
Structural changes in the crystal packing of highly hindered symmetrical vicinal bis-amides
7
Structural changes in the crystal packing of highly hindered symmetrical vicinal bis-amides Claudio A. Jime ´nez,* a Julio B. Belmar, a Fernando S. Delgado, bc Miguel Julve d and Catalina Ruiz-Pe ´rez b Received 4th December 2006, Accepted 20th April 2007 First published as an Advance Article on the web 17th May 2007 DOI: 10.1039/b617605d The crystal structures of 1,4-bis(3,5-di-tert-butyl-2-hydroxybenzamido)butane (1) and 1,3-bis(3,5- di-tert-butyl-2-hydroxybenzamido)-2,2-dimethylpropane (2) have being characterized by single crystal X-ray diffraction. Their crystal packing is discussed in terms of the different interactions they exhibit. A brief discussion, based on hydrogen bonds, on the structural features of bis-(3,5- di-tert-butyl-2-hydroxybenzamide) compounds is carried out. Introduction The complementary recognition between functional groups in molecules is the first step in crystallization. Because molecular recognition is complementary in nature, 1 a functional group approach to crystal engineering is, at least currently, not possible, 2 and prediction of crystal structures is correspond- ingly difficult. 3 Current systematic synthetic strategies in purely organic species attempt to tackle this issue by identifying systems where the crystal construction may be taken up in a modular fashion and where interaction inter- ference is minimal. Thus the effect of any particular func- tionality can be identified. 4 Indeed, this is one of the well accepted goals of crystal engineering. Functional groups exert both steric and electronic effects on the structure and reactivity, but even in solution, where the functional group approach is broadly applicable, separating these two effects is quite difficult. 5 This issue is more or less intractable in the solid state because the effects of the functional groups are in themselves implicitly connected to the nature and position of the other functional groups in the molecule. In the context of crystal engineering, steric and electronic effects are synonymous with geometrical and chemical effects in the crystal packing. 6 While the origin of all intermolecular interactions is electrostatic 7 and there is no rigorous basis for such a distinction, it is still convenient to distinguish these components on the basis of the distance dependence of the corresponding interactions. 8 In this framework, amide functions are the robust synthons of wide consensus and they have become the choice of a great number of research groups. 9 This functional group has numerous applications. In particular, bulky amides and their lithium salts are widely used as deprotonating reagents in organic synthesis due to their relatively low nucleophilicity and strong Brønsted basicity, 10,11 as well as reagents for asymmetric synthesis of compounds containing C–C bonds. 12 Furthermore, amides have been found to be promising precursors to synthesize a variety of complexes with main group elements, transition metal ions, and lanthanide cations, many of them well-characterized. 13 Networking of molecules through interactions such as hydrogen bonds and metal coordination can be exploited to generate a wide range of fascinating solid-state structures. 14,15 In this contribution, we wish to present the changes in the crystal packing when a conformational modification in the structure is incorporated. The crystal structures of 1,4-bis(3,5- di-tert-butyl-2-hydroxybenzamido)butane (1) and 1,3-bis(3,5- di-tert-butyl-2-hydroxybenzamido)- 2,2- dimethylpropane (2) (Scheme 1) show the occurrence of different chain motifs which are mainly monitored by a different hydrogen bonding pattern. Their structures are analyzed in terms of hydrogen bond and weaker supramolecular interactions and they are compared with the crystal structures of homologous series which were already reported. 16 Experimental Ligands The full synthesis of 1,4-bis(3,5-di-tert-butyl-2-hydroxybenz- amido)butane (1) and 1,3- bis(3,5-di-tert-butyl-2-hydroxybenz- amido)-2,2-dimethylpropane (2) are already reported. 17 X-Ray quality crystals of 1 and 2 were obtained by slow evaporation of their ethanolic solution. a Dpto. Quı ´mica Orga ´nica. Facultad de Ciencias Quı ´micas. Universidad de Concepcio ´n. Casilla 160-C, Concepcio ´n, Chile. E-mail: [email protected] b Laboratorio de Rayos X y Materiales Moleculares. Dpto. de Fı ´sica Fundamental II. Facultad de Fı ´sica. Universidad de La Laguna. Avda. Astrofı ´sico Francisco Sa ´nchez s/n, 3820-La Laguna, Tenerife, Spain c BM16 – LLS European Synchrotron Radiation Facility 6 Rue Jules Horowitz - BP 220 38043 Grenoble CEDEX 9, France d ICMol/Departament de Quı ´mica Inorga `nica de la Universitat de Vale `ncia, Polı ´gono La Coma s/n, 46980 Paterna (Vale `ncia), Spain Scheme 1 Structures of 1,4-bis(3,5-di-tert-butyl-2-hydroxybenz- amido)butane (1) and 1,3-bis(3,5-di-tert-butyl-2-hydroxybenzamido)- 2,2-dimethylpropane (2). PAPER www.rsc.org/crystengcomm | CrystEngComm 746 | CrystEngComm, 2007, 9, 746–752 This journal is ß The Royal Society of Chemistry 2007
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Structural changes in the crystal packing of highly hindered symmetrical vicinal bis-amides

Structural changes in the crystal packing of highly hindered symmetricalvicinal bis-amides
Claudio A. Jimenez,*a Julio B. Belmar,a Fernando S. Delgado,bc Miguel Julved and Catalina Ruiz-Perezb
Received 4th December 2006, Accepted 20th April 2007
First published as an Advance Article on the web 17th May 2007
DOI: 10.1039/b617605d
The crystal structures of 1,4-bis(3,5-di-tert-butyl-2-hydroxybenzamido)butane (1) and 1,3-bis(3,5-
di-tert-butyl-2-hydroxybenzamido)-2,2-dimethylpropane (2) have being characterized by single
crystal X-ray diffraction. Their crystal packing is discussed in terms of the different interactions
they exhibit. A brief discussion, based on hydrogen bonds, on the structural features of bis-(3,5-
di-tert-butyl-2-hydroxybenzamide) compounds is carried out.
Introduction
The complementary recognition between functional groups in
molecules is the first step in crystallization. Because molecular
recognition is complementary in nature,1 a functional group
approach to crystal engineering is, at least currently, not
possible,2 and prediction of crystal structures is correspond-
ingly difficult.3 Current systematic synthetic strategies in
purely organic species attempt to tackle this issue by
identifying systems where the crystal construction may be
taken up in a modular fashion and where interaction inter-
ference is minimal. Thus the effect of any particular func-
tionality can be identified.4 Indeed, this is one of the well
accepted goals of crystal engineering.
Functional groups exert both steric and electronic effects on
the structure and reactivity, but even in solution, where the
functional group approach is broadly applicable, separating
these two effects is quite difficult.5 This issue is more or less
intractable in the solid state because the effects of the
functional groups are in themselves implicitly connected to
the nature and position of the other functional groups in the
molecule. In the context of crystal engineering, steric and
electronic effects are synonymous with geometrical and
chemical effects in the crystal packing.6 While the origin of
all intermolecular interactions is electrostatic7 and there is no
rigorous basis for such a distinction, it is still convenient to
distinguish these components on the basis of the distance
dependence of the corresponding interactions.8
In this framework, amide functions are the robust synthons
of wide consensus and they have become the choice of a great
number of research groups.9 This functional group has
numerous applications. In particular, bulky amides and their
lithium salts are widely used as deprotonating reagents in
organic synthesis due to their relatively low nucleophilicity
and strong Brønsted basicity,10,11 as well as reagents for
asymmetric synthesis of compounds containing C–C bonds.12
Furthermore, amides have been found to be promising
precursors to synthesize a variety of complexes with main
group elements, transition metal ions, and lanthanide cations,
many of them well-characterized.13 Networking of molecules
through interactions such as hydrogen bonds and metal
coordination can be exploited to generate a wide range of
fascinating solid-state structures.14,15
In this contribution, we wish to present the changes in the
crystal packing when a conformational modification in the
structure is incorporated. The crystal structures of 1,4-bis(3,5-
di-tert-butyl-2-hydroxybenzamido)butane (1) and 1,3-bis(3,5-
di-tert-butyl-2-hydroxybenzamido)- 2,2- dimethylpropane (2)
(Scheme 1) show the occurrence of different chain motifs
which are mainly monitored by a different hydrogen bonding
pattern. Their structures are analyzed in terms of hydrogen
bond and weaker supramolecular interactions and they are
compared with the crystal structures of homologous series
which were already reported.16
Experimental
Ligands
The full synthesis of 1,4-bis(3,5-di-tert-butyl-2-hydroxybenz-
amido)butane (1) and 1,3- bis(3,5-di-tert-butyl-2-hydroxybenz-
amido)-2,2-dimethylpropane (2) are already reported.17 X-Ray
quality crystals of 1 and 2 were obtained by slow evaporation
of their ethanolic solution.
aDpto. Quımica Organica. Facultad de Ciencias Quımicas. Universidadde Concepcion. Casilla 160-C, Concepcion, Chile.E-mail: [email protected] de Rayos X y Materiales Moleculares. Dpto. de FısicaFundamental II. Facultad de Fısica. Universidad de La Laguna. Avda.Astrofısico Francisco Sanchez s/n, 3820-La Laguna, Tenerife, SpaincBM16 – LLS European Synchrotron Radiation Facility 6 Rue JulesHorowitz - BP 220 38043 Grenoble CEDEX 9, FrancedICMol/Departament de Quımica Inorganica de la Universitat deValencia, Polıgono La Coma s/n, 46980 Paterna (Valencia), Spain
Scheme 1 Structures of 1,4-bis(3,5-di-tert-butyl-2-hydroxybenz-
amido)butane (1) and 1,3-bis(3,5-di-tert-butyl-2-hydroxybenzamido)-
2,2-dimethylpropane (2).
PAPER www.rsc.org/crystengcomm | CrystEngComm
746 | CrystEngComm, 2007, 9, 746–752 This journal is � The Royal Society of Chemistry 2007

Crystal data collection and structure determination
Single crystals of 1 and 2 were mounted on a Bruker-Nonius
KappaCCD diffractometer. Orientation matrix and lattice
parameters were obtained by least-squares refinement of the
reflections obtained by a h–x scan (Dirax/lsq method).
Diffraction data for all compounds were collected at 293(2) K
using graphite-monochromated Mo Ka radiation (l =
0.71073 A). Data collection and data reduction were done
with the COLLECT18 and EVALCCD19 programs. Empirical
absorption corrections were carried out using SADABS20 for
all compounds. The indexes of data collection were 212 ¡ h
¡ 12, 214 ¡ k ¡ 14 and 220 ¡ l ¡ 20 for 1 and 226 ¡ h ¡
16, 226 ¡ k ¡ 29 and 211 ¡ l ¡ 14 for 2. Of the 7124 (1)
and 5432 (2) measured independent reflections in the h range
4.14–27.50 (1) and 6.48–24.50 (2), 3915 (1) and 3845 (2) have
I ¢ 2s(I). All calculations for data reduction, structure
solution, and refinement were done by standard procedures
(WINGX).21 The structures were solved by direct methods and
refined with full-matrix least-squares technique on F2 using the
SHELXS-97 and SHELXL-97 programs.22 All hydrogen
atoms bound to carbon and those of the phenol groups were
placed in calculated positions and refined isotropically. The
hydrogen atoms for all compounds were refined with isotropic
temperature factors. The final Fourier-difference maps
showed maximum and minimum height peaks of 0.297 and
20.225 e A23 (1) and 0.134 and 20.116 e A23 (2). A summary
of the crystallographic data and structure refinement is
given in Table 1. The final geometrical calculations and the
graphical manipulations were carried out with PARST9723
and DIAMOND24 and MERCURY25 programs, respectively.
Supramolecular interactions are listed in Tables 2 (1) and 4 (2)
whereas the main bond distances and angles are given in
Tables 3 (1) and 5 (2).
CCDC reference numbers 629529 (1) and 629530 (2. For
crystallographic data in CIF or other electronic format see
DOI: 10.1039/b617605d
Results and discussion
The structure of 1 is built up from neutral bis-amide units
which are held together by means of hydrogen bonds, C–H…p
and CH3…CH3 hydrophobic interactions affording a 3-D
structure. Intramolecular hydrogen bonds involving the phenol
and amide oxygens help to stabilize each branch of the
molecule (Fig. 1a and Table 4) and they make the amide and
phenol groups roughly coplanar. The values of the dihedral
angle between the mean planes of the amide group and the
phenyl ring within each half of the molecule are 12.7(2) [C(1)–
C(6)/O(3)–C(7)–N(1)] and 7.49(15)u [C(13)–C(18)/O(4)–C(12)–
N(2)]. The separation between the amide nitrogen atoms
N(1)…N(2) is 5.127(3) A, a value which is longer than that
reported for other related compounds11,16,26 because of the
longer size of the organic substituent in 1 compared to that
reported previously. Within the bis-amide unit, the values of
the centroid…centroid separation and that of the dihedral
angle between the planes defined by phenyl rings from each
half of the molecule are 9.423(5) A and 60.79(7)u, respectively.
The torsion angle of the alkyl bridge is 265.5(8)u [N(1)–C(8)–
C(12)–N(2)]. It deserves to be noted that the bis-amide group
adopts the anti conformation, as observed in other related
compounds.16,26 Each bis-amide unit is connected to other
two centrosymmetrically related molecules through hydrogen
Table 1 Crystallographic data for compounds 1 and 2
Compound 1 2
Formula C34H52N2O4 C35H54N2O4
M 552.78 566.80Crystal system Triclinic OrthorhombicSpace group P1 Iba2a/A 10.0092(6) 23.029(4)b/A 11.2895(7) 25.201(5)c/A 15.5109(8) 12.087(3)a/u 98.90(4) —b/u 102.04(4) —c/u 104.00(4) —V/A3 1623.85(16) 7015(3)Z 2 8rcalc(Mg m23) 1.131 1.073m(Mo Ka)/mm21 0.71073 0.71073l(Mo Ka)/mm21 0.073 0.069R1a, I . 2s(I) (all) 0.065 (0.132) 0.046(0.087)wR2b, I . 2s(I) (all) 0.147 (0.175) 0.085(0.098)Measured reflections 15426 15705Independent reflections (Rint) 7124 (0.029) 5432 (0.042)a R1 = S | |Fo| 2 |Fc| |/S |Fo| b wR2 = {S [w(Fo
2 2 Fc2)2]/S
[w(Fo2)2]}1/2; w = 1/[s2(Fo)2 + (aP)2 + bP], where P = [2Fc
2 +Max(Fo
2,0)]/3.
Fig. 1 Supramolecular association in 1 mediated by N–H…O
interactions (a). It leads to the formation of chains along the a-axis
(b). The atom numbering is also shown.
This journal is � The Royal Society of Chemistry 2007 CrystEngComm, 2007, 9, 746–752 | 747

bonds, involving the amide groups: 2.990(3) A for N(1)…O(4)i
[(i) 2x 2 1, 2y + 1, 2z + 1] and 3.003(3) A for N(2)…O(3)ii
[(ii) 2x, 2y + 1, 2z + 1.] leading to regular belt-like chains
that run along the a-axis (Fig. 1b). These chains are held
together by means of C–H…p interactions [H…centroid
separations ranging from 2.94(5) to 3.54(5) A] along the
b- and c-axes (see Fig. 2), affording a 3-D network. In
addition, weak CH3…CH3 hydrophobic interactions contri-
bute to stabilize the whole structure, the shortest C…C
distances varying in the range 3.678(5)–3.895(3) A (Table 2).
The crystal packing in 1 (Fig. 3) is very similar to that observed
in the related bis-amide compounds with an n-alkyl bridge
connecting the two halves:11 hydrogen bonds in the a direction
and CH…p type interactions in the bc-plane, build up the 3-D
structure (see Fig. S1).
As in 1, the structure of 2 consists of neutral bis-amide units
which are held together by means of hydrogen bonds, C–H…p
and CH3…CH3 interactions leading to a 3-D structure.
Similarly to 1, intramolecular hydrogen bonds involving the
phenol and amide oxygens help to stabilize each branch of the
molecule (Fig. 4 and Table 5). However in 2, contrary to 1,
Table 2 Interaction data for compounds (1)
H Bond Typea D…A/A H…Aa/A D-H…A/u
O(1)–H(1)O…O(3) 2.520(3) 1.63(3) 157(3)O(2)–H(2)O…O(4) 2.519(3) 1.57(4) 158(3)N(1)–H(1)N…O(4)i 2.990(3) 2.21(2) 149(2)N(2)–H(2)N…O(3)ii 3.003(3) 2.20(3) 152(2)
CH…pb Catom C…Cg/A H…Cg/A C– H…Cg/u
C(26)– H(26)A…Cg1iii C(26)iii 3.634(4) 2.99(5) 150 (2)C(32)–H(32A)…Cg2iv C(18)iv 3.928(5) 3.04(5) 149 (3)C(25)–H(25A)…Cg2iii C(14)iii 3.916(4) 3.54(5) 114 (2)
CH3…CH3 C…C/A
(Hydrophobic C(22)…C(32)v 3.863(5)Interactions) C(28)…C(20)vi 3.678(5)
C(30)…C(25)vii 3.895(3)C(34)…C(20)viii 3.830(7)
a Symmetry operator: (i) 2x 2 1, 2y + 1, 2z + 1; (ii) 2x, 2y + 1,2z + 1; (iii) 2x, 2y + 2, 2z + 1; (iv) 2x, 2y + 1, 2z + 2; (v) x 21, y, z 2 1; (vi) 2x 2 1, 2y + 2, 2z + 1; (vii) x, y, z + 1; (viii) 2x,2y + 2, 2z + 1. b Centroids: Cg1:C(1)–C(6); Cg2:C(13)–C(18).
Fig. 2 Chains of 1 projected on the bc -plane showing its connection
to four adjacent ones through C–H…p type interactions (broken lines).
Table 3 Selected bond lengths (A) and angles (u) for 1
C(1)–C(2) 1.399(3) C(2)–C(1)–C(7) 119.1(2)C(1)–C(7) 1.483(3) O(1)–C(2)–C(1) 120.8(2)C(2)–O(1) 1.355(2) O(3)–C(7)–N(1) 119.9(2)C(7)–O(3) 1.251(2) O(3)–C(7)–C(1) 121.0(2)C(7)–N(1) 1.332(3) N(1)–C(7)–C(1) 119.1(2)C(8)–N(1) 1.462(3) N(1)–C(8)–C(9) 113.9(2)C(8)–C(9) 1.522(4) C(10)–C(9)–C(8) 114.0(2)C(9)–C(10) 1.497(4) C(9)–C(10)–C(11) 113.4(2)C(10)–C(11) 1.517(4) N(2)–C(11)–C(10) 114.4(2)C(11)–N(2) 1.457(3) O(4)–C(12)–N(2) 119.5(2)C(12)–O(4) 1.252(2) O(4)–C(12)–C(13) 121.1(2)C(12)–N(2) 1.332(3) N(2)–C(12)–C(13) 119.4(2)C(12)–C(13) 1.479(3) C(14)–C(13)–C(12) 119.4 (2)C(13)–C(14) 1.405(3) O(2)–C(14)–C(13) 120.6(2)C(14)–O(2) 1.355(2)
Fig. 3 View of the packing of 1 along the a-axis.
Fig. 4 The molecular structure of 2 with labelling scheme of relevant
atoms showing intra and intermolecular H-bonds
748 | CrystEngComm, 2007, 9, 746–752 This journal is � The Royal Society of Chemistry 2007
there exist intramolecular bifurcated hydrogen bonds invol-
ving the nitrogen and oxygen atoms from the amide groups
which favor the bending of the molecule (Table 3).
Table 4 Interaction data for compounds (2)
H Bond Typea D…A/A H…Aa/A D–H…A/u
N(1)–H(1)N…O(4) 2.943(3) 2.14(3) 154(3)O(1)–H(1)O…O(3) 2.541(3) 1.51(4) 153(3)O(2)–H(2)O…O(4) 2.613(3) 1.90(3) 147(3)N(2)–H(2)N…O(3)i 2.876(3) 1.97(2) 164(2)
CH…pb Catom C…Cg/A H…Cg/A C–H…Cg/u
C(22)–H(22A)…Cg1ii C(6)ii 3.772(4) 3.40(7) 112(2)C(27)–H(27C)…Cg1iii C(2)iii 3.724(5) 2.96(8) 145(3)C(30)–H(30C)…Cg2iv C(15)iv 3.876(4) 3.35(6) 128(2)
CH3…CH3 C…C/A
(Hydrophobic C(33)…C(21)v 3.811(5)Interactions)a Symmetry operator: (i) 2x + 1/2, 2y + 1/2, z + 1/2; (ii) x, 2y, z 21/2; (iii) x, 2y, z + 1/2; (iv) x, y, z + 1/2; (v) 2x + 1/2, y + 1/2, z.b Centroids: Cg1:C(1)–C(6); Cg2:C(14)–C(19).
Table 5 Selected bond lengths (A) and angles (u) for 2
C(1)–C(2) 1.399(3) C(2)–C(1)–C(7) 118.4(2)C(1)–C(7) 1.478(3) O(1)–C(2)–C(1) 120.5(2)C(2)–O(1) 1.353(3) O(3)–C(7)–N(1) 120.8(2)C(7)–O(3) 1.252(3) O(3)–C(7)–C(1) 120.9(2)C(7)–N(1) 1.327(3) N(1)–C(7)–C(1) 118.2(2)C(8)–N(1) 1.450(3) N(1)–C(8)–C(9) 114.4(2)C(8)–C(9) 1.528(3) C(12)–C(9)–C(8) 109.9(2)C(9)–C(12) 1.527(3) N(2)–C(12)–C(9) 114.5(2)C(12)–N(2) 1.454(3) O(4)–C(13)–N(2) 121.1(2)C(13)–O(4) 1.248(3) O(4)–C(13)–C(14) 121.2(2)C(13)–N(2) 1.330(3) N(2)–C(13)–C(14) 117.7(2)C(13)–C(14) 1.476(3) C(15)–C(14)–C(13) 119.1(2)C(14)–C(15) 1.394(3) O(2)–C(15)–C(14) 120.8(2)C(15)–O(2) 1.355(3)
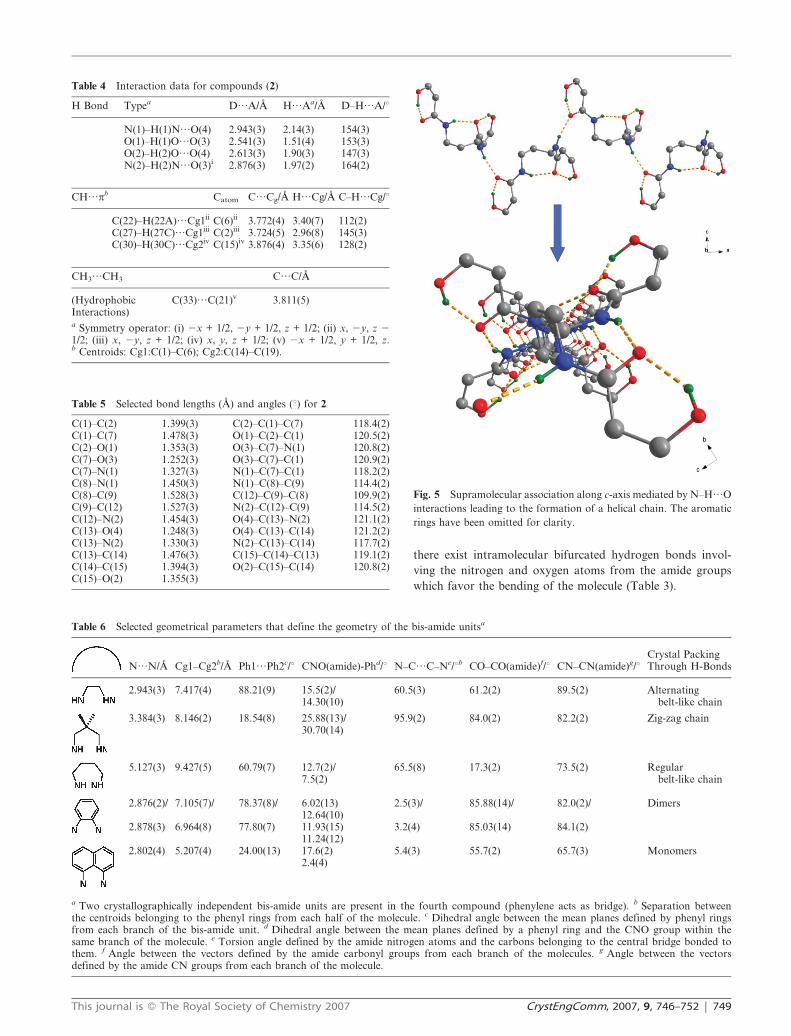
Fig. 5 Supramolecular association along c-axis mediated by N–H…O
interactions leading to the formation of a helical chain. The aromatic
rings have been omitted for clarity.
Table 6 Selected geometrical parameters that define the geometry of the bis-amide unitsa
N…N/A Cg1–Cg2b/A Ph1…Ph2c/u CNO(amide)-Phd/u N–C…C–Ne/ub CO–CO(amide)f/u CN–CN(amide)g/uCrystal PackingThrough H-Bonds
2.943(3) 7.417(4) 88.21(9) 15.5(2)/ 60.5(3) 61.2(2) 89.5(2) Alternatingbelt-like chain14.30(10)
3.384(3) 8.146(2) 18.54(8) 25.88(13)/ 95.9(2) 84.0(2) 82.2(2) Zig-zag chain30.70(14)
5.127(3) 9.427(5) 60.79(7) 12.7(2)/ 65.5(8) 17.3(2) 73.5(2) Regularbelt-like chain7.5(2)
2.876(2)/ 7.105(7)/ 78.37(8)/ 6.02(13) 2.5(3)/ 85.88(14)/ 82.0(2)/ Dimers12.64(10)
2.878(3) 6.964(8) 77.80(7) 11.93(15) 3.2(4) 85.03(14) 84.1(2)11.24(12)
2.802(4) 5.207(4) 24.00(13) 17.6(2) 5.4(3) 55.7(2) 65.7(3) Monomers2.4(4)
a Two crystallographically independent bis-amide units are present in the fourth compound (phenylene acts as bridge). b Separation betweenthe centroids belonging to the phenyl rings from each half of the molecule. c Dihedral angle between the mean planes defined by phenyl ringsfrom each branch of the bis-amide unit. d Dihedral angle between the mean planes defined by a phenyl ring and the CNO group within thesame branch of the molecule. e Torsion angle defined by the amide nitrogen atoms and the carbons belonging to the central bridge bonded tothem. f Angle between the vectors defined by the amide carbonyl groups from each branch of the molecules. g Angle between the vectorsdefined by the amide CN groups from each branch of the molecule.
This journal is � The Royal Society of Chemistry 2007 CrystEngComm, 2007, 9, 746–752 | 749

The separation between nitrogen atoms N(1)…N(2)
[3.383(3) A] is shorter than in 1, as expected, due to the
shorter length of the organic substituent in 2 in comparison to
that of 1. Within the bis-amide unit, the values of the dihedral
angles between the mean planes of the amide group and the
phenyl ring within each half of the molecule are 25.70(13)u[C(1)–C(6)/O(3)–C(7)–N(1)] and 30.70(15)u [C(14)–C(19)/
O(4)–C(13)–N(2)]. The value of the centroid…centroid separa-
tion and that of the dihedral angle between the planes defined
by phenyl rings from each branch of the bis-amide unit are
8.146(2) A and 18.54(8)u, respectively. The torsion angle of the
alkyl bridge is 295.9(2)u [N(1)–C(8)–C(12)–N(2)]. As in 1, the
bis-amide group adopts the anti conformation. Each bis-amide
unit is connected to other ones through hydrogen bonds
[N(2)…O(3)i; (i) x 2 0.5, 2y + 0.5, z + 0.5] affording helical
chains that run along the c-axis (Fig. 5). The period of the
helical chain is 12.087(4) A [C(2)…C(2)(x + 1, y, z)]. In
addition each helix is interconnected with other four ones
through C–H…p interactions [the values of the H…centroid
separations ranging from 2.96(8) to 3.40(7) A] (Fig. 6a). These
interactions run in the same directions that the hydrogen
bonded helix that is along the c-axis leading to a zip-like struc-
ture (see Fig. 6b). Finally methyl…methyl hydrophobic inter-
actions connect adjacent helixes and contribute to stabilize a
3-D packing, the CH3…CH3 separations being 3.811(5) A.
Let us carry out a little survey of the structural features of
the bis-(3,5-di-tert-butyl-2-hydroxybenzamides) focusing on
hydrogen bonds. Some structural parameters of compounds
1 and 2 together with those of the previously reported related
compounds16 are compared in Table 6. Scheme 2 shows the
different hydrogen patterns exhibited by these compounds.
It deserved to be noted that ligands are able to pack,
affording hydrogen bonding chains only when the central
organic bridge is non-aromatic. The aromatic groups make the
molecule more rigid (see the values of the N–C…C–N torsion
angles in Table 6) and introduce steric effects that preclude the
formation of chains through hydrogen bonds.
Fig. 6 Two adjacent chains of 2 running along the c-axis showing (a)
the C–H…p interactions and (b) space-filling.
Scheme 2 Packing through hydrogen bonds of the bis-amide units for
each compound belonging to bis-(3,5-di-tert-butyl-2-hydroxybenz-
amides). Hydrogen bonds are drawn as blue broken lines. For the
sake of clarity, tert-butyl groups have been omitted.
750 | CrystEngComm, 2007, 9, 746–752 This journal is � The Royal Society of Chemistry 2007

Concerning the intramolecular weak interactions, in all
complexes, the phenol and amide oxygens are involved in
hydrogen bonds within each moiety of the molecule, making
the phenyl and amide groups roughly coplanar [the dihedral
angle between the two groups ranging from 2.4(4)–30.70(14)u].On the other hand, when the central organic bridge is short
enough, amide nitrogens are involved in intramolecular
interactions connecting the two moieties of the bis-amide
molecules. The size of the organic bridge is directly related
with the N…N distance and the separation between the
centroids belonging to the aromatic rings of each branch of the
molecule. It deserves to be noted that the shortest N…N
separation is observed for the naphthylidene group although
there are two shorter bridges (ethylene and phenylene groups).
The explanation for this feature is the formation of a different
intramolecular hydrogen bond, involving the two amide
nitrogens in this compound. In the case of 2, the long N…N
separation avoids the formation of intramolecular interactions
involving the amide nitrogens, thus being the key factor in the
formation of the hydrogen–bonded regular belt-like chains in
this compound, because of the freedom of the two amide
groups to form intermolecular interactions. Also in 2, the bis-
amide groups seem to arrange in a quasi-parallel fashion, the
angle between the carbonyl groups belonging to the two
different moieties of the molecules being 17.3(2)u. It deserves to
be pointed out that this angle ranges from 55.7(2) to 85.88(14)ufor the other compounds where regular belt-like pattern have
not been observed and it has a value for 7.55(11) and 30.6(2)ufor two related compounds18 [N,N9-bis(benzoyl)-(1R,2R)-
diaminocyclohexane and N,N9-bis(isonicotinoyl)-(1R,2R)-dia-
minocyclohexane, respectively].
Regarding intermolecular hydrogen bonds, amide nitrogen
and oxygen atoms are always involved. Interestingly, belt-like
hydrogen bonds involving amide nitrogens and oxygens are
the main intermolecular pattern that appears in these families
of compounds. The steric effects, due to the nature of the
central organic substituent, are responsible for the non-
formation of this hydrogen pattern in the naphthylidene and
phenylene compounds. As commented above, a 1-D hydrogen
pattern appears when the central organic bridge is non-
aromatic. When the ethylene group acts as an organic bridge,
the phenol oxygen participates in the intermolecular interac-
tions leading to alternating belt-like chains. For the 2,29-
dimethylpropylene compound, zigzag chains are observed.
Finally, for the tetramethylene compound regular belt-like
chains are observed as for other related compounds where no
intramolecular interactions connecting the amide branch’s of
the molecule appear.26
In conclusion, the nature and the size of the central moiety
which bridges the amide branches became a key factor for the
formation of intra- and intermolecular hydrogen bonds and
consequently is the main factor responsible for the crystal
packing of the molecule.
Summary
In this paper, we tried to present the changes in the crystal
packing issued from a conformational modification in the
structure. In some cases, one may predict the connectivity
pattern and thus the geometry, topology, and directionality of
the molecular networks. Our experience showed that, as one
may expect, with our present level of knowledge and under-
standing, we are unable to predict the conformation
adopted by flexible ligands. However, as stated by Dunitz,1b
by considering a crystal as a ‘‘supermolecule par excellence’’,
using concepts and principles developed in crystal engineer-
ing,3 one may predict some of the connectivity patterns and
thus the formation of molecular networks, through the control
of intermolecular interactions. After many years of intense
research efforts by several groups, nowadays one may design
molecular networks in the crystalline phase with an acceptable
degree of precision. So far the description of molecular
networks is based on geometrical features. A further important
issue is obviously to use this knowledge for the design of
molecular networks with predicted properties. In other terms,
the molecular tectonics approach must extend structural
networks to functional networks. This aspect is of outstanding
importance in terms of applications and it remains a challenge.
Acknowledgements
This work was supported by University of Concepcion (PDI
205.023.041-1.0 and 205.023.042-1.0). CONICYT provided a
doctoral scholarship for Mr. C.A.J. Financial support from
the Spanish Ministerio de Educacion y Ciencia through
projects MAT2004-03112, CTQ2004-03633 and ‘‘Factorıa de
Cristalizacion’’ (Consolider-Ingenio2010, CSD2006-0015) and
the Generalitat Valenciana (Grupos 03/197) is gratefully
acknowledged. F.S.D acknowledges a postdoctoral fellowship
(‘‘Grandes Instalaciones’’) from the Spanish Ministerio de
Educacion y Ciencia.
References
1 (a) E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985; (b) L. Paulingand M. Delbruck, Science, 1940, 92, 77; (c) J. D. Dunitz, PureAppl. Chem., 1991, 63, 177; (d) J. D. Dunitz, Perspectives inSupramolecular Chemistry. The Crystal as a Supramolecular Entity,ed. Desiraju G. R., Wiley, Chichester, 1995, vol. 2, p. 1.
2 (a) G. R. Desiraju, Stimulating Concepts in Chemistry, ed.F. Vogtle, J. F. Stoddart and M. Shibasaki, Wiley-VCH,Weinheim, 2000, p. 293; (b) G. R. Desiraju and A. Nangia, Top.Curr. Chem., 1998, 198, 57.
3 (a) G. R. Desiraju, Nat. Mater., 2002, 1, 77; (b) J. D. Dunitz, Chem.Commun., 2003, 545; (c) J. A. R. P. Sarma and G. R. Desiraju,Cryst. Growth Des., 2002, 2, 93; (d) T. Beyer, T. Lewis andS. L. Price, CrystEngComm, 2001, 44, 1; (e) W. D. S. Motherwell,H. L. Ammon, J. D. Dunitz, A. Dzyabchenko, P. Erk,A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen, J. P. M.Lommerse, W. T. M. Mooij, S. L. Price, H. Scheraga, B. Schweizer,M. U. Schmidt, B. P. van Eijck, P. Verwer and D. E. Williams,Acta Crystallogr., Sect. B, 2002, 58, 647; (f) J. P. M. Lommerse,W. D. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Gavezzotti,D. W. M. Hofmann, F. J. J. Leusen, W. T. M. Mooij, S. L. Price,B. Schweizer, M. U. Schmidt, B. P. van Eijck, P. Verwer andD. E. Williams, Acta Crystallogr., Sect. B, 2000, 56, 697.
4 (a) S. Hanessian, R. Saladino, R. Margarita and M. Simard,Chem.–Eur. J., 1999, 5, 2169; (b) C. B. Aakeroy, A. M. Beatty andB. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240; (c)C. M. Reddy, A. Nangia, C.-K. Lam and T. C. W. Mak,CrystEngComm, 2002, 4, 323; (d) C. B. Aakeroy, A. M. Beatty andB. A. Helfrich, J. Am. Chem. Soc., 2002, 124, 14425.
5 L. Williams and M. N. Paddon-Row, J. Chem. Soc., Chem.Commun., 1994, 353.
This journal is � The Royal Society of Chemistry 2007 CrystEngComm, 2007, 9, 746–752 | 751

6 (a) G. R. Desiraju and A. Nangia, Acta Crystallogr., Sect. A, 1998,54, 934; (b) K. E. Schwiebert, D. N. Chin, J. C. MacDonald andG. M. Whitesides, J. Am. Chem. Soc., 1996, 118, 4018; (c)R. E. Melendez and A. D. Hamilton, Design of Organic Solids, ed.Weber E., Springer, Berlin, 1998, p. 97; (d) K. T. Holman,A. M. Pivovar, J. A. Swift and M. D. Ward, Acc. Chem. Res., 2001,34, 107; (e) M. J. Zaworotko, Chem. Commun., 2001, 1; (f)S. V. Kolotuchin, E. E. Fenlon, S. R. Wilson, C. J. Loweth andS. C. Zimmerman, Angew. Chem., Int. Ed. Engl., 1995, 34, 2654; (g)D. Braga, G. R. Desiraju, J. S. Miller, A. G. Orpen and S. L. Price,CrystEngComm, 2002, 4, 500; (h) J. Martz, E. Graf, A. D. Cian andM. W. Hosseini, Crystal Design.Structure and Function,Perspectives in Supramolecular Chemistry, ed. G. R. Desiraju,Wiley, Chichester, 2003, vol. 7, pp. 177–209; (i) L. R. MacGillivrayand J. L. Atwood, Angew. Chem., Int. Ed., 1999, 38, 1018; (j)L. Brammer, Crystal Design. Structure and Function, Perspectivesin Supramolecular Chemistry, ed. G. R. Desiraju, Wiley,Chichester, 2003, vol. 7, p. 1.
7 (a) K. Morokuma, J. Phys. Chem., 1971, 55, 1236–1244; (b)K. Morokuma, Acc. Chem. Res., 1977, 10, 294–300.
8 G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1989, 179–180.9 (a) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015;
(b) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173; (c) R. P. Cheng,S. H. Gellman and W. F. Degrado, Chem. Rev., 2001, 101, 3219.
10 (a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1988, 27, 1624; (b)P. Beak, A. Basu, D. J. Gallagher, Y. S. Park andS. Thayumanavan, Acc. Chem. Res., 1996, 29, 552.
11 (a) D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 36,2282; (b) V. Snieckus, Chem. Rev., 1990, 90, 879; (c) P. J. Cox andN. S. Simpkins, Tetrahedron: Asymmetry, 1991, 2, 1.
12 (a) P. J. O’Brien, J. Chem. Soc., Perkin Trans. 1, 1998, 1439; (b)P. J. O’Brien, J. Chem. Soc., Perkin Trans. 1, 2001, 95.
13 (a) C. Eaborn, P. B. Hitchcock, J. D. Smith and S. E. Sozerli,Organometallics, 1997, 16, 5653; (b) V. C. Gibson, P. J. Maddox,C. Newton, C. Redshaw, G. A. Solan, A. J. P. White and
D. J. Williams, Chem. Commun., 1998, 1651; (c) B. Qian, D. L.Ward and M. R. Smith, III, Organometallics, 1998, 17, 3070; (d)P. L. Holland and W. B. Tolman, J. Am. Chem. Soc., 1999, 121,7270; (e) C. C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer,H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274.
14 (a) J. L. Atwood, J. E. D. Davies, J.-M. Lehn, D. D. MacNicol andF. Vogtle, Comprehensive Supramolecular Chemistry, Pergamon,Oxford, UK, 1996; (b) H.-J. Schneider and A. K. Yatsimirsky,Principles and Methods in Supramolecular Chemistry, Wiley,Chichester, U.K., 2000.
15 (a) C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev.,1997, 97, 2005; (b) B. Moulton and M. J. Zaworotko, Chem. Rev.,2001, 101, 1629.
16 C. A. Jimenez, J. B. Belmar, J. Alderete, F. S. Delgado, M. Lopez-Rodrıguez, O. Pena, M. Julve and C. Ruiz-Perez, Dalton Trans.,2007, DOI: 10.1039/b617604f.
17 C. Jimenez and J. Belmar, Tetrahedron, 2005, 61, 3933.18 R. W. W. Hooft, COLLECT. Nonius BV, Delft, 1999.19 A. J. M. Duisenberg, L. M. J. Kroon-Batenburg and A. M. M.
Schreurs, J. Appl. Crystallogr., 2003, 36, 220 (EVALCCD).20 SADABS, version 2.03. Bruker AXS Inc., Madison, WI, 2000.21 L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 (WINGX).22 G. M. Sheldrick, SHELX97, Programs for Crystal Structure
Analysis (Release 97-2), Institut fur Anorganische Chemie derUniversitat:Gottingen, Germany, 1998.
23 M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659.24 DIAMOND 2.1d, Crystal Impact GbR, CRYSTAL IMPACT, K,
Brandenburg & H. Putz GbR, Bonn, Germany, 2000.25 Mercury: visualization and analysis of crystal structures
C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock,G. P. Shields, R. Taylor, M. Towler and J. van de Streek,J. Appl. Crystallogr., 2006, 453, 39.
26 S. P. Anthony, K. Basavaiah and T. P. Radhakrishnan, Cryst.Growth Des., 2005, 5, 1663.
752 | CrystEngComm, 2007, 9, 746–752 This journal is � The Royal Society of Chemistry 2007




















