







Bahasa
Halaman
Hukum


This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2203
Cite this: Phys. Chem. Chem. Phys., 2012, 14, 2203–2215
Spectroscopic IR, EPR, and operando DRIFT insights into surface
reaction pathways of selective reduction of NO by propene over
the Co–BEA zeolite
Piotr Pietrzyk,*a Christophe Dujardin,b Kinga Gora-Marek,a Pascal Grangerb and
Zbigniew Sojkaa
Received 25th September 2011, Accepted 8th November 2011
DOI: 10.1039/c1cp23038g
Interaction of a Co–BEA catalyst with individual components (NO, C3H6, CO, O2) and mixtures
simulating the real feed of the selective catalytic reduction (SCR) of nitric oxide in static and pulse
experiments at variable temperatures was investigated by means of IR, EPR, and operando
DRIFT spectroscopy coupled with QMS/GC analysis of the products. Speciation of cobalt active
sites into Co(II), mono- and polynuclear oxo-cobalt species as well as CoO clusters was quantified
by IR using CO and NO as probe molecules. The key intermediates, by-products, and final
products of the SCR reaction were identified and their spectroscopic signatures ascertained.
Based on the spectroscopic operando results, a concise mechanistic scheme of the selective catalytic
reduction of nitric oxide by propene, triggered by a two-electron Co(II)/Co(0) redox couple, was
developed. It consists of a complex network of the sequential/parallel selective reduction steps that
are interlocked by the rival nonselective oxidation of the intermediates and their thermal
decomposition. It has been shown that the SCR process is initiated by the chemoselective capture
of NO from the reaction mixture by the cobalt active sites leading to the cobalt(II) dinitrosyls,
which in the excess of oxygen are partially oxidized to surface nitrates and nitrites. N2O is produced
by semi-decomposition of the dinitrosyl intermediates on the mononuclear centers, whereas NO2 via
NO oxidation on the polynuclear oxo-cobalt sites. Cyanide and isocyanate species, formed together
with propene oxygenates in the course of the CQC bond scission, are the mechanistically pivotal
reaction intermediates of C3H6 interaction with the dinitrosyles and NO3�/NO2
� surface species.
Dinitrogen is produced by three main reaction routes involving oxidation of cyanides by NO/NO2,
reduction of dinitrosyls, nitrates, and nitrites by propene oxygenates (medium temperature range)
or their reduction by carbon monoxide (high temperature range).
I. Introduction
Removal of NOx from exhaust gases by its selective catalytic
reduction (SCR) or direct decomposition (deNOx) over porous
materials functionalized with transition-metal ions (TMI) involves
active centers of specific electron and magnetic structure that are
able to constitute suitable redox couples triggered by electron and
spin transfer processes occurring within the NO–TMI moiety.1,2
Indeed, the chemical reactivity of the coordinated NO is featured
by removal of the unpaired electron from the 2p* antibonding
orbital to form a nitrosonium NO+ cation or addition of an
electron to the 2p* orbital, giving rise to a nitroside anion NO� of
distinctly different reactivity.3 The other pathway leading to the
changes in the valence state of the active sites consists of an
oxygen transfer step associated with the oxidation of NO to
NO2 or mild oxidation of the hydrocarbon reducing agent to
oxygenates by the active sites containing reactive extra-lattice
oxygen (ELO). Obviously the occurrence of both pathways
depends on the nature of the active site, and electron transfer is
characteristic of the bare cations, whereas in the case of the
mono or polynuclear oxo-cation species oxygen transfer plays
a decisive role.4 Pronounced spin redistribution on passing
from nitric oxide to dinitrogen is another noteworthy issue in
the mechanistic studies of NOx abatement, and this constraint
can be surmounted owing to the active sites constituted by
transition-metal ions (TMI).1,3
All those aspects of the catalytic chemistry of NOx can be
investigated using zeolites exchanged with TMI, which are
important model and practical materials extensively studied in
the SCR context.1,2,5–7 Zeolitic catalysts provide relatively
a Faculty of Chemistry, Jagiellonian University, ul. Ingardena 3,30-060 Krakow, Poland. E-mail: [email protected];Fax: +48 12 634-05-15; Tel: +48 12 663-22-24
bUnite de Catalyse et de Chimie du Solide, Universite de Lille1,Sciences et Technologies, UMR CNRS 8181, 59655 Villeneuved’Ascq Cedex, France
PCCP Dynamic Article Links
www.rsc.org/pccp PAPER
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
GView Article Online / Journal Homepage / Table of Contents for this issue

2204 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 This journal is c the Owner Societies 2012
wide temperature windows for SCR and, depending on the
nature and nuclearity of the dispersed metal ions, high catalytic
activity. In particular, cobalt-exchanged high-silica zeolites,
such as ZSM-5 and BEA, have been recognized as the efficient
catalysts in SCR of NOx with methane7–9 and with larger
hydrocarbons.10–12
The nature of the cobalt active sites is, however, still debating.
In general two types of cobalt centers have been distinguished:
isolated mononuclear cobalt(II) cations located in the exchange-
able positions13–15 and cobalt mono- and polynuclear clusters
bearing extra-lattice oxygen of poorly defined stoichiometry
CoxOy.16–19 To some extent, the concentration of both types
of cobalt centers, i.e., the isolated and clustered ones, can be
controlled by adjusting the zeolite type, its Si/Al ratio, concen-
tration of cobalt ions, and the nature of the solvent used during
the ion exchange process. It has been recognized that isolated
and clustered oxo-centers differ in reactivity toward NOx
abatement. As shown by Smeets et al.,7 isolated Co(II) sites
have been the most active in N2O decomposition, whereas
oxo-forms have been found less active or even inactive.
From the practical point of view, removal of noxious gases
emitted by internal combustion engines operating under O2-rich
conditions (diesel and lean-burn gasoline engines), where the
noble-metal three way catalysts are not effective, is a major
challenge for environmental catalysis. The exhaust gases produced
under such conditions include nitrogen oxides (NOx), unburned
carbon monoxide, hydrocarbons and oxygenates. However,
despite the considerable progress made in the last decade,
development of an efficient and robust catalyst for SCR of
NOx by hydrocarbons (HC-SCR) still remains an open issue,
mainly due to the incomplete understanding of the reaction
mechanism.20 Mechanistic investigations of NOx-SCR over
Co-exchanged zeolites revealed the appearance of –CN and
–NCO species.8,21 They can be directly oxidized to N2 by
NOx8,21 or in an alternative contentious account, hydrolyzed
by water to NH3 and CO2 leading to higher SCR activity.12
Addition of oxygen leads to a significant enhancement of the
SCR efficiency by controlling formation of the highly reactive
oxygenates used as efficient reductants,22 yet this point has not
been elucidated definitely.
The objectives of the present study comprise quantification
of the cobalt cationic and oxo species in the Co–BEA zeolite,
assessment of its reactivity with single and mixed gas reactants,
identification of the reaction intermediates appearing in the
selective catalytic reduction of NO with CO and propene, and
evaluation of their mechanistic role. Combination of static IR
and EPR studies complemented by operando (DRIFT/gas-phase
IR/QMS) measurements was employed to provide a detailed
insight into the chemical nature of both the metal active centers
and the adsorbed species.
II. Experimental
Co–BEA samples were obtained by a standard ion exchange
procedure from the parent ammonium form of the BEA
zeolite with Si/Al = 12 (Tricat Zeolites). The mixture of
0.1 M Co(NO3)2 water solution in proportion 1 g zeolite per
100 cm3 was stirred for 20 h at 323 K, and the final pH of the
solution varied around 5. After drying at 353 K the obtained
samples were characterized by an ICP method, revealing
2.04 wt% of Co and 2.45 wt% of Al content. Adsorption and
reaction under static conditions were monitored with a Bruker
ELEXSYS-E500 X-band EPR spectrometer and a Bruker
Tensor 27 FTIR spectrometer equipped with an MCT detector
and with a spectral resolution of 2 cm�1. Computer simulations of
the EPR spectra were carried out with the EPRsim32 software.23
Prior to adsorption the samples were evacuated in vacuum
(B10�5 Pa) at 773 K (6 K min�1) for 3 h. Before adsorption
the gas reactants, NO and NO2 (Aldrich, 98.5 and 99.5%,
respectively), were purified by the freeze–pump–thaw technique,
whereas O2, CO, and propene (Aldrich, 99.95%) were adsorbed
directly without any prior purification. Adsorption was carried
out at 77 K or at room temperature and the samples were next
exposed to various temperatures.
In the case of the operando DRIFT experiments, Co–BEA
samples (B20 mg) were treated in a helium flow (13 mL min�1)
with the heating rate 6 K min�1 followed by final activation at
673 K for 2 hours. Prior to the measurements, the samples were
cooled down to 423 K. A typical temperature-programmed
surface reaction (TPSR) experiment consisted of a sequence of
pulses of the reactants followed by flow of the gas mixture (30min)
at 423 K and 503 K. After switching off the reactant flux, the
temperature was raised up to 673 K (6 K min�1) and during
that time a TPD-QMS experiment was carried out using a
quadruple mass spectrometer (Balzers Omnistar). The sample
was cooled down again to 423 K and the adsorption sequence
was repeated. For the SCR reaction, the reactant gas mixture
(744 ppm NO, 1388 ppm C3H6, 1388 ppm CO, and 0–3–10%
O2) was passed through the sample, and temperature was
increased with the heating rate of 6 K min�1. In the course
of those experiments, IR spectra of the gas-phase and DRIFT
spectra of the catalyst surface were recorded with 4 cm�1
resolution using a Thermo 380 and a Thermo 460 Protege
instrument, respectively. Both spectrometers were equipped
with MCT detectors.
Additional temperature-programmed catalytic activity
measurements were conducted in a fixed bed flow reactor with
a temperature gradient of 5 K min�1. The same gas mixture
composition as in operando TPSR was used with variable
amounts of oxygen 0–3–10% O2. 360 mg of the catalyst in
powder form was exposed to a total flow rate of 18 L h�1. The
catalyst was dehydrated under He at 673 K prior to the catalytic
measurements. The inlet and outlet gas mixtures were analyzed
using a mGC Varian CP-4900 chromatograph equipped with
two thermal conductivity detectors. The reactants and products
were separated on 5 A molecular sieves and poraplot Q columns.
III. Results and discussion
1. Spectroscopic characterization of cobalt active centers and
their interaction with single SCR reactants
1.1. Speciation of cobalt active sites.The nature and diversity
of the cobalt sites were examined by IR using CO and NO as
probe molecules. The IR spectra recorded upon CO and NO
absorption on the activated Co–BEA sample are shown in Fig. 1.
Adsorption of the CO probe leads to a complex broad IR band
appearing in the range of 2220–2160 cm�1 with three clear
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online
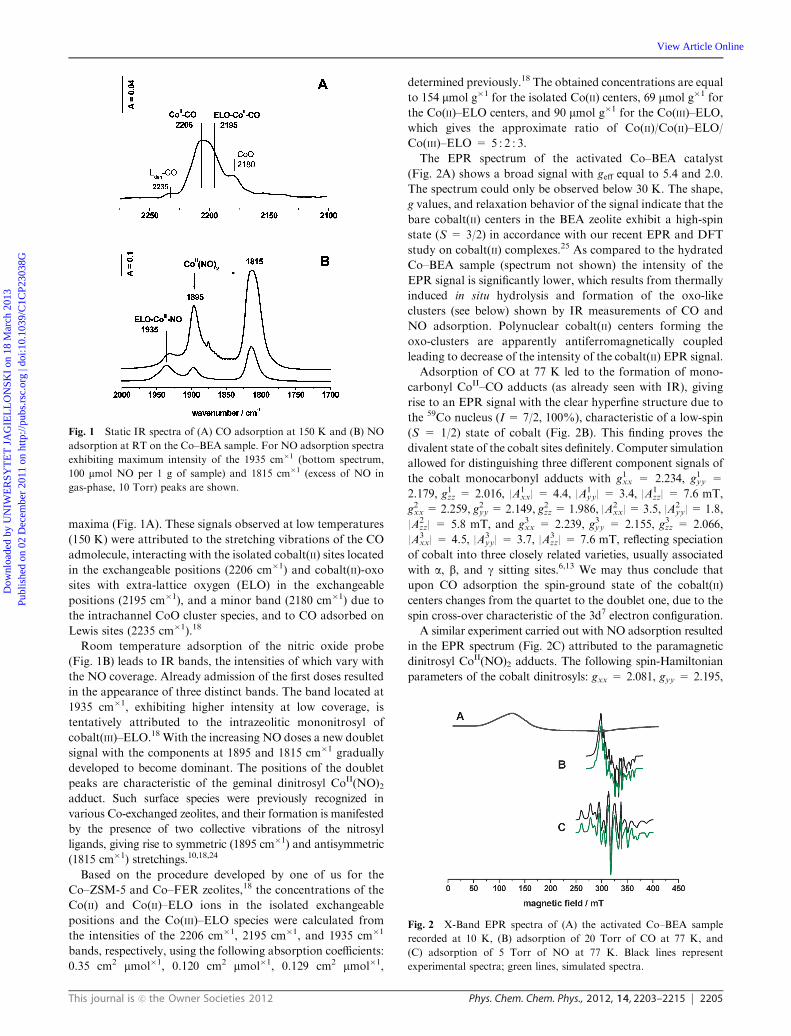
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2205
maxima (Fig. 1A). These signals observed at low temperatures
(150 K) were attributed to the stretching vibrations of the CO
admolecule, interacting with the isolated cobalt(II) sites located
in the exchangeable positions (2206 cm�1) and cobalt(II)-oxo
sites with extra-lattice oxygen (ELO) in the exchangeable
positions (2195 cm�1), and a minor band (2180 cm�1) due to
the intrachannel CoO cluster species, and to CO adsorbed on
Lewis sites (2235 cm�1).18
Room temperature adsorption of the nitric oxide probe
(Fig. 1B) leads to IR bands, the intensities of which vary with
the NO coverage. Already admission of the first doses resulted
in the appearance of three distinct bands. The band located at
1935 cm�1, exhibiting higher intensity at low coverage, is
tentatively attributed to the intrazeolitic mononitrosyl of
cobalt(III)–ELO.18 With the increasing NO doses a new doublet
signal with the components at 1895 and 1815 cm�1 gradually
developed to become dominant. The positions of the doublet
peaks are characteristic of the geminal dinitrosyl CoII(NO)2adduct. Such surface species were previously recognized in
various Co-exchanged zeolites, and their formation is manifested
by the presence of two collective vibrations of the nitrosyl
ligands, giving rise to symmetric (1895 cm�1) and antisymmetric
(1815 cm�1) stretchings.10,18,24
Based on the procedure developed by one of us for the
Co–ZSM-5 and Co–FER zeolites,18 the concentrations of the
Co(II) and Co(II)–ELO ions in the isolated exchangeable
positions and the Co(III)–ELO species were calculated from
the intensities of the 2206 cm�1, 2195 cm�1, and 1935 cm�1
bands, respectively, using the following absorption coefficients:
0.35 cm2 mmol�1, 0.120 cm2 mmol�1, 0.129 cm2 mmol�1,
determined previously.18 The obtained concentrations are equal
to 154 mmol g�1 for the isolated Co(II) centers, 69 mmol g�1 for
the Co(II)–ELO centers, and 90 mmol g�1 for the Co(III)–ELO,
which gives the approximate ratio of Co(II)/Co(II)–ELO/
Co(III)–ELO = 5 : 2 : 3.
The EPR spectrum of the activated Co–BEA catalyst
(Fig. 2A) shows a broad signal with geff equal to 5.4 and 2.0.
The spectrum could only be observed below 30 K. The shape,
g values, and relaxation behavior of the signal indicate that the
bare cobalt(II) centers in the BEA zeolite exhibit a high-spin
state (S = 3/2) in accordance with our recent EPR and DFT
study on cobalt(II) complexes.25 As compared to the hydrated
Co–BEA sample (spectrum not shown) the intensity of the
EPR signal is significantly lower, which results from thermally
induced in situ hydrolysis and formation of the oxo-like
clusters (see below) shown by IR measurements of CO and
NO adsorption. Polynuclear cobalt(II) centers forming the
oxo-clusters are apparently antiferromagnetically coupled
leading to decrease of the intensity of the cobalt(II) EPR signal.
Adsorption of CO at 77 K led to the formation of mono-
carbonyl CoII–CO adducts (as already seen with IR), giving
rise to an EPR signal with the clear hyperfine structure due to
the 59Co nucleus (I = 7/2, 100%), characteristic of a low-spin
(S = 1/2) state of cobalt (Fig. 2B). This finding proves the
divalent state of the cobalt sites definitely. Computer simulation
allowed for distinguishing three different component signals of
the cobalt monocarbonyl adducts with g1xx = 2.234, g1yy =
2.179, g1zz = 2.016, |A1xx| = 4.4, |A1
yy| = 3.4, |A1zz| = 7.6 mT,
g2xx=2.259, g2yy =2.149, g2zz=1.986, |A2xx| = 3.5, |A2
yy| = 1.8,
|A2zz| = 5.8 mT, and g3xx = 2.239, g3yy = 2.155, g3zz = 2.066,
|A3xx| = 4.5, |A3
yy| = 3.7, |A3zz| = 7.6 mT, reflecting speciation
of cobalt into three closely related varieties, usually associated
with a, b, and g sitting sites.6,13 We may thus conclude that
upon CO adsorption the spin-ground state of the cobalt(II)
centers changes from the quartet to the doublet one, due to the
spin cross-over characteristic of the 3d7 electron configuration.
A similar experiment carried out with NO adsorption resulted
in the EPR spectrum (Fig. 2C) attributed to the paramagnetic
dinitrosyl CoII(NO)2 adducts. The following spin-Hamiltonian
parameters of the cobalt dinitrosyls: gxx = 2.081, gyy = 2.195,
Fig. 1 Static IR spectra of (A) CO adsorption at 150 K and (B) NO
adsorption at RT on the Co–BEA sample. For NO adsorption spectra
exhibiting maximum intensity of the 1935 cm�1 (bottom spectrum,
100 mmol NO per 1 g of sample) and 1815 cm�1 (excess of NO in
gas-phase, 10 Torr) peaks are shown.
Fig. 2 X-Band EPR spectra of (A) the activated Co–BEA sample
recorded at 10 K, (B) adsorption of 20 Torr of CO at 77 K, and
(C) adsorption of 5 Torr of NO at 77 K. Black lines represent
experimental spectra; green lines, simulated spectra.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online

2206 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 This journal is c the Owner Societies 2012
gzz=2.086, |Axx| = 18.2, |Ayy| = 10.2 and |Azz| = 3.1 mT were
obtained by computer simulation. Formation of this intrazeolitic
complex can be rationalized in terms of a spin pairing process
between the quartet 4CoII(3d7) sites and two spin doublet NO
(2P1/2) radical ligands: CoII (4A2) + 2 NO (2P1/2)-
2Co(NO)2.
The resultant spin density is largely located on the metal center
(r3d E 80%), implying the following magnetic structure of the
dinitrosyl intermediates NO(km)mCoII(mk)NO. The actual
oxidation state of cobalt depends on the localization of both
electron pairs (km) on Co or NO moieties, which is dictated by
the relative position of the 2p* antibonding orbital of NO with
respect to the 3d manifold of Co (vide infra).
1.2. Mononuclear oxo and dinuclear oxo-cobalt sites. Apart
from the isolated cobalt(II) ions in the exchangeable positions
of the BEA zeolite, polynuclear oxo-bridged cobalt species
detected by both EPR and IR low-temperature adsorption
experiments were identified (in agreement with H2-TPR10,26,27
and other adsorption18,28 studies) and quantified. Their molecular
models, obtained by cutting off the fragments from the model of
the BEA zeolite (Materials Studio, Accelrys) and saturating the
resulting dangling bonds with hydrogen atoms, are shown in
Fig. 3. Because of their high charge, the bare CoIII ions cannot be
accommodated in the exchangeable positions of the high-silica
zeolites such as BEA. Thus, they are most probably associated
with the mono- and polynuclear cobalt–ELO species giving rise
to CoxOyn+ (n = 1, 2) entities. Our earlier IR and DFT study29
showed that during dehydration and activation of cobalt-zeolite
samples, the intrazeolitic Co2+(H2O)6 complexes hydrolyze,
producing a hydroxo (CoOH)+aq and a H+ species, counter-
balancing separately the negative charges of the zeolite frame-
work (Si–O–Al)�, according to the Pauling local electroneutrality
principle. They are precursors for the formation of the mono-
nuclear CoO+ and polynuclear oxo-centers detected by low-
temperature CO adsorption (see above). Possible reactions leading
to the formation of dinuclear centers involve subsequent olation
and oxolation steps:
(CoII–OH)+ + CoII–OH2 - (CoII–OH–CoII)3+ + H2O,
(1)
(CoII–OH)+ + (CoII–OH)+ - (CoII–O–CoII)2+ + H2O.
(2)
With the temperature increase, intrazeolite redox processes
may occur to produce mixed-valence cobalt sites, according to
the reaction such as:
2(CoII–OH)+ - (HO–CoIII–O–CoII)2+ + 1/2 H2. (3)
Proton release improves the stabilization of both resultant
cationic species within the zeolite channels:
(HO–CoIII–O–CoII)2+ - (O–CoIII–O–CoII)+ + H+zeol, (4)
where (O–CoIII–O–CoII)+ actually represents the polynuclear
CoxOyn+ (n = 1, 2) entities of undefined structure at present.
Similar steps have been suggested in the literature earlier.16,17
A molecular model of the last complex attached to the zeolite
wall is shown in Fig. 3C. Reactivity of the proposed dimeric
centers toward NO is discussed below.
2. Evaluation of putative partial reaction pathways of the SCR
process
2.1. Interaction with C3H6 and oxygen–carboxylic inter-
mediates. The interaction of the Co–BEA with C3H6 was
monitored after adsorption of propene at 298 K and sub-
sequent reaction with dioxygen at 673 K (Fig. 4). The main
feature in the IR spectrum of propene adsorbed in the
Co–BEA zeolite is a pronounced shift of the CQC stretching
band from the position of 1646 cm�1, typical for the gas-phase
C3H6, or 1630 cm�1, characteristic of propene interacting with
Si–(OH)–Al sites, into the region of 1595–1610 cm�1. This
effect is connected with p-backdonation between cobalt 3d
electrons and carbon–carbon p bond, proving a direct inter-
action between the propene CQC moiety and the cobalt active
centers. After reaction with O2 (Fig. 4B), the bands due to the
coordinated propene disappear, and a new intense band at
1590 cm�1 attributable to cobalt(II) carboxylate species can be
observed, indicating a straightforward catalytic oxidation of
the adsorbed propene by dioxygen to acetates or formates.30–32
The broad, intense band at 1675 cm�1 can be assigned to the
vibrations of the carboxyl groups and deformation vibration
of the water by-product formed upon oxidation of propene.
Its higher energy tail (1700 cm�1) is characteristic of
Fig. 3 Schematic representation of the molecular models of (A) the
isolated CoII site in the BEA zeolite (cluster model of [Si5Al2O8(OH)12]2�
stoichiometry cut-off from the structure of BEA and terminated with H
atoms), (B) (CoIII–O)+ species, and (C) one of the possible dimeric
oxide-like species with a terminal oxo moiety (O–CoIII–O–CoII)+
(cluster model of [Si8Al2O11(OH)17]2� stoichiometry).
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online
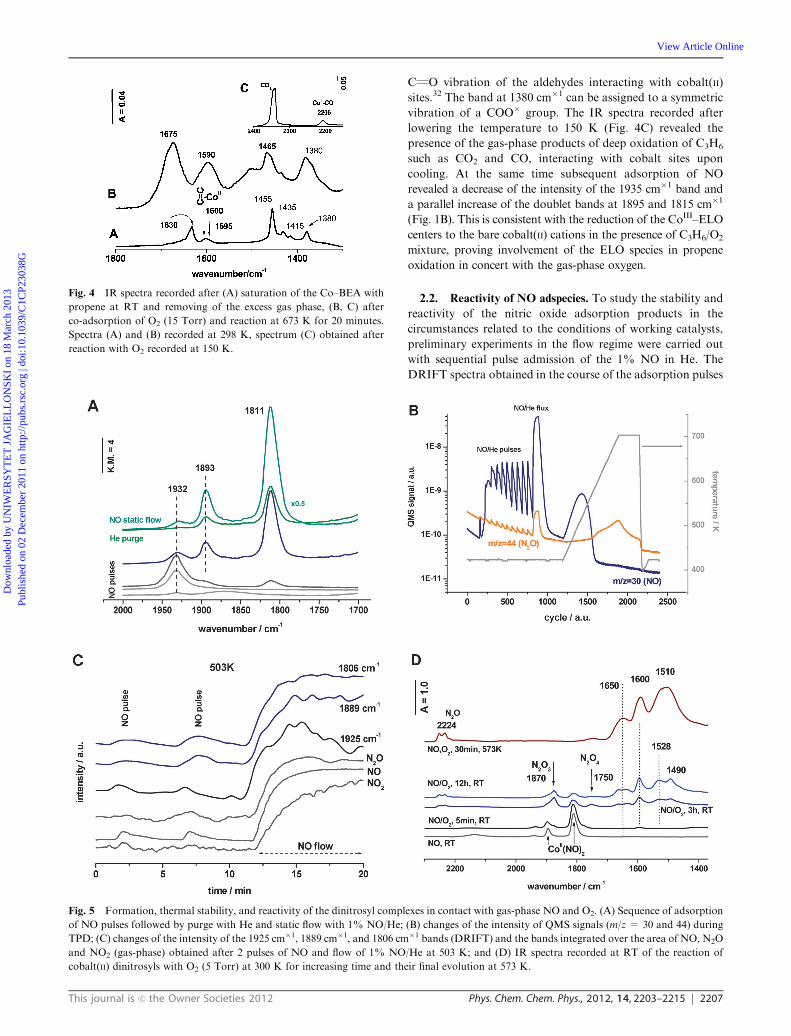
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2207
CQO vibration of the aldehydes interacting with cobalt(II)
sites.32 The band at 1380 cm�1 can be assigned to a symmetric
vibration of a COO� group. The IR spectra recorded after
lowering the temperature to 150 K (Fig. 4C) revealed the
presence of the gas-phase products of deep oxidation of C3H6
such as CO2 and CO, interacting with cobalt sites upon
cooling. At the same time subsequent adsorption of NO
revealed a decrease of the intensity of the 1935 cm�1 band and
a parallel increase of the doublet bands at 1895 and 1815 cm�1
(Fig. 1B). This is consistent with the reduction of the CoIII–ELO
centers to the bare cobalt(II) cations in the presence of C3H6/O2
mixture, proving involvement of the ELO species in propene
oxidation in concert with the gas-phase oxygen.
2.2. Reactivity of NO adspecies. To study the stability and
reactivity of the nitric oxide adsorption products in the
circumstances related to the conditions of working catalysts,
preliminary experiments in the flow regime were carried out
with sequential pulse admission of the 1% NO in He. The
DRIFT spectra obtained in the course of the adsorption pulses
Fig. 4 IR spectra recorded after (A) saturation of the Co–BEA with
propene at RT and removing of the excess gas phase, (B, C) after
co-adsorption of O2 (15 Torr) and reaction at 673 K for 20 minutes.
Spectra (A) and (B) recorded at 298 K, spectrum (C) obtained after
reaction with O2 recorded at 150 K.
Fig. 5 Formation, thermal stability, and reactivity of the dinitrosyl complexes in contact with gas-phase NO and O2. (A) Sequence of adsorption
of NO pulses followed by purge with He and static flow with 1% NO/He; (B) changes of the intensity of QMS signals (m/z = 30 and 44) during
TPD; (C) changes of the intensity of the 1925 cm�1, 1889 cm�1, and 1806 cm�1 bands (DRIFT) and the bands integrated over the area of NO, N2O
and NO2 (gas-phase) obtained after 2 pulses of NO and flow of 1% NO/He at 503 K; and (D) IR spectra recorded at RT of the reaction of
cobalt(II) dinitrosyls with O2 (5 Torr) at 300 K for increasing time and their final evolution at 573 K.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online

2208 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 This journal is c the Owner Societies 2012
at 423 K are shown in Fig. 5A. After the first pulse the only
band observed at 1932 cm�1 was assigned to the NO–CoIII–ELO
intrazeolite species. The intensity of this signal passed through a
maximum to gradually disappear at the end of the third pulse.
At the same time, the doublet signal of the dinitrosyl species with
the components at 1894 and 1810 cm�1 (identified in static IR
experiments) developed to become dominant. During the next
pulses their intensity decreased moderately, but at the end of the
pulse sequence they were the only remaining bands that are
associated with the adsorbed NO. Upon the helium flush the
band at 1932 cm�1 disappeared, in contrast to the doublet
signal, indicating that only the dinitrosyl species are stable under
such conditions.
Subsequent steady flow of the NO/He mixture led to a sharp
increase of the intensity of the dinitrosyl bands, whereas the
1932 cm�1 band recovered only 10–20% of its initial intensity.
Thermal stability of the cobalt(II) dinitrosyls was assessed in
the subsequent TPD-QMS-IR experiment. The maximum of
the NO desorption peak appeared at 503 K (Fig. 5B), whereas
at 593 K the dinitrosyls disappeared completely. During the
desorption no new bands such as that of cobalt(II) mononitrosyl
were produced at their expense. However, the gas-phase IR
and QMS showed the presence of N2O at 703 K—the product
of semi-decomposition of the dinitrosyls (in contrast to com-
plete decomposition leading to elemental N2), schematically
represented as
CoII(NO)2 - CoII–ELO + N2O. (5)
The sequence of the NO pulses followed by a continuous
flow of NO/He was repeated at the temperature of maximum
NO release (503 K) due to decomposition of the dinitrosyl
species (Fig. 5C). At this temperature, admission of the NO/
He pulses resulted in the formation of the bands characteristic
of the CoII(NO)2 and NO–CoIII–ELO species, with the dinitrosyl
signal being the predominant one. In the case of the
NO–CoIII–ELO band, a slight red-shift from 1932 to 1925 cm�1
was observed upon passing to the higher temperatures. It could
indicate a ‘‘pure’’ temperature effect on band position33 or thermal
activation of the coordinated NO reactant (change in the
metal–nitrosyl distance resulting in loosening of the N–O bond).
To unravel the reactivity of the observed species, the evolution
of the 1925 cm�1 band intensity due to the NO–CoIII–ELO
species was compared with the changes in the gas-phase NO2
band intensity (integrated over the area of 1658–1567 cm�1)
registered after the NO/He flux at 503 K (Fig. 5C). The results
indicate that the production of NO2(g) is related to a decline of
the intrazeolitic NO–CoIII–ELO species, whereas the intensity of
the dinitrosyl bands remains nearly constant with the elapsing
time at this temperature. The integral intensity plots for NO2
and N2O show that these two by-products are apparently
formed with different rates, indicating that the oxidation of
NO(g) to NO2(g) by Co–ELO species is more difficult than the
semi-decomposition of CoII(NO)2 into N2O(g) and ELO.
The reactivity of the dinitrosyl species with dioxygen was
investigated in static IR measurements and the results are
shown in Fig. 5D. The bands of the dinitrosyl CoII(NO)2intermediates decrease, and apart from the bands at 1870 cm�1
and 1750 cm�1 due to gas phase N2O3 and N2O4, respectively,
new IR signals in the 1650–1350 cm�1 region assigned to
NO2� and NO3
� species develop.24 This indicates that the
nitrosyls can be easily oxidized to the surface nitrates and
nitrites upon contact with O2 already at 300 K. They were
stable under the evacuation even at higher temperatures. The
position of these bands strongly depends on the NO2�/NO3
�
structure (speciation into monodentate, bidentate, or bridged
entities), and on the kind of the cation to which they are
bonded. The bands in the region 1660–1560 cm�1 are supposed
to originate form the bidentate nitrates NO3�. The mono-
dentate NO2� species were detected at lower frequency (1528,
1510 cm�1).24 Heating the reaction mixture at 573 K led to
a complete disappearance of the dinitrosyl complexes and N2O3/
N2O4 nitrogen oxides, which was accompanied by a further
development of the IR bands due to NO2�/NO3
� species, with
the nitrites (1510 cm�1) peak being the dominant one.
The above observations show that the cobalt(II) dinitrosyl
adducts can easily be thermally semi-decomposed into nitrous
oxide and cobalt–ELO species, whereas in the presence of
oxygen they can be oxidized to the surface nitrates and nitrites.
The CoxOyn+ centers are the reactive sites towardNO oxidation
to NO2 via an oxygen transfer pathway. We can postulate the
following stoichiometry of the redox processes explaining the
observed cobalt reactivity leading to the formation of NO2:
(O–CoIII–O–CoIII)2+ + NO - (CoII–O–CoII)2+ + NO2,
(6)
or taking into account the well known equilibrium reaction
2NO + 1/2 O2 + 2H+zeol " 2NO+ + H2O,
(O–CoIII–O–CoII)+ + NO+ - (CoII–O–CoII)2+ + NO2.
(7)
The real mechanism depends obviously on the actual structure
of the CoxOyn+ centers. The mononuclear Co–ELO centers
most likely can oxidize NO in a similar way but this process is
expected to be energetically less favorable.
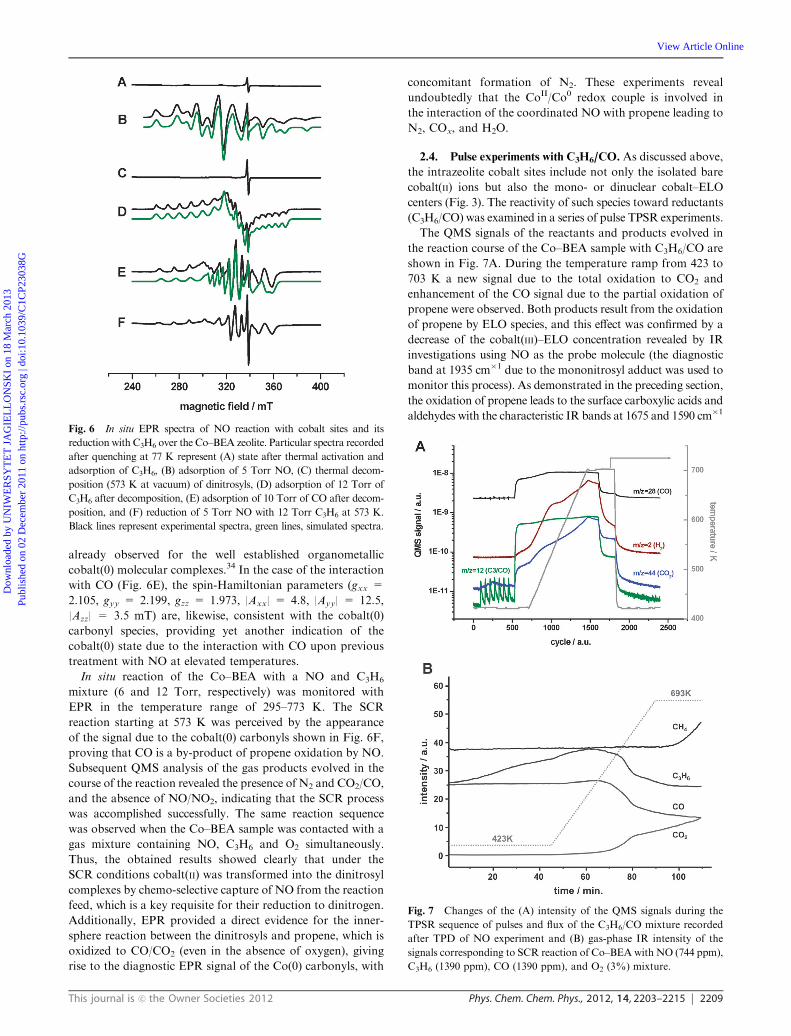
2.3. Interaction of C3H6 with dinitrosyls. The reactivity of
the intermediate dinitrosyl complexes of cobalt(II) was next
tested in the presence of C3H6, and monitored with in situ EPR
measurements. This allows for probing the reaction progress
from the cobalt center view point. The resultant spectra are
shown in Fig. 6. At first, a blank adsorption of propene on the
activated Co–BEA sample was carried out. No new EPR
signal was observed at room temperature (Fig. 6A). In the
next experiment the sample with the preadsorbed nitric oxide
(Fig. 6B) was evacuated at temperatures increasing from 373
to 573 K. The resulting spectrum showed destruction of the
dinitrosyl complex without formation of any new EPR signal
(Fig. 6C). After such treatment the sample was probed sepa-
rately with CO and C3H6, which gave rise to appearance of
EPR signals (Fig. 6D and E) completely different from that
previously observed for the reference cobalt(II) carbonyl
complex (Fig. 2B). Reaction of C3H6 (12 Torr, 443 K) with
the cobalt sites generated upon decomposition of the dinitrosyl
intermediates at 573 K led to a new EPR signal (Fig. 6D)
characterized by well-resolved cobalt hyperfine structure with
gxx = 2.096, gyy = 1.924, gzz = 2.297, |Axx| = 1.2, |Ayy| =
5.2, |Azz| = 9.9 mT. Such EPR parameters are similar to those
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2209
already observed for the well established organometallic
cobalt(0) molecular complexes.34 In the case of the interaction
with CO (Fig. 6E), the spin-Hamiltonian parameters (gxx =
2.105, gyy = 2.199, gzz = 1.973, |Axx| = 4.8, |Ayy| = 12.5,
|Azz| = 3.5 mT) are, likewise, consistent with the cobalt(0)
carbonyl species, providing yet another indication of the
cobalt(0) state due to the interaction with CO upon previous
treatment with NO at elevated temperatures.
In situ reaction of the Co–BEA with a NO and C3H6
mixture (6 and 12 Torr, respectively) was monitored with
EPR in the temperature range of 295–773 K. The SCR
reaction starting at 573 K was perceived by the appearance
of the signal due to the cobalt(0) carbonyls shown in Fig. 6F,
proving that CO is a by-product of propene oxidation by NO.
Subsequent QMS analysis of the gas products evolved in the
course of the reaction revealed the presence of N2 and CO2/CO,
and the absence of NO/NO2, indicating that the SCR process
was accomplished successfully. The same reaction sequence
was observed when the Co–BEA sample was contacted with a
gas mixture containing NO, C3H6 and O2 simultaneously.
Thus, the obtained results showed clearly that under the
SCR conditions cobalt(II) was transformed into the dinitrosyl
complexes by chemo-selective capture of NO from the reaction
feed, which is a key requisite for their reduction to dinitrogen.
Additionally, EPR provided a direct evidence for the inner-
sphere reaction between the dinitrosyls and propene, which is
oxidized to CO/CO2 (even in the absence of oxygen), giving
rise to the diagnostic EPR signal of the Co(0) carbonyls, with
concomitant formation of N2. These experiments reveal
undoubtedly that the CoII/Co0 redox couple is involved in
the interaction of the coordinated NO with propene leading to
N2, COx, and H2O.
2.4. Pulse experiments with C3H6/CO. As discussed above,
the intrazeolite cobalt sites include not only the isolated bare
cobalt(II) ions but also the mono- or dinuclear cobalt–ELO
centers (Fig. 3). The reactivity of such species toward reductants
(C3H6/CO) was examined in a series of pulse TPSR experiments.
The QMS signals of the reactants and products evolved in
the reaction course of the Co–BEA sample with C3H6/CO are
shown in Fig. 7A. During the temperature ramp from 423 to
703 K a new signal due to the total oxidation to CO2 and
enhancement of the CO signal due to the partial oxidation of
propene were observed. Both products result from the oxidation
of propene by ELO species, and this effect was confirmed by a
decrease of the cobalt(III)–ELO concentration revealed by IR
investigations using NO as the probe molecule (the diagnostic
band at 1935 cm�1 due to the mononitrosyl adduct was used to
monitor this process). As demonstrated in the preceding section,
the oxidation of propene leads to the surface carboxylic acids and
aldehydes with the characteristic IR bands at 1675 and 1590 cm�1
Fig. 6 In situ EPR spectra of NO reaction with cobalt sites and its
reduction with C3H6 over the Co–BEA zeolite. Particular spectra recorded
after quenching at 77 K represent (A) state after thermal activation and
adsorption of C3H6, (B) adsorption of 5 Torr NO, (C) thermal decom-
position (573 K at vacuum) of dinitrosyls, (D) adsorption of 12 Torr of
C3H6 after decomposition, (E) adsorption of 10 Torr of CO after decom-
position, and (F) reduction of 5 Torr NO with 12 Torr C3H6 at 573 K.
Black lines represent experimental spectra, green lines, simulated spectra.
Fig. 7 Changes of the (A) intensity of the QMS signals during the
TPSR sequence of pulses and flux of the C3H6/CO mixture recorded
after TPD of NO experiment and (B) gas-phase IR intensity of the
signals corresponding to SCR reaction of Co–BEAwith NO (744 ppm),
C3H6 (1390 ppm), CO (1390 ppm), and O2 (3%) mixture.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online
2210 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 This journal is c the Owner Societies 2012
(Fig. 4B). At elevated temperatures such species can easily
undergo decarboxylation leading to the secondary gas-phase
by-products such as dihydrogen and methane observed by QMS
and IR, following the reactions: CH3COOH - CH4 + CO2
and H2CO - H2 + CO (Fig. 7A and B). The literature data
indicate that the decarboxylation rates at 473 K (1.81� 10�8 s�1)
and 573 K (8.17 � 10�8 s�1) are sufficiently high owing to the
low activation energy of 8.1 kcal mole�1 only.35 Both by-products
(H2 and CH4) were detected during the pulse TPSR experiments,
whereas methane was also observed during the reaction with the
SCR mixture composed of NO and C3H6/CO with O2 (Fig. 7B).
In the latter case the absence of H2 and the decrease in CO
concentration are obviously connected with their oxidation by
dioxygen, proving again that both, the gas-phase and ELO
oxygens, are involved in the SCR process.
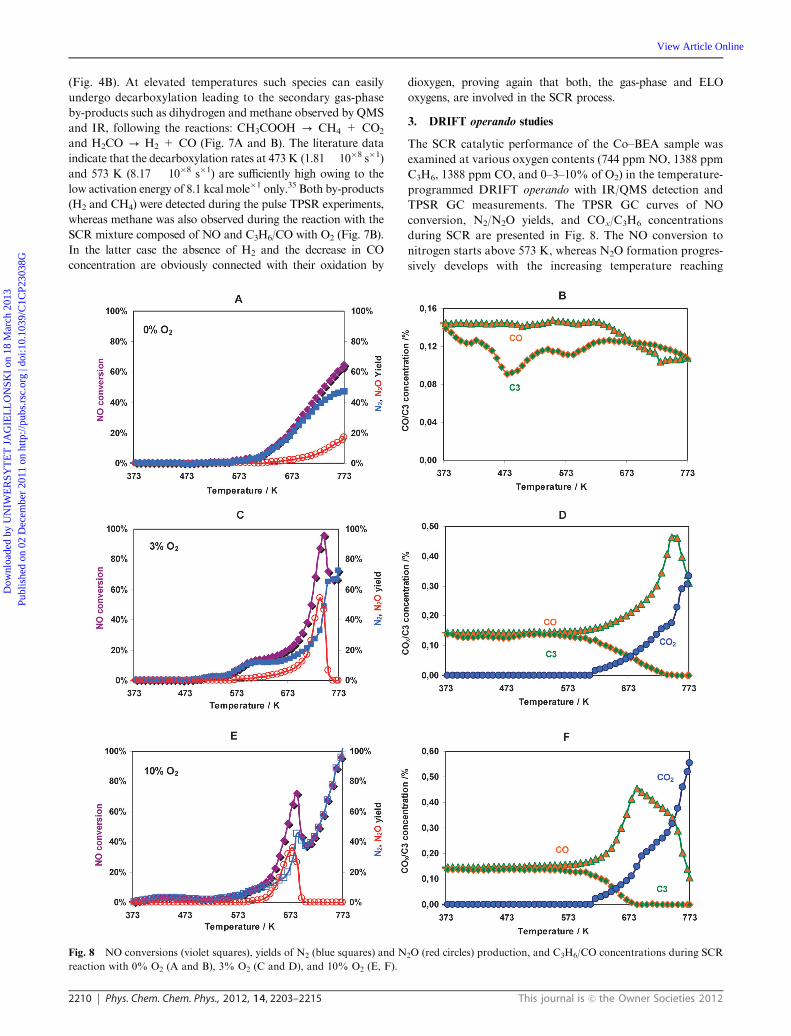
3. DRIFT operando studies
The SCR catalytic performance of the Co–BEA sample was
examined at various oxygen contents (744 ppm NO, 1388 ppm
C3H6, 1388 ppm CO, and 0–3–10% of O2) in the temperature-
programmed DRIFT operando with IR/QMS detection and
TPSR GC measurements. The TPSR GC curves of NO
conversion, N2/N2O yields, and COx/C3H6 concentrations
during SCR are presented in Fig. 8. The NO conversion to
nitrogen starts above 573 K, whereas N2O formation progres-
sively develops with the increasing temperature reaching
Fig. 8 NO conversions (violet squares), yields of N2 (blue squares) and N2O (red circles) production, and C3H6/CO concentrations during SCR
reaction with 0% O2 (A and B), 3% O2 (C and D), and 10% O2 (E, F).
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online
This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2211
the level of 20%. The NO conversion remains incomplete
(65%) at 773 K in the absence of oxygen (Fig. 8A). Addition
of O2 changes dramatically the overall SCR behavior, and
plays
a significant beneficial role for NO conversion. The N2O
production starts atB573 K and upon reaching the maximum
at 673–743 K (depending on the oxygen content) declines
rapidly, which is accompanied by the parallel increase of the
N2 yield (Fig. 8C and E). Inspection of the GC profiles
indicates that conversion into N2O vanishes with depletion
of C3H6 from the reaction mixture. Once N2O and propene
disappeared, formation of N2 was accompanied by dramatic
decline of the CO concentration. The latter was initially
produced by decomposition of the propene oxygenates (see
below), and then upon reaching the maximum content at the
zero propene level, oxidized in two different consecutive
processes inferred from two slopes shown in Fig. 8D and F.
Thus, we may conclude that at the highest temperatures CO is
the main reductant. The lack of C3H6 in the feed triggers
reduction of the surface sites by CO via oxygen transfer
resulting in pronounced CO2 formation, as it can be deduced
from the increased slope of the corresponding concentration
profile and simultaneous sharp decline of the
CO profile (Fig. 8D and F). The amount of oxygen available
during SCR shifts the local maximum of NO conversion to
desired N2 by controlling the concentration of the C3H6
reductor and its oxygenated derivatives. Higher the O2 level
faster the oxidation of C3H6 and enhanced conversion to N2 at
the high temperature stage.
3.1. SCR in the absence of O2. In the case of reduction of
NO with a C3H6/CO mixture without oxygen predominant
formation of N2O is related to the already observed decom-
position of the cobalt(II) dinitrosyl adducts, and production of
ELO species (eqn (5)). The latter are important in the high-
temperature stage of the reaction (above 633 K) where they are
responsible for the oxidation of CO as revealed by the decline
of the carbon monoxide content (Fig. 8B). At the highest
temperatures, above 723 K, formation of N2O by semi-
decomposition of the dinitrosyls is accelerated with respect to
SCR conversion into N2 (Fig. 8A). Formation of N2 is connected
with the interaction of the dinitrosyls with propene and CO. The
concentration of C3H6 drops already at the beginning of the
reaction mostly due to the sorption by the catalyst but the
minimum at 573 K, correlated with the onset of the N2 appear-
ance, is clearly connected with its chemical transformation.
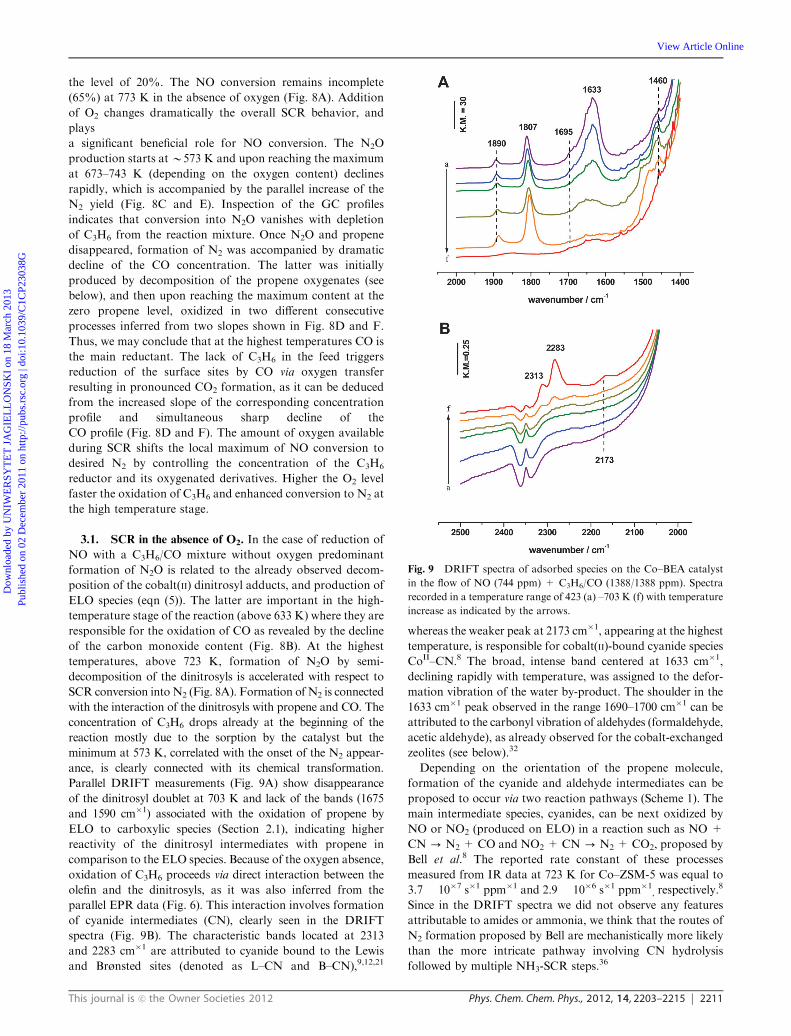
Parallel DRIFT measurements (Fig. 9A) show disappearance
of the dinitrosyl doublet at 703 K and lack of the bands (1675
and 1590 cm�1) associated with the oxidation of propene by
ELO to carboxylic species (Section 2.1), indicating higher
reactivity of the dinitrosyl intermediates with propene in
comparison to the ELO species. Because of the oxygen absence,
oxidation of C3H6 proceeds via direct interaction between the
olefin and the dinitrosyls, as it was also inferred from the
parallel EPR data (Fig. 6). This interaction involves formation
of cyanide intermediates (CN), clearly seen in the DRIFT
spectra (Fig. 9B). The characteristic bands located at 2313
and 2283 cm�1 are attributed to cyanide bound to the Lewis
and Brønsted sites (denoted as L–CN and B–CN),9,12,21
whereas the weaker peak at 2173 cm�1, appearing at the highest
temperature, is responsible for cobalt(II)-bound cyanide species
CoII–CN.8 The broad, intense band centered at 1633 cm�1,
declining rapidly with temperature, was assigned to the defor-
mation vibration of the water by-product. The shoulder in the
1633 cm�1 peak observed in the range 1690–1700 cm�1 can be
attributed to the carbonyl vibration of aldehydes (formaldehyde,
acetic aldehyde), as already observed for the cobalt-exchanged
zeolites (see below).32
Depending on the orientation of the propene molecule,
formation of the cyanide and aldehyde intermediates can be
proposed to occur via two reaction pathways (Scheme 1). The
main intermediate species, cyanides, can be next oxidized by
NO or NO2 (produced on ELO) in a reaction such as NO +
CN- N2 + CO and NO2 + CN - N2 + CO2, proposed by
Bell et al.8 The reported rate constant of these processes
measured from IR data at 723 K for Co–ZSM-5 was equal to
3.7 � 10�7 s�1 ppm�1 and 2.9 � 10�6 s�1 ppm�1, respectively.8
Since in the DRIFT spectra we did not observe any features
attributable to amides or ammonia, we think that the routes of
N2 formation proposed by Bell are mechanistically more likely
than the more intricate pathway involving CN hydrolysis
followed by multiple NH3-SCR steps.36
Fig. 9 DRIFT spectra of adsorbed species on the Co–BEA catalyst
in the flow of NO (744 ppm) + C3H6/CO (1388/1388 ppm). Spectra
recorded in a temperature range of 423 (a) –703 K (f) with temperature
increase as indicated by the arrows.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online

2212 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 This journal is c the Owner Societies 2012
Summarizing, in the absence of oxygen the reaction starts
with formation of the key cobalt(II) dinitrosyl intermediates.
They can be semi-decomposed into N2O and ELO or they can
attack the double bond of the C3H6 reductant to produce
aldehyde and cyanide. The latter reduces NO/NO2 to dinitrogen,
whereas the ELO species are depleted by CO at elevated
temperatures restoring the active sites. Methane and hydrogen
indicated in Scheme 1, detected in the TPSR and partial
reaction experiments (Section 2.4), are diagnostic products
for thermal decomposition of CH3CHO and H2CO.
3.2. SCR in the presence of O2.OperandoDRIFT spectra of
Co–BEA after exposure to NO (744 ppm), C3H6 (1390 ppm),
CO (1390 ppm), and 3% O2 mixture (in He balance) were
collected at temperatures increasing from 423 K up to 703 K,
and then the reaction was monitored under the steady-state
conditions. The spectra in the range characteristic of the
nitrosyls and the products of their oxidation, nitrites and
nitrates (2000–1400 cm�1), are shown in Fig. 10A. At the
temperatures up to 540 K, the growing bands at 1807 and
1890 cm�1 of the CoII(NO)2 intermediates were observed.
Under these conditions, the band due to NO–CoIII–ELO was
not detected. At the same time the signals located at 1575, 1500,
and 1464 cm�1, indicating formation of the surface nitrates and
nitrites, were observed. Part of this spectral region was obscured
by the 1633 cm�1 bending band of water, and as expected, its
intensity declined with temperature disappearing at 550 K (see
Section 3.1). At higher temperatures a new, relatively broad,
intense signal centered at 1668 cm�1 developed, and its presence
is associated with the surface oxygenates of propene, such as
acetates and formates (see the reference spectrum in Fig. 4). In
the earlier studies of nitromethane decomposition over
Co–ZSM-5,10,37,38 the band at 1665 cm�1 has been assigned
to the species responsible for catalyst’s deactivation such as
s-triazine and melamine.37 This peak has also been assigned to
oxime intermediates by Beutel et al.39 but involvement of such
odd species leads to an excessive complexity of the postulated
reaction mechanism and is in variance with recent spectroscopic
results.36
In the spectral range of 2150–2350 cm�1 characteristic of
NQCO and CRN stretching vibrations,30 operando DRIFT
spectra exhibited well developed multicomponent bands
shown in Fig. 10B. A distinct peak observed at 2173 cm�1 is
characteristic of cyanide CoII–CN species.8 It has been found
previously that during NO reduction with CH4 they were
the most abundant N-containing intermediates.8 This peak
was associated with the additional bands at the positions 2314
and 2287 cm�1, assigned to the cyanides bound to the Lewis
and Brønsted sites, respectively.9,12,21 The cyanides attached to
the acidic sites appeared to be more stable and less reactive
than those attached to cobalt, since their diagnostic bands
were still present under the steady-state conditions at 703 K
(Fig. 10B). This observation is in agreement with the recent
Scheme 1
Fig. 10 DRIFT spectra in the steady state of adsorbed species on
CoBEA catalyst in the flow of NO (744 ppm), C3H6/CO (1388/1388 ppm),
and O2 (3%). Spectra recorded in a temperature range of 423 K
(a)–703 K (g) as indicated by the arrows.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2213
kinetic IR studies showing that the consumption rate of the
cyanide intermediates during CH4-SCR follows the order of
k(CoII–CN) 4 k(L–CN) 4 k(B–CN).21 In addition, the rate of
the reaction of CoII–CN with NO2 has been found to be an
order of magnitude higher than the reaction rate of NO with
O2.8 For heavier hydrocarbons, like propene, isocyanate species
were also observed during SCR, and their two characteristic
bands at 2262 cm�1 and 2239 cm�1 can be seen easily in
Fig. 10B. The first one was attributed to isocyanates bound to
the Lewis sites (L–NCO).8,12,37,40 The second one was situated
in the spectral range 2235–2190 cm�1 that can be connected with
the presence of the cobalt-bound isocyanates CoII–NCO.12,37,41,42
The position of this band is very sensitive to the valence state of
the metal, as it has been shown for Cu–ZSM-5 zeolites,40 for
which reduction of the metal site leads to the pronounced (up
to 50 cm�1) blue-shift. Typical positions of the CoII–NCO
band are around 2200 cm�1,12,37 which in comparison to
2235 cm�1 found in this study can indicate reduction of the
cobalt sites in agreement with EPR experiments (see above).
The intensities of the cyanide and isocyanate bands showed
interesting evolution with the temperature ramp. Already at
423 K, at the beginning of the SCR reaction both intermediates,
CoII–CN and L–NCO, appeared. However, as the temperature
increased, the band centered at 2173 cm�1 (CoII–CN) evolved
passing through maximum intensity at 515 K to disappear
completely at 580 K, which is a typical behavior for the reaction
intermediates. At the same time the bands at 2262, 2287,
and 2239 cm�1 diagnostic for surface isocyanates developed,
showing the maximum intensity at about 673 K, and then
slightly declined.
Because stabilization of the –CN and –NCO groups involved
the acid sites of both Lewis and Brønsted nature, the corres-
ponding spectral changes in the region characteristic of –OH
groups were also investigated (Fig. 10C). Initial stages of the
SCR reaction led to the consumption of the OH groups
attached to extra-framework aluminium (3667 cm�1), which
next reacted with the cyanides and isocyanates giving rise to
the L–CN and L–NCO species (compare Fig. 10B). At 700 K
the free acidic Si–OH–Al groups (3599 cm�1) were partially
restored (Fig. 10C). Their appearance was additionally accom-
panied by the acid sites interacting with propene, as indicated
by the strongly red-shifted band centered at 3380 cm�1. The
C–H stretching vibrations of the propene admolecules can be
seen in the range 3000–2855 cm�1 (Fig. 10C). The broad band
located at 3225 cm�1, observed at the lowest temperatures
(i.e. 455 K), was attributed to the HNCO bonded with water
molecules43 inside the channels of the BEA zeolite.
The SCR process in the presence of oxygen is featured by the
simultaneous presence of the cobalt dinitrosyls and the nitrite
and nitrate surface species (Fig. 10A), which are the central
intermediates responsible for the initial oxidation of propene.
As shown in Scheme 2B, this process leads to the scission of
the CQC double bond and formation of CH3COOH and
CH3CHO oxygenates together with surface cyanide and iso-
cyanate species. The latter can be oxidized with O2, NO2, and
NO to N2, N2O, CO2, and CO (Scheme 2C). The oxygenates,
such as acetic or oxalic acids, interacting with the nitrates
and nitrites are the principal intermediates of the medium
temperature range of SCR (Scheme 2D) associated with the
local maximum of N2 yield (Fig. 8C and E). Parallel increase
Scheme 2
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online

2214 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 This journal is c the Owner Societies 2012
of CO and CO2 observed in Fig. 8D and F can be associated
with the thermal decomposition of oxalic acid produced
according to Scheme 2D. The generated carbon monoxide is
used in the last stage of the SCR reaction as a terminal
reductant depleting residual nitrates, nitrites, and ELO oxygen
from the cobalt sites (Scheme 2E), which is illustrated by the
sudden increase of CO2 production at the expense of CO with
concomitant increase of the N2 yield (Fig. 8). The EPR spectra
shown in Fig. 6 provide direct evidence for CO generation in
the course of SCR and its coordination to the cobalt active
sites. The SCR pathways described above compete with two
non-selective routes (shown in Scheme 2F) of combustion of
the propene oxygenates and their thermal decomposition into
methane, CO, and CO2 fragments (detected by QMS/IR
techniques, Section 2.4).
As a result we may distinguish five principal steps of the
propene NO SCR process. (1) The key step initiating the whole
reaction is the formation of the dinitrosyl complexes by
selective capture of NO from the reaction mixture by divalent
cobalt centers. (2) The next step consists of the formation of
the carbon–nitrogen bond in a form of the CN and NCO
intermediates, and concomitant formation of the propene
oxygenates. (3) The CN and NCO species can be oxidized
by NO, NO2 (produced on polynuclear oxo-cobalt sites) or by
dioxygen to N2. (4) An alternative pathway of N2 formation,
dominant in the medium temperature range, consists of
reduction of the nitrates and nitrites by the oxygenates.
(5) At the highest temperatures reduction of nitrogen oxides,
residual NOx�, and Co–ELO sites involves CO as the principal
reductor.
IV. Conclusions
By means of combined use of IR, EPR, and operando DRIFT
spectroscopies and catalytic tests principal steps of the catalytic
selective reduction of NOx by propene over the Co–BEA
catalyst were explored in detail. Model studies of the inter-
actions of the cobalt active sites with individual reactants and
their mixtures provided spectroscopic signatures for the reaction
intermediates and identification of the main by-products and
final products. Static and operando measurements allowed for
establishing the principal mechanistic events of the SCR process,
triggered by the two-electron Co(II)/Co(0) redox couple. The
key step of the reaction consists of the prompt chemoselective
adsorption of NO on the cobalt(II) sites initiating the catalytic
cycle. In the absence of dioxygen, the dinitrosyl route leads to
the N2O intermediate reduced next by CO to N2. It is paralleled
by the reaction of propene with Co(NO)2 leading to the cyanide
(CN) and aldehyde (CH3CHO, H2CO) intermediates. They
are produced by oxidative insertion of the bound NO to the
CQC double bond. In the presence of dioxygen, the cobalt(II)
dinitrosyls are largely oxidized to the nitrates and nitrites
already in the low temperature range. The next step involves
formation of CN and NCO intermediates, associated with the
production of the propene oxygenates (carboxylic acids and
aldehydes), and occurs in the medium temperature range. In
this catalytic route dinitrogen is produced by direct interaction
of the CN and NCO species with NO and NO2. In parallel,
dinitrogen can also be produced during the reduction of the
nitrates and nitrites by the propene oxygenates. Upon depletion
of propene at the highest temperatures, carbon monoxide
becomes the main reducing agent for the remaining nitrates
and nitrites.
Acknowledgements
P.P. is grateful to the Universite des Sciences et Technologies de
Lille for the invited professorship. Partial financial support
from the statutory funds of Faculty of Chemistry, Jagiellonian
University, is acknowledged. The work was carried out within
the framework of The International Group of Research (GDRI).
Part of the equipment used in this study was purchased thanks to
the financial support of the European Regional Development
Fund in the framework of the Polish Innovation Economy
Operational Program (contract no. POIG.02.01.00-12-023/08).
References
1 P. Pietrzyk and Z. Sojka, Chem. Commun., 2007, 1930–1932.2 P. Pietrzyk, K. Podolska and Z. Sojka, J. Phys. Chem. C, 2011,115, 13008–13015.
3 P. Pietrzyk and Z. Sojka, Stud. Surf. Sci. Catal., 2007, 171, 27–65.4 P. Da Costa, B. Moden, G. D. Meitzner, D. K. Lee and E. Iglesia,Phys. Chem. Chem. Phys., 2002, 4, 4590–4601.
5 Y. Traa, B. Burger and J. Weitkamp, Microporous MesoporousMater., 1999, 30, 3–41.
6 B. Wichterlova, Top. Catal., 2004, 28, 131–140.7 P. J. Smeets, Q. Meng, S. Corthals, H. Leeman andR. A. Schoonheydt, Appl. Catal., B, 2008, 84, 505–513.
8 L. J. Lobree, A. W. Aylor, J. A. Reimer and A. T. Bell, J. Catal.,1997, 169, 188–193.
9 C. Resini, T. Montanari, L. Nappi, G. Bagnasco, M. Turco,G. Busca, F. Bregani, M. Notaro and G. Rocchini, J. Catal.,2003, 214, 179–190.
10 S.-K. Park, H. Choo and L. Kevan, Phys. Chem. Chem. Phys.,2001, 3, 3247–3253.
11 X.-Y. Chen, S.-C. Shen, H.-H. Chen and S. Kawi, J. Catal., 2004,221, 137–147.
12 A. Shichi, T. Hattori and A. Satsuma, Appl. Catal., B, 2007, 77,92–99.
13 J. Dedecek, D. Kaucky and B. Wichterlova, MicroporousMesoporous Mater., 2000, 35–36, 483–494.
14 V. Schwartz, R. Prins, X. Wang and W. M. H. Sachtler, J. Phys.Chem. B, 2002, 106, 7210–7217.
15 T. Montanari, O. Marie, M. Daturi and G. Busca, Catal. Today,2005, 110, 339–344.
16 A. Martınez-Hernandez and G. A. Fuentes, Appl. Catal., B, 2005,57, 167–174.
17 C. Chupin, A. C. van Veen, M. Konduru, J. Despres andC. Mirodatos, J. Catal., 2006, 241, 103–114.
18 K. Gora-Marek, B. Gil and J. Datka, Appl. Catal., A, 2009, 353,117–122.
19 X. Wang, H.-Y. Chen and W. M. H. Sachtler, Appl. Catal., B,2000, 26, L227–L239.
20 R. Burch, Catal. Rev. Sci. Eng., 2004, 46, 271–333.21 X. Chen, X. Yang, A. Zhu, C. T. Au and C. Shi, J. Mol. Catal. A:
Chem., 2009, 312, 31–39.22 S. Dzwigaj, J. Janas, W. Rojek, L. Stievano, F. E. Wagner and
F. Averseng, Appl. Catal., B, 2009, 86, 45.23 T. Spalek, P. Pietrzyk and Z. Sojka, J. Chem. Inf. Model., 2005, 45,
18–29.24 K. I. Hadjiivanov, Catal. Rev. Sci. Eng., 2000, 42, 71–144.25 P. Pietrzyk, M. Srebro, M. Radon, Z. Sojka and A. Michalak,
J. Phys. Chem. A, 2011, 115, 2316–2324.26 X. Wang, H.-Y. Chen and W. M. H. Sachtler, J. Catal., 2001, 197,
281–291.27 Q. Tang, Q. Zhang, P. Wang, Y. Wang and H. Wan,
Chem. Mater., 2004, 16, 1967–1976.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online

This journal is c the Owner Societies 2012 Phys. Chem. Chem. Phys., 2012, 14, 2203–2215 2215
28 T. Montanari, O. Marie, M. Daturi and G. Busca, Catal. Today,2005, 110, 339–344.
29 B. Gil, P. Pietrzyk, J. Datka, P. Kozyra and Z. Sojka, Stud. Surf.Sci. Catal., 2005, 158, 893–900.
30 L. H. Little, Infrared spectra of adsorbed species, Academic Press,London, New York, 1966.
31 C. He and K. Kohler, Phys. Chem. Chem. Phys., 2006, 8,898–905.
32 K. Gora-Marek and J. Datka, Catal. Today, 2008, 137, 466–470.33 A. Zecchina, S. Coluccia and C. Morterra, Appl. Spectrosc. Rev.,
1985, 21, 259–310.34 N. G. Connelly, W. E. Geiger, G. A. Lane, S. J. Raven and
P. H. Rieger, J. Am. Chem. Soc., 1986, 108, 6219–6224.35 Y. K. Kharaka, W. W. Carothers and R. J. Rosenbauer, Geochim.
Cosmochim. Acta, 1983, 47, 397–402.
36 J. Long, Z. Zhang, Z. Ding, R. Ruan, Z. Li and X. Wang, J. Phys.Chem. C, 2010, 114, 15713–15727.
37 A. Satsuma, A. D. Cowan, N. W. Cant and D. L. Trimm, J. Catal.,1999, 181, 165–169.
38 S. A. Beloshapkin, E. A. Paukshtis and V. A. Sadykov, J. Mol.Catal. A: Chem., 2000, 158, 355–359.
39 T. Beutel, B. Adelman and W. M. H. Sachtler, Catal. Lett., 1996,37, 125–130.
40 F. Solymosi and T. Bansagi, J. Catal., 1995, 156, 75–84.41 S. S. Goryashenko, Y. K. Park, D. S. Kim and S.-E. Park,
Res. Chem. Intermed., 1998, 24, 933–951.42 F. Lonyi, J. Valyon, L. Gutierrez, M. A. Ulla and E. A. Lombardo,
Appl. Catal., B, 2007, 73, 1–10.43 M. S. Lowenthal, R. K. Khanna and M. H. Moore, Spectrochim.
Acta, Part A, 2002, 58, 73–78.
Dow
nloa
ded
by U
NIW
ER
SYT
ET
JA
GIE
LL
ON
SKI
on 1
8 M
arch
201
3Pu
blis
hed
on 0
2 D
ecem
ber
2011
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
1CP2
3038
G
View Article Online
Top Related
Copyright © 2022 FDOKUMEN

