







Bahasa
Halaman
Hukum


doi:10.1016/j.jmb.2005.02.038 J. Mol. Biol. (2005) 348, 575–588
Human, Drosophila, and C. elegans TDP43: Nucleic AcidBinding Properties and Splicing Regulatory Function
Youhna M. Ayala1, Sergio Pantano2,3, Andrea D’Ambrogio1
Emanuele Buratti1, Antonia Brindisi1, Caterina Marchetti1
Maurizio Romano4 and Francisco E. Baralle1*
1International Centre forGenetic Engineering andBiotechnology (ICGEB) 34012Trieste, Italy
2International School forAdvanced Studies (ISAS) andINFM, DEMOCRITOSModeling Center for Research inAtomistic Simulation, ViaBeirut 2-4, 34014 TriesteItaly
3Venetian Institute of MolecularMedicine (VIMM), Via Orus 235129 Padua, Italy
4Department of Physiology andPathology, University of TriesteVia A. Fleming 22, 34127 TriesteItaly
0022-2836/$ - see front matter q 2005 E
Abbreviations used: TDP43, TARprotein; hnRNP, heterogeneous nucribonucleoprotein; dm, Drosophila mCaenorhabditis elegans; RRM, RNA rCFTR, cystic fibrosis transmembranregulator; GST, glutathione S-transfelectromobility shift assay; DTT, ditborate–EDTA buffer; IPTG, isopropthiogalactopyranoside.E-mail address of the correspond
TAR DNA binding protein (TDP43), a highly conserved heterogeneousnuclear ribonucleoprotein, was found to down-regulate splicing of theexon 9 cystic fibrosis transmembrane conductance regulator (CFTR)through specific binding to a UG-rich polymorphic region upstream ofthe 3 0 splice site. Despite the emergence of new information regarding theprotein’s nuclear localization and splicing regulatory activity, TDP43’s rolein cells remains elusive. To investigate the function of human TDP43 and itshomologues, we cloned and characterized the proteins from Drosophilamelanogaster and Caenorhabditis elegans. The proteins from human, fly, andworm show striking similarities in their nucleic acid binding specificity. Wefound that residues at two different positions, which show a strongconservation among TDP43 family members, are linked to the tightrecognition of the target sequence. Our three-dimensional model of TDP43in complex with a (UG)m sequence predicts that these residues make aminoacid side-chain to base contacts. Moreover, our results suggest thatDrosophila TDP43 is comparable to human TDP43 in regulating exonsplicing. On the other hand, C. elegans TDP43 has no effect on exonrecognition. TDP43 from C. elegans lacks the glycine-rich domain found atthe carboxy terminus of the other two homologues. Mutants of human andfly TDP43 devoid of the C-terminal domain are likewise unable to affectsplicing. Our studies suggest that the glycine-rich domain is essential forsplicing regulation by human and fly TDP43.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: TDP43; RNA–protein interaction; splicing regulation; hnRNP;RNA recognition motif
*Corresponding authorIntroduction
In higher eukaryotes, splice sites are defined by acombination of cis and trans-acting elements duringthe removal of intronic sequences from pre-mRNA.Sequences acting in cis include loosely definedintronic elements (5 0 and 3 0 splice sites and branch
lsevier Ltd. All rights reserve
DNA bindinglearelanogaster; ce,ecognition motif;e conductanceerase; EMSA,hiothreitol; TBE, Tris–yl-beta-D-
ing author:
point), and sequences located in both exons andintrons that act as splicing enhancer or silencingsignals.1,2 On the other hand, trans-acting elementscommonly are proteins that associate with thespecific pre-mRNA sequence directly or as part ofa complex to promote or repress exon inclusion.Aberrant splicing of mRNA is caused by theintroduction or removal of a cis or trans-actingfactor that tips the balance of the delicate splicingprocess. The abnormal processing of pre-mRNA, inmany cases, results in disease.3,4
A polymorphic region in intron 8 of the cysticfibrosis transmembrane conductance regulator gene(CFTR) modifies the levels of exon 9 inclusion.Transcripts devoid of exon 9 translate into a non-functional protein.5,6 This region located near the 3 0
splice site contains a (TG)m(T)n sequence, where ahigh number of TG repeats correlates with the
d.

576 Characterization of Human, Fly and Worm TDP43
skipping of exon 9 in the presence of T5.7,8 This
polymorphic sequence is associated with a strongpenetrance of disease in patients suffering from theabsence of the vas deference, a non-canonical formof cystic fibrosis.7,9 Buratti and colleagues isolatedTAR DNA binding protein (TDP43) while searchingfor a splicing modulator that inhibited exon 9inclusion through the binding of (UG)12.
10 Thesame protein had been identified earlier as atranscriptional repressor upon association to theTARDNA region in HIV-1.11 TDP43 binds to the UGrepeats specifically and is able to inhibit exon 9inclusion in HeLa cells.10,12 Moreover, in vitrosplicing studies have recently shown that thepresence of TDP43 in nuclear extracts specificallyinhibits exon splicing in the presence of a highnumber (12) of UG repeats.13
TDP43 belongs to the family of heterogeneousnuclear ribonucleoproteins (hnRNPs), character-ized for binding RNA and in some cases DNAsequences, through a common nucleotide bindingdomain known as the RNA recognition motif(RRM). Proteins of the hnRNP family participatein a variety of processes such as transport,stabilization, and modification of RNA, pre-mRNA splicing, and transcriptional regulation.The diversity in hnRNP function is largely due tothe presence of modular domains that mediatespecific protein–protein interactions.14–16 The RNAbinding domains contain two octameric andhexameric consensus sequences: RNP1 and RNP2,respectively. TDP43 contains two RNA bindingdomains of which RRM1 is necessary and sufficientfor nucleotide binding.12 Two residues in the RNP1consensus site of RRM1, Phe147 and Phe149, wereshown to be essential for the recognition of RNAand DNA. The carboxy-terminal RRM (RRM2), onthe other hand, is not required for (UG)m/(TG)mrecognition.
TDP43 is highly conserved throughout evolution;amino acid sequence comparison of the proteinfrom human, mouse, Drosophila, and Caenorhabditiselegans shows a striking degree of similarity.12,17 Atleast four and two alternatively spliced forms ofhuman and Drosophila TDP43 are expressed,respectively.17,18 However, no information aboutthe structural and functional similarity of theseproteins is available up to date. Here we presentevidence that TDP43 from distantly related organ-isms share similar nucleic acid binding specificity.We, moreover, constructed a homology-basedmodel of the TDP43–RNA complex to extend ourknowledge on the basis of RNA sequence specificrecognition. The theoretical 3D model allowed theidentification of crucial residues for the specificrecognition as confirmed by site-directed muta-genesis. Our experimentally validated modeltogether with a comparison of the naturalvariability among the different species and otherhnRNPs offer new information on the protein–RNA recognition interface and delimitate theminimal specific (UG)m sequence recognized byTDP43. We also show thatDrosophila TDP43, but not
TDP43 from C. elegans, inhibits exon splicing. Theefficient regulation of splicing by human andDrosophila TDP43 requires the presence of thecarboxy-terminal domain, which in the caseC. elegans TDP43, is considerably shorter anddevoid of a sequence rich in glycine. In fact,mutants of human and Drosophila TDP43 that lackthis region fail to regulate exon recognition.
Results
Sequence comparison of the longest isoforms ofthe human, Drosophila, and C. elegans TDP43proteins shows that the highest degree of similarityamong the three proteins resides in the regioncorresponding to the putative RNA recognitionmotifs (RRM1 and RRM2) (Figure 1(a)). In fact, localpair-wise alignment of this region indicates 59% ofamino acid identity between human and DrosophilaTDP43, and 38% in the case of human and C. elegansproteins. Figure 1(b) is a cartoon representation ofthe three proteins to highlight the length differencesbetween the different domains in all three proteins.
The longest form of the D. melanogaster TDP43(dmTDP43), previously denominated TBPH-2,18
was isolated from embryonic Drosophila cell cDNAand cloned for bacterial expression. The cDNAcorresponding to the C. elegans protein was insteadcloned from a total RNA preparation as describedinMaterials andMethods. Upon sequencing severalclones from C. elegans, we identified the presence oftwo splicing variants differing in the presence ofnine nucleotides. The isoforms are likely generatedfrom the usage of two alternative 3 0 splice sites ofthe sixth exon. The longer form (ceTDP43-L)contains three extra amino acid residues (SLQ) atposition 251 with respect to the shorter C. elegansTDP43 variant (ceTDP43) (Figure 1(a)). All clonedproducts were expressed as glutathione S-transfer-ase (GST) and histidine-tagged proteins. The pre-dicted molecular masses of dmTDP43 and ceTDP43lacking purification tags are 58.1 kDa and 45.5 kDa,respectively.
Nucleic acid binding specificity of human, fly,and worm TDP43
The RNA binding specificity of dmTDP43 andboth ceTDP43 isoforms was initially determined bycompetition experiments using UV light in vitrocross-linking as shown in Figure 2 (shown areresults pertaining to ceTDP43). Like human TDP43,the Drosophila and C. elegans proteins bound UGrepeats specifically. Association of the proteins withlabeled UG repeats was inhibited only by increasingamounts of cold (UG)12. Equal amounts of a non-related oligonucleotide, (UCUU)3, showed no com-petitive activity (Figure 2(a)). Both isoforms ofceTDP43 competed equally well with hTDP43 forbinding to labeled (UG)12; shown are the results forceTDP43 lacking the three amino acid insertion. Infact, hTDP43 association to its specific RNA could

Figure 1. Amino acid sequence comparison between human, fly, and worm TDP43. (a) Alignment of human (hTDP43), Drosophila (dmTDP43), and C. elegans (ceTDP43)proteins. The longest isoforms of human and fly proteins were used for this analysis (Genbank accession no. NP031401, BAA34421 and NP495921, respectively). Amino acidsidentical or similar among the three proteins are depicted in red or orange, respectively. The boxed areas contain the RNA recognition domains, RRM1 and RRM2. Underlinedare the RNP consensus sequences 2 and 1 contained in each RRM. The position of the three amino acid insertion in ceTDP43 is also indicated with an asterisk (*). (b) Schematicrepresentation of the relative size corresponding to each domain in the three homologues including the amino acid position of C-terminal truncated mutants.

Figure 2. UV cross-linking competition analysis usingrecombinant ceTDP43. (a) His-ceTDP43 (3.75 mg) wascross-linked to 2 ng of radiolabeled (UG)12, in the absence(first lane) and in the presence of cold (UG)12 and(UCUU)3 competitor at a five and tenfold molar excesswith respect to labeled RNA. (b) 100 ng of recombinantGST-hTDP43 was cross-linked to 2 ng of labeled (UG)12with increasing amounts of His-ceTPD43 (0, 3, 7.5, 15, and30-fold excess of competitor ceTDP43 was added to thereaction). The last lane shows the binding of ceTDP43 to(UG)12 in the absence of hTDP43. Reactions wereseparated on SDS-10% (w/v) PAGE following incubation,the corresponding molecular masses are shown on theleft.
578 Characterization of Human, Fly and Worm TDP43
be displaced by increasing amounts of the homo-logous recombinant ceTDP43 (Figure 2(b)).Identical results were obtained using dmTDP43(data not shown). For these experiments, we usedGST-tagged hTDP43 and His-tagged ceTDP43,hence, hTDP43 corresponds to the slower migrating
Table 1. Oligonucleotide sequences corresponding to the RN
Nucleotide sequence (50–3 0)
GUGUGUAAAAAAAAAGUGUGUGUAAAAAAAGUGUGUGUGUAAAAAAGUGUGUGUGUGUUGUGUGUGUGUGUGUGUGUGUGUGTGTGTGTGTGTGTGTGTGTGTGTGACUCUCUCUCUCUCTGCTTTTTGCCTGTACTGGGTCTCTCTGGTTAGACCAGATCTGAAGGACCUGAUGGAGAAGGUCCGCGACGGACGGAAAGACCCCUAUCCGUCGCG
ssDNA and RNA oligonucleotides used for EMSA. NS1 and NS2 cor
bands. Once we established the similarities betweenthe two ceTDP43 isoforms in terms of their RNAbinding activity, all subsequent experiments werecarried out using the shorter form, ceTDP43.
Electromobility shift assay (EMSA) studiesfurther showed that the proteins from Drosophilaand C. elegans specifically recognize UG/TG con-taining RNA and DNA oligonucleotides (Figure 3).The pattern of sequence specificity is equivalent tothat of the human homologue described by Buratti& Baralle.12 All three proteins recognize a minimumnumber of four UG repeats (Figure 3(a)), althoughthe affinity for (UG)4 is significantly reduced withrespect to (UG)5, (UG)6, and (UG)12. In analogy toprevious observations made with the humanvariant,12 dmTDP43 and ceTDP43 strongly bound(TG)12-DNA (Figure 3(b)), did not interact withdouble-stranded DNA (data not shown), andshowed little or no binding to RNA and DNAsequences lacking UG or TG repeats, including theTAR DNA sequence (Figure 3(b)). Likewise,Figure 3(b) shows that TDP43 homologues did notbind RNA or DNA dinucleotide repeats other thanTG repeats (shown are the results for (UC)6 RNA).The sequences of the oligonucleotides used in thebinding experiments are described in Table 1.
As in the case of hTDP43, the C-terminal domainin Drosophila and C. elegans TDP43 was not requiredfor the interaction with DNA or RNA. The deletionof the C-terminal fragment that immediatelyfollows RRM2 in all three proteins did not affectthe binding to (UG)6 (data not shown). As indicatedin Figure 1, these mutants were truncated atpositions 260, 269, and 344 in hTDP43, dmTDP43,and ceTDP43, respectively. Analogous results wereobtained with (UG)5, (UG)12, and (TG)12 oligo-nucleotides (data not shown). In all band shiftexperiments, whether we compared RNA bindingof wild-type or C-terminal truncated mutants,bands corresponding to ceTDP43–nucleic acidcomplexes had lower electrophoretic mobility thanRNA–human/Drosophila TDP43 complexes(Figure 3). This variation in migration patterncould not be attributed to differences in the intrinsicmolecular mass of the proteins, since dmTDP43 isactually larger than hTDP43 or ceTDP43. Theseresults were confirmed using recombinant TDP43proteins fused to histidine tags instead of GST tags
A and DNA probes used in the binding experiments
Oligo
(UG)3(UG)4(UG)5(UG)6(UG)12(TG)12(UC)6
G TARNS1NS2
respond to non-specific sequences used as a control.
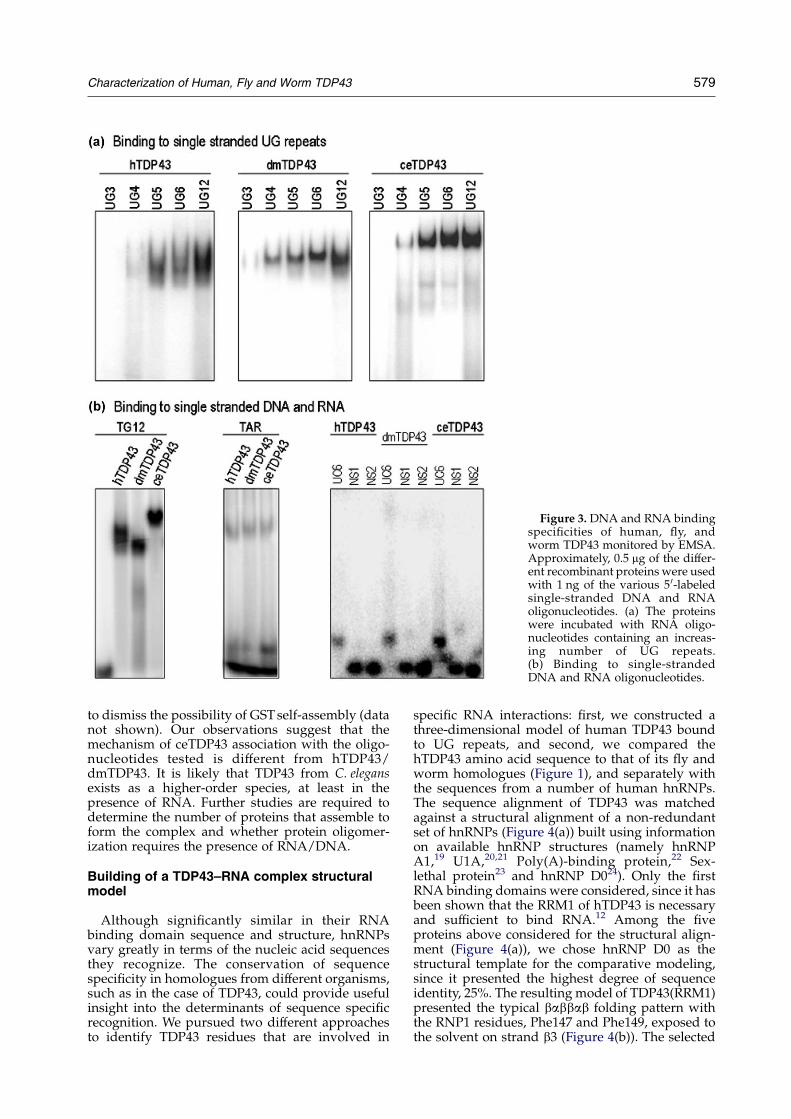
Figure 3. DNA and RNA bindingspecificities of human, fly, andworm TDP43 monitored by EMSA.Approximately, 0.5 mg of the differ-ent recombinant proteins were usedwith 1 ng of the various 5 0-labeledsingle-stranded DNA and RNAoligonucleotides. (a) The proteinswere incubated with RNA oligo-nucleotides containing an increas-ing number of UG repeats.(b) Binding to single-strandedDNA and RNA oligonucleotides.
Characterization of Human, Fly and Worm TDP43 579
to dismiss the possibility of GSTself-assembly (datanot shown). Our observations suggest that themechanism of ceTDP43 association with the oligo-nucleotides tested is different from hTDP43/dmTDP43. It is likely that TDP43 from C. elegansexists as a higher-order species, at least in thepresence of RNA. Further studies are required todetermine the number of proteins that assemble toform the complex and whether protein oligomer-ization requires the presence of RNA/DNA.
Building of a TDP43–RNA complex structuralmodel
Although significantly similar in their RNAbinding domain sequence and structure, hnRNPsvary greatly in terms of the nucleic acid sequencesthey recognize. The conservation of sequencespecificity in homologues from different organisms,such as in the case of TDP43, could provide usefulinsight into the determinants of sequence specificrecognition. We pursued two different approachesto identify TDP43 residues that are involved in
specific RNA interactions: first, we constructed athree-dimensional model of human TDP43 boundto UG repeats, and second, we compared thehTDP43 amino acid sequence to that of its fly andworm homologues (Figure 1), and separately withthe sequences from a number of human hnRNPs.The sequence alignment of TDP43 was matchedagainst a structural alignment of a non-redundantset of hnRNPs (Figure 4(a)) built using informationon available hnRNP structures (namely hnRNPA1,19 U1A,20,21 Poly(A)-binding protein,22 Sex-lethal protein23 and hnRNP D024). Only the firstRNA binding domains were considered, since it hasbeen shown that the RRM1 of hTDP43 is necessaryand sufficient to bind RNA.12 Among the fiveproteins above considered for the structural align-ment (Figure 4(a)), we chose hnRNP D0 as thestructural template for the comparative modeling,since it presented the highest degree of sequenceidentity, 25%. The resulting model of TDP43(RRM1)presented the typical babbab folding pattern withthe RNP1 residues, Phe147 and Phe149, exposed tothe solvent on strand b3 (Figure 4(b)). The selected

Figure 4. Structural model of the hTDP43 RRM1 domain complexed with a nine nucleotide-long UG repeat sequence.(a) Primary sequence alignment of the hnRNP structural templates. Red boxes indicate the two conserved RNP motifs.Arrows at the bottom indicate important residues for RNA interactions revealed by theoretical analysis and confirmedby site-directed mutagenesis. The secondary structure elements of the TDP43 model are indicated at the top. (b) Cartoonrepresentation of a three-dimensional model of TDP43. (c) Model of the protein–RNA complex; amino acid side-chainsare in green. (d)–(f) Close-ups of the specific interactions of Arg151, Phe147/149 and Trp113, respectively.
580 Characterization of Human, Fly and Worm TDP43
structure exhibited a good Ramachandran map,with 93.0% of the residues in most favored regions,7% in allowed regions, and no residues indisallowed zones.
The side-chains of Phe147 and Phe149 couldinteract with either uracil or guanosine, thus twoalternative models were available (see Materialsand Methods). One of the models, corresponding tothe RNA filament with a U placed on Phe147 and aG on Phe149, resulted in lower intermolecularinteraction energy. Therefore, we focused on thedescription of this model only, although, giventhe limited accuracy of the theoretical approach, thealternative arrangement cannot be absolutely ruledout. The 3D structure of the complex is shown inFigure 4(c), where the RNA filament follows the 5 0
to 3 0 sense from right to left. Among the largenumber of intramolecular interactions observed inthe model, we focused only on those retained to
strongly contribute to the specificity of the RNA–protein interaction. The first intermolecular inter-action involved Arg151. The side-chain of thisresidue participates in H-bond interactions withU1 and G2 (Figure 4(d)). Addition of nucleotides inthe 5 0 direction did not result in further protein–RNA interactions, suggesting that Arg151 is the firstinteracting residue in the b-sheet plane of TDP43.Only the Sex-lethal protein bears an arginineresidue in this position in its second RNA bindingdomain (Figure 4(a)). G2 and U3 are (by construc-tion) stacked on the aromatic moieties of Phe149and Phe147, respectively. G4, U5, G6 and U7 formedstacking interactions with each other (G4-U5, andG6-U7) and were involved in a complicatednetwork of intra- and intermolecular H-bondinteractions (Figure 4(c)). Notably, G8 formed astrong stacking interaction with Trp113 (Figure 4(f)).This interaction fixed the position of U9 that made a
Characterization of Human, Fly and Worm TDP43 581
H-bond with the amine moiety of Lys114. Thisfilament of nine nucleotides spanned the entireb-sheet and part of the short loop 1, suggesting thatfurther addition of nucleotides may result inintermolecular interactions between the UG repeatsand loop 3, as reported for the close homologuehnRNP D0.24 However, due to the high flexibility ofboth RNA and the long loop, these cannot bereliably described with the currently availablemodeling methods.
Trp113 and Arg151 are conserved among TDP43family members with the exception of TDP43 fromC. elegans, where Trp is substituted by Phe atposition 113. At the same time, these positions arehighly variable among hnRNPs. Residues in RRM1that are conserved among members of the TDP43family, but not among hnRNPs, are likely respon-sible for the specific recognition of UG repeats bythe human, fly, and worm TDP43 homologues.
Site-directed mutagenesis and affinitydetermination
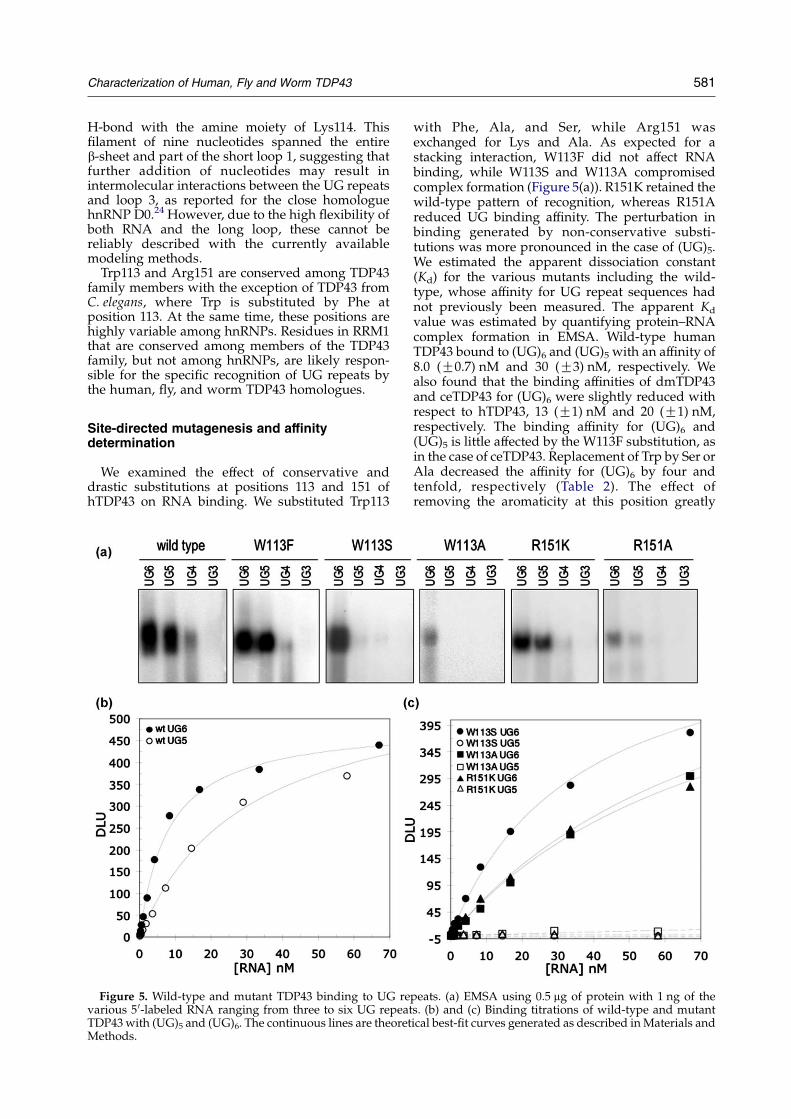
We examined the effect of conservative anddrastic substitutions at positions 113 and 151 ofhTDP43 on RNA binding. We substituted Trp113
Figure 5. Wild-type and mutant TDP43 binding to UG revarious 5 0-labeled RNA ranging from three to six UG repeatTDP43 with (UG)5 and (UG)6. The continuous lines are theoretMethods.
with Phe, Ala, and Ser, while Arg151 wasexchanged for Lys and Ala. As expected for astacking interaction, W113F did not affect RNAbinding, while W113S and W113A compromisedcomplex formation (Figure 5(a)). R151K retained thewild-type pattern of recognition, whereas R151Areduced UG binding affinity. The perturbation inbinding generated by non-conservative substi-tutions was more pronounced in the case of (UG)5.We estimated the apparent dissociation constant(Kd) for the various mutants including the wild-type, whose affinity for UG repeat sequences hadnot previously been measured. The apparent Kd
value was estimated by quantifying protein–RNAcomplex formation in EMSA. Wild-type humanTDP43 bound to (UG)6 and (UG)5 with an affinity of8.0 (G0.7) nM and 30 (G3) nM, respectively. Wealso found that the binding affinities of dmTDP43and ceTDP43 for (UG)6 were slightly reduced withrespect to hTDP43, 13 (G1) nM and 20 (G1) nM,respectively. The binding affinity for (UG)6 and(UG)5 is little affected by the W113F substitution, asin the case of ceTDP43. Replacement of Trp by Ser orAla decreased the affinity for (UG)6 by four andtenfold, respectively (Table 2). The effect ofremoving the aromaticity at this position greatly
peats. (a) EMSA using 0.5 mg of protein with 1 ng of thes. (b) and (c) Binding titrations of wild-type and mutantical best-fit curves generated as described inMaterials and

Table 2. Kd values for the binding of wild-type andhTDP43 mutants to (UG)6 and (UG)5
Kd (nM)
(UG)6 (UG)5
Wild-type hTDP43 8.0G0.7 30G3W113F 15G1 63G6W113S 35G3 N.A.W113A 80G9 N.A.R151K 40G3 130G9R151A 100G9 N.A.
582 Characterization of Human, Fly and Worm TDP43
compromised the binding of (UG)5 (Figure 5(a)). Infact, the Kd value for (UG)5 binding to W113S andW113A could not be determined, as no complexformation was observed under the conditionstested. Substitution of Arg151 for a Lys slightlydecreased the affinity for both oligonucleotides,while the mutation R151A led to a significant loss inthe recognition of both oligonucleotides (Table 2).(UG)6 was recognized 13-fold less efficiently byR151A compared to wild-type, and as in the case ofW113S andW113A, the Kd value for (UG)5 could notbe estimated (Figure 5(c)). Our results suggest thatthe TDP43 interaction with UG repeats favors anaromatic residue at 113 and a positive charge atposition 151, suggesting that electrostatic inter-actions play a role in binding. We cannot rule out,however, the presence of hydrogen bond inter-actions between Arg/Lys at position 151 and theRNA sequence. In fact, salt concentration seems tohave little effect in the association of hTDP43 with(UG)6 even at 600 mM NaCl (data not shown).
In vitro splicing assays
The association of TDP43 with UG repeats at the3 0 splice site is fundamental for the protein’s abilityto inhibit exon inclusion. It was interesting to testwhether the homologous proteins from fly andworm, comparable in their ability to recognize(UG)m, may also substitute human TDP43 insplicing regulation. In vitro splicing assays wereused to monitor the effect of the proteins on exonrecognition (Figure 6). A transcript containingexons 2 and 3 of a-tropomyosin, separated by asynthetic intron with the 5 0 end partially corre-sponding to the human b-globin gene (Py7wt), wasgenerated in vitro from the Py7 plasmid25
(Figure 6(a)). Processing of the Py7wt transcriptshowed the appearance of spliced exons 2 and 3,and the intron lariat (Figure 6(b), lane 1). As shownby Buratti et al., insertion of the (TG)12T5 sequencedownstream of the intron’s branch point(PY7(UG)12U5) blocked the recognition of the exondue to the specific binding of TDP43 to its targetsequence13 (lane 2). Lanes 3 and 4 show thatdepletion of TDP43 from the human nuclear extractrecovered exon splicing, despite the presence of UGrepeats. Western blot analysis of the complete anddepleted nuclear extract using a specific antibodyagainst TDP43 determined that greater than 90% ofthe endogenous protein had been removed.13
Addition of recombinant hTDP43 to the depletednuclear extract again prevented exon splicing. Asshown in Figure 6, lane 6, addition of recombinantdmTDP43 to the TDP43-depleted nuclear extractinhibited transcript splicing. On the other hand,reconstitution of the depleted extract with C. elegansTDP43 did not preclude formation of the splicedproduct (lane 7). These results were confirmed withrecombinant proteins fused to histidine or GST tags.
The inability of ceTDP43 to control exon splicingin our in vitro system suggested that the proteindiffers from the human and Drosophila homologuesin its capacity to interact with the splicing regulat-ory complex. The C-terminal region following theRRMs is the most divergent portion among thethree sequences. To test whether the lack of thecarboxy-terminal domain in ceTDP43 was respon-sible for the control of exon recognition, we studiedthe effect of TDP43 truncated mutants from humanand fly lacking this region (hTDP43DC anddmTDP43DC). Lanes 8 and 9 of Figure 6 showthat neither of the mutants inhibited exon splicingeven though, as in the case of ceTDP43, their (UG)mbinding activity is equivalent to the wild-typeproteins (data not shown). Our results suggestthat the C-terminal domains of hTPD43 anddmTDP43, perhaps due to their glycine-rich nature,mediate relevant interactions with proteinsinvolved in splicing regulation.
Discussion
We compared the nucleic acid binding propertiesand splicing regulatory function of TDP43 proteinsfrom human, Drosophila, and C. elegans. Despiterecent observations on the protein’s cellularlocalization and nucleic acid binding function,12,26
little is known regarding the role of human TDP43.Even less is known regarding the Drosophila andC. elegans proteins, although the pattern of develop-mental expression of their genes suggests that theyplay a role in the development of the respectiveorganisms. Expression of Drosophila TDP43 isconcentrated in the embryonic and pupal stages,whereas in C. elegans, higher levels of the transcriptare detected in oocytes and in the first larval stagesof the nematode.18,27 Amino acid sequence analysesindicate that the human protein is more similar tothe Drosophila TDP43 than to the worm protein.Nevertheless, our observations indicate that astructural conservation exists in all three proteinsthat translates into equivalent RNA and DNAsequence specificity. In fact, our EMSA experimentswith RNA probes of various lengths show that allthree proteins have similar RNA binding affinityand require at least four UG repeats to interact withthe oligonucleotides. Probes lacking UG/TGrepeats, including the TAR DNA sequence, showlittle or no recognition. Our observations indicatethat TDP43’s affinity for UG sequences does notvary greatly with the length of the oligonucleotideand that a similar binding pattern is exhibited for
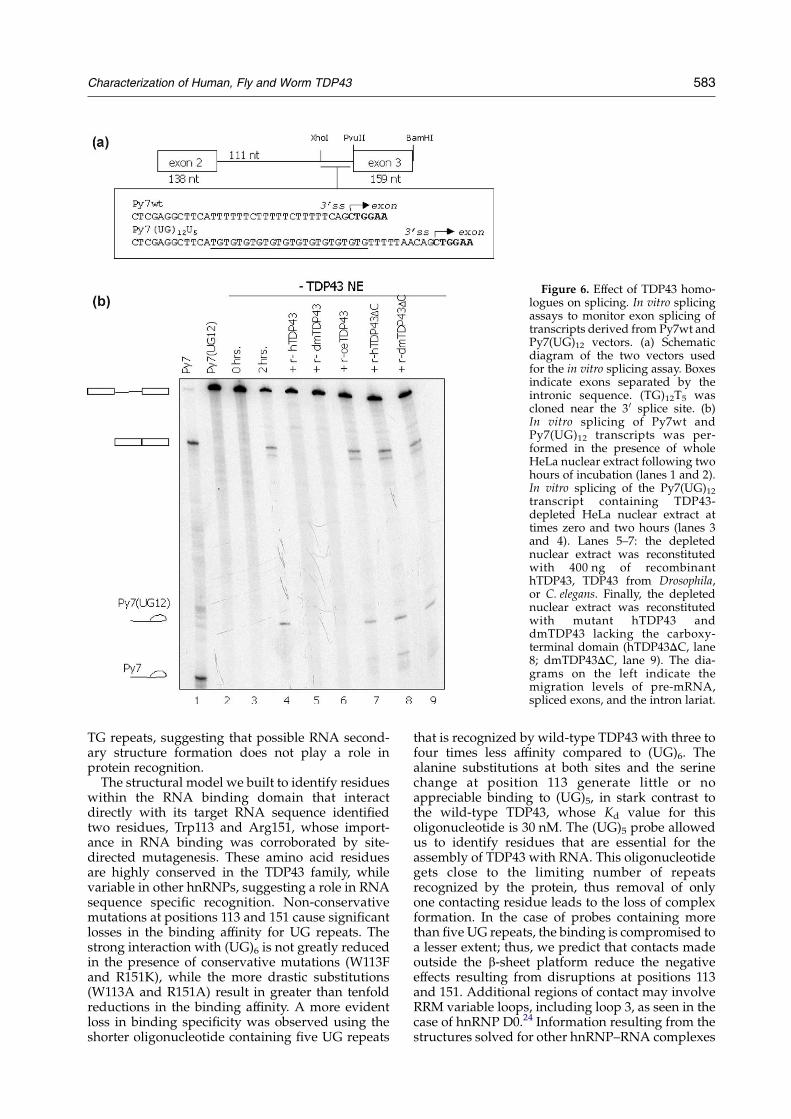
Figure 6. Effect of TDP43 homo-logues on splicing. In vitro splicingassays to monitor exon splicing oftranscripts derived from Py7wt andPy7(UG)12 vectors. (a) Schematicdiagram of the two vectors usedfor the in vitro splicing assay. Boxesindicate exons separated by theintronic sequence. (TG)12T5 wascloned near the 3 0 splice site. (b)In vitro splicing of Py7wt andPy7(UG)12 transcripts was per-formed in the presence of wholeHeLa nuclear extract following twohours of incubation (lanes 1 and 2).In vitro splicing of the Py7(UG)12transcript containing TDP43-depleted HeLa nuclear extract attimes zero and two hours (lanes 3and 4). Lanes 5–7: the depletednuclear extract was reconstitutedwith 400 ng of recombinanthTDP43, TDP43 from Drosophila,or C. elegans. Finally, the depletednuclear extract was reconstitutedwith mutant hTDP43 anddmTDP43 lacking the carboxy-terminal domain (hTDP43DC, lane8; dmTDP43DC, lane 9). The dia-grams on the left indicate themigration levels of pre-mRNA,spliced exons, and the intron lariat.
Characterization of Human, Fly and Worm TDP43 583
TG repeats, suggesting that possible RNA second-ary structure formation does not play a role inprotein recognition.
The structural model we built to identify residueswithin the RNA binding domain that interactdirectly with its target RNA sequence identifiedtwo residues, Trp113 and Arg151, whose import-ance in RNA binding was corroborated by site-directed mutagenesis. These amino acid residuesare highly conserved in the TDP43 family, whilevariable in other hnRNPs, suggesting a role in RNAsequence specific recognition. Non-conservativemutations at positions 113 and 151 cause significantlosses in the binding affinity for UG repeats. Thestrong interaction with (UG)6 is not greatly reducedin the presence of conservative mutations (W113Fand R151K), while the more drastic substitutions(W113A and R151A) result in greater than tenfoldreductions in the binding affinity. A more evidentloss in binding specificity was observed using theshorter oligonucleotide containing five UG repeats
that is recognized by wild-type TDP43 with three tofour times less affinity compared to (UG)6. Thealanine substitutions at both sites and the serinechange at position 113 generate little or noappreciable binding to (UG)5, in stark contrast tothe wild-type TDP43, whose Kd value for thisoligonucleotide is 30 nM. The (UG)5 probe allowedus to identify residues that are essential for theassembly of TDP43 with RNA. This oligonucleotidegets close to the limiting number of repeatsrecognized by the protein, thus removal of onlyone contacting residue leads to the loss of complexformation. In the case of probes containing morethan five UG repeats, the binding is compromised toa lesser extent; thus, we predict that contacts madeoutside the b-sheet platform reduce the negativeeffects resulting from disruptions at positions 113and 151. Additional regions of contact may involveRRM variable loops, including loop 3, as seen in thecase of hnRNP D0.24 Information resulting from thestructures solved for other hnRNP–RNA complexes

584 Characterization of Human, Fly and Worm TDP43
shows that position 113, found in the first loop ofRRM1, may occupy a strategic location to specifi-cally interact with RNA. The structures of varioushnRNPs complexed with their RNA targetsequence have shown amino acid side-chain–basespecific interactions at positions equivalent to W113in TDP43. Such is the case of the Sex-lethal protein(Sxl) of D. melanogaster residue Q134 (RRM1),23
adenosine–uridine-rich element binding proteinHuD residue Q48,28 and U1A spliceosomal proteinresidue E19.20 Fewer examples are available forinteractions formed by residues homologous toR151 in TDP43. Arg258 occupies an equivalentposition in the RRM2 of Sxl and was shown to forma hydrogen bond and a salt bridge with U2 and U10of the 12 nucleotide sequence derived from the trapolypyrimidine tract.23 Also, N58 of the poly(A)-binding protein acts as a hydrogen bond donor andacceptor for one of the adenine bases in the RNAsequence.22 Our observations and the evolutionaryconservation of W113 and R151 indicate that theseresidues in TDP43 are important for the specificrecognition of RNA. Extensive scanning muta-genesis studies as well as further structural infor-mation regarding the TDP43-(UG)m complex will berequired to identify additional interactions that playessential roles in recognition.
In addition to characterizing the TDP43 inter-action with RNA, we wanted to determine whetherthe similarities among TDP43 family membersextend to equivalent functions in the regulation ofsplicing. Human TDP43 has been shown to inhibitexon 9 CFTR inclusion in HeLa cells.10 Moreover,recently reported in vitro splicing assays indicatethat splicing is specifically inhibited by hTDP43 inthe presence of UG repeats near the 3 0 splice site of atwo exon–one intron transcript.13 Removal ofendogenous TDP43 from human nuclear extractresults in exon splicing while addition of therecombinant protein to the same nuclear extractblocks exon recognition. We now show thatdmTDP43 can substitute hTDP43 in a TDP43-depleted human nuclear extract to prevent exonsplicing, whereas ceTDP43 has no effect on exonrecognition. Our results indicate that the TDP43homologues from human and fly are well con-served in their ability to specifically recognize aregulatory sequence in pre-mRNA and inhibit thesplicing machinery. Most likely, ceTDP43 fails toinhibit exon recognition because it is unable toassemble factors that are required for the regulationof splicing. C. elegans TDP43 binds UG repeats withhigh affinity and its primary structure is signifi-cantly similar to hTDP43 and dmTDP43 in the RRMregions. What distinguishes the worm protein fromhTDP43, dmTDP43, and TDP43 homologues fromother organisms is a considerably shorter carboxylterminus. In fact, like ceTDP43, human andDrosophila mutants that lack this region recognizeRNA efficiently but no longer inhibit exon splicing.Among the TDP43 homologues studied heredmTPD43 presents the longest C-terminal domain,while the shortest belongs to ceTDP43 (see Figure 1).
The region displays no sequence conservation andin terms of glycine residue content, human andDrosophila TDP43 are 24% and 15% rich in glycineresidues within their C-terminal end, respectively.The best-studied glycine-rich domain among RRM-class proteins belongs to hnRNP A1 and iscomparable in size to that of hTDP43 althoughhigher in glycine content (O40%). Further studieswill be required to ascertain that the role of theC-terminal region in TDP43-mediated splicinginhibition is determined by high glycine content.We speculate, however, that this domain function-ally resembles glycine-rich regions found in othersplicing factors.
Domains rich in glycine are present at the aminoor carboxy terminus of various hnRNPs in additionto hnRNPA1 (e.g. U1snRNP,7029 U2AF,35 DrosophilaP element splicing regulator30) and are generallydescribed to mediate the association of glycine-richdomain-containing proteins. The interactionthrough this domain has been shown to play arole in splicing for a number of cases, includinghnRNP A1 repression of exon recognition.31–33 Thestrong presence of glycine in hnRNPA1 is requiredfor cooperative association on the nucleic acid,interaction with other proteins, and for its RNA andDNA-strand annealing activity.34–40 In addition tohnRNP A1, glycine-rich domain-modulatedassembly of hnRNPs has been determined for Sxlassociation with hnRNP L and with the Drosophilahomologue of hnRNP A/B.50
The presence or absence of the C-terminaldomain in TDP43 variants might determine proteinfunction. Upon cloning of D. melanogaster TDP43,two isoforms, varying in the presence of thecarboxy-terminal domain, were found.18 Likewise,recent work detected the presence of alternativelyspliced TDP43 variants of mouse and human thatlack the glycine-rich domain.17,26 Experimentsusing a mini-gene system in 293 cells indicatedthat the longer mouse TDP43 variant, but not theC-terminal truncated isoform prevents CFTR exon 9splicing.17 The existence in nature of TDP43isoforms with truncated C termini,17,18 recentfindings,26 and our present results all suggest thatthe different protein variants perform distinctfunctions. As of yet, TDP43 has been shown toplay roles in the control of alternative splicing andtranscription.10,11,13,26 Splicing regulation may bereserved for TDP43 isoforms containing the longer,glycine-rich C-terminal domain, whereas theshorter variants (including ceTDP43) may playroles in transcription and other aspects of RNA/DNA metabolism that still need to be explored.
In conclusion, the cloning and characterization ofTDP43 homologues from Drosophila and C. eleganshas provided novel information regarding theconservation and divergence among species ofstructural and functional domains of this highlyconserved protein. More in general, our analysis ofthe molecular determinants for nucleic acid recog-nition by TDP43 provide further insight into themechanism of RNA recognition by hnRNPs. The

Characterization of Human, Fly and Worm TDP43 585
striking conservation of sequence specificity andthe interchangeability of the human and Drosophilaproteins in an in vitro splicing system point to animportant role of TDP43 in RNA metabolism.Further investigation into the factors that associatewith these proteins might elucidate functionalaspects of TDP43 as well as common and distinctmechanisms of splicing regulation in widelydifferent organisms.
Materials and Methods
RNA preparation from C. elegans
Nematodes were grown on NGM (nematode growthmedium) plates seeded with OP50 bacteria. A mixedpopulation of worms was collected from the plates,pelleted, and washed with phosphate-buffered saline(PBS). Approximately, 5 g of nematodes were placed intoliquid nitrogen in amortar and ground to a powder. Then,the worm powder was resuspended in 1 ml of RNAzol Breagent (Biotec Laboratories) and total RNA wasextracted according to manufacturer instructions.Poly(dT) cDNA was synthesized using M-MLV reversetranscriptase (Invitrogen).
Construct preparation
Constructs for protein expression and in vitro transcrip-tion were prepared by standard cloning protocols.41 The1245 nt Q20414 coding sequence of C. elegans TDP43(Genbank accession number Z49910.1) was amplifiedby PCR for 35 cycles (95 8C for 30 seconds, 60 8C for30 seconds, 72 8C for one minute) using primersceTDP20414BamHI-5 0 and ceTDP20414HindIII-3 0. Inorder to confirm the identity of the PCR products and toexclude the presence of mutations, the amplicons werecloned into a pUC19 SmaI-cut vector. The variantscorresponding to the two alternative spliced productswere excised from pUC19 and cloned between BamHI/HindIII into the pQE-30 bacterial expression vector(Qiagen) using oligonucleotides C4 and C5. dmTDP43was amplified from Quick clone cDNA from D. melano-gaster, embryo cells obtained from Clontech with primersD1 and D2. The shorter form of C. elegans TDP43,ceTDP43, was amplified by PCR with oligonucleotidesC1 and C2 from pQE-30/ceTDP43 and the product wascloned into a pGEX-3X expression vector (AmershamBiosciences) between BamHI and SmaI. Constructslacking the C-terminal domains of human, Drosophila,and C. elegans TDP43 were made by PCR extension usingprimers T1, T2, D1, D3, C1, and C3. The fragments werecloned into pGEX-3X using the same restriction sites usedto clone the corresponding wild-type sequences.The oligonucleotides used for amplification were
synthesized by Sigma Genosis and the sequences are asfollows: D1, 5 0-TTCCCCCGGGGATTTCGTTCAAGTGTCGGA-3 0; D2, 5 0-CCGGAATTCAAGAAAGTTTGACTTCTCCGCGGC-3 0; D3, 5 0-CCGGAATTCTATCTGTTCTGCTCGGCCTTTGG-3 0; C1, 5 0-CGCGGATCCTCGCCGACGAAACGCCGAAGGTTAAAAC-3 0; C2, 5 0-TTCCCCCGGGTCACCATCCTGGTCCTCTCGAG-3 0; C3, 5 0-TCCCCCGGGCTATTGATTATTCTCTTCTCGAGG-3 0;C4, 5 0-TCACGGATCCATGGCCGACGAAACGCCGAA-3 0; C5, 5 0-ACGTAAGCTTTCACCATCCTGGTCCTCTCGA-3 0; T1, 5 0-GGGCTGGCAAGCCACGTTTGGT-3 0;T2, 5 0-CCGGAATTCCCTTCGGCATTGGATATAT
GAACGC-3 0; ceTDP20414BamHI-5 0, 5 0-TCACGGATCCATGGCCGACGAAACGCCGAA-3 0; ceTDP20414HindIII-3 0, 5 0-ACGTAAGCTTTCACCATCCTGGTCCTCTCGA.TDP43 mutants were constructed using the Quik-
change kit (Stratagene) according to the manufacturer’sinstructions. The pGEX-3X vector encoding for the wild-type sequence was used as vector with the followingoligonucleotides: W113FRV, 5 0-CTTTCAGGTCCTGTTCGGTTGTTTTGAATGGGAGACCCAACAC-3 0; W113FFW,5 0-GTGTTGGGTCTCCCATTCAAAACAACCGAACAGGACCTGAAAG-3 0; W113SR, 5 0-CTTTCAGGTCCTGTTCGGTTGTTTTAGATGGGAGACCCAACAC-30; W113SFW,5 0-GTGTTGGGTCTCCCATCTAAAACAACCGAACAGGACCTGAAAG-3 0; W113ARV, 5 0-CTTTCAGGTCCTGTTCGGTTGTTTTCGCTGGGAGACCCAACAC-3 0;W113AFW, 5 0-GTGTTGGGTCTCCCAGCGAAAACAACCGAACAGGACCTGAAAG-3 0; R151KRV, 5 0-TGTTTCATATTCCGTAAATTTAACAAAGCCAAACCCCTTTG-3 0; R151KFW, 5 0-AAGGGGTTTGGCTTTGTTAAATTTACGGAATATGAAACAC-3 0; R151ARV, 5 0-TGTTTCATATTCCGTAAACGCAACAAAGCCAAACCCCTTTG-3 0 R151AFW, 5 0-AAGGGGTTTGGCTTTGTTGCGTTTACGGAATATGAAACAC-3 0.
Recombinant protein expression
Expression and purification of the GST-tagged proteinswere performed as described.12 The histidine-taggedproteins were expressed in BL21(DE3) bacteria (Novagen)under the induction of 0.5 mM IPTG. Purification of theproteins was carried out according to the manufacturer’sinstructions using imidazole buffers for protein elution.
UV cross-linking and competition analysis
Transcription of cold and labeled RNA and UV cross-linking analysis were carried out as reported.10,42 UVcross-linking assays were performed with histidine-tagged ceTDP43, ceTDP43L, dmTDP43, and GST-taggedhTDP43. (UCUU)3 was transcribed from a pB1uescript SKplasmid as described.10
Binding assays
Protein–nucleic acid binding activity was measuredwith EMSA, on a native polyacrylamide gel (5%). Thebinding buffer contained 10 mM NaCl, 10 mM Tris(pH 8.0), 2 mM MgCl2, 5% (v/v) glycerol, 1 mM DTT.DNA oligonucleotides were synthesized by SigmaGenosis, whereas RNA oligonucleotides were made byIntegrated DNA Technologies. A description of thenucleotide sequence of the different oligonucleotides isprovided in Table 1. The lyophilized forms of thesynthesized oligonucleotides were resuspended inwater to a concentration of 1 mg/ml. Labeling of theseoligonucleotides was carried out as indicated.12 Thebinding reactions were incubated at room temperaturefor 15–20 minutes before loading. The gels were run at100–120 V at 4 8C. The proteins expressed as GST fusionswere used for EMSA, unless otherwise specified.
Amino acid sequence comparison
The multiple and global amino acid sequence align-ments for hTDP43, dmTDP43, and ceTDP43 were carriedout with CLUSTALW version 1.8243 using the Gonnet 250matrix. Local pair-wise sequence alignments were

586 Characterization of Human, Fly and Worm TDP43
performed with the BLOSUM62 matrix with gap openingand gap extension penalties of 11 and 1, respectively. Inthe case of hTDP43 alignments with other humanhnRNPs we considered TDP43 residues 106–175. A non-redundant set of five hNRNPs whose structure has beendetermined experimentally was used for the sequencealignment (PDB code, 1B7F;23 PDB code, 1HD0,24; PDBcode, 1CVJ,22; PDB code, 1HA1;19 PDB code, 1G2E,28).The RRMs pertaining to all five proteins were structurallyaligned with the Homology module of Insight II andthose exhibiting the highest sequence identity wereselected. The primary sequence of TDP43 was thenaligned to those of the potential templates. The structureof hnRNP D0 protein, featuring the highest sequenceidentity (50%) was chosen as the structural template.
Construction of the 3D model
The structure of hnRNP D0 protein was chosen as thestructural template. The coordinate mapping and refine-ment of the backbone and side-chains of the model wereperformed with the program Modeller V6.2.44 The beststructural model was selected based on Modeller’srestraints violation and structural analysis performedwith PROCHECK.45 For the construction of the modeldescribing the TDP43–RNA complex we followed aniterative approach following structural informationinferred from the literature. (i) hnRNPs bind RNA as asingle chain with its bases facing the beta sheet plane ofthe protein.46 (ii) Site-directed mutagenesis with non-aromatic residues on Phe147/149 confirmed that theseresidues form stacking interactions with RNA bases.12
(iii) Taking into account TDP43’s specificity for UGm,either, a UG or a GU pair must stack on those aromaticresidues on TDP43. We placed two bases parallel with thearomatic rings of Phe147 and 149 at a distance of 3.4 A.The phosphate backbone atoms of the dinucleotide weregenerated avoiding sterical clashes. This proceduregenerated two models, one with a U stacked on Phe147and a G stacked on Phe149 and another with the invertedconfiguration of the nucleotides. Both models underwentan energy minimization protocol (see below). Additionalnucleotides were then added in canonical conformation(either G or U, according to the sequence) at theextremities of the previous energy-minimized modelsminimizing the energy after every inclusion. Energyminimization (EM) protocol: to avoid anomalous confor-mations, each protein–oligonucleotide interaction under-went EM followed by 100 ps of molecular dynamics (MD)simulations followed by 100 of EM. EM and MDsimulations were carried out in the presence of implicitsolvent within the generalized Born framework with acutoff of 18 A and a salt concentration of 0.15 M, asimplemented in the Amber7 package. The simulationsused the Amber94 force field47 at 100 K applying 0.5 kcal/mol harmonic constraints to the protein backbone atoms.This setup was intended to (i) initially remove badcontacts and spurious torsional tensions (initial minimiz-ation), (ii) allow the systems to escape from local minima(MD), and (iii) cool down the system to low energyconformation.
Estimation of binding affinities
Limiting protein concentrations were used for thedetermination of the apparent dissociation constants.Oligonucleotide concentration was varied while theprotein concentration was kept constant. A proteinconcentration of approximately 50 nM was used for
wild-type and mutants in 10 mM NaCl, 10 mM Tris (pH8.0), 2 mM MgCl2, 5% glycerol, 1 mM DTT. The bindingreactions were carried out as described above using 5 0-labeled RNA. The quantification of protein–RNA com-plex formation was performed with OptiQuant imageanalysis software (Packard Instrument Co.). The bindingcurves were analyzed according to:
yZx=Kd
1Cx=Kd
where y is the quantified intensity coming from theprotein–RNA complex and x is the oligonucleotideconcentration. Background subtraction was taken intoaccount for each single point.
In vitro transcription and splicing reactions
The protocols and reagent preparation for the tran-scription and splicing reactions were commonly adaptedfrom methods described.48 32P-labeled transcripts weregenerated from the Py7wt25 and PY7(UG12U5) vectors.Construction of PY7(UG12U5) along with transcriptpreparation were as described.13 For the in vitro splicingreaction, approximately 100 mg of HeLa nuclear extract(Biotech) were used in a total volume of 10 ml. Thereactions contained nuclear extract in 50 mM Hepes–KOH (pH 7.3), 3.2 mM MgCl2, 1 ml of 25! ATP/creatinephosphate mixture, 20 fmol of 32P-labeled transcripts, and3.3% (w/v) polyvinyl alcohol. The reaction was incubatedat 30 8C for up to two hours and stopped with theaddition of sodium acetate, SDS, and tRNA. The RNAwas then phenol-extracted and ethanol-precipitated. Atotal of 400 ng of the recombinant proteins were used forthe add back experiments.Depletion of the HeLa nuclear extract of endogenous
TDP43 was carried out as described.12 Concentration ofdepleted extracts was determined by Bradford assays.49
Depletion of TDP43 was monitored by Western blotanalysis according to standard protocols using a 1 : 1000dilution of the primary antibody and developed usingECL (Amersham Biosciences).
Acknowledgements
Y.M.A. is a National Science FoundationMinority Research fellow. This work wassupported by Telethon grants GGP02453 and FIRBRBNE01W9PM (to F.E.B.).
References
1. Burge, C. B., Tuschl, T. & Sharp, P. A. (1999). In TheRNA World II (Gesteland, R. F., Cech, T. R. & Atkins,J. F., eds), pp. 525–560, Cold Spring Harbor Labora-tory Press, Cold Spring Harbor, NY.
2. Cartegni, L., Chew, S. L. & Krainer, A. R. (2002).Listening to silence and understanding nonsense:exonic mutations that affect splicing. Nature Rev.Genet. 3, 285–298.
3. Faustino, N. A. & Cooper, T. A. (2003). Pre-mRNAsplicing and human disease. Genes Dev. 17, 419–437.
4. Pagani, F. & Baralle, F. E. (2004). Genomic variants inexons and introns: identifying the splicing spoilers.Nature Rev. Genet. 5, 389–396.

Characterization of Human, Fly and Worm TDP43 587
5. Delaney, S. J., Rich, D. P., Thomson, S. A., Hargrave,M. R., Lovelock, P. K., Welsh, M. J. &Wainwright, B. J.(1993). Cystic fibrosis transmembrane conductanceregulator splice variants are not conserved and fail toproduce chloride channels. Nature Genet. 4, 426–431.
6. Strong, T. V., Wilkinson, D. J., Mansoura, M. K., Devor,D. C., Henze, K., Yang, Y. et al. (1993). Expression of anabundant alternatively spliced form of the cysticfibrosis transmembrane conductance regulator(CFTR) gene is not associated with a cAMP-activatedchloride conductance. Hum. Mol. Genet. 2, 225–230.
7. Cuppens, H., Lin, W., Jaspers, M., Costes, B., Teng, H.,Vankeerberghen, A. et al. (1998). Polyvariant mutantcystic fibrosis transmembrane conductance regulatorgenes. The polymorphic (Tg)m locus explains thepartial penetrance of the T5 polymorphism as adisease mutation. J. Clin. Invest. 101, 487–496.
8. Niksic, M., Romano, M., Buratti, E., Pagani, F. &Baralle, F. E. (1999). Functional analysis of cis-actingelements regulating the alternative splicing of humanCFTR exon 9. Hum. Mol. Genet. 8, 2339–2349.
9. Groman, J. D., Hefferon, T. W., Casals, T., Bassas, L.,Estivill, X., Des Georges, M. et al. (2004). Variation in arepeat sequence determines whether a commonvariant of the cystic fibrosis transmembrane conduct-ance regulator gene is pathogenic or benign. Am.J. Hum. Genet. 74, 176–179.
10. Buratti, E., Dork, T., Zuccato, E., Pagani, F., Romano,M. & Baralle, F. E. (2001). Nuclear factor TDP-43 andSR proteins promote in vitro and in vivo CFTR exon 9skipping. EMBO J. 20, 1774–1784.
11. Ou, S. H., Wu, F., Harrich, D., Garcia-Martinez, L. F. &Gaynor, R. B. (1995). Cloning and characterization of anovel cellular protein, TDP-43, that binds to humanimmunodeficiency virus type 1 TAR DNA sequencemotifs. J. Virol. 69, 3584–3596.
12. Buratti, E. & Baralle, F. E. (2001). Characterization andfunctional implications of the RNA bindingproperties of nuclear factor TDP-43, a novel splicingregulator of CFTR exon 9. J. Biol. Chem. 276,36337–36343.
13. Buratti, E., Brindisi, A., Pagani, F. & Baralle, F. E.(2004). Nuclear factor TDP-43 binds to the poly-morphic TG repeats in CFTR intron 8 and causesskipping of exon 9: a functional link with diseasepenetrance. Am. J. Hum. Genet. 74, 1322–1325.
14. McAfee, J. G., Huang, M., Soltaninassab, S., Rech, J. E.,Iyengar, S. & Lesturgeon, W. M. (1997). In EukaryoticmRNA Processing (Krainer, A. R., ed.), pp. 68–102,Oxford University Press, Oxford/New York.
15. Krecic, A. M. & Swanson, M. S. (1999). hnRNPcomplexes: composition, structure, and function.Curr. Opin. Cell Biol. 11, 363–371.
16. Burd, C. G. & Dreyfuss, G. (1994). Conservedstructures and diversity of functions of RNA-bindingproteins. Science, 265, 615–621.
17. Wang, H. Y., Wang, I. F., Bose, J. & Shen, C. K. (2004).Structural diversity and functional implications of theeukaryotic TDP gene family. Genomics, 83, 130–139.
18. Lukacsovich, T., Asztalos, Z., Juni, N., Awano, W. &Yamamoto, D. (1999). The Drosophila melanogaster 60Achromosomal division is extremely dense with func-tional genes: their sequences, genomic organization,and expression. Genomics, 57, 43–56.
19. Shamoo, Y., Krueger, U., Rice, L. M., Williams, K. R. &Steitz, T. A. (1997). Crystal structure of the two RNAbinding domains of human hnRNP A1 at 1.75 Aresolution. Nature Struct. Biol. 4, 215–222.
20. Oubridge, C., Ito, N., Evans, P. R., Teo, C. H. & Nagai,
K. (1994). Crystal structure at 1.92 A resolution of theRNA-binding domain of the U1A spliceosomalprotein complexed with an RNA hairpin. Nature,372, 432–438.
21. Nagai, K., Oubridge, C., Jessen, T. H., Li, J. & Evans,P. R. (1990). Crystal structure of the RNA-bindingdomain of the U1 small nuclear ribonucleoprotein A.Nature, 348, 515–520.
22. Deo, R. C., Bonanno, J. B., Sonenberg, N. & Burley,S. K. (1999). Recognition of polyadenylate RNA by thepoly(A)-binding protein. Cell, 98, 835–845.
23. Handa, N., Nureki, O., Kurimoto, K., Kim, I.,Sakamoto, H., Shimura, Y. et al. (1999). Structuralbasis for recognition of the tra mRNA precursor bythe Sex-lethal protein. Nature, 398, 579–585.
24. Nagata, T., Kurihara, Y., Matsuda, G., Saeki, J., Kohno,T., Yanagida, Y. et al. (1999). Structure and interactionswith RNA of the N-terminal UUAG-specific RNA-binding domain of hnRNP D0. J. Mol. Biol. 287,221–237.
25. Deirdre, A., Scadden, J. & Smith, C. W. (1995).Interactions between the terminal bases of mam-malian introns are retained in inosine-containing pre-mRNAs. EMBO J. 14, 3236–3246.
26. Wang, I. F., Reddy, N. M. & Shen, C. K. (2002). Higherorder arrangement of the eukaryotic nuclear bodies.Proc. Natl Acad. Sci. USA, 99, 13583–13588.
27. Kim, S. K., Lund, J., Kiraly, M., Duke, K., Jiang, M.,Stuart, J. M. et al. (2001). A gene expression map forCaenorhabditis elegans. Science, 293, 2087–2092.
28. Wang, X. & Tanaka Hall, T. M. (2001). Structural basisfor recognition of AU-rich element RNA by the HuDprotein. Nature Struct. Biol. 8, 141–145.
29. Theissen, H., Etzerodt, M., Reuter, R., Schneider, C.,Lottspeich, F., Argos, P. et al. (1986). Cloning of thehuman cDNA for the U1 RNA-associated 70Kprotein. EMBO J. 5, 3209–3217.
30. Krainer, A. R., Mayeda, A., Kozak, D. & Binns, G.(1991). Functional expression of cloned humansplicing factor SF2: homology to RNA-bindingproteins, U1 70K, and Drosophila splicing regulators.Cell, 66, 383–394.
31. Del Gatto-Konczak, F., Olive, M., Gesnel, M. C. &Breathnach, R. (1999). hnRNPA1 recruited to an exonin vivo can function as an exon splicing silencer. Mol.Cell. Biol. 19, 251–260.
32. Mayeda, A. & Krainer, A. R. (1992). Regulation ofalternative pre-mRNA splicing by hnRNP A1 andsplicing factor SF2. Cell, 68, 365–375.
33. Mayeda, A., Munroe, S. H., Caceres, J. F. & Krainer,A. R. (1994). Function of conserved domains ofhnRNP A1 and other hnRNP A/B proteins. EMBO J.13, 5483–5495.
34. Cartegni, L., Maconi, M., Morandi, E., Cobianchi, F.,Riva, S. & Biamonti, G. (1996). hnRNP A1 selectivelyinteracts through its Gly-rich domain with differentRNA-binding proteins. J. Mol. Biol. 259, 337–348.
35. Casas-Finet, J. R., Smith, J. D., Jr, Kumar, A., Kim, J. G.,Wilson, S. H. & Karpel, R. L. (1993). Mammalianheterogeneous ribonucleoprotein A1 and its con-stituent domains. Nucleic acid interaction, structuralstability and self-association. J. Mol. Biol. 229, 873–889.
36. Kumar, A. & Wilson, S. H. (1990). Studies of thestrand-annealing activity of mammalian hnRNPcomplex protein A1. Biochemistry, 29, 10717–10722.
37. Munroe, S. H. & Dong, X. F. (1992). Heterogeneousnuclear ribonucleoprotein A1 catalyzes RNA. RNAannealing. Proc. Natl Acad. Sci. USA, 89, 895–899.
38. Nadler, S. G., Merrill, B. M., Roberts, W. J., Keating,

588 Characterization of Human, Fly and Worm TDP43
K. M., Lisbin, M. J., Barnett, S. F. et al. (1991).Interactions of the A1 heterogeneous nuclear ribo-nucleoprotein and its proteolytic derivative, UP1,with RNA and DNA: evidence for multiple RNAbinding domains and salt-dependent binding modetransitions. Biochemistry, 30, 2968–2976.
39. Pontius, B. W. & Berg, P. (1990). Renaturation ofcomplementary DNA strands mediated by purifiedmammalian heterogeneous nuclear ribonucleoproteinA1 protein: implications for a mechanism for rapidmolecular assembly. Proc. Natl Acad. Sci. USA, 87,8403–8407.
40. Portman, D. S. & Dreyfuss, G. (1994). RNA annealingactivities in HeLa nuclei. EMBO J. 13, 213–221.
41. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989).Molecular Cloning: A Laboratory Manual, Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
42. Pagani, F., Buratti, E., Stuani, C., Romano, M.,Zuccato, E., Niksic, M. et al. (2000). Splicing factorsinduce cystic fibrosis transmembrane regulator exon 9skipping through a nonevolutionary conservedintronic element. J. Biol. Chem. 275, 21041–21047.
43. Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994).CLUSTAL W.: improving the sensitivity of pro-gressive multiple sequence alignment through
sequence weighting, position-specific gap penaltiesand weight matrix choice. Nucl. Acids Res. 22,4673–4680.
44. Sali, A. & Blundell, T. L. (1993). Comparative proteinmodelling by satisfaction of spatial restraints. J. Mol.Biol. 234, 779–815.
45. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallog. 26, 283–291.
46. Kranz, J. K. & Hall, K. B. (1999). RNA recognition bythe human U1A protein is mediated by a network oflocal cooperative interactions that create the optimalbinding surface. J. Mol. Biol. 285, 215–231.
47. Case, D. A., Pearlman, D. A., Caldwell, J. W.,Cheatham, T. E., III, Ross, W. S., Simmerling, C. L.et al. (1997). AMBER 5.0 edit., University of California,San Francisco.
48. Akila, M. & Krainer, A. R. (1999). In Methods inMolecular Biology (Haynes, S., ed.), vol. 118, pp. 315–321, Humana Press, Totowa, NJ.
49. Bradford, M. M. (1976). A rapid and sensitive methodfor the quantitation of microgram quantities ofprotein utilizing the principle of protein–dye binding.Anal. Biochem. 72, 248–254.
Edited by D. E. Draper
(Received 2 December 2004; received in revised form 17 February 2005; accepted 20 February 2005)
Top Related
Copyright © 2022 FDOKUMEN

