
![Photophysical properties and computational investigations of tricarbonylrhenium(I)[2-(4-methylpyridin-2-yl)benzo[ d]-X-azole]L and tricarbonylrhenium(I)[2-(benzo[ d]-X-azol-2-yl)-4-methylquinoline]L](https://static.fdokumen.com/doc/165x107/631cc3eea906b217b9071d28/photophysical-properties-and-computational-investigations-of-tricarbonylrheniumi2-4-methylpyridin-2-ylbenzo.jpg)






Bahasa
Halaman
Hukum


Synthesis Structural Characterization and Ab Initio Study ofCu5+δIn2+xSb2minusx A New B8-Related Structure TypeCarola J Mullerdagger Sven Lidindagger Susana Ramos de DebiaggiDaggersect Crispulo E Deluque ToroDagger
and Armando Fernandez Guillermetsectperp
daggerPolymer and Materials Chemistry Lund University Lund SwedenDaggerDepartamento de Fiacutesica Facultad de Ingenieriacutea Universidad Nacional del Comahue Neuquen Province ArgentinasectCONICET Santa Fe ArgentinaperpUnidad de Actividad de Fiacutesica Fiacutesica de Metales Centro Atomico Bariloche S C de Bariloche Argentina
S Supporting Information
ABSTRACT A new ternary orthorhombic compound with the formulaCu5+δIn2+xSb2minusx crystallizing in the space group Cmc21 with 36 atoms perunit cell [a = 101813(4) Aring b = 84562(4) Aring c = 73774(2) Aring Z = 4] hasbeen synthesized by conventional high-temperature methods The structureis based on the B8 archetype (NiAsNi2In) and features InSb ordering aswell as ordering of interstitial copper Details of the experimental study andthe structural parameters of this compound are reported in the first part ofthe work In the second part ab initio calculations based on the densityfunctional theory and the projector augmented-wave method are used tocharacterize the structural thermodynamic and phase-stability properties ofthe new ternary phase The present calculations include the latticeparameters molar volume bulk modulus and its pressure derivative theenergy of formation from the elements and the electronic density of statesMoreover the present ab initio method is used to investigate thethermodynamic properties of the anti-structure Cu5Sb2In2 compound obtained by exchanging the In and Sb Wyckoffsymmetric positions
1 INTRODUCTION
The study of the stable phases in the CuminusInminusSb system is ofinterest in connection with the contact metallurgy of InSb anarrow gap semiconductor compound with a very high electronmobility that is widely used in a variety of electronic devices Itis also of relevance for the design of lead-free soldering alloys1
In spite of the practical relevance of the CuminusInminusSb alloys thephase diagram and the properties of the compounds occurringin this system have not yet been accurately determined In factthe most recent attempts to establish the ternary phase diagraminvolve extrapolations based on thermodynamic models23
While this may yield useful insights into pseudobinary systembehavior it will not predict the occurrence and constitution oftrue ternary phases The purpose of the present paper is toreport a combined experimental and theoretical study of aternary CuminusInminusSb compound that was detected in theframework of a study of the η phase a common equilibriumphase of both the CuminusIn45 and CuminusSn binary systems6minus8
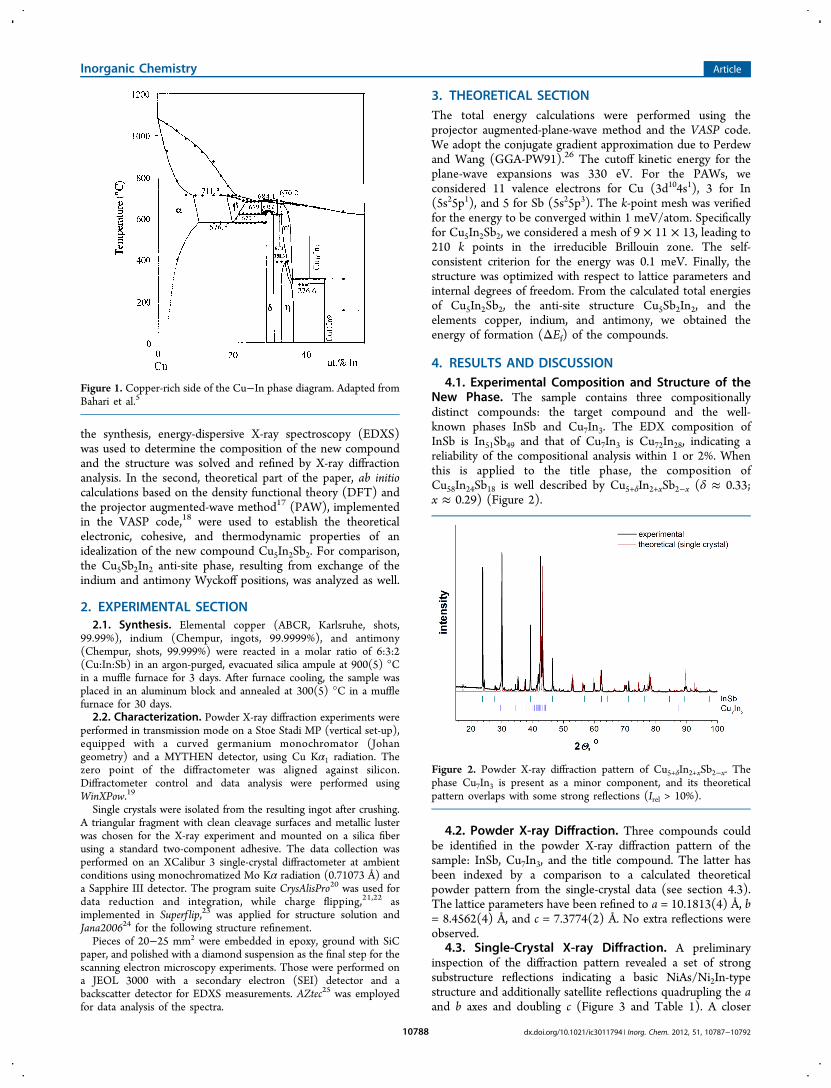
The B8 phase field (NiAsNi2In structure types) in thebinary system CuminusIn is complex and comprises at least fourwell-defined regions (Figure 1) The indium-rich side of thephase field contains the line compounds Cu11In9 and Cu10In7while the (internal) η-phase field is centered around thecomposition Cu2In The copper-rich side is dominated by the
phase Cu7In3 which has a rather broad existence interval(Cu72minusδIn28+δ 0 le δ le 04) The structure of the η-phase fieldin the CuminusIn binary system has not yet been fully resolvedalthough it is accepted that it involves at least one high-temperature (HT) phase (ηprime) and at least one at lowtemperature (LT η)9
The ηprimeη transition temperature depends upon thecomposition10 The HT structure is of the type NiAsNi2Inwith random partial occupation of the Cu sites with 2dsymmetry whereas the LT phase presents modulated super-structures based on the B8minusNiAsNi2In prototype with anordered distribution of defects at sites (2d)11
On the other hand there are no B8-type binary CuminusSbphases The copper-rich side of the phase diagram consists ofCu11Sb3
12 Cu10Sb313 and HT1-Cu3Sb
14 which are coloredhexagonal close packings While HT2-Cu3Sb
15 adopts the BiF3structure type Cu2Sb crystallizes in a structure featuringpuckered slabs of colored cubic close packing16 Full solidsolubility in the ternary system is impossibleIn this paper we report on the synthesis and characterization
of a new ternary phase in the CuminusInminusSb system Subsequent to
Received June 2 2012Published October 2 2012
Article
pubsacsorgIC
copy 2012 American Chemical Society 10787 dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus10792
the synthesis energy-dispersive X-ray spectroscopy (EDXS)was used to determine the composition of the new compoundand the structure was solved and refined by X-ray diffractionanalysis In the second theoretical part of the paper ab initiocalculations based on the density functional theory (DFT) andthe projector augmented-wave method17 (PAW) implementedin the VASP code18 were used to establish the theoreticalelectronic cohesive and thermodynamic properties of anidealization of the new compound Cu5In2Sb2 For comparisonthe Cu5Sb2In2 anti-site phase resulting from exchange of theindium and antimony Wyckoff positions was analyzed as well
2 EXPERIMENTAL SECTION21 Synthesis Elemental copper (ABCR Karlsruhe shots
9999) indium (Chempur ingots 999999) and antimony(Chempur shots 99999) were reacted in a molar ratio of 632(CuInSb) in an argon-purged evacuated silica ampule at 900(5) degCin a muffle furnace for 3 days After furnace cooling the sample wasplaced in an aluminum block and annealed at 300(5) degC in a mufflefurnace for 30 days22 Characterization Powder X-ray diffraction experiments were
performed in transmission mode on a Stoe Stadi MP (vertical set-up)equipped with a curved germanium monochromator (Johangeometry) and a MYTHEN detector using Cu Kα1 radiation Thezero point of the diffractometer was aligned against siliconDiffractometer control and data analysis were performed usingWinXPow19
Single crystals were isolated from the resulting ingot after crushingA triangular fragment with clean cleavage surfaces and metallic lusterwas chosen for the X-ray experiment and mounted on a silica fiberusing a standard two-component adhesive The data collection wasperformed on an XCalibur 3 single-crystal diffractometer at ambientconditions using monochromatized Mo Kα radiation (071073 Aring) anda Sapphire III detector The program suite CrysAlisPro20 was used fordata reduction and integration while charge flipping2122 asimplemented in Superf lip23 was applied for structure solution andJana200624 for the following structure refinementPieces of 20minus25 mm2 were embedded in epoxy ground with SiC
paper and polished with a diamond suspension as the final step for thescanning electron microscopy experiments Those were performed ona JEOL 3000 with a secondary electron (SEI) detector and abackscatter detector for EDXS measurements AZtec25 was employedfor data analysis of the spectra
3 THEORETICAL SECTIONThe total energy calculations were performed using theprojector augmented-plane-wave method and the VASP codeWe adopt the conjugate gradient approximation due to Perdewand Wang (GGA-PW91)26 The cutoff kinetic energy for theplane-wave expansions was 330 eV For the PAWs weconsidered 11 valence electrons for Cu (3d104s1) 3 for In(5s25p1) and 5 for Sb (5s25p3) The k-point mesh was verifiedfor the energy to be converged within 1 meVatom Specificallyfor Cu5In2Sb2 we considered a mesh of 9 times 11 times 13 leading to210 k points in the irreducible Brillouin zone The self-consistent criterion for the energy was 01 meV Finally thestructure was optimized with respect to lattice parameters andinternal degrees of freedom From the calculated total energiesof Cu5In2Sb2 the anti-site structure Cu5Sb2In2 and theelements copper indium and antimony we obtained theenergy of formation (ΔEf) of the compounds
4 RESULTS AND DISCUSSION41 Experimental Composition and Structure of the
New Phase The sample contains three compositionallydistinct compounds the target compound and the well-known phases InSb and Cu7In3 The EDX composition ofInSb is In51Sb49 and that of Cu7In3 is Cu72In28 indicating areliability of the compositional analysis within 1 or 2 Whenthis is applied to the title phase the composition ofCu58In24Sb18 is well described by Cu5+δIn2+xSb2minusx (δ asymp 033x asymp 029) (Figure 2)
42 Powder X-ray Diffraction Three compounds couldbe identified in the powder X-ray diffraction pattern of thesample InSb Cu7In3 and the title compound The latter hasbeen indexed by a comparison to a calculated theoreticalpowder pattern from the single-crystal data (see section 43)The lattice parameters have been refined to a = 101813(4) Aring b= 84562(4) Aring and c = 73774(2) Aring No extra reflections wereobserved
43 Single-Crystal X-ray Diffraction A preliminaryinspection of the diffraction pattern revealed a set of strongsubstructure reflections indicating a basic NiAsNi2In-typestructure and additionally satellite reflections quadrupling the aand b axes and doubling c (Figure 3 and Table 1) A closer
Figure 1 Copper-rich side of the CuminusIn phase diagram Adapted fromBahari et al5
Figure 2 Powder X-ray diffraction pattern of Cu5+δIn2+xSb2minusx Thephase Cu7In3 is present as a minor component and its theoreticalpattern overlaps with some strong reflections (Irel gt 10)
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210788
inspection of the intensity distribution of the satellite reflectionsclearly indicated a lowering of the symmetry from hexagonal toorthorhombic Consequently a twinned orthorhombic sym-metry compatible with the extinction conditions and theallowed groupminussubgroup relations needed to be identifiedMoreover the C-centering generated by the transformation
from hexagonal to orthorhombic symmetry is violated bysuperstructure reflections A new centering is introduced in theaugmented 2 times 1 times 2 orthorhombic supercell resulting in apattern that is characteristic for the space group BbmmHowever this group is disallowed by the groupminussubgrouprelations since the doubling of the hexagonal c axis will violatethe c glide The maximal space group is therefore uniquelydetermined to Bb21m (Cmc21 in the conventional setting) Thecorresponding groupminussubgroup relations are shown in Scheme1 together with the accompanying orbit splittingIt is notable that the In position (2c) and the partially
occupied Cu position (2d) are both split into threeindependent positions (8b 4a and 4a) with the relative
multiplicities 211 This allows perfect InSb order andprovides a site that corresponds to a 14 occupancy of thesecond Cu position The Wyckoff position that is fully occupiedby copper in the basic model splits into two equivalent orbits(Cu1 on 8b and Cu2 on 8b)A starting model was generated using Superf lip23 This
refined satisfactorily to a fully ordered structure Cu5In2Sb2 inthe space group Cmc21 (Figure 4)In this ordered model antimony and indium are arranged in
such a way as to avoid homoatomic contacts Together theyform a hexagonal close packing ABAB where alternating layersare composed exclusively of antimony (A) or indium (B) Thiscreates only one kind of octahedral interstices (all filled bycopper) but two different kinds of trigonal-bipyramidalinterstices one with apical antimony and equatorial indiumand another in which these roles are reversed Only half of theformer are filled by copper while the latter are empty Fromstructural images it is obvious that the apical antimony relaxessubstantially away from the occupied sites and toward thevacant sites As an outcome the inter-apex distance in emptytrigonal-bipyramidal interstices is 48 Aring while the correspond-ing value for filled interstices is 54 AringReturning to the results of the EDX analysis it is perspicuous
that the fully ordered model is not completely correct Clearlythe position assigned to contain only antimony must alsocontain a small amount of indium Given the small difference inthe scattering power between these two elements it isunderstandably meaningless to try to refine this Further theslight overoccupancy of copper must reside somewhere andthe most obvious place to look is in the empty 4a and 8bpositions There is no support for any occupancy in the 8bposition but a copper atom in the 4a position with the idealcoordinates (12
78112) refines to an occupancy of 0175 and
yields a total copper content close to 13 times that of the sumof indium and antimony The trigonal bipyramids with indiumapexes are completely unoccupied while for those with apexesthat are predominantly antimony every second interstice isfully occupied while the remaining are randomly occupied toabout 16The data pertinent to the structural work may be found in
the Supporting Information44 Ab Initio Calculated Properties The ordering of
indium and antimony in Cu5In2Sb2 is reasonable from achemical point of view but the evidence is not so strong giventhe similarity of their scattering factors for X-rays It is thereforevaluable to compare the relative stability of this compound withthe InSb site-inverted compound Cu5Sb2In2 In Table 2 wepresent the ab initio results for the equilibrium volume (V0)lattice parameters bulk modulus (B0) and its pressurederivative (B0) and energy of formation (ΔEf) obtained inthe generalized gradient approximation (GGA) for theelements copper indium and antimony in their equilibriumstructures for Cu5In2Sb2 and for the Cu5Sb2In2 anti-structurecompound resulting from exchange of the indium andantimony Wyckoff symmetric positions The experimentallattice parameters and B0 of the elements are reproducedwithin 3 and 18 respectively For antimony the presentcalculations correctly predict that the most stable structure at 0K is the R3 m as found experimentally at LT The calculatedlattice parameters of Cu5In2Sb2 differ by less than 12 fromthe experimental ones The compound Cu5In2Sb2 is thermo-dynamically stable with respect to the elements while wepredict the Cu5Sb2In2 anti-site structure to be unstable In
Figure 3 Reconstructed reciprocal lattice layer hk0 (in hexagonalsetting c is vertical and a is horizontal) Note the presence of weaksatellites doubling c and quadrupling a Reflections with odd l indicesin the fundamental B8-type structure are constitutionally weak
Table 1 Data Collection and Refinement Details forCu5+δIn2+xSb2minusx
chemical formula fromrefinement from EDXS
Cu533In2Sb2 Cu533In229Sb171
space group Z Cmc21 (No 36)lattice parameters frompowder Aring
a = 101813(4) b = 84562(4) c = 73774(2)(from powder X-ray diffraction)
volume Aring3 63516(3)temperature K 295(1)density (calcd) gcm3 83019λ Aring 071073reflectionsparameter 117654μ mm2 323Robs wRobs Rall wRall 0060 0058 0090 0063twin law (1 0 0 0 minus12 minus 34 0 1 minus12)
(1 0 0 0 minus1234 0 minus1 minus12)
twin fractions 011(3) 014(3)
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210789
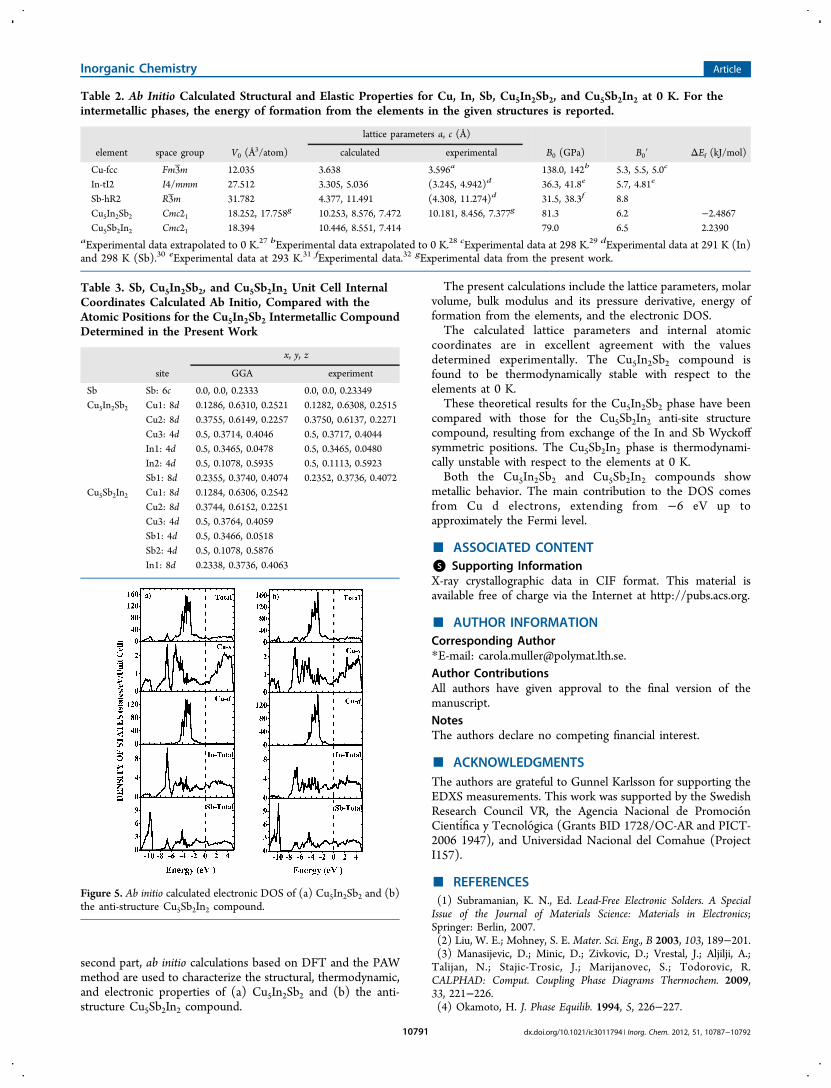
Table 3 the internal atomic positions for Sb Cu5In2Sb2 andCu5In2Sb2 are presented The calculated positions are in verygood agreement with those obtained in the experimentIn Figure 5 the electronic density of states (DOS) calculated
for the ab initio predicted equilibrium structures of Cu5In2Sb2and its anti-structure Cu5Sb2In2 compound are depicted BothCu5In2Sb2 and its anti-structure Cu5Sb2In2 have metalliccharacter with a relatively low value of the DOS at the Fermilevel The main contribution to the DOS comes from the dstates of copper The copper d band extends from minus6 eV up to
approximately the Fermi level At lower energies two bands canbe distinguished The first band extending from minus115 to minus95eV is mainly determined by antimony states whereas indiumstates dominate the second band extending from minus90 to minus60eV Between the two bands a pseudogap is located at about minus9eV When the DOS of both compounds Cu5In2Sb2 andCu5Sb2In2 are compared it is found that there is a difference inthe location of the second band However for the anti-structureCu5Sb2In2 the contribution of copper s states as well as indiumand antimony s and p states is shifted to higher energies withrespect to Cu5In2Sb2 This shift causes a larger pseudogapwhich extends from minus95 to minus8 eV and could in principleexplain the vast difference in the energies of formation of bothcompounds For energies above the Fermi level the electronicstates of all species contribute to the determination of the shapeof the DOS Generally the shapes of the DOS for bothcompounds are similar and follow the general trends that havebeen recently discussed for other structurally related binaryCuminusIn and CuminusSn compounds32minus34
5 SUMMARY AND CONCLUDING REMARKSA new ternary orthorhombic compound with the formulaCu533In229Sb171 crystallizing in the space group Cmc21 andwith 36 atoms per unit cell has been synthezised Details of theexperimental study and the structural parameters of thiscompound are reported in the first part of the work In the
Scheme 1 GroupminusSubgroup Relations between the Fundamental Space Group for NiAsNi2In P63mmc and the UniquelyDetermined Subgroup Bb21m
Figure 4 Idealized structure of Cu5In2Sb2 thermal ellipsoidprobability of 50
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210790
second part ab initio calculations based on DFT and the PAWmethod are used to characterize the structural thermodynamicand electronic properties of (a) Cu5In2Sb2 and (b) the anti-structure Cu5Sb2In2 compound
The present calculations include the lattice parameters molarvolume bulk modulus and its pressure derivative energy offormation from the elements and the electronic DOSThe calculated lattice parameters and internal atomic
coordinates are in excellent agreement with the valuesdetermined experimentally The Cu5In2Sb2 compound isfound to be thermodynamically stable with respect to theelements at 0 KThese theoretical results for the Cu5In2Sb2 phase have been
compared with those for the Cu5Sb2In2 anti-site structurecompound resulting from exchange of the In and Sb Wyckoffsymmetric positions The Cu5Sb2In2 phase is thermodynami-cally unstable with respect to the elements at 0 KBoth the Cu5In2Sb2 and Cu5Sb2In2 compounds show
metallic behavior The main contribution to the DOS comesfrom Cu d electrons extending from minus6 eV up toapproximately the Fermi level
ASSOCIATED CONTENTS Supporting InformationX-ray crystallographic data in CIF format This material isavailable free of charge via the Internet at httppubsacsorg
AUTHOR INFORMATIONCorresponding AuthorE-mail carolamullerpolymatlthseAuthor ContributionsAll authors have given approval to the final version of themanuscriptNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors are grateful to Gunnel Karlsson for supporting theEDXS measurements This work was supported by the SwedishResearch Council VR the Agencia Nacional de PromocionCientifica y Tecnologica (Grants BID 1728OC-AR and PICT-2006 1947) and Universidad Nacional del Comahue (ProjectI157)
REFERENCES(1) Subramanian K N Ed Lead-Free Electronic Solders A SpecialIssue of the Journal of Materials Science Materials in ElectronicsSpringer Berlin 2007(2) Liu W E Mohney S E Mater Sci Eng B 2003 103 189minus201(3) Manasijevic D Minic D Zivkovic D Vrestal J Aljilji ATalijan N Stajic-Trosic J Marijanovec S Todorovic RCALPHAD Comput Coupling Phase Diagrams Thermochem 200933 221minus226(4) Okamoto H J Phase Equilib 1994 5 226minus227
Table 2 Ab Initio Calculated Structural and Elastic Properties for Cu In Sb Cu5In2Sb2 and Cu5Sb2In2 at 0 K For theintermetallic phases the energy of formation from the elements in the given structures is reported
lattice parameters a c (Aring)
element space group V0 (Aring3atom) calculated experimental B0 (GPa) B0prime ΔEf (kJmol)
Cu-fcc Fm3m 12035 3638 3596a 1380 142b 53 55 50c
In-tI2 I4mmm 27512 3305 5036 (3245 4942)d 363 418e 57 481e
Sb-hR2 R3m 31782 4377 11491 (4308 11274)d 315 383f 88Cu5In2Sb2 Cmc21 18252 17758g 10253 8576 7472 10181 8456 7377g 813 62 minus24867Cu5Sb2In2 Cmc21 18394 10446 8551 7414 790 65 22390
aExperimental data extrapolated to 0 K27 bExperimental data extrapolated to 0 K28 cExperimental data at 298 K29 dExperimental data at 291 K (In)and 298 K (Sb)30 eExperimental data at 293 K31 fExperimental data32 gExperimental data from the present work
Table 3 Sb Cu5In2Sb2 and Cu5Sb2In2 Unit Cell InternalCoordinates Calculated Ab Initio Compared with theAtomic Positions for the Cu5In2Sb2 Intermetallic CompoundDetermined in the Present Work
x y z
site GGA experiment
Sb Sb 6c 00 00 02333 00 00 023349Cu5In2Sb2 Cu1 8d 01286 06310 02521 01282 06308 02515
Cu2 8d 03755 06149 02257 03750 06137 02271Cu3 4d 05 03714 04046 05 03717 04044In1 4d 05 03465 00478 05 03465 00480In2 4d 05 01078 05935 05 01113 05923Sb1 8d 02355 03740 04074 02352 03736 04072
Cu5Sb2In2 Cu1 8d 01284 06306 02542Cu2 8d 03744 06152 02251Cu3 4d 05 03764 04059Sb1 4d 05 03466 00518Sb2 4d 05 01078 05876In1 8d 02338 03736 04063
Figure 5 Ab initio calculated electronic DOS of (a) Cu5In2Sb2 and (b)the anti-structure Cu5Sb2In2 compound
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210791
(5) Bahari Z Dichi E Legendre B Dugue J Thermochim Acta2003 401 131minus138(6) Saunders N Miodownik A P Bull Alloy Phase Diagrams 199011 278minus287(7) Kattner U JOM 2002 54 45minus51(8) Shim J-H Oh Ch-S Lee B-J Lee D N Z Metallkd 1996 8205minus212(9) Massalski T B Ed Binary Alloy Phase Diagrams 2nd ed ASMInternational Metal Park OH 1996 pp 1424minus1426(10) Elding-Ponten M Stenberg L Lidin S J Alloys Compd 1997261 162minus171(11) Lidin S Larsson A K J Solid State Chem 1995 118 313minus322(12) Jandali M Z Rajasekharan T P Schubert K Z Metallkd1982 73 463minus467(13) Gunzel E Schubert K Z Metallkd 1958 49 124minus133(14) Schubert K Burkhardt W Esslinger P Gunzel E MeissnerH G Schutt W Wegst J Wilkens M Naturwissenschaften 1956 43248minus249(15) Hofmann W Z Metallkd 1941 33 373(16) Nuss J Jansen M Z Anorg Allg Chem 2002 628 1152minus1157(17) Blochl P E Phys Rev B Condens Matter Mater Phys 1994 5017953minus17979(18) Kresse G Furthmuller J Comput Mater Sci 1996 6 15minus50(19) WinXPow Powder Diffraction Software version 31 Stoe amp CieDarmstadt Germany 2011(20) CrysAlisPro version 1171356 Oxford Diffraction LtdAbington UK2011(21) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2004 60 134minus141(22) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2005 61 147minus152(23) Palatinus L Chapuis G J Appl Crystallogr 2007 40 786minus790(24) Petricek V Dusek M Palatinus L Jana2006 The crystallo-graphic computing system Institute of Physics Praha Czech Republic2006(25) Aztec version 10 Oxford Instruments Nanotechnology ToolsLtd Oxford UK 2010minus2011(26) Perdew J P Wang Y Phys Rev B Condens Matter 1991 4413298minus13307(27) Straumanis M E Yu L S Acta Crystallogr Sect A Cryst PhysDiffr Theor Gen Crystallogr 1969 25 676minus682(28) Overton W C Jr Gaffney J Phys Rev 1955 98 969minus977(29) Steinberg D J J Phys Chem Solids 1982 43 1173minus1175(30) Villars P Pearsons Handbook Desk Edition ASM InternationalMaterials Park OH 1997(31) Takemura K Phys Rev B Condens Matter Mater Phys 199144 545minus549(32) Kittel C Introduction to solid state physics 7th ed Wiley NewYork 1996(33) Ramos de Debiaggi S Cabeza G F Deluque Toro C MontiA M Sommadossi S Fernandez Guillermet A J Alloys Compd2011 509 3238minus3245(34) Ramos de Debiaggi S Deluque Toro C Cabeza G FFernandez Guillermet A J Alloys Compd 2012 httpdxdoiorg101016jjallcom201206138
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210792
the synthesis energy-dispersive X-ray spectroscopy (EDXS)was used to determine the composition of the new compoundand the structure was solved and refined by X-ray diffractionanalysis In the second theoretical part of the paper ab initiocalculations based on the density functional theory (DFT) andthe projector augmented-wave method17 (PAW) implementedin the VASP code18 were used to establish the theoreticalelectronic cohesive and thermodynamic properties of anidealization of the new compound Cu5In2Sb2 For comparisonthe Cu5Sb2In2 anti-site phase resulting from exchange of theindium and antimony Wyckoff positions was analyzed as well
2 EXPERIMENTAL SECTION21 Synthesis Elemental copper (ABCR Karlsruhe shots
9999) indium (Chempur ingots 999999) and antimony(Chempur shots 99999) were reacted in a molar ratio of 632(CuInSb) in an argon-purged evacuated silica ampule at 900(5) degCin a muffle furnace for 3 days After furnace cooling the sample wasplaced in an aluminum block and annealed at 300(5) degC in a mufflefurnace for 30 days22 Characterization Powder X-ray diffraction experiments were
performed in transmission mode on a Stoe Stadi MP (vertical set-up)equipped with a curved germanium monochromator (Johangeometry) and a MYTHEN detector using Cu Kα1 radiation Thezero point of the diffractometer was aligned against siliconDiffractometer control and data analysis were performed usingWinXPow19
Single crystals were isolated from the resulting ingot after crushingA triangular fragment with clean cleavage surfaces and metallic lusterwas chosen for the X-ray experiment and mounted on a silica fiberusing a standard two-component adhesive The data collection wasperformed on an XCalibur 3 single-crystal diffractometer at ambientconditions using monochromatized Mo Kα radiation (071073 Aring) anda Sapphire III detector The program suite CrysAlisPro20 was used fordata reduction and integration while charge flipping2122 asimplemented in Superf lip23 was applied for structure solution andJana200624 for the following structure refinementPieces of 20minus25 mm2 were embedded in epoxy ground with SiC
paper and polished with a diamond suspension as the final step for thescanning electron microscopy experiments Those were performed ona JEOL 3000 with a secondary electron (SEI) detector and abackscatter detector for EDXS measurements AZtec25 was employedfor data analysis of the spectra
3 THEORETICAL SECTIONThe total energy calculations were performed using theprojector augmented-plane-wave method and the VASP codeWe adopt the conjugate gradient approximation due to Perdewand Wang (GGA-PW91)26 The cutoff kinetic energy for theplane-wave expansions was 330 eV For the PAWs weconsidered 11 valence electrons for Cu (3d104s1) 3 for In(5s25p1) and 5 for Sb (5s25p3) The k-point mesh was verifiedfor the energy to be converged within 1 meVatom Specificallyfor Cu5In2Sb2 we considered a mesh of 9 times 11 times 13 leading to210 k points in the irreducible Brillouin zone The self-consistent criterion for the energy was 01 meV Finally thestructure was optimized with respect to lattice parameters andinternal degrees of freedom From the calculated total energiesof Cu5In2Sb2 the anti-site structure Cu5Sb2In2 and theelements copper indium and antimony we obtained theenergy of formation (ΔEf) of the compounds
4 RESULTS AND DISCUSSION41 Experimental Composition and Structure of the
New Phase The sample contains three compositionallydistinct compounds the target compound and the well-known phases InSb and Cu7In3 The EDX composition ofInSb is In51Sb49 and that of Cu7In3 is Cu72In28 indicating areliability of the compositional analysis within 1 or 2 Whenthis is applied to the title phase the composition ofCu58In24Sb18 is well described by Cu5+δIn2+xSb2minusx (δ asymp 033x asymp 029) (Figure 2)
42 Powder X-ray Diffraction Three compounds couldbe identified in the powder X-ray diffraction pattern of thesample InSb Cu7In3 and the title compound The latter hasbeen indexed by a comparison to a calculated theoreticalpowder pattern from the single-crystal data (see section 43)The lattice parameters have been refined to a = 101813(4) Aring b= 84562(4) Aring and c = 73774(2) Aring No extra reflections wereobserved
43 Single-Crystal X-ray Diffraction A preliminaryinspection of the diffraction pattern revealed a set of strongsubstructure reflections indicating a basic NiAsNi2In-typestructure and additionally satellite reflections quadrupling the aand b axes and doubling c (Figure 3 and Table 1) A closer
Figure 1 Copper-rich side of the CuminusIn phase diagram Adapted fromBahari et al5
Figure 2 Powder X-ray diffraction pattern of Cu5+δIn2+xSb2minusx Thephase Cu7In3 is present as a minor component and its theoreticalpattern overlaps with some strong reflections (Irel gt 10)
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210788
inspection of the intensity distribution of the satellite reflectionsclearly indicated a lowering of the symmetry from hexagonal toorthorhombic Consequently a twinned orthorhombic sym-metry compatible with the extinction conditions and theallowed groupminussubgroup relations needed to be identifiedMoreover the C-centering generated by the transformation
from hexagonal to orthorhombic symmetry is violated bysuperstructure reflections A new centering is introduced in theaugmented 2 times 1 times 2 orthorhombic supercell resulting in apattern that is characteristic for the space group BbmmHowever this group is disallowed by the groupminussubgrouprelations since the doubling of the hexagonal c axis will violatethe c glide The maximal space group is therefore uniquelydetermined to Bb21m (Cmc21 in the conventional setting) Thecorresponding groupminussubgroup relations are shown in Scheme1 together with the accompanying orbit splittingIt is notable that the In position (2c) and the partially
occupied Cu position (2d) are both split into threeindependent positions (8b 4a and 4a) with the relative
multiplicities 211 This allows perfect InSb order andprovides a site that corresponds to a 14 occupancy of thesecond Cu position The Wyckoff position that is fully occupiedby copper in the basic model splits into two equivalent orbits(Cu1 on 8b and Cu2 on 8b)A starting model was generated using Superf lip23 This
refined satisfactorily to a fully ordered structure Cu5In2Sb2 inthe space group Cmc21 (Figure 4)In this ordered model antimony and indium are arranged in
such a way as to avoid homoatomic contacts Together theyform a hexagonal close packing ABAB where alternating layersare composed exclusively of antimony (A) or indium (B) Thiscreates only one kind of octahedral interstices (all filled bycopper) but two different kinds of trigonal-bipyramidalinterstices one with apical antimony and equatorial indiumand another in which these roles are reversed Only half of theformer are filled by copper while the latter are empty Fromstructural images it is obvious that the apical antimony relaxessubstantially away from the occupied sites and toward thevacant sites As an outcome the inter-apex distance in emptytrigonal-bipyramidal interstices is 48 Aring while the correspond-ing value for filled interstices is 54 AringReturning to the results of the EDX analysis it is perspicuous
that the fully ordered model is not completely correct Clearlythe position assigned to contain only antimony must alsocontain a small amount of indium Given the small difference inthe scattering power between these two elements it isunderstandably meaningless to try to refine this Further theslight overoccupancy of copper must reside somewhere andthe most obvious place to look is in the empty 4a and 8bpositions There is no support for any occupancy in the 8bposition but a copper atom in the 4a position with the idealcoordinates (12
78112) refines to an occupancy of 0175 and
yields a total copper content close to 13 times that of the sumof indium and antimony The trigonal bipyramids with indiumapexes are completely unoccupied while for those with apexesthat are predominantly antimony every second interstice isfully occupied while the remaining are randomly occupied toabout 16The data pertinent to the structural work may be found in
the Supporting Information44 Ab Initio Calculated Properties The ordering of
indium and antimony in Cu5In2Sb2 is reasonable from achemical point of view but the evidence is not so strong giventhe similarity of their scattering factors for X-rays It is thereforevaluable to compare the relative stability of this compound withthe InSb site-inverted compound Cu5Sb2In2 In Table 2 wepresent the ab initio results for the equilibrium volume (V0)lattice parameters bulk modulus (B0) and its pressurederivative (B0) and energy of formation (ΔEf) obtained inthe generalized gradient approximation (GGA) for theelements copper indium and antimony in their equilibriumstructures for Cu5In2Sb2 and for the Cu5Sb2In2 anti-structurecompound resulting from exchange of the indium andantimony Wyckoff symmetric positions The experimentallattice parameters and B0 of the elements are reproducedwithin 3 and 18 respectively For antimony the presentcalculations correctly predict that the most stable structure at 0K is the R3 m as found experimentally at LT The calculatedlattice parameters of Cu5In2Sb2 differ by less than 12 fromthe experimental ones The compound Cu5In2Sb2 is thermo-dynamically stable with respect to the elements while wepredict the Cu5Sb2In2 anti-site structure to be unstable In
Figure 3 Reconstructed reciprocal lattice layer hk0 (in hexagonalsetting c is vertical and a is horizontal) Note the presence of weaksatellites doubling c and quadrupling a Reflections with odd l indicesin the fundamental B8-type structure are constitutionally weak
Table 1 Data Collection and Refinement Details forCu5+δIn2+xSb2minusx
chemical formula fromrefinement from EDXS
Cu533In2Sb2 Cu533In229Sb171
space group Z Cmc21 (No 36)lattice parameters frompowder Aring
a = 101813(4) b = 84562(4) c = 73774(2)(from powder X-ray diffraction)
volume Aring3 63516(3)temperature K 295(1)density (calcd) gcm3 83019λ Aring 071073reflectionsparameter 117654μ mm2 323Robs wRobs Rall wRall 0060 0058 0090 0063twin law (1 0 0 0 minus12 minus 34 0 1 minus12)
(1 0 0 0 minus1234 0 minus1 minus12)
twin fractions 011(3) 014(3)
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210789
Table 3 the internal atomic positions for Sb Cu5In2Sb2 andCu5In2Sb2 are presented The calculated positions are in verygood agreement with those obtained in the experimentIn Figure 5 the electronic density of states (DOS) calculated
for the ab initio predicted equilibrium structures of Cu5In2Sb2and its anti-structure Cu5Sb2In2 compound are depicted BothCu5In2Sb2 and its anti-structure Cu5Sb2In2 have metalliccharacter with a relatively low value of the DOS at the Fermilevel The main contribution to the DOS comes from the dstates of copper The copper d band extends from minus6 eV up to
approximately the Fermi level At lower energies two bands canbe distinguished The first band extending from minus115 to minus95eV is mainly determined by antimony states whereas indiumstates dominate the second band extending from minus90 to minus60eV Between the two bands a pseudogap is located at about minus9eV When the DOS of both compounds Cu5In2Sb2 andCu5Sb2In2 are compared it is found that there is a difference inthe location of the second band However for the anti-structureCu5Sb2In2 the contribution of copper s states as well as indiumand antimony s and p states is shifted to higher energies withrespect to Cu5In2Sb2 This shift causes a larger pseudogapwhich extends from minus95 to minus8 eV and could in principleexplain the vast difference in the energies of formation of bothcompounds For energies above the Fermi level the electronicstates of all species contribute to the determination of the shapeof the DOS Generally the shapes of the DOS for bothcompounds are similar and follow the general trends that havebeen recently discussed for other structurally related binaryCuminusIn and CuminusSn compounds32minus34
5 SUMMARY AND CONCLUDING REMARKSA new ternary orthorhombic compound with the formulaCu533In229Sb171 crystallizing in the space group Cmc21 andwith 36 atoms per unit cell has been synthezised Details of theexperimental study and the structural parameters of thiscompound are reported in the first part of the work In the
Scheme 1 GroupminusSubgroup Relations between the Fundamental Space Group for NiAsNi2In P63mmc and the UniquelyDetermined Subgroup Bb21m
Figure 4 Idealized structure of Cu5In2Sb2 thermal ellipsoidprobability of 50
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210790
second part ab initio calculations based on DFT and the PAWmethod are used to characterize the structural thermodynamicand electronic properties of (a) Cu5In2Sb2 and (b) the anti-structure Cu5Sb2In2 compound
The present calculations include the lattice parameters molarvolume bulk modulus and its pressure derivative energy offormation from the elements and the electronic DOSThe calculated lattice parameters and internal atomic
coordinates are in excellent agreement with the valuesdetermined experimentally The Cu5In2Sb2 compound isfound to be thermodynamically stable with respect to theelements at 0 KThese theoretical results for the Cu5In2Sb2 phase have been
compared with those for the Cu5Sb2In2 anti-site structurecompound resulting from exchange of the In and Sb Wyckoffsymmetric positions The Cu5Sb2In2 phase is thermodynami-cally unstable with respect to the elements at 0 KBoth the Cu5In2Sb2 and Cu5Sb2In2 compounds show
metallic behavior The main contribution to the DOS comesfrom Cu d electrons extending from minus6 eV up toapproximately the Fermi level
ASSOCIATED CONTENTS Supporting InformationX-ray crystallographic data in CIF format This material isavailable free of charge via the Internet at httppubsacsorg
AUTHOR INFORMATIONCorresponding AuthorE-mail carolamullerpolymatlthseAuthor ContributionsAll authors have given approval to the final version of themanuscriptNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors are grateful to Gunnel Karlsson for supporting theEDXS measurements This work was supported by the SwedishResearch Council VR the Agencia Nacional de PromocionCientifica y Tecnologica (Grants BID 1728OC-AR and PICT-2006 1947) and Universidad Nacional del Comahue (ProjectI157)
REFERENCES(1) Subramanian K N Ed Lead-Free Electronic Solders A SpecialIssue of the Journal of Materials Science Materials in ElectronicsSpringer Berlin 2007(2) Liu W E Mohney S E Mater Sci Eng B 2003 103 189minus201(3) Manasijevic D Minic D Zivkovic D Vrestal J Aljilji ATalijan N Stajic-Trosic J Marijanovec S Todorovic RCALPHAD Comput Coupling Phase Diagrams Thermochem 200933 221minus226(4) Okamoto H J Phase Equilib 1994 5 226minus227
Table 2 Ab Initio Calculated Structural and Elastic Properties for Cu In Sb Cu5In2Sb2 and Cu5Sb2In2 at 0 K For theintermetallic phases the energy of formation from the elements in the given structures is reported
lattice parameters a c (Aring)
element space group V0 (Aring3atom) calculated experimental B0 (GPa) B0prime ΔEf (kJmol)
Cu-fcc Fm3m 12035 3638 3596a 1380 142b 53 55 50c
In-tI2 I4mmm 27512 3305 5036 (3245 4942)d 363 418e 57 481e
Sb-hR2 R3m 31782 4377 11491 (4308 11274)d 315 383f 88Cu5In2Sb2 Cmc21 18252 17758g 10253 8576 7472 10181 8456 7377g 813 62 minus24867Cu5Sb2In2 Cmc21 18394 10446 8551 7414 790 65 22390
aExperimental data extrapolated to 0 K27 bExperimental data extrapolated to 0 K28 cExperimental data at 298 K29 dExperimental data at 291 K (In)and 298 K (Sb)30 eExperimental data at 293 K31 fExperimental data32 gExperimental data from the present work
Table 3 Sb Cu5In2Sb2 and Cu5Sb2In2 Unit Cell InternalCoordinates Calculated Ab Initio Compared with theAtomic Positions for the Cu5In2Sb2 Intermetallic CompoundDetermined in the Present Work
x y z
site GGA experiment
Sb Sb 6c 00 00 02333 00 00 023349Cu5In2Sb2 Cu1 8d 01286 06310 02521 01282 06308 02515
Cu2 8d 03755 06149 02257 03750 06137 02271Cu3 4d 05 03714 04046 05 03717 04044In1 4d 05 03465 00478 05 03465 00480In2 4d 05 01078 05935 05 01113 05923Sb1 8d 02355 03740 04074 02352 03736 04072
Cu5Sb2In2 Cu1 8d 01284 06306 02542Cu2 8d 03744 06152 02251Cu3 4d 05 03764 04059Sb1 4d 05 03466 00518Sb2 4d 05 01078 05876In1 8d 02338 03736 04063
Figure 5 Ab initio calculated electronic DOS of (a) Cu5In2Sb2 and (b)the anti-structure Cu5Sb2In2 compound
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210791
(5) Bahari Z Dichi E Legendre B Dugue J Thermochim Acta2003 401 131minus138(6) Saunders N Miodownik A P Bull Alloy Phase Diagrams 199011 278minus287(7) Kattner U JOM 2002 54 45minus51(8) Shim J-H Oh Ch-S Lee B-J Lee D N Z Metallkd 1996 8205minus212(9) Massalski T B Ed Binary Alloy Phase Diagrams 2nd ed ASMInternational Metal Park OH 1996 pp 1424minus1426(10) Elding-Ponten M Stenberg L Lidin S J Alloys Compd 1997261 162minus171(11) Lidin S Larsson A K J Solid State Chem 1995 118 313minus322(12) Jandali M Z Rajasekharan T P Schubert K Z Metallkd1982 73 463minus467(13) Gunzel E Schubert K Z Metallkd 1958 49 124minus133(14) Schubert K Burkhardt W Esslinger P Gunzel E MeissnerH G Schutt W Wegst J Wilkens M Naturwissenschaften 1956 43248minus249(15) Hofmann W Z Metallkd 1941 33 373(16) Nuss J Jansen M Z Anorg Allg Chem 2002 628 1152minus1157(17) Blochl P E Phys Rev B Condens Matter Mater Phys 1994 5017953minus17979(18) Kresse G Furthmuller J Comput Mater Sci 1996 6 15minus50(19) WinXPow Powder Diffraction Software version 31 Stoe amp CieDarmstadt Germany 2011(20) CrysAlisPro version 1171356 Oxford Diffraction LtdAbington UK2011(21) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2004 60 134minus141(22) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2005 61 147minus152(23) Palatinus L Chapuis G J Appl Crystallogr 2007 40 786minus790(24) Petricek V Dusek M Palatinus L Jana2006 The crystallo-graphic computing system Institute of Physics Praha Czech Republic2006(25) Aztec version 10 Oxford Instruments Nanotechnology ToolsLtd Oxford UK 2010minus2011(26) Perdew J P Wang Y Phys Rev B Condens Matter 1991 4413298minus13307(27) Straumanis M E Yu L S Acta Crystallogr Sect A Cryst PhysDiffr Theor Gen Crystallogr 1969 25 676minus682(28) Overton W C Jr Gaffney J Phys Rev 1955 98 969minus977(29) Steinberg D J J Phys Chem Solids 1982 43 1173minus1175(30) Villars P Pearsons Handbook Desk Edition ASM InternationalMaterials Park OH 1997(31) Takemura K Phys Rev B Condens Matter Mater Phys 199144 545minus549(32) Kittel C Introduction to solid state physics 7th ed Wiley NewYork 1996(33) Ramos de Debiaggi S Cabeza G F Deluque Toro C MontiA M Sommadossi S Fernandez Guillermet A J Alloys Compd2011 509 3238minus3245(34) Ramos de Debiaggi S Deluque Toro C Cabeza G FFernandez Guillermet A J Alloys Compd 2012 httpdxdoiorg101016jjallcom201206138
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210792

inspection of the intensity distribution of the satellite reflectionsclearly indicated a lowering of the symmetry from hexagonal toorthorhombic Consequently a twinned orthorhombic sym-metry compatible with the extinction conditions and theallowed groupminussubgroup relations needed to be identifiedMoreover the C-centering generated by the transformation
from hexagonal to orthorhombic symmetry is violated bysuperstructure reflections A new centering is introduced in theaugmented 2 times 1 times 2 orthorhombic supercell resulting in apattern that is characteristic for the space group BbmmHowever this group is disallowed by the groupminussubgrouprelations since the doubling of the hexagonal c axis will violatethe c glide The maximal space group is therefore uniquelydetermined to Bb21m (Cmc21 in the conventional setting) Thecorresponding groupminussubgroup relations are shown in Scheme1 together with the accompanying orbit splittingIt is notable that the In position (2c) and the partially
occupied Cu position (2d) are both split into threeindependent positions (8b 4a and 4a) with the relative
multiplicities 211 This allows perfect InSb order andprovides a site that corresponds to a 14 occupancy of thesecond Cu position The Wyckoff position that is fully occupiedby copper in the basic model splits into two equivalent orbits(Cu1 on 8b and Cu2 on 8b)A starting model was generated using Superf lip23 This
refined satisfactorily to a fully ordered structure Cu5In2Sb2 inthe space group Cmc21 (Figure 4)In this ordered model antimony and indium are arranged in
such a way as to avoid homoatomic contacts Together theyform a hexagonal close packing ABAB where alternating layersare composed exclusively of antimony (A) or indium (B) Thiscreates only one kind of octahedral interstices (all filled bycopper) but two different kinds of trigonal-bipyramidalinterstices one with apical antimony and equatorial indiumand another in which these roles are reversed Only half of theformer are filled by copper while the latter are empty Fromstructural images it is obvious that the apical antimony relaxessubstantially away from the occupied sites and toward thevacant sites As an outcome the inter-apex distance in emptytrigonal-bipyramidal interstices is 48 Aring while the correspond-ing value for filled interstices is 54 AringReturning to the results of the EDX analysis it is perspicuous
that the fully ordered model is not completely correct Clearlythe position assigned to contain only antimony must alsocontain a small amount of indium Given the small difference inthe scattering power between these two elements it isunderstandably meaningless to try to refine this Further theslight overoccupancy of copper must reside somewhere andthe most obvious place to look is in the empty 4a and 8bpositions There is no support for any occupancy in the 8bposition but a copper atom in the 4a position with the idealcoordinates (12
78112) refines to an occupancy of 0175 and
yields a total copper content close to 13 times that of the sumof indium and antimony The trigonal bipyramids with indiumapexes are completely unoccupied while for those with apexesthat are predominantly antimony every second interstice isfully occupied while the remaining are randomly occupied toabout 16The data pertinent to the structural work may be found in
the Supporting Information44 Ab Initio Calculated Properties The ordering of
indium and antimony in Cu5In2Sb2 is reasonable from achemical point of view but the evidence is not so strong giventhe similarity of their scattering factors for X-rays It is thereforevaluable to compare the relative stability of this compound withthe InSb site-inverted compound Cu5Sb2In2 In Table 2 wepresent the ab initio results for the equilibrium volume (V0)lattice parameters bulk modulus (B0) and its pressurederivative (B0) and energy of formation (ΔEf) obtained inthe generalized gradient approximation (GGA) for theelements copper indium and antimony in their equilibriumstructures for Cu5In2Sb2 and for the Cu5Sb2In2 anti-structurecompound resulting from exchange of the indium andantimony Wyckoff symmetric positions The experimentallattice parameters and B0 of the elements are reproducedwithin 3 and 18 respectively For antimony the presentcalculations correctly predict that the most stable structure at 0K is the R3 m as found experimentally at LT The calculatedlattice parameters of Cu5In2Sb2 differ by less than 12 fromthe experimental ones The compound Cu5In2Sb2 is thermo-dynamically stable with respect to the elements while wepredict the Cu5Sb2In2 anti-site structure to be unstable In
Figure 3 Reconstructed reciprocal lattice layer hk0 (in hexagonalsetting c is vertical and a is horizontal) Note the presence of weaksatellites doubling c and quadrupling a Reflections with odd l indicesin the fundamental B8-type structure are constitutionally weak
Table 1 Data Collection and Refinement Details forCu5+δIn2+xSb2minusx
chemical formula fromrefinement from EDXS
Cu533In2Sb2 Cu533In229Sb171
space group Z Cmc21 (No 36)lattice parameters frompowder Aring
a = 101813(4) b = 84562(4) c = 73774(2)(from powder X-ray diffraction)
volume Aring3 63516(3)temperature K 295(1)density (calcd) gcm3 83019λ Aring 071073reflectionsparameter 117654μ mm2 323Robs wRobs Rall wRall 0060 0058 0090 0063twin law (1 0 0 0 minus12 minus 34 0 1 minus12)
(1 0 0 0 minus1234 0 minus1 minus12)
twin fractions 011(3) 014(3)
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210789
Table 3 the internal atomic positions for Sb Cu5In2Sb2 andCu5In2Sb2 are presented The calculated positions are in verygood agreement with those obtained in the experimentIn Figure 5 the electronic density of states (DOS) calculated
for the ab initio predicted equilibrium structures of Cu5In2Sb2and its anti-structure Cu5Sb2In2 compound are depicted BothCu5In2Sb2 and its anti-structure Cu5Sb2In2 have metalliccharacter with a relatively low value of the DOS at the Fermilevel The main contribution to the DOS comes from the dstates of copper The copper d band extends from minus6 eV up to
approximately the Fermi level At lower energies two bands canbe distinguished The first band extending from minus115 to minus95eV is mainly determined by antimony states whereas indiumstates dominate the second band extending from minus90 to minus60eV Between the two bands a pseudogap is located at about minus9eV When the DOS of both compounds Cu5In2Sb2 andCu5Sb2In2 are compared it is found that there is a difference inthe location of the second band However for the anti-structureCu5Sb2In2 the contribution of copper s states as well as indiumand antimony s and p states is shifted to higher energies withrespect to Cu5In2Sb2 This shift causes a larger pseudogapwhich extends from minus95 to minus8 eV and could in principleexplain the vast difference in the energies of formation of bothcompounds For energies above the Fermi level the electronicstates of all species contribute to the determination of the shapeof the DOS Generally the shapes of the DOS for bothcompounds are similar and follow the general trends that havebeen recently discussed for other structurally related binaryCuminusIn and CuminusSn compounds32minus34
5 SUMMARY AND CONCLUDING REMARKSA new ternary orthorhombic compound with the formulaCu533In229Sb171 crystallizing in the space group Cmc21 andwith 36 atoms per unit cell has been synthezised Details of theexperimental study and the structural parameters of thiscompound are reported in the first part of the work In the
Scheme 1 GroupminusSubgroup Relations between the Fundamental Space Group for NiAsNi2In P63mmc and the UniquelyDetermined Subgroup Bb21m
Figure 4 Idealized structure of Cu5In2Sb2 thermal ellipsoidprobability of 50
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210790
second part ab initio calculations based on DFT and the PAWmethod are used to characterize the structural thermodynamicand electronic properties of (a) Cu5In2Sb2 and (b) the anti-structure Cu5Sb2In2 compound
The present calculations include the lattice parameters molarvolume bulk modulus and its pressure derivative energy offormation from the elements and the electronic DOSThe calculated lattice parameters and internal atomic
coordinates are in excellent agreement with the valuesdetermined experimentally The Cu5In2Sb2 compound isfound to be thermodynamically stable with respect to theelements at 0 KThese theoretical results for the Cu5In2Sb2 phase have been
compared with those for the Cu5Sb2In2 anti-site structurecompound resulting from exchange of the In and Sb Wyckoffsymmetric positions The Cu5Sb2In2 phase is thermodynami-cally unstable with respect to the elements at 0 KBoth the Cu5In2Sb2 and Cu5Sb2In2 compounds show
metallic behavior The main contribution to the DOS comesfrom Cu d electrons extending from minus6 eV up toapproximately the Fermi level
ASSOCIATED CONTENTS Supporting InformationX-ray crystallographic data in CIF format This material isavailable free of charge via the Internet at httppubsacsorg
AUTHOR INFORMATIONCorresponding AuthorE-mail carolamullerpolymatlthseAuthor ContributionsAll authors have given approval to the final version of themanuscriptNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors are grateful to Gunnel Karlsson for supporting theEDXS measurements This work was supported by the SwedishResearch Council VR the Agencia Nacional de PromocionCientifica y Tecnologica (Grants BID 1728OC-AR and PICT-2006 1947) and Universidad Nacional del Comahue (ProjectI157)
REFERENCES(1) Subramanian K N Ed Lead-Free Electronic Solders A SpecialIssue of the Journal of Materials Science Materials in ElectronicsSpringer Berlin 2007(2) Liu W E Mohney S E Mater Sci Eng B 2003 103 189minus201(3) Manasijevic D Minic D Zivkovic D Vrestal J Aljilji ATalijan N Stajic-Trosic J Marijanovec S Todorovic RCALPHAD Comput Coupling Phase Diagrams Thermochem 200933 221minus226(4) Okamoto H J Phase Equilib 1994 5 226minus227
Table 2 Ab Initio Calculated Structural and Elastic Properties for Cu In Sb Cu5In2Sb2 and Cu5Sb2In2 at 0 K For theintermetallic phases the energy of formation from the elements in the given structures is reported
lattice parameters a c (Aring)
element space group V0 (Aring3atom) calculated experimental B0 (GPa) B0prime ΔEf (kJmol)
Cu-fcc Fm3m 12035 3638 3596a 1380 142b 53 55 50c
In-tI2 I4mmm 27512 3305 5036 (3245 4942)d 363 418e 57 481e
Sb-hR2 R3m 31782 4377 11491 (4308 11274)d 315 383f 88Cu5In2Sb2 Cmc21 18252 17758g 10253 8576 7472 10181 8456 7377g 813 62 minus24867Cu5Sb2In2 Cmc21 18394 10446 8551 7414 790 65 22390
aExperimental data extrapolated to 0 K27 bExperimental data extrapolated to 0 K28 cExperimental data at 298 K29 dExperimental data at 291 K (In)and 298 K (Sb)30 eExperimental data at 293 K31 fExperimental data32 gExperimental data from the present work
Table 3 Sb Cu5In2Sb2 and Cu5Sb2In2 Unit Cell InternalCoordinates Calculated Ab Initio Compared with theAtomic Positions for the Cu5In2Sb2 Intermetallic CompoundDetermined in the Present Work
x y z
site GGA experiment
Sb Sb 6c 00 00 02333 00 00 023349Cu5In2Sb2 Cu1 8d 01286 06310 02521 01282 06308 02515
Cu2 8d 03755 06149 02257 03750 06137 02271Cu3 4d 05 03714 04046 05 03717 04044In1 4d 05 03465 00478 05 03465 00480In2 4d 05 01078 05935 05 01113 05923Sb1 8d 02355 03740 04074 02352 03736 04072
Cu5Sb2In2 Cu1 8d 01284 06306 02542Cu2 8d 03744 06152 02251Cu3 4d 05 03764 04059Sb1 4d 05 03466 00518Sb2 4d 05 01078 05876In1 8d 02338 03736 04063
Figure 5 Ab initio calculated electronic DOS of (a) Cu5In2Sb2 and (b)the anti-structure Cu5Sb2In2 compound
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210791
(5) Bahari Z Dichi E Legendre B Dugue J Thermochim Acta2003 401 131minus138(6) Saunders N Miodownik A P Bull Alloy Phase Diagrams 199011 278minus287(7) Kattner U JOM 2002 54 45minus51(8) Shim J-H Oh Ch-S Lee B-J Lee D N Z Metallkd 1996 8205minus212(9) Massalski T B Ed Binary Alloy Phase Diagrams 2nd ed ASMInternational Metal Park OH 1996 pp 1424minus1426(10) Elding-Ponten M Stenberg L Lidin S J Alloys Compd 1997261 162minus171(11) Lidin S Larsson A K J Solid State Chem 1995 118 313minus322(12) Jandali M Z Rajasekharan T P Schubert K Z Metallkd1982 73 463minus467(13) Gunzel E Schubert K Z Metallkd 1958 49 124minus133(14) Schubert K Burkhardt W Esslinger P Gunzel E MeissnerH G Schutt W Wegst J Wilkens M Naturwissenschaften 1956 43248minus249(15) Hofmann W Z Metallkd 1941 33 373(16) Nuss J Jansen M Z Anorg Allg Chem 2002 628 1152minus1157(17) Blochl P E Phys Rev B Condens Matter Mater Phys 1994 5017953minus17979(18) Kresse G Furthmuller J Comput Mater Sci 1996 6 15minus50(19) WinXPow Powder Diffraction Software version 31 Stoe amp CieDarmstadt Germany 2011(20) CrysAlisPro version 1171356 Oxford Diffraction LtdAbington UK2011(21) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2004 60 134minus141(22) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2005 61 147minus152(23) Palatinus L Chapuis G J Appl Crystallogr 2007 40 786minus790(24) Petricek V Dusek M Palatinus L Jana2006 The crystallo-graphic computing system Institute of Physics Praha Czech Republic2006(25) Aztec version 10 Oxford Instruments Nanotechnology ToolsLtd Oxford UK 2010minus2011(26) Perdew J P Wang Y Phys Rev B Condens Matter 1991 4413298minus13307(27) Straumanis M E Yu L S Acta Crystallogr Sect A Cryst PhysDiffr Theor Gen Crystallogr 1969 25 676minus682(28) Overton W C Jr Gaffney J Phys Rev 1955 98 969minus977(29) Steinberg D J J Phys Chem Solids 1982 43 1173minus1175(30) Villars P Pearsons Handbook Desk Edition ASM InternationalMaterials Park OH 1997(31) Takemura K Phys Rev B Condens Matter Mater Phys 199144 545minus549(32) Kittel C Introduction to solid state physics 7th ed Wiley NewYork 1996(33) Ramos de Debiaggi S Cabeza G F Deluque Toro C MontiA M Sommadossi S Fernandez Guillermet A J Alloys Compd2011 509 3238minus3245(34) Ramos de Debiaggi S Deluque Toro C Cabeza G FFernandez Guillermet A J Alloys Compd 2012 httpdxdoiorg101016jjallcom201206138
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210792

Table 3 the internal atomic positions for Sb Cu5In2Sb2 andCu5In2Sb2 are presented The calculated positions are in verygood agreement with those obtained in the experimentIn Figure 5 the electronic density of states (DOS) calculated
for the ab initio predicted equilibrium structures of Cu5In2Sb2and its anti-structure Cu5Sb2In2 compound are depicted BothCu5In2Sb2 and its anti-structure Cu5Sb2In2 have metalliccharacter with a relatively low value of the DOS at the Fermilevel The main contribution to the DOS comes from the dstates of copper The copper d band extends from minus6 eV up to
approximately the Fermi level At lower energies two bands canbe distinguished The first band extending from minus115 to minus95eV is mainly determined by antimony states whereas indiumstates dominate the second band extending from minus90 to minus60eV Between the two bands a pseudogap is located at about minus9eV When the DOS of both compounds Cu5In2Sb2 andCu5Sb2In2 are compared it is found that there is a difference inthe location of the second band However for the anti-structureCu5Sb2In2 the contribution of copper s states as well as indiumand antimony s and p states is shifted to higher energies withrespect to Cu5In2Sb2 This shift causes a larger pseudogapwhich extends from minus95 to minus8 eV and could in principleexplain the vast difference in the energies of formation of bothcompounds For energies above the Fermi level the electronicstates of all species contribute to the determination of the shapeof the DOS Generally the shapes of the DOS for bothcompounds are similar and follow the general trends that havebeen recently discussed for other structurally related binaryCuminusIn and CuminusSn compounds32minus34
5 SUMMARY AND CONCLUDING REMARKSA new ternary orthorhombic compound with the formulaCu533In229Sb171 crystallizing in the space group Cmc21 andwith 36 atoms per unit cell has been synthezised Details of theexperimental study and the structural parameters of thiscompound are reported in the first part of the work In the
Scheme 1 GroupminusSubgroup Relations between the Fundamental Space Group for NiAsNi2In P63mmc and the UniquelyDetermined Subgroup Bb21m
Figure 4 Idealized structure of Cu5In2Sb2 thermal ellipsoidprobability of 50
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210790
second part ab initio calculations based on DFT and the PAWmethod are used to characterize the structural thermodynamicand electronic properties of (a) Cu5In2Sb2 and (b) the anti-structure Cu5Sb2In2 compound
The present calculations include the lattice parameters molarvolume bulk modulus and its pressure derivative energy offormation from the elements and the electronic DOSThe calculated lattice parameters and internal atomic
coordinates are in excellent agreement with the valuesdetermined experimentally The Cu5In2Sb2 compound isfound to be thermodynamically stable with respect to theelements at 0 KThese theoretical results for the Cu5In2Sb2 phase have been
compared with those for the Cu5Sb2In2 anti-site structurecompound resulting from exchange of the In and Sb Wyckoffsymmetric positions The Cu5Sb2In2 phase is thermodynami-cally unstable with respect to the elements at 0 KBoth the Cu5In2Sb2 and Cu5Sb2In2 compounds show
metallic behavior The main contribution to the DOS comesfrom Cu d electrons extending from minus6 eV up toapproximately the Fermi level
ASSOCIATED CONTENTS Supporting InformationX-ray crystallographic data in CIF format This material isavailable free of charge via the Internet at httppubsacsorg
AUTHOR INFORMATIONCorresponding AuthorE-mail carolamullerpolymatlthseAuthor ContributionsAll authors have given approval to the final version of themanuscriptNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors are grateful to Gunnel Karlsson for supporting theEDXS measurements This work was supported by the SwedishResearch Council VR the Agencia Nacional de PromocionCientifica y Tecnologica (Grants BID 1728OC-AR and PICT-2006 1947) and Universidad Nacional del Comahue (ProjectI157)
REFERENCES(1) Subramanian K N Ed Lead-Free Electronic Solders A SpecialIssue of the Journal of Materials Science Materials in ElectronicsSpringer Berlin 2007(2) Liu W E Mohney S E Mater Sci Eng B 2003 103 189minus201(3) Manasijevic D Minic D Zivkovic D Vrestal J Aljilji ATalijan N Stajic-Trosic J Marijanovec S Todorovic RCALPHAD Comput Coupling Phase Diagrams Thermochem 200933 221minus226(4) Okamoto H J Phase Equilib 1994 5 226minus227
Table 2 Ab Initio Calculated Structural and Elastic Properties for Cu In Sb Cu5In2Sb2 and Cu5Sb2In2 at 0 K For theintermetallic phases the energy of formation from the elements in the given structures is reported
lattice parameters a c (Aring)
element space group V0 (Aring3atom) calculated experimental B0 (GPa) B0prime ΔEf (kJmol)
Cu-fcc Fm3m 12035 3638 3596a 1380 142b 53 55 50c
In-tI2 I4mmm 27512 3305 5036 (3245 4942)d 363 418e 57 481e
Sb-hR2 R3m 31782 4377 11491 (4308 11274)d 315 383f 88Cu5In2Sb2 Cmc21 18252 17758g 10253 8576 7472 10181 8456 7377g 813 62 minus24867Cu5Sb2In2 Cmc21 18394 10446 8551 7414 790 65 22390
aExperimental data extrapolated to 0 K27 bExperimental data extrapolated to 0 K28 cExperimental data at 298 K29 dExperimental data at 291 K (In)and 298 K (Sb)30 eExperimental data at 293 K31 fExperimental data32 gExperimental data from the present work
Table 3 Sb Cu5In2Sb2 and Cu5Sb2In2 Unit Cell InternalCoordinates Calculated Ab Initio Compared with theAtomic Positions for the Cu5In2Sb2 Intermetallic CompoundDetermined in the Present Work
x y z
site GGA experiment
Sb Sb 6c 00 00 02333 00 00 023349Cu5In2Sb2 Cu1 8d 01286 06310 02521 01282 06308 02515
Cu2 8d 03755 06149 02257 03750 06137 02271Cu3 4d 05 03714 04046 05 03717 04044In1 4d 05 03465 00478 05 03465 00480In2 4d 05 01078 05935 05 01113 05923Sb1 8d 02355 03740 04074 02352 03736 04072
Cu5Sb2In2 Cu1 8d 01284 06306 02542Cu2 8d 03744 06152 02251Cu3 4d 05 03764 04059Sb1 4d 05 03466 00518Sb2 4d 05 01078 05876In1 8d 02338 03736 04063
Figure 5 Ab initio calculated electronic DOS of (a) Cu5In2Sb2 and (b)the anti-structure Cu5Sb2In2 compound
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210791
(5) Bahari Z Dichi E Legendre B Dugue J Thermochim Acta2003 401 131minus138(6) Saunders N Miodownik A P Bull Alloy Phase Diagrams 199011 278minus287(7) Kattner U JOM 2002 54 45minus51(8) Shim J-H Oh Ch-S Lee B-J Lee D N Z Metallkd 1996 8205minus212(9) Massalski T B Ed Binary Alloy Phase Diagrams 2nd ed ASMInternational Metal Park OH 1996 pp 1424minus1426(10) Elding-Ponten M Stenberg L Lidin S J Alloys Compd 1997261 162minus171(11) Lidin S Larsson A K J Solid State Chem 1995 118 313minus322(12) Jandali M Z Rajasekharan T P Schubert K Z Metallkd1982 73 463minus467(13) Gunzel E Schubert K Z Metallkd 1958 49 124minus133(14) Schubert K Burkhardt W Esslinger P Gunzel E MeissnerH G Schutt W Wegst J Wilkens M Naturwissenschaften 1956 43248minus249(15) Hofmann W Z Metallkd 1941 33 373(16) Nuss J Jansen M Z Anorg Allg Chem 2002 628 1152minus1157(17) Blochl P E Phys Rev B Condens Matter Mater Phys 1994 5017953minus17979(18) Kresse G Furthmuller J Comput Mater Sci 1996 6 15minus50(19) WinXPow Powder Diffraction Software version 31 Stoe amp CieDarmstadt Germany 2011(20) CrysAlisPro version 1171356 Oxford Diffraction LtdAbington UK2011(21) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2004 60 134minus141(22) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2005 61 147minus152(23) Palatinus L Chapuis G J Appl Crystallogr 2007 40 786minus790(24) Petricek V Dusek M Palatinus L Jana2006 The crystallo-graphic computing system Institute of Physics Praha Czech Republic2006(25) Aztec version 10 Oxford Instruments Nanotechnology ToolsLtd Oxford UK 2010minus2011(26) Perdew J P Wang Y Phys Rev B Condens Matter 1991 4413298minus13307(27) Straumanis M E Yu L S Acta Crystallogr Sect A Cryst PhysDiffr Theor Gen Crystallogr 1969 25 676minus682(28) Overton W C Jr Gaffney J Phys Rev 1955 98 969minus977(29) Steinberg D J J Phys Chem Solids 1982 43 1173minus1175(30) Villars P Pearsons Handbook Desk Edition ASM InternationalMaterials Park OH 1997(31) Takemura K Phys Rev B Condens Matter Mater Phys 199144 545minus549(32) Kittel C Introduction to solid state physics 7th ed Wiley NewYork 1996(33) Ramos de Debiaggi S Cabeza G F Deluque Toro C MontiA M Sommadossi S Fernandez Guillermet A J Alloys Compd2011 509 3238minus3245(34) Ramos de Debiaggi S Deluque Toro C Cabeza G FFernandez Guillermet A J Alloys Compd 2012 httpdxdoiorg101016jjallcom201206138
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210792
second part ab initio calculations based on DFT and the PAWmethod are used to characterize the structural thermodynamicand electronic properties of (a) Cu5In2Sb2 and (b) the anti-structure Cu5Sb2In2 compound
The present calculations include the lattice parameters molarvolume bulk modulus and its pressure derivative energy offormation from the elements and the electronic DOSThe calculated lattice parameters and internal atomic
coordinates are in excellent agreement with the valuesdetermined experimentally The Cu5In2Sb2 compound isfound to be thermodynamically stable with respect to theelements at 0 KThese theoretical results for the Cu5In2Sb2 phase have been
compared with those for the Cu5Sb2In2 anti-site structurecompound resulting from exchange of the In and Sb Wyckoffsymmetric positions The Cu5Sb2In2 phase is thermodynami-cally unstable with respect to the elements at 0 KBoth the Cu5In2Sb2 and Cu5Sb2In2 compounds show
metallic behavior The main contribution to the DOS comesfrom Cu d electrons extending from minus6 eV up toapproximately the Fermi level
ASSOCIATED CONTENTS Supporting InformationX-ray crystallographic data in CIF format This material isavailable free of charge via the Internet at httppubsacsorg
AUTHOR INFORMATIONCorresponding AuthorE-mail carolamullerpolymatlthseAuthor ContributionsAll authors have given approval to the final version of themanuscriptNotesThe authors declare no competing financial interest
ACKNOWLEDGMENTSThe authors are grateful to Gunnel Karlsson for supporting theEDXS measurements This work was supported by the SwedishResearch Council VR the Agencia Nacional de PromocionCientifica y Tecnologica (Grants BID 1728OC-AR and PICT-2006 1947) and Universidad Nacional del Comahue (ProjectI157)
REFERENCES(1) Subramanian K N Ed Lead-Free Electronic Solders A SpecialIssue of the Journal of Materials Science Materials in ElectronicsSpringer Berlin 2007(2) Liu W E Mohney S E Mater Sci Eng B 2003 103 189minus201(3) Manasijevic D Minic D Zivkovic D Vrestal J Aljilji ATalijan N Stajic-Trosic J Marijanovec S Todorovic RCALPHAD Comput Coupling Phase Diagrams Thermochem 200933 221minus226(4) Okamoto H J Phase Equilib 1994 5 226minus227
Table 2 Ab Initio Calculated Structural and Elastic Properties for Cu In Sb Cu5In2Sb2 and Cu5Sb2In2 at 0 K For theintermetallic phases the energy of formation from the elements in the given structures is reported
lattice parameters a c (Aring)
element space group V0 (Aring3atom) calculated experimental B0 (GPa) B0prime ΔEf (kJmol)
Cu-fcc Fm3m 12035 3638 3596a 1380 142b 53 55 50c
In-tI2 I4mmm 27512 3305 5036 (3245 4942)d 363 418e 57 481e
Sb-hR2 R3m 31782 4377 11491 (4308 11274)d 315 383f 88Cu5In2Sb2 Cmc21 18252 17758g 10253 8576 7472 10181 8456 7377g 813 62 minus24867Cu5Sb2In2 Cmc21 18394 10446 8551 7414 790 65 22390
aExperimental data extrapolated to 0 K27 bExperimental data extrapolated to 0 K28 cExperimental data at 298 K29 dExperimental data at 291 K (In)and 298 K (Sb)30 eExperimental data at 293 K31 fExperimental data32 gExperimental data from the present work
Table 3 Sb Cu5In2Sb2 and Cu5Sb2In2 Unit Cell InternalCoordinates Calculated Ab Initio Compared with theAtomic Positions for the Cu5In2Sb2 Intermetallic CompoundDetermined in the Present Work
x y z
site GGA experiment
Sb Sb 6c 00 00 02333 00 00 023349Cu5In2Sb2 Cu1 8d 01286 06310 02521 01282 06308 02515
Cu2 8d 03755 06149 02257 03750 06137 02271Cu3 4d 05 03714 04046 05 03717 04044In1 4d 05 03465 00478 05 03465 00480In2 4d 05 01078 05935 05 01113 05923Sb1 8d 02355 03740 04074 02352 03736 04072
Cu5Sb2In2 Cu1 8d 01284 06306 02542Cu2 8d 03744 06152 02251Cu3 4d 05 03764 04059Sb1 4d 05 03466 00518Sb2 4d 05 01078 05876In1 8d 02338 03736 04063
Figure 5 Ab initio calculated electronic DOS of (a) Cu5In2Sb2 and (b)the anti-structure Cu5Sb2In2 compound
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210791
(5) Bahari Z Dichi E Legendre B Dugue J Thermochim Acta2003 401 131minus138(6) Saunders N Miodownik A P Bull Alloy Phase Diagrams 199011 278minus287(7) Kattner U JOM 2002 54 45minus51(8) Shim J-H Oh Ch-S Lee B-J Lee D N Z Metallkd 1996 8205minus212(9) Massalski T B Ed Binary Alloy Phase Diagrams 2nd ed ASMInternational Metal Park OH 1996 pp 1424minus1426(10) Elding-Ponten M Stenberg L Lidin S J Alloys Compd 1997261 162minus171(11) Lidin S Larsson A K J Solid State Chem 1995 118 313minus322(12) Jandali M Z Rajasekharan T P Schubert K Z Metallkd1982 73 463minus467(13) Gunzel E Schubert K Z Metallkd 1958 49 124minus133(14) Schubert K Burkhardt W Esslinger P Gunzel E MeissnerH G Schutt W Wegst J Wilkens M Naturwissenschaften 1956 43248minus249(15) Hofmann W Z Metallkd 1941 33 373(16) Nuss J Jansen M Z Anorg Allg Chem 2002 628 1152minus1157(17) Blochl P E Phys Rev B Condens Matter Mater Phys 1994 5017953minus17979(18) Kresse G Furthmuller J Comput Mater Sci 1996 6 15minus50(19) WinXPow Powder Diffraction Software version 31 Stoe amp CieDarmstadt Germany 2011(20) CrysAlisPro version 1171356 Oxford Diffraction LtdAbington UK2011(21) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2004 60 134minus141(22) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2005 61 147minus152(23) Palatinus L Chapuis G J Appl Crystallogr 2007 40 786minus790(24) Petricek V Dusek M Palatinus L Jana2006 The crystallo-graphic computing system Institute of Physics Praha Czech Republic2006(25) Aztec version 10 Oxford Instruments Nanotechnology ToolsLtd Oxford UK 2010minus2011(26) Perdew J P Wang Y Phys Rev B Condens Matter 1991 4413298minus13307(27) Straumanis M E Yu L S Acta Crystallogr Sect A Cryst PhysDiffr Theor Gen Crystallogr 1969 25 676minus682(28) Overton W C Jr Gaffney J Phys Rev 1955 98 969minus977(29) Steinberg D J J Phys Chem Solids 1982 43 1173minus1175(30) Villars P Pearsons Handbook Desk Edition ASM InternationalMaterials Park OH 1997(31) Takemura K Phys Rev B Condens Matter Mater Phys 199144 545minus549(32) Kittel C Introduction to solid state physics 7th ed Wiley NewYork 1996(33) Ramos de Debiaggi S Cabeza G F Deluque Toro C MontiA M Sommadossi S Fernandez Guillermet A J Alloys Compd2011 509 3238minus3245(34) Ramos de Debiaggi S Deluque Toro C Cabeza G FFernandez Guillermet A J Alloys Compd 2012 httpdxdoiorg101016jjallcom201206138
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210792

(5) Bahari Z Dichi E Legendre B Dugue J Thermochim Acta2003 401 131minus138(6) Saunders N Miodownik A P Bull Alloy Phase Diagrams 199011 278minus287(7) Kattner U JOM 2002 54 45minus51(8) Shim J-H Oh Ch-S Lee B-J Lee D N Z Metallkd 1996 8205minus212(9) Massalski T B Ed Binary Alloy Phase Diagrams 2nd ed ASMInternational Metal Park OH 1996 pp 1424minus1426(10) Elding-Ponten M Stenberg L Lidin S J Alloys Compd 1997261 162minus171(11) Lidin S Larsson A K J Solid State Chem 1995 118 313minus322(12) Jandali M Z Rajasekharan T P Schubert K Z Metallkd1982 73 463minus467(13) Gunzel E Schubert K Z Metallkd 1958 49 124minus133(14) Schubert K Burkhardt W Esslinger P Gunzel E MeissnerH G Schutt W Wegst J Wilkens M Naturwissenschaften 1956 43248minus249(15) Hofmann W Z Metallkd 1941 33 373(16) Nuss J Jansen M Z Anorg Allg Chem 2002 628 1152minus1157(17) Blochl P E Phys Rev B Condens Matter Mater Phys 1994 5017953minus17979(18) Kresse G Furthmuller J Comput Mater Sci 1996 6 15minus50(19) WinXPow Powder Diffraction Software version 31 Stoe amp CieDarmstadt Germany 2011(20) CrysAlisPro version 1171356 Oxford Diffraction LtdAbington UK2011(21) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2004 60 134minus141(22) Oszlanyi G Suto A Acta Crystallogr Sect A FoundCrystallogr 2005 61 147minus152(23) Palatinus L Chapuis G J Appl Crystallogr 2007 40 786minus790(24) Petricek V Dusek M Palatinus L Jana2006 The crystallo-graphic computing system Institute of Physics Praha Czech Republic2006(25) Aztec version 10 Oxford Instruments Nanotechnology ToolsLtd Oxford UK 2010minus2011(26) Perdew J P Wang Y Phys Rev B Condens Matter 1991 4413298minus13307(27) Straumanis M E Yu L S Acta Crystallogr Sect A Cryst PhysDiffr Theor Gen Crystallogr 1969 25 676minus682(28) Overton W C Jr Gaffney J Phys Rev 1955 98 969minus977(29) Steinberg D J J Phys Chem Solids 1982 43 1173minus1175(30) Villars P Pearsons Handbook Desk Edition ASM InternationalMaterials Park OH 1997(31) Takemura K Phys Rev B Condens Matter Mater Phys 199144 545minus549(32) Kittel C Introduction to solid state physics 7th ed Wiley NewYork 1996(33) Ramos de Debiaggi S Cabeza G F Deluque Toro C MontiA M Sommadossi S Fernandez Guillermet A J Alloys Compd2011 509 3238minus3245(34) Ramos de Debiaggi S Deluque Toro C Cabeza G FFernandez Guillermet A J Alloys Compd 2012 httpdxdoiorg101016jjallcom201206138
Inorganic Chemistry Article
dxdoiorg101021ic3011794 | Inorg Chem 2012 51 10787minus1079210792
Top Related
Copyright © 2022 FDOKUMEN

