
Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells
24
Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells Guillaume J. Filion, 1,5 Joke G. van Bemmel, 1,5 Ulrich Braunschweig, 1,5 Wendy Talhout, 1 Jop Kind, 1 Lucas D. Ward, 3,4,6 Wim Brugman, 2 Ine ˆ s J. de Castro, 1,7 Ron M. Kerkhoven, 2 Harmen J. Bussemaker, 3,4 and Bas van Steensel 1, * 1 Division of Gene Regulation 2 Central Microarray Facility Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands 3 Department of Biological Sciences, Columbia University, 1212 Amsterdam Avenue, New York, NY 10027, USA 4 Center for Computational Biology and Bioinformatics, Columbia University, 1130 St. Nicholas Avenue, New York, NY 10032, USA 5 These authors contributed equally to this work 6 Present address: Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA 02139, USA 7 Present address: Genome Function Group, MRC Clinical Sciences Centre, Imperial College School of Medicine, Hammersmith Hospital Campus, Du Cane Road, London W12 0NN, UK *Correspondence: [email protected] DOI 10.1016/j.cell.2010.09.009 SUMMARY Chromatin is important for the regulation of transcrip- tion and other functions, yet the diversity of chromatin composition and the distribution along chromo- somes are still poorly characterized. By integrative analysis of genome-wide binding maps of 53 broadly selected chromatin components in Drosophila cells, we show that the genome is segmented into five principal chromatin types that are defined by unique yet overlapping combinations of proteins and form domains that can extend over > 100 kb. We identify a repressive chromatin type that covers about half of the genome and lacks classic heterochromatin markers. Furthermore, transcriptionally active eu- chromatin consists of two types that differ in molec- ular organization and H3K36 methylation and regu- late distinct classes of genes. Finally, we provide evidence that the different chromatin types help to target DNA-binding factors to specific genomic regions. These results provide a global view of chro- matin diversity and domain organization in a meta- zoan cell. INTRODUCTION Chromatin consists of DNA and all associated proteins. The scaffold of chromatin is formed by nucleosomes, which are histone octamers in a tight complex with DNA. This scaffold serves as the docking platform for hundreds of structural and regulatory proteins. Furthermore, histones carry a variety of posttranslational modifications that form recognition sites for specific proteins (Berger, 2007; Rando and Chang, 2009). The local composition of chromatin is a major determinant of the transcriptional activity of a gene; some chromatin proteins enhance transcription, whereas others have repressive effects. Traditionally, chromatin was divided into heterochromatin and euchromatin. There is now ample evidence that a finer classifica- tion is required. For example, in Drosophila, at least two types of heterochromatin exist that have distinct regulatory functions and consist of different proteins. The first type is marked by Poly- comb group (PcG) proteins and methylation of lysine 27 of histone H3 (H3K27). PcG chromatin forms large continuous domains; it is a repressive type of chromatin that primarily regu- lates genes with developmental functions (Sparmann and van Lohuizen, 2006). The second type is marked by heterochromatin protein 1 (HP1) and several associated proteins, combined with methylation of H3K9. This type of heterochromatin can also cover large genomic segments, particularly around centro- meres. Reporter genes integrated in or near HP1 heterochro- matin tend to be repressed, but paradoxically, many genes that are naturally bound by HP1 are transcriptionally active (Hediger and Gasser, 2006). Direct comparison of genome- wide binding maps indicates that PcG and HP1 heterochromatin are nonoverlapping (de Wit et al., 2007). HP1 and PcG chromatin illustrate two important principles of chromatin organization: each type is marked by unique combi- nations of proteins and can cover long stretches of DNA. But are there other major types of chromatin that follow these same principles? For example, is euchromatin also organized into domains with distinct protein compositions? Are there additional types of repressive chromatin that have remained unnoticed? In order to address these questions, we generated genome- wide location maps of 53 broadly selected chromatin proteins and four key histone modifications in Drosophila cells, providing 212 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
Transcript of Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells

Systematic Protein LocationMapping Reveals Five PrincipalChromatin Types in Drosophila CellsGuillaume J. Filion,1,5 Joke G. van Bemmel,1,5 Ulrich Braunschweig,1,5 Wendy Talhout,1 Jop Kind,1 Lucas D. Ward,3,4,6
Wim Brugman,2 Ines J. de Castro,1,7 Ron M. Kerkhoven,2 Harmen J. Bussemaker,3,4 and Bas van Steensel1,*1Division of Gene Regulation2Central Microarray Facility
Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands3Department of Biological Sciences, Columbia University, 1212 Amsterdam Avenue, New York, NY 10027, USA4Center for Computational Biology and Bioinformatics, Columbia University, 1130 St. Nicholas Avenue, New York, NY 10032, USA5These authors contributed equally to this work6Present address: Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge,
MA 02139, USA7Present address: Genome Function Group, MRC Clinical Sciences Centre, Imperial College School of Medicine,
Hammersmith Hospital Campus, Du Cane Road, London W12 0NN, UK*Correspondence: [email protected]
DOI 10.1016/j.cell.2010.09.009
SUMMARY
Chromatin is important for the regulation of transcrip-tionandother functions, yet thediversity of chromatincomposition and the distribution along chromo-somes are still poorly characterized. By integrativeanalysis of genome-wide binding maps of 53 broadlyselected chromatin components in Drosophila cells,we show that the genome is segmented into fiveprincipal chromatin types that are defined by uniqueyet overlapping combinations of proteins and formdomains that can extend over > 100 kb. We identifya repressive chromatin type that covers about halfof the genome and lacks classic heterochromatinmarkers. Furthermore, transcriptionally active eu-chromatin consists of two types that differ in molec-ular organization and H3K36 methylation and regu-late distinct classes of genes. Finally, we provideevidence that the different chromatin types help totarget DNA-binding factors to specific genomicregions. These results provide a global view of chro-matin diversity and domain organization in a meta-zoan cell.
INTRODUCTION
Chromatin consists of DNA and all associated proteins. The
scaffold of chromatin is formed by nucleosomes, which are
histone octamers in a tight complex with DNA. This scaffold
serves as the docking platform for hundreds of structural and
regulatory proteins. Furthermore, histones carry a variety of
posttranslational modifications that form recognition sites for
212 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
specific proteins (Berger, 2007; Rando and Chang, 2009). The
local composition of chromatin is a major determinant of the
transcriptional activity of a gene; some chromatin proteins
enhance transcription, whereas others have repressive effects.
Traditionally, chromatin was divided into heterochromatin and
euchromatin. There is now ample evidence that a finer classifica-
tion is required. For example, in Drosophila, at least two types of
heterochromatin exist that have distinct regulatory functions and
consist of different proteins. The first type is marked by Poly-
comb group (PcG) proteins and methylation of lysine 27 of
histone H3 (H3K27). PcG chromatin forms large continuous
domains; it is a repressive type of chromatin that primarily regu-
lates genes with developmental functions (Sparmann and van
Lohuizen, 2006). The second type is marked by heterochromatin
protein 1 (HP1) and several associated proteins, combined with
methylation of H3K9. This type of heterochromatin can also
cover large genomic segments, particularly around centro-
meres. Reporter genes integrated in or near HP1 heterochro-
matin tend to be repressed, but paradoxically, many genes
that are naturally bound by HP1 are transcriptionally active
(Hediger and Gasser, 2006). Direct comparison of genome-
wide binding maps indicates that PcG and HP1 heterochromatin
are nonoverlapping (de Wit et al., 2007).
HP1 and PcG chromatin illustrate two important principles of
chromatin organization: each type is marked by unique combi-
nations of proteins and can cover long stretches of DNA. But
are there other major types of chromatin that follow these
same principles? For example, is euchromatin also organized
into domains with distinct protein compositions? Are there
additional types of repressive chromatin that have remained
unnoticed?
In order to address these questions, we generated genome-
wide location maps of 53 broadly selected chromatin proteins
and four key histone modifications in Drosophila cells, providing

A
C
B
Principal component analysis
Hidden Markov model
53 ch
rom
atin
prot
eins
16000 16200 16400 16600 16800 17000
Position on chr2L (kb)
PC1PC2PC3
type
16000 16200 16400 16600 16800 17000
Position on chr2L (kb)
MRG15SU(VAR)3−7SU(VAR)3−9
HP6HP1LHR
CAF1ASF1
MUS209TOP1
RPII18SIR2
RPD3CDK7DSP1DF31MAX
PCAFASH2HP1cCtBPJRA
BRMECRBCD
MED31SU(VAR)2−10
LOLALGAF
CG31367ACT5C
TIP60MNT
SIN3ATBP
DWGPHOLPROD
BEAF32bSU(HW)
LAMD1H1
SUUREFFIAL
GROPHO
CTCFPC
E(Z)PCLSCE
Genes+-
−20
−10
010
20PC
1
−15 −10 −5 0 5 10 15PC2
−15 −10 −5 0 5 10 15
−15
−10
−50
510
PC2PC
3
Figure 1. Overview of Protein Binding Profiles and Derivation of the Five-Type Chromatin Segmentation
(A) Sample plot of all 53 DamID profiles (log2 enrichment over Dam-only control). Positive values are plotted in black and negative values in gray for contrast.
Below the profiles, genes on both strands are depicted as lines with blocks indicating exons.
(B) Two-dimensional projections of the data onto the first three principal components. Colored dots indicate the chromatin type of probed loci as inferred by
a five-state HMM.
(C) Values of the first three principal components along the region shown in (A), with domains of the different chromatin types after segmentation by the five-state
HMM highlighted by the same colors as in (B).
See also Figure S1 and Table S1.
a rich description of chromatin composition along the genome.
By integrative computational analysis, we identified, aside from
PcG and HP1 chromatin, three additional principal chromatin
types that are defined by unique combinations of proteins. One
of these is a type of repressive chromatin that covers �50% of
the genome. In addition, we identified two types of transcription-
ally active euchromatin that are bound by different proteins and
harbor distinct classes of genes.
RESULTS
Genome-wide Location Maps of 53 Chromatin ProteinsWe constructed a database of high-resolution binding profiles of
53 chromatin proteins in the embryonicDrosophila melanogaster
cell line Kc167 (Figure 1A and Figure S1A available online). In
order to obtain a representative cross-section of the chromatin
proteome, we selected proteins from most known chromatin
protein complexes, including a variety of histone-modifying
enzymes, proteins that bind specific histone modifications,
general transcription machinery components, nucleosome re-
modelers, insulator proteins, heterochromatin proteins, struc-
tural components of chromatin, and a selection of DNA-binding
factors (DBFs) (Table S1). For�40 of these proteins, full-genome
high-resolution binding maps have not previously been reported
in any Drosophila cell type or tissue. Though chromatin immuno-
precipitation (ChIP) is widely used to map protein-genome inter-
actions (Collas, 2009), large-scale application of this method is
hampered by the limited availability of highly specific antibodies.
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. 213

Moreover, at least for some chromatin proteins, ChIP results can
greatly depend on the choice of crosslinking reagents (Wang
et al., 2009) and can be unreliable for proteins with short resi-
dence times (Gelbart et al., 2005; Schmiedeberg et al., 2009).
We therefore used the DamID technology, which does not
require crosslinking or antibodies. With DamID, DNA adenine
methyltransferase (Dam) fused to a chromatin protein of interest
deposits a stable adenine-methylation ‘‘footprint’’ in vivo at the
interaction sites of the chromatin protein so that even transient
interactions may be detected (van Steensel et al., 2001). Note
that the fusion protein is expressed at very low levels, averting
overexpression artifacts. The DamID profiles of all 53 proteins
were generated in duplicate under standardized conditions
and were detected using oligonucleotide microarrays that query
the entire fly genome at�300 bp intervals. Comparisons to pub-
lished and new ChIP data confirm the overall reliability of the
DamID data (Figure S1B), which was also reported in previous
comparative studies (Moorman et al., 2006; Negre et al., 2006).
For reference purposes, we also generated ChIP maps of
histone H3 and the histone marks H3K4me2, H3K9me2,
H3K27me3, and H3K79me3 on the same array platform.
Most of the Fly Genome Interacts with NonhistoneChromatin ProteinsComparison of the DamID profiles for all 53 proteins shows
a variety of binding patterns (Figure 1A). Nevertheless, several
sets of proteins exhibit profiles that are similar. Some similarities
were anticipated, such as for PC, PCL, SCE, and E(Z), which are
all PcG proteins (Sparmann and van Lohuizen, 2006), and for
HP1, SU(VAR)3-9, LHR, and HP6, which are part of classic
HP1-type heterochromatin (Greil et al., 2007). We also observe
extensive colocalization of Lamin (LAM), histone H1 (H1),
Effete (EFF), Suppressor of Underreplication (SUUR), and the
AT-hook protein D1, which have not been linked previously
except for LAM and SUUR (Pindyurin et al., 2007). There is a
prominent overlap in the binding patterns of a large set of �30
proteins, including histone-modifying enzymes (e.g., RPD3 and
SIR2), components of the basal transcription machinery (e.g.,
CDK7 and TBP), and others detailed below.
In order to identify target and nontarget loci for each protein,
we applied a two-state hidden Markov model (HMM) to each
individual binding map (Extended Experimental Procedures).
This method identifies themost likely segmentation into ‘‘bound’’
and ‘‘unbound’’ probed loci. According to the resulting binary
classifications, the genome-wide occupancy by individual
proteins varies broadly, ranging from about 2% (GRO) to 79%
(IAL). Of interest, 99.99% of the probed loci are bound by at least
one protein and 99.6% by at least three proteins. This indicates
that, at least at the resolution of our maps, essentially no part of
the fly genome is permanently in a configuration that consists of
nucleosomes only. Approximately 1% of the genome shows
extremely high protein occupancy, being bound by 36–44 of
the 53 mapped proteins.
Principal Chromatin Types Defined by Combinationsof ProteinsNext, we used a computational classification strategy to identify
themajor types of chromatin, defined as distinct combinations of
214 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
proteins that are recurrent throughout the genome. To identify
such combinations, we initially performed principal component
analysis on the 53 quantitative DamID profiles to reduce the
dimensionality of the data. We then focused on the first three
principal components, which together account for 57.7% of
the total variance. By projecting the genomic sites on the prin-
cipal components, we could distinguish five distinct lobes in
the three-dimensional scatter plot (Figure 1B). No additional
distinct lobes could be observed upon further inspection of
higher-level principal components. Importantly, the five groups
were also clearly separated when using the previously defined
binary target definitions (Figure S1C), showing that this result is
robust to different quantification methods.
Having established that classification into five types properly
summarizes the data, we fitted a five-state HMM onto the first
three principal components. Thus, every probed sequence in
the genome was assigned one of five exclusive chromatin types
(Extended Experimental Procedures). To avoid semantic confu-
sion, and in line with the Greek word chroma (color), we labeled
each of the five protein signatures with a color (BLUE, GREEN,
BLACK, RED, and YELLOW). The HMM classification produced
a mosaic pattern of chromosomal domains that vary widely in
length (Figure 1C). We emphasize that this segmentation is
purely data driven, without using any other knowledge besides
the 53 DamID profiles. The segmentation is generally robust:
removal of any of the proteins except for PC still yields a five-
state classification that is, on average, 96.7% identical to the
model obtained with all 53 proteins. A detailed analysis of the
robustness is summarized in Figure S1D.
Domain Organization of Chromatin TypesThe five types of chromatin differ substantially in their genome
coverage, numbers of domains, and numbers of genes (Fig-
ure 2A). We identified a total of 8428 domains that typically range
from �1 to 52 kb (5th–95th percentiles) with a median length of
6.5 kb, although the size distribution depends on the chromatin
type (Figure 2B). 441 domains are larger than 50 kb, and 155
are larger than 100 kb, with the largest domain being 737 kb.
Many individual domains include multiple neighboring genes
(Figure 2C), the largest number of which within a single domain
is 139 (for a centromere-proximal GREEN domain). Taken
together, these data indicate that the fly genome is generally
organized into large regions that are covered by specific combi-
nations of proteins.
BLUE and GREEN Chromatin Correspond to KnownHeterochromatin TypesVisualization of the protein occupancy in each of the five chro-
matin types (Figure 3A) shows that most proteins are not
confined to a single chromatin type. Rather, the five chromatin
types are defined by unique combinations of proteins. Impor-
tantly, BLUE and GREEN chromatin closely resemble previously
identified chromatin types. GREEN chromatin corresponds to
classic heterochromatin that is marked by SU(VAR)3-9, HP1,
and the HP1-interacting proteins LHR and HP6. As described
previously (Ebert et al., 2006; Greil et al., 2007), this type of chro-
matin is prominent in pericentric regions and on chromosome 4
(Figure S2A). To further validate this classification, we conducted

A
B C D
Genome coverage
117 Mb
Number of domains
8428 domains
All genes
15145 genes
Silent genes
4229 silent genes
Length of domains
040
800
200
020
0
coun
t0
200
Domain length (kb)0 10 20 30 40
025
0
>50
Number of genes per domain
020
00
600
030
0
coun
t0
200
Number of genes / domain
050
0
0 1 2 3 4 5 6 7 8 9 10 >10
mRNA expression
020
400
400
015
00
gene
cou
nt
040
00
100
−1 0 1 2 3 4
log10(RNA tag count)
no ta
gs
Figure 2. Characteristics of the Five Chromatin Types
(A) Coverage and gene content of chromatin domains of each type. The chromatin type of a gene is defined as the chromatin type at its transcription start site
(TSS). Gray sectors correspond to geneswhose TSSmaps at the transition between two chromatin types. Silent genes have an average RNA tag count less than 1
per million total tags (see D).
(B) Length distribution of chromatin domains, i.e., genomic segments covered contiguously by one chromatin type.
(C) Distribution of the number of genes per chromatin domain. Because some genes overlap with more than one domain, genes are assigned to a chromatin type
based on the type at the transcription start site.
(D) Histogram of mRNA expression determined by RNA tag profiling. Data are represented as log10 (tags per million total tags).
Dashed vertical lines in (B)–(D) indicate medians.
genome-wide ChIP of H3K9me2, a histone mark that is predom-
inantly generated by SU(VAR)3-9 and bound by HP1 (Hediger
and Gasser, 2006) . Indeed, H3K9me2 is highly and specifically
enriched in GREEN chromatin (Figure 3B).
BLUE chromatin corresponds to PcG chromatin, as shown by
the extensive binding by the PcG proteins PC, E(Z), PCL, and
SCE. Indeed, well-known PcG target loci such as the Hox
gene clusters are localized in BLUE domains (Figure S2B).
Furthermore, genome-wide ChIP of H3K27me3, the histone
mark that is generated by E(Z) and recognized by PC (Sparmann
and van Lohuizen, 2006), is highly enriched in BLUE chromatin
(Figure 3B). We emphasize that these histone modification
profiles serve as independent validation because they were not
used in the five-state HMMclassification. The fact that twomajor
well-known chromatin types were faithfully recovered indicates
that our chromatin classification strategy is biologically mean-
ingful.
Of interest, we identified several additional proteins that mark
BLUE or GREEN chromatin, or both. For example, moderate
degrees of occupancy of the histone deacetylase (HDAC)
RPD3 occur in both BLUE and GREEN chromatin, in accordance
with known biochemical and genetic interactions of RPD3 with
PcG proteins as well as SU(VAR)3-9 (Czermin et al., 2001; Tie
et al., 2003). The presence of EFF in BLUE chromatin is consis-
tent with a reported role of this protein in PcG-mediated silencing
(Fauvarque et al., 2001).
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. 215

BA
MRG15SIR2
RPD3DF31
RPII18BEAF32B
TOP1SIN3AASH2MAX
ASF1DSP1PCAFCDK7HP1C
JRACTBPCAF1
MUS209TIP60
TBPMNTDWG
PHOLSU(VAR)3−7
PRODACT5C
GROPHO
MED31BCD
SU(VAR)2−10GAF
CG31367LOLAL
ECRBRM
CTCFIALH1D1
SUURLAMEFF
SU(HW)PC
E(Z)PCLSCEHP6LHRHP1
SU(VAR)3−9
0 0.5 1
Fraction of bound loci
−10
12
H3K9me2
log2
(H3K
9me2
ChI
P / H
3 C
hIP)
−3−2
−10
1
H3K27me3
log2
(H3K
27m
e3 C
hIP
/ H3
ChI
P)−2
−10
12
3
H3K79me3
log2
(H3K
79m
e3 C
hIP
/ H3
ChI
P)
−2−1
01
23
H3K4me2
log2
(H3K
4me2
ChI
P / H
3 C
hIP)
−1.0
−0.5
0.0
0.5
Histone H3
log2
(H3
ChI
P / i
nput
)
Figure 3. Chromatin Types Are Characterized by Distinctive Protein Combinations and Histone Modifications
(A) Fraction of all probed genomic loci within each chromatin type that is bound by each protein. Bound loci were determined separately for each protein as
described in the text.
(B) Levels of histone H3 and four histone modifications as determined by genome-wide ChIP. The distribution of values is shown as ‘‘violin plots,’’ which are
symmetrized density plots of binding values per chromatin type: the wider the violin, the more data points are associated to that value. Dashed horizontal lines
indicate the median binding value for each chromatin type. Histone modification ChIP data were normalized to H3 occupancy.
See also Figure S2.
BLACK Chromatin Is the Prevalent Typeof Repressive ChromatinBLACK chromatin covers 48%of the probed genome and is thus
by far the most abundant type (Figure 2A). With a median size of
17 kb and with 134 domains larger than 100 kb, BLACK chro-
matin domains tend to be longer than domains of the four other
types (Figure 2B). BLACK chromatin is overall relatively gene
poor (Figure 2A; compare genome coverage and number of
genes), but it nevertheless harbors 4162 genes. By mRNA
high-throughput sequencing, we detected no transcriptional
activity (<1 mRNA molecule per 10 million) for 66% of the genes
in BLACK chromatin, whereas the remaining 34% have very low
activity (Figure 2D). This is in agreement with the low coverage of
BLACK chromatin by RPII18, a subunit shared by all three RNA
polymerases (Figure 3A), and a lack of the active histone marks
H3K4me2 and H3K79me3 as detected by ChIP (Figure 3B). We
note that the majority of silent genes in the genome are located
216 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
in BLACK chromatin (Figure 2A). Thus, BLACK chromatin is
a distinctively silent type of chromatin that covers a large part
of the genome.
BLACK chromatin is almost universally marked by four of the
53 mapped proteins: histone H1, D1, IAL, and SUUR, whereas
SU(HW), LAM, and EFF are also frequently present (Figure 3A).
Close-up views show that H1, D1, IAL, SUUR, and LAM have
a broad distribution within BLACK domains, whereas SU(HW)
exhibits a distinct, more focal pattern (Figure 4A).
Given that genes in BLACK chromatin are expressed at very
low levels, we asked whether BLACK chromatin actively
represses transcription or merely forms secondary to a lack of
transcription. In the former model, transgenes inserted into
BLACK chromatin may exhibit reduced transcription, whereas,
in the latter model, transgenes should be unaffected. To test
this, we examined a data set of 2852 random P element inser-
tions that carry a mini-white eye color reporter gene. For each

A
C
B D
GREEN BLUE BLACK RED YELLOW
HC
NE
s pe
r M
B0
2040
6080
100
log 2
(Dam
−H
1/D
am)
H1
−3
−1
1
log 2
(Dam
−S
UU
R/D
am)
SUUR
−1.
50.
5
log 2
( Dam
−D
1/D
am)
D1
−1.
50.
5
log 2
( Dam
−La
m/D
am)
LAM
−1
01
log 2
(Dam
−E
FF
/Dam
) EFF
−1.
00.
0
log 2
(Dam
−IA
L/D
am)
IAL
−1.
00.
0
log 2
(Dam
−S
U(H
W)/
D.) SU(HW)
−1
12
Genes
Type
16000 16100 16200 16300 16400 16500
Position on chr2R (kb)
GREEN BLUE BLACK RED YELLOW
Fra
ctio
n of
sile
nced
tran
sgen
es
0.0
0.1
0.2
0.3
0.4
26 302 307 841 1345
weakmediumstrong
Silencing:
S2 cellslarval tubulelarval salivary glandlarval midgutlarval hindgutlarval trachea
larval CNSlarval fat body
larval carcasstubulesalivary glandcropmidguthindgut
fat bodyvirgin spermathecamated spermatheca
heart
headeyebrainthorac. ganglionovarytestismale access. glandscarcasswhole fly
difference fromgenome mean
(log10)
−2 0 2
4,086 BLACK genes
Figure 4. Properties of BLACK Chromatin
(A) Sample plots of binding profiles of the six proteins that are the most prevalent in BLACK chromatin. Genes on both strands, as well as chromatin types, are
depicted below the profiles. Gray blocks in the background correspond to BLACK chromatin domains.
(B) Silencing of a white reporter gene in 2852 P element insertions in adult eyes (Babenko et al., 2010) separated by chromatin type in Kc cells. The fraction of
silenced insertions is higher among those overlapping with BLACK regions than in the rest of the genome (p < 2.2*10�16, chi-square test).
(C) Relative expression levels (log10 scale, normalized to genome-wide average) of BLACK genes in various tissues (Chintapalli et al., 2007).
(D) Density of highly conserved noncoding elements (HCNEs) per chromatin type.
of these insertions, the expression level was previously scored
and the integration site mapped (Babenko et al., 2010). Strik-
ingly, of 307 insertions located in BLACK regions, 36% exhibited
various degrees of w silencing, compared to 13% genome wide
(Figure 4B). Moreover, repression of transgene insertions in
BLACK chromatin ismore pronounced than in BLUE andGREEN
chromatin. This result strongly indicates that BLACK chromatin
has an active role in transcriptional silencing.
Developmental Regulation of Genes in BLACKChromatinNot all genes in BLACK regions are expected to remain silenced
in various tissues. Indeed, a survey of tissue expression profiling
data (Chintapalli et al., 2007) indicates that genes in BLACK
chromatin can become active, although their expression tends
to be restricted to a few tissues only (Figure 4C). This suggests
that BLACK chromatin domains, as defined in Kc167 cells, can
be remodeled into a different chromatin type in some cell types.
Consistent with this dynamic regulation, BLACK chromatin is
particularly rich in highly conserved noncoding elements
(HCNEs) (Figure 4D), which are thought to mediate gene regula-
tion (Engstrom et al., 2007). The density of HCNEs in BLACK
chromatin is comparable to that in BLUE chromatin, which
harbors many developmentally regulated genes (Tolhuis et al.,
2006), and is much higher than in the other three chromatin
types. Together, these data suggest that BLACK chromatin is,
at least in part, under developmental control.
YELLOW and RED Chromatin Are Two Distinct Typesof EuchromatinIn contrast to BLACK and BLUE chromatin, RED and YELLOW
chromatin have hallmarks of transcriptionally active euchro-
matin. Most genes in these two chromatin types produce
substantial amounts of mRNA (Figure 2D), and levels of RNA
polymerase (Figure 3A), H3K4me2, and H3K79me3 are typically
high, whereas levels of H3K9me2 and H3K27me3 are low
(Figure 3B).
RED andYELLOWchromatin share various chromatin proteins
(Figure 3A). Among these are the HDACs RPD3 and SIR2, as well
as the RPD3-interacting protein SIN3A. HDACs have recently
also been found in transcriptionally active chromatin in human
cells (Wang et al., 2009). Other proteins that are highly abundant
in both RED and YELLOW chromatin include DF31, a little-
studied protein that drives chromatin decondensation in vitro
(Crevel et al., 2001); ASH2, a homolog of a subunit of a H3K4
methyltransferase complex in yeast and vertebrate cells (Nagy
et al., 2002); and MAX, a DBF that is part of the MYC network of
regulators of growth and proliferation (Orian et al., 2003).
Aside from these similarities, RED and YELLOW chromatin
display striking differences. RED chromatin is abundantly
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. 217
A B
C
D
−0.
50.
51.
01.
52.
0
−5 0 5
Relative position from 5' end (kb)
log 2
(Dam
−M
RG
15D
am)
−5 0 5
Relative position from 3' end (kb)
−1
01
23
4
H3K36me3
−5 0 5
Relative position from 5' end (kb)
log 2
(H3K
36m
e3in
put)
−5 0 5
Relative position from 3' end (kb)
MRG15
−10
12
34
ORC binding
OR
C e
nric
hmen
t
−20
−10
010
20
Replication timing
log2
(Ear
ly /
Late
)
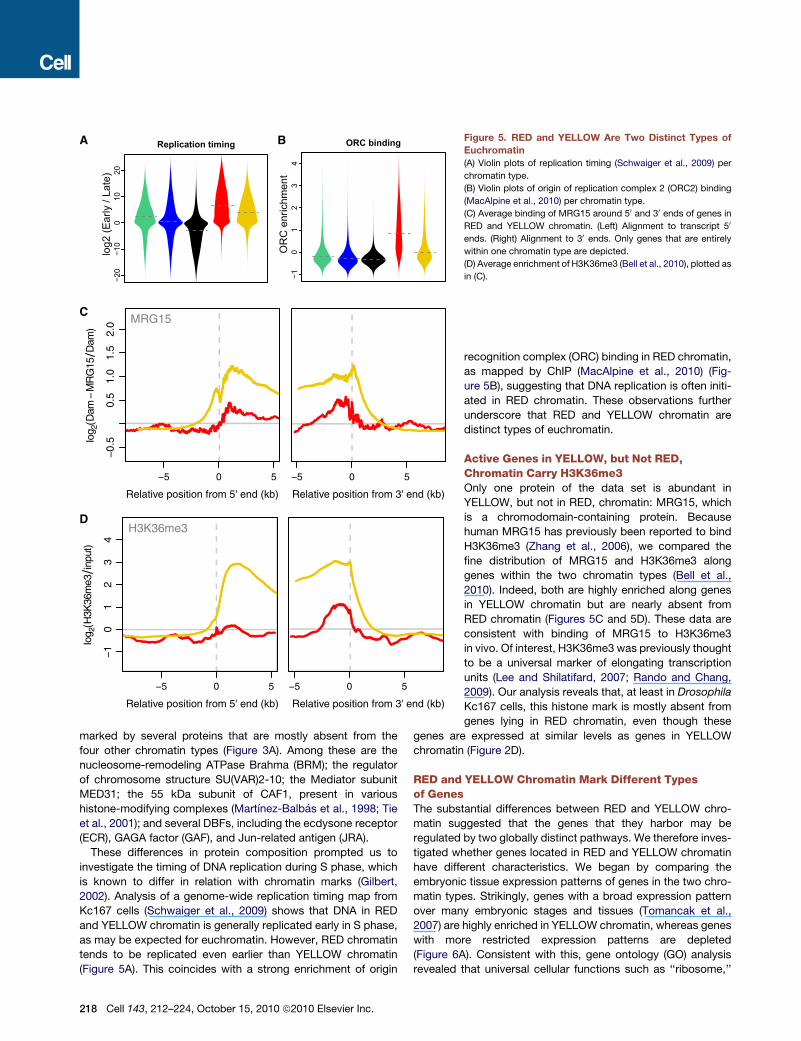
Figure 5. RED and YELLOW Are Two Distinct Types of
Euchromatin
(A) Violin plots of replication timing (Schwaiger et al., 2009) per
chromatin type.
(B) Violin plots of origin of replication complex 2 (ORC2) binding
(MacAlpine et al., 2010) per chromatin type.
(C) Average binding of MRG15 around 50 and 30 ends of genes in
RED and YELLOW chromatin. (Left) Alignment to transcript 50
ends. (Right) Alignment to 30 ends. Only genes that are entirely
within one chromatin type are depicted.
(D) Average enrichment of H3K36me3 (Bell et al., 2010), plotted as
in (C).
marked by several proteins that are mostly absent from the
four other chromatin types (Figure 3A). Among these are the
nucleosome-remodeling ATPase Brahma (BRM); the regulator
of chromosome structure SU(VAR)2-10; the Mediator subunit
MED31; the 55 kDa subunit of CAF1, present in various
histone-modifying complexes (Martınez-Balbas et al., 1998; Tie
et al., 2001); and several DBFs, including the ecdysone receptor
(ECR), GAGA factor (GAF), and Jun-related antigen (JRA).
These differences in protein composition prompted us to
investigate the timing of DNA replication during S phase, which
is known to differ in relation with chromatin marks (Gilbert,
2002). Analysis of a genome-wide replication timing map from
Kc167 cells (Schwaiger et al., 2009) shows that DNA in RED
and YELLOW chromatin is generally replicated early in S phase,
as may be expected for euchromatin. However, RED chromatin
tends to be replicated even earlier than YELLOW chromatin
(Figure 5A). This coincides with a strong enrichment of origin
218 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
recognition complex (ORC) binding in RED chromatin,
as mapped by ChIP (MacAlpine et al., 2010) (Fig-
ure 5B), suggesting that DNA replication is often initi-
ated in RED chromatin. These observations further
underscore that RED and YELLOW chromatin are
distinct types of euchromatin.
Active Genes in YELLOW, but Not RED,Chromatin Carry H3K36me3Only one protein of the data set is abundant in
YELLOW, but not in RED, chromatin: MRG15, which
is a chromodomain-containing protein. Because
human MRG15 has previously been reported to bind
H3K36me3 (Zhang et al., 2006), we compared the
fine distribution of MRG15 and H3K36me3 along
genes within the two chromatin types (Bell et al.,
2010). Indeed, both are highly enriched along genes
in YELLOW chromatin but are nearly absent from
RED chromatin (Figures 5C and 5D). These data are
consistent with binding of MRG15 to H3K36me3
in vivo. Of interest, H3K36me3 was previously thought
to be a universal marker of elongating transcription
units (Lee and Shilatifard, 2007; Rando and Chang,
2009). Our analysis reveals that, at least in Drosophila
Kc167 cells, this histone mark is mostly absent from
genes lying in RED chromatin, even though these
genes are expressed at similar levels as genes in YELLOW
chromatin (Figure 2D).
RED and YELLOW Chromatin Mark Different Typesof GenesThe substantial differences between RED and YELLOW chro-
matin suggested that the genes that they harbor may be
regulated by two globally distinct pathways. We therefore inves-
tigated whether genes located in RED and YELLOW chromatin
have different characteristics. We began by comparing the
embryonic tissue expression patterns of genes in the two chro-
matin types. Strikingly, genes with a broad expression pattern
over many embryonic stages and tissues (Tomancak et al.,
2007) are highly enriched in YELLOW chromatin, whereas genes
with more restricted expression patterns are depleted
(Figure 6A). Consistent with this, gene ontology (GO) analysis
revealed that universal cellular functions such as ‘‘ribosome,’’
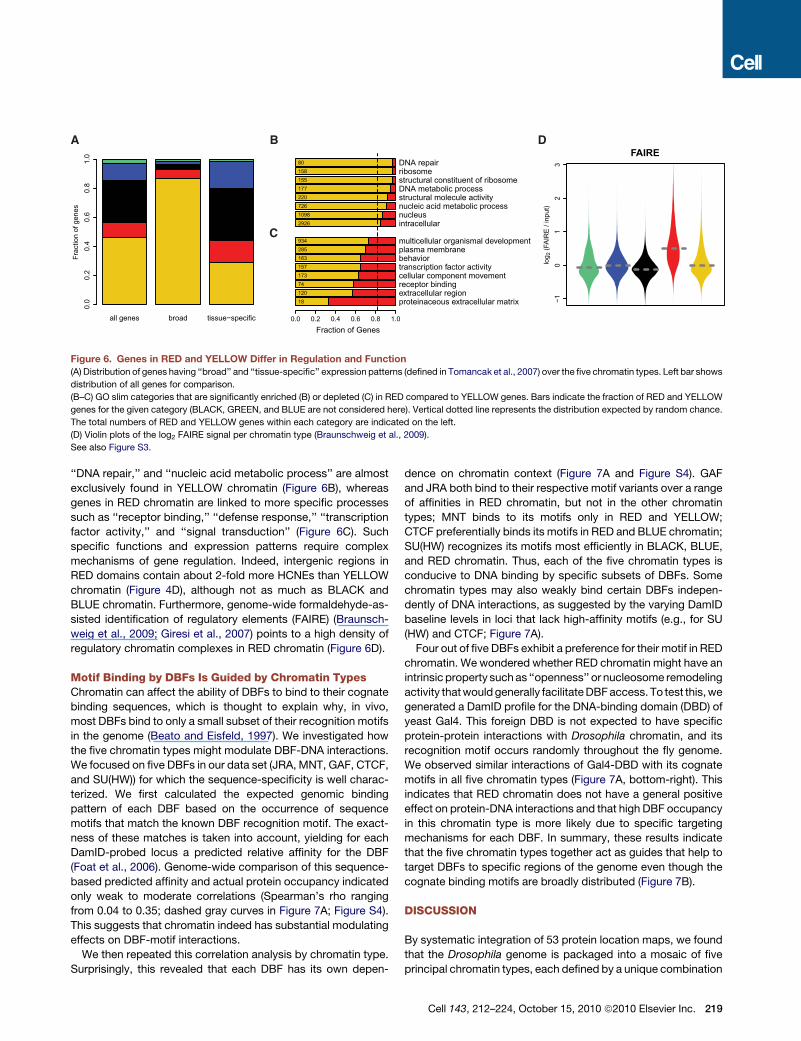
proteinaceous extracellular matrixextracellular regionreceptor bindingcellular component movementtranscription factor activitybehaviorplasma membranemulticellular organismal development
intracellularnucleusnucleic acid metabolic processstructural molecule activityDNA metabolic processstructural constituent of ribosomeribosomeDNA repair
Fraction of Genes0.0 0.2 0.4 0.6 0.8 1.0
1812074173157163285934
2926109872622017715515880
all genes broad tissue−specific
Frac
tion
of g
enes
0.0
0.2
0.4
0.6
0.8
1.0
A B
C
D
−10
12
3
FAIRE
log 2
(FAI
RE
/ inp
ut)
Figure 6. Genes in RED and YELLOW Differ in Regulation and Function
(A) Distribution of genes having ‘‘broad’’ and ‘‘tissue-specific’’ expression patterns (defined in Tomancak et al., 2007) over the five chromatin types. Left bar shows
distribution of all genes for comparison.
(B–C) GO slim categories that are significantly enriched (B) or depleted (C) in RED compared to YELLOW genes. Bars indicate the fraction of RED and YELLOW
genes for the given category (BLACK, GREEN, and BLUE are not considered here). Vertical dotted line represents the distribution expected by random chance.
The total numbers of RED and YELLOW genes within each category are indicated on the left.
(D) Violin plots of the log2 FAIRE signal per chromatin type (Braunschweig et al., 2009).
See also Figure S3.
‘‘DNA repair,’’ and ‘‘nucleic acid metabolic process’’ are almost
exclusively found in YELLOW chromatin (Figure 6B), whereas
genes in RED chromatin are linked to more specific processes
such as ‘‘receptor binding,’’ ‘‘defense response,’’ ‘‘transcription
factor activity,’’ and ‘‘signal transduction’’ (Figure 6C). Such
specific functions and expression patterns require complex
mechanisms of gene regulation. Indeed, intergenic regions in
RED domains contain about 2-fold more HCNEs than YELLOW
chromatin (Figure 4D), although not as much as BLACK and
BLUE chromatin. Furthermore, genome-wide formaldehyde-as-
sisted identification of regulatory elements (FAIRE) (Braunsch-
weig et al., 2009; Giresi et al., 2007) points to a high density of
regulatory chromatin complexes in RED chromatin (Figure 6D).
Motif Binding by DBFs Is Guided by Chromatin TypesChromatin can affect the ability of DBFs to bind to their cognate
binding sequences, which is thought to explain why, in vivo,
most DBFs bind to only a small subset of their recognition motifs
in the genome (Beato and Eisfeld, 1997). We investigated how
the five chromatin types might modulate DBF-DNA interactions.
We focused on five DBFs in our data set (JRA, MNT, GAF, CTCF,
and SU(HW)) for which the sequence-specificity is well charac-
terized. We first calculated the expected genomic binding
pattern of each DBF based on the occurrence of sequence
motifs that match the known DBF recognition motif. The exact-
ness of these matches is taken into account, yielding for each
DamID-probed locus a predicted relative affinity for the DBF
(Foat et al., 2006). Genome-wide comparison of this sequence-
based predicted affinity and actual protein occupancy indicated
only weak to moderate correlations (Spearman’s rho ranging
from 0.04 to 0.35; dashed gray curves in Figure 7A; Figure S4).
This suggests that chromatin indeed has substantial modulating
effects on DBF-motif interactions.
We then repeated this correlation analysis by chromatin type.
Surprisingly, this revealed that each DBF has its own depen-
dence on chromatin context (Figure 7A and Figure S4). GAF
and JRA both bind to their respective motif variants over a range
of affinities in RED chromatin, but not in the other chromatin
types; MNT binds to its motifs only in RED and YELLOW;
CTCF preferentially binds its motifs in RED and BLUE chromatin;
SU(HW) recognizes its motifs most efficiently in BLACK, BLUE,
and RED chromatin. Thus, each of the five chromatin types is
conducive to DNA binding by specific subsets of DBFs. Some
chromatin types may also weakly bind certain DBFs indepen-
dently of DNA interactions, as suggested by the varying DamID
baseline levels in loci that lack high-affinity motifs (e.g., for SU
(HW) and CTCF; Figure 7A).
Four out of five DBFs exhibit a preference for their motif in RED
chromatin. We wondered whether RED chromatin might have an
intrinsic property suchas ‘‘openness’’ or nucleosome remodeling
activity thatwouldgenerally facilitateDBFaccess. To test this,we
generated a DamID profile for the DNA-binding domain (DBD) of
yeast Gal4. This foreign DBD is not expected to have specific
protein-protein interactions with Drosophila chromatin, and its
recognition motif occurs randomly throughout the fly genome.
We observed similar interactions of Gal4-DBD with its cognate
motifs in all five chromatin types (Figure 7A, bottom-right). This
indicates that RED chromatin does not have a general positive
effect on protein-DNA interactions and that high DBF occupancy
in this chromatin type is more likely due to specific targeting
mechanisms for each DBF. In summary, these results indicate
that the five chromatin types together act as guides that help to
target DBFs to specific regions of the genome even though the
cognate binding motifs are broadly distributed (Figure 7B).
DISCUSSION
By systematic integration of 53 protein location maps, we found
that the Drosophila genome is packaged into a mosaic of five
principal chromatin types, each defined by a unique combination
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. 219

0 20 40 60 80 100
0.0
0.5
1.0
1.5GAF
0 20 40 60 80 100
−0.2
0.0
0.2
0.4
0.6
0.8
1.0 MNT
70 75 80 85 90 95 100
0.0
0.5
1.0
CTCF
70 75 80 85 90 95 100
−0.5
0.0
0.5
1.0
1.5
2.0 SU(HW)
0 20 40 60 80 100
0.0
0.5
1.0
1.5 JRA
0 20 40 60 80 100
−0.4
−0.2
0.0
0.2
0.4
0.6 GAL4
A
B
Sequence affinity (rank %) Sequence affinity (rank %) Sequence affinity (rank %)
Dam
ID s
core
Dam
ID s
core
Dam
ID s
core
Figure 7. Binding of DBFs to Their Cognate Motifs Is Differentially Guided by Chromatin Types(A) Correlations between predicted DNA affinity and actual binding detected by DamID, genome-wide (gray dashed lines), or for each chromatin type (solid lines)
for six DBFs as indicated. Curves are loess-fitted lines; raw data are shown in Figure S4.
(B) Cartoon model depicting the specific guidance of DBFs to their cognate motifs in only certain chromatin types, illustrated for CTCF and MNT. DBF binding to
its cognate motif (gray box) is guided by protein-protein interactions. The presence of specific interactors (colored shapes) only in some chromatin types may
account for targeting.
See also Figure S4.
of proteins. Extensive evidence demonstrates that the five types
differ in a wide range of characteristics aside from protein
composition, such as biochemical properties, transcriptional
activity, histone modifications, replication timing, and DBF tar-
geting, as well as sequence properties and functions of the
embedded genes. This validates our classification by indepen-
dent means and provides important insights into the functional
properties of the five chromatin types.
The Number of Chromatin StatesIdentifying five chromatin states out of the binding profiles of 53
proteins comes out as a surprisingly low number (one can form
�1016 subsets of 53 elements). We emphasize that the five chro-
220 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
matin types should be regarded as the major types. Some may
be further divided into subtypes, depending on how fine-grained
one wishes the classification to be. For example, within each of
the transcriptionally active chromatin types, promoters and 30
ends of genes exhibit (mostly quantitative) differences in their
protein composition (data not shown) and thus could be re-
garded as distinct subtypes. However, these local differences
are minor relative to the differences between the five principal
types that we describe here. We cannot exclude that the accu-
mulation of binding profiles of additional proteins would reveal
other novel chromatin types. We also anticipate that the pattern
of chromatin types along the genome will vary between cell
types. For example, many genes that are embedded in BLACK

chromatin (defined in Kc167 cells) are activated in some other
cell types (Figure 4C). Thus, the chromatin of these genes is likely
to switch to an active type.
Whereas the integration of data for 53 proteins provides
substantial robustness to the classification of chromatin along
the genome, a subset of only five marker proteins (histone H1,
PC, HP1, MRG15, and BRM), which together occupy 97.6% of
the genome, can recapitulate this classification with 85.5%
agreement (Figure S1E). Assuming that no unknown additional
principal chromatin types exist in some cell types, DamID or
ChIP of this small set of markers may thus provide an efficient
means to examine the distribution of the five chromatin types
in various cells and tissues, with acceptable accuracy.
BLACK Chromatin: A Distinct Type of RepressiveChromatinPrevious work on the expression of integrated reporter genes
(Handler and Harrell, 1999; Kelley and Kuroda, 2003; Markstein
et al., 2008) had suggested that most of the fly genome is tran-
scriptionally repressed, contrasting with the low coverage of
PcG and HP1-marked chromatin. BLACK chromatin, which
consists of a previously unknown combination of proteins and
covers about half of the genome, may account for these obser-
vations. Essentially all genes in BLACK chromatin exhibit
extremely low expression levels, and transgenes inserted in
BLACK chromatin are frequently silenced, indicating that BLACK
chromatin constitutes a strongly repressive environment. Impor-
tantly, BLACK chromatin is depleted of PcG proteins, HP1, SU
(VAR)3-9, and associated proteins and is also the latest to
replicate, underscoring that it is different from previously charac-
terized types of heterochromatin (here identified as BLUE and
GREEN chromatin).
The proteins that mark BLACK domains provide important
clues to the molecular biology of this type of chromatin. Loss
of LAM, EFF, or histone H1 causes lethality during Drosophila
development (Cenci et al., 1997; Lenz-Bohme et al., 1997; Lu
et al., 2009). Extensive in vitro and in vivo evidence has sug-
gested a role for H1 in gene repression, most likely through
stabilization of nucleosome positions (Laybourn and Kadonaga,
1991; Wolffe and Hayes, 1999; Woodcock et al., 2006). The
enrichment of LAM points to a role of the nuclear lamina in
gene regulation in BLACK chromatin (Pickersgill et al., 2006),
consistent with the long-standing notion that peripheral chro-
matin is silent (Towbin et al., 2009). Depletion of LAM causes
derepression of several LAM-associated genes (Shevelyov
et al., 2009), whereas artificial targeting of genes to the nuclear
lamina can reduce their expression (Finlan et al., 2008; Reddy
et al., 2008), suggesting a direct repressive contribution of the
nuclear lamina in BLACK chromatin. D1 is a little-studied protein
with 11 AT-hook domains. Overexpression of D1 causes ectopic
pairing of intercalary heterochromatin (Smith and Weiler, 2010),
suggesting a role in the regulation of higher-order chromatin
structure. SUUR specifically regulates late replication on poly-
tene chromosomes (Zhimulev et al., 2003), which is of interest
because BLACK chromatin is particularly late replicating. EFF
is highly similar to the yeast and mammalian ubiquitin ligase
Ubc4 that mediates ubiquitination of histone H3 (Liu et al.,
2005; Singh et al., 2009), raising the possibility that nucleosomes
in BLACK chromatin may carry specific ubiquitin marks. These
insights suggest that BLACK chromatin is important for chromo-
some architecture as well as gene repression and provide
important leads for further study of this previously unknown yet
prevalent type of chromatin.
RED and YELLOW: Distinct Types of EuchromatinIn RED and YELLOW chromatin, most genes are active, and
the overall expression levels are similar between these two
chromatin types. However, RED and YELLOW chromatin differ
in many respects. One of the conspicuous distinctions is the
disparate levels of H3K36me3 at active transcription units. This
histone mark is thought to be laid down in the course of tran-
scription elongation and may block the activity of cryptic
promoters inside of the transcription unit (Li et al., 2007). Why
active genes in RED chromatin lack H3K36me3 remains to be
elucidated.
The remarkably high protein occupancy in RED chromatin
suggests that RED domains are ‘‘hubs’’ of regulatory activity.
This may be related to the predominantly tissue-specific expres-
sion of genes in RED chromatin, which presumably requires
many regulatory proteins. We note that our DamID assay inte-
grates protein binding events over nearly 24 hr, so it is likely
that not all proteins bind simultaneously; some proteins may
bind only during a specific stage of the cell cycle. It is highly
unlikely that the high protein occupancy in RED chromatin
originates from an artifact of DamID, e.g., caused by a high
accessibility of RED chromatin. First, all DamID data are cor-
rected for accessibility using parallel Dam-only measurements.
Second, several proteins, such as EFF, SU(VAR)3-9, and histone
H1, exhibit lower occupancies in RED than in any other chro-
matin type. Third, ORC also shows a specific enrichment in
RED chromatin even though it was mapped by ChIP, by another
laboratory, and on another detection platform (MacAlpine et al.,
2010). Fourth, DamID of Gal4-DBD does not show any enrich-
ment in RED chromatin.
RED chromatin resembles DBF binding hot spots that were
previously discovered in a smaller-scale study inDrosophila cells
(Moorman et al., 2006). Discrete genomic regions targeted by
many DBFs have recently also been found in mouse ES cells
(Chen et al., 2008); hence, it is tempting to speculate that an
equivalent of RED chromatin may also exist in mammalian cells.
Housekeeping and dynamically regulated genes in budding
yeast also exhibit a dichotomy in chromatin organization (Tirosh
and Barkai, 2008), which may be related to our distinction
between YELLOW and RED chromatin. The observations that
RED chromatin is generally the earliest to replicate and is
strongly enriched in ORC binding suggest that this chromatin
type may be not only involved in transcriptional regulation, but
also in the control of DNA replication.
Chromatin Types as Guides for DBF TargetingOur analysis of DBF binding indicates that the five chromatin
types together act as a guidance system to target DBFs to
specific genomic regions. This system directs DBFs to certain
genomic domains even though the DBF recognition motifs are
more widely distributed. We propose that targeting specificity
is, at least in part, achieved through interactions of DBFs with
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. 221

particular partner proteins that are present in some of the five
chromatin types, but not in others (Figure 7B). The observation
that yeast Gal4-DBD binds its motifs with nearly equal efficiency
in all five chromatin types suggests that differences in compac-
tion among the chromatin types represent overall a minor factor
in the targeting of DBFs. Although additional studies will be
needed to further investigate the molecular mechanisms of
DBF guidance, the identification of five principal types of chro-
matin provides a firm basis for future dissection of the roles of
chromatin organization in global gene regulation.
EXPERIMENTAL PROCEDURES
Constructs
DamID constructs used for this study are listed in Table S1. New constructs
were cloned by TOPO cloning and GATEWAY recombination as described
(Braunschweig et al., 2009) or by Cre-mediated recombination. For the latter,
we generated an acceptor vector containing the Hsp70 promoter upstream of
myc-epitope tagged Dam, using the Creator Acceptor Vector Construction Kit
(Clontech, 631618). Chromatin protein open reading frames from pDNR-Dual
donor vectors (Drosophila Genomics Resource Center, Bloomington) were
cloned into the acceptor vector using the Creator DNA Cloning Kit (Clontech
PT3460-1). Nuclear localization was checked for all Dam-fusion proteins by
immunofluorescence microscopy with the 9E10 anti-Myc antibody (Santa
Cruz Biotechnology) after heat shock-induced expression as described (Greil
et al., 2007). Only MNT, GRO, and IAL gave weak nuclear signals but were not
discarded because MNT and GRO were successfully mapped by DamID in
previous studies (Bianchi-Frias et al., 2004; Orian et al., 2003) and IAL binds
metaphase chromosomes (Giet and Glover, 2001).
DamID, ChIP, and Microarrays
DamID assays were carried out under standardized conditions as described
previously (Moorman et al., 2006), with a minor modification: proteins were
grouped in sets sharing the same Dam-only controls for hybridization
purposes. For each group, three to five DamID assays on Dam alone were
carried out in parallel, the product of which was pooled before labeling.
ChIP and subsequent linear amplification reactions were done as described
(Kind et al., 2008) using anti-H3K27me3 (07-449) and anti-H3K4me2
(07-030) from Upstate Biotechnology; anti-H3K9me2 (1220) and anti-H3
(1791) from Abcam; affinity-purified anti-H1 serum (Braunschweig et al.,
2009); and anti-H3K79me3 (Schubeler et al., 2004) kindly provided by Fred
van Leeuwen. Fluorescent labeling of DamID and ChIP samples and two-color
hybridizations on custom-designed 385k NimbleGen arrays (Braunschweig
et al., 2009) were performed according to NimbleGen’s array users guide,
version 4.0. Arrays were scanned at 5 mm resolution, and raw data were
extracted using NimbleScan software. The identity of the hybridized material
was tracked by the presence of unique oligonucleotide spikes in each sample.
Furthermore, because the Dam-fusion expression vectors are produced in
Dam-positive bacteria, small amounts of the transfected plasmids are coam-
plified in the methylation-specific amplification protocol. This leads to a strong
signal in the open reading frame of the mapped protein, which allows us to
verify the identity of the used vector from the microarray data alone. This
open reading frame was masked before further data analysis.
Digital Gene Expression
Total RNA was isolated from growing Kc cells using TriZOL (Invitrogen), and
remaining DNA was degraded by shearing and DNaseI digestion. Poly(A)
RNA tag sequencing was carried out on an Illumina Solexa GAII using the
tag profiling kit with DpnII. Two RNA samples yielded 7.4 and 9.0 million reads.
Tags were mapped by BLAST, requiring at most two mismatches and eleven
consecutively matching bases. Only the tags mapping to the last GATC of
a transcript (FlyBase release 5.8) were counted and represented 70.3% and
69.4% of the total number of reads, respectively. Counts were normalized to
the total number of reads, and replicates were averaged.
222 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
Data Availability and Analysis
Computational methods are described in the Extended Experimental Proce-
dures.
ACCESSION NUMBERS
DamID, ChIP, and expression data, as well as binarized DamID data and a list
of the coordinates of all identified chromatin domains are available from
NCBI’s Gene Expression Omnibus, accession number GSE22069.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Extended Experimental Procedures,
five figures, and one table and can be found with this article online at
doi:10.1016/j.cell.2010.09.009.
ACKNOWLEDGMENTS
We thank Francesco Russo for help with vector cloning; Marja Nieuwland and
Arno Velds for help with RNA tag sequencing; Dirk Schubeler’s laboratory for
sharing H3K36 methylation data prior to publication; and Reuven Agami, Fred
van Leeuwen, Wouter Meuleman, Ludo Pagie, and Aleksey Pindyurin for help-
ful suggestions. Supported by an EMBO long-term fellowship to J.K.; National
Institutes of Health grants T32GM082797, R01HG003008, and U54CA121852
to L.D.W. and H.J.B.; and grants from the Netherlands Genomics Initiative,
NWO-ALW VICI, and an EURYI Award to B.v.S.
Received: June 10, 2010
Revised: August 2, 2010
Accepted: August 27, 2010
Published online: September 30, 2010
REFERENCES
Babenko, V.N., Makunin, I.V., Brusentsova, I.V., Belyaeva, E.S., Maksimov,
D.A., Belyakin, S.N., Maroy, P., Vasil’eva, L.A., and Zhimulev, I.F. (2010).
Paucity and preferential suppression of transgenes in late replication domains
of the D. melanogaster genome. BMC Genomics 11, 318.
Beato, M., and Eisfeld, K. (1997). Transcription factor access to chromatin.
Nucleic Acids Res. 25, 3559–3563.
Bell, O., Schwaiger, M., Oakeley, E.J., Lienert, F., Beisel, C., Stadler, M.B., and
Schubeler, D. (2010). Accessibility of the Drosophila genome discriminates
PcG repression, H4K16 acetylation and replication timing. Nat. Struct. Mol.
Biol. 17, 894–900.
Berger, S.L. (2007). The complex language of chromatin regulation during
transcription. Nature 447, 407–412.
Bianchi-Frias, D., Orian, A., Delrow, J.J., Vazquez, J., Rosales-Nieves, A.E.,
and Parkhurst, S.M. (2004). Hairy transcriptional repression targets and
cofactor recruitment in Drosophila. PLoS Biol. 2, E178.
Braunschweig, U., Hogan, G.J., Pagie, L., and van Steensel, B. (2009). Histone
H1 binding is inhibited by histone variant H3.3. EMBO J. 28, 3635–3645.
Cenci, G., Rawson, R.B., Belloni, G., Castrillon, D.H., Tudor, M., Petrucci, R.,
Goldberg, M.L., Wasserman, S.A., and Gatti, M. (1997). UbcD1, a Drosophila
ubiquitin-conjugating enzyme required for proper telomere behavior. Genes
Dev. 11, 863–875.
Chen, X., Xu, H., Yuan, P., Fang, F., Huss,M., Vega, V.B.,Wong, E., Orlov, Y.L.,
Zhang, W., Jiang, J., et al. (2008). Integration of external signaling pathways
with the core transcriptional network in embryonic stem cells. Cell 133,
1106–1117.
Chintapalli, V.R., Wang, J., and Dow, J.A. (2007). Using FlyAtlas to identify
better Drosophila melanogaster models of human disease. Nat. Genet. 39,
715–720.
Collas, P. (2009). The state-of-the-art of chromatin immunoprecipitation.
Methods Mol. Biol. 567, 1–25.

Crevel, G., Huikeshoven, H., and Cotterill, S. (2001). Df31 is a novel nuclear
protein involved in chromatin structure in Drosophila melanogaster. J. Cell
Sci. 114, 37–47.
Czermin, B., Schotta, G., Hulsmann, B.B., Brehm, A., Becker, P.B., Reuter, G.,
and Imhof, A. (2001). Physical and functional association of SU(VAR)3-9 and
HDAC1 in Drosophila. EMBO Rep. 2, 915–919.
de Wit, E., Greil, F., and van Steensel, B. (2007). High-resolution mapping
reveals links of HP1 with active and inactive chromatin components. PLoS
Genet. 3, e38.
Ebert, A., Lein, S., Schotta, G., and Reuter, G. (2006). Histonemodification and
the control of heterochromatic gene silencing in Drosophila. Chromosome
Res. 14, 377–392.
Engstrom, P.G., Ho Sui, S.J., Drivenes, O., Becker, T.S., and Lenhard, B.
(2007). Genomic regulatory blocks underlie extensive microsynteny conserva-
tion in insects. Genome Res. 17, 1898–1908.
Fauvarque, M.O., Laurenti, P., Boivin, A., Bloyer, S., Griffin-Shea, R., Bourbon,
H.M., and Dura, J.M. (2001). Dominant modifiers of the polyhomeotic extra-
sex-combs phenotype induced by marked P element insertional mutagenesis
in Drosophila. Genet. Res. 78, 137–148.
Finlan, L.E., Sproul, D., Thomson, I., Boyle, S., Kerr, E., Perry, P., Ylstra, B.,
Chubb, J.R., and Bickmore, W.A. (2008). Recruitment to the nuclear periphery
can alter expression of genes in human cells. PLoS Genet. 4, e1000039.
Foat, B.C., Morozov, A.V., and Bussemaker, H.J. (2006). Statistical mechan-
ical modeling of genome-wide transcription factor occupancy data by
MatrixREDUCE. Bioinformatics 22, e141–e149.
Gelbart, M.E., Bachman, N., Delrow, J., Boeke, J.D., and Tsukiyama, T. (2005).
Genome-wide identification of Isw2 chromatin-remodeling targets by localiza-
tion of a catalytically inactive mutant. Genes Dev. 19, 942–954.
Giet, R., and Glover, D.M. (2001). Drosophila aurora B kinase is required for
histone H3 phosphorylation and condensin recruitment during chromosome
condensation and to organize the central spindle during cytokinesis. J. Cell
Biol. 152, 669–682.
Gilbert, D.M. (2002). Replication timing and transcriptional control: beyond
cause and effect. Curr. Opin. Cell Biol. 14, 377–383.
Giresi, P.G., Kim, J., McDaniell, R.M., Iyer, V.R., and Lieb, J.D. (2007). FAIRE
(Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active
regulatory elements from human chromatin. Genome Res. 17, 877–885.
Greil, F., de Wit, E., Bussemaker, H.J., and van Steensel, B. (2007). HP1
controls genomic targeting of four novel heterochromatin proteins in
Drosophila. EMBO J. 26, 741–751.
Handler, A.M., and Harrell, R.A., 2nd. (1999). Germline transformation of
Drosophila melanogaster with the piggyBac transposon vector. Insect Mol.
Biol. 8, 449–457.
Hediger, F., and Gasser, S.M. (2006). Heterochromatin protein 1: don’t judge
the book by its cover! Curr. Opin. Genet. Dev. 16, 143–150.
Kelley, R.L., and Kuroda, M.I. (2003). The Drosophila roX1 RNA gene can
overcome silent chromatin by recruiting the male-specific lethal dosage
compensation complex. Genetics 164, 565–574.
Kind, J., Vaquerizas, J.M., Gebhardt, P., Gentzel, M., Luscombe, N.M.,
Bertone, P., and Akhtar, A. (2008). Genome-wide analysis reveals MOF as
a key regulator of dosage compensation and gene expression in Drosophila.
Cell 133, 813–828.
Laybourn, P.J., and Kadonaga, J.T. (1991). Role of nucleosomal cores and
histone H1 in regulation of transcription by RNA polymerase II. Science 254,
238–245.
Lee, J.S., and Shilatifard, A. (2007). A site to remember: H3K36 methylation
a mark for histone deacetylation. Mutat. Res. 618, 130–134.
Lenz-Bohme, B., Wismar, J., Fuchs, S., Reifegerste, R., Buchner, E., Betz, H.,
and Schmitt, B. (1997). Insertional mutation of the Drosophila nuclear
lamin Dm0 gene results in defective nuclear envelopes, clustering of nuclear
pore complexes, and accumulation of annulate lamellae. J. Cell Biol. 137,
1001–1016.
Li, B., Gogol, M., Carey, M., Pattenden, S.G., Seidel, C., and Workman, J.L.
(2007). Infrequently transcribed long genes depend on the Set2/Rpd3S
pathway for accurate transcription. Genes Dev. 21, 1422–1430.
Liu, Z., Oughtred, R., and Wing, S.S. (2005). Characterization of E3Histone,
a novel testis ubiquitin protein ligase which ubiquitinates histones. Mol. Cell.
Biol. 25, 2819–2831.
Lu, X., Wontakal, S.N., Emelyanov, A.V., Morcillo, P., Konev, A.Y., Fyodorov,
D.V., and Skoultchi, A.I. (2009). Linker histone H1 is essential for Drosophila
development, the establishment of pericentric heterochromatin, and a normal
polytene chromosome structure. Genes Dev. 23, 452–465.
MacAlpine, H.K., Gordan, R., Powell, S.K., Hartemink, A.J., and MacAlpine,
D.M. (2010). Drosophila ORC localizes to open chromatin and marks sites of
cohesin complex loading. Genome Res. 20, 201–211.
Markstein, M., Pitsouli, C., Villalta, C., Celniker, S.E., and Perrimon, N. (2008).
Exploiting position effects and the gypsy retrovirus insulator to engineer
precisely expressed transgenes. Nat. Genet. 40, 476–483.
Martınez-Balbas, M.A., Tsukiyama, T., Gdula, D., and Wu, C. (1998).
Drosophila NURF-55, a WD repeat protein involved in histone metabolism.
Proc. Natl. Acad. Sci. USA 95, 132–137.
Moorman, C., Sun, L.V., Wang, J., de Wit, E., Talhout, W., Ward, L.D., Greil, F.,
Lu, X.J., White, K.P., Bussemaker, H.J., and van Steensel, B. (2006). Hotspots
of transcription factor colocalization in the genome of Drosophila mela-
nogaster. Proc. Natl. Acad. Sci. USA 103, 12027–12032.
Nagy, P.L., Griesenbeck, J., Kornberg, R.D., and Cleary, M.L. (2002). A tri-
thorax-group complex purified from Saccharomyces cerevisiae is required
for methylation of histone H3. Proc. Natl. Acad. Sci. USA 99, 90–94.
Negre, N., Hennetin, J., Sun, L.V., Lavrov, S., Bellis, M., White, K.P., and
Cavalli, G. (2006). Chromosomal distribution of PcG proteins during
Drosophila development. PLoS Biol. 4, e170.
Orian, A., van Steensel, B., Delrow, J., Bussemaker, H.J., Li, L., Sawado, T.,
Williams, E., Loo, L.W., Cowley, S.M., Yost, C., et al. (2003). Genomic binding
by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes
Dev. 17, 1101–1114.
Pickersgill, H., Kalverda, B., de Wit, E., Talhout, W., Fornerod, M., and
van Steensel, B. (2006). Characterization of the Drosophila melanogaster
genome at the nuclear lamina. Nat. Genet. 38, 1005–1014.
Pindyurin, A.V., Moorman, C., de Wit, E., Belyakin, S.N., Belyaeva, E.S.,
Christophides, G.K., Kafatos, F.C., van Steensel, B., and Zhimulev, I.F.
(2007). SUUR joins separate subsets of PcG, HP1 and B-type lamin targets
in Drosophila. J. Cell Sci. 120, 2344–2351.
Rando, O.J., and Chang, H.Y. (2009). Genome-wide views of chromatin
structure. Annu. Rev. Biochem. 78, 245–271.
Reddy, K.L., Zullo, J.M., Bertolino, E., and Singh, H. (2008). Transcriptional
repression mediated by repositioning of genes to the nuclear lamina. Nature
452, 243–247.
Schmiedeberg, L., Skene, P., Deaton, A., and Bird, A. (2009). A temporal
threshold for formaldehyde crosslinking and fixation. PLoS ONE 4, e4636.
Schubeler, D., MacAlpine, D.M., Scalzo, D., Wirbelauer, C., Kooperberg, C.,
van Leeuwen, F., Gottschling, D.E., O’Neill, L.P., Turner, B.M., Delrow, J.,
et al. (2004). The histone modification pattern of active genes revealed
through genome-wide chromatin analysis of a higher eukaryote. Genes Dev.
18, 1263–1271.
Schwaiger, M., Stadler, M.B., Bell, O., Kohler, H., Oakeley, E.J., and
Schubeler, D. (2009). Chromatin state marks cell-type- and gender-specific
replication of the Drosophila genome. Genes Dev. 23, 589–601.
Shevelyov, Y.Y., Lavrov, S.A., Mikhaylova, L.M., Nurminsky, I.D., Kulathinal,
R.J., Egorova, K.S., Rozovsky, Y.M., and Nurminsky, D.I. (2009). The B-type
lamin is required for somatic repression of testis-specific gene clusters.
Proc. Natl. Acad. Sci. USA 106, 3282–3287.
Singh, R.K., Kabbaj, M.H., Paik, J., and Gunjan, A. (2009). Histone levels are
regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat.
Cell Biol. 11, 925–933.
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. 223

Smith, M.B., and Weiler, K.S. (2010). Drosophila D1 overexpression induces
ectopic pairing of polytene chromosomes and is deleterious to development.
Chromosoma 119, 287–309.
Sparmann, A., and van Lohuizen, M. (2006). Polycomb silencers control cell
fate, development and cancer. Nat. Rev. Cancer 6, 846–856.
Tie, F., Furuyama, T., Prasad-Sinha, J., Jane, E., and Harte, P.J. (2001).
The Drosophila Polycomb Group proteins ESC and E(Z) are present in
a complex containing the histone-binding protein p55 and the histone
deacetylase RPD3. Development 128, 275–286.
Tie, F., Prasad-Sinha, J., Birve, A., Rasmuson-Lestander, A., and Harte, P.J.
(2003). A 1-megadalton ESC/E(Z) complex from Drosophila that contains
polycomblike and RPD3. Mol. Cell. Biol. 23, 3352–3362.
Tirosh, I., and Barkai, N. (2008). Two strategies for gene regulation by promoter
nucleosomes. Genome Res. 18, 1084–1091.
Tolhuis, B., deWit, E., Muijrers, I., Teunissen, H., Talhout, W., van Steensel, B.,
and van Lohuizen, M. (2006). Genome-wide profiling of PRC1 and PRC2
Polycomb chromatin binding in Drosophila melanogaster. Nat. Genet. 38,
694–699.
Tomancak, P., Berman, B.P., Beaton, A., Weiszmann, R., Kwan, E.,
Hartenstein, V., Celniker, S.E., and Rubin, G.M. (2007). Global analysis of
patterns of gene expression during Drosophila embryogenesis. Genome
Biol. 8, R145.
224 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.
Towbin, B.D., Meister, P., and Gasser, S.M. (2009). The nuclear envelope—
a scaffold for silencing? Curr. Opin. Genet. Dev. 19, 180–186.
van Steensel, B., Delrow, J., and Henikoff, S. (2001). Chromatin profiling using
targeted DNA adenine methyltransferase. Nat. Genet. 27, 304–308.
Wang, Z., Zang, C., Cui, K., Schones, D.E., Barski, A., Peng, W., and Zhao, K.
(2009). Genome-wide mapping of HATs and HDACs reveals distinct functions
in active and inactive genes. Cell 138, 1019–1031.
Wolffe, A.P., and Hayes, J.J. (1999). Chromatin disruption and modification.
Nucleic Acids Res. 27, 711–720.
Woodcock, C.L., Skoultchi, A.I., and Fan, Y. (2006). Role of linker histone in
chromatin structure and function: H1 stoichiometry and nucleosome repeat
length. Chromosome Res. 14, 17–25.
Zhang, P., Du, J., Sun, B., Dong, X., Xu, G., Zhou, J., Huang, Q., Liu, Q., Hao,
Q., and Ding, J. (2006). Structure of human MRG15 chromo domain and its
binding to Lys36-methylated histone H3. Nucleic Acids Res. 34, 6621–6628.
Zhimulev, I.F., Belyaeva, E.S., Makunin, I.V., Pirrotta, V., Volkova, E.I.,
Alekseyenko, A.A., Andreyeva, E.N., Makarevich, G.F., Boldyreva, L.V.,
Nanayev, R.A., and Demakova, O.V. (2003). Influence of the SuUR gene on
intercalary heterochromatin in Drosophila melanogaster polytene chromo-
somes. Chromosoma 111, 377–398.

Supplemental Information
EXTENDED EXPERIMENTAL PROCEDURES
Data AnalysisMicroarray Data
Microarray data normalization and analysis were performed with R (R Development Core Team, 2009). All DamID data were sub-
jected to loess normalization. The median correlation between independent replicate experiments was 0.72.
The DamID procedure relies on laying amethylation ‘‘footprint’’ on the DNA at GATC sites, themotif that is recognized by Dam. The
methylation is subsequently assayed by the enzyme DpnI that specifically cuts DNA on methylated GATC sites. For those reasons,
the target material in DamID experiments consists of DNA fragments flanked by GATC sites. Those fragments are typically 200-300
bp in size, but can be longer and hybridize to several probes of the array. To avoid this complication, the main text refers to them as
‘‘probed loci.’’ For the sake of clarity, they will be referred to as ‘GATC fragments’ in the present document.
The array platform covers 183,258GATC fragments (FlyBase release 5) on six chromosome arms.When several probesmapped to
the same GATC fragment, we averaged their normalized log2 ratio to obtain a single score per GATC fragment.
Alignment Plots
CustomR scripts were used to calculate average binding profiles around 5’ and 3’ ends of genes. Genomic locations were converted
to coordinates relative to the nearest 5’ (respectively 3’) end of a gene, before applying a running median with a window covering 2%
of the plotted data. To ensure that points are aligned only once, windows around the end of a gene range from the midpoint of the
gene to the mid distance to the next gene. For 5’ (respectively 3’) alignments, genes that had an upstream (downstream) neighbor in
tandem orientation closer than 500 bp were excluded. This was done to avoid that features in 3’ (5’) of a gene would influence the
alignment plot.
Highly Conserved Noncoding Elements
Highly conserved noncoding elements (HCNEs) were defined as sequences of minimal length 50 bp and having at least 98% identity
with another species (Engstrom et al., 2007). Accordingly, we mapped HCNEs by aligning the introns and intergenic regions of
Drosophila melanogaster (FlyBase release 5.17) to the genome of D. mojavensis (FlyBase release 1) by exonerate (Slater and Birney,
2005).
Mapping Digital Gene Expression Tags
Mapping of Digital Gene Expression tagswas carried out by BLAST.We prepared a database of tags consisting of the 23 nucleotides
downstream of the last GATC site of every annotated transcript. Using the BLAST default parameters to map the Solexa reads
against this database, we could map 70.3% and 69.4% of the total number of reads from both experiments, respectively. All hits
had at most two mismatches. Counts per gene were computed by adding up the counts of all the transcripts of a given gene. Counts
were then normalized to the total number of reads per experiment and replicates were averaged. Gene mapping informations were
taken from D. melanogaster FlyBase release 5.8.
GO Analysis
The GO Slim terms for D. melanogaster were downloaded from http://www.geneontology.org/GO_slims/archived_GO_slims/
goslim_Drosophila.0200. The obsolete terms were updated according to the data obtained from http://www.geneontology.org/
ontology/obo_format_1_2/gene_ontology_ext.obo (date: 09:11:2009 14:57). Gene associations were downloaded from http://
www.geneontology.org/cgi-bin/downloadGOGA.pl/gene_association.fb.gz (CVS version 1.159). Genes were further associated
with all the terms higher in the GO hierarchy as indicated by the ‘‘is a’’ and ‘‘part of’’ keywords. Enrichment or depletion for GO terms
were tested against the hypergeometric distribution, at alpha level 0.01 (correcting for multiple testing by the Bonferroni method).
FlyAtlas Data
We took the log10 of the intensity reads from the FlyAtlas database (Chintapalli et al., 2007) and mean normalized them. 4086 genes
from the database were annotated as BLACK in our study. The genes were clustered by using the hclust and dist functions from R
with default parameters.
Analysis of DNA-Binding Factor Targeting
First we obtained a position-specific affinity matrix (PSAM) for each of the DBFs in our compendium. For each separate chromatin
type, we subtracted the type-specific mean DamID log2 ratio from all GATC fragments assigned to it. This was done to prevent the
inferred PSAMs from converging to a motif with vastly different abundance among the chromatin types. For each DBF, we then used
the OptimizePSAM tool from the REDUCE suite (http://bussemakerlab.org/software/REDUCE/) to fit a PSAM to the 10,000 GATC
fragments with the highest DamID signal, expected to cover an adequate range of binding affinities. A 2 kb window around the center
of the GATC fragment was associated with each DamID log2 ratio. To seed the fit, we used a consensus motif based on the known
binding preferences of each factor (AGGTGGCGC for CTCF; GAGAG for GAF; CGGN(11)CCG for GAL4; TGMSWMA for JRA;
CACGTG for MNT; and GCATAYYY for SU(HW)). Optimal relative affinity parameters were determined for each nucleotide position
with this consensus, as well as 3–4 flanking nucleotide positions on each side.
We used the inferred PSAM corresponding to each DBF to predict its relative in vitro binding affinity to the 2 kb region using the
AffinityProfile tool. To analyze the trend between predicted affinity and DamID log2 ratio, affinities were first converted to ranks, and
the loess function from the R language was used (using span = 1/10) to fit a smooth curve for each chromatin type. Statistical
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. S1

significance of the association between predicted affinity and DamID log2 ratios was determined using a rank correlation (imple-
mented in the function cor.test(method=‘‘spearman’’) in R).
Hidden Markov ModelsDamID Data Structure
DamID log2 ratios (Dam fusion protein over Dam only) of biological replicates were loess-normalized and averaged to give a probe-
wise estimated log2 ratio. Since several probes of the array canmap to the sameGATC fragment of index k, we averaged the normal-
ized log2 ratios of those probes into a single score xk. In our experience, after loess normalization, the Student’s t represents an excel-
lent approximation of the distribution of xk, whereas the Gaussian does not (see Figure S5A).
The DamID profile of protein FBpp0074431 used in Figure S5A showed no specific target. As a result, the distribution of the scores
xk is unimodal. Note that when a protein binds specific targets, the distribution of scores is actually a mixture of two Student’s t distri-
butions. Another peak centered around a higher average score corresponds to the score distribution of the bound loci.
Target Identification
To identify the binding sites of the protein of interest, we assume the existence of two states: ‘unbound’ and ‘bound.’ Intuitively,
a GATC fragment will be more likely in state ‘bound,’ say, if its DamID score is close to the mean score of all GATC fragments in
the state ‘bound,’ and if its immediate neighbors are also in the state ‘bound.’ Hidden Markov Models (HMMs) provide a statistical
framework to integrate the information of probes along with their neighbors and provide tractable algorithms to find the most likely
segregation into ‘unbound’ and ‘bound,’ i.e. the optimal set of targets (Cappe et al., 2005).
The fundamental concept of HMMs is to use the linear ordering of the observations to decompose their probability into a transition
term and an emission term. The transition term is the probability of going from one state to another at any single jump, for example the
probability of going from ‘unbound’ to ‘bound’ between two consecutive GATC fragments. Those probabilities are defined and can
be estimated. The emission termdescribes the probability of the observation given a state, for example the probability that the DamID
score would equal xk given that the GATC fragment is in state ‘bound’. Parameter estimation in the HMM framework is typically
carried out through the Baum-Welch algorithm, which is a particular case of EM (Estimation-Maximization) algorithm (Dempster
et al., 1976). Each iteration of the algorithm consists in computing the expected likelihood of the model and then finding the values
of the parameters for which this quantity is maximal. The form of the likelihood function allows to carry out the computation indepen-
dently for the transition parameters, through the Forward-Backward algorithm, and the emission parameters. Computing and opti-
mizing the expected likelihood of the emission parameters depends on the distribution.
In the case of the Student’s t distribution, this can be done by an EM algorithm. Because the Baum-Welch algorithm is itself an EM
algorithm, their iterations can be combined in a single EM algorithm optimizing both transition and emission parameters. However,
the convergence of the EM algorithm is very slow in the case of the Student’s t distribution, motivating the use of an alternative algo-
rithm termed MCECM (Liu and Rubin, 1995). This algorithm, specifically designed for the Student’s t distribution, features a 4-step
iteration. First, the degree of freedom n is fixed and the expected likelihood is computed and maximized as a function of the other
parameters (m and s2). Then m and s2 are fixed and the expected likelihood is computed and maximized as a function of n. We
combined the MCECM, and the Baum-Welch algorithms into an algorithm allowing efficient parameter estimation of HMMs with
Student’s t distribution.
Detailed Derivation
We model the series of DamID scores xk ðk = 1;. ;NÞ by a two-state HMM with emissions following a Student’s t distribution. The
states ‘unbound’ and ‘bound’ are denoted S0 and S1, respectively. Target identification amounts to inferring themost likely sequence
of states ðS1;. ;SNÞ. Given Sk =Siði = 0; 1Þ, xk follows a Student’s t distribution with mean mi, variance s2i and n degrees of freedom.
Note that n is the same for both states. The transition between the states are described by the 232 matrix Q. We will refer to the
probability of transition from state Si to state Sj by QðSi;SjÞ.We will denote qðtÞ = ðmðtÞ
0 ;mðtÞ1 ;s
2ðtÞ0 ;s
2ðtÞ1 ; nðtÞ;QðtÞÞ the vector of parameters at iteration t of the algorithm. Following the Baum-Welch
procedure, we will write the full log-likelihood of the observations (i.e., the likelihood of the observed and unobserved variables) and
iteratively maximize its expected value conditional on the observed data and the current values of the parameters.
The likelihood of the Student’s t distribution is best described as that of a Gaussian random variable with random variance. More
specifically, X � tðm; s2; nÞif and only if X � Nðm;s2=tÞ and t � c2ðnÞ, where X and t are independent. Importantly, t is not observed.
Note that ~s2, the variance of X, is not s2, but rather a function of ðs2; nÞ. The complete log-likelihood of X is decomposed as a sum of
two terms: the log-likelihood of the Gaussian variable, ‘N, and that of the c2 variable, ‘c2 .
‘�m; s2; n
��x; t�= ‘N�m; s2
��x; t�+ ‘c2ðnjtÞ (1)
= � 1
2log2p� logs+
ðx � mÞ2t2s2
+
�n=2log2 � logGðn=2Þ � t=2+ ðn=2� 1Þlogt
From this, we can write the complete log-likelihood of the data as:
S2 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.

‘�m0;m1;s
20; s
21; n;Q
��ðS1; x1; t1Þ;.; ðSN; xN; tNÞ�
= logðProbðS1ÞÞ+PNk = 2
logQðSk�1;SkÞ+PNk = 1
‘Nðm0;m1;s20; s
21jSk ; xk ; tkÞ+
PNk = 1
‘c2ðnjtkÞ: (2)
The conditional expectation of the full log-likelihood can be computed separately for all the terms of the sum. The term
EqðtÞ
(logðProbðS1ÞÞ+
XNk = 2
logQðSk�1;SkÞ��x1;.; xN
)
corresponds to the transition parameters and can be computed by the Forward-Backward algorithm. Using results and notations
from Cappe et al. (2005), this yields the estimate of Qðt + 1Þ as:
Qðt +1Þ�Si;Sj
�=
PNk = 1fk�1:kjnði; jÞPN
k = 1
Pl fk�1:kjnði; lÞ
; (3)
where fkðiÞ=PqðtÞðSk =Sijx1;.; xNÞ, i.e. the probability that the GATC fragment of index k is in state Si ði = 0; 1Þ given the whole
sequence of observations provided by the Forward-Backward algorithm.
The second term can be expressed as a function of moments, which makes subsequent computations easier.
EqðtÞ(XN
k =1
‘N�m0;m1; s
20; s
21
��Sk ; xk ; tk���x1;.; xN
)=Constant � EqðtÞ
(Xi
XNk = 1
1fSk =Sig
logsi +
ðxk � miÞ2tk2s2
i
!��x1;.; xN
)=Constant
�Xi
logsiEqðtÞ
(XNk = 1
1fSk =Sig��x1;.; xN
)+Xi
1
2s2i
EqðtÞ
(XNk = 1
1fSk =Sigtkx2k
��x1;.; xN
)+Xi
mi
s2i
EqðtÞ
(XNk = 1
1fSk =Sigtkxk��x1;.; xN
)
�Xi
m2i
2s2i
EqðtÞ
(XNk = 1
1fSk =Sigtk��x1;.; xN
): (4)
Here, 1 Sk =Si ð,Þg�is the indicator function of the set Sk =Sig�
. It can be shown by direct computation that the conditional distri-
bution of tk given ðx1;. ; xnÞ, Sk =Sig�and qðtÞ is GððnðtÞ + 1Þ=2; ðnðtÞ + ðxk � m
ðtÞi Þ2=s2ðtÞÞ=2Þ, so that
EqðtÞ
n1fSk =Sigtk
���x1;. ; xn
o=fkðiÞwkðiÞ;where (5)
wkðiÞ= nðtÞ + 1
nðtÞ +�xk � m
ðtÞi
�2=s2ðtÞ
: (6)
In other terms, we define weights wkðiÞ at index k and for state Si that are the expected values of the unobserved tk conditional on
the observations and the current values of the parameters. Note that the weights are small for outliers (i.e. for ðxk � mðtÞi Þ2=s2ðtÞ high),
so that those observations contribute little to the final estimate of the mean and variance. By substituting (5) into (4), one obtains the
following values for the expected sufficient statistics.
EqðtÞ
(XNk = 1
1fSk =Sigtk��x1;.; xN
)=XNk = 1
fkðiÞwkðiÞ;
EqðtÞ
(XNk = 1
1fSk =Sigtkxk��x1;.; xN
)=XNk = 1
fkðiÞwkðiÞxk ;
EqðtÞ
(XNk = 1
1fSk =Sigtkx2k
��x1;.; xN
)=XNk = 1
fkðiÞwkðiÞx2k :
The expected sufficient statistics turn out to beweightedmoments of the observations. The combinedweights fkðiÞwkðiÞ show that
observations unlikely to be in state Si (i.e. fkðiÞsmall) or being an outlier for state Si (i.e. wkðiÞsmall) contribute little to the estimation of
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. S3

mean and variance for state Si. The updated values of m0;m1; s20 and s21 are the ones that maximize the expected log-likelihood. They
are obtained by differentiating (4) with respect to the parameters:
mðt + 1Þi =
PNk =1fkðiÞwkðiÞxkPNk = 1fkðiÞwkðiÞ
; (7)
s2ðt + 1Þi =
PNk =1fkðiÞwkðiÞ
�xk � m
ðt + 1Þi
�2PN
k = 1fkðiÞ: (8)
Following the EMprocedure, one could update nðtÞ by differentiating the last term of (2). However this procedure proves to give slow
convergence. Instead, we used the idea of the MCECM algorithm (Liu and Rubin, 1995), which is substantially faster than the EM in
terms of number of cycles. This involves performing another Forward-Backward pass with parameters~qðtÞ= ðmðt +1Þ
0 ;mðt + 1Þ1 ; s
2ðt +1Þ0 ;s
2ðt + 1Þ1 ; nðtÞ;Qðt + 1Þ; Þ and computing the weights wkðiÞ again through formula (6) before differentiating the
last term of (2). Let
~wk =Xi
fkðiÞwkðiÞ: (9)
The weights ~wk are the weighted averages of the weights wkðiÞ and are designed to be small for observations that are outliers in
every state. The updated value nðt + 1Þ is then the value n such that
1� j�n2
�+ log
�n2
�+1
N
XNk =1
log�~wk
�� ~wk +1
N
XNk =1
j
�n+ 1
2
� log
�n+ 1
2
= 0; (10)
where jðzÞ is the digamma function i.e. jðzÞ=dlogGðzÞ=dz. The solution of this equation is computed numerically (by dichotomy).
Here is a summary of the different steps carried out at each cycle of the algorithm, where CM stands for ‘constrained’ or ‘condi-
tional’ maximization (Liu and Rubin, 1995).
E-step 1: Compute the Forward-Backward smoothing estimates using qðtÞ and compute the weights wkðiÞ through (6).
CM-step 1: Update the transition probabilities through (3), the estimated means through (7), and the estimated variances through
(8).
E-step 2: Compute the Forward-Backward smoothing estimates using ~qðtÞ
and compute the weights again through (6). Compute
the averaged weights ~wk through (9).
CM-step 2: Compute nðt +1Þ by solving (13).
After multiple cycles, the estimates usually converge to stable values, at which point the algorithm is stopped. The most likely
sequence of states ðS1;. ;SNÞ is then computed through the Viterbi algorithm.
Virtual Bridging
Gaps in the array design are filled by virtual positions for which emissions are not available (NA). Gaps longer than 2D are bridged by
L=ðD� 1Þ virtual positions, where D is the mean distance between two consecutive GATC fragment ‘start’ positions and L is the gap
size.
The emission probability of those virtual positions is set to 1 for every state. As a consequence, virtual bridging directly influences
the estimation of the transition probabilities Q in the modified Baum-Welch algorithm presented above because it modifies the
spacing between reads.
Multidimensional Inference
The modified Baum-Welch algorithm presented above can easily be applied to the case of multidimensional measurements. These
are typically different binding profiles, or different linear combinations of binding profiles.
Assume thatmeasurements xk ðk = 1;. ;N) are now p -dimensional vectors, such that xk;p denotes the value of the k th read in the p
th dimension. The distribution of xk given Sk =Si is a multidimensional Student’s t variable with mean vector mi, and mean variance
matrix ~Si and n degrees of freedom.
Again, the likelihood of the multidimensional Student’s t distribution is described as that of a multidimensional Gaussian random
variable with random variance. X � Tðm;S; nÞif and only if X � Nðm;S=tÞ and t � c2ðnÞ, where X and t are independent. Again ~S, the
variance matrix of X, is not S, but rather a function of ðS; nÞ.Formula (6) giving the weights is replaced by
wkðiÞ= nðtÞ +p
nðtÞ +�xk � m
ðtÞi
�2=s2ðtÞ
: (11)
S4 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.

The sufficient statistics now include the cross-products of xk, and equation (8) should be replaced by
Sðt + 1Þi ðp;qÞ=
PNk = 1fkðiÞwkðiÞ
�xk;p � m
2ðt + 1Þi;p
��xk;q � m
2ðt + 1Þi;q
�PN
k = 1fkðiÞ: (12)
Equation (10) giving the updated value of n is replaced by
1� j�n2
�+ log
�n2
�+1
N
XNk = 1
log�~wk
�� ~wk +1
N
XNk =1
j
�n+p
2
� log
�n+p
2
= 0: (13)
TheMCECMprocedure outlined at the end of ‘‘Detailed Derivation’’ can be applied to identify targets in amultidimensional setting.
Also, the procedure simply extends to the case of more than two states. Ifm is the number of states, the matrix Q is anm3mmatrix
and equation (3) is still valid. The bounds of the sum in equation (4) and (9) have been left out on purpose so that all the formulas hold
irrespective of the number of states.
Definition of the Chromatin TypesDetermining the Number of States
By compiling the DamID scores of the 53 profiles, the probed genome can be viewed as a cloud of points in a 53-dimensional space.
In this space, two GATC fragments are close to each other if they are bound by the same proteins (irrespective of their genomic coor-
dinates). Principal Component Analysis (PCA) gives the approximation of the cloud in lower dimensions that best preserves the over-
all spread (i.e. the variance) of the cloud. Each recurrent protein binding signature should appear as an individual lobe of the projected
cloud. Importantly, one expects the projected coordinates to be distributed as a Student’s t variable because they are weighted aver-
ages of the DamID binding scores, meaning that the projected cloud can be described by the multi-state multi-dimensional HMM
presented above.
We performed scaled PCA on the 53 profiles and examined the projection of the cloud on the first principal components. It imme-
diately appeared that at least three components are required to give a qualitative representation of the data, because a distinct sub-
cloud separates in the third component. The projection on the first 3 principal components reveals 5 distinct lobes. Whereas the plot
shown in Supplementary Figure 5B suggests that the fourth component might be included in the analysis, none of the 5 clouds forks
into multiple sub-clouds in the fourth or higher order principal components. Therefore, we reduced the data to its first 3 principal
components.
A substantial part of the variance in tiling array experiments is noise. For example, discretizing a DamID binding profile by a two-
state HMM accounts for approximately 40% of the total variance (the median score out of 53 profiles is 39.4%). In comparison, the
total variance captured by the first 3 principal components is 57.7%. This figure suggests that the procedure efficiently removes the
technical variation from the dataset without causing a strong loss of information.
Mapping the Chromatin Types
Fitting an HMM to the original dataset would involve estimating 7,441 parameters (53 4 transition parameters, 53 53means, 53 53
variances, 53 1374 covariances and 1 degree of freedom). In contrast, if the dataset is replaced by its projections on the first 3 prin-
cipal components, the number of parameters drops to 66 (5 3 4 transition parameters, 53 3 means, 53 3 variances, 53 3 covari-
ances and 1 degree of freedom), representing a substantial gain of robustness.
To map the chromatin types in the fly genome, we used the method described in ‘‘Multidimensional Inference’’ with 5 states. The
initial degree of freedom was set to 6. The initial means of the modified Baum-Welch algorithm were determined visually. The initial
covariance matrices were set to the covariance matrix of the 3-dimensional cloud. Note that within each lobe, the projections on the
principal components may be correlated, even if their correlation is null on the whole dataset. Thus, after the first iteration the terms in
equation (12) are not expected to be 0 for psq.
The output of the modified Baum-Welch algorithm showed some sensitivity to initial parameter values, mainly to the means. For
some initial values, the modified Baum-Welch algorithm failed to identify 5 clearly separated states. However, there was only one
fitted model where the states were clearly separated, showing that the segmentation is robust to initial conditions.
The initial DamID binding values and final state calls are provided as supplementary files. An implementation of the procedure as an
R package (R Development Core Team, 2009) is available upon request.
SUPPLEMENTAL REFERENCES
Bushey, A.M., Ramos, E., and Corces, V.G. (2009). Three subclasses of a Drosophila insulator show distinct and cell type-specific genomic distributions. Genes
Dev. 23, 1338–1350.
Cappe, O., Moulines, E., and Ryden, T. (2005). Inference in Hidden Markov Models (Springer), pp. 369.
Chintapalli, V.R., Wang, J., and Dow, J.A. (2007). Using FlyAtlas to identify better Drosophila melanogastermodels of human disease. Nat. Genet. 39, 715–720.
Dempster, A.P., Laird, M., and Rubin, D.B. (1976). Maximum likelihood from Incomplete Data via the EM Algorithm. J.R. Stat. Soc., B 39, 1–38.
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. S5

Engstrom, P.G., Ho Sui, S.J., Drivenes, O., Becker, T.S., and Lenhard, B. (2007). Genomic regulatory blocks underlie extensive microsynteny conservation in
insects. Genome Res. 17, 1898–1908.
Liu, C., and Rubin, D.B. (1995). ML estimation of the t distribution using EM and its extensions, ECM and ECME. Statist. Sinica 5, 19–39.
R Development Core Team. 2009. R: A Language and Environment for Statistical Computing. http://www.R-project.org
Slater, G.S., and Birney, E. (2005). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31.
S6 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.

AMRG15
SU(VAR)3−7SU(VAR)3−9
HP6HP1LHR
CAF1ASF1
MUS209TOP1
RPII18SIR2
RPD3CDK7DSP1DF31MAX
PCAFASH2HP1cCtBPJRA
BRMECRBCD
MED31SU(VAR)2−10
LOLALGAF
CG31367ACT5C
TIP60MNT
SIN3ATBP
DWGPHOLPROD
BEAF32bSU(HW)
LAMD1H1
SUUREFFIAL
GROPHO
CTCFPC
E(Z)PCLSCE
16400 16500 16600 16700 16800 16900Postition on chr2L (kb)
Type
Genes
B C
D
log 2
(Dam
−H
1/D
am)
H1 DamID
−4
−2
0
log 2
(H1
ChI
P/in
put)
H1 ChIP
−1.
00.
00.
5
Genes
11650 11750 11850 11950 12050 12150 12250 12350 12450 12550 12650
Position on chr2L (kb)
log 2
(Dam
−S
U(H
W)/
Dam
) SU(HW) DamID
−1
01
23
log 2
(SU
(HW
) C
hIP
/inpu
t) SU(HW) ChIP
−1
12
34
Genes
6800 6900 7000 7100 7200 7300 7400 7500 7600 7700 7800
Position on chr2R (kb)
log 2
(Dam
−C
TC
F/D
am) CTCF DamID
−0.
51.
52.
5
log 2
(CT
CF
ChI
P/in
put) CTCF ChIP
−1
12
34
Genes
400 500 600 700 800 900 1000 1100 1200 1300 1400
Position on chr2L (kb)
E
−6 −4 −2 0
−4−2
02
46
PC1
−6 −4 −2 0 2 4 6
−4−2
02
46
PC2
PC3
−5 0
05
1015
PC1
−5 0 5
−20
24
68
PC2
PC3
35 40 45 50
020
4060
8010
0
Robustness of the classification
Subsample size
Estim
ated
per
cent
age
% Faithful segmentations% Overlap with current segmentation
5PC2
2 4 6PC2
Figure S1. Validation of DamID and Robustness Analysis of the Five-Type segmentation, Related to Figure 1
(A) Magnification of the 53 DamID profiles as depicted in Figure 1. Traces show log2 DamID ratios, and width of black bars represents the length of DpnI restriction
fragments.
(B) Comparisons of DamID (Braunschweig et al., 2009) and ChIP for histone H1 (this study), SU(HW), and CTCF (Bushey et al., 2009). All data sets were subjected
to running mean smoothing with a window of three data points. Genes on the top and bottom strand are depicted as lines with blocks indicating exons.
(C) The five-state classification is robust to the quantification method. DamID profiles for each protein were binarized before projection onto the first three prin-
cipal components. The shape of the cloud is different from the one shown inmain Figure 1B, but the five types still form separated clouds in the first three principal
components.
(D) Sensitivity analysis of the segmentation. The principal component analysis and HMM procedure was applied as in Figure 1 to data sets where one or more
proteins were left out. It is expected that smaller sets will not always exhibit all five states; for example, a subset that lacks the four PcG proteins will not identify
the BLUE state. In that case, the loci formerly assigned to BLUE will be assigned to another color by the HMM. We call such a segmentation ‘‘unfaithful.’’ On the
contrary, ‘‘faithful’’ segmentations show no replacement of a color by another. The percentage of overlap between the current segmentation (based on the
complete data set) and unfaithful segmentations is irrelevant: it mostly reflects the size of the type that has been replaced. For example if BLUE has been replaced
by another color at least 20% of the calls will differ (i.e., all the former BLUE calls). For subsets of 35–51 proteins, 50 samples were drawn at random without
replacement. For subsets of 52 proteins, all possible 53 subsets were tested. For each subsample size, the percentage of faithful segmentations (thin lines)
and their mean percentage of overlap with the current segmentation (thick lines) were determined. Vertical bars represent ± standard deviations. The plot shows
that larger sets of proteins give an increasingly reliable discovery of the five states. Faithful segmentations show substantial agreement with the current definition,
even for smaller sample sizes. Thus, identification of the states is sensitive to protein choice, but the classification itself is robust.
(E) Minimal set of proteins defining the five types. A carefully selected set of five proteins (histone H1, PC, HP1, MRG15, and BRM) summarizes the segmentation
in 5 types. The data points were projected on their first three principal components, showing that the types are clearly visible with this minimal set of proteins. The
coloring was obtained by applying the HMM to the first three principal components, exactly as was done for the 53 proteins. The agreement with the 53 profile
segmentation is 85.5%.
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. S7
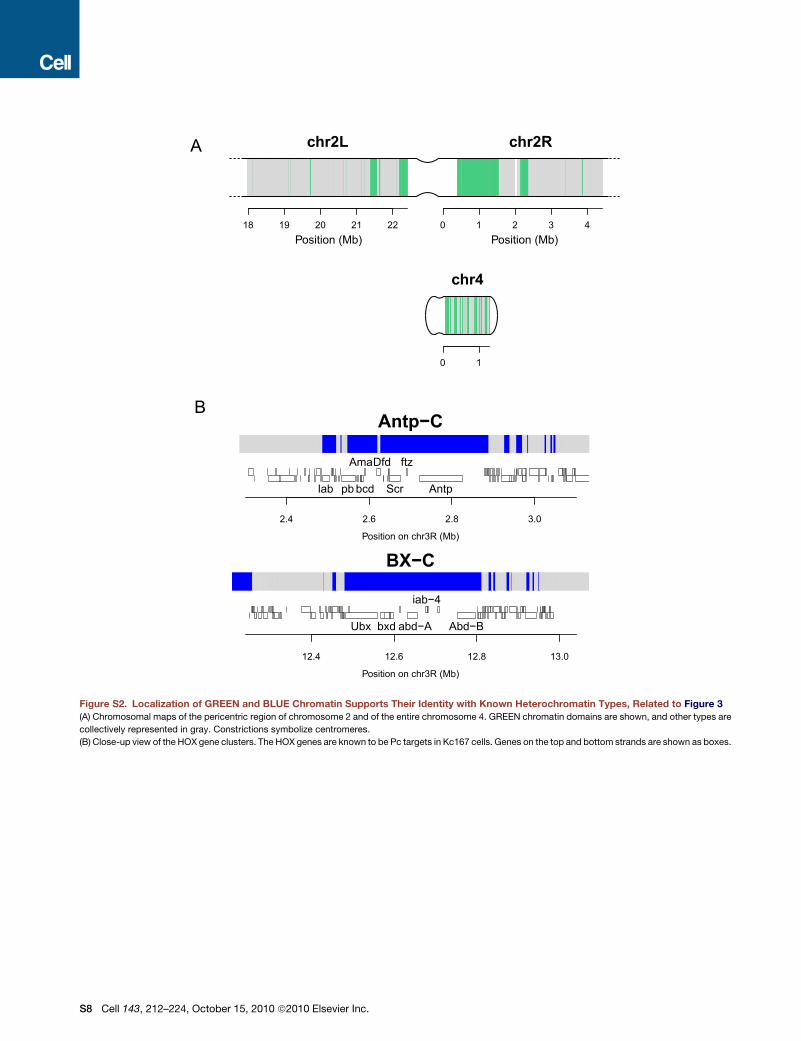
18 19 20 21 22Position (Mb)
chr2L chr2R
0 1 2 3 4Position (Mb)
0 1
chr4
A
BAntp−C
AmaDfd ftz
lab pb bcd Scr Antp
2.4 2.6 2.8 3.0
Position on chr3R (Mb)
BX−C
iab−4
Ubx bxd abd−A Abd−B
12.4 12.6 12.8 13.0
Position on chr3R (Mb)
Figure S2. Localization of GREEN and BLUE Chromatin Supports Their Identity with Known Heterochromatin Types, Related to Figure 3
(A) Chromosomal maps of the pericentric region of chromosome 2 and of the entire chromosome 4. GREEN chromatin domains are shown, and other types are
collectively represented in gray. Constrictions symbolize centromeres.
(B) Close-up view of the HOX gene clusters. The HOX genes are known to be Pc targets in Kc167 cells. Genes on the top and bottom strands are shown as boxes.
S8 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.

A
B
16.2 16.4 16.6 16.8 17.0 17.2
Position on chr2L (Mb)
P
rote
ins
boun
d
018
36
Protein count per locus
Freq
uenc
y
015
000
Freq
uenc
y
040
010
00
Freq
uenc
y
015
00
Freq
uenc
y
030
00
Freq
uenc
y
060
014
00
0 10 20 30 40Number of bound proteins (out of 53 proteins)
Figure S3. Chromatin Types Differ Widely in Their Total Protein Occupancy, Related to Figure 6
(A) Sample plot of the total occupancy (out of 53 mapped proteins) on a 1Mb segment of chromosome 2L. The height of each vertical line indicates the number of
proteins bound to a locus. The color of the line indicates the local chromatin type.
(B) Histograms of total occupancy distributions per chromatin type. Gray vertical dashed lines indicate median values.
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. S9

Position Position Position Position Position Position
Dam
ID s
core
Dam
ID s
core
Dam
ID s
core
Dam
ID s
core
Dam
ID s
core
Dam
ID s
core
A
B
0 10020 40 60 80 0 10020 40 60 80 0 10020 40 60 80 0 10020 40 60 80 0 10020 40 60 80 0 10020 40 60 80
Figure S4. Chromatin Types Guide DBF Binding to Their Motifs, Related to Figure 7
(A) Optimized position-specific affinity matrices for 5 Drosophila DBFs and Gal4-DBD (see Extended Experimental Procedures for details).
(B) Distributions of relative affinites of GATC fragments mapping within each of the five types. For each chromatin type, DamID scores of GATC fragments ranked
by the predicted affinity of a 2 kb window around its center. Gray lines represent the loess fit. Last row: superimposed loess fits for the five chromatin types.
Colored numbers indicated the Spearman’s rank correlation coefficients of DamID value with predicted affinities.
S10 Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc.

−2 −1 0 1 2
0.0
0.5
1.0
1.5
2.0
2.5
Distribution of x scores
x score
Den
sity
Observed x scoresFitted Student's tFitted Gaussian
1 4 7 10 14 18 22 26 30 34 38 42 46 50
Principal Components
Frac
tion
of to
tal v
aria
nce
0.0
0.1
0.2
0.3
0.4
A B
Figure S5. Addendum to the Analytical Methods, Related to Extended Experimental Procedures
(A) The scores x follow a Student’s t distribution. The plot shows the density of loess-normalized and averaged per GATC fragments log2 ratios of the DamID
profile of protein FBpp0074431 (black line). DamID for this protein shows no specific signal, and no targets were detected. The red line is the density of Student’s
t distribution fitted by maximum likelihood, and the blue line is the density of a fitted Gaussian distribution.
(B) Variance accounted for by individual principal components.
Cell 143, 212–224, October 15, 2010 ª2010 Elsevier Inc. S11




















