
2007-jinorchemistry
21
Accepted Manuscript
-
Upload
univ-montp1 -
Category
Documents
-
view
2 -
download
0
Transcript of 2007-jinorchemistry

Accepted Manuscript
TEM study of the morphology of Mn2+-doped calcium hydroxyapatite and ß-
tricalcium phosphate
I. Mayer, F.J.G. Cuisinier, S. Gdalya, I. Popov
PII: S0162-0134(07)00262-0
DOI: 10.1016/j.jinorgbio.2007.09.004
Reference: JIB 8082
To appear in: Journal of Inorganic Biochemistry
Received Date: 25 April 2007
Revised Date: 4 July 2007
Accepted Date: 13 September 2007
Please cite this article as: I. Mayer, F.J.G. Cuisinier, S. Gdalya, I. Popov, TEM study of the morphology of Mn2+-
doped calcium hydroxyapatite and ß-tricalcium phosphate, Journal of Inorganic Biochemistry (2007), doi: 10.1016/
j.jinorgbio.2007.09.004
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers
we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and
review of the resulting proof before it is published in its final form. Please note that during the production process
errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.

ACCEPTED MANUSCRIPT
TEM study of the morphology of Mn2+-doped calcium hydroxyapatite and ß-tricalcium phosphate.
I. Mayera*, F.J.G. Cuisinierb, S. Gdalyaa and I. Popovc
a Department of Inorganic and Analytical Chemistry, Hebrew University, Jerusalem 91904, Israel
b INSERM U 424, Centre de Recherches Odontologique 1 place de l’Hospital, F-6700 Strasbourg, France
c Unit Nanoscopic Characterization, Hebrew University, Jerusalem 91904, Israel Abstract Mn-doped carbonated hydroxyapatites (HA) were prepared by precipitation method. Ca-deficient HA samples were obtained by this method with the characteristic hexagonal apatite structure. Scanning Transmission Electron Microscopy (STEM) of two HA samples with two different Mn content has shown that their morphology depends on their Mn content. In case of relatively low (0.73%) Mn content (HAMn1), platelet crystals about micron size and needle-like crystals up to 100 nm were observed, while with 1.23% Mn (HAMn2) crystals were smaller, needle-like and with sizes up to 400 nm only. Mn-doped TCP samples were prepared by two methods. In one case it was obtained by direct solid state reaction with the characteristic rhombohedral structure of �-TCP and with composition of Ca2.7Mn0.3(PO4)2. TEM pictures of crystals of this sample were tens of micron and submicron size with visible faces. Crystals of ß-TCP obtained by high temperature partial transformation of sample HAMn2 to ß-TCP. were found by TEM to be smaller, micron sized, drop-like shaped, sensitive to beam radiation. These results indicate. that the morphology of Mn doped �-tricalcium phosphate samples depends on the method of their preparation. Morphological properties of HA and TCP are discussed and it is suggested that the smaller and less perfect HA crystals with the higher Mn-content as well as the less perfect TCP crystals obtained by transformation of HA to TCP might be of more biocompatible character. Keywords: Hydroxyapatite. Tricalcium phosphate. Manganese. TEM. Morphology. Introduction Synthetic calcium hydroxyapatite (HA), because of its analogy to the mineral components of bones and teeth, is one of the most important implantable materials due to its biocompatibility, bioactivity and osteoconductivity. HA is widely used in dentistry and orthopedics, to repair bone defects and as coating material for metallic implants [1]. In order to make synthetic HA comparable to that of natural bone tissues, cationic or anionic substituents have been added to them. Considering the anionic substituents CO3
2−is found in natural apatites in substantial amounts, ~4% in teeth and ~6% in bones. The position in which the carbonate group enters in the crystalline structure of the apatite is variable, with higher content B type (CO3
2− substituting in PO4
3− sites) or A-type (CO32− substituting in OH− sites) in young bone.
Trace amounts of F- and Cl- can be incorporated in HA [2] and enhance its thermal stability [3]. Trace amounts of Na+, Zn2+, Fe3+, Cu2+, Pb2+, Sr2+ and Si are found as well in bones and teeth [4]. Two of these ions Cu2+ and Pb2+ are toxic, cumulative in

ACCEPTED MANUSCRIPT
animals and humans. Doping HA with Mg2+ improves activity of biological cells [5] and co-substitution with Na+ and Mg2+ ions influences its thermal stability [6]. ��
The importance of morphology, size and crystallinity of implanted HA materials was shown in a study in which HA was implanted into a male rat tibia [7].The morphology and size of the implanted HA affected the intrusion of bone marrow derived cells among HA material, and this cell intrusion was important for regulating the newly formed bone mass. High crystalline sintered HA was stable and remained in its initial shape for long periods. However, low crystalline HA was absorbed and disappeared during bone remodeling. ß-tricalcium phosphate (ß-TCP) is as well biocompatible and was found in many cases advantageous compared to HA [8, 9]. and was also studied in HA/TCP composite materials [10, 11]. The influence of dopants, like Mg2+, Zn2+ and Si on the physical, mechanical, and biological properties of ß-TCP, used with implants, was investigated [12, 13, 14]. HA and ß-TCP differ in their crystal structure and can be identified by their powder X-ray diffraction patterns. HA crystallizes in a hexagonal and ß-TCP in a rhombohedral structure [15, 16]. �
In previous studies [17, 18] Mn2+-containing HA and TCP were studied. The motivation for the addition of Mn2+ ions to HA was due to the fact that divalent Mn2+ has been linked to the activation of integrins [19], a family of receptors which mediate cellular interactions with extracellular matrix and cell surface ligands. In the presence of Mn2+ ions the ligand affinity of integrin increases and cell adhesion is promoted. In further works the beneficial effect of Mn-doped HA thin films was demonstrated [20,21]. When human osteoblasts were cultured on the surfaces of such thin films deposited on etched Ti substrates, biological tests have shown that the Mn-doped HA coatings favour osteoblasts proliferation, activation of their metabolism and differentiation.
EPR spectroscopy has proved that manganese is divalent in the sintered samples and seems to partly occupy the Ca2+ sites in the ß-TCP structure. The present work was aimed to determine morphological properties of Mn-containing HA and TCP samples by TEM. HA samples with different Mn content (0.73 and 1.23%) and ß-TCP samples prepared in two ways: (a) via the heat treatment of Mn containing HA and (b) by a direct high temperature solid state reaction of the mixtures of CaCO3, (NH4)2HPO4 and Mn(NO3)2 were selected for this study. Experimental
a. Synthesis of Mn-containing HA samples
Samples of HA with Mn (0.1-4.0%) and with carbonate (1.0-3.0%) were prepared by a precipitation method [22]. A phosphate solution [3.7 g (NH4)2HPO4 in 200 ml triple distilled water (TDW)] was added dropwise to a calcium solution [9.47 g Ca(NO3)2.4H2O in 200 ml TDW]. Carbonate was added from a 1M NaHCO3 stock solution and Mn from a 0.025 M MnC�2 solution. For the preparation of HA samples with Mn2+ the pH of 5.8-6.0 was chosen, because it was found that at this pH Mn2+ is not oxidized to a higher valence state. The pH was maintained constant during precipitation by adding NaOH solution using a Mettler pH-stat automatic titrator. The

ACCEPTED MANUSCRIPT
precipitation was carried out during 2 h under controlled temperature (85°C), and following this the temperature was raised to boiling point and the system was refluxed for 2 h. The sample was then washed with TDW and dried overnight in air at 120ºC.
b. Synthesis of Mn containing ß-TCP. 1. Synthesis of ß-TCP by heating precipitated HA samples.
Precipitated HA samples (as described above) were heated in a porcelain crucible during 5 h up to 800ºC in an electric furnace. HA at this temperature transforms partially or almost completely to ß-TCP.
2. Synthesis of ß-TCP by high temperature solid-state reaction. Ca2.7Mn0.3(PO4)2 was prepared from a stoichiometric mixture of CaCO3, (NH4)2HPO4 and Mn(NO3)2 according to the chemical reaction: 2.7CaCO3(s) + 2(NH4)2H(PO4)(s) + 0.3Mn(NO3)2(s) � � Ca2.7Mn0.3(PO4)2+ 2.7CO2(g) + 4NH3(g) + 3H2O(g) + 0.3N2O5(g) The mixture was thoroughly mixed in an agate mortar, heated in an alumina crucible at 300ºC for three hours, crushed, mixed and then heated at 1100ºC overnight. The gaseous products of the reaction leave the system after the formation of Ca2.7Mn0.3(PO4)2.
c. Characterization methods
The Ca, P and Mn contents of the samples were determined by ICP-atomic emission spectroscopy with a precision of ±0.1, ±0.5, and ±0.3% for Ca, P and Mn respectively.
Crystal phase was determined by powder X-ray diffraction (XRD) with a Philips Automatic Diffractometer using CuK� radiation. The samples were scanned in the 2� range of 20-55º. The lattice parameters were calculated by a least-square computer program. Maximum deviation of the lattice constants was ±0.003Å. Infrared spectroscopy (IR) characterization was made by a Nicolet FTIR spectrometer using samples (~1 mg) pressed into pellets with KBr (~150 mg). The carbonate content of the samples was estimated by IR analysis using the extinction ratio of the carbonate (1420 cm-1) and phosphate (575 cm-1) bands. Carbonate is determined by this method with an accuracy of ±5% [23]. Samples were also examined by Scanning Transmission Electron Microscopy (STEM). For this purpose the samples were ground and sonicated in ethanol. The obtained suspension was deposited onto 400 mesh carbon coated copper grid. All the study was performed with Tecnai F20 G2 (FEI Company) operated at 200 kV and equipped with STEM HAADF (high angle annular dark field) detector. Combination of TEM and STEM techniques allowed us to characterize morphology, microstructure (down to atomic level) and crystalline structure of the samples having nanoscale-to-submicron size features.

ACCEPTED MANUSCRIPT
Results
a. Mn containing HA
Two of the Mn containing HA samples were selected for the TEM investigation, HAMn1 (0.73% Mn, 3.6% carbonate, Ca/P=1.61) and HAMn2 (1.23% Mn, 2.1% carbonate, Ca/P=1.48)). Molar Ca/P ratios of the samples were lower than 1.67, indicating Ca-deficient HA with the general chemical formula Ca10-x(HPO4)x(PO4)5-
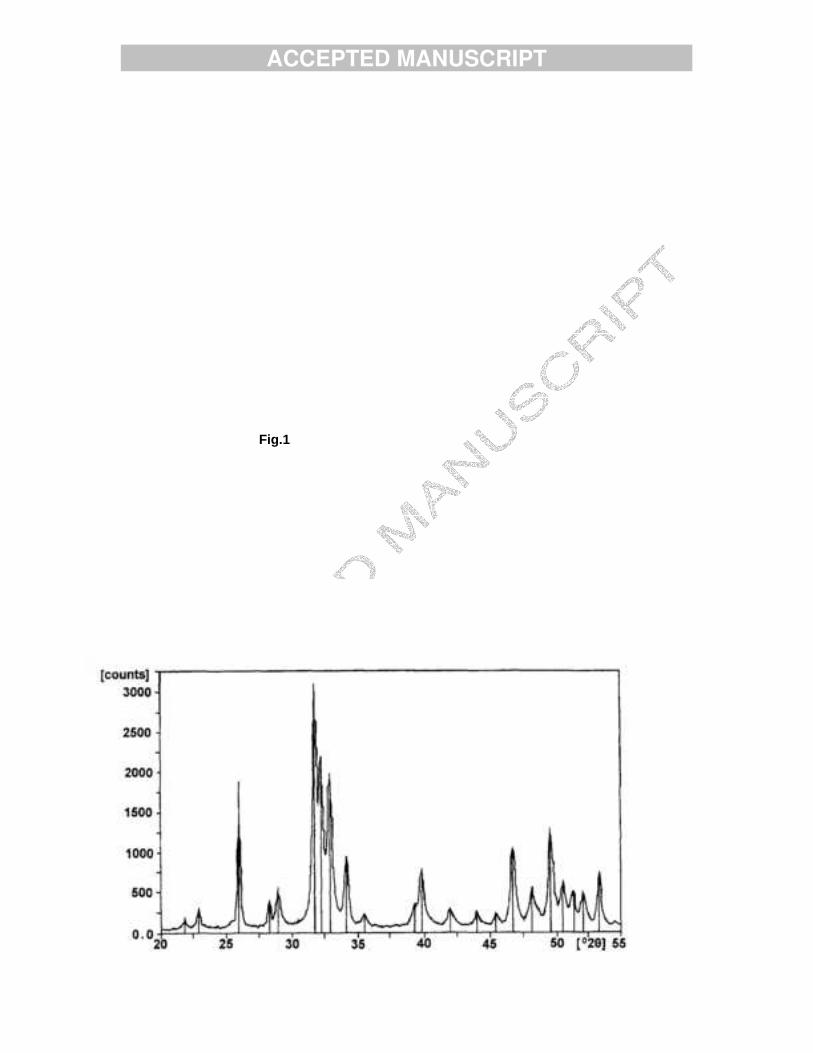
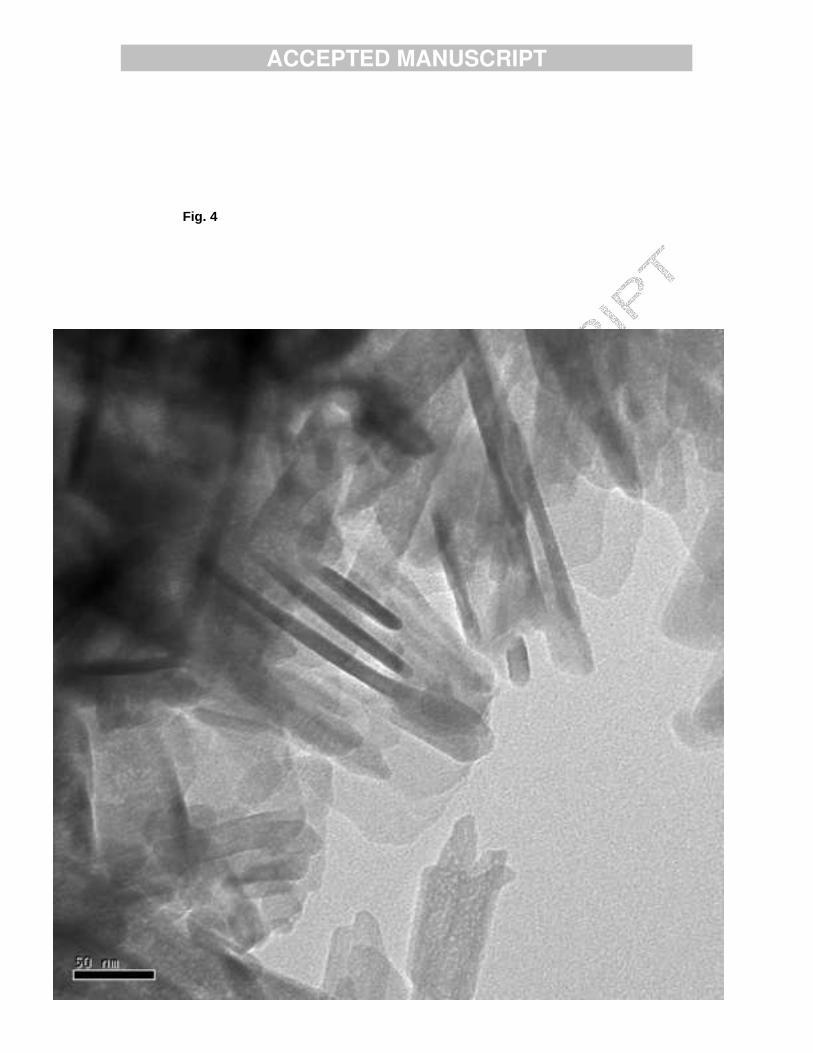
x(OH)2-x. The Ca/P ratio decreased with the increase of the Mn content of the samples. Powder XRD patterns of the samples consisted of well defined reflections with the characteristic P63/m hexagonal structure of apatites and with no additional crystal phases. Fig. 1 demonstrates such a powder pattern obtained for sample HAMn2. The calculated lattice constants are close to those known for pure HA. IR spectra of the samples displayed the characteristic phosphate, hydroxide and water bands of HA. The carbonate bands appeared around 1420 and 1470 cm-1, indicating the presence of B-type carbonate. TEM results show that sample HAMn1 consists of crystallites of various sizes. Selected Area Diffraction (SAD) patterns were observed of both rings and diffraction points, thus confirming the presence of large (1 micron) and small (less than 100 nm) size crystallites. Fig.2 shows the diffraction pattern of a crystal of sample HAMn1 observed along the [1-1.10] zone axis and indexes of a few reflections. Indexes and d values calculated are in good agreement with the hexagonal structure of HA. The crystallites with needle-like morphology were in the range of tens to hundreds nanometers and platelets of micron size (Fig. 3). All the crystallites, large and small, are beam sensitive; beam damage appears within several tens of seconds and causes the defragmentation of the crystallites. TEM pictures of Mn-rich HAMn2 sample show crystallites of much lower crystallinity. Needle-like crystals of 400 nm or less only were observed (Fig. 4). Their diffraction patterns contain rings only (Fig. 5), proving also the presence of crystals less than micron size. The d values of two of these diffraction rings were calculated and were found to correspond to two of the strongest reflections of HA. Beam damage of this sample was faster and finally caused holes in the crystallites. b. Mn-containing ß-TCP.
Sample TCPMn1 obtained by heat treatment of sample HAMn2 at 800ºC
The XRD pattern of sample HAMn2 after heating in the 800-1000ºC region contained reflections of two crystal phases; reflections of the original hexagonal HA phase [15] and reflections of the rhombohedric ß-TCP [16]. Most of the HA (around 75 %) transformed to ß-TCP. The degree of transformation of HA to ß-TCP has been roughly estimated by the relative peak heights of the strongest HA (211) and ß-TCP (0.2.10) reflections. Carbonate content of the samples decomposed during heating, no carbonate bands appeared on the IR spectra of the heated samples.

ACCEPTED MANUSCRIPT
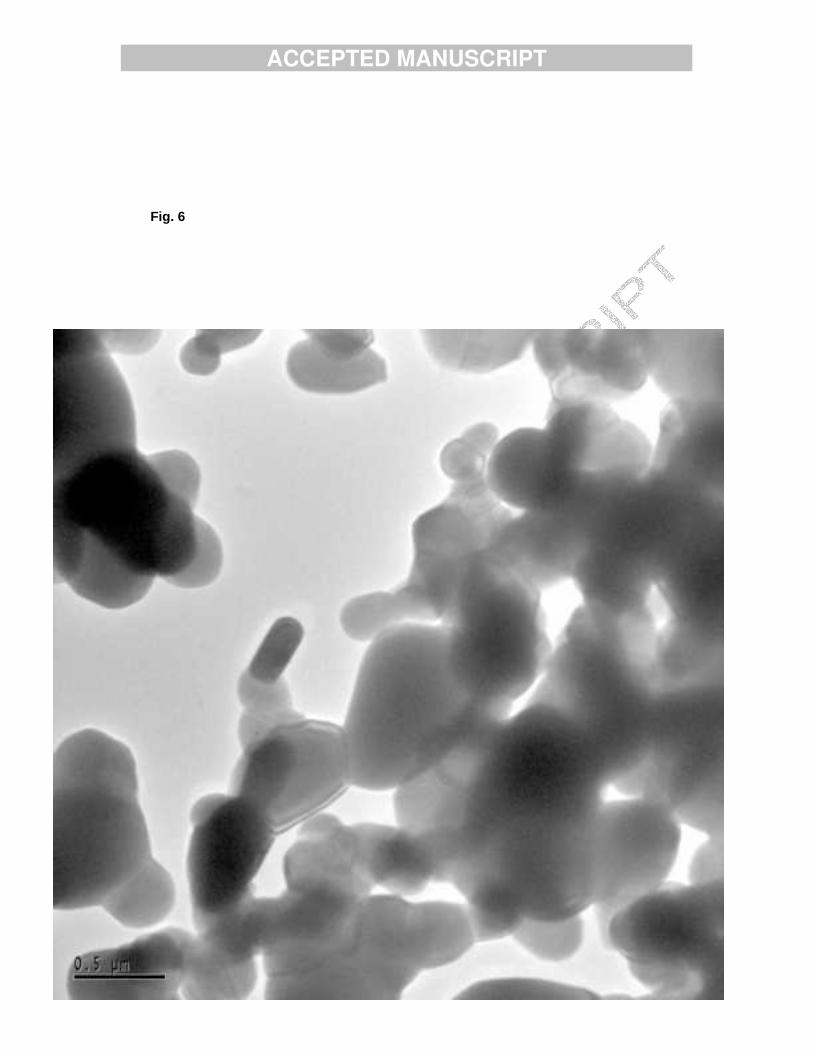
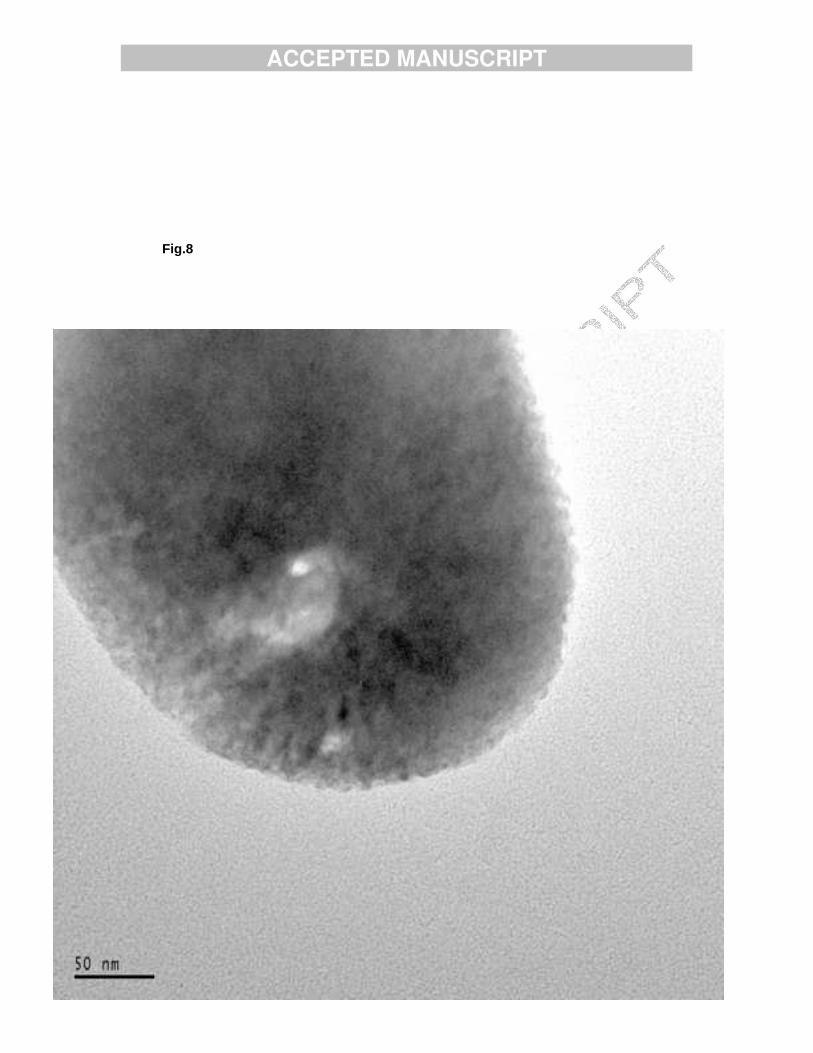
TEM pictures show that the crystals of sample TCPMn1 are rounded, on the whole, drop-like shaped (Fig. 6). The smallest crystals were of 100 nm in size, but in general they were larger, up to tens of microns. The small crystals have polyhedral shape in most cases, while small round shaped crystals were also observed. These crystals were not transparent enough to observe their lattice, and not stable enough for SAD tilt series acquisition. Still a few good lattice images (Fig. 7) were obtained and Fourier filtered. The d-spacings found in these images fitted well with the structure of TCP [16]. The crystallites decompose under the electron beam; in TEM images small holes formed in the crystallites (Fig. 8) This might indicate that the material is composed of nanometer size crystalline flakes, which loose their mutual order under the radiation but do not disappear completely. Comparison of the above results with previous TEM results of nonheated HAMn2 [17] shows that following heat treatment the morphology changes from thin plate-like polygons to three dimensional crystals.
Ca2.7Mn0.3(PO4)2 prepared by high temperature solid-state reaction
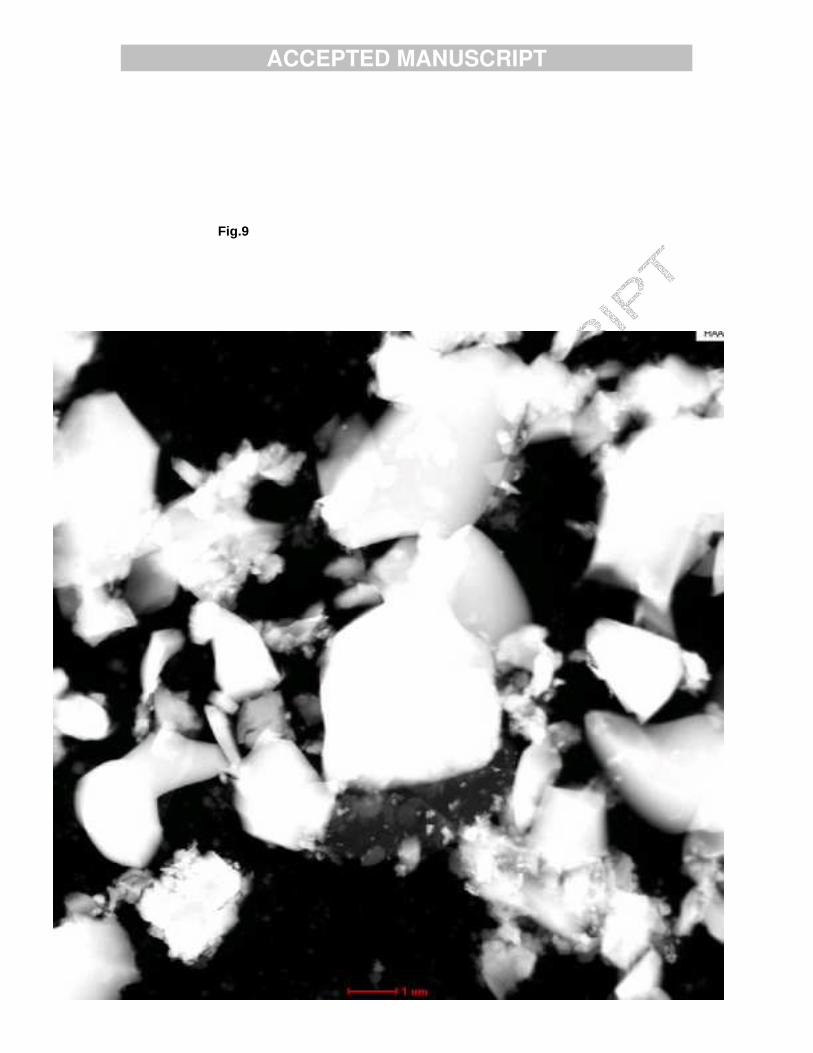
The Ca2.7Mn0.3(PO4)2 sample was pink in colour and analytical results verified its Mn content. XRD pattern of the sample reveals that it crystallizes in the R3c rhombohedral structure characteristic for ß-TCP. No reflections of impurity phase were present. The lattice constants calculated, a=10.38 Å; c=37.32 Å, are smaller than those known for Ca3(PO4)2 due to the substitution of Ca2+ by the smallerMn2+ ions. Fig. 9 is the HAADF STEM image of Ca2.7Mn0.3(PO4)2 showing crystallites with sizes varying from tens of nanometers to microns. All the observed crystallites have the same dominating morphological features of broken ceramics with outer contours obtained by glass-like cleavage. Owing to this morphology even thick, micron size crystallites have thin wedge-like edges and allowed the study of the electron diffraction pattern (Fig. 10) and to observe the lattice structures at HR TEM mode (Fig. 11). Discussion The results (XRD, IR and ICP) proved that when HA was prepared by precipitation Ca-deficient Mn-doped HA samples were obtained. The two samples prepared differ in their Ca/P ratio; the sample relatively poor in Mn, HAMn1 (0.73% Mn), has Ca/P ratio of 1.61, while Ca/P of sample HAMn2 (1.23% Mn) was found to be 1.48. The decrease of the Ca/P ratio with the increase of the Mn content of the samples was already reported in our previous work [18]. Both samples had the structural characteristics of the hexagonal apatite structure and their IR spectra contained the bands of carbonate-containing HA. The low Ca/P ratio of sample HAMn2 (1.48) indicates an enhanced substitution of HPO4
2- ions into the HA lattice, thus generating Ca2+ vacancies. At room temperature this sample with Ca/P ratio even below 1.5 still crystallizes in the hexagonal apatite structure and favors the transformation to ß-TCP. XRD analyses prove that heating to 800ºC led to a biphasic mixture of HA and ß-TCP; around 75% of HA transformed to the rhombohedric ß-TCP structure. Ca2.7Mn0.3(PO4)2 prepared by high temperature solid-state reaction has the rhombohedric ß-TCP structure as well. The lattice constants and volume are smaller

ACCEPTED MANUSCRIPT
than that of pure ß-TCP indicating the formation of a solid solution by partial replacement of Ca2+ by the smaller Mn2+ ions in the ß-TCP lattice. TEM results of HA samples show their dependence on their Mn content. In the case of HAMn1 with lower, 0.73% Mn content both platelets and needle-like crystals were observed, while in HAMn2 (1.23% Mn) only needle-like crystals were found. The morphology of HA crystals is first pre-determined by the parameters of the unit cell (a = 9.418 A; c = 6.884 A) [15], which do define formation of non-aqui-axial crystallites of possible rod-like and plate-like morphology even under equilibrium growth conditions and ideal stoichiometry. But, kinetic factors like presence of Mn in the growth medium, are expected to affect the crystal habit as well, since accommodation of this additional element within the crystalline structure of apatite (even in the interstitial positions) could limit or decrease the growth rate in one or more directions, thus providing needle-like shape crystals. In the case of Ca2.7Mn0.3(PO4)2 formed by high temperature solid state reaction as a rule, larger crystallites were formed due to the high temperature of the reaction in preference to needles. It is of interest to point out that less perfect crystals were observed in two cases: in HA sample with the higher Mn content, and in TCP Mn doped in a biphasic mixture with HA. Accordingly it seems that these two types of samples are good candidates for obtaining resorption sites in vivo and in vitro environments. Conclusions
1. HA samples with 0.71 and 1.23% Mn content were synthesized by precipitation method having Ca/P ratio 1.61 and 1.48 respectively.
2. Morphology of the above samples by TEM analysis revealed larger plate and needle like crystals with the Mn-poor and smaller only needle like crystals with the Mn-rich sample. Both were sensitive to irradiation.
3. TCP samples with Mn were obtained as a biphasic phase by HA heated to 800ºC and as a pur phase by a direct solid state reaction.
4. The morphology of TCP in the biphasic phase was of droplet-like crystals and of micron size. TCP obtained by solid state reaction was composed of big ten of micron size three dimensional crystals.

ACCEPTED MANUSCRIPT
RErrReferences
[1] M. Wallet Regi, J. Chem.Soc. Dalton Trans. (2001) 97-108.��
[2] S. Kannan, A. Rebelo, J.M.F. Ferreira, J. Inorg. Biochem. 100 (2006) 1692-1697. [3] S. Kannan, A. F. Lemos, J.M.F. Ferreira, Chem. Mater. 18 (2006) 2181-2186. [4] M.E.J. Curzon, T.W. Cutress, Trace Elements and Dental Disease, John Wright, Boston, 1983. [5] E. Landi, A. Tampieri, M. Mattioli-Belmonte, G. Celotti, M.Sandri, A. Gigante, P. Fava, G. Biagini, J. Europ. Ceram. Soc. 26 (2006) 2593-2601. [6] S. Kannan, J.H.G. Rocha, J.M.F. Ferreira, Mater. Chem.16 (2006) 286-291. ����T. Matsumoto , K. Tamine , R. Kagawa , Y. Hamada , M. Okazaki, J. Takahashi J. Ceram. Soc. Japan, 114 (2006) 760-762. [8] A. Ogose, T. Hotta, H. Kawashima, N. Kondo, W. Gu, T. Kamura, N. Endo, J. Biomed. Mater. Res., Part B, Appl. Biomater. 72B (2005) 94-101. [9] M. Aizawa, K. Itatani, I. Okada, J. Ceram. Soc. Japan 113 (2005) 245-251. [10] K.B. Fleckenstein, M. F. Cuenin, M. E. Peacock, M.A. Billman, G. D. Swiec, T. B. Buxton, B. B. Singh, James C. Mc. Pherson, J. Periodont, 77 (2006) 39-45. [11] H.Y. Yashuda, M. Aoki, Y. Fujita, W. Fujitani, Y. Umakoshi, A. Takaoka, Mater. Trans. 47 (2006) 2368-2372. [12] A. Bandyopadhyay, Sh. Bernard, W. Xue, S. Bose, J. Amer. Ceram. Soc. 89 (2006) 2675-2688. [13] S. Kannan, I.A.F. Lemos, J.H.G. Rocha, J.M.F. Ferreira, . Solid State Chem. 178 (2005) 3190-3196. [14] S. Kannan, S. Pina and J.M.F. Ferrerra, J. Amer. Ceram. Soc. 89 (2006) 3277-3280. [15] International Centre for Diffraction Data. Powder Diffraction File 2005, 09-0432.
[16] International Centre for Diffraction Data. Powder Diffraction File 2005, 09-0169 (2001). [17] I. Mayer, O. Jacobsohn, T. Niazov, J. Werckman, M. Iliescu, M. Richard-Plouet, O. Burghaus and D. Reinen, Eur. J. Inorg. Chem. (2003) 1445-1451.

ACCEPTED MANUSCRIPT
[18] I. Mayer, F.J.G. Cuisinier, I. Popov, Y. Schleich, S. Gdalya, O. Burghaus and D. Reinen, Eur. J. Inorg. Chem. 2006, 1460-1465. [19] A. Armulik, G. Svineng, K. Wennerberg, R. Faessler, S. Johansson, Experimental Cell Res. 254 (2000) 55-63. [20] E. György, P. Toricelli, G. Socol, M. Iliescu, I. Mayer, I.N. Mihailescu, A. Bigi, and J. Werckman, J. Biomed. Mater. Res. Part A, 71A (2004) 353-358. [21] A. Bigi, B. Bracci, F. Cusinier, R. Elkaim, M. Fini, I. Mayer, I.N. Mihailescu, G. Socol, L. Sturba and P. Toricelli, Biomaterials 26 (2005) 2381-2389. [22] J.D.B. Featherstone, I. Mayer, F.C.M. Driessens, R.M.H. Verbeeck and M. Heijligers, Calcif. Tissue Int. 35 (1983) 169-171. [23] J.D.B. Featherstone, S. Pearson, R.Z. and LeGeros, Caries Res. 8 (1984) 63-66.
��

ACCEPTED MANUSCRIPT
Figure captions
Fig. 1. X-ray powder diffraction pattern of sample HAMn2.
Fig. 2. Selected area diffraction pattern of sample HAMn1.
Fig. 3. Needle like crystallites and hexagon plates of sample HAMn1.
Fig. 4. Needle like crystals of sample HAMn2
Fig. 5. Selected area diffraction pattern of sample HAMn2.
Fig. 6. Assembly of drop like shaped crystals of sample TCPMn1.
Fig. 7 Lattice image of sample TCPMn1.
Fig. 8. Beam damage of sample TCPMn1.
Fig. 9. Small and large crystals of Ca2.7Mn0.3(PO4)2.
Fig.10. Selected area diffraction pattern of sample Ca2.7Mn0.3(PO4)2.
Fig.11. Lattice image of sample Ca2.7Mn0.3(PO4)2.
ACCEPTED MANUSCRIPT
Fig.1

ACCEPTED MANUSCRIPT
Fig. 2

ACCEPTED MANUSCRIPT
Fig. 3
ACCEPTED MANUSCRIPT
Fig. 4

ACCEPTED MANUSCRIPT
Fig. 5
ACCEPTED MANUSCRIPT
Fig. 6

ACCEPTED MANUSCRIPT
Fig.7
ACCEPTED MANUSCRIPT
Fig.8
ACCEPTED MANUSCRIPT
Fig.9

ACCEPTED MANUSCRIPT
Fig.10

ACCEPTED MANUSCRIPT
Fig.11




















