
A locus of group A streptococcus involved in invasive disease and DNA transfer: sil , streptococcal...
13
Molecular Microbiology (2002) 46 (1), 87–99 © 2002 Blackwell Publishing Ltd Blackwell Science, LtdOxford, UKMMIMolecular Microbiology 0950-382X Blackwell Science, 200245Original Article sil, streptococcal invasive locusC. Hidalgo-Grass et al. Accepted 24 June, 2002. *For correspondence. E-mail hanski@ cc.huji.ac.il; Tel. ( + 972) 2 6758196; Fax ( + 972) 2 6434170. A locus of group A streptococcus involved in invasive disease and DNA transfer Carlos Hidalgo-Grass, 1 Miriam Ravins, 1 Mary Dan-Goor, 1 Joseph Jaffe, 1 Allon E. Moses 2 and Emanuel Hanski 1 1 Department of Clinical Microbiology, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel. 2 Department of Clinical Microbiology and Infectious Diseases, Hadassah University Hospital, Jerusalem 91120, Israel. Summary Group A streptococcus (GAS) causes diseases rang- ing from benign to severe infections such as necro- tizing fasciitis (NF). The reasons for the differences in severity of streptococcal infections are unex- plained. We developed the polymorphic-tag-lengths- transposon-mutagenesis (PTTM) method to identify virulence genes in vivo . We applied PTTM on an emm 14 strain isolated from a patient with NF and screened for mutants of decreased virulence, using a mouse model of human soft-tissue infection. A mutant that survived in the skin but was attenuated in its ability to reach the spleen and to cause a lethal infection was identified. The transposon was inserted into a small open reading frame (ORF) in a locus termed sil , s treptococcal i nvasion l ocus. sil contains at least five genes ( sil A-E) and is highly homologous to the quorum-sensing competence regulons of Streptococcus pneumoniae . sil A and sil B encode a putative two-component system whereas sil D and sil E encode two putative ABC transporters. sil C is a small ORF of unknown function preceded by a combox promoter. Insertion and deletion mutants of sil had a diminished lethality in the animal model. Virulence of a deletion mutant of sil C was restored when injected together with the avirulent emm 14- deletion mutant, but not when these mutants were injected into opposite flanks of a mouse. DNA transfer between these mutants occurred in vivo but could not account for the complementation of virulence. DNA exchange between the emm 14-deletion mutant and mutants of sil occurred also in vitro, at a frequency of ~ 10 - 8 for a single antibiotic marker. Whereas sil C and sil D mutants exchanged markers with the emm 14 mutant, sil B mutant did not. Thus, we identified a novel locus, which controls GAS spreading into deeper tissues and could be involved in DNA transfer. Introduction Group A streptococcus (GAS) is a Gram-positive patho- gen globally responsible for human morbidity and mortal- ity. GAS causes extremely variable clinical manifestations ranging from mild self-limiting superficial infections of the skin and mucosa, to life-threatening invasive infections such as septicaemia, toxic shock syndrome and necrotiz- ing fasciitis (NF). The organism is also responsible for acute rheumatic fever and acute glomerulonephritis (Bisno and Stevens, 1996; Musser and Krause, 1998). Group A streptococcus infections have increased in fre- quency and severity in the last decade in the United States and Europe (Cunningham, 2000). Group A streptococcus infection is defined as invasive when bacteria are isolated from blood or other sterile tissues. A prospective population-based study (PPBS) in Ontario, Canada, showed that the annual incidence of GAS invasive infections is 1.5 cases per 100 000 persons (Davies et al ., 1996). Recently we have conducted a PPBS study in Israel, showing that the overall annual incidence of invasive GAS infections is even higher, reach- ing 3.7 cases per 100 000 persons (Moses et al ., 2002). Epidemiological studies revealed that most outbreaks of invasive diseases in the world were mainly caused by strains of M1 and M3 serotypes (Stromberg et al ., 1991; Low et al ., 1998; Efstratiou, 2000). The emergence of exceptionally virulent strains of these serotypes (Musser et al ., 1991; Cleary et al ., 1992; Musser et al ., 1995) sug- gested that the increased virulence might be associated with specific genetic elements that could be acquired by horizontal transmission, but no such evidence has been provided. Other studies demonstrated that addi- tional serotypes caused invasive infections (Norgren et al ., 1992; Stanley et al ., 1995; Chaussee et al ., 1996; Cockerill et al ., 1997; Muotiala et al ., 1997). Furthermore,
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of A locus of group A streptococcus involved in invasive disease and DNA transfer: sil , streptococcal...

Molecular Microbiology (2002)
46
(1), 87–99
© 2002 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology 0950-382X Blackwell Science, 200245Original Article
sil, streptococcal invasive locusC. Hidalgo-Grass et al.
Accepted 24 June, 2002. *For correspondence. E-mail [email protected]; Tel. (
+
972) 2 6758196; Fax (
+
972) 2 6434170.
A locus of group A streptococcus involved in invasive disease and DNA transfer
Carlos Hidalgo-Grass,
1
Miriam Ravins,
1
Mary Dan-Goor,
1
Joseph Jaffe,
1
Allon E. Moses
2
and Emanuel Hanski
1
1
Department of Clinical Microbiology, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel.
2
Department of Clinical Microbiology and Infectious Diseases, Hadassah University Hospital, Jerusalem 91120, Israel.
Summary
Group A streptococcus (GAS) causes diseases rang-ing from benign to severe infections such as necro-tizing fasciitis (NF). The reasons for the differencesin severity of streptococcal infections are unex-plained. We developed the polymorphic-tag-lengths-transposon-mutagenesis (PTTM) method to identifyvirulence genes
in vivo
. We applied PTTM on an
emm
14 strain isolated from a patient with NF andscreened for mutants of decreased virulence, usinga mouse model of human soft-tissue infection. Amutant that survived in the skin but was attenuatedin its ability to reach the spleen and to cause a lethalinfection was identified. The transposon was insertedinto a small open reading frame (ORF) in a locustermed
sil
,
s
treptococcal
i
nvasion
l
ocus.
sil
containsat least five genes (
sil
A-E) and is highly homologousto the quorum-sensing competence regulons of
Streptococcus pneumoniae
.
sil
A and
sil
B encode aputative two-component system whereas
sil
D and
sil
E encode two putative ABC transporters.
sil
C isa small ORF of unknown function preceded by acombox promoter. Insertion and deletion mutants of
sil
had a diminished lethality in the animal model.Virulence of a deletion mutant of
sil
C was restoredwhen injected together with the avirulent
emm
14-deletion mutant, but not when these mutants wereinjected into opposite flanks of a mouse. DNA transferbetween these mutants occurred
in vivo
but could notaccount for the complementation of virulence. DNA
exchange between the
emm
14-deletion mutant andmutants of
sil
occurred also
in vitro,
at a frequencyof ~~~~
10
----
8
for a single antibiotic marker. Whereas
sil
Cand
sil
D mutants exchanged markers with the
emm
14mutant,
sil
B mutant did not. Thus, we identified anovel locus, which controls GAS spreading intodeeper tissues and could be involved in DNA transfer.
Introduction
Group A streptococcus (GAS) is a Gram-positive patho-gen globally responsible for human morbidity and mortal-ity. GAS causes extremely variable clinical manifestationsranging from mild self-limiting superficial infections of theskin and mucosa, to life-threatening invasive infectionssuch as septicaemia, toxic shock syndrome and necrotiz-ing fasciitis (NF). The organism is also responsible foracute rheumatic fever and acute glomerulonephritis(Bisno and Stevens, 1996; Musser and Krause, 1998).Group A streptococcus infections have increased in fre-quency and severity in the last decade in the UnitedStates and Europe (Cunningham, 2000).
Group A streptococcus infection is defined as invasivewhen bacteria are isolated from blood or other steriletissues. A prospective population-based study (PPBS) inOntario, Canada, showed that the annual incidence ofGAS invasive infections is 1.5 cases per 100 000 persons(Davies
et al
., 1996). Recently we have conducted aPPBS study in Israel, showing that the overall annualincidence of invasive GAS infections is even higher, reach-ing 3.7 cases per 100 000 persons (Moses
et al
., 2002).Epidemiological studies revealed that most outbreaks
of invasive diseases in the world were mainly caused bystrains of M1 and M3 serotypes (Stromberg
et al
., 1991;Low
et al
., 1998; Efstratiou, 2000). The emergence ofexceptionally virulent strains of these serotypes (Musser
et al
., 1991; Cleary
et al
., 1992; Musser
et al
., 1995) sug-gested that the increased virulence might be associatedwith specific genetic elements that could be acquired byhorizontal transmission, but no such evidence hasbeen provided. Other studies demonstrated that addi-tional serotypes caused invasive infections (Norgren
et al
., 1992; Stanley
et al
., 1995; Chaussee
et al
., 1996;Cockerill
et al
., 1997; Muotiala
et al
., 1997). Furthermore,

88
C. Hidalgo-Grass
et al.
© 2002 Blackwell Publishing Ltd,
Molecular Microbiology
,
46
, 87–99
the clonal M1T1 strains could be isolated from patientswith severe and non-severe invasive infections (Chatellier
et al
., 2000). These reports emphasize the role of hostresponse in determining the severity of GAS invasivediseases.
The hallmark of severe invasive GAS infections such asNF, is an aggressive and rapid spread of the bacteria.Ashbaugh
et al
. (1998) developed a murine model resem-bling human NF. Mice inoculated subcutaneously with aNF-causing strain, developed a spreading tissue necrosis,became bacteraemic and subsequently died (Ashbaugh
et al
., 1998). In this model, M protein and hyaluronic acidcapsule (HAC) are absolutely required for mouse mortal-ity. Furthermore, HAC production is induced upon intro-duction of GAS into the pharynx of baboons or intoperitoneum of mice (Gryllos
et al
., 2001). Also, it wasdemonstrated that cysteine protease (SpeB), streptolysinS (SLS) and C5a peptidase also contribute to tissuenecrosis and bacterial spreading (Lukomski
et al
., 1997;1999; Kuo
et al
., 1998; Betschel
et al
., 1998; Ji
et al
.,1996).
Pathogenic bacteria modulate virulence gene expres-sion in response to the rapidly changing environmentsencountered during the course of infection (Chiang
et al
.,1999). Group A streptococcus can survive and multiply indiverse anatomic sites, particularly during aggressive in-vasive diseases. To identify genes contributing to GASability to spread, we developed the polymorphic-tag-lengths-transposon-mutagenesis (PTTM) method. Thismethod resembles the original signature-tagged mutage-nesis (STM) method, which has been successfully usedboth in Gram-negative and Gram-positive pathogens toidentify genes that contribute to virulence
in vivo
(Mecsas,2002). Using the murine model we identified a mutant ofa strain causing NF (
emm
14 type) that was severely atten-uated in the ability to spread from the skin to the spleenand to cause a lethal infection. The mutant contained atransposon insertion in a locus that we termed
sil
, for
s
treptococcal
i
nvasion
l
ocus.
sil
exhibits a high homologyto the quorum-sensing competence regulon
com
and tothe peptide-sensing system
blp
of
Streptococcus pneu-moniae
(Hui
et al
., 1995; de Saizieu
et al
., 2000). Wedemonstrate that
sil
is also involved in DNA transfer.
Results
Lethality of NF-causing GAS strains in the murine model
We conducted a PPBS of invasive GAS infections in Israelduring a 2 year period, and identified 409 patients. In 125cases (31%), GAS was isolated from blood. The mostcommon illnesses were soft tissue infection (63%) andprimary bacteraemia (14%). Twenty-eight patients (7%)had toxic shock syndrome and 13 (3.4%) had NF (Moses
et al
., 2002). We examined the NF-causing strains in themurine model and found that all 13 strains produced anarea of spreading tissue necrosis extending from the siteof inoculation, as previously observed for other NF strains(Ashbaugh
et al
., 1998; Ravins
et al
., 2000; Engleberg
et al
., 2001). However, only three strains, JS20, JS95 andJS198, were lethal when injected under the surface of theskin at inocula that ranged from 4.0
¥
10
7
to 8.0
¥
10
8
colony-forming units (CFU). These three strains were of
emm
14 type as determined by
emm
sequencing (Facklam
et al
., 1999). We used strain JS95 for further studies.
Polymorphic-tag-lengths-transposon-mutagenesis (PTTM)
To identify genes that may contribute to the lethal pheno-type of JS95, we developed the PTTM system strategy(Fig. 1A). The frequency of transposition of the trans-posons IS
256pts
harbouring tags of different lengths intoJS95 ranged from 10
3
to 10
4
per
m
g DNA, whereas thefrequency of integration of the entire plasmids wasnegligible.
The PTTM strategy (Fig. 1A) resembles that of the orig-inal STM (Hensel
et al
., 1995) except for the followingdifferences: (i) the DNA tags are derived from a singlesequence but are different in size, whereas in STM thetags are of the same size but have different sequences;(ii) comparison of the output pools to the input pools isconducted by gel electrophoresis instead of DNA hybrid-ization and, (iii) the
E. coli
origin of replication in IS
256pts
allows easy identification of the insertion sites because itpermits replication of self-ligated chromosomal fragmentscontaining the transposon in
E. coli
. To achieve a satisfac-tory resolution, particularly between tags of close sizes,polymerase chain reaction (PCR) products were sepa-rated on a sequencing gel. A separation of tags isolatedfrom the skin and the spleen of a mouse challenged witha pool of 21 different mutants (harbouring tags of 21different sizes) is shown in Fig. 1B. In this experiment, theinoculum of the input pool was 2
¥
10
6
CFU. The mousewas sacrificed 4 days after inoculation and the outputpools from the skin and the spleen were compared. Oneof the mutants, designated JS95::
pttm
112, survived in theskin but failed to spread into the spleen. JS95::
pttm
112was subjected to further analysis.
Characterization of JS95::
pttm
112 in the murine model
We compared the ability of JS95 and that ofJS95::
pttm
112 to cause a fatal infection using a high doseof
~
5
¥
10
8
CFU. Mice challenged with JS95 died at arate that was significantly higher (
n
=
8;
P
=
0.001, LifeTable analysis, Logrank test, two groups comparison) than

sil
, streptococcal invasive locus
89
© 2002 Blackwell Publishing Ltd,
Molecular Microbiology
, 46, 87–99
Fig. 1. Identification of an attenuated mutant of JS95 by PTTM.A. The structure of IS256pt and a schematic presentation of PTTM strategy. IS256pt contains left and right inverted repeats (IRL and IRR), transposase, spectinomycin resistant gene, aad9, a tag and E. coli origin of replication (ori). Primers A and B amplify the tags and primers C and D are used to identify the insertion site. Detection of a missing tag in the output pool allows the identification of a mutant that is unable to grow in a specific tissue.B. Chromatograms of PCR amplified tags isolated from a mouse inoculated with an inoculum of ~ 2 ¥ 106 CFU containing 21 IS256pts mutants. One tag that is present in the skin (upper panel) is missing in the spleen (indicated by an arrow in the lower panel).
A
90 C. Hidalgo-Grass et al.
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
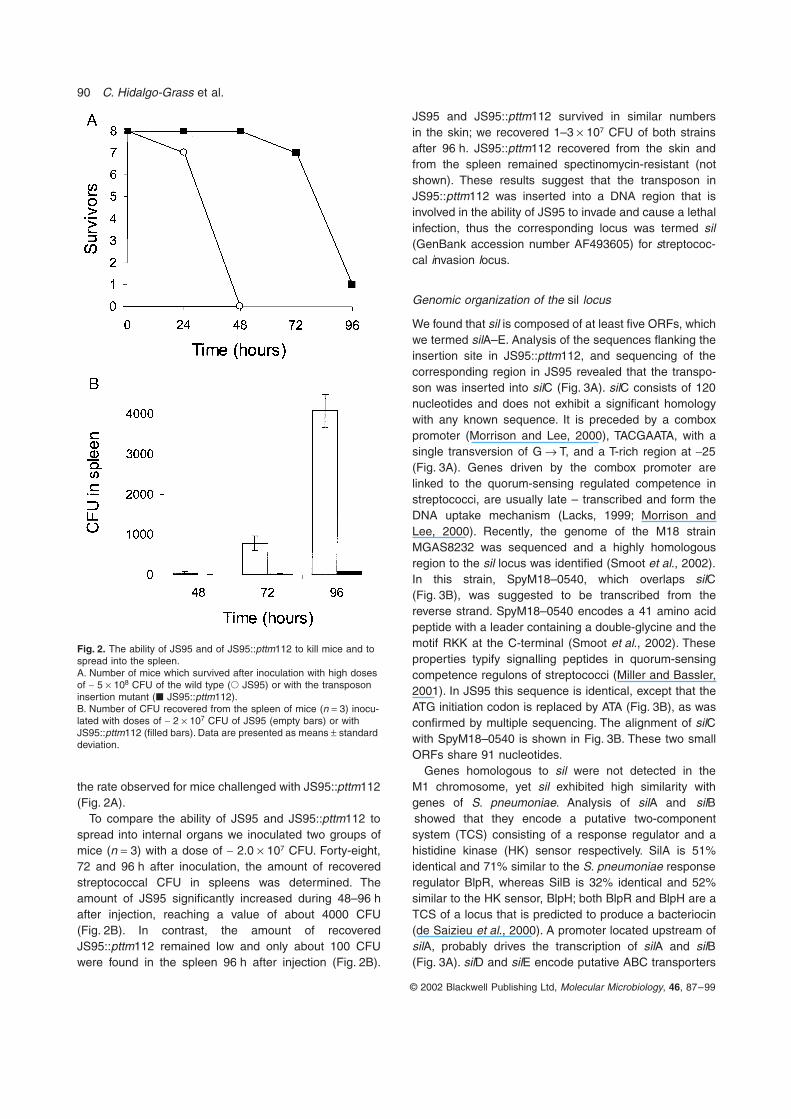
the rate observed for mice challenged with JS95::pttm112(Fig. 2A).
To compare the ability of JS95 and JS95::pttm112 tospread into internal organs we inoculated two groups ofmice (n = 3) with a dose of ~ 2.0 ¥ 107 CFU. Forty-eight,72 and 96 h after inoculation, the amount of recoveredstreptococcal CFU in spleens was determined. Theamount of JS95 significantly increased during 48–96 hafter injection, reaching a value of about 4000 CFU(Fig. 2B). In contrast, the amount of recoveredJS95::pttm112 remained low and only about 100 CFUwere found in the spleen 96 h after injection (Fig. 2B).
JS95 and JS95::pttm112 survived in similar numbersin the skin; we recovered 1–3 ¥ 107 CFU of both strainsafter 96 h. JS95::pttm112 recovered from the skin andfrom the spleen remained spectinomycin-resistant (notshown). These results suggest that the transposon inJS95::pttm112 was inserted into a DNA region that isinvolved in the ability of JS95 to invade and cause a lethalinfection, thus the corresponding locus was termed sil(GenBank accession number AF493605) for streptococ-cal invasion locus.
Genomic organization of the sil locus
We found that sil is composed of at least five ORFs, whichwe termed silA–E. Analysis of the sequences flanking theinsertion site in JS95::pttm112, and sequencing of thecorresponding region in JS95 revealed that the transpo-son was inserted into silC (Fig. 3A). silC consists of 120nucleotides and does not exhibit a significant homologywith any known sequence. It is preceded by a comboxpromoter (Morrison and Lee, 2000), TACGAATA, with asingle transversion of G Æ T, and a T-rich region at -25(Fig. 3A). Genes driven by the combox promoter arelinked to the quorum-sensing regulated competence instreptococci, are usually late – transcribed and form theDNA uptake mechanism (Lacks, 1999; Morrison andLee, 2000). Recently, the genome of the M18 strainMGAS8232 was sequenced and a highly homologousregion to the sil locus was identified (Smoot et al., 2002).In this strain, SpyM18–0540, which overlaps silC(Fig. 3B), was suggested to be transcribed from thereverse strand. SpyM18–0540 encodes a 41 amino acidpeptide with a leader containing a double-glycine and themotif RKK at the C-terminal (Smoot et al., 2002). Theseproperties typify signalling peptides in quorum-sensingcompetence regulons of streptococci (Miller and Bassler,2001). In JS95 this sequence is identical, except that theATG initiation codon is replaced by ATA (Fig. 3B), as wasconfirmed by multiple sequencing. The alignment of silCwith SpyM18–0540 is shown in Fig. 3B. These two smallORFs share 91 nucleotides.
Genes homologous to sil were not detected in theM1 chromosome, yet sil exhibited high similarity withgenes of S. pneumoniae. Analysis of silA and silBshowed that they encode a putative two-componentsystem (TCS) consisting of a response regulator and ahistidine kinase (HK) sensor respectively. SilA is 51%identical and 71% similar to the S. pneumoniae responseregulator BlpR, whereas SilB is 32% identical and 52%similar to the HK sensor, BlpH; both BlpR and BlpH are aTCS of a locus that is predicted to produce a bacteriocin(de Saizieu et al., 2000). A promoter located upstream ofsilA, probably drives the transcription of silA and silB(Fig. 3A). silD and silE encode putative ABC transporters
Fig. 2. The ability of JS95 and of JS95::pttm112 to kill mice and to spread into the spleen.A. Number of mice which survived after inoculation with high doses of ~ 5 ¥ 108 CFU of the wild type (� JS95) or with the transposon insertion mutant (� JS95::pttm112).B. Number of CFU recovered from the spleen of mice (n = 3) inocu-lated with doses of ~ 2 ¥ 107 CFU of JS95 (empty bars) or with JS95::pttm112 (filled bars). Data are presented as means ± standard deviation.

sil, streptococcal invasive locus 91
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
that are highly homologous to BlpB/ComB, and to ComA(Hui et al., 1995; de Saizieu et al., 2000). SilD exhibits56% identity and 76% similarity with BlpB, and 33% iden-tity and 55% similarity with ComB. A homologue of silD inthe strain MGAS8232 is truncated and composed of twoORFs, SpyM18–0541 and SpyM18–0542 (Smoot et al.,2002). SilE shares 67% identity and 83% similarity withComA.
Downstream of the sil locus we found an ORF homol-ogous to S. pneumoniae blpM; a gene that encodes apredicted bacteriocin peptide (de Saizieu et al., 2000),which we named blpMH (Fig. 3C). By sequence alignmentwe identified a truncated blpM homologue (blpMT) locatedupstream of SPy0484 in the M1 chromosome (Fig. 3C).To characterize the region further downstream of sil wedesigned primers hybridizing to blpMH in JS95 and to
Fig. 3. The genomic organization of sil.A. The sil locus includes the putative TCS genes silA and silB, silC that is preceded by a combox promoter, and the two putative ABC transporters genes, silD and silE. The insertion of IS256pt in JS95::pttm112 disrupts silC. The small arrow upstream of silA represents a promoter. IS1562 and blpM-homologue (BlpMH.) are located upstream and downstream of sil respectively. Bent arrows represent primers that are described in Table 3.B. silC and SpyM18–0540 are transcribed in oppo-site directions. The relative positions of silC and SpyM18–0540 transcripts are shown in the upper part. The indicated StyI and BspEI sites were used for replacement of silC by the spectinomycin resis-tance gene, aad9. The alignment of silC with SpyM18–0540, which encodes a putative compe-tence stimulating peptide in the M18 MGAS8232 strain, is shown in the lower part. A point mutation in JS95 at position 124 destroys the start codon of the ORF predicted by Smoot et al. (2002).C. sil is located in JS95 in a region that corre-sponds to section 36 of M1 chromosome. The black arrows in the M1 chromosome designate genes encoding peptides with a double-glycine leader. blpMT is homologous to blpMH but is trun-cated in M1. The bent arrows represent primers used to identify the upstream and downstream regions of sil.
ATGACAAGTTTAAAAAATGACAAGTTTAAAAA
TATCTCCACCAATCACTTTAAGTAATTCAGATTCAGATAATGTTGAAAAGTATCTCCACCAATCACTTTAAGTAATTCAGATTCAGATAATGTTGAAAAG
TTATTTTTTGTTTTTTTATTGTTTTATTTTTTGTTTTTTTATTGTT
ATAT
A
B
C
92 C. Hidalgo-Grass et al.
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
SPy0488 and mutR in M1 (Fig. 3C). The amplified frag-ments in JS95 (blpMH-SPy0488 and blpMH-mutR) wereidentical in size to those expected for the M1 chromo-some, indicating that the region downstream of the sillocus in JS95 is homologous to section 36 of the M1.Upstream of silA we found the IS1562 (Berge et al., 1998).To identify the region further upstream to IS1562 wedesigned primers annealing to IS1562 in JS95, and toera and SPy0479 in the M1 chromosome respectively(Fig. 3C). The PCR analysis showed that, in JS95, sillocus is also flanked upstream by sequences presentin section 36 of M1 chromosome, but JS95 containsadditional regions of 2 kb located between IS1562 andSPy0479 and 4 kb between era and SPy0479.
The role of sil in virulence
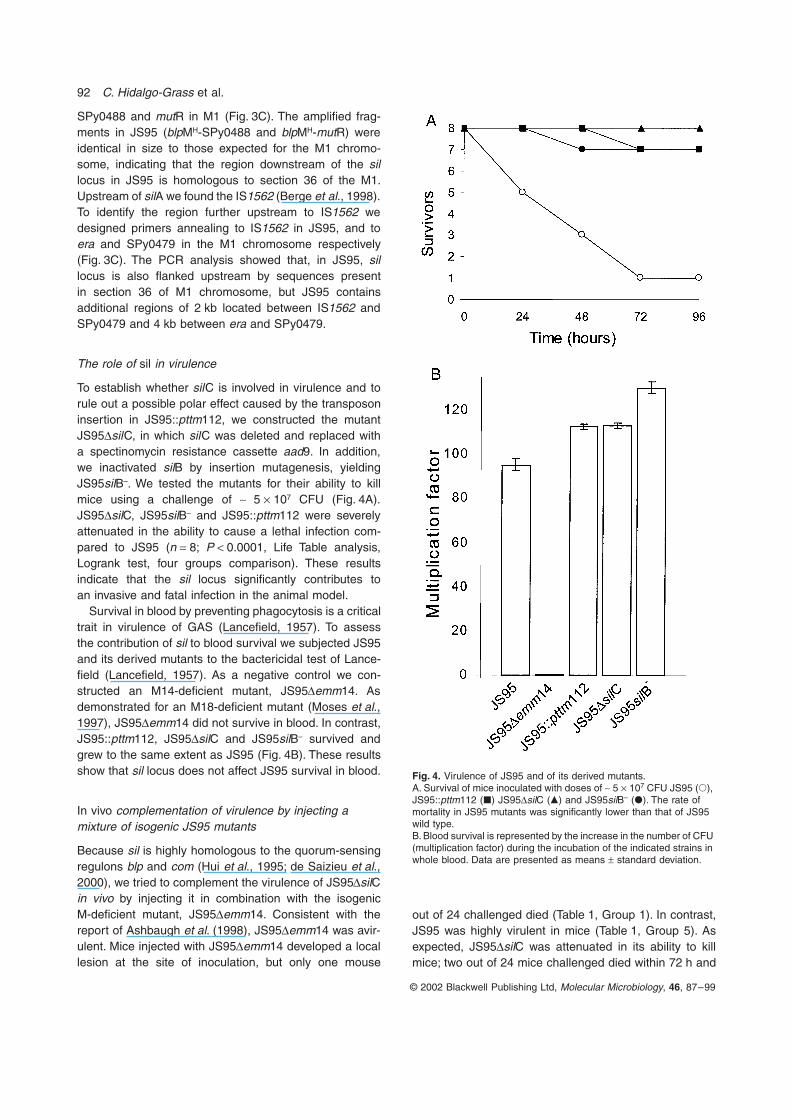
To establish whether silC is involved in virulence and torule out a possible polar effect caused by the transposoninsertion in JS95::pttm112, we constructed the mutantJS95DsilC, in which silC was deleted and replaced witha spectinomycin resistance cassette aad9. In addition,we inactivated silB by insertion mutagenesis, yieldingJS95silB–. We tested the mutants for their ability to killmice using a challenge of ~ 5 ¥ 107 CFU (Fig. 4A).JS95DsilC, JS95silB– and JS95::pttm112 were severelyattenuated in the ability to cause a lethal infection com-pared to JS95 (n = 8; P < 0.0001, Life Table analysis,Logrank test, four groups comparison). These resultsindicate that the sil locus significantly contributes toan invasive and fatal infection in the animal model.
Survival in blood by preventing phagocytosis is a criticaltrait in virulence of GAS (Lancefield, 1957). To assessthe contribution of sil to blood survival we subjected JS95and its derived mutants to the bactericidal test of Lance-field (Lancefield, 1957). As a negative control we con-structed an M14-deficient mutant, JS95Demm14. Asdemonstrated for an M18-deficient mutant (Moses et al.,1997), JS95Demm14 did not survive in blood. In contrast,JS95::pttm112, JS95DsilC and JS95silB– survived andgrew to the same extent as JS95 (Fig. 4B). These resultsshow that sil locus does not affect JS95 survival in blood.
In vivo complementation of virulence by injecting a mixture of isogenic JS95 mutants
Because sil is highly homologous to the quorum-sensingregulons blp and com (Hui et al., 1995; de Saizieu et al.,2000), we tried to complement the virulence of JS95DsilCin vivo by injecting it in combination with the isogenicM-deficient mutant, JS95Demm14. Consistent with thereport of Ashbaugh et al. (1998), JS95Demm14 was avir-ulent. Mice injected with JS95Demm14 developed a locallesion at the site of inoculation, but only one mouse
out of 24 challenged died (Table 1, Group 1). In contrast,JS95 was highly virulent in mice (Table 1, Group 5). Asexpected, JS95DsilC was attenuated in its ability to killmice; two out of 24 mice challenged died within 72 h and
Fig. 4. Virulence of JS95 and of its derived mutants.A. Survival of mice inoculated with doses of ~ 5 ¥ 107 CFU JS95 (�), JS95::pttm112 (�) JS95DsilC (�) and JS95silB– (�). The rate of mortality in JS95 mutants was significantly lower than that of JS95 wild type.B. Blood survival is represented by the increase in the number of CFU (multiplication factor) during the incubation of the indicated strains in whole blood. Data are presented as means ± standard deviation.

sil, streptococcal invasive locus 93
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
another two died between 72 h and 96 h (Table 1, Group2). However, when mice were injected at one site witha premixed inoculum of JS95DsilC and JS95Demm14, alethal infection developed rapidly. Out of 32 challengedmice, 23 died within 72 h and another six between 72 hand 96 h (Table 1, Group 3). In contrast, when mice werechallenged with the same mutants, but each mutant wasinjected separately into opposite flanks, lethality was onlyslightly higher than in mice challenged with JS95DsilCalone. Two out of 16 mice challenged died after 72 h andanother three died between 72 h and 96 h (Table 1, Group4). A mixed model analysis of variance was performedin which ‘Group’ was a fixed effect, ‘Experiment’ was arandom effect, and the response variable was percentsurvival at 72 h and 96 h respectively. ‘Group’ had a sig-nificant effect on mortality at both of these times(P = 0.0012, 72 h; P < 0.0001, 96 h). A pair-wise compar-ison between each of the five ‘Groups’ was performed,using the Bonferonni correction for multiple comparisons.The difference in the rate of mortality of Groups 3 and 5compared to Groups 1, 2 and 4 was statistically significantat both 72 h and 96 h.
We examined the number of GAS CFU in the spleenof mice in Groups 3 and 4. Seventy-two hours afterinoculation the amount of CFU in Group 3 (n = 3) was25 305 ± 2269, whereas in Group 4 (n = 3) only1720 ± 120. The complementation of the full virulentphenotype observed only for the premixed inocula,suggests that signalling occurs between JS95DsilC andJS95Demm14.
In vivo and in vitro DNA transfer
We tested which of the strains in the premixed inocula(Table 1, Group 3) reached the spleen. Most of the colo-nies present in the spleen were JS95DsilC (spectinomy-cin-resistant), and only very few were JS95Demm14(kanamycin-resistant) probably because the latter do notsurvive in whole blood (Fig. 4B). In one of the four miceexamined, we recovered some colonies that lost antibioticresistance completely. We analysed nine of these coloniesby PCR, and one spectinomycin-resistant colony as a
control. All 10 colonies possessed an intact emm14 gene(Fig. 5 upper panel). As expected, the spectinomycin-resistant colony possessed an intact aad9 cassette andthus produced a larger PCR product equal in size tothat of JS95DsilC (Fig. 5 lower panel). All nine antibiotic-
Table 1. Complementation of virulence.
Mortality (%)a
Group Strain InjectionAnimals(n) Experiments
Inoculum(108 CFU) 72 h 96 h
1 JS95Demm14 One site 24 3 1.6–29.0 4.1 4.12 JS95DsilC One site 24 3 1.0–15 8.3 16.63 JS95Demm14 and JS95DsilC (premixed) One site 32 4 1.2–2.2 71.8 90.64 JS95Demm14 and JS95DsilC Two sitesb 16 2 1.2–4.1 12.5 31.25 JS95 One site 21 3 1.0–2.3 76.2 90.5
a. The difference in the rate of mortality of groups 3 and 5 compared to groups 1, 2 and 4 was statistically significant at both 72 and 96 h.b. Injected into opposite flanks
Fig. 5. In vivo DNA transfer between JS95Demm14 and JS95DsilC. PCR analyses were performed on 10 colonies recovered from the spleen of a mouse challenged with a mixture of both mutants. Nine of these colonies lost antibiotic resistance and one colony was spec-tinomycin resistant (colony 10). The upper panel shows emm14 anal-ysis using primers allM-f and allM-r (Table 3). The lower panel shows silC analysis using primers SilB-f1 and SilD-r1 (Fig. 3A). The indi-cated strains served as controls for PCR analyses. M represents a 100 bp ladder.

94 C. Hidalgo-Grass et al.
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
sensitive colonies possessed an intact silC (Fig. 5 lowerpanel). These data show that DNA transfer occurredbetween JS95DsilC and JS95Demm14 during the in-fection in vivo, and that this transfer led to the regenerationof wild-type JS95. Because of the low frequency ofDNA transfer that was discovered in only one mouse,it is probably not accountable for complementation ofvirulence.
The DNA transfer between JS95DsilC andJS95Demm14 was further examined in vitro. The mutantswere grown separately to an OD650 of 0.16; samples werewithdrawn, mixed together for 60 min and plated on THY(Todd-Hewitt medium supplemented with 0.2% yeastextract) plates containing both spectinomycin and kana-mycin. This was repeated every hour during the growth ofthe mutants. We found that there was antibiotic markerexchange between these mutants, which peaked duringmid-log to late-log phase (Fig. 6). Antibiotic resistancemarker exchange was verified by PCR analyses usingtwo sets of primers, aad9-f and aad9-r and A11 and C5(Table 3). These sets amplify the spectinomycin and theWkm-2 cassettes respectively. As expected, the strainsthat were resistant to kanamycin and spectinomycin pos-sessed both cassettes. The rate of marker transfer wasas low as ~ 10-8, which is much lower than that observedfor naturally transformable S. pneumoniae (Lacks, 1999).When the experiment was repeated with JS95silD– (aninsertion mutant of silD) similar results were obtained (notshown). However, no exchange of markers was detectedbetween JS95Demm14 and JS95silB–. These results,suggest that the TCS SilA/B is involved in the DNA trans-fer of markers observed in vitro.
Discussion
We identified the locus sil in GAS strain JS95, whichcontributes to GAS virulence by enhancing GAS spread-ing and invasion of internal organs. Mutants of sil wereattenuated in their ability to disseminate from the skin intothe spleen and to cause a lethal infection in a mouse-model of human NF. We found that the sil locus is presentin three different M14 strains isolated from patients withNF, which were also lethal in the mouse model. Recently,the genome of an M18-type strain associated with acuterheumatic fever was sequenced (Smoot et al., 2002), andit contains a locus highly homologous to sil. Thus, silmay regulate virulence genes in GAS strains of differentgenetic backgrounds causing diverse types of humandiseases.
In Gram-positive bacteria, the regulation of genes byquorum sensing is density-dependent and is mediated bypeptides (Miller and Bassler, 2001). Mutants of silB andsilC were severely attenuated in their ability to reach thespleen and kill mice. The avirulent mutant JS95Demm14complemented the virulence of JS95DsilC in vivo, onlywhen the two mutants were injected together in the samesite. Based on the mechanism of competence regulationin S. pneumoniae (Morrison and Lee, 2000), it is possiblethat JS95Demm14 produces a signalling peptide, whichreaches a critical density and activates the TCS SilA/B in JS95DsilC. This in turn, activates genes required forGAS spreading and mouse lethality. The identification ofJS95::pttm112 was carried out using a low inoculum of2 ¥ 106 CFU (Fig. 1B), thus strengthening the notion thatthe complementation of virulence is a density dependentphenomenon.
The TCS SilA/B appears to be involved also in DNAtransfer as silB mutant failed to exchange antibiotic resis-tance markers with JS95Demm14. As in S. pneumoniae(Morrison and Lee, 2000), it is conceivable that the failureto signal through SilA/B abrogates DNA transfer. However,at this stage we cannot completely exclude that theexchange of markers might have occurred via other mech-anisms such as transduction or conjugation.
Genes involved in DNA binding and uptake are usuallyexpressed only during the competence period, and arecharacterized by the combox promoter (Morrison and Lee,2000). In the M1 chromosome there are 18 genes pre-ceded by a combox promoter, most of an unknown func-tion (Opdyke et al. 2001). The expression of such genesis controlled by a secondary sigma factor (Morrison andLee, 2000). Such a factor has recently been purified fromGAS and was shown to function in vitro (Opdyke et al.,2001). Further studies are required to demonstratewhether silC is expressed from the forward or the reversestrands and whether it plays a role in the process of DNAuptake.
Fig. 6. In vitro exchange of DNA between JS95Demm14 and JS95DsilC. Measuring OD at 650 nm monitored the growth of JS95Demm14 (�) and of JS95DsilC (�). Transformation frequency (�) is the number of mutants resistant to both kanamycin and spec-tinomycin divided by the sum of CFU of the mutants in the mixture.

sil, streptococcal invasive locus 95
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
The M1 chromosome contains a significant portion ofthe genes required for competence-mediated transforma-tion (Ferretti et al., 2001). However, DNA uptake hasnever been reported for GAS, and it could not be deter-mined whether these genes had ever mediated such anevent (Ferretti et al., 2001). The inability to mediate com-petence has been attributed to the lack of the crucialgenes comAB in the M1 chromosome (Ferretti et al.,2001). The identification of the sil locus, which containshighly homologous genes to comAB and to comDE, andthe discovery that sil affects DNA exchange in vitro, pro-vides the first indication for DNA transfer in GAS.
There are several lines of evidence suggesting that silmight have been acquired by a horizontal transmissionduring GAS evolution. The G + C content of the individualsil ORFs (A to E) are: 33.1%, 29.7%, 20.5%, 33.0%, and33.0%, respectively, thus sil has a markedly lower G + Ccontent (average 32%) than that of the M1 chromosome(38.5%, Ferretti et al., 2001). As indicated above sil exhib-its a high degree of homology with the blp and com lociof S. pneumoniae. Among the 14 TCSs of S. pneumoniae,only ComD/E (Lange et al., 1999) and BlpR/H (de Saizieuet al., 2000) display unusually low G + C content whereasthe content of the other TCSs is similar to that of the S.pneumoniae chromosome (39.7%) (Tettelin et al., 2001).Thus, sil, like com and blp loci, might have been acquiredfrom the same bacterial strain of low G + C content. silcontains IS1562, which is found in most GAS strains indifferent locations and thus has been suggested to play arole in the genomic plasticity of GAS (Berge et al., 1998).Comparison of the blpM homologue sequence locateddownstream of silE in JS95 with that present on the M1SF370 chromosome reveals that the latter is truncated,suggesting that the sil locus might have been lost from theM1 chromosome. These data taken together, suggest thatsil could reside on a mobile element. Further study of sil
will shed light on the contributions of quorum-sensing andDNA transfer to GAS virulence.
Experimental procedures
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study arelisted in Table 2. Group A streptococcus (GAS) strains weregrown in Todd-Hewitt medium supplemented with 0.2%yeast extract (THY) and E. coli was grown in Luria–Bertani(LB). Antibiotics used for GAS: 250 mg ml-1 kanamycin,50 mg ml-1 spectinomycin, 500 mg ml-1 streptomycin, and1 mg ml-1 erythromycin and for E. coli: 100 mg ml-1 ampicillin,50 mg ml-1 spectinomycin and 750 mg ml-1 erythromycin.
A murine model of invasive soft-tissue infection
The ability of GAS to cause lethal infection in 10 g BALB/cfemale mice (Harlan Laboratories, Jerusalem, Israel) wasassessed as described before (Ravins et al., 2000). In brief,bacteria grown in THY to mid-log phase (OD600 of 0.4) werewashed in phosphate-buffered saline (PBS), concentrated ordiluted, and injected under the surface of the skin. The num-ber of CFU injected was verified for each experiment byplating bacteria on THY-blood agar plates and counting. Micewere daily observed for death. To determine the presence ofbacteria in skin and spleen, mice were sacrificed; a wide areasurrounding the skin lesions was excised, spleens wereremoved, and organs were homogenized in 10 or 1 ml ofsterile PBS respectively. Aliquots were plated and streptococ-cal colonies were counted. The Institutional Ethics Commit-tee for animal care approved all animal procedures.
DNA manipulation and primers
All manipulations of chromosomal and plasmid DNA wereperformed by standard techniques (Sambrook et al., 1989).The primers used are listed in Table 3. Cloning and pri-
Table 2. Bacterial strains and plasmids.
Strain/plasmid Description Reference/source
JS95 GAS strain isolated from a patient with NF (Moses et al., 2002)JS95::pttm112 silC PTTM-inactivated derivative of JS95 This studyJS95DsilC silC deletion mutant of JS95 This studyJS95silB– silB insertional-inactivated derivative of JS95 This studyJS95silD– silD insertional-inactivated derivative of JS95 This studyJS95Demm14 emm14 deletion mutant of JS95 This studypMGC57 A plasmid containing IS256 with a unique EcoRV site (Lyon et al., 1998)pGEM-T-Easy Cloning vector Promega, Madison, WisconsinpUC19 Cloning vector New England Biolabs, Inc. Beverly, MApCHOP-10 pCRII (Invitrogen, Carlsbad, California) containing 677 bp
of the Mouse chop-10 mRNAA gift of Dr Y. Dor (The Hebrew University,
Jerusalem, Israel)pFW11 A streptococcal shuttle vector containing the aad9
spectinomycin resistance gene(Podbielski et al., 1996)
pIS256pts Plasmids containing tags of different lengths This studypORI280 A vector for gene replacement in GAS (Leenhouts et al., 1996)pJRS233 Streptococcus-E. coli temperature sensitive shuttle vector (Perez-Casal et al., 1993)

96 C. Hidalgo-Grass et al.
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
mers were designed using VectorNTI software (InforMax,Bethesda, Maryland). Alignment analyses were done byBLAST <http://www.ncbi.nlm.nih.gov/blast> and FASTA (GCG,University of Wisconsin).
The emm typing was conducted according to Facklamet al. (1999) and confirmed by the National Centers forDisease Control, Biotechnology Core Facility Comput-ing Laboratory <http://www.cdc.gov/ncidod/biotech/strep/strepblast.html>
Polymorphic-tag-lengths-transposon-mutagenesis (PTTM)
Construction of pIS256pts. IS256 from pMGC57 was ampli-fied using universal and reverse primers, and cloned intopGEM-T-easy. The resulting plasmid was digested with XbaIand SphI and the fragment containing IS256 was cloned intopUC19, generating pC256. A fragment of 677 bp of themouse gene chop-10, which displays no homology with theM1 GAS genome, was amplified from pCHOP-10 using uni-versal and reverse primers, and the product was cloned intopGEM-T-easy. The resulting plasmid was digested withEcoRV and XbaI and the XbaI-SmaI fragment containinga spectinomycin-resistant gene, aad9, from pFW11 was
cloned. Following digestion with BglI and BglII the fragmentcontaining aad9, the chop-10 and the E. coli origin of repli-cation, ColE1, was cloned into the EcoRV site of pC256. Theampicillin-resistance gene was removed by digestion withMunI and PvuI, generating pIS256pt. To produce a set ofplasmids with different tag lengths, pIS256pt was digestedwith NheI (Fig. 1A) and subjected to a bidirectional deletionusing Erase-a-Base system (Promega, Madison, Wisconsin).The resulting set of plasmids was termed pIS256pts.
GAS mutagenesis. JS95 was transformed with pIS256pts(21 tags of different lengths) as described previously (Lyonet al., 1998) and spectinomycin resistant clones wereselected.
In vivo screening of mutants
A library of mutants each containing a tag of a different length(input pool), was grown to an OD600 of 0.4, adjusted to theappropriate inoculum and injected into mice (Ravins et al.,2000). Output pools of mutants were isolated from the skinand from the spleen and subjected to PCR analysis usingprimers A and B (Fig. 1A). Primer A was fluorescentlylabelled with 6FAM (Biosource international, Camarillo, CA).The tags were resolved by electrophoresis on a sequencing
Table 3. PCR and sequencing primers.
Name Sequence Description/Location Reference
Universal 5¢-GTAAAAAACGACGGCCAGT-3¢ M13/pUC sequencing primer (-20) New England Biolabs,Inc. Beverly, MA
Reverse (A) 5¢-AACAGCTATGACCATG-3¢ M13/pUC reverse sequencing primer(-21) Forward primer for tagamplification
New England Biolabs,Inc. Beverly, MA
B 5¢-AGCAGTTCGTAGTTATCTTG-3¢ Reverse primer for tag a amplification This studyC 5¢-TTATCAGCAATAAACCAGC-3¢ Inverse PCR primer from IRR This studyD 5¢-AAAGTCCTCCTGGGTATG-3¢ Inverse PCR primer from IRL This studyE 5¢-TTTGGCAGCTTTGACGATGC-3¢ Inverse PCR primer from 3¢ of silE This studyF 5¢-TCTTCAAGCAGCTGATTGGG-3¢ Inverse PCR primer from 5¢ of silA This studySilB-f1 5¢-GGAGTTGGTTTATCAAATGTCAG-3¢ 2598–2620 in sil This studySilD-r1 5¢-ATCTGCCACAAAGACTGATCAAG-3¢ 3213–3235 in sil This studySilB-f2 5¢-TTATTGGATCGGAACTTACGC-3¢ 2013–2033 in sil This studySilD-r2 5¢-TGCTTCCCAACAACTTACCAC-3¢ 3554–3574 in sil This studySilB-f3 5¢-GCTCGCTATAGTAAGCAAATCG-3¢ 2088–2109 in sil This studySilE-r 5¢-CAGCGATTAAGCATTGAC-3¢ 5871–5888 in sil This studyHK-f 5¢- ACGAAAGGTCAATGGTTCAC-3¢ 1616–1634 in sil This studyHK-r 5¢- AGGTATGGATAAGCGTTGAG-3¢ 2338–2357 in sil This studyABC1-f 5¢-ATGACACTTGTTACACGTCC-3¢ 3120–3138 in sil This studyABC1-r 5¢- ACTAGTCAGCTTGACGAACTTC-3¢ 3873–3894 in sil This studyPrimer1 5¢-TATTCGCTTAGAAAATTAA-3¢ emm typing forward primer (Whatmore and Kehoe, 1994)Primer2 5¢-GCAAGTTCTTCAGCTTGTTT-3¢ emm typing reverse primer (Whatmore and Kehoe, 1994)aad9-f 5¢ CCATGGTCCTCGAGCTCTAGATCTTAAG 3¢ aad9 forward primer This studyaad9-r 5¢ CTGCAGGCGCTTACCAATTAGAATG 3¢ aad9 reverse primer This studyBlpM-f 5¢-TCGATATGGAGATAAAGAAACTGG-3¢ 6873–6896 in JS95 sil, 5096–5119
in M1This study
SPy0488-r 5¢-AACAGTGCTTTCAGGAACTCCT-3¢ 6804–6825 in M1 section 36 This studyMutR-r 5¢-CTAGGTGCAATTGAGGAGTCAA-3¢ 10031–10052 in M1 section 36 This studyIS1562-r 5¢-TCCTCGCACTGTTCCAATAG-3¢ 20–43 in JS95 sil, 7287–7306
section 152 in M1This study
SPy0479-f 5¢-AGGTGGTGTTGGAGCAGGTA-3¢ 3580–3599 in M1 section 36 This studyera-f 5¢-AAGAAGTGGTCCCAATTTCTG-3¢ 1545–1565 in M1 section 36 This studyallM-f 5¢-CCTGAAAATGAGGATCCTTCCTAAAAAACG-3¢ Forward all M primer with BamHI site This studyallM-r 5¢-GGGGGGCTGCAGAGCTTAGTTTTCTTCTTTGCG-3¢ Reverse all M primer with PstI site (Berkower et al., 1999)A11 5¢- GATTCCAGAAGCGATTATTG-3¢ 3¢ of mga (Podbielski, 1993)C5 5¢-AATGGCAAGTTTATCAAATGG Leader peptide region of scpA (Podbielski, 1993)

sil, streptococcal invasive locus 97
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
gel and analysed using ABI-Prism 3.2 GeneScan software(Perkin Elmer, Wellesley, MA); TAMRA 1000 (Biosourceinternational, Camarillo, CA) was used as a size marker. Tagsizes of mutants present in the input and output pool werecompared.
Identification of the insertion site in JS95::pttm112 and mapping of sil
JS95::pttm112 DNA was digested with MluI, which does notcut inside IS256pt. The digested DNA was self-ligated andtransformed into E. coli. Plasmids derived from coloniesresistant to spectinomycin were sequenced using primersC and D (Fig. 1A). The corresponding region in JS95 wasamplified using primers SilB-f2 and SilD-r2 and sequencedusing primers SilB-f1 and SilD-r1. The regions upstream anddownstream of the insertion site were amplified by inversePCR and sequenced (Ochman et al., 1990).
Construction of JS95 mutants
JS95DsilC. To construct JS95DsilC we amplified from JS95a 1572-bp fragment containing silC, 695 bp upstream and511 bp downstream, using the primers SilB-f2 and SilD-r2(Fig. 3A). The PCR product was cloned into pORI280. A StyI-BspEI fragment encompassing silC was replaced by aad9.The resulting plasmid was electroporated into JS95, andmutants resistant to spectinomycin but not to erythromycinwere selected. Fidelity of the replacement and the integrityof silB and silD were confirmed in one mutant, JS95DsilC byPCR using primers SilB-f2 and SilD-r2 and by sequencing.
JS95Demm14. For deletion of the emm14 gene, we clonedinto pJRS233 a fragment of 409 bp upstream of emm14, theWKm-2 and a fragment of 450 bp downstream to emm14. Theresulting plasmid was introduced into JS95, and erythromy-cin and kanamycin double resistant transformants wereselected at the permissive temperature (30∞C). Growth oftransformants at the non-permissive temperature (37∞C)resulted in a single crossover insertion. A second crossoverwas obtained as described previously (Ashbaugh et al.,1998). Growth of transformants in the presence of kanamycinbut not erythromycin indicated that a second recombinationhad occurred. Fidelity of the replacement was confirmed byPCR using primers allM-f and allM-r and by Western blotusing anti-M14 sera.
JS95silB –. For insertion inactivation of silB, an intragenicfragment of silB was amplified using primers HK-f and HK-r,and cloned into pJRS233. The resulting plasmid was intro-duced into JS95 and erythromycin-resistant transformantswere isolated. The disruption of silB was verified by PCRanalysis using primers SilB-f3 and SilD-r2.
JS95silD–. For the insertion inactivation of silD, a 754 bpintragenic region of this ORF was amplified using the primersABC1-f and ABC1-r, and cloned into pFW11. The resultingplasmid was introduced into JS95 by electroporation andspectinomycin-resistant transformants were isolated. Thedisruption of silD was verified by PCR analysis using primersSilB-f1 and SilD-r2.
Blood survival assay
The ability of GAS to survive in human blood was tested bythe direct bactericidal test of Lancefield as described by uspreviously (Moses et al., 1997). In brief, 0.6 ml of freshlydrawn heparinized human blood was mixed with 0.2 ml ofGAS grown to OD600 of 0.16 and diluted in THY 1:40 000 sothat the initial number of bacteria in the assay was approxi-mately 100–300 CFU. Plating bacterial culture on THY platesand counting CFU determined the exact number of bacteriaadded to the blood. The mixture was incubated at 37∞C for3 h with end-over-end rotation, after which, bacteria wereplated on THY plates and CFU were counted. Multiplicationfactor was calculated by dividing the number of CFU after 3 hwith the initial CFU added to blood.
In vitro transfer of DNA
JS95Demm14 and sil-derived mutants of JS95 were grownseparately without antibiotics at 37∞C to an OD650 of 0.16. Atthe indicated time equal volumes (7 ml) of each culture weremixed and further incubated for 60 min. Fifteen samples of0.2 ml each were plated on THY plates containing kanamy-cin and spectinomycin, and streptococcal colonies werecounted.
Acknowledgements
This work was supported by a grant from the Center for theStudy of Emerging Diseases (E.H.), and grants from theIsraeli Science Foundation administrated by the Israel Acad-emy of Science and Humanities (E.H. and A.E.M.). C.H.G.was supported by The Golda Meir Fellowship Fund. We thankZinaida Korenman from the Ministry of Health, the Strepto-coccal Reference Laboratory, Jerusalem, Israel for perform-ing T-typing and for the donation of anti-M14 sera. We wouldlike also to thank Dr M. Korner from the Hebrew Universityfor her help in resolving the tags in PTTM.
References
Ashbaugh, C.D., Warren, H.B., Carey, V.J., and Wessels,M.R. (1998) Molecular analysis of the role of the group Astreptococcal cysteine protease, hyaluronic acid capsule,and M protein in a murine model of human invasive soft-tissue infection. J Clin Invest 102: 550–560.
Berge, A., Rasmussen, M., and Bjorck, L. (1998) Identifica-tion of an insertion sequence located in a region encodingvirulence factors of Streptococcus pyogenes. Infect Immun66: 3449–3453.
Berkower, C., Ravins, M., Moses, A.E., and Hanski, E. (1999)Expression of different group A streptococcal M proteins inan isogenic background demonstrates diversity in adher-ence to and invasion of eukaryotic cells. Mol Microbiol 31:1463–1475.
Betschel, S.D., Borgia, S.M., Barg, N.L., Low, D.E., and DeAzavedo, J.C. (1998) Reduced virulence of group A strep-tococcal Tn916 mutants that do not produce streptolysinS. Infect Immun 66: 1671–1679.

98 C. Hidalgo-Grass et al.
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
Bisno, A.L., and Stevens, D.L. (1996) Streptococcal infec-tions of skin and soft tissues. N Engl J Med 334: 240–245.
Chatellier, S., Ihendyane, N., Kansal, R.G., Khambaty, F.,Basma, H., Norrby-Teglund, A., et al. (2000) Genetic relat-edness and superantigen expression in group A strepto-coccus serotype M1 isolates from patients with severeand nonsevere invasive diseases. Infect Immun 68: 3523–3534.
Chaussee, M.S., Liu, J., Stevens, D.L., and Ferretti, J.J.(1996) Genetic and phenotypic diversity among isolates ofStreptococcus pyogenes from invasive infections. J InfectDis 173: 901–908.
Chiang, S.L., Mekalanos, J.J., and Holden, D.W. (1999) Invivo genetic analysis of bacterial virulence. Annu RevMicrobiol 53: 129–154.
Cleary, P.P., Kaplan, E.L., Handley, J.P., Wlazlo, A., Kim,M.H., Hauser, A.R., and Schlievert, P.M. (1992) Clonalbasis for resurgence of serious Streptococcus pyogenesdisease in the 1980s. Lancet 339: 518–521.
Cockerill, F.R., III, MacDonald, K.L., Thompson, R.L.,Roberson, F., Kohner, P.C., Besser-Wiek, J., et al. (1997)An outbreak of invasive group A streptococcal diseaseassociated with high carriage rates of the invasive cloneamong school-aged children. JAMA 277: 38–43.
Cunningham, M.W. (2000) Pathogenesis of group A strepto-coccal infections. Clin Microbiol Rev 13: 470–511.
Davies, H.D., McGeer, A., Schwartz, B., Green, K., Cann, D.,Simor, A.E., and Low, D.E. (1996) Invasive group Astreptococcal infections in Ontario, Canada. OntarioGroup A Streptococcal Study Group. N Engl J Med 335:547–554.
Efstratiou, A. (2000) Group A streptococci in the 1990s. JAntimicrob Chemother 45 (Suppl.): 3–12.
Engleberg, N.C., Heath, A., Miller, A., Rivera, C., and DiRita,V.J. (2001) Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenesresult in enhanced virulence in a murine model of skin andsoft tissue infection. J Infect Dis 183: 1043–1054.
Facklam, R., Beall, B., Efstratiou, A., Fischetti, V., Johnson,D., Kaplan, E., et al. (1999) emm typing and validation ofprovisional M types for Group A streptococci. Emerg InfectDis 5: 247–253.
Ferretti, J.J., McShan, W.M., Ajdic, D., Savic, D.J., Savic, G.,Lyon, K., et al. (2001) Complete genome sequence of anM1 strain of Streptococcus pyogenes. Proc Natl Acad SciUSA 98: 4658–4663.
Gryllos, I., Cywes, C., Shearer, M.H., Cary, M., Kennedy,R.C., and Wessels, M.R. (2001) Regulation of capsulegene expression by group A Streptococcus during pharyn-geal colonization and invasive infection. Mol Microbiol 42:61–74.
Hensel, M., Shea, J.E., Gleeson, C., Jones, M.D., Dalton, E.,and Holden, D.W. (1995) Simultaneous identification ofbacterial virulence genes by negative selection. Science269: 400–403.
Hui, F.M., Zhou, L., and Morrison, D.A. (1995) Competencefor genetic transformation in Streptococcus pneumoniae:organization of a regulatory locus with homology to twolactococcin A secretion genes. Gene 153: 25–31.
Ji, Y., McLandsborough, L., Kondagunta, A., and Cleary, P.P.
(1996) C5a peptidase alters clearance and trafficking ofgroup A streptococci by infected mice. Infect Immun 64:503–510.
Kuo, C.F., Wu, J.J., Lin, K.Y., Tsai, P.J., Lee, S.C., Jin, Y.T.,Lei, H.Y., and Lin, Y.S. (1998) Role of streptococcal pyro-genic exotoxin B in the mouse model of group A strepto-coccal infection. Infect Immun 66: 3931–3935.
Lacks, S.A. (1999) DNA Uptake by Transformable Bacteria.Cambridge, UK: Cambridge University Press.
Lancefield, R.C. (1957) Differentiation of group A strepto-cocci with a common R antigen into three serological typeswith special reference to the bactericidal test. J Exp Med107: 525–544.
Lange, R., Wagner, C., de Saizieu, A., Flint, N., Molnos, J.,Stieger, M., et al. (1999) Domain organization and molec-ular characterization of 13 two-component systems identi-fied by genome sequencing of Streptococcus pneumoniae.Gene 237: 223–234.
Leenhouts, K., Buist, G., Bolhuis, A., ten Berge, A., Kiel, J.,Mierau, I., et al. (1996) A general system for generatingunlabelled gene replacements in bacterial chromosomes.Mol Gen Genet 253: 217–224.
Low, D.E., Schwartz, B., and McGeer, A. (1998) The re-emergence of severe group a streptococcal disease: anevolutionary perspective. Washington, DC: AmericanSociety for Microbiology Press.
Lukomski, S., Montgomery, C.A., Rurangirwa, J., Geske,R.S., Barrish, J.P., Adams, G.J., and Musser, J.M. (1999)Extracellular cysteine protease produced by Streptococcuspyogenes participates in the pathogenesis of invasive skininfection and dissemination in mice. Infect Immun 67:1779–1788.
Lukomski, S., Sreevatsan, S., Amber, C., Reichardt, W.,Woischnik, M., Podbielski, A., and Musser, J.M. (1997)Inactivation of Streptococcus pyogenes extracellularcysteine protease significantly decreases mouse lethalityof serotype M3 and M49 strains. J Clin Invest 99: 2574–2580.
Lyon, W.R., Gibson, C.M., and Caparon, M.G. (1998) Arole for trigger factor and an rgg-like regulator in thetranscription, secretion and processing of the cysteineproteinase of Streptococcus pyogenes. EMBO J 17: 6263–6275.
Mecsas, J. (2002) Use of signature-tagged mutagenesis inpathogenesis studies. Curr Opin Microbiol 5: 33–37.
Miller, M.B., and Bassler, B.L. (2001) Quorum sensing inbacteria. Annu Rev Microbiol 55: 165–199.
Morrison, D.A., and Lee, M.S. (2000) Regulation of compe-tence for genetic transformation in Streptococcus pneumo-niae: a link between quorum sensing and DNA processinggenes. Res Microbiol 151: 445–451.
Moses, A.E., Goldberg, S., Korenman, Z., Ravins, M.,Hanski, E., and Shapiro, M. (2002) Invasive group Astreptococcal infection in Israel. Emerg Infect Dis 8: 421–426.
Moses, A.E., Wessels, M.R., Zalcman, K., Alberti, S.,Natanson-Yaron, S., Menes, T., and Hanski, E. (1997)Relative contributions of hyaluronic acid capsule and Mprotein to virulence in a mucoid strain of the group AStreptococcus. Infect Immun 65: 64–71.
Muotiala, A., Seppala, H., Huovinen, P., and Vuopio-Varkila,

sil, streptococcal invasive locus 99
© 2002 Blackwell Publishing Ltd, Molecular Microbiology, 46, 87–99
J. (1997) Molecular comparison of group A streptococci ofT1M1 serotype from invasive and noninvasive infections inFinland. J Infect Dis 175: 392–399.
Musser, J.M., Hauser, A.R., Kim, M.H., Schlievert, P.M.,Nelson, K., and Selander, R.K. (1991) Streptococcuspyogenes causing toxic–shock–like syndrome and otherinvasive diseases: clonal diversity and pyrogenic exotoxinexpression. Proc Natl Acad Sci USA 88: 2668–2672.
Musser, J.M., Kapur, V., Szeto, J., Pan, X., Swanson, D.S.,and Martin, D.R. (1995) Genetic diversity and relationshipsamong serotype M1 strains of Streptococcus pyogenes.Dev Biol Stand 85: 209–213.
Musser, J.M., and Krause, R.M. (1998) The Revival of Groupa Streptococcal Diseases, with a Commentary on Staphy-lococcal Toxic Shock Syndrome. San Diego: AcademicPress.
Norgren, M., Norrby, A., and Holm, S.E. (1992) Genetic diver-sity in T1M1 group A streptococci in relation to clinicaloutcome of infection. J Infect Dis 166: 1014–1020.
Ochman, H., Ajioka, J.W., Garza, D., and Hartl, D.L. (1990)Inverse polymerase chain reaction. Biotechnology 8: 759–760.
Opdyke, J.A., Scott, J.R., and Moran, C.P., Jr (2001) A sec-ondary RNA polymerase sigma factor from Streptococcuspyogenes. Mol Microbiol 42: 495–502.
Perez-Casal, J., Price, J.A., Maguin, E., and Scott, J.R.(1993) An M protein with a single C repeat prevents phago-cytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequencesto the chromosome of S. pyogenes. Mol Microbiol 8: 809–819.
Podbielski, A. (1993) Three different types of organization ofthe vir regulon in group A streptococci. Mol Gen Genet237: 287–300.
Podbielski, A., Spellerberg, B., Woischnik, M., Pohl, B., andLutticken, R. (1996) Novel series of plasmid vectors for
gene inactivation and expression analysis in group A strep-tococci (GAS). Gene 177: 137–147.
Ravins, M., Jaffe, J., Hanski, E., Shetzigovski, I., Natanson-Yaron, S., and Moses, A.E. (2000) Characterization of amouse-passaged, highly encapsulated variant of group Astreptococcus in in vitro and in vivo studies. J Infect Dis182: 1702–1711.
de Saizieu, A., Gardes, C., Flint, N., Wagner, C., Kamber,M., Mitchell, et al. (2000) Microarray-based identificationof a novel Streptococcus pneumoniae regulon controlledby an autoinduced peptide. J Bacteriol 182: 4696–4703.
Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989) In Molec-ular Cloning a Laboratory Manual: Cold Spring Harbor:Cold Harbor Laboratory Press.
Smoot, J.C., Barbian, K.D., Van-Gompel, J.J., Smoot, L.M.,Chaussee, M.S., Sylva, G.L., et al. (2002) Genomesequence and comparative microarray analysis of serotypeM18 group A Streptococcus strains associated with acuterheumatic fever outbreaks. Proc Natl Acad Sci USA 99:4668–1673.
Stanley, J., Linton, D., Desai, M., Efstratiou, A., and George,R. (1995) Molecular subtyping of prevalent M serotypes ofStreptococcus pyogenes causing invasive disease. J ClinMicrobiol 33: 2850–2855.
Stromberg, A., Romanus, V., and Burman, L.G. (1991)Outbreak of group A streptococcal bacteremia in Sweden:an epidemiologic and clinical study. J Infect Dis 164: 595–598.
Tettelin, H., Nelson, K.E., Paulsen, I.T., Eisen, J.A., Read,T.D., Peterson, S., et al. (2001) Complete genomesequence of a virulent isolate of Streptococcus pneumo-niae. Science 293: 498–506.
Whatmore, A.M., and Kehoe, M.A. (1994) Horizontal genetransfer in the evolution of group A streptococcal emm- likegenes: gene mosaics and variation in Vir regulons. MolMicrobiol 11: 363–374.




















