
下載 - 台灣胸腔暨重症加護醫學會
65
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of 下載 - 台灣胸腔暨重症加護醫學會


原著
不同手術治療方式對於Boerhaave’s Syndrome癒後的影響:文獻回顧及本院經驗 .........................168~174李佳穎,李章銘,黃培銘,李元麒
病例報告
肺部分化良好的胎兒型腺癌―三個病例報告 ....................................................................................175~183鍾政錦,周德盈,許文虎
以葉克膜成功的治療因氣管-無名動脈瘻管所造成的致命併發症:病例報告 ..................................184~189吳南鈞,郭進榮,鄭伯智
肺原始性神經外胚層腫瘤併杵狀指―個案報告 .................................................................................190~196蔡昇翰,張漢煜
罕見胃癌併發肺轉移之影像學表現擬肺泡細胞癌:病例報告 ...........................................................197~202林冠群,羅啟文,余秉真
犬心絲蟲造成孤立肺部結節:病例報告與台灣病例整理 ..................................................................203~210李育松,黃建達,余志騰,王志偉,郭漢彬
以喘鳴為最初表現的聲帶白黴菌病―病例報告 .................................................................................211~215林延淞,涂智彥,陳家弘,劉奕亨,廖偉志,沈煥庭
惡性肋膜間皮瘤合併瀰漫性肺轉移於化學治療後併發自發性氣胸一病例報告 ..................................216~221王保山,朱國安,林秀玲,吳銘庭,賴瑞生
免疫正常的人在麴菌氣管支氣管炎時成功的使用抗黴菌治療 ...........................................................222~229鄭人豪,曾若琦,陳俊叡,劉育志
中華民國九十九年八月 第二十五卷 第四期
Vol.25 No.4 August 2010

中華民國九十九年八月 第二十五卷 第四期
Vol.25 No.4 August 2010
Orginial ArticlesSurgical Outcomes of Different Approaches in Treating Boerhaave’s Syndrome ...........................168~174Chia-Ying Li, Jang-Ming Lee, Pei-Ming Huang, Yung-Chie Lee
Case ReportsWell-Differentiated Fetal Adenocarcinoma of the Lung: Three Case Reports .................................175~183Cheng-Ching Chung, The-Ying Chou, Wen-Hu Hsu
Successful Treatment of a Potentially Fatal Complication of Tracheoinnominate ArteryFistula with Extracorporeal Life Support: Report of a Case.............................................................184~189Nan-Chun Wu, Jinn-Rung Kuo, Bor-Chih Cheng
Pulmonary Primitive Neuroectodermal Tumor Associated with Digital Clubbing – A Case Report .................................................................................................................................190~196Sheng-Han Tsai, Han-Yu Chang
A Rare Radiological Pattern of Pulmonary Metastasis of Gastric Cancer Mimicking Bronchioloalveolar Carcinoma: A Case Report ...............................................................................197~202Kuan-Chun Lin, Chi-Wen Lo, Ping-Chen Yu
Solitary Pulmonary Nodule Due to Dirofilariasis: A Case Report and Case Review in Taiwan .......203~210Yu-Song Lee, Chien-Da Huang, Chih-Teng Yu, Chih-Wei Wang, Han-Pin Kuo
Vocal Cord Mucormycosis: An Unusual Cause of Stridor – Case Report .......................................211~215Yen-Sung Lin, Chih-Yen Tu, Chia-Hung Chen, Yi-Heng Liu, Wei-Chih Liao, Huan-Ting Shen
Spontaneous Pneumothorax Following Chemotherapy for Malignant Pleural Mesothelioma with Diffuse Pulmonary Metastasis – A Case Report ......................................................................216~221Pao-Shan Wang, Kuo-An Chu, Shong-Ling Lin, Ming-Ting Wu, Ruay-Sheng Lai
Aspergillus Tracheobronchitis in an Immunocompetent Patient Successfully Treated with Voriconazole ....................................................................................................................................222~229Jen-Hao Cheng, Jo-Chi Tseng, Jim-Ray Chen, Yu-Chih Liu

168
Thorac Med 2010. Vol. 25 No. 4
Surgical Outcomes of Different Approaches in Treating Boerhaave’s Syndrome
Chia-Ying Li, Jang-Ming Lee, Pei-Ming Huang, Yung-Chie Lee
Background: Boerhaave’s syndrome is a rare, devastating disease that results from spontaneous esophageal rupture. There is no consensus on the optimal treatment. We reviewed the literature and the data of our patients to determine the more optimal treatments for different conditions.
Methods: We retrospectively reviewed the clinical results of patients with Boerhaave’s syndrome undergoing surgical intervention at National Taiwan University Hospital from 2000 to 2006.
Results: The patients comprised 2 females and 6 males, ranging in age from 51 to 87 years. Four patients received cervical esophageal exclusion with a cervical T-tube, video-assisted thoracoscopic surgery decortication and gastrostomy, 2 underwent esophageal exclusion by esophagostomy and gastrostomy, and 1 had primary esophageal repair. Another patient underwent chest tube thoracostomy, gastrostomy and jejunostomy only, due to a generally poor condition. Two patients died of profound sepsis because of delayed intervention.
Conclusions: Patients with Boerhaave’s syndrome can be successfully treated with early diagnosis and adequate surgical intervention. Cervical T-tube esophageal drainage can provide a 1-stage operation for temporary saliva exclusion and a satisfactory long-term outcome in selected cases. (Thorac Med 2010; 25: 168-174)
Key words: esophagus, esophageal perforation, thoracic surgery
Division of Thoracic Surgery, Department of Surgery, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei, TaiwanChia-Ying Li and Jang-Ming Lee contributed equally to the manuscript.Address reprint requests to: Dr. Yung-Chie Lee, Department of Surgery, National Taiwan University Hospital and National Taiwan University College of Medicine, No.7, Chung-Shan South Rd., Taipei, Taiwan 100
Introduction
Esophageal perforation is a critical condi-tion with a high mortality rate. Boerhaave’s syndrome accounts for 30-40% of all esopha-geal perforations [1]. It was first described by Dr. Hermann Boerhaave in 1724. The patho-
genesis is thought to be a baro-rupture caused by a rapid rise of intra-esophageal pressure as-sociated with vomiting or exertion [2]. Because of the rarity and mortality, there is no consensus on the most suitable treatment. We present our results in the management of 8 patients during the past 6 years and review past reports in order

169Surgery for Boerhaave’s Syndrome
胸腔醫學:民國99年25卷4期����
to determine the more optimal managements under different clinical conditions.
Materials and Methods
From January 2000 to December 2006, 8 patients (6 males, mean age: 67.1 years, range: 51-87 years) were diagnosed with Boerhaave’s syndrome in our unit. We reviewed the clinical conditions and results retrospectively. Diagno-sis was made using history, imaging studies and endoscopic or intra-operative findings.
Initial manifestations included pleural effu-sion (8 patients), chest pain (6 patients), sepsis-related symptoms (2 patients), subcutaneous emphysema (2 patients) and pneumomedi-astinum (1 patient). The 2 patients with sepsis suffered from fever, altered consciousness and shock as initial manifestations. There was a his-tory of vomiting preceding the onset of symp-toms in 6 patients. The perforation was located at the upper thoracic esophagus in 1 patient, middle in 1 and lower in 6 patients. The clinical data of these patients are summarized in Table 1.
Five patients received surgical intervention within 48 hours after perforation. Two patients were initially misdiagnosed as having pneu-monia-related sepsis; diagnosis of Boerhaave’s syndrome was finally made, but surgical inter-vention had been delayed for 10 and 18 days after symptom onset. Another patient received chest tube drainage initially because of a poor condition and the operation was delayed for 14 days.
All patients received surgical intervention, the modes of which could be categorized into 4 groups: 1. Thoracotomy for primary repair of esophageal defect, decortication and chest tube drainage in 1 patient with a fresh perforation. 2. Cervical T-tube esophageal exclusion, drain-
ing tube gastrostomy, feeding jejunostomy and decortication by video-assistant thoracoscopic surgery (VATS) or thoracotomy in 4 patients. 3. Cervical esophagostomy, draining tube gastros-tomy, feeding jejunostomy and decortication by VATS or thoracotomy in 2 patients; one of these 2 patients received esophageal stripping at the same time. 4. Palliative draining tube gastrostomy, feeding jejunostomy and chest tube drainage in 1 patient 14 days after esopha-geal perforation, due to a poor condition. All patients received parenteral nutritional support and broad-spectrum antibiotics during the peri-operative period.
We chose different surgical approaches based on the esophageal status and extent of inflammation. One patient (group 1) received primary repair of the esophageal defect because the esophageal wall was healthy. Cervical T-tube esophageal exclusion was performed in 4 patients (group 2) who had an esophageal defect with local inflammatory changes. Two other patients (group 3) received a traditional esophagostomy because severe tissue reaction and necrosis were noted around the perforation site and the esophageal wall was fragile.
Cervical T-tube esophageal exclusion is a procedure for temporary esophageal division, performed through an incision in the left neck. The lower end of the T-tube is closed by silk ligation and the T-tube is inserted into the cer-vical esophagus as a splint and to drain saliva from the proximal esophagus. The lower arm is tied circumferentially to the esophageal wall us-ing chromic catgut or dexon. Since the tie will be absorbed and lose tension after 2-3 weeks, the T-tube can be removed easily and the patient does not need a second operation. The wound heals 3-4 days after tube removal [3-4].

170 Chia-Ying Li, Jang-Ming Lee, et al.
Thorac Med 2010. Vol. 25 No. 4
Results
The surgical procedures and results are summarized in Table 2. The overall mortality rate was 25% (2 mortalities). All of the 5 pa-tients who had surgical intervention within 48 hours survived. But in the group for which the operation was delayed for more than 48 hours after esophageal perforation, 2 of 3 patients died. One fatality was a 78-year-old female who was misdiagnosed as having intractable pneu-monia and and so underwent surgical interven-tion 18 days later with cervical T-tube esopha-geal exclusion, draining tube gastrostomy, feed-ing jejunostomy, decortication by thoracotomy and chest tube drainage. She expired on the 23rd post-operative day because of profound sepsis. Another 80-year-old male was transferred from another hospital and received conservative medical treatment with chest tube drainage at first, due to his poor condition. Palliative drain-ing tube gastrostomy and feeding jejunostomy were performed 14 days after esophageal per-foration. He passed away on the 20th postopera-tive day because of sepsis.
After surgery, 2 patients suffered from per-sistent empyema and underwent a 2nd VATS decortication or open window, individually, lat-er on. Another patient had an enterocutaneous fistula after removal of the jejunostomy tube a year later; he received primary closure of the fistula.
The 2 patients who received cervical eso-phagostomy, draining tube gastrostomy and feeding jejunostomy were lost to follow-up 1-2 years later, so they did not receive secondary reconstruction. The other 4 patients (3 cervical T-tube esophageal exclusions and 1 primary closure) had fair swallowing functions during the clinical follow-up.
Discussion
The common presentations of Boerhaave’s syndrome involve chest pain and subcutane-ous emphysema with a history of vomiting [2]. Pleural effusion was another common finding in our experience. However, some cases suffer only from sepsis-like manifestations. The rup-ture site is in the lower third of the esophagus in 90% of cases [1-2]. In our unit, 75% of cases were ruptured in this area.
Esophageal perforation causes extravasation of saliva or gastric acid into the mediastinum or pleural space, which may result in mediastinitis, empyema and eventually profound sepsis [4]. Most mortalities result from delayed treatment. The mortality rate is 10-25% if the interven-tion begins within 24 hours after perforation; however, it could rise to 40-60% if treatment is delayed for more than 48 hours [5]. The diag-nosis of Boerhaave’s syndrome is often made later than that of non-spontaneous esophageal perforation (perforation related to malignancy, foreign body ingestion or iatrogenic proce-dures) [2]. The delayed diagnosis can lead to a catastrophic outcome. In our experience, early diagnosis and surgical intervention are the keys to survival. Of those patients who had surgical intervention within 48 hours, all survived. But if the operation were delayed for more than 48 hours, the mortality rate would rise to 67%.
No matter the procedure performed, certain elements must be included in the treatment, such as effective drainage to stop further leak-age, adequate nutritional support and broad spectrum antibiotic coverage [2, 5]. In most cases, surgical intervention was more effective than conservative therapy, especially with an early diagnosis [2].
However, conservative therapy could be

171Surgery for Boerhaave’s Syndrome
胸腔醫學:民國99年25卷4期����
Table 1. Clinical data of the 8 patients
Caseno.
Age/Sex
Initial manifestations*History of vomiting
DiagnosticTools**
Timeinterval***
1 62/F Sepsis-related symptomsChest pain
Subcutaneous emphysemaPneumomediastinum
- Endoscopy 8 days
2 51/M Hematemesis + Endoscopy/Esophagography
2 days
3 80M Chest painFever
+ Endoscopy 14 days
4 87M FeverTachypnea
+ Endoscopy/Esophagography
2 days
5 71M Chest pain + CT/Intra-operative
findings
1 days
6 78F Sepsis-related symptoms Chest painSubcutaneous emphysema
- CT/Intra-operative
findings
>14 days
7 54M Chest pain + CT/Intra-operative
findings
2 days
8 54M Chest painFever
+ Endoscopy 1 days
CT = computerized tomography* All patients presented with pleural effusion** The examination used to make a definite diagnosis.*** Time interval from initial manifestations to surgical intervention
considered under some conditions, such as intramural perforation, perforation with well-encapsulated extravasation or perforation with delayed diagnosis and minimal symptoms [2, 6-7]. During conservative therapy, if the condi-tions deteriorate, surgical intervention should be performed immediately [6]. Endoscopic treatment with a metal stent has been used in some reports, but the success rates and definite therapeutic roles remain controversial [2, 8-10].
Several surgical approaches are available for the management of Boerhaave’s syndrome. Esophageal status, time interval from perfora-tion to intervention, location of rupture, and extent of inflammation should be considered
in making a choice [11]. When perforation is diagnosed early and the esophageal wall is healthy, primary repair of the defect with ad-equate drainage is the optimal choice [5, 11-12]. Diversion of saliva with esophageal exclu-sion may be preferable in patients who have an inflammatory or fragile esophageal wall. For cervical esophageal exclusion, both traditional esophagostomy and temporary esophageal T-tube exclusion can be considered, based on the different conditions [4]. Because the absorbable ligation that seals the upper esophagus and low-er arm of theT-tube will lose its tension within 2-3 weeks, T-tube exclusion is preferable for a smaller esophageal defect with moderate in-

172 Chia-Ying Li, Jang-Ming Lee, et al.
Thorac Med 2010. Vol. 25 No. 4
flammatory changes that is predicted to recover within 2 weeks [4]. The advantages of T-tube exclusion include simplicity and an absence of subsequent esophageal reconstructive surgery. However, when the esophageal defect is larger and the esophageal wall is fragile because of se-vere inflammation, traditional cervical esopha-gostomy is safer. In our reported experience, 6 patients received cervical esophageal exclusion (4 with temporary esophageal T-tube exclusion
and 2 with traditional esophagostomy). One pa-tient who received esophageal T-tube exclusion 18 days after esophageal perforation passed away because of profound sepsis. Two patients who underwent traditional esophagostomy were lost to follow-up 1-2 years later, so they did not receive secondary reconstruction. The other 3 patients who underwent cervical T-tube esopha-geal exclusion had fair swallowing functions during the clinical follow-up.
Table 2. Surgical procedures and results for 8 patients with Boerhaave’s syndrome
Surgical procedure Patient number Intervention timing(early / late)*
Mortality
Primary repair 1 1 / 0 0Cervical esophageal exclusion withT-tube drainage, gastrostomy and jejunostomy 4
2 / 21**
Esophagostomy, gastrostomy and jejunostomy 2*** 2 / 0 0Palliative gastrostomy and jejunostomy 1 0 /1 1All except the last patient received decortication and chest tube drainage during operation.* Intervention timing after perforation: early <48 hours; late >48 hours.** The patient who died had a delayed intervention of more than 14 days.*** One patient received esophageal stripping at the same time.
Fig. 1. Treatment algorithm for Boerhaave’s syndrome.

173Surgery for Boerhaave’s Syndrome
胸腔醫學:民國99年25卷4期����
Esophageal resection with reconstruction is rare because most patients with esophageal perforation are too weak to undergo such ag-gressive procedures. In those patients with a delayed diagnosis (>48 hours), both conser-vative medical treatment and diversion with esophageal exclusion can be considered. There is no consensus currently as to which therapy is preferable, because of the high mortality rates [5-6, 11].
Based on the literature review and our ex-perience, we have proposed an algorithm for the treatment of Boerhaave’s syndrome (Figure 1). In conclusion, the time interval from esophageal perforation to diagnosis is the key to successful treatment. Delayed diagnosis and intervention leads to a high mortality rate, even with surgical management. When diagnosis is made within 48 hours after perforation, primary repair or esophageal exclusion by esophagostomy or T-tube placement with draining tube gastrostomy and feeding jejunostomy can be effective and life-saving, depending on the esophageal status.
References
1. Lawrence DR, Ohri SK, Moxon RE, et al. Primary esophageal repair for Boerhaave’s syndrome. Ann Thorac Surg 1999; 67: 818-20.
2. De Schipper JP, Pull ter Gunne AF, Oostvogel HJ, et al. Spontaneous rupture of the oesophagus: Boerhaave’s syndrome in 2008: literature review and treatment algori-
thm. Dig Surg 2009; 26: 1-6. 3. Lee YC, Lee ST, Chu SH. New technique of esophageal
exclusion for chronic esophageal perforation. Ann Thorac Surg 1991; 51: 1020-2.
4. Lee YC, Luh SP, Wu RM, et al. A rational surgical appro-ach for intra-thoracic esophageal perforation. Int Surg 1993; 78: 307-10.
5. Gupta NM, Kaman L. Personal management of 57 con-secutive patients with esophageal perforation. Am J Surg 2004; 187: 58-63.
6. Altorjay A, Kiss J, Voros A, et al. Nonoperative manage-ment of esophageal perforations: is it justified? Ann Surg 1997; 225: 415-21.
7. Vogel SB, Rout WR, Martin TD, et al. Esophageal perfor-ation in adults: aggressive, conservative treatment lowers morbidity and mortality. Ann Surg 2005; 241: 1016-21.
8. Fisher A, Thomusch O, Benz S, et al. Nonoperative treatment of 15 benign esophageal perforations with self-expandable covered metal stents. Ann Thorac Surg 2006; 81: 467-73.
9. Siersema PD, Homs MY, Haringsma J, et al. Use of large-diameter metallic stents to seal traumatic nonmalignant perforations of the oesophagus. Gastrointest Endosc 2003; 58: 356-61.
10. Chung MG, Kang DH, Park DK, et al. Successful treat-ment of Boerhaave’s syndrome with endoscopic insertion of a self-expandable metallic stent: report of three cases and a review of the literature. Endoscopy 2001; 33: 894-7.
11. Kiernan PD, Sheridan MJ, Hettrick V, et al. Thoracic esophageal perforation: one surgeon’s experience. Dis Esophagus 2006; 19: 24-30.
12. Sung SW, Park JJ, Kim YT, et al. Surgery in thoracic esophageal perforation: primary repair is feasible. Dis Esoph 2002; 15: 204-9.

174 Chia-Ying Li, Jang-Ming Lee, et al.
Thorac Med 2010. Vol. 25 No. 4
不同手術治療方式對於Boerhaave’s Syndrome癒後的影響:文獻回顧及本院經驗
李佳穎 李章銘 黃培銘 李元麒
前言:Boerhaave’s syndrome是指自發性的食道破裂,是一種及少見但致命的疾病 。目前對於此疾病
的治療方式尚未有一致的共識。本文回顧近幾年的文獻以及整理本院治療此疾病之經驗,以期能找到在
不同狀況下較適合的治療方式。
方法:本文回溯性回顧台大醫院於2000年至2006間被診斷為Boerhaave’s syndrome並接受手術的病人
之術式、癒後及影響癒後之相關因素。
結果:共有兩位女性及六位男性病患被診斷為Boerhaave’s syndrome。其中四位病患接受T型管頸部食
道排除(exclusion)、胸腔鏡膿胸剝除(decortication)及引流性胃造口手術。兩位病人接受食道排除手
術(藉由頸部食道造口及胃造口)。一位病患接受直接食道修補手術(primary repair)。另一位病患因病
況較差,只接受胸管引流及引流性胃造口手術。所有病患中有兩位病患因為延遲的手術治療後來因敗血
症而死亡。
結論:只要早期診斷再加上適當的手術治療,Boerhaave’s syndrome是一個可以被成功治療的疾病。
T型管頸部術在食道排除手適合的病人身上是一個可以暫時提供唾液排除引流而且只需要一階段手術的另
一個選擇。(胸腔醫學 2010; 25: 168-174)
關鍵詞:食道,食道破裂,胸腔手術
國立台灣大學附設醫院 外科部
索取抽印本請聯絡:李元麒醫師,國立台灣大學附設醫院外科部 胸腔外科,台北市中山南路7號

175
胸腔醫學:民國99年25卷4期��������
Well-Differentiated Fetal Adenocarcinoma of the Lung: Three Case Reports
Cheng-Ching Chung*,***,****, The-Ying Chou**, Wen-Hu Hsu*,***
Well-differentiated fetal adenocarcinoma (WDFA) is a rare malignant neoplasm of the lung, and was first described by Kradin et al., in 1982. Since then, there have been about 65 cases reported in the literature. It is important to identify this rare variant of adenocarcinoma with low-grade malignancy and low associated mortality. We reported 3 young adult patients (all less than 50 years old) with WDFA. They all received surgical resection without adjuvant therapy, and all of them were still alive and disease-free at 11 years, 8 years and 32 months, respectively. (Thorac Med 2010; 25: 175-183)
Key words: well-differentiated fetal adenocarcinoma, pulmonary blastoma, lung cancer
*Division of Thoracic Surgery, Department of Surgery, **Department of Pathology, Taipei Veterans General Hospital***School of Medicine, National Yang-Ming University, Taipei, Taiwan****School of Medicine, Taipei Medical University, Taipei, TaiwanAddress reprint requests to: Dr. Wen-Hu Hsu, Division of Thoracic Surgery, Department of Surgery; Taipei Veterans General Hospital, No. 201, Sec. 2, Shih-Pai Road, Taipei, Taiwan
Introduction
Well-differentiated fetal adenocarcinoma (WDFA) was first described by Kradin et al. [1] in 1982. Histologically, it resembles pulmonary blastoma, and is thought to be a subtype thereof. In 1991, Koss et al. [2], classified pulmonary blastoma into 2 subtypes: 1. Biphasic blastoma consisting of fetal lung adenocarcinomatous features and sarcomatous features 2. Monopha-sic blastoma, lacking sarcomatous features and referred to as WDFA. Pulmonary blastomas are also divided into 3 histopathologic groups: WDFA, biphasic pulmonary blastoma (BPB), and pleuropulmonary blastoma (PPB) [3]. In the most recent classification by the World Health Organization in 1999, WDFA tumors
were removed from the pulmonary blastoma category and classified as a variant of adenocar-cinoma of the lung [4]. WDFA simulates fetal lung tubules in the canalicular to pseudoglandu-lar stages of development [1-2, 5-6]. The clas-sic age range seems to be clear, with a majority of patients presenting in the 3rd to 4th decades [1-2, 5, 7]. However, there are some reported cases in the pediatric age range, defined as less than 20 years old [8]. WDFA is a low-grade malignancy, and its prognosis tends to be bet-ter than that of biphasic blastoma and common pulmonary adenocarcinoma. Complete surgical resection is essential for long-term survival [9]. In this report, we present our experience in our hospital with 3 young adult patients with his-topathologically confirmed well-differentiated

176 Cheng-Ching Chung, The-Ying Chou, et al.
Thorac Med 2010. Vol. 25 No. 4
fetal adenocarcinoma of the lung. Two patients had an excellent outcome after surgical resec-tion (survived without tumor recurrence for 11 years and 8 years respectively). The tumor in the third patient demonstrated a special slow-growth pattern, and was still at stage IA as of this writing, despite having been first noted 3 years previous.
Case Reports
Case 1A 42-year-old female smoker was referred
to our hospital due to cough with bloody spu-tum. Chest X-ray and computed tomography (CT) of the chest revealed a 4.0 x 3.0 cm round soft tissue mass in the left upper lobe of the lung, without hilar or mediastinal lymphadenop-athy. Bronchoscopic biopsy was performed and carcinoid tumor was suspected. We performed a left upper lobectomy with lymph node dissec-tion. The pathology revealed well-differentiated fetal adenocarcinoma without regional lymph node metastasis. The pathological staging was T2aN0M0, stage IB. The patient did not receive adjuvant therapy and was well with no evidence of disease 11 years after resection.
Case 2A 30-year-old man, a non-smoker, had an
incidental finding of a pulmonary nodule dur-ing a physical examination at a local hospital. Chest X-ray and CT revealed a homogeneous well-defined nodule about 3.0 x 2.0 cm in size, without calcification in the right upper lobe and without hilar or mediastinal lymphadenopathy (Figures 1-3). Hamartoma or sclerosing he-mangioma was suspected. After referral to our hospital, wedge resection of the right upper lobe was performed. The lesion showed adeno-
carcinoma in the frozen section. Then, complete lobectomy of the right upper lobe with lymph node dissection was carried out. The pathology revealed adenocarcinoma composed of well-differentiated non-ciliated tall columnar cells arranged in a glandular pattern that resembled fetal lung tubules. Morules were also noted. The pathological staging was T1bN0M0, stage
Fig. 1. Chest X-ray shows a pulmonary nodule about 3 cm in diameter with central calcification in the right upper lobe of the lung in case 2.
Fig. 2. The CT scan of the chest shows a homogeneous well-defined nodule in the right upper lobe, approximately 3 x 2 cm size in case 2.

177Well-Differentiated Fetal Adenocarcinoma of Lung
胸腔醫學:民國99年25卷4期����
IA. The patient received no further treatment and was well and with no evidence of disease 8 years after resection.
Case 3A 49-year-old female non-smoker was
found to have a pulmonary nodule about 0.5 cm in diameter in the left lung in a CT scan of the chest during a physical check-up (Figure 4). She did not pay attention to it until another physical examination 3 years later, when whole body MRI revealed that the pulmonary nodule had enlarged to 2.5 x 2.0 cm in size. A different diagnosis including lung cancer, hemangioma, or vascular malformation was suspected. Chest
X-ray and chest CT revealed a homogenous well-defined nodule about 2.0 x 2.0 cm without obvious enhancement in the left upper lobe of the lung (Figures 5-7). Sclerosing hemangioma or other slow-growing tumor was suspected, but malignancy could not be ruled out. She was then admitted to our hospital, where a pre-op-
Fig. 3. Lung window of the chest CT scan.
Fig. 4. The CT scan of the chest in 2004 shows a pulmonary nodule about 0.5 cm in diameter in the left upper lobe of the lung in case 3.
Fig. 5. Chest X-ray shows a pulmonary nodule about 2 cm in diameter in the left upper lobe of the lung in case 3.
Fig. 6. The CT scan of the chest in 2007 shows a homogeneous well-defined nodule about 2 x 2 cm in size in the left upper lobe of the lung in case 3.

178 Cheng-Ching Chung, The-Ying Chou, et al.
Thorac Med 2010. Vol. 25 No. 4
erative survey showed no endobronchial lesion or distal metastasis. The patient underwent left thoracotomy and wedge resection of the left up-per lobe. A frozen section of the lesion revealed adenocarcinoma. Then complete lobectomy of the left upper lobe with radical lymph node dis-section was performed. Grossly, the tumor was gray-white and soft, about 2.8 x 2.5 x 2.0 cm in size (Figure 8). Under light microscopy, the tu-mor revealed a picture of fetal adenocarcinoma, well-differentiated and consisting of branching tubules in a cribriform pattern lined by pseudo-stratified, non-ciliated columnar cells and some morules beneath the glandular epithelium (Figure 9). The tumor was immunoreactive
for CK and TTF-1, focally positive for CD56, synaptophysin, and chromogranin A and nega-tive for CK7 and CK20 (Figure 10). No pleural angiolymphatic or perineural invasion or lymph node metastases were seen. The pathological staging was T1bN0M0, stage IA. We performed mutation analysis of the EGFR and K-ras genes and found a polymorphism at EGFR exon 20 leading to no change in the amino acid se-quence, such as, glutamine (cag) → glutamine (caa), and no mutation was found in K-ras. The patient received no other treatment, and was well without recurrence of disease 32 months after resection.
The characteristics of these 3 patients are summarized in Table 1.
Discussion
Pulmonary blastoma is rare, and accounts for only about 0.5% of all primary malignant lung tumors [1-3, 6]. The glycogen-rich noncili-ated tubules and embryonic stroma resemble those of the fetal lung at 10 to 15 weeks of ges-tation [4-5]. These blastomas are classified as
Fig. 7. Lung window of the chest CT scan.
Fig. 8. Surgical specimen: a gray-white soft tumor about 2.8 x 2.5 x 2.0 cm in size
Fig. 9. Histology section shows branching tubules in a cribriform pattern lined by pseudostratified, nonciliated columnar cells and some squamoid morules (arrow) beneath the glandular epithelium (H & E stain 40X)

179Well-Differentiated Fetal Adenocarcinoma of Lung
胸腔醫學:民國99年25卷4期����
Fig. 10. The tumor cells were immunoreactive to CK (a, 100X) and TTF-1 (b, 400X), as well as focally positive for CD56 (c, 400X), synaptophysin (d, 400X), chromogranin A (e, 400X)
(a) (b)
(c) (d)
(e)

180 Cheng-Ching Chung, The-Ying Chou, et al.
Thorac Med 2010. Vol. 25 No. 4
WDFA, BPB, and PPB [3]. However, in 1999, the World Health Organization classified BPB and PPB as variants of sarcomatoid carcinoma [4, 10]. WDFA now is included as a histological subtype of adenocarcinoma and is distinguished from BPB and PPB in having a much better prognosis [2-4, 10].
BPB and WDFA have a strong relationship to smoking [2-3] however, among our patients, only 1 of 3 had a smoking history. The male-to-female ratio is equal and the classic age range is 30 to 40 years [1-2, 5, 7]. Morules, gatherings of polygonal cells, are present in the gland in approximately 86% of cases [2-3].
In the imaging findings, the tumor has a homogeneous solid appearance, and necrosis and hemorrhage are rare. Usually, there is a well-defined lobulated margin due to expanding tumor growth, and the presentation of a soli-tary peripheral or mid-lung mass. Adenopathy and pleural effusion are rare [2-3]. Patients are usually asymptomatic or complain of cough,
hemoptysis, fever, and chest pain [2-3]. Among our patients, only 1 of 3 had the symptom of hemoptysis. The tumor demonstrated intense focal uptake on FDG-PET scanning [11].
The differential diagnosis of this tumor in-cludes high-grade adenocarcinoma of the fetal lung or clear cell adenocarcinoma with fetal lung features [12].
Immunohistochemical analysis showed positive staining for CD56, synaptophysin, and chromogranin A, indicating that the tumor cells showed neuroendocrine differentiation [13]. Little information is available on the ge-netic changes and lack of p53 mutation seen in WDFA, whereas the presence of mutation in the p53 gene in BPB (42%) has been reported [2]. Aberrant cytoplasmic expression of β-catenin has also been demonstrated in WDFA [14]. One study suggested that a β-catenin gene alteration might be involved in the development of WDFA [13]. In our case 3, we found a polymorphism at EGFR exon 20 and no mutation in K-ras.
Table 1. Patient characteristics
Patient 1 2 3Age 42 30 49
Gender Female Male FemaleSmoking history Yes No No
Symptoms Hemoptysis No NoTumor size (cm) 4 x 3 3 x 2 2 x 2Tumor location LUL RUL LUL
Impression Carcinoid HamartomaSclerosing hemangioma
Hemangioma, vascular malformation
Operation Lobectomy Lobectomy LobectomyDiagnosis WDFA WDFA WDFA
Stage T2aN0M0, IB T1bN0M0, IA T1bN0M0, IAAdjuvant therapy Nil Nil Nil
Outcome Alive and disease-free for 11 years
Alive and disease-free for 8 years
Alive and disease-free for 32 months

181Well-Differentiated Fetal Adenocarcinoma of Lung
胸腔醫學:民國99年25卷4期����
According to Koss et al. [2], the prognosis of WDFA is better than that of other subtypes of adenocarcinoma and biphasic blastoma, and the 5-year survival rate of WDFA was 80%, while that of BPB was 30% [2-3]. In a review of repor- ted cases in Japan by Sato et al. [9], the 5-year survival rate with WDFA was 90% in stage I, and the prognosis was favorable. Among patients with stage II or greater, however, no long-term survival has been reported, and che-motherapy or radiotherapy has seldom been ef-fective [2, 6]. A review of the literature reveals a mean survival of 14.7 months in advanced inoperable cases that receive chemotherapy and radiotherapy. [15]. For these reasons, complete surgical resection is considered essential for long-term survival [9, 16]; this can be seen in our case 1 and case 2, in which long-term dis-ease-free survival was achieved after complete resection.
The slow growth pattern of WDFA differs from that of other subtypes of adenocarcinoma. This special slow-growth pattern in young patients and the radiological features (homo-geneous and well-defined low-density solid nodule without enhancement) may mimic some benign lung tumors, such as hamartoma, scle-rosing hemangioma, vascular malformation, and carcinoid. Among each of our 3 patients, the first impression was benign tumor, and the 3 patients were all diagnosed as having malig-nancy after operation. The tumor in our case 3 presented as a slow-growing pulmonary nodule for about 3 years, and it was still at stage IA after 3 years of follow-up. A similar case was reported by Kiyomi et al in 2008 [17], in which the patient was followed up for 2 years without lymph node metastasis and was living unevent-fully at 24 months after surgery.
In summary, WDFA is a rare and unique
subtype of adenocarcinoma with low-grade ma-lignancy and a special slow-growth pattern. It is important to identify this rare kind of tumor due to its excellent prognosis after complete resec-tion, compared to BPB and common pulmonary adenocarcinoma. Physicians should be alert to young patients with slow-growing pulmonary nodules because they might be malignant, and with surgical resection, the patients could have a good outcome.
References
1. Kradin RL, Kirkham SE, Young RH, et al. Pulmonary blastoma with arygyrophil cells and lacking sarcomatous features (pulmonary endodermal tumor resembling fetal lung). Am J Surg Pathol 1982; 6: 165-72.
2. Koss MN, Hochholzer L, O’Leary T. Pulmonary blasto-mas. Cancer 1991; 67: 2368-81.
3. Colby TV, Koss MN, Travis WD. Tumors of the Lower Respiratory Tract. Atlas of Tumor Pathology. Fascide 13. 3rd ed. Washington, DC: Armed Forces Institute of Pathology; 1995: 395-410.
4. Travis WD, Colby TV, Corrin B. Histological Typing of Lung and Pleural Tumors. World Health Organization, International Histological Classification of Tumors. 3rd ed. Berlin: Springer Verlag, 1999; 43-4.
5. Kodama T, Shimosato Y, Watanabe S, et al. Six cases of well differentiated adenocarcinoma simulating fetal lung tubules in the pseudoglandular stage. Am J Surg Pathol 1984; 8: 735-44.
6. Francis D, Jacobsen M. Pulmonary blastoma. Curr Top Pathol 1983; 73: 265-94.
7. Nakatani Y, Kitamura H, Inayama Y, et al. Pulmonary adenocarcinoma of the fetal lung type. A clinicopathologic study indicating differences in histology, epidemiology, and natural history of low-grade and high-grade forms. Am J Surg Pathol 1998; 22: 399-411.
8. DiFurio MJ, Auerbach A, Kaplan KJ. Well-differentiated fetal adenocarcinoma: rare tumor in the pediatric popu-lation. Pediatr Dev Pathol 2003; 6: 564-7.
9. Sato K, Maeda K, Ichinose Y, et al. A case report of well differentiated fetal adenocarcinoma. J Jpn Assoc Thorac

182 Cheng-Ching Chung, The-Ying Chou, et al.
Thorac Med 2010. Vol. 25 No. 4
Surg 1992; 40: 1792-6.10. Carey FA. Pulmonary adenocarcinoma. Curr Diagn Pa-
thol 2001; 7: 187-93.11. Paull DE, Moezzi J, Katz N, et al. Positron emission
tomography in well differentiated fetal adenocarcinoma of the lung. Clin Nucl Med 2006; 31: 213-4.
12. Sheehan KM, Curran J, Kay EW, et al. Well differentiated fetal adenocarcinoma of the lung in a 29 year old woman. J Clin Pathol 2003; 56: 478-9.
13. Mori S, Sekido Y, Shigemitsu K, et al. A case of well differentiated fetal adenocarcinoma with a mutation of the BETA-catenin gene. Lung cancer 2003; 43: 351-5.
14. Nakatani Y, Masudo K, Miyagi Y, et al. Aberrant nuclear localization and gene mutation of β-catenin in low grade
adenocarcinoma of fetal lung type: up-regulation of the Wnt signaling pathway may be a common denominator for the development of tumours that form morules. Mod Pathol 2002; 15: 617-24.
15. Christina M, Maximo L, Shamji S, et al. A 50-year-old male with fetal type adenocarcinoma of the lung. Chest 2008; 134: 27002S.
16. Shozo F, Yoshikuni A, Takaaki K, et al. Case report - well-differentiated fetal adenocarcinoma of lung. Lung cancer 1995; 13: 311-6.
17. Kiyomi F, Kotaro Y, Sadanori T, et al. Well-differentiated fetal adenocarcinoma of the lung: early-phase sequential high-resolution computed tomographic findings. J Com-put Assist Tomogr 2008; 32: 806-9.

183Well-Differentiated Fetal Adenocarcinoma of Lung
胸腔醫學:民國99年25卷4期����
肺部分化良好的胎兒型腺癌─三個病例報告
鍾政錦*,***,**** 周德盈** 許文虎*,***
肺部分化良好的胎兒型腺癌(WDFA)是一種罕見的肺部惡性腫瘤。這是由Kradin於1982年初次描
述。自此之後,文獻上約有65個案例被報告出來。確認這種罕見的肺腺癌變形是重要的,因為這是一種
低度惡性且致死率低的腫瘤。本篇文章中,我們報告三個罹患WDFA的年輕病患(皆未滿50歲)。這三位病
患全都接受手術切除,並且至今都還存活著,沒有復發的現象分別維持了十一年,八年及三十二個月。
(胸腔醫學 2010; 25: 175-183)
關鍵詞:肺部分化良好之胎兒型腺癌,肺母細胞癌,肺癌
*台北榮民總醫院外科部胸腔外科,**病理部,***國立陽明大學醫學院,****台北醫學大學醫學院
索取抽印本請聯絡:許文虎醫師,台北榮民總醫院外科部 胸腔外科,台北市石牌路二段201號

184
Thorac Med 2010. Vol. 25 No. 4
Successful Treatment of a Potentially Fatal Complication of Tracheoinnominate Artery Fistula with
Extracorporeal Life Support: Report of a Case
Nan-Chun Wu, Jinn-Rung Kuo*, Bor-Chih Cheng
Tracheoinnominate artery fistula is a rare disease with an extremely fatal course. Without aggressive surgical treatment, the mortality rate is nearly 100%. We report a 14-year-old male who suffered from acute respiratory distress syndrome as the complication of tracheoinnominate artery fistula. We concluded that ligation of the innominate artery plus iliofemoral arterial bypass and tracheal repair could cure this life-threatening disease, without recurrence. Furthermore, extracorporeal membrane oxygenation can provide adequate pulmonary and cardiac support to help these patients get through the critical postoperative condition. (Thorac Med 2010; 25: 184-189)
Key words: tracheoinnominate artery fistula, percutaneous dilated tracheostomy, extracorporeal membrane oxygenation
Division of Cardiovascular Surgery, Division of Neurosurgery*, Department of Surgery, Chi-Mei Foundation Hospital, Tainan, Taiwan, R.O.C.Address reprint requests to: Dr. Bor-Chih Cheng, Division of Cardiovascular Surgery, Department of Surgery, Chi-Mei Foundation Hospital, No. 911, Chung-Hua Rd., Yung-Kang 710, Tainan, Taiwan, R.O.C.
Introduction
Tracheoinnominate artery (TIF) is an un-common but usually fatal complication follow-ing a tracheostomy. Without surgery, the mor-tality rate is nearly 100% [1]. Even with proper surgical intervention, the survival rate remains low [1-2]. Most patients die of postoperative complications such as aspiration pneumonia, sepsis, acute respiratory distress syndrome (ARDS) and ischemic brain injury [2-5]. We re-port a 14-year-old young man who had massive bleeding from a tracheostomy. A partial sterno-tomy with innominate artery ligation, tracheal
repair and extra-anatomical iliosubclavian arte-rial bypass and intracranial pressure monitor insertion were performed. Postoperative ARDS was evident on plain chest X-ray films, and ar-terial blood gases indicated hypoxemia. The pa-tient was placed on venovenous (VV) mode ex-tracorporeal life support (ECLS) on postopera-tive day (POD) 3, and was successfully weaned from it on POD 10. The patient recovered with-out neurological sequelae. The patency of the bypass graft was verified by Doppler ultrasound and angiography. Only with a prompt, accurate diagnosis, aggressive surgical management, and intensive postoperative care could the patient

185Tracheoinnominate Artery Fistula
胸腔醫學:民國99年25卷4期����
have survived.
Case Report
A 14-year-old male sustained a head in-jury with traumatic left temporal and bilateral occipital epidural hemorrhage on October 12, 2007. He underwent craniectomy for in-tracranial decompression, and installation of a ventricular peritoneal (V-P) shunt following hydrocephalus. Two weeks later, a percutane-ous dilated tracheostomy (PTD) was performed for airway toilet and long-term pulmonary care. He had no other diseases and had managed the tracheostomy with a routine exchange of tra-cheostomy tubing at the referring hospital dur-ing the previous 7 months, until the day before admission when a sudden onset of bleeding at the tracheostomy was noted. Angiography showed no significant abnormalities, similar to the result of the contrast-enhanced chest com-puted tomography (CT). A thoracic surgeon was consulted to perform a fiberoptic bronchoscopy (FOB), which revealed only minor bleeding from the granulation tissue around the tracheo-stomy. Unfortunately, massive tracheostomy bleeding occurred after he was sent back to the ward. Overinflating the tracheostomy tube cuff temporarily stopped the bleeding. The patient was rushed into the operating room after an emergency consultation with the cardiovascular surgeon. An upper partial sternotomy was per-formed under general anesthesia. The innomi-nate artery was isolated and ligated proximally and distally (Figure 1). A 7.5-mm orotracheal tube was inserted with the assistance of a FOB to replace the in-situ tracheostomy tube. The tracheal perforation was debrided and repaired. The tracheostomy showed downward erosion, and was also debrided and repaired, leaving a
10-mm stoma (Figure 2). The cuff of the orotra-cheal tube was inflated and fixed just between the carina and the repaired tracheal wall to pre-vent barotraumas from mechanical ventilation. The tracheostomy wound was left open for wet dressing. The specimen of the resected innomi-nate artery was examined, and the fistula tract was easily penetrated with a probe (Figures 3 and 4).
When we performed the innominate artery ligation, damping of the arterial line waveform of the right radial artery was observed, as well as dilation of the right pupil, without light re-flex. An extra-anatomical right-sided femoro-subclavian arterial bypass was performed using
Fig. 1. The TIF lesion was revealed after innominate arteriotomy. The proximal innominate artery has been suture ligated.
Fig. 2. Repairing the tracheal defect of the tracheostomy.

186 Nan-Chun Wu, Jinn-Rung Kuo, et al.
Thorac Med 2010. Vol. 25 No. 4
a 6-mm expanded polytetrafluoroethylene graft for the prevention of right cerebral ischemia. After bypass surgery, the right pupil returned to a size equal to the left. An intracranial pressure (ICP) probe was implanted for ICP monitoring. The patient was then sent to the ICU for post-operative care.
On POD 1, bilateral diffused bronchoalveo-lar infiltrations were visible on the plain chest X-ray film. Arterial desaturation (SaO2<90%) was recorded, even with high ventilatory set-tings. Many blood clots were evacuated with aggressive airway toilet using FOB. On POD 3, the patient developed severe hypoxia and shock. Emergency ECLS was set up in the ICU, with VV mode extracorporeal membrane oxy-genation (ECMO) via the right internal jugular vein and common femoral vein. The initial blood flow was 2.7 L/min and the FiO2 was 100%. The patient’s condition improved during the following week. The patient was successful-ly weaned from ECMO on POD 10. The patient regained consciousness and his motor function
returned to the level before surgery. The follow-up FOB revealed good healing of the repaired tracheal lesion. The orotracheal tube was re-placed on POD 14 and the cuff was fixed proxi-mal to the repaired tracheal wall. Throughout the entire ICU course, the ICP was maintained at 3-8 mmHg. No sternotomy wound infec-tion occurred. Follow-up angiography showed patency of the prosthetic, and CT of the brain showed no cerebral ischemia. The patient was transferred to the ordinary ward later and dis-charged uneventfully on POD 40.
Discussion
TIF is a life-threatening complication of tracheostomy. It was first described by Körte in 1879. The approximate incidence ranges from 0.1% to 1%, with peak incidence about 7-14 days after tracheostomy [6-7].
About 72% of patients present TIF within the first 3 weeks after tracheostomy [5, 8]. A
Fig. 4. The tracheal aspect of the TIF with probe penetration.
Fig. 3. The arterial aspect of the innominate artery, which shows the TIF lesion.

187Tracheoinnominate Artery Fistula
胸腔醫學:民國99年25卷4期����
higher incidence (10% vs. 0.6%) has been re-ported in patients with head injuries [9]. TIF occurs due to either persistent cuff inflation as a result of long-term ventilator use, causing ischemic necrosis of the tracheal membrane and subsequent erosion of the tracheal wall, or re-peated infection causing tracheal mucosal ero-sion [9-10]. TIF occurred in our patient about 7 months after tracheostomy, which is a longer period than that for the patients described in the published articles. Our patient required occasional ventilator support due to repeated tracheobronchitis, which might have caused the TIF.
When tracheostomy bleeding is encoun-tered, a suspicion of TIF will lead to the neces-sary prompt actions being taken. If the bleeding ceases with overinflation of the cuff, a brachio-cephalic artery fistula is most likely present. Flexible bronchoscopy performed through the tracheostomy to identify the fistula is not al-ways satisfactory, because bleeding into the tra-chea impairs the visual field [7,10]. However, FOB can exclude other pathologies, such as an oozing granuloma, and allows direct vision of a clear blood-free airway [7]. Rigid bronchosco-py is recommended, but it may not be possible to perform when the hemorrhage is massive and the condition is an emergency [1, 5, 7]. Angiog-raphy is inapplicable due to its low diagnostic accuracy (about 8%) and the time constraints of the situation [1, 5]. In our patient, preoperative FOB revealed only oozing from the granulation tissue and the angiography was unremarkable.
Without surgical intervention for TIF, the mortality rate is nearly 100% [1, 7]. With proper surgical treatment, about 25% to 60% of patients survive [5]. The operation should be performed with access by sternotomy. An upper sternotomy, angled toward the 3rd inter-
costal space, ought to be done to prevent sternal wound infection [11]. The innominate artery is best excised and both ends closed with running vascular sutures and covered with robust flaps of healthy tissue, such as pedicled strap muscle, thymus, or omentum [4, 12]. Because the risk of delayed septic rupture is high [10], primary closure and patching of the perforation is con-traindicated for repair of the arterial defects.
Reconstruction of the innominate artery is not necessary in every case. Very few patients developed neurological deficits in the published reports [1-2, 10, 14]. Rapid replacement of blood volume to maintain blood pressure is im-portant to permit adequate collateral circulation before clamping of the innominate artery [13]. In our patient, damping of the arterial wave-form of the right radial artery occurred after clamping of the innominate artery. The right pu-pil also dilated at the same time; thus, we sus-pected inadequate cerebral and extremity perfu-sion and performed an extra-anatomical arterial bypass from the right external iliac artery to the subclavian artery. Because vascular reconstruc-tion through an infected operative field was contraindicated, extra-anatomical bypass was advised [10]. Immediately after bypass surgery, the right pupil returned to its normal size, with an intact light reflex. Ramesh and colleagues reported that if carotid stump pressure was less than 50 mmHg, a bypass graft should be con-sidered [1, 14].
Many patients die of complications of TIF, such as aspiration pneumonia, ARDS (due to massive blood transfusion, septic shock, as-piration of blood), neurological sequelae, and surgical site infections that cause sepsis [2-5]. Among patients with ARDS, ECMO provides temporary cardiopulmonary support by manag-ing oxygen supply and CO2 removal to help

188 Nan-Chun Wu, Jinn-Rung Kuo, et al.
Thorac Med 2010. Vol. 25 No. 4
patients get through this critical condition. A re-view of the published reports revealed no article mentioning the clinical application of ECLS for TIF complicated by ARDS. In our experience, ECMO successfully supported the patient until his lungs recovered, without the complications of coagulopathy and sepsis.
In conclusion, TIF is an uncommon life-threatening complication of trachestomy. Bleed-ing from a tracheostomy dictates an aggressive diagnostic approach and immediate surgical intervention. Innominate artery ligation plus extra-anatomical bypass can effectively prevent hemorrhage recurrence and neurological defi-cits. Furthermore, ECMO can provide cardiac and pulmonary support to help patients recover from postoperative ARDS.
References
1. Schaefer OP, Irwin RS. Tracheoarterial fistula: an unusual complication of tracheostomy. J Inten Care Med 1995; 10: 64-75.
2. Nelems JMB. Tracheo-innominate artery fistula. Am J Surg 1981; 141: 526-7.
3. Lu KT, Yang SF, Ting MC. Tracheo-innominate artery fistula after tracheostomy—A case report. Acta Anaesthe-siol Taiwan 2005; 43: 237-41.
4. Roh JL, Na MH, Kim KH. Treating tracheo-innominate artery fistula with interposition of a pectoralis major myocutaneous flap. Eur Arch Otorhinolaryngol 2006; 263: 180-2.
5. Jones JW, Reynolds M, Hewitt R, et al. Tracheo-inno-minate artery erosion: successful surgical management of a devastating complication. Ann Surg 1976; 184: 194-204.
6. Hazarika P, Kamath SG, Balakrishnan R, et al. Tracheo-innominate artery fistula: a rare complication in a laryn-gectomized patient. J Laryngol Otol 2002; 116: 562-4.
7. Grant CA, Dempsey G, Harrison J, et al. Tracheo-inno-minate artery fistula after percutaneous tracheostomy: three case reports and a clinical review. Br J Anaesth 2006; 96: 127-31.
8. Cokis C, Towler S. Tracheo-innominate artery fistula after initial percutaneous tracheostomy. Anaesth Intensive Care 2000; 28: 566-9.
9. Mehalic TF, Farhat SM. Tracheoarterial fistula: a com-plication of tracheostomy in patients with brain stem injury. J Trauma 1972; 12: 140-3.
10. Copper JD. Trachea-innominate artery fistula: successful management of 3 consecutive patients. Ann Thorac Surg 1977; 24: 439-47.
11. Wood DE, Mathisen DJ. Late complications of tracheos-tomy. Clin Chest Med 1991; 12: 597-609.
12. Nakanishi R, Shimazu A, Mitsudomi T, et al. Successful management of tracheo-innominate artery fistula using interposition of a thymus pedicle flap. J Laryngol Otol 1995; 109: 161-2.
13. Allan JS, Wright CD. Tracheoinnominate fistula: diag-nosis and management. Chest Surg Clin N Am 2003; 13: 331-41.
14. Ramesh M, Gazzaniga AB. Management of tracheo-innominate artery fistula. J Thorac Cardiovascular Surg 1978; 75: 138-40.

189Tracheoinnominate Artery Fistula
胸腔醫學:民國99年25卷4期����
以葉克膜成功的治療因氣管-無名動脈瘻管所造成的致命併發症:病例報告
吳南鈞 郭進榮* 鄭伯智
氣管-無名動脈瘻管是一個相當罕見的疾病,但是卻有極高的致死率。如果沒有積極的以外科手
術治療,死亡率幾乎是百分之百。我們報告一位十四歲的男性因為氣管-無名動脈瘻管,以外科手術將
無名動脈切除,同時做氣管修補與右側髂動脈到鎖骨下動脈繞道手術,術後因為合併急性呼吸窘迫症候
群,而使用葉克膜支持,成功的治療此一致命的疾病與其併發症,且至目前為止並無復發的現象,亦無
神經學方面的後遺症。(胸腔醫學 2010; 25: 184-189)
關鍵詞:氣管-無名動脈瘻管,經皮擴張氣管造口術,葉克膜
奇美醫院 心臟血管外科,神經外科*索取抽印本請聯絡:鄭伯智醫師,奇美醫院外科部 心臟血管外科,台南縣永康市中華路911號

190
Thorac Med 2010. Vol. 25 No. 4
Pulmonary Primitive Neuroectodermal Tumor Associated with Digital Clubbing – A Case Report
Sheng-Han Tsai, Han-Yu Chang
Digital clubbing, one of the syndromes of hypertrophic osteoarthropathy, is associated with many types of medical illness, including infectious, inflammatory disease, cyanotic heart disease and neoplasm. Classically, digital clubbing has been thought to be associated with lung cancer. The incidence of clubbing fingers in lung cancer is about 10-29%, and it is more associated with non-small cell lung cancer than small cell lung cancer. We present a rare case of pulmonary primitive neuroectodermal tumor with clubbing fingers. A 56-year-old man had suffered from progressive dyspnea on exertion, accompanied with cough, abdominal fullness and body weight loss of 10 kg in the most recent 5 months. On examination, obvious digital clubbing was found in both hands. Imaging study demonstrated a huge left lung tumor. Sonography-guided biopsy was performed and the pathology report suggested primitive neuroectodermal tumor. The patient received chemotherapy with doxorubicin, decarbazine and ifosfamide, and began gradually feeling less dyspneic after chemotherapy. (Thorac Med 2010; 25: 190-196)
Key words: nail, lung tumor, hypertrophic osteoarthropathy
Division of Chest Medicine, Department of Internal Medicine, National Cheng-Kung University Medical College and Hospital, Tainan, TaiwanAddress reprint requests to: Dr. Han-Yu Chang, Division of Chest Medicine, Department of Internal Medicine, National Cheng Kung University Medical College and Hospital, No. 138, Sheng-Li Rd., Tainan, 704, Taiwan
Introduction
Digital clubbing is the thickening of the soft tissue beneath the proximal nail plate which re-sults in sponginess and thickening of the distal digit [1]. Clubbing fingers can be an isolated finding or occur as part of the syndrome of hy-pertrophic osteoarthropathy (HOA), which also includes arthralgia and periostitis [2]. Aside from the rare primary form of HOA, pachyder-moperiostosis, secondary HOA is associated with extrapulmonary or pulmonary disorders,
especially lung carcinoma [3-4]. The incidence of clubbing fingers in lung cancer is about 10-29%, and it is more commonly associated with non-small cell lung cancer than small cell lung cancer [5]. Primitive neuroectodermal tumor (PNET) is a malignant tumor of neural crest origin and arises outside the central nervous system. PNET very rarely manifests as primary lung tumor. Our search of the literature revealed that clubbing fingers had never been mentioned in a patient with pulmonary PNET. Herein, we report the case of a huge pulmonary PNET

191Pulmonary Tumor with Digital Clubbing
胸腔醫學:民國99年25卷4期����
combined with digital clubbing.
Case Report
The presented case is that of a 56-year-old male patient without a medical history, although he had a smoking history of about 20 years. About 5 months before admission, he suffered from progressive shortness of breath during exercise. The associated symptoms included cough with some sputum, and abdominal dis-tension, especially after intake or during a peri-od when he felt dyspneic. He also reported poor appetite and body weight loss of 10 kg within the most recent 5 months. Both the dyspnea and abdominal distension could be relieved by rest. He denied orthopnea, paroxysmal nocturnal dyspnea, chest pain or hemoptysis. The symp-toms progressed, and finally, he felt dyspneic even when walking. He visited a local hospital and abdominal sonography revealed a left up-per abdominal mass. Gastric lymphoma was suspected and he was referred to our hospital.
On admission, no fever was noted and the respiratory rate was 18 per minute, with no re-spiratory distress noted. Physical examination
showed decreased breathing sounds and dull percussion in the lower half of the left lung. Obvious digital clubbing of both hands (Figure 1) and leg edema were also found. Hemogram showed mild normocytic anemia (Hb = 12.8 mg/dl). Biochemistry data revealed normal liver function, renal function and electrolytes. Arte-rial blood gas showed metabolic alkalosis with respiratory compensation. The chest radiogra-phy (Figure 2) showed nearly total opacity of the left hemithorax with mediastinum and heart shifting to the right side. Panendoscopy was performed for abdominal discomfort and re-vealed external compression with posterior wall indentation of the stomach. Chest computed to-mography (CT) scan (Figure 3) showed a huge heterogeneous mass with focal necrosis and pleural effusion in the left thoracic cavity. There was also compression of the mediastinum, liver and stomach. Mediastinal lymph node metas-tasis was also suspected. The chest CT favored
Fig. 1. Marked clubbing of the fingers of both hands with a shiny appearance
Fig. 2. Chest radiograph shows a left opaque lesion with a midline structure shift to the right side

192 Sheng-Han Tsai, Han-Yu Chang
Thorac Med 2010. Vol. 25 No. 4
an intrapulmonary origin of the tumor. We per-formed intermittent thoracentesis for relief of the dyspnea. The pleural effusion was reddish and the exudate was lymphocyte-predominant (54%) in character. Malignant pleural effusion was suspected, but no malignant cells could be found by effusion cytology. The pathology obtained by sonography-guided biopsy showed diffuse small blue cells with hyperchromatic nuclei. In the immunohistochemical study, the tumor cells were positive for CD99 and synap-tophysin, and negative for cytokeratin, LCA, S-100, CD34 and TTF-1 (Figure 4). The final pathologic diagnosis was PNET.
The pulmonary function test demonstrated a severe restrictive ventilatory defect. Bone scan revealed a focal hot spot at the left 11th costo-vertebral junction and bone involvement was considered. Because of advanced disease with a huge tumor and suspicious metastases to the lymph node and bone, surgical intervention was not indicated. He then underwent chemotherapy with a regimen of doxorubicin, dacarbazine and ifosfamide. No obvious side effect was noted
after the treatment and the patient felt less dys-pneic.
Discussion
Digital clubbing, the most ancient sign of medicine, was first documented by Hippocrates about 2500 years ago as “water accumulation” in a patient with empyema [6]. Alternatively, it is also called Hippocratic finger or drumstick fingers. Four grades or stages have been de-scribed as follows: fluctuation and softening of nail bed; an increase in the normal 160o angle between the nail bed and proximal nailfold; the development of a clubbing appearance; shiny nail and periungual skin and longitudinal ridg-ing of the nail [7]. Researchers have described several methods for reaching an accurate diag-nosis, such as the profile sign; modified profile angle; hyponychial angle; and Schamroth sign [8]. Initially, digital clubbing was considered an obvious physical sign associated with internal illness. With the use of radiologic imaging, it was discovered that digital clubbing is usually
(A) (B)
Fig. 3. (A) Chest CT scan shows a tumor in the left lingual and lower lobe with pleural effusion and subcarinal lymphadenopathy. (B) At the lower level, the tumor occupies almost the whole left thoracic cavity with the heart and great vessel deviating to the right side.

193Pulmonary Tumor with Digital Clubbing
胸腔醫學:民國99年25卷4期����
accompanied with periosteal proliferation of the tubular bone. The triad of digital clubbing, arth-ralgia, and periostitis comprises HOA. Current-ly, many authors consider that digital clubbing and HOA represent different stages of the same syndrome [6, 9]. In this case, the clubbing was so obvious that at least grade 4 clubbing was considered. However, the patient did not have arthralgia and no evidence of HOA was noted.
Digital clubbing is an ominous sign with which we are familiar; however, we appear to know more about it than we really do [10]. There are many reports describing the associa-tion between clubbing and numerous diseases,
and many hypotheses of the pathophysiology have been proposed, but no single theory could well explain all the disease entities. The most acceptable mechanism has been proposed by Dickinson and Martin [11]. When the normal pulmonary capillary network is disrupted or by-passed (such as by chronic lung inflammation, carcinoma or intracardiac right-to-left shunt), the megakaryocytes, which escape being nor-mally fragmented in the lung, can enter sys-temic circulation and impact fingertip circula-tion. These cells release platelet-derived growth factor (PDGF), which can promote blood flow, vascular permeability and connective tissue
Fig. 4. (A) Histological finding shows diffuse small blue cells with areas of tumor necrosis and focal fibrous tissue. (B) Immunohistochemically, the tumor cells show positive staining for CD99 and (C) synaptophysin.
(A) (B)
(C)

194 Sheng-Han Tsai, Han-Yu Chang
Thorac Med 2010. Vol. 25 No. 4
change, subsequently resulting in the clubbing. Recently, observation has suggested that vascu-lar endothelial growth factor (VEGF) may be more associated with clubbing [12]. However, the exact mechanism is not fully understood. Clubbing fingers also indicate a poor prognostic sign, as the underlying disease has reached an advanced stage [6]. Some reports have demon-strated improved clubbing after treatment of the underlying disease [4, 13]. In the present case, operation was not feasible due to the advanced stage, so the patient received chemotherapy. However, the patient did not follow up in our hospital, so we could not know whether the digital clubbing was reversible or not.
Peripheral PNET is classified as part of the Ewing’s sarcoma family of tumors which also includes Ewing’s sarcoma of bone, extraosseous Ewing’s sarcoma, and Askin’s tumor [14]. They all represent small round cell malignan-cies of neural crest origin and arise outside the central nervous system [15]. PNET can be dis-tinguished from Ewing’s sarcoma by evidence of neuronal differentiation. Peripheral PNET is rare in adults and most cases originate from the chest wall (Askin’s tumor), abdomen, pelvis, and extremities [16]. To date, there have been only occasional case reports of primary pulmo-nary PNET [15, 17-19]. In these reports, the common presentation included cough, dyspnea, hemoptysis, and chest pain, and digital clubbing was never mentioned as a physical sign. PNET is generally considered to have a poor prognosis and needs to be treated with systemic chemo-therapy. Nevertheless, the mean life expectancy is less than 1 year.
In summary, we have presented a case of pulmonary PNET associated with clubbing fingers. Due to its aggressive behavior, early diagnosis and appropriate treatment are neces-
sary. Clubbing can be the only or the preceding sign of underlying malignant disease, thus we underscore the importance of a detailed physi-cal examination which may lead to the correct diagnosis.
References
1. Robert S, Thomas M, Linford S, et al. Nail abnormalities: clues to systemic disease. Am Fam Physician 2004; 69: 1417-24.
2. Gorospe I, Fernandez-Gil MA, Torres I, et al. Misleading lead: inflammatory pseudotumor of the mediastinum with digital clubbing. Med Ped Oncol 2000; 35: 484-7.
3. Pichler G, Eber E, Thalhammer G, et al. Arthralgia and digital clubbing in a child: hypertrophic osteoarthropathy with inflammatory pseudotumour of the lung. Scand J Rheumatol 2004; 33: 189-91.
4. Kozak KR, Milne GL, Morrow JD, et al. Hypertrophic osteoarthropathy pathogenesis: a case highlighting the potential role for cyclo-oxygenase-2-derived prostaglan-din E2. Nat Clin Pract Rheumatol 2006; 8: 452-6.
5. Sridhar KS, Lobo CF, Altman RD. Digital clubbing and lung cancer. Chest 1998; 114: 1535-7.
6. Martinez-Lavin M. Exploring the cause of the most ancient clinical sign of medicine: finger clubbing. Semin Arthritis Rheum 2004; 36: 380-5.
7. Altman RD, Tenenbaum J. Hypertrophic osteoarthropathy. In: Ruddy S, Harris EPJ, Sledge CB, eds. Kelly’s Text-book of Rheumatology. 6th ed. Philadelphia: WB Saun-ders, 2001: 1589.
8. Kerith E, Joseph C. Clubbing: an update on diagnosis, differential diagnosis, pathophysiology, and clinical relevance. J Am Acad Dermatol 2005; 52: 1020-8.
9. Martinez-Lavin M, Vargas A, Rivera-Vinas M. Hypertro-phic osteoarthropathy: a palindrome with a pathogenic connotation. Curr Opin Rheumatol 2008; 20: 88-91.
10. Brouwers AAM, Vermeij-Keers C, van Zoelen EJ, et al. Clubbed fingers: the claws we lost? Medical Hypotheses 2004; 62: 321-4.
11. Dickinson CJ, Martin JF. Megakaryocytes and platelet clumps as the cause of finger clubbing. Lancet 1987; 2: 1434-5.
12. Atkinson S, Fox SB. Vascular endothelial growth factor

195Pulmonary Tumor with Digital Clubbing
胸腔醫學:民國99年25卷4期����
(VEGF)-A and platelet-derived growth factor (PDGF) play a central role in the pathogenesis of digital clubbing. J Pathol 2004; 203: 721-8.
13. Staalman CR, Umans U. Hypertrophic osteoarthropathy in childhood malignancy. Med Pediatr Oncol 1993; 21: 676-9.
14. Carvajal R, Meyers P. Ewing’s sarcoma and primitive neuroectodermal family of tumors. Hematol Oncol Clin N Am 2005; 19: 501-25.
15. Imamura F, Funakoshi T, Nakamura SI, et al. Primary primitive neuroectodermal tumor of the lung: report of two cases. Lung Cancer 2000; 27: 55-60.
16. Schmidt D, Herrman C, Jurgens J, et al. Malignant peri-
pheral neuroectodermal tumor and its necessary distin-ction from Ewing’s sarcoma. Cancer 1991; 68: 2251-9.
17. Fritz J, Bassam O, Samuel W. Primitive neuroectodermal tumor of the pulmonary hilum in an adult. Ann Thorac Surg 2001; 72: 285-7.
18. Shah ASM, Mohamed Z, Abdullah A, et al. Primitive neuroectodermal tumor of the lung with pericardial extension: a case report. Cardiovasc Pathol 2007; 16: 351-3.
19. Mikami Y, Nakajima M, Hashimoto H, et al. Primary pulmonary primitive neuroectodermal tumor (PNET): a case report. Pathol Res Pract 2001; 197: 113-9.

196 Sheng-Han Tsai, Han-Yu Chang
Thorac Med 2010. Vol. 25 No. 4
肺原始性神經外胚層腫瘤併杵狀指─個案報告
蔡昇翰 張漢煜
杵狀指是肥厚性骨頭關節病變的其中一種表徵,並與許多種類的內科疾病包括感染、發炎、發紺性
心臟病及腫瘤等有相關。傳統上認為杵狀指與肺癌有相關聯。在肺癌中杵狀指的發生率約10-29%,相較
於小細胞癌更容易發生於非小細胞癌。我們在此報告一個罕見的肺部原始性神經外胚層腫瘤合併有杵狀
指的個案。一個56歲男性在最近的五個月內發生了漸進的呼吸性氣促,伴隨有咳嗽,腹脹及體重減輕10公斤。經檢查發現兩手有明顯的杵狀指。影像學檢查顯示了左肺有巨大的腫瘤。經實行超音波導引切片
術後病理報告為原始性神經外胚層腫瘤。病人接受了包含Doxorubicin, Decarbazine及Ifosfamide的化學治
療。病人在治療後感覺氣促的症狀有漸漸改善。(胸腔醫學 2010; 25: 190-196)
關鍵詞:指甲,肺腫瘤,肥厚性骨頭關節病變
成功大學附設醫院內科部 胸腔內科
索取抽印本請聯絡:張漢煜醫師,成大醫院內科部 胸腔內科,台南市勝利路138號

197
胸腔醫學:民國99年25卷4期��������
A Rare Radiological Pattern of Pulmonary Metastasis of Gastric Cancer Mimicking Bronchioloalveolar
Carcinoma: A Case Report
Kuan-Chun Lin, Chi-Wen Lo, Ping- Chen Yu*
This report describes a patient with pulmonary metastasis of gastric cancer who unexpectedly presented with bilateral consolidation, air bronchogram and ground-glass opacities on chest radiography. All imaging findings and clinical symptoms suggested bronchioloalveolar carcinoma. This was a rare radiological pattern that has only occasionally been reviewed. (Thorac Med 2010; 25: 197-202)
Key words: pulmonary metastasis, bronchioloalveolar carcinoma, gastric cancer
Division of Critical Care Medicine, Department of Internal Medicine, Chia-Yi Hospital*Division of Chest Medicine, Department of Internal Medicine, Chia-Yi HospitalAddress reprint requests to: Dr. Kuan-Chun Lin, Division of Critical Care Medicine, Department of Internal Medicine, Chia-Yi Hospital, No. 312, Beigang Rd, West District, Chiayi City 600, Taiwan
Introduction
Bronchioloalveolar carcinoma (BAC) ac-counts for 4% of lung cancers. The radiographic presentation features single or multiple areas of consolidation, and ground-glass or ill-defined opacities [1-3]. The characteristic pathologic feature is malignant cells using the surrounding alveolar walls as a scaffold. Because the cells produce mucus, expectoration of large quanti-ties of mucoid sputum is always the chief com-plaint. This report describes a 51-year-old male with a history of gastric cancer status post-sub-total gastrectomy 2 years before, who presented with dyspnea and cough with much sputum. The bronchoscopic biopsy showed metastatic gastric cancer, rather than BAC, although imag-ing findings and clinical symptoms suggested
BAC. This is a rare radiological pattern of pul-monary metastasis of adenocarcinoma of the stomach, with a lack of evidence of recurrence at the primary site and local organs.
Case Report
A 51-year-old male presented with a 3- month history of progressive dyspnea. Obvious orthopnea with severe persistent hypoxemia under room air and a tendency toward the prone position were found on admission. Initial chest X-rays (CXRs) (Figure 1) showed bilateral pleural effusion and multiple areas of consoli-dation. Chest computed tomography (CT) (Fig-ure 2) demonstrated bilateral consolidation, air bronchogram and ground-glass opacities. There was no imaging finding of liver or intra-abdom-

198 Kuan-Chun Lin, Chi-Wen Lo, et al.
Thorac Med 2010. Vol. 25 No. 4
inal metastasis (Figure 3). Diffuse lymphangitic permeation or nodular thickening of the pulmo-nary septa was absent, which excluded the sus-picion of lymphangitis carcinomatosa. Based on imaging findings and clinical symptoms, BAC was highly suspected. Bronchoscopy (Figure 4) revealed no endobronchial lesion or bleeding. Trans-bronchial lung biopsy (TBLB) (Figure 5) demonstrated signet-ring cells from bron-chial mucosa. The immunohistochemical study
showed positive staining of pan-cytokeratin, but negative staining for TTF-1. Lung metastasis of gastric cancer was confirmed. The upper gas-trointestinal (GI) endoscopy (Figure 6) showed subtotal gastrectomy and no evidence of cancer recurrence at the operation site.
Discussion
The latest World Health Organization clas-sification divides lung adenocarcinoma mainly into adenocarcinoma mixed subtype, acinar adenocarcinoma, papillary adenocarcinoma,
Fig. 1. CXR showed bilateral pleural effusion and multiple areas of consolidation
Fig. 2. Chest CT demonstrated bilateral consolidation, air bron-chogram and ground-glass opacities
Fig. 3. There was no imaging finding of liver or intra-abdominal metastasis
Fig. 4. Bronchoscopy revealed no endobronchial lesion or bleeding

199Pulmonary Metastasis of Gastric Cancer
胸腔醫學:民國99年25卷4期����
BAC, and solid adenocarcinoma with mucin production [4]. Although pure BAC accounts for 4% of lung cancers, tumors with histologi-cally mixed BAC and adenocarcinoma account for >20% of all lung cancers [5-7], and the inci-dence of BAC might be increasing. BAC is for-mally classified into 3 subtypes, nonmucinous, mucinous, and mixed nonmucinous-mucinous [4, 8].
The radiographic presentation that distin-guishes BAC from other types of lung cancer is that the lesion takes the form of single or mul-tiple areas of consolidation or ill-defined opaci-ties. A variety of radiographic, CT, and high resolution CT appearances are seen with this form of the disease [1-3]: ill-defined consolida-tion, resembling pneumonia, pure ground-glass opacity, ground-glass opacity surrounding a core of soft tissue density, mixed ground-glass opacity and denser consolidation, homogenous consolidation of 1 or more lobes, and multifo-cal patchy consolidation or multiple ill-defined nodules spread widely through multiple lobes in 1 or both lungs. Air bronchograms may be an obvious feature, and are particularly well dem-onstrated by CT [9].
Differentiating between the consolidative form of BAC and various non-neoplastic condi-tions, such as pulmonary infection, organizing pneumonia, aspiration, or pulmonary edema, depends on knowing the clinical findings and appreciating the lack of response to treatment. Pleural effusions are seen in up to one-third of patients.
The incidence of pulmonary metastasis varies with the primary tumor and the stage of disease. In an autopsy series, the most common sources included tumors of the breast, colon, kidney, uterus, prostate, head and neck [10]. The hallmark of blood-borne metastasis to the lung on imaging is 1 or more oval or spherical, discrete pulmonary nodules, maximal in the outer portions of the lungs [11-13]. The nodules vary in size, from microscopic to many centi-meters in diameter, are usually multiple, and have well or moderately well-defined, smooth or irregular outlines [14-15]. A variety of other patterns are encountered. On occasion, particu-larly when there is metastatic adenocarcinoma, or if the metastases have bled into the surround-ing lung [16-17], the nodules show irregular
Fig. 6. The upper GI endoscopy showed subtotal gastrectomy and no evidence of cancer recurrence at the operation site
Fig. 5. Trans-bronchial lung biopsy demonstrated signet-ring cells from bronchial mucosa

200 Kuan-Chun Lin, Chi-Wen Lo, et al.
Thorac Med 2010. Vol. 25 No. 4
or ill-defined edges or the features of airspace shadowing [18-19]. A less common picture is that of diffuse lymphangitic permeation, which may produce the clinical and radiologic feature of lymphangitis carcinomatosa [20].
Initially, we performed bronchoscopy to survey if there was an endobronchial lesion or bleeding. Both findings were negative. There-fore, a TBLB was performed. The pathology showed signet-ring cells from bronchial muco-sa, which stained positive for cytokeratin. The lung metastasis of gastric cancer was docu-mented at last. There was no typical radiologi-cal feature of metastasis such as 1 or more oval or spherical, discrete pulmonary nodules and lymphangitis carcinomatosa, but ill-defined consolidation, mixed ground-glass opacity and dense consolidation of more lobes were revealed from chest CT. Besides, a large amount of spu-tum was expectorated by the patient. Dyspnea on exertion, orthopnea, and a tendency toward the prone position were the chief complaints on admission. The laboratory data revealed elevat-ed carcinoembryonic antigen in the pleural ef-fusion and serum. Persistent severe hypoxemia under room air prompted the patient to undergo oxygen therapy throughout the hospital course. Though all clinical symptoms and signs sug-gested the diagnosis of BAC, the results on the bronchoscopic biopsy report were surprising. The upper GI endoscopy demonstrated subtotal gastrectomy and no evidence of cancer recur-rence at the operation site, and there was no imaging finding of liver or intra-abdominal me-tastasis. No enlarged lymph node was palpable on physical examination.
References
1. Shah RM, Balsara G, Webster M, et al. Bronchioloal-
veolar carcinoma: impact of histology on dominant CT pattern. J Thorac Imaging 2000; 15: 180-6.
2. Bonomo L, Storto ML, Ciccotosto C, et al. Bronchioloal-veolar carcinoma of the lung. Eur Radiol 1998; 8: 996-1001.
3. Akira M, Atagi S, Kawahara M, et al. High-resolution CT findings of diffuse bronchioloalveolar carcinoma in 38 patients. AJR Am J Roentgenol 1999; 173: 1623-9.
4. Travis WD, Brambilla E, Muller-Hermelink HK, et al. World Health Organization classification. In: Tumors of the Lung, Pleura, Thymus, and Heart. Lyon, France: IARC Press, 2004.
5. Zell JA, Ou SH, Ziogas A, et al. Epidemiology of bron-chioloalveolar carcinoma: improvement in survival after release of the 1999 WHO classification of lung tumors. J Clin Oncol 2005; 23: 8396-405.
6. Barsky SH, Cameron R, Osann KE, et al. Rising incid-ence of bronchioloalveolar lung carcinoma and its unique clinicopathologic features. Cancer 1994; 73: 1163-70.
7. Auerbach O, Garfinkel L. The changing pattern of lung carcinoma. Cancer 1991; 68: 1973-7.
8. Travis WD, Garg K, Franklin WA, et al. Evolving con- cepts in the pathology and computed tomography ima-ging of lung adenocarcinoma and bronchioloalveolar car-cinoma. J Clin Oncol 2005; 23: 3279-87.
9. Adler B, Padley S, Miller RR, et al. High-resolution CT of bronchioloalveolar carcinoma. AJR Am J Roentgenol 1992; 159: 275-7.
10. Coppage L, Shaw C, Curtis AM. Metastatic disease to the chest in patients with extrathoracic malignancy. J Thorac Imagimg 1987; 2: 24-37.
11. Crow J, Slavin G, Kreel L. Pulmonary metastasis: a pathologic and radiologic study. Cancer 1981; 47: 2595-602.
12. Davis SD. CT evaluation for pulmonary metastases in patients with extrathoracic malignancy. Radiology 1991; 180: 1-12.
13. Gross BH, Glazer GM, Bookstein FL. Multiple pulmo-nary nodules detected by computed tomography: diagnos-tic implications. J Comput Assist Tomogr 1985; 9: 880-5.
14. Hirakata K, Nakata H, Haratake J. Appearance of pulmo-nary metastases on high-resolution CT scans: comparison with histopathologic findings from autopsy specimens. AJR Am J Roentgenol 1993; 161: 37-43.
15. Hirakata K, Nakata H, Nakagawa T. CT of pulmonary

201Pulmonary Metastasis of Gastric Cancer
胸腔醫學:民國99年25卷4期����
metastases with pathological correlation. Semin Ultra-sound CT MR 1995; 16: 379-94.
16. Benditt JO, Farber HW, Wright J, et al. Pulmonary hemorrhage with diffuse alveolar infiltrates in men with high-volume choriocarcinoma. Ann Intern Med 1988; 109: 674-5.
17. Nirenberg A, Meikle GR, Goldstein D, et al. Metastatic carcinoma infiltrating lung mimicking BOOP. Australas Radiol 1995; 39: 405-7.
18. Caeta M, Volta S, Scribano E, et al. Air space pattern in lung metastasis from adenocarcinoma of GI tract. J Comput Assist Tomogr 1996; 20: 300-4.
19. Herold CJ, Bankier AA, Fleischmann D. Lung metastases. Eur Radiol 1996; 6: 596-606.
20. Janower ML, Blennerhassett JB. Lymphangitic spread of metastatic cancer to the lung. A radiographic-pathologic classification. Radiol 1971; 101: 267-73.

202 Kuan-Chun Lin, Chi-Wen Lo, et al.
Thorac Med 2010. Vol. 25 No. 4
罕見胃癌併發肺轉移之影像學表現擬肺泡細胞癌: 病例報告
林冠群 羅啟文 余秉真*
肺泡細胞癌的比率佔所有肺癌中的4%,其影像學之表現為單一或多處的肺實質性變化及毛玻璃狀。
典型的病理學為惡性細胞利用周圍的肺泡細胞壁築成一個像鷹架的組織。本文描述一位胃癌經胃全切除
術後之患者,因咳嗽併痰多,持續呼吸困難,端坐呼吸及嚴重低血氧而住院。胸部X光呈現肺廣泛浸潤及
兩側肋膜積水,胸部電腦斷層顯示兩側肺實質性病變及空氣支氣管造影,毛玻璃狀;且沒發現任何肝臟
或腹腔內轉移之影像。所有的影像學檢查及臨床症狀均傾向為肺泡細胞癌。支氣管鏡檢查顯示無氣管內
病灶或是出血情形,切片病理報告表示是由胃癌所轉移而不是肺泡細胞癌。這是一個經由影像學證實由
胃腺癌轉移至肺部的罕見病例。(胸腔醫學 2010; 25: 197-202)
關鍵詞:肺轉移,肺泡細胞癌,胃癌
行政院衛生署嘉義醫院 內科部胸腔暨重症醫學科,*內科部胸腔內科
索取抽印本請聯絡:林冠群醫師,行政院衛生署嘉義醫院 胸腔暨重症醫學科,嘉義市西區北港路312號

203
胸腔醫學:民國99年25卷4期��������
Solitary Pulmonary Nodule Due to Dirofilariasis: A Case Report and Case Review in Taiwan
Yu-Song Lee, Chien-Da Huang, Chih-Teng Yu, Chih-Wei Wang*, Han-Pin Kuo
Solitary pulmonary nodule caused by Dirofilariasis immitis (dog heartworm), a filarial nematode, is a rare lung parasitic infection. Man is a “dead end” host of D. immitis. The greatest problem that D. immitis creates in the human body is focal pulmonary infarction with a secondary granulomatous and fibrotic reaction after necrosis of the worm. We reported a 53-year-old male patient with a solitary pulmonary nodule in the left upper lung caused by dirofilariasis. The solitary nodule caused by human pulmonary dirofilariasis is easily confused with cancer. Clinicians should be alert to the possibility of pulmonary dirofilariasis when a solitary, noncalcified pulmonary nodule is noted. (Thorac Med 2010; 25: 203-210)
Key words: pulmonary dirofilariasis, solitary pulmonary nodule, lung parasitic infection
Department of Thoracic Medicine; *Department of Pathology, Chang Gung Memorial Hospital, Chang Gung University College of Medicine, Taipei, TaiwanAddress reprint requests to: Dr. Chien-Da Huang, Department of Thoracic Medicine, Chang Gung Memorial Hospital, 5 Fushing Street, Gueishan Shiang, Taoyuan, Taiwan 333
Introduction
Pulmonary dirofilariasis is a rare zoonotic disease, caused by Dirofilariasis immitis (dog heartworm), a filarial nematode [1]. Man is a “dead end” host of D. immitis. It is believed that D. immitis has no way to leave the human body in its natural life cycle, and thus dead filaria are washed out by the circulation, resulting in the lodging of the worms in the distal pulmonary arterial circulation, where they cause a small infarct lesion [2-5]. The spherical rather than wedge-formed shape of the infarct is the result of a secondary granulomatous and fibrotic reac-tion after necrosis of the worm [6].
Human pulmonary dirofilariasis resulting
in pulmonary infarction was first documented in 1961 [7]. Cases of pulmonary dirofilariasis have since been reported worldwide, including America, Japan, Italy, and Taiwan [2, 8-12]. In Taiwan, the first case of pulmonary dirofilari-asis was reported in 1993 [11]. Subsequently, 2 cases of asymptomatic pulmonary dirofi-lariasis have been reported (Table 1) [10, 12]. The prevalence of canine dirofilariasis has increased over the past years, and the overall prevalence of adult worms in the dog popula-tion was reported to be 57% in Taiwan in 2003 [13]. Therefore, the recognition of this entity is important in Taiwan. We herein report a symp-tomatic 53-year-old male patient with a solitary pulmonary nodule in the left upper lung caused

204 Yu-Song Lee, Chien-Da Huang, et al.
Thorac Med 2010. Vol. 25 No. 4
Table 1. Reported cases of pulmonary dirofilariasis in Taiwan
Yang et al. [11] Tsung et al. [10] Hung et al. [12] Current caseYear, reported 1993 2003 2008 2010Age, yr 56 69 66 53Sex Female Male Female MaleSmoking No ND ND NoAlcohol No ND ND NoDog-breeder history No ND ND YesOccupation ND ND ND VeteranAssociated disease Hypertension No No NoSymptoms Asymptomatic Asymptomatic Asymptomatic Dry cough for 2
monthsLocation RUL Between RUL and
RMLRUL and RLL LUL
Nodule Solitary Solitary Double SolitaryCXR finding A 2.5x3 cm coin
nodule in the right middle lung field
A noncalcified nodular lesion in the right lung field
An abnormal shadow LUL nodular density
Bronchoscopy No endobronchial lesion
ND ND No endobronchial lesion
Chest CT finding A solitary and well-defined soft tissue density in the RUL, adjacent to the pleural surface
A well-defined noncalcified nodule 2.0 cm in diameter between the RUL and RML
A well-defined nodule 2.0 cm in diameter with a cavity in the RUL and a smaller nodule in the RLL adjacent to the diaphragm
A nodule (3.3x1.6 cm) in the anterior segment (B3) of the LUL abutting the left pleura.
Pathology An immature male worm located in a pulmonary arteriole
An area of coagulation necrosis; several degenerating nematodes observed within the lumen of a thrombosed pulmonary artery.
Pulmonary dirofilariasis
7 necrotic dirofilarial organisms lodged in a partially necrotic pulmonary artery. Worms could be seen at the center of the necrosis, surrounded by fibrosis and granulation tissue.
Treatment Right exploratory thoracotomy with wedge resection of the RUL
Right thoracotomy with removal of the nodule
VATS VATS with wedge resection of the LUL for tumor resection
ND=not determined; RUL=right upper lobe; RML=right middle lobe; RLL=right lower lobe; LUL=left upper lobe; CXR=chest X-ray; CT=computed tomography; VATS=video-assisted thoracoscopic surgery

205Pulmonary Dirofilariasis
胸腔醫學:民國99年25卷4期����
by dirofilariasis, and review the 3 reported cases of pulmonary dirofilariasis in Taiwan.
Case Report
A 53-year-old male patient came to our hospital with the chief complaint of dry cough for 2 months. He denied the presence of fever, chest pain, hemoptysis, or body weight loss. The patient had not had any major systemic diseases before. He was a veteran with a history of dog breeding during his military service. On physical examination, he was afebrile with a body temperature of 36.5°C, pulse of 83 beats per minute and respiratory rate of 17 breaths per minute. Lung auscultation revealed clear breathing sounds symmetrically in the bilateral lung fields. No skin nodules or tender points were palpable over the entire body. The hemo-globin level was 15.4 g/dl and the leukocyte count was 8000/ul with a normal differential. The chemistry profile was also within normal limits.
A chest X-ray (Figure 1A) showed a nodu-lar density in the left upper lung field. Com-puted tomography (CT) (Figure 1B) revealed a nodular density in the anterior segment of the left upper lobe (3.3 x 1.6 cm) abutting the left pleura. Bronchoscopy was performed for airway evaluation, but revealed no endobron-chial lesions. A lesion, however, was found in the left B3 segment by endobronchial ultra-sound (EBUS), which yielded heterogeneous echogenicity with an irregular margin (Figure 2A). Transbronchial lung biopsy (TBLB) of the lesion was performed for tissue diagnosis. However, only chronic inflammation was seen. Under the impression of a solitary pulmonary tumor, the patient underwent wedge resection of the left upper lobe by video-assisted tho-
racoscopic surgery (VATS) for the purpose of further tissue diagnosis and tumor resection. Pulmonary dirofilariasis was diagnosed by pathological examination (Figure 2B). The pa-tient was discharged uneventfully after surgical intervention without residual consequences or
(A)
(B)
Fig. 1. (A) Chest X-ray showed a nodular density in the left upper lung field. (B) Computed tomography showed a nodular density (3.3 x 1.6 cm) in the anterior segment (B3) of the left upper lobe abutting the left pleura.

206 Yu-Song Lee, Chien-Da Huang, et al.
Thorac Med 2010. Vol. 25 No. 4
symptoms during the follow-up period.
Discussion
Solitary pulmonary nodule caused by D. immitis (dog heartworm) is a rare lung parasitic infection. Heartworms are common in dogs, and are spread by Aedes, Culex, and Anopheles mosquitoes; humans who are bitten by larvae-carrying mosquitoes become accidental hosts [2, 4-6]. When D. immitis enters the human body, which rarely occurs, it cannot leave. Because its life cycle ceases in the human body, humans are called “dead end” hosts for D. immitis. An oc-casional larva may reach the right ventricle, and
develop into a sexually immature form before death and embolization to the lungs. The most common problem that D. immitis creates in the human body is focal pulmonary infarction with granulomatous formation. The cycle can also be arrested in the subcutaneous tissue, resulting in a nodular formation. Cutaneous, periorbital or premasseteric nodules in extrapulmonary or-gans have been reported, and were believed to be caused by D. immitis or D. repens [14-16].
Typically, patients with pulmonary diro-filariasis experience symptoms that include thoracic pain, cough, fever, hemoptysis, and dyspnea, but most reported cases have been as-ymptomatic and were discovered during routine
Fig. 2. (A) A lesion was found in the left B3 segment by endobronchial ultrasound (EBUS), which yielded heterogeneous echogenicity with an irregular margin. (B) Transverse sections of 7 necrotic dirofilarial organisms lodged in a partially necrotic pulmonary artery. Worms were seen in the center of the necrosis, surrounded by fibrosis and granulation tissue. (C) Thick cuticles and the internal structures consisting of abundant somatic muscles and tubular elements of the worms were noted. The adjacent lung tissue showed coagulative necrosis.
(A)
(B)
(C)

207Pulmonary Dirofilariasis
胸腔醫學:民國99年25卷4期����
chest X-ray check-ups [3-4, 10-12]. Our patient had complained of a dry cough for 2 months. In addition, systemic eosinophilia was believed to be a common finding in parasitic infections, but was in only 7.5% to 15% of cases [2-4, 6, 17]. However, no eosinophilia was noted in the he-mogram test in our case. Serological detection of human antibody to dirofilariasis by enzyme-linked immunosorbent assay (ELISA) has been used for confirming the diagnosis. However, the clinical application is limited, since no further data of sensitivity and specificity are available [2, 8]. No laboratory test specific for dirofilari-asis was performed in our case. Since there are no helpful laboratory tests for the definite diag-nosis, pulmonary dirofilariasis was diagnosed in the main after biopsy by invasive procedures such as needle aspiration or surgical resection in the reported cases [2].
The differential diagnosis for single pul-monary dirofilariasis found on chest X-film includes malignancy, metastatic cancer, benign tumors, mycotic infections such as histoplasmo-sis and tuberculoma, Wegener’s granulomato-sis, rheumatoid nodules, and pulmonary throm-boemboli [4, 6]. The solitary nodules caused by human pulmonary dirofilariasis have been reported to be easily confused with cancer [3]. Many reported patients with pulmonary diro-filariasis were admitted to the hospital due to concerns of lung malignancy in imaging studies [3, 9]. Our male patient had a solitary pulmo-nary nodule in the left upper lung field found on chest X-ray, and was admitted for further stud-ies because pulmonary malignancy was first suspected.
In our case, the chest CT study yielded a soft tissue nodule 3 cm in diameter adjacent to the pleural surface of the left upper lobe. Pul-monary dirofilariasis causes lung lesions with
different radiographic presentations on CT [2]. Typically, the nodules are well-circumscribed, solitary, and non-calcified round or oval in shape, and are predominantly peripherally lo-cated; there can also be subpleural nodules [3]. However, multiple nodules seen on CT have also been reported, and were distributed in uni-lateral or bilateral lung fields [4, 6]. Most of the lesions have been reported to be less than 3 cm in diameter [2-4, 6, 18]. Two review articles showed that pulmonary dirofilariasis lesions were predominantly located in the upper lobes [18-19], but 2 other articles reported that the lesions in their cases were found in the lower lobes [2, 4]. The presentation of pulmonary dirofilariasis in CT imaging has been reported to be round or spherical with characteristics of central high attenuation and peripheral ground glass attenuation within the lesion, which dif-fers from that of the typical wedge-shaped image presentation in classical pulmonary in-farction. In thinner-section CT, the nodules of pulmonary dirofilariasis also present as running vessels into the apex of wedge-shaped infarc-tion lesions, the so-called vascular sign in clas-sic thromboemboli [2, 20].
Typical pathological findings of pulmo-nary dirofilariasis may include a grossly well-circumscribed firm nodule with grayish color, as well as microscopically infarction-like coagulative necrosis with granulomatous cap-sules composed of lymphocytes, plasma cells, eosinophils, epithelioid histiocytes and Langer-hans’ giant cells [4, 6]. The central necrotic area is attributed to the homogeneous low attenua-tion area in contrast-enhanced CT [2]. Larvae may be found, but often only after a thorough search in serial microscopic transverse sections. Transverse sections of dead nematodes can be found on microscopic examination of arteries at

208 Yu-Song Lee, Chien-Da Huang, et al.
Thorac Med 2010. Vol. 25 No. 4
the center of the necrotic area [6, 18, 20]. In our case, 7 necrotic dirofilarial organisms lodged in a partially necrotic pulmonary artery were found. Worms were seen at the center of the necrosis, surrounded by fibrosis and granulation tissue. Thick cuticles and internal structures consisting of abundant somatic muscles and tu-bular elements of worms were also noted.
Pulmonary dirofilariasis diagnosed by nee-dle aspiration has been previously reported [19]; however, later articles suggested that local re-section is unavoidable for diagnosis unless the pulmonary nodule can be reliably identified [4, 20]. In Japan, VATS is believed to be the best method for diagnosing pulmonary dirofilariasis [21]. Bronchoscopy with transbronchial lung biopsy (TBLB) of the lesion was performed initially for tissue diagnosis in this study, but no malignancy was noted. This patient under-went a wedge resection of the left upper lobe by VATS with a final diagnosis of pulmonary diro-filariasis.
Humans are the accidental and “dead end” hosts for pulmonary dirofilariasis, and no fur-ther therapy is needed [3-4, 6, 18]. Usually there are neither residual consequences nor symptoms after surgical resection in the report-ed cases [3, 10-11, 18]. This patient was dis-charged uneventfully after surgical intervention, and had a smooth course during the follow-up period.
In conclusion, pulmonary dirofilariasis is easily confused with cancer. Clinicians should be alert to the possibility of pulmonary dirofi-lariasis when a solitary, noncalcified pulmonary nodule is found.
Conflict of Interest Statement
None of the authors has any potential fi-
nancial conflict of interest related to this manu-script.
References
1. Kuzucu A. Parasitic diseases of the respiratory tract. Curr Opin Pulm Med 2006; 12(3): 212-21.
2. Oshiro Y, Murayama S, Sunagawa U, et al. Pulmonary dirofilariasis: computed tomography findings and corre-lation with pathologic features. J Comput Assist Tomogr 2004; 28(6): 796-800.
3. Lagrotteria DD, Crowther MA, Lee CH, et al. A 44-year-old woman with dry cough and solitary nodule. CMAJ 2003; 169(7): 696-7.
4. Milanez de Campos JR, Barbas CS, Filomeno LT, et al. Human pulmonary dirofilariasis: analysis of 24 cases from Sao Paulo, Brazil. Chest 1997; 112(3): 729-33.
5. Moore W, Franceschi D. PET findings in pulmonary dirofilariasis. J Thorac Imaging 2005; 20(4): 305-6.
6. Breitenbucher A, Gayer R, Giachino D, et al. What is your diagnosis. Multiple pulmonary nodules. Respiration 1998; 65(1): 91-4.
7. Dashiell GF. A case of dirofilariasis involving the lung. Am J Trop Med Hyg 1961; 10: 37-8.
8. Angeli L, Tiberio R, Zuccoli R, et al. Human dirofila-riasis: 10 new cases in Piedmont, Italy. Int J Dermatol 2007; 46(8): 844-7.
9. Shah MK. Human pulmonary dirofilariasis: review of the literature. South Med J 1999; 92(3): 276-9.
10. Tsung SH, Liu CC. Human pulmonary dirofilariasis in Taiwan. J Formos Med Assoc 2003; 102(1): 42-5.
11. Yang SS, Wang LS, Fahn HJ, et al. Solitary pulmonary nodule caused by Dirofilaria immitis. J Surg Assoc ROC 1993; 26(2): 1713-8.
12. Hung JJ, Yen MH, Liu JS, et al. Human pulmonary diro- filariasis: a case report. Thoracic Medicine 2008; 23(6, Supplement (2008 Annual Meeting of TSPCCM, Abstract: P107)): 111.
13. Wu CC, Fan PC. Prevalence of canine dirofilariasis in Taiwan. J Helminthol 2003; 77(1): 83-8.
14. Beden U, Hokelek M, Acici M, et al. A case of orbital dirofilariasis in Northern Turkey. Ophthal Plast Reconstr Surg 2007; 23(4): 329-31.
15. Radmanesh M, Saberi A, Maraghi S, et al. Dirofilaria

209Pulmonary Dirofilariasis
胸腔醫學:民國99年25卷4期����
repens: a dog parasite presenting as a paranasal subcuta-neous nodule. Int J Dermatol 2006; 45(4): 477-8.
16. Latifoglu O, Ozmen S, Sezer C, et al. Dirofilaria repens presenting as a premasseteric nodule. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 94(2): 217-20.
17. Flieder DB, Moran CA. Pulmonary dirofilariasis: a clini-copathologic study of 41 lesions in 39 patients. Hum Pathol 1999; 30(3): 251-6.
18. Jarratt M. Solitary pulmonary nodule in a 62-year-old man. Chest 1995; 107(1): 271-3.
19. Hawkins AG, Hsiu JG, Smith RM, et al. Pulmonary diro-filariasis diagnosed by fine needle aspiration biopsy. A case report. Acta Cytol 1985; 29(1): 19-22.
20. Wand A, Kasirajan LP, Sridhar S. Solitary pulmonary nodule due to dirofilariasis. J Thorac Imaging 2000; 15(3): 198-200.
21. Miyoshi T, Tsubouchi H, Iwasaki A, et al. Human pulmo-nary dirofilariasis: a case report and review of the recent Japanese literature. Respirology 2006; 11(3): 343-7.

210 Yu-Song Lee, Chien-Da Huang, et al.
Thorac Med 2010. Vol. 25 No. 4
犬心絲蟲造成孤立肺部結節:病例報告與台灣病例整理
李育松 黃建達 余志騰 王志偉* 郭漢彬
犬心絲蟲(Dirofilariasis immitis)造成孤立性肺部結節(Solitary pulmonary nodule)在感染人類的
寄生蟲病例中屬於少見。對犬心絲蟲而言,人類是終宿主;而其在人類肺部導致的主要問題是局部的肺
部栓塞,與蟲體凋亡之後併發的肉芽腫和纖維化。在本文中,我們報告一位53歲男性病患,因肺部感染
犬心絲蟲造成的孤立性結節。臨床上,因為犬心絲蟲造成的肺部結節在影像學檢查不易與惡性腫瘤作區
別,所以建議臨床醫師對於孤立性肺部結節必須考慮犬心絲蟲感染的可能性。(胸腔醫學 2010; 25: 203-210)
關鍵詞:犬心絲蟲,孤立性肺部結節,肺部寄生蟲
長庚大學暨林口長庚紀念醫院 胸腔內科,*病理科
索取抽印本請聯絡:黃建達醫師,長庚大學暨林口長庚紀念醫院 胸腔內科,桃園縣龜山鄉復興街5號

211
胸腔醫學:民國99年25卷4期��������
Vocal Cord Mucormycosis: An Unusual Cause of Stridor – Case Report
Yen-Sung Lin, Chih-Yen Tu, Chia-Hung Chen, Yi-Heng Liu, Wei-Chih Liao, Huan-Ting Shen
Mucormycosis is an opportunistic fungal infection. It occurs in patients with diabetes mellitus, malignancy or long-term steroid use, and in those who are immunocompromised. Usually, it infects the rhinocerebral, pulmonary, cutaneous or gastrointestinal systems [1]. Pulmonary mucormycosis commonly occurs in immunocompromised patients or those with malignancies, but vocal cord mucormycosis is rare and the standard treatment is still inconclusive. We reported a patient with acute myeloid leukemia and type 2 diabetes mellitus suffering from vocal cord mucormycosis infection with the initial presentation of stridor. We treated the patient by intravenous amphotericin B for more than 1 month because he refused surgical intervention. But the patient still expired due to disseminated mucormycosis infection. (Thorac Med 2010; 25: 211-215)
Key words: mucormycosis, acute myeloid leukemia, vocal cord, stridor
Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, China Medical University Hospital, Taichung, TaiwanAddress reprint requests to: Dr. Chih-Yen Tu, Department of Internal Medicine, China Medical University Hospital, No. 2, Yude Road, Taichung, Taiwan
Introduction
Mucormycosis is a rare fungal infection. It occurs in patients that are immunocom-promised and in those with poorly controlled diabetes mellitus (DM), malignancy, or long-term steroid use. Immunocompromised patients with mucormycosis infection have mortality rates up to 80% [1]. The common presentations of mucormycosis infection are rhinocerebral, pulmonary or disseminated disease [2]. The infection is rapidly progressive, locally spread and systemically involved. The first case of pul-monary mucormycosis was described in 1876
by Furbinger et al. [3] and endobronchial mu-cormycosis was reported in 1958 [4]. In 1999, Lee et al. [5] reviewed the cases of 87 patients with primary pulmonary mucormycosis and found most patients (39%, 34/87) had an endo-bronchial lesion. Only 1 patient (1%) had vocal cord involvement. We report a patient with type 2 DM and acute myeloid leukemia (AML) who suffered from vocal cord mucormycosis with the initial presentation of stridor after he had received chemotherapy for his AML.

212 Yen-Sung Lin, Chih-Yen Tu, et al.
Thorac Med 2010. Vol. 25 No. 4
Case Report
A 55-year-old male had had type 2 DM, under control with an oral hyperglycemic agent, since 2005. He was diagnosed with AML in January 2007 and was treated with chemo-therapy beginning at that time. After the 3rd course of chemotherapy, the patient suffered from pancytopenia with neutropenic fever. He received filgrastim plus broad spectrum anti-biotics during the neutropenic fever episode. With this treatment, his pancytopenia and neu-tropenic fever subsided and he was discharged from the hospital to prepare for the next cycle of chemotherapy for his AML, set to begin on 28 May 2007. Unfortunately, he suffered from shortness of breath and progressive dyspnea 1 day after hospital discharge. He came to our emergency room where physical examinations revealed stridor during respiration. Chest X-ray revealed upper airway narrowing with a left
upper lung nodular lesion (Figure 1), and his pulmonary function test showed fixed upper airway obstruction (Figure 2). Bronchoscopic examination revealed yellowish-gray lesions on the bilateral vocal cords complicated with airway narrowing (Figure 3). The pathology of the bronchoscopy-guided vocal cord biopsy re-vealed unseptated hyphae and spores (Figure 4), which was compatible with mucormycosis in-fection. Surgical intervention was suggested for his vocal cord mucormycosis, but the patient refused due to the high risk, so he was treated
Fig. 1. Chest X-ray showed upper airway narrowing (white arrow) and left upper lobe infiltration (black arrow).
Fig. 2. Flow-volume curve revealed fixed upper airway obstruction.
Fig. 3. Bronchoscopy demonstrated a bilateral yellowish-gray lesion at the bilateral vocal cords complicating airway narrowing (white arrow).

213Vocal Cord Mucormycosis
胸腔醫學:民國99年25卷4期����
Fig. 5. Magnetic resonance imaging of the brain revealed a left nasal cavity mucormycosis fungal ball (white arrow).
with intravenous amphotericin B therapy. How-ever, the follow-up bronchoscopy revealed a new left nasal mucormycosis lesion, and mag-netic resonance imaging of the brain revealed a left nasal cavity mucormycosis fungal ball (Figure 5). Unfortunately, the patient expired 3 weeks later because of his uncontrolled dis-seminated mucormycosis infection.
Discussion
Mucormycosis is an infection by fungus of the order Mucorales, belonging to the class Zygomycetes [6]. The most commonly identi-fied genus is Rhizopus, followed by Mucor and Cunninghamella [7]. Humans inhale the spores, which are usually found on soil, dung and veg-etable matter. They form characteristic broad aseptate hyphae with branching at right angles in the tissues [6]. The main risk factors are DM, hematologic malignancies, renal insufficiency, and organ transplantation. In immunocompro-mised states, pulmonary mucormycosis is an uncommon, but life-threatening infection [5]. Fungal balls [8], fatal hemoptysis [9] and recur-rent pneumonia [10] have been reported when pulmonary mucormycosis infection occurs after inhalation of airborne fungal spores. The preva-lence of mucormycosis infection is about 8% in autopsied patients with leukemia at post mor-tem and 2% in allogenic bone marrow trans-plant patients [13]. Patients with hematologic malignancies or bone marrow transplantation have an increased incidence of mucormycosis infection [14]. Pulmonary mucormycosis is a high mortality disease and aggressive therapy by surgical resection plus intravenous amphot-ericin B injection [7, 12] is necessary. Tedder et al. [7] reported that the outcomes of pulmonary mucormycosis in both non-neutropenic and neutropenic patients treated by surgical therapy were better than in those treated with intrave-nous amphotericin.
Upper airway obstruction is a life-threat-ening disease and the most common causes are a foreign body in the upper intrathoracic airway or esophagus, the status post-exubation, acquired lesions of the airway, and congenital lesions in children. Airway or vocal cord fun-
Fig. 4. Microphotograph of bronchoscopy-guided vocal cord biopsy shows broad aseptate fungal hyphae with right-angled branching (black arrow). (Periodic acid-Schiff stain)

214 Yen-Sung Lin, Chih-Yen Tu, et al.
Thorac Med 2010. Vol. 25 No. 4
gal infection is very rare. However, vocal cord paralysis due to endobronchial mucormycosis has been reported previously [5, 11]. In our reported case, the patient had type 2 DM and AML, which confer a high risk for acquired fungal infection, especially when the patient is in a pancytopenic status after chemotherapy. Based on our case, we suggest that if an immu-nocompromised patient presents with stridor, an immediate bronchoscopic exam is necessary to survey whether vocal cord fungal infection has occurred. Moreover, prompt surgical interven-tion should be recommended for those patients with endobronchial mucormycosis, because it is a high mortality disease under intravenous anti-fungal agent treatment.
References
1. Harald B, Monika V, Manfred W, et al. Postoperative relapse of pulmonary mucormycosis: The difficulty of imaging recurrent disease in the neuropenic patient. European Journal of Radiology 2006; 60: 51-4.
2. Omer W, Ziv G, Leonor L, et al. Tracheal mucormycosis presented as an intraluminal soft tissue mass. Head & neck 2004; 26: 541-3.
3. Furbinger P. Beobachtungen uber lungenmycose bein meschen. Arch Pathol Anat Physiol Klin Med 1876; 66: 330-65.
4. Dillon ML, Sealy MC, Fetter BF. Mucormycosis of the bronchus successfully treated by lobectomy. J Thoracic
Surgery 1958; 35: 464-8. 5. Lee FYW, Mossad SB, Adal KA. Pumonary mucormy-
cosis: the last 30 years. Arch Inter Med 1999; 159: 1301-9.
6. Rippon JW. Zycomycosis. In Medical Mycology: The pathogenic fungii and the pathogenic actinomycetes. Philadelphia, WB Saunders 1988: 681-713.
7. Sentochnik DE, Eliopoulos GM. Infection and Diabetes. In Ronald Kahn C, Weir GC (ed.) Joslin’s Diabetes Mellitus. Baltimore Waverly International 1998: 867-88.
8. Lahiri TK, Agarwal D, Reddy GE, et al. Pulmonary mucoraceous fungal ball. Indian J Chest Diab Allied Sci 2001; 43: 107-10.
9. Gupta KL, Khullar DK, Behera D, et al. Pulmonary mucormycosis presenting as fatal massive hemoptysis in a renal transplant patient. Nephrol Dial Transplant 1998; 13: 3258-60.
10. Bhowmik D, Dhinda AK, Khilnani GC, et al. Pulmonary mucormycosis in a diabetic renal transplant patient. Indian J Chest Dis Allied Sci 2002; 44: 275-7.
11. Suresh V, Bhansali A, Sridhar C, et al. Pulmonary mu-cormycosis presenting with recurrent laryngeal nerve palsy. J Assoc Physicians India. 2003 Sep; 51: 912-3.
12. Bigby TD, Serota ML, Tierney LM, et al. Clinical spec-trum of pulmonary mucormycosis. Chest 1986; 89: 420-3.
13. Viswam D, Gopinathan VP, Indira K, et al. Medical ablation of endobronchial mucormycosis with ampho-tericin-B. J Assoc Physicians India. 2007 Dec; 55: 861-5.
14. Marr KA, Carter RA, Crippa F, et al. Epidemiology and outcome of mould infections in hematopoietic stem cell transplant recipients. Clin Infect Dis. 2002; 34: 909-17.

215Vocal Cord Mucormycosis
胸腔醫學:民國99年25卷4期����
以喘鳴為最初表現的聲帶白黴菌病─病例報告
林延淞 涂智彥 陳家弘 劉奕亨 廖偉志 沈煥庭
白黴菌病是一種伺機性黴菌感染。它通常發生在有糖尿病、免疫不全、癌症和長期使用類固醇的病
人身上。通常侵犯鼻腦部、肺部、皮膚和腸胃道。肺部白黴菌通常發生在糖尿病和癌症病人。聲帶侵犯
的病例則很少被報告也尚未有標準治療方式的結論。我們提出一個急性骨髓性白血病合併有糖尿病的病
人,以喘鳴為最初表現的聲帶白黴菌病歷報告。因為病人拒絕開刀介入,我們以靜脈注射amphotericin B超過一個月。最後病人仍然因為瀰漫性白黴菌病感染而死亡。(胸腔醫學 2010; 25: 211-215)
關鍵詞:白黴菌,急性骨髓性白血病,聲帶,喘鳴
中國醫藥大學附設醫院內科部 胸腔暨重症系
索取抽印本請聯絡:涂智彥醫師,中國醫藥大學附設醫院內科部 胸腔暨重症系,台中市育德路2號

216
Thorac Med 2010. Vol. 25 No. 4
Spontaneous Pneumothorax Following Chemotherapy for Malignant Pleural Mesothelioma with Diffuse
Pulmonary Metastasis – A Case Report
Pao-Shan Wang, Kuo-An Chu, Shong-Ling Lin*, Ming-Ting Wu**, Ruay-Sheng Lai
Malignant pleural mesothelioma (MPM) is an asbestos-associated neoplasm that arises from mesothelial surfaces of the pleural cavities. Contralateral pulmonary metastasis in MPM, although reported, is unusual. Spontaneous pneumothorax (SP) following chemotherapy for malignancy is relatively rare, but has been reported in patients with a variety of tumors. To the best of our knowledge, SP occurring as a complication of chemotherapy in patients with MPM has not been reported before. In this report, we described a 54-year-old man with right-sided MPM who underwent combination chemotherapy with cisplatin and pemetrexed. Seven days after chemotherapy, he presented with an acute onset of worsening dyspnea with left pneumothorax. A computed tomography (CT) scan of the chest revealed right pleural-based masses, numerous newly developed bilateral pulmonary nodules and multiple subpleural nodules of the left lung with pneumothorax. The new development of numerous bilateral pulmonary nodules in this patient was believed to have been caused by the MPM, including contralateral pulmonary metastasis. Based on the temporal relationship to chemotherapy and the multiple subpleural nodules demonstrated by chest CT, chemotherapy-induced pneumothorax was considered. A chest tube was inserted and the dyspnea improved. Chemical pleurodesis was performed after complete expansion of the left lung. At that point, the patient opted for palliative care and refused further chemotherapy. He died less than 3 months later without recurrence of SP. In patients with pulmonary metastasis from MPM, the acute onset of dyspnea following chemotherapy should alert clinicians to the possibility of SP. Chest tube insertion and subsequent pleurodesis should be arranged properly and immediately. (Thorac Med 2010; 25: 216-221)
Key words: chemotherapy, malignant pleural mesothelioma, spontaneous pneumothorax
Division of Chest Medicine, Department of Internal Medicine, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan; *Department of Pathology, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan**Department of Radiology, Kaohsiung Veterans General Hospital, Kaohsiung, TaiwanAddress reprint requests to: Dr. Kuo-An Chu, Division of Chest Medicine, Department of Internal Medicine, Kaohsiung Veterans General Hospital, 386 Ta-Chung 1st Road, Kaohsiung 813, Taiwan

217Pneumothorax Following Chemotherapy for Malignant Pleural Mesothelioma
胸腔醫學:民國99年25卷4期����
Introduction
Malignant pleural mesothelioma (MPM) is a rare and asbestos-associated neoplasm that arises from mesothelial surfaces of the pleural cavities. It commonly spreads by direct inva-sion of surrounding tissues. Distant metastasis is generally a very late finding [1]. Its involve-ment of the contralateral lung, although report-ed, is very unusual [2-3]. Most patients with MPM initially experience the insidious onset of shortness of breath or chest pain. Spontaneous pneumothorax (SP) is a very rare presenting feature of MPM [4]. To the best of our knowl-edge, SP occurring as a complication of chemo-therapy in patients with MPM has not been re-ported before. In this report, we describe a case of SP after cytotoxic chemotherapy in a patient with diffuse pulmonary metastasis from MPM.
Case Report
A 54-year-old man without a history of prior lung disease was admitted to our hospital because of progressive shortness of breath for 3 weeks. He was a nonsmoker but had had occu-pational exposure to asbestos for 30 years. The initial chest radiograph showed right pleural effusion. Analysis of the pleural fluid revealed lymphocyte-predominant exudates. Three sepa-rate pleural fluid cytologic studies were nega-tive for malignant cells. Pleural fluid acid-fast stains also revealed negative results. A comput-ed tomography (CT) scan of the chest disclosed right pleural effusion with pleural thickness without a pulmonary nodule or mass.
Since the pleural effusion was of undeter-mined etiology, a video-assisted thoracoscopic (VATS) biopsy was performed. The patho-logical examination of the right pleural biopsy
specimen showed epithelioid neoplastic meso-thelial cells with desmoplastic stroma and focal invasion to adipose tissue. The tumor cells were positive for cytokeratin and calretinin immuno- histochemical staining, and negative for CEA immunohistochemical staining. These patho-logical findings confirmed the diagnosis of ep-ithelioid-type MPM. The patient then received 5 courses of combination chemotherapy with cisplatin and pemetrexed, with stable chest ra-diographs and no significant side effects.
Seven days after the 6th course of chemo-therapy, he came to our emergency department due to an acute onset of worsening dyspnea. He had no fever or trauma. Chest auscultation re-vealed bilaterally diminished breathing sounds. A chest radiograph showed right lobulated pleu-ral thickness and left pneumothorax (Figure 1). Screening laboratory tests were normal except for a hemoglobin level of 10.5 g/dl. Chest CT revealed extensive irregular lobulated pleural-based masses in the right chest, numerous no-
Fig. 1. Chest radiograph showing right lobulated pleural thickness and left pneumothorax (arrows)

218 Pao-Shan Wang, Kuo-An Chu, et al.
Thorac Med 2010. Vol. 25 No. 4
Fig. 2. Chest CT showing numerous nodules of variable sizes over bilateral lung parenchyma and subpleural nodules (arrows) of the left lung with pneumothorax.
dules of various sizes in the bilateral lung pa-renchyma and multiple subpleural nodules in the left lung with left pneumothorax (Figure 2). Suction was performed after inserting a pigtail catheter into the left hemithorax, and re-expan-sion of the left lung occurred immediately. After complete expansion of the left lung, the patient underwent chemical pleurodesis with minocy-cline. The patient was discharged and followed up at the outpatient department.
One month after the occurrence of SP, follow-up chest CT revealed progression of the multiple metastatic nodules of the bilateral lung parenchyma. At that point, the patient opted for palliative care and refused further chemother-apy. He died less than 3 months later, with no recurrence of SP.
Discussion
MPM is an asbestos-associated neoplasm and is usually diagnosed in the 5th to 7th decades of life [5]. The diagnosis of MPM is a challenge for clinicians. Its differential diagnosis includes both benign and malignant processes. Cytologic
diagnosis of MPM from pleural fluid is unreli-able, since reactive mesothelial cells and cells from other malignant tumors such as sarcomas and adenocarcinomas are often difficult to dis-tinguish from malignant mesothelial cells [6]. Surgical intervention, via VATS biopsy or open thoracotomy, is often necessary to firmly estab-lish the diagnosis [7].
MPM generally spreads by local invasion to adjacent tissues, the involved pleura progres-sively thicken, and finally, the lung becomes encased by the tumor. Contralateral pulmonary metastasis has seldom been described with MPM [2-3]. In our patient, the initial chest CT demonstrated right pleural effusion with pleural thickness without a pulmonary nodule or mass and right-sided MPM was diagnosed by VATS biopsy. Subsequent chest CT showed, in addi-tion to the previously noted right pleural chang-es, numerous newly developed bilateral pulmo-nary nodules. These newly developed nodules were believed to have been caused by the MPM with contralateral pulmonary metastasis, which is uncommon in MPM.
Most patients with MPM are not candidates for surgical or radiotherapy treatment, and chemotherapy is their only option. The current standard of care for first-line chemotherapy in MPM patients with good performance is a combination treatment with cisplatin and pem-etrexed [8]. The prognosis of MPM is poor and the median survival time from symptom onset is approximately 1 year [1]. Death is rarely a result of metastatic disease and is usually due to infection, invasion of adjacent structures, or re-spiratory failure secondary to lung encasement by the tumor itself [1].
Pneumothorax is classified as spontane-ous, traumatic or iatrogenic [9]. SP can occur secondarily to a variety of lung diseases such as

219Pneumothorax Following Chemotherapy for Malignant Pleural Mesothelioma
胸腔醫學:民國99年25卷4期����
chronic obstructive pulmonary disease, pulmo-nary tuberculosis, idiopathic pulmonary fibrosis and cancer [9]. Cancer-related SP is unusual [10]. SP is a very rare manifestation of MPM. Hillerdal in 1982 reviewed 4710 published cas-es of malignant mesothelioma and found only 1 case that presented with pneumothorax [11].
SP following cytotoxic chemotherapy is relatively rare, but has been reported in patients with a variety of tumors. Most of these tumors were osteosarcoma or germ cell tumors with pulmonary metastasis [12]. Several pathophysi-ologic mechanisms for these chemotherapy-associated pneumothoraces have been pro-posed: (1) rupture of chemosensitive peripheral or subpleural nodules; (2) enlargement of a rapidly necrotizing tumor; (3) chemotherapy-induced impairment of the repair process; and (4) increased intrathoracic pressure following emetogenic chemotherapy [10, 12-13]. To the best of our knowledge, the occurrence of SP as a complication of chemotherapy in patients with MPM has not been reported before. Our patient was a nonsmoker and had no previous history of lung disease. Based on the temporal relationship to chemotherapy and the multiple subpleural metastatic nodules demonstrated by chest CT, we postulated that combination chemotherapy with cisplatin and pemetrexed induced cell lysis and necrosis of subpleurally located metastatic nodules, leading to pneu-mothorax.
Chemotherapy-associated SP is a potential-ly life-threatening event, because lung function in these patients is usually compromised, and it should be managed with immediate chest tube insertion. In cases of pneumothorax with persis-tent air leak despite closed chest tube drainage, surgical intervention should be attempted [10]. Chemical pleurodesis should be considered for
patients with recurrent or bilateral SP associated with chemotherapy [10, 12].
Although rare, it is possible that bilateral pulmonary metastasis will be found during the course of management of MPM, as in our patient. In addition, the acute onset of dysp-nea following chemotherapy for pulmonary metastasis from MPM should alert clinicians to the possibility of SP. A chest tube should be inserted immediately for this potentially life-threatening event and pleurodesis may also be needed to prevent recurrent SP.
References
1. Pistolesi M, Rusthoven J. Malignant pleural mesothe-lioma: update, current management, and newer therapeu-tic strategies. Chest 2004; 126: 1318-29.
2. Ohishi N, Oka T, Fukuhara T, et al. Extensive pulmonary metastases in malignant pleural mesothelioma: a rare clinical and radiographic presentation. Chest 1996; 110: 296-8.
3. Fares M, Abbas O, Jamaleddine G, et al. Metastases in malignant pleural mesothelioma: a new radiological appearance. Respirology 2008; 13: 746-7.
4. Alkhuja S, Miller A, Mastellone AJ, et al. Malignant pleural mesothelioma presenting as spontaneous pneumo-thorax: a case series and review. Am J Ind Med 2000; 38(2): 219-23.
5. Antman KH. Natural history and epidemiology of mali-gnant mesothelioma. Chest 1993; 103: 373-6.
6. Henderson DW, Shilkin KB, Whitaker D, et al. Reactive mesothelial hyperplasia vs. mesothelioma, including mesothelioma in situ: a brief review. Am J Clin Pathol 1998; 110: 397-404.
7. Boutin C, Rey F, Gouvernet J, et al. Thoracoscopy in ple-ural malignant mesothelioma: a prospective study of 188 consecutive patients. Cancer 1993; 72: 389-93.
8. Vogelzang NJ, Rusthoven JJ, Symanowski J, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003; 21: 2636-44.
9. Sahn SA, Heffner JE. Spontaneous pneumothorax. N

220 Pao-Shan Wang, Kuo-An Chu, et al.
Thorac Med 2010. Vol. 25 No. 4
Engl J Med 2000; 342: 868-74.10. Upadya A, Amoateng-Adjepong Y, Haddad RG, et al.
Recurrent bilateral spontaneous pneumothorax compli-cating chemotherapy for metastatic sarcoma. South Med J 2003; 96(8): 821-3.
11. Sheard JDH, Taylor W, Soorae A, et al. Pneumothorax and malignant mesothelioma in patient over the age of 40. Thorax 1991; 46: 584-5.
12. Bini A, Zompatori M, Ansaloni L, et al. Bilateral recur-rent pneumothorax complicating chemotherapy for pul-monary metastatic breast ductal carcinoma: report of a case. Surg Today 2000; 30(5): 469-72.
13. Stein ME, Haim N, Drumea K, et al. Spontaneous pneu-mothorax complicating chemotherapy for metastatic semi- noma: a case report and a review of the literature. Cancer 1995; 75(11): 2710-3.

221Pneumothorax Following Chemotherapy for Malignant Pleural Mesothelioma
胸腔醫學:民國99年25卷4期����
惡性肋膜間皮瘤合併瀰漫性肺轉移於化學治療後併發 自發性氣胸一病例報告
王保山 朱國安 林秀玲* 吳銘庭** 賴瑞生
惡性肋膜間皮瘤是原發於肋膜腔間皮細胞的腫瘤,與石棉的曝露有關。惡性肋膜間皮瘤合併對側肺
部轉移在文獻查證上非常罕見。接受化學治療後併發自發性氣胸的病例是少見的,但在多種腫瘤病人曾
被報導過。惡性肋膜間皮瘤的病人接受化學治療後併發自發性氣胸在文獻上尚未被報導。本文描述一名
54歲男性患有右側惡性肋膜間皮瘤病人,接受化學治療七天後出現呼吸困難惡化的症狀。胸部電腦斷層
影像顯示新形成的雙側瀰漫性肺結節及左側多發性肋膜下結節併左側氣胸。根據化學治療的時間關聯性
及胸部電腦斷層影像之證據,診斷為惡性肋膜間皮瘤合併瀰漫性肺部轉移,經化學治療後併發自發性氣
胸。經胸管引流及後續肋膜沾黏治療後氣胸的症狀解除。此時,病人選擇支持性療法而不願再接受化學
治療。病患於三個月後死亡而無復發自發性氣胸。此病例提醒臨床醫師,惡性肋膜間皮瘤合併肺部轉移
的病人接受化學治療後,若發生急性呼吸困難的症狀,應考慮自發性氣胸之可能性。(胸腔醫學 2010; 25: 216-221)
關鍵詞:化學治療,惡性肋膜間皮瘤,自發性氣胸
高雄榮民總醫院內科部 胸腔內科,*高雄榮民總醫院 病理部,**高雄榮民總醫院 放射部
索取抽印本請聯絡:朱國安醫師,高雄榮民總醫院內科部 胸腔內科,高雄市左營區大中一路386號

222
Thorac Med 2010. Vol. 25 No. 4
Aspergillus Tracheobronchitis in an Immunocompetent Patient Successfully Treated with Voriconazole
Jen-Hao Cheng, Jo-Chi Tseng, Jim-Ray Chen*, Yu-Chih Liu
Tracheobronchial aspergillosis is a rare, unique variant of invasive aspergillosis. Aspergillus tracheobronchitis is a type of tracheobronchial aspergillosis. Most reported cases are of immunocompromised patients receiving chemotherapy or immunosuppressive agents, and symptoms may include fever, hemoptysis, cough, chest discomfort, and unilateral wheezing. This entity is not detectable by chest roentgenogram or computed tomography scan, but may be diagnosed by careful bronchoscopy. Treatment options for tracheobronchial aspergillosis include surgical resection of the lesion or antifungal therapy, such as voriconazole. In this report, we described a 73-year-old immunocompetent patient who presented with recurrent, excessive hemoptysis and required intubation and management in the intensive care unit. Bronchoscopy revealed an obstructing yellow plug in the superior segmental lumen of the left lower lobe. Microscopic examination of a biopsy specimen was diagnostic for Aspergillus tracheobronchitis. Voriconazole was administered and resolution of the lesion was followed by serial bronchoscopy monitoring. (Thorac Med 2010; 25: 222-229)
Key words: invasive aspergillosis, Aspergillus tracheobronchitis, voriconazole, bronchoscopy
Department of Thoracic Medicine; *Department of Pathology, Chang Gung Memorial Hospital at Keelung, Chang Gung University College of Medicine, Taoyuan, TaiwanAddress reprint requests to: Dr. Yu-Chih Liu, Department of Thoracic Medicine, Chang Gung Memorial Hospital at Keelung, 222, Mai-jin Rd., Anle District, Keelung, Taiwan
Introduction
Aspergillosis is a large spectrum of diseases caused by members of the genus Aspergillus. The most common forms are allergic broncho-pulmonary aspergillosis, pulmonary aspergillo-ma, and invasive aspergillosis. Colonization of the respiratory tract is also common. Invasive aspergillosis can cause a serious lung infec-tion that can spread to other parts of the body in immunocompromised patients. The mortal-ity rate with invasive aspergillosis is as high as
50-100%. An invasive Aspergillus infection in the lungs often causes cough, fever, chest pain, difficulty breathing, and severe hemoptysis, and in some cases fatal bleeding. Aspergillosis tracheobronchitis is a unique variant of invasive Aspergillus infection that is primarily limited to the tracheobronchial trees. We herein report a patient with Aspergillus tracheobronchitis that presented with persistent severe hemoptysis. Bronchoscopic examination revealed a round, yellow aspergilloma situated in the superior segmental bronchial lumen of the left lower

223Aspergillus Tracheobronchitis in Immunocompetent Patient
胸腔醫學:民國99年25卷4期����
lobe (LB6). The lesion was successfully treated with voriconazole for several weeks, and the entire course of treatment was monitored by re-peated bronchoscopy.
Case Report
A 73-year-old man was admitted due to progressive shortness of breath and moderate hemoptysis in September 2008. He worked as a coal miner and had pulmonary tuberculosis approximately 7 years prior to this admission that was treated with a 6-month course of anti-tuberculous drugs. Sequelae of the tuberculosis were left lung destruction and right upper lobe collapse (Figure 1).
Six months prior to this admission, he had been admitted due to hemoptysis and dysp-nea. At that time, computed tomography (CT) of the chest revealed a cavity at the left upper lobe and destruction of the right upper and left
lower lung (Figures 2A, 2B). Bronchoscopy showed no endobronchial lesion, and the source of bleeding was not identified at that time. Repeated sputum cultures for M. tuberculosis were all negative. After antibiotic treatment, his symptoms gradually improved and he was dis-charged in good condition.
During the current admission, he was ini-tially treated for bronchiectasis with secondary infection. Massive hemoptysis developed on the 5th day of admission, and he was intubated for airway protection and transferred to the in-tensive care unit (ICU). Bronchoscopy revealed narrowed, irregular, and swollen mucosa in the
Fig. 1. Chest roentgenogram showed bilateral upper lung destruction and left upper lobe cavity formation 4 months before this admission.
(A)
(B)
Figs. 2A, 2B. A cavity at the left upper lobe. Right upper lobe and left lower lobe destruction.
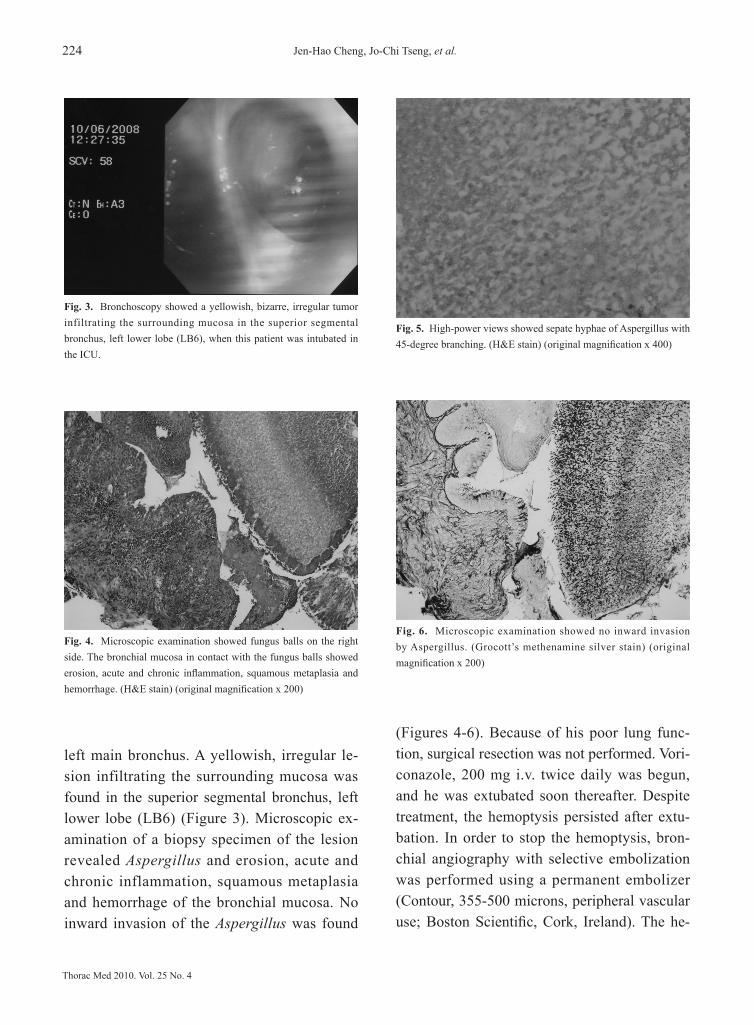
224 Jen-Hao Cheng, Jo-Chi Tseng, et al.
Thorac Med 2010. Vol. 25 No. 4
left main bronchus. A yellowish, irregular le-sion infiltrating the surrounding mucosa was found in the superior segmental bronchus, left lower lobe (LB6) (Figure 3). Microscopic ex-amination of a biopsy specimen of the lesion revealed Aspergillus and erosion, acute and chronic inflammation, squamous metaplasia and hemorrhage of the bronchial mucosa. No inward invasion of the Aspergillus was found
(Figures 4-6). Because of his poor lung func-tion, surgical resection was not performed. Vori-conazole, 200 mg i.v. twice daily was begun, and he was extubated soon thereafter. Despite treatment, the hemoptysis persisted after extu-bation. In order to stop the hemoptysis, bron-chial angiography with selective embolization was performed using a permanent embolizer (Contour, 355-500 microns, peripheral vascular use; Boston Scientific, Cork, Ireland). The he-
Fig. 3. Bronchoscopy showed a yellowish, bizarre, irregular tumor infiltrating the surrounding mucosa in the superior segmental bronchus, left lower lobe (LB6), when this patient was intubated in the ICU.
Fig. 4. Microscopic examination showed fungus balls on the right side. The bronchial mucosa in contact with the fungus balls showed erosion, acute and chronic inflammation, squamous metaplasia and hemorrhage. (H&E stain) (original magnification x 200)
Fig. 5. High-power views showed sepate hyphae of Aspergillus with 45-degree branching. (H&E stain) (original magnification x 400)
Fig. 6. Microscopic examination showed no inward invasion by Aspergillus. (Grocott’s methenamine silver stain) (original magnification x 200)

225Aspergillus Tracheobronchitis in Immunocompetent Patient
胸腔醫學:民國99年25卷4期����
Fig. 9. Six months after voriconazole 400 mg per day had been discontinued, there was a clear lumen.
moptysis ceased immediately after the selective bronchial embolization. Bronchoscopy 1 month after voriconazole therapy was begun showed a yellowish ball with clean mucosa, indicating the aspergillosis was improving (Figure 7). Oral voriconazole, 400 mg per day, was then sub-stituted for intravenous therapy. Two months after voriconazole treatment was begun, bron-choscopy revealed normal LB6 mucosa and the aspergilloma was no longer present (Figure 8). Voriconazole was discontinued, and bronchos-copy 6 months after discontinuing voriconazole revealed no evidence of aspergillosis (Figure 9).
Discussion
Aspergillus is a genus of fungi that is very common in the environment and is found in soil, on plants, and in decaying plant matter. There many types of Aspergillus, the common ones being Aspergillus fumigatus, Aspergillus flavus, Aspergillus terreus, Aspergillus nidu-lans, and Aspergillus niger. The most frequently
isolated species causing invasive aspergillosis is A. fumigatus [1]. Invasive aspergillosis most commonly infects the lungs via inhalation, and can spread throughout the body causing infec-tion in many other organs, including the central nervous and cardiovascular systems [1]. Tra-cheobronchial aspergillosis is a unique variant of invasive aspergillosis and its prevalence is unknown, though it is probably rare because
Fig. 7. Bronchoscopy showed a smaller, round, yellowish ball with clean mucosa 1 month after voriconazole therapy 400 mg per day.
Fig. 8. The yellowish ball was not seen after 2 months of treatment with voriconazole 400 mg per day. There was light reflection interference when taking the picture.

226 Jen-Hao Cheng, Jo-Chi Tseng, et al.
Thorac Med 2010. Vol. 25 No. 4
only sporadic cases have been reported in im-munocompetent patients. A review of our hospi-tal records indicated no cases of endobronchial aspergillosis during the past 20 years. To the best of our knowledge, endobronchial aspergil-losis has not been described by the Pulmonary and Critical Care Society of Taiwan.
Tracheobronchial aspergillosis is classi-fied into ulcerative, pseudomembranous, and obstructing types [1, 3]. The ulcerative type presents a focal, ulcerative or plaque-like pro-cess under bronchoscopic observation, and the pathology shows invasion of the bronchial mu-cosa or cartilage by Aspergillus hyphae [3]. The pseudomembranous type includes extensive Aspergillus infiltrates throughout the lumen that appear as an exudative coating [3]. The defini-tion of obstructing bronchial aspergillosis is Aspergillus plugs blocking the airway with little surrounding tissue invasion or inflammation, and no evidence of allergic reaction [2]. How-ever, the presence of a fourth type, Aspergillus tracheobronchitis, has been suggested in cases in which there is evidence of bronchial and/or tracheal inflammation and excessive mucus production, and in which Aspergillus is the only pathogen without evidence of invasion of the bronchial mucosa on biopsy [2]. The fourth variant differs from the obstructing type due to the presence of tracheobronchial inflammation, though neither exhibits Aspergillus bronchial invasion. In our case, there was evidence of acute and chronic inflammation with squamous cell metaplasia without Aspergillus airway inva-sion. Thus, our case was most likely Aspergillus tracheobronchitis.
Invasive aspergillosis generally affects in-dividuals with compromised immune systems, such as those who have had a bone marrow or solid organ transplant, who are taking high dos-
es of corticosteroids, and who are undergoing chemotherapy. Rarely do individuals with ad-vanced human immunodeficiency virus acquire the infection [1-3]. Chest roentgenogram and CT scan are non-diagnostic for tracheobronchi-al aspergillosis. Sputum exam is also not suf-ficient to reach a diagnosis. A double-sandwich enzyme-linked immunosorbent assay (ELISA) for detecting Aspergillus galactomannan (GM) was prospectively evaluated for the diagnosis of invasive aspergillosis [4]. The rate of false-pos-itive results was around 12%. The GM ELISA appears useful in assessing invasive aspergil-losis and in following the efficacy of antifungal treatment; however, this test was not available in our hospital. Bronchoscopic evaluation is the best method to detect tracheobronchial aspergil-losis, and a precise diagnosis can be made by pathological examination of biopsy specimens [5]. According to the treatment guideline pub-lished by the Infectious Diseases Society of America (IDSA), the treatment strategy is simi-lar to that for invasive aspergillosis. If there is no early diagnosis and immediate treatment, the mortality rate is high [5].
Only 5 cases of obstructing bronchial asper-gillosis in immunocompetent patients have been reported in the Korean and Indian medical literature during the last 10 years [6-10]. He-moptysis was the initial presentation in 4 of the cases, and the other presented with pneumonia. In these cases, imaging studies were not useful in arriving at a diagnosis; bronchoscopy was performed in all patients and a ball-like mass occluding the lumen of the bronchial tree was found. The fungal balls were removed during bronchoscopy in 4 of the cases, and surgical removal was required in 1 patient because the fungal ball was too friable to be removed by bronchoscopy. In all cases, microscopic exami-

227Aspergillus Tracheobronchitis in Immunocompetent Patient
胸腔醫學:民國99年25卷4期����
nation revealed branching septate hyphae of Aspergillus with acute angulations.
In a review of 8 cases of obstructing asper-gillosis from 1993 to 2005 [11], the most com-mon symptom was hemoptysis (100%), fol-lowed by cough (63%), excessive sputum (25%), and chest discomfort (25%). Recurrent and moderate to large hemoptysis was the unique symptom that was found in this review, and it was both common and serious. Pulmonary tuberculosis (75%), bronchiectasis (25%), and small cell lung cancer (13%) were the common underlying diseases. Five patients received sur-gery and their outcomes were good. A patient who refused surgery experienced persistent, in-termittent hemoptysis. Two patients died of the disease; 1 patient received an organ transplant and the other was complicated by acute pneu-monia, diabetes mellitus, and end-stage renal disease. The conclusion of the author was that surgery may be the first choice of treatment in immunocompetent patients.
As in the cases described in the literature review, our case of obstructing bronchial asper-gillosis had developed in an immunocompetent patient. The patient had pulmonary tuberculo-sis 7 years prior with resulting bronchiectasis. He presented with repeated, large to massive episodes of hemoptysis, and was intubated and admitted to the ICU. Imaging studies were non-diagnostic, and the diagnosis of tracheobron-chial aspergillosis required bronchoscopy. Like other reported cases, we found a yellowish ball-like lesion stuck in the left lower lobe bronchial tree with surrounding tissue reaction during bronchoscopy. We could not remove it because it was too friable. Microscopic examination of a biopsy specimen revealed Aspergillus without mucosal invasion. Surgical intervention was not considered because of the patient’s poor lung
function. Bronchial angiography was performed with successful selective embolization using a permanent embolizer. Voriconazole, 400 mg per day, was administered based on the guide-lines of the IDSA. Follow-up bronchoscopic examinations were performed until the lesion had completely resolved. Removing the ob-structing bronchial Aspergillus lesion may be a better choice because long-term medication is not needed in a patient who is immunocompe-tent; however, removal was not possible in our patient. Our case did not receive surgical resec-tion, and his outcome was better compared with other reported cases in which the lesion was not resectable. This may be due to management with a permanent embolizer and voriconazole antifungal therapy, which were not mentioned in the other cases.
In conclusion, tracheobronchial aspergillo-sis is not commonly seen in immunocompetent patients and the obstructing bronchial type is rare. Bronchoscopy combined with pathological examination of biopsy specimens is likely the best diagnostic tool. Surgical resection or bron-choscopic removal of the lesion is appropriate management of obstructing tracheobronchial aspergillosis. Long-term voriconazole therapy with bronchoscopy monitoring is an option for patients in whom surgical resection or broncho-scopic removal of the lesion is not possible.
References
1. Walsh TJ, Anaissie EJ, Denning DW, et al. Treatment of aspergillosis: clinical practice guidelines of the Infectious Disease Society of America. Clin Infect Dis 2008; 46: 327-60.
2. Denning DW. Commentary: unusual manifestations of aspergillosis. Thorax 1995; 50: 812-3.
3. Karnak D, Avery RK, Gildea TR, et al. Endobronchial fungal disease: an under-recognized entity. Respiration

228 Jen-Hao Cheng, Jo-Chi Tseng, et al.
Thorac Med 2010. Vol. 25 No. 4
2007; 74: 88-104. 4. S. Bretagne, A. Marmorat-Khuong, M. Kuentz, et al.
Serum Aspergillus galactomannan antigen testing by sandwich ELISA: Practical use in neutropenic patients. J of Infection 1997 Jul; 35(1): 7-15.
5. S. Tasci , A. Glasmacher, S. Lentini, et al. Pseudomem-branous and obstructive Aspergillus tracheobronchitis - optimal diagnostic strategy and outcome, Mycoses 2006 Jan; 49(1): 37-42.
6. Kim JS, Rhee Y, Kang SM, et al. A case of endobronchial aspergilloma. Yonsei Med J 2000 Jun; 41(3): 422-5.
7. Dar KA, Shah NN, Bhargava R, et al. Endobronchial aspergilloma in a 30-year-old man. Journal of Broncho-logy 2007 July; 14(3): 207-9.
8. Shammeem M, Bhargava R, Ahmad Z, et al. Endobron-chial aspergilloma- presenting as solitary pulmonary nodule. Respiratory Medicine CME (2009, article in press)
9. Eom WY, Kim NI, Kim SW, et al. A case of endobron-chial aspergilloma in patient with collapse of right middle lobe. Korean J Med 2006 Feb; 70(2): 221-5.
10. Kim SJ, Lee EJ, Lee TH, et al. A case of endobronchial aspergilloma. Tuberc Respir Dis 2006 Jul; 61(1): 60-4.
11. Kim D, Chang Y, Kim H, et al. Endobronchial asper-gilloma: A retrospective case review. Chest 2005 Oct; 128(4): 374s.

229Aspergillus Tracheobronchitis in Immunocompetent Patient
胸腔醫學:民國99年25卷4期����
免疫正常的人在麴菌氣管支氣管炎時成功的 使用抗黴菌治療
鄭人豪 曾若琦 陳俊叡* 劉育志
氣管支氣管麴菌感染是一種侵入性麴菌感染的特殊形態而且發生率可能是少見的,麴菌氣管支氣管
炎則又是氣管支氣管麴菌感染中的一種形態,大多數的病人都是免疫不全的病人,像是接受化學治療或
免疫抑制劑的病人並且症狀可能有發燒、咳血、咳嗽、胸部不適、甚至單邊哮喘。這種疾病無法使用胸
部放射檢查或是電腦斷層掃瞄而得知,但是可輕易的由支氣管鏡小心檢查而診斷。麴菌氣管支氣管炎的
治療方式包含了手術切除病灶處或是抗黴菌藥物,像是黴飛(voriconazole)。在此我們報告一個73歲免
疫正常的病人因反覆大量咳血,而插管住入加護病房,支氣管檢查發現有一個黃色的異物塞住左下肺的
上分枝腔內,經切片檢查而診斷為麴菌氣管支氣管炎;我們使用黴飛做為治療併用支氣管鏡追蹤直到完
全消失。(胸腔醫學 2010; 25: 222-229)
關鍵詞:侵入性麴菌症,麴菌氣管支氣管炎,黴飛,支氣管鏡
基隆長庚紀念醫院內科部 胸腔內科,基隆長庚紀念醫院 病理科*,長庚大學醫學院
索取抽印本請聯絡:劉育志醫師,基隆長庚紀念醫院內科部 胸腔內科,基隆市麥金路222號




















