







Bahasa
Halaman
Hukum


BỘ GIÁO DỤC VÀ ĐÀO TẠO VIỆN HÀN LÂM KHOA HỌC
VÀ CÔNG NGHỆ VIỆT NAM
HỌC VIỆN KHOA HỌC VÀ CÔNG NGHỆ
-----------------------------
TỔNG HỢP VẬT LIỆU COMPOSITE
LiFe1-xMxPO4/GRAPHENE LÀM CATHODE ĐỂ CẢI
THIỆN TÍNH NĂNG ĐIỆN HÓA PIN SẠC
LITHIUM-ION
LUẬN ÁN TIẾN SĨ
TP. HCM – 2019

VIỆN HÀN LÂM KHOA HỌC VÀ CÔNG NGHỆ VIỆT NAM
HỌC VIỆN KHOA HỌC VÀ CÔNG NGHỆ
……..….***…………
TỔNG HỢP VẬT LIỆU COMPOSITE
LiFe1-xMxPO4/GRAPHENE LÀM CATHODE ĐỂ CẢI
THIỆN TÍNH NĂNG ĐIỆN HÓA PIN SẠC
LITHIUM-ION
LUẬN ÁN TIẾN SĨ
Ma ngành: 9440119
Mã NCS: 16903037026
Khóa học: 2016-2019
Người hướng dẫn khoa học:
PGS.TS Nguyễn Nhị Trự
PGS.TS Lê Mỹ Loan Phụng
TP. HCM-2019

i
LỜI CAM ĐOAN
Tôi xin cam đoan luận án này là công trình nghiên cứu của riêng tôi
và không trùng lặp với các công trình khoa học khác đã công bố.
Số liệu kết quả nghiên cứu trong luận án hoàn toàn trung thực, một số
kết quả là kết quả chung của nhóm nghiên cứu trong đề tài C2015-20-
25 Đại học Quốc gia Tp.HCM.
Tôi xin cam đoan rằng các thông tin trích dẫn trong luận án này có
nguồn gốc rõ ràng.
TP. HCM, ngày tháng năm 2019
Tác giả luận án

ii
LỜI CẢM ƠN
Tôi xin bày tỏ lòng biết ơn sâu sắc và đầy kính trọng đến thầy, cô hướng dẫn
đã dẫn dắt tôi từ ngày đầu nghiên cứu đầy bỡ ngỡ và nhiều thiếu sót, thầy cô hướng
dẫn tận tâm cho tôi suốt cả một chặng đường dài trong suốt quá trình nghiên cứu đề
tài luận án. Thầy, cô luôn tạo mọi cơ hội và điều kiện tốt nhất để tôi có thể thực hiện
và hoàn thành các thí nghiệm trong điều kiện thiết bị máy móc ở Việt Nam còn
nhiều khó khăn.
Tôi xin trân trọng cám ơn Ban lãnh đạo cơ sở đào tạo: Viện Khoa học Vật
liệu ứng dụng, Học viện Khoa học và Công nghệ Việt Nam-Viện Hàn lâm Khoa
học và Công nghệ Việt Nam đã tạo điều kiện cho tôi hoàn thành khóa học và bảo vệ
luận án.
Tôi xin thành thật biết ơn tới lãnh đạo trường Đại học Sư Phạm Kỹ Thuật
Vĩnh Long, Khoa Khoa học cơ bản, phòng Tổ chức-Hành chính…đã quan tâm giúp
đỡ và hỗ trợ tối đa để tôi hoàn thành khóa học.
Tôi gửi lời cám ơn sâu sắc đến chủ nhiệm đề tài C2015-20-25,
107/2016/HĐ-SKHCN và C2018-18-11 đã hỗ trợ một phần hóa chất và thiết bị
trong quá trình thực hiện luận án.
Lời cám ơn tiếp theo tôi xin cám ơn Khoa Công nghệ Vật liệu trường
ĐHBK-ĐHQG Tp.HCM, Phòng thí nghiệm Hóa lý ứng dụng trường Đại học
KHTN-ĐHQG Tp.HCM, Viện Công nghệ Nano là những nơi tôi thực hiện đề tài
luận án.
Cuối cùng, tôi bày tỏ lòng biết ơn sâu sắc tới đồng nghiệp, bạn bè và gia đình
những người luôn chia sẻ và động viên tôi vượt qua mọi thử thách, tiếp thêm sức
mạnh nghị lực để hoàn thành luận án.

iii
DANH MỤC CÁC CHỮ VIẾT TẮT
AA Ascorbic acid
AAS Atomic Absorption Spectrophotometry: phổ hấp phụ nguyên tử
AC Citric acid
BG Benzyl alcohol
CVs Cyclic Voltammetry: quét thế vòng tuần hoàn
CNTs Carbon Nanotubes: carbon dạng ống nano
CTAB Cetyltrimethylammonium bromide
DMC Dimethylene carbonate
EDS Energy-dispersive X-ray Spectroscopy: quang phổ X-quang tán xạ
năng lượng
EIS Electrochemical Impedance Spectroscopy: phổ tổng trở điện hóa
EG Ethylene glycol
EC Ethylene carbonate
FESEM Field-Emission Scanning Electron Microscopy: hiển vi điện tử quét
trường phát xạ
Gr Graphene
HRTEM High-Resolution Transmission Electron Microscopy: hiển vi điện tử
truyền qua độ phân giải cao
Li-ion Lithium-ion
LIBs Li-ion batteries: pin Li-ion
LFP LiFePO4
LMO Lithium manganese oxide
LFNP LiFeNiPO4
MO Molecular orbital: vân đạo phân tử
rGO Reduction of graphene oxide: graphene được khử từ graphene oxide
SEM Scanning Electron Microscopy: hiển vi điện tử quét
TEM Transmission Electron Microscopy: hiển vi điện tử truyền qua
TGA Thermogravimetric analysis: phân tích nhiệt trọng lượng
VB Valence bond: liên kết cộng hóa trị
XRD X-ray Diffraction: nhiễu xạ tia X
XPS X - ray Photoelectron Spectroscopy: phổ quang điện tử tia X

iv
MỤC LỤC
LỜI CẢM ƠN ........................................................................................................ ii
DANH MỤC CÁC CHỮ VIẾT TẮT .................................................................... iii
DANH MỤC CÁC HÌNH VẼ.............................................................................. vii
DANH MỤC CÁC BẢNG ................................................................................... xi
MỞ ĐẦU .............................................................................................................. 01
CHƯƠNG 1: TỔNG QUAN .............................................................................. 08
1.1. Sơ lược về pin sạc Li-ion và vật liệu điện cực olivine ............................. 08
1.2. Cơ chế hoạt động và cấu tạo của pin sạc Li-ion ....................................... 09
1.2.1. Cơ chế hoạt động pin Li-ion ...................................................................... 09
1.2.2. Các thông số tính năng của pin .................................................................. 10
1.2.3. Cấu tạo pin Li-ion ..................................................................................... 11
1.2.3.1. Vật liệu cathode ...................................................................................... 12
1.2.3.2. Vật liệu anode ........................................................................................ 14
1.2.3.3. Chất điện giải .......................................................................................... 15
1.2.3.4. Màng ngăn ............................................................................................... 15
1.3. Cấu trúc vật liệu cathode ............................................................................ 16
1.3.1. Cấu trúc dạng lớp (layers) .......................................................................... 16
1.3.2. Cấu trúc dạng spinel .................................................................................. 17
1.3.3. Nhóm hợp chất polyanion cấu trúc olivine ................................................ 18
1.4. Nghiên cứu cải thiện tính năng điện hóa của vật liệu LFP ..................... 21
1.4.1. Giảm kích thước hạt ................................................................................... 22
1.4.2. Pha tạp kim loại .......................................................................................... 25
1.4.3. Phủ carbon .................................................................................................. 28
1.5. Các phương pháp tổng hợp pin điện cực sạc Li-ion ................................ 35
1.5.1. Phương pháp keo hóa (sol-gel) .................................................................. 37
1.5.2. Phương pháp vi sóng (microwave) ............................................................ 37
1.5.3. Phương pháp nghiền bi năng lượng (mechanochemical method).............. 38
1.5.4. Phương pháp pha rắn (solid state) .............................................................. 38
1.5.5. Phương pháp phun nhiệt (spray pyrolysis) ................................................ 38
1.5.6. Phương pháp đồng kết tủa (co-precipitation) ............................................. 39
1.5.7. Phương pháp thủy nhiệt (hydrothermal) .................................................... 39

v
1.5.8. Phương pháp nhiệt dung môi (solvothermal) ............................................. 40
CHƯƠNG 2: THỰC NGHIỆM ........................................................................ 41
2.1. Tổng hợp vật liệu điện cực ......................................................................... 41
2.1.1. Hóa chất, thiết bị ........................................................................................ 41
2.1.2. Tổng hợp LFP ............................................................................................ 43
2.1.3. Pha tạp kim loại M (Ni, Mn, Y) LiFe1-xMxPO4 ......................................... 45
2.1.4. Composite hóa LiFe1-xMxPO4/graphene .................................................... 47
2.1.5. Tạo màng điện cực và quy trình lắp pin mô hình Swagelok ..................... 48
2.2. Chuẩn độ xác định hàm lượng Fe2+ và carbon trong mẫu ...................... 49
2.2.1. Xác định hàm lượng Fe2+ bằng phương pháp chuẩn độ ............................. 49
2.2.2. Xác định hàm lượng carbon bằng phương pháp hóa học & nhiệt ............. 50
2.3. Phương pháp nghiên cứu và đánh giá tính chất vật liệu ......................... 50
2.3.1. Nhiễu xạ tia X (X-ray Diffraction) ............................................................ 50
2.3.2. Quang phổ Raman ...................................................................................... 52
2.3.3. Phương pháp phân tích nhiệt (TGA) ......................................................... 53
2.3.4. Phổ quang điện tử (XPS) ............................................................................ 54
2.3.5. Phổ hấp thụ nguyên tử (AAS) .................................................................... 55
2.3.6. Phổ tán sắc năng lượng tia X (EDS) .......................................................... 56
2.3.7. Hiển vi điện tử quét (SEM, FESEM) ......................................................... 57
2.3.8. Hiển vi điện tử truyền qua TEM ................................................................ 57
2.3.9. Phương pháp điện hóa. ............................................................................... 58
2.3.9.1. Quét thế vòng tuần hoàn (CV) ................................................................ 59
2.3.9.2. Phóng sạc dòng cố định .......................................................................... 60
2.3.9.3. Phổ tổng trở (EIS) ................................................................................... 61
CHƯƠNG 3: KẾT QUẢ VÀ THẢO LUẬN .................................................... 64
3.1. Vật liệu điện cực LFP……………………………………………………..64
3.1.1. Xác định hàm lượng Fe và Fe2+ có trong mẫu LFP………………………64
3.1.2. Cấu trúc tinh thể và thành phần pha ........................................................... 66
3.1.2.1. Ảnh hưởng tỉ lệ tiền chất đến cấu trúc LFP ............................................ 66
3.1.2.2. Ảnh hưởng dung môi đến cấu trúc LFP .................................................. 68
3.1.2.3. Ảnh hưởng nhiệt độ nung đến kết tinh của vật liệu ................................ 70
3.1.2.4. Hiệu suất phản ứng (H), khối lượng kết tinh (T) của mẫu ...................... 70

vi
3.1.3. Thành phần nguyên tố hóa học vật liệu ..................................................... 76
3.1.4. Hình thái vật liệu ....................................................................................... 80
3.1.5. Tính chất điện hóa ...................................................................................... 81
3.1.5.1. Đánh giá khả năng đan cài ion Li+ .......................................................... 82
3.1.5.2. Tính năng phóng sạc của vật liệu ............................................................ 84
3.1.5.3.Độ dẫn điện vật liệu ................................................................................. 85
3.2. Pha tạp kim loại LiFe1-xMxPO4 .................................................................. 88
3.2.1. Cấu trúc tinh thể và thành phần pha ........................................................... 88
3.2.2. Thành phần nguyên tố hóa học ................................................................. 95
3.2.3 Hình thái hạt. ............................................................................................... 98
3.2.4. Tính chất điện hóa ...................................................................................... 103
3.3. Vật liệu composite LiFe1-xMxPO4/Graphene ............................................ 109
3.3.1. Cấu trúc tinh thể và thành phần pha ........................................................... 106
3.3.2. Thành phần hóa học và xác định sự có mặt graphene ............................... 114
3.3.3. Khả năng bao phủ và độ phân tán mẫu ...................................................... 115
3.3.4. Tính chất điện hóa ...................................................................................... 119
KẾT LUẬN VÀ KIẾN NGHỊ ........................................................................... 125
NHỮNG ĐÓNG GÓP MỚI CỦA ĐỀ TÀI ...................................................... 127
CÔNG TRÌNH ĐÃ CÔNG BỐ ......................................................................... 128
TÀI LIỆU THAM KHẢO ................................................................................. 129
PHỤ LỤC ............................................................................................................ 142

vii
DANH MỤC CÁC HÌNH VẼ
Hình 1.1 Các lĩnh vực ứng dụng của pin sạc Li-ion [Tr.01]
Hình 1.2 Cơ chế hoạt động của pin Li-ion (a) [Tr.08]
Cơ chế đan cài và phóng thích ion Li+ (b) [Tr.08]
Hình 1.3 Cấu tạo của pin Li-ion [Tr.10]
Hình 1.4 Sự ảnh hưởng vật liệu đến điện thế oxi hóa-khử [Tr.12]
Hình 1.5 Cấu trúc lớp của vật liệu LiMO2 [Tr.15]
Hình 1.6 Cấu trúc spinel của LiM2O4 [Tr.16]
Hình 1.7 Cấu trúc tinh thể olivine LixMy(XO4)z [Tr.16]
Hình 1.8 Cấu trúc olivine LiFePO4 [Tr.17]
Hình 1.9 So sánh đường cong phóng/nạp của các vật liệu LMPO4 [Tr.19]
Hình 1.10 Phân bố điện tử theo thuyết trường tinh thể trên vân đạo 3d [Tr.19]
Hình 1.11 Tinh thể vật liệu LFP: xốp (a), tấm (b), que (c) [Tr.21]
Hình 1.12 Kích thước hạt ảnh hưởng đến biên độ oxi hóa-khử [Tr.22]
Hình 1.13 Biên độ oxi hóa - khử phụ thuộc vào kích thước hạt [Tr.23]
Hình 1.14 Ảnh hưởng của cấu trúc nano đến nồng độ ion Li+ [Tr.23]
Hình 1.15 Đường CV thể hiện vùng peak thế oxi khử trong LiMnFePO4 [Tr.23]
Hình 1.16 So sánh thế oxi hóa khử trước và sau khi pha tạp [Tr.26]
Hình 1.17 Quá trình hình thành carbon in-situ [Tr.26]
Hình 1.18 Vai trò của carbon trong quá trình phản ứng [Tr.27]
Hình 1.19 Một số dạng cấu trúc carbon [Tr.28]
Hình 1.20 Cấu trúc dạng tổ ong của graphene (2D) [Tr.31]
Hình 1.21 Các loại liên kết trong cấu trúc graphene [Tr.31]
Hình 1.22 Vai trò carbon in-situ và ex-situ [Tr.32]
Hình 1.23 So sánh dung lượng xả LFP/graphene và LFP/carbon black [Tr.33]
Hình 2.1 Sơ đồ quy trình tổng hợp LFP bằng phương pháp NDM [Tr.42]
Hình 2.2 Quy trình pha tạp các nguyên tố M (Ni, Mn, Y) vào mẫu LFP [Tr.45]
Hình 2.3 Sơ đồ quy trình tổng hợp vật liệu composite LiFe1-xMxPO4/Gr [Tr.47]
Hình 2.4 Mô tả quy trình tổng hợp vật liệu composite [Tr.48]
Hình 2.5 Dung dịch chứa carbon sau khi phá mẫu [Tr.49]
Hình 2.6 Giản đồ phổ Raman của LFP [Tr.54]
Hình 2.7 Thế oxi hóa-khử vật liệu bằng phương pháp CV [Tr.60]

viii
Hình 2.8 Vectơ Fresnel trong mặt phẳng M [Tr.62]
Hình 2.9 Mạch điện tương đương của chất điện môi [Tr.63]
Hình 2.10 Diễn biến quá trình động học phản ứng dựa trên đồ thị tổng trở [Tr.64]
Hình 2.11 Mô hình chuẩn Swagelok [Tr.65]
Hình 3.1 Giản đồ XRD của LFP so sánh với phổ chuẩn thương mại (a) [Tr.67]
Xác định các mặt nhiễu xạ bằng phần mềm Match!2 (b) [Tr.67]
Kiểm chứng cấu trúc pha LFP bằng chức năng Retveld (c) [Tr.68]
Hình 3.2 Giản đồ XRD của LFP với tỉ lệ tiền chất khác nhau [Tr.70]
Hình 3.3 Giản đồ XRD của các mẫu ST01 và ST00 (không sử dụng [Tr.70]
ascorbic acid với tốc độ quét 5o /phút góc quét từ 0<2 <90o [Tr.71]
Hình 3.4 Giản đồ phân tích nhiệt biểu thị biến thiên giữa nhiệt độ % TG [Tr.72]
Hình 3.5 Giản đồ XRD so sánh ảnh hưởng nhiệt độ nung pha vật liệu [Tr.72]
Hình 3.6 Phổ Raman của vật liệu LFP trong khoảng tần số thấp [Tr.75]
Hình 3.7 Phổ Raman của các mẫu ST01, ST02, ST03 [Tr.75]
Hình 3.8 Giản đồ EDS xác định các liên kết trong mẫu ST01 [Tr.77]
Hình 3.9 Giản đồ năng lượng XPS của ST01 sau khi nung 550oC [Tr.78]
Hình 3.10 Xác định sự có mặt của Fe2+ bằng phổ XPS [Tr.78]
Hình 3.11 SEM của LFP trước khi nung [Tr.79]
Hình 3.12 SEM của LFP ở các nhiệt độ khác nhau [Tr.80]
Hình 3.13 FESEM của LFP (ST01) sau khi nung 550 oC [Tr.80]
Hình 3.14 Biểu đồ so sánh kích thước hạt LFP trước, sau khi nung [Tr.82]
Hình 3.15 Đường cong CV của LFP (ST01) với tốc độ khác nhau (a) [Tr.83]
So sánh CV mẫu ST01 với ST00 tại 80µV/s (b) [Tr.83]
Biên độ peak oxi hóa khử của LFP với tốc độ 10 μV/s (c) [Tr.84]
Giản đồ hệ số khuếch tán của LFP các mẫu ST01 và ST00 (d) [Tr.85]
Hình 3.16 Đường cong phóng/ sạc sau 20 chu kì của LFP mẫu ST00 (a)
Đường cong phóng/ sạc sau 20 chu kì của LFP mẫu ST01 [Tr.85]
Hình 3.17 Phổ trở kháng và mạch tương đương LFP [Tr.87]
Hình 3.18 Phân tích tạp mẫu STN2 (a); STM2 (b); STY2 (b) bằng
phần mềm Match!2 [Tr.90]
Hình 3.19 Khảo sát sự dịch chyển pha khi pha tạp Ni (a), Y (b), Mn (c) [Tr.91]
Hình 3.20 Giản đồ phân tích nhiệt các mẫu ST01, STM2, STN2 (a) [Tr.92]

ix
Giản đồ Heat Flow của các mẫu STM2, STN2 (b) [Tr.93]
Hình 3.21 Đồ thị phổ Raman của vật liệu LiFe1-xMxPO4 (a) [Tr.94]
Phổ Raman sự dịch chuyển dao động sau khi pha tạp (b) [Tr.95]
Hình 3.22 Giãn đồ XRD phân tích thành phần mẫu STM2 [Tr.96]
Hình 3.23 Sự phân bố của LiFe1-xYxPO4 (STY2) bởi EDS-M (a) [Tr.97]
Giãn đồ XRD phân tích thành phần mẫu STM2 (b) [Tr.98]
Hình 3.24 Giản đồ XPS của (STN2) (a); STM3 (b) [Tr.99]
Hình 3.25 SEM của mẫu LiFe1-xNixPO4 (STN2) [Tr.101]
Hình 3.26 FESEM của mẫu LiFe1-xNixPO4 (STN2) [Tr.101]
Hình 3.27 FESEM của mẫu LiFe1-xYxPO4 (STY2) [Tr.102]
Hình 3.28 FESEM của mẫu LiFe1-xMnxPO4 (STM2) [Tr.102]
Hình 3.29 TEM của mẫu LiFe1-xMnxPO4 (STM2) [Tr.102]
Hình 3.30 TEM của mẫu LiFe1-xYxPO4 (STY2) [Tr.102]
Hình 3.31 TEM của mẫu STY2, xác định kích thước hạt thủ công [Tr.103]
Hình 3.32 CV của LiFe1-xNixPO4 (STN2) (a); LiFe1-xMnxPO4 (STM2)
ở nhiều tốc độ khác nhau [Tr.103]
Hình 3.33 Peak oxi hóa khử của kim loại pha tạp [Tr.105]
Hình 3.34 Đường cong phóng sạc LiFe1-xMnxPO4 (STM2) (a),
LiFe1-xYxPO4 (STY2) (b), LiFe1-xNixPO4 (STN2) (c)
và hiệu suất phóng sạc LiFe1-xMxPO4 trong 20 chu kì (d) [Tr.106]
Hình 3.35 Phổ tổng trở của LiFe1-xMxPO4 (M: Mn, Ni, Y) (a) [Tr.107]
Hệ số Warburg thông qua tổng trở Warburg mẫu STN2 [Tr.108]
Hình 3.36 Giản đồ XRD so sánh pha phủ 5% và 10% grpaphene (a) [Tr.110]
Giản đồ XRD phân tích thành phần pha STM2-G1 [Tr.110]
Giản đồ XRD xác định cấu trúc olivine của LiFe1-xMxPO4/Gr
bằng chức năng Rietveld Refinement (c)
Hình 3.37 Định tính sự có mặt graphene bằng phổ Raman (a) [Tr.110]
Định tính vùng dao động có mặt kim loại pha tạp (b) [Tr.113]
Kiểm chứng vùng dao động của graphene thương mại (c) [Tr.114]
Hình 3.38 Định lượng thành phần các nguyên tố trong mẫu sau khi STN2-G1,
STY2-G1, STM2-G1 bằng EDS [Tr.115]
Hình 3.39 TEM của LiFe1-xNixPO4/Gr (STN2-G1) (a) [Tr.116]

x
TEM của LiFe1-xYx PO4/Gr (STY2-G1) (b) [Tr.116]
TEM của LiFe1-xMnx PO4/Gr (STM2-G1) (c) [Tr.116]
Hình 3.40 Ảnh SEM-EDS-Mapping thể hiện sự phân bố, và thành phần
các nguyên tố trong vật liệu LiFe1-x MxPO4 (STM3-G2) [Tr.118]
Hình 3.41 Ảnh HRTEM xác định khả năng bao phủ mảng graphene [Tr.118]
Hình 3.42 Đường cong CV của LiFe0.9Ni0.05PO4/5% Gr (STN2-G2) (a) [Tr.119]
LiFe0.8Mn0.2PO4/5% Gr (STM2-G2) (c) [Tr.119]
Hình 3.43 Đường phóng sạc của LiFe0.9Ni0.05PO4/10% Gr STN2-G1(a) [Tr.120]
LiFe0.98 Y0.02PO4/10% Gr (STY2-G1)(b) [Tr.120]
LiFe0.8Mn0.2PO4/10% Gr STM2-G1 (c) [Tr.120]
Hình 3.44 Đường phóng sạc của LiFe0.9Ni0.05PO4/5% Gr STN2-G2) (a) [Tr.120]
LiFe0.98 Y0.02PO4/5% Gr (STY2-G2) (b) [Tr.120]
LiFe0.8Mn0.2PO4/5% Gr (STM2-G2) (c) [Tr.122]
Hình 3.45 Dung lượng sạc LiFe1-xMnx PO4/Gr biến thiên sau 150 chu kì [Tr.122]
Hình 3.46 Đường cong phóng/ sạc và dung lượng theo chu kì với
nhiều tốc độ phóng/ sạc mẫu LiFe0.8Mn0.2PO4/5%Gr. [Tr.123]
Hình 3.47 So sánh phổ tổng trở (EIS) trước và khi phủ graphene [Tr.124]

xi
DANH MỤC CÁC BẢNG
Bảng 1.1 Các lĩnh vực ứng dụng của pin sạc Li-ion [Tr.11]
Bảng 1.2 Độ dẫn điện, hệ số khuếch tán và mật độ một số vật liệu [Tr.18]
Bảng 1.3 Thông số mạng và thể tích ô mạng LiFePO4 và FePO4 [Tr.21]
Bảng 1.4 Khảo sát ảnh hưởng pha tạp đến hiệu suất điện hóa [Tr.24]
Bảng 1.5 So sánh hằng số và thể tích mạng trước, sau khi pha tạp [Tr.25]
Bảng 2.1 Ảnh hưởng phương pháp tổng hợp đến kích thước và hiệu
suất phóng sạc pin Li-ion [Tr.34]
Bảng 2.2 Hóa chất và một số thông số hóa lý [Tr.41]
Bảng 2.3 Khối lượng và thể tích của các tiền chất tổng hợp LFP [Tr.42]
Bảng 2.4 Tên thiết bị và thông số kỹ thuật [Tr.45]
Bảng 2.5 Khối lượng tiền chất cần tổng hợp LFP, pha tạp kim loại M [Tr.46]
Bảng 2.6 Khối lượng cần để tổng hợp LiFe1-xMxPO4/Gr [Tr.47]
Bảng 3.1a Hàm lượng % Fe2+ trong LFP được xác định XPS [Tr.47]
Bảng 3.1b Định lượng Fe2+ trong mẫu bằng phương pháp chuẩn độ [Tr.66]
Bảng 3.2 Định lượng Fe trong mẫu bằng phương pháp AAS [Tr.67]
Bảng 3.3 Thông số mạng và thể tích ô mạng của LFP [Tr.68]
Bảng 3.4 Sự biến thiến khối lượng bằng phương pháp phân tích nhiệt [Tr.71]
Bảng 3.5 Hiệu suất phản ứng và % hàm lượng tinh khiết pha tạp [Tr.73]
Bảng 3.6 Độ dẫn ion của LFP dựa trên phổ EIS [Tr.86]
Bảng 3.7a Một số thống số hóa lí của các kim loại pha tạp [Tr.92]
Bảng 3.7b So sánh thông số và thể tích mạng trước và sau khi pha tạp [Tr.92]
Bảng 3.8 Định lượng hệ số khuếch tán (phương pháp CV)
và độ dẫn điện (phổ EIS) [Tr.97]
Bảng 3.11 So sánh độ dẫn điện của vật liệu composite LiFe1-xMxPO4/Gr với LFP
.

1
MỞ ĐẦU
1. Tính cấp thiết của đề tài
Hiện nay nguồn năng lượng hóa thạch hay còn gọi năng lượng không tái tạo
được dần trở nên cạn kiệt dẫn đến mất cân bằng sinh thái và ô nhiễm môi trường,
gây ra mối lo ngại lớn cho sự thay đổi khí hậu trái đất. Điều này đã đẩy nhanh sự
phát triển của năng lượng tái tạo như: mặt trời, gió, thủy triều... để giảm thiểu vấn
đề khai thác các nguồn năng lượng hóa thạch và đảm bảo tính thân thiện môi
trường. Tuy nhiên, các dạng năng lượng này cung cấp không liên tục và phụ thuộc
vào điều kiện thời tiết, đã gây cản trở cho việc tận dụng khai thác sử dụng trong
thực tế.
Do đó, sự phát triển các thiết bị lưu trữ năng lượng, tái tạo năng lượng,
chẳng hạn như: pin nhiên liệu, pin sạc, hệ thống đèn LED, sinh khối (biomass)...
đã trở nên có vai trò đặc biệt quan trọng trong chiến lược phát triển năng lượng.
Công nghệ pin sạc lithium-ion (Li-ion) ra đời đã mang lại những thành tựu
lớn cho lĩnh vực khoa học ứng dụng góp phần giảm thiểu ô nhiễm môi trường và
cạn kiệt tài nguyên thiên nhiên. Pin sạc đang chiếm lĩnh thị trường thiết bị cầm tay
vì có lợi thế: sức điện động pin lớn, mật độ năng lượng cao, tuổi thọ bền, nhẹ,
cũng như tính linh hoạt trong thiết kế. Từ các ưu điểm nổi bật trên thì pin sạc Li-
ion đã thay thế pin dự phòng truyền thống (Ni-Cd, Ni-NH…).
Hiện nay pin Li-ion chủ yếu ứng dụng cho ba lĩnh vực chính: thiết bị điện
tử cầm tay, xe điện và động cơ điện lai (Hình 1.1) với tổng giá trị đạt 22,5 tỉ USD
vào năm 2016 và hướng tới sẽ sử dụng cho trạm phát điện có công suất lớn đạt
77,5 tỉ USD năm 2024.
Hình 1.1. Các lĩnh vực ứng dụng của pin sạc Li-ion [1]

2
Đến nay, vật liệu làm điện cực cathode cho pin sạc Li-ion được nghiên cứu
chủ yếu phân loại theo ba nhóm cấu trúc chính:
- Nhóm thứ nhất là nhóm cấu trúc lớp (layers), tiêu biểu là vật liệu LiCoO2
(có dung lượng lý thuyết 248 mAh.g-1) được hãng Sony Energitech
thương mại hóa năm 1992 với nhiều ưu điểm so với các thiết bị lưu trữ
khác, nhưng vẫn chưa đáp ứng đa dạng ứng dụng do hạn chế về công
suất chưa đủ lớn, tính ổn định và mức độ an toàn cũng như giá thành
cao.
- Nhóm thứ hai là vật liệu cấu trúc spinel (lập phương, tám mặt) với dạng
công thức tổng quát AB2O4, cụ thể như vật liệu LiMn2O4 có dung lượng
lý thuyết tương đối thấp 120 mAh.g-1, kém bền với nhiệt và dễ hòa tan
trong môi trường dung dịch điện giải ở nhiệt độ cao mặc dù ưu điểm lớn
của vật liệu này là giá thành rẻ, thân thiện với môi trường và vùng thế
hoạt động tương đối lớn.
- Nhóm thứ ba được đánh giá là vật liệu làm điện cực tối ưu nhất có cấu
trúc bền olivine thuộc hệ trực thoi (nhóm không gian Pbnm), công thức
tổng quát LiMz(PO4)z trong đó M chủ yếu là các kim loại chuyển tiếp.
Trong hai thập kỷ gần đây, nhóm vật liệu này đang được giới nghiên
cứu quan tâm với nhờ ưu thế: dung lượng cao, thế phẳng, ổn định và an
toàn. Vật liệu LiFePO4 (LFP) là một ứng cử viên sáng giá cho vật liệu
điện cực thuộc cấu trúc olivine. Tuy nhiên, trong cấu trúc vật liệu này,
ion Li+ chỉ khuếch tán theo hướng [010] trong cấu trúc đường hầm
olivine nên hệ số khuếch tán và độ dẫn điện kém. Ngoài ra ion Fe2+
trong LFP dễ bị oxi hóa, đây cũng là thách thức lớn để thương mại hóa
vật liệu này.
Các loại vật liệu LiFePO4, LiFexM1-xPO4 và LiFePO4/graphene đã được các
công trình trên thế giới công bố cho thấy rằng pha tạp hoặc phủ carbon nói chung
và graphene nói riêng có thể cải tiến tính năng điện hóa. Tuy nhiên, chưa có công
trình nào công bố vật liệu nano composite dạng LiFexM1-xPO4/graphene (pha tạp
và phủ đồng thời).
Từ tính cấp thiết của thị trường pin sạc Li-ion thương mại các nhà nghiên
cứu đã không ngừng cải tiến tối ưu vật liệu pin để nâng cao tuổi thọ, giảm thiểu ô

3
nhiễm môi trường và mở rộng phạm vi ứng dụng điều hết sức cần thiết cho lĩnh
vực lưu trữ năng lượng. Đó là lý do chọn đề tài luận án: “Tổng hợp vật liệu
composite LiFe1-xMxPO4/graphene làm cathode để cải thiện tính năng điện hóa
pin lithium-ion”. Với mong muốn hướng đến mục tiêu cải thiện và nâng cao tính
năng điện hóa của vật liệu LiFePO4 (LFP) làm điện cực cathode cho pin sạc Li-
ion. Vật liệu LFP là một trong những vật liệu tiềm năng ứng dụng trong xe điện
với giá thành thấp và thân thiện môi trường.
2. Mục tiêu của luận án
Tổng hợp thành công vật liệu composite LiFe1-xMxPO4/Gr có kích hạt nano,
đơn pha kết tinh tốt nhằm cải thiện các thông số điện hóa: độ dẫn điện, hệ số
khuếch tán và dung lượng tốt hơn vật liệu LFP tổng hợp bằng phương pháp nhiệt
dung môi (solvothermal) cụ thể hóa bằng các mục mục tiêu nhỏ sau:
- Nghiên cứu quy trình chế tạo vật liệu cấu trúc olivine LFP bằng phương
pháp solvothermal.
- Tổng hợp thành công vật liệu nanocomposite LiFe1-xMxPO4/Gr, nghiên
cứu về cấu trúc, hình thái học và thành phần hóa học của vật liệu.
- Nghiên cứu ảnh hưởng quá trình động học đan cài ion Li+ sau khi pha tạp
ion kim loại Mn+ (Mn, Ni, Y) với nhiều tỉ lệ pha tạp khác nhau và phủ màng
graphene lên vật liệu nền LFP.
- Nghiên cứu đánh giá tính liên kết giữa vật liệu với màng mỏng graphene,
cấu trúc LiFe1-xMxPO4/Gr cũng như vai trò graphene tác động tính chất dẫn điện
và tính chất điện hóa lên vật liệu.
- Đánh giá tính năng điện hóa (dung lượng, độ bền phóng sạc) của vật liệu
điện cực LiFe1-xMxPO4/Gr trong mô hình bán pin cúc áo CR2032...
3. Đối tượng và phạm vi nghiên cứu
3.1. Đối tượng nghiên cứu
Tổng hợp vật liệu nanocomposite LiFe1-xMxPO4/Gr bằng phương pháp
nhiệt dung môi sử dụng làm điện cực cathode cho pin sạc Li-ion và khảo sát và
đánh giá mức độ ảnh hưởng vật liệu đối đến tính chất điện hóa khi thay đổi thành
phần từ đó mở rộng phạm vi ứng dụng.

4
3.2. Phạm vi nghiên cứu
3.2.1. Phương pháp tổng hợp
Chọn phương pháp nhiệt dung môi để tổng hợp vật liệu LiFe1-
xMxPO4/Gr vì phương pháp này tiết kiệm chi phí, pha sạch và có tính khả thi
cao. Ngoài ra, phương pháp solvothermal được quan tâm và thay thế cho các
phương pháp truyền thống trước đó bởi sản phẩm tạo ra ít tạp và kiểm soát kích
thước hạt.
3.2.2. Phương pháp nghiên cứu đánh giá
- Phương pháp đánh giá cấu trúc, định tính và định lượng thành phần vật liệu:
nhiễu xạ tia X (XRD), phổ Raman, phổ tán sắc năng lượng (EDS), phổ hấp thụ
nguyên tử (AAS), phân tích nhiệt trọng lượng (TGA), phổ quang điện tử tia X
(XPS).
- Phương pháp đánh giá hình thái hạt: hiển vi điện tử quét (SEM), hiển vi phát xạ
trường (FESEM), hiển vi điện tử truyền qua (TEM-HRTEM), SEM-EDS-
Mapping.
- Phương pháp đánh giá về tính chất điện hóa: phương pháp quét thế vòng tuần
hoàn (CV), phương pháp đo chu kỳ phóng sạc, tổng trở điện hóa (EIS).
4. Cơ sở lý luận và tình hình nghiên cứu
4.1. Cơ sở lý luận
Vật liệu cấu trúc olivine LFP được xem là vật liệu lý tưởng sử dụng làm
điện cực cathode cho pin sạc Li-ion vì tính ổn định, an toàn thân thiện với môi
trường, dung lượng tương đối cao.
LFP có thể tổng hợp bằng nhiều phương pháp khác nhau chẳng hạn như:
hóa keo (sol-gel), pha rắn (solid state), vi sóng (microwave), đồng kết tủa (co-
precipitation), nhiệt dung môi (solvothermal), thủy nhiệt (hydrothermal)… Mỗi
phương pháp đều có ưu nhược điểm khác nhau.
Phương pháp nhiệt dung môi là một phương pháp được đánh giá cao bởi
dung môi hòa tan tiền chất là các chất hữu cơ có độ nhớt do đó có thể kiểm soát
kích thước hạt, hạn chế sự kết tụ hạt trong quá trình kết tinh. Ngoài ra, thời gian
tổng hợp dài là tín hiệu tốt để pha hình thành ổn định và hạn chế tạp chất, đây là
nhưng yếu tố quyết định cải tiến hiệu suất tiết kiệm chi phí tổng hợp vật liệu.

5
Màng mỏng graphene có cấu trúc đặc biệt; khả năng kết dính, độ dẫn điện
tốt; diện tích tiếp xúc bề mặt lớn; cách nhiệt và độ đàn hồi tốt; siêu nhẹ… chính vì
vậy, graphene được giới nghiên cứu quan tâm nghiên cứu quan tâm làm vật liệu
phủ được quan tâm.
Với sự phát triển của Khoa học và Công nghệ ứng dụng đáp ứng nhiều thiết
bị, máy móc hiện đại đủ độ tin cậy và chính xác cao dùng để phân tích xác định
thành phần vật liệu composite có cấu trúc tương đối phức tạp.
4.2. Tình hình nghiên cứu
Tình hình nghiên cứu trên thế giới
Vật liệu điện cực cathode LiCoO2 thương mại hóa (1992) với dung lượng
thực tế đạt 155 mAh.g-1 so với dung lượng lý thuyết 248 mAh.g-1. Vật liệu này có
cấu trúc lớp nên không ổn định cấu trong quá trình phóng sạc dẫn đến dung lượng
giảm nhanh.
Thực tế, cobalt có hàm lượng chiếm trong vỏ trái đất thấp nên giá thành vật
liệu này cao (450 USD/kWh). Hơn nữa, vật liệu này có tính độc nên sau khi thải ra
nguy hại đến môi trường.
Vật liệu LFP có cấu trúc ổn định olivine được J. B. Goodenough cùng cộng
sự đề xuất năm 1997 với dung lượng lý thuyết 170 mAh.g-1, vùng thế phẳng, giá
thành trung bình và thân thiện môi trường. Tuy nhiên, Fe dễ bị oxi hóa, độ dẫn
điện và hệ số khuếch tán thấp nên cải tiến tối ưu vật liệu là thách thức lớn cho giới
nghiên cứu.
Các hướng đề cập cải tiến hiện nay bao gồm: giảm kích thước hạt, pha tạp
kim loại, phủ carbon, thay đổi phương pháp tổng hợp, thay đổi thông số hóa lý
trong quá trình tổng hợp.
Theo các báo cáo công bố gần đây vật liệu LFP đã được thương mại hóa
với dung lượng đạt 145 -155 mAh.g-1 tại tốc độ phóng sạc C/10 và ổn định 100
chu kì. Dung lượng này tương đối thấp so với dung lượng lý thuyết.
Một thập kỉ gần đây, các nhóm nghiên cứu đã tập trung cải thiện vật liệu
cathode chẳng hạn như phủ carbon (graphene oxide, carbon black, CNTs,
graphene) chế tạo được vật liệu LFP/C đạt dung lượng 150-160 mAh.g-1, các công
bố cho thấy rằng với tính chất hóa lí đặc biệt của graphene vì vậy khi phủ

6
graphene với các tỉ lệ 5- 12% graphene mang lại hiệu quả tốt hơn các loại thù hình
carbon khác.
Một số kết quả công bố khác hướng tới pha tạp các kim loại giàu số oxi hóa
và vùng thế hoạt động cao với mong muốn cải thiện hệ số khuếch tán và độ dẫn
điện. Các kim loại được sử dụng pha tạp chủ yếu: Mn, Ni, La, Co, Ti, Yb… Kết
quả sau khi pha tạp với nhiều tỉ lệ khác nhau 5-20% thành phần kim loại pha tạp
thì độ dẫn điện, hệ số khuếch tán tăng 102-104 lần so với vật liệu nền LFP.
Tình hình nghiên cứu trong nước
Từ năm 2000 trở lại đây, vật liệu làm điện cực cho pin sạc được quan tâm
nghiên cứu như một số nhóm nghiên cứu thuộc Viện Hàn lâm Khoa học và Công
nghệ Việt Nam (VAST), Đại học Quốc gia Tp.HCM... Các công trình đã công bố
tập trung vào vật liệu dạng LiMxOy: Li0.9Mn2−xFexO4, LiNi0.5Mn1.5O4,
Li0.9Mn2−xFexO4 và LiMn2-xFexO4, NaMxOy.
Năm 2002, nhóm nghiên cứu do tác giả Nguyễn Tiến Tài (VAST) kết hợp
với Viện Công nghệ tiên tiến Nhật Bản đã công bố pha tạp Fe vào vật liệu
Li0.9Mn2−xFexO4 và LiMn2-xFexO4 (cấu trúc spinel) nhằm làm cân bằng nhiệt độ để
ứng dụng làm điện cực cathode cho pin Li-ion.
Năm 2010, tác giả Lê Hà Chi (VAST) và cộng sự trường Đại học
Basilicata Italia nghiên cứu vật liệu cathode LiNi0.5Mn1.5O4 với nhiều phương
pháp khác nhau.
Năm 2013, công trình nghiên cứu về các sợi silic phủ Al2O3 dùng để chế
tạo điện cực anode cho pin Li-ion được công bố do nhóm tác giả Nguyễn Trần
Hùng Viện Hóa học-Vật liệu.
Năm 2014, nhóm nghiên cứu phòng thí nghiệm Hóa lý ứng dụng trường
Đại học KHTN-ĐHQG Tp.HCM đã nghiên cứu tổng hợp và cải tiến tính năng
điện hóa vật liệu LiNi0.5Mn1.5O4 làm cathode cho pin sạc Li-ion.
Hướng nghiên cứu cải thiện vật liệu
Trong phạm vi thông tin truy cập được chưa thấy công bố trong nước nào
liên quan đến chế tạo LFP, pha tạp và composite của nó với mục đích sử dụng làm
vật liệu cathode cho pin sạc Li-ion.
Chế tạo vật liệu nano composite LiFe1-xMxPO4/Graphene là sự kết hợp phối
trộn giữa vật liệu nền LFP đồng thời phủ và pha tạp kim loại Mn, Ni, Y làm tăng

7
độ dẫn điện, hệ số khuếch tán. Ngoài ra, graphene tạo cầu nối dẫn điện giữa các
đơn tinh thể, ngăn cản vật liệu tiếp xúc với không khí và ăn mòn điện cực.
5. Các đóng góp mới, ý nghĩa lý luận và ý nghĩa thực tiễn
Thiết lập điều chỉnh quy trình tổng hợp để chế tạo thành công vật liệu
composite LiFe1-xMxPO4/Gr ở kích thước hạt nano. Khảo sát đánh giá vai trò của
pha tạp kim loại và phủ graphene. Sự thành công của luận án có ý nghĩa về mặt
khoa học cũng như ý nghĩa lý luận thực tiễn như sau:
5.1. Ý nghĩa khoa học
Chế tạo thành công vật liệu LiFe1-xMxPO4/Gr, đây là vật liệu mới có thể
ứng dụng làm vật liệu điện cực cho pin sạc Li-ion có tính năng điện hóa tốt hơn
vật liệu truyền thống LiFePO4 được tổng hợp cùng phương pháp.
Đánh giá vai trò ion kim loại Mn+ sau khi pha tạp ảnh hưởng đến cấu trúc,
hình thái và tính năng điện hóa vật liệu. Xác định rõ mối quan hệ giữa cấu trúc và
tính chất điện hóa của vật liệu; từ đó góp phần giúp cải tiến quy trình tổng hợp,
thành phần kim loại pha tạp…
Làm sáng tỏ vấn đề quá trình khống chế động học hay quá trình khống chế
khuếch tán Warburg ảnh hưởng tới sự di chuyển ion Li+ trong cấu trúc olivine
nanocomposite LiFe1-xMxPO4/Gr; quá trình này ảnh hưởng gì đến tính chất điện
hóa của vật liệu.
Ý nghĩa lý luận và thực tiễn
Sự thành công của đề tài sẽ là kết quả kế thừa của các nghiên cứu về vật
liệu điện cực cathode cho lĩnh vực pin sạc Li-ion trong nước cũng như quốc tế. Từ
đó hướng tới mở rộng phạm vi ứng dụng vật liệu rút ngắn khoảng cách thương mại
hóa vật liệu LiFe1-xMxPO4/Gr, góp phần thúc đẩy và phát triển Công nghệ vật liệu
pin sạc Li-ion với công suất lớn tạo nguồn năng lượng sạch, giảm thiểu ô nhiễm và
hạn chế khai thác tài nguyên thiên nhiên.

8
CHƯƠNG 1: TỔNG QUAN
1.1. Sơ lược về pin sạc Li-ion và vật liệu điện cực olivine
Pin hóa học sơ cấp (không sạc lại) đầu tiên được phát minh bởi A. Volta
vào năm 1800, phát minh này đã đem lại nhiều lợi ích thiết thực ứng dụng vào lĩnh
vực thiết bị điện gia đình và công nghiệp [1].
Năm 1970, M.S. Whittingham và cộng sự đã khởi đầu nghiên cứu phát triển
pin sạc Li-ion với cấu trúc Li/Li+/LixTiS2 mang lại hướng đi đầy hứa hẹn cho công
nghệ lưu trữ năng lượng. Vấn đề an toàn là rào cản lớn nhất của thế hệ pin thứ cấp
này để hướng tới thương mại hóa. Nguyên nhân chính là kim loại Li lắng đọng
trong quá trình sạc gây đoản mạch dẫn đến không ổn định và dễ cháy nổ [1, 2].
Sau hai thập kỷ, năm 1990 Tập đoàn Sony Energitech đã thương mại hóa
pin Li- ion với cấu trúc LixC6/Li+/Li1-xCoO2 ứng dụng trong một số lĩnh vực: điện
thoại di động, máy tính, máy ảnh kỹ thuật số, xe điện và xe điện lai...
Tuy nhiên, pin Li-ion thương mại LiCoO2 bị khuyến cáo về vấn đề môi
trường vì nguyên tố cobalt có tính độc hại khi thải ra sẽ tác động tiêu cực đến môi
trường. Ngoài ra, vật liệu LiCoO2 có cấu trúc lớp nên không có bền nhiệt, giá
thành pin tương đối cao do trữ lượng cobalt trong vỏ quả đất rất thấp cũng là
nguyên cản trở pin sạc này thâm nhập vào thị trường. Vì vậy, khắc phục nhược
điểm để tối ưu hóa vật liệu vẫn là thách thức lớn cho giới nghiên cứu [3, 4].
Để cải thiện tính năng điện hóa của pin các nhà nghiên cứu đã đề nghị thay
đổi thành phần cấu trúc chất nền vật liệu cobalt trong Li1-xCoO2 bằng các kim loại
chuyển tiếp khác như: Fe, Ni, Mn hoặc các nguyên tố đất hiếm với từ tính và tính
dẫn điện cao nhằm đạt được hiệu suất phóng sạc cao và ổn định.
Năm 1997, J. B. Goodenough et al. đã công bố đề xuất LiFePO4 làm vật
liệu điện cực cathode cho pin sạc Li-ion với cấu trúc bền olivine thuộc hệ trực
thoi, dung lượng riêng lý thuyết tương đối lớn (170 mAh.g-1) vùng thế phẳng,
trọng lượng riêng nhỏ so với các vật liệu khác, vùng nhiệt độ hoạt động rộng và
không gây nguy hại đến môi trường. Vật liệu này được giới nghiên cứu cho rằng
đây là vật liệu lý tưởng cho pin sạc Li-ion. Kể từ đó, vật liệu này được giới khoa
học tập trung nghiên cứu [3-5].

9
Pin sạc LFP hoạt động dựa trên có chế phản ứng thuận nghịch của quá trình
oxi hóa khử Fe2+/Fe3+ (LiFePO4/FePO4). Trong quá trình sạc, ion Fe3+ có tính oxi
hóa mạnh hơn ion Li+ cho nên hạn chế sự lắng cặn Li khi sạc pin Li-ion. Nhờ vào
tính thuận nghịch nên cấu trúc vật liệu LFP ổn định trong quá trình phóng sạc
(charge- discharge) đây chính là chìa khóa để cải thiện công suất điện hóa của pin
và chính nhờ vào cấu trúc ổn định tạo nên vùng thế phóng sạc phẳng [4-6]. Sự có
mặt nhóm có dạng tứ diện PO43-, đặc biệt là liên kết P-O đóng vai trò điều chỉnh
thế của cặp thế oxi hóa- khử M3+/M2+. Tuy nhiên, công trình gần đây nhất công bố
rằng sự chuyển pha LiFePO4-FePO4 biến mất khi kích thước hạt nhỏ hơn 5 nm [7-
8]. Đây là cơ sở để nghiên cứu kiểm soát kích thước hạt nhỏ tối thiểu.
Hơn nữa, LFP làm vật liệu cathode có giá thành rẻ hơn nhờ nguồn sắt (Fe)
dồi dào (chiếm 6,3% hàm lượng vỏ trái đất) từ đó tiết kiệm chi phí chế tạo. Fe là
nguyên tố không có tính độc hại nên thân thiện môi trường đây cũng là lợi thế của
vật liệu LFP so với LiCoO2 [8, 9].
Xét về tính chất hóa lý, vật liệu LFP có vùng nhiệt độ hoạt động rộng hơn
nhóm vật liệu spinel và nhóm có cấu trúc lớp (layers). Đây là tiêu chí được xem
xét về mặt an toàn vì trong quá trình phóng điện nhiệt độ của pin sẽ tăng lên, vượt
qua ngưỡng nhiệt độ cho phép dễ dẫn đến cháy nổ và tiêu hao dung lượng. Bên
cạnh đó, LFP khối lượng riêng nhỏ, dung lượng và năng lượng riêng lớn [10-11].
1.2. Cơ chế hoạt động và cấu tạo của pin Li-ion
1.2.1. Cơ chế hoạt động của pin Li-ion
Pin sạc Li-ion là dạng chuyển hóa từ năng lượng hóa học sang điện năng
thông qua quá trình chuyển hóa thuận nghịch. Dòng điện được sản sinh nhờ ion tự
do Li+ di chuyển cùng lúc với phản ứng oxi hóa khử Fe3+/Fe2+. Để đạt được hiệu
suất độ bền phóng sạc cao, thì sự di chuyển của ion Li+ trong cấu trúc chủ cathode
và anode phải không làm thay đổi cấu trúc vật liệu nền [5, 11].
Cơ chế phóng sạc là quá trình phản ứng điện hóa của hai pha
LiFePO4/FePO4 của vật liệu LiFePO4 khác với các vật liệu cathode truyền thống
có cấu trúc spinel hoặc cấu trúc lớp như LiCoO2 và LiNiO2. Quá trình sạc, ion Li+
được tách ra từ LiFePO4, đồng thời ion Fe2+ bị mất electron chuyển thành Fe3+ tạo
thành pha FePO4, quá trình gọi là quá trình oxi hóa [13-14] (Hình 1.2).

10
Về mặt điện cực trong quá trình sạc, ion Li+ bị tách ra khỏi cathode, di
chuyển trong dung dịch điện li qua màng ngăn, sau đó được xen cài vào trong cấu
trúc của anode [15-16]
Quá trình phóng, ion Li+ đi vào cấu trúc FePO4, ion Fe3+ nhận electron và bị
khử thành Fe2+. Do đó ion Li+ đan cài/phóng thích (intercalation/extraction) trên
hai pha LiFePO4/FePO4 [16].
Phản ứng xảy ra tại cathode:
LiFePO4 xFePO4 (1-x) LiFePO4 xLi+ xe (1)
Quá trình sạc xảy ra ngược lại khi pin gắn nguồn tiêu thụ thì hóa năng
chuyển sang điện năng.
Phản ứng xảy ra tại cực anode:
xFePO4 (1-x) LiFePO4 xLi+ xe LiFePO4 (2)
Hình 1.2. Cơ chế hoạt động của pin Li-ion [16]
1.2.2. Các thông số tính năng của pin
1.2.2.1. Sức điện động của pin
Xem xét pin sạc Li-ion bao gồm một điện cực anode thường là kim loại Li
và điện cực cathode gồm các vật liệu: LiMx(PO4)y hoặc LiMxOy. Nếu ta xét hệ điện
cực Li/LiC6/LiCoO2 thì tại bề mặt điện cực và dung dịch điện giải, phản ứng trong
quá trình phóng như sau:
Tại cực anode: Li → Li+ + e Eanode= Eo
Li + )a
aln(
nF
RT
Li
Li (3)

11
Trong đó: Eo
Li là thế oxy hóa khử tiêu chuẩn của cặp oxi hóa khử Li+/Li so
với điện cực tiêu chuẩn hydro (-3,04 V so với SHE), T là nhiệt độ Kelvin (K), R là
hằng số khí lý tưởng (8,314 J.mol1.K-1), F là hằng số Faraday (96500 C.mol-1), a
là hoạt độ.
Tại cathode, quá trình đan cài Li+ theo phương trình:
Li+ + e + CoO2 LiCoO2
Ecathode = EoCoO 2 /LiCoO
2+ )
a*a
aln(
nF
RT
2
2
CoOLi
LiCoO
(4)
Phương trình cân bằng của hai bán phản ứng là:
Li + CoO2 LiCoO2
Trường hợp đan cài một phần của Li+ vào trong vật liệu điện cực, phương
trình cân bằng được mô tả:
xLi + CoO2 LixCoO2
Sức điện động của pin (Epin) là hiệu điện thế giữa thế điện cực âm (3) và thế
điện cực dương (4).
pin cathode anodeE E - E (5)
Dựa vào định luật Faraday m = ))((Z
M
F
Qsuy ra các công thức sau:
1.2.2.2. Điện lượng chuyển qua dung dịch điện giải (dung lượng Q)
Q = t
0
d )( I (6)
1.2.2.3. Dung lượng riêng Q (Specific capacity):
nF
MQ (7)
Dung lượng lý thuyết LiFePO4 là 170 mAh.g-1, n: số điện tử trao đổi;
F= 96500 C/mol, M: Khối lượng mol
1.2.2.4. Hiệu suất phóng sạc (H)
H= 100Qx
Qs (9)
Với Qs: dung lượng sạc; Qx là dung lượng còn lại sau khi phóng

12
1.2.3. Cấu tạo pin Li-ion
Cấu tạo của pin gồm các thành phần chính: cathode, anode, dung dịch điện
giải (electrolyte) và màng ngăn (separator) [17-19]:
Li (-)| LiPF6 (1M) | LMO hoặc LMPO4 (+).
Trong đó, LiPF6 (1M)/ EC-DMC (1:1) là dung dịch điện giải và Li là anode
(cực âm). Điện thế xảy ra phản ứng ở cathode thường từ 3,5 - 4,1 V. LiFePO4
hoặc LMO đóng vai trò điện cực cathode (Hình 1.3).
Hình 1.3. Cấu tạo của pin Li-ion [5]
1.2.3.1. Vật liệu điện cathode
Vật liệu điện cực cathode lý tưởng nên có các đặc tính sau đây: có khả năng
đan cài/thoát ion Li+ trong cấu trúc, phản ứng thuận nghịch đan cài/phóng thích
Li+, độ ổn định cấu trúc tốt trong quá trình đan cài/phóng thích lithi và năng lượng
của quá trình đan cài thấp. Hơn nữa, độ dẫn điện tốt, an toàn và chi phí sản xuất
thấp [19, 20].
Các oxít kim loại chuyển tiếp thường được sử dụng làm vật liệu điện cực
cathode vì đa hóa trị giàu số oxi hóa để phản ứng thuận nghịch có thể xảy ra dễ
dàng chẳng hạn như: V5+/V4+, Mn4+/Mn3+, Fe3+ /Fe2+, Co4+/Co3+ Ni3+/Ni2+ [21-22].
Đối với nhóm vật liệu cấu trúc lớp và spinel các hợp chất sử dụng làm vật
liệu điện cực cụ thể như: LixCoO2, LixV2O5, LiMn2O4, LiNiMnCoO2…; nhóm có
cấu trúc olivine được nghiên cứu chi tiết như: LixFePO4, LixMnyPO4, LixCoyPO4.
Trong đó, LixMnyPO4, LixCoyPO4 có vùng điện thế hoạt động điện thế cao (3,9 -
13
4V), tuy nhiên ở nhiệt độ cao chúng dễ bị oxi hóa vì cấu trúc không ổn định [22-
25] (Hình 1.4).
Trong đó, các vật liệu có cấu trúc tinh thể cho phép quá trình đan cài thuận
nghịch của ion Li+ có thể kể đến là cấu trúc lớp -NaFeO2, cấu trúc spinel LiM2O4
và cấu trúc olivine LiMPO4 (M = Fe, Co...) [26-29].
Vật liệu điện cực cathode LFP với cặp oxi hóa khử Fe2+/Fe3+ có số oxi hóa
thấp nhưng trong quá trình oxi hóa nó có thể làm tăng năng lượng khử tại cực âm
(thế khử âm) và thế điện cực thấp vì thế nó có cấu trúc bền, ổn định. Để phân biệt
cặp oxi hóa khử Fe2+/Fe3+ thuộc cấu trúc nào ta có thể dựa vào vùng thế hoạt động
Fe2O3, Fe2(SO4)3, LFP tương ứng lần; lượt với vùng thế hoạt động 2,5 V, 3,6 V, và
3,45 V [29-31] (Hình 1.4).
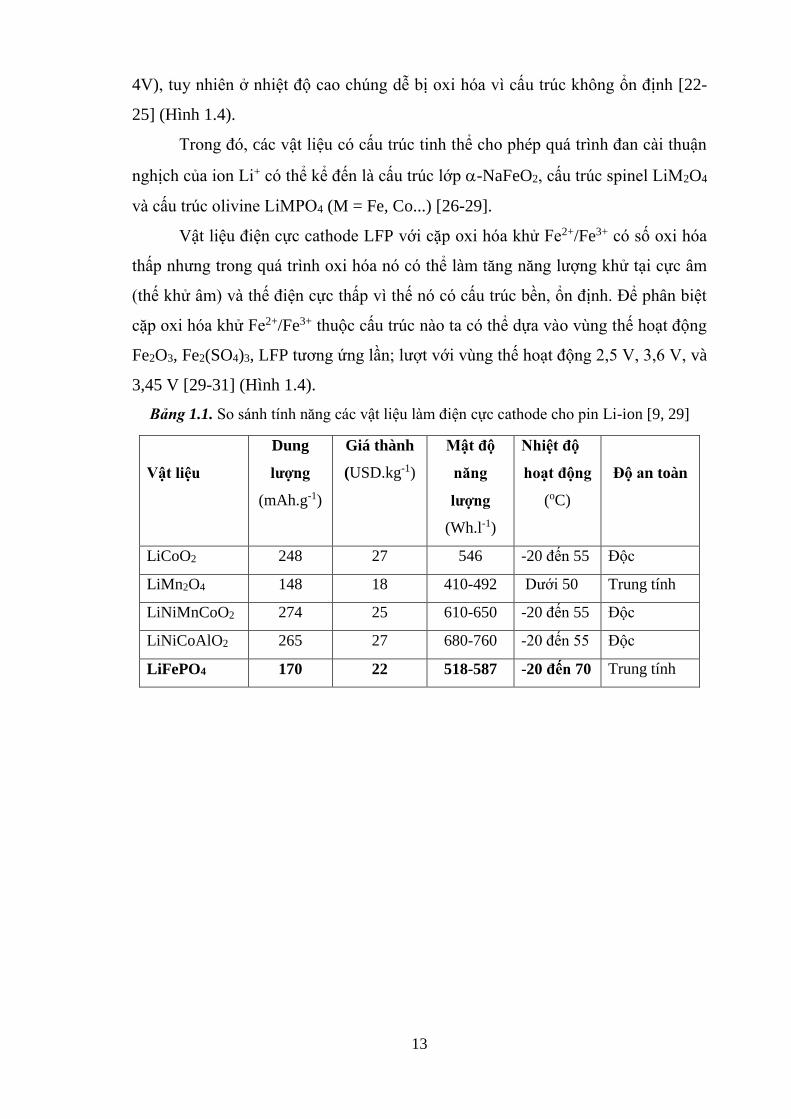
Bảng 1.1. So sánh tính năng các vật liệu làm điện cực cathode cho pin Li-ion [9, 29]
Vật liệu
Dung
lượng
(mAh.g-1)
Giá thành
(USD.kg-1)
Mật độ
năng
lượng
(Wh.l-1)
Nhiệt độ
hoạt động
(oC)
Độ an toàn
LiCoO2 248 27 546 -20 đến 55 Độc
LiMn2O4 148 18 410-492 Dưới 50 Trung tính
LiNiMnCoO2 274 25 610-650 -20 đến 55 Độc
LiNiCoAlO2 265 27 680-760 -20 đến 55 Độc
LiFePO4 170 22 518-587 -20 đến 70 Trung tính

14
Hình 1.4. Sự ảnh hưởng vật liệu đến điện thế oxi – khử [11]
Trong hình 1.4 cũng thể hiện được rằng các mối liên kết trong olivine Fe-
O-X-O-Fe; X-O (Mo-O; P-O; S-O) đều thuộc nhóm polyanion olivine. Tuy nhiên,
liên kết cộng hóa trị X-O càng lớn thì mật độ điện tử tập trung trên Fe-O sẽ giảm
đi hay nói cách khác ảnh hưởng chính đến năng lượng oxi hóa-khử Fe2+/Fe3+. X-O
mạnh thì Fe-O yếu và ngược lại. Nếu so sánh Fe2(MoO4)3 với LiFePO4 thì liên kết
Mo-O mạnh hơn P-O do đó vùng thế Fe2(MoO4)3 nhỏ hơn vùng thế của LFP là
0,45V. Đây là lý giải cho việc tại sao chọn polyanion [PO43-]. Bên cạnh đó, dung
lượng lý thuyết tương đối cao (170 mAh.g-1) với vùng thế phóng điện phẳng, LFP
là ứng cử viên sáng giá nhất cho vật liệu điện cực dương thay thế LiCoO2 [29-32].
1.2.3.2. Vật liệu điện anode
Vật liệu điện cực anode thường sử dụng là các kim loại trong đó chủ yếu là
các kim loại có thế oxi hóa-khử thấp điển hình như kim loại Li (ELi+/Li = -3,05 V).
Một số vật liệu anode khác thì sử dụng carbon đã được nghiên cứu chủ yếu
carbon dạng graphite nhiệt độ cao hoặc carbon phi graphite thu được ở nhiệt độ
thấp. Sử dụng những vật liệu này làm điện cực anode có khả năng cho đan/cài và
phóng thích ion Li+ thuận nghịch ở điện thế thấp so với Li+/Li khoảng 0,0-0,2 V.
Dẫn chứng cụ thể hợp chất graphite (LiC6) với dung lượng lý thuyết là 372 mAh.g-
1 tránh được Li tẻ nhánh gây ra hiện tượng đoản mạch bởi quá trình đảo ngược Li
xen kẽ do đó tạo tính ổn định và an toàn [11, 29, 32].
Ngoài vật liệu carbon, một số kim loại khác kim loại có khả năng hình
thành hợp kim với lithium là vật liệu anode đầy hứa hẹn: Si, Sn, Sb, Al, Mg, Bi,

15
In, Zn, Pb, Ag, Pt, Au… vì nó khả năng phóng điện thấp 0,006V so với Li/Li+ và
có dung lượng lý thuyết cao nhất khoảng 4200 mAh.g-1 [30, 32].
1.2.3.3. Chất điện giải
Chất điện giải là chất dẫn điện ion cung cấp môi trường cho việc truyền ion
giữa cực dương và cực âm. Chất điện giải thường là dung dịch lỏng chủ yếu muối
hòa tan, axít, kiềm (alkalies) để truyền dẫn độ dẫn ion. Chất điện giải phải có độ
dẫn ion tốt nhưng không dẫn điện tử, vì điều này có thể gây ra ngắn mạch.
Trong pin sạc Li-ion thương mại chất điện giải được sử dụng chủ yếu các
muối dạng lỏng tan tốt có chứa gốc ion Li+: lithium hexafluorophosphate (LiPF6),
lithium tetrafluoroborate (LiBF4), lithium trifluoromethanesulfonate (LiCF3SO3)
và lithium hexafluoroarsenate (LiAsF6). Các muối hữu cơ khác có độ phân cực tốt
cũng được sử dụng làm chất điện giải: propylen cacbonat (PC) ethylene carbonate
(EC), dimetyl carbonate (DMC), diethyl carbonate (DEC), ethyl methyl carbonate
(EMC) [15, 32].
Chất điện giải lỏng cần có độ dẫn ion cao nhất để đem lại hiệu suất cao pin.
Tuy nhiên, do các dung môi hữu cơ có áp suất hơi cao dễ cháy, vì vậy việc sử
dụng chúng có thể gây ra những rủi ro nghiêm trọng. Các chất lỏng với nhiệt độ
nóng chảy tối đa khoảng dưới 100 oC được coi như lý tưởng là không cháy, không
bị ăn mòn ổn định nhiệt và áp suất không đáng kể [16, 29, 32].
Chất điện giải dạng rắn là dạng vật liệu điện cực tối ưu nhất hiện nay vì nó
đáp ứng được các yêu cầu của pin sạc Li-ion [32, 33].
1.2.3.4. Màng ngăn
Giữa điện cathode và anode phải được cách điện bởi một màng ngăn để
ngăn chặn hiện tượng ngắn mạch khi các điện cực tiếp xúc với nhau.
Màng ngăn thường là các dạng vật liệu cách điện, các loại vật liệu này được
chế tạo từ nhiều loại hợp chất thiên nhiên, vô cơ và hữu cơ có chứa các lỗ xốp với
kích thước từ 50-100 Å, độ dày <25 µm.
Các vật liệu như sợi không dệt (nylon, cotton, polyester, thủy tinh), màng
dạng cao phân tử polymer: polyethylene, polypropylen, tetrafluoroethylene, poly
vinyl chloride, và những hợp chất thiên nhiên (cao su, gỗ, amiang) được sử dụng
làm màng ngăn cách vi xốp trong các pin sạc hoạt động ở nhiệt độ thấp (<100 oC).

16
Loại vật liệu phân cách này bao gồm chất nền rắn và pha điện giải lỏng,
được giữ lại trong cấu trúc vi xốp bằng lực mao dẫn. Để hoạt động hiệu quả, chất
lỏng trong các màng vi xốp không được hòa tan trong chất điện giải, tính ổn định
hóa học cao và cung cấp ion dẫn đầy đủ. Một số loại polymer như polypropylene,
polysulfone, poly tetrafloroethylene và cellulose acetate đã được sử dụng cho chất
nền xốp trong màng hỗ trợ chất lỏng. PVdF phủ màng vi xốp polyolefin dùng
trong pin sạc Li-ion dùng chất điện giải polymer dạng gel (gel-polymerelectrolyte-
GPE) thuộc loại này [33, 34].
1.3. Cấu trúc vật liệu điện cực cathode
Hiện nay, có ba loại cấu trúc vật liệu cathode chính:vật liệu cấu trúc lớp,
spinels, và dạng cấu trúc olivine (polyanion). Các loại vật liệu cathode khác cũng
đã được đề xuất, nhưng chưa thương mại hóa, bao gồm các chất hữu cơ liên hợp,
lưu huỳnh, không khí.
1.3.1. Cấu trúc dạng lớp
Vật liệu dạng lớp phổ biến nhất được sử dụng làm vật liệu cathode cho pin
Li-ion được thương mại điển hình là LiCoO2 (10-3 S.cm-1; 248 mAh.g-1, 3,88 V)
với công thức chung LiMO2, trong đó M có thể là một hoặc nhiều kim loại chuyển
tiếp (M = Ni, Co, V, Mn…) [34].
Tuy nhiên, có nhiều vấn đề liên quan đến sự không ổn định của cấu trúc và
hoạt tính xúc tác cao đối với quá trình oxi hóa điện li, đặc biệt ở trạng thái số oxi
hóa cao hoặc điện thế cao của của oxít kim loại chuyển tiếp. Sự kém ổn định của
cấu trúc lớp liên quan đến vận chuyển ion Li+ trong quá trình đan cài dẫn đến dung
lượng thấp. Vật liệu lớp bị mất oxi ở nhiệt độ cao vấn đề này dẫn đến hiện tượng
tạo áp suất nội phân tử gây cháy nổ [20-22, 34](Hình 1.5).
Hình 1.5. Cấu trúc lớp của vật liệu LiMO2 [11]

17
Ngoài ra, nguyên tố cobalt có trữ lượng thấp trong vỏ trái đất và độc tính
nên đây cũng là một vấn đề trở ngại lớn. Do đó, nghiên cứu tập trung vào các hợp
chất cấu trúc lớp là dẫn xuất của LiCoO2, Co được một phần thay thế bởi các ion
kim loại chuyển tiếp thân thiện với môi trường và vùng nhiệt độ rộng hơn, như Ni
và Mn, Fe, Al… Chẳng hạn như: hợp chất LiNi0.5Mn0.5O2 có hiệu suất phóng sạc
tốt ở độ cao nhiệt độ, LiFePO4 có cấu trúc ổn định, bền và thân thiện môi trường.
Nghiên cứu về vật liệu điện cực đã cho thấy sự tổng tổng hợp thành công chất mới
là LiCoNi Mn1-x-yO2 [23, 34].
Những công trình công bố gần đây đã đề xuất các cấu trúc Li[LixM]O2, đây
là sự xếp chồng của lớp LiMO2 và Li2MO3. Tuy nhiên, sự ổn định điện thế làm
nghèo mật độ eletron trong quá trình phóng sạc. Ví dụ, LiNi0.5Mn0.5O2 và
LiCo1/3Ni1/3Mn1/3O2 là vật liệu có hiệu suất tốt nhưng kém bền nhiệt (< 55 oC)
1.3.2. Cấu trúc dạng Spinel
Cấu trúc của LiMn2O4 thuộc hệ spinel AB2O4 nhóm không gian F 3d m. Các
nguyên tử oxi phân bố dựa trên qui luật xếp cầu lập phương (trùng với mạng lập
phương tâm mặt). LiMn2O4 xuất hiện cấu trúc “đường ống” và khung ba chiều cho
phép các ion Li+ dễ dàng di chuyển giữa các vật liệu.
Các nguyên tử oxi coi như các quả cầu xếp chặt tạo ra các vị trí trống bát
diện và vị trí trống tứ diện. Ion Li+ chỉ chiếm vị trí trống tứ diện và ion Mn4+
chiếm vị trí trống bát diện (Hình 1.6).
Vật liệu LiMn2O4 có tiềm năng thương mại cao đặc biệt nó cho các ứng
dụng với chi phí thấp hoặc yêu cầu ổn định khi quá tải và dung lượng thấp (120
mAh.g-1), điện thế cao (4,0 V) nhưng dung lượng giảm rất nhanh ở nhiệt độ cao do
hiện tượng hòa tan các hợp chất trong môi trường điện giải trên 60 °C [ 27, 34].
Hình 1.6. Cấu trúc spinel của LiM2O4

18
1.3.3. Nhóm hợp chất polyanion có cấu trúc olivine
Nhóm vật liệu lithium polyanion dạng LixMy(XO4)z (X: P, S, As, Mo, W, M:
Ni, Co, Mn, Fe) là kiểu cấu trúc olivine với thông số mạng a, b, c thỏa mãn: a ≠ b
≠ c, trực thoi (orthorhombic, D2h16, Pmnb) dạng đường hầm, với nhóm không gian
Pnma, và được gọi là triphylite gồm 2 mặt bát diện M1 và M2 với mặt M1 là mặt
đối xứng và M2 phản đối xứng. Ion Li+ nằm trên mặt M1 và ion Mn+ nằm trên mặt
M2 (Hình 1.7) [19, 28, 35].
Hình 1.7. Cấu trúc tinh thể olivine LixMy(XO4)z [5]
Trong số đó, LFP là thành viên gia đình họ olivine được xem là vật liệu lí
tưởng kết tinh với cấu trúc olivine. Các thông số mạng là a = 10,33 Å, b = 6,01 Å
và c =4,69 Å; thể tích ô mạng cơ sở là V = 291,2 Å3 [5, 34, 35].
Các nguyên tử oxi được sắp xếp theo sáu phương xếp chặt bất đối xứng,
nguyên tử P chiếm các mặt tứ diện để tạo thành tứ diện [PO4]-. Ion Li+ và Fe2+
được điền vào các khoảng trống của các bát diện chứa oxi. Trong đó, ion Li+
chiếm các mặt cạnh bát diện chung M1 (100), các ion Fe2+ chiếm tám mặt góc
thông thường M2 (010) tương ứng để tạo thành các khối tám mặt [LiO6] và [FeO6],
việc sắp xếp luân phiên bát diện [LiO6]-bát diện [FeO6]-tứ diện [PO4] tạo thành
cấu trúc lớp.
Các bát diện [FeO6] liền kề liên kết bằng cách chia sẻ các nguyên tử oxi ở
đỉnh để tạo thành lớp [FeO6] trên mặt bc. Tại các lớp [FeO6], các bát diện [LiO6]
liền kề sử dụng hai nguyên tử oxi dọc theo cạnh nhau để tạo thành một chuỗi theo

19
trục b, một khối tứ diện [PO4] và một bát diện [FeO6] dùng chung hai nguyên tử
oxi dọc theo cạnh (010) [30, 32, 35] (Hình 1.8).
Một số nghiên cứu cho rằng liên kết Fe-O ảnh hưởng đến tính chất điện hóa
nhiều hơn so với P-O. Chính vì vậy, việc pha tạp có thể cải thiện kích thước Fe-O
ngoài việc tăng mật độ điện tích. Ngoài ra, liên kết P-O dạng 3D bền nên vật liệu
LFP rất bền ở nhiệt độ cao hơn 200 oC. Li có khối lượng nhỏ nên quá trình phóng
sạc không ảnh hưởng đến cấu trúc [32, 35].
Hình 1.8. Cấu trúc olivine LiFePO4 [3]
Mặc dù LFP có nhiều ưu điểm hơn so với các vật liệu cathode chẳng hạn
như: LiCoO2, LiMn2O4…Tuy nhiên, LFP có cấu trúc olivine bền nên ion Li+ kém
linh động và ion Li+ chỉ di chuyển theo một hướng duy nhất [010] là nguyên nhân
dẫn đến vật liệu này có độ dẫn điện kém và hệ số khuếch tán thấp so với độ dẫn
điện các vật liệu khác [25, 27, 35] (Bảng 1.2).
Bảng 1.2. Độ dẫn điện, hệ số khuếch tán và khối lượng riêng một số vật liệu [29, 35]
Vật liệu Độ dẫn điện
(S.cm-1)
Hệ số khuếch tán
(cm2.s-1)
Khối lượng riêng
(g.cm-1)
LiCoO2 10-3-10-4 10-7-10-9 5,05
LiMnO2 10-5-10-6 10-9-10-12 4,15
LiFePO4 10-9-10-10 10-12-10-14 3,60
20
Nguyên nhân dẫn điện kém của LFP có thể được lý giải như sau: P5+ có
nhiều điện tử trống, tính hóa trị cao thu hút mạnh O2- kết quả là tính chất cộng hóa
trị của liên kết Fe-O bị giảm là việc rào cản dẫn điện LFP.
Để cải tiến hiệu suất của pin về mặt động học cần xem xét các yếu tố sau:
cấu trúc pha, cơ chế phản ứng và cơ chế dẫn ion Li+ trong mạng tinh thể olivine và
thành phần vật liệu.
Ví dụ, một số nghiên cứu đã thay thế hoàn toàn Fe (LFP) bằng một số kim
loại chuyển tiếp: Ni, Co, Mn... tương ứng vật liệu điện cực dương là: LiNiPO4;
LiCoPO4; LiMnPO4 đồng thời điện thế của điện cực cũng tăng lên đáng kể từ 3,45
tăng vùng 4,0- 4,5 V [10, 29, 35].
Xem xét về mặt cấu trúc, những vật liệu này đều giống nhau ở gốc anion là
polyanion [PO4] - vậy thì yếu tố ảnh hưởng ở đây chính là liên kết cộng hóa trị M-
O (M là kim loại chuyển tiếp, Mn: 3d54s2, Fe:3d64s2, Co: 3d74s2...). Năng lượng
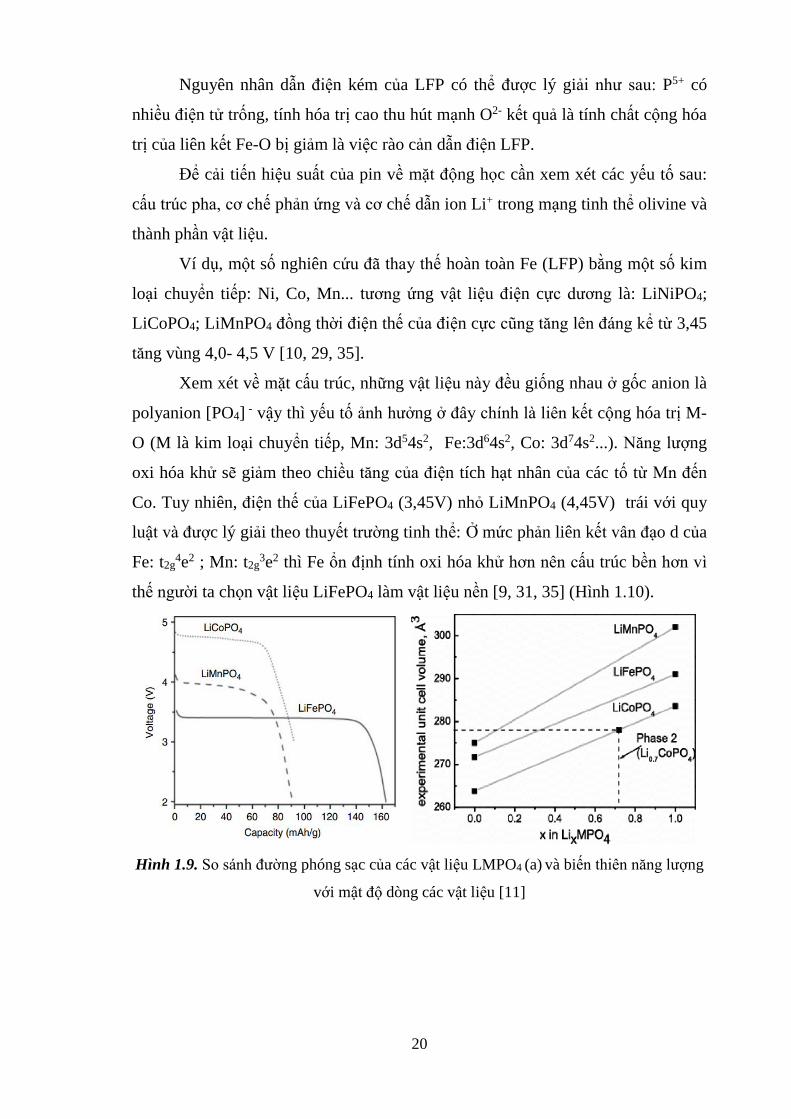
oxi hóa khử sẽ giảm theo chiều tăng của điện tích hạt nhân của các tố từ Mn đến
Co. Tuy nhiên, điện thế của LiFePO4 (3,45V) nhỏ LiMnPO4 (4,45V) trái với quy
luật và được lý giải theo thuyết trường tinh thể: Ở mức phản liên kết vân đạo d của
Fe: t2g4e2 ; Mn: t2g
3e2 thì Fe ổn định tính oxi hóa khử hơn nên cấu trúc bền hơn vì
thế người ta chọn vật liệu LiFePO4 làm vật liệu nền [9, 31, 35] (Hình 1.10).
Hình 1.9. So sánh đường phóng sạc của các vật liệu LMPO4 (a) và biến thiên năng lượng
với mật độ dòng các vật liệu [11]

21
Hình 1.10. Phân bố điện tử theo thuyết trường tinh thể trên vân đạo nguyên tử 3d thì các
vân đạo nguyên tử có spin cao (các điện tử độc thân [11]
Kết luận: Với những ưu điểm của vật liệu LFP có cấu trúc olivine bền và
vùng thế phóng điện phẳng nên không bị thay đổi trong quá trình phóng sạc giúp
cho tuổi thọ của pin cao và an toàn. Bên cạnh đó, giá thành tương đối thấp, thân
thiện với trường. Cuối cùng, vùng nhiệt độ hoạt động của vật liệu này rộng hơn
các vật liệu khác, khối lượng riêng nhỏ và dung lượng lý thuyết tương đối cao.
Tuy nhiên, vật liệu này dễ bị oxi hóa do ion Fe2+kém bền, ion Li+ chỉ di
chuyển được theo một hướng duy nhất [010] trong cấu trúc đường hầm olivine
nên độ dẫn điện và hệ số khuếch tán thấp là rào cản vật liệu này mở rộng quy mô
ứng dụng của vật liệu này. Với những ưu nhược điểm trên, chúng tôi chọn vật liệu
nền LFP và nghiên cứu các hướng cải thiện nhược điểm để nhằm tăng độ dẫn điện
cũng như hệ số khuếch tán để khắc nhược điểm vật liệu này.
1.4. Nghiên cứu cải thiện tính năng điện hóa của vật liệu LFP
Tính năng điện hóa luôn là yếu tố được xem xét chủ yếu trong quá trình chế
tạo pin sạc Li-ion. Tính năng điện hóa được nghiên cứu và đánh giá thông qua các
giá trị cụ thể như: dung lượng tuổi thọ và hiệu suất phóng sạc dựa trên các phương
pháp quét thế tuần hoàn (CV), phương pháp phóng sạc cố định dòng.

22
Thực tế, dung lượng càng lớn thì khả năng lưu trữ càng cao; độ dẫn điện là
yếu tố cũng được quan tâm vì độ dẫn điện cải thiện tốc độ phóng sạc của pin.
Phương pháp CV giúp xác định được dung lượng và số chu kỳ. Phương pháp tổng
trở điện hóa (EIS) là phương pháp đo độ dẫn phổ biến nhất, thông qua giá trị tổng
trở suất (vật liệu + dây dẫn) tính được độ dẫn điện. Tính ổn định và độ bền phóng
sạc được nghiên cứu qua đường cong phóng sạc, thực hiện lắp ráp trong pin mô
hình Swagelok [8, 9, 29, 31, 35].
Vật liệu LFP được cho là vật liệu điện cathode có nhiều ưu điểm hơn so với
các vật liệu khác. Mật độ năng lượng và dung lượng riêng lớn, tính ổn định động
học, tuổi thọ cao so với các thế hệ pin sơ cấp đặc biệt giá thành thấp và thân thiện
môi trường [29-31, 35].
Từ các nghiên cứu chúng tôi nhận hấy rằng, giảm kích thước hạt có thể làm
tăng khả năng khuếch tán ion Li+; rút ngắn khoảng cách biên độ thế oxi hóa khử
giúp quá trình sạc của pin nhanh hơn. Hơn nữa, pha tạp kim loại nhất là các kim
loại chuyển tiếp có tương đồng về cấu trúc điện tử với nguyên tử Fe và giàu số oxi
hóa nhất là các kim loại tính oxi hóa mạnh hơn Fe ngoài việc làm giàu mật độ
điện tử làm tăng độ dẫn điện và hệ số khuếch tán giúp ion Li+di chuyển tốt còn
góp phần hạn chế Fe2+bị oxi hóa. Thêm vào đó, phủ carbon (in-situ và ex-situ) sẽ
hỗ trợ vật liệu LFP kết tinh tốt, kiểm soát kích thước hạt đồng thời là cầu nối dẫn
điện cho ion Li+ di chuyển giữa các tinh thể.
1.4.1. Giảm kích thước hạt
1.4.1.1. Khái niệm về cấu trúc nano tinh thể (nanocrystal)
Cấu trúc tinh thể của vật liệu LFP ở kích thước nano tồn tại ba dạng: đơn
tinh thể (single crystal) hoặc đa tinh thể (polycrystal) và dạng vô định hình
(amorphous).
Vật liệu có cấu trúc là một mạng lưới các hạt tinh thể sắp xếp trật tự liên tục
và không bị gián đoạn có tính dị hướng. Cấu trúc tinh thể gồm một ô cơ sở và rất
nhiều các nguyên tử sắp xếp đặc biệt; vị trí của chúng được lặp lại một cách tuần
hoàn trong không gian ba chiều theo một mạng Bravais. Kích thước của ô mạng
theo các chiều khác nhau được gọi là các thông số mạng hay hằng số mạng.
Tùy thuộc vào tính chất đối xứng của ô mạng mà tinh thể đó thuộc vào một
trong các nhóm không gian khác nhau. Cấu trúc và đối xứng của tinh thể có vai trò

23
rất quan trọng với các tính chất liên kết, tính chất điện, tính chất quang... của tinh
thể. Ngoài ra, đơn tinh thể có tính dị hướng (nhiều hướng khác nhau) dẫn đến tính
chất hóa lý khác nhau của vật liệu.
Cấu trúc đa tinh thể (polycrystal) là dạng cấu trúc chuổi được liên kết bởi
nhiều đơn tinh thể. Đặc điểm của chúng có tính đẳng hướng được chia 3 dạng chủ
yếu: xốp (porous), tấm (nanoplate), que (like-rod).
Hình 1.12. Tinh thể LiFePO4 dạng xốp (a); tấm (b); que (c) [2, 11]
1.4.1.2. Ảnh hưởng cấu trúc nano đến tính chất khuếch tán và điện thế hoạt động
Kích thước hạt dạng nano hứa hẹn cải thiện được nhược điểm của vật liệu
LFP nói riêng vì những lý do sau: kích thước nhỏ và diện tích bề mặt lớn của vật
liệu nano làm tăng diện tích tiếp xúc lớn hơn giữa vật liệu điện cực và chất điện
giải.
Ngoài ra, biên độ khoảng cách thế oxi hóa-khử được rút ngắn dẫn đến
khoảng cách các ion Li phải khuếch tán qua điện cực được rút ngắn. Do đó, tốc độ
phóng/sạc nhanh hơn [29, 31, 34, 35].
Về mặt lý thuyết người ta đã chứng minh rằng tỉ lệ giữa diện tích/thể tích
càng lớn thì khả năng chứa ion Li+ càng lớn có nghĩa phụ thuộc vào kích thước
của hạt vật liệu [32, 35].
Bảng 1.3. Thông số mạng và thể tích ô mạng LiFePO4 và FePO4
Vật liệu
Hằng số mạng (Å) Thể tích
(Å3)
Nhóm
không gian ‘a ‘b ‘c
LiFePO4 10,334 6,008 4,693 291,392 Pbnm
FePO4 9,821 5,792 4,788 273,352 Pbnm
Dung lượng của pin giảm là một phần là do kích thước các hạt lớn dẫn điện
diện tích tiếp xúc bề mặt nhỏ ảnh hưởng đến sự vận chuyển Li qua giao diện

24
LiFePO4/FePO4 nguyên nhân chính do ảnh hưởng sự khuếch tán ion Li+ [30-32,
35] (Hình 1.13; 1.14). Dẫn chứng so sánh cụ thể với ba kích thước hạt 40, 80 và
200 nm, kết quả là ở kích thước 40 nm biên độ vùng cấm Fe2+/Fe3+ ngắn nhất.
Hình 1.13. Kích thước hạt ảnh hưởng tới biên độ oxi hóa và khử [35]
Như vậy, các vật liệu được tổng hợp có cùng thành phần hóa học và cấu
trúc tinh thể nếu giảm kích thước hạt đến giới hạn nhất định khoảng nm dẫn đến
giảm độ dài khuếch tán thì mật độ ion Li+càng lớn và thời gian phóng sạc điện
nhanh.
Damian Burch et al., 2009 [36] đề ra hai cơ chế chung để ngăn chặn sự
phân tách pha trong các hạt nano. Ranh giới giai đoạn khuếch tán trở nên hạn chế
bởi hình học hạt; và thuyết hiệu ứng bề mặt, được dự đoán bởi các động học phản
ứng phụ hóa học phụ thuộc vào tiềm ẩn hóa học, trong đó các phản ứng chèn/tách
ổn định sự thành phần gần các bề mặt cân bằng với môi trường.
Hình 1.14. Khoảng cách biên độ peak oxi hóa-khử phụ thuộc vào kích thước hạt [36]

25
Hình 1.15. Ảnh hưởng của cấu trúc nano đến nồng độ ion Li+ [36]
Phương pháp tổng hợp, điều kiện tiền chất có thể ảnh hưởng đến kết quả
kích thước và hình thái của tinh thể: tỉ lệ tiền chất, phương pháp tổng hợp [37, 38].
1.4.2. Pha tạp kim loại
Pha tạp kim loại có thể làm giàu mật độ điện tử trong vật liệu, tăng điện thế
hoạt động của vật liệu dẫn đến hiệu suất cathode được cải thiện [39-44].
Tuy vật liệu LiFePO4 (10-9 S.cm-1, 3,45 V) có độ dẫn điện và vùng thế hoạt
động thấp hơn so với các vật liệu olivine có thế phẳng khác chẳng hạn như
LiCoPO4 cho pin sạc Li-ion thương mại trong tương lai [45-47] (Bảng 1.4.). Vai
trò nguyên tố trung tâm kim loại chuyển tiếp M có vai trò quyết định đến điện thế
hoạt động [48-51]. Do đó, để tối ưu vật liệu LFP thì pha tạp kim loại là một hướng
khả thi để rút ngắn khoảng cách mở rộng ứng dụng thương mại vật liệu LFP
Bảng 1.4. Khảo sát ảnh hưởng pha tạp đến hiệu suất điện hóa
Pha tạp kim loại
vào cấu trúc LFP
Phương pháp
Kích thước: KT (nm)
Điện thế hoạt động: E (V)
Dung lượng riêng: Q (mAh.g-1)
TLTK
LiFe1-xMnxPO4
(x = 0.15, 0.2)
20% là tốt nhất
Pha rắn
KT < 100
E: 2,5-4,5
Q: 162 (C/10), 103 (C/20)
[10]
LiFe1-xNixPO4
Nhiệt dung môi KT: <100
E: 2,5-4,5
Q: 170 (C/5); 150 (10C)
[45]
LiFe0.95Ni0.02Mn0.03PO4/C Nhiệt dung môi KT: 100 -2000
E: 2,5- 4,0
Q: 115,2 (10C); 164 (C/10)
[56]
LiFe1-xTixPO4
Nghiền bi KT: 200-800
E: 2,4-4,0
Q:125 (C/10), 150 (2C)
[54]

26
LiFe0.99Y0.01 PO4
x = 0.01-5% (1% tốt nhất)
Nhiệt dung môi E: 2,0- 4,0
C: Sau 70 chu kì còn 27,3
[8]
Li0.99Fe0.98Y0.01Ni0.01PO4/C Pha rắn KT: 100-300
E: 2,5-4,0
Q: 155 (C/5); 55 (30C)
[51]
LiFe0.99La0.01PO4
Pha rắn KT< 200
E: 2,8-4,2
C: 156 (C/5) so với LFP (104)
[55]
LiFe1-xPdxPO4/C
(x=0.02; 0.04)
Hóa keo KT < 200
E: 2,5-4,2
C:150 (x = 0.02); 165 (x =
0.04) tại C/5
[10]
Lựa chọn kim loại pha tạp phải đáp ứng được yêu cầu không làm ảnh
hưởng đến cấu trúc pha của LFP. Ngoài ra, tỉ lệ pha tạp cũng được quan tâm vì
nếu hàm lượng pha tạp >20% sẽ thay thế lượng sắt hàm lượng Fe lớn có trong cấu
trúc sẽ dẫn đến khuyết tật mạng vì thế người ta thường chọn các kim loại nhóm d
chẳng hạn như: pha tạp Ni, Mn, Co… để tương đồng về mặt cấu trúc so với Fe
nhóm vân đạo (orbital) d nguyên tử có nhiều orbital trống [51-53].
Khi thay thế các ion kim loại M2+ có bán kính nhỏ hơn Fe ta có thể giải
thích sẽ làm giảm kích thước hằng số ô mạng giảm dẫn đến thể tích ô mạng cơ sở
(V = abc) giảm đi đây là yếu tố tăng động học khuếch tán ion Li+ và làm tăng độ
dẫn điện kép tăng lên 105 -107 lần [54-55] (Bảng 1.5).
Bảng 1.5. So sánh hằng số và thể tích mạng trước, sau khi pha tạp [46 -55]
Pha tạp
Độ dẫn điện
(S.cm-1)
Hằng số mạng, thể tích mạng
‘a ‘b ‘c V [Å3]
Vật liệu LFP 5,6.10-9 10,353 6,110 4,691 296,6
Ni_LFP 2,1.10-1 10,347 6,015 4,678 291,1
Mn_LFP 2,8.10-6 10,332 5,991 4,698 290,75
Co_LFP 2,9.10-6 10,350 5,992 4,707 291,91
La_LFP 2,4.10-6 10,342 5,989 4,701 291,17
Ce_LFP 1,5.10-6 10,337 5,979 4,698 290,36
Ne_LFP 4,5.10-5 10,333 5,987 4,700 290,75
27
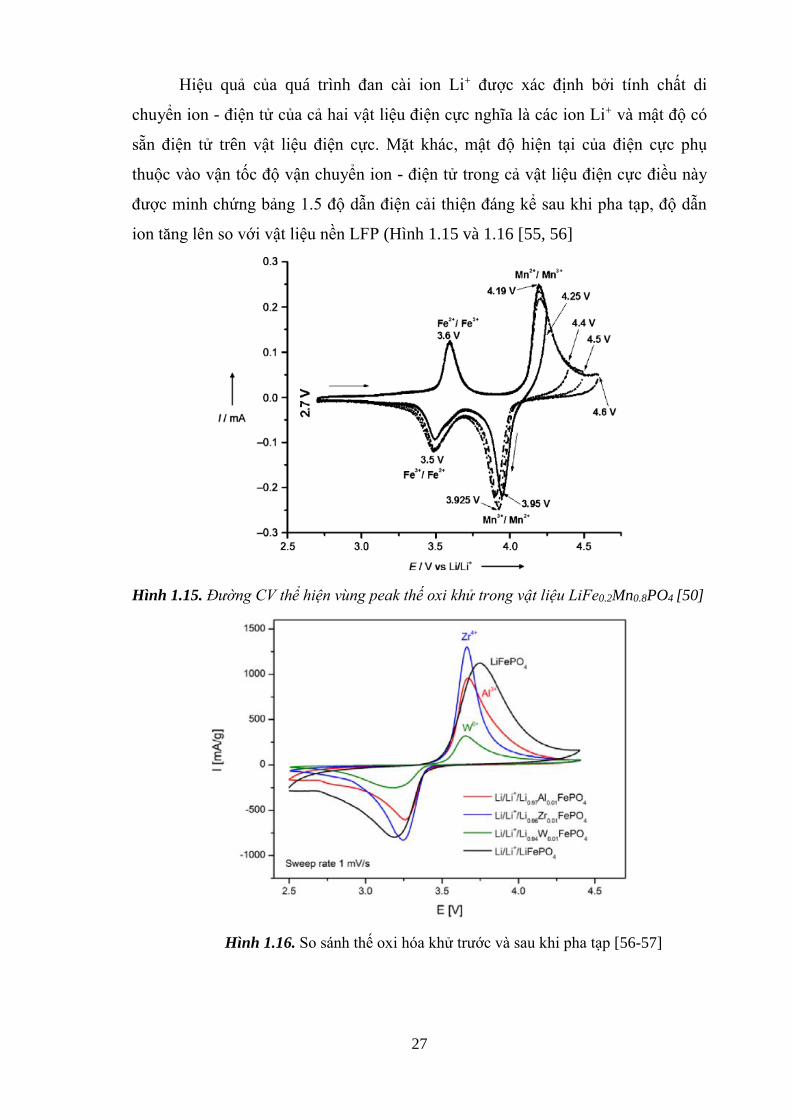
Hiệu quả của quá trình đan cài ion Li+ được xác định bởi tính chất di
chuyển ion - điện tử của cả hai vật liệu điện cực nghĩa là các ion Li+ và mật độ có
sẵn điện tử trên vật liệu điện cực. Mặt khác, mật độ hiện tại của điện cực phụ
thuộc vào vận tốc độ vận chuyển ion - điện tử trong cả vật liệu điện cực điều này
được minh chứng bảng 1.5 độ dẫn điện cải thiện đáng kể sau khi pha tạp, độ dẫn
ion tăng lên so với vật liệu nền LFP (Hình 1.15 và 1.16 [55, 56]
Hình 1.15. Đường CV thể hiện vùng peak thế oxi khử trong vật liệu LiFe0.2Mn0.8PO4 [50]
Hình 1.16. So sánh thế oxi hóa khử trước và sau khi pha tạp [56-57]

28
Như vậy, từ kết quả công trình công bố là cơ sở để đi đến kết luận sơ bộ
rằng: Pha tạp kim loại có thể cải thiện hiệu suất của pin về dung lượng, độ dẫn
điện và chu kỳ phóng sạc.
1.4.3. Phủ carbon (coating carbon)
Các phương pháp thông thường để tổng hợp LFP thường sử dụng các muối
Fe2+ dễ tan có độ phân cực lớn tuy nhiên muối này dễ bị oxi hóa [33, 37, 58]. Lớp
phủ carbon hỗ trợ giảm sự tiếp xúc trực tiếp giữa vật liệu điện cực và chất điện
phân để giảm thiểu sự ăn mòn của điện cực [59]. Việc kết hợp sử dụng giảm kích
thước tinh thể và lớp phủ cacbon đã cải thiện đáng kể tính năng điện hóa và do đó
thực tế của LiMPO4 [60, 61].
Tuy nhiên, sự giảm kích thước tinh thể olivine LiMPO4 và sử dụng lớp phủ
carbon giống như màng bảo vệ có thể đạt hiệu quả ở một mức độ nhất định. Nếu
các tinh thể giảm kích thước nhanh kết hợp với việc sử dụng carbon mật độ thấp
không hoạt động điện hóa có thể làm giảm đáng kể thể tích mật độ năng lượng
(tính theo đơn vị thể tích ô mạng) của vật liệu composite nano-carbon LiMPO4
[62, 63].
Để đánh giá vai trò ảnh hưởng của nano carbon tác động lên vật liệu LFP
được phân thành 2 dạng: dạng phủ carbon bên trong (in-situ) là dạng carbon sẽ
tham gia trực tiếp trong quá trình tổng hợp vật liệu chủ yếu lấy từ nguồn hữu cơ
(citric acid, acid ascorbic, glucose, sucrose…) với vai trò tác nhân ngừa oxi hóa
hoặc dung môi; dạng thứ hai là carbon phủ bên ngoài (ex-situ) vật liệu nền LFP
bằng phương pháp vật lí thông thường là carbon black, carbon nanotube (CNTs),
graphene…
1.4.3.1. Ảnh hưởng của carbon (in-situ)
Carbon in-situ coating được thêm vào trong tiền chất hoặc sử dụng thêm tác
chất hữu cơ đóng vai trò tác nhân khử hoặc chất ngừa oxi hóa.
- Vai trò tác nhân khử: chủ yếu các a xít hữu cơ có tính khử
Fe3+ thành Fe2+ vì Fe2+ dễ bị oxi hóa thành Fe3+ trong môi trường kiềm
(pH> 7,5) nhưng nó lại bền trong môi trường axít có pH khoảng 5,0-7,0. Vì vậy
thông qua kiểm soát độ pH trong dung dịch có thể kiểm được ion Fe2+ [58-64].

29
Một số công trình công bố rằng pH 5,0-7,0 hỗ trợ Fe2+ bền trong môi trường axít
[62-64]. Điều này được lý giải như sau:
Fe2+ + 6H2O Fe[(H2O)6]2+ (màu xanh rêu)
Nếu 5<pH<8 (kiềm nhẹ) thì phức Fe2+ sẽ chuyển thành phức Fe2+ trung hòa.
Fe[(H2O)6]2+ (màu xanh rêu) + 2OH- Fe[(H2O)4(OH)2]
Nếu pH >8, nghĩa là môi trường kiềm mạnh, thì phức Fe2+ sẽ chuyển thành phức
Fe3+ màu đỏ cam: Fe[(H2O)6]2+ (màu xanh rêu) + 3OH- Fe[(H2O)6]3+
Vai trò tác nhân ngừa oxi hóa: sử dụng các hợp chất hữu cơ giàu carbon
và dễ bị nhiệt phân, sau khi nung lượng carbon hữu cơ sẽ bị nhiệt phân chuyển hóa
carbon dạng rắn và bao phủ xung quanh vật liệu LFP. Lớp phủ ngăn cản quá trình
vật liệu tiếp xúc với không khí (Hình 1.17).
Hình 1.17. Quá trình hình thành carbon in-situ [61]
Để thu được pha LFP tinh khiết thì hầu hết các phương pháp đều khử bằng
nhiệt độ cao trong môi trường khí trơ lúc này các dạng carbon hữu cơ chuyển về
dạng graphite ngăn ngừa oxy hóa về Fe3O4 và cấu trúc vật liệu xốp hơn [59, 65]
(Hình 1.18).
Hình 1.18. Vai trò của carbon trong quá trình phản ứng [63]
Hiệu quả phóng sạc của vật liệu phụ thuộc vào phương pháp tổng hợp và
các thông số có thể kiểm soát như nhiệt độ, pH, dung môi...vì đây là yếu tố trực

30
tiếp kiểm soát kích thước và sự phân bố của các thành phần nanocomposite [40,
63, 64].
Carbon (in-situ) được xem như lớp vỏ bao bọc bên ngoài và vật liệu LFP là
bên trong, chính vì lớp carbon bao bọc bên ngoài vật liệu này có thể hạn chế ăn
mòn điện cực và ngăn Fe tiếp xúc với không khí [61-64]. Ngoài ra, carbon nguồn
từ acid hữu cơ chẳng hạn như lauric, oleic acid làm cầu nối dẫn điện và hạn chế
phát triển kích thước hạt [64] (Hình 1.19).
Tổng hợp LFP kết hợp đường làm tác nhân, nhóm nghiên cứu K. Wang
[64] công bố rằng nếu thêm glucose vào tiền chất dung lượng xả đạt 174 mAh.g-1
tại C/10 và 117 mAh.g-1 tại 20C sau 50 chu kì hiệu suất giảm 10%. Một số nghiên
cứu khác cũng cho thấy rằng sử dụng các acid hữu cơ, hoặc dung môi hữu cơ
mang lại kết quả tốt hơn [64-68].
1.4.3.2. Ảnh hưởng carbon vô cơ (ex-situ)
Carbon đơn chất là nguyên tố đóng vai trò quan trọng trong công nghệ vật
liệu điện cực vì nó có khả năng dẫn điện tốt, nhẹ và xốp. Nguyên tử carbon, các
electron lớp ngoài cùng có thể hình thành nên nhiều kiểu lai hóa. Do đó khi các
nguyên tử này liên kết lại với nhau chúng cũng có khả năng tạo nên nhiều dạng
cấu trúc tinh thể như: ba chiều (3D), hai chiều (2D), một chiều (1D) và không
chiều (0D). Điều này được thể hiện thông qua sự phong phú về các dạng thù hình
của vật liệu carbon là: kim cương, graphite, graphene, nanotubes và fullerens.
(Hình 1.19).
Hình 1.19. Một số dạng cấu trúc carbon

31
Những năm gần đây, carbon dạng fullerene, CNTs và graphene đang rất
được quan tâm, năm 2005 số lượng công bố liên quan đến ba vật liệu trên chỉ đạt
khoảng 5000 công trình, đến năm 2016 đã tăng lên 160000 công trình. Trong đó,
xu hướng nghiên cứu về graphene đã tăng mạnh từ khi giải Nobel về graphene
được công bố năm 2010, nhất là lĩnh vực vật liệu nano làm cathode pin sạc Li-ion.
Carbon ứng dụng vào vật liệu cho pin sạc bằng cách phủ phía bên ngoài vật
liệu nhằm làm cầu nối dẫn electron tăng mật độ điện tử tự do, cải thiện diện tích bề
mặt ngoài việc cải thiện hiệu suất điện hóa mà còn kiểm soát kích thước hạt và hạn
chế khả năng kết tụ hạt cũng như ngăn cản quá trình Fe2+ bị oxi hóa thành Fe3+ và
ngăn ngừa quá trình tiếp xúc với khí quyển [67, 68]. Thực tế, LFP có tính dẫn điện
kém là một trong những nguyên nhân đó là các electron chỉ tập trung di chuyển về
một hướng vì thế để kiểm soát chúng bằng cách phủ carbon ở nhiều cấu trúc khác
nhau trên điện cực cathode.
- Carbon than chì (Graphite)
Carbon dạng graphite sử dụng làm anode thay thế kim loại Li vì có thế oxi
hóa khử thấp, dung lượng lý thuyết lớn và hạn chế ăn mòn điện cực. Sau đó, nhiều
nhóm nghiên cứu thử nghiệm thay graphite bằng cách phủ carbon black [67-69].
Ngoài các công bố về ứng dụng carbon làm điện cực anode, các công trình
khác đã nghiên cứu tối ưu hóa vật liệu điện cực cathode bằng cách phối trộn LFP
với than chì (graphite). Các lớp cấu trúc của than chì ngoài khả năng làm tăng độ
dẫn điện cao của nó ngăn ngừa quá trình tập hợp các hạt nano [70].
Dựa trên các công trình nghiên cứu đã đưa đến kết luận lớp phủ carbon làm
tăng độ dẫn điện vì nó làm tăng hệ số khuếch tán của vật liệu điện cực cho pin sạc
Li-ion [67-72]. Từ đó đã mở ra hướng nghiên cứu mới về sử dụng graphite làm
thành phần phối trộn cực dương.
Một yếu tố cần quan tâm đó là hàm lượng carbon cần phối trộn, cải thiện
công suất xả của pin Li-ion. Cụ thể là khi hàm lượng carbon tăng lên đến 6 % khối
lượng, pin xả công suất tăng lên, trong khi tiếp tục gia tăng hàm lượng carbon đến
10 % khối lượng kết quả trong giảm công suất phóng [70-72].
Kết quả kiểm chứng, hàm lượng carbon ảnh hưởng tới tính năng điện hóa
của pin Li-ion này. Thật vậy, so với LFP (10-9-10-10 S.cm-1) thì độ dẫn điện LFP/C

32
(2,86.10-2 S.cm-1) sau khi trộn carbon cải thiện rất nhiều. Dung lượng của LFP/C
đạt 164,33 mAh.g-1 (C/10) và 149,12 mAh.g-1 (C/1).
- Carbon ống (CNTs)
CNTs được phát hiện từ năm 1991 bởi S. Iijima có độ dẫn điện, độ dẫn điện
khối tương đối cao; diện tích bề mặt tương đối lớn được ứng dụng rộng rãi trọng
ngành vật liệu điện, điện tử và cảm biến… Ngoài ra, CNTs cũng được coi là vật
liệu hứa hẹn để tăng độ dẫn điện và tăng cường vận chuyển ion cho LFP [71-73]
(Bảng 1.5). Toprakci và cộng sự [73] đã tổng hợp thành công LFP/CNTs/C bằng
phương pháp hóa keo (sol-gel), kết quả minh chứng cho cải thiện rõ rệt về dung
lượng trước và sau khi trộn CNT: LFP/CNTs/C, LFP lần lượt dung lượng đạt 169
mAh.g-1; 150 mAh.g-1 với tốc độ C/10.
- Graphene
Về mặt cấu trúc, màng graphene được tạo thành từ các nguyên tử carbon
sắp xếp theo cấu trúc lục giác trên cùng một mặt phẳng được gọi cấu trúc tổ ong.
Carbon có 6 điện tử do đó có bốn điện tử phân bố ở trạng thái 2s và 2p đóng vai
trò quan trọng trong việc liên kết hóa học giữa các nguyên tử với nhau (Hình
1.20).
Các trạng thái 2s và 2p của nguyên tử lai hóa với nhau tạo thành 3 vân đạo
lai hóa sp2, các trạng thái này định hướng theo ba phương tạo với nhau một góc
120o. Mỗi trạng thái sp của nguyên tử carbon này xen phủ với một trạng thái sp
của nguyên tử cacbon khác hình thành nên liên kết cộng hóa trị ( )bền vững.
Chính vì các liên kết bền này quy định cấu trúc mạng tinh thể graphene ở hình
dạng tổ ong và lý giải tại sao graphene rất bền vững về mặt cơ học và trơ về mặt
hóa học trong mặt phẳng mạng. Ngoài các liên kết ( ), giữa hai nguyên tử cacbon
lân cận còn tồn tại một liên kết pi (π) khác kém bền vững hơn hình thành do sự
xen phủ của các orbitan pz không bị lai hóa với các orbital s. Do liên kết π này yếu
và có định hướng không gian vuông góc với các orbital sp nên các điện tử tham
gia liên kết này rất linh động và quy định tính chất điện và quang của graphene
[72, 76-79].
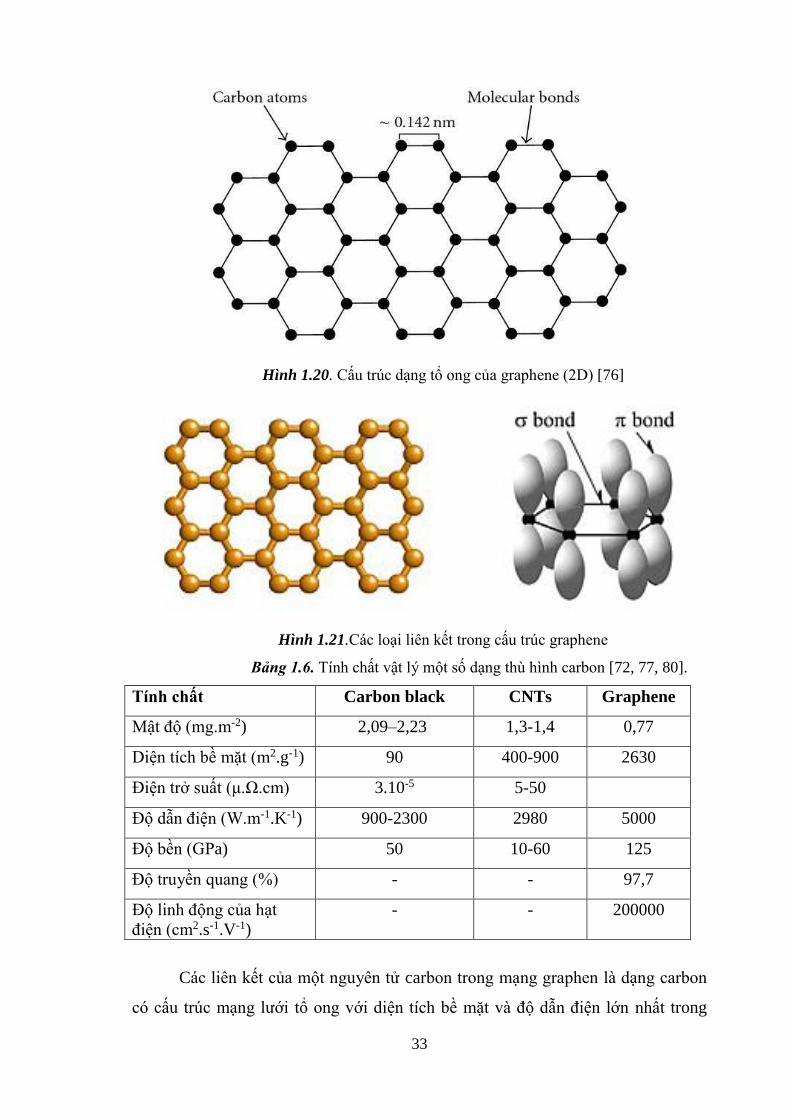
33
Hình 1.20. Cấu trúc dạng tổ ong của graphene (2D) [76]
Hình 1.21.Các loại liên kết trong cấu trúc graphene
Bảng 1.6. Tính chất vật lý một số dạng thù hình carbon [72, 77, 80].
Tính chất Carbon black CNTs Graphene
Mật độ (mg.m-2) 2,09–2,23 1,3-1,4 0,77
Diện tích bề mặt (m2.g-1) 90 400-900 2630
Điện trở suất (μ.Ω.cm) 3.10-5 5-50
Độ dẫn điện (W.m-1.K-1) 900-2300 2980 5000
Độ bền (GPa) 50 10-60 125
Độ truyền quang (%) - - 97,7
Độ linh động của hạt
điện (cm2.s-1.V-1)
- - 200000
Các liên kết của một nguyên tử carbon trong mạng graphen là dạng carbon
có cấu trúc mạng lưới tổ ong với diện tích bề mặt và độ dẫn điện lớn nhất trong

34
các dạng thù hình được giới nghiên cứu quan tâm đưa vào sử dụng nghiên cứu cho
pin sạc Li-ion [81-85].
Cấu trúc của nó có tính ổn định và dẫn điện tốt, bền, diện tích tiếp xúc bề
mặt lớn, siêu nhẹ nên rất được quan tâm trong lĩnh vực nghiên cứu vật liệu điện
cực cathode cho pin sạc Li-ion [88-90].
Graphene đóng vai trò như một chất kết dính thay thế cho các polymer [88,
91]. Hơn nữa, graphene có độ dẫn cao, diện tích tiếp xúc lớn làm cầu nối cho ion
Li+ chạy liên tục trong đường hầm olivine có thể cải thiện hiệu suất của pin. Mặc
dù phủ CNTs hay graphene không làm thay đổi thế nhưng mật độ dòng tăng lên
chứng tỏ độ dẫn điện tăng [92].
Dung lượng xả LFP/Gr cao hơn, hiệu suất phóng xạc ổn định hơn so với
LFP/carbon black [93-96].
Hai là, phủ graphene làm hạn chế quá trình phát triển tinh thể vì ngoài ra
sức mạnh cơ học của graphene có tiềm năng hấp thu một số sự giãn nở và co lại
của các hạt nano trong quá trình oxi-khử [90, 97].
Trên 5% khối lượng phủ graphene trực tiếp cải thiện tính chất điện hóa tăng
cường quá trình oxi hóa-khử (redox) làm tăng các đỉnh nhiễu xạ phổ XRD thấp
hơn.
Nghiên cứu mới nhất công bố với một lượng nhỏ graphene có tác động rất
lớn đan cài của Li (intercalation/de-intercalation) trong quá trình phóng sạc.
Cuối năm 2017, Tập đoàn Sony Engitech công bố rằng phủ một lượng nhỏ
graphene có thể cách nhiệt tốt vật liệu bền ở nhiệt độ 60 oC so với vật liệu trước đó
ổn định ở nhiệt độ 60 oC; Ngoài ra thời gian sạc của pin Li-ion chỉ còn 12 phút.
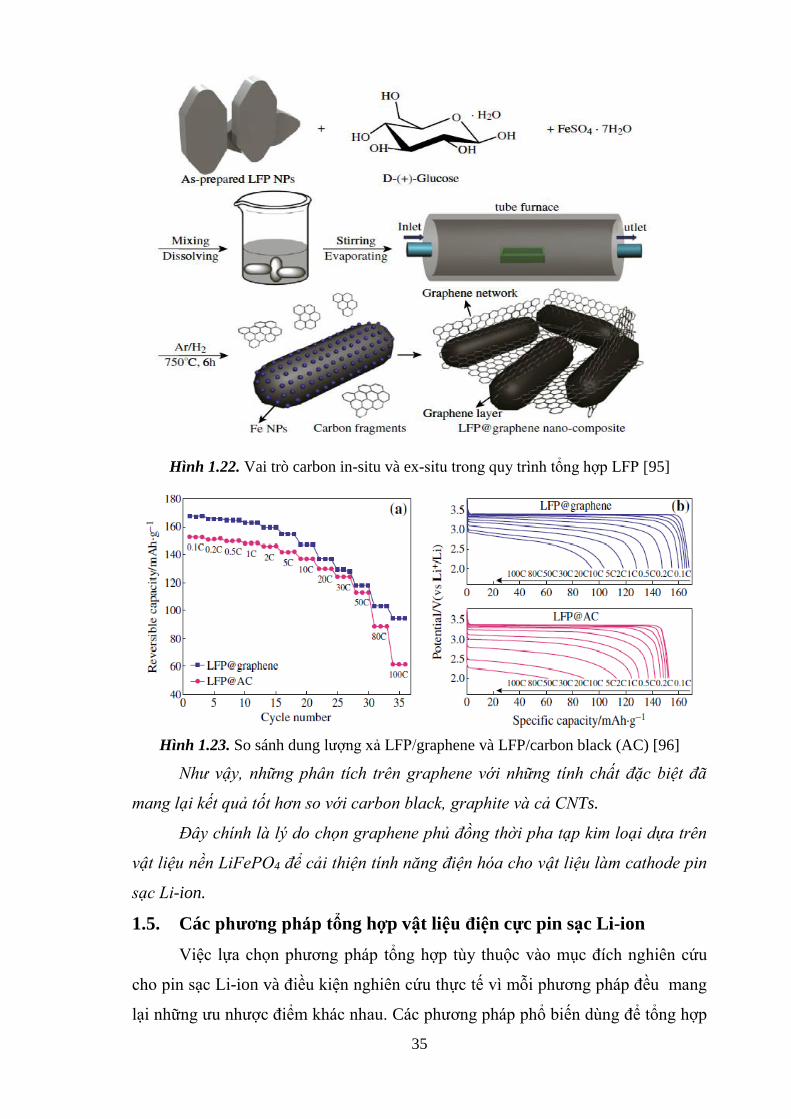
35
Hình 1.22. Vai trò carbon in-situ và ex-situ trong quy trình tổng hợp LFP [95]
Hình 1.23. So sánh dung lượng xả LFP/graphene và LFP/carbon black (AC) [96]
Như vậy, những phân tích trên graphene với những tính chất đặc biệt đã
mang lại kết quả tốt hơn so với carbon black, graphite và cả CNTs.
Đây chính là lý do chọn graphene phủ đồng thời pha tạp kim loại dựa trên
vật liệu nền LiFePO4 để cải thiện tính năng điện hóa cho vật liệu làm cathode pin
sạc Li-ion.
1.5. Các phương pháp tổng hợp vật liệu điện cực pin sạc Li-ion
Việc lựa chọn phương pháp tổng hợp tùy thuộc vào mục đích nghiên cứu
cho pin sạc Li-ion và điều kiện nghiên cứu thực tế vì mỗi phương pháp đều mang
lại những ưu nhược điểm khác nhau. Các phương pháp phổ biến dùng để tổng hợp
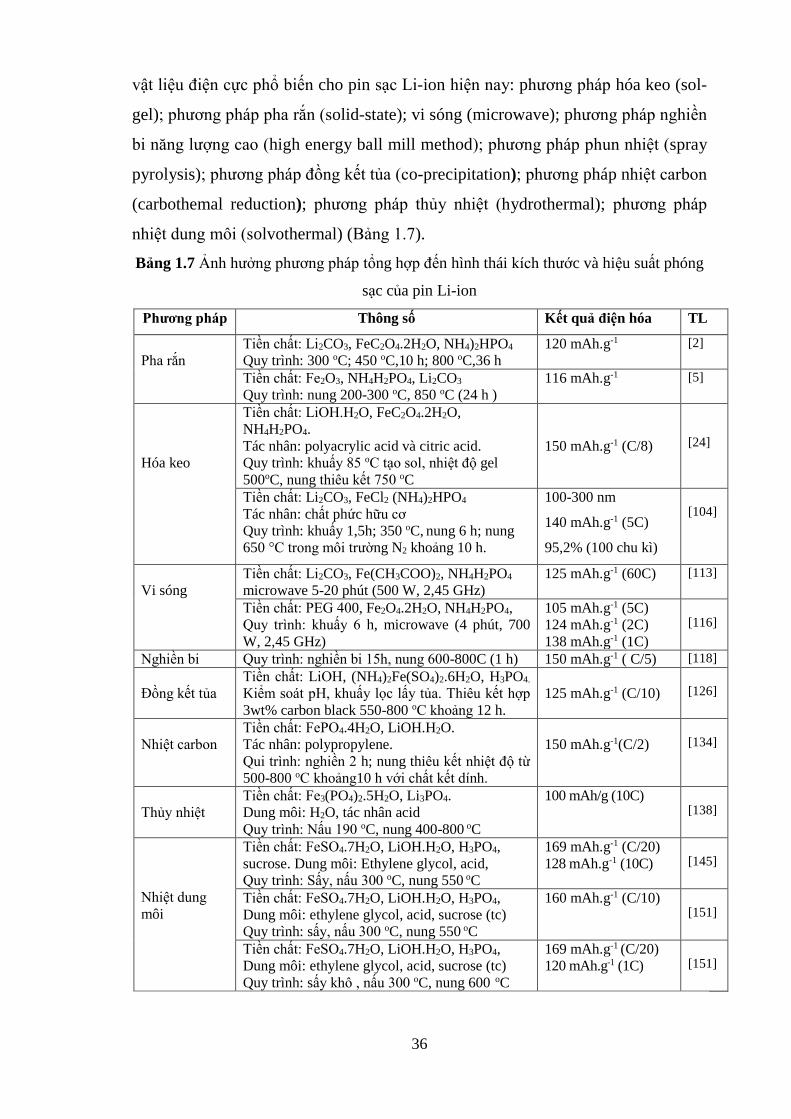
36
vật liệu điện cực phổ biến cho pin sạc Li-ion hiện nay: phương pháp hóa keo (sol-
gel); phương pháp pha rắn (solid-state); vi sóng (microwave); phương pháp nghiền
bi năng lượng cao (high energy ball mill method); phương pháp phun nhiệt (spray
pyrolysis); phương pháp đồng kết tủa (co-precipitation); phương pháp nhiệt carbon
(carbothemal reduction); phương pháp thủy nhiệt (hydrothermal); phương pháp
nhiệt dung môi (solvothermal) (Bảng 1.7).
Bảng 1.7 Ảnh hưởng phương pháp tổng hợp đến hình thái kích thước và hiệu suất phóng
sạc của pin Li-ion
Phương pháp Thông số Kết quả điện hóa TL
Pha rắn
Tiền chất: Li2CO3, FeC2O4.2H2O, NH4)2HPO4
Quy trình: 300 oC; 450 oC,10 h; 800 oC,36 h
120 mAh.g-1 [2]
Tiền chất: Fe2O3, NH4H2PO4, Li2CO3
Quy trình: nung 200-300 oC, 850 oC (24 h )
116 mAh.g-1 [5]
Hóa keo
Tiền chất: LiOH.H2O, FeC2O4.2H2O,
NH4H2PO4.
Tác nhân: polyacrylic acid và citric acid.
Quy trình: khuấy 85 oC tạo sol, nhiệt độ gel
500oC, nung thiêu kết 750 oC
150 mAh.g-1 (C/8)
[24]
Tiền chất: Li2CO3, FeCl2 (NH4)2HPO4
Tác nhân: chất phức hữu cơ
Quy trình: khuấy 1,5h; 350 oC, nung 6 h; nung
650 °C trong môi trường N2 khoảng 10 h.
100-300 nm
140 mAh.g-1 (5C)
95,2% (100 chu kì)
[104]
Vi sóng
Tiền chất: Li2CO3, Fe(CH3COO)2, NH4H2PO4
microwave 5-20 phút (500 W, 2,45 GHz)
125 mAh.g-1 (60C) [113]
Tiền chất: PEG 400, Fe2O4.2H2O, NH4H2PO4,
Quy trình: khuấy 6 h, microwave (4 phút, 700
W, 2,45 GHz)
105 mAh.g-1 (5C)
124 mAh.g-1 (2C)
138 mAh.g-1 (1C)
[116]
Nghiền bi Quy trình: nghiền bi 15h, nung 600-800C (1 h) 150 mAh.g-1 ( C/5) [118]
Đồng kết tủa
Tiền chất: LiOH, (NH4)2Fe(SO4)2.6H2O, H3PO4.
Kiểm soát pH, khuấy lọc lấy tủa. Thiêu kết hợp
3wt% carbon black 550-800 oC khoảng 12 h.
125 mAh.g-1 (C/10)
[126]
Nhiệt carbon
Tiền chất: FePO4.4H2O, LiOH.H2O.
Tác nhân: polypropylene.
Qui trình: nghiền 2 h; nung thiêu kết nhiệt độ từ
500-800 oC khoảng10 h với chất kết dính.
150 mAh.g-1(C/2)
[134]
Thủy nhiệt
Tiền chất: Fe3(PO4)2.5H2O, Li3PO4.
Dung môi: H2O, tác nhân acid
Quy trình: Nấu 190 oC, nung 400-800 oC
100 mAh/g (10C)
[138]
Nhiệt dung
môi
Tiền chất: FeSO4.7H2O, LiOH.H2O, H3PO4,
sucrose. Dung môi: Ethylene glycol, acid,
Quy trình: Sấy, nấu 300 oC, nung 550 oC
169 mAh.g-1 (C/20)
128 mAh.g-1 (10C)
[145]
Tiền chất: FeSO4.7H2O, LiOH.H2O, H3PO4,
Dung môi: ethylene glycol, acid, sucrose (tc)
Quy trình: sấy, nấu 300 oC, nung 550 oC
160 mAh.g-1 (C/10)
[151]
Tiền chất: FeSO4.7H2O, LiOH.H2O, H3PO4,
Dung môi: ethylene glycol, acid, sucrose (tc)
Quy trình: sấy khô , nấu 300 oC, nung 600 oC
169 mAh.g-1 (C/20)
120 mAh.g-1 (1C)
[151]

37
Mỗi phương pháp đều có những ưu nhược điểm riêng, tùy theo điều kiện cơ
sở vật chất, mong muốn sản phẩm và giá thành vật liệu có thể lựa chọn phương
pháp thích hợp nhưng chúng đều dựa trên cơ chế phản ứng sau.
1.5.1. Phương pháp hóa keo (sol-gel)
Là phương pháp tổng hợp vùng nhiệt độ thấp, phản ứng hóa học xảy ra ở
dạng huyền phù sử dụng để điều chế vật liệu dạng rắn. Cơ chế phản ứng là dựa
trên quá trình tách pha dẫn đến sự hình thành sol (một chất ổn định keo ổn định
của các hạt rắn trong một dung môi) và gelation (bộ xương cứng cáp kết nối với lỗ
xốp được tạo thành từ các hạt keo) [99-105]. Để có được các sản phẩm bột cuối
cùng, tất cả chất lỏng cần phải được loại bỏ khỏi bề mặt của các lỗ bằng nhiệt
được thực hiện ở nhiệt độ cao đây là nguyên nhân làm giảm số lượng các lỗ nối
kết nối, gọi là mật độ [106, 107].
Phương pháp sol-gel chi phí tương đối thấp, không cần nhiệt độ cao, thiết bị
đơn giản nhưng dễ tạo thành nhiều pha trong quá trình phản ứng. Các tiền chất
thường là hợp chất muối tan tốt trong một dung môi: hydroxide, acetate, nitrate
của kim loại kết hợp với tác nhân acid để kiểm soát pH [108, 109]. Cuối cùng là
quá trình làm khô vật liệu ở nhiệt độ (70-80 oC).
1.5.2. Phương pháp vi sóng (microwave)
Phương pháp này thực hiện dựa trên tốc độ gia nhiệt được kiểm soát bởi tần
số dao động (300 MHz -300 GHz) của lò vi sóng và sự tản nhiệt xảy ra thông qua
bề mặt vật liệu vì thế một số nhóm nghiên cứu đề xuất và sử dụng phương pháp
này tổng hợp vật liệu LFP (microwave heating) [110-113]. So với các phương
pháp khác, phương pháp này thời gian thực hiện phản ứng nhanh (15-20 phút), có
thể điều chỉnh nhiệt độ thích hợp theo ý muốn tối ưu vật liệu, kích thước hạt tương
đối nhỏ và mịn hơn, tiếp xúc nhiệt tốt nên bề mặt mịn, ngoài ra chi phí thấp vì thời
gian tổng hợp ngắn [114-116]. Tuy nhiên, nhược điểm là do thời gian thực hiện
nhanh nên quá trình hình thành pha chưa tốt, giá thành khá cao, khối lượng sản
phẩm tổng hợp nhỏ gây khó khăn cho việc phân tích [117].
Tiền chất được hòa tan trong dung môi (ethanol hoặc ethylene glycol...) sấy
khô ở nhiệt độ phòng (60-70 oC) sau đó ép thành viên. Mỗi viên bỏ trong len thủy
tinh và đặt trong vỏ nhôm có nắp đậy trong lò vi sóng gia nhiệt với tốc độ quét

38
theo mong muốn. Năng lượng điện sẽ hấp thụ đồng thời toàn bộ vật liệu để gia
nhiệt ổn định trong thời gian ngắn.
1.5.3. Phương pháp nghiền bi năng lượng (mechanochemical method)
Nguyên tắc là sử dụng năng lượng được tạo ra bởi các hạt quay vòng tốc độ
cao để thúc đẩy phản ứng giữa nguyên liệu thô. Các hạt được phá vỡ thông qua lực
cơ học, làm tăng diện tích tiếp xúc của chất phản ứng [117, 118]. Tuy nhiên do
pha vỡ cấu trúc dẫn đến tinh thể khuyết tật mạng, biến dạng mạng, nhiệt độ hoạt
động cao, chi phí khá tốn kém.
Phản ứng của phương pháp nghiền bi được điều khiển bởi năng lượng sinh
ra từ các quả bóng thép ở tốc độ cao. Sản phẩm tổng hợp được nghiền từ 10 ~ 24 h
sau đó đi nung 1h [118]. Do nhược điểm phương pháp này không ổn định cấu trúc
nên hiệu suất phóng sạc giảm nhanh [119]
1.5.4. Phương pháp pha rắn (solid-state)
Phương pháp tổng hợp pha rắn thiết bị đơn giản, chi phí thấp, khối lượng
tổng hợp nhiều có thể nâng cấp quy mô công nghiệp. Cơ chế phản ứng chủ yếu
dựa vào nhiệt độ để kiểm soát phản ứng và phá vỡ liên kết. Tuy nhiên, bất lợi của
phương pháp này mang lại thành phần hạt không đồng nhất, kích thước hạt to khó
kiểm soát sự kết tụ hạt và thời gian thực hiện dài [119].
Các tiền chất được trộn và nghiền sau đó chất ép viên sau đó xử lí nhiệt trải
qua hai giai đoạn: giai đoạn sấy khô (550-600 oC). Các viên nén được nghiền và ép
một lần nữa chúng sẽ phản ứng ở nhiệt độ cao giữa 700-1200˚C [119]. Nhóm F.Q.
Chen sử dụng thêm lauric acid làm tác nhân (2 nm) công suất xả 141,2 mAh.g-1
(1C); 130,9 mAh.g-1 (10C).
Để khắc phục nhược điểm của phương pháp pha rắn (solid state) kết hợp
Microwave hỗ trợ LFP được tổng hợp thông qua một phản ứng trạng thái rắn hỗ
trợ với phương pháp solvothermal [120].
1.5.5. Phương pháp phun nhiệt (spray pyrolysis)
Phương pháp nhiệt phân phun là một kỹ thuật rất hiệu quả để sản xuất các
hạt gốm mịn với tinh thể tinh khiết và kết tinh tốt giai đoạn trong một thời gian
ngắn, ưu điểm của phương pháp này tạo ra bột mịn, độ tinh khiết cao [121-123].
Trong phương pháp này, hệ thống tạo giọt là một bước quan trọng bởi vì
những giọt hoạt động như các trung tâm tạo mầm và cuối cùng phát triển thành các

39
hạt tinh thể, dày đặc và tinh khiết. Bột được sản xuất bằng phương pháp này có
kích thước hạt nhỏ (<1 μm), phân bố kích thước hẹp (1-2 μm), diện tích bề mặt lớn
và độ tinh khiết cao [124, 125]. Dung dịch hỗn hợp tiền chất phun vào lò nhiệt
phân từ 400-600 °C dưới dạng giọt nhờ dòng khí. Bột vật liệu được nung nhiệt độ
khoảng 700-800 °C. Giống như nhiều phương pháp khác, carbon nguồn có thể
được thêm vào trong quá trình nhiệt phân phun để có hạt nhỏ hơn kích thước bột
LiFePO4/C làm tăng diện tích tiếp xúc bề mặt bột [126].
1.5.6. Phương pháp đồng kết tủa (co-precipitation)
Phương pháp đồng kết tủa là phương pháp dựa trên cơ chế phản phứng trao
đổi các ion trong dung dịch. Các tiền chất chủ yếu là các muối có độ phân cực lớn
tan tốt trong nước, pha trộn các tiền chất hòa tan và muối lithium trong nước, điều
chỉnh giá trị pH trong môi trường khí N2 và khuấy cho đến khi hỗn hợp phản ứng
với kết tủa LFP. Lọc rửa sạch thu kết tủa và sấy khô thu được LFP, loại bỏ tạp
chất nhiệt độ cao (700-800 oC) trong môi trường chân không [127-129].
Phương pháp này dễ thực hiện thời gian tổng hợp ngắn nên cấu trúc pha
chưa ổn định, không kiểm soát được kích thước hạt, sản phẩm thu được khá ít.
Để cải thiện tính năng điện hóa phương pháp này kết hợp phủ carbon dung
lượng xả LFP/Gr (160 mAh.g-1 tại C/5) tăng lên so với LFP (128 mAh.g-1) [129].
1.5.7. Phương pháp thủy nhiệt (hydrothermal)
Phương pháp thủy nhiệt là phương pháp mà sản phẩm được kết tinh trong
môi trường nước kết hợp với gia nhiệt (hydrothermal) [135-140].
Các tiền chất được hòa tan trong autoclave (nồi hấp) đảm bảo giữ nhiệt độ
ổn định và áp suất cao để các chất phản ứng không hòa tan có thể được phân tán.
Nhiệt độ dùng để tổng hợp tương đối thấp (100-150 oC) và cho phép kiểm soát
hình thái học với hình dạng khác nhau, như hình cầu, khối, sợi, dạng tấm và tốt
tinh thể các hạt với kích thước khác nhau từ 1 nm- 10 m [135-143].
Y.Y. Liu và đồng nghiệp [140] tổng hợp LFP bằng phương pháp thủy nhiệt
kết hợp glucose (phản ứng trong autoclave 180 oC ~3 h; nung chân không 750 oC)
thu được dung lượng xả 160,1 mAh.g-1 tại C/10; 90,8 mAh.g-1 tại 20C).
Nhóm nghiên cứu cho thấy kỹ thuật khuấy cũng ảnh hưởng đến hình thái
hạt: Khuấy cơ thông thường thu được hạt tinh thể hình que; khuấy tốc độ cao tạo
ra hạt hình cầu.
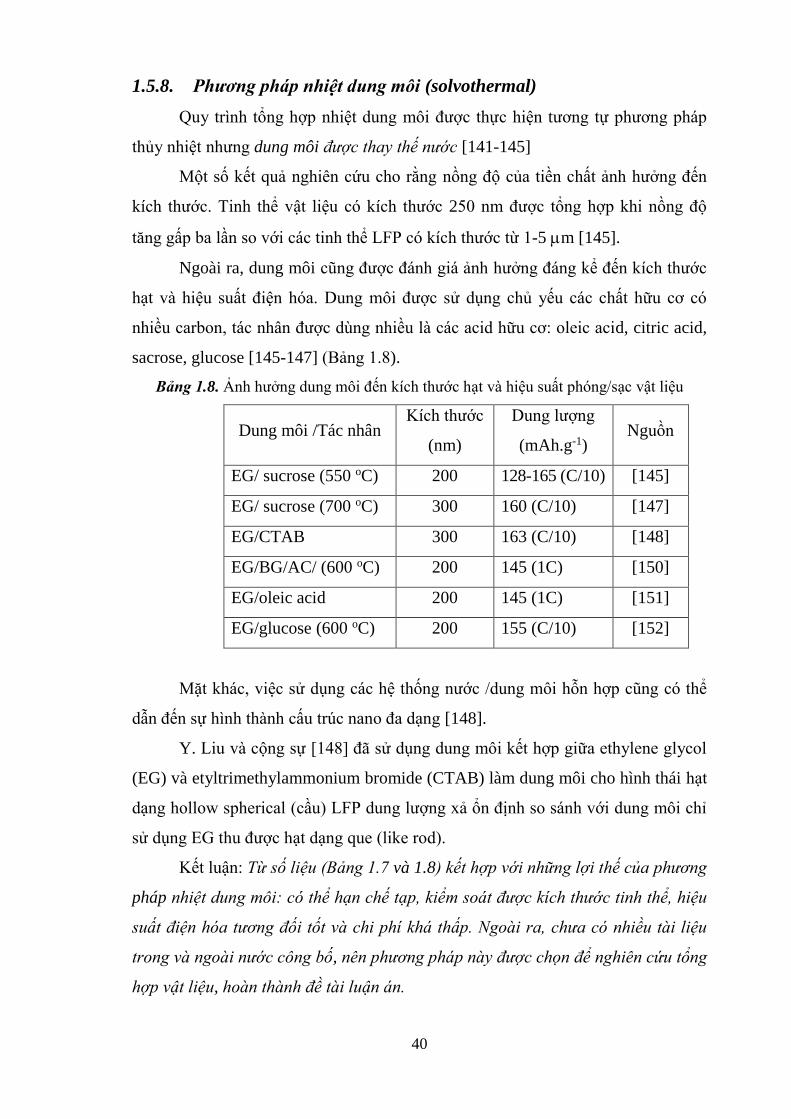
40
1.5.8. Phương pháp nhiệt dung môi (solvothermal)
Quy trình tổng hợp nhiệt dung môi được thực hiện tương tự phương pháp
thủy nhiệt nhưng dung môi được thay thế nước [141-145]
Một số kết quả nghiên cứu cho rằng nồng độ của tiền chất ảnh hưởng đến
kích thước. Tinh thể vật liệu có kích thước 250 nm được tổng hợp khi nồng độ
tăng gấp ba lần so với các tinh thể LFP có kích thước từ 1-5 m [145].
Ngoài ra, dung môi cũng được đánh giá ảnh hưởng đáng kể đến kích thước
hạt và hiệu suất điện hóa. Dung môi được sử dụng chủ yếu các chất hữu cơ có
nhiều carbon, tác nhân được dùng nhiều là các acid hữu cơ: oleic acid, citric acid,
sacrose, glucose [145-147] (Bảng 1.8).
Bảng 1.8. Ảnh hưởng dung môi đến kích thước hạt và hiệu suất phóng/sạc vật liệu
Dung môi /Tác nhân Kích thước
(nm)
Dung lượng
(mAh.g-1) Nguồn
EG/ sucrose (550 oC) 200 128-165 (C/10) [145]
EG/ sucrose (700 oC) 300 160 (C/10) [147]
EG/CTAB 300 163 (C/10) [148]
EG/BG/AC/ (600 oC) 200 145 (1C) [150]
EG/oleic acid 200 145 (1C) [151]
EG/glucose (600 oC) 200 155 (C/10) [152]
Mặt khác, việc sử dụng các hệ thống nước /dung môi hỗn hợp cũng có thể
dẫn đến sự hình thành cấu trúc nano đa dạng [148].
Y. Liu và cộng sự [148] đã sử dụng dung môi kết hợp giữa ethylene glycol
(EG) và etyltrimethylammonium bromide (CTAB) làm dung môi cho hình thái hạt
dạng hollow spherical (cầu) LFP dung lượng xả ổn định so sánh với dung môi chỉ
sử dụng EG thu được hạt dạng que (like rod).
Kết luận: Từ số liệu (Bảng 1.7 và 1.8) kết hợp với những lợi thế của phương
pháp nhiệt dung môi: có thể hạn chế tạp, kiểm soát được kích thước tinh thể, hiệu
suất điện hóa tương đối tốt và chi phí khá thấp. Ngoài ra, chưa có nhiều tài liệu
trong và ngoài nước công bố, nên phương pháp này được chọn để nghiên cứu tổng
hợp vật liệu, hoàn thành đề tài luận án.

41
CHƯƠNG 2: THỰC NGHIỆM VÀ PHƯƠNG PHÁP NGHIÊN CỨU
2.1. Tổng hợp vật liệu điện cực
2.1.1. Hóa chất, thiết bị
2.1.1.1. Hóa chất
Bảng 2.1. Hóa chất và độ tinh khiết và hãng sản xuất
TT Công thức Tên Thông số Xuất xứ
01 LiOH.H2O Lithium
hydroxide
Trạng thái: rắn (g), PA: ≥
99,99% Fisher
02 FeSO4.7H2O Iron(II) sulfate
heptahydrate
Trạng thái: rắn (g)
PA: 99,99% Fisher
03
Ni(NO3)2.6H2O
Nickel nitrate
hexahydrate
Trạng thái: rắn (g)
PA: 99,99%
Nhiệt độ sôi: 136,7 oC; tan tốt
MercK
04 C Graphene Trạng thái: rắn (g) (2-10 nm); = 8.104 S.m-1; S= 40 m2.g-1 MercK
05 C2H6O2 Ethylene glycol
Trạng thái: lỏng (l); PA
Nhiệt độ sôi: 197,6 oC Fisher
06 C6H8O6 Ascorbic acid
Trạng thái: rắn (g)
Nhiệt độ sôi: 553 oC MercK
07 H3PO4
Phosphoric acid
Trạng thái: lỏng (l)
Tỷ trọng: 1,88 g.ml-1
Nhiệt độ sôi: 337 oC
MercK
08 H2SO4
Sulfuric acid
Trạng thái: lỏng(l), PA: > 98%
Tỷ trọng: 1,84 g.ml-1
Nhiệt độ sôi: 337 oC
MercK
9 Y(NO3)3.6H2O Yttrium nitrate
hexahydrate
Trạng thái: rắn (g)
PA: 98,99%
Mật độ: 2,682 g.ml-1 tại 25°C
Merck
10 Mn(NO3)2.4H2O
Manganese(II)
nitrate tetra
hidrat
Trạng thái: rắn (g)
Nhiệt độ sôi: 100 oC
PA: 98,99%
Merck
2.1.1.2. Thiết bị
a. Thiết bị tổng hợp vật liệu
Thiết bị sử dụng tổng hợp vật liệu được thực hiện tại Khoa Công nghệ Vật
liệu trường Đại học Bách khoa TpHCM, phòng thí nghiệm Hóa lý ứng dụng
trường Đại học Khoa học Tự nhiên TpHCM và Viện Công nghệ Nano
b. Thiết bị phân tích vật liệu:
Thiết bị kỹ thuật hỗ trợ phân tích vật liệu tại các cơ sở trong và ngoài nước
như: Viện Công nghệ Nano (INT), Trung tâm Nghiên cứu Vật liệu Cấu trúc nano
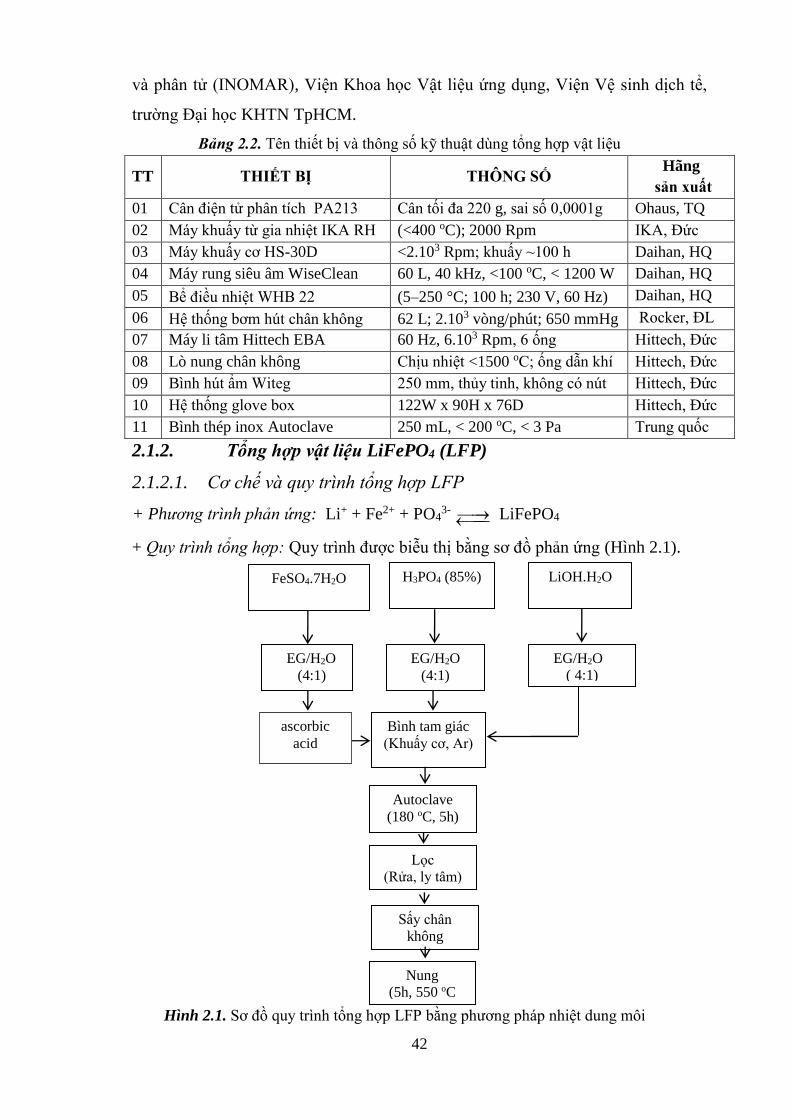
42
và phân tử (INOMAR), Viện Khoa học Vật liệu ứng dụng, Viện Vệ sinh dịch tể,
trường Đại học KHTN TpHCM.
Bảng 2.2. Tên thiết bị và thông số kỹ thuật dùng tổng hợp vật liệu
TT THIẾT BỊ THÔNG SỐ Hãng
sản xuất
01 Cân điện tử phân tích PA213 Cân tối đa 220 g, sai số 0,0001g Ohaus, TQ
02 Máy khuấy từ gia nhiệt IKA RH (<400 oC); 2000 Rpm IKA, Đức
03 Máy khuấy cơ HS-30D <2.103 Rpm; khuấy ~100 h Daihan, HQ
04 Máy rung siêu âm WiseClean 60 L, 40 kHz, <100 oC, < 1200 W Daihan, HQ
05 Bể điều nhiệt WHB 22 (5–250 °C; 100 h; 230 V, 60 Hz) Daihan, HQ
06 Hệ thống bơm hút chân không 62 L; 2.103 vòng/phút; 650 mmHg Rocker, ĐL
07 Máy li tâm Hittech EBA 60 Hz, 6.103 Rpm, 6 ống Hittech, Đức
08 Lò nung chân không Chịu nhiệt <1500 oC; ống dẫn khí Hittech, Đức
09 Bình hút ẩm Witeg 250 mm, thủy tinh, không có nút Hittech, Đức
10 Hệ thống glove box 122W x 90H x 76D Hittech, Đức
11 Bình thép inox Autoclave 250 mL, < 200 oC, < 3 Pa Trung quốc
2.1.2. Tổng hợp vật liệu LiFePO4 (LFP)
2.1.2.1. Cơ chế và quy trình tổng hợp LFP
+ Phương trình phản ứng: Li+ + Fe2+ + PO43- LiFePO4
+ Quy trình tổng hợp: Quy trình được biễu thị bằng sơ đồ phản ứng (Hình 2.1).
Hình 2.1. Sơ đồ quy trình tổng hợp LFP bằng phương pháp nhiệt dung môi
ascorbic
acid
Autoclave
(180 oC, 5h)
Lọc
(Rửa, ly tâm)
Sấy chân
không
(10h, 70 oC)
Nung
(5h, 550 oC
Ar)
Bình tam giác
(Khuấy cơ, Ar)
FeSO4.7H2O
H3PO4 (85%) LiOH.H2O
EG/H2O
(4:1)
EG/H2O
(4:1)
EG/H2O
( 4:1)

43
2.1.2.2. Mô tả chi tiết và điều kiện khảo sát
Tiến hành tổng hợp bốn mẫu kí hiệu ST00, ST01, ST02, ST00 (Bảng 2.3).
Mẫu ST00 không sử dụng ascorbic acid, dung môi chỉ dùng nước cất. Ba mẫu
ST01, ST02, ST03 sử dụng dung môi gồm ethylene glycol và H2O (4:1), tuy nhiên
ba mẫu này thay đổi các thông số tỉ lệ tiền chất.
Khảo sát các mẫu với tỉ lệ Li thay đổi, đem các mẫu tổng hợp phân tích
XRD. Tỉ lệ nào cho kết quả pha sạch, không xuất hiện tạp thì sử dụng tỉ lệ này để
thực hiện các thí nghiệm tiếp theo.
Bảng 2.3. Khối lượng và thể tích của các tiền chất tổng hợp LFP
Kí
hiệu
mẫu
LiOH.H2O
(g)
FeSO4.7H2O
(g)
C6H8O6
(g)
H3PO4
(g)
C2H4(OH)2
(mL)
H2O
(mL)
Tỉ lệ
Li: Fe: P
A 41,96 278,01 176,12 97,99 - - -
ST01 1,2588 2,7801 0,2202 1,1596 80 20 3,0:1:1
ST02 1,1329 2,7801 0,2202 1,1596 80 20 2,7:1:1
ST03 1,0490 2,7801 0,2202 1,1596 80 20 2,5:1:1
ST00 1,2588 2,7801 - 1,1596 - 100 3,0:1:1
A: Khối lượng mol; sai số khối lượng 5.10-4
- Mô tả chi tiết quy trình tổng hợp
Bước 1: pha trộn tiền chất
Các tiền chất theo tỉ lệ (Bảng 2.3) được cân bằng cân phân tích 4 số lẻ. Lấy
số 0,03 mol của LiOH.H2O làm chuẩn, dựa vào tỉ lệ cân khối lượng các
chất còn lại.
Pha trộn hỗn hợp dung môi gồm 80 mL EG và 20 mL H2O cất phân tích với
tỉ lệ 4:1 chia làm hai phần bằng nhau cho vào bình tam giác có hai nút đậy.
Đem 40 mL EG/H2O và LiOH.H2O cho vào bình tam giác có nút đậy, sau
đó rung siêu âm cho tới khi dung dịch đồng nhất, tiếp tục cho dung dịch
H3PO4 khuấy từ gia nhiệt để tăng tốc độ phản ứng thu được hỗn hợp có chất
rắn tủa trắng đục. 3
4 3 4Li + PO Li PO
Cho 40 mL dung môi EG/H2O vào bình tam giác chứa FeSO4.7H2O có nút
đậy, tiếp tục cho ascorbic acid vào thu được dung dịch có màu xanh trong
suốt. Khuấy từ trong môi trường khí argon (5 mL/phút) khoảng 30 phút thu
được dung dịch màu xanh nhạt.

44
Nhỏ từ từ hệ dung dịch LiOH/H3PO4/EG/H2O vào dung dịch FeSO4/AA/
EG/H2O được khuấy liên tục trong môi trường argon khoảng 30 phút thì
thu được dung dịch có màu xanh xám, cho toàn bộ vào bình Teflon 250 mL
và đặt vào bình bọc thép (autoclave).
Li+ + PO43- + Fe2+
=> LiFePO4
Bước 2: thực hiện phản ứng nhiệt dung môi
Dung dịch được đựng trong autoclave, đậy kín nắp để tránh vật liệu tiếp xúc
với không khí. Đem bình bọc thép đó đặt vào trong bể điều nhiệt chứa
glycerin ở 180 oC khoảng 5 -7 h sau đó lấy ra để nguội nhiệt độ phòng, đem
ly tâm rửa sạch thu được chất rắn màu xanh nhạt.
Bước 3: sấy khô bột LFP
Để làm mất hơi nước, dung môi sau khi ly tâm đem bột đi sấy trong tủ sấy
chân không hoặc hệ thống bơm hút chân không cho tới khi bột hoàn toàn
khô (10-12 h) và bảo quản vật liệu trong bình hút ẩm. Vật liệu nano
composite LFP/C thu được có màu xanh xám.
Bước 4: nung thiêu kết vật liệu composite LFP/C
Bột vật liệu sấy khô đem đi nung trong môi trường argon 5h, 550 oC. Vật
liệu được để vào thuyển nhỏ bằng sứ có tráng men sau đó đưa vào lò nung
điều chỉnh áp suất khí 5 Pa/s, nhiệt độ nung được gia tăng 100-550 oC. Đây
là giai đoạn cuối cùng của quá trình tổng hợp. Nung thiêu kết loại bỏ tạp
chất dạng rắn và giúp carbon phủ bên ngoài hạt vật liệu LFP.
2.1.3. Pha tạp kim loại (Mn, Ni, Y): LiFe1-xMxPO4
2.1.3.1. Quy trình và cơ chế tổng hợp
Phương trình phản ứng dạng ion rút gọn:
Li+ + xFe2+ + yM2++ PO43-
LiFexMyPO4 + e
Fe2+ và ion kim loại pha tạp M2+ có thể bị oxi hóa thành Fe3+
3xFe2+ + 3yM2+ + 2H2O + O2 (FexMy)3 O4 + 4H+ + (9x+6y -4)e
ascorbic acid có vai trò khử lượng ion kim loại dư (M2+) và Fe3+ đã bị khử trở về
trạng thái ion Fe2+ và M2+:
(FexMy)3O4 + 4H+ + C + 3Li+ + 3PO43- + 2e 3LiFexMyPO4 + CO2 + 2H2O
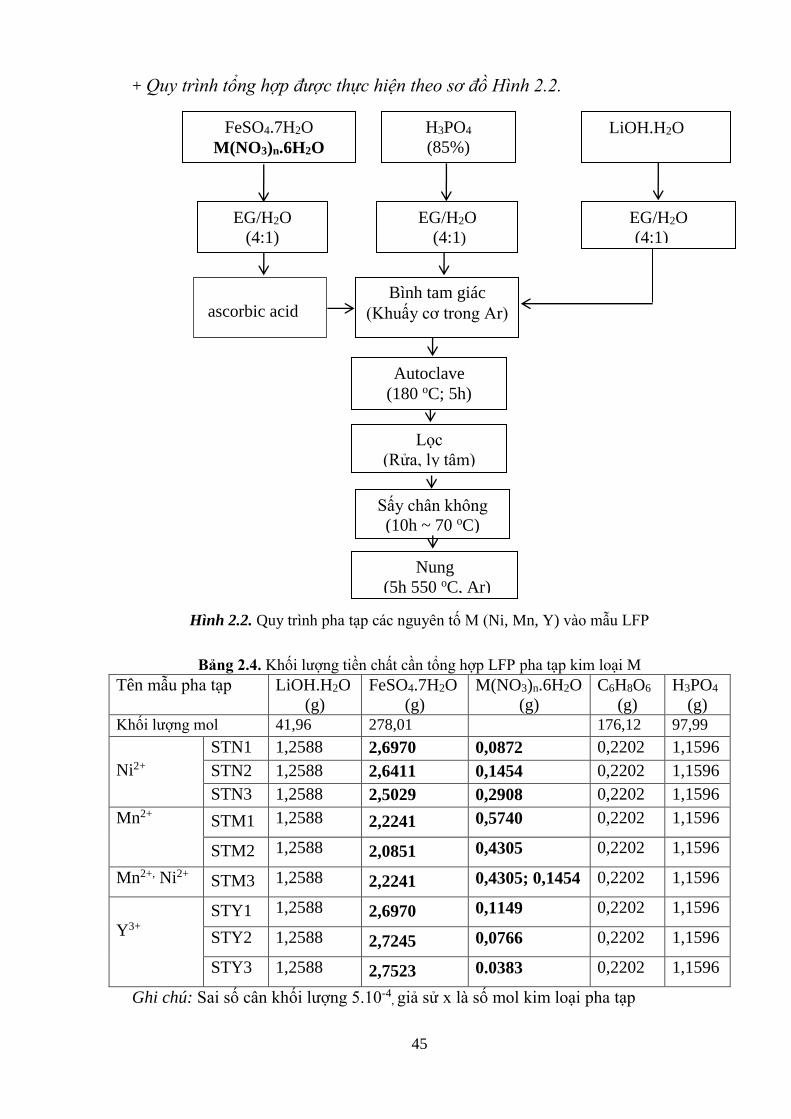
45
+ Quy trình tổng hợp được thực hiện theo sơ đồ Hình 2.2.
Hình 2.2. Quy trình pha tạp các nguyên tố M (Ni, Mn, Y) vào mẫu LFP
Bảng 2.4. Khối lượng tiền chất cần tổng hợp LFP pha tạp kim loại M
Tên mẫu pha tạp LiOH.H2O
(g)
FeSO4.7H2O
(g)
M(NO3)n.6H2O
(g)
C6H8O6
(g)
H3PO4
(g) Khối lượng mol 41,96 278,01 176,12 97,99
Ni2+
STN1 1,2588 2,6970 0,0872 0,2202 1,1596
STN2 1,2588 2,6411 0,1454 0,2202 1,1596
STN3 1,2588 2,5029 0,2908 0,2202 1,1596
Mn2+ STM1 1,2588 2,2241 0,5740 0,2202 1,1596
STM2 1,2588 2,0851 0,4305 0,2202 1,1596
Mn2+, Ni2+ STM3 1,2588 2,2241 0,4305; 0,1454 0,2202 1,1596
Y3+
STY1 1,2588 2,6970 0,1149 0,2202 1,1596
STY2 1,2588 2,7245 0,0766 0,2202 1,1596
STY3 1,2588 2,7523 0.0383 0,2202 1,1596
Ghi chú: Sai số cân khối lượng 5.10-4, giả sử x là số mol kim loại pha tạp
ascorbic acid
Autoclave
(180 oC; 5h)
Lọc
(Rửa, ly tâm)
Sấy chân không
(10h ~ 70 oC)
Nung
(5h 550 oC, Ar)
Bình tam giác
(Khuấy cơ trong Ar)
FeSO4.7H2O
M(NO3)n.6H2O
H3PO4
(85%) LiOH.H2O
EG/H2O
(4:1)
EG/H2O
(4:1)
EG/H2O
(4:1)

46
Trong bảng 2.4 các số liệu lấy số 0,03 mol LiOH.H2O làm chuẩn, số liệu
các tiền chất được cân đựa trên tỉ lệ do đó tỉ lệ Li:Fe:M:P = 0,03:(0,01-x): x:0.01.
Tùy theo số mol x kim loại pha tạp thì số mol Fe2+ cũng sẽ bị biến đổi.
2.1.3.2. Tổng hợp LiFe1-xMxPO4 (pha tạp Ni)
Quy trình thực hiện theo mục 2.1.2.2
Tương tự tổng hợp LiFePO4 tuy nhiên chọn tỉ Li:Fe: P = 3:1:1 (có kết quả
phân tích tốt nhất) với trộn với 3, 5, 10% Ni lần lượt kí hiệu.
Mẫu 5: LiFe0.97Ni0.03PO4 (STN1)
Mẫu 6: LiFe0.95Ni0.05PO4 (STN2)
Mẫu 7: LiFe0.90Ni0.1PO4 (STN3)
Chọn mẫu có kết quả điện hóa tốt nhất (cấu trúc pha và tính chất điện hóa)
đem phối trộn với graphene.
2.1.3.3. Tổng hợp LiFe1-xMnxPO4 (pha tạp Mn, Ni)
Theo khảo sát một số công trình tỉ lệ pha tạp Fe:Mn = 80:20 cho kết quả tốt
nhất. Vì thế trong công trình này thực hiện tổng hợp 2 mẫu với với Fe:Mn =75:15
và 80:20 và 01 mẫu pha tạp đồng thời 2 kim loại Mn, Ni (Fe: Mn: Ni = 80:15:5)
đánh giá vai trò ảnh hưởng pha tạp Mn và Ni.
STM1: LiFe0.75Mn0.15PO4
STM2: LiFe0.8Mn0.2PO4
STM3: LiFe0.8Mn0.15 Ni0.05 PO4
Chọn mẫu có kết quả điện hóa tốt nhất đem đi trộn với graphene
2.1.3.4. Tổng hợp LiFe1-xYxPO4 (pha tạp ytri)
Nguyên tố hiếm giá thành cao và với tính từ và dẫn điện cao nên thông
thường chọn tỉ lệ pha tạp nhỏ mang lại hiệu suất tốt và không làm biến đổi cấu trúc
olivine [52-57]. Trong đề tài luận án này, chọn pha tạp ytri với tỉ lệ 1, 2, 3%:
STY1: LiFe0.97Y0.03PO4
STY2: LiFe0.98Y0.02PO4
STY3: LiFe0.97Y0.03PO4

47
2.1.4. Composite hóa LiFe1-xMxPO4/graphene
2.1.4.1. Phương trình phản ứng và quy trình tổng hợp
a) Phương trình phản ứng
Li+ +Fe2+ + Mn+ + PO43- LiFexMyPO4 + ne (với Mn+: Mn2+, Ni2+, Y3+)
Quy trình phản ứng:
Graphene được hòa tan trong dung môi EG/H2O sau đó rung siêu âm để phân tán
graphene đều tạo thành hệ Gr/EG/H2O. Quy trình tương tự mục 2.1.2.2
2.1.4.2. Tiến hành tổng hợp
Bảng 2.5. Khối lượng cần để tổng hợp LiFe1-xMxPO4/Gr
Mẫu LiOH.H2O
(g)
FeSO4.7H2O
(g)
M(NO3)n.6H2O
(g)
C6H8O6
(g)
H3PO4
(g)
Graphene
(g)
M 41,96 278,01 (*) 176,12 97,99 12
STN1-G1 1,2588 2,6411 0,1454 0,2202 1,1596 0,0861
STN2-G2 1,2588 2,6411 0,1454 0,2202 1,1596 0,1721
STM1-G1 1,2588 2,2241 0,5740 0,2202 1,1596 0,0859
STM2-G2 1,2588 2,2241 0,5740 0,2202 1,1596 0,1718
STY1-G1 1,2588 2,7245 0,0766 0,2202 1,1596 0,0863
STY2-G2 1,2588 2,7245 0,0766 0,2202 1,1596 0,1727
STM3-G1 1,2588 2,2241 0,4305 0,1454 0,2202 1,1596 0,0845
(*) Ni(NO3)2.6H2O = 290,8; Mn(NO3)2.6H2O = 287,04; Y(NO3)3.6H2O = 383
Lấy số 0,03 mol LiOH.H2O làm chuẩn, dựa trên tỉ lệ
Li:Fe:M:P=0,03:(0,01-x):x:0,01 và LFMP:Gr = 90:10 (10% Gr); LFMP:Gr = 95:5
(5% Gr). Do vậy, số liệu bảng 2.5 cho thấy sau khi pha tạp thì có mối tương quan:
khối lượng muối kim loại pha tạp tăng thì khối lượng muối Fe giảm.
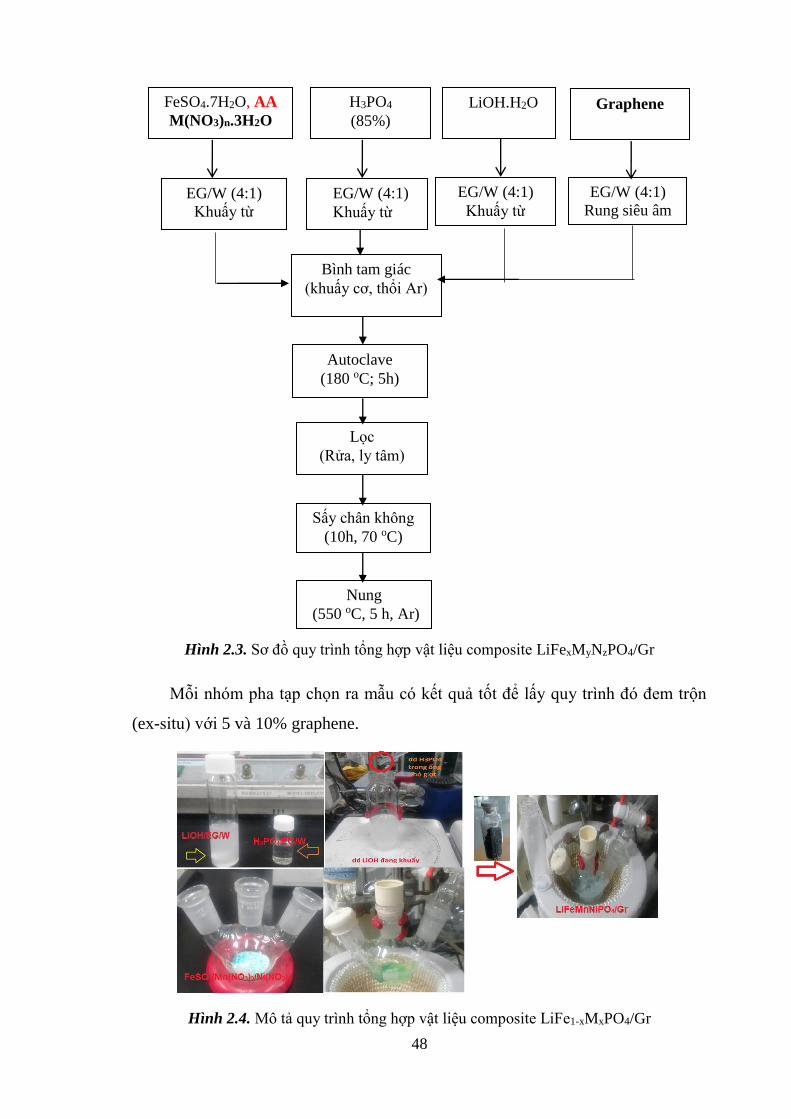
48
Hình 2.3. Sơ đồ quy trình tổng hợp vật liệu composite LiFexMyNzPO4/Gr
Mỗi nhóm pha tạp chọn ra mẫu có kết quả tốt để lấy quy trình đó đem trộn
(ex-situ) với 5 và 10% graphene.
Hình 2.4. Mô tả quy trình tổng hợp vật liệu composite LiFe1-xMxPO4/Gr
Autoclave
(180 oC; 5h)
Lọc
(Rửa, ly tâm)
Sấy chân không
(10h, 70 oC)
Nung
(550 oC, 5 h, Ar)
Bình tam giác
(khuấy cơ, thổi Ar)
FeSO4.7H2O, AA
M(NO3)n.3H2O
H3PO4
(85%)
LiOH.H2O
EG/W (4:1)
Khuấy từ
EG/W (4:1)
Khuấy từ
EG/W (4:1)
Khuấy từ
Graphene
EG/W (4:1)
Rung siêu âm

49
2.2. Xác định hàm lượng Fe2+ và carbon trong mẫu
2.2.1. Xác định hàm lượng Fe2+ bằng phương pháp chuẩn độ
Sử dụng dung dịch 100 mL H2SO4/KMnO4 0,0625 M (cân 4,900 g KMnO4
vào 50 mL H2O) tỉ lệ 1:1. Cân chính xác 0,1000 g mẫu pha loãng trong 50 mL
dung dịch vừa pha 50 mL H2SO4. Nguyên tắc chuẩn độ khi dư một giọt KMnO4
thì làm dung dịch mẫu đổi màu tím hết hàm lượng Fe2+ trong mẫu phân tích:
Phương trình phản ứng: 2 3 2
4 25Fe MnO 8H 5Fe Mn 4H O
Khối lượng Fe xác định: 24 4
3
KMnO KMnOFem 5C V 56 10
Nếu gọi a (mol) của Fe có trong tiền chất, x là tỉ pha tạp của ion kim loại
Mn+, tỉ lệ mol pha trộn của tiền chất Li:Fe:M:P = 3:1:1 = 3a:a(1-x):ax:a
thì % Fe2+ theo lý thuyết trong mẫu được tính:
1 4
56a (1-x)% Fe (LiFe M PO ) 100
7 3a+[56a (1-x) M ax] +95ax x
Chi tiết hơn, nếu pha tạp 20% Mn thì hàm lượng Fe trong mẫu theo lý
thuyết sẽ là:
0.8 0.2 4
56a (1-0,2)% Fe (LiFe M PO ) 100
7 3a +[56a (1-0,2) 55 0,2a] +95a
0.8 0.2 4% Fe (LiFe M PO ) 26,07%
2.2.2. Xác định hàm lượng carbon bằng phương pháp hóa học và nhiệt
Mẫu sau khi nung và bảo quản trong bình hút ẩm, vật liệu cân 0,1000 g sau
đó hòa tan trong dung dịch H2SO4 đặc khuấy tan hết rồi thu được dung dịch có
chứa các hạt rắn màu đen chính là carbon (Hình 2.5).
Lọc, rửa chất rắn bằng ethanol, đem sấy và cân. Khối lượng chất rắn có
trong mẫu chính là khối lượng carbon trong mẫu.
Phương trình thể hiện tách carbon ra khỏi vật liệu bằng H2SO4
LiFePO4/C (rắn) + 4H+
Fe2+ + C (rắn) + PO43- + 1/2H2

50
Hình 2.5. Dung dịch chứa carbon sau khi phá mẫu
2.3. Phương pháp nghiên cứu và đánh giá tính chất vật liệu
2.3.1. Nhiễu xạ X-ray (XRD)
Nhiễu xạ tia X (X-rays Diffraction) là một công cụ phân tích cấu trúc tinh
thể vật liệu hiệu quả. Bên cạnh đó, phương pháp này có thể xác định sự hiện diện
của các tạp chất vì tia X có bước sóng rất nhỏ khoảng 0,01-10 nm, có khả năng
đâm xuyên cao nên được dùng để nghiên cứu cấu trúc vật liệu [150].
Khi một chùm tia X có bước sóng λ và cường độ I đi qua vật liệu, nếu tia
tới thay đổi phương truyền và thay đổi năng lượng gọi là tán xạ không đàn hồi.
Khi tia tới thay đổi phương truyền nhưng không thay đổi năng lượng gọi là tán xạ
đàn hồi [150-153].
Mối quan hệ giữa bước sóng của chùm tia X tới với khoảng cách d giữa các
mặt mạng (hkl) và góc θ, hiện tượng xảy ra nếu thỏa mãn hệ thức Vufl-Bragg:
2d.sinθ = n.λ (với n = 1, 2, 3…) (2.6)
Trong đó, d: khoảng cách giữa hai mặt phẳng tinh thể trong một họ mặt; θ:
góc giữa chùm tia tới và tia phản xạ; n: bậc nhiễu xạ; λ: bước sóng của tia X.
Từ thông số nhiễu xạ của XRD có thể xác định kích thước đơn tinh thể
(monocrystaline) thể bằng phương trình Scherrer [152].
cos2
L
K (2.7)
là bề số của một nửa của cường độ cực đại (FHWM), K là yếu tố hình dạng
không có thứ nguyên, có giá trị gần bằng phần tử đơn vị. Yếu tố hình dạng có một
giá trị điển hình khoảng 0,9 nhưng thay đổi theo hình dạng thực tế của các tinh
thể; λ là bước sóng của tia X; θ là góc nhiễu xạ Bragg 0 < θ< 90˚.

51
Vật liệu LFP, LiFe1-xMxPO4, LiFe1-xMxPO4/Gr được phân tích trên máy
nhiễu xạ tia X với bức xạ của Cu-Kα, λ =1,5406 Å, góc quét 2 = 0-90o; tốc độ
quét: 0,25 -1,00°.s-1.
Để xác định thành phần cấu trúc pha của vật liệu thông qua thông số mạng
của mặt nhiễu xạ bằng cách số sánh số lượng vạch, vị trí vạch và cường độ của các
vạch nhiễu xạ tia X đo được bằng thực nghiệm với số liệu phổ chuẩn LFP.
Vật liệu được đánh giá pha kết tinh tốt khi quan sát giản đồ nhiễu xạ tia X
có nền phổ thấp, cường độ mạnh, các peak nhọn và 5 peak có cường độ mạnh
trùng khớp với phổ chuẩn thì có thể đánh giá bước đầu pha được hình thành [154].
Các phần mềm phần mềm Match!2, phần mềm Xpert’s High Score được
được sử dụng để phân tích dữ liệu XRD trong luận án này.
2.3.2. Quang phổ Raman
Khi chiếu một nguồn sáng đơn sắc vào mẫu và cho chùm ánh sáng tán xạ
thường được quan sát theo phương vuông góc với chùm ánh sáng tới. Các photon
này tạo ra sự tán xạ không đàn hồi của các photon từ chùm tia tới trên phân tử.
Tại mỗi dịch chuyển dao động, cường độ hấp thụ I được xác định bởi định
luật Lambert -Beer:
cd
oI I e (2.8)
Với: I và Io lần lượt là cường độ của chùm ánh sáng tới và chùm ánh sáng
truyền qua, là hệ số hấp thụ phân tử; c và d lần lượt là nồng độ của mẫu và bề
rộng của mẫu.
Phổ Raman là kỹ thuật giúp xác định thành phần các dao động đặc trưng
trong cấu trúc vật liệu thông qua tần số dao động của các liên kết. Đặc biệt,
phương pháp này giúp xác định sự có mặt graphene và định tính tương đối kích
thước lớp màng mỏng grahene. Thiết bị Horiba Jobin Yvon LabRAM HR300
được dùng để đo phổ Raman được trang bị một thiết bị cảm biến điện tích làm mát
bằng chất lỏng nitơ CCD (cooled charge-coupled device), kính hiển vi đầu dò
Olympus BX41 với độ phóng đại 5-100 với công suất 10-25 mW và bước sóng
514,5 nm, độ phân giải của hệ đo là 2 cm-1. Dải phổ Raman thường 100-3000 cm-
1). Hệ đo sử dụng hai tia laser kích thích và 4 cách từ (600 dòng/mm đến 2400
dòng/mm). Việc điều khiển tốc độ gia nhiệt hoàn toàn chủ động bằng phần mềm
với sai số khoảng ± 0,1oC, vật liệu đo ở dạng bột [154].
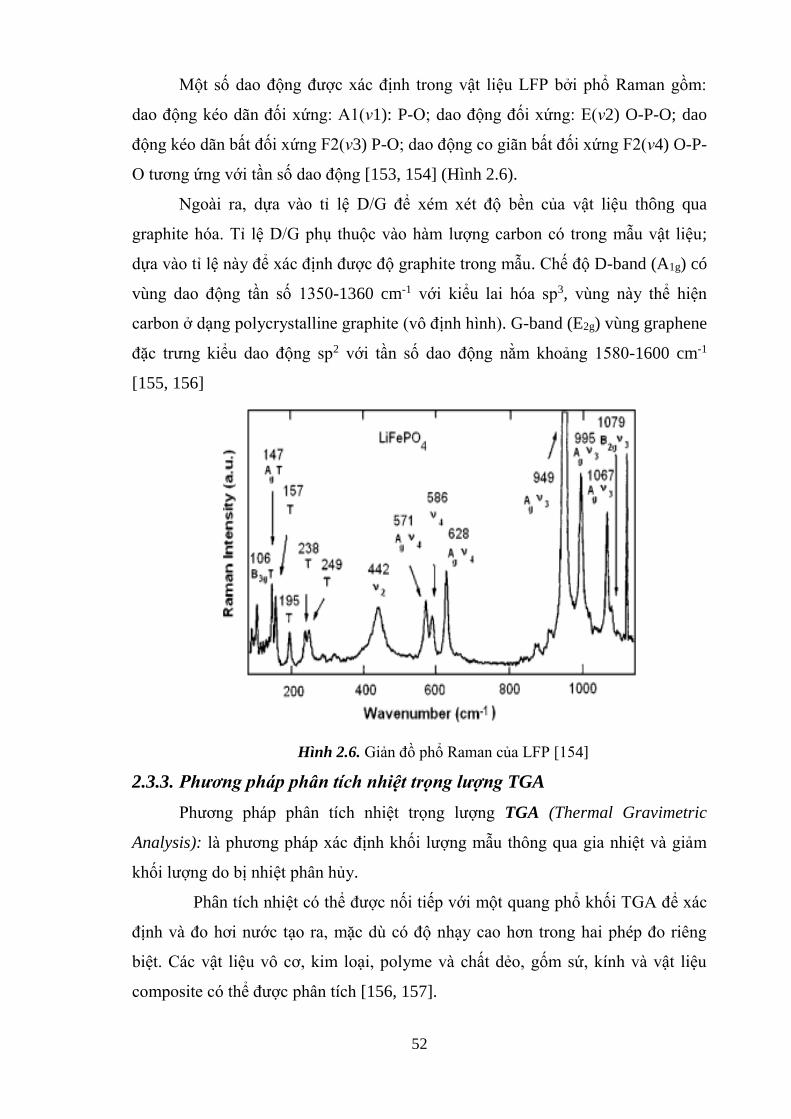
52
Một số dao động được xác định trong vật liệu LFP bởi phổ Raman gồm:
dao động kéo dãn đối xứng: A1(ν1): P-O; dao động đối xứng: E(ν2) O-P-O; dao
động kéo dãn bất đối xứng F2(ν3) P-O; dao động co giãn bất đối xứng F2(ν4) O-P-
O tương ứng với tần số dao động [153, 154] (Hình 2.6).
Ngoài ra, dựa vào tỉ lệ D/G để xém xét độ bền của vật liệu thông qua
graphite hóa. Tỉ lệ D/G phụ thuộc vào hàm lượng carbon có trong mẫu vật liệu;
dựa vào tỉ lệ này để xác định được độ graphite trong mẫu. Chế độ D-band (A1g) có
vùng dao động tần số 1350-1360 cm-1 với kiểu lai hóa sp3, vùng này thể hiện
carbon ở dạng polycrystalline graphite (vô định hình). G-band (E2g) vùng graphene
đặc trưng kiểu dao động sp2 với tần số dao động nằm khoảng 1580-1600 cm-1
[155, 156]
Hình 2.6. Giản đồ phổ Raman của LFP [154]
2.3.3. Phương pháp phân tích nhiệt trọng lượng TGA
Phương pháp phân tích nhiệt trọng lượng TGA (Thermal Gravimetric
Analysis): là phương pháp xác định khối lượng mẫu thông qua gia nhiệt và giảm
khối lượng do bị nhiệt phân hủy.
Phân tích nhiệt có thể được nối tiếp với một quang phổ khối TGA để xác
định và đo hơi nước tạo ra, mặc dù có độ nhạy cao hơn trong hai phép đo riêng
biệt. Các vật liệu vô cơ, kim loại, polyme và chất dẻo, gốm sứ, kính và vật liệu
composite có thể được phân tích [156, 157].

53
Vật liệu bị nung nóng thì khối lượng của chúng bị mất đi từ các quá trình
như bay hơi nước, phân hủy các gốc hữu cơ. Một số vật liệu có thể nhận được khối
lượng khi phản ứng với không khí trong môi trường kiểm tra. Khoảng nhiệt độ sử
dụng khi phân tích tùy thuộc vào từng loại vật liệu. Môi trường bao quanh có thể
là khí trơ hoặc khí tích cực. Khi phân tích kết quả, đường đạo hàm thường được sử
dụng để chỉ các điểm mất khối lượng hoặc để tăng độ phân giải.
Nhiệt độ thông thường phân tích từ 25-1200 °C. Nhiệt độ tối đa là
1400°C trên thiết bị. Trọng lượng mẫu có thể dao động từ 1-150 mg. Độ nhạy thay
đổi trọng lượng 0,01 mg. Thời gian thực hiện tối đa 50 phút, có thể thực hiện trong
môi trường khí trơ để đảm bảo khối lượng không bị ảnh hưởng do một số chất bị
oxi hóa. Các mẫu có thể được phân tích ở dạng bột hoặc các miếng nhỏ và tốc độ
quét 5-10 oC/ phút [157-158].
Để phân tích khối lượng tinh khiết và hàm lượng tạp chất trọng mẫu bằng
thiết bị Seratam LABSYS Evo TG-DSC (0-1600 oC) sử dụng phân tích nhiệt với
tốc độ gia nhiệt 5 oC/phút; 230 kV, 60 Hz) trong môi trường khí trơ. Dựa theo
nhiệt độ gia nhiệt tăng dần thì hàm lượng mẫu biến thiến theo giãn đồ phân tích
nhiệt %TG.
2.3.4. Phổ quang điện tử XPS
Phổ XPS (X-ray Photoelectron Spectroscopy) hay còn gọi ESCA (Electron
Spectroscopy for Chemical Analysis) được sử dụng để nghiên cứu thành phần hóa
học trong vật liệu thông qua năng lượng liên kết [159].
Do sự tương tác của điện tử với chùm photon hoặc electron năng lượng cao
điện tử phát xạ cấp 1 (điện tử quang điện). Một điện tử khác ở mức cao hơn nhảy
vào lỗ trống giải phóng năng lượng, truyền cho điện tử khác điện tử Auger. Cho
nên điện tử bật ra từ lớp điện tử hóa trị hoặc từ lớp điện tử trong cùng. Năng lượng
E của lớp này được cho bởi biểu thức [159, 160]:
E = h -BE- (2.10)
Năng lượng liên kết là đại lượng đặc trưng cho nguyên tử, từ đó có thể nhận
được một số thông tin quang trọng về mẫu nghiên cứu như các nguyên tố có mặt
Với h: hằng số Planck : tần số của photon, BE: năng lượng liên kết điện tử
: Công thoát điện tử

54
trong mẫu; hàm lượng phần trăm của mỗi nguyên tố và trạng thái hóa học của các
nguyên tố có mặt và đặc biệt xác định % hàm lượng Fe2+ và Fe3+ có trong mẫu.
Hệ thống Perkin-Elmer XPS, được trang bị máy phân tích hình trụ và Al
đơn sắc cao Kα (1486,6 eV) nguồn tia X. Viên nén nhỏ gọn của các vật liệu này
được chế tạo bằng thủy lực nhấn ở áp suất cao và gắn trên ngăn chứa mẫu bằng
băng keo hai mặt. Các buồng được duy trì ở áp suất xấp xỉ 10-9 torr trong thử
nghiệm. Các quan sát trong điều kiện năng lượng của mỗi phần tử được xác định
với tham chiếu đến cơ sở dữ liệu Perkin-Elmer [160].
Máy phổ quang điện tử tia X (XPS) AXIS-NOVA (Kratos) tại Trường Đại
học DGIST (Hàn Quốc) với bộ lọc AlK (hυ = 1486,6 eV), công suất nguồn 150 W,
áp suất là 2,6 × 10-9 Torr dùng để xác định thành phần liên kết hóa học có trong vật
liệu LFP và LiFe1-xMxPO4/Gr. Số liệu được xử lí trên phần mềm CASEXPS và vẽ
lại giản đồ bằng phần mềm Origin 8.5.
2.3.5. Phương pháp phổ hấp thụ nguyên tử AAS
Phổ AAS (Atomic Absorption Spectrometric) chủ yếu dựa trên sự phát xạ
hay hấp thu bức xạ của nguyên tử hoặc ion, tương ứng là các kĩ thuật phân tích
phổ hấp thu và phổ phát xạ. Đối tượng chủ yếu của phân tích phổ nguyên tử là các
nguyên tố kim loại do tín hiệu phổ nằm trong vùng có thể dễ quan sát và ghi nhận.
Các nguyên tố phi kim thường cho tín hiệu ra khỏi vùng tử ngoại nên ứng dụng
hạn chế hơn [161].
Phương pháp F-AAS sử dụng ngọn lửa đèn khí từ hỗn hợp acetylen –
không khí, N2O-acetylen hay hydro-acetylen. Nếu không ngọn lửa sử dụng dòng
điện có cường độ cao để tạo ra nhiệt làm hóa hơi mẫu. Thông thường quá trình này
xảy ra trong lò graphit và phương pháp tương ứng được gọi là phổ hấp thu nguyên
tử kĩ thuật lò graphit (GF-AAS) [161,162].
Trong đề tài này, thiết bị AA-6800 (Shimadzu, Nhật Bản) được sử dụng
phân tích phổ AAS với bức xạ kích thích có bước sóng 253,7 nm và độ nhạy là
0,1ppm, chế độ đo là CV-AAS.
Sử dụng phương pháp đường chuẩn để phân tích phổ hấp thụ nguyên tử
(AAS) kiểm chứng hàm lượng các nguyên tố kim loại có trong vật liệu. Vật liệu
được hòa tan bằng dung dịch HNO3. Khối lượng các ion kim loại trong dung dịch
tính toán theo công thức (mg/L):

55
Me = A(C + B)
Với Me: khối lượng ion kim loại
A: nồng độ kim loại được xác định theo đường chuẩn mg/L.
B: lượng dung môi pha loãng (mL).
C: định mức mẫu phân tích (mL).
2.3.6. Phổ tán sắc năng lượng tia X (EDS)
Nhiễu xạ điện tử EDS (Energy-Dispersive X-ray) là hiện tượng sóng điện
tử nhiễu xạ trên các mặt tinh thể của chất rắn dựa trên thuyết sóng của de Broglie.
Có thể giải thích hình ảnh nhiễu xạ điện tử và hiển vi điện tử khi chúng ta hiểu
được chùm hạt nhiễu xạ Bragg. Điện tử là các hạt điện tử vừa mang tính hạt vừa
mang tính chất sóng [163]. Xét một chùm điện tử có năng lượng cao dựa trên quy
tắc cơ học lượng tử sẽ tương đương với một sóng vật chất ta có phương trình tính
bước sóng điện từ
2
h h
p m eUo
(2.11)
Với: là hằng số Planck h; Khối lượng tĩnh v; Thế tăng tốc điện tử E; c=106 m/s tốc
độ ánh sáng; điện trường cao thế U. Thông thường máy EDS có U>100kV (TEM-
EDS); U>10kV (SEM-EDS).
Hiện nay phương pháp này dùng xác định thành phần nguyên tố trong vật
liệu và hàm lượng tham gia liên kết trong phân tử. EDS-Hitachi SU6600 (điện thế
6 kV, áp suất <10 pA, dòng thời gian 30-40 s, độ phân giải 127 eV) dùng để đo vật
liệu dạng bột. Số liệu được xử lí trên phần mềm EDS Edax Team.
SEM-EDS-Mapping là kỹ thuật hỗ trợ xác định sự có mặt các nguyên tố và
sự phân tán các nguyên tố trong mẫu [164].
2.3.7. Hiển vi điện tử quét SEM-FESEM
Hiển vi điện tử quét SEM (Scanning Electron Microscopy) hoặc hiển vi
điện tử quét phát xạ trường FeSEM (Field Emission Scanning Electron
Microscopy) là kỹ thuật để nghiên cứu hình thái bề mặt và đặc điểm cấu trúc vi mô
của mẫu như cũng như phân tích nguyên tố của vật liệu [165].
Khi một chùm tia tập trung các electron đơn năng bị ảnh hưởng trên một
mẫu, nó tạo ra các điện tử thứ cấp, các điện tử được phản chiếu lại tia X, ánh sáng

56
và nhiệt. Các điện tử thứ cấp và ghi lại được sử dụng cho hình ảnh và tia X được
sử dụng cho thành phần cơ bản của mẫu hiệu điện thế với 25-30 KV. Chùm
electron không chỉ tương tác với bề mặt mẫu mà còn có thể đi sâu vào thể tích
mẫu. Năng lượng của điện tử càng lớn thì thể tích tương tác càng lớn và quãng
đường điện tử đi được trong mẫu càng dài [166].
Trong công trình sử dụng máy Hitachi S-4800 (5 kV; 8,5 mm x 20,0 k) và
FESEM - Hitachi SU6600 (10 kV; 9,8 mm x 20,0 k).
2.3.8. Hiển vi điện tử truyền qua TEM và Hiển vi điện tử truyền qua phân
giải cao (HR-TEM)
Hiển vi điện tử truyền qua TEM (Transmission Electron Microscopy) là
một thiết bị nghiên cứu vi cấu trúc vật rắn sử dụng một chùm electron đơn sắc
(100-400 kV) để chụp ảnh, nơi mà các electron được truyền qua mẫu vật.
TEM hoạt động dựa trên nguyên tắc cơ bản của kính hiển vi quang học
nhưng do bước sóng rất nhỏ (Sóng de Broglie) của các electron năng lượng cao,
hình ảnh thu được ở độ phân giải cao so với SEM rất nhiều lần [166, 167].
Các chùm electron truyền đi, mang thông tin về cấu trúc vi mô của mẫu,
bao gồm cả hai nhóm electron phân tán trung tâm và Bragg. Các electron không bị
phân tán trung tâm được sử dụng để chụp ảnh trường sáng và các electron phân tán
Bragg được sử dụng hình ảnh trường tối [167].
Trong luận án này chủ yếu sử dụng máy TEM-JEM-1400 với độ phân giải
ảnh từ 0,20-0,38 nm; điện áp tăng từ 40-100 kW đa dạng độ phóng đại, hiệu suất
phân giải cỡ 2-3 Å. Trước khi đo TEM, bột vật liệu được phân tán trong dung môi
hữu cơ dễ bay hơi, sau đó mẫu ở dạng dung dịch được đưa vào máy, trong máy có
hệ thống sấy chân không tự động đảm bảo dung môi bay hết.
Hiển vi điện tử truyền qua độ phân giải cao (HR-TEM) là một chế độ ghi
ảnh của kính hiển vi điện tử truyền qua cho phép quan sát ảnh vi cấu trúc của vật
rắn với độ phân giải rất cao, đủ quan sát được sự tương phản của các lớp nguyên
tử trong vật rắn có cấu trúc tinh thể tới cấp độ nguyên tử.
Khác với các ảnh TEM thông thường có độ tương phản chủ yếu là tương
phản biên độ do hiệu ứng hấp thụ thì kỹ thuật High-Resolution Transmission
Electron Microscopy (HRTEM) hoạt động dựa trên nguyên lý tương phản pha, tức
là ảnh tạo ra nhờ sự giao thoa giữa chùm tia thẳng góc và chùm tia tán xạ. Khi

57
chùm điện tử chiếu qua mẫu (có chiều dày, độ sạch và sự định hướng thích hợp) sẽ
bị tán xạ nhiều hướng khác nhau và sóng tán xạ sẽ ghi lại thông tin về cấu trúc, vị
trí các nguyên tử... Vật kính phải có độ quang sai đủ nhỏ và có độ phân giải điểm
đủ lớn để hội tụ các chùm tán xạ này, thực hiện việc giao thoa với chùm chiếu
thẳng góc để tạo ra ảnh có độ phân giải cao [166, 167].
2.3.9. Phương pháp xác định các thông số điện hóa
Hiệu suất điện hóa được phân tích và đo lường bằng các phương pháp: Quét
thế vòng tuần hoàn (CV); Phổ tổng trở (EIS). Xác định tốc độ sạc xả cố định dòng
(charge-discharge).
2.3.9.1. Phương pháp quét thế vòng tuần hoàn (CV)
Quét thế vòng tuần hoàn là phương pháp dùng để xác định điện thế theo sự
biến đổi cường độ dòng điện tuyến tính theo thời gian. Mối quan hệ điện hóa giữa
chất oxy hóa (Ox) và chất khử (Red) qua phương trình thế oxi hóa-khử Nerst:
ln( )o
o
R
CRTE E
nF C (2.13)
Thế được quét từ giá trị đầu đến giá trị thế thứ hai, sau đó quay ngược lại
giá trị thế ban đầu hay về một giá trị cuối nào đó. Khi quét thế về phía âm, điện
cực nhận điện tử và trở thành chất khử mạnh. Chất oxi hóa sẽ bị khử trở lại chất
khử ban đầu cho giá trị dòng cathode. Khi quét thế về phía dương, chất khử bị oxi
hoá, điện cực đóng vai trò là chất oxi hóa. Tính oxi hóa của điện cực ngày càng
mạnh khi áp thế càng dương. Kết quả nhận được dòng anode. Sự hình thành của
chất oxi hóa và chất khử trên bề mặt điện cực sẽ cung cấp thông tin về nhiệt động
học của cặp oxi hóa khử và động học của phản ứng điện hóa [168, 169].
Từ đường cong CV, xác định các thông số mật độ dòng cathode (Ipc), mật
độ dòng anode (Ipa), thế oxi hóa khử của vật liệu ở nhiều tốc độ. Dựa trên phương
trình Randles-Sevcik, có thể xây dựng mối quan hệ tuyến tính giữa cường độ dòng
cathode (Ipc) theo căn bậc hai tốc độ quét thế (v1/2) và từ đó giúp xác định hệ số
khuếch tán của ion Li+ (DLi+) trong cấu trúc của vật liệu điện cực dương:
5 3/2 1/2 1/2 *
P (2,69 10 )n AD Ci
Với cường độ dòng điện của đỉnh cathode (A), số electron trao đổi của cặp
oxy hóa (với ion Li+ là 1), tốc độ quét thế (V.s-1), diện tích điện cực (cm2), Li+ hệ số
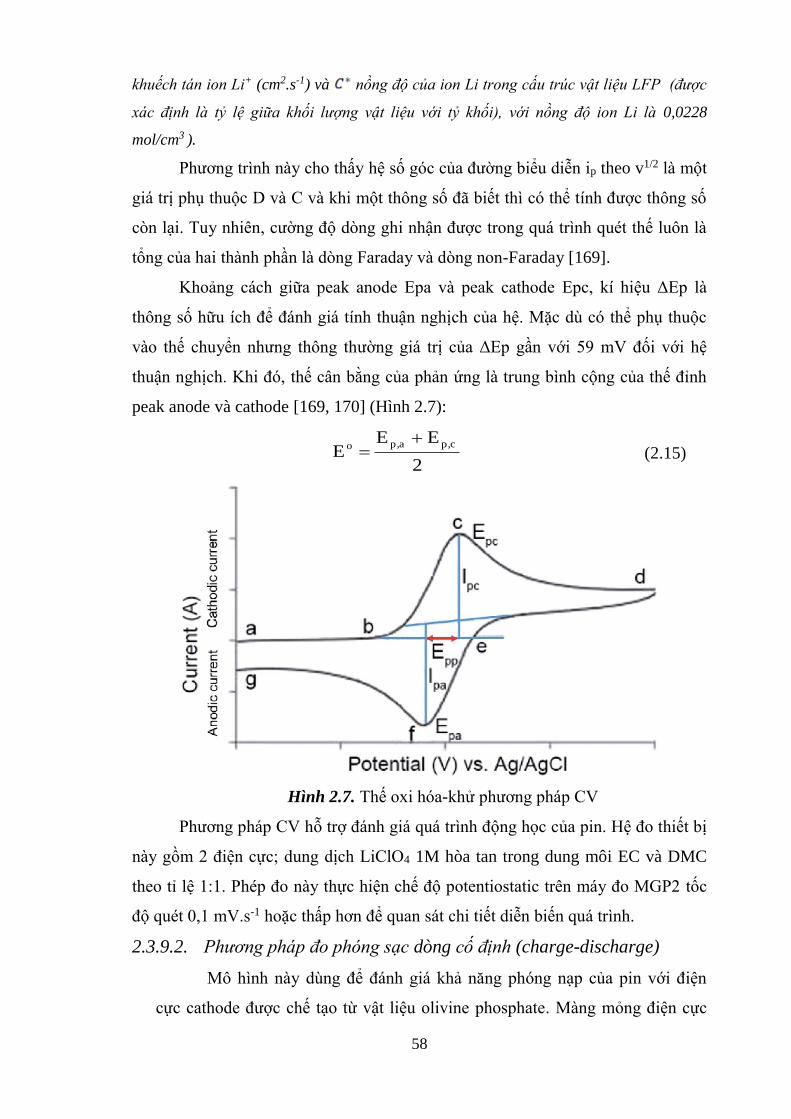
58
khuếch tán ion Li+ (cm2.s-1) và nồng độ của ion Li trong cấu trúc vật liệu LFP (được
xác định là tỷ lệ giữa khối lượng vật liệu với tỷ khối), với nồng độ ion Li là 0,0228
mol/cm3 ).
Phương trình này cho thấy hệ số góc của đường biểu diễn ip theo v1/2 là một
giá trị phụ thuộc D và C và khi một thông số đã biết thì có thể tính được thông số
còn lại. Tuy nhiên, cường độ dòng ghi nhận được trong quá trình quét thế luôn là
tổng của hai thành phần là dòng Faraday và dòng non-Faraday [169].
Khoảng cách giữa peak anode Epa và peak cathode Epc, kí hiệu ΔEp là
thông số hữu ích để đánh giá tính thuận nghịch của hệ. Mặc dù có thể phụ thuộc
vào thế chuyển nhưng thông thường giá trị của ΔEp gần với 59 mV đối với hệ
thuận nghịch. Khi đó, thế cân bằng của phản ứng là trung bình cộng của thế đỉnh
peak anode và cathode [169, 170] (Hình 2.7):
2
EEE
cp,ap,o
(2.15)
Hình 2.7. Thế oxi hóa-khử phương pháp CV
Phương pháp CV hỗ trợ đánh giá quá trình động học của pin. Hệ đo thiết bị
này gồm 2 điện cực; dung dịch LiClO4 1M hòa tan trong dung môi EC và DMC
theo tỉ lệ 1:1. Phép đo này thực hiện chế độ potentiostatic trên máy đo MGP2 tốc
độ quét 0,1 mV.s-1 hoặc thấp hơn để quan sát chi tiết diễn biến quá trình.
2.3.9.2. Phương pháp đo phóng sạc dòng cố định (charge-discharge)
Mô hình này dùng để đánh giá khả năng phóng nạp của pin với điện
cực cathode được chế tạo từ vật liệu olivine phosphate. Màng mỏng điện cực

59
được chế tạo với đường kính khoảng 6-10 mm, hệ điện giải gồm LiPF6 và dung
môi EC-DMC (1:1). Điện cực anode gồm kim loại lithium đường kính 10 mm.
Giữa 2 điện cực có màng ngăn làm bằng thủy tinh. Pin được ráp trong glove
box. Thiết bị MGP2 dùng để đo và lập trình phần mềm chuyên dụng. Nguyên lí
đo đạc là áp dòng và áp thế trong vùng 3,6-4,2 V tại C/10.
Q dung lượng lí thuyết Q =26802 26802
170Ah/kgM 158
xC
= 170 mAh.g-1 (2.16)
Với M: khối lượng Mol, x là số điện tử trao đổi
Phương pháp đo phóng sạc dòng cố định là kỹ thuật điện hóa dùng để đánh
giá hiệu năng pin hoặc siêu tụ điện hóa. Kỹ thuật này áp một dòng điện cố định
vào hệ điện hóa đang khảo sát và theo dõi sự thay đổi thế theo thời gian. Thế sẽ bị
giới hạn trong một khoảng giá trị nhất định, thông thường là khoảng thế so với
lithi hoặc natri. Khi thế đạt đến một giá trị cố định đã được thiết lập trước, dòng
điện sẽ đổi chiều và chu trình sẽ lặp đi lặp lại cho đến khi hoàn thành số chu kỳ
cần khảo sát.
Điện lượng Q ở mỗi chu kỳ phóng sạc được xác định thông qua tích số giữa
thời gian t và dòng cố định I trên một đơn vị khối lượng vật liệu đang khảo sát. Q
thường được biểu diễn bằng đơn vị mAh.g-1. Đối với vật liệu đan cài đã biết rõ
công thức hợp chất có thể tính được dung lượng lý thuyết của vật liệu. Mật độ
dòng là một thông số quan trọng để đánh giá pin. Mật độ dòng C/N nghĩa là quá
trình phóng hoặc sạc của hệ điện hóa sẽ hoàn thành trong N giờ. Để đánh giá đúng
tính năng điện hóa trước sau trộn graphene và pha tạp kim loại (Mn, Ni, Y) các
mẫu vật liệu tổng hợp cùng điều kiện nhiệt động, hóa chất và phương pháp từ đó
khảo sát và so sánh [169, 170]
2.3.9.3. Phương pháp tổng trở (EIS)
Phương pháp phổ tổng trở điện hóa EIS (Electrochemical Impedance
Spectroscopy) là phương pháp nghiên cứu cơ chế phản ứng điện hóa bao gồm quá
trình động học, lớp kép, phản ứng hóa học…; xác định được tổng trở và tìm mối
liên quan giữa tổng trở và độ dẫn điện [171].
Phương pháp EIS dựa trên tín hiệu điện áp đến một điện cực hóa có dạng
sin và lệch pha với dao động đặt vào. Phản ứng điện cực với nhiễu loạn điện áp có

60
tần số giống như nhiễu loạn và thường được dịch chuyển giai đoạn. Các phép đo
EIS trong nghiên cứu này được thực hiện bằng cách sử dụng hệ thống đo lường
(EIS 300) ở dải tần số từ 0,1 đến 100 kHz với biên độ AC 10 mV [171, 172].
Nguyên lý của phương pháp này là đặt một tín hiệu xoay chiều hình sin
V()=V0sin(t) với tần số góc f=/2 lên hai cực của mẫu và thu được dòng điện
I() = I0sin(t+) với là góc lệch pha giữa điện áp và dòng điện [173].
Trở kháng (Z) được xác định bằng biểu thức (Hình 2.8):
jj eZe
I
V
I
VZ
0
0
)(
)()( (2.17)
M
| Z |
| Z
| s
in
-Im
(Z)
| Z | cos Re(Z)
H×nh II.5.1.BiÓu diÔn vector Fresnel trong mÆt ph¼ng phøc.
Hình 2.8.Vectơ Fresnel trong mặt phẳng M [171]
Trở kháng tổng của hệ ghép nối tiếp bằng tổng các trở kháng thành phần
trong mạch được biễu diễn phương trình sau (1.19):
2
2 2( ) [R ] [ ]
1 ( ) 1 ( )
ct ctS
ct ct
R R CZ j Z jZ
R C R C
(2.18)
Mối quan hệ giữa Z và Z’’ phương trình sau:
2 2 2[( ) ] ( ) ( )2 2
ct cts
R RZ R Z (2.19)
Phương trình bán nguyệt tâm nằm trên trục thực (Rs + Rct /2, 0) và bán kính
Rct/2. Đường cong bán nguyệt cắt trục thực tại điểm Rs khi và điểm
(Rs+Rct) khi 0. Độ lớn cực đại max / 2ctZ R dẫn đến RctC = 1.
Bởi vậy, nếu xác định 0 tại đỉnh của bán nguyệt ta sẽ tìm được giá trị của
tụ điện C Warburg (Hình 2.9):
C = 1 / 0 Rct (2.20)

61
Hình 2.9. Mạch điện tương đương của chất điện môi
Phản ứng điện cực chất rắn/điện cực kim loại theo cơ chế khuếch tán xảy ra ở
vùng tần số thấp. Chúng được mô tả bởi trở kháng của quá trình khuếch tán Zdiff
được xác định từ định luật Fick II:
2
2 ),(),(
x
txcD
t
txc
(2.21)
Với D là hệ số khuếch tán, c(x, t) là gradient nồng độ tại điểm cách phân
biên chất điện phân/điện cực một khoảng bằng x.
Trong trường hợp lớp khuếch tán có chiều dài bán vô hạn, giải phương trình
Fick II sẽ nhận được biểu thức của trở kháng Warburg [172, 173]
Zj
w
( )1 (2.22)
Trong đó: RT
n F c D2 2
1
2 (2.23)
Trở kháng Warburg biểu diễn trên mặt phẳng Nyquist bao gồm một bán
nguyệt và một đường thẳng nghiêng một góc 45o so với trục trong vùng tần số cao.
Từ bán nguyệt này người ta có thể tìm được các giá trị của trở kháng liên quan đến
phản ứng khuếch tán và phản ứng truyền điện tích.
Kết quả là, hệ số khuếch tán lithium-ion của vật liệu có thể được tính toán
dựa theo phương trình sau:
2 4 4 2 2
RTD
2A n F C (2.24)
Với: R hằng số khí lí tưởng (8,314 J.mol-1K-1), T là nhiệt độ (K), A là diện tích điện
cực, n là số điện tử trao đổi. F là hằng số Faraday (96500 C.mol-1), C là nồng độ
mol ion Li+, hệ số Warburg.
Như vậy: Một điện cực của pin Li-ion được mô hình hóa bằng mạch tương
đương gồm có RS (điện trở chưa được bù), điện trở chuyển điện tích nối tiếp Rct
với tổng trở khuếch tán ZW (tổng trở Warburg) (Hình 2.10).
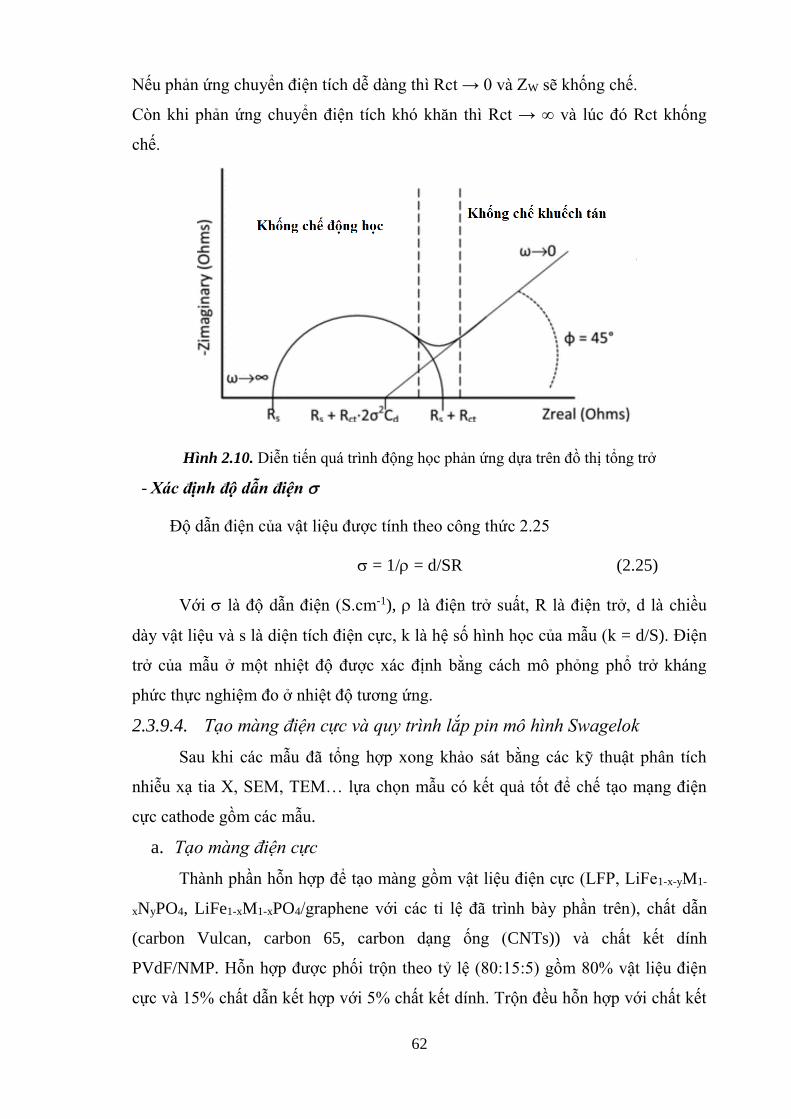
62
Nếu phản ứng chuyển điện tích dễ dàng thì Rct → 0 và ZW sẽ khống chế.
Còn khi phản ứng chuyển điện tích khó khăn thì Rct → ∞ và lúc đó Rct khống
chế.
Hình 2.10. Diễn tiến quá trình động học phản ứng dựa trên đồ thị tổng trở
- Xác định độ dẫn điện
Độ dẫn điện của vật liệu được tính theo công thức 2.25
= 1/ = d/SR (2.25)
Với là độ dẫn điện (S.cm-1), là điện trở suất, R là điện trở, d là chiều
dày vật liệu và s là diện tích điện cực, k là hệ số hình học của mẫu (k = d/S). Điện
trở của mẫu ở một nhiệt độ được xác định bằng cách mô phỏng phổ trở kháng
phức thực nghiệm đo ở nhiệt độ tương ứng.
2.3.9.4. Tạo màng điện cực và quy trình lắp pin mô hình Swagelok
Sau khi các mẫu đã tổng hợp xong khảo sát bằng các kỹ thuật phân tích
nhiễu xạ tia X, SEM, TEM… lựa chọn mẫu có kết quả tốt để chế tạo mạng điện
cực cathode gồm các mẫu.
a. Tạo màng điện cực
Thành phần hỗn hợp để tạo màng gồm vật liệu điện cực (LFP, LiFe1-x-yM1-
xNyPO4, LiFe1-xM1-xPO4/graphene với các tỉ lệ đã trình bày phần trên), chất dẫn
(carbon Vulcan, carbon 65, carbon dạng ống (CNTs)) và chất kết dính
PVdF/NMP. Hỗn hợp được phối trộn theo tỷ lệ (80:15:5) gồm 80% vật liệu điện
cực và 15% chất dẫn kết hợp với 5% chất kết dính. Trộn đều hỗn hợp với chất kết

63
dính rồi tiến hành cán màng trên mặt kính thủy tinh với màng nhôm. Ép màng
bằng máy với áp suất ép 50 MPa, thời gian ép 10 phút/ mẫu. Màng được sấy chân
không ở 100 oC trong 15 h sau đó được đục thành các màng nhỏ với đường kính
10 mm được thao tác trong buồng glovebox với áp suất khí argon < 2 ppm H2O.
b. Quy trình lắp pin mô hình Swagelok
Pin lắp ráp theo mô hình Swagelok được sử dụng cho các phép đo điện
hóa. Điện cực cathode là màng mỏng hỗn hợp chứa vật liệu tổng hợp được tạo
thành LFP, LiFexMxPO4/graphene sau khi đã chế tạo màng (mục a) được cắt
thành hình tròn với đường kính 10 mm, bề dày màng 2 mm. Pin mô hình được lắp
ráp trong buồng chân không (glovebox) để tránh không khí và oxygen (Hình 2.11).
Hệ điện giải gồm muối LiPF6 nồng độ 1M được pha trong hỗn hợp dung
môi EC:DMC:PC theo tỉ lệ 1:1:1 (tỉ lệ dung môi tính theo thể tích). Điện cực
cathode là kim loại lithi được cắt thành hình tròn với đường kính 10 mm. Màng
ngăn là màng sợi thủy tinh Whatman tẩm chất điện giải có đường kính được cắt là
12 mm.
Hình 2.11. Mô hình pin chuẩn Swagelok
Thiết bị kỹ thuật hiện đại là những công cụ hỗ trợ đặc lực phân tích vật liệu
nhất là vật liệu composite với kích thước hạt nhỏ và cấu trúc phức tạp.
Với các kỹ thuật tiên tiến sử dụng đáp ứng phân tích được cấu trúc vật liệu
và tính năng điện hóa để tối ưu vật liệu điện cực cathode cho pin Li-ion.

64
Chương 3. KẾT QUẢ VÀ THẢO LUẬN
3.1. VẬT LIỆU ĐIỆN CỰC LiFePO4 (LFP)
3.1.1. Xác định hàm lượng Fe và Fe2+ trong cấu trúc LFP
Pin sạc Li-ion hoạt động dựa trên cơ chế đan cài ion Li+ trên giao diện
Fe2+/Fe3+ thông qua quá trình oxi khử thuận nghịch. Bên cạnh đó, vật liệu LFP ổn
định khi hàm lượng Fe2+>12% khối lượng mẫu vật liệu LFP, LFMP hoặc
LFMP/Gr. Vì thế, đây là yếu tố giới hạn cho hàm lượng kim loại pha tạp phải đảm
bảo sao cho ion Fe2+ không thay thế quá nhiều làm ảnh hưởng đến cấu trúc chính
của vật liệu. Tuy nhiên, ion Fe2+ (LFP) không bền, dễ bị oxi hóa do đó, việc xác
định hàm lượng trong mẫu vật liệu là hết sức cần thiết vì nếu như hàm lượng Fe2+
quá chênh lệch so với lý thuyết (các mẫu LFP) dẫn đến ảnh hưởng sự ổn định của
cấu trúc trong quá trình phóng sạc.
Có nhiều phương pháp để xác định hàm lượng Fe2+ cụ thể như: phương
pháp chuẩn độ, phương pháp phổ khối lượng, chính xác hơn là phương pháp XPS.
Phương pháp chuẩn độ tương đối dễ thực hiện, dựa vào nguyên tắc lượng Fe2+ hiện
diện trong mẫu được khử bởi chất oxi hóa mạnh KMnO4 và H2SO4 đậm đặc.
Xác định % hàm lượng Fe (Fe2+, Fe3+) có trong mẫu có thể sử dụng phương
pháp AAS [26, 37], EDS [45, 56, 64] hoặc XPS [172, 173].
Để khảo sát đánh giá hàm lượng Fe2+ có trong mẫu vật liệu tiến hành quan
sát màu sắc mẫu sau khi tổng hợp nếu vật liệu có màu nâu vàng chứng tỏ đây là
màu của muối Fe3+, có thể kết luận sơ bộ vật liệu kết tinh kém và không nên tiếp
tục phân tích các kĩ thuật khác.
Các mẫu vật liệu chỉ đem chuẩn độ sau khi nung để loại tạp chất và kiểm
chứng kết quả XRD tốt. Ngoài ra, phương pháp AAS và EDS xác định hàm lượng
tổng từng nguyên tố kim loại có trong mẫu.
3.1.1.1. Phương pháp chuẩn độ
Chuẩn độ là phương pháp hóa học thông dụng hỗ trợ xác định sự có mặt
của Fe2+ trong vật liệu bằng chất oxi hóa KMnO4 và H2SO4 đậm đặc. Dựa vào số
liệu Bảng 3.1 để phân tích sự biến đổi % hàm lượng Fe2+ theo tính toán lý thuyết
và dựa vào số liệu của phương pháp chuẩn độ.

65
Tiến hành chuẩn độ mỗi mẫu ba lần lấy kết quả trung bình mẫu ST01 có
hàm lượng %Fe2+ chiếm 30,53% trong khi các mẫu không dùng ascorbic acid thì
hàm lượng Fe2+ thấp nhất chỉ chiếm 26,60%, các mẫu thay đổi tỉ lệ tiền chất ST02,
ST03 có hàm lượng Fe2+ cao hơn mẫu ST00 nhưng lại thấp hơn mẫu ST01 (Bảng
3.1b). Từ kết quả chuẩn độ ta thấy sự biến thiên tăng lần hàm lượng Fe2+
ST00<ST03<ST02<ST01
Như vậy, từ kết quả trên có thể thấy rằng hàm lượng Fe2+ phụ thuộc vào tác
nhân khử, cụ thể trong công trình này, ascorbic acid ngoài việc có chứa liên kết
C=O linh động đóng vai trò khử Fe3+ về Fe2+ [93, 94]. Mặt khác, môi trường 5 <
pH < 6,5 (axít) bền cho ion Fe2+.
2Fe3+ + C6H8O6 (ascorbic) => 2Fe2+ + 2H+ + C6H6O6
Phương XPS cho kết quả hàm lượng Fe2+ tham gia liên kết trong vật liệu
chiếm 14,9% phù hợp tài liệu [163].
Bảng 3.1a. Hàm lượng % Fe2+ trong LFP được xác định XPS
Mẫu
% Hàm lượng
Li1s O1s P2p Fe 2p3 (+2) C
ST00 5,1 61,0 15,0 13,9 5,0
ST01 5,3 60,0 15,1 14,9 4,9
Nguyên nhân hàm lượng Fe2+ chênh lệch giữa lý thuyết và kết quả phân tích
có thể ion Fe2+ bị oxi hóa thành Fe3+ vì điều kiện bảo quản mẫu chưa tốt hoặc có
thể do trong quá trình nung ở nhiệt độ cao một số muối hữu cơ bị phân hủy tạo ra
gốc oxi tự do kết hợp với hơi nước tạo thành một lượng nhỏ tạo thành Fe3O4.
2Fe2+ + O2 + 2H2O =>2Fe3+ (màu vàng) + 4OH-
3Fe2+ + 3O2 + 6OH- 570 ot CFe3O4 + 3H2O (Hơi nước)
Ngoài ra, sự chênh lệch % hàm lượng Fe2+ giữa chuẩn độ và lý thuyết có
thể một phần Fe2+ đã bị oxi hóa thành Fe3+ và do sai số chuẩn độ ngay điểm tương
đương.

66
Bảng 3.1b. Định lượng Fe2+ trong mẫu bằng phương pháp chuẩn độ
Mẫu V (ml)
KMnO4/H2SO4 (1:1)
Khối lượng
Fe có trong mẫu
% m Fe2+
chuẩn độ
% m Fe
lý thuyết
Nhân tố
tác động
ST00 15,2 0,2660 26,600 32,56 Tác nhân
ST01 17,5 0,3063 30,6250 32,56 Tỉ lệ tiền
chất khác
nhau ST02 16,3 0,2854 28,5250 32,96
ST03 15,7 0,2748 27,4750 33,23
3.1.2. Cấu trúc tinh thể và thành phần pha
3.1.2.1. Ảnh hưởng tỉ lệ tiền chất đến cấu trúc vật liệu LFP
Nhiễu xạ tia X (XRD) là phương pháp phân tích cấu trúc vật liệu được sử
dụng nhiều nhất vì ưu điểm nó là giúp xác định cấu trúc tinh thể, phát hiện các
pha tạp và dự đoán kích thước hạt.
Vật liệu LFP có cấu trúc bền olivine thuộc hệ trực thoi (nhóm không gian
Pbnm) với thông số mạng (a≠b≠c) và các góc thỏa mãn: 90o . Mặt khác
thể tích ô mạng được tính theo công thức:
22 21 2cos( )cos( )cos( ) cos ( ) cos ( ) cos ( )V abc abc
Sử dụng phần mềm phân tích dữ liệu XRD gồm Xpert’s high score và
Match!2. Kết quả là các mẫu vật liệu ST00, ST01, ST02 và ST03 có vạch rõ
ràng, đỉnh nhiễu xạ nhọn và có 26 peak trùng khớp với phổ LFP (JPCDS 96-101-
1112) (Hình 3.1b). Tiêu biểu một số đỉnh nhiễu xạ có cường độ lớn: [200]; [101];
[111]; [211]; [311] tương ứng 2 = 36o, 30o, 26o, 15o (Hình 3.1b).
Cấu trúc olivine của LFP tổng hợp bằng phương pháp Nhiệt dung môi
được kiểm chứng bằng chức năng Retveld Refinement (Hình 3.1c) để xác định
chính xác sự trùng lặp của các mặt nhiễu xạ, kết quả thu được được thành phần
pha hoàn tòn phù hợp với phổ chuẩn LFP, có một vài vị trí peak lạ năm trong
khoảng ứng 2 = 25-30o là vùng có mặt nhiễu xạ của carbon [002], tuy nhiên đây
sự dự đoán vì cường độ peak rất nhỏ. Kết quả này thích hợp với hiệu suất phản
ứng <90 % vì pha chưa hoàn toàn sạch.

67
Ngoài ra, từ dữ liệu XRD hỗ trợ xác định thông số mạng, từ kết quả này
có thể xác định thể tích ô mạng olivine của LFP (Bảng 3.3), số liệu về thể tích và
thông số mạng phù hợp số liệu [5, 46].
Hình 3.1a. Giản đồ XRD của LFP so sánh với phổ chuẩn LFP
Hình 3.1b. Xác định các mặt nhiễu xạ bằng phần mềm Match!2
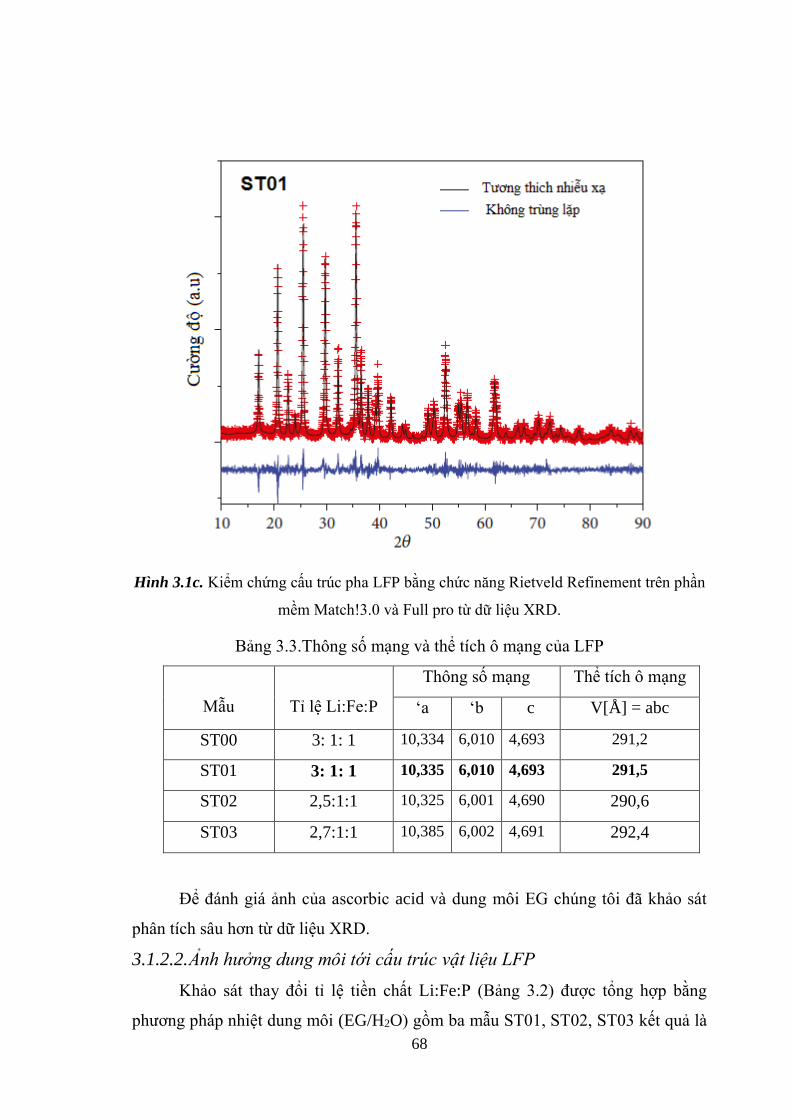
68
Hình 3.1c. Kiểm chứng cấu trúc pha LFP bằng chức năng Rietveld Refinement trên phần
mềm Match!3.0 và Full pro từ dữ liệu XRD.
Bảng 3.3.Thông số mạng và thể tích ô mạng của LFP
Mẫu
Tỉ lệ Li:Fe:P
Thông số mạng Thể tích ô mạng
‘a ‘b c V[Å] = abc
ST00 3: 1: 1 10,334 6,010 4,693 291,2
ST01 3: 1: 1 10,335 6,010 4,693 291,5
ST02 2,5:1:1 10,325 6,001 4,690 290,6
ST03 2,7:1:1 10,385 6,002 4,691 292,4
Để đánh giá ảnh của ascorbic acid và dung môi EG chúng tôi đã khảo sát
phân tích sâu hơn từ dữ liệu XRD.
3.1.2.2. Ảnh hưởng dung môi tới cấu trúc vật liệu LFP
Khảo sát thay đổi tỉ lệ tiền chất Li:Fe:P (Bảng 3.2) được tổng hợp bằng
phương pháp nhiệt dung môi (EG/H2O) gồm ba mẫu ST01, ST02, ST03 kết quả là

69
cường độ peak rõ và mạnh, nền phổ thấp chứng tỏ pha kết tinh tốt và không phát
hiện tạp so với mẫu ST00 không sử dụng dung môi hữu cơ. Điều này hoàn toàn
phù hợp với khuynh hướng giải thích dung môi làm tăng độ nhớt do đó kiểm soát
được kích thước hạt, dung môi hữu cơ EG không linh động bằng H2O nên sẽ
không tham gia quá trình phản ứng do đó, hạn chế xuất hiện pha lạ ngoài mong
muốn [141].
Với thang chia cường độ counts mẫu ST01 cường độ peak dao động 0-150
trong khi đó mẫu ST02 (0-70) mẫu ST03 (0-50) điều này chứng tỏ rằng thay đổi
tiền chất sẽ ảnh hưởng quá trình hình thành pha dẫn đến cường độ peak khác nhau
[13] (Hình 3.2). Như vậy tỉ lệ Li:Fe:P = 3:1:1 cho kết quả tổng hợp vật liệu tốt hơn
so với các tỉ lệ khác so sánh kiểm chứng với các tài liệu [13, 19].
Mẫu ST00 (không dùng AA và EG) làm mẫu đối chứng so sánh với các
mẫu trên để đánh giá vai trò tác nhân và dung môi. Từ hình 3.3 có thể suy luận
rằng mẫu ST00 các đỉnh nhiễu xạ có cường độ thấp nhất trong bốn mẫu và số
lượng peak cũng ít hơn so với các mẫu còn lại chứng tỏ mẫu ST00 kết tinh kém,
nền phổ cao và còn tạp Li3PO4 ứng với 36 và 43o; Fe3(PO4)2.3H2O tại 28o; FeO2
tại 24o. So sánh với mẫu ST00 với ST01 rõ ràng cường độ mẫu ST01 mạnh hơn
gấp hai lần.
Dung dịch Fe2+ (FeSO4) không bền trong môi trường kiềm và tiếp xúc với
không khí ẩm nên dễ bị oxi hóa thành Fe3+, để khử Fe3+ về Fe2+ sử dụng ascorbic
acid làm tác nhân khử.
2Fe3+ + C6H8O6 => 2Fe2+ + 2H+ + C6H6O6
Như vậy: Dung môi EG/H2O và tác chất khử ascorbic acid có vai trò hỗ trợ
vật liệu kết tinh tốt và hạn chế tạp.
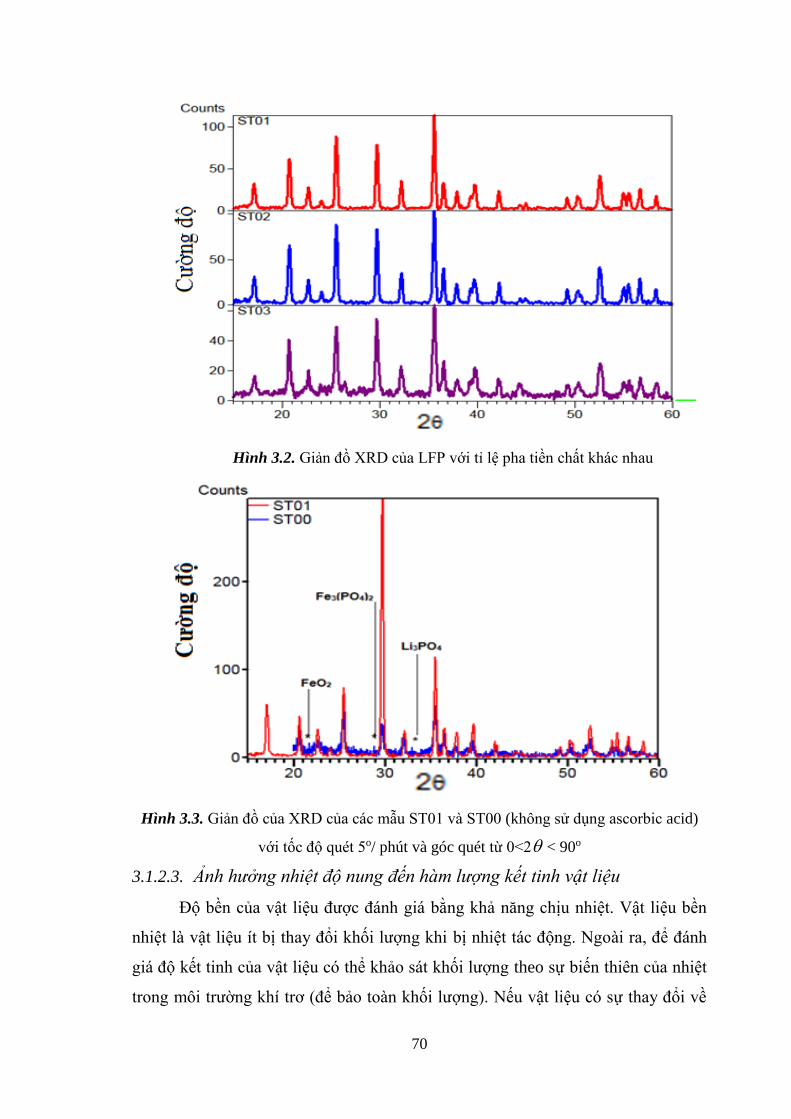
70
Hình 3.2. Giản đồ XRD của LFP với tỉ lệ pha tiền chất khác nhau
Hình 3.3. Giản đồ của XRD của các mẫu ST01 và ST00 (không sử dụng ascorbic acid)
với tốc độ quét 5o/ phút và góc quét từ 0<2 < 90o
3.1.2.3. Ảnh hưởng nhiệt độ nung đến hàm lượng kết tinh vật liệu
Độ bền của vật liệu được đánh giá bằng khả năng chịu nhiệt. Vật liệu bền
nhiệt là vật liệu ít bị thay đổi khối lượng khi bị nhiệt tác động. Ngoài ra, để đánh
giá độ kết tinh của vật liệu có thể khảo sát khối lượng theo sự biến thiên của nhiệt
trong môi trường khí trơ (để bảo toàn khối lượng). Nếu vật liệu có sự thay đổi về

71
khối lượng trong vật liệu có thể có những pha khác khối lượng tương đối nhỏ hơn
5% thì XRD không phát hiện tạp. Đối với vật liệu LiFePO4 được đánh giá là vật
liệu bền nhiệt từ 300-600 oC) [32], tuy nhiên các muối Fe3(PO4)2 bị nhiệt phân ở
nhiệt độ 200-230 oC. Hơi nước, các muối hữu cơ sẽ chuyển pha về dạng hơi nước,
carbon và CO2 từ 100-300 oC. Đây là phương pháp dùng để dự đoán nhiệt độ kết
tinh làm cơ sở nung vật liệu. Từ giản đồ nhiệt biến thiên hàm lượng %TG theo
nhiệt độ dùng đánh giá hàm lượng tạp có trong mẫu từ đó tính khối lượng tinh
khiết của vật liệu.
% Hàm lượng kết tinh của pha LFP = 1
2
100 %TGm
m (3.2)
m1: Khối lượng đem phân tích TGA; m2: Khối lượng sau phân tích TG
- Giai đoạn từ 30-300 oC, khối lượng LFP giảm mạnh (18-20%) điều này có thể
giải do mất hơi nước và các gốc hữu cơ bị phân hủy chuyển thành CO2 và
H2O.
- Giai đoạn 300-500 oC khối lượng giảm nhẹ (2-3%) các gốc anion SO42-, OH-
(bazơ) hoặc muối Fe3(PO4)2 bị nhiệt phân.
- Giai đoạn 550- 900 oC khối lượng giảm không đáng kể. Hàm lượng kết tinh
vật liệu LFP kết tinh là 85 % phù hợp với kết quả XRD và tài liệu [32, 109].
Như vậy, vùng nhiệt độ vật liệu kết tinh ổn định nhiệt độ nằm khoảng 500-650 oC.
Từ giản đồ TGA kết hợp XRD thay đổi nhiệt độ nung vật liệu (Hình 3.5) cho thấy:
vật liệu bắt đầu kết tinh từ 300 oC và pha tinh khiết tốt > 500 oC. Hàm lượng kết
tinh chiếm <80% (Hình 3.4).

72
Hình 3.4. Giản đồ phân tích nhiệt biến thiên nhiệt độ và %TG của mẫu ST01
Hình 3.5. Giản đồ XRD so sánh ảnh hưởng nhiệt độ nung đến thành phần pha
Bảng 3.4. Sự biến thiến khối lượng bằng phương pháp phân tích nhiệt

73
ST01-Lần 1 ST01-Lần 2
Nhiệt độ TG (%) % m Nhiệt độ TG (%) % m
28,83 100,00 -
31,03 100,00 -
184,22 90,34 9,64 156,22 90,88 9,12
300,78 82,92 17,08 300,76 80,76 19,24
400,05 82,72 17,28 400,06 80,64 19,36
500,05 82,33 17,67 500,05 80,44 19,56
550,06 82,01 17,99 550,04 80,04 19,96
650,04 81,08 18,92 650,01 79,60 20,40
750,04 80,83 18,17 750,04 79,80 20,20
900,06 81,40 19,60 900,4 79,90 20,10
3.1.2.4. Hiệu suất phản ứng (H), khối lượng kết tinh (T)của mẫu
Trong quá trình hình thành pha LFP sẽ có những pha lạ hình thành đồng
thời hình thành vì thế vật liệu sẽ không đảm bảo 100% khối lượng lý thuyết
(mLiFePO4 = mLi + mFe + mPO4 = 1,720 g nếu số mol Li = 0,03 mol và tỉ lệ
Li:Fe:P=3:1:1). Do đó, khối lượng kết tinh thực tế = khối lượng lý thuyết – khối
lượng tạp.
Hiệu suất phản ứng là yếu tố quan trọng để tính giá thành của vật liệu cũng
hiệu quả phương pháp tổng hợp. Hiệu suất được tính như sau:
2H= 100o
m
m (3.3)
Hiệu suất phản các ứng có khuynh hướng tăng sau khi pha tạp (< 80%)
chứng tỏ quy trình này mang lại hiệu suất khá tốt nhất là các mẫu pha tạp hiệu suất
đạt > 86%; trong đó một số mẫu hiệu suất cao nổi trội STN2; STM2; STY2. Hiệu
suất phản ứng sau khi pha tạp tăng lên là dấu hiệu khởi đầu rất tốt có thể giải thích
các kim loại pha tạp có tính oxi hóa mạnh hơn Fe2+ hạn chế bị oxi hóa.
M2+ + Fe3+ M3+ + Fe2+
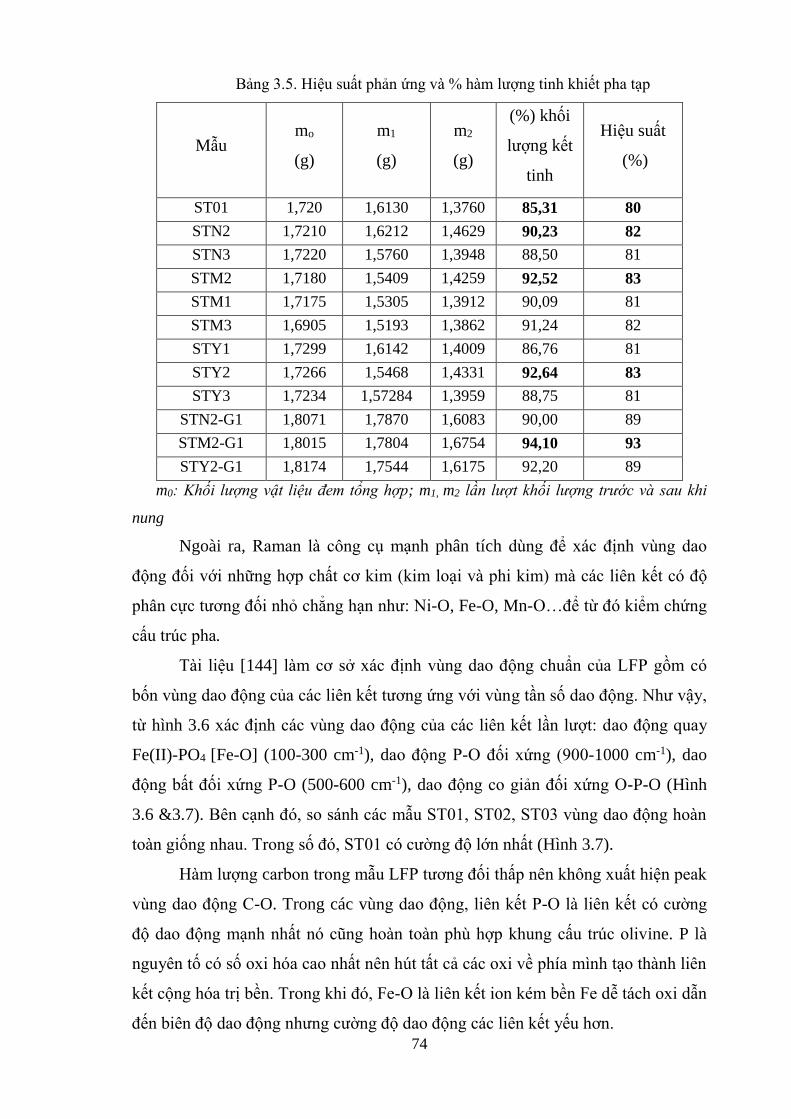
74
Bảng 3.5. Hiệu suất phản ứng và % hàm lượng tinh khiết pha tạp
Mẫu mo
(g)
m1
(g)
m2
(g)
(%) khối
lượng kết
tinh
Hiệu suất
(%)
ST01 1,720 1,6130 1,3760 85,31 80
STN2 1,7210 1,6212 1,4629 90,23 82
STN3 1,7220 1,5760 1,3948 88,50 81
STM2 1,7180 1,5409 1,4259 92,52 83
STM1 1,7175 1,5305 1,3912 90,09 81
STM3 1,6905 1,5193 1,3862 91,24 82
STY1 1,7299 1,6142 1,4009 86,76 81
STY2 1,7266 1,5468 1,4331 92,64 83
STY3 1,7234 1,57284 1,3959 88,75 81
STN2-G1 1,8071 1,7870 1,6083 90,00 89
STM2-G1 1,8015 1,7804 1,6754 94,10 93
STY2-G1 1,8174 1,7544 1,6175 92,20 89
m0: Khối lượng vật liệu đem tổng hợp; m1, m2 lần lượt khối lượng trước và sau khi
nung
Ngoài ra, Raman là công cụ mạnh phân tích dùng để xác định vùng dao
động đối với những hợp chất cơ kim (kim loại và phi kim) mà các liên kết có độ
phân cực tương đối nhỏ chẳng hạn như: Ni-O, Fe-O, Mn-O…để từ đó kiểm chứng
cấu trúc pha.
Tài liệu [144] làm cơ sở xác định vùng dao động chuẩn của LFP gồm có
bốn vùng dao động của các liên kết tương ứng với vùng tần số dao động. Như vậy,
từ hình 3.6 xác định các vùng dao động của các liên kết lần lượt: dao động quay
Fe(II)-PO4 [Fe-O] (100-300 cm-1), dao động P-O đối xứng (900-1000 cm-1), dao
động bất đối xứng P-O (500-600 cm-1), dao động co giản đối xứng O-P-O (Hình
3.6 &3.7). Bên cạnh đó, so sánh các mẫu ST01, ST02, ST03 vùng dao động hoàn
toàn giống nhau. Trong số đó, ST01 có cường độ lớn nhất (Hình 3.7).
Hàm lượng carbon trong mẫu LFP tương đối thấp nên không xuất hiện peak
vùng dao động C-O. Trong các vùng dao động, liên kết P-O là liên kết có cường
độ dao động mạnh nhất nó cũng hoàn toàn phù hợp khung cấu trúc olivine. P là
nguyên tố có số oxi hóa cao nhất nên hút tất cả các oxi về phía mình tạo thành liên
kết cộng hóa trị bền. Trong khi đó, Fe-O là liên kết ion kém bền Fe dễ tách oxi dẫn
đến biên độ dao động nhưng cường độ dao động các liên kết yếu hơn.
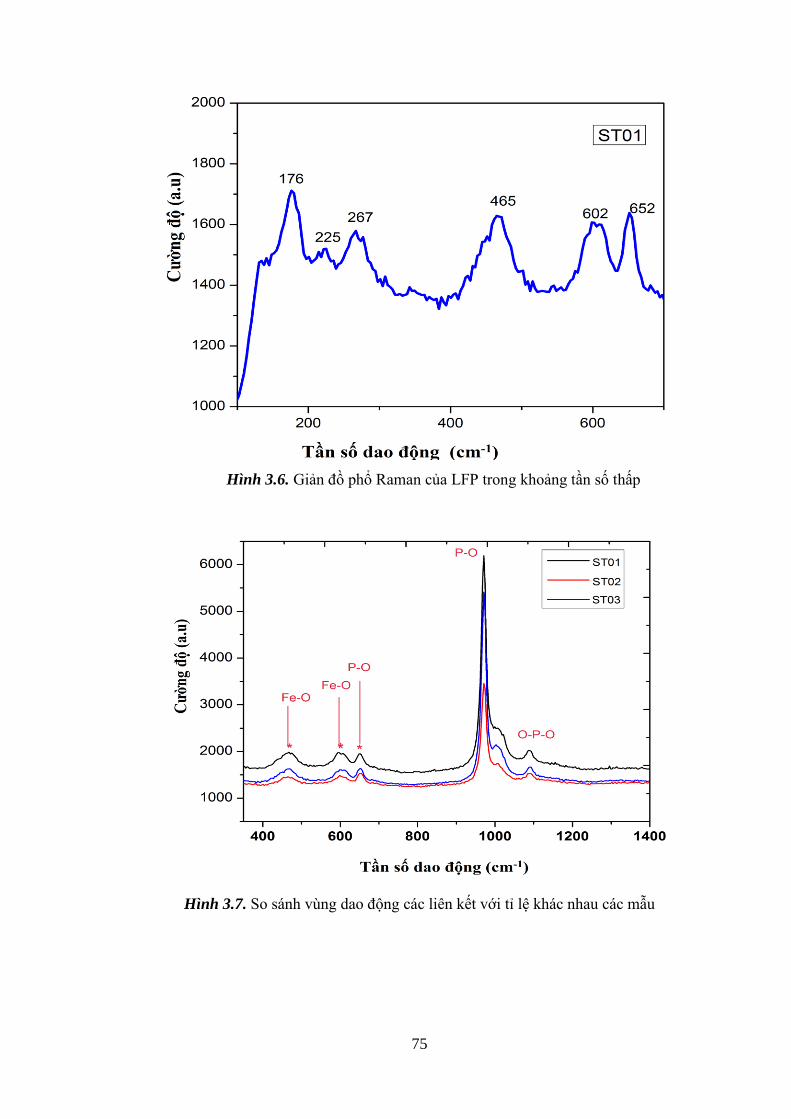
75
Hình 3.6. Giản đồ phổ Raman của LFP trong khoảng tần số thấp
Hình 3.7. So sánh vùng dao động các liên kết với tỉ lệ khác nhau các mẫu

76
Như vậy:
- Cấu trúc tinh thể olivine LFP được xác định bằng nhiễu xạ tia X, kết quả mẫu
phân tích có 5 đỉnh có cường độ mạnh trùng phổ chuẩn JPCDS 96-101-1112 và
được kiểm chứng sự có mặt các liên kết: Fe-O, P-O, P-O-P.. Mặt khác, được
kiểm chứng cấu trúc pha bằng chức năng Retveld.
- Trong các tỉ lệ đem tổng hợp thì tỉ lệ Li:Fe:P=3:1:1 đem lại kết quả thành phần
pha tốt nhất: peak nhọn, nền phổ thấp và không phát hiện tạp.
- Nhiệt độ nung tốt nhất để tối ưu vật liệu khoảng 550-600 oC thông qua giãn đồ
XRD và biểu đồ phân tích nhiệt TGA, trong khoảng nhiệt độ này pha của vật
liệu ổn định, loại bỏ hầu hết tạp.
- ascorbic acid là tác nhân khử có tác động rất lớn đến độ bền của vật liệu (ổn
định Fe2+, khả năng kết tinh).
3.1.3. Thành phần nguyên tố hóa học của vật liệu
Kỹ thuật SEM-EDS, phương pháp EDS hỗ trợ định lượng về thành phần
nguyên tố trong vật liệu để khảo sát vật liệu ở nhiều vùng khác nhau, quét khảo sát
bốn vùng ngẫu nhiên của vật liệu, sau đó lấy số liệu kết quả trung bình để định
lượng thành phần nguyên tố và phương pháp này có thể thấy hình thái bề mặt của
vật liệu [55, 56, 163].
Kết quả phân tích EDS chứa thành nguyên tố trong vật liệu như: Fe, C, O, P
chiếm hàm lượng lớn, tuy nhiên trong mẫu phân tích xuất hiện nguyên tố Pt vì nó
dùng đề làm điện cực cách vật liệu với màng silicon. Chi tiết hơn, % hàm lượng
các nguyên tố trong vật liệu LFP (ST01) được xác định tương đối phù hợp lý
thuyết: O (38-42%); Fe (15-18%); P (15-18%); C (6-9%) (Hình 3.8). Kết quả này
phù hợp kết quả tài liệu [163]. Tuy nhiên, phương pháp EDS, định lượng hàm
lượng mang tính tương đối vì vùng vật liệu nghiên cứu nhỏ, nếu vật liệu không
phân tán đồng đều thì sẽ ảnh hưởng đến hàm lượng các nguyên tố.
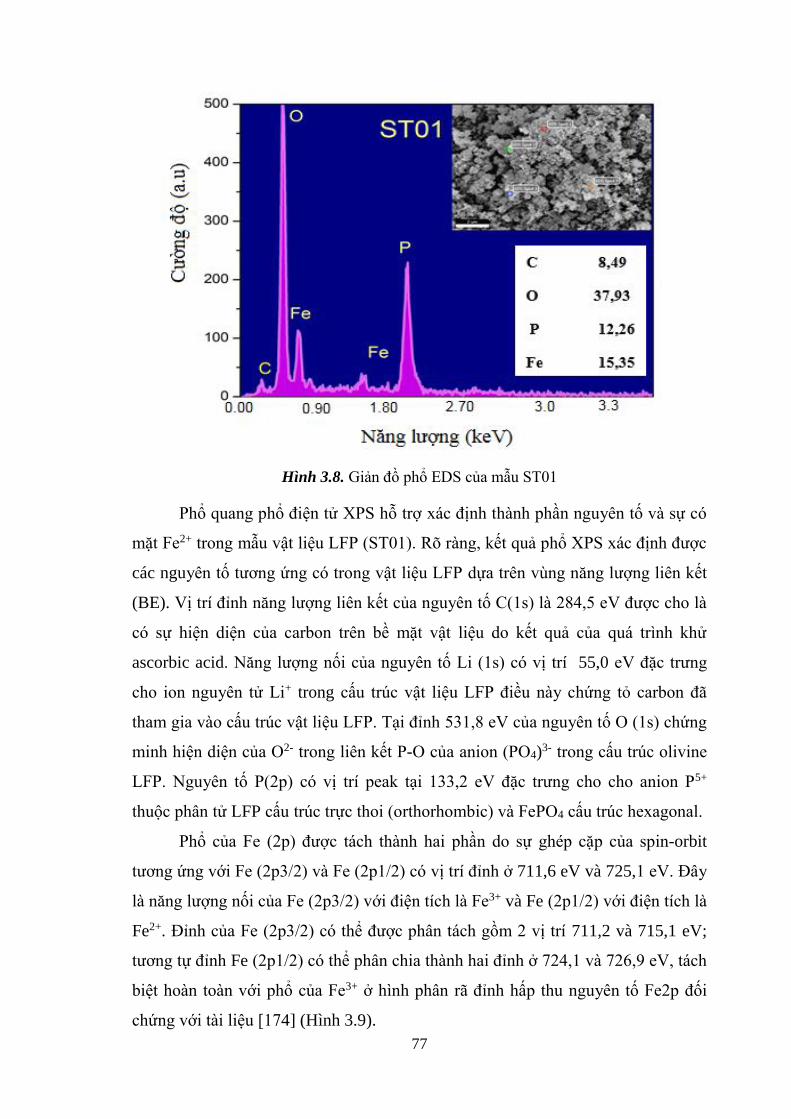
77
Hình 3.8. Giản đồ phổ EDS của mẫu ST01
Phổ quang phổ điện tử XPS hỗ trợ xác định thành phần nguyên tố và sự có
mặt Fe2+ trong mẫu vật liệu LFP (ST01). Rõ ràng, kết quả phổ XPS xác định được
các nguyên tố tương ứng có trong vật liệu LFP dựa trên vùng năng lượng liên kết
(BE). Vị trí đỉnh năng lượng liên kết của nguyên tố C(1s) là 284,5 eV được cho là
có sự hiện diện của carbon trên bề mặt vật liệu do kết quả của quá trình khử
ascorbic acid. Năng lượng nối của nguyên tố Li (1s) có vị trí 55,0 eV đặc trưng
cho ion nguyên tử Li+ trong cấu trúc vật liệu LFP điều này chứng tỏ carbon đã
tham gia vào cấu trúc vật liệu LFP. Tại đỉnh 531,8 eV của nguyên tố O (1s) chứng
minh hiện diện của O2- trong liên kết P-O của anion (PO4)3- trong cấu trúc olivine
LFP. Nguyên tố P(2p) có vị trí peak tại 133,2 eV đặc trưng cho cho anion P5+
thuộc phân tử LFP cấu trúc trực thoi (orthorhombic) và FePO4 cấu trúc hexagonal.
Phổ của Fe (2p) được tách thành hai phần do sự ghép cặp của spin-orbit
tương ứng với Fe (2p3/2) và Fe (2p1/2) có vị trí đỉnh ở 711,6 eV và 725,1 eV. Đây
là năng lượng nối của Fe (2p3/2) với điện tích là Fe3+ và Fe (2p1/2) với điện tích là
Fe2+. Đỉnh của Fe (2p3/2) có thể được phân tách gồm 2 vị trí 711,2 và 715,1 eV;
tương tự đỉnh Fe (2p1/2) có thể phân chia thành hai đỉnh ở 724,1 và 726,9 eV, tách
biệt hoàn toàn với phổ của Fe3+ ở hình phân rã đỉnh hấp thu nguyên tố Fe2p đối
chứng với tài liệu [174] (Hình 3.9).
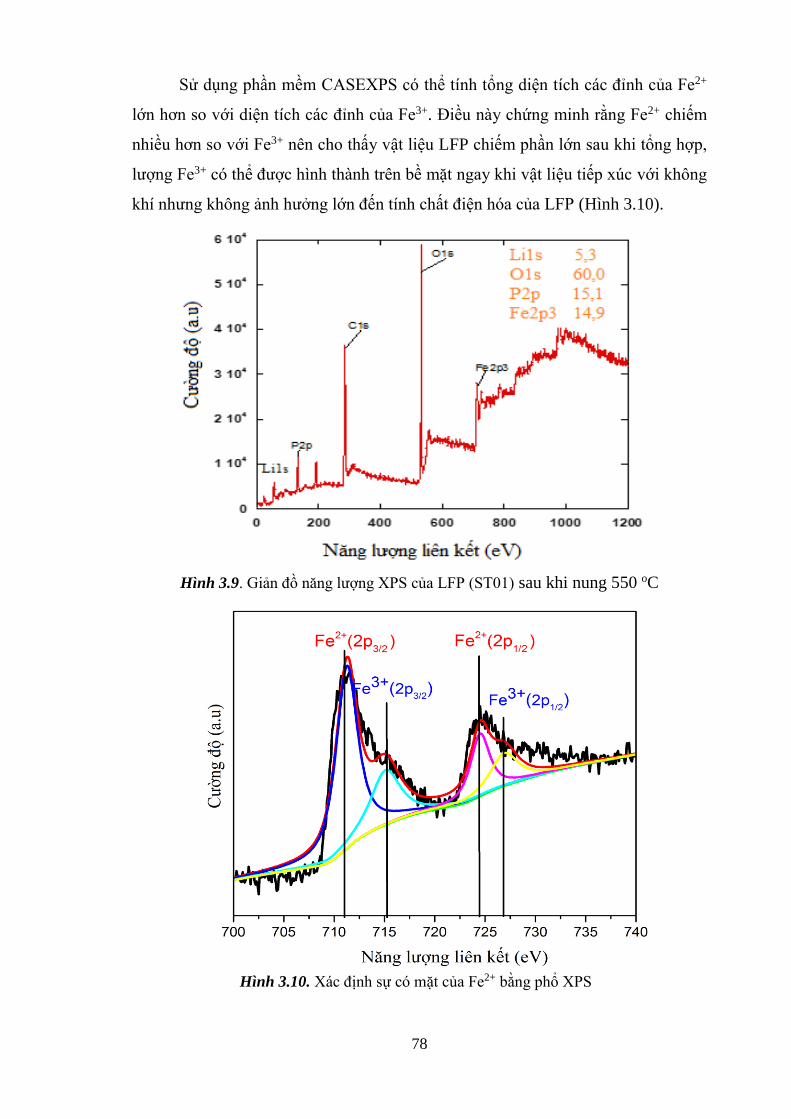
78
Sử dụng phần mềm CASEXPS có thể tính tổng diện tích các đỉnh của Fe2+
lớn hơn so với diện tích các đỉnh của Fe3+. Điều này chứng minh rằng Fe2+ chiếm
nhiều hơn so với Fe3+ nên cho thấy vật liệu LFP chiếm phần lớn sau khi tổng hợp,
lượng Fe3+ có thể được hình thành trên bề mặt ngay khi vật liệu tiếp xúc với không
khí nhưng không ảnh hưởng lớn đến tính chất điện hóa của LFP (Hình 3.10).
Hình 3.9. Giản đồ năng lượng XPS của LFP (ST01) sau khi nung 550 oC
Hình 3.10. Xác định sự có mặt của Fe2+ bằng phổ XPS

79
3.1.4. Hình thái vật liệu LFP
3.1.4.1. Hình dạng tinh thể
Hình dạng tinh thể của vật liệu LFP phụ thuộc điều kiện tiền chất, quá trình
khuấy tiền chất và nhiệt nung: porous [8], nanorod [13, 15]. Các công bố có nhận
định chung là dạng que (nanorod) mạng lại tính chất điện hóa tốt hơn các hình thái
hạt khác, do đó các công trình thường kiểm soát hạt nanorod [8, 13, 15]. Ngoài ra,
tài liệu [13, 56] khảo sát chi tiết ảnh hưởng của tỉ lệ tiền chất và quá trình nung
đến hình thái hạt.
Trong luận này, với phương pháp tổng hợp nhiệt dung môi sử dụng tổng
hợp LFP thay đổi tỉ lệ tiền chất và dung môi. Hình ảnh SEM của các mẫu ST00,
ST01, ST02 trước khi nung (Hình 3.11) thể hiện tinh thể vật liệu dạng hạt
(nanoparticles), các hạt có khuynh hướng kết tụ thành mảng [13, 15], trong các
mẫu thì mẫu ST01 đồng đều và ít kết tụ hạt hơn các mẫu còn lại.
Hình 3.11. SEM của LFP tổng hợp bằng phương pháp nhiệt dung môi trước khi nung
Tinh thể vật liệu LFP thay đổi theo nhiệt độ nung tăng dần 100-550 oC, các
hạt tinh thể tách rời, đồng đều và to hơn các mẫu trước khi chưa nung (Hình 3.12)
phù hợp kết quả công bố tài liệu [13].
Như vậy, nhiệt độ nung cũng ảnh hưởng đến hình thái kích thước hạt, nhiệt
độ nung càng lớn vật liệu bị giãn nở do đó, kích thước hạt thường lớn hơn.Tuy
nhiên, nung nhiệt độ cao carbon ở dạng hữu cơ bị thiêu kết bao bọc ngoài vật liệu
làm cho các tinh thể vật liệu LFP tách rời ra hạn chế các hạt kết tụ gây cản trở ion
Li+ di chuyển trong cấu trúc olivine [56] (Hình 3.12).

80
Hình 3.12. SEM của LFP nung nhiệt độ khác nhau: sấy (a); 400oC (b); 550oC (c).
Ảnh SEM (Hình 3.12c) và ảnh FESEM (Hình 3.13) đều cho kết quả tương
đối giống nhau: hạt hình que, đồng đều và rõ ràng. Kích thước to hơn rất nhiều các
mẫu trước khi nung. Kích thước hạt lớn hơn 50-150 nm và được tính cụ thể mục
3.1.4.2.
Hình 3.13. FESEM của LFP ở nhiệt độ 550oC.
3.1.4.2. Kích thước hạt
Kích thước hạt đơn tinh thể vật liệu LFP dự đoán ~ 50 nm được tính dựa
trên công thức Scherrer và dữ liệu FHWM của XRD.
cosFWHM
154,09,0
cos
k
D (3.4)
D là kích thước tinh thể thường nhỏ hơn kích thước hạt; β: bề rộng của peak (FWHM =
full–width–at–half–maximum length of the diffraction); 0<2θ<90o: góc quét (độ); λ=
0,154 nm là bước sóng của tia X; K= 0,9: hằng số thứ nguyên.
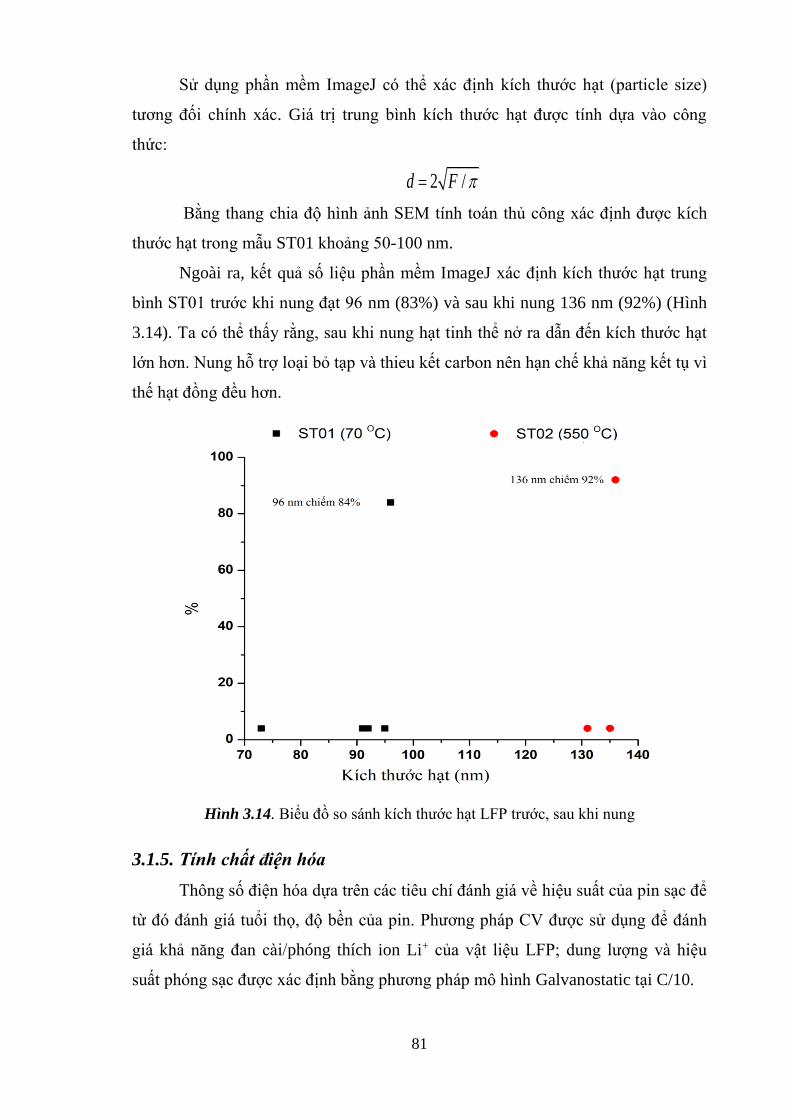
81
Sử dụng phần mềm ImageJ có thể xác định kích thước hạt (particle size)
tương đối chính xác. Giá trị trung bình kích thước hạt được tính dựa vào công
thức:
2 /d F
Bằng thang chia độ hình ảnh SEM tính toán thủ công xác định được kích
thước hạt trong mẫu ST01 khoảng 50-100 nm.
Ngoài ra, kết quả số liệu phần mềm ImageJ xác định kích thước hạt trung
bình ST01 trước khi nung đạt 96 nm (83%) và sau khi nung 136 nm (92%) (Hình
3.14). Ta có thể thấy rằng, sau khi nung hạt tinh thể nở ra dẫn đến kích thước hạt
lớn hơn. Nung hỗ trợ loại bỏ tạp và thieu kết carbon nên hạn chế khả năng kết tụ vì
thế hạt đồng đều hơn.
Hình 3.14. Biểu đồ so sánh kích thước hạt LFP trước, sau khi nung
3.1.5. Tính chất điện hóa
Thông số điện hóa dựa trên các tiêu chí đánh giá về hiệu suất của pin sạc để
từ đó đánh giá tuổi thọ, độ bền của pin. Phương pháp CV được sử dụng để đánh
giá khả năng đan cài/phóng thích ion Li+ của vật liệu LFP; dung lượng và hiệu
suất phóng sạc được xác định bằng phương pháp mô hình Galvanostatic tại C/10.

82
3.1.5.1. Đánh giá khả năng đan cài ion Li+ bằng phương pháp CV
Phương pháp quét thế vòng tuần hoàn (CV) của vật liệu composite LFP có
cấu trúc olivine được quét với nhiều tốc độ khác nhau từ 0-100 µV/s để nghiên
cứu khả năng đan cài ion Li+ vào vật liệu. Kết quả phương pháp này cho thấy xuất
hiện peak Fe3+/ Fe2+ rõ ràng, những đặc trưng về hình dạng và tỷ lệ tương đối giữa
các peak thể hiện rõ ràng. Các peak xuất hiện trên đường CV là của quá trình oxi
hóa- khử thuận nghịch Fe3+/Fe2+ tương ứng với sự phóng thích/đan cài ion Li+ vào
cấu trúc với khoảng thế 3,4-3,5 V, không có xuất hiện các peak khác, do đó, không
có sự hình thành pha mới trong quá trình đan cài (Hình 3.15a và 3.15b).
Quan sát hình 3.14a, 3.14b, so sánh giữa hai nhóm mẫu ST01 (2 mẫu) và
ST00 cùng quét với tỉ lệ tốc độ (20, 40, 60, 80, 100 µV/s) cho thấy rằng mẫu ST01
(ST01-L1 và ST01-L2) có cường độ đỉnh oxi hóa khử cao hơn nó nghĩa là mật độ
dòng lớn đồng nghĩa việc dự đoán dung lượng lớn hơn. Chẳng hạn, tại tốc độ 80
µV/s mẫu ST00 có mật độ dòng xấp xỉ 0,038 I/mAh.g-1 trong khi đó ST01-L1 và
ST01-L2 tương ứng khoảng 0,056 I/mA.g-1.
Ngoài ra, đường cong CV (Hình 3.15c) cho thể hiện biên độ oxi hóa- khử
của mẫu ST00 lớn hơn biên độ oxi hóa-khử các mẫu còn lại ST01-L1 và ST01-L2,
dẫn đến quá trình khuếch tán ion Li+ trong mẫu vật liệu diễn ra chậm hơn các mẫu
còn lại. Vấn đề này được làm rõ và cụ thể hơn trong phổ tổng trở (EIS). Nguyên
nhân có thể giải thích do kích hạt [36] mẫu ST00 còn tạp nên ảnh hưởng đến quá
trình khuếch tán ion Li+ vào cấu trúc. Điều này được minh chứng bằng số liệu hệ
số khuếch tán.
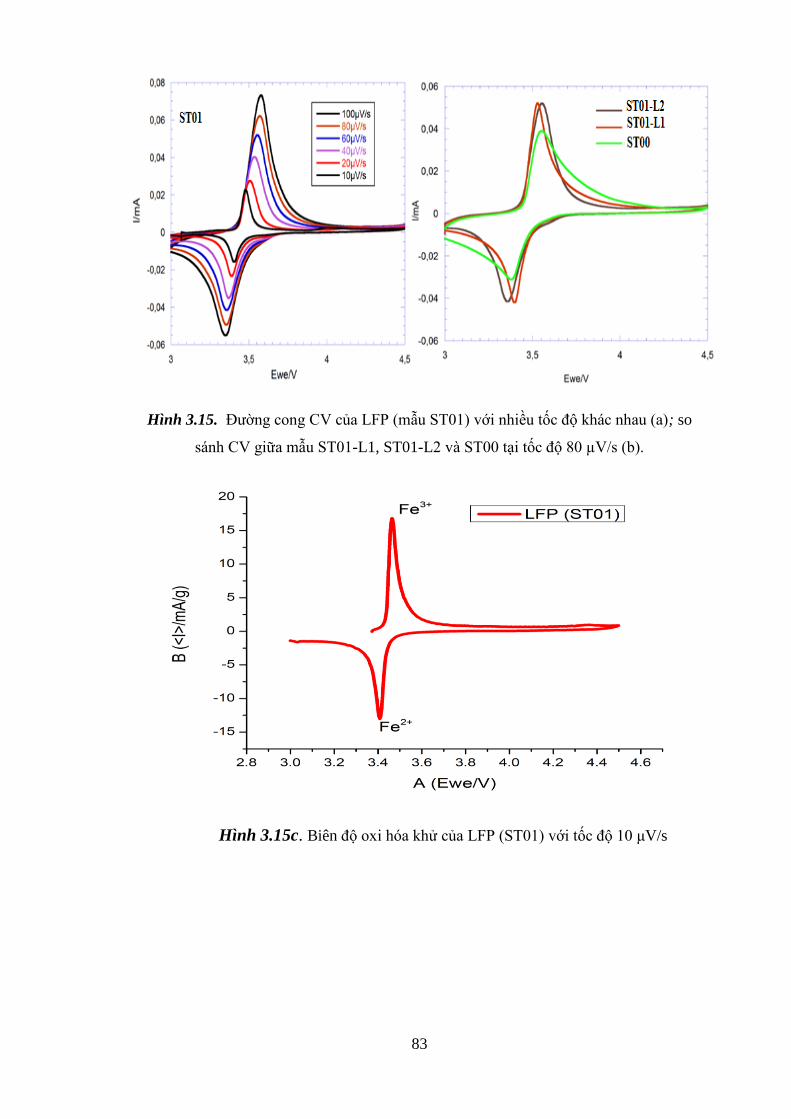
83
Hình 3.15. Đường cong CV của LFP (mẫu ST01) với nhiều tốc độ khác nhau (a); so
sánh CV giữa mẫu ST01-L1, ST01-L2 và ST00 tại tốc độ 80 µV/s (b).
Hình 3.15c. Biên độ oxi hóa khử của LFP (ST01) với tốc độ 10 μV/s
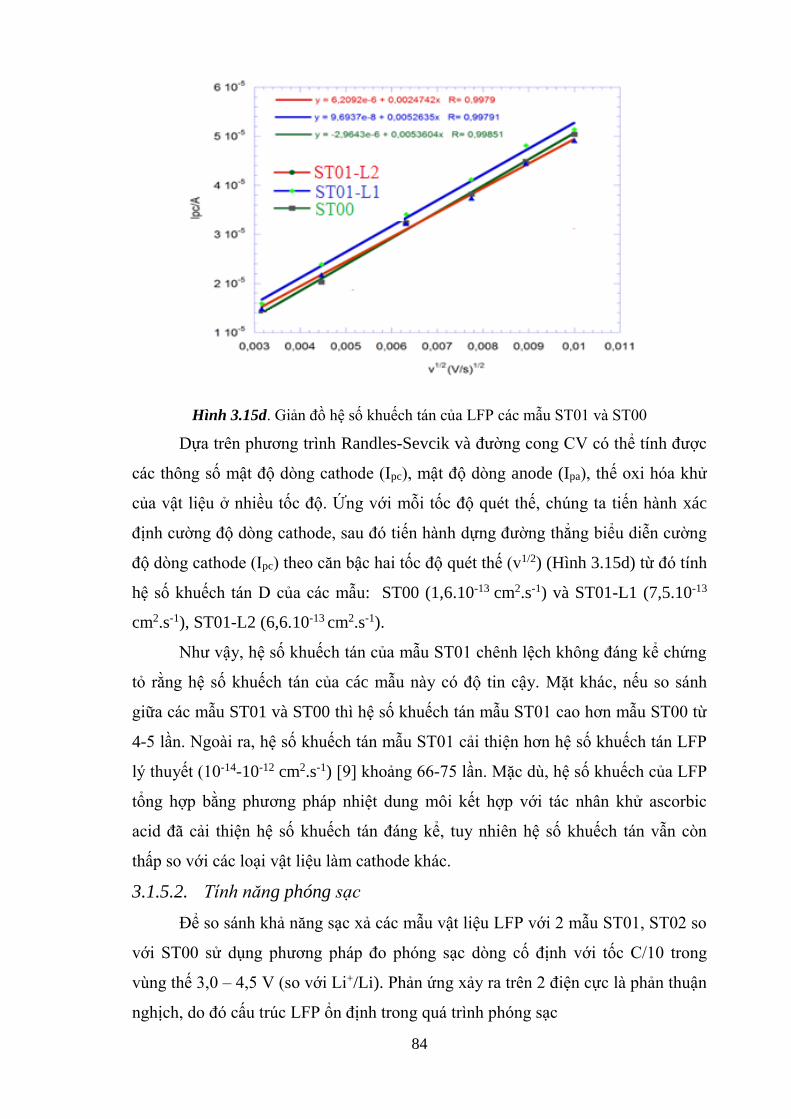
84
Hình 3.15d. Giản đồ hệ số khuếch tán của LFP các mẫu ST01 và ST00
Dựa trên phương trình Randles-Sevcik và đường cong CV có thể tính được
các thông số mật độ dòng cathode (Ipc), mật độ dòng anode (Ipa), thế oxi hóa khử
của vật liệu ở nhiều tốc độ. Ứng với mỗi tốc độ quét thế, chúng ta tiến hành xác
định cường độ dòng cathode, sau đó tiến hành dựng đường thẳng biểu diễn cường
độ dòng cathode (Ipc) theo căn bậc hai tốc độ quét thế (v1/2) (Hình 3.15d) từ đó tính
hệ số khuếch tán D của các mẫu: ST00 (1,6.10-13 cm2.s-1) và ST01-L1 (7,5.10-13
cm2.s-1), ST01-L2 (6,6.10-13 cm2.s-1).
Như vậy, hệ số khuếch tán của mẫu ST01 chênh lệch không đáng kể chứng
tỏ rằng hệ số khuếch tán của các mẫu này có độ tin cậy. Mặt khác, nếu so sánh
giữa các mẫu ST01 và ST00 thì hệ số khuếch tán mẫu ST01 cao hơn mẫu ST00 từ
4-5 lần. Ngoài ra, hệ số khuếch tán mẫu ST01 cải thiện hơn hệ số khuếch tán LFP
lý thuyết (10-14-10-12 cm2.s-1) [9] khoảng 66-75 lần. Mặc dù, hệ số khuếch của LFP
tổng hợp bằng phương pháp nhiệt dung môi kết hợp với tác nhân khử ascorbic
acid đã cải thiện hệ số khuếch tán đáng kể, tuy nhiên hệ số khuếch tán vẫn còn
thấp so với các loại vật liệu làm cathode khác.
3.1.5.2. Tính năng phóng sạc
Để so sánh khả năng sạc xả các mẫu vật liệu LFP với 2 mẫu ST01, ST02 so
với ST00 sử dụng phương pháp đo phóng sạc dòng cố định với tốc C/10 trong
vùng thế 3,0 – 4,5 V (so với Li+/Li). Phản ứng xảy ra trên 2 điện cực là phản thuận
nghịch, do đó cấu trúc LFP ổn định trong quá trình phóng sạc
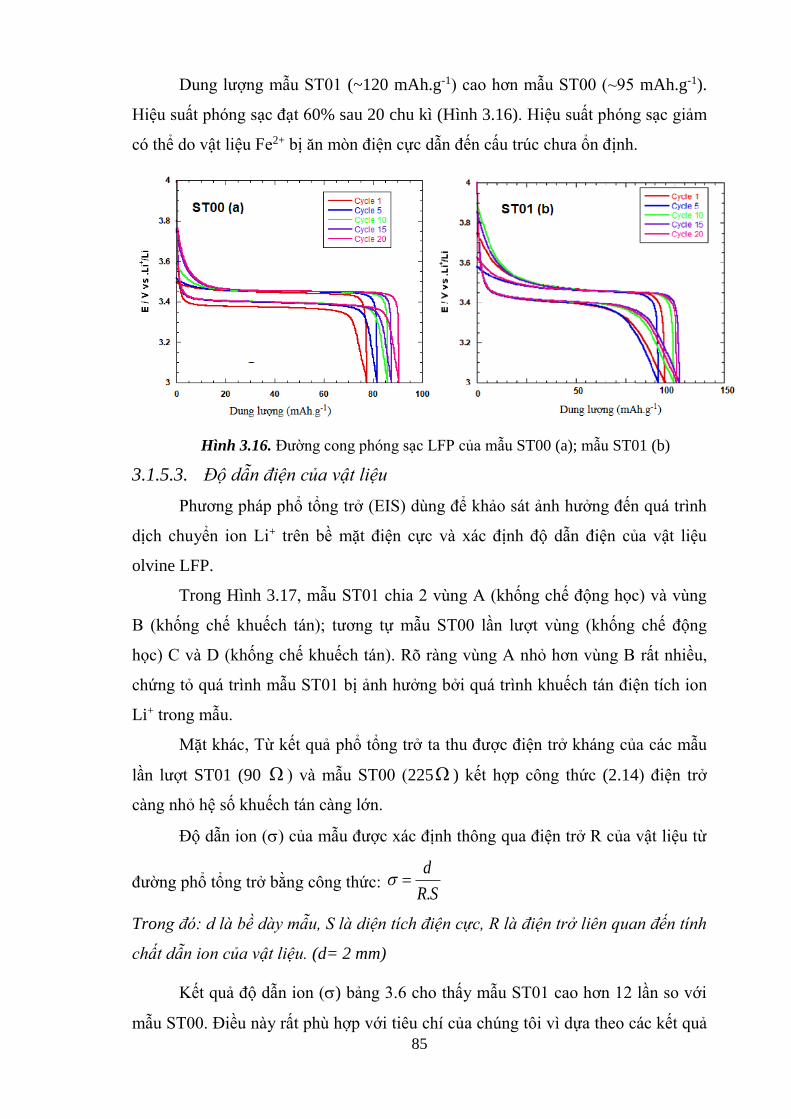
85
Dung lượng mẫu ST01 (~120 mAh.g-1) cao hơn mẫu ST00 (~95 mAh.g-1).
Hiệu suất phóng sạc đạt 60% sau 20 chu kì (Hình 3.16). Hiệu suất phóng sạc giảm
có thể do vật liệu Fe2+ bị ăn mòn điện cực dẫn đến cấu trúc chưa ổn định.
Hình 3.16. Đường cong phóng sạc LFP của mẫu ST00 (a); mẫu ST01 (b)
3.1.5.3. Độ dẫn điện của vật liệu
Phương pháp phổ tổng trở (EIS) dùng để khảo sát ảnh hưởng đến quá trình
dịch chuyển ion Li+ trên bề mặt điện cực và xác định độ dẫn điện của vật liệu
olvine LFP.
Trong Hình 3.17, mẫu ST01 chia 2 vùng A (khống chế động học) và vùng
B (khống chế khuếch tán); tương tự mẫu ST00 lần lượt vùng (khống chế động
học) C và D (khống chế khuếch tán). Rõ ràng vùng A nhỏ hơn vùng B rất nhiều,
chứng tỏ quá trình mẫu ST01 bị ảnh hưởng bởi quá trình khuếch tán điện tích ion
Li+ trong mẫu.
Mặt khác, Từ kết quả phổ tổng trở ta thu được điện trở kháng của các mẫu
lần lượt ST01 (90 ) và mẫu ST00 (225 ) kết hợp công thức (2.14) điện trở
càng nhỏ hệ số khuếch tán càng lớn.
Độ dẫn ion () của mẫu được xác định thông qua điện trở R của vật liệu từ
đường phổ tổng trở bằng công thức: SR
d
.
Trong đó: d là bề dày mẫu, S là diện tích điện cực, R là điện trở liên quan đến tính
chất dẫn ion của vật liệu. (d= 2 mm)
Kết quả độ dẫn ion () bảng 3.6 cho thấy mẫu ST01 cao hơn 12 lần so với
mẫu ST00. Điều này rất phù hợp với tiêu chí của chúng tôi vì dựa theo các kết quả

86
nghiên cứu sử dụng dung môi và tác nhân khử để hạn chế Fe2+ bị oxi hóa, ngoài ra
dung môi tạo độ nhớt hạn chế kết tụ nên ion Li+ trở nên linh động hơn.
Tuy nhiên độ dẫn điện các LFP mẫu ST01 (2.9.10-3 S.cm-1) của chúng tôi
có kết quả độ dẫn cao hơn các mẫu có phủ carbon của các công trình công bố 10-5
S.cm-1 xấ xỉ 1000 lần có thể do quá trình tạo màng có sử dụng carbon làm chất
trộn so sánh với tài liệu [52, 54] và cao hơn LFP 104 -105 lần tài liệu công bố
(5,9.10-9) [55].
Bảng 3.6 Độ dẫn ion của LFP dựa trên phổ EIS
Mẫu R
( )
S
(cm2)
d
(cm)
(S.cm-1)
D (EIS)
(cm2.s-1)
D (CV)
(cm2.s-1)
ST01 90 0,0785 0,2 2,9.10-3 7.10-13 7,5.10-13
ST00 225 0,0785 0,2 1,1.10-3 1,29.10-14 1,6.10-13
Từ số liệu của phổ tổng trở ta cũng có thể tính hệ số khuếch tán dựa trên
thông số sau:
Vậy hệ số khuếch tán của mẫu ST00 là 1,29.10-14 cm2.s-1
Trong đó:
R: hằng số khí lí tưởng (8,314 J.mol-1K-1), T: nhiệt độ (K)
A: diện tích điện cực n: số điện tử trao đổi
F: là hằng số Faraday (96500 C.mol-1). C: là nồng độ mol ion Li+
: hệ số Warburg
14 2
2 4 4 2 2 2 4 2
RT 8,31 298D (ST00) 1,29.10 ( / )
2A n F 2 0,785 96500 0,0228 3,454cm S
C
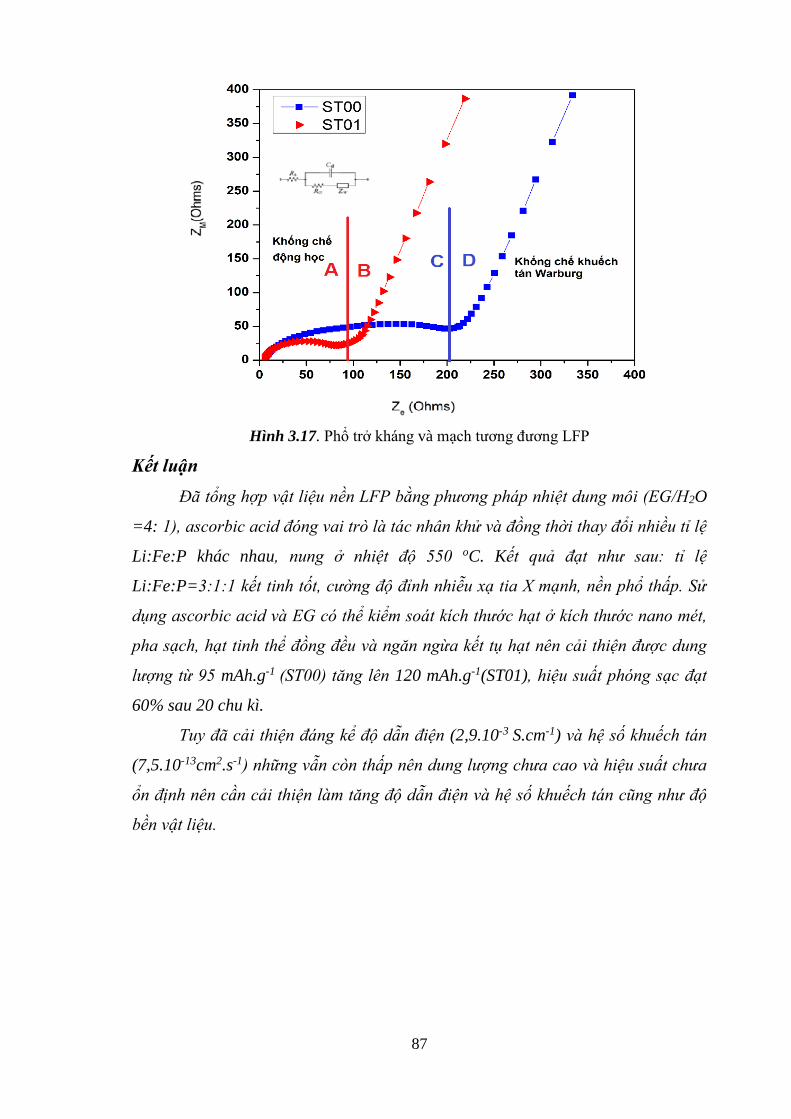
87
Hình 3.17. Phổ trở kháng và mạch tương đương LFP
Kết luận
Đã tổng hợp vật liệu nền LFP bằng phương pháp nhiệt dung môi (EG/H2O
=4: 1), ascorbic acid đóng vai trò là tác nhân khử và đồng thời thay đổi nhiều tỉ lệ
Li:Fe:P khác nhau, nung ở nhiệt độ 550 oC. Kết quả đạt như sau: tỉ lệ
Li:Fe:P=3:1:1 kết tinh tốt, cường độ đỉnh nhiễu xạ tia X mạnh, nền phổ thấp. Sử
dụng ascorbic acid và EG có thể kiểm soát kích thước hạt ở kích thước nano mét,
pha sạch, hạt tinh thể đồng đều và ngăn ngừa kết tụ hạt nên cải thiện được dung
lượng từ 95 mAh.g-1 (ST00) tăng lên 120 mAh.g-1(ST01), hiệu suất phóng sạc đạt
60% sau 20 chu kì.
Tuy đã cải thiện đáng kể độ dẫn điện (2,9.10-3 S.cm-1) và hệ số khuếch tán
(7,5.10-13cm2.s-1) những vẫn còn thấp nên dung lượng chưa cao và hiệu suất chưa
ổn định nên cần cải thiện làm tăng độ dẫn điện và hệ số khuếch tán cũng như độ
bền vật liệu.
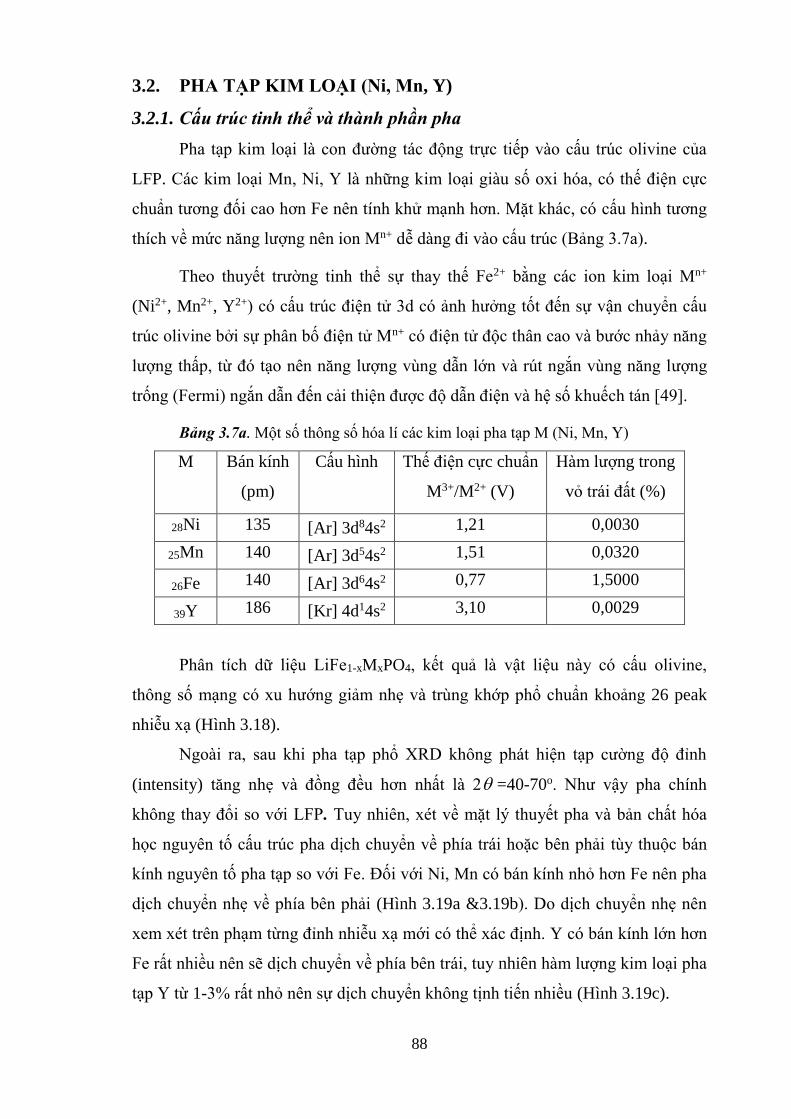
88
3.2. PHA TẠP KIM LOẠI (Ni, Mn, Y)
3.2.1. Cấu trúc tinh thể và thành phần pha
Pha tạp kim loại là con đường tác động trực tiếp vào cấu trúc olivine của
LFP. Các kim loại Mn, Ni, Y là những kim loại giàu số oxi hóa, có thế điện cực
chuẩn tương đối cao hơn Fe nên tính khử mạnh hơn. Mặt khác, có cấu hình tương
thích về mức năng lượng nên ion Mn+ dễ dàng đi vào cấu trúc (Bảng 3.7a).
Theo thuyết trường tinh thể sự thay thế Fe2+ bằng các ion kim loại Mn+
(Ni2+, Mn2+, Y2+) có cấu trúc điện tử 3d có ảnh hưởng tốt đến sự vận chuyển cấu
trúc olivine bởi sự phân bố điện tử Mn+ có điện tử độc thân cao và bước nhảy năng
lượng thấp, từ đó tạo nên năng lượng vùng dẫn lớn và rút ngắn vùng năng lượng
trống (Fermi) ngắn dẫn đến cải thiện được độ dẫn điện và hệ số khuếch tán [49].
Bảng 3.7a. Một số thông số hóa lí các kim loại pha tạp M (Ni, Mn, Y)
M Bán kính
(pm)
Cấu hình
Thế điện cực chuẩn
M3+/M2+ (V)
Hàm lượng trong
vỏ trái đất (%)
28Ni 135 [Ar] 3d84s2 1,21 0,0030
25Mn 140 [Ar] 3d54s2 1,51 0,0320
26Fe 140 [Ar] 3d64s2 0,77 1,5000
39Y 186 [Kr] 4d14s2 3,10 0,0029
Phân tích dữ liệu LiFe1-xMxPO4, kết quả là vật liệu này có cấu olivine,
thông số mạng có xu hướng giảm nhẹ và trùng khớp phổ chuẩn khoảng 26 peak
nhiễu xạ (Hình 3.18).
Ngoài ra, sau khi pha tạp phổ XRD không phát hiện tạp cường độ đỉnh
(intensity) tăng nhẹ và đồng đều hơn nhất là 2 =40-70o. Như vậy pha chính
không thay đổi so với LFP. Tuy nhiên, xét về mặt lý thuyết pha và bản chất hóa
học nguyên tố cấu trúc pha dịch chuyển về phía trái hoặc bên phải tùy thuộc bán
kính nguyên tố pha tạp so với Fe. Đối với Ni, Mn có bán kính nhỏ hơn Fe nên pha
dịch chuyển nhẹ về phía bên phải (Hình 3.19a &3.19b). Do dịch chuyển nhẹ nên
xem xét trên phạm từng đỉnh nhiễu xạ mới có thể xác định. Y có bán kính lớn hơn
Fe rất nhiều nên sẽ dịch chuyển về phía bên trái, tuy nhiên hàm lượng kim loại pha
tạp Y từ 1-3% rất nhỏ nên sự dịch chuyển không tịnh tiến nhiều (Hình 3.19c).
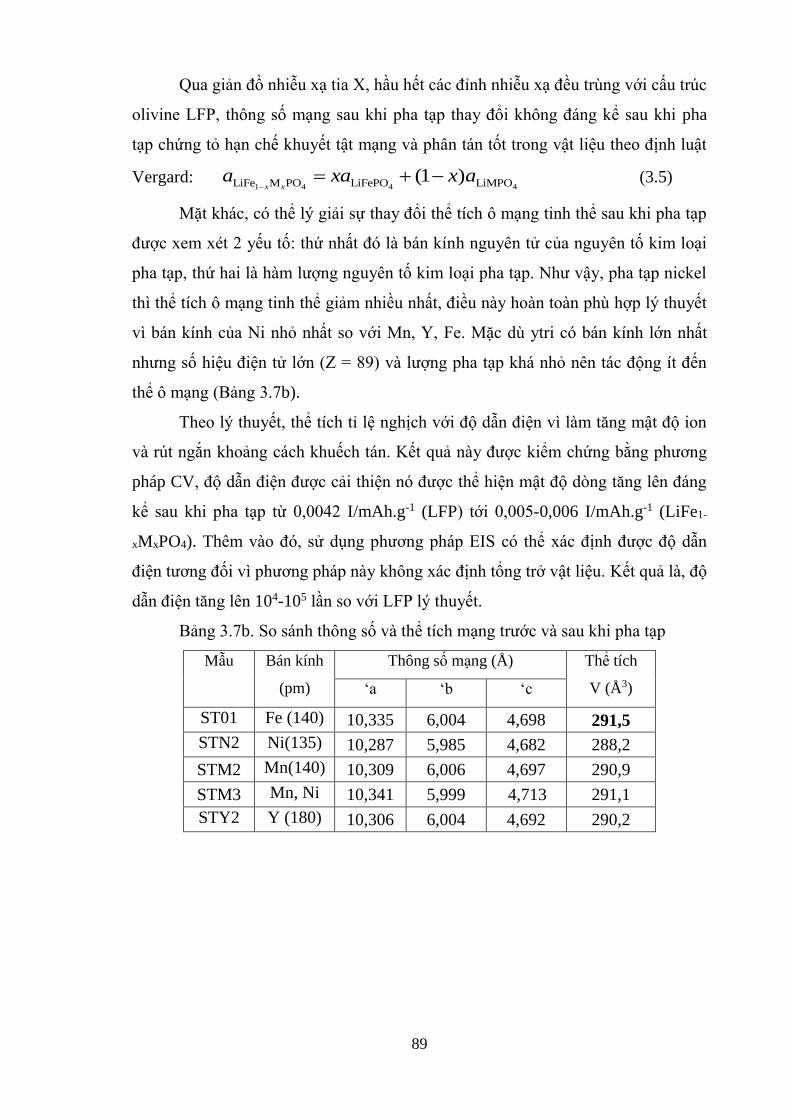
89
Qua giản đồ nhiễu xạ tia X, hầu hết các đỉnh nhiễu xạ đều trùng với cấu trúc
olivine LFP, thông số mạng sau khi pha tạp thay đổi không đáng kể sau khi pha
tạp chứng tỏ hạn chế khuyết tật mạng và phân tán tốt trong vật liệu theo định luật
Vergard: 1 4 4 4LiFe M PO LiFePO LiMPO(1 )
x xa xa x a
(3.5)
Mặt khác, có thể lý giải sự thay đổi thể tích ô mạng tinh thể sau khi pha tạp
được xem xét 2 yếu tố: thứ nhất đó là bán kính nguyên tử của nguyên tố kim loại
pha tạp, thứ hai là hàm lượng nguyên tố kim loại pha tạp. Như vậy, pha tạp nickel
thì thể tích ô mạng tinh thể giảm nhiều nhất, điều này hoàn toàn phù hợp lý thuyết
vì bán kính của Ni nhỏ nhất so với Mn, Y, Fe. Mặc dù ytri có bán kính lớn nhất
nhưng số hiệu điện tử lớn (Z = 89) và lượng pha tạp khá nhỏ nên tác động ít đến
thể ô mạng (Bảng 3.7b).
Theo lý thuyết, thể tích tỉ lệ nghịch với độ dẫn điện vì làm tăng mật độ ion
và rút ngắn khoảng cách khuếch tán. Kết quả này được kiểm chứng bằng phương
pháp CV, độ dẫn điện được cải thiện nó được thể hiện mật độ dòng tăng lên đáng
kể sau khi pha tạp từ 0,0042 I/mAh.g-1 (LFP) tới 0,005-0,006 I/mAh.g-1 (LiFe1-
xMxPO4). Thêm vào đó, sử dụng phương pháp EIS có thể xác định được độ dẫn
điện tương đối vì phương pháp này không xác định tổng trở vật liệu. Kết quả là, độ
dẫn điện tăng lên 104-105 lần so với LFP lý thuyết.
Bảng 3.7b. So sánh thông số và thể tích mạng trước và sau khi pha tạp
Mẫu Bán kính
(pm)
Thông số mạng (Å) Thể tích
V (Å3) ‘a ‘b ‘c
ST01 Fe (140) 10,335 6,004 4,698 291,5
STN2 Ni(135) 10,287 5,985 4,682 288,2
STM2 Mn(140) 10,309 6,006 4,697 290,9
STM3 Mn, Ni 10,341 5,999 4,713 291,1
STY2 Y (180) 10,306 6,004 4,692 290,2
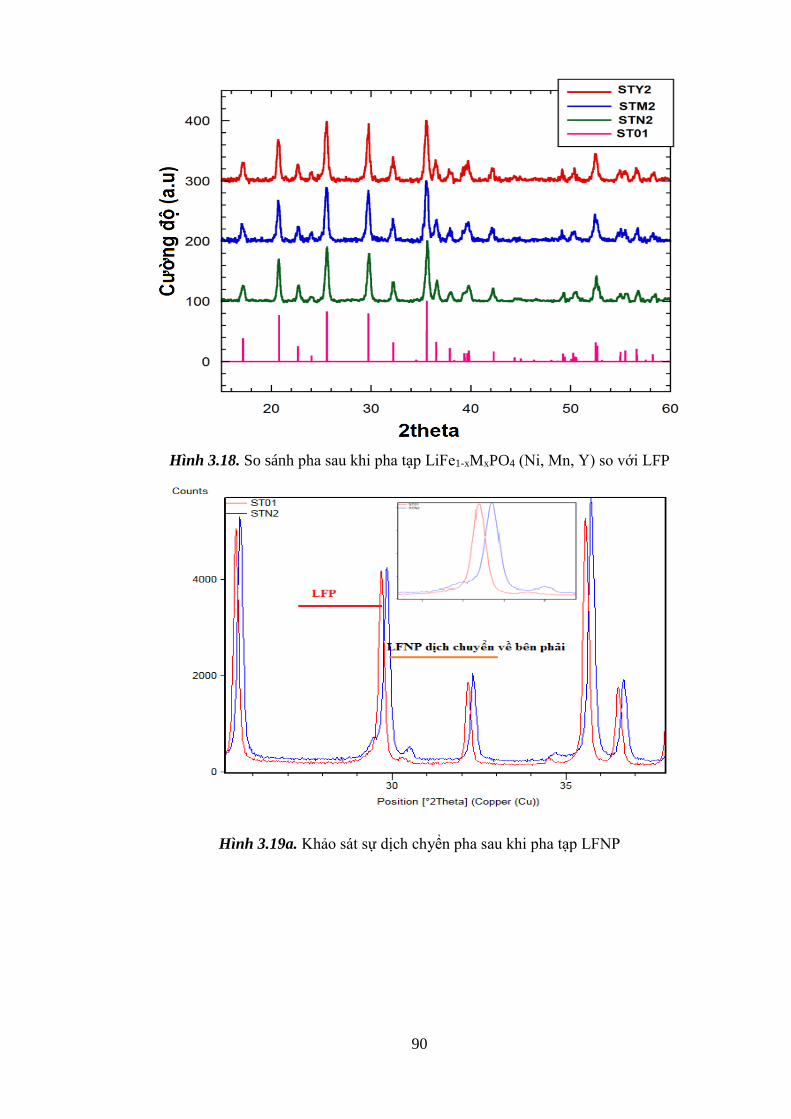
90
Hình 3.18. So sánh pha sau khi pha tạp LiFe1-xMxPO4 (Ni, Mn, Y) so với LFP
Hình 3.19a. Khảo sát sự dịch chyển pha sau khi pha tạp LFNP
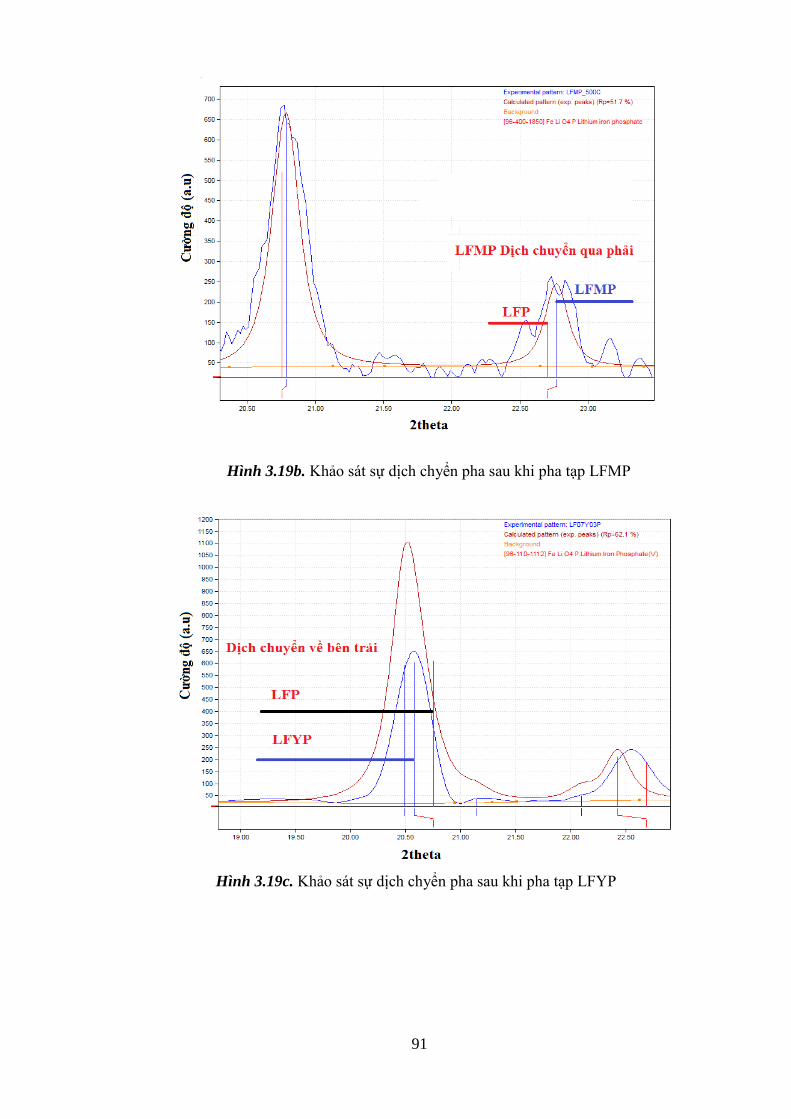
91
Hình 3.19b. Khảo sát sự dịch chyển pha sau khi pha tạp LFMP
Hình 3.19c. Khảo sát sự dịch chyển pha sau khi pha tạp LFYP
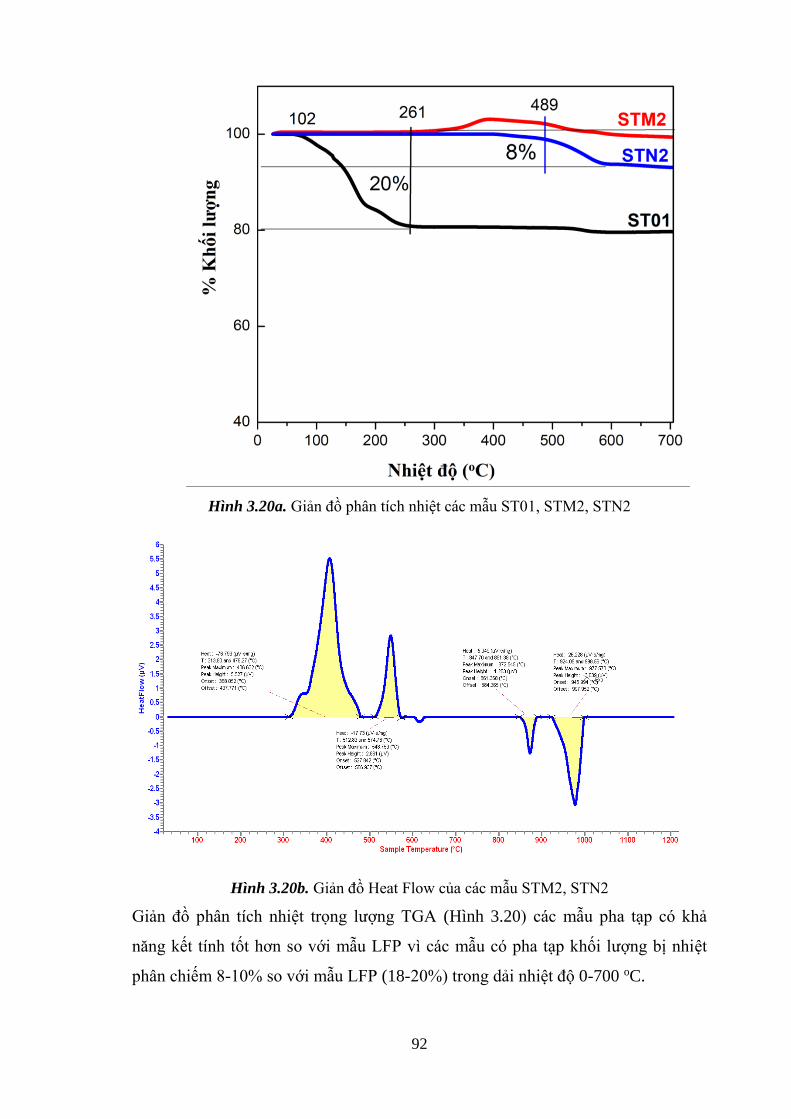
92
Hình 3.20a. Giản đồ phân tích nhiệt các mẫu ST01, STM2, STN2
Hình 3.20b. Giản đồ Heat Flow của các mẫu STM2, STN2
Giản đồ phân tích nhiệt trọng lượng TGA (Hình 3.20) các mẫu pha tạp có khả
năng kết tính tốt hơn so với mẫu LFP vì các mẫu có pha tạp khối lượng bị nhiệt
phân chiếm 8-10% so với mẫu LFP (18-20%) trong dải nhiệt độ 0-700 oC.

93
Nguyên nhân giảm khối lượng có thể bốc hơi H2O (trên 100 oC), gốc nước
dạng phức (200-300 oC), lượng dư dung môi EG bị chuyển pha phân hủy thành
carbon (300-600oC), muối Fe3(PO4)2 (nhiệt phân ở 230-400 oC). Tuy nhiên, bản
chất hóa học LiFePO4 bền nhiệt, trong quá trình tạo kết tinh LFP có thể tồn tại một
lượng nhỏ muối Fe (II) phophate (Fe3(PO4)2) chính vì lượng này hình thành khác
nhau nên các mẫu STM2 và STN2 có % khối lượng (%TG) không tương đồng.
Sự chuyển pha được thể hiện cụ thể đường Heat Flow (Hình 3.20b). Giai
đoạn nhiệt độ 300-400 oC có sự tỏa nhiệt chứng tỏ có sự thay đổi pha trong vật
liệu và %TG tăng có thể là do lượng carbon sinh ra 5-7%, tuy nhiên tiếp tục gia
nhiệt (500-600 oC) thì %TG giảm.
Một là, lượng carbon sinh ra kết hợp với hàm lượng O2 có sẵn trong luồng
khí thổi đưa argon vào (không khí di chuyển từ vùng nhiệt độ thấp sang vùng nhiệt
độ cao) hoặc trong khí argon chưa hoàn toàn sạch nên tạo CO2.
C + O2 (có lẫn trong luồng khí argon thổi vào) -> CO2 (Khí)
Hai là, %TG giảm là do trong vật liệu còn chứa tạp như Fe3(PO4)2, lượng
dư LiOH.H2O… bị nhiệt phân nên khối lượng sụt giảm, trong khi đó lượng carbon
vẫn được bảo toàn. Yếu tố này phù hợp với kết quả Raman vì các mẫu STM2,
STN2, STY2 có chứa vùng dao động tần số (1500-1600 cm-1) (Hình 3.21b).
Raman là công cụ dùng để xác định thành phần nguyên tố thông qua các
vùng dao động các liên kết có độ phân cực thấp. Cụ thể, vùng tần số dao động
200-450 cm-1 và 1200-1300 cm-1 là vùng dao động của Fe-O; từ 500-800 cm-1 là
các dao động M–O lần lượt Ni–O; Mn–O còn lại 900-1100 cm-1 là vùng dao động
P–O. Cường độ dao động mạnh khu vực Fe–O và P–O điều này chứng minh được
Fe, O, P chiếm hàm lượng lớn trong mẫu (Hình 3.21a) [154, 155].
Ngoài ra, từ giản đồ dao động theo tần số của Raman chứng minh được
vùng dao động Fe–O có cường độ dao động mạnh đỉnh phổ nhọn hơn rất nhiều so
với Ni–O, Mn–O và Y–O, điều này phù hợp với hàm lượng thành phần khối lượng
các nguyên tố trong vật liệu. Tuy nhiên, lượng Y pha tạp tương đối nhỏ nên dao
động Y-O khó xác định nhưng được kiểm chứng bằng kỹ thuật EDS-M (Hình
3.22a).
Bên cạnh đó, sự có mặt các nguyên tố pha tạp cường độ dao động vùng tần
số thấp chiếm ưu thế (300-700 cm-1), vùng này tập trung các dao động động Fe–O,

94
Mn–O, Ni–O, Y–O; trong khi đó, phổ raman của LFP thì dao động O–P–O chiếm
ưu thế, đây là một tín hiệu tốt biểu hiện mật độ điện tích Mn+ được tăng lên đáng
kể so với trước khi pha tạp (Hình 3.21b). Chính vì giàu mật độ điện tích với các
ion kim loại có tính khử mạnh hơn ion Fe2+ nên cấu trúc LiFePO4 ổn định hơn,
mặt khác, pha hình thành nhanh hơn nên hạn chế hình thành pha lạ đây là cơ sở
minh chứng hạn chế tạp và giải thích tại sao %TG giảm đáng kể.
Hình 3.21a. Phổ Raman của vật liệu LiFe1-xMxPO4
Hình 3.21b. Phổ Raman sự dịch chuyển dao động sau khi pha tạp

95
Điều đáng lưu ý, khi pha tạp vùng dao động dịch chuyển nhẹ về vùng tần số
cao (1500-1600 cm-1) là vùng có sự xuất hiện carbon, điều này hoàn toàn phù hợp
với kết quả phân tích TGA có khoảng 5-7% carbon hình thành, trong vùng này hầu
như không có sự tham gia của liên kết C-O.
3.2.2. Thành phần nguyên tố hóa học của vật liệu
Để định tính thành phần nguyên tố trong mẫu vật liệu sau khi pha tạp LiFe1-
xMxPO4 tương đối phức tạp vì phải xác định tỉ lệ pha tạp và thành phần nguyên tố
tham gia liên kết trong vật liệu. Do đó, tác giả đã kết hợp nhiều phương pháp để có
độ chính xác và tin cậy về mặt khoa học. Phương pháp AAS xác định % hàm
lượng kim loại có trong mẫu. EDS và XPS hỗ trợ xác định % hàm các nguyên tố
có trong mẫu chính xác các nguyên tố này có tham gia trong cấu trúc vật liệu. EDS
-Map xác định khả năng phân bố vật liệu và các nguyên tố có mặt trong cấu trúc.
Hàm lượng kim loại Fe2+ có trong các mẫu pha tạp được xác định bằng
phương pháp chuẩn độ. Từ số liệu bảng 3.8 cho thấy rằng hàm lượng Fe2+ có sự
giảm sau khi pha tạp tạp chứng tỏ kim loại pha tạp đã đi vào cấu trúc thay thế cho
kim loại Fe. Tuy nhiên, trong dung dịch chứa đa ion kim loại vì thế cần xem xét
thứ tự phản ứng: về bản chất, thứ tự phản ứng thế điện cực lớn xảy ra phản ứng
trước: Ni3+/Ni2+ (2,1 V)>Mn3+/Mn2+ (1,51 V)>Fe3+/Fe2+ (0,77 V). Tuy nhiên, các
Mn3+, Ni3+ kết hợp với gốc anion tạo muối không bền vì thế trong môi trường a xít
chủ yếu tồn tại Mn2+, Ni2+. Do vậy, không sử dụng KMnO4 để nhận biết ion Mn2+,
Ni2+, hay nói cách khác các ion này không tham gia quá trình phản ứng làm mất
màu KMnO4 /H2SO4 (đ). Tuy nhiên, kết quả chuẩn độ xác định ion Fe2+ chỉ để
tham khảo và được tham chiếu kết quả phân tích AAS và XPS.
Phương pháp phổ hấp thụ nguyên tử AAS dùng để định lượng các kim loại
pha tạp có trong mẫu và từ đó có thể tính được tỉ lệ pha tạp so với lý thuyết (Bảng
3.8). Cụ thể, mẫu STM2 (Fe: Mn = 80:20), kết quả AAS (Fe: Mn = 79.1: 21.1) so
sánh tài liệu [37].
Kết quả phân tích phổ AAS làm căn cứ để tính được tỉ lệ pha tạp các ion
kim loại Ni, Mn, Y phù hợp với tỉ lệ đem trộn. Chẳng hạn, mẫu LiFe0.8Mn0.2PO4
kết quả AAS thu được LiFe0.791Mn0.21PO4 (Bảng 3.9). Ngoài ra, phần mềm
Match!3.0 hỗ trợ phân tích từ dữ liệu XRD thu được Li0.91Fe0.791Mn0.21PO4 (JPDS
[96-152-4251]) (Hình 3.22)
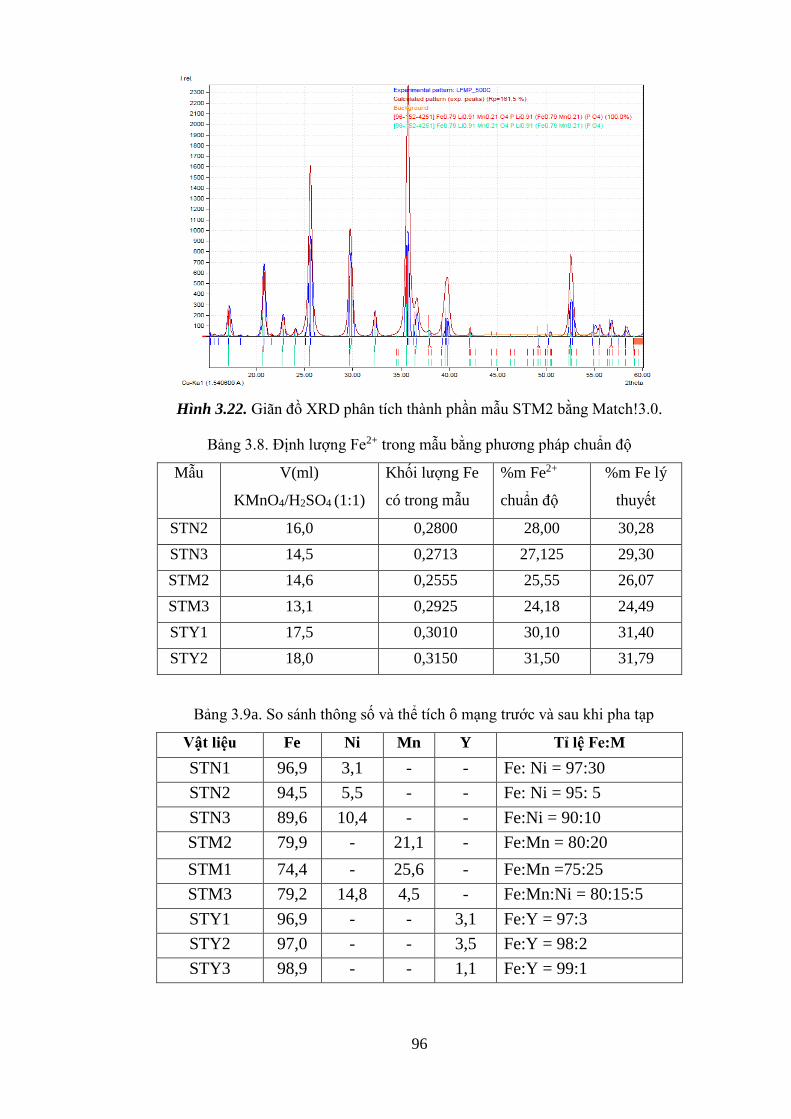
96
Hình 3.22. Giãn đồ XRD phân tích thành phần mẫu STM2 bằng Match!3.0.
Bảng 3.8. Định lượng Fe2+ trong mẫu bằng phương pháp chuẩn độ
Mẫu V(ml)
KMnO4/H2SO4 (1:1)
Khối lượng Fe
có trong mẫu
%m Fe2+
chuẩn độ
%m Fe lý
thuyết
STN2 16,0 0,2800 28,00 30,28
STN3 14,5 0,2713 27,125 29,30
STM2 14,6 0,2555 25,55 26,07
STM3 13,1 0,2925 24,18 24,49
STY1 17,5 0,3010 30,10 31,40
STY2 18,0 0,3150 31,50 31,79
Bảng 3.9a. So sánh thông số và thể tích ô mạng trước và sau khi pha tạp
Vật liệu Fe Ni Mn Y Tỉ lệ Fe:M
STN1 96,9 3,1 - - Fe: Ni = 97:30
STN2 94,5 5,5 - - Fe: Ni = 95: 5
STN3 89,6 10,4 - - Fe:Ni = 90:10
STM2 79,9 - 21,1 - Fe:Mn = 80:20
STM1 74,4 - 25,6 - Fe:Mn =75:25
STM3 79,2 14,8 4,5 - Fe:Mn:Ni = 80:15:5
STY1 96,9 - - 3,1 Fe:Y = 97:3
STY2 97,0 - - 3,5 Fe:Y = 98:2
STY3 98,9 - - 1,1 Fe:Y = 99:1

97
Hình ảnh SEM-EDS-Mapping xác định khả năng phân tán các nguyên tố
trong mẫu và cũng xác định thành phần các nguyên tố có trong mẫu. Cụ thể, để
kiểm chứng lại sự có mặt ytri trong vật liệu LiFe0.98Y0.02PO4 (STY2). Bằng
phương pháp này thu được kết quả thể hiện rõ trong Hình 3.22 sự phân tán các
nguyên tố Fe, O, P, Y dàn trải khắp các vùng trong mẫu.
Bảng 3.9b. Kết quả phân tích % khối lượng các nguyên tố bằng EDS và XPS
MẪU PP C O P Fe Ni Y Mn
STM2 EDS 4,96 41,08 15,10 19,04 - - 6,10
XPS 5,21 41,25 15,02 10,50 - - 6,25
STN2 EDS 6,27 40,99 14,01 20,03 3,10 - -
XPS 5,49 42,05 16,20 21,00 3,10
STY2 EDS 5,69 40,78 18,80 20,22 - 0,01 -
So sánh 2 mẫu STN2 và STY2 cho thấy cường độ nguyên tố C, O, P hầu
như không thay đổi. Fe, hai mẫu khác nhau, Fe trong mẫu STY2 cao hơn mẫu
STN2 hoàn toàn phù hợp lý thuyết vì pha tạp nguyên tố Y thấp hơn Ni nên lượng
Fe trong mẫu STY2 bị thay thế ít hơn (Hình 3.23).
Kết quả XPS xác định hàm lượng Fe2+ có trong các mẫu pha tạp chiếm 9,5-
10,7% trong khi mẫu LFP thì Fe2+ chiếm 14,9%. Điều này chứng tỏ rằng sự giảm
sụt 4-5% hàm lượng Fe2+ có trong mẫu vật liệu sau khi pha tạp đã bị thay thế bằng
kim loại pha tạp (Hình 3.24).
Đối với mẫu STN2 đã xuất hiện nguyên tố Ni2p3 (850-900 eV) với hàm
lượng 1% điều này khẳng định lại thêm là Ni đã tham gia vào cấu trúc so với tài
liệu [45]; tương tự mẫu STM3 xác định vùng năng lượng liên kết có mặt nguyên tố
Ni2p3 (850-900 eV), Mn2p3 (950-1000 eV). Thêm vào đó, dựa các vùng năng
lượng liên kết khác có thể xác định sự có mặt chính xác hơn thành phần nguyên tố
gồm Li, Ni, O, Fe, Mn [52].
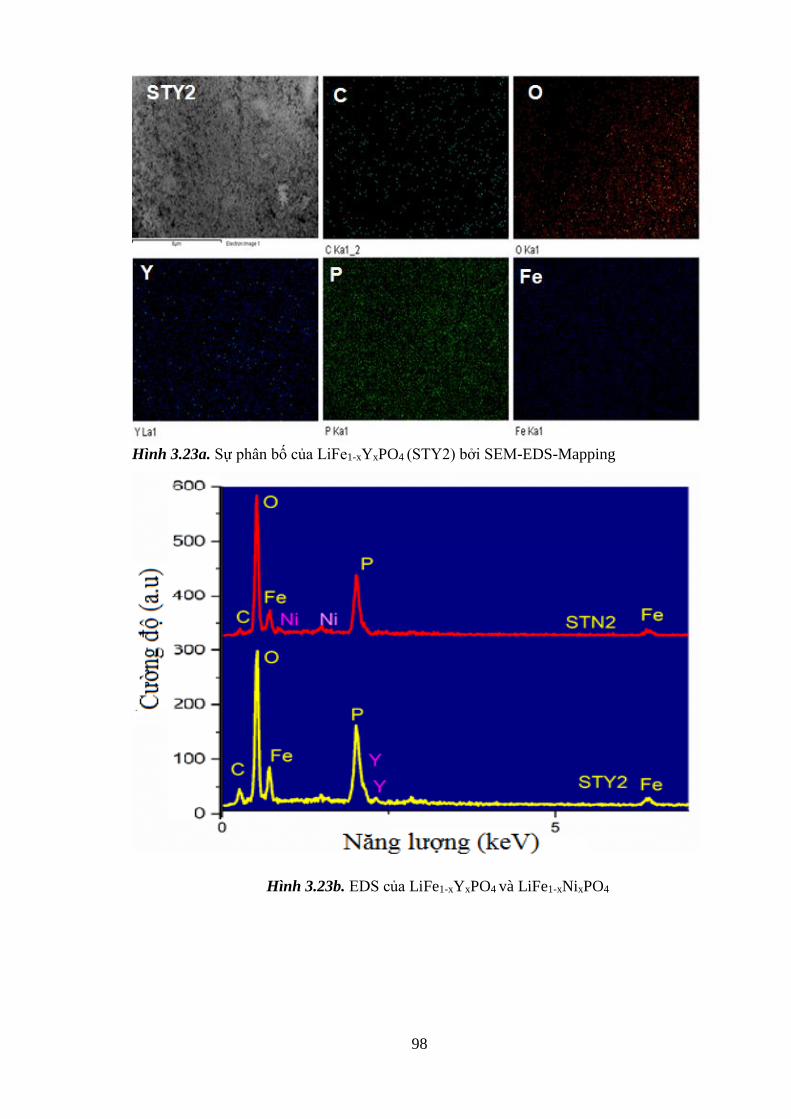
98
Hình 3.23a. Sự phân bố của LiFe1-xYxPO4 (STY2) bởi SEM-EDS-Mapping
Hình 3.23b. EDS của LiFe1-xYxPO4 và LiFe1-xNixPO4
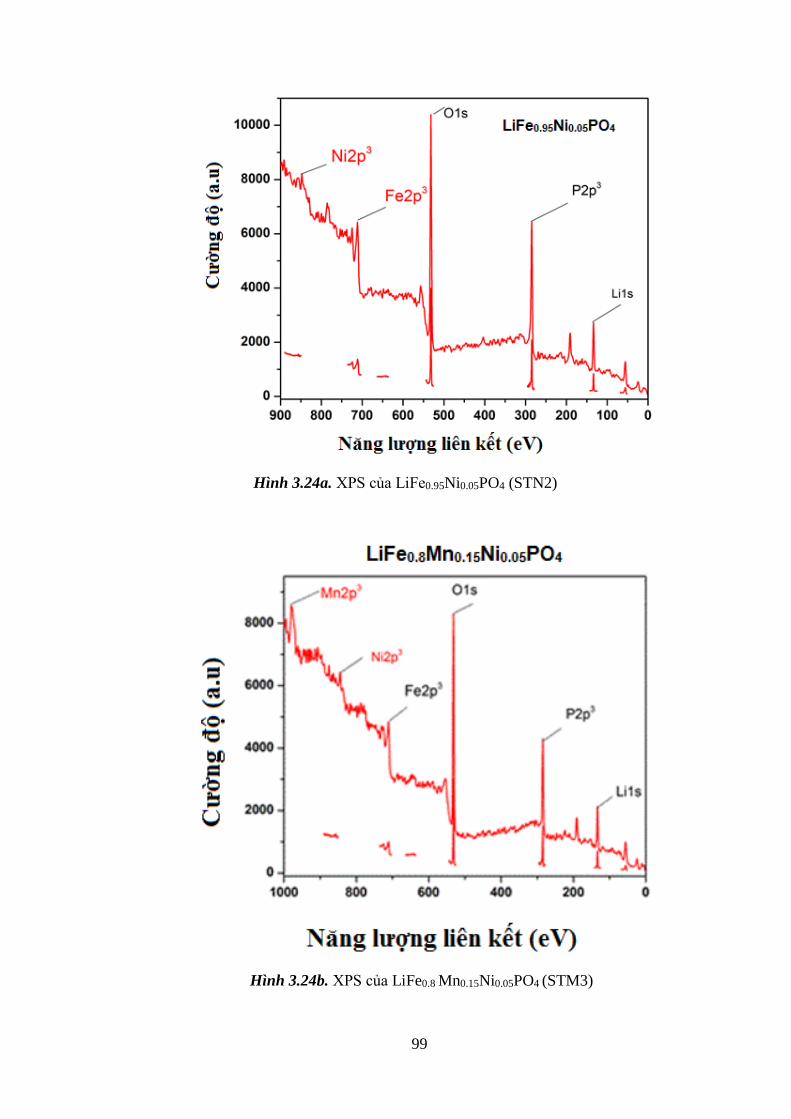
99
Hình 3.24a. XPS của LiFe0.95Ni0.05PO4 (STN2)
Hình 3.24b. XPS của LiFe0.8 Mn0.15Ni0.05PO4 (STM3)

100
3.2.3. Hình thái vật liệu
Kỹ thuật SEM và FESEM là những công cụ hỗ trợ xác định hình dạng tinh
thể dạng m tới nm. Kích thước hạt đơn tinh thể được xác định bằng công thức
Scherrer, kích thước hạt trung bình được tính toán bằng phần mềm ImageJ và tính
thủ công bằng thang chia kích thước trên hình ảnh SEM, TEM.
Nhìn chung sau khi pha tạp kim loại M (Ni, Mn, Y) vào cấu trúc LFP thì
tạo thành vật liệu nano có kích thước hạt đồng đều hơn vật liệu LFP. Bên cạnh đó,
các hạt có khuynh hướng tách rời không kết tụ. Từ kết quả Raman, TGA thấy rằng
trong vật liệu sau khi pha tạp đã có mặt carbon trong vật liệu nhiều hơn mẫu LFP,
có thể giải thích do các ion kim loại tham gia vào cấu trúc làm giàu mật độ điện
tích dương tạo ra tương tác bứt tách carbon ra khỏi các chất hữu cơ nhờ tác chất
nhiệt độ.
3 mẫu pha tạp Ni, khích thước hạt đạt (70-100 nm), đồng đều, dạng hình
que (nanorod) rõ rệt và các hạt được tách rời hơn so với các mẫu của vật liệu LFP
phù hợp tài liệu [52,142]. Trong đó mẫu STN2 cho kết quả pha tạp tốt hơn các tỉ lệ
khác Ni (Hình 3.25, 3.26).
Đối với các mẫu pha tạp Y, Mn kích thước đạt khoảng 70-150 nm (Hình
3.27 và 3.28), hình ảnh SEM, FESEM cho thấy các độ đồng đều về kích thước
chưa tốt so với STN2. Lựa các mẫu tốt nhất đại diện các kim loại pha tạp STN2,
STM2, STY2 đem chụp TEM và so sánh. Hình ảnh TEM (Hình 3.29, 3.30, 3.31)
thể hiện rõ và cụ thể hơn về hình dạng (hình que, kích thước hạt 50-100 nm) và độ
đồng đều của các hạt vì độ phân giải của kỹ thuật TEM tốt hơn. So sánh về độ
đồng đều, cả FESEM và TEM đều cho kết quả độ đều mẫu này (STN2) tốt nhất
trong các 3 mẫu STN2, STM2, STY2.
Pha tạp Ni có kết quả kích thước hạt nhỏ và đồng đều có thể lý giải như
sau: bán kính nguyên tử Ni (135 pm) nhỏ hơn Fe (140 pm), khi thay thế Ni làm
cho thể tích ô mạng nhỏ hơn dẫn đến kích thước hạt được giảm trong khi đó bán
kính Mn (140 pm) và Y(186 pm) lơn hơn hoặc bằng bán kính Fe nên thể ô mạng
không thay đổi sau khi pha tạp dẫn đến kích thước hạt không cải thiện tốt hơn Ni;
yếu tố thứ 2 do yếu tố động học phản ứng, trong dãy hoạt động hóa học Ni có tính
khử mạnh hơn Fe ngược lại Mn và Y tính khử yếu hơn Fe do đó, độ linh động ion

101
khi pha tạp Ni mạnh hơn, khi pha tạp tốc độ phản ứng xảy ra nhanh hơn dẫn đến
phản diễn ra tốt hơn.
Tuy nhiên, theo các công bố khác cho thấy kích thước 100 -300 nm thì kết
quả điện hóa tốt hơn [8, 11]. Kết quả điện hóa phụ thuộc vào nhiều yếu tố nhưng
đây cũng là cơ sở thấy rằng mặc dù pha tạp Ni kích thước hạt nhỏ hơn Mn, Y
nhưng hạt quá nhỏ có khả năng ảnh hưởng đến quá trình vận chuyển ion Li+ trong
cấu trúc olivine.
Hình 3.25. SEM của LiFe0.95Ni0.5PO4 (STN2) sau khi nung 550 oC
Hình 3.26. FESEM của LiFe1-xNixPO4 (STN2) sau khi nung 550 oC
Hình 3.27. FESEM của LiFe1-xYxPO4 (STY2)

102
Hình 3.28. SEM của LiFe1-xMnxPO4 (STM2)
Hình 3.29. TEM của LiFe1-xMnxPO4 (STM2)
Hình 3.30. TEM của LiFe1-xYxPO4 (STY2)
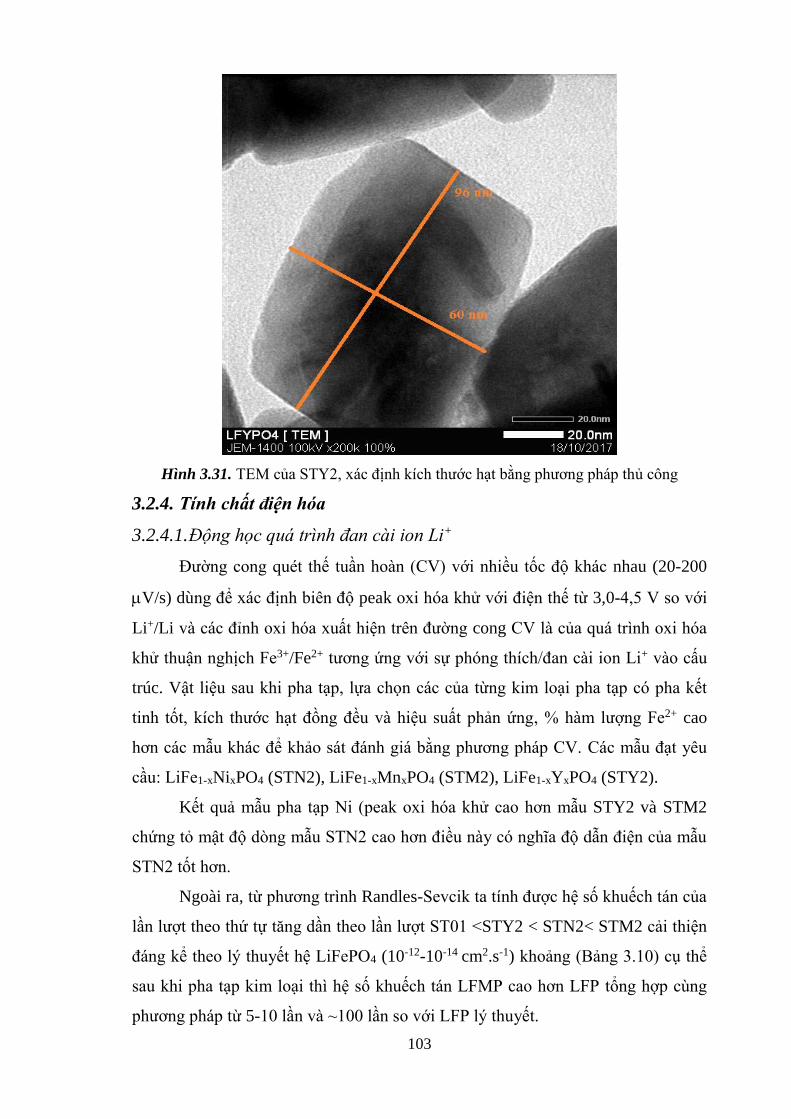
103
Hình 3.31. TEM của STY2, xác định kích thước hạt bằng phương pháp thủ công
3.2.4. Tính chất điện hóa
3.2.4.1. Động học quá trình đan cài ion Li+
Đường cong quét thế tuần hoàn (CV) với nhiều tốc độ khác nhau (20-200
V/s) dùng để xác định biên độ peak oxi hóa khử với điện thế từ 3,0-4,5 V so với
Li+/Li và các đỉnh oxi hóa xuất hiện trên đường cong CV là của quá trình oxi hóa
khử thuận nghịch Fe3+/Fe2+ tương ứng với sự phóng thích/đan cài ion Li+ vào cấu
trúc. Vật liệu sau khi pha tạp, lựa chọn các của từng kim loại pha tạp có pha kết
tinh tốt, kích thước hạt đồng đều và hiệu suất phản ứng, % hàm lượng Fe2+ cao
hơn các mẫu khác để khảo sát đánh giá bằng phương pháp CV. Các mẫu đạt yêu
cầu: LiFe1-xNixPO4 (STN2), LiFe1-xMnxPO4 (STM2), LiFe1-xYxPO4 (STY2).
Kết quả mẫu pha tạp Ni (peak oxi hóa khử cao hơn mẫu STY2 và STM2
chứng tỏ mật độ dòng mẫu STN2 cao hơn điều này có nghĩa độ dẫn điện của mẫu
STN2 tốt hơn.
Ngoài ra, từ phương trình Randles-Sevcik ta tính được hệ số khuếch tán của
lần lượt theo thứ tự tăng dần theo lần lượt ST01 <STY2 < STN2< STM2 cải thiện
đáng kể theo lý thuyết hệ LiFePO4 (10-12-10-14 cm2.s-1) khoảng (Bảng 3.10) cụ thể
sau khi pha tạp kim loại thì hệ số khuếch tán LFMP cao hơn LFP tổng hợp cùng
phương pháp từ 5-10 lần và ~100 lần so với LFP lý thuyết.

104
Cm LFP (Li+) = 24 23
4
291,5.10 6,023.10A
n Z
V N V
= 0,0228 mol/cm3
(Với Z: số nguyên tử Lithi trong 1 ô mạng, V thể tích ô mạng, NA số Avogadro)
Bảng 3.10. Định lượng hệ số khuếch tán (phương pháp CV) và độ dẫn điện (phổ EIS)
Mẫu ST00 ST01 STN2 STM2 STY2
D (cm2.s-1) 10-14 7,5.10-13 1,1.10-12 7,66.10-12 4,13.10-12
(S.cm-1) 1,1.10-3 2,9.10-3 5,1.10-3 5,0.10-3 5,1.10-3
Cm (mol/cm3) 0,0228 0,0228 0,0230 0,0228 0,0228
Hình 3.32. CV của LFMP với nhiều tốc độ khác nhau
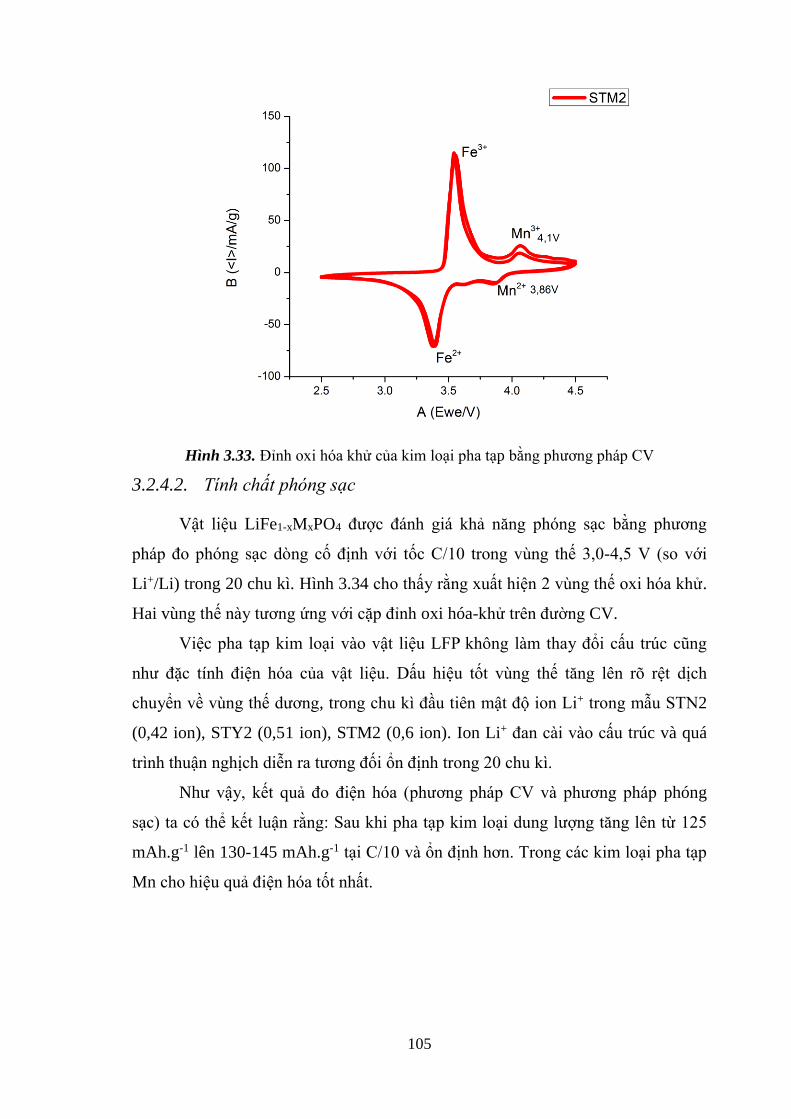
105
Hình 3.33. Đỉnh oxi hóa khử của kim loại pha tạp bằng phương pháp CV
3.2.4.2. Tính chất phóng sạc
Vật liệu LiFe1-xMxPO4 được đánh giá khả năng phóng sạc bằng phương
pháp đo phóng sạc dòng cố định với tốc C/10 trong vùng thế 3,0-4,5 V (so với
Li+/Li) trong 20 chu kì. Hình 3.34 cho thấy rằng xuất hiện 2 vùng thế oxi hóa khử.
Hai vùng thế này tương ứng với cặp đỉnh oxi hóa-khử trên đường CV.
Việc pha tạp kim loại vào vật liệu LFP không làm thay đổi cấu trúc cũng
như đặc tính điện hóa của vật liệu. Dấu hiệu tốt vùng thế tăng lên rõ rệt dịch
chuyển về vùng thế dương, trong chu kì đầu tiên mật độ ion Li+ trong mẫu STN2
(0,42 ion), STY2 (0,51 ion), STM2 (0,6 ion). Ion Li+ đan cài vào cấu trúc và quá
trình thuận nghịch diễn ra tương đối ổn định trong 20 chu kì.
Như vậy, kết quả đo điện hóa (phương pháp CV và phương pháp phóng
sạc) ta có thể kết luận rằng: Sau khi pha tạp kim loại dung lượng tăng lên từ 125
mAh.g-1 lên 130-145 mAh.g-1 tại C/10 và ổn định hơn. Trong các kim loại pha tạp
Mn cho hiệu quả điện hóa tốt nhất.
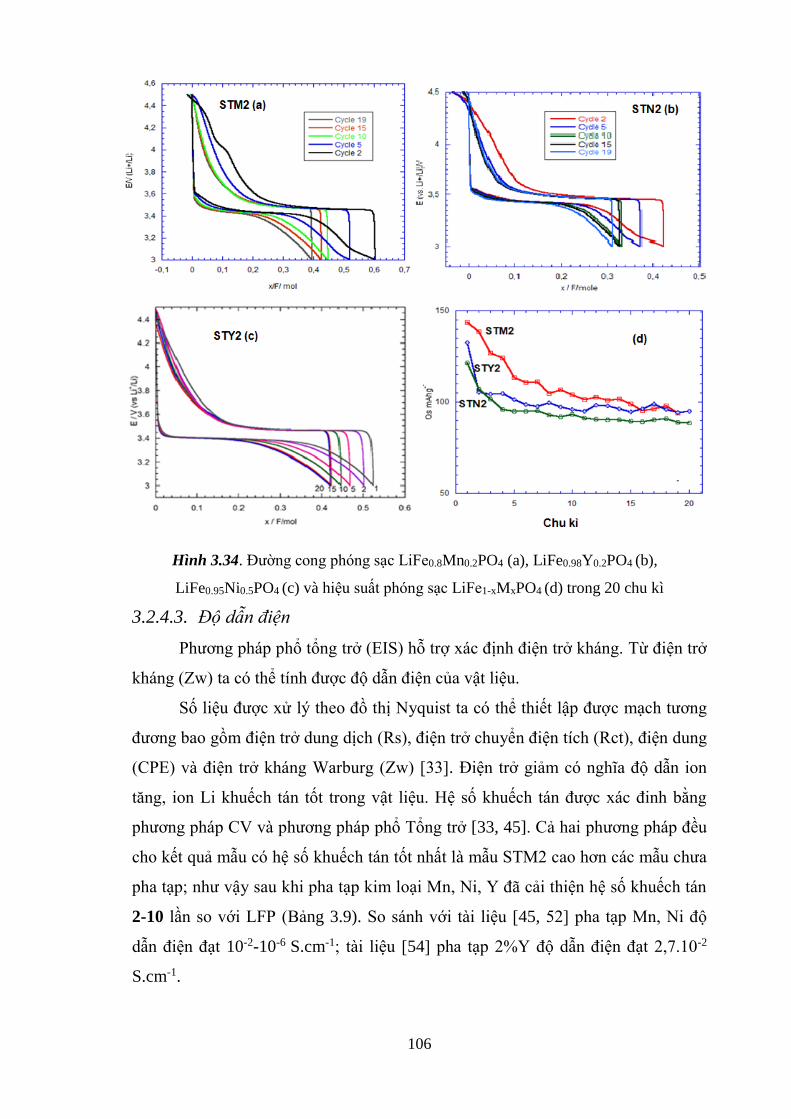
106
Hình 3.34. Đường cong phóng sạc LiFe0.8Mn0.2PO4 (a), LiFe0.98Y0.2PO4 (b),
LiFe0.95Ni0.5PO4 (c) và hiệu suất phóng sạc LiFe1-xMxPO4 (d) trong 20 chu kì
3.2.4.3. Độ dẫn điện
Phương pháp phổ tổng trở (EIS) hỗ trợ xác định điện trở kháng. Từ điện trở
kháng (Zw) ta có thể tính được độ dẫn điện của vật liệu.
Số liệu được xử lý theo đồ thị Nyquist ta có thể thiết lập được mạch tương
đương bao gồm điện trở dung dịch (Rs), điện trở chuyển điện tích (Rct), điện dung
(CPE) và điện trở kháng Warburg (Zw) [33]. Điện trở giảm có nghĩa độ dẫn ion
tăng, ion Li khuếch tán tốt trong vật liệu. Hệ số khuếch tán được xác đinh bằng
phương pháp CV và phương pháp phổ Tổng trở [33, 45]. Cả hai phương pháp đều
cho kết quả mẫu có hệ số khuếch tán tốt nhất là mẫu STM2 cao hơn các mẫu chưa
pha tạp; như vậy sau khi pha tạp kim loại Mn, Ni, Y đã cải thiện hệ số khuếch tán
2-10 lần so với LFP (Bảng 3.9). So sánh với tài liệu [45, 52] pha tạp Mn, Ni độ
dẫn điện đạt 10-2-10-6 S.cm-1; tài liệu [54] pha tạp 2%Y độ dẫn điện đạt 2,7.10-2
S.cm-1.
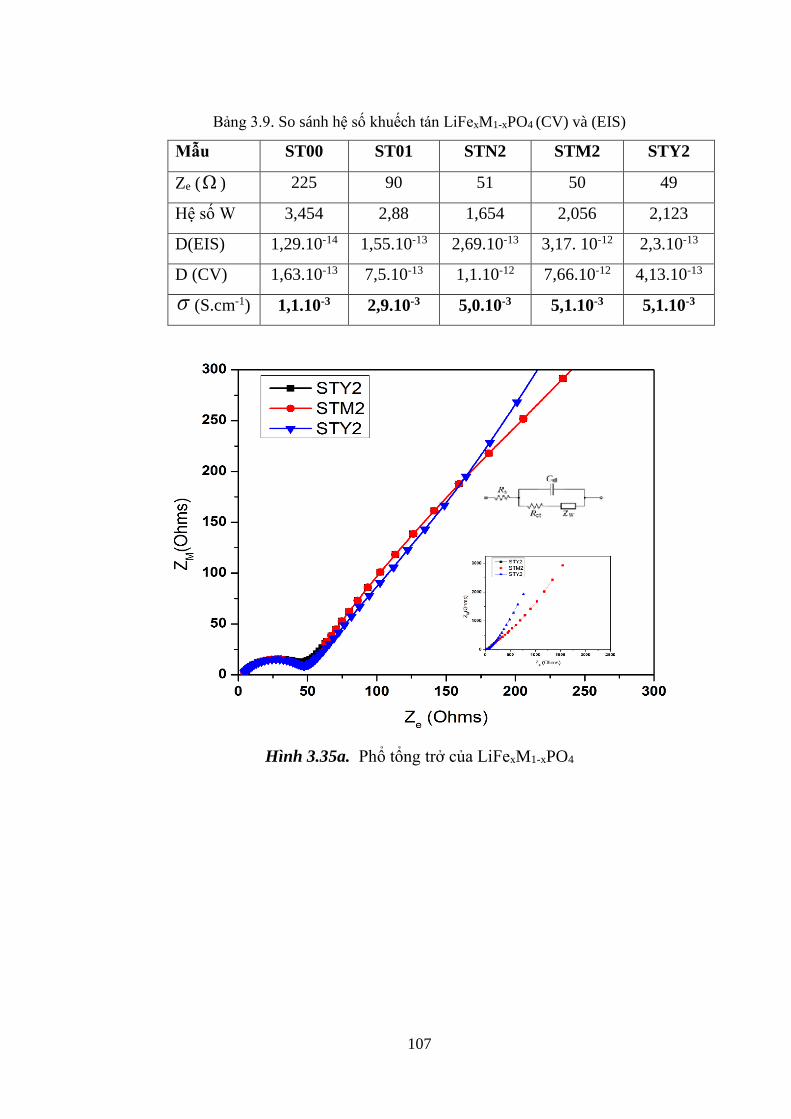
107
Bảng 3.9. So sánh hệ số khuếch tán LiFexM1-xPO4 (CV) và (EIS)
Hình 3.35a. Phổ tổng trở của LiFexM1-xPO4
Mẫu ST00 ST01 STN2 STM2 STY2
Ze ( ) 225 90 51 50 49
Hệ số W 3,454 2,88 1,654 2,056 2,123
D(EIS) 1,29.10-14 1,55.10-13 2,69.10-13 3,17. 10-12 2,3.10-13
D (CV) 1,63.10-13 7,5.10-13 1,1.10-12 7,66.10-12 4,13.10-13
(S.cm-1) 1,1.10-3 2,9.10-3 5,0.10-3 5,1.10-3 5,1.10-3

108
Hình 3.35b. Hệ số Warburg thông qua tổng trở Warburg mẫu STN2
3.2.5. Kết luận
Pha tạp thành công các kim loại Ni, Mn, Y với nhiều tỉ lệ khác nhau tuy
nhiên các mẫu LiFe0.9Ni0.1PO4 (STN2), LiFe0.98Y0.02PO4 (STY2), LiFe0.8Mn0.2PO4
(STM2) là những tỉ lệ cho kết quả tốt hơn các tỉ lệ khác.
Trong 3 mẫu STN2, STM2, STY2 kích thước hạt khoảng 70-150 nm có khuynh
hướng lớn hơn vật liệu LFP (50-100 nm), hình que, phân bố đồng đều và hạn chế
hết tụ hơn so với LFP.
Sau khi pha tạp không thay đổi cấu trúc chính so với vật liệu nền nhưng có
khuynh hướng dịch chuyển tuyến tính thỏa mãn định luật Vergard.
Hệ số khuếch tán được xác định thông qua 2 phương pháp EIS và CV cho
thấy rằng pha tạp kim loại cải thiện đáng kể tăng 5-10 lần so với LFP tổng hợp
cùng phương pháp. Mặt khác, độ dẫn điện LFMP 2 lần so với LFP tổng hợp cùng
phương pháp nhưng cải thiện hơn rất nhiều so với LFP lý thuyết (~106 lần). Độ
dẫn điện tăng hơn khoảng.
Dung lượng tại tốc độ C/10 của các mẫu lần lượt STN2 đạt 120 mAh.g-1;
STM2 đạt 135 mAh.g-1 và STY2 đạt 130 mAh.g-1; hiệu suất phóng giảm 20% sau
20 chu kì.

109
3.3. VẬT LIỆU NANOCOMPOSITE LiFe1-XMXPO4/Gr
Quy trình tổng hợp LFP kết hợp với phủ carbon và pha tạp kim loại M cũng
chính là tổng hợp LiFe1-xMxPO4/Gr vì ion Mn+ và graphene được đưa vào trong
qua trình tổng hợp LFP vì đưa vào tiền chất có khả năng M đi vào cấu trúc tốt hơn
và graphene phân tán tốt hơn.
3.3.1. Cấu trúc tinh thể và thành phần pha
Hình 3.36a minh chứng được rằng sau khi pha tạp và phủ graphene (5, 10,
15%) thì pha không thay đổi, cường độ peak nhiễu xạ tia X mạnh, đỉnh nhọn và
nền phổ thấp hơn so với các mẫu ST00 và ST01.
Hơn nữa, dữ liệu XRD ngoài việc xác định được pha của LFP thì có thể xác
định được sự có mặt carbon trong pha với hàm lượng pha tạp graphene lớn hơn
5%. Cụ thể, từ hình 3.36b mẫu có trộn graphene xuất hiện được mặt nhiễu xạ
[002], tuy nhiên để khẳng định cụ thể đây là dạng graphene (nano sheet 2D) được
minh chứng TEM và SEM-EDS-Mapping. Rõ ràng, cường độ peak nhiễu xạ
carbon [002] pha tạp Ytri có cường độ lớn hơn pha tạp Mn mặc dù tỉ lệ %
graphene. Điều này cũng có thể giải thích do tỉ lệ pha tạp Mn nhiều hơn Ytri nên
quá trình động học Mn diễn ra nhanh hơn và chiếm chỗ cho sự hình thành pha
carbon.
Thật vậy, các mẫu phủ graphene nhiệt độ nung 550 oC hình thành pha tốt
hơn cường độ các peak thấp được cải thiện. Có hai khả năng tác động đến sự hình
và kết tinh vật liệu. Một là do, graphene bao phủ nên bền nhiệt hạn chế khả năng
oxi hóa so với tất cả các mẫu khác nung ở cùng nhiệt độ. Hai là, nhiệt độ cao
cường độ peak mạnh hơn, vô định hình ít hơn vì một số tạp đã bị phân hủy. Từ đó,
cho thấy rằng graphene có vai trò rất lớn đối với quá trình phản ứng, chính vì thế
hiệu suất phản ứng các mẫu có phủ graphene tăng lên >90% (Bảng 3.3).
Sử dụng chức năng Rietveld Refinement từ phần mềm match!3.0 và Full
pro để kiểm chứng cấu trúc olivine của LiFe1-xMxPO4/Gr. Rõ ràng, sau khi đồng
thời pha tạp kim loại và phủ carbon (graphene và carbon từ nguồn hữu cơ) không
làm thay đổi cấu trúc (Hình 3.36c) nhưng giữa pha chuẩn và mẫu có peak có sự
chênh lệch là do kim loại đi vào cấu trúc làm dịch chuyển pha. Tuy nhiên các tỉ lệ
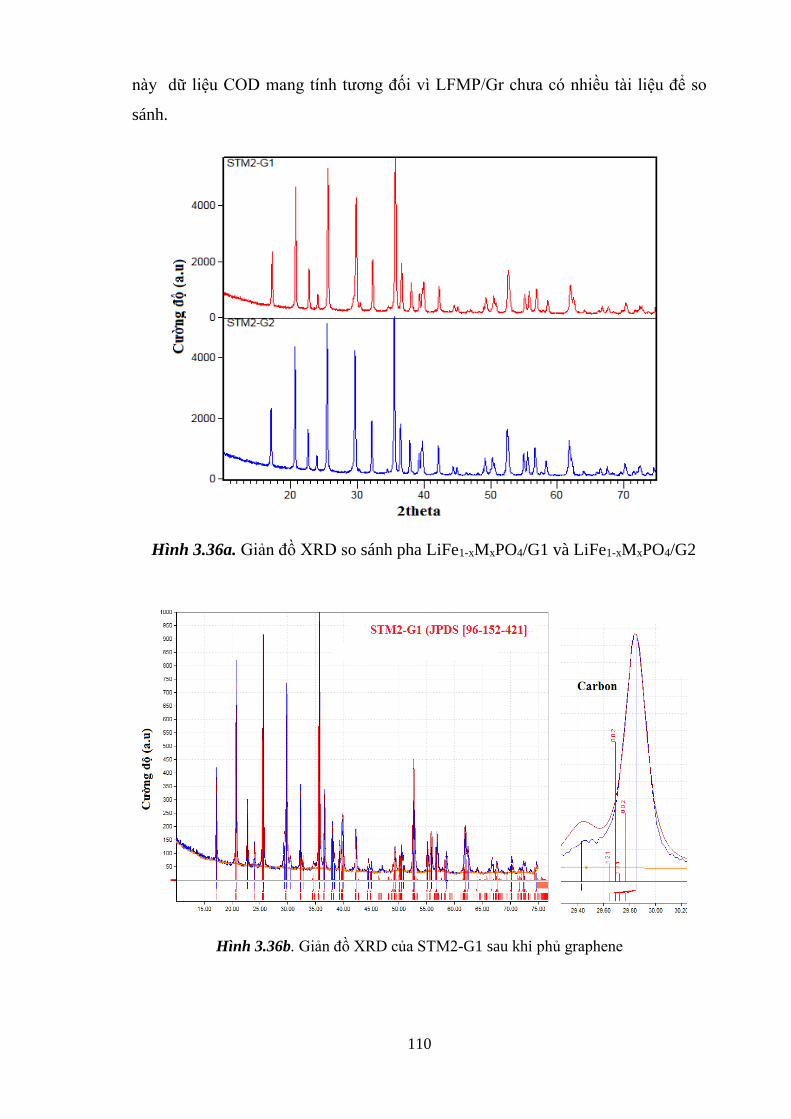
110
này dữ liệu COD mang tính tương đối vì LFMP/Gr chưa có nhiều tài liệu để so
sánh.
Hình 3.36a. Giản đồ XRD so sánh pha LiFe1-xMxPO4/G1 và LiFe1-xMxPO4/G2
Hình 3.36b. Giản đồ XRD của STM2-G1 sau khi phủ graphene

111
Hình 3.36c. Giản đồ XRD xác định cấu trúc olivine của LiFe1-xMxPO4/Gr bằng chức
năng Rietveld Refinement từ phần mềm match!3.0 và Full pro.
Khảo sát sự ảnh hưởng nhiệt độ nung đến sự kết tinh pha và Raman được
dùng để thành phần nguyên tố trong các mẫu LiFe1-xMxPO4/Gr. Ngoài ra, kỹ thuật
EDS đã xác định thành phần nguyên tố hóa học phù hợp với thành phần pha và có
tỉ lệ tương đối phù hợp với tỉ lệ tiền chất ban đầu.
Vấn đề đặt ra là TGA, EDS, EDS-Mapping chưa xác định được carbon ở
dạng thù hình nào vì vậy, trong công trình này để xác định được sự có mặt của
graphene phải kết hợp các kỹ thuật như TEM, HRTEM kết hợp với Raman.
Raman hỗ trợ xác định sự có mặt graphene (C-C) trong khoảng vùng dao
động D-band (1300 cm-1) và G-band (1588 cm-1), nếu vùng dao động G-band vượt
qua tần số này C-C dạng graphite hoặc carbon black [153, 154]. Trong đó, D
(disordered)-band (A1g) vùng dao động kiểu lai hóa SP3 (C-C). G (Graphene)-

112
band (E2g) kiểu lai hóa SP2 của carbon (C=O) kiểu lai hóa của graphene. Nguồn
graphene thương mại với I đỉnh phổ mạnh và không có đỉnh pha lạ nhiễu phổ
(Hình 3.37a).
Từ giản đồ phổ Raman tính được tỉ lệ I (D-band/G- band) để kiểm chứng
khả năng graphite hóa là chỉ số đánh giá độ bền của mẫu. Nếu I càng nhỏ thì mẫu
hạn chế bị nhiễu và khuyết tật. Rõ ràng, các mẫu pha tạp 5% graphene (STN2-G1,
STM2-G1, STM3-G1) có chỉ số I nhỏ hơn mẫu chưa pha tạp và phủ graphene.
(Bảng 3.10 và Hình 3.37b, 3.37c).
Ngoài ra, giá trị của tỷ lệ ID /IG đã được sử dụng để ước tính bề dày (La) của
graphene phủ lên vật liệu LiFe1-xMxPO4 bằng công thức [155]:
1
4
I560L ( )
E I
Da
lazer G
Trong đó, La: bề dày lớp phủ graphene, E = 2,54 eV là năng lượng Laser
kích thích Raman từ đó tính được La xấp xỉ 16 nm, số lớp của graphene (sheet)
khoảng 3-4 sheet [82]. Kết quả phù hợp và được kiểm chứng bởi HRTEM (3
sheet).
Mặt khác, nếu so sánh (Hình 3.37a và 3.37b) có một sự khác biệt là đối với
hình Raman LFP (vùng dao động tập trung từ biên độ 300-1000 cm-1); Sau khi phủ
graphene vùng dao động dịch chuyển tới 2700 cm-1 điều này khẳng định sự có mặt
của graphene. Ngoài ra, vùng 2684 cm-1 dùng để xác định số lớp (nano sheet) của
graphene so sánh với tài liệu [155, 156] vùng nano sheet graphite ở dao động 2710
cm-1 và 2719 cm-1.
Bảng 3.10. Độ graphite hóa của các mẫu bằng phổ Raman
Mẫu STN2-G1 STM2-G1 STM3-G1 ST01 Graphene
D- band 1310 1300 1302 973 1330
G-band 1588 1576 1576 1009 1578
ID/G 0,825 0,825 0,824 0,964 0,843
La (nm) 16,3 16,3 16,3 15,89

113
Hình 3.37a. Định tính sự có mặt graphene trong mẫu bằng Raman (Vùng 1)
Hình 3.37b. Định tính các vùng dao động có mặt kim loại pha tạp (Vùng 2)
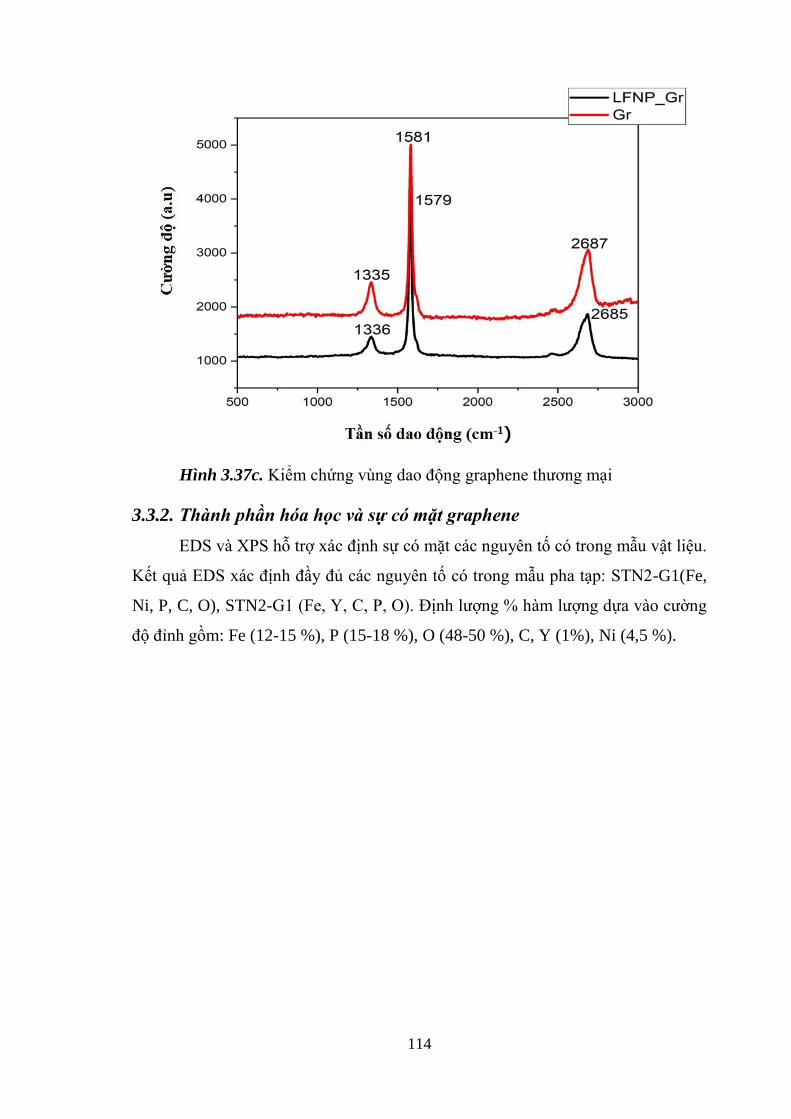
114
Hình 3.37c. Kiểm chứng vùng dao động graphene thương mại
3.3.2. Thành phần hóa học và sự có mặt graphene
EDS và XPS hỗ trợ xác định sự có mặt các nguyên tố có trong mẫu vật liệu.
Kết quả EDS xác định đầy đủ các nguyên tố có trong mẫu pha tạp: STN2-G1(Fe,
Ni, P, C, O), STN2-G1 (Fe, Y, C, P, O). Định lượng % hàm lượng dựa vào cường
độ đỉnh gồm: Fe (12-15 %), P (15-18 %), O (48-50 %), C, Y (1%), Ni (4,5 %).
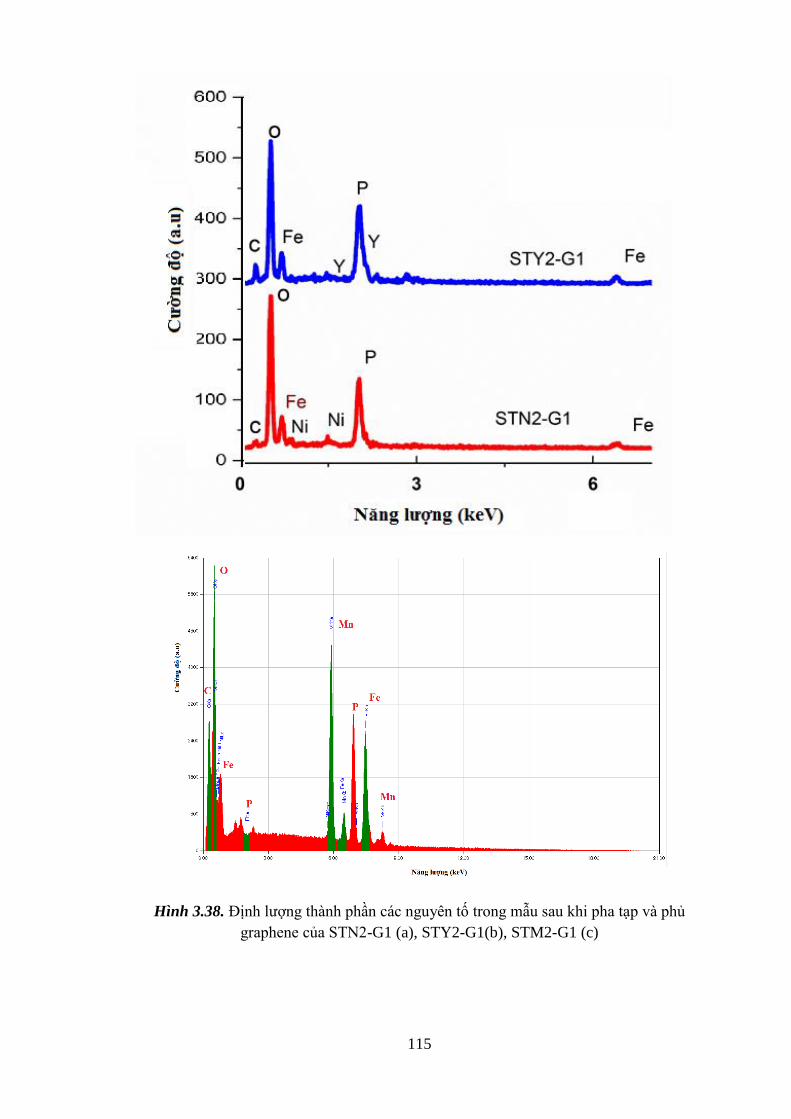
115
Hình 3.38. Định lượng thành phần các nguyên tố trong mẫu sau khi pha tạp và phủ
graphene của STN2-G1 (a), STY2-G1(b), STM2-G1 (c)

116
3.3.3. Xác định khả năng bao phủ graphene và độ phân tán của mẫu
Bản chất graphene 2D là dạng màng mỏng, các hạt tinh thể LiFe1-xMxPO4
có thể hình cầu hoặc các hạt que kết nối với nhau tạo dạng hạt (nanorod) vì thế
TEM sẽ hỗ trợ xác định chính xác hình thái của vật liệu và vùng graphene.
Graphene thương mại do quá bảo quản lâu nên có chứa hơi nước do đó
trước khi tổng hợp graphene được nung chân không 600 oC khoảng 3 h, quan sát
thấy các hạt tơi xốp làm nguội nhiệt độ phòng và đem chụp TEM để kiểm chứng
số lớp và thù hình. Qua đây, một lần nữa khẳng định được chính xác graphene về
mặt cấu trúc làm cơ sở so sánh trước và sau phủ.
Từ kết quả TEM và FESEM, các hạt tinh thể LFP rất lớn và các hạt kết tụ
với nhau vấn đề này ảnh hưởng rất lớn mật độ diện tích cũng như sự di chuyển ion
Li+. Sau khi phủ, các hạt tách rời nhau và nhỏ hơn khoảng 10 lần, màng mỏng
graphene là cầu nối liên kết giữa các tinh thể.
Kích thước hạt được xác định chính xác hơn bằng hình ảnh TEM vì độ
phóng đại của kính hiển vi truyền qua TEM rất lớn. Cụ thể, từ hình 3.39 các hạt
tương đối đều, được bao phủ bởi lớp màng mỏng graphene cỡ 20-200 nm, độ phân
tán mẫu STM3-G1 (LFMNP/Gr) phân tán đều nhất và mật độ hạt mẫu STY2-G1
(LFYP/Gr) tốt nhất. EDS-Mapping hỗ trợ xác định khu vực xác suất có mặt các
nguyên tố trong mẫu vật liệu (Hình 3.40).
Mỗi tinh thể LiFe1-xMxPO4 đính trên màng graphene được giải thích theo cơ
chế điện tích: Tinh thể LiFe1-xMxPO4 giàu oxi tích điện âm ( ) đính trên mỗi ô
mạng lục giác của graphene có nối chứa liên kết giàu mật độ điện tích dương
( ) tạo ra hiệu ứng cảm giữa mạng lúc giác graphene và LFMP tạo nên vật liệu
composite có cấu trúc bền vững và tính chất điện hóa tốt.
HRTEM là công cụ phân tích chuyên sâu về cấu trúc bề mặt vật liệu cho
chúng ta thông tin chính xác. Kết quả HRTEM cụ thể hóa sự có mặt grahene độ
dày của màng 5 -10 nm và 2-3 lớp (sheet) grpahene phù hợp với kết quả của
Raman. Bên cạnh đó, các mặt nhiễu xạ của vật liệu được thể hiện [100], [101]…
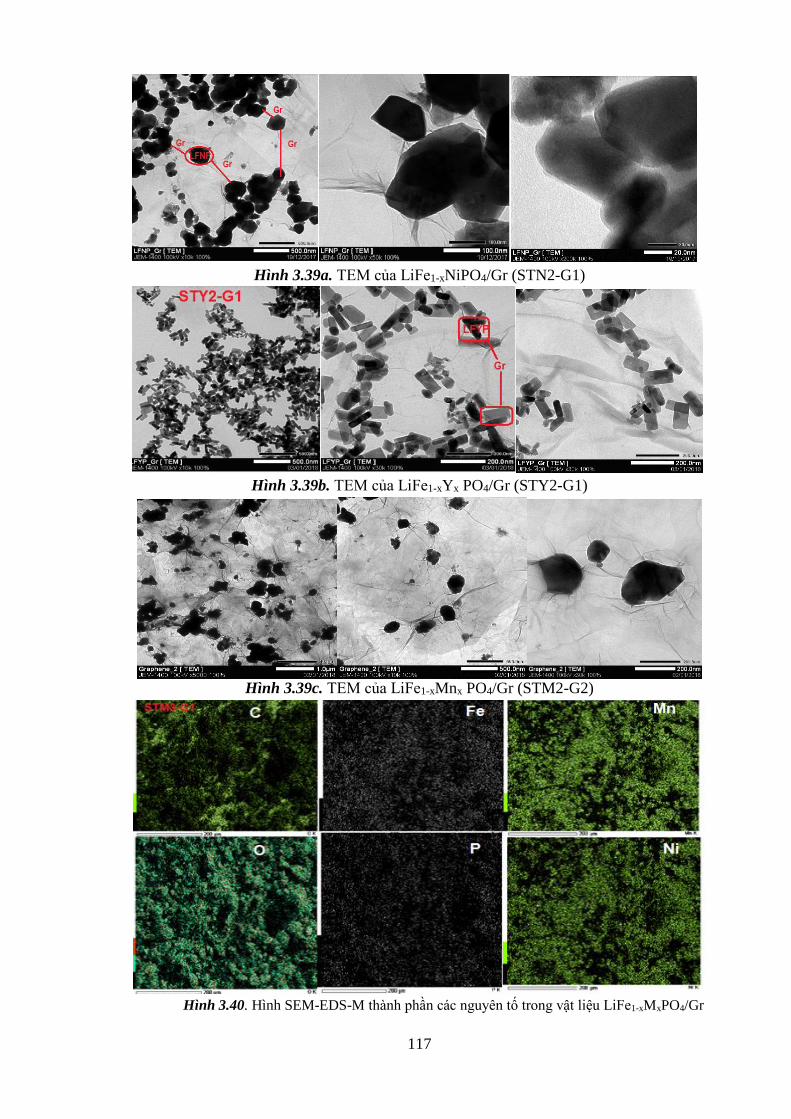
117
Hình 3.39a. TEM của LiFe1-xNiPO4/Gr (STN2-G1)
Hình 3.39b. TEM của LiFe1-xYx PO4/Gr (STY2-G1)
Hình 3.39c. TEM của LiFe1-xMnx PO4/Gr (STM2-G2)
Hình 3.40. Hình SEM-EDS-M thành phần các nguyên tố trong vật liệu LiFe1-xMxPO4/Gr
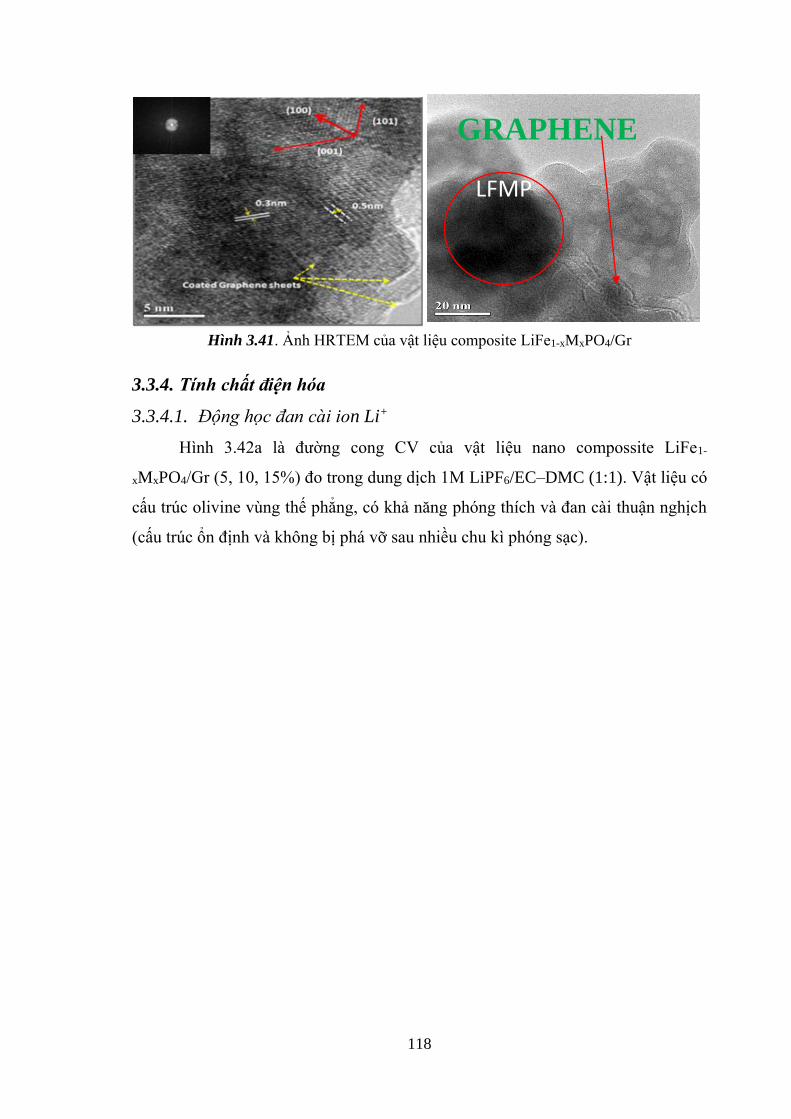
118
Hình 3.41. Ảnh HRTEM của vật liệu composite LiFe1-xMxPO4/Gr
3.3.4. Tính chất điện hóa
3.3.4.1. Động học đan cài ion Li+
Hình 3.42a là đường cong CV của vật liệu nano compossite LiFe1-
xMxPO4/Gr (5, 10, 15%) đo trong dung dịch 1M LiPF6/EC–DMC (1:1). Vật liệu có
cấu trúc olivine vùng thế phẳng, có khả năng phóng thích và đan cài thuận nghịch
(cấu trúc ổn định và không bị phá vỡ sau nhiều chu kì phóng sạc).
GRAPHENE
LFMP
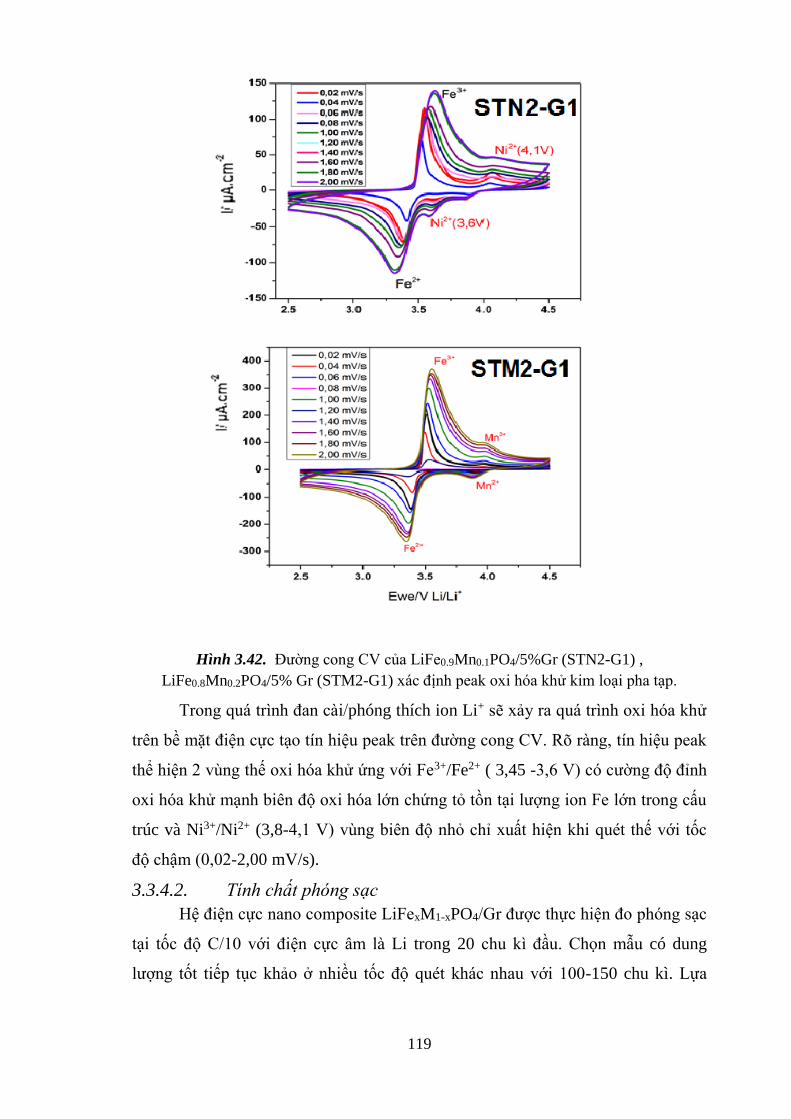
119
Hình 3.42. Đường cong CV của LiFe0.9Mn0.1PO4/5%Gr (STN2-G1) ,
LiFe0.8Mn0.2PO4/5% Gr (STM2-G1) xác định peak oxi hóa khử kim loại pha tạp.
Trong quá trình đan cài/phóng thích ion Li+ sẽ xảy ra quá trình oxi hóa khử
trên bề mặt điện cực tạo tín hiệu peak trên đường cong CV. Rõ ràng, tín hiệu peak
thể hiện 2 vùng thế oxi hóa khử ứng với Fe3+/Fe2+ ( 3,45 -3,6 V) có cường độ đỉnh
oxi hóa khử mạnh biên độ oxi hóa lớn chứng tỏ tồn tại lượng ion Fe lớn trong cấu
trúc và Ni3+/Ni2+ (3,8-4,1 V) vùng biên độ nhỏ chỉ xuất hiện khi quét thế với tốc
độ chậm (0,02-2,00 mV/s).
3.3.4.2. Tính chất phóng sạc
Hệ điện cực nano composite LiFexM1-xPO4/Gr được thực hiện đo phóng sạc
tại tốc độ C/10 với điện cực âm là Li trong 20 chu kì đầu. Chọn mẫu có dung
lượng tốt tiếp tục khảo ở nhiều tốc độ quét khác nhau với 100-150 chu kì. Lựa
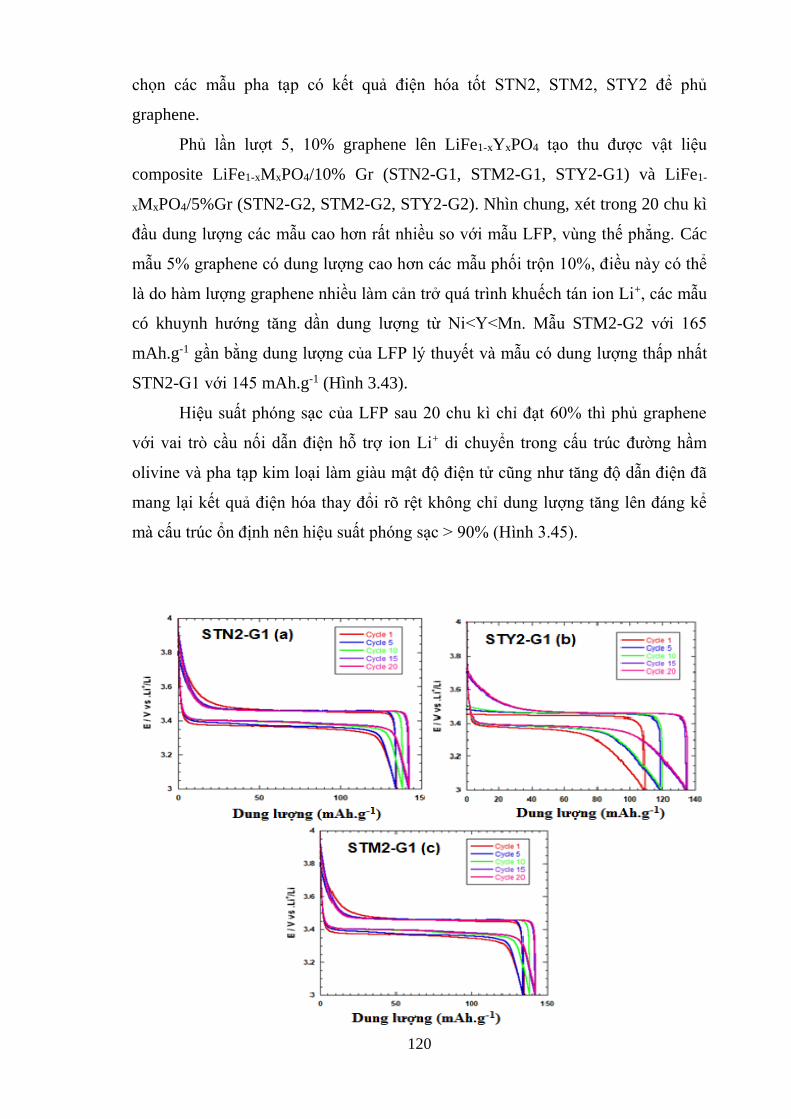
120
chọn các mẫu pha tạp có kết quả điện hóa tốt STN2, STM2, STY2 để phủ
graphene.
Phủ lần lượt 5, 10% graphene lên LiFe1-xYxPO4 tạo thu được vật liệu
composite LiFe1-xMxPO4/10% Gr (STN2-G1, STM2-G1, STY2-G1) và LiFe1-
xMxPO4/5%Gr (STN2-G2, STM2-G2, STY2-G2). Nhìn chung, xét trong 20 chu kì
đầu dung lượng các mẫu cao hơn rất nhiều so với mẫu LFP, vùng thế phẳng. Các
mẫu 5% graphene có dung lượng cao hơn các mẫu phối trộn 10%, điều này có thể
là do hàm lượng graphene nhiều làm cản trở quá trình khuếch tán ion Li+, các mẫu
có khuynh hướng tăng dần dung lượng từ Ni<Y<Mn. Mẫu STM2-G2 với 165
mAh.g-1 gần bằng dung lượng của LFP lý thuyết và mẫu có dung lượng thấp nhất
STN2-G1 với 145 mAh.g-1 (Hình 3.43).
Hiệu suất phóng sạc của LFP sau 20 chu kì chỉ đạt 60% thì phủ graphene
với vai trò cầu nối dẫn điện hỗ trợ ion Li+ di chuyển trong cấu trúc đường hầm
olivine và pha tạp kim loại làm giàu mật độ điện tử cũng như tăng độ dẫn điện đã
mang lại kết quả điện hóa thay đổi rõ rệt không chỉ dung lượng tăng lên đáng kể
mà cấu trúc ổn định nên hiệu suất phóng sạc > 90% (Hình 3.45).

121
Hình 3.43. Đường phóng sạc của mẫu STN2-G1 (a), STY2-G1 (b), STM2-G1 (c)
Hình 3.44. Đường phóng sạc của mẫu STN2-G2 (a), STY2-G2 (b), STM2-G2 (c)
Để hướng tới đưa vật liệu LiFe1-xMxPO4/Gr vào ứng dụng làm điện cực
cathode cho pin sạc Li-ion chúng tôi đã chọn mẫu pha tạp 20% Mn có kí hiệu
STM2-G (5,10%) đạt tính chất điện hóa tốt nhất để khảo sát hiệu suất phóng sạc
150-200 chu kì (Hình 3.45). Kết quả là, hiệu suất phóng sạc đạt > 96% sau 150
chu kì.
So sánh với các báo cáo về vật liệu thương mại LiCoO2 thì đạt dung lượng
đạt 155 mAh.g-1 và các công trình công bố về vật liệu LFP đạt 155-160 mAh.g-1
thì có thể thấy rằng vật liệu mới LiFe1-xMxPO4/Gr có tính khả quan áp dụng làm
vật liệu điện cực.
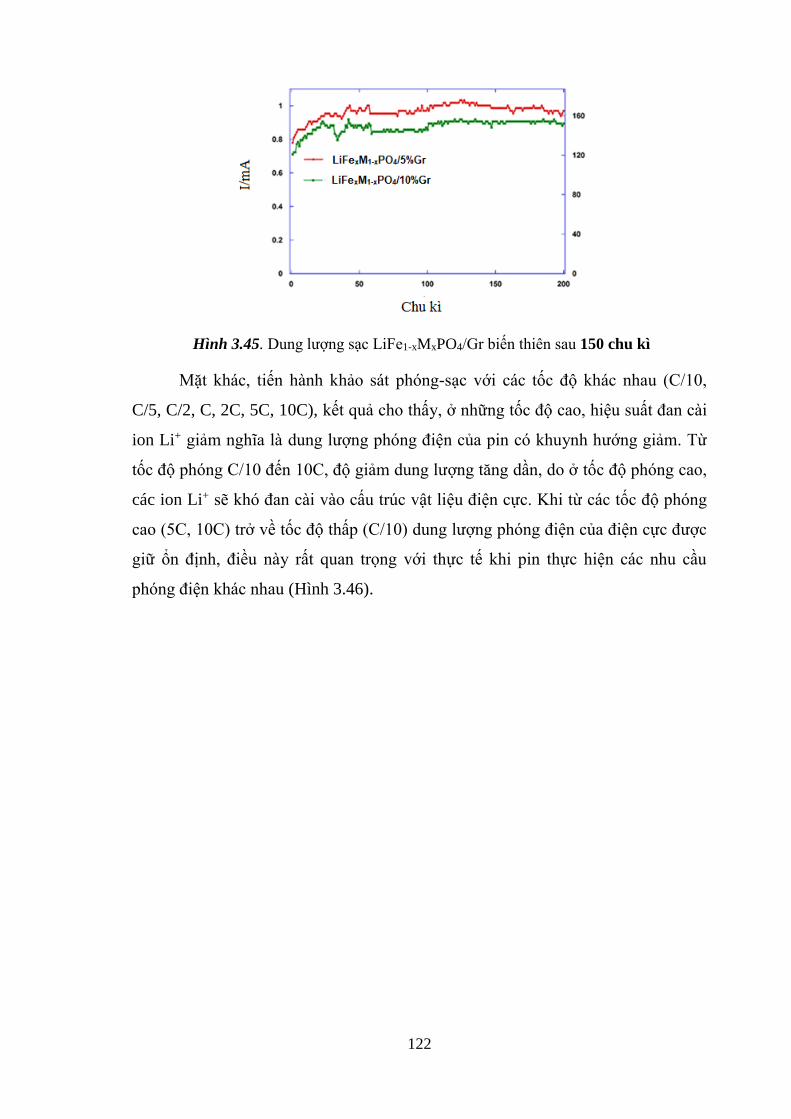
122
Hình 3.45. Dung lượng sạc LiFe1-xMxPO4/Gr biến thiên sau 150 chu kì
Mặt khác, tiến hành khảo sát phóng-sạc với các tốc độ khác nhau (C/10,
C/5, C/2, C, 2C, 5C, 10C), kết quả cho thấy, ở những tốc độ cao, hiệu suất đan cài
ion Li+ giảm nghĩa là dung lượng phóng điện của pin có khuynh hướng giảm. Từ
tốc độ phóng C/10 đến 10C, độ giảm dung lượng tăng dần, do ở tốc độ phóng cao,
các ion Li+ sẽ khó đan cài vào cấu trúc vật liệu điện cực. Khi từ các tốc độ phóng
cao (5C, 10C) trở về tốc độ thấp (C/10) dung lượng phóng điện của điện cực được
giữ ổn định, điều này rất quan trọng với thực tế khi pin thực hiện các nhu cầu
phóng điện khác nhau (Hình 3.46).
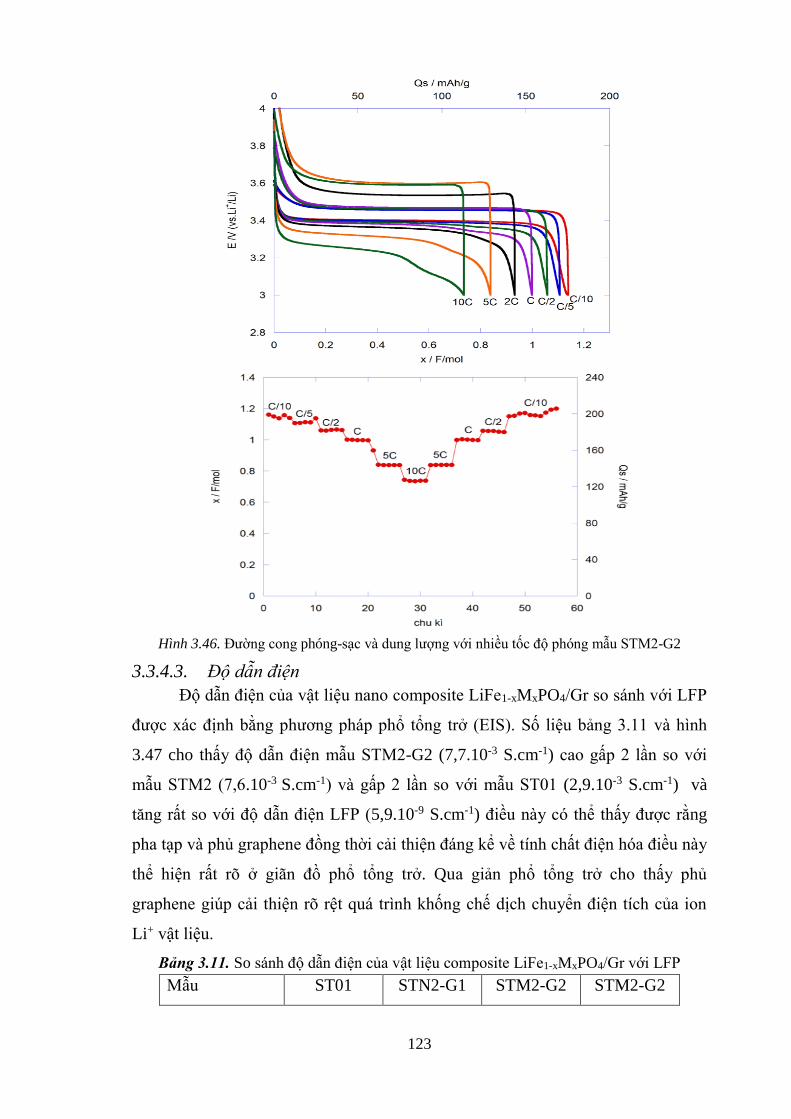
123
Hình 3.46. Đường cong phóng-sạc và dung lượng với nhiều tốc độ phóng mẫu STM2-G2
3.3.4.3. Độ dẫn điện
Độ dẫn điện của vật liệu nano composite LiFe1-xMxPO4/Gr so sánh với LFP
được xác định bằng phương pháp phổ tổng trở (EIS). Số liệu bảng 3.11 và hình
3.47 cho thấy độ dẫn điện mẫu STM2-G2 (7,7.10-3 S.cm-1) cao gấp 2 lần so với
mẫu STM2 (7,6.10-3 S.cm-1) và gấp 2 lần so với mẫu ST01 (2,9.10-3 S.cm-1) và
tăng rất so với độ dẫn điện LFP (5,9.10-9 S.cm-1) điều này có thể thấy được rằng
pha tạp và phủ graphene đồng thời cải thiện đáng kể về tính chất điện hóa điều này
thể hiện rất rõ ở giãn đồ phổ tổng trở. Qua giản phổ tổng trở cho thấy phủ
graphene giúp cải thiện rõ rệt quá trình khống chế dịch chuyển điện tích của ion
Li+ vật liệu.
Bảng 3.11. So sánh độ dẫn điện của vật liệu composite LiFe1-xMxPO4/Gr với LFP
Mẫu ST01 STN2-G1 STM2-G2 STM2-G2

124
(S/cm) 2,9.10-3 7,0.10-3 7,7.10-3 4.10-3
Cm (mol/cm3) 0,0228 0,0228 0,0228 0,0228
Hình 3.47. Phổ tổng trở (EIS) của vật liệu LFP trước và khi phủ graphene
Kết luận:
Hình ảnh TEM, HRTEM kết hợp với vùng tần số dao động Raman có thể
xác định được sự hiện diện của graphene trong cấu trúc vật liệu với 2-3 lớp
(sheet), bề dày 5-10 nm là cầu nối dẫn điện, kiểm soát kích thước hạt và ngừa oxi
hóa. Do vậy, tính chất điện hóa như độ dẫn điện, hệ số khuếch tán và dung lượng
được cải thiện: dung lượng 145-165 mAh.g-1 tại C/10 và độ dẫn điện tăng lên 1,5-
2 lần so với LFP tổng hợp cùng phương pháp nhưng tăng hơn 104-105 lần so với
độ dẫn điện lý thuyết.
Để hướng tới đưa vật liệu vào ứng dụng làm điện cực cathode đã khảo sát
hiệu suất phóng sạc các mẫu STM2-G1 và STM2-G2 sau 150 chu kì.

125
KẾT LUẬN VÀ KIẾN NGHỊ
Luận án này nhằm cải thiện tính chất điện hóa cụ thể là tăng độ dẫn điện và
hệ số khuếch tán nhằm làm tăng dung lượng vật liệu cũng như hiệu suất phóng
sạc: với độ dẫn điện lý thuyết, hệ số khuếch tán của vật liệu LFP bằng cách:
giảm kích thước hạt, pha tạp kim loại và phủ graphene.
1. Vật liệu điện cực olivine LFP
- Đã chế tạo vật liệu LFP bằng phương pháp nhiệt dung môi. Kết quả thu được
vật liệu có độ tinh khiết đạt 80%, đơn pha có cấu trúc olivine thuộc nhóm
tryphylite Pnma. Kích thước hạt vật liệu đạt 50-100 nm, độ dẫn điện 2,9.10-3
S.cm-1, hệ số khuếch tán 7,5.10-13cm2.s-1, dung lượng đạt 120 mAh.g-1 tại C/10,
hiệu suất phóng sạc sau 20 chu kì còn lại 60%.
- Tổng hợp được vật liệu LFP nung ở nhiệt độ 550 oC, khảo sát với nhiều với
nhiều tỉ lệ tiền chất khác nhau nhưng tỉ lệ Li:Fe:P = 3:1:1 đem lại pha sạch và
hiệu suất điện hóa tốt hơn, độ dẫn điện tăng 2-3 lần, hệ số khuếch tán 60-70 lần.
- Sử dụng arcosbic acid làm tác nhân khử ngăn ngừa Fe2+ bị oxi hóa và cũng là
tác nhân ngừa oxi hóa vì tạo ra nguồn carbon hữu cơ vật liệu bền với nhiệt và
vật liệu có kích thước đồng đều hơn.
2. Pha tạp kim loại
- Đã pha tạp thành công kim loại M (Ni, Y, Mn) theo nhiều tỉ lệ khác nhau thu
được vật liệu LiFe1-xMxPO4 pha sạch, hiệu suất phản ứng đạt > 90%. Ion kim
loại M phân bố đều trong cấu trúc vật liệu và ion kim loại Mn+ đã đi vào tham
gia cấu trúc thay thế một lượng nhỏ Fe2+ dẫn đến hàm lượng Fe bị sụt giảm với
quy luật tăng hàm lượng pha tạp thì hàm lượng Fe bị giảm. Tuy sự pha tạp
không thay đổi thành phần chính của pha mà chỉ có khuynh hướng làm chuyển
dịch peak nhiễu xạ về phía bên phải đối với Mn, Ni và bên trái đối với Y tương
ứng với sự thay đổi kích thước ô mạng.
- Sự pha tạp giúp tăng vùng thế oxi hóa-khử lên và đường cong CV xuất hiện 2
peak với cường khác nhau trong đó cặp thế V (Fe3+/Fe2+) có cường độ đỉnh oxi
hóa khử mạnh và rõ ràng, vùng thế Mm+/Mn+ xuất hiện vùng thế cao hơn nhưng
cường độ peak oxi hóa khử yếu (cụ thể là các vùng thế tương ứng 3,8-4,1 V
(Ni3+/N2+); 4,0-4,2 V (Mn3+/Mn2+); 3,45-3,6 V (Fe3+/Fe2+)) so với Li+/Li.

126
- Sự pha tạp các ion kim loại Mn+ vào vật liệu LFP làm độ dẫn điện (4.10-3 -
7,66.10-3 S.cm-1) tăng gấp 1,5-2 lần so với vật liệu LFP tổng hợp cùng tỉ lệ và
phương pháp tổng hợp, tăng 104-105 lần so với LFP (5,9.10-9 S.cm-1) lý thuyết.
Mặt khác, sau khi pha tạp các kim loại Ni, Mn, Y thì độ dẫn điện cao hơn một
số công trình đã công bố: Co (2,9.10-6 S.cm-1), La (2,4.10-6 S.cm-1).
- Dung lượng tại tốc độ phóng sạc C/10 với các mẫu LFNP (125-135 mAh.g-1),
LFYP (120-130 mAh.g-1); LFMP (140-145 mAh.g-1); hiệu suất phóng sạc giảm
30% sau 20 chu kì.
Như vậy, tính chất điện hóa tăng theo quy luật các mẫu vật STN2<
STYN2<STM3< STM2 có nghĩa là, trong nghiên cứu này pha tạp Mn mang lại
hiệu quả tốt hơn Ni và Y.
3. Vật liệu nano composite LiFe1-xMxPO4/Gr
- Chế tạo thành công vật liệu nanocomposite LiFe1-xMxPO4/Gr với tỉ lệ 5, 10%
graphene. Với 5% graphene cho kết quả tốt nhất; các tinh thể vật liệu LiFexM1-
xPO4 đính trên màng mỏng graphene với bề dày 5-10 nm (2-3 sheet), các ion Li+
linh động hơn nhờ màng graphene làm cầu nối dẫn điện cho in Li+ di chuyển và
hạn chế ăn mòn điện cực. Mỗi tinh thể LiFe1-xMxPO4 đính trên màng graphene
được giải thích theo cơ chế điện tích: Tinh thể LiFe1-xMxPO4 giàu oxi tích điện
âm ( ) đính trên mỗi ô mạng lục giác của graphene có nối chứa liên kết
giàu mật độ điện tích dương ( ) tạo ra hiệu ứng cảm giữa mạng lúc giác
graphene và LFMP tạo nên vật liệu composite có cấu trúc bền vững và tính chất
điện hóa tốt.
- Đồng thời pha tạp kim loại và phủ graphene mạng lại kết quả điện hóa với độ
dẫn điện tăng hơn 1,5 -2,0 lần so với mẫu LFP tổng hợp cùng phương pháp
nhưng tăng 104-105 lần so với LFP lý thuyết, dung lượng tăng hơn 1,5 -1,8 lần
(STN2-G2: 140 mAh.g-1); (STY2-G2: 150 mAh.g-1); STM2-G2: 165 mAh.g-1)
tại C/10. Hiệu suất phóng sạc ổn định duy trì 96% sau 150 chu kì.

127
KIẾN NGHỊ
Vật liệu nano composite LiFe1-xMxPO4/Gr với M (Y, Ni, Mn) được
tổng hợp bằng phương pháp nhiệt dung môi có thể đưa vào ứng dụng làm vật
liệu điện cực cathode cho pin sạc Li-ion.
Hướng phát triển đề tài: tiếp tục khảo sát vật liệu này trên 500 chu kì để
đánh giá tuổi thọ của pin và rút ngắn khoảng cách thương mại hóa và mở rộng
phạm vi ứng dụng.

128
NHỮNG ĐÓNG GÓP MỚI CỦA ĐỀ TÀI
1. Pha tạp nhiều kim loại khác nhau (Mn, Ni, Y..) và đồng thời sử dụng
carbon hữu cơ (in-situ) kết hợp graphene (ex-situ) chế tạo nên vật liệu nano
composite với các tinh thể vật liệu LiFe1-xMx PO4/Gr của mạng lưới tổ ong
graphene dựa trên chênh lệch về độ âm điện của các nguyên tử tạo ra hiệu
ứng hút điện tích giữa mạng lục giác graphene và LFMP tạo nên vật liệu
composite có cấu trúc bền vững và tính chất điện hóa tốt. Điểm mới so với
các công trình đã công bố trước đó chỉ thực hiện riêng rẽ phủ graphene lên
LiFePO4 tạo vật liệu LiFePO4/Gr hoặc pha tạp kim loại M vào cấu trúc vật
liệu LiFe1-xMx PO4.
2. Đã chứng minh được quy luật: sau khi pha tạp kim loại M, cấu trúc vật liệu
dịch chuyển tuyến tính tuân thủ theo định luật Vegard và không làm ảnh
hưởng đến cấu trúc olivine.
3. Luận án làm sáng tỏ vấn đề pha tạp kim loại đồng thời phủ graphene đã tạo
nên vật liệu trong đó quá trình khống chế khuếch tán Warburg chiếm ưu thế
hơn quá trình khống chế động học. Ion Li+ di chuyển trong cấu trúc olvine
phụ thuộc chủ yếu vào sự khuếch tán trong vật liệu. Đây là cơ sở để lý giải
tại sao cải thiện được hệ số khuếch tán và độ dẫn điện (độ dẫn điện 104-105
lần so với độ dẫn điện lý thuyết của vật liệu LiFePO4) dẫn đến việc hiệu
suất điện hóa cải thiện rõ rệt: dung lượng đạt 155-165 mAh.g-1 tại C/10.
4. Bằng phương pháp nhiệt dung môi, đã chế tạo thành công vật liệu
composite LiFe0.8Mn0.2PO4/5%Gr có dung lượng gần bằng với dung lượng
lý thuyết của LFP ổn định trong 150 chu kì. Như vậy, có thể sử dụng tỉ lệ
hàm lượng 20% Mn và 5% graphene để chế tạo vật liệu LiFe1-xMx PO4/C
làm cathode pin sạc Li-ion và từ đó định hướng mở rộng phạm vi ứng dụng
ở quy mô công suất cao hơn.

129
CÔNG TRÌNH ĐÃ CÔNG BỐ
1. Huynh Le Thanh Nguyen, Nguyen Thi My Anh, Tran Van Man, La Thi Hang,
Tran Thu Trang, Tran Thi Thuy Dung, Electrode composite LiFePO4@carbon:
structure and electrochemical performances, Journal of Nanomaterials,2019, 1-10.
2. La Thi Hang, Nguyen Thi My Anh, Nguyen Nhi Tru, Huynh Le Thanh Nguyen,
Le My Loan Phung - Modification of nano-sized LiFePO4 via nickel doping and
graphene coating, International Journal of Nanotechnology, 2019, 914-924.
3. Dinh Duc Thanh, Nguyen Thi My Anh, Nguyen Nhi Tru, La Thi Hang, Le My
Loan Phung, The impact of carbon additives on lithium ion diffusion kinetic of
LiFePO4/C composites, The Science and Technology Development Journal, 22(1)
2019, 173-179.
4. La Thi Hang, Nguyen Nhi Tru, Le My Loan Phung, Olivine structured
LiFexYyPO4/C composite synthesized via solvothermal route as cathode material
for lithium batteries, Vietnam Journal of Chemistry, 56(6E2), 2018, 267-271.
5. Bui Thi Thao Nguyen, Doan Thi Kim Bong, La Thi Hang, Nguyen Nhi Tru,
Hoang Xuan Tung, Nguyen Thi My Anh, -Modification of Ketjenblack EC-600JD
carbon as filler in cathode material for lithium-ion battery, Vietnam Journal of
Chemistry, 56(6E2), 2018, 262-266.
6. Nguyen Thi My Anh, Doan Luong Vu, Nguyen Thai Hoa, Le My Loan Phung,
Nguyen Ba Tai, La Thi Hang, Nguyen Ngoc Trung, Nguyen Nhi Tru-
Characterization of LiFePO4 nanostructures synthesized by solvothermal method.
Journal of Science and Technology, Technical universities 118, 2017, 45-50.
7. La Thi Hang, Le My Loan Phung, Nguyen Thi My Anh, Hoang Xuan Tung,
Doan Phuc Luan, NguyenNhi Tru- Enhancement of li–ion battery capacity using
nickel doped LiFePO4 as cathode material. Journal of Science and Technology
55_1B, 2017, 267-283.
8. La Thi Hang, Nguyen Nhi Tru, Nguyen Thi My Anh, Le My Loan Phung, Doan
Luong Vu, Doan Phuc Luan, –Microwave-assisted solvothermal synthesis of
LiFePO4/C nanostrutures for lithium ion batteries, Proceedings of the 5th Asian
Materials Data Symposium. HaNoi10, 2016, 343-352.

130
TÀI LIỆU THAM KHẢO
1. M.S. Whittingham, Materials challenges facing electrical energy storage, MRS
Bull., 2008, 33(4), 411–419.
2. A.S. Andersson, J.O. Thomas, The source of first-cycle capacity loss in
LiFePO4, J. Power Sources, 2001, 97-98, 498-502.
3. J.M. Tarascon, M. Armand, Issues and challenges facing rechargeable lithium
batteries, Nature, 2001, 414, 359–367.
4. G.R.E. Dahlin, K.E. Strom, Lithium batteries, Research and Applications,
Publishers: Nova Science. Inc., 2016, New York.
5. A.K. Padhi, K.S. Nanjundaswamy, J.B. Goodenough, Phospho-olivines as
positive-electrode materials for rechargeable lithium batteries, J. Electrochem.
Soc., 1997, 144, 1188-1194.
6. N. Meethong, H.-Y.S. Huang, S.A. Speakman, W.C. Carter, Y.M. Chiang,
Strain accommodation during phase transformations in olivine-based cathodes
as a materials selection criterion for high-power rechargeable batteries, Adv.
Funct. Mater., 2007, 17(7), 1115-1123.
7. M.K. Satam, R. Natarajan, S. Kobi, M.K. Jangid, Y. Krishnan, A.
Mukhopadhyay, Growth induced phase transformation during
delithiation/lithiation of ‘ultra-small’ LiFePO4 nanoparticles decorating carbon
nanotubes, Scr. Mater., 2016, 124, 1-5.
8. A. Eftekhari, LiFePO4/C nanocomposites for lithium-ion batteries, J. Power
Sources, 2017, 343, 395-411.
9. V. Mathew, J. Gim, E. Kim et al., A rapid polyol combustion strategy towards
scalable synthesis of nanostructured LiFePO4/C cathodes for Li-ion batteries,
J. Solid State Electrochem., 2014, 1557–1567.
10. J.O. Herrera, H. Camacho-Montes, L.E. Fuentes, L.Alvarez-Contreras,
LiMnPO4: Review on synthesis and electrochemical properties, J. Mater. Sci,
Chem. Eng., 2015, 3, 54-64.
11. C. Daniel, Materials and processing for lithium-ion batteries, JOM, 2008,
60(9), 43–48.
12. L.W. Ji, Z. Lin, M. Alcoutlabi, X.W. Zhang, Recent developments in
nanostructured anode materials for rechargeable lithium-ion batteries, Energy
& Environ. Sci., 2011, 3, 2682-2699.
13. C.Y. Nan, J. Lu, L.H. Li, Q. Peng, Y.D. Li, Size and shape control of LiFePO4
nanocrystals for better lithium ion battery cathode materials, Nano Res., 2013,
6(7), 469-477.
14. F.Q. Cheng, W. Wan, Z. Tan, Y.Y. Huang, H.H. Zhou, J.L. Chen, X.X.
Zhang, High power performance of nano-LiFePO4/C cathode material
synthesized via lauric acid-assisted solid-state reaction, Electrochim. Acta,
2011, 56, 2999-3005.

131
15. H. Ghafarian-Zahmatkesh, M. Javanbakht, M. Ghaemi, Ethylene glycol-
assisted hydrothermal synthesis and characterization of bow-tie-like lithium
iron phosphate nanocrystals for lithium-ion batteries, J. Power Sources, 2015,
284, 339-348.
16. P. P. Prosini, Iron phosphate materials as cathodes for lithium batteries, The
use of environmentally friendly iron in lithium batteries, Springer, 2011
(ebook)
17. S.I. Lee, K.H. Yoon, M. Song, H. Peng, K.A. Page, C.L. Soles, D.Y. Yoon,
Structure and properties of polymer electrolyte membranes containing
phosphonic acids for anhydrous fuel cells, Chem. Mater., 2011, 24, 115-222.
18. M. Kumar, S.S. Sekhon, Ionic conductance behaviour of plasticized polymer
electrolytes containing different plasticizers, Ionics, 2002, 8(3-4), 223-233.
19. G. Dahlin, K. Strom. Lithium batteries: Research, technology and
applications, Nova Science, 2010, New York.
20. G. Venugopal, A. Hunt, F. Alamgir, Nanomaterials for Energy Storage in
Lithium-ion Battery Applications, Material Matters, 2010, 5, 42.
21. Y. Miao, P. Hynan, A. von Jouanne, A. Yokochi, Current Li-Ion battery
technologies in electric vehicles and opportunities for advancements,
Energies, 2019, 12, 1074; doi:10.3390/en12061074
22. B. Dunn, H. Kamath, J.M. Tarascon, Electrical energy storage for the grid: A
battery of choices, review, Science, 2011, 334, 928-935
23. M. Nakayama, M. Kaneko, M. Wakihara, First-principles study of lithium ion
migration in lithium transition metal oxides with spinel structure, Chem. Phys.,
2012, 14, 13963-13970.
24. M.M. Thackeray, A. de Kock, M.H. Rossouw, D. Liles, R. Bittihn, D. Hoge,
Spinel electrodes from the Li‐Mn‐O system for rechargeable Lithium battery
applications, J. Electrochem. Soc., 1992, 139(2), 363-366.
25. G.X. Wang, S. Bewlay, J. Yao, J.H. Ahn, S.X. Dou, H.K. Liu,
Characterization of LiMxFe1-x PO4 (M=Mg, Zr, Ti) cathode materials prepared
by the sol-gel method, Electrochem. Solid-state Lett., 2004, 7(12), A503-
A506.
26. A. Manthiram, W. Choi, Suppression of Mn dissolution in spinel cathodes by
trapping the protons within layered oxide cathodes, Electrochem. Solid State
Lett., 2007, 10(9), A228-231.
27. V. Aravindan, K. Karthikeyan, K.S. Kang, W.S. Yoon, W.S. Kim, Y.S. Lee,
Influence of carbon towards improved lithium storage properties of
Li2MnSiO4 cathodes, J. Mater. Chem., 2011, 21, 2470-2475.
28. D. Gryzlov, S. Novikova, T. Kulova, A. Skundin, A. Yaroslavtsev, Behavior
of LiFePO4/CPVDF/Ag-based cathode materials obtained using
polyvinylidene fluoride as the carbon source, Mater. Des., 2016, 104, 95-101.
29. R. Yazami, Nanomaterials for Li-ion batteries: Fundamental and Applications,
Pan Stanford Pubishers. 2014 (eBook).

132
30. L. Wang, X.M. He, W.T. Sun, J.L. Wang, Y.D. Li, S. Fan, Crystal orientation
tuning of LiFePO4 nanoplates for high rate lithium battery cathode materials,
Nano Lett. 2012, 12(11), 5632–5636.
31. A. Odani, A. Nimberger, B. Markovsky, E. Sominski, E. Levi, V.G. Kumar,
Development and testing of nano materials for rechargeable lithium batteries,
J. Power Sources, 2003, 119–121, 517-521.
32. J. Świder, M. Świętosławski, M. Molenda, R. Dziembaj, A novel concept for
the synthesis of nanometric LiFePO4 by co-precipitation method in an
anhydrous environment, Procedia Eng., 2014, 98, 36 – 41.
33. Y.T. Dai, L. Ma, H. Zhang, Y.H. Chen, Y.L. Chen, W. Wang, Hydrothermal
synthesis of spindle-like nanostructured LiFePO4 powders mediated by
organic acid, Int. J. Electrochem. Sci., 2016, 11, 1730 – 1737.
34. G. Arnold, J. Garche, R. Hemmer, S. Strobele, C. Vogler, M. Wohlfahrt-
Mehrens, Fine-particle lithium iron phosphate LiFePO4 synthesized by a new
low-cost aqueous precipitation technique, J. Power Sources., 2003, 119-121,
247–251.
35. T.V.S.L. Satyavani, A.S. Kumar, P.S.V.S. Rao, Methods of synthesis and
performance improvement of lithium iron, JESTECH, 2016, 19(1), 178–188.
36. D. Burch, M.Z. Bazant, Size-dependent spinodal and miscibility gaps for
intercalation in nanoparticles, Nano Lett., 2009, 9(11), 3795–3800.
37. S.M. Oh, H.G. Jung, C.S. Yoon, S.T. Myung, Z.H. Chen, K. Amine, Y.K.
Sun, Enhanced electrochemical performance of carbon-LiMn1-xFexPO4
nanocomposite cathode for lithium-ion batteries, J. Power Sources, 2011,196,
6924-6928.
38. A. Yamada, S.C. Chung, K. Hinokuma, Optimized LiFePO4 for Lithium
battery cathodes, J. Electrochem. Soc., 2001, 148, A224-A229.
39. J.F. Qian, M. Zhou, Y.L. Cao, X.P. Ai, H.X. Yang, Template-free
hydrothermal synthesis of nanoembossed mesoporous LiFePO4 microspheres
for high-performance lithium-ion batteries, J. Phys. Chem. C, 2010, 114,
3477-3482.
40. H.L. Wang, Y. Yang, Y.Y. Liang, L.F. Cui, H.S. Casalongue , Y.G. Li ,
G.S. Hong, Y. Cui , H.J. Dai, LiMn1-xMxPO4, Nanorods grown on graphene
sheets for ultrahigh‐rate‐performance lithium ion batteries, Angew. Chem. Int.
Ed., 2011, 50, 1-6.
41. K. Bazzi, M. Nazri, V.M. Naik, V.K. Garg, A.C. Oliveira, P.P. Vaishnava,
G.A. Nazri, R. Naik, Enhancement of electrochemical behavior of
nanostructured LiFePO4/carbon cathode material with excess Li, J. Power
Sources, 2016, 306, 17-23.
42. R. Ramachadran, S. M. Chen, Modern development of novel electrode
materials as effective electrode catalyst for lithium ion batteries, Int. J.
Electrochem. Sci., 2015, 10, 9488 – 9512.

133
43. Y. Xiao, F. C. Zhang, J. I. Han, Electrical structures, magnetic polaron and
lithium ion dynamics in three transition metal doped LiFe1-xMxPO4 (M = Mn,
Co and La) cathode material for Li ion batteries from density functional theory
study, Solid State Ion., 2016, 294, 73–81.
44. T. Muraliganth, A. Manthiram, Understanding the shifts in the redox
potentials of olivine LiM1-YMYPO4 (M= Fe, Mn, Co, and Mg) solid solution,
J. Phys. Chem. C, 2010, 114(36), 15530.
45. Y.C. Ge, X.D. Yan, J. Liu, X.F. Zhang, J.W. Wang, X.G. He, R.S. Wang,
H.M. Xie, An optimized Ni doped LiFePO4/C nanocomposite with excellent
rate performance, Electrochimica Acta, 2010, 55, 5886–5890.
46. B. Ding, P. Xiao, G. Ji, Y. Ma, L. Lu, J.Y. Lee, High-performance Lithium-
ion cathode LiMn0.3Fe0.7PO4/C and the mechanism of performance
enhancements through Fe substitution. ACS Appl. Mater. Interfaces 2013,
5(22), 12120-12126.
47. T. Shiratsuchi, S. Okada, T. Doi, J.I. Yamaki, Cathodic performance of
LiMn1-xM xPO4 (M= Ti, Mg, Zr) annealed in an inert atmosphere, Electrochim.
Acta, 2009, 54, 3145–3151
48. H.H. Yi, C.L. Hu, H.S. Fang, B. Yang, Y.C. Yao, W.H. Ma, Y.N. Dai,
Optimized electrochemical performance of LiMn0.9Fe0.9-xMgxPO4/C for
Lithium ion batteries. Electrochim. Acta, 2011, 56(11), 4052-4057.
49. J. Molenda, A. Kulka, A. Milewska, W. Zając and K. Świerczek, Structural,
transport and electrochemical properties of LiFePO4 substituted in lithium
and iron sublattices (Al, Zr, W, Mn, Co and Ni), Materials, 2013, 6(5),
1656-1687.
50. S.K. Martha, J. Grinblat, O. Haik, LiMn0.8Fe0.2PO4: An advanced cathode
material for rechargeable Lithium batteries. Angew. Chem. Int. Ed., 2009, 48,
8559-8563.
51. A. A. Chekannikov, A. A. Kuz’mina, T. L. Kulova, A. M. Skundin, S. A.
Novikova, I. A. Stenina, A. B. Yaroslavtsev, Development of Lithium-ion
battery of the “Doped lithium iron phosphate–Doped lithium titanate” system
for power applications, Int. J. Electrochem. Sci., 2017, 12, 4417 – 4427.
52. J.Z. Hu, J. Xie, X.B. Zhao, H.M. Yu, X. Zhou, G.S. Cao, J.P. Tu, Doping
effects on electronic conductivity and electrochemical performance of
LiFePO4, J. Mater. Sci. Technol., 2009, 25(3), 405–409.
53. S.H. Wu, M.S. Chen, C.J. Chen, Y.P. Fu, Preparation and characterization of
Ti4+– doped LiFePO4 cathode materials for lithium-ion batteries, J. Power
Sources, 2009,189(1), 440-444.
54. H. Göktepe, Electrochemical performance of Yb–doped Li– LiFePO4/C
composites as cathode materials for lithium–ion batteries, Res. Chem.
Intermediates, 2013, 39, 2979–2987.

134
55. Y.D. Cho, G.T-K. Fey., H. M. Kao, Physical and electrochemical properties
of La–doped LiFePO4/C composites as cathode materials for lithium–ion
batteries, J. Solid State Electrochem., 2008, 12(7-8), 815–823.
56. H.B. Shu, X.Y. Wang, Q. Wu, B.N. Hu, X.K. Yang, Q.L. Wei, Q.Q. Liang,
Y.S. Bai, M. Zhou, C. Wu, M.F. Chen, A.W. Wang, L.L. Jiang, Improved
electrochemical performance of LiFePO4/C cathode via Ni and Mn co-doping
for Lithium ion batteries, J. Power Sources, 2013, 237, 149–155.
57. J.H. Gim, J.J. Song D. Nguyen, M.H. Alfaruqi, S.J. Kim, J.W. Kang, A.K.
Rai, V. Mathew, J.K. Kim, A two-step solid state synthesis of LiFePO4/C
cathode with varying carbon contents for Li-Ion batteries, Ceram. Int., 2014,
40, 1561-1567.
58. J.J. Wang, J.L. Yang, Y.J. Tang, R.Y. Li, G.X. Liang, T.K. Sham, X.L. Sun,
Surface aging at olivine LiFePO4: a direct visual observation of iron
dissolution and the protection role of nano-carbon coating, J. Mater. Chem. A,
2013, 1, 1579-1586.
59. C.G. Son, D.R. Chang, H.S. Kim, Y.S. Lee, Synthesis and electrochemical
properties of nano crystalline LiFePO4 obtained by different methods, J.
Electrochem. Sci. Technol., 2011, 2(2), 103-109.
60. K. Bazzi, B.P. Mandal, M. Nazri, V.M. Naik, V.K. Garg, A.C. Oliveira, P.P.
Vaishnava, G.A. Nazri , R. Naik, Effect of surfactants on the electrochemical
behavior of LiFePO4 cathode material for lithium ion batteries, J. Power
Sources, 2014, 265, 67-74
61. T. Kozawa, N. Kataoka, A. Kondo, E. Nakamura, H. Abe, M. Naito, One-step
mechanical synthesis of LiFePO4/C composite granule under ambient
atmosphere, Ceram. Int. 2014, 40, 16127-16131.
62. K. Wang, R. Cai, T. Yuan, X. Yu, R. Ran, Z.P. Shao, Process investigation,
electrochemical characterization and optimization of LiFePO4/C composite
from mechanical activation using sucrose as carbon source, Electrochim. Acta
2009, 54(10), 2861-2868.
63. S.X. Deng, H. Wang, H. Liu, J.B. Liu, H. Yan, Review, Research progress in
improving the rate performance of LiFePO4 cathode materials, Nano-Micro
Lett., 2014, 6(3), 209-226.
64. Y. D. Cho, G. T. K. Fey, H. M. Kao, ơ, J. Power Sources, 2009, 189(1), 256-
262.
65. X. Huang, X. He, C. Jiang, G. Tian, Morphology evolution and impurity
analysis of LiFePO4 nanoparticles via a solvothermal synthesis process, RSC
Adv., 2014, 4(99), 56074–56083.
66. K. Vediappan, A. Guerfi, V. Gariepy, G.P. Demopoulos, P. Hovington, J.
Trottier, A.Mauger, C.M. Julien, K. Zaghib, Stirring effect in hydrothermal
synthesis of nano C-LiFePO4, J. Power Sources, 2014, 266, 99-106.
67. K. Oyaizu, Polymers for high-density and fast charging batteries, Organic
battery days, 2017, Uppsala, Sweden.

135
68. L. Castro, R. Dedryvère, J.-B. Ledeuil, J. Bréger, C. Tessier, D. Gonbeau,
Aging mechanisms of LiFePO4/Graphite cells studied by XPS: Redox reaction
and electrode/electrolyte interfaces, J. Electrochem. Soc., 2012, 159, A357-
363.
69. H.C. Shin, W.I. Cho, H. Jang, Electrochemical properties of carbon-coated
LiFePO4 cathode using graphite, carbon black and acetylene black,
Electrochim. Acta,2006, 52, 1472–1476.
70. Y.Z. Dong, Y.M. Zhao, Y.H. Chen, Z.F. He, Q. Kuang. Optimized carbon-
coated LiFePO4 cathode material for lithium-ion batteries, Mater. Chem.
Phys., 2009, 115(1), 245-250.
71. R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Carbon nanotubes--the
route toward applications, Science, 2002, 297, 787-792.
72. Phan Ngọc Minh, Vật liệu cacbon cấu trúc nano và các ứng dụng tiềm năng,
NXB Khoa học tự nhiên và công nghệ, 2014, Hà Nội.
73. O. Toprakci, H. A. Toprakci, L. Ji, G. Xu, Z. Lin and X. Zhang, Carbon
nanotube-loaded electrospun LiFePO4/carbon composite nanofibers as stable
and binder-free cathodes for rechargeable lithium-ion batteries, ACS Appl.
Mater. Interfaces, 2012, 4(3), 1273-1280.
74. J. Wang, X. Sun, Understanding and recent development of carbon coating on
LiFePO4 cathode materials for lithium-ion batteries, Ener. Environ. Sci., 2012,
5(1), 5163-5185.
75. R. Saito, M. Fujita, G. Dresselhaus, and M. S Dresselhaus, Electronic structure
of chiral graphene tubules, Appl. Phys. Lett., 1992, 60(18), 2203-2206.
76. T. Ohta, A. Bostwick, T. Seyller, K. Horn, E. Rotenberg, Controlling the
electronic structure of bilayer graphene, Science, 2006, 313, 951-954.
77. K.S. Novoselov, A.K. Geim, S.V. Morozov, D. Jiang, Y. Zhang, S.V.
Dubonos, I.V. Grigorieva, A.A. Firsov, Electric field effect in atomically thin
carbon films, Science, 2004, 306(5696), 666–669.
78. A.K. Geim, P. Kim, Carbon wonderland, Sci. Am., 2008, 298(4), 90–97.
79. Y.-W. Tan, Y. Zhang, K. Bolotin, Y. Zhao, S. Adam, E. H. Hwang, S. Das
Sarma, H. L. Stormer, P. Kim, Measurement of scattering rate and minimum
conductivity in graphene, Phys. Rev. Lett., 2007, 99, 246803-1:246803-4 .
80. B. Marinho, M. Ghislandi, E.Tkalya, C. E. Koning, G. With, Electrical
conductivity of compacts of graphene, multi-wall carbon nanotubes, carbon
black, and graphite powder, Powder Technol., 2012, 221, 351-358.
81. Y.W. Zhu, S. Murali, W.W. Cai, X.S. Li, J.W. Suk, J.R. Potts, R.S. Ruoff,
Graphene and graphene oxide: Synthesis, properties, and applications, Adv.
Mater., 2010, 22(35), 3906–3924.
82. W.B. Choi, J.W. Lee, Graphene synthesis and applications, Carbon, 2010, 48,
2127-2150.

136
83. D.C. Marcano, D. V. Kosynkin, J.M. Berlin, A. Sinitskii, Z.Z. Sun, A.
Slesarev, L.B. Alemany, W.Lu, J. M. Tour, Improved synthesis of graphene
oxide, ACS Nano, 2010, 4, 4806-4814.
84. N.I. Zaaba, K.L. Foo, U. Hashim, S.J. Tan, Wei-Wen Liu, C.H. Voon,
Synthesis of graphene oxide using modified Hummers method: Solvent
influence, Procedia Eng., 2017, 184, 469 – 477.
85. S. Park, J. An, J.R. Potts, A.Velamakanni, S.Murali, R.S.Ruoff. Hydrazine-
reduction of graphite- and graphene oxide. Carbon, 2011, 49, 3019 – 3023.
86. B. Rajagopalan, J. S. Chung, Reduced chemically modified graphene oxide for
supercapacitor electrode, Nanoscale Res. Lett. 2014, 9, 535
87. W.Z. Shen, Y.M. Wang, J. Yan, H.X. Wu, S.W. Guo, Enhanced
electrochemical performance of lithium iron (II) phosphate modified
cooperatively via chemically reduced graphene oxide and polyaniline,
Electrochim. Acta, 2015, 173, 310–315.
88. S.J.R. Neale, E.P. Randviir, A.S.A. Dena, C.E. Banks, An overview of recent
applications of reduced graphene oxide as a basis of electron analytical
sensing platforms, Appl. Mater. Today, 2018, 10, 218–226.
89. J.G. Radich, P.J. McGinn, P.V. Kamat, Graphene-based composites for
electrochemical energy storage, Electrochem. Soc. Interface, 2011, 20(1), 63-
66.
90. A. Abouimrane, O. C. Compton, K. Amine, S.T. Nguyen, Non annealed
graphene paper as a binder-free anode for lithium-ion batteries, J. Phys. Lett.
C, 2010, 114(29), 12800-12804.
91. X.L. Lei, H.Y. Zhang, Y.M. Chen, W.G. Wang, Y.P. Ye, C.C. Zheng, P.
Deng, Z.C. Shi, A three-dimensional LiFePO4/carbon nanotubes/graphene
composite as a cathode material for lithium-ion batteries with superior high-
rate performance, J. Alloys Compd, 2015, 626, 280-286.
92. R. Raccichini, A.Varzi, S. Passerini, B. Scrosti, The role of graphene for
electrochemical energy storage, Nature Mater., 2015, 14, 271–279.
93. G.H. Yuan, J.T. Bai, T.N.L. Doan, P. Chen, Synthesis and electrochemical
properties of LiFePO4/graphene composite as a novel cathode material for
rechargeable hybrid aqueous battery, Mater. Lett., 2015, 158, 248–251.
94. Y. Zhang, Y.D. Huang, X.C. Wang, Y. Guo, D.Z. Jia, X.C. Tang. Improved
electrochemical performance of lithium iron phosphate in situ coated with
hierarchical porous nitrogen-doped graphene-like membrane. J. Power
Sources, 2016, 305, 122-127.
95. H.X. Wu, Q.J. Liu, Sh. W. Guo, Composites of graphene and LiFePO4 as
cathode materials for lithium-ion battery, Nano-Micro Lett., 2014, 6(4), 316–
326.
96. S.A. Hong, S.J. Kim, J. Kim, B.G. Lee, K.Y. Chung, Y.W. Lee, Carbon
coating on lithium iron phosphate (LiFePO4 ): comparison between continuous

137
supercritical hydrothermal method and solid-state method, Chem. Eng. J.,
2012, 198-199, 318–326.
97. P.P. Prosini, M. Carwska, F. Maroni, R. Tossici, F. Nobili, A lithium-ion
battery based on LiFePO4 and silicon/reduced graphene oxide nanocomposite,
Solid State Ion., 2015, 283, 145–151.
98. M. Köhler, W. Fritzsche, Nanotechnology- an introduction to nanostructuring
techniques, Wiley-VCH Verlag GmbH & Co. KGaA., 2007.
99. M. Barghamadi, A. Kapoor, C. Wen, A review on li-s batteries as a high
efficiency rechargeable lithium battery, J. Electrochem. Soc., 2013, 160,
A1256-A126.
100. C.J. Brinker, G.W. Scherer, Sol-gel Science, The Physics and Chemistry of
Sol-gel Processing, Academic Press Inc., 1990, San Diego, CA.
101. K. Rana, A. Sil, S. Ray, Synthesis of ribbon type carbon nanostructure using
LiFePO4 catalyst and their electrochemical performance, Mater. Res. Bull.,
2009, 44, 2155–2159.
102. R. Dominko, J.M. Goupil, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel,
Impact of LiFePO4/C composites porosity on their electro chemical
performance, J. Electrochem. Soc., 2005, 152(5), A858–A863.
103. L.L. Hench, J.K. West, The sol-gel process, Chem. Rev. 1990, 90(1), 33-37.
104. M.Y. Gao, N.Q. Liu, Zh.Y. Li, W.K. Wang, Ch.M. Li, H. Zhang, Y.L. Chen ,
Z.B. Yu, Y.Q. Huang, A gelatin-based sol–gel procedure to synthesize the
LiFePO4/C nanocomposite for lithium ion batteries. Solid state Ion., 2014,
258, 8–12.
105. Z.L. Gong, Y. Yang, Recent advances in the research of polyanion-type
cathode materials for Li-ion batteries, Ener. Environ. Sci., 2011, 4, 3223-
3242.
106. P.C. Smecellato, Alternative route for LiFePO4 synthesis: Carbothermal
reduction combined with microwave-assisted solid-state reaction, Mater.
Res. Bull., 2017, 86, 209-214.
107. B. Scrosati, S. Panero, P. Reale, D. Satolli, Y. Aihara, Investigation of new
types of lithium ion battery materials, J. Power Sources, 2002, 105(2), 161–
168.
108. T. Nakamura, Y. Miwa, M. Tabuchi, Y. Yamada, Structural and Surface
Modifications of LiFePO4 olivine particles and their electrochemical
properties. J. Electrochem. Soc., 2006, 153, A1108-A1114.
109. F. Teng, S. Santhanagopalan, A. Asthana, X.B. Geng, S.I. Mho, R.
Shahbazian-Yassar, Self-assembly of LiFePO4 nanodendrites in a novel
system of ethylene glycol–water, J. Cryst. Growth, 2010, 312(23), 3493-
3502.
110. Y.J. Zhu, T. Gao, X.L. Fan, F.D. Han, C.S. Wang, Electrochemical
techniques for intercalation electrode materials in rechargeable batteries, Acc.
Chem. Res., 2017, 50(4), 1022-1031.

138
111. Y.V. Bykov, K.I. Rybakov, V.E. Semenov, High temperature microwave
processing of materials, J. Phys. D: Appl. Phys., 2001, 34(13), R55–R75.
112. H. Zou, G. Zhang, P.K. Shen, Intermittent microwave heating synthesized
high performance spherical LFP/C for Li-ion batteries, Mater. Res. Bull.,
2010, 45, 149–152.
113. M. Higuchi, K. Katayama, Y. Azuma, M. Yukawa, M. Suhara, Synthesis of
LiFePO4 cathode material by microwave processing, J. Power Sources
2003,119–121, 258–261.
114. S. Beninati, L. Damen, M. Mastragostino, MW assisted synthesis of LiFePO4
for high power applications, J. Power Sources, 2008, 180, 875–79.
115. A.V. Murugan, T. Muraliganth, A. Manthiram, Rapid microwave
solvothermal synthesis of phosphor olivine nanorods and their coating with a
mixedconducting polymer for lithium ion batteries, Electrochem. Commun.,
2008, 10, 903–906.
116. Y.H. Wang, R. Mei, X. Yang, Enhanced electrochemical properties of
LiFePO4/C synthesized with two kinds of carbon sources, PEG-4000
P(inorganic) and Super, Ceram. Int., 2014, 40(6), 8439-8444.
117. C. Calderón, J.E. Thomas, G. Lener, D.E. Barraco, A.Visintin,
Electrochemical comparison of LiFePO4 synthesized by a solid-state
method using either microwave heating or a tube furnace, J. Appl.
Electrochem., 2017, 47, 1179–1188.
118. H.C. Shin, W.I. Cho, H. Jang, J.P. Souvern, Electrochemical properties of the
carbon-coated LiFePO4 as a cathode material for lithium-ion secondary
batteries, J. Power Sources, 2006, 159(2), 1383–1389.
119. M. Konarova, I. Taniguchi, Synthesis of carbon-coated
LiFePO4 nanoparticles with high rate performance in lithium secondary
batteries, J. Power Sources, 2010, 195, 3661-3667.
120. H.C. Kang, D.K. Jun, B. Jin, E. Jin, K.H. Park, H.B. Gu, K.W. Kim,
Optimized solid-state synthesis of LiFePO4 cathode materials using ball-
milling, J. Power Sources, 2008, 179(1), 340-346.
121. V. Palomares, T. Rojo, Synthesis processes for Li-Ion battery electrodes –
From solid state reaction to solvothermal self-assembly methods, Chapter
from book: Lithium ion batteries: New developments 2012, doi:
10.5772/27496[book]
122. M. R. Yang, Y. H. Teng & S. H. Wu. LiFePO4/carbon cathode materials
prepared by ultrasonic spray pyrolysis, J. Power Sources, 2006, 159, 307-
311.
123. J.H. Lee, K.Y. Jung, S.B. Park, Modification of titani a particles by
ultrasonic spray pyrolysis of colloid, J. Mater. Sci., 1999, 34, 4089–4093
124. S.H. Ju, Y.C. Kang, LiFePO4/C cathode powders prepared by spray pyrolysis
from the colloidal spray solution containing nanosized carbon black, Mater.
Chem. Phys., 2008, 107, 328–333.

139
125. J.K. Kim, Supercritical synthesis in combination with a spray process for 3D
porous microsphere lithium iron phosphate, Cryst. Eng. Comm, 2014, 16,
2818–2822.
126. K.S. Park, K.T. Kang, S.B. Lee, Synthesis of LiFePO4 with fine particle by
co-precipitation method, Mater. Res. Bull., 2004, 39, 1803-1810
127. J.C. Zheng, X.H. Li, Z.X. Wang, H.J. Guo, S.Y. Zhou, LiFePO4 with
enhanced performance synthesized by a novel synthetic route, J. Power
Sources, 2008, 184, 574–577.
128. Y. Ding, Y. Jiang, F. Xu, J. Yin, H. Ren, Q. Zhuo, Preparation of
nanostructured LiFePO4/graphene composites by co-precipitation method,
Electrochem. Communs., 2010, 12, 10–13.
129. C.C. Huang, D.S. Ai, L. Wang, X.M. He, Rapid synthesis of LiFePO4 by co-
precipitation, Chem. Lett., 2013, 42(10), 1191–1193.
130. C.C. Huang, D.S. Ai, L. Wang, X.M. He, LiFePO4 crystal growth during co-
precipitation, Int. J. Electrochem. Sci., 2016, 11, 754 – 762.
131. J. Barker, M.Y. Saidi, J.L. Swoyer, Lithium iron(II) phosphoolivines
prepared by a novel carbothermal reduction method, Electrochem. Solid State
Lett. 2003, 6, A53–A55.
132. W.Y. Yang, Z.S Wang, L. Chen, Y. Chen, L. Zhang, Y.B.Lin, J.X Lia and
Zh.G Huang, Suppression of degradation for lithium iron phosphate
cylindrical batteries by nano silicon surface modification, RSC Adv., 2017,
7, 33680.
133. B.Q. Zhu, X.H. Li, Y.X. Wang, H.J. Guo, Novel synthesis of LiFePO4 by
aqueous precipitation and carbothermal reduction, Chem. Phys.,2006, 98,
373-6.
134. C.H. Mi, X.B. Zhao, G.S. Cao, In situ synthesis and properties of carbon-
coated LiFePO4 as Li-ion battery cathodes, J. Electrochem Soc, 2005, 152,
483-487.
135. S, Franger, F. Le Cras, C. Bourbon, H. Rouault, LiFePO4 synthesis routes for
enhanced electrochemical performance, Electrochem. Solid State Lett., 2002,
5(10), A231-A233
136. M. Zhou, J.F. Qian, Y.L. Cao, H.X. Yang., Low temperature Hydrothermal
synthesis and electrochemical performances as a cathode material for lithium
ion batteries, New Ener. Mater., 2012, 57(32), 4164- 4169.
137. S. Tajimi, Y. Ikeda, K. Uematsu, K. Toda, M. Sato, Enhanced
electrochemical performance of LiFePO4 prepared by hydrothermal reaction,
Solid State Ion., 2004, 175(1-4), 287-290.
138. H. Nakano, K. Dokko, S. Koizumi, H. Tannai, K.Kanamura, Hydrothermal
synthesis of carbon-coated LiFePO4 and its application to Lithium polymer
battery. J. Electrochem. Soc., 2008, 155, A909-A914.
139. Kanamura, K. & Koizumi, S. Hydrothermal synthesis of LiFePO4 as a
cathode material for lithium batteries. J. Mater. Sci., 2008, 43, 2138-2142.

140
140. C. Nan, J.Lu, C.Chen, Q.Peng, Y.D.Li, Solvothermal synthesis of lithium
iron phosphate nanoplates, J. Mater. Chem., 2011, 21(27), 9994-9996.
141. Y.Y. Liu, J.J. Gu, J.L. Zhang, J. Wang, N. Nie, Y. Fu, W. Li, F. Yu,
Controllable synthesis of nano-sized LiFePO4/C via a high shear mixer
facilitated hydrothermal method for high rate Li-ion batteries, Electrochim.
Acta, 2015, 173, 448-45.
142. A. Vadivel Murugan, T. Muraliganth, A. Manthiram. Comparison of
microwave assisted solvothermal and hydrothermal syntheses of LFP/C
nanocomposite cathodes for lithium ion batteries, J. Phys. Chem. C, 2008,
112, 14665–14671.
143. Y. Liu, J. Zhang, Y. Li , Y. Hu, W.Li, M.Y. Zhu , P.F. Hu, S.L. Chou,
G.X.Wang, Solvothermal synthesis of a hollow micro-sphere LiFePO4/C
composite with a porous interior structure as a cathode material for
lithium ion batteries, Nanomaterials, 2017, 7(11), 368.
144. Y. Long, Y. Shu, X.H. Ma, M.X. Ye, In-situ synthesizing superior high-rate
LiFePO4-C nanorods embedded in graphene matrix, Electrochim. Acta, 2014,
117, 105-112.
145. H.X. Gong, Y. Yu, T. Li, T. Mei, Z. Xing, Y.C. Zhu, Y.T. Qian, X.Y. Shen,
Solvothermal synthesis of LiFePO4-C nanopolyhedrons and microellipsoids
and their performance in lithium-ion batteries, Mater. Lett., 2012, 66(1), 374-
376.
146. D. Rangappa, K.Sone, T. Kudo, I. Honma, Directed growth of
nanoarchitectured LiFePO4 electrode by solvothermal synthesis and their
cathode properties, J. Power Sources, 2010, 195(18), 6167-6171.
147. S.L. Yang, X.F. Zhou, J.G. Zhang, Z.P. Liu, Morphology-controlled
solvothermal synthesis of LiFePO4 as cathode material for lithium-ion
batteries. J. Mater. Chem., 2010, 20, 8086-809.
148. T. Tsuji, M. Nagao, Y. Yamamura, Nguyen Tien Tai, Structural and thermal
properties of LiMn2O4 substituted for manganese by iron, Solid State Ion.,
2002, 154–155, 381–386.
149. S. Kasap, Elements of X-ray Diffration by crystals, An Ebooklet, 1990-2001.
150. A.Monshi, M.Reza Foroughi, M.R. Monshi, Modified Scherrer Equation to
estimate more accurately nano-crystallite size using XRD, World J. Nano
Sci. Eng. (WJNSE), 2012, 2, 154-160.
151. M.F.C. Ladd, R.A.Palmer, Structure Determination by X-ray
Crystallography, 3rd ed., Plenum Press, 1993, New York.
152. S.A. Speakman, Estimating Crystallite Size Using XRD, MIT Center for
Materials Science and Engineering, http://prism.mit.edu/xray, 12/2018.
153. A. Kwiatkowski, M. Gnyba, J. Smulko, P. Wierzba, Algorithms of chemicals
detection using Raman spectra, Metrol. Meas. Syst., 2010, 12, 549–560.
154. J.R. Ferraro, K. Nakamoto, Introductory Raman Spectroscopy, Academic
Press, 2003, San Diego.

141
155. R. Baddour-Hadjean, J.-P. Perreira-Ramos, Raman microspectrometry
applied to the study of electrode materials for lithium batteries, Chem. Rev.
2010, 110, 1278–1319.
156. K.S. Kulwinder, B.P.Mandal, K. Bazzi, M.W.Lin, M. Nazri, G.A. Nazri,
V.M. Naik, V.K.Garg, A.C. Oliveira, P. Vaishnava, R. Naik, Z.X. Zhou,
Enhanced electrochemical performance of graphene modified LiFePO4
cathode material for lithium ion batteries, Solid State Ion., 2013, 253, 94–
100.
157. D.A. Skoog, F.J. Holler, C.R. Crouch, Principles of instrumental analysis,
Thomson Brooks/Cole Publ, 1999.
158. Thermogravimetric Analysis (TGA), PerkinElmer Instrument Training,
USA, 2015, http://las.perkinelmer.com/Trainings/Courses.htm.
159. N. Kamarulzaman, M.H. Jaafar, Synthesis and stoichiometric analysis of Li-
ion battery cathode material, Chapter in the book Stoichiometry and
Materials Science-When numbers matter, Intech Publication, 2012. DOI:
10.5772/33189.
160. D. Briggs, J.T. Grant, Surface Analysis by Auger and X-ray Photoelectron
Spectroscopy, IM Publications and SurfaceSpectra Ltd., 2003.
161. J.F. Moulder, W.F. Stickle, P.E. Sobol, K.D. Bomben, Handbook of X-ray
Photoelectron Spectroscopy, 1992.
162. R. García, A.P. Báez, Atomic Absorption Spectrometry (AAS), InTech,
2012.
163. H. Takahara, H. Miyauchi, M. Tabuchi, T. Nakamura, Elemental Distribution
Analysis of LiFePO4/Graphite Cells Studied with Glow Discharge Optical
Emission Spectroscopy, J. Electrochem. Soc., 2013, 160(2), A272-A278.
164. A.F. Orliukas, K.-Z. Fung, V. Venckutė, V. Kazlauskienė, J. Miškinis, A.
Dindune, Z. Kanepe, J. Ronis, A. Maneikis, T. Šalkus, A. Kežionis,
SEM/EDX, XPS and impedance spectroscopy of LiFePO4 and LiFePO4
ceramics, Lithuanian J. Phys., 2014, 54(2), 106–113.
165. J. Goldstein, D.E. Newbury, D.C. Joy, C.E. Lyman, P. Echlin, E. Lifshin,
L.C. Sawyer, J.R. Michael. Scanning Electron Microscopy and X-ray
Microanalysis. Springer, 2003.
166. M. de Graef, Intoduction to Conventional Transmission Electron
Microscopy. Cambridge University Press. ISBN 0-521-62995, 2003.
167. Noémie.E, K.J. Rountree, B.D. McCarthy, E.S. Rountree, T.T. Eisenhart,
J.L. Dempsey, A Practical Beginner’s Guide to Cyclic Voltammetry,
American Chemical Society and Division of Chemical Education, 2017,
doi:10.1021/acs.jchemed.7b00361
168. F. Hoffart, Charging and discharging methods that extend Li-ion battery life,
Design features, https://www.rcgroups.com/forums/showatt.
169. T. Osaka, D. Mukoyama, H. Nara, Development of diagnostic process for
commercially available batteries, especially lithium ion battery by

142
electrochemical impedance spectroscopy, J. Electrochem. Soc.,
2015, 162(14), A2529-A2537.
170. C.R. Birkl, D.A. Howey, Model identification and parameter estimation for
LiFePO4 batteries, IET Hybrid and Electric Vehicles Conference (HEVC),
2013, DOI http://dx.doi.org/10.1049/cp.2013.1889
171. A. Lasia, Electrochemical Impedance Spectroscopy and its Applications,
from book: Modern Aspects of Electrochemistry, Kluwer Academic/Plenum
Publishers, 1999, 32, 143-248.
172. E. Barsoukov and J. R. Macdonald, Impedance Spectroscopy: Theory,
Experiment, and Applications, Wiley-Interscience, 2005.
173. J.M. Atebamba, J.Moskon, S.Pejovnik, M. Gabersceka, On the interpretation
of measured impedance spectra of insertion cathodes for Lithium-ion
batteries, J. Electrochem. Soc., 2010, 157, A1218.
174. R. Dedryvere, M. Maccario, L. Croguennec, F. Le Cras, C. Delmas, D.
Gonbeau, X-ray photoelectron spectroscopy investigations of carbon-coated
LixFePO4 materials, Chem. Mater., 2008, 20, 7164-7170.
175. M. Köhler, W. Fritzsche, Nanotechnology- an introduction to
nanostructuring techniques, Wiley-VCH Verlag GmbH & Co. KGaA., 2007.
176. M.Vergani, Electronic Instrumentation for Electrochemical Cell Monitoring
in Lab-on-Chip Devices, Doctoral Dissertation, Polytechnical University of
Milan (Politechnico di Milano), 2012.
177. Trần Đại Lâm, Các phương pháp hóa lý vật liệu, Nxb KHTN&CN, 2017, Hà
Nội.
178. Mai Thanh Tùng, Kỹ thuật nguồn điện, Nxb ĐHBK Hà Nội, 2016, Hà Nội
179. Le Ha Chi, Nguyen Nang Dinh, S. Brutti, B. Scrosati, Synthesis,
characterization and electrochemical properties of 4.8V LiNi0.5Mn1.5O4
cathode material in lithium-ion batteries, Electrochim. Acta, 2010, 55(18),
5110–5116.
180. Hung Tran Nguyen, M.R.Zamfir, Loc Dinh Duong, Y.H. Lee, P.Bondavalli,
D.Pribat, Alumina-coated silicon-based nanowire arrays for high quality Li-
ion battery anodes, J. Mater. Chem., 2012, 22, 24618-24626
181. Thang Van Le, Ha Tran Nguyen, Anh Tuan Luu, Van Man Tran, Phung
Loan My Le, LiMn2O4/CNTs and LiNi0.5Mn1.5O4 nanocomposite as High
performance cathode materials for lithium batteries, Acta Metall. Sin., 2015,
28(10), 122-128.
182. Nguyen Thi My Anh, Doan Luong Vu, Nguyen Thai Hoa, Le My Loan
Phung, Nguyen Ba Tai, La Thi Hang, Nguyen Ngoc Trung, Nguyen Nhi Tru,
Characterization of LiFePO4 nanostructures synthesized by solvothermal
method. J. Sci.Technol. - Technical Universities, 2017, 118, 45-50.
183. G. Berckmans, M. Messagie, J. Smekens, N. Omar, L. Vanhaverbeke, L. van
Mierlo, Cost projection of state of the art Lithium-ion batteries for electric
vehicles up to 2030, Energies, 2017, 10, 1314.

143
PHỤ LỤC
1. Hình ảnh bột vật liệu
- Bột LFP sau khi nung
- Bột vật liệu LFe1-xMxPO4/Gr
2. Hình ảnh màng điện cực vật liệu
3. Giản đồ XRD
144
- XRD CỦA LFP (ST00, ST01)
- XRD của LFP (ST01)

145
- XRD của mẫu STM2
146
- STY2-G2
- XRD của STN2-G2

147
- XRD của STM2-G2
148
4. TGA
- TGA của LFP (ST01)
5. EDS
- LFP

149
- LiFe0.98Y0.02PO4/5%Gr
- LiFe0.95Y0.05PO4/5%Gr
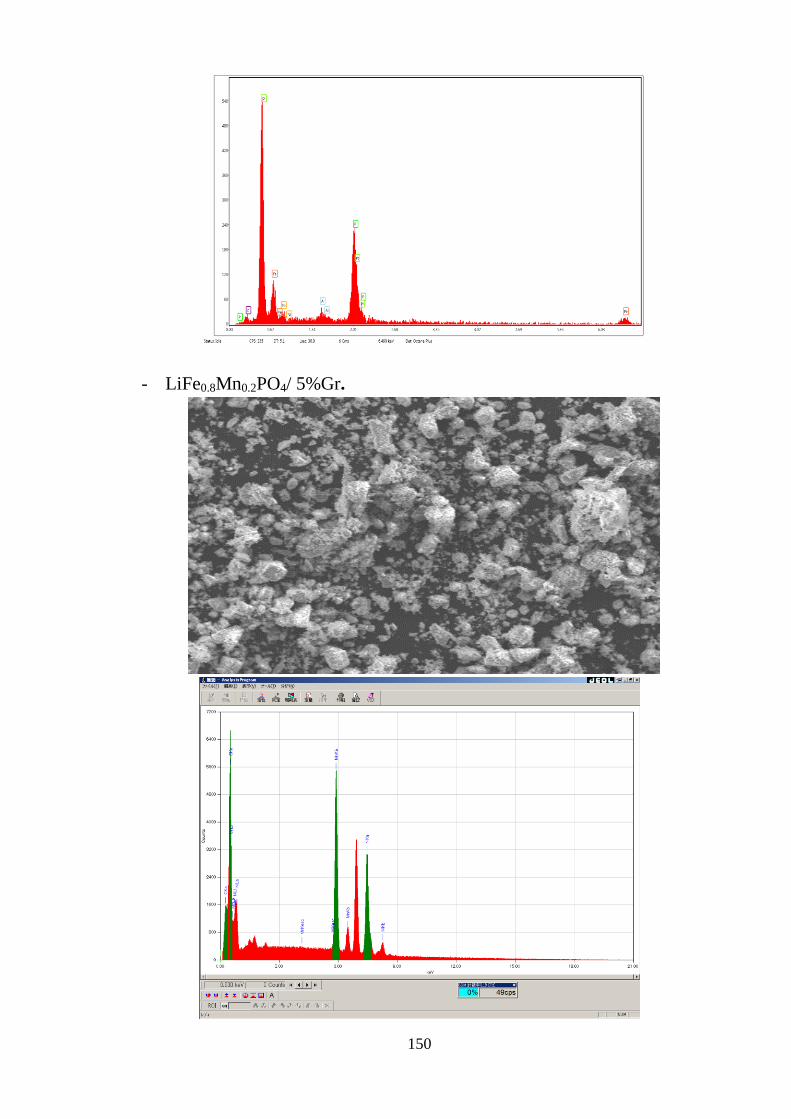
150
- LiFe0.8Mn0.2PO4/ 5%Gr.
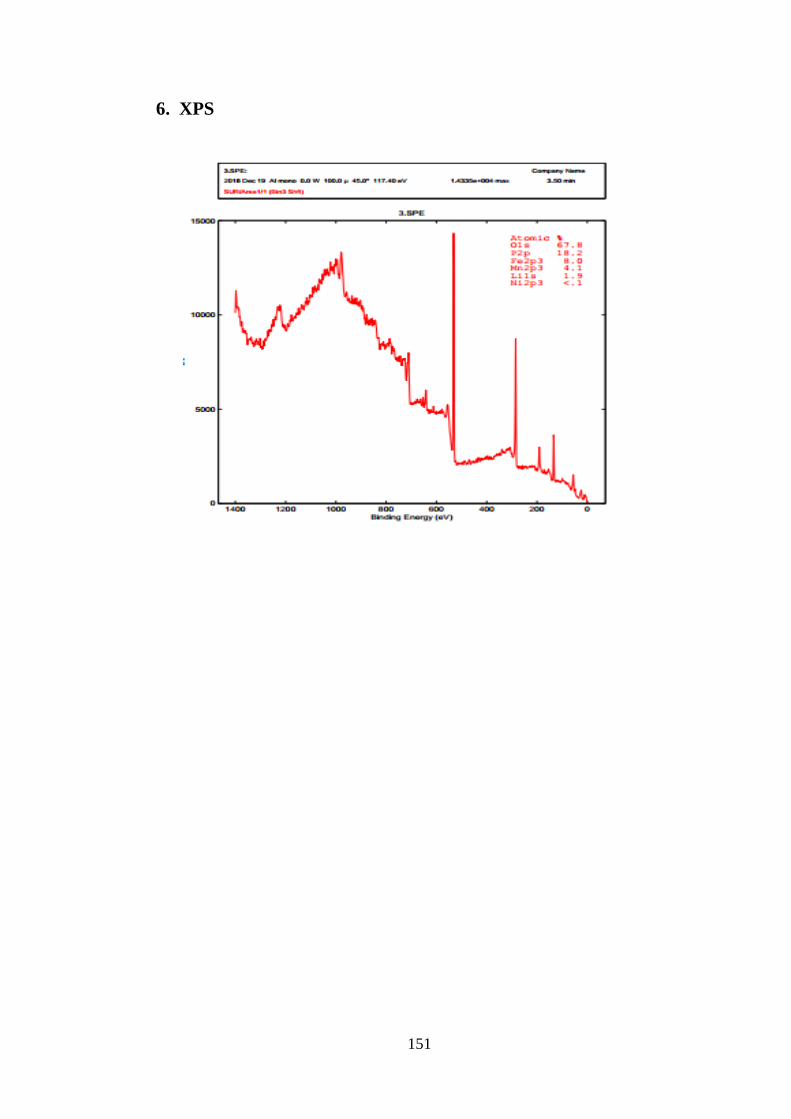
151
6. XPS
Top Related
Copyright © 2022 FDOKUMEN

