
static second hyperpolarizability of lamda shapped alkaline earth metal complexes
12
Static second hyperpolarizability of ¤ shaped alkaline earth metal complexes Kaushik Hatua and Prasanta K. Nandi * Department of Chemistry Indian Institute of Engineering Science and Technology Shibpur, Howrah 711 103, India * nandi _ [email protected] Received 21 April 2014 Accepted 11 June 2014 Published A number of shaped complexes of alkaline earth metals Be, Mg and Ca with varying terminal groups have been considered for the theoretical study of their second hyperpolarizability. The chosen complexes are found to be su±ciently stable and for a chosen ligand the stability decreases in the order: Be-complex > Ca-complex > Mg-complex. The calculated results of second hyperpolarizability obtained at di®erent DFT functionals for the 6-311þþG(d,p) basis set are found to be fairly consistent. The shaped ligands upon complex formation with metals lead to strong enhancement of second hyperpolarizability. The highest magnitude of cubic polarizability has been predicted for the metal complex having > C(C 2 H 5 ) 2 group. For a chosen ligand, the magnitude of second hyperpolarizability increases in the order Be-complex < Mg-complex < Ca-complex which is the order of increasing size and electropositive character of the metal. The variation of second hyperpolarizability among the investigated metal complexes has been explained in terms of the transition energy and transition moment associated with the most intense electronic transition. Keywords: shaped complexes; ground state structure and stability; second hyperpolariz- ability; two state model. 1. Introduction Increasing demand for electro-optic materials in photonic application, optical data transmission, data processing leads to tremendous growth of research interest in the ¯eld of nonlinear optics. 1–4 There have been several attempts to optimize the NLO response of materials ranging from the isolated gas phase molecules to the bulk phase. Most of the theoretical investigations have been carried out by considering isolated molecules for the purpose of designing new kind of NLO phores having de¯nite structure-property correlation. In the very early days, investigation on nonlinear optical materials was mostly con¯ned to various kinds of donor–acceptor *Corresponding author. June 26, 2014 5:01:16pm WSPC/178-JTCC 1450039 ISSN: 0219-6336 1stReading 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 Journal of Theoretical and Computational Chemistry Vol. 13, No. 4 (2014) 1450039 (12 pages) # . c World Scienti¯c Publishing Company DOI: 10.1142/S0219633614500394 1450039-1
Transcript of static second hyperpolarizability of lamda shapped alkaline earth metal complexes

Static second hyperpolarizability of ¤ shaped
alkaline earth metal complexes
Kaushik Hatua and Prasanta K. Nandi*
Department of ChemistryIndian Institute of Engineering Science and Technology
Shibpur, Howrah 711 103, India*[email protected]
Received 21 April 2014
Accepted 11 June 2014Published
A number of � shaped complexes of alkaline earth metals Be, Mg and Ca with varying terminal
groups have been considered for the theoretical study of their second hyperpolarizability.
The chosen complexes are found to be su±ciently stable and for a chosen ligand the stabilitydecreases in the order: Be-complex > Ca-complex > Mg-complex. The calculated results of
second hyperpolarizability obtained at di®erent DFT functionals for the 6-311þþG(d,p) basis
set are found to be fairly consistent. The � shaped ligands upon complex formation with metals
lead to strong enhancement of second hyperpolarizability. The highest magnitude of cubicpolarizability has been predicted for the metal complex having >C(C2H5)2 group. For a chosen
ligand, the magnitude of second hyperpolarizability increases in the order Be-complex <
Mg-complex < Ca-complex which is the order of increasing size and electropositive character ofthe metal. The variation of second hyperpolarizability among the investigated metal complexes
has been explained in terms of the transition energy and transition moment associated with the
most intense electronic transition.
Keywords: � shaped complexes; ground state structure and stability; second hyperpolariz-
ability; two state model.
1. Introduction
Increasing demand for electro-optic materials in photonic application, optical data
transmission, data processing leads to tremendous growth of research interest in the
¯eld of nonlinear optics.1–4 There have been several attempts to optimize the NLO
response of materials ranging from the isolated gas phase molecules to the bulk
phase. Most of the theoretical investigations have been carried out by considering
isolated molecules for the purpose of designing new kind of NLO phores having
de¯nite structure-property correlation. In the very early days, investigation on
nonlinear optical materials was mostly con¯ned to various kinds of donor–acceptor
*Corresponding author.
June 26, 2014 5:01:16pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Journal of Theoretical and Computational Chemistry
Vol. 13, No. 4 (2014) 1450039 (12 pages)
#.c World Scienti¯c Publishing CompanyDOI: 10.1142/S0219633614500394
1450039-1
substituted � conjugated systems5–14 for optimizing NLO responses. During the
last few years, with the aid of modern quantum chemical approach, other kind of
novel materials have emerged which have interesting electronic structure and
superior electric response property. Amongst these organometallic complexes
where suitable manipulation of electronic environment by metal atom may result
in larger NLO responses that deserve special attention. Di®use electron sys-
tem,15,16 Li–Calixpyrrole,17 highly deformed halofullernes,18,19 nitrogen edge-
doped of single walled CNT,20 lithiated silicon cage,21 alkali–alkaline earth metal
cluster and alkalides,22 mixed �-conjugated bridge in carbon nanotube-based
X
Z
Y
>CH2 >CMe2
MeN< >CO
>SO2
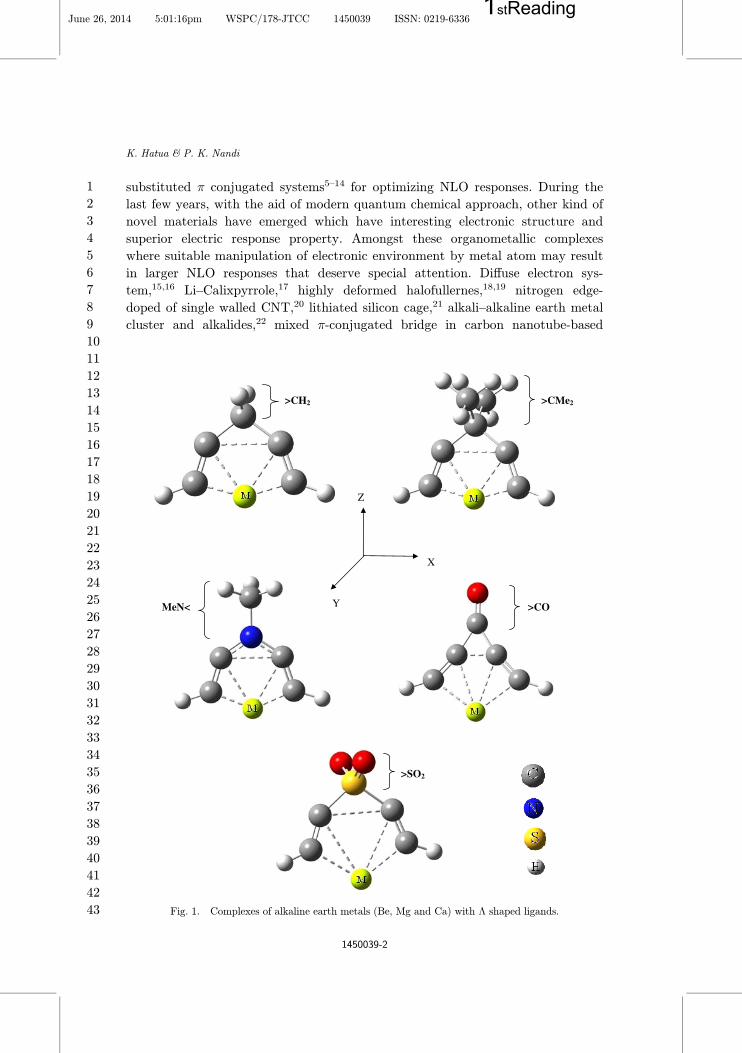
Fig. 1. Complexes of alkaline earth metals (Be, Mg and Ca) with � shaped ligands.
June 26, 2014 5:01:16pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
K. Hatua & P. K. Nandi
1450039-2

system,23 alkali metal atom-aromatic ring,24 alkaline earth-based alkalides,25 �
and W shaped sandwich metallocarborane-containing chromophores26 are impor-
tant examples to be worth mentioned in the context of modern approach for NLO
enhancement. Interestingly most of these studies involve the enhancement and
optimization of the ¯rst hyperpolarizability (�) while the second hyperpolariz-
ability (�) has been less explored. In our previous work, we intended to optimize
the second hyperpolarizability by considering small Be based organometallic27–30
compounds. Molecular design of NLO phores becomes a state of art today. It was
noted from our previous study27 that acceptor hydrocarbon complex of beryllium
metal has more ground state charge polarization compared to that of the ligand
itself. The second hyperpolarizability has been found to be considerably enhanced
when charge transfer from metal to ligand is delocalized rather than accumulated
in the vicinity of the metal. When Be atom has been replaced by the more elec-
tropositive metal like Mg and Ca, the larger extent of ground state polarization
occurs which in turn results larger magnitude of the longitudinal component of
second hyperpolarizability of Mg and Ca complexes. With this idea in mind, we
have designed few �-shaped molecular complexes of alkaline earth metals (Be, Mg
and Ca). The molecular structure consists of di®erent donor/acceptor group X
(>CH2, >C(CH3)2 (>CMe2), >C(C2H5)2 (>CEt2), >NMe2, >CO and >SO2)
which are covalently bonded with two adjacent –C�C–H units (Fig. 1). The larger
component of second hyperpolarizability is associated with the larger ground state
polarization which leads to strong electronic coupling between the ground and the
low lying excited state. We have rationalized the variation of NLO property by
the calculated NPA charges and the excited state electronic parameters obtained
by using the time dependent density functional theory (TDDFT) formalism.
2. Computational Methods
Geometry of the chosen � shaped ligands and complexes have been fully optimized
using the B3LYP31 functional. The basis set employed consists of triple zeta quality
(GTO) with two sets of polarization functions augmented by di®use functions on
heavy atom as well as over hydrogen atom i.e. 6-311þþG(d,p). Vibrational analysis
under harmonic approximation has been carried out at the same level of theory
which con¯rmed the optimized geometry as a stationary point in the respective
potential energy hypersurface. The atomic charges have been calculated by using
the natural population analysis (NPA) scheme at the B3LYP/6-311þþG(d,p)
level. Interaction energy (�E) of the complexes has been calculated by subtracting
the electronic energy of the ligand and metal from that of the complex ð�E ¼Ecomplex � Eligand � EmetalÞ. The basis set superposition error (BSSE) has been
eliminated by employing the counterpoise method. Also the zero point energy (ZPE)
correction has been incorporated in the calculation of interaction energy.
During the interaction of matter with the static radiation ¯eld (F0), an induce
dipole moment is generated which can be expanded in Taylor series. The
June 26, 2014 5:01:18pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Static second hyperpolarizability of � shaped alkaline earth metal complexes
1450039-3

hyperpolarizabilities have been calculated in the static limit F0.
� ¼ �0 þ �F0 þ1
2!�F 2
0 þ 1
3!�F 3
0 � � � ð1Þ
�0 is the permanent dipole moment and the coe±cients �, � and � are called
polarizability, ¯rst hyperpolarizability and second hyperpolarizability, respectively.
The average second hyperpolarizability has been calculated by using the following
expression.
�av ¼1
5½�xxxx þ �yyyy þ �zzzz þ 2ð�xxyy þ �yyzz þ �xxzzÞ�: ð2Þ
The Cartesian components of � have been calculated by the numerical di®erentiation
of the analytically evaluated ¯rst-hyperpolarizability obtained at di®erent DFT
functionals: B3LYP, BHHLYP32 and long range corrected CAM-B3LYP.33 Recently
developed double hybrid functional B2PLYP34 which has MP2 like electron corre-
lation has also been used for the sake of comparison with the conventional DFT
functionals.
In our earlier investigations,27–29 it was noted that the 6-311þþG(d,p) basis set
can reproduce the result obtained with the aug-cc-pVXZ (X¼D,T) basis set with
reasonable computational cost. Furthermore, the double augmentation by another
set of di®use function i.e. d-aug-cc-pVDZ does not enhance the longitudinal com-
ponent of second hyperpolarizability appreciably.30 Thus the basis set dependence of
second hyperpolarizability is expected to be insigni¯cant and has not been considered
in the present study. All calculations have been carried out by using the G09
quantum chemistry program package.35
3. Results and Discussion
3.1. Ground state equilibrium structure
Important equilibrium structural parameters, natural atomic charges and the metal
ligand interaction energy have been presented in Table 1. An inspection of Table 1
reveals that C�C bond distance in ligands is nearly constant (� 1.20�A) but in
Table 1. B3LYP/6-311þþG(d,p) calculated equilibrium C�C triple bond length (rCC , �A) in the ligand and in the
complex, the nearest metal–carbon bond distance (rC-M , �A), natural atomic charges (q, a.u.) and the metal ligand
interaction energy (�E, Kcal/mol).
rCC rC-M q �E
�X Ligand Be Mg Ca Be Mg Ca Be Mg Ca Be Mg Ca
>CH2 1.201 1.298 1.255 1.264 1.647 2.254 2.462 0.842 0.671 0.910 �84.194 �36.114 �43.822
>CMe2 1.202 1.298 1.256 1.267 1.646 2.236 2.437 0.832 0.695 0.966 �123.050 �73.734 �82.220
>CEt2 1.202 1.298 1.256 1.266 1.645 2.246 2.446 0.817 0.667 0.927 �158.139 �108.855 �117.002
>NMe 1.203 1.315 1.274 1.284 1.649 2.175 2.406 0.898 0.840 1.073 �103.268 �52.283 �60.927
>CO 1.203 1.328 1.337 1.347 1.682 2.100 2.332 0.889 1.407 1.670 �38.343 �18.558 �48.316
>SO2 1.202 1.291 1.271 1.281 1.663 2.152 2.336 0.848 1.063 1.516 �90.089 �41.390 �63.321
June 26, 2014 5:01:20pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
K. Hatua & P. K. Nandi
1450039-4

complexes, it varies signi¯cantly which may be due to the di®erent extent of charge
transfer (CT) interaction. From Table 2, it can be seen that with the exception of
>SO2, the <CXC angle of ligands decreases signi¯cantly upon complex formation
with metals. The amount of CT from a chosen metal depends on the nature of the
end group of the ligand. Electron transfer from a metal is smaller with electron
donating group. On the other hand, the extent of CT enhances when electron
withdrawing groups (>CO and >SO2) are present. Owing to the greater electro-
positive character Mg and Ca prefer to form ionic complexes. The greatest amount
of charge transfer from metal occurs when the ligand contains the >CO group
which is re°ected in the signi¯cant polarization (elongation) of C�C bond length
the extent of which increases with the amount of CT varying in the order
Be < Mg < Ca. As can be seen from Table 2 that this exceptionally larger extent of
charge transfer from alkaline earth metals is also accompanied by substantial re-
duction of the <CXC angle. It should be noted that C–Be distance remains almost
same (� 1.65�A) except for the complex with >CO group. The possible reason
behind the smallest interaction energy obtained for the chosen metal complexes
having >CO group may be rather signi¯cant increase of angular strain due to the
shortening of <CXC angle with respect to the free ligand. The metal–carbon dis-
tance increases with increasing size of the metal atom. The calculated interaction
energy has been found to be negative which suggest that the metal–ligand CT
interaction stabilizes the complexes with respect to free metal and ligand. The
order of stability of Be and Mg complexes decreases in the order >CEt2, >CMe2,
>NMe, >SO2, >CH2 and >CO. This order slightly di®ers from that obtained for
Ca complexes.
The relative order of stability of metal complexes with a particular end
group (>X) can be qualitatively understood from the second-order stabilization
energy (see Table 1A in the Supplementary Material section) associated with
metal–ligand CT interactions. The Be metal forms stronger covalent interactions
with \C�C" moieties while Mg and Ca uses valence s and p orbitals in the
weaker � interaction with acetylene moieties. This accounts for substantially
Table 2. B3LYP/6-311þþG(d,p) calculated
angles (deg.).
<CXC
>X Ligand Be Mg Ca
>CH2 113.82 105.54 107.27 110.26>CMe2 109.52 101.98 104.09 107.18
>CEt2 108.69 100.03 102.93 105.27
>NMe 120.41 111.78 115.29 117.96
>CO 115.14 70.36 63.55 65.40>SO2 100.81 101.98 103.78 114.81
June 26, 2014 5:01:21pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Static second hyperpolarizability of � shaped alkaline earth metal complexes
1450039-5

higher stabilization energy of beryllium complexes compared to the magnesium
and calcium complexes.
3.2. Second hyperpolarizability and TDDFT analysis
The dominant component of the ground state dipole moment of the chosen ligands
and metal complexes lies along the Z axis. The calculated results of all the axial
components of � along with the mean second hyperpolarizability obtained at the
B3LYP/6-311þþG(d,p) level are presented in Table 3. The results obtained with
other DFT functional are reported in Table 2A (Supplementary Materials sec-
tion). The z-component of second hyperpolarizability (�zzzz) is found to be the
major axial component. It is evident that �zzzz value of Mg and Ca complexes has
been predicted larger than that of the ligands by an order of magnitude. This can
be attributed to the enhanced CT interaction taking place in these metal com-
plexes. For a chosen ligand, the magnitude of second hyperpolarizability of the
complexes increases in the order Be < Mg < Ca. This trend follows a nice corre-
lation with the <CXC angle of the complexes which increases gradually in the
order Be < Mg < Ca and approaches the angle of free ligands. It can be noted that
the B3LYP and B2PLYP calculated �zzzz values obtained for Be-complexes are
found to lie within a narrow margin. Likewise, the BHHLYP and CAM-B3LYP
calculated result of �zzzz di®ers within a close margin. However, in the case of Mg
and Ca complexes B3LYP and BHHLYP calculated �zzzz values obtained for the
¯rst three ligands lies within a small range while the BHHLYP and CAM-B3LYP
values of Mg-complexes having >CO and >SO2 groups show an excellent agree-
ment. For a chosen alkaline earth metal, the maximum value of �zzzz has been
predicted for complexes having >CEt2 group. However, the magnitude of �zzzz and
�av obtained for complexes having >CEt2 and >CMe2 groups di®ers by a narrow
margin. In the case of Be-complexes, the �zzzz values obtained with >CMe2 and
>NMe groups are almost identical. Longitudinal component of second hyperpo-
larizability obtained for ligands with di®erent >X varies in the order >CEt2>>
CMe2>>NMe>>CH2>> SO2>>CO for magnesium and calcium complexes, and
in the order >CEt2>NMe �>CMe2>>CH2>>SO2>>CO for beryllium com-
plexes. It should be noted that at the B3LYP level, the �zzzz value of complexes
with >CMe2 ð>CEt2Þ group has been predicted larger by 0.22% (7.12%) for Be,
26.13% (28.03%) for Mg and 28.37% (32.80%) for Ca than the corresponding
complexes with >NMe group. It is interesting to note that replacement of two H
atoms of >CH2 group with two methyl (ethyl) groups enhances the magnitude of
�zzzz by 64.38% (75.68%) in Be, 51.92% (59.94%) in Mg, and 60.80% (67.28%) in
Ca at the B3LYP level. On the other hand, complexes containing electron with-
drawing groups >CO and >SO2 involve greater amount of charge transfer from
the metal atom (Table 1) but have smaller values of second hyperpolarizability
which may be due to the higher angular strain arising from the substantial
shortening and widening of <CXC angle, respectively.
June 26, 2014 5:01:22pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
K. Hatua & P. K. Nandi
1450039-6

Tab
le3.
Axialcom
pon
entsof
secondhyperpolarizab
ility(�
iiii,104a.u.)an
dav
erag
esecondhyperpolarizab
ility(�
av,104a.u.)of
thechosen
metal
complexes
obtained
attheB3L
YP/6-311þþ
G(d,p)level.
�zzzz
�yyyy
�xxxx
�av
>X
Ligan
dBe
Mg
Ca
Ligan
dBe
Mg
Ca
Ligan
dBe
Mg
Ca
Ligan
dBe
Mg
Ca
>CH
22.06
58.99
635
.498
77.781
1.10
13.07
5.30
822
.253
0.92
33.40
520
.521
92.796
1.33
35.23
920
.974
64.420
>CMe 2
2.56
414
.788
53.930
125.779
2.03
44.41
06.25
622
.451
2.35
74.19
720
.076
86.337
2.37
47.65
127
.797
84.058
>CEt 2
2.95
515
.805
56.774
130.115
2.59
41.57
813
.646
20.615
2.34
02.94
84.98
441
.808
2.76
57.37
028
.570
87.091
>NMe
4.10
914
.755
44.343
97.979
2.23
13.37
94.64
214
.960
2.25
83.90
113
.555
56.964
2.97
07.10
320
.774
60.622
>CO
1.60
73.17
011
.161
20.766
1.33
62.66
23.10
05.04
50.96
21.23
52.69
54.15
20.67
12.06
15.21
99.47
9
>SO
22.49
82.67
917
.419
70.928
0.95
01.61
21.65
63.43
20.69
21.10
44.70
96.90
31.29
11.75
37.36
223
.271
June 26, 2014 5:01:24pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Static second hyperpolarizability of � shaped alkaline earth metal complexes
1450039-7

In order to explain the relative variation of second hyperpolarizability, the
following two state model (TSM) has been invoked
� 2Lzzzz �
� 2gn��2
gn
�E 3gn
� �4gn
�E 3gn
� �; ð3Þ
where �Egn, �gn and ��gn are the transition energy, transition dipole moment and
dipole moment di®erence between the ground \g" and the crucial excited state \n".
It should be noted that for the crucial excited state \n", �zzzz depends strongly on the
magnitude of �Egn, �gn and ��gn terms. For a nonpolar or weakly polar molecule,
the ¯rst term (dipolar contribution) is generally insigni¯cant due to very small
magnitude of ��gn but the second term may be signi¯cant. Hence, considering only
the magnitude of the nondipolar contribution, the above equation can be further
approximated to the following expression.
� 2Lzzzz �
�4gn
�E 3gn
��������: ð4Þ
Here, it is worthwhile to mention that Eq. (3) is the simpli¯ed SOS expression of
the longitudinal component of second hyperpolarizability and contains the spectro-
scopic properties of the crucial state corresponding to the most intense linear tran-
sition. However, the exact SOS expression of � also involves two and three photon
electronic transitions encompassing many higher lying excited states. Thus the neg-
ative sign of second hyperpolarizability cannot be ascertained even the second term of
Eq. (3) is larger. Nevertheless, the later term may be useful in the evolution of second
hyperpolarizability. In our previous study,29 it has been noted that the overall sign of
SOS � obtained by considering about 150 excited states is positive even though the
one photon contribution (OPC) is largely negative. Also if the sign is discarded, the
larger magnitude of OPC corresponds to the larger magnitude of �. The spectroscopic
parameters in Eq. (4) has been calculated by employing the time dependent DFT
(TDDFT) method for the B3LYP functional and 6-311þþG(d,p) basis set for 50
lowest lying singlet excited states. The most dominant electronic transition has been
determined by the larger magnitude of �gn associated with the signi¯cant oscillator
strength which are given in Table 4. The comparable but larger values of �zzzzobtained for Be complexes having >CMe2, >CEt2 and >NMe groups may arise from
the relatively smaller transition energy and higher transition moment. In the case of
Mg and Ca complexes, the highest value of second hyperpolarizability obtained with
>CEt2 group may be explained for notably smaller transition energy and higher
transition moment. Also for a chosen ligand, the increasing trend of second hyper-
polarizability with increasing electropositive character of metal primarily originates
from the decreasing transition energy gap.
The electronic transitions in metal complexes having >CMe2 group are displayed
in Fig. 2. As can be seen from the orbital picture, overlapping interaction is signif-
icant in both HOMO and LUMOþ3 of Be complex. In the case of Mg complex, the
June 26, 2014 5:01:24pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
K. Hatua & P. K. Nandi
1450039-8

orbital overlap in HOMO is similar to that of Be complex but the overlap of metal
orbitals in LUMO is rather poor resulting in spreading of electron density which
accounts for the notably larger transition moment. For the more electropositive
alkaline earth metal Ca, the charge distribution in both transition orbitals are
comparable showing no orbital interaction between the metal atom and the ligand
which is consistent with the electrostatic interaction. Owing to the greater electron
withdrawing power of the polar functional groups >CO and >SO2, the corre-
sponding metal complexes have highly polar ground state which may result poor
orbital overlap with the metal atom (having higher positive charge) (Table 1) and
result in larger transition energy gap which in turn substantially lowers the
Mg-CMe2 Ca-CMe2Be-CMe2
Fig. 2. Major electronic transition of complexes of Be, Mg and Ca containing the >CMe2 group.
Table 4. Transition energy (�Egn, eV), transition moment (�gn, a.u.), oscillator strength (f0, a.u.) and the major
transitiona involved in the crucial excitation for the chosen complexes obtained at TD-B3LYP/6-311þþG(d,p) level.
Be-complexes Mg-complexes Ca-complexes
X �Egn �gn f0
Major
transition �Egn �gn f0
Major
transition �Egn �gn f0
Major
transition
>CH2 4.499 1.051 0.121 H ! Lþ 3 3.431 1.875 0.296 H ! Lþ 1 1.673 1.976 0.160 H ! L
>CMe2 4.464 1.214 0.161 H ! Lþ 3 3.043 2.527 0.476 H ! L 1.587 2.136 0.177 H ! L
>CEt2 4.416 0.921 0.092 H ! Lþ 3 3.028 2.541 0.479 H ! L 1.581 2.177 0.183 H ! L
>NMe 4.385 1.352 0.196 H ! Lþ 1 3.085 2.169 0.355 H ! L 1.828 2.269 0.230 H ! L
>CO 5.827 1.148 0.188 H� 4 ! L 3.929 1.502 0.217 H� 1 ! Lþ 2 3.042 0.853 0.054 H� 1 ! Lþ 2
>SO2 6.051 0.946 0.133 H ! Lþ 3 3.328 2.063 0.347 H ! L 1.922 0.819 0.031 H ! L
aH and L stand for highest occupied MO and lowest unoccupied MO, respectively.
June 26, 2014 5:01:27pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Static second hyperpolarizability of � shaped alkaline earth metal complexes
1450039-9

magnitude of cubic polarizability. Thus it emerges that apart from the ground state
polarization, the energy gap between the ground and crucial excited states plays a
key role in the modulation of second hyperpolarizability of the chosen � shaped
molecular complexes.
4. Conclusion
In the present investigation, a number of complexes between the alkaline earth
metals Be, Mg and Ca and � shaped ligands with varying functional groups have
been considered for the theoretical study of the ground state structure and second
hyperpolarizability by employing di®erent DFT functionals and 6-311þþG(d,p)
basis set. The � shaped complexes are found to be thermally stable. For a particular
choice of ligand having a functional group X, the order of stability of complexes with
metal varies in the order Be > Ca > Mg except for the >CO group. For a chosen
metal, the maximum stability has been found for the ligand having>C(C2H5)2 group
and also for each metal, the maximum value of second hyperpolarizability is also
predicted for the same ligand. One of the most interesting ¯nding of the present work
is the preference of bulky functional group rather than the polar electron with-
drawing group while optimizing the second hyperpolarizability. The gradual opening
of <CXC angle on substitution with larger alkaline earth metals plays an important
role in enhancing the CT interaction and second hyperpolarizability of the chosen �
shaped complexes. The variation of NLO property of the chosen � shaped metal
complexes have been explained satisfactorily in terms of the transition energy and
transition moment corresponding to the most intense electronic transition.
Acknowledgments
(PKN) acknowledges the grant from UGC, Government of India under the Major
Research Project (F. No. 42-339/2013 (SR) for carrying out this research work. The
authors are grateful to the valuable comments of the Reviewer to improve the quality
of the manuscript.
References
1. Prasad PN, Williams DJ, Introduction to Nonlinear Optical E®ects in Molecules andPolymers, Wiley-Interscience, New York, 1991.
2. Mayers F, Marder S, Perry JW, Introduction to the Nonlinear Optical Properties ofOrganic Materials in Chemistry of Advanced Materials, Wiley-VCH, New York, 1998.
3. Papdopoulos MG, Sadlej AJ, Leszczysnki J, Non-Linear Optical Properties of Matter,Challenges and Advances in Computational Chemistry and Physics, Springer, 2006.
4. Maroulis G, Atoms, Molecules and Clusters in Electric Field, Imperial College Press,World Scienti¯c, 2006.
5. Suponitsky KY, Liao Y, Masunov AE, Electric hyperpolarizabilities for donor–acceptormolecules with long conjugated bridges: Calculations versus experiment, J Phys Chem A13:10994–11001, 2009.
June 26, 2014 5:01:28pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
K. Hatua & P. K. Nandi
1450039-10

6. Ji Y, Qian Y, Lu W, E±cient � bridges based on ¯ve-membered heterocyclic rings forsecond-order NLO properties of push-pull type molecules, J Mat Chem 22:12376–12380,2012.
7. Capobiaco A, Centore R, Noce C, Peluso A, Molecular hyperpolarizabilities of push-pullchromophores: A comparison between theoretical and experimental results, Chem PhysLett 411:11–16, 2013.
8. Ramirez MA, Cuadro AM, Builla JV, Castano O, Andres JL, Mendicuti F, Clays K,Asselberghs I, Vaquero JJ, Donor-(�-bridge)-azinium as D-�-Aþ one dimensional andD-�-Aþ-�-D multidimensional V-shaped chromophores, Org Biomol Chem 10:1659–1669, 2012.
9. Hatua K, Nandi PK, Relationship between di®erent order polarizabilities and groundstate dipole moment, J Theor Comput Chem 12:1250099, 2012.
10. Marcono E, Squitieri E, Murgich J, Soscun H, Theoretical investigation of the static(dynamic) polarizability of DAAD quadrupolar push-pull molecules. A Comparisonamong HF (TD-HF), DFT (TD-B3LYP), and MP2 (TD-MP2) methods, Comp TheoChem 985:72–79, 2012.
11. Nandi PK, Panja N, Kar T, Hyperpolarizabilities of hetero-cycle based chromophores: Asemi-quantitative SOS scheme, Chem Phys Lett 444:366–374, 2007.
12. Nandi PK, Panja N, Ghanty TK, Heterocycle-based isomeric chromophores with sub-stantially varying NLO properties: A new structure-property correlation study, J PhysChem A 112:4844–4852, 2008.
13. Nandi PK, Panja N, Ghanty TK, Kar T, Theoretical study of the e®ect of structuralmodi¯cations on the hyperpolarizabilities of indigo derivatives, J Phys Chem A113:2623–2631, 2009.
14. Panja N, Ghanty TK, Nandi PK, A sum-over-state scheme of analysis of hyperpolariz-abilities and its application to spiroconjugated molecular system, Theor Chem Acc126:323–337, 2010.
15. Li ZJ,Wang FF, Li ZR, XuHL, Huang XR,WuD, ChenW, YuGT, Gu FL, Aoki Y, Largestatic ¯rst and second hyperpolarizabilities dominated by excess electron transition forradical ion pair saltsMþ
2 TCNQ� (M¼Li, Na,K),Phys ChemChemPhys 11:402–408, 2009.16. Zhong RL, Xu HL, Muhammad S, Zhang J, Su ZM, The stability and nonlinear optical
properties: Encapsulation of an excess electron compound LiCN� � �Li within boron nitridenanotubes, J Mater Chem 22:2196, 2012.
17. Chen W, Li ZR, Wu D, Li Y, Sun CC, Gu FL, The structure and the large non linearoptical properties of Li–Calix[4]pyrrole, J Am Chem Soc 127:10977–10981, 2005.
18. Tang SW, Feng JD, Qing YQ, Sun H, Wang FD, Su ZM, Chang YF, Wang RS,Thermochemical stabilities, electronic, and optical properties of C56X10 (X¼H, F and Cl)fullerene compounds, J Comput Chem 32:658–667, 2011.
19. Tang SW, Feng JD, Qiu YQ, Sun H, Wang FD, Chang YF, Wang RS, Electronic stru-cuture and optical properties of highly deformed halofullerenes C3V C60F18 and D3d
C60Cl30, J Comput Chem 31:2650–2657, 2010.20. Xu HL, Wang FF, Li ZR, Wang BQ, Wu D, Chen W, Tao G, Gu FL, Aoki Y, The
nitrogen edge-doped e®ect on the static ¯rst hyperpolarizability of the supershot single-walled carbon nanotube, J Comput Chem 30:1128–1134, 2009.
21. Koukaras EN, Zdetsis AD, Karamanis P, Pouchan C, Avramopoulos A, PapadopoulosMG, Structural and static electric response properyies of hughly symmetric silicon cages:Theoretical predictions, J Comput Chem 33:1068–1079, 2012.
22. Oscar J, Halla J, Matito E, Blancafort L, Robles J, Sola M, Tuning aromaticity in trigonalalkaline earth metal clusters and their alkali metal salts, J Comput Chem 30:2764–2776,2009.
June 26, 2014 5:01:28pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
Static second hyperpolarizability of � shaped alkaline earth metal complexes
1450039-11

23. Yu G, Zhao X, Niu M, Huang X, Zhang H, Chen W, Constructing a mixed �-conjugatedbridge: A simple and e®ective approach to realize a large ¯rst hyperpolarizability incarbon nanotube-based systems, J Mater Chem C 1:3833–3841, 2013.
24. Yu G, Huang XR, Chen W, Sun CC, Alkali metal atom-aromatic ring: A novel inter-action mode realizes large ¯rst hyperpolarizabilities of M–AR (M¼Li, Na, andK, AR¼Pyrrole, Indole, Thiophene and Benzene), J Comput Chem 32:2005–2011, 2011.
25. Wang YF, Huang J, Zhou G, Coordination number e®ect of nitrogen atom on thestructures and NLO responses: Alkaline earth-based alkalides, Struct Chem 24(5):1545–1553, 2013.
26. Ma NN, Yang GC, Sun SL, Liu CG, Qiu YQ, Computational study of second ordernonlinear optical properties of a novel class of two-dimensional �- and W-shapedsandwich metallocarborane-containing chromophores, J Organometallic Chem 696:2380–2387, 2011.
27. Hatua K, Nandi PK, Interaction of beryllium with acceptor hydrocarbon: Electronicstructure and second hyperpolarizability, J Theor Comp Chem 12:1350046, 2013.
28. Hatua K, Nandi PK, Third order NLO properties of beryllium-pyridyne complexes,J Theor Comp Chem 12:1350075, 2013.
29. Hatua K, Nandi PK, Theoretical study of electronic structure and third-order opticalproperties of beryllium–hydrocarbon complexes, Comp Theo Chem 996:82–90, 2012.
30. Hatua K, Nandi PK, Beryllium-cyclobutadiene multidecker inverse sandwiches: Elec-tronic structure and second-hyperpolarizability, J Phys Chem A 117:16340–16346, 2013.
31. Becke AD, Density-functional thermochemistry. III. The role of exact exchange, J ChemPhys 98:5648–5652, 1993.
32. Becke AD, A new mixing of Hartree-Fock and local density-functional theories, J ChemPhys 98:1372–1377, 1993.
33. Yanai T, Tew D, Handy N, A new hybrid exchange-correlation functional using theCoulomb-attenuating method (CAM-B3LYP), Chem Phys Lett 393:51–57, 2004.
34. Grimme S, Semiempirical hybrid density functional with perturbative second-ordercorrelation, J Chem Phys 124:034108, 2006.
35. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GW, Robb MA, Cheeseman JR, ScalmaniG, Barone V, Mennucci B, Petersson GA, Nakatsuji H et al., Gaussian 09, Revision A.02,Gaussian Inc., Wallingford CT, 2009.
June 26, 2014 5:01:29pm WSPC/178-JTCC 1450039 ISSN: 0219-63361stReading
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
K. Hatua & P. K. Nandi
1450039-12

















![CO[sub 2] capture properties of alkaline earth metal oxides and hydroxides: A combined density functional theory and lattice phonon dynamics study](https://static.fdokumen.com/doc/165x107/633319fdb6829c19b80c41e7/cosub-2-capture-properties-of-alkaline-earth-metal-oxides-and-hydroxides-a-combined.jpg)


