly occupied orbitals are the 3b1 and 2a2
orbitals for the 2B1 and 2A2 states, respectively,and the fractionally occupied orbitals are the 9b2
and 12a1 orbitals for both state symmetries. Eventhough, no symmetry constraints were imposed onthe orbitals during REKS calculations the calcu-
lations converged naturally to the C2v symmetricspecies.
A completely di�erent picture emerges fromthe UKS approach. Although for the bicy-clo[3.2.0]hepta-1,3,5-triene the UKS/BLYP calcu-lation yields virtually the same molecular structureas the REKS(3,3), neither of the two states of themonocyclic species can be obtained with UKS.The UKS breaks the spatial symmetry of themonocyclic species and instead of the C2v struc-tures collapses to the Cs symmetric structures (seeFig. 5). Furthermore, UKS does not distinguish
Fig. 5. Molecular structures ��A� and relative energies (kcal/mol) of the 2B1 (a) and 2A2 (b) states of cycloheptatriene-2,5,7-triyl tri-
radical and of the ground 2B1 (c) state bicyclo[3.2.0]hepta-1,3,5-triene radical. The MR-(S)DCI data are given with a boldface font, the
REKS(3,3) with a normal font and the UKS data are italicized.
M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419 417

between the 2B1 and 2A2 states, and converges tothe same Cs symmetric structure for both states,irrespective of the starting points (geometries, ini-tial guesses).
The reason for such a behavior is simple.Within the spin-unrestricted approach (UKS orUHF) a broken single bond is described by a wavefunction where a spin-up electron resides on oneatom and spin-down electron on another. If anadditional unpaired electron with spin up is pre-sent, this odd electron will experience an exchangeinteraction with one side of the molecule and nonewith the other side of the molecule. Thus, the totaldensity will be arti®cially polarized. It should beemphasized that even at the starting C2v symmetricmolecular geometry the UKS method yields thedistorted Cs symmetric density. Such a densitydistortion is not con®rmed by the CASSCF norMR-(S)DCI calculations and is certainly an arti-fact of the UKS method. Additional tests (BP86,B3LYP, B3PW91) show the same spatial symme-try breaking. This is an inherent property of thespin polarized form in UKS that is recti®ed byREKS(3,3).
5. Conclusions
We have presented REKS(3,3); a computa-tional scheme based on an ensemble approach indensity functional theory which provides an ade-quate treatment of the strong non-dynamic cor-relation in radicals. REKS(3,3) removes thearti®cial symmetry breaking which appears indensity functional and conventional ab initio cal-culations based on the spin-unrestricted referencewavefunction. Despite several approximations, thenew method provides a reasonably good descrip-tion of the strong non-dynamic correlation intriradicaloid species. A comparison with the high-level MR-(S)DCI calculations shows that theREKS(3,3) yields results which are of essentiallythe same quality as sophisticated multireferenceapproaches.
Like its predecessor, REKS(2,2), the newREKS(3,3) scheme is independent of the choice ofdensity functional. It can be used together withany existing density functional, pure or hybrid.
Acknowledgements
This work is supported in part by the RobertSzold Fund and the German±Israeli Foundation(GIF).
Appendix A. Bond dissociation in CH3 radical
Let us consider the C±H bond stretching in CH3
radical. The stretched bond is along the z-axis andthe radical is in the xz-plane. The active orbitalsare (see also Fig. 1): the C±H bonding orbital /r,the C±H antibonding orbital /s and the singlyoccupied p-type orbital /y. The ®rst two orbitalsbelong to the a1 representation in C2v group, whilethe last one belongs to the b1 representation. Fromthe three orbitals four doublet states of b1 sym-metry may be constructed:
W1 � 1���2p /r/r/y
�� �ÿ ÿ /s/s/y
�� ��;
W2 � 1���2p /r/r/y
�� �ÿ � /s/s/y
�� ��;
W3 � 1���2p /r/s/y
�� �ÿ � /s/r/y
�� ��;
and
W4 � 1���6p 2 /r/s/y
�� �ÿ ÿ /r/s/y
�� �� /s/r/y
�� ��:
Near the equilibrium C±H bond length the largesto�-diagonal matrix element in the corresponding4� 4 secular problem is the W1h jH W2j i elementgiven in Eq. (5) (with /r replaced by /y). Thus, theenergy of the CH3 radical in this region may beapproximated by Eq. (6) with nr � 2 and ns � 0.
At large C±H distance, most of the o�-diagonalmatrix elements of the Hamiltonian vanish. Thenon-vanishing elements are the hW1jH jW4i andhW2jH jW3i elements. Now, the 4� 4 secularproblem reduces to the two uncoupled 2� 2problems, �W1;W4� and �W2;W3�; which describe apair of purely covalent and a pair of purely ionicdoublet states of dissociated CH3. The lowest en-ergy covalent state is � ���3p =2�W1 ÿ �1=2�W4 and itsenergy equals the energy of the quartet statej/r/s/yi. In Section 2, the �W1;W2� secular prob-lem is considered and its solution for the com-
418 M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419

pletely dissociated C±H bond in CH3 is repre-sented by W1. Although W1 still dominates in thecorrect dissociated state, its energy, given by Eq.(6) with nr � ns � 1, is �1=2�E�/r/s/y� � �1=2�E�/r/s/y�. Thus, the correct energetic description ofthe bond dissociation may be imposed by addinga term �1=2�E�/r/s/y�ÿ�1=2�E�/r/s/y� to thebracketed term in Eq. (6).
References
[1] P.-O. L�owdin, Adv. Chem. Phys. 14 (1969) 283.
[2] W. Chen, H.B. Schlegel, J. Chem. Phys. 101 (1994) 5957.
[3] H. Yuan, D. Cremer, Chem. Phys. Lett. 324 (2000) 389.
[4] E.R. Davidson, W.T. Borden, J. Phys. Chem. 87 (1983)
4783.
[5] A.F. Voter, W.A. Goddard, Chem. Phys. 57 (1981) 253.
[6] P.Y. Ayala, H.B. Schlegel, J. Chem. Phys. 108 (1998) 7560.
[7] C.F. Jackels, E.R. Davidson, J. Chem. Phys. 64 (1976)
2908.
[8] T.D. Crawford, J.F. Stanton, J. Chem. Phys. 112 (2000)
7873.
[9] W. Kohn, L.J. Sham, Phys. Rev. 140 (1965) A1133.
[10] C.D. Sherrill, M.S. Lee, M. Head-Gordon, Chem. Phys.
Lett. 302 (1999) 425.
[11] A.D. Becke, Phys. Rev. A 38 (1988) 3098.
[12] C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37 (1988) 785.
[13] J.P. Perdew, Phys. Rev. B 33 (1986) 8822.
[14] J.P. Perdew, Phys. Rev. B 34 (1986) 7406(E).
[15] J.P. Perdew, Y. Wang, Phys. Rev. B 45 (1992) 13244.
[16] J.P. Perdew, J.A. Chevary, S.H. Vosko, K.A. Jackson,
M.R. Pederson, D.J. Singh, C. Fiolhais, Phys. Rev. B 46
(1992) 6671.
[17] J.P. Perdew, J.A. Chevary, S.H. Vosko, K.A. Jackson,
M.R. Pederson, D.J. Singh, C. Fiolhais, Phys. Rev. B 48
(1993) 4978(E).
[18] P.J. Stevens, F.J. Devlin, C.F. Chablowski, M.J. Frisch,
J. Phys. Chem. 98 (1994) 11623.
[19] A.D. Becke, J. Chem. Phys. 98 (1993) 1372.
[20] C.W. Bauschlicher, D.M. Hudgins, L.J. Allamandola,
Theor. Chem. Acc. 103 (1999) 154.
[21] M. Alcami, O. M�o, M. Y�a~nez, I.L. Cooper, J. Chem. Phys.
112 (2000) 6131.
[22] R. Bauernschmitt, R. Ahlrichs, J. Chem. Phys. 104 (1996)
9047.
[23] D.M. Chipman, Theor. Chim. Acta 82 (1992) 93.
[24] P.R.T. Schipper, O.V. Gritsenko, E.J. Baerends, Theor.
Chem. Acc. 99 (1998) 329.
[25] P.R.T. Schipper, O.V. Gritsenko, E.J. Baerends, J. Chem.
Phys. 111 (1999) 4056.
[26] M.M. Valiev, G.W. Fernando, Phys. Rev. B 52 (1995)
10697.
[27] E.H. Lieb, Int. J. Quantum Chem. 24 (1983) 243.
[28] H. Englisch, R. English, Phys. Stat. Sol. (b) 123 (1984)
711.
[29] H. Englisch, R. English, Phys. Stat. Sol. (b) 124 (1984)
373.
[30] M. Filatov, S. Shaik, J. Chem. Phys. 110 (1999) 116.
[31] M. Filatov, S. Shaik, Chem. Phys. Lett. 304 (1999) 429.
[32] M. Filatov, S. Shaik, J. Phys. Chem. A 104 (2000) 6628.
[33] U. von Barth, Phys. Rev. A 20 (1979) 1693.
[34] R. Neumann, R.H. Nobes, N.C. Handy, Mol. Phys. 87
(1996) 1.
[35] R.D. Amos, et al., CADPAC5, The Cambridge Analytic
Derivatives Package, Cambridge, UK, 1992.
[36] M.J. Frisch, et al., GAUSSIANAUSSIAN98, Gaussian, Inc., Pitts-
burgh, PA, 1998.
[37] E.R. Davidson, Adv. Quantum Chem. 6 (1972) 235.
[38] T.H. Dunning, Jr., J. Chem. Phys. 90 (1989) 1007.
[39] W.J. Hehre, L. Radom, P. von R.Schleyer, J.A. Pople,
Ab Initio Molecular Orbital Theory, Wiley-Interscience,
New York, 1986.
M. Filatov, S. Shaik / Chemical Physics Letters 332 (2000) 409±419 419




















