







Bahasa
Halaman
Hukum


Life Sciences 76 (2004) 445–459www.elsevier.com/locate/lifescie
Interaction between the polyol pathway and non-enzymatic
glycation on aortic smooth muscle cell migration and
monocyte adhesion
Qinghong Dana, Rachel Wonga, Sookja K. Chungb, Stephen S.M. Chungb,
Karen S.L. Lama,*
aDepartment of Medicine, The University of Hong Kong, Queen Mary Hospital, 102 Pokfulam Road, Hong Kong SAR, ChinabInstitute of Molecular Biology, The University of Hong Kong, Hong Kong, China
Received 30 April 2004; accepted 10 August 2004
Abstract
We investigated for the interaction between the polyol pathway and enhanced non-enzymatic glycation, both
implicated in the pathogenesis of diabetic atherosclerosis, in the activation of aortic smooth muscle cell (SMC)
function. Mouse aortas and primary cultures of SMCs from wildtype (WT) mice and transgenic (TG) mice
expressing human aldose reductase (AR) were studied regarding changes in AR activity, and SMC gene activation,
migration and monocyte adhesion, in response to advanced glycation end-product modified BSA (AGE-BSA).
Results showed that AGE-BSA increased AR activity in both WT and TG aortas, with greater increments (p b
0.05) in TG aortas which, basally, had elevated AR activity (2.8 fold of WT). These increments were attenuated by
zopolrestat, an AR inhibitor. Similar AGE-induced increments in AR activity were observed in primary cultures of
aortic SMCs from WT and TG mice (60% and 100%, respectively, P b 0.01). Such increments were accompanied
by increases in intercellular adhesion molecule-1 (ICAM-1) and monocyte chemoattractant protein-1 (MCP-1)
mRNA levels (both P b 0.05), activation of membrane-associated PKC-h1 (P b 0.05) as well as increased SMC
migration and Tamm-Horsfall protein (THP)-1 monocyte adhesion to SMCs (both p b 0.01), with all changes
being significantly greater in TG SMCs (P b 0.05) and suppressible by either zopolrestat or transfection with an
AR antisense oligonucleotide. Our findings suggest that the effects of AGEs on SMC activation, migration and
monocyte adhesion are mediated partly through the polyol pathway and, possibly, PKC activation. The greater
0024-3205/$ -
doi:10.1016/j.l
* Correspo
E-mail add
see front matter D 2004 Elsevier Inc. All rights reserved.
fs.2004.09.010
nding author. Tel.: +86 852 2855 4769; fax: +86 852 2816 2187.
ress: [email protected] (K.S.L. Lam).

Q. Dan et al. / Life Sciences 76 (2004) 445–459446
AGE-induced changes in the TG SMCs have provided further support for the dependency of such changes on
polyol pathway hyperactivity.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Aldose reductase gene; Advanced glycation end-products (AGEs); Diabetic atherosclerosis; Transgenic mouse
Introduction
Diabetes mellitus is associated with accelerated atherosclerosis, leading to the increased incidence of
coronary artery disease and stroke (Pyorala et al., 1997). The mechanisms underlying the initiation and
progression of the atherosclerotic lesions are probably complex and involve multiple pathways. While
conventional cardiovascular risk factors, such as hypertension, hyperlipidemia and obesity are known to
be more prevalent in diabetic patients, recent interventional studies have provided compelling evidence
that hyperglycemia itself also plays an important contributory role in both type 1 and type 2 diabetes
(Nathan et al., 2003; Stratton et al., 2000). Various biochemical abnormalities induced by hyper-
glycemia, such as activation of the polyol pathway, enhanced non-enzymatic glycation with formation of
advanced glycation end-products (AGEs), and altered protein kinase C (PKC) activities have been
implicated in the pathogenesis of diabetic microangiopathic complications. Recent evidence suggests
that they are also involved in the pathogenesis of diabetic atherosclerosis (Massi-Benedetti and Federici,
1999; Kasuya et al., 1999; Ruef et al., 2000; Cerami et al., 1985; Bucala and Vlassara, 1995; Berg et al.,
1997; Ross, 1999; Higashi et al., 1997; Renier et al., 2003).
The aldose reductase (AR) gene, coding for the first and rate-limiting enzyme in the polyol pathway,
has been implicated in the etiology of diabetic atherosclerosis. Inhibition of AR prevents intimal
thickening in coronary arteries of galactose-fed beagle dogs (Kasuya et al., 1999), and mitogen-induced
DNA synthesis and cell proliferation in vascular smooth muscle cells (SMCs) (Ruef et al., 2000; Ramana
et al., 2002). Mitogen stimulation of cultured human vascular SMCs is associated with upregulation of
AR expression, which is also observed in the neointima of balloon-injured rat carotid arteries (Ruef et
al., 2000). These findings suggest that the activation of AR, known to be present in patients with
diabetes, can potentially contribute to the development of diabetic atherosclerosis.
Increased non-enzymatic glycation has also been proposed to contribute to the development of
vascular complications in diabetes (Cerami et al., 1985). AGE deposits are found in atherosclerotic
plaques and myocardium of patients with diabetes (Bucala and Vlassara, 1995), and increased
circulating plasma AGEs is associated with heart stiffness in patients with type 1 diabetes (Berg et al.,
1997). SMC migration from the media to the intima, and the adhesion of circulating monocytes to
endothelial cells and SMCs are thought to be early events in the development of atherosclerosis (Ross,
1993). Incubation of AGE-BSA results in increased rabbit aortic SMC migration (Higashi et al., 1997),
and increased monocyte adhesion to bovine endothelium (Renier et al., 2003), supporting a role of AGEs
in the early stages of diabetic atherosclerosis.
There is evidence that the various biochemical pathways, activated in the presence of hyperglycemia,
may interact in potentiating the hyperglycemia-induced tissue damage. The inhibition of AR has been
shown to prevent the activation of PKC induced by high glucose and growth factors (Keogh et al., 1997;
Ishii et al., 1998; Ramana et al., 2003a). In addition to its interaction with the PKC pathway, the polyol
pathway may also promote diabetic complications through the increased production of AGEs. In both

Q. Dan et al. / Life Sciences 76 (2004) 445–459 447
diabetic rats and humans, AR inhibitors have been reported to reduce the formation of AGEs (Suarez et al.,
1988; Hamada et al., 2000; Nakamura et al., 2003). On the other hand, AGE-BSA has been shown to
increase the expression of the AR gene in cultured humanmicrovascular endothelial cells (Nakamura et al.,
2000). More recently, it was reported that methylglyoxal, a precursor of AGEs, could induce AR mRNA,
protein and activity in rat aortic SMCs (Chang et al., 2002), suggesting another mechanism whereby the
polyol pathway and non-enzymatic glycation may interact in promoting diabetic atherosclerosis.
In the present study, we investigated whether AGE-BSA could also up-regulate the expression of AR
in aortic SMCs, using aortas and primary cultures of SMCs from wildtype and human AR (hAR)
transgenic mice. The effects of interaction of the two pathways on the gene expression of intercellular
adhesion molecule-1 (ICAM-1) and monocyte chemoattractant protein-1 (MCP-1), PKC-h1 activation,
as well as SMC migration and THP-1 monocyte adhesion, were also examined.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), phosphate buffer saline (PBS), penicillin/streptomycin
solution, trypin, fetal bovine serum (FBS), bovine serum albumin (BSA, fraction V, very low endotoxin),
Trizol Reagent, random primer labeling kit and Lipofectin AMINE Plus were purchased from GIBCO
BRL Life Technologies (Gaithersburg, MD, USA). Zopolrestat (Zop) was obtained from Pfizer (Groton,
CT, USA). Mouse monoclonal anti-PKC-h1 antibody, horseradish peroxidase-conjugated anti-rabbit andanti-mouse IgG antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, Calif., USA).
Tamm-Horsfall protein (THP)-1 cells, a human monocyte line, were purchased from the American Type
Culture Collection (ATCC, Rockville, MD, USA). Phosphorothioate AR antisense oligonucleotide (5V-CCTGGGCGCAGTCAATGTGG-3V) andmismatched control (Scrambled) oligonucleotide (5V-GGTGA-TAGCTGACGCGGTCC-3V) from Invitrogen (Gaithersburg, MD, USA). Other reagents used in Western
blot analysis and AR activity were obtained from Sigma Chemical (St. Louis, MO, USA).
TG mice expressing hAR gene in SMCs
TG mice were generated using modifications of a protocol previously used for generating TG mice
expressing hAR in the lens (Lee et al., 1995), another of our genetically manipulated mice for studying
the pathogenesis of diabetic complications. The entire 1.4 kb hAR cDNA was inserted into the pAL1
vector (Wu et al., 1994) which contains human scavenger receptor regulatory elements. A DNA
fragment containing the human scavenger receptor-hAR hybrid gene was microinjected into oocytes
from CBA egg donors fertilized by C57BL males. Mice carrying the hAR transgene were identified by
PCR amplification of tail DNA and further confirmed by Southern blot analyses. It was anticipated that
the transgene will be expressed in various cell types which normally express the scavenger receptor, such
macrophages, renal epithelial and mesangial cells, fibroblasts and SMCs, which are known to express
scavenger receptors (Pitas, 1990; Pitas et al., 1992). PCR was performed using primers specific for hAR
(Yamaoka et al., 1995). All procedures involving animals were carried out in compliance with the Guide
for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH
Publication No. 85-23, revised 1996).

Q. Dan et al. / Life Sciences 76 (2004) 445–459448
Preparation of AGE-modified bovine serum albumin (BSA)
AGE-BSAwas prepared by incubating 20% BSA and 1.67 M glucose in 0.5 M phosphate buffer (PH:
7.4) for 12 weeks at 37 8C under sterile conditions. Unincorporated sugar was removed by dialysis
against PBS. Control non-glycated BSA was obtained by incubation of the same solution in the same
conditions without glucose. AGE formation was confirmed by measurement of fluorescence at 440nm of
wavelength. Contamination of BSA by endotoxin was checked using a commercial kit for endotoxin.
(Limulus J Single Test; Wako, Osaka, Japan). Both non-glycated BSA and AGE-BSA used in this study
contained b 0.2 ng/ml of endotoxin.
Isolation of mouse thoracic aortas and primary culture of SMCs
WT and TG mice thoracic aortas and aortic SMCs were isolated by an explant method as previously
described (Yasuda et al., 2000). SMCs were maintained in DMEM containing 10% FBS supplemented
with 100 U/ml penicillin and 100 Ag/ml streptomycin. SMCs were identified by their typical bhill-and-valleyQ growth pattern, and by positive fluorescence with antibodies against a-smooth muscle actin but
no fluorescence with antibodies against factor VIII antigen and cytokeratin.
Ex vivo study
Ex vivo study was conducted as previously described (Ramana et al., 2003b). The thoracic aorta
dissected from WT and TG mice was cut into six 5 mm strips. Aortic strips with 5 to 6 animals were
pooled and divided into groups with 6 random pieces in each group. The aortic strips were incubated in
DMEM supplemented with 10% FBS containing 100 Ag/ml of AGE-BSA or control BSA for 24 hours,
in the presence or absence of 10 AM of Zop. After treatment, the samples were washed with ice-cold
PBS and homogenized in 1 ml of 0.1 M phosphate (PH: 7.4) containing proteinase inhibitor cocktail. AR
activity was measured as described below.
AR activity measurement
Tissues or cells were homogenized in 1 ml of 0.1 M phosphate (PH: 7.4) containing protease cocktail.
AR activity was measured using glyceraldehyde as substrate as described previously (Nishinaka and
Yabe-Nishimura, 2001).
RNA extraction, Northern blot analysis and reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from isolated aortas and aortic SMCs with Trizol Reagent according to the
manufacturer’s instructions. Heat-denatured RNAwas transferred onto Hybond-N nylon membrane. hAR
cDNA against hAR exon 10 was labelled with [a-32P]-dCTP using a random primer labeling kit. For RT-
PCR, 2 Ag of each total RNA sample was converted into cDNA with Superscript II RNase H reverse
transcriptase (RT) and random hexamer primers according to the manufacturer’s instructions. Equal
amounts of cDNAwere amplified by PCR. The sense and antisense primers for the amplification of each
fragment were as follows: mouse endogenous AR (mAR): sense: 5V-CCCAGGTGTACCAGAATGAGA-3V; antisense: TGGCTGCAATTGCTTTGATCC-3V; expected 580 bp; hAR: sense: 5V-GCCGTAT-

Q. Dan et al. / Life Sciences 76 (2004) 445–459 449
CCTGCTCAACAAC-3V; antisense: 5V-ACC ACAGCCTCAAAACTCTTC-3V; expected: 252 bp.
ICAM-1: sense: 5V-GGAGCAAGACTGTGAACACG-3V; antisense: 5V-GAGAACCACTGCTAGTC-CAC-3V; expected: 435 bp; MCP-1: sense:5V-ACTGAAGCCAGCTCTCTCTTCCTC-3V; antisense: 5V-TTCCTTCTTGGGGTCAGCACAGAC-3V; expected: 274 bp; GAPDH: sense:5V-TGATGACATCAA-GAAGGTGGTGAAG-3V, antisense:5V-CCTTGGAGGCCATGTAGGCCAT-3V, expected 239 bp. PCR
conditionswere as follows: 94 8C for 1min, 57 8C for 1min, and 72 8C for 1min. A final extension of 72 8Cfor 10 min also was included. 28 cycles were performed for ICAM-1, MCP-1 and GAPDH, and 35 cycles
for hAR and mAR. Primers for GAPDH were used as the internal standard.
Immunoblot for hAR
Western blot was performed as previously described (Nakamura et al., 2001). SMCs were lysed in lyses
buffer. Equal amount of proteins were subjected to 10% SDS-polyacrylamide gel electrophoresis and
transferred onto polyvinylidene diffuoride (PVDF) membranes. Membranes were blocked overnight with
ovalbumin and reacted with anti-hAR polyclonal antibody diluted to 1:500 overnight at 4 8C and then
incubated with horseradish peroxidase-conjugated anti-rabbit antibodies diluted to 1:5000 for 2 h at room
temperature. Immunoreactive bands were detected using DAB peroxidase substrate developing reagent.
Cell fractionation and PKC-b1 immunoblots
PKC activation was determined by the method of Nakamura et al (Nakamura et al., 2001). Briefly,
SMCs were washed, resuspended in buffer, sonicated, and homogenates were ultra-centrifuged to isolate
the plasma membrane fractions. Equal amounts of membrane protein levels in each sample were loaded
and separated by SDS-PAGE under reducing conditions, and the membrane PKC isoform of PKC-h1were visualized by Western blot using specific monoclonal antibody against PKC-h1.
SMC migration and monocyte adhesion assays
The rate of migration of SMCs was determined using a scrape/wound assay as described previously
(Hamuro et al., 2002). Briefly, SMCs from WT and TG mice were grown in 35 mm dishes until
subconfluent. SMCs monolayer was rinsed with PBS and disrupted with a sterile rubber policeman to
create a cell-free zone. The cells were washed and treated with AGE-BSA (100 Ag/ml) in the presence or
absensc of Zop (10 AM) for 24 hours. In some experiments, instead of the addition of Zop, SMCs were
transfected with AR antisense or AR scrambled oligonucleotides (1.0 AM) followed by disruption. At the
end of the incubation, the cells were washed with PBS, fixed in methnol and stained with Giema stain
(Sigma). Stained cells were examined on an inverted microscope. SMC migration was quantified by
measuring the number of migrating cells that crossed the 2.0-mm length of wounded borderline into
denuded area. Measurements were performed at three different areas per dish.
Adhesion of THP-1 monocytic cell to SMCs was assessed as previously detailed (Lynn et al., 2000).
SMCs from WT and TG mice were cultured 24 hours in 35 mm dishes. After treatment, an aliquot of
THP-1 cells (2 � 105) was added to each dish, and cells were further incubated for 1 hour. After 3
washes with complete medium, cells were fixed with 1% glutaraldehyde. The number of attached THP-1
was determined by counting 10 random fields per dish under light microscopy (�100 magnification).
Co-cultured SMCs and THP-1 cells were identified by morphology (large, spindle shaped and small,

Q. Dan et al. / Life Sciences 76 (2004) 445–459450
spherical, respectively), as well as by staining with hematoxylin. Hematoxylin-stained nuclei of SMCs
were light blue, while THP-1 nuclei were dark blue/black.
Transfection with antisense oligonucleotides (Ramana et al., 2003a)
WT and TG MCs grown to 60–70% confluency in DMEM were washed with serum-free DMEM
medium 3 times, 60 minutes before transfection. The cells were incubated with 1.0 AM AR antisense or
scrambled control oligonucleotides using Lipofect AMINE plus (15 Ag/ml) as the transfection reagent as
suggested by the supplier. After 12 hours, the medium was replaced with fresh DMEM (containing 10%
FBS) for another 12 hours followed by 24 hours of incubation in serum-free DMEM before stimulation
by AGE-BSA. The effect of AR ablation on AGE-BSA induced MCP-1 and ICAM-1 mRNA
expression, SMC migration and THP-1 monocyte adhesion to SMCs were assessed by incubating the
transfected cells with AGE-BSA (100 Ag/ml) for 24 hours.
Statistical analysis
Data are expressed as mean F SEM. Student’s t test or analysis of variance (ANOVA) in conjunction
with the Newman-Keuls test was employed as applicable. Differences between groups were considered
statistically significant when p b 0.05.
Results
AR expression in WT and TG aorta and aortic SMCs
Northern blot analysis employing a cDNA probe directed against exon 10 of hAR, specific for
the hAR gene, demonstrated the presence of hAR mRNA in the aortas and aortic SMCs of TG
mice, but not in that of WT mice (Fig. 1A). The protein expression of hAR gene products in
Fig. 1. A, B. hAR expression in aorta strips and aortic SMCs of transgenic mice. (A). Northern blot analysis showing a distinct
1.4 kb band representing the human AR transcript only in TG aortas and TG aortic SMCs. (B). Western blot analysis showing
the human AR gene product with an approximate molecular weight of 39 kDa in TG SMCs.
Q. Dan et al. / Life Sciences 76 (2004) 445–459 451
primary cultures of TG mice aortic SMCs was also confirmed using anti-hAR polyclonal antibody
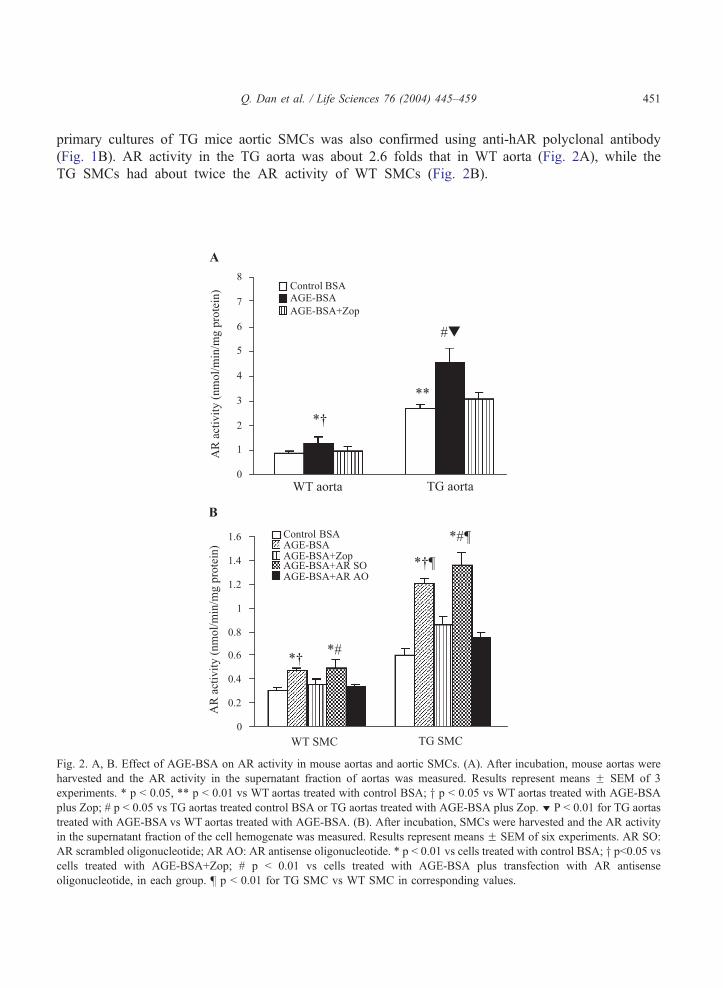
(Fig. 1B). AR activity in the TG aorta was about 2.6 folds that in WT aorta (Fig. 2A), while the
TG SMCs had about twice the AR activity of WT SMCs (Fig. 2B).
Fig. 2. A, B. Effect of AGE-BSA on AR activity in mouse aortas and aortic SMCs. (A). After incubation, mouse aortas were
harvested and the AR activity in the supernatant fraction of aortas was measured. Results represent means F SEM of 3
experiments. * p b 0.05, ** p b 0.01 vs WT aortas treated with control BSA; y p b 0.05 vs WT aortas treated with AGE-BSA
plus Zop; # p b 0.05 vs TG aortas treated control BSA or TG aortas treated with AGE-BSA plus Zop. z P b 0.01 for TG aortas
treated with AGE-BSA vs WT aortas treated with AGE-BSA. (B). After incubation, SMCs were harvested and the AR activity
in the supernatant fraction of the cell hemogenate was measured. Results represent means F SEM of six experiments. AR SO:
AR scrambled oligonucleotide; AR AO: AR antisense oligonucleotide. * p b 0.01 vs cells treated with control BSA; y pb0.05 vscells treated with AGE-BSA+Zop; # p b 0.01 vs cells treated with AGE-BSA plus transfection with AR antisense
oligonucleotide, in each group. b p b 0.01 for TG SMC vs WT SMC in corresponding values.
Q. Dan et al. / Life Sciences 76 (2004) 445–459452
Effect of AGE-BSA on AR activity in mice aortas and aortic SMCs
After exposure to 100 Ag/ml of AGE-BSA for 24 hours, significant increases in AR activity
were observed in the mice aortas (p b 0.05) and SMCs (p b 0.01). The increment was signi-
ficantly greater in TG aortas and SMCs compared to their WT counterparts (p b 0.01). To further
elucidate the role of AR, we examined the effect of an AR specific inhibitor, zopolrestat (Zop), on
AGE-stimulated AR activity. Our preliminary data showed that Zop at concentration of 1 AM, 10
AM and 100 AM inhibited AR activity by 37%, 76% and 88%, respectively in WT SMCs.
However, cell toxicity was observed when 100 AM Zop was used. The increment in AR activity in
response to AGE-BSA was largely abolished by 10 AM of Zop in both WT and TG aortas and
SMCs (Fig. 2).
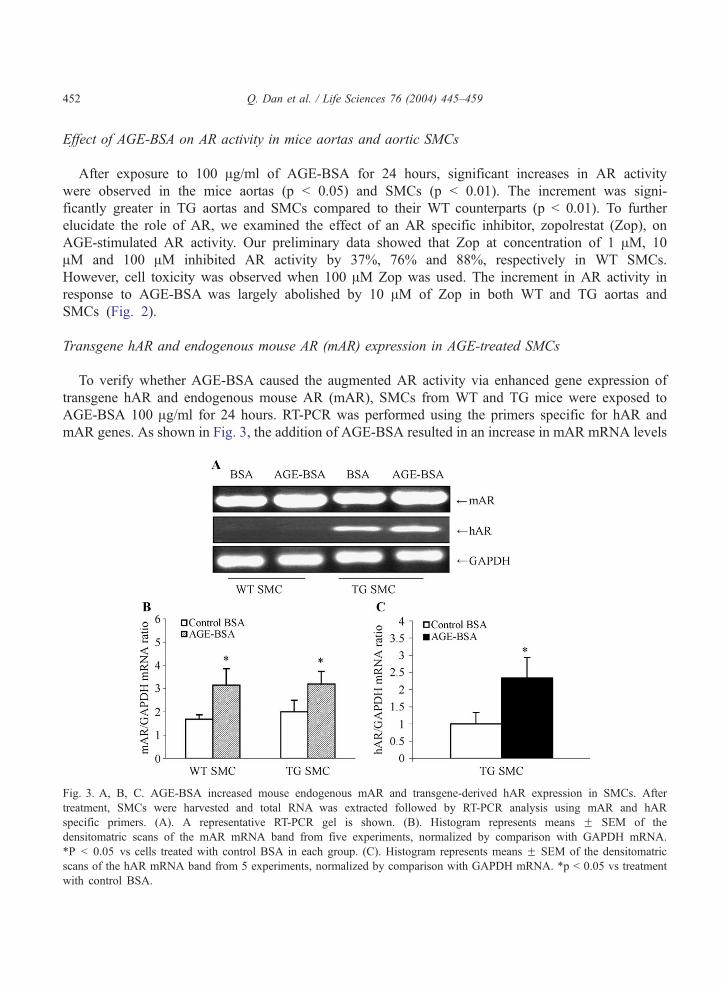
Transgene hAR and endogenous mouse AR (mAR) expression in AGE-treated SMCs
To verify whether AGE-BSA caused the augmented AR activity via enhanced gene expression of
transgene hAR and endogenous mouse AR (mAR), SMCs from WT and TG mice were exposed to
AGE-BSA 100 Ag/ml for 24 hours. RT-PCR was performed using the primers specific for hAR and
mAR genes. As shown in Fig. 3, the addition of AGE-BSA resulted in an increase in mAR mRNA levels
Fig. 3. A, B, C. AGE-BSA increased mouse endogenous mAR and transgene-derived hAR expression in SMCs. After
treatment, SMCs were harvested and total RNA was extracted followed by RT-PCR analysis using mAR and hAR
specific primers. (A). A representative RT-PCR gel is shown. (B). Histogram represents means F SEM of the
densitomatric scans of the mAR mRNA band from five experiments, normalized by comparison with GAPDH mRNA.
*P b 0.05 vs cells treated with control BSA in each group. (C). Histogram represents means F SEM of the densitomatric
scans of the hAR mRNA band from 5 experiments, normalized by comparison with GAPDH mRNA. *p b 0.05 vs treatment
with control BSA.

Q. Dan et al. / Life Sciences 76 (2004) 445–459 453
in both WT and TG SMCs (p b 0.05). hAR mRNA levels were also significantly increased (by 2.2 fold)
in TG SMCs (p b 0.05; Fig. 3). As expected, no hAR mRNA could be demonstrated in WT SMCs,
before or after AGE-BSA treatment (Fig. 3A).
MCP-1 and ICAM-1 mRNA expression in AGE-treated SMCs
We next examined the effect of AGE-BSA on the mRNA levels of MCP-1 and ICAM-1 in SMCs.
Exposure of SMCs to 100 Ag/ml of AGE-BSA for 24 hours resulted in increased MCP-1 and ICAM-1
mRNA expression (both p b 0.05), with a greater increment being observed in TG SMCs (p b 0.05
versus WT SMCs; Fig. 4). The AR inhibitor, Zop (10 AM) attenuated the AGE-BSA induced MCP-1
and ICAM-1 mRNA increments in both WT and TG SMCs.
Fig. 4. A, B, C. Expression of MCP-1 and ICAM-1 mRNA in AGE-treated SMCs. After treatment, SMCs were harvested and
total RNA was extracted followed by RT-PCR analysis. AR SO: AR scrambled oligonucleotide; AR AO: AR antisense
oligonucleotide. (A). A representive gel is shown. Lanes 1–5, WT SMCs; lanes 6–10, TG SMCs. The results shown are the ratio
of the integrated absorbance of ICAM-1 (B) and MCP-1 (C) band and the corresponding GAPDH band in arbitrary units, and
are the means F SEM for 5 independent experiments. * p b 0.05 vs cells treated with control BSA; y p b 0.05 vs cells treated
with AGE-BSA+Zop; # p b 0.05 vs cells treated with AGE-BSA plus transfection with AR AO, in each group. b p b 0.05 for
TG SMC vs WT SMC in corresponding parameters.

Q. Dan et al. / Life Sciences 76 (2004) 445–459454
SMC migration and monocyte adhesion in AGE-treated SMCs
To determine whether the enhanced MCP-1 and ICAM-1 mRNA levels were associated with
significant effects on SMC migration and monocyte adhesion in response to AGE-BSA, we tested the
effects of AGE-BSA on these two parameters in SMCs. As shown in Fig. 5, AGE-BSA treatment
significantly increased SMC migration and monocyte adhesion (both p b 0.01), with TG SMCs showing
significantly greater increments in SMC migration and monocyte adhesion (p b 0.01 versus WT SMCs
for both changes). AGE-BSA-induced SMC migration and monocyte adhesion to SMCs were also
attenuated by the addition of zopolrestat.
Fig. 5. A, B. Effect of AGE-BSA on SMC migration and monocyte adhesion to SMCs. After treatment, THP-1 monocyte
adhesion (A) and SMC migration (B) were measured. AR SO: AR scrambled oligonucleotide; AR AO: AR antisense
oligonucleotide. * p b 0.01 vs cells treated with control BSA; y p b 0.01 vs cells treated with AGE-BSA + Zop; # p b 0.01 vs
cells treated with AGE-BSA plus transfection with AR AO, in each group. b p b 0.01 for TG SMC vs WT SMC, in
corresponding values.
Q. Dan et al. / Life Sciences 76 (2004) 445–459 455
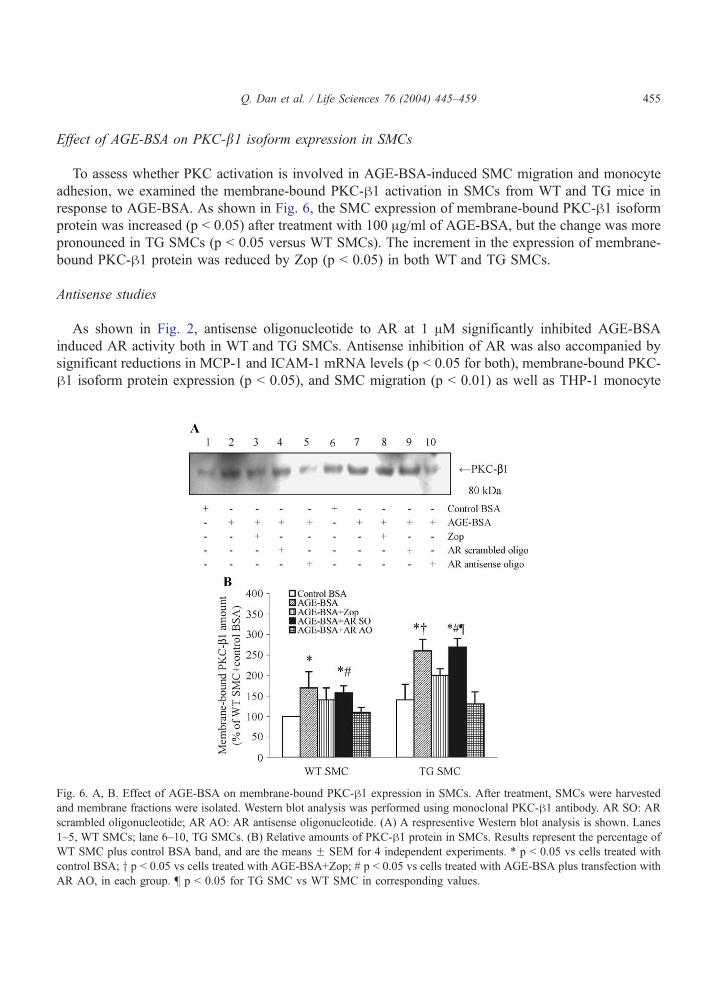
Effect of AGE-BSA on PKC-b1 isoform expression in SMCs
To assess whether PKC activation is involved in AGE-BSA-induced SMC migration and monocyte
adhesion, we examined the membrane-bound PKC-h1 activation in SMCs from WT and TG mice in
response to AGE-BSA. As shown in Fig. 6, the SMC expression of membrane-bound PKC-h1 isoform
protein was increased (p b 0.05) after treatment with 100 Ag/ml of AGE-BSA, but the change was more
pronounced in TG SMCs (p b 0.05 versus WT SMCs). The increment in the expression of membrane-
bound PKC-h1 protein was reduced by Zop (p b 0.05) in both WT and TG SMCs.
Antisense studies
As shown in Fig. 2, antisense oligonucleotide to AR at 1 AM significantly inhibited AGE-BSA
induced AR activity both in WT and TG SMCs. Antisense inhibition of AR was also accompanied by
significant reductions in MCP-1 and ICAM-1 mRNA levels (p b 0.05 for both), membrane-bound PKC-
h1 isoform protein expression (p b 0.05), and SMC migration (p b 0.01) as well as THP-1 monocyte
Fig. 6. A, B. Effect of AGE-BSA on membrane-bound PKC-h1 expression in SMCs. After treatment, SMCs were harvested
and membrane fractions were isolated. Western blot analysis was performed using monoclonal PKC-h1 antibody. AR SO: AR
scrambled oligonucleotide; AR AO: AR antisense oligonucleotide. (A) A respresentive Western blot analysis is shown. Lanes
1–5, WT SMCs; lane 6–10, TG SMCs. (B) Relative amounts of PKC-h1 protein in SMCs. Results represent the percentage of
WT SMC plus control BSA band, and are the means F SEM for 4 independent experiments. * p b 0.05 vs cells treated with
control BSA; y p b 0.05 vs cells treated with AGE-BSA+Zop; # p b 0.05 vs cells treated with AGE-BSA plus transfection with
AR AO, in each group. b p b 0.05 for TG SMC vs WT SMC in corresponding values.

Q. Dan et al. / Life Sciences 76 (2004) 445–459456
adhesion (p b 0.01), in response to AGE-BSA (Figs. 4, 5 and 6). However, sense oligonucleotide to AR
(1 AM) had no effect on the above parameters.
Discussion
This study has demonstrated, for the first time that AGEs can upregulate the gene expression of AR in
SMCs, leading to increased AR activity in cultured SMCs as well as in incubated aortic strips. In the
cultured SMCs, this increase in AR expression was accompanied by increases in membrane PKC-h1protein expression, MCP-1 and ICAM-1 gene expression, as well as SMC migration and monocyte
adhesion. The attenuation of the above AGE-induced SMC activations by zopolrestat, an AR inhibitor,
or transfection with an AR antisense oligonucleotide, suggest that the stimulatory effects of AGEs on
SMC cytokine expression and migration are mediated, at least in part, through activation of the polyol
pathway. The much greater AGE-induced enhancement of SMC cytokine expression and migration
observed in the TG SMCs, in which the elevated basal AR activity showed a further two-fold rise in the
presence of AGEs, has provided additional evidence that these AGE-induced SMC changes, commonly
seen in the early stages of atherosclerosis, are dependent on the level of activity of the polyol pathway.
Previous studies have shown that AGE-BSA can induce oxidative stress in the vascular wall (Wautier
et al., 1994) and also activate various vascular cells through stimulating the activity of the extracellular
signal-regulated kinase (ERK) pathway (Treins et al., 2001; Lander et al., 1997). It has also been
reported that activation of the ERK pathway plays a major role in the upregulation of AR expression in
rat vascular SMCs under oxidative stress (Nishinaka and Yabe-Nishimura, 2001). Similar mechanisms,
mediated through AGE-induced oxidative stress, may be responsible for the enhancement of mAR gene
expression and activity in the mouse SMCs observed in this study. Indeed, the induction of AR activity
in human vascular endothelial cells by AGE-BSA was found to be associated with the enhancement of
specific DNA binding activity for AP-1 consensus sequence in these cells (Nakamura et al., 2000). The
authors suggested that the AGE-induced increase in AR activity may be mediated through AP-1, a
transcription factor implicated in the expression of various genes in response to oxidative stress, the
binding site of which is present in the AR gene promoter (Ko et al., 1997). In this study, AGE-BSA also
significantly increased the expression of hAR mRNA in the TG SMCs, suggesting that the AGE-induced
AR hyperactivity was also mediated through mechanisms independent of the endogenous mouse AR
gene promoter. In macrophages, AGE-BSA has been shown to increase type A scavenger receptor
mRNA expression (Iwashima et al., 2000). It is possible that, in this study, AGE-BSA also increased AR
activity in the TG SMCs indirectly via stimulation of the scavenger receptor promoter linked to the hAR
transgene. Nevertheless, our observations that AGE-BSA also increased AR activity and other responses
in the WT tissues (Figs. 2–6) suggest that the AR and AGE pathways interact even in non-TG mice in
which the AR gene is not coupled to the scavenger receptor promoter.
Atherosclerosis is now recognized as an inflammatory disorder (Ross, 1999) in which the adhesion of
monocytes to the vascular endothelium and SMCs and their subsequent migration into the vessel wall
are pivotal early events in pathogenesis (Ross, 1993). Experimental evidence strongly implicates
adhesion molecules including intercellular adhesion molecule ICAM-1 and inflammatory cytokines and
chemokines including monocyte chemoattractant peptide (MCP)-1, as mediators of the subintimal
monocyte accumulation in atherosclerosis (Ross, 1993, 1999). PKC activation has been reported to
induce ICAM-1 and MCP-1 expression in endothelial cells (Vielma et al., 2003) and macrophages (Nitti

Q. Dan et al. / Life Sciences 76 (2004) 445–459 457
et al., 2002) respectively. On the other hand, it has been shown that polyol pathway hyperactivity
contributes to glucose-induced SMC proliferation in cultured rat aortic SMCs through the activation of
PKC (Nakamura et al., 2001). In the present study, membrane-bound PKC-h1 activation was observed
in mouse SMCs in response to AGE-BSA, with greater activation being found in the TG SMCs. Thus
PKC activation, consequent to enhanced activity of the polyol pathway, could represent one plausible
mechanism for the changes in ICAM-1 and MCP-1 gene expression, and subsequent SMC migration and
monocyte adhesion to SMCs. This was supported by the finding of a complete normalization of the
PKC-h1 activation in the presence of the AR antisense oligonucleotide. On the other hand, the partial
inhibition of PKC-h1 activation by Zop was probably related to the incomplete suppression of AR
activity by this AR inhibitor at the dosage used.
In conclusion, there is evidence from these ex vivo and in vitro studies of an interaction between the
polyol pathway and non-enzymatic glycation, at the level of the aortic SMCs, in mediating the early
events of diabetic atherosclerosis. These events, which include enhancement of the polyol pathway by
AGEs, activation of membrane-bound PKC-h1, induction of inflammatory mediators such as ICAM-1
and MCP-1, and stimulation of SMC migration and monocyte adhesion to SMCs, may be more
pronounced in individuals with an underlying cause of increased AR gene activation, either
environmental or genetic.
Acknowledgments
This work was supported by a grant from the Hong Kong Research Grant council (HKU7270/98M).
We thank Dr Christopher K Glass for the generous gift of the pAL1 vector containing human scavenger
receptor regulatory elements.
References
Berg, T.J., Bangsted, H.J., Torjesen, P.A., Osterby, R., Bucala, R., Hanssen, K.F., 1997. Advanced glycation end products in serum
predict changes in the kidney morphology of patients with insulin-dependent diabetes mellitus. Metabolism 46, 661–665.
Bucala, R., Vlassara, H., 1995. Advanced glycosylation end products in diabetic renal and vascular disease. American Journal
of Kidney Diseases 26, 875–888.
Cerami, A., Vlassara, H., Brownlee, M., 1985. Protein glycosylation and the pathogenesis of atherosclerosis. Metabolism 34,
37–42.
Chang, KC., Paek, H.S., Kim, H.J., Lee, Y.S., Yabe-Nishimura, Y., Seo, H.G., 2002. Substrate-induced up-regulation of aldose
reductase by methylglyoxal, a reactive oxoaldehyde elevated in diabetes. Molecular Pharmacology 61, 1184–1191.
Hamada, Y., Nakamura, J., Naruse, K., Komori, T., Kato, K., Kasuya, Y., Nagai, R., Horiuchi, S., Hotta, N., 2000. Epalrestat, an
aldose reductase inhibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in
erythrocytes from diabetic patients. Diabetes Care 23, 1539–1544.
Hamuro, M., Polan, J., Natarajan, M., Mohan, S., 2002. High glucose induced nuclear factor kappa B mediated inhibition of
endothelial cell migration. Atherosclerosis 162, 277–287.
Higashi, T., Sano, H., Saishoji, T., Ikeda, K., Jinnouchi, Y., Kanzaki, T., Morisaki, N., Rauvala, H., Shichiri, M., Horiuchi, S.,
1997. The receptor for advanced glycation end products mediates the chemotaxis of rabbit smooth muscle cells. Diabetes 46,
463–472.
Ishii, H., Tada, H., Isogai, S., 1998. An aldose reductase inhibitor prevents glucose-induced increase in transforming growth
factor-beta and protein kinase C activity in cultured mesangial cells. Diabetologia 41, 362–364.

Q. Dan et al. / Life Sciences 76 (2004) 445–459458
Iwashima, Y., Eto, M., Hata, A., Kaku, K., Horiuchi, S., Ushikubi, F., Sano, H., 2000. Advanced glycation end products-
induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. Biochemical and
Biophysical Research Communications 277, 368–380.
Kasuya, Y., Ito, M., Nakamura, J., Hamada, Y., Nakayama, M., Chaya, S., Komori, T., Naruse, K., Nakashima, E., Kato, K.,
Koh, N., Hotta, N., 1999. An aldose reductase inhibitor prevents the intimal thickening in coronary arteries of galactose-fed
beagle dogs. Diabetologia 42, 1404–1409.
Keogh, R.J., Dunlop, M.E., Larkins, R.G., 1997. Effect of inhibition of aldose reductase on glucose flux, diacylglycerol
formation, protein kinase C, and phospholipase A2 activation. Metabolism 46, 41–47.
Ko, B.C., Ruepp, B., Bohren, K.M., Gabbay, K.H., Chung, S.S., 1997. Identification and characterization of multiple osmotic
response sequences in the human aldose reductase gene. Journal of Biological Chemistry 272, 16431–16437.
Lander, H.M., Tauras, J.M., Ogiste, J., Hori, O., Moss, R.A., Schmidt, A.M., 1997. Activation of the receptor for advanced
glycation end products triggers a p21ras-dependent mitogen-activated protein kinase pathway regulated by oxidant stress.
Journal of Biological Chemistry 272, 17810–17814.
Lee, A.Y.W., Chung, S.K., Chung, S.S.M., 1995. Demonstration that polyol accumulation is responsible for diabetic cataract by
the use of transgenic mice expressing the aldose reductase gene in the lens. Proceedings of the National Academy of
Sciences of the United States of America 92, 2780–2784.
Lynn, E.G., Siow, Y.L., Karmin, O., 2000. Very low-density lipoprotein stimulates the expression of monocyte chemoattractant
protein-1 in mesangial cells. Kidney Internal 57, 1472–1483.
Massi-Benedetti, M., Federici, M.O., 1999. Cardiovascular risk factors in type 2 diabetes: the role of hyperglycaemia.
Experimental and Clinical Endocrinology & Diabetes 107 (Suppl 4), S120–S123.
Nakamura, N., Obayshi, H., Fuji, M., Fukui, M., Yoshimori, K., Ogata, M., Hasegawa, G., Shigeta, H., Kitagawa, Y.,
Yoshikawa, T., Kondo, M., Ohta, M., Nishimura, M., Nishinaka, T., Nishimura, C.Y., 2000. Induction of aldose reductase in
cultured human microvascular endothelial cells by advanced glycation end products. Free Radical Biology & Medicine 29,
17–25.
Nakamura, J., Kasuya, Y., Hamada, Y., Nakashima, E., Naruse, K., Yasuda, Y., Kato, K., Hotta, N., 2001. Glucose-induced
hyperproliferation of cultured rat aortic smooth muscle cells through polyol pathway hyperactivity. Diabetologia 44,
480–487.
Nakamura, N., Yamazaki, K., Satoh, A., Urakaze, M., Kobayashi, M., Yamabe, H., Osawa, H., Shirato, K., Sugawara, T.,
Nakamura, M., Tamaura, M., Okumua, K., 2003. Effects of eparlestat on plasma levels of advanced glycation end products
in patients with type 2 diabetes. In vivo 17, 177–180.
Nathan, D.M., Lachin, J., Cleary, P., Orchard, T., Britton, D.J., Backlund, J.Y., OTLeary, D.H., Genuth, S., 2003. Intensivediabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. New England Journal of Medicine 348,
2294–2303.
Nishinaka, T., Yabe-Nishimura, C., 2001. EGF receptor-ERK pathway is the major signaling pathway that mediates
upregulation of aldose reductase expression under oxidative stress. Free Radical Biology & Medicine 31, 205–216.
Nitti, M., Domenicotti, C., dTAbramo, C., Assereto, S., Cottalasso, D., Melloni, E., Poli, G., Bias, F., Marinari, U.M., Pronzato,
M.A., 2002. Activation of PKC-beta isoforms mediates NE-induced MCP-1 release by macrophages. Biochemical and
Biophysical Research Communications 294, 547–552.
Pitas, R.E., 1990. Expression of the acetyl low-density-lipoprotein receptor by rabbit fibroblasts and smooth-muscle cells: up-
regulation by phorbol esters. Journal of Biological Chemistry 265, 12722–12727.
Pitas, R.E., Friera, A., McGuire, J., Dejager, S., 1992. Further characterization of the acetyl-LDL (scavenger) receptor
expression by rabbit smooth muscle cells and fibroblasts. Arteriosclerosis and Thrombosis 12, 1235–1244.
Pyorala, K., Laakso, M., Uutisitupa, M., 1997. Diabetes and atherosclerosis: an epidemiologic view. Diabetes & Metabolism
3, 463–524.
Ramana, K.V., Chandra, D., Srivastava, S., Bhatnagar, A., Aggarwal, B.B., Srivastava, S.K., 2002. Aldose reductase mediates
mitogenic signaling in vascular smooth muscle cells. Journal of Biological Chemistry 277, 32063–32070.
Ramana, K.V., Friedrich, B., Bhatnagar, A., Srivastava, S.K., 2003a. Aldose reductase mediates cytotoxic signals of
hyperglycemia and TNF-a in human lens epithelial cells. The FASEB Journal 17, 315–317.
Ramana, K.V., Chandra, D., Srivastava, S., Bhatangar, A., Srivastava, S.K., 2003b. Nitric oxide regulates the polyol pathway of
glucose metabolism in vascular smooth muscle cells. FASEB Journal 17, 417–425.
Renier, G., Mamputu, J.C., Desfaits, A.C., Serri, O., 2003. Monocyte adhesion in diabetic angiopathy effects of ferr-radical
scavenging. Journal of Diabetes and its Complications 17, 20–29.

Q. Dan et al. / Life Sciences 76 (2004) 445–459 459
Ross, R., 1993. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature (Landon) 362, 801–809.
Ross, R., 1999. Atherosclerosis: an inflammatory disease. New England Journal of Medicine 340, 115–126.
Ruef, J., Liu, S.Q., Bode, C., Tocchi, M., Srivastava, S., Runge, M.S., Bhatnagar, A., 2000. Involvement of aldose reductase
in vascular smooth muscle cell growth and lesion formation after arterial injury. Arteriosclerosis, Thrombosis and Vascular
Biology 20, 1745–1752.
Stratton, I.M., Adler, A.I., Neil, H.A., Mattews, D.R., Manley, S.E., Cull, C.A., Hadden, D., Turner, R.C., Holman, R.R., 2000.
Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35); prospective
observational study. British Medical Journal 321, 405–412.
Suarez, G., Rajaram, R., Bhuyan, K.C., Oronsky, A.L., Goidl, J.A., 1988. Administration of an aldose reductase inhibitor
induces a decrease of collagen fluorescence in diabetic rats. Journal of Clinical Investigation 82, 624–627.
Treins, C., Giorgett-Peraldi, S., Murdaca, J., Van Obberghen, E., 2001. Regulation of vascular endothelial growth factor
expression by advanced glycation end products. Journal of Biological Chemistry 276, 43836–43841.
Vielma, S.A., Krings, G., Lopes-Virella, M.F., 2003. Chlamydophila pneumoniae induces ICAM-1 expression in human aortic
endothelial cells via protein kinase C-dependent activation of nuclear factor-kappaB. Circulation Research 92, 1130–1137.
Wautier, J.L., Wautier, MP., Schmidt, A.M., Anderson, G.M., Hori, O., Zoukourian, C., Capron, L., Chappey, O., Yan, S.D.,
Brett, J., Guillauseau, P.J., Stern, D., 1994. Advanced glycation end products (AGEs) on the surface of diabetic erythrocytes
bind to the vessel wall via a specific receptor inducing oxidant stress in the vasculature: a link between surface-associated
AGEs and diabetic complications. Proceedings of the National Academy of Sciences of the United States of America 91,
7742–7746.
Wu, H., Moulton, K., Horvai, A., Parik, S., Glass, C.K., 1994. Combinatorial interactions between AP-1 and ets domain proteins
contribute to the developmental regulation of the macrophage scavenger receptor gene. Molecular and Cell Biology 14,
2129–2139.
Yamaoka, T., Nishimura, C., Yamashita, K., Itakura, M., Yamada, T., Fujimoto, J., Kokai, Y., 1995. Acute onset of diabetic
pathological changes in transgenic mice with human aldose reductase cDNA. Diabetologia 38, 255–261.
Yasuda, O., Zhang, S.H., Miyamoto, Y., Maeda, N., 2000. Differential expression of the alpha 1 type VIII collagen gene by
smooth muscle cells from atherosclerotic plaques of apolipoprotein-E-deficient mice. Journal of Vascular Research 37,
158–169.
Top Related
Copyright © 2022 FDOKUMEN

