







Bahasa
Halaman
Hukum


BRAINA JOURNAL OF NEUROLOGY
Abnormal recruitment of extracellular matrixproteins by excess Notch3ECD: a newpathomechanism in CADASILMarie Monet-Lepretre,1,2 Iman Haddad,3 Celine Baron-Menguy,1,2 Maı Fouillot-Panchal,1,2
Meriem Riani,1,2 Valerie Domenga-Denier,1,2 Claire Dussaule,1,2 Emmanuel Cognat,1,2
Joelle Vinh3 and Anne Joutel1,2
1 INSERM, U740, Paris, F-75010, France
2 Univ Paris Diderot, Sorbonne Cite, UMR S740, Paris, F-75010, France
3 CNRS ESPCI ParisTech, USR3149, SMBP, Paris France
Correspondence to: Anne Joutel
Faculte de Medecine Paris Diderot,
site Villemin,
10 av de Verdun,
75010 Paris,
France
E-mail: [email protected]
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, or CADASIL, one of the most
common inherited small vessel diseases of the brain, is characterized by a progressive loss of vascular smooth muscle cells and
extracellular matrix accumulation. The disease is caused by highly stereotyped mutations within the extracellular domain of the
NOTCH3 receptor (Notch3ECD) that result in an odd number of cysteine residues. While CADASIL-associated NOTCH3 muta-
tions differentially affect NOTCH3 receptor function and activity, they all are associated with early accumulation of Notch3ECD-
containing aggregates in small vessels. We still lack mechanistic explanation to link NOTCH3 mutations with small vessel
pathology. Herein, we hypothesized that excess Notch3ECD could recruit and sequester functionally important proteins within
small vessels of the brain. We performed biochemical, nano-liquid chromatography-tandem mass spectrometry and immuno-
histochemical analyses, using cerebral and arterial tissue derived from patients with CADASIL and mouse models of CADASIL
that exhibit vascular lesions in the end- and early-stage of the disease, respectively. Biochemical fractionation of brain and
artery samples demonstrated that mutant Notch3ECD accumulates in disulphide cross-linked detergent-insoluble aggregates in
mice and patients with CADASIL. Further proteomic and immunohistochemical analyses identified two functionally important
extracellular matrix proteins, tissue inhibitor of metalloproteinases 3 (TIMP3) and vitronectin (VTN) that are sequestered into
Notch3ECD-containing aggregates. Using cultured cells, we show that increased levels or aggregation of Notch3 enhances the
formation of Notch3ECD–TIMP3 complex, promoting TIMP3 recruitment and accumulation. In turn, TIMP3 promotes complex
formation including NOTCH3 and VTN. In vivo, brain vessels from mice and patients with CADASIL exhibit elevated levels of
both insoluble cross-linked and soluble TIMP3 species. Moreover, reverse zymography assays show a significant elevation of
TIMP3 activity in the brain vessels from mice and patients with CADASIL. Collectively, our findings lend support to a Notch3ECD
cascade hypothesis in CADASIL disease pathology, which posits that aggregation/accumulation of Notch3ECD in the brain
vessels is a central event, promoting the abnormal recruitment of functionally important extracellular matrix proteins that
may ultimately cause multifactorial toxicity. Specifically, our results suggest a dysregulation of TIMP3 activity, which could
contribute to mutant Notch3ECD toxicity by impairing extracellular matrix homeostasis in small vessels.
doi:10.1093/brain/awt092 Brain 2013: 136; 1830–1845 | 1830
Received October 3, 2012. Revised January 21, 2013. Accepted February 20, 2013. Advance Access publication May 6, 2013
� The Author (2013). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved.
For Permissions, please email: [email protected]
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

Keywords: CADASIL; Notch3; protein aggregation; extracellular matrix proteins; cerebrovasculature
Abbreviations: CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy;Notch3ECD = NOTCH3 extracellular domain; RIPA = radio-immunoprecipitation assay
IntroductionSmall vessel disease of the brain is a major contributor to stroke
and a leading cause of vascular cognitive impairment in human
adults. Clinical manifestations result from the occurrence of
multiple subcortical lacunar infarctions and extensive white
matter injuries (Dichgans, 2007; Pantoni, 2010). CADASIL is the
most common causative diagnosis of hereditary small vessel dis-
ease of the brain caused by dominant mutations in the NOTCH3
receptor (Joutel et al., 1996; Chabriat et al., 2009). The under-
lying vasculopathy involves primarily the leptomeningeal and small
penetrating arteries and is characterized by progressive loss of
vascular smooth muscle cells, prominent thickening of the vessel
wall by various types of collagens and extracellular accumulation
of the so-called granular osmiophilic material (Ruchoux et al.,
1995; Tikka et al., 2009; Dong et al., 2012).
NOTCH3 receptor exists at the plasma membrane as a hetero-
dimer, which consists of a 210 kDa ectodomain (Notch3ECD) con-
taining 34 epidermal growth factor-like repeats, non-covalently
attached to a 97 kDa membrane tethered intracellular domain
(Notch3TMIC) (Joutel et al., 2000). In response to ligand binding,
NOTCH3, like the other Notch receptors, undergo sequential pro-
teolytic cleavages that release the Notch intracellular domain,
which translocates to the nucleus where it binds to the transcrip-
tion factor RBPJ and co-activators to activate the transcription of
genes (Kopan and Ilagan, 2009). Notch3 is predominantly
expressed in vascular smooth muscle cells of small arteries and
pericytes of brain capillaries. Studies in the mouse have demon-
strated a critical role for Notch3 in mural cell investment, arterial
differentiation and maturation of vascular smooth muscle cells of
small arteries (Domenga et al., 2004; Liu et al., 2010; Fouillade
et al., 2012).
Patients with CADASIL harbour highly stereotyped NOTCH3
mutations. The vast majority of mutations are missense mutations
and virtually all mutations hitherto reported result in an odd
number of cysteine residues within a given EGF-like repeat
(Joutel et al., 1997; Peters et al., 2005). Yet, the connection be-
tween NOTCH3 mutations and small vessel pathology is un-
known. On one hand, cell-based systems and in vivo studies
have shown that mutations differentially affect NOTCH3–RBPJ
activity, with a few mutations behaving as loss of function or
hypomorphic mutations and many others appearing not to
impair NOTCH3 receptor function and canonical signalling
(Joutel et al., 2004; Peters et al., 2004; Monet et al., 2007,
2009; Arboleda-Velasquez et al., 2011). Moreover, whether
impaired NOTCH3 activity might contribute to the disease pheno-
type is still a matter of debate because total loss of NOTCH3
in the mouse is not associated with a CADASIL phenotype
and genotype–phenotype correlation analyses suggest that
CADASIL-associated loss of function mutations are associated
with an attenuated clinical phenotype (Domenga et al., 2004;
Monet-Lepretre et al., 2009). On the other hand, the common
denominator to CADASIL-associated mutations is the presence of
non-fibrillar deposits of Notch3ECD and granular osmiophilic ma-
terial in the vessel wall. Notch3ECD accumulates as microscopic
extracellular aggregates around vascular smooth muscle cells and
brain pericytes and has been recently recognized as a component
of granular osmiophilic material (Joutel et al., 2000, 2001; Lesnik
Oberstein et al., 2003; Ishiko et al., 2006; Monet et al., 2007,
2009; Tikka et al., 2009; Joutel et al., 2010). The observation in
transgenic mouse models that Notch3ECD accumulation is one of
the earliest events in pathogenesis suggests that it might be the
proximate cause of cellular pathology (Monet et al., 2007, 2009;
Joutel et al., 2010). Herein we tested the hypothesis that excess
Notch3ECD can recruit and sequester functionally important pro-
teins. To achieve this, we used a combination of cerebral and
arterial tissue derived from patients with CADASIL and mouse
models of CADASIL to perform biochemical, nano-liquid chroma-
tography-tandem mass spectrometry (nanoLC-MS/MS) and
immunohistochemical analyses. Post-mortem CADASIL brain tis-
sues exhibit robust pathology, although in the end-stage of the
disease process, with prominent vascular smooth muscle cell
degeneration and vessel fibrosis, whereas material derived from
CADASIL mouse models shows fewer lesions, but, in the early
stage of the disease.
Materials and methodsAdditional information is provided in the Supplementary material.
Brain and artery samples
Human brain tissue
We used frozen or paraffin embedded samples (frontal, temporal or
occipital lobes) from seven deceased elderly patients with CADASIL
(mean age 62.8, range 49–70 years) with molecular genetically
confirmed NOTCH3 mutations (R153C, C1261R, R169C, R133C and
R110C) and nine deceased elderly control subjects (mean age 68,
range 48–86 years) with no known cerebrovascular disorders. All
human samples were stored and handled in accordance with the
French bioethics laws and the study was approved by the
Institutional Review Board of INSERM.
Human brain vessels
Microvessels were isolated from occipital or frontal lobes as described
previously (Yousif et al., 2007). Briefly, brain tissue was weighed
(1–2 g), minced with a scalpel and homogenized in cold PBS (20 ml)
with 20–40 up-and-down strokes in a glass homogenizer.
Homogenate was mixed with an equal volume of cold PBS containing
30% dextran. The suspension was then centrifuged at 6000g for
20 min at 4�C. The supernatant was discarded and the pellet was
resuspended in PBS containing 1% bovine serum albumin. The sus-
pension was then poured through a 40 mm nylon mesh, triturated and
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1831
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

abundantly washed with PBS. Microvessels were collected in PBS by
inversion of the nylon mesh and pelleted by centrifugation. Purity of
microvessel preparations was monitored by phase-contrast microscopy.
Murine brain vessels
We used TghNotch3(WT), TghNotch3(R90C) and TghNotch3(C428S)
mice, which express a human NOTCH3 transgene, with the wild-type
or mutant sequence, in the C57BL/6J background (Monet et al., 2007,
2009). The transgene in the TghNotch3(WT) and TghNotch3(R90C)
mice is expressed at the homozygous state. TgNotch3WT and
TgNotch3R169C are transgenic mice overexpressing the wild-type or
mutant rat Notch3 locus, respectively, at the heterozygous state, in
the FVB/N background (Joutel et al., 2010). Cerebral arteries, includ-
ing arteries of the circle of Willis and the medium-sized branches, were
dissected under microscope, immediately snap frozen in liquid nitrogen
and stored at �80�C until use. The experimental procedures con-
formed to the national guidelines for the use of animals in research
and were approved by the Ethics committee on animal experiment
(Local committee of University Paris Diderot, Lariboisiere-Villemin).
Biochemical proceduresAll extraction buffers contained a mixture of protease inhibitors
(CompleteTM
protease inhibitor mixture; Roche). For experiments
using quantitative amounts of protein, total protein concentration
was determined with the BCA protein assay kit (Pierce) using bovine
serum albumin as a standard.
Total vessel lysate
Brain vessels were homogenized using a ground glass homogenizer in
SDS buffer (2% SDS, 50 mM Tris, pH 7.4) with �30 strokes and
rotated at room temperature for 1 h.
Sequential biochemical fractionation
Human brain tissue samples (1.5–2.0 g) were homogenized on ice in
10 ml/g of buffer A (2 M NaCl, 10 mM HEPES/NaOH pH 7.4, 1 mM
EDTA, 10 mM N-methylmaleimide) using a Potter-Elvehjem glass/glass
homogenizer (30 strokes) and sedimented by ultracentrifugation at
100 000g for 1 h. The supernatant was saved as the high-salt (S1)
fraction and the resulting pellet (P1) was then homogenized in
10 ml/g of buffer B (100 mM NaCl, 10 mM HEPES/NaOH pH 7.4,
1 mM EDTA, 10 mM N-methylmaleimide) containing 2% SDS and
rotated at room temperature for 1 h before sedimentation at
100 000g at 15�C for 1 h. The supernatant was saved as the SDS
(S2)-soluble fraction and the remaining pellet (P2) was re-extracted
in SDS-Laemmli buffer (0.44 ml/g) with 10% 2-mercaptoethanol for
at least 24 h before centrifugation at 25 000g for 30 min at 12�C and
saved as the ‘SDS + ß-mercaptoethanol’ fraction. In another set of
experiments, the pellet P1 was resuspended in buffer B containing
either 1% TritonTM X-100 or 4 M urea or 6 M guanidine hydrochloride
or a combination of 7 M urea, 2 M thiourea and 3% CHAPS or was
extracted in 99% formic acid. All resuspended P1 were rotated for 1 h
and sedimented at 100 000g for 1 h. The supernatants were saved as
the S2 fractions and a final extraction using SDS-Laemmli buffer with
10% 2-mercaptoethanol was conducted on the resulting pellets (P2).
Human or murine brain vessels were homogenized on ice using a
ground glass homogenizer in radio-immunoprecipitation assay (RIPA)
buffer (150 mM NaCl, 50 mM Tris–HCl, 1% NP-40, 0.1% SDS, 0.5%
sodium deoxycholate supplemented with 10 mM N-methylmaleimide)
and centrifuged at 25 000g for 30 min at 4�C. The supernatant was
retained (S1), and the resultant insoluble pellet (P1) was extracted for
several hours in SDS buffer (3% SDS, 125 mM Tris–HCl, 10 mM
N-methylmaleimide) and centrifuged at 25 000g for 30 min at 11�C.
The supernatant was saved as the S2 fraction and a final extraction
using SDS-Laemmli buffer with 10% 2-mercaptoethanol or 20 mM
dithiothreitol was conducted on the resulting pellet (P2) before centri-
fugation at 25 000g for 30 min at 11�C. S1 and S2 fractions were
concentrated by acetone precipitation.
Western blot analyses
Samples were mixed with SDS-Laemmli buffer (4% SDS, 125 mM Tris–
HCl, pH 6.8, 20% glycerol) containing 10% 2-mercaptoethanol or
20 mM DTT, passed through a 29 gauge needle 15–20 times, clarified
by centrifugation at 25 000g for 30 min at 11�C and heated for 5 min
at 95�C before SDS-PAGE. Protein extracts were electrophoresed on a
6% Tris–glycine SDS-PAGE or 4–12% Bis-tricine NUPAGE (Invitrogen)
and transferred to a nitrocellulose membrane. The following primary
antibodies were used: mouse monoclonal anti-Notch3ECD (clone 5E1,
dilution 1:500; clone 11A1, dilution 1:1000; Joutel et al., 2000),
mouse monoclonal anti-vitronectin (Clone BV2, Chemicon, dilution
1:200), rabbit monoclonal anti-TIMP3 (clone D74B10, Cell Signaling,
dilution 1:2500), mouse monoclonal anti-smooth muscle alpha actin
(clone 1A4, Dako, dilution 1:25 000) and mouse monoclonal anti-ß
actin (clone AC15, Sigma, dilution 1:10 000). The blots were incubated
with the primary antibody at 4�C overnight followed by secondary
polyclonal anti-mouse or anti-rabbit immunoglobulins conjugated
with horseradish peroxidase and enhanced chemoluminescence detec-
tion (Thermo Fisher Scientific). Densitometric quantification of band
intensity was performed using ImageJ (version 10.2, NIH).
Reverse zymography assay
Equal amounts of total brain vessel lysates (8 mg) were mixed with
non-reducing Laemmli sample buffer and loaded onto a 12% SDS-
PAGE gel containing 1 mg/ml gelatin and conditioned medium from
baby hamster kidney cells expressing gelatinase A (MMP2), which is
inhibited by all four TIMP proteins (a generous gift from Dylan
Edwards). The gel was washed, incubated for 24 h in rinse buffer
(50 mM Tris pH 7.5, 5 mM CaCl2, 2.5% TritonTM X-100) at room
temperature, then incubated for 18 h in regenerating buffer (50 mM
Tris pH 7.5, 5 mM CaCl2) at 37�C, washed in water and stained with
Coomassie blue. Lysates of cells transfected with human TIMP1,
TIMP2 or TIMP3 were used as controls. Under these conditions,
TIMPs inhibit gelatin digestion by activated gelatinase A, producing
dark blue bands against a lighter background. Identical samples were
run in parallel onto a 4–12% NUPAGE gel, transferred and immuno-
blotted with ß-actin antibody. Densitometric quantification of band
intensity was performed using ImageJ and normalized against ß-actin
signal.
Sample preparation and massspectrometry analysisHuman brain samples and murine brain artery samples were fractio-
nated using RIPA and then SDS-containing buffer. Each murine sample
was prepared with vessels pooled from four transgenic mice. The final
pellet was extracted in 40 ml of Laemmli buffer containing 20 mM
dithiothreitol between 20–48 h and clarified by centrifugation. Thirty
microlitres of protein extracts were denatured by incubation at 95�C
for 5 min, electrophoresed on a 4–12% NUPAGE Bis-tricine gel run for
12 min at 200 V. The gel was recovered, washed in water, stained with
Coomassie blue (Bio-Rad) and extensively washed in water for 2 to 3 h
at room temperature (three changes). Each lane was divided in three
(murine samples) or five (human samples) parts based on size, cut into
1832 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

1 mm cubes and put into 1% acetic acid. Hence, each sample further
comprised three or five subsamples.
In-gel tryptic digestion method was used on the purified samples as
described (Shevchenko, 2001). Briefly, after reduction–alkylation
(5 mM dithiothreitol in 50 mM NH4HCO3, 30 min at 56�C; 25 mM
iodoacetamide in 50 mM NH4HCO3, 20 min in dark at room tempera-
ture), gel pieces were digested by incubation with 12.5 ng/ml Trypsin
(modified sequencing grade, Roche) in sodium carbonate, overnight at
37�C with gentle shaking. The reaction was stopped with one volume
(50 ml) 5% formic acid. Subsamples were sonicated for 10 min in an
ultrasonic bath at room temperature and processed for nanoLC-MS/
MS analysis as described in the Supplementary material or stored at
�20�C until use.
Immunohistochemical analysesThe following antibodies (mouse monoclonal anti-vitronectin, Clone
BV2, Chemicon, dilution 1:200; mouse monoclonal anti-TIMP3,
Clone 13613H4, Chemicon, dilution 1:1000) were applied to paraffin
sections of human brain as described in the Supplementary material.
The following antibodies (rabbit polyclonal anti-vitronectin, Oxford
biomedical research, dilution 1:1000 and Genway, dilution 1:1000;
mouse monoclonal anti-TIMP3, Clone 13613H4, Chemicon, dilution
1:200; mouse monoclonal anti-Notch3ECD, clone 5E1, dilution 1:2;
polyclonal anti-Notch3ECD, BC2, dilution 1:1000) were used on
frozen brain sections as described in the Supplementary material.
Immuno-electron microscopy analysisFrozen brain sections were incubated with the mouse monoclonal anti-
TIMP3 (Clone 13613H4, dilution 1:50) and processed as described in
the Supplementary material.
In situ hybridizationA 951 bp human TIMP3 probe (nucleotides, 3964–4915 gi: 75905820)
was made by PCR amplification and cloned into pBluescript�. In situ
hybridization was performed on brain paraffin sections from patients
with CADASIL (n = 3) and control individuals (n = 3) as described in
the Supplementary material.
Expression plasmids, cell culture andco-immunoprecipitation analysis
Expression plasmids
Full-length wild-type NOTCH3, NOTCH3 deletion and point muta-
tions, full-length TIMP3 and deletion mutants, and vitronectin con-
structs (Supplementary Fig. 5) were generated as described in the
Supplementary material.
Cell culture and transfection
HEK 293T cells were grown in GlutamaxTM supplemented with 10%
foetal bovine serum and 1% penicillin–streptomycin. Human coronary
artery smooth muscle cells (Cascade Biologics) were grown in Medium
231 supplemented with Smooth Muscle Growth Supplement (Cascade
Biologics). Plasmids were transfected into subconfluent 293T cells by
using the calcium phosphate precipitation method as described previ-
ously (Joutel et al., 2004). For co-immmunoprecipitation, 293T cells
were grown in 50 cm2 plates and transfected with 20 ng of haem-
agglutinin-tagged TIMP3, 1 mg of haemagglutinin or V5-tagged
vitronectin and 1 mg of FLAG�-tagged NOTCH3 plasmids. For
co-expression experiments, 293T cells were transfected with 200 ng
haemagglutinin-tagged TIMP3 and 1 mg of FLAG�-tagged NOTCH3
plasmids. At 48 h after transfection, the cells were harvested and the
extracts prepared for downstream assays.
Co-immunoprecipitation, cellular fractions and immu-noblot analyses
293T cells were lysed in immunoprecipitation buffer (0.5% TritonTM
X-100, 10 mM TrisCl pH 8, 140 mM NaCl) supplemented with a cock-
tail of protease inhibitors. Immunoprecipitation was performed using
anti-FLAG M2, anti-haemagglutinin or anti-V5 affinity gel (Sigma-
Aldrich). Endogenous NOTCH3 and TIMP3 were immunoprecipitated
from human coronary artery smooth muscle cells with anti-Notch3ECD
rabbit polyclonal antibody (BC2) (Joutel et al., 2000), rabbit immuno-
globulins were used as a control. To prepare cellular fractions, 293T
cells were dislodged from the culture plate in Ca2 + , Mg2 + free PBS,
harvested and lysed in immunoprecipitation or RIPA buffer. The cul-
ture plate was then rinsed several times in PBS, incubated in PBS
containing 5 mM EDTA for 15 min, followed by a final rinse in PBS
and the extracellular matrix was scraped in a small volume of Laemmli
buffer.
Protein extracts were separated on 4–12% NUPAGE gels (Invitrogen)
and transferred onto nitrocellulose membranes. Epitope tags were de-
tected with rabbit polyclonal anti-FLAG� (Sigma-Aldrich dilution,
1:50,000), rabbit polyclonal anti-haemagglutinin (Sigma-Aldrich, dilu-
tion 1:5000) or rabbit polyclonal anti-c-myc, peroxydase conjugate
(Sigma-Aldrich, dilution 1:5,000), vitronectin with rabbit polyclonal
anti-vitronectin (GenWay, dilution 1:5000), followed by secondary poly-
clonal anti-rabbit or anti-mouse immunoglobulins conjugated with
horseradish peroxidase and enhanced chemoluminescence detection
(Thermo Fisher Scientific). Samples of extracellular matrix were run in
parallel on 4–12% NUPAGE gels stained with silver stain (Pierce Silver
stain, Thermo Fisher Scientific). Densitometric quantification of band in-
tensity was performed using ImageJ.
Statistical analysisData are expressed as mean � standard error of the mean (SEM).
Two-group comparisons were analysed by the two-tailed t-test for
independent samples. Multiple comparisons were evaluated by one-
factor ANOVA followed by post hoc test. Results with a P-value
50.05 were considered statistically significant.
Results
Mutant Notch3ECD accumulates indisulphide cross-linkeddetergent-insoluble aggregatesWe first analysed the biochemical properties of Notch3ECD in
human derived material. We performed serial extractions on
brain samples from patients with CADASIL and control subjects
using buffers of increasing protein extraction strength (Fig. 1A).
Consistent with our previous report (Joutel et al., 2000),
Notch3ECD was almost undetectable in the control whereas it ro-
bustly accumulated in the CADASIL sample. In the patient, little or
no Notch3ECD was detected in the high salt and SDS-soluble frac-
tions, wheras a substantial amount was present in the SDS plus
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1833
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from
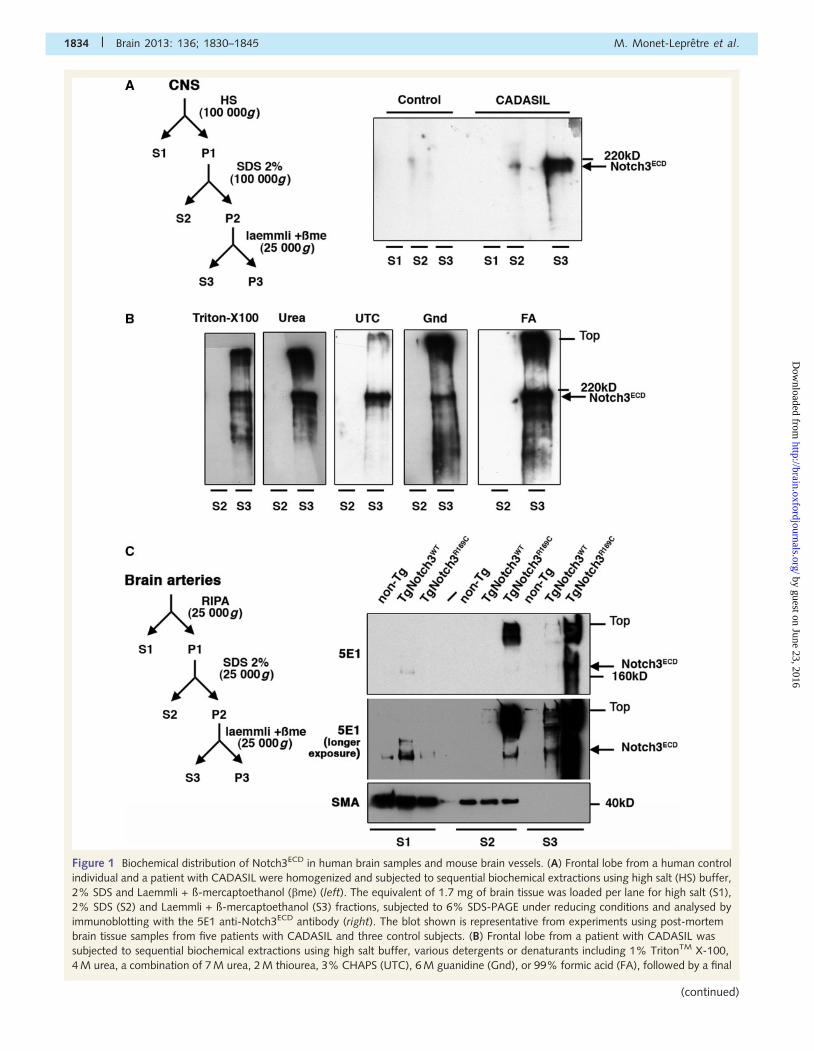
Figure 1 Biochemical distribution of Notch3ECD in human brain samples and mouse brain vessels. (A) Frontal lobe from a human control
individual and a patient with CADASIL were homogenized and subjected to sequential biochemical extractions using high salt (HS) buffer,
2% SDS and Laemmli + ß-mercaptoethanol (bme) (left). The equivalent of 1.7 mg of brain tissue was loaded per lane for high salt (S1),
2% SDS (S2) and Laemmli + ß-mercaptoethanol (S3) fractions, subjected to 6% SDS-PAGE under reducing conditions and analysed by
immunoblotting with the 5E1 anti-Notch3ECD antibody (right). The blot shown is representative from experiments using post-mortem
brain tissue samples from five patients with CADASIL and three control subjects. (B) Frontal lobe from a patient with CADASIL was
subjected to sequential biochemical extractions using high salt buffer, various detergents or denaturants including 1% TritonTM X-100,
4 M urea, a combination of 7 M urea, 2 M thiourea, 3% CHAPS (UTC), 6 M guanidine (Gnd), or 99% formic acid (FA), followed by a final
1834 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
(continued)
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

reducing agent (Laemmli + ß-mercaptoethanol) fraction (Fig. 1A).
The data were confirmed using brain samples from two other
control subjects and four patients (Supplementary Fig. 1). We
then assessed buffers containing other detergents and denatur-
ants, including TritonTM X-100 (1%), formic acid, urea (4 M),
guanidine (6 M), or a mixture of urea (7 M), thiourea (2 M) and
CHAPS (3%), for their ability to extract Notch3ECD. Strikingly, we
found that none of these detergent/denaturants could solubilize
Notch3ECD whereas Notch3ECD was recovered from the resultant
pellet in SDS buffer containing a reducing agent (Fig. 1B).
We extended our analysis to isolated brain arteries of CADASIL
mouse models. TgNotch3WT and TgNotch3R169C mice overexpress
a rat Notch3 transgene �4-fold that of the endogenous Notch3
(Joutel et al., 2010). In TgNotch3WT mice, a low amount of
Notch3ECD was equally extractable in RIPA and SDS containing
ß-mercaptoethanol buffers. In contrast, in TgNotch3R169C mice,
there was a dramatic accumulation of Notch3ECD in the SDS con-
taining ß-mercaptoethanol fraction and to a lesser extent in the
SDS-soluble fraction whereas Notch3ECD was barely extractable in
RIPA buffer alone (Fig. 1C).
We also analysed biochemical properties of Notch3ECD in
TghNotch3(R90C) and TghNotch3(C428S) mutant mice and the
control TghNotch3(WT) mice that express physiological levels of a
human NOTCH3 transgene (�1.5-fold over the endogenous
mouse Notch3). R90C and C428S are active and inactive
NOTCH3–RPBJ mutants, respectively (Joutel et al., 2004; Monet
et al., 2007, 2009). Of interest, the transgene in these mice can
be specifically recognized using a monoclonal antibody specific to
human Notch3ECD (Ruchoux et al., 2003). We found that, in wild-
type arteries, Notch3ECD was predominantly recovered in RIPA
buffer and that the majority of both human and endogenous
Notch3ECD was detected in this fraction (Supplementary Fig. 2A
and B). By contrast, in both TghNotch3(R90C) and
TghNotch3(C428S) mice, mutant Notch3ECD was poorly extract-
able in RIPA buffer whereas it accumulated in the SDS + ß-mer-
captoethanol fraction (Supplementary Fig. 2B). Taken together,
the data suggest that mutant Notch3ECD is incorporated into
higher order multimers, cross-linked by disulphide bonds.
TIMP3 and vitronectin are enriched inthe Notch3ECD-enriched fraction ofCADASIL samplesNext, we determined the proteome of the Notch3ECD-enriched frac-
tion, hypothesizing that proteins sequestered by Notch3ECD should
be enriched in this fraction. To achieve this, we performed a sensitive
and semi-quantitative proteomic analysis using nanoLC-MS/MS and
the spectral counting approach (Liu et al., 2004). We first conducted
a pilot proteomic study using post-mortem brain tissue from one
patient with CADASIL and one control subject. Consistent with the
claim that we were surveying the Notch3ECD enriched fraction, the
CADASIL sample contained 18 distinct peptides of NOTCH3, exclu-
sively derived from the Notch3ECD sequence, whereas the control
sample contained none. We identified a total of 323 and 281 proteins
in the control and CADASIL samples, respectively, 104 of which were
enriched in the CADASIL sample. Among these, 72 proteins, includ-
ing predominantly extracellular matrix proteins, were present in the
CADASIL sample and absent in the control (Supplemental Table 1).
To identify proteins that were differentially expressed at the
early stage of the disease process, we repeated the proteomic ana-
lysis using brain arteries from 10–12 month old wild-type
TghNotch3(WT) and mutant transgenic TghNotch3(R90C) mice.
Proteomic experiments conducted on three pairs of wild-type and
mutant samples identified 39 and 48 proteins in wild-type and
mutant samples, respectively, proteins being identified by the con-
sensus of two or more unique peptides in at least two of three bio-
logical replicates (Supplementary Table 2). Again, we identified eight
distinct peptides of NOTCH3, exclusively derived from the
Notch3ECD sequence, in the mutant samples and none in the wild-
type samples. Importantly, two proteins, TIMP3 and VTN, identified
as strongly enriched in the human CADASIL sample, were present in
the mutant arteries and almost absent in the control arteries
(Supplementary Table 2 and Supplementary Fig. 3).
Immunoblot analysis confirmed that TIMP3 and VTN were en-
riched in the SDS plus reducing agent (Laemmli + ß-mercap-
toethanol) fraction of human CADASIL brains when compared
with analogous fraction of control brains (Supplementary Fig. 4A
and B). Likewise, Laemmli + ß-mercaptoethanol fractions prepared
from arterial samples derived from 10–12 month old mice showed
an accumulation of TIMP3 and VTN in the mutant mice compared
with control mice (Supplementary Fig. 4C and D).
TIMP3 and vitronectin are incorporatedinto Notch3ECD-containing depositsWe next evaluated the expression pattern of TIMP3 and VTN in
brain tissue from patients and control subjects and from mouse
models by immunohistochemistry. Examination of CADASIL brain
tissue revealed robust and granular immunoreactivity in the media
of pial and penetrating white matter arteries, with both anti-vitro-
nectin (Fig. 2B and C) and anti-TIMP3 (Fig. 3B and C) antibodies,
bearing a striking resemblance to that seen with anti-Notch3ECD
Figure 1 Continuedextraction with Laemmli + ß-mercaptoethanol. The detergent/denaturant (S2) and corresponding Laemmli + ß-mercaptoethanol (S3)
fractions were analysed by immunoblotting with the 5E1 antibody. The equivalent of 34 mg of brain tissue was loaded per lane for the
formic acid-soluble and insoluble fractions and equivalent of 1.7 mg of brain tissue for all other fractions. (C) Brain arteries from non-
transgenic (non-Tg), TgNotch3WT and TgNotch3R169C mice aged 6 months were subjected to sequential biochemical extractions using
RIPA, 2% SDS and Laemmli + ß-mercaptoethanol (left). Ten micrograms of proteins were loaded per lane for RIPA fractions (S1), 4 mg for
2% SDS fractions (S2) and the resultant 2% SDS-insoluble/Laemmli + ß-mercaptoethanol-extractable fractions (S3) were loaded directly,
and subjected to immunoblot with the 5E1 antibody (upper and middle panels) and smooth muscle actin antibody (SMA, lower panel). A
representative blot from at least two independent experiments is shown.
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1835
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

antibody (Joutel et al., 2000). In contrast, vessels of control sub-
jects exhibited a weak and homogeneous staining pattern (Figs 2A
and 3A). Likewise, immunofluorescence analysis revealed the pres-
ence of VTN accumulation in the form of dot-like structures within
the brain vessels of TgNotch3R169C mice that was absent in control
TgNotch3WT mice (Fig. 2D–L). Granular VTN immunoreactivity
was detectable in the pial and intracerebral arteries as early as
12 months of age (data not shown), but was marked at 20
months of age (Fig. 2G), whereas it was already obvious in the
brain capillaries at 12 months of age (Fig. 2J). Of note, Notch3ECD
starts to form microscopic aggregates earlier in these mice,
between 1 and 6 months of age (Joutel et al., 2010).
Importantly, we found significant co-localization of VTN and
TIMP3 with Notch3ECD-positive aggregates in brain vessels of
TgNotch3R169C mice and patients with CADASIL, respectively
(Fig. 2I, L and 3F). Owing to the lack of suitable antibodies, we
Figure 2 VTN deposits colocalize with Notch3ECD aggregates. (A–C) Paraffin brain sections from control (A) and CADASIL subjects (B and
C) were stained with the monoclonal anti-VTN antibody (�-Vtn), which robustly detects VTN deposits into the media of CADASIL brain
vessels. The images shown are representative of at least two sections from an experiment using four patients with CADASIL and four
control subjects. (D–L) Frozen brain sections from TgNotch3WT (D–F) and TgNotch3R169C mice (G–L) were stained with anti-Vitronectin
and anti-Notch3ECD antibodies and analysed by immunofluorescence. Representative sections of a pial artery from a 20-month-old
TgNotch3WT mouse (D–F), of two adjacent pial arteries cut longitudinally from a 20-month-old TgNotch3R169C mouse (G–I) and from
capillaries of a 12-month-old TgNotch3R169C mouse (J–L) are shown. Panels are representative of at least six sections from two experi-
ments using three mutant and three control mice. Scale bars: 50 mm (A–C), 26 mM (D–I) and 15 mM (J–L).
1836 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

could not analyse TIMP3 distribution in murine tissues, nor
could we examine Notch3ECD and VTN co-localization in human
tissues.
To assess the relationship between Notch3ECD and TIMP3
deposits in more detail, brain sections were processed for
immuno-electron microscopy analysis. Fine localization of VTN
by this technique was hampered by a lack of suitable antibodies.
As shown in Fig. 3H, immunogold labelling of Notch3ECD was
visible at the plasma membrane of vascular smooth muscle cells
in the vicinity of granular osmiophilic material and also within the
granular osmiophilic material deposits, consistent with a recent
report suggesting that Notch3ECD can accumulate into the granu-
lar osmiophilic material (Ishiko et al., 2006). Notably, TIMP3
immunogold labelling was detected in the granular osmiophilic
material deposits, although the silver-enhanced gold particles
were seen mainly at the perimeters of granular osmiophilic mater-
ial and almost absent from the core area (Fig. 3G). Nevertheless,
we acknowledge that the pre-embedding method used for this
Figure 3 TIMP3 and Notch3ECD are incorporated into granular osmiophilic material deposits. (A–C) Paraffin brain sections from control
(A) and CADASIL subjects (B and C) were stained with the monoclonal anti-TIMP3 antibody, which reliably detects TIMP3 deposits in
CADASIL brain vessels. The images shown are representative of at least two sections from an experiment using four patients with
CADASIL and four control subjects. (D–F) Frozen brain sections from CADASIL subjects were stained with anti-TIMP3 and anti-Notch3ECD
antibodies and analysed by immunofluorescence. The images shown are representative of at least four sections from two experiments
using material from three patients with CADASIL. (G and H) Frozen brain sections from a CADASIL subject were processed for immuno-
electron microscopy with anti-TIMP3 (G) or anti-Notch3ECD (H) antibodies. Asterisks indicate granular osmiophilic material deposits.
Immunolabelling appears as black dots (arrows). Panels are representative of at least three tissue blocks from two experiments.
SMC = smooth muscle cell. Scale bars: 50 mm (A–C), 60 mM (D–F) and 1 mM (G and H).
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1837
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

experiment may have restricted the accessibility of the antibody
and may have not provided with the optimal signal. These results,
taken together, indicate that VTN and TIMP3 are recruited into
CADASIL deposits in vivo.
Increased levels or aggregation ofNOTCH3 enhances NOTCH3–TIMP3complex formation and TIMP3 promotescomplex formation including NOTCH3and VTNNext, we investigated whether TIMP3 and VTN complex with
Notch3ECD, using co-immunoprecipitation assays. We found that a
robust amount of haemagglutinin-tagged TIMP3 (TIMP3-HA) was
pulled down by anti-FLAG antibody in the presence of FLAG-tagged
wild-type NOTCH3 (Flag-N3-WT) but not in the presence of the
NOTCH3 deletion mutant lacking all 34 EGF-like repeats (Flag-
N3�EGFR1-34) (Fig. 4A). Similar results were obtained in the reverse
co-immunoprecipitation experiments (Fig. 4D). Significantly, the
CADASIL-associated mutations in NOTCH3 (R90C, C212S and
C428S) complexes with TIMP3, in a manner similar to wild-type
NOTCH3 (Fig. 4B). In contrast, a very low amount of wild-type
and mutant NOTCH3 co-immunoprecipitated with V5-tagged vitro-
nectin (VTN-V5) using anti-V5 antibody (Fig. 4C). Moreover, no
Flag-N3-WT co-immunoprecipitated with haemagglutinin-tagged
vitronectin (VTN-HA) using the anti-haemagglutinin (HA) antibody
whereas a large amount of Flag-N3-WT co-immunoprecipitated
with TIMP3-HA, confirming that VTN had a much lower affinity
for Notch3ECD than TIMP3 in this assay (Fig. 4D).
We then tested the possibility that TIMP3 complexes with VTN.
We found that anti-V5 could pull down TIMP3-HA in the pres-
ence of VTN-V5 but not in its absence, (Fig.4E). Importantly, Flag-
N3FL-WT could pull down a larger amount of VTN-V5 in the
presence of exogenous TIMP3-HA (Fig. 4E).
It is worth considering that there is an inevitable accumulation of
intracellular Notch3 aggregates in cells transfected with wild-type or
mutant Notch3, even at low expression levels (Opherk et al., 2009).
To determine whether NOTCH3 and TIMP3 associate in the same
complex when expressed at physiological levels, we performed co-
immunoprecipitation of cultured human coronary smooth muscle
cells using anti-Notch3ECD antibody; we were unable to perform
the reverse co-immunoprecipitation assay owing to the lack of suit-
able TIMP3 antibodies. We found that Notch3ECD was able to co-
immunoprecipitate a tiny amount of TIMP3 (Fig. 4F). Collectively the
results suggest that increased levels or aggregation of Notch3ECD
enhances NOTCH3–TIMP3 complex formation and that TIMP3 pro-
motes complex formation including NOTCH3 and VTN. Hence,
TIMP3 was selected for further studies.
Increased levels or aggregation ofNotch3ECD promote TIMP3 recruitmentand accumulationWe next asked whether increased levels or aggregation of
Notch3ECD could influence the expression level and subcellular
localization of TIMP3 using HEK293T cells expressing TIMP3-HA
alone or in combination with FLAG-tagged NOTCH3. It is
important to consider that in this assay NOTCH3 is primarily over-
expressed intracellularly (data not shown). Upon overexpression of
full-length wild-type NOTCH3 we found that the steady-state
level of TIMP3 protein was increased in the cell lysate by 2.6-
fold relative to cells expressing TIMP3 alone (Fig. 5A). TIMP3 is
a secreted protein that distinguishes itself from TIMP1, TIMP2 and
TIMP4 by its ability to bind the extracellular matrix (Lee et al.,
2007). Quantification of TIMP3 bound to the extracellular matrix,
where Notch3ECD is not present (data not shown), revealed a 2.3-
fold decrease in cells overexpressing NOTCH3 versus cells express-
ing TIMP3 alone (Fig. 5B). Significantly, the familial linked
CADASIL R90C mutant modulated TIMP3 protein levels in a
manner similar to wild-type NOTCH3 (Fig. 5A and B).
Interestingly, NOTCH3 protein level was unchanged on overex-
pression of TIMP3 (Supplementary Fig. 6). Importantly, we
checked that transiently transfected cells expressed comparable
levels of TIMP3-HA messenger RNA in these assays (Fig. 5A
and data not shown). Thus, these data indicate that excess
NOTCH3 can promote the recruitment and accumulation of
TIMP3 protein, in the cellular compartment where it is
overexpressed.
We next examined whether NOTCH3-induced TIMP3 protein
level changes required NOTCH3–TIMP3 complex formation.
TIMP3 protein level was unaffected on overexpression of the
NOTCH3 deletion mutant, N3�EGFR1-34, which is unable to
complex with TIMP3 (Fig. 5C). On the other hand, a NOTCH3
construct containing the ectodomain only (Notch3ECD) strongly
upregulated the levels of TIMP3 (Supplementary Fig. 7).
Moreover, a TIMP3 mutant lacking the N-terminal half (TIMP-
3�NTD-HA), which is unable to complex with NOTCH3
(Supplementary Fig. 8), had unchanged expression on overexpres-
sion of NOTCH3 (Fig. 5D), whereas the steady-state level of a
TIMP3 mutant lacking the C-terminal half (TIMP-3�CTD-HA),
which retains the ability to form a complex with NOTCH3, was
increased on NOTCH3 overexpression (Fig. 5E and Supplementary
Fig. 8). Hence, these data suggest that NOTCH3-induced
TIMP3 accumulation requires NOTCH3–TIMP3 complex forma-
tion and that NOTCH3 signalling activity is dispensable for this
effect.
TIMP3 protein level and biologicalactivity in CADASIL brain vesselsWe assessed whether Notch3ECD accumulation/aggregation af-
fects TIMP3 expression and activity in vivo, in brain vessels.
First, microvessels were isolated from post-mortem brain samples
of patients with CADASIL and control subjects and analysed by
western blot. Quantification of total levels of TIMP3 in lysates
from brain vessels revealed a 14.4-fold increase in patients with
CADASIL compared with control subjects. Notably, there was a
dramatic accumulation of high molecular weight TIMP3 species
ranging from 30 kDa to the top of the gel, which likely correspond
to TIMP3 aggregates that were not detected in control subjects
(Fig. 6A). Sequential extractions in RIPA buffer, then SDS buffers
1838 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from
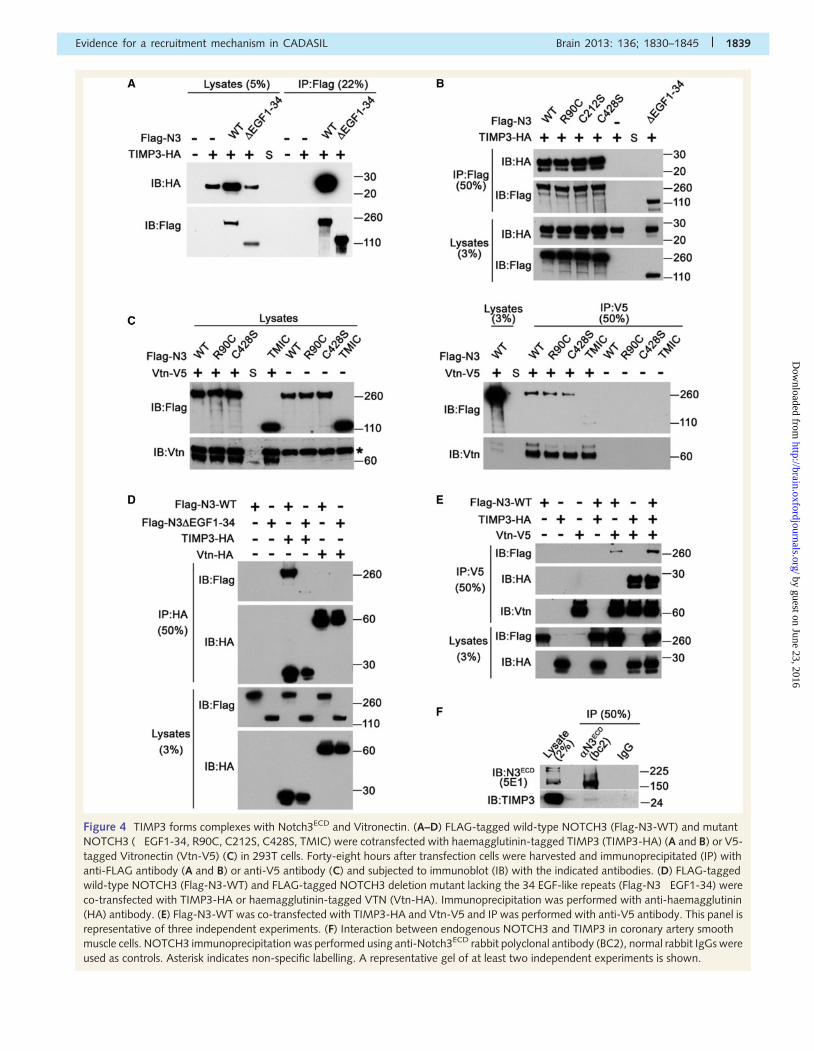
Figure 4 TIMP3 forms complexes with Notch3ECD and Vitronectin. (A–D) FLAG-tagged wild-type NOTCH3 (Flag-N3-WT) and mutant
NOTCH3 (�EGF1-34, R90C, C212S, C428S, TMIC) were cotransfected with haemagglutinin-tagged TIMP3 (TIMP3-HA) (A and B) or V5-
tagged Vitronectin (Vtn-V5) (C) in 293T cells. Forty-eight hours after transfection cells were harvested and immunoprecipitated (IP) with
anti-FLAG antibody (A and B) or anti-V5 antibody (C) and subjected to immunoblot (IB) with the indicated antibodies. (D) FLAG-tagged
wild-type NOTCH3 (Flag-N3-WT) and FLAG-tagged NOTCH3 deletion mutant lacking the 34 EGF-like repeats (Flag-N3�EGF1-34) were
co-transfected with TIMP3-HA or haemagglutinin-tagged VTN (Vtn-HA). Immunoprecipitation was performed with anti-haemagglutinin
(HA) antibody. (E) Flag-N3-WT was co-transfected with TIMP3-HA and Vtn-V5 and IP was performed with anti-V5 antibody. This panel is
representative of three independent experiments. (F) Interaction between endogenous NOTCH3 and TIMP3 in coronary artery smooth
muscle cells. NOTCH3 immunoprecipitation was performed using anti-Notch3ECD rabbit polyclonal antibody (BC2), normal rabbit IgGs were
used as controls. Asterisk indicates non-specific labelling. A representative gel of at least two independent experiments is shown.
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1839
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

showed, as expected, an accumulation of TIMP3 in the SDS-in-
soluble fraction from brain vessels of patients with CADASIL.
Strikingly, we also detected elevated levels of RIPA and
SDS-soluble forms of TIMP3 in the vessels of patients with
CADASIL (Fig. 6B). Of note, in these later vessels, Notch3ECD
accumulated exclusively in the SDS-insoluble fraction.
In situ hybridization on human brain sections from control indi-
viduals revealed that vascular cells are the predominant source of
TIMP3 messenger RNA in the brain, the highest expression being
detected in the small penetrating vessels. Importantly, we found
no overt difference in the level of TIMP3 messenger RNA in the
vessels from patients with CADASIL versus vessels from control
individuals indicating that the observed increase in TIMP3 protein
levels was unlikely due to a transcriptional effect but rather
resulted from increased translational efficiency or compromised
protein degradation (Fig. 6C).
Figure 5 NOTCH3 influences TIMP3 protein levels in a manner requiring NOTCH3–TIMP3 complex formation. (A and B)
Haemagglutinin-tagged TIMP3 (TIMP3-HA) was transfected alone or in combination with wild-type (Flag-N3-WT) or mutant R90C
FLAG-tagged NOTCH3 (Flag-N3-R90C). Forty-eight hours after transfection cells (A) and extracellular matrix (B) were harvested. Cell
lysates and extracellular matrix were subjected to immunoblot analysis with the indicated antibodies or to silver staining (top panels).
Relative TIMP3-HA messenger RNA and TIMP3-HA protein levels were determined (bottom panels). Shown are relative TIMP3-HA
messenger RNA levels normalized to G6PD (A, left), relative TIMP3-HA protein levels in the cell lysate normalized to ß-actin (A, middle) or
NOTCH3 (A, right) and relative TIMP3-HA protein levels in the extracellular matrix normalized to total protein content (B) (n = 3) (S,
molecular weight standard). (C) FLAG-tagged NOTCH3 deleted of the 34 EGF-like repeats (Flag-N3�EGF1-34) does not affect TIMP3-
HA steady-state levels in the cell lysate compared with cells transfected with TIMP3-HA alone and to cells co-expressing TIMP3-HA and
Flag-N3-WT. Bottom: Relative TIMP3-HA messenger RNA levels normalized to G6PD (C, left) and relative TIMP3-HA protein levels in the
cell lysate normalized to ßactin (C, right) (n = 3). (D and E) Flag-N3-WT does not affect TIMP3�NTD-HA steady-state levels (D) but
increases TIMP3�CTD-HA steady-state levels (E). Bottom panels, relative messenger RNA levels of TIMP3�NTD-HA (D, left) and of
TIMP3�CTD-HA (E, left) normalized to G6PD and relative intracellular protein levels of TIMP3�NTD-HA (D, right) and of TIMP3�CTD-
HA (E, right) normalized to ß-actin (n = 3). *P50.05, **P5 0.01 and ***P50.001, ANOVA with Tukey post hoc analysis (A and C) or
student’s test (D and E). ns = not significant.
1840 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from
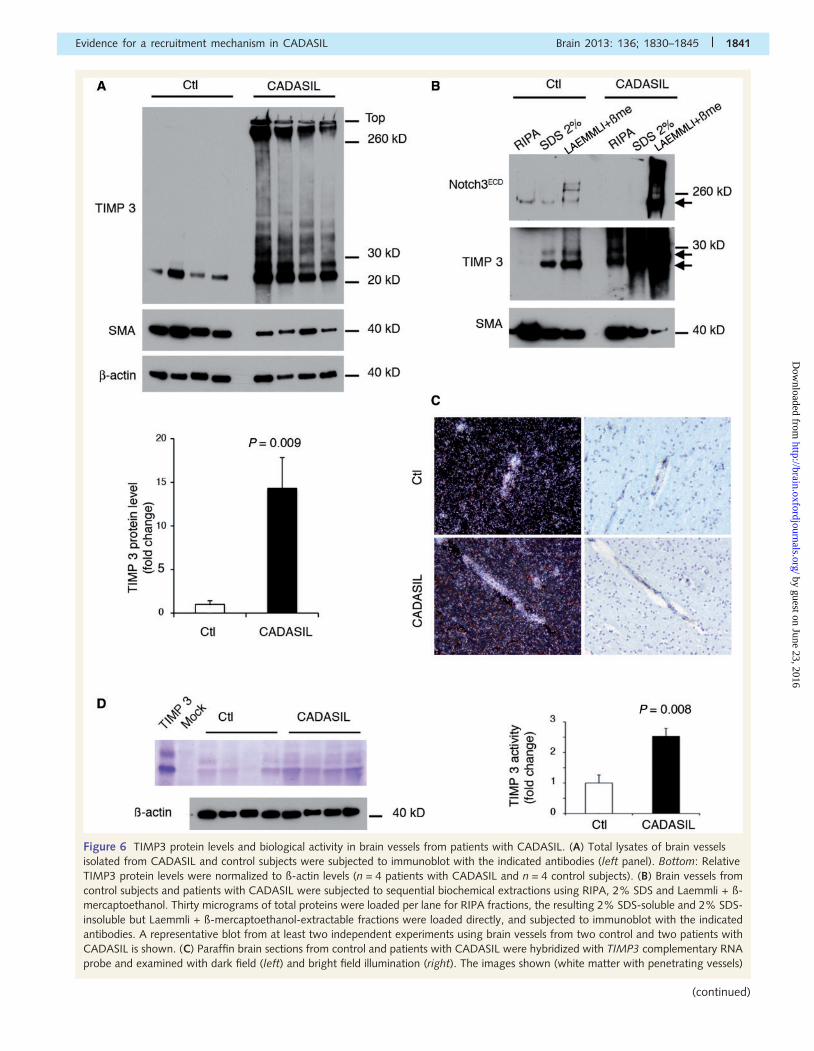
Figure 6 TIMP3 protein levels and biological activity in brain vessels from patients with CADASIL. (A) Total lysates of brain vessels
isolated from CADASIL and control subjects were subjected to immunoblot with the indicated antibodies (left panel). Bottom: Relative
TIMP3 protein levels were normalized to ß-actin levels (n = 4 patients with CADASIL and n = 4 control subjects). (B) Brain vessels from
control subjects and patients with CADASIL were subjected to sequential biochemical extractions using RIPA, 2% SDS and Laemmli + ß-
mercaptoethanol. Thirty micrograms of total proteins were loaded per lane for RIPA fractions, the resulting 2% SDS-soluble and 2% SDS-
insoluble but Laemmli + ß-mercaptoethanol-extractable fractions were loaded directly, and subjected to immunoblot with the indicated
antibodies. A representative blot from at least two independent experiments using brain vessels from two control and two patients with
CADASIL is shown. (C) Paraffin brain sections from control and patients with CADASIL were hybridized with TIMP3 complementary RNA
probe and examined with dark field (left) and bright field illumination (right). The images shown (white matter with penetrating vessels)
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1841
(continued)
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

We further analysed the TIMP3 activity levels in human brain
vessels by reverse zymography, which measures the ability of
TIMP3 to inhibit matrix metalloproteinases (Stetler-Stevenson,
2008). Strikingly, in human brain vessels, TIMP3 accounts for
the majority of gelatinase inhibitory activity (Fig. 6D and data
not shown). Importantly, TIMP3 activity was 2.5-fold higher in
patients with CADASIL compared with controls (Fig. 6D).
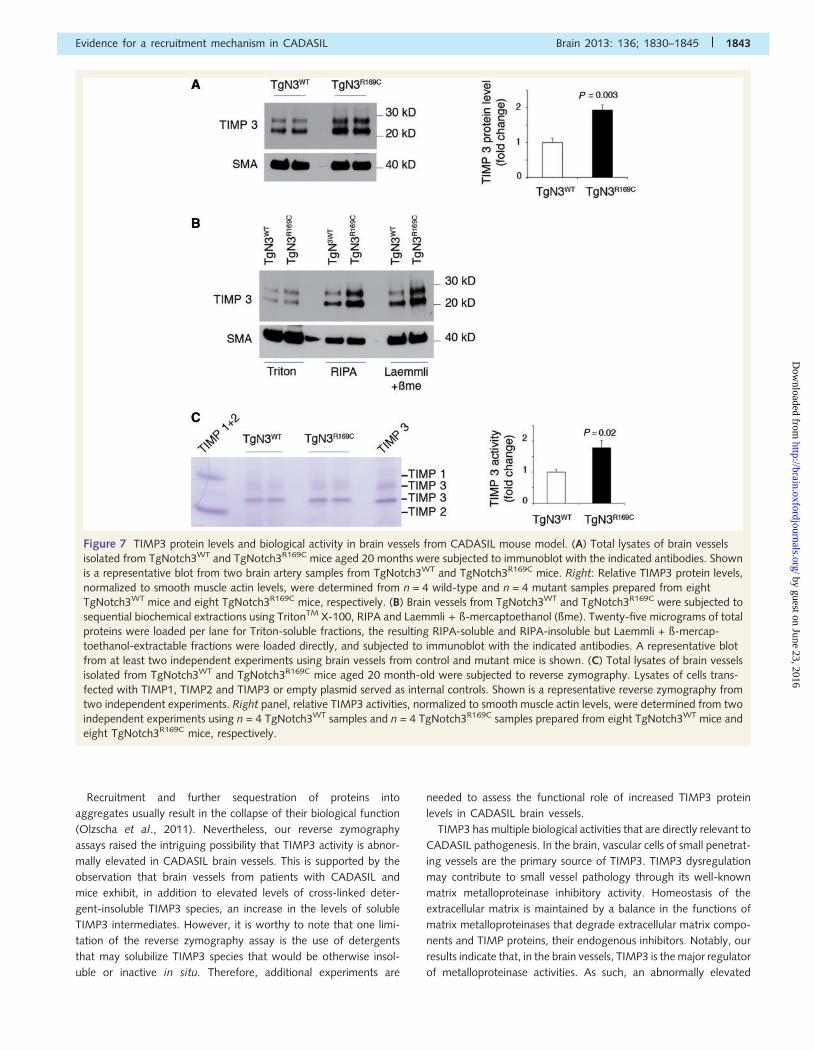
Next, we repeated the same analyses using brain arteries
isolated from TgNotch3R169C and control TgNotch3WT mice.
Immunoblot analysis revealed a 1.9-fold increase in total levels
of TIMP3 in TgNotch3R169C mice compared with TgNotch3WT
control mice (Fig. 7A). Notably, TgNotch3WT and non-transgenic
mice express comparable protein levels of TIMP3 (Supplementary
Fig. 9). Sequential extractions in TritonTM X-100, RIPA then
SDS + ß-mercaptoethanol buffers revealed more TIMP3 in all
three fractions, including the TritonTM X-100 and RIPA-soluble
fractions, in the TgNotch3R169C mice (Fig. 7B). Reverse zymogra-
phy assay showed that TIMP3 is the predominant endogenous
matrix metalloproteinases inhibitor also in murine brain arteries.
Significantly, gelatinase inhibitory activity was 1.8-fold higher in
TgNotch3R169C mice compared with TgNotch3WT mice (Fig. 7C).
Collectively, these data demonstrate that brain vessels from pa-
tients and mice with CADASIL accumulate a range of insoluble
and soluble TIMP3 species and suggest that this is associated
with an abnormally elevated TIMP3 activity.
DiscussionHere we demonstrate that, in brain vessels from patients with
CADASIL and transgenic mice, Notch3ECD accumulates in aggre-
gates that contain functionally important matricellular proteins,
including TIMP3 and VTN. Importantly, TIMP3 and VTN have
not been previously implicated in CADASIL and were not known
as interaction partners of NOTCH3. By focusing our further
analysis on these two proteins, we could decipher some of the
mechanisms of CADASIL deposit formation and the possible con-
sequences of their abnormal recruitment.
Previously, we and others have reported that the addition of
both detergent and reducing agent were necessary to solubilize
Notch3ECD from CADASIL brains (Joutel et al., 2000; Duering
et al., 2011). Moreover, in vitro studies using purified recombin-
ant NOTCH3 fragments containing the first five EGF-like repeats
showed that fragments bearing a cysteine mutation spontaneously
form high order multimers, cross-linked by disulphide bonds
(Duering et al., 2011). Here, our biochemical analyses using
brain and artery samples from patients with CADASIL and
mouse models extend these findings by showing that mutant
Notch3ECD is incorporated into denaturant and detergent-insoluble
aggregates, from which it can be recovered by the addition of a
reducing agent. Notably, NOTCH3 molecules bearing either RBPJ-
active or -inactive mutations behave similarly. Thus, these data
strongly argue that cysteine modifications are critical determinants
of the altered conformation of mutant Notch3ECD molecules,
which ultimately results in the formation of microscopically dis-
cernible Notch3ECD aggregates. Yet, the exact pairing and contri-
bution of individual cysteine residues in this process remains to be
determined. The possibility that Notch3ECD aggregates occur first
in vivo receives some support from the observations in transgenic
mutant mice that Notch3ECD aggregates can be detected as early
as 1 month of age and that aggregation of TIMP3 and VTN occurs
afterward. Next, our experimental results showed that increased
levels or aggregation of Notch3ECD, rather than the unpaired cyst-
eine residue within mutant Notch3ECD, strongly enhanced TIMP3–
Notch3ECD complex formation, resulting in the recruitment and
accumulation of TIMP3, which in turns promotes complex forma-
tion including NOTCH3 and VTN. This suggests that aggregated
or excess levels of Notch3ECD may form a platform or act as a
seed that influences interactions with extracellular matrix proteins
like TIMP3, that in turn can bind and recruit more and more
proteins, like VTN, generating new interaction surfaces and mag-
nifying the toxic potential of aggregates in a snowball effect. This
is highly reminiscent of what occurs in many neurodegenerative
diseases with inappropriate deposition of protein aggregates
(Wolfe and Cyr, 2011).
So far, only two other proteins, clusterin, an extracellular chap-
erone, and endostatin, a naturally-occurring proteolytic fragment
derived from the collagen alpha-1(XVIII) chain, have been identi-
fied as granular osmiophilic material components using laser cap-
ture microdissection on post-mortem brain tissue from patients
with CADASIL (Arboleda-Velasquez et al., 2011). Interestingly,
our proteomic analyses revealed additional proteins, including
mostly extracellular matrix proteins, which were enriched in the
CADASIL samples that warrant further investigation. It is import-
ant to note that the human-derived CADASIL sample was strongly
enriched with clusterin and endostatin, supporting the validity and
value of our approach. Nonetheless, abundance of these two pro-
teins seemed comparable in wild-type and mutant brain arteries of
transgenic mice, suggesting that clusterin and endostatin may ac-
cumulate at an advanced stage of the disease. This may apply to
many other proteins identified through our proteomic analyses, a
finding that would be consistent with the snowball model pro-
posed above. Thus, comparative proteomic analysis of the
Notch3ECD enriched fraction prepared from mutant and wild-
type samples, at various stages of the disease process, may rep-
resent an interesting approach to identifying proteins that co-ag-
gregate with Notch3ECD during the pathogenesis.
Figure 6 Continuedare representative of at least four sections from an experiment using three patients with CADASIL and three control subjects. (D) Lysates
of brain vessels isolated from four patients with CADASIL and four control subjects were subjected in parallel to reverse zymography and
to immunoblot with ß-actin. Lysates of cells transfected with TIMP3 or empty plasmid served as internal controls for reverse zymography.
Shown is a representative reverse zymogram from two independent experiments. Right: Relative TIMP3 activities, normalized to ß-actin
levels, were determined from n = 4 patients with CADASIL and n = 4 control subjects. SMA = smooth muscle actin.
1842 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from
Recruitment and further sequestration of proteins into
aggregates usually result in the collapse of their biological function
(Olzscha et al., 2011). Nevertheless, our reverse zymography
assays raised the intriguing possibility that TIMP3 activity is abnor-
mally elevated in CADASIL brain vessels. This is supported by the
observation that brain vessels from patients with CADASIL and
mice exhibit, in addition to elevated levels of cross-linked deter-
gent-insoluble TIMP3 species, an increase in the levels of soluble
TIMP3 intermediates. However, it is worthy to note that one limi-
tation of the reverse zymography assay is the use of detergents
that may solubilize TIMP3 species that would be otherwise insol-
uble or inactive in situ. Therefore, additional experiments are
needed to assess the functional role of increased TIMP3 protein
levels in CADASIL brain vessels.
TIMP3 has multiple biological activities that are directly relevant to
CADASIL pathogenesis. In the brain, vascular cells of small penetrat-
ing vessels are the primary source of TIMP3. TIMP3 dysregulation
may contribute to small vessel pathology through its well-known
matrix metalloproteinase inhibitory activity. Homeostasis of the
extracellular matrix is maintained by a balance in the functions of
matrix metalloproteinases that degrade extracellular matrix compo-
nents and TIMP proteins, their endogenous inhibitors. Notably, our
results indicate that, in the brain vessels, TIMP3 is the major regulator
of metalloproteinase activities. As such, an abnormally elevated
Figure 7 TIMP3 protein levels and biological activity in brain vessels from CADASIL mouse model. (A) Total lysates of brain vessels
isolated from TgNotch3WT and TgNotch3R169C mice aged 20 months were subjected to immunoblot with the indicated antibodies. Shown
is a representative blot from two brain artery samples from TgNotch3WT and TgNotch3R169C mice. Right: Relative TIMP3 protein levels,
normalized to smooth muscle actin levels, were determined from n = 4 wild-type and n = 4 mutant samples prepared from eight
TgNotch3WT mice and eight TgNotch3R169C mice, respectively. (B) Brain vessels from TgNotch3WT and TgNotch3R169C were subjected to
sequential biochemical extractions using TritonTM X-100, RIPA and Laemmli + ß-mercaptoethanol (ßme). Twenty-five micrograms of total
proteins were loaded per lane for Triton-soluble fractions, the resulting RIPA-soluble and RIPA-insoluble but Laemmli + ß-mercap-
toethanol-extractable fractions were loaded directly, and subjected to immunoblot with the indicated antibodies. A representative blot
from at least two independent experiments using brain vessels from control and mutant mice is shown. (C) Total lysates of brain vessels
isolated from TgNotch3WT and TgNotch3R169C mice aged 20 month-old were subjected to reverse zymography. Lysates of cells trans-
fected with TIMP1, TIMP2 and TIMP3 or empty plasmid served as internal controls. Shown is a representative reverse zymography from
two independent experiments. Right panel, relative TIMP3 activities, normalized to smooth muscle actin levels, were determined from two
independent experiments using n = 4 TgNotch3WT samples and n = 4 TgNotch3R169C samples prepared from eight TgNotch3WT mice and
eight TgNotch3R169C mice, respectively.
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1843
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

TIMP3 activity is anticipated to result in vessel fibrosis. Fibrotic
thickening of the arteriolar walls has been amply demonstrated in
CADASIL and recently, an increase in types I, III, IV and VI collagens
has been documented in all calibres of brain vessels (Dong et al.,
2012). Noteworthy, a number of non-extracellular matrix molecules
are also potential substrates of matrix metalloproteinases, whereby
elevated TIMP3 activity could exert a deleterious effect. TIMP3 dys-
regulation could also contribute to pathology through its anti-angio-
genic activity (Qi et al., 2003; Ebrahem et al., 2011). Particularly,
TIMP3 can suppress vascular endothelial growth factor-mediated
angiogenesis independently of its matrix metalloproteinases inhibi-
tory activity. Previously, we have documented a substantial reduc-
tion of capillary density in the TgNotch3R169C CADASIL mouse
model. Also, TIMP3 is the only TIMP that can inhibit ADAM17/
TNF converting enzyme, which mediates ectodomain shedding of
transmembrane receptors and ligands including TNF and ligands of
the epidermal growth factor receptor (Nagase et al., 2006). Of inter-
est, recent work suggests a role for heparin-binding EGF-like growth
factor in the pericyte recruitment process that leads to vessel matur-
ation and stabilization (Stratman et al., 2010). Finally, mutations in
TIMP3 cause Sorsby fundus dystrophy, an adult-onset hereditary
macular degenerative disease characterized by abnormal deposition
of TIMP3, macular atrophy and choroidal neovascularization (Weber
et al., 1994; Fariss et al., 1998; Qi et al., 2003). Disturbed homeo-
stasis in extracellular matrix remodelling is likely involved in Sorsby
fundus dystrophy pathology, although the mechanisms remain un-
clear. On the other hand, it’s conceivable that the diverse biological
activities of TIMP3 are differentially affected by TIMP3 accumula-
tion. Further studies are required to test these hypotheses.
Likewise, abnormal recruitment and sequestration of VTN
may impair its functions. While VTN is known as an abundant
circulating plasma protein, we found that, in the brain vessels,
VTN is abundantly transcribed (unpublished) and is a normal com-
ponent of the extracellular matrix. By its ability to bind to integrin-
type cell adhesion receptors, urokinase receptor and extracellular
matrix proteins, VTN has the ability to regulate cell adhesion, sig-
nalling and cytoskeletal reorganization. Moreover, by its ability to
bind and stabilize plasminogen activation inhibitor type 1, VTN can
modulate the balance of the fibrinolytic system, and consequently,
the extracellular matrix homeostasis (Preissner and Reuning, 2011).
Further studies are required to examine whether and how focal
VTN deposits can alter these biological activities.
In summary our results lend support to a Notch3ECD cascade hy-
pothesis, which posits that the aggregation of Notch3ECD in the brain
vessels is a central event in CADASIL disease pathology, promoting
the abnormal recruitment and potential dysregulation of functionally
important proteins of the extracellular matrix that may ultimately
cause multifactorial toxicity. Identifying the CADASIL–Notch3ECD
interactome may represent an interesting approach to further delin-
eate the molecular mechanisms of this toxicity.
AcknowledgementsWe are grateful to the laboratory of Dr. Dylan Edwards for the
TIMP activity assay, to Drs. Hannu Kalimo, Francoise Chapon,
Jean-Jacques Haw, Catherine Godfraind and the GIE-NeuroCeb
brain bank (Paris, France) for human brain samples, and to
Barbara Lemaire-Carrette for technical assistance. We thank
Electron microscopy Core at Institut du Fer-a-Moulin and
TAAM-Orleans (Karine Jambou) for animal housing.
FundingThis work was supported by grants from the French National
Research Agency (grant number ANR Genopath 2009-
RAE09011HSA), National Institutes of Health (grant number R01
NS054122) and the Fondation Leducq (Transatlantic Network of
Excellence on the Pathogenesis of Small Vessel Disease of the
Brain) to A.J. M.R. is a recipient of a fellowship from the
European community (Marie Curie Initial Training Network). FT
ICR MS acquisition was supported by investment grant from the
contrat de plan Etat-Region (CPER) Fonds Recherche et
Technologie (2002-06). Financial support from the TGE FT-ICR
for conducting the research is gratefully acknowledged.
Supplementary materialSupplementary material is available at Brain online.
ReferencesArboleda-Velasquez JF, Manent J, Lee JH, Tikka S, Ospina C,
Vanderburg CR, et al. Hypomorphic Notch 3 alleles link Notch signal-
ing to ischemic cerebral small-vessel disease. Proc Natl Acad Sci USA
2011; 108: E128–35.
Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG.
Cadasil. Lancet Neurol 2009; 8: 643–53.
Dichgans M. Genetics of ischaemic stroke. Lancet Neurol 2007; 6:
149–61.Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT,
et al. Notch3 is required for arterial identity and maturation of vascular
smooth muscle cells. Genes Dev 2004; 18: 2730–5.Dong H, Blaivas M, Wang MM. Bidirectional encroachment of collagen
into the tunica media in cerebral autosomal dominant arteriopathy
with subcortical infarcts and leukoencephalopathy. Brain Res 2012;
1456: 64–71.
Duering M, Karpinska A, Rosner S, Hopfner F, Zechmeister M, Peters N,
et al. Co-aggregate formation of CADASIL-mutant NOTCH3: a single-
particle analysis. Hum Mol Genet 2011; 20: 3256–65.
Ebrahem Q, Qi JH, Sugimoto M, Ali M, Sears JE, Cutler A, et al.
Increased neovascularization in mice lacking tissue inhibitor of metal-
loproteinases-3. Invest Ophthalmol Vis Sci 2011; 52: 6117–23.
Fariss RN, Apte SS, Luthert PJ, Bird AC, Milam AH. Accumulation of
tissue inhibitor of metalloproteinases-3 in human eyes with Sorsby’s
fundus dystrophy or retinitis pigmentosa. Br J Ophthalmol 1998; 82:
1329–34.
Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch sig-
nalling in smooth muscle cells during development and disease.
Cardiovasc Res 2012; 95: 138–46.
Ishiko A, Shimizu A, Nagata E, Takahashi K, Tabira T, Suzuki N. Notch3
ectodomain is a major component of granular osmiophilic material
(GOM) in CADASIL. Acta Neuropathol 2006; 112: 333–9.Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, et al.
The ectodomain of the Notch3 receptor accumulates within the cere-
brovasculature of CADASIL patients. J Clin Invest 2000; 105: 597–605.
1844 | Brain 2013: 136; 1830–1845 M. Monet-Lepretre et al.
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from

Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al.Notch3 mutations in CADASIL, a hereditary adult-onset condition
causing stroke and dementia. Nature 1996; 383: 707–10.
Joutel A, Favrole P, Labauge P, Chabriat H, Lescoat C, Andreux F, et al.
Skin biopsy immunostaining with a Notch3 monoclonal antibody forCADASIL diagnosis. Lancet 2001; 358: 2049–51.
Joutel A, Monet M, Domenga V, Riant F, Tournier-Lasserve E.
Pathogenic mutations associated with cerebral autosomal dominant
arteriopathy with subcortical infarcts and leukoencephalopathy differ-ently affect Jagged1 binding and Notch3 activity via the RBP/JK sig-
naling pathway. Am J Hum Genet 2004; 74: 338–47.
Joutel A, Monet-Lepretre M, Gosele C, Baron-Menguy C, Hammes A,Schmidt S, et al. Cerebrovascular dysfunction and microcirculation rar-
efaction precede white matter lesions in a mouse genetic model of
cerebral ischemic small vessel disease. J Clin Invest 2010; 120: 433–45.
Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C,et al. Strong clustering and stereotyped nature of Notch3 mutations in
CADASIL patients. Lancet 1997; 350: 1511–5.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding
the activation mechanism. Cell 2009; 137: 216–33.Lee MH, Atkinson S, Murphy G. Identification of the extracellular matrix
(ECM) binding motifs of tissue inhibitor of metalloproteinases (TIMP)-3
and effective transfer to TIMP-1. J Biol Chem 2007; 282: 6887–98.
Lesnik Oberstein SA, van Duinen SG, van den Boom R, Maat-Schieman ML, van Buchem MA, van Houwelingen HC, et al.
Evaluation of diagnostic NOTCH3 immunostaining in CADASIL. Acta
Neuropathol (Berl) 2003; 106: 107–11.Liu H, Sadygov RG, Yates JR III. A model for random sampling and
estimation of relative protein abundance in shotgun proteomics. Anal
Chem 2004; 76: 4193–201.
Liu H, Zhang W, Kennard S, Caldwell RB, Lilly B. Notch3 is critical forproper angiogenesis and mural cell investment. Circ Res 2010; 107:
860–70.
Monet M, Domenga V, Lemaire B, Souilhol C, Langa F, Babinet C, et al.
The archetypal R90C CADASIL-NOTCH3 mutation retains NOTCH3function in vivo. Hum Mol Genet 2007; 16: 982–92.
Monet-Lepretre M, Bardot B, Lemaire B, Domenga V, Godin O,
Dichgans M, et al. Distinct phenotypic and functional features ofCADASIL mutations in the Notch3 ligand binding domain. Brain
2009; 132: 1601–12.
Nagase H, Visse R, Murphy G. Structure and function of matrix metal-
loproteinases and TIMPs. Cardiovasc Res 2006; 69: 562–73.Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH,
Tartaglia GG, et al. Amyloid-like aggregates sequester numerous
metastable proteins with essential cellular functions. Cell 2011; 144:
67–78.Opherk C, Duering M, Peters N, Karpinska A, Rosner S, Schneider E,
et al. CADASIL mutations enhance spontaneous multimerization of
NOTCH3. Hum Mol Genet 2009; 18: 2761–7.
Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical
characteristics to therapeutic challenges. Lancet Neurol 2010; 9:
689–701.
Peters N, Opherk C, Bergmann T, Castro M, Herzog J, Dichgans M.
Spectrum of mutations in biopsy-proven CADASIL: implications for
diagnostic strategies. Arch Neurol 2005; 62: 1091–4.
Peters N, Opherk C, Zacherle S, Capell A, Gempel P, Dichgans M.
CADASIL-associated Notch3 mutations have differential effects both
on ligand binding and ligand-induced Notch3 receptor signaling
through RBP-Jk. Exp Cell Res 2004; 299: 454–64.
Preissner KT, Reuning U. Vitronectin in vascular context: facets of a
multitalented matricellular protein. Semin Thromb Hemost 2011; 37:
408–24.
Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M,
et al. A novel function for tissue inhibitor of metalloproteinases-3
(TIMP3): inhibition of angiogenesis by blockage of VEGF binding to
VEGF receptor-2. Nat Med 2003; 9: 407–15.
Ruchoux MM, Domenga V, Brulin P, Maciazek J, Limol S, Tournier-
Lasserve E, et al. Transgenic mice expressing mutant Notch3 develop
vascular alterations characteristic of cerebral autosomal dominant
arteriopathy with subcortical infarcts and leukoencephalopathy. Am J
Pathol 2003; 162: 329–42.
Ruchoux MM, Guerouaou D, Vandenhaute B, Pruvo JP, Vermersch P,
Leys D. Systemic vascular smooth muscle cell impairment in cerebral
autosomal dominant arteriopathy with subcortical infarcts and leu-
koencephalopathy. Acta Neuropathol (Berl) 1995; 89: 500–12.
Shevchenko A. Evaluation of the efficiency of in-gel digestion of proteins
by peptide isotopic labeling and MALDI mass spectrometry. Anal
Biochem 2001; 296: 279–83.
Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell sig-
naling: metalloproteinase-independent biological activities. Sci Signal
2008; 1: re6.
Stratman AN, Schwindt AE, Malotte KM, Davis GE. Endothelial-derived
PDGF-BB and HB-EGF coordinately regulate pericyte recruitment
during vasculogenic tube assembly and stabilization. Blood 2010;
116: 4720–30.Tikka S, Mykkanen K, Ruchoux MM, Bergholm R, Junna M,
Poyhonen M, et al. Congruence between NOTCH3 mutations and
GOM in 131 CADASIL patients. Brain 2009; 132: 933–39.Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue
inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s
fundus dystrophy. Nat Genet 1994; 8: 352–6.Wolfe KJ, Cyr DM. Amyloid in neurodegenerative diseases: friend or foe?
Semin Cell Dev Biol 2011; 22: 476–81.
Yousif S, Marie-Claire C, Roux F, Scherrmann JM, Decleves X. Expression
of drug transporters at the blood-brain barrier using an
optimized isolated rat brain microvessel strategy. Brain Res 2007;
1134: 1–11.
Evidence for a recruitment mechanism in CADASIL Brain 2013: 136; 1830–1845 | 1845
by guest on June 23, 2016http://brain.oxfordjournals.org/
Dow
nloaded from
Top Related
Copyright © 2022 FDOKUMEN

