
A Novel Heavy-Atom Label for Side-Specific Peptide Iodination: Synthesis, Membrane Incorporation and...
10
DOI: 10.1002/cphc.200900241 A Novel Heavy-Atom Label for Side-Specific Peptide Iodination: Synthesis, Membrane Incorporation and X-ray Reflectivity Philipp E. Schneggenburger, [a] AndrȖ Beerlink, [b] Brigitte Worbs, [a] Tim Salditt,* [b] and Ulf Diederichsen* [a] 1. Introduction In recent years, an increasing need in biomedical and biophysi- cal research for techniques and tools capable of probing the structure and conformation of membrane-active proteins and peptides has arisen. Hence, it is essential to investigate ion channels, pores or active surface proteins in the native fluid state of lipid membranes. [1] X-ray diffraction on membrane surfaces has proven to be a powerful method providing access to a variety of membrane parameters like the membrane thickness D, the water content between stacked bilayers, the repeat spacing, the mosaicity and the bilayer integrity (Figure 1). [2] Depending on the experi- mental setup, the structural information can either be related to the lateral membrane dimensions (GISAXS = grazing-inci- dence small-angle X-ray scattering) or to the z-direction (reflec- tivity) of the lipid bilayer. [3, 4] Investigating membrane-active proteins or peptides, the change in the membrane geometry upon peptide insertion contains information about a successful reconstitution of the peptide species within membrane lipid bilayers. However, the membrane response does not reveal the depth of peptide in- sertion, nor does it allow distinguishing between a membrane- spanning orientation or a membrane-surface anchoring. This question is better addressed with two-dimensional X-ray scat- tering and grazing-incidence diffraction (GID) using labeled lipids in combination with reciprocal space mapping or with fluorescence emission and FRET experiments respectively. [5, 6] A shift in the fluorescence emission bands of amino acid residues like the indole ring of tryptophane can be observed upon membrane insertion, but this experiment is solely appropriate to distinguish between a hydrophobic and a hydrophilic envi- ronment. [7, 8] FRET experiments on the other hand rely on fluo- rescence labeling of the lipid molecules and are often prob- lematic in case of lipid mixtures. [9] In addition, FRET experi- ments are usually performed in vesicle membrane systems with a limitation in resolution caused by the relatively large- scaled Fçrster radii of the fluorescence probes. Applying fluo- rescence microscopy with a resolution typically at the micron scale down to a few hundred nanometers at best, impedes a geometrical analysis of transmembrane peptide domains. [10, 11] On the other hand, just as in crystallography, heavy-atom la- beling in combination with anomalous X-ray scattering can provide important structural constraints, for example, to locate an individual amino acid in the lipid bilayer with respect to the Structural parameters, such as conformation, orientation and penetration depth of membrane-bound peptides and proteins that may function as channels, pores or biocatalysts, are of persistent interest and have to be probed in the native fluid state of a membrane. X-ray scattering in combination with heavy-atom labeling is a powerful and highly appropriate method to reveal the position of a certain amino acid residue within a lipid bilayer with respect to the membrane normal axis up to a resolution of several ĸngstrøm. Herein, we report the synthesis of a new iodine-labeled amino acid building block. This building block is intended for peptide incorporation to provide high intensities for electron density difference anal- ysis of X-ray reflectivity data and improve the labeling poten- tial for the lipid bilayer head-group and water region. The novel building block as well as the commercially available non- iodinated analogue, required for X-ray scattering, was imple- mented in a transmembrane peptide motif via manual solid- phase peptide synthesis (SPPS) following the fluorenylmethyl- oxycarbonyl (Fmoc)-strategy. The derived peptides were recon- stituted in lipid vesicles as well as in highly aligned multilamel- lar lipid stacks and investigated via circular dichroism (CD) and X-ray reflectivity. Thereby, it has been revealed that the bulky iodine probe neither causes conformational change of the peptide structure nor lamellar disordering of the membrane complexes. [a] P. E. Schneggenburger, B. Worbs, Prof. Dr. U. Diederichsen Institut fɒr Organische und Biomolekulare Chemie Georg-August-UniversitȨt Gçttingen Tammannstr. 2, D-37077 Gçttingen (Germany) Fax: (+ 49) 551-39322944 E-mail : [email protected] [b] A. Beerlink, Prof. Dr. T. Salditt Institut fɒr Rçntgenphysik Georg-August-UniversitȨt Gçttingen Friedrich-Hund-Platz 1, D-37077 Gçttingen (Germany) Fax: (+ 49) 551-399430 E-mail : [email protected] Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cphc.200900241. ChemPhysChem 2009, 10, 1567 – 1576 # 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1567
Transcript of A Novel Heavy-Atom Label for Side-Specific Peptide Iodination: Synthesis, Membrane Incorporation and...

DOI: 10.1002/cphc.200900241
A Novel Heavy-Atom Label for Side-Specific PeptideIodination: Synthesis, Membrane Incorporation and X-rayReflectivityPhilipp E. Schneggenburger,[a] Andr� Beerlink,[b] Brigitte Worbs,[a] Tim Salditt,*[b] andUlf Diederichsen*[a]
1. Introduction
In recent years, an increasing need in biomedical and biophysi-cal research for techniques and tools capable of probing thestructure and conformation of membrane-active proteins andpeptides has arisen. Hence, it is essential to investigate ionchannels, pores or active surface proteins in the native fluidstate of lipid membranes.[1]
X-ray diffraction on membrane surfaces has proven to be apowerful method providing access to a variety of membraneparameters like the membrane thickness D, the water contentbetween stacked bilayers, the repeat spacing, the mosaicityand the bilayer integrity (Figure 1).[2] Depending on the experi-mental setup, the structural information can either be relatedto the lateral membrane dimensions (GISAXS = grazing-inci-dence small-angle X-ray scattering) or to the z-direction (reflec-tivity) of the lipid bilayer.[3, 4]
Investigating membrane-active proteins or peptides, thechange in the membrane geometry upon peptide insertioncontains information about a successful reconstitution of thepeptide species within membrane lipid bilayers. However, themembrane response does not reveal the depth of peptide in-sertion, nor does it allow distinguishing between a membrane-spanning orientation or a membrane-surface anchoring. Thisquestion is better addressed with two-dimensional X-ray scat-tering and grazing-incidence diffraction (GID) using labeledlipids in combination with reciprocal space mapping or withfluorescence emission and FRET experiments respectively.[5, 6] Ashift in the fluorescence emission bands of amino acid residueslike the indole ring of tryptophane can be observed upon
membrane insertion, but this experiment is solely appropriateto distinguish between a hydrophobic and a hydrophilic envi-ronment.[7, 8] FRET experiments on the other hand rely on fluo-rescence labeling of the lipid molecules and are often prob-lematic in case of lipid mixtures.[9] In addition, FRET experi-ments are usually performed in vesicle membrane systemswith a limitation in resolution caused by the relatively large-scaled Fçrster radii of the fluorescence probes. Applying fluo-rescence microscopy with a resolution typically at the micronscale down to a few hundred nanometers at best, impedes ageometrical analysis of transmembrane peptide domains.[10, 11]
On the other hand, just as in crystallography, heavy-atom la-beling in combination with anomalous X-ray scattering canprovide important structural constraints, for example, to locatean individual amino acid in the lipid bilayer with respect to the
Structural parameters, such as conformation, orientation andpenetration depth of membrane-bound peptides and proteinsthat may function as channels, pores or biocatalysts, are ofpersistent interest and have to be probed in the native fluidstate of a membrane. X-ray scattering in combination withheavy-atom labeling is a powerful and highly appropriatemethod to reveal the position of a certain amino acid residuewithin a lipid bilayer with respect to the membrane normalaxis up to a resolution of several �ngstrøm. Herein, we reportthe synthesis of a new iodine-labeled amino acid buildingblock. This building block is intended for peptide incorporationto provide high intensities for electron density difference anal-ysis of X-ray reflectivity data and improve the labeling poten-
tial for the lipid bilayer head-group and water region. Thenovel building block as well as the commercially available non-iodinated analogue, required for X-ray scattering, was imple-mented in a transmembrane peptide motif via manual solid-phase peptide synthesis (SPPS) following the fluorenylmethyl-oxycarbonyl (Fmoc)-strategy. The derived peptides were recon-stituted in lipid vesicles as well as in highly aligned multilamel-lar lipid stacks and investigated via circular dichroism (CD) andX-ray reflectivity. Thereby, it has been revealed that the bulkyiodine probe neither causes conformational change of thepeptide structure nor lamellar disordering of the membranecomplexes.
[a] P. E. Schneggenburger, B. Worbs, Prof. Dr. U. DiederichsenInstitut f�r Organische und Biomolekulare ChemieGeorg-August-Universit�t GçttingenTammannstr. 2, D-37077 Gçttingen (Germany)Fax: (+ 49) 551-39322944E-mail : [email protected]
[b] A. Beerlink, Prof. Dr. T. SaldittInstitut f�r RçntgenphysikGeorg-August-Universit�t GçttingenFriedrich-Hund-Platz 1, D-37077 Gçttingen (Germany)Fax: (+ 49) 551-399430E-mail : [email protected]
Supporting information for this article is available on the WWW underhttp://dx.doi.org/10.1002/cphc.200900241.
ChemPhysChem 2009, 10, 1567 – 1576 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 1567

membrane normal axis (Figure 1).[12] With this technique, thealignment of the membrane-spanning helical hairpin formedby the SARS coronavirus E protein (SARS = severe acute respi-ratory syndrome) could be elucidated using the commerciallyavailable building block Fmoc-PheACHTUNGTRENNUNG(4-I)-OH as heavy-atom labelto locate the Phe23 adjacent to the lipid head-group region.[13–15]
Instead of determining the certain iodine position by a com-parison of the deduced electron density profiles 1(z) of thesamples with unlabeled to those of labeled peptides, the re-sults often appear more conclusive, if obtained from an iso-morphic sample by using several photon energies of a syn-chrotron radiation source around the iodine LIII absorptionedge at E = 4.5545 keV.[12]
For a fast in-house analysis, electron difference analysis is aunique method to obtain high-resolution information aboutthe localization of a certain structural element within a lipid bi-layer membrane. As shown in previous studies for the doublehelical homodimer motif of membrane-active peptides with ad,l-alternating configuration, this method is even capable ofevidencing a peptide orientation within the lipid bilayer up toa few �ngstrøm resolution via labeling at diverse positions.[16]
The applied peptide–lipid complexes composed of highly-aligned, oriented lipid bilayer stacks were derived via self-as-sembling at controlled peptide-to-lipid (P/L) ratios on siliconsubstrates. These samples were swollen under controlled hu-midity (water vapor) at temperatures above the lipid meltingtransition (tm) in the fluid La-phase.[17] To detect a single iodinelabel via difference analysis of the electron density curves of la-beled and unlabeled peptides, very high peptide-to-lipid ratiosare often required—in the range of P/L�1/10.[13, 16]
The necessity for an iodine label improvement has been rec-ognized at least for two reasons. First, there are many trans-membrane peptides, especially those connected to an innerand outer membrane domain, that are not at all or only hardlyreconstitutable within the membrane environment at suchhigh P/L ratios. Secondly, it is more difficult to detect the par-
ticular electron density signal from the iodine label at positionswhere the membrane background itself exhibits relatively highelectron densities, for example, in the head-group region orthe adjacent water layers. Therefore, the synthesis of Fmoc-diiodo-allylglycine-OH (1) is reported (Figure 2). This labeled
amino acid is appropriate to be used for the proposed applica-tions and can be compared to the non-iodinated commerciallyavailable analogue Fmoc-allylglycine-OH (2). These buildingblocks were proven to be suited for solid-phase peptide syn-thesis (SPPS) following the Fmoc strategy and were incorporat-ed in membrane-active peptides without affecting the mem-brane integrity, that is, no causing of large perturbation associ-ated with strain fields, lipid fluctuation or curvature. This is im-portant, as X-ray scattering analysis of lipid membrane stackscan only be accomplished with a low membrane-surface mo-saicity.[18]
2. Results and Discussion
2.1. Synthesis of the Iodine-Labeled Amino Acid
For the synthesis of a novel heavy-atom peptide label, anamino acid side-chain modification was selected that yielded abuilding block compatible with the Fmoc-strategy SPPS. Ingeneral, the building block can be inserted in the peptidebackbone or be used for on-resin side-chain functionalizationof amine groups available for example, in lysine or ornithine.In order to strongly affect the electron density by iodine label-ing, a doubly iodinated compound with iodine atoms posi-tioned as close as possible to each other was chosen. More-over, the effect of an increase within the electron density dif-ference profiles of iodinated and non-iodinated analoguesshould be accompanied by a maximum of resolution in the z-direction. Otherwise, different higher-iodinated amino acidbuilding blocks could be conceived like tri-substituted phenyl-alanines, although causing a loss in accuracy according to thez-position (depth) within the membrane where the labeledamino acid is located. For spectroscopy, an attachment of theiodine atoms, close to the peptide backbone with a limiteddegree of freedom seems to be desirable. Nevertheless, thesterical demand of the iodine atoms should not affect theSPPS and conformation of the generated peptides.
A starting point for the synthesis of the iodinated aminoacid 1 was Boc-Asp-OtBu (3, Figure 3). The carboxylic acid side
Figure 1. Left : Schematic representation of the scattering geometry for X-rayreflectivity experiments at highly-aligned multilamellar lipid samples with in-corporated membrane spanning peptides. Right: Associated electron densityprofile and difference curves used in heavy atom labeling analysis.
Figure 2. Structural representation of the novel, doubly-iodinated peptidebuilding block Fmoc-diiodo-allylglycine-OH (1) for application in SPPS andthe commercially available non-labeled analogue Fmoc-allylglycine-OH (2)for electron density difference analysis.
1568 www.chemphyschem.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2009, 10, 1567 – 1576
T. Salditt, U. Diederichsen et al.

chain was quantitatively converted to the Weinreb amide 4using N,O-dimethylhydroxylamine hydrochloride, N-ethylmor-pholine (NEM) and propane phosphonic acid anhydride(PPA).[19] Without prior purification the protected aldehyde 5was derived by dibutylaluminium hydride (DIBAH) reduction ofthe tert-butyl protected Weinreb amide 4 in 75 % yield.[19–21]
The aldehyde 5 was then treated with iodoform under Wittig-conditions to yield the (S)-tert-butyl-2-(tert-butoxycarbonylami-no)-5,5-diiodopent-4-enoate (6). Removal of the Boc- and tBu-protecting groups was accomplished by treatment with TFA/water (95:5 v/v) and was directly followed by Fmoc-protectionusing Fmoc-Cl in aqueous sodium carbonate/dioxane solutionto yield the desired product Fmoc-5-diiodo-allylglycine-OH (1)with 86 % yield over two steps.
2.2. Transmembrane Peptide Design and Solid-PhasePeptide Synthesis
For a reconstitution of labeled as well as unlabeled referencepeptides in lipid model membranes, the recently publishedpeptide motif H-(FY)5WW-OH (7, Figure 4) was selected (under-lined amino acids in the one-letter code indicate d-aminoacids).[16] The motif derived from gramicidin A is characterizedby a membrane-spanning b5.6-helical dimer of antiparallel ori-ented and d,l-alternating configurated peptides. The tubularstructures are supposed to anchor at the polar–hydrophobichead-group interface with their C-terminal tryptophane moiet-ies spanning the hydrophobic membrane core without com-plete penetration of the lipid head-group region(Figure 4).[16, 23] Therefore, the termini are sensitive to structuralor charge modifications and supposed to be located at theelectron-rich head-group interface. The water-insoluble pep-tide motif can be incorporated into lipid complexes nearly in-dependent of the peptide-to-lipid ratio (up to P/L = 0.1) with-out causing lamellar disordering.[16] Thus, incorporation of theiodine-labeled peptides proves whether the double iodinelabel is qualified to serve as heavy atom probe within a lipidbilayer environment, even at high ratios. Hence, a P/L ratio of1/10, equivalent with a pore-to-lipid ratio of 1/20 and aniodine atom-to-lipid ratio of 1/5 was applied. In combination
with a peptide applicable in high P/L-ratios, this label couldalso be used to increase electron density of lipid bilayers pro-ducing an electron contrast often required for the X-ray analy-sis of single lipid bilayers and usually achieved by using bromi-nated lipids.[5, 23]
The labeled amino acid Fmoc-5-diiodo-allylglycine-OH (1) aswell as the commercially available building block Fmoc-allylgly-cine-OH (2) were incorporated into peptides at the N-terminusvia manual SPPS. In previous studies these peptide motifs havealready been labeled with the mono-iodine label Fmoc-Phe-ACHTUNGTRENNUNG(4-I)-OH.[16] Coupling using HATU [ = 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate]/HOAt(= 1-Hydroxy-7-Azabenzotriazole)/DIPEA (= N,N-diisopropyleth-yl-amine) activation provided the labeled peptide H-diiodoallyl-glycine-(YF)4YWW-OH (8) and its unlabeled analogue H-allylgly-cine-(YF)4YWW-OH (9) (Figure 5). As antiparallel orienteddouble helices, the peptide dimers exhibit nearly symmetricalpores carrying the iodine building blocks at both ends. Todemonstrate that amino acid 1 can be incorporated not onlyas the terminal part of a peptide chain, two lysine branchedpeptides were subsequently generated and used for a differentpurpose. The side-chain-labeled oligomers H-K ACHTUNGTRENNUNG(K-H)-K-diiodoal-lylglycine-G3-OH (10) and H-K ACHTUNGTRENNUNG(K-H)-K-allylglycine-G3-OH (11)were synthesized accordingly.
2.3. Secondary Structure Analysis of Peptide–LipidComplexes
The synthesized peptides 8 and 9 were reconstituted in lipidcomplexes of 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC,12:0) that exhibit bilayers with a hydrophobic core of about26 � in diameter (glycerol–carbonyl distance). This bilayer iswell suited for membrane-spanning insertion of b5.6-doublehelical peptides 8 and 9 with an overall length of about25.7 �.[24] This has already been demonstrated in comparison
Figure 3. Structural representation of the synthesis route for Fmoc-diiodoal-lylglycine-OH (1), starting from the commercially available building blockBoc-Asp-OtBu 3. Conversion to the Weinreb amide 4 and reduction withDIBAH led to the semialdehyde 5. Treatment with iodoform under Wittig-conditions followed by protection group operations yielded in the desiredproduct 1.
Figure 4. Recently proposed structure (left : side view, right: view along thehelical axis) of the membrane-active peptide H-(FY)5-WW-OH (7), based onPDB 1S1O, after energy minimization (Amber*, MMCM).[22]
ChemPhysChem 2009, 10, 1567 – 1576 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1569
Side-Specific Peptide Iodination
with lipid bilayers of 1,2-dimiristoyl-sn-glycero-3-phosphocho-line (DMPC, 14:0).[16]
The impact of iodine label insertion into the double helicalsecondary structure of peptides 8 and 9 was revealed by CD-spectroscopy within large unilamellar vesicles (LUVs) of DLPCwith a 50 nm radius derived from extrusion techniques. TheCD spectra of peptides 8 and 9 were compared to the CDspectra of the recently reported peptide motif H-(FY)5WW-OH(7) (Figure 4). The constitution of the peptide termini that pre-sumably interact via coulomb forces (pH 7.0 in the vesiclebuffer medium) is an important parameter for the helix pro-pensity. The secondary structure of the b5.6-helical antiparalleldimer is characterized by maxima at 225 nm and 203 nm, aCotton effect at 198 nm and a global minimum below 190 nm(Figure 6). Furthermore, the secondary structure depends onmembrane curvature and is considerably diminished for meas-urements in small unilamellar vesicles (SUVs, data not shown).
The typical Cotton effects for a b5.6-helix can be recognizedwith dodecamer 7 providing the zwitterionic termini. In con-trast, the CD signals of the sequence analogue 12 with a C-ter-minal amide showed significantly lower intensity in accordancewith diminished helix formation. In case of the N-terminal acy-lated peptide amide 13, the b-helical secondary structurecould not be revealed any more. It can be concluded that thecharged termini are an important structural prerequisite for ab5.6-helix in DLPC bilayers. Incorporation of the iodine label asN-terminal amino acid 1 in peptide 8 was compared to thenon-iodinated peptide analogue 9 and oligomer 7. The pep-tide secondary structure incorporated in the membrane wasalmost not affected by the artificial amino acids 1 and 2 or theiodine label despite the bulkiness of the iodinated side chain.
The CD spectra of these peptides show Cotton effects atidentical wavelengths and with the same relative intensities.Compared with oligomer 7 for peptides 8 and 9, the helixhandedness is inverted in accordance with the configurationof the terminal amino acid; starting the peptide sequence witha d-amino acid at the C-terminus results in the right-handedhelix.
2.4. Membrane Incorporationof b5.6-Helices and X-rayReflectivity
In contrast to CD analysis fromvesicle solutions, the X-ray re-flectivities were recorded usingsolid supported multilamellarlipid stacks. Parallel orientedlipid membranes on silicon sub-strates were obtained fromspreading techniques at temper-atures above the lipid meltingtemperature (Tm) in the fluid La-phase.[25] In this way, a few thou-sand bilayers can be perfectlyaligned at the substrate surfacesand swollen by water vaporunder controlled osmotic pres-
sure (relative humidity fixed by salt solutions) and at definedP/L-ratios.[17] Instead of using chloroform/trifluoroethanol (TFE)for spreading the hydrophobic substrates, the lipid/peptidemixtures were solely spread from isopropanol. The multilamel-
Figure 5. Structural representation of the synthesized, terminally iodine-labeled peptide (8) and the unlabeled an-alogue (9) forming an antiparallel b5.6-double helix as shown in the comparative CD study.
Figure 6. Top: CD spectra of the membrane-active oligomer 7 with zwitter-ionic termini and the sequentially identical peptides 12 and 13 with differ-ently charged states at the terminal ends. Bottom: Comparison of peptide 7with peptides 8 and 9 containing non-proteinogenic amino acids 1 and 2(indicated as 1’ and 2’).
1570 www.chemphyschem.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2009, 10, 1567 – 1576
T. Salditt, U. Diederichsen et al.

lar lipid films of pure DLPC appear as homogeneous sampleswith a surface of low mosaicity (Figure 7). For samples with in-corporated peptides the optical microscope images evenwhen taken in the dry state of the lipid membrane show defi-nite surfaces with only slight defect structures of the few top-most bilayers, but with differing characteristics. For the P/L =
1/100 and P/L = 1/50 ratios these superficial defects can beconsidered as marginal and are equally dispersed throughoutthe whole surface (Figure 7). In case of higher peptide ratioswith a 1/20 or a 1/10 peptide–lipid composition, the defectstructures appear less homogeneously dispersed and formmajor defect compartments. This observation might reflect anaggregating effect of the peptide motif that was also observedduring reverse-phase chromatographic purification and hasbeen elucidated by X-ray structural analysis of the relatedwater-soluble peptide H-(YY)4K-OH.[26] The membrane surfacesobserved by optical microscopy already indicated the need ofa new electron-rich and site-specific label for X-ray reflectivityanalysis of membrane-bound species because such labelsallow for the application of lower P/L-ratio samples, yieldingenhanced quality of the sample itself and of the deduced ana-lytical data.
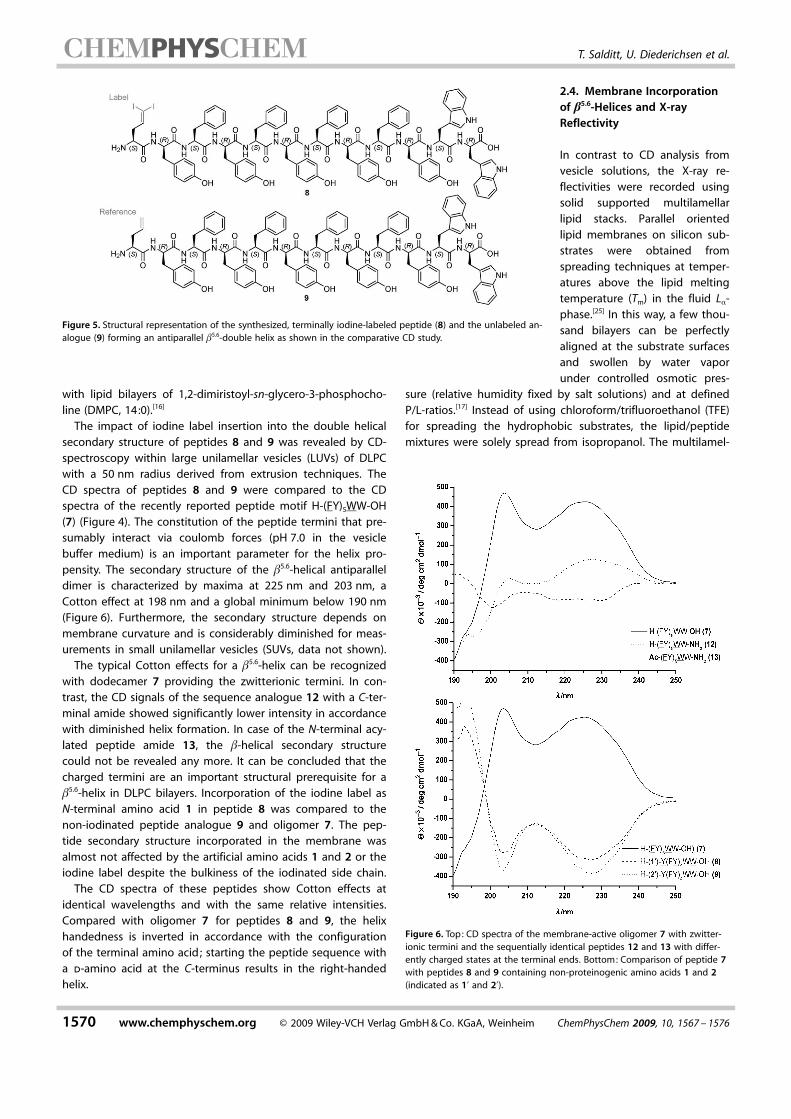
The reflectivity curves of the multilamellar peptide-lipidcomplexes of peptides 8 and 9 embedded in DLPC-stacks illus-trate the characteristics of highly-oriented multilamellar filmswith the plateau of total reflection at small qz and a set of in-tense, equidistant Bragg peaks (Figure 8, reflectivity curves areshifted for clarity). Up to five lamellar orders were obtained atthe commercial diffractometer for the samples with a peptide-to-lipid ratio of 1/20 or smaller. For the sample with a P/L-ratioof 1/10 only four equidistant Bragg peaks were observed witha beginning disorder characterized by the occurance of an ad-ditional aperiodic reflectivity signal at high qz. The shouldersattached to the Bragg reflexes especially at high qz might becaused by peptide aggregation (superstructures) or the coinci-dental existance of a second lipid phase. This observation is inline with the changes of the surface properties revealed fromoptical microscopy (Figure 7). The intensities of the Bragg re-flexes decrease monotonously with increasing peptide concen-
tration and only the sample of peptide 9 with P/L = 1/100 devi-ates from the monotonous dependence.
The obtained centrosymmetric electron density profiles aredepicted in Figure 8 (right, on arbitrary scale). The profilemaxima correspond to the lipid head-group regions, while theglobal minimum is correlated with the acyl–acyl contact of theopposing hydrocarbon chains. The adjacent water layers arerepresented by the side minima. The phase coefficients for thereconstruction of the electron density has been estimated viaa one-dimensional swelling method applied to the lamellarlipid phase (see the Experimental Section). Upon peptide inser-tion, the electron-density profiles show a slight increase of thelipid bilayer thickness of about 0.6 � from 29.4 � for pureDLPC to 30.0 � for peptide 9 at P/L = 1/10. The trend of bilayerthickness rise with increasing peptide-to-lipid ratio is quiteconvincing for peptide 9 (Figure 8, bottom right). The X-ray re-flectivity data resulting in case of the membrane/peptide com-plex of peptide 8 show two outliers with the samples P/L =
1/100 and 1/10, but an increase in membrane thickness canclearly be observed and might result from hydrophobic mis-matching.[26–32] Considering that the membrane thickness is afunction of distance between the anchoring peptide elementsat the membrane polar/unpolar interface, a slight increase inmembrane thickness is in line with a peptide b-helical struc-ture that fits the carbonyl–glycerol distance of a DLPC bilay-er.[24, 33–38] For the samples with incorporated peptides, a de-crease in the electron-density contrast of the side to the mainminima (shape parameter E) is observed as in most peptide/lipid systems. The decrease can be explained by beginninglipid disorder that evens the density profile by increased densi-ty in the water layer and/or an enlarged area per headgroup.[39] A loss in electron density resulting from the lipidhead-groups and the growth of water-layer-associated electrondensity with respect to the z-direction seems to be induced bythe peptide incorporation and might be an indicator for a lat-eral membrane expansion. A lateral membrane expansioncould be verified via measuring the acyl chain peak by in plainX-ray scattering under grazing incidence.[40, 41]
The difference analysis of the iodine-labeled and the non-labeled samples at P/L = 1/10 and P/L = 1/50 concentrations
Figure 7. Optical microscopy images of the highly-aligned peptide–lipid complexes on silicon surfaces (amplification: 100 � ) in the dry state at room tempera-ture. Top row (from left to right): pure DLPC; oligomer 8/DLPC = 1/10, 1/20, 1/50, 1/100. Bottom row (from left to right): pure DLPC; oligomer 9/DLPC = 1/10,1/20, 1/50, 1/100.
ChemPhysChem 2009, 10, 1567 – 1576 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1571
Side-Specific Peptide Iodination
are depicted in Figure 9. With a total bilayer thickness ofabout 36.8 � (inflexion point distance to the adjacent water
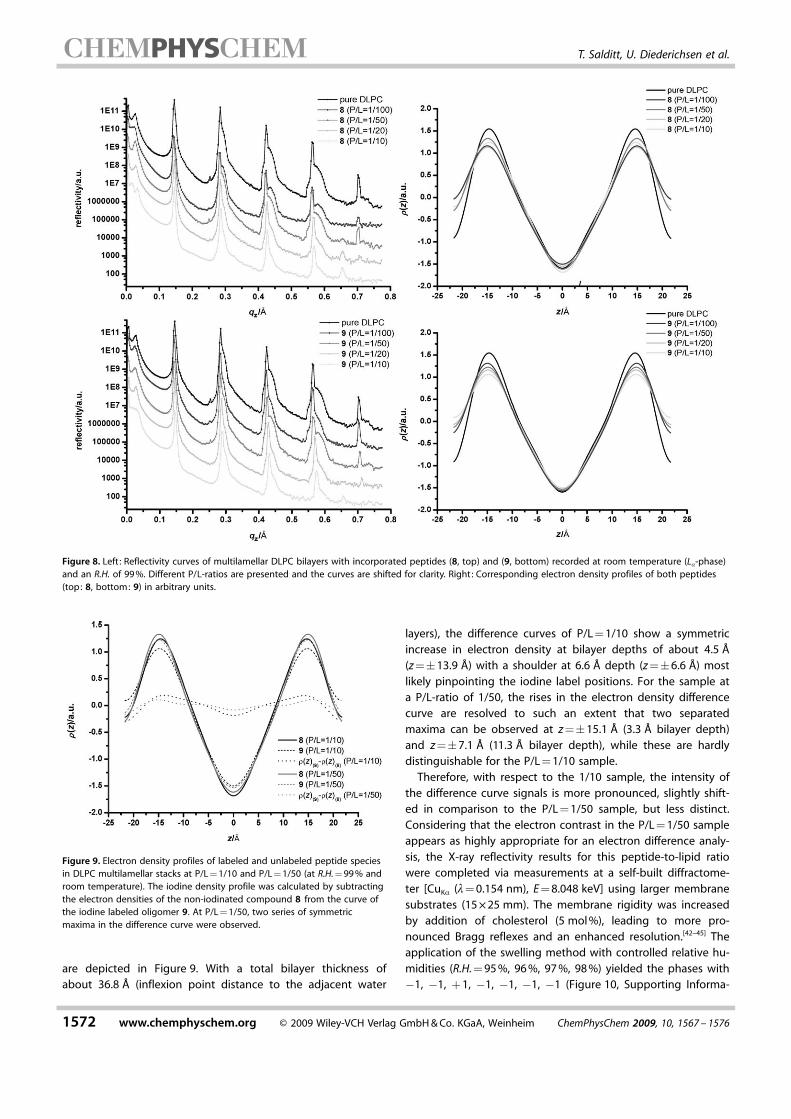
layers), the difference curves of P/L = 1/10 show a symmetricincrease in electron density at bilayer depths of about 4.5 �(z =�13.9 �) with a shoulder at 6.6 � depth (z =�6.6 �) mostlikely pinpointing the iodine label positions. For the sample ata P/L-ratio of 1/50, the rises in the electron density differencecurve are resolved to such an extent that two separatedmaxima can be observed at z =�15.1 � (3.3 � bilayer depth)and z =�7.1 � (11.3 � bilayer depth), while these are hardlydistinguishable for the P/L = 1/10 sample.
Therefore, with respect to the 1/10 sample, the intensity ofthe difference curve signals is more pronounced, slightly shift-ed in comparison to the P/L = 1/50 sample, but less distinct.Considering that the electron contrast in the P/L = 1/50 sampleappears as highly appropriate for an electron difference analy-sis, the X-ray reflectivity results for this peptide-to-lipid ratiowere completed via measurements at a self-built diffractome-ter [CuKa (l= 0.154 nm), E = 8.048 keV] using larger membranesubstrates (15 � 25 mm). The membrane rigidity was increasedby addition of cholesterol (5 mol %), leading to more pro-nounced Bragg reflexes and an enhanced resolution.[42–45] Theapplication of the swelling method with controlled relative hu-midities (R.H. = 95 %, 96 %, 97 %, 98 %) yielded the phases with�1, �1, + 1, �1, �1, �1, �1 (Figure 10, Supporting Informa-
Figure 8. Left : Reflectivity curves of multilamellar DLPC bilayers with incorporated peptides (8, top) and (9, bottom) recorded at room temperature (La-phase)and an R.H. of 99 %. Different P/L-ratios are presented and the curves are shifted for clarity. Right: Corresponding electron density profiles of both peptides(top: 8, bottom: 9) in arbitrary units.
Figure 9. Electron density profiles of labeled and unlabeled peptide speciesin DLPC multilamellar stacks at P/L = 1/10 and P/L = 1/50 (at R.H. = 99 % androom temperature). The iodine density profile was calculated by subtractingthe electron densities of the non-iodinated compound 8 from the curve ofthe iodine labeled oligomer 9. At P/L = 1/50, two series of symmetricmaxima in the difference curve were observed.
1572 www.chemphyschem.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2009, 10, 1567 – 1576
T. Salditt, U. Diederichsen et al.
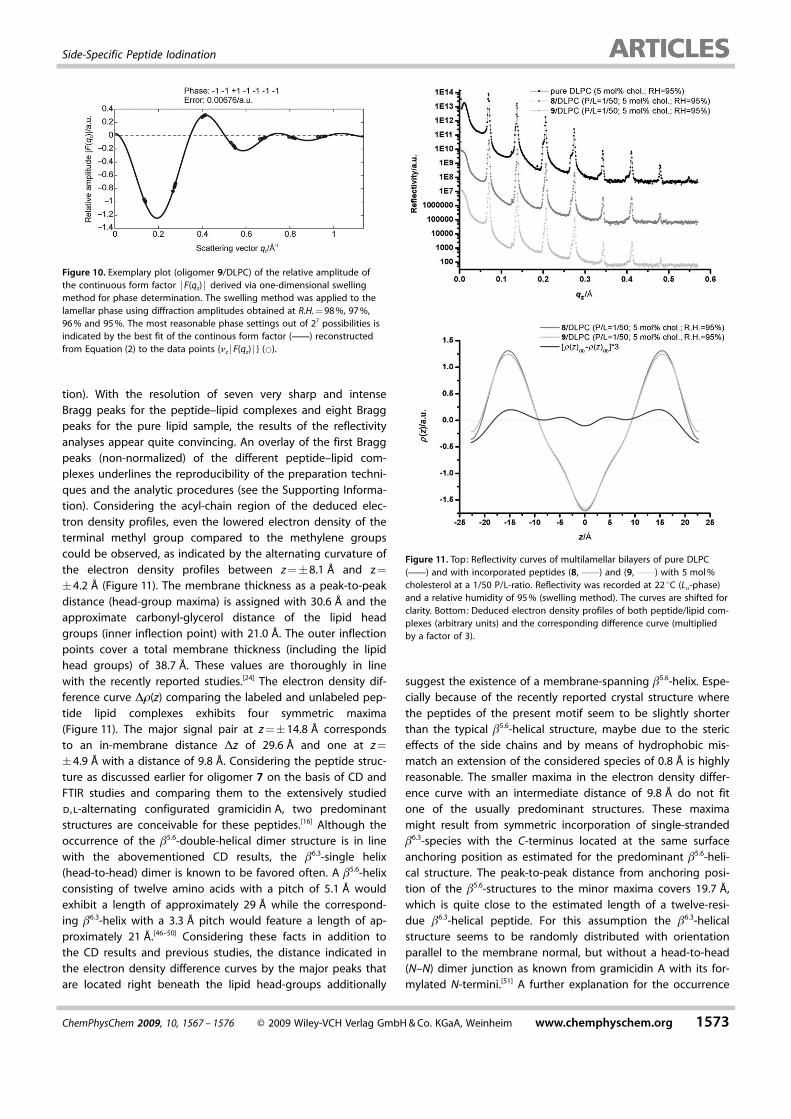
tion). With the resolution of seven very sharp and intenseBragg peaks for the peptide–lipid complexes and eight Braggpeaks for the pure lipid sample, the results of the reflectivityanalyses appear quite convincing. An overlay of the first Braggpeaks (non-normalized) of the different peptide–lipid com-plexes underlines the reproducibility of the preparation techni-ques and the analytic procedures (see the Supporting Informa-tion). Considering the acyl-chain region of the deduced elec-tron density profiles, even the lowered electron density of theterminal methyl group compared to the methylene groupscould be observed, as indicated by the alternating curvature ofthe electron density profiles between z =�8.1 � and z =
�4.2 � (Figure 11). The membrane thickness as a peak-to-peakdistance (head-group maxima) is assigned with 30.6 � and theapproximate carbonyl-glycerol distance of the lipid headgroups (inner inflection point) with 21.0 �. The outer inflectionpoints cover a total membrane thickness (including the lipidhead groups) of 38.7 �. These values are thoroughly in linewith the recently reported studies.[24] The electron density dif-ference curve D1(z) comparing the labeled and unlabeled pep-tide lipid complexes exhibits four symmetric maxima(Figure 11). The major signal pair at z =�14.8 � correspondsto an in-membrane distance Dz of 29.6 � and one at z =
�4.9 � with a distance of 9.8 �. Considering the peptide struc-ture as discussed earlier for oligomer 7 on the basis of CD andFTIR studies and comparing them to the extensively studiedd,l-alternating configurated gramicidin A, two predominantstructures are conceivable for these peptides.[16] Although theoccurrence of the b5.6-double-helical dimer structure is in linewith the abovementioned CD results, the b6.3-single helix(head-to-head) dimer is known to be favored often. A b5.6-helixconsisting of twelve amino acids with a pitch of 5.1 � wouldexhibit a length of approximately 29 � while the correspond-ing b6.3-helix with a 3.3 � pitch would feature a length of ap-proximately 21 �.[46–50] Considering these facts in addition tothe CD results and previous studies, the distance indicated inthe electron density difference curves by the major peaks thatare located right beneath the lipid head-groups additionally
suggest the existence of a membrane-spanning b5.6-helix. Espe-cially because of the recently reported crystal structure wherethe peptides of the present motif seem to be slightly shorterthan the typical b5.6-helical structure, maybe due to the stericeffects of the side chains and by means of hydrophobic mis-match an extension of the considered species of 0.8 � is highlyreasonable. The smaller maxima in the electron density differ-ence curve with an intermediate distance of 9.8 � do not fitone of the usually predominant structures. These maximamight result from symmetric incorporation of single-strandedb6.3-species with the C-terminus located at the same surfaceanchoring position as estimated for the predominant b5.6-heli-cal structure. The peak-to-peak distance from anchoring posi-tion of the b5.6-structures to the minor maxima covers 19.7 �,which is quite close to the estimated length of a twelve-resi-due b6.3-helical peptide. For this assumption the b6.3-helicalstructure seems to be randomly distributed with orientationparallel to the membrane normal, but without a head-to-head(N–N) dimer junction as known from gramicidin A with its for-mylated N-termini.[51] A further explanation for the occurrence
Figure 10. Exemplary plot (oligomer 9/DLPC) of the relative amplitude ofthe continuous form factor jF(qz) j derived via one-dimensional swellingmethod for phase determination. The swelling method was applied to thelamellar phase using diffraction amplitudes obtained at R.H. = 98 %, 97 %,96 % and 95 %. The most reasonable phase settings out of 27 possibilities isindicated by the best fit of the continous form factor (c) reconstructedfrom Equation (2) to the data points {nz jF(qz) j } (*).
Figure 11. Top: Reflectivity curves of multilamellar bilayers of pure DLPC(c) and with incorporated peptides (8, c) and (9, c) with 5 mol %cholesterol at a 1/50 P/L-ratio. Reflectivity was recorded at 22 8C (La-phase)and a relative humidity of 95 % (swelling method). The curves are shifted forclarity. Bottom: Deduced electron density profiles of both peptide/lipid com-plexes (arbitrary units) and the corresponding difference curve (multipliedby a factor of 3).
ChemPhysChem 2009, 10, 1567 – 1576 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1573
Side-Specific Peptide Iodination

of the minor maxima might be given by a change in themethyl–methyl group intersections as a response to peptideinsertion. The terminal methyl groups of the lipid chains mightbe affected by the planar aromatic side-chains that are perpen-dicular oriented to the membrane normal and produce a sym-metric change in the electron density around the bilayercenter.
Certainly, the existing diversities of two non-isomorphic sam-ples may also contribute to the differences in the jointly com-pared electron density profiles, and therefore, are not fullyconclusive. This is in fact the case if an isomorphic sample isconsidered and studied via anomalous X-ray reflectivity byusing several photon energies of a synchrotron radiation asmentioned. For this application the new heavy-atom label hasbeen designed and was tested in these in-house measure-ments.
3. Conclusions
A novel iodine heavy atom peptide label 1 has been synthe-sized by modifying a commercially available amino-acid build-ing block in five straightforward synthetic steps including theapplication of the protecting group pattern for Fmoc-SPPS.The amino acid label 1 was incorporated via SPPS into a pore-forming peptide motif of d,l-alternating configuration. As re-vealed by CD spectroscopic analyses of the vesicle bound pep-tide, the peptide’s secondary structure is not affected by thebulky label. By reconstitution of the labeled peptide as well asa non-labeled analogue in multilamellar membrane stacks ofDLPC, clean stack surface only with usual defect structureswere obtained. For this aspect the likewise commercially avail-able Fmoc-allylglycine-OH 2 can serve as a reference buildingblock.
From X-ray reflectivity studies of solid supported peptide–lipid complexes up to eight lamellar orders could be obtainedat an in-house diffractometer. Even at lower peptide-to-lipidratios (P/L = 1/50) the derived electron density difference analy-sis showed maxima in the z-direction of the lipid bilayer, pin-pointing the iodine label position. Compared to the formerlyused samples with a 1/10 P/L-ratio, the resolution could be en-hanced for these higher-quality samples with plain membranesurfaces (Figure 7).[16] The cholesterol addition also suppressesundulations by increasing the membrane rigidity.[42–45] From CDstudies and X-ray reflectivity using the novel heavy atom label,the b5.6-double helical dimer structure could be confirmed forthe considered peptides.
In upcoming experiments at a synchrotron radiation sourcethe novel label will be used to allocate the assumedly nearhead-group located functionalization side of a membraneactive hairpin structure that is intended to serve as a bilayeranchor for molecular recognition moieties. Performing anoma-lous X-ray scattering at a synchrotron source (different photonenergies) using an isomorphic sample will further extend thepresent study. With the novel label building block it mighteven be possible to obtain data from single bilayer reflectivityat high P/L-ratios on an absolute scale or to clarify inter-pep-tide distances in the plane of lipid bilayers.
Experimental Section
For material and methods, building block synthesis, peptide syn-thesis and purification and the analytical data please see the Sup-porting Information.
Preparation of Lipid–Peptide Complexes: For CD spectroscopy, sol-utions of large unilamellar vesicles (LUVs) were prepared from aphosphate buffer solution (1 mm NaH2PO4/Na2HPO4, pH 7.0) follow-ing a method from MacDonald et al.[52] For preparation of the lipidfilms the DLPC dissolved in CHCl3 (10 mg mL�1) and the peptidesdissolved in EtOH (concentration estimated via UV absorption)were mixed yielding in less than 1 mL of a one-to-one solution ofCHCl3/EtOH. This solution was dried in a nitrogen steam underheating at temperatures above the lipid melting temperature ofDLPC (Tm�2 8C) to yield in an almost clear film attached to thetest tube walls. After further drying under reduced pressure atT>Tm the lipid films were rehydrated with the appropriate amountof buffer solution. After 1 h of incubation at T>Tm the hydratedlipid films were vortexed in five cycles each with 30 s of vortexingand 5 min of incubation. The milky suspensions were extruded21 times through a polycarbonate membrane with a 100 nm poresize using a Mini-Extruder (Liposofast, Avestin, Ottawa, Canada) toproduce an almost clear solution. For CD measurements, the vesi-cles were deposited in precision cells (light path 1 cm, Quartz Su-prasil, Hellma, M�hlheim, Germany).
For X-ray scattering the preparation of peptide–lipid complexes fol-lowed the procedure described by Seul and Sammon.[25] DLPC wasdissolved in isopropanol at a concentration of 40 mg mL�1, whilethe peptide was dissolved at 5 mg mL�1 concentration. Peptideand lipid stock solutions were mixed to yield a final concentrationfor the lipid of 10 mg mL�1 with the desired peptide-to-lipid ratio(P/L). Silicon wafers (10 � 15 mm or 15 � 25 mm, 100 orientation, Sil-chem, Freiberg, Germany), cleaned in methanol by 15 min cycles inan ultrasonic bath, followed by 15 min cycles in ultrapure waterwere used as substrates. A droplet of 80 mL (10 � 15 mm substrates)or 150 mL (15 � 25 mm substrates) of each mixed peptide–lipid sol-ution was spread on a strictly horizontal positioned Si-wafer. Thespread solution was allowed to dry very slowly overnight atnormal pressure, covered by a watch-glass to prevent film ruptureand fast dewetting. The samples were exposed to low pressure(<1 mbar) for 12 h to ensure a complete removal of the solventtraces followed by rehydration to yield a film thickness in therange of D�2–5 mm. This procedure produces well-aligned multila-mellar stacks with a typical mosaicity of less than the instrumentalresolution (0.018).[53]
X-ray Reflectivity Measurements: The reflectivity experiments werecarried out at two different diffractometers. The first one is a com-mercial AXS D8 Advance diffractometer (Bruker, Karlsruhe, Germa-ny) with a sealed tube of MoKa (l= 0.0709 nm, E = 17.48 keV) radia-tion, equipped with a V/V-goniometer, a collimating Gçbel mirrorsystem, automated attenuators and a fast NaI-scintillation countingdetector. The beam size in the reflectivity plane was defined by en-trance slits to 50 mm beamsize. The collimating Gçbel mirrors,50 mm beam size in scattering plane, and large divergence in thehorizontal plane optimize signal-to-noise ratio due to shortsource–sample distances. Prior to X-ray reflectivity measurements,the resulting multilamellar stacks were inserted in a closed humidi-ty controlled chamber with rubber sealing. The chamber wasmounted on the z-axis diffractometer with samples oriented hori-zontally. The chamber consists of a rectangular stainless steel cylin-der with kapton windows and areservoir filled with ultrapure waterto achieve full hydration of the membrane stacks from water vapor
1574 www.chemphyschem.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2009, 10, 1567 – 1576
T. Salditt, U. Diederichsen et al.

swelling (R.H. = 99 %). The hydration state and measurement roomtemperature assured the bilayers to be in the fluid La-state. Thesecond used diffractometer (swelling method) is a self-built instru-ment operating with a sealed tube (line focus) generating CuKa ra-diation (8.048 keV). The beam is collimated by a Gçbel mirrorsystem and controlled by three motorized pairs of slits (entrance,guard, detector). The sample is positioned on a z-axis diffractome-ter (Huber). The reflectivity signal was measured by a fast scintilla-tion counter (Cyberstar, Oxford-Danfysik) using an automated at-tenuator system. For instrument control the Spec software (Certi-fied Scientific Software Inc.) was used. For application of the swel-ling method the sample substrates were mounted vertically withina round stainless steel cylinder with kapton windows. The chamberwas equipped with a relative humidity sensor, inlets and outletsfor flushing the chamber with a humidity adjusted gas-flow andwith mass flow controllers. The humidity set up is PID-controlledand interfaced with the diffractometer control software. Also thereflectivity measurements under swelling conditions were accom-plished at approximately 22 8C in the fluid La-phase of DLPC. Amore specified description of the humidity control and swellingmethod is given in ref. [45] . The resulting reflectivity is plotted as afunction of the vertical momentum transfer qz, after subtraction ofthe diffuse scattering (offset scan) and illumination correction. Theelectron density profiles were calculated by applying an empiricalFourier Synthesis (FS) scheme, exploiting the area under Braggpeak intensities In, as it is used for such multilamellar lipid mem-branes.[54–57] In simple terms the one-dimensional electron densityprofile 1(z) was obtained via Fourier synthesis from the integratedpeak intensities, using a Lorentz correction factor 1/qz
2 and thephases �1, �1, + 1, �1 or �1, �1, + 1, �1, �1, �1, �1, respective-ly. The electron density profile 1(z) normal to the interface is com-puted by N0 Fourier coefficients fn = Inqz and Equation (1):
1ðzÞ ¼XN0
n¼1
un
ffiffiffiffiffiffinIn
pcos
2pnzd
� �ð1Þ
The factor n in front of the Bragg peak intensity In follows from anempirical Gauss-like correction factor to calculate the nth Fouriercoefficient. The curves have been normalized by scaling higher-order Bragg peaks to the area under the first Bragg peak. Thephases nn are reduced to positive/negative signs due to the mirrorplane symmetry and were reconstructed in accordance to the onedimensional swelling method approach (Figure 10) using Equa-tion (2) to obtain the continuous-form factor F ~Qz
� �with its rela-
tive amplitude F ~Ql
� �and the phase nl. The scattering vectors are
represented by Qz and Ql, whereas d is the membrane periodici-ty.[45]
Fð~QzÞ ¼X
nF ~Qsincd2
Qz � Qlð Þ� �
ð2Þ
General aspects of reflectivity experiments are discussed inref. [58].
Acknowledgements
We thank Tobias Reusch and Britta Weinhausen (Salditt group)for providing their software tools for X-ray data analysis and forassistance in operating the WENDI. Generous financial support ofthe Deutsche Forschungsgemeinschaft (SFB 803) is gratefully ac-knowledged.
Keywords: heavy-atom labeling · lipids · membranes ·peptides · X-ray diffraction
[1] T. Heimburg, Tutorials in Biophysics—Thermal Biophysics of Membranes,Wiley-VCH, Weinheim, 2007.
[2] T. J. McIntosh, Methods Mol. Biol. 2007, 398, 221–230.[3] T. Salditt, Curr. Opin. Struct. Biol. 2003, 13, 467–478.[4] C. Li, D. Constantin, T. Salditt, J. Phys. Condens. Matter 2004, 16, S2439–
S2453.[5] S. Qian, W. Wang, L. Yang, H. W. Huang, Proc. Natl. Acad. Sci. USA 2008,
105, 17379–17383.[6] D. Constantin, G. Brotons, A. Jarre, T. Salditt, Biophys. J. 2007, 92, 3978–
3987.[7] D. T. Bong, A. Janshoff, C. Steinem, R. Ghadiri, Biophys. J. 2000, 78, 839–
845.[8] W. C. Wimley, S. H. White, Biochemistry 2000, 39, 4432–4442.[9] L. A. Chung, J. D. Lear, W. F. DeGrado, Biochemistry 1992, 31, 6608–6616.
[10] B. BilgiÅer, K. Kumar, Proc. Natl. Acad. Sci. USA 2004, 101, 15 324–15 329.[11] J. Korlach, P. Schwille, W. Webb, G. Feigenson, Proc. Natl. Acad. Sci. USA
1999, 101, 15 324–15 329; W. C. Wimley, S. H. White, Biochemistry 2000,39, 4432–4442.
[12] Z. Khattari, G. Brotons, E. Arbely, I. T. Arkin, T. H. Metzger, T. Salditt, Phys.B 2005, 357, 34–38.
[13] A. Arbely, Z. Khattari, G. Brotons, M. Akkawi, T. Salditt, I. T. Arkin, J. Mol.Biol. 2004, 341, 769–779.
[14] Z. Khattari, G. Brotons, M. Akkawi, E. Arbely, I. T. Arkin, T. Salditt, Biophys.J. 2006, 90, 2038–2050.
[15] Z. Khattari, I. T. Arkin, T. Salditt, Eur. Biophys. J. 2006, 36, 45–55.[16] A. K�sel, Z. Khattari, P. E. Schneggeburger, A. Banerjee, T. Salditt, U. Die-
derichsen, ChemPhysChem 2007, 8, 2336–2343.[17] T. J. McIntosh, A. D. Magid, S. A. Simon, Biochemistry 1989, 28, 17–25.[18] T. Salditt, C. M�nster, J. Lu, M. Vogel, W. Fenzl, A. Souvorov, Phys. Rev. E
1999, 60, 7285–72899.[19] D. Wernic, J. DiMaio, J. Adams, J. Org. Chem. 1989, 54, 4224–4228.[20] C. Douat, A. Heitz, J. Martinez, J. A. Fehrentz, Tetrahedron Lett. 2001, 42,
3319–3321.[21] T. Bayer, C. Riemer, H. Kessler, J. Pept. Sci. 2001, 7, 250–261.[22] E. Navarro, E. Fenude, B. Celda, Biopolymers 2002, 64, 198–209.[23] T. J. McIntosh, P. W. Holloway, Biochemistry 1987, 26, 1783–1788.[24] N. Kucerka, Y. Liu, N. Chu, H. I. Petrache, S. Tristam-Nagle, J. F. Nagle, Bio-
phys. J. 2005, 88, 2626–2637.[25] M. Seul, M. J. Sammon, Thin Solid Films 1990, 185, 287–28.[26] E. Alexopoulos, A. K�sel, G. M. Sheldrick, U. Diederichsen, I. Us�n, Acta
Crystallogr. Sect. D 2004, 60, 1971–1980.[27] J. A. Killian, Biochim. Biophys. Acta Rev. Biomembr. 1998, 1376, 401–415.[28] M. R. R. de Planque, B. B. Bonev, J. A. A. Demmers, D. V. Greathouse, R. E.
Koeppe II, F. Separovic, A. Watts, J. A. Killian, Biochemistry 2003, 42,5341–5348.
[29] J. A. Killian, I. Salemink, M. R. R. de Planque, G. Lindblom, R. E. Koeppe II,D. V. Greathouse, Biochemistry 1996, 35, 1037–1045.
[30] M. R. R. de Planque, J.-W. P. Boots, D. T. S. Rijkers, R. M. J. Liskamp, D. V.Greathouse, J. A. Killian, Biochemistry 2002, 41, 8396–8404.
[31] T. A. Harroun, W. T. Heller, T. M. Weiss, L. Yang, H. W. Huang, Biophys. J.1999, 76, 937–945.
[32] T. A. Harroun, W. T. Heller, T. M. Weiss, L. Yang, H. W. Huang, Biophys. J.1999, 76, 3176–3185.
[33] J. A. Killian, M. R. R. de Planque, Mol. Membr. Biol. 2003, 20, 271–284.[34] M. R. R. de Planque, J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V.
Greathouse, R. E. Koeppe II, B. de Kruijff, J. A. Killian, J. Biol. Chem. 1999,274, 20839–20846.
[35] J. A. Killian, G. von Heijne, Trends Biochem. Sci. 2000, 25, 429–434.[36] E. Strandberg, S. Morein, D. T. S. Rijkers, R. M. J. Liskamp, P. C. A. van der
Wel, J. A. Killian, Biochemistry 2002, 41, 7190–7198.[37] J. A. Killian, FEBS Lett. 2003, 555, 134–138.[38] S. Morein, J. A. Killian, M. M. Sperotto, Biophys. J. 2002, 82, 1405–1417.[39] L. Greenspan, J. Res. Nat. Bur. Stand. Sect. A 1977, 81, 89–96.[40] K. He, S. J. Ludke, Y. Wu, H. W. Huang, Biophys. J. 1993, 64, 157–162.[41] K. He, S. J. Ludke, Y. Wu, H. W. Huang, J. Phys. IV France 1993, C8-265-
C8-270.
ChemPhysChem 2009, 10, 1567 – 1576 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 1575
Side-Specific Peptide Iodination

[42] R. A. Cooper, J. Supramol. Struct. 1978, 8, 413–430.[43] M. J. Carmena, C. Hueso, L. G. Guijarro, J. C. Prieto, Regul. Pept. 1991, 33,
287–297.[44] C. M. Yip, A. A. Darabie, J. McLaurin, J. Mol. Biol. 2002, 318, 97–107.[45] S. Aeffner, T. Reusch, B. Weinhausen, T. Salditt, Eur. Phys. J. , accepted.[46] B. M. Burkhart, R. M. Gassmann, D. A. Langes, W. A. Pangborn, W. L.
Duax, V. Pletnev, Biopolymers 1999, 51, 129–144.[47] W. R. Veatch, E. T. Fossel, E. R. Blout, Biochemistry 1974, 13, 5249–5256.[48] B. A. Wallace, Biophys. J. 1986, 49, 295–306.[49] B. A. Wallace, J. Struct. Biol. 1998, 121, 123–141.[50] S. Fahsel, E.-M. Pospiech, M. Zein, T. L. Hazlet, E. Gratton, R. Winter, Bio-
phys. J. 2002, 83, 334–344.[51] B. A. Wallace, Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 127–157.[52] M. Gude, J. Ryf, P. D. White, Lett. Pept. Sci. 2002, 9, 203–206.
[53] R. C. MacDonald, R. I. MacDonald, B. P. Menco, K. Takeshita, N. K. Subbar-ao, L. R. Hu, Biochim. Biophys. Acta 1991, 1061, 297–303.
[54] C. M�nster, J. Lu, S. Schinzel, B. Bechinger, T. Salditt, Eur. Biophys. J.2000, 28, 683–688.
[55] A. Spaar, C. M�nster, T. Salditt, Biophys. J. 2004, 87, 396–407.[56] a) C. R. Cantor, P. R. Schimmel, Biophysical Chemistry, Part II : Techniques
for the study of biological structure and function, W. H. Freeman, NewYork, 1980.
[57] Y. Wu, K. He, S. J. Ludtke, H. W. Huang, Biophys. J. 1995, 68, 2361–2369.[58] T. Salditt, C. Li, A. Spaar, U. Mennicke, Eur. Phys. J. E 2002, 7, 105–116.
Received: March 26, 2009
1576 www.chemphyschem.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2009, 10, 1567 – 1576
T. Salditt, U. Diederichsen et al.




















