
Plasticity of human dental pulp stromal cells with bioengineering platforms: a versatile tool for...
14
Micron 67 (2014) 155–168 Contents lists available at ScienceDirect Micron j ourna l ho me page: www.elsevier.com/locate/micron Plasticity of human dental pulp stromal cells with bioengineering platforms: A versatile tool for regenerative medicine Serena Barachini a,∗ , Serena Danti b , Simone Pacini a , Delfo D’Alessandro b , Vittoria Carnicelli b , Luisa Trombi a , Stefania Moscato a , Claudio Mannari a , Silvia Cei b , Mario Petrini a a Department of Clinical and Experimental Medicine, University of Pisa, Italy b Department of Surgical, Medical, Molecular Pathology and Emergency Medicine, University of Pisa, Italy a r t i c l e i n f o Article history: Received 24 March 2014 Received in revised form 15 July 2014 Accepted 20 July 2014 Available online 27 July 2014 Keywords: Stem cells Dental pulp Differentiation Bioengineering Regenerative medicine a b s t r a c t In recent years, human dental pulp stromal cells (DPSCs) have received growing attention due to their characteristics in common with other mesenchymal stem cells, in addition to the ease with which they can be harvested. In this study, we demonstrated that the isolation of DPSCs from third molar teeth of healthy individuals allowed the recovery of dental mesenchymal stem cells that showed self-renewal and multipotent differentiation capability. DPSCs resulted positive for CD73, CD90, CD105, STRO-1, neg- ative for CD34, CD45, CD14 and were able to differentiate into osteogenic and chondrogenic cells. We also assayed the angiogenic potential of DPSCs, their capillary tube-like formation was assessed using an in vitro angiogenesis assay and the uptake of acetylated low-density lipoprotein was measured as a marker of endothelial function. Based on these results, DPSCs were capable of differentiating into cells with phe- notypic and functional features of endothelial cells. Furthermore, this study investigated the growth and differentiation of human DPSCs under a variety of bioengineering platforms, such as low frequency ultrasounds, tissue engineering and nanomaterials. DPSCs showed an enhanced chondrogenic differ- entiation under ultrasound application. Moreover, DPSCs were tested on different scaffolds, poly(vinyl alcohol)/gelatin (PVA/G) sponges and human plasma clots. We showed that both PVA/G and human plasma clot are suitable scaffolds for adhesion, growth and differentiation of DPSCs toward osteoblastic lineages. Finally, we evaluated the interactions of DPSCs with a novel class of nanomaterials, namely boron nitride nanotubes (BNNTs). From our investigation, DPSCs have appeared as a highly versatile cellular tool to be employed in regenerative medicine. © 2014 Elsevier Ltd. All rights reserved. Abbreviations: DPSCs, dental pulp stromal cells; MSC, mesenchymal stromal cells; SHEDs, stromal cells from human exfoliated deciduous; BM-MSCs, bone marrow-derived mesenchymal stromal cells; NTFs, neurotrophic factors; NGF, nerve growth factor; BDNF, brain-derived neurotrophic factor; PPP, platelet poor plasma; US, ultrasounds; LIUS, low intensity US; LFUS, low frequency US; PVA, poly(vinyl alcohol); G, gelatin; BNNTs, boron nitride nanotubes; -MEM, minimum essen- tial medium alpha modification; FBS, fetal bovine serum; PDT, population doubling time; CPs, chondrogenic pellets; HUVECs, human umbilical vein endothelial cells; SEM, scanning electron microscopy; TEM, transmission electron microscopy. ∗ Corresponding author at: Hematology Division, Department of Clinical and Experimental Medicine, University of Pisa, Via Roma 56, 56100 Pisa, Italy. Tel.: +39 050 993484; fax: +39 050830162. E-mail address: [email protected] (S. Barachini). 1. Introduction Among mesenchymal stromal cells (MSCs) of autologous origin, human dental pulp stromal cells (DPSCs) have received growing attention in recent years firstly due to the easy accessibility of the tissue in any adult individual. Dental pulp tissue is thought to be derived from migratory neural crest during development (Peters and Balling, 1999). Den- tal pulp is a soft connective tissue and its main functions are to produce dentin, and to maintain the biological and physiological vitality of the dentin. DPSCs are multipotent stromal cells derived from neural crest and mesenchyme and have the capacity to dif- ferentiate into multiple cell lineages. Firstly, Gronthos et al. (2000, 2002) have isolated post-natal stromal cells from the human den- tal pulp of permanent teeth. Other stromal cell populations from surrounding tissues of the tooth have been isolated from periodon- tal ligament, human exfoliated deciduous teeth, apical papilla and http://dx.doi.org/10.1016/j.micron.2014.07.003 0968-4328/© 2014 Elsevier Ltd. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Plasticity of human dental pulp stromal cells with bioengineering platforms: a versatile tool for...

Pp
SVSa
b
a
ARRAA
KSDDBR
cmgUattS
ET
h0
Micron 67 (2014) 155–168
Contents lists available at ScienceDirect
Micron
j ourna l ho me page: www.elsev ier .com/ locate /micron
lasticity of human dental pulp stromal cells with bioengineeringlatforms: A versatile tool for regenerative medicine
erena Barachinia,∗, Serena Dantib, Simone Pacinia, Delfo D’Alessandrob,ittoria Carnicelli b, Luisa Trombia, Stefania Moscatoa, Claudio Mannaria,ilvia Ceib, Mario Petrinia
Department of Clinical and Experimental Medicine, University of Pisa, ItalyDepartment of Surgical, Medical, Molecular Pathology and Emergency Medicine, University of Pisa, Italy
r t i c l e i n f o
rticle history:eceived 24 March 2014eceived in revised form 15 July 2014ccepted 20 July 2014vailable online 27 July 2014
eywords:tem cellsental pulpifferentiationioengineeringegenerative medicine
a b s t r a c t
In recent years, human dental pulp stromal cells (DPSCs) have received growing attention due to theircharacteristics in common with other mesenchymal stem cells, in addition to the ease with which theycan be harvested. In this study, we demonstrated that the isolation of DPSCs from third molar teeth ofhealthy individuals allowed the recovery of dental mesenchymal stem cells that showed self-renewaland multipotent differentiation capability. DPSCs resulted positive for CD73, CD90, CD105, STRO-1, neg-ative for CD34, CD45, CD14 and were able to differentiate into osteogenic and chondrogenic cells. Wealso assayed the angiogenic potential of DPSCs, their capillary tube-like formation was assessed using anin vitro angiogenesis assay and the uptake of acetylated low-density lipoprotein was measured as a markerof endothelial function. Based on these results, DPSCs were capable of differentiating into cells with phe-notypic and functional features of endothelial cells. Furthermore, this study investigated the growthand differentiation of human DPSCs under a variety of bioengineering platforms, such as low frequencyultrasounds, tissue engineering and nanomaterials. DPSCs showed an enhanced chondrogenic differ-entiation under ultrasound application. Moreover, DPSCs were tested on different scaffolds, poly(vinyl
alcohol)/gelatin (PVA/G) sponges and human plasma clots. We showed that both PVA/G and humanplasma clot are suitable scaffolds for adhesion, growth and differentiation of DPSCs toward osteoblasticlineages. Finally, we evaluated the interactions of DPSCs with a novel class of nanomaterials, namelyboron nitride nanotubes (BNNTs). From our investigation, DPSCs have appeared as a highly versatilecellular tool to be employed in regenerative medicine.© 2014 Elsevier Ltd. All rights reserved.
Abbreviations: DPSCs, dental pulp stromal cells; MSC, mesenchymal stromalells; SHEDs, stromal cells from human exfoliated deciduous; BM-MSCs, bonearrow-derived mesenchymal stromal cells; NTFs, neurotrophic factors; NGF, nerve
rowth factor; BDNF, brain-derived neurotrophic factor; PPP, platelet poor plasma;S, ultrasounds; LIUS, low intensity US; LFUS, low frequency US; PVA, poly(vinyllcohol); G, gelatin; BNNTs, boron nitride nanotubes; �-MEM, minimum essen-ial medium alpha modification; FBS, fetal bovine serum; PDT, population doublingime; CPs, chondrogenic pellets; HUVECs, human umbilical vein endothelial cells;EM, scanning electron microscopy; TEM, transmission electron microscopy.∗ Corresponding author at: Hematology Division, Department of Clinical andxperimental Medicine, University of Pisa, Via Roma 56, 56100 Pisa, Italy.el.: +39 050 993484; fax: +39 050830162.
E-mail address: [email protected] (S. Barachini).
ttp://dx.doi.org/10.1016/j.micron.2014.07.003968-4328/© 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Among mesenchymal stromal cells (MSCs) of autologous origin,human dental pulp stromal cells (DPSCs) have received growingattention in recent years firstly due to the easy accessibility of thetissue in any adult individual.
Dental pulp tissue is thought to be derived from migratoryneural crest during development (Peters and Balling, 1999). Den-tal pulp is a soft connective tissue and its main functions are toproduce dentin, and to maintain the biological and physiologicalvitality of the dentin. DPSCs are multipotent stromal cells derivedfrom neural crest and mesenchyme and have the capacity to dif-ferentiate into multiple cell lineages. Firstly, Gronthos et al. (2000,
2002) have isolated post-natal stromal cells from the human den-tal pulp of permanent teeth. Other stromal cell populations fromsurrounding tissues of the tooth have been isolated from periodon-tal ligament, human exfoliated deciduous teeth, apical papilla and
1 icron
dSftcsift2tc2ii2etiern(
hteelptppfiMecotg
s2dpoa2Btosmd
aeabbTTpodd
56 S. Barachini et al. / M
ental follicle precursor cells (Miura et al., 2003; Seo et al., 2004;onoyama et al., 2006). These post-natal populations have MSC-likeeatures, namely the capacity for self-renewal and the potentialo differentiate into multiple lineages including osteoblasts andhondroblasts (Huang et al., 2009). However, DPSCs show higherelf-renewal ability, immunomodulatory capacity and proliferationn vitro than bone marrow-mesenchymal stromal cells (BM-MSCs);urthermore, they preferentially differentiate to osteoblasts ratherhan into adipocytes (Gronthos et al., 2000; Pierdomenico et al.,005). Although the majority of studies have focused their atten-ion on the ability of DPSCs to differentiate into odontoblast-likeells (Almushayt et al., 2006; Cordeiro et al., 2008; Paula-Silva et al.,009) or osteoblasts (Laino et al., 2005, 2006; D’Aquino et al., 2007),
t has also been discovered that they are capable of differentiatingnto other cell types, including smooth muscle cells (Kerkis et al.,006; Gandia et al., 2008) and neurons (Arthur et al., 2008; Kadart al., 2009). DPSCs express nestin and glial fibrillary acidic pro-ein and, under appropriate stimuli, are capable of differentiatingnto functionally active neurons (Arthur et al., 2008), influencingndogenous recruitment of neural stem cells and generating neu-ospheres (Sasaki et al., 2008). Neural stem cell markers, such asestin, expressed in DPSCs reflect the neural origin of dental pulpKerkis et al., 2006; Estrela et al., 2011).
Recently, we have investigated specific molecular profiles ofuman stromal stem cell populations derivated from differentissues, particularly with regard to the global HOX gene familyxpression profile (HOX code) and their three amino acid loopxtension co-factor subfamilies (Picchi et al., 2013). The differentevels of HOX expression detected in stromal cells with differentotency strongly suggest that HOX genes may not only reflect posi-ional and embryological cell identity, but also indicate the cellularosition within the stem cell hierarchy. Such considerations sup-ort the growing evidence that HOX code provides a “biologicalngerprint” to distinguish stem cell populations (Chang et al., 2002;oens and Selleri, 2006). Our previous study shows that DPSCs
xhibit extremely low levels of expression of a few HOX genes,onfirming previous findings, and in line with the neuroectodermalrigin of DPSCs (Couly et al., 2002; D’Antò et al., 2006). It is knownhat neural crests are HOX negative and indeed the few active HOXenes in DPSCs are expressed at barely detectable levels.
MSCs appear to exert paracrine trophic effects through theecretion of bioactive molecules (Caplan and Dennis, 2006; Caplan,007) as the neurotrophic factors (NTFs). In particular, brain-erived neurotrophic factor (BDNF) and nerve growth factor (NGF)roduced by DPSCs, have been shown to have a crucial influencever neurons in the central nervous system such as motor neuronsnd dopaminergic neurons of the substantia nigra (Nosrat et al.,001, 2004). In our previous study, we found that high levels ofDNF, NGF transcripts were constitutively expressed by DPSCs andhat the neuroprotective effect of DPSCs against two neurotoxinsn an in vitro model of Parkinson’s could be due to soluble factors,uch as BDNF and NGF, released by DPSCs (Nesti et al., 2010). DPSCsay thus be an alternative source in cell therapy for neurological
iseases.Recent highlights have started considering stem cells not only
s the final therapeutic product, but rather as part of complex bio-ngineered therapeutic strategies. In this view, MSCs can be fullyppreciated as a versatile tool in combination with biomaterials andiomedical devices to guide their commitment in various humanody tissues (Kshitiz et al., 2012; Kinney and McDevitt, 2013).herefore, stem cell plasticity thus represents a master feature.he potential use of stem cells in bioengineering-based thera-
ies includes tissue engineering and nanotechnology. As a prooff concept, we tested the biological response of DPSCs in threeifferent bioengineering platforms. As an example of biomedicalevice-assisted platforms for tissue regeneration, ultrasounds (US)67 (2014) 155–168
represent a non-invasive and versatile tool that has been shown topromote tissue repair, such as cartilage healing in animal modelsof articular cartilaginous defects (Cook et al., 2001). Studies on theunderlying mechanism leading to cell response to US stimuli areongoing, however preliminary evidence in rat chondrocyte cultureshave highlighted that US can enhance the synthesis of proteogly-cans depending on their application cycles and intensity (Parviziet al., 1999). In line with these observations, recent reports havepointed out that low intensity US (LIUS) can exert a favorable effecton the proliferation, extracellular matrix synthesis and chondro-genic differentiation of MSCs in vitro (Lee et al., 2006; Shah et al.,2013). Differently from LIUS, used for diagnostic purposes, low fre-quency US (LFUS) (range 20–100 kHz) is used in sonophoresis anddentistry since they transfer higher mechanical energy. In our studywe tested the efficacy of LFUS on DPSC pellets under chondrogenicdifferentiation with respect to traditional pellet culture.
Moreover, we investigated DPSCs for tissue engineering appli-cations, using biocompatible three-dimensional spongy materialsto assess their differentiation capability. Poly(vinyl alcohol) (PVA)has been among one of first synthetic macromolecules employed inboth implantable and non-implantable medical devices (e.g., con-tact lenses, artificial meniscus) due to its characteristics includinglow protein adsorption, biocompatibility, high hydrophilicity, easyprocessability and chemical inertia (Alves et al., 2011). Additionally,PVA can be easily added to biologic molecules to obtain bioartificialmatrices with specific architectural features (Cascone et al., 2004).Among which, gelatin (G) is a natural protein derived from colla-gen able to promote cell adhesion (Dubruel et al., 2007). Spongyscaffolds based on PVA and G have been successfully tested withgingival fibroblasts, being reported as very promising substratesfor tissue engineering (Moscato et al., 2008). In the present study,PVA/G sponges were prepared and investigated in vitro for three-dimensional growth and osteogenic differentiation of DPSCs.
Finally, nanomedicine is emerging as a powerful source of inno-vative cell-targeted-therapies based on nanoengineered materials,whose toxicological risks have yet to be disclosed. The balance ofthese features has thus become an intriguing challenge for suc-cessful development of therapeutically effective nanosystems. Inparticular, due to their small size, nanoparticles can enter the cellmembrane and be used as non-viral vectors. Boron nitride nano-tubes (BNNTs) are a class of ceramic nanoparticles with a tubularshape that have attracted recent attention due to their excel-lent physical properties and putative biocompatibility (Ciofani andDanti, 2012). In this study, we report our preliminary findings onthe interactions of BNNTs with DPSCs, aimed at supporting thefuture disclosement of novel nanomedicine-based therapies.
In summary, the specific aims of the current study are to improveunderstanding of the biological properties of human DPSCs. More-over, we have investigated DPSC growth and differentiation indifferent bioengineering applications, evaluating their multipotentpotential in regenerative medicine.
2. Materials and methods
2.1. Extraction and isolation of human DPSCs
Human dental pulps were obtained from molars of healthysubjects (n = 10) 18–35 years of age, after informed consent (OralSurgery Department, Santa Chiara Hospital, Pisa, Italy) according toa protocol approved by the local University committee on Ethics inMedicine. Each subject, before extraction, was checked for systemic
and oral diseases and pre-treated a week before with professionaldental hygiene. Before extraction, the dental crown was rinsed witha 0.2% chlorexidin gel (Dentosan, Johnson & Johnson Medical S.p.A.,Rome, Italy) for 2 min. Radicular dental pulps were obtained, using
icron
awmPaaw1Itm9teUpunNwt
2
2
fm(manfltpcrw(t(PSB
c
2
DetAtaKcaccEdwwbfi
S. Barachini et al. / M
Gracey curette, from healthy and non-carious teeth. The samplesere placed inside sterile tube containing 20 ml of proliferationedium, transported to the laboratory and immediately processed.
roliferation medium consisted of minimum essential mediumlpha modification (�-MEM; Sigma–Aldrich, St. Louis, MO, USA)nd 10% fetal bovine serum (FBS; Sigma–Aldrich) supplementedith 100 IU/ml penicillin (Pharmacia & Upjohn S.p.A., Milan, Italy),
00 IU/ml streptomycin (Bristol-Myers Squibb S.p.A., Sermoneta,taly) and 2 mM l-glutamine (Lonza, Wolkersville, MD, USA). Pulpissue explants were placed in tissue flasks with the proliferation
edium and were cultured at 37 ◦C in 5% CO2 concentration at5% humidity. Medium changes were carried out twice a week. Onhe confluence, the cells were detached with 0.05% trypsin–0.02%thylenediaminetetraacetic acid (Life Technologies, Carlsbad, CA,SA) for further expansions. Cell growth was analyzed after the firstassage (P1) by direct cell counts to determine the cumulative pop-lation doublings (PDs) and the population doubling time (PDT). PDumber was calculated using the formula log10(N)/log10(2), where
= cells harvested/cells seeded ratio (Kern et al., 2006). Resultsere expressed as cumulative PDs. PDTs were calculated dividing
otal culture time in hours (h) by cumulative PDS.
.2. Characterization of DPSCs
.2.1. Immunophenotypic analysisThe phenotypical analysis was performed on cells detached
rom passages 2 through 6, after incubation of the DPSCs withonoclonal antibodies (mAbs). A total of 100 �l of cell suspension
5 × 105 cells) was aliquoted per tube and appropriately labeledAbs were added for multicolor analysis and incubated for 30 min
t 4 ◦C; then, samples were washed twice in MACSQuant run-ing buffer (Miltenyi Biotech, Bergisch Gladbach, Germany). Theow cytometer instrument was set using cells stained with iso-ypic controls. Cells were gated on a forward versus side scatterlot to eliminate debris. Acquisition was performed using 10,000ells that were analyzed with an MACSQuant cytofluorimeterunning the MACSQuantify software (Miltenyi Biotech). DPSCsere stained using mAbs specific for CD105 PE-Cy7-conjugated
BioLegend, San Diego, CA, USA), CD73 PE-Cy7-conjugated (Mil-enyi Biotech), CD90 PerCP-Cy5.5 (BioLegend), CD31 PE-conjugatedMiltenyi Biotech), CD146 PE-conjugated (BioLegend), CD309E-conjugated (BioLegend), SSEA-4 AlexaFluor®488 (BioLegend),TRO-1 AlexaFluor®647 (BioLegend), CD45 APC-Vio770 (Miltenyiiotech) and CD34 VioBlue (Miltenyi Biotech).
If not indicated, further experiments were performed in tripli-ate.
.2.2. Neurotrophin quantificationIn order to evaluate BDNF and NGF levels in the medium of the
PSC cultures, the whole supernatant medium from three distinctxperiments was collected weekly in duplicate. After centrifuga-ion to remove particulates, 2 ml aliquots were stored at −20 ◦C.fter thawing at room temperature, 0.5 ml were collected from
he aliquots and analyzed with an enzyme-linked immunosorbentssay (ELISA). NGF was detected with an ELISA kit (Koma Biotech,orea). Briefly, NGF pre-coated 96-well plate was incubated withell supernatants and NGF standards for 2 h. After, it was washednd incubated with a biotinylated rabbit anti-human NGF poly-lonal antibody, washed again, and then incubated with avidinonjugated to horseradish peroxidase. BDNF was detected with anLISA kit (Promega, USA). According to manufacturing recommen-ations, 96-well plate was coated with anti-BDNF mAb and blocked
ith the blocking buffer provided, then samples and standardsere added. After sample incubation and washing, plate was incu-ated with anti-human BDNF polyclonal antibody, washed, andnally incubated with anti-IgG antibody conjugated to horseradish
67 (2014) 155–168 157
peroxidase. For both neurotrophins, the plates were incubated witha tetramethylbenzidine (TMB) solution and absorbance measuredat 450 nm using a microplate reader (Model 550, BioRad Laborato-ries, Hercules, CA, USA).
2.3. Differentiation of DPSCs
2.3.1. Endothelial differentiationWhen 80–90% of confluence, DPSCs were detached by trypsin
digestion, re-plated at a density of 1 × 104 cells/cm2 on 5 �g/cm2
fibronectin BioCoatTM (Becton Dickinson, San Jose, CA, USA) in6-well plates. The cells were cultured in EGM-2 media (Lonza,Wolkersville, MD, USA) for 15 days, replacing the medium twicea week. An undifferentiated control group was cultured in prolif-eration medium. To evaluate endothelial differentiation, cells wereprocessed for immunophenotype analysis, as described in the pre-vious section, and PECAM1 (CD31) gene expression, as describedlater.
Assessment of in vitro capillary tube-like formation was carriedout using Matrigel (Becton Dickinson Bioscience, San Jose, CA, USA):an in vitro assay that shows the angiogenetic potential of cells. TheMatrigel was thawed overnight at 4 ◦C and mixed to homogeneityusing cooled pipette tips. Aliquots of Matrigel (50 �l) were dis-tributed as a thin layer on the bottom of a 12-well cell culture platesand left for polymerization at 37 ◦C for 30 min. Differentiated DPSCswere detached and resuspended in EGM-2 (Lonza) to give a finalconcentration of 5 × 105 cells/ml, plated onto the Matrigel-coatedsurface and incubated for 1 h in a 37 ◦C humidified incubator. Undif-ferentiated cells were cultured in proliferation medium. Tube-likestructure formation was examined via light microscopy and imagedafter 12 h. The uptake of Ac-LDL was investigated as an endothelialmarker. It was assessed incubating cell samples for 4 h at 37 ◦C with5 �g/ml AlexaFluor-488®-conjugated Ac-LDL (Life Technologies).Finally, specimens were observed under fluorescence microscopy.
2.3.2. Osteogenic differentiationTo promote osteogenic differentiation, the cells were seeded
at a density of 4 × 103 cells/cm2 in 6-well plates (CoStar Group,Bethesda, MD, USA) and cultured in basal medium, �-MEMsupplemented with 10% FBS, antibiotics and l-glutamine, untilthey reached 60–70% confluence. As soon as subconfluence wasreached, osteogenic differentiation of cells was induced cultur-ing them for 2.5 weeks with osteogenic induction medium,consisting of 100 �M dexamethasone (Sigma–Aldrich), 10 mM �-glycerophosphate (Sigma–Aldrich), 100 �M ascorbic acid (VitaminC, Roche, Indianapolis, IN, USA) and 10% FBS in �-MEM. Mediumwas changed twice a week. Cells kept in basal medium were usedas negative controls. Osteogenic differentiation was investigatedusing the von Kossa and Alizarin S (Sigma–Aldrich) staining, todemonstrate the deposition of a mineralized matrix. The cells werefixed with 1% formalin (Bio-Optica) for 10 min at 4 ◦C and stainedfor 15 min with 1% silver nitrate (Fluka, Millwaukee, WI, USA, andSigma–Aldrich). The stain was developed incubating the cells in0.5% pyrogallol (Fluka) and then fixed with 5% sodium thiosul-fate (Fluka) for 5 min. Finally, the cells were counterstained with0.1% nuclear fast red (Fluka). The samples were then dehydratedand mounted with DPX mountant (Fluka). Mineral deposition wasevaluated as black granules using optical light microscopy.
2.3.3. Chondrogenic differentiation under US stimulationDPSCs at passage 2 were trypsinized, counted and placed in 5 ml
tubes at a density of 250,000 cells/tube, centrifuged at 1200 rpm for
7 min to obtain chondrogenic pellets (CPs), which were washed insterile PBS. Chondrogenic culture medium was thus added, con-sisting of DMEM/F12, 1.25 �g/ml bovine serum albumin (BSA),5.35 �g/ml linoleic acid, 50 �g/ml ascorbic acid, 100 �g/ml sodium
158 S. Barachini et al. / Micron
Table 1Primer pairs designed for detection of PECAM1 and ACTB mRNA.
NCBI ref# Gene name Primer pair Ampliconlength (bp)
NM 000442 PECAM1 ForwardGAACCTGTCCTGCTCCATC
231
Reverse TCAAACTGGGCAT-CATAAGAAAT
NM 001101.3 ACTB ForwardCGCCGCCAGCTCACCATG
101
pm(rsOfsw
2
ebyHsRsa(MPscC2t
2
2
etpT0w((bhe4dp4
2
oB
Reverse CACGATG-GAGGGGAAGACGG
yruvate, insulin–transferrin–selenium (ITS premix), 10−7 M dexa-ethasone and 10 ng/ml transforming growth factor beta 1
TGF-�1; PeproTech, Rocky Hill, NJ). If not otherwise specified, alleagents were purchased from Sigma–Aldrich. CPs were cultured intandard cell culture conditions (i.e., 5/95% CO2/air, >90% humidity).ne set of CPs was daily treated (3 times for 5 s) with LFUS (at 40 kHz
requency and 20 W output power) using a sonicator bath (Bran-onic 2510; Bransonic, Danbury, CT, USA). After 3 weeks, samplesere processed for histologic procedures.
.3.4. Quantitative RT-PCRDPSCs from three independent samples were cultured in prolif-
ration or in EGM-2 medium for 15 days. Cells were then harvestedy trypsin–EDTA digestion and processed for gene expression anal-sis. Total RNA was extracted using RNeasy Mini Kit (Qiagen GmbH,ilden, Germany), as indicated by manufacturer’s protocol. DNa-
eI digestion was performed on column. One microgram of eachNA sample was retrotranscripted with QuantiTect Reverse Tran-cription Kit (Qiagen GmbH) and two-fold dilutions of cDNAs werenalyzed by quantitative RT-PCR on iCycler-iQ5 Optical SystemBioRad Laboratories, Hercules, CA), using SsoFast EvaGreen Super-
ix (BioRad). All samples were run in duplicate. Primers fromECAM1 and ACTB genes (Table 1) were designed from codingequences published on GenBank database with the help of Bea-on Designer v.7 Software (Premier Biosoft International, Palo Alto,A, USA). Relative quantitative analysis was performed following−��Ct Livak’s method (Livak and Schmittgen, 2001). Normaliza-ion was made with ACTB housekeeping gene.
.4. DPSC interaction with different biomaterial platforms
.4.1. DPSCs cultured inside human plasma clotsPlasmas for the clot preparation were obtained from periph-
ral blood of healthy adult donors. The blood was collected intoubes containing EDTA as anticoagulant and the platelet poorlasma (PPP) was obtained by centrifugation (1700 × g for 20 min).he DPSCs were resuspended in PPP at three different densities,.25 × 105, 0.5 × 105 and 1.0 × 105 cells/sample, and seeded in 6-ell plates (CoStar Group). To produce DPSC/clot constructs, CaCl2
7 mM; Dade Berhing, Marburg, Germany) was added to each wellTrombi et al., 2008; Barachini et al., 2009). After 30 min of incu-ation at 37 ◦C in 5% humidified CO2 atmosphere, constructs werearvested and cultured for 7 days in osteogenic medium or prolif-ration medium as control. After 7 days the constructs were fixed in% neutral buffered formalin and processed for histological proce-ures. As an osteoblastic marker, calcium deposition was tested onaraffin sections by means of von Kossa staining. Cell viability after8 h, was assessed using alamarBlue® (Life Technologies) assay.
.4.2. DPSCs cultured on PVA/G scaffoldsPVA/G scaffolds (weight composition ratio of 80/20) were
btained via emulsion and freeze-drying (Moscato et al., 2008).riefly, an aqueous solution of PVA (Mw = 85,000–124,000, from
67 (2014) 155–168
Sigma–Aldrich) and G (gelatin type B from bovine skin, 75bloom, from Sigma–Aldrich) was obtained at 50 ◦C under stir-ring and further added with sodium lauryl sulfate (SLS, fromSigma–Aldrich) to obtain a dense foam that was quenched in liquidnitrogen and lyophilized. Dried foams were stabilized by crosslink-ing with glutaraldehyde (GTA; grade II, from Sigma–Aldrich)vapors, cut into cylinders (5 mm diameter, 1.5 mm thickness)and subsequently treated with glycine (Sigma–Aldrich) solutionto block GTA unreacted binding sites. The resulting sponge is ahighly porous, biocompatible, hydrophilic and bio-stable material,suitable for tissue engineering applications. The scaffolds were ster-ilized with absolute ethanol, washed with sterile 2× PEN-Strep(Sigma–Aldrich)/Diflucan (Pfizer Italia, Latina, Italy) saline andfinally rinsed with phosphate buffered saline (PBS, Sigma–Aldrich)prior to cell seeding. Twice passaged DPSCs were seeded on PVA/Gscaffolds at a density of 500,000 cells/sample. After 24 h, sets ofcell/scaffold constructs were committed either to the osteogenicor to the chondrogenic lineage replacing the proliferation culturemedium with appropriate differentiative media. Differentiationswere performed for 21 days using osteogenic and chondrogenic cul-ture medium, as reported in the previous sections. Undifferentiatedcontrols of DPSC/scaffold constructs were also carried out culturingthe specimens in proliferation medium until the endpoint. Alongthe culture time, viability of the constructs was assayed, while at theendpoint samples were processed for qualitative (scanning elec-tron microscopy, histology) and quantitative (viability test, DNAquantification, glycosaminoglycan content) analyses.
2.4.3. DPSCs cultured with BNNTsBNNTs, supplied by the Australian National University, Can-
berra, Australia, were produced by a ball-milling and annealingmethod (Yu et al., 2005). Details of sample purity and compo-sition, as provided by the supplier, comprise yield > 80%, boronnitride > 97 wt%, residual metal catalysts (Fe and Cr) ∼1.5 wt% andabsorbed O2 ∼1.5 wt%. Stable aqueous dispersions of BNNTs wereobtained using 0.1% poly-l-lysine (PLL, Mw 70,000–150,000; Fluka,Buchs, Switzerland) as described in a previous study (Ciofani andDanti, 2012). Excess PLL was removed by repeated ultracentrifu-gation (Allegra 64R; Beckman Coulter, Fullerton, CA, USA). StablePLL-BNNT dispersions were thus obtained by the non-covalentcoating of the nanotube walls with PLL. The final concentration ofBNNTs in the dispersion was quantified using a UV/Vis/NIR spec-trophotometer (LIBRA S12, Biochrom, Cambridge, UK), while theresidual PLL concentration was assessed using the bicinchoninicacid (BCA) method, as previously reported (Ciofani and Danti,2012). For cytotoxicological investigations, DPSCs were seeded on96-well plates (20,000 cells/well) in proliferation medium. After24 h, the medium was replaced with a modified medium, contain-ing the following PLL-BNNT concentrations: 0, 5, 10 and 15 �g/ml.Cultures were performed for 72 h to monitor cell viability. To eval-uate cellular uptake of the nanotubes, the DPSCs were seeded onT75 flasks (800,000 cells/flask) and, after 24 h, they were incubatedovernight with medium containing PLL-BNNTs at 10 �g/ml to besubsequently treated for transmission electron microscopy (TEM;Zeiss 902 microscope, Zeiss, Oberkochen, Germany).
2.4.4. DPSC viabilityViability of DPSCs cultured with different biomaterial plat-
forms was investigated using the alamarBlue® assay. Data wereacquired according to manufacturer’s instructions, and expressedad percentage of reduced alamarBlue® (%ABred). This bioassayincorporates a REDOX indicator resulting in color change of the
culture medium according to cell metabolism and can be per-formed multiple times on the same samples due to its negligibletoxicity. Samples (n = 3 and n = 6, for cell/scaffold construct andPLL-BNNT experiments, respectively) and controls, including blank
icron
ccwrpasftwpaf
2c
elaptwpilEBavtsUwudetra
2
i,4oWodo64fiod(fp(ipSMth
S. Barachini et al. / M
ontrols (i.e., scaffolds with no cells, and empty well plates forell/scaffold construct and PLL-BNNT experiments, respectively),ere incubated for 4 h with the dye according to the manufacturer’s
ecommendations. Samples were assayed at different culture time-oints: 1, 11, 21 days to assess cell/scaffold construct viability,nd 24, 48, 72 h to assess PLL-BNNT cytotoxicity. After each assay,upernatants were removed from the cultures and replaced withresh culture medium. Samples were analyzed with a spectropho-ometer (BioRad Laboratories, Hercules, CA, USA) under a doubleavelength reading (570 nm and 600 nm). Finally, dye reductionercentage was calculated using dye molar extinction coefficientsnd appropriate absorbance equations as provided by the manu-acturer.
.4.5. Double stranded (ds)-DNA and glycosaminoglycan (GAG)ontents in PVA/G constructs
At the endpoint (21 days), un-, osteo-, and chondro-diff-rentiated cell/scaffold constructs were either fixed for morpho-ogical and histologic evaluation, or stored in enzymatic solutiont −80 ◦C for quantitative assays. Digestive enzymes consisted ofroteinase K, pepstatin A and iodoacetamide (Sigma–Aldrich) solu-ion in phosphate-buffered EDTA (Park et al., 2005). Cell lysatesere obtained using a freeze/thaw/vortex treatment of the sam-les. Contents of double ds-DNA and GAGs could be thus quantified
n cascade on the same specimens (n = 3). ds-DNA content in cellysates was measured using the PicoGreen kit (Molecular Probes,ugene, OR, USA), as previously reported (Ciofani and Danti, 2012).riefly, working buffer and PicoGreen dye solution were preparedccording to the manufacturer’s instructions using reagents pro-ided within the kit. After 10 min incubation in the dark at roomemperature, the fluorescence intensity of the samples was mea-ured on a plate reader (Victor3; PerkinElmer, Waltham, MA,SA), using an excitation wavelength of 485 nm and an emissionavelength of 535 nm. To determine GAG content, the constructsnderwent digestion in a 60 ◦C water bath for 16 h, then theimethylmethylene blue dye (DMMB) assay was performed (Parkt al., 2005). Absorbance was measured at 520 nm on a plate spec-rophotometer (BioRad Laboratories). GAG contents were finallyeported per microgram of ds-DNA, as obtained from the PicoGreenssay.
.4.6. Histologic analysisThree-dimensional cultures of DPSCs (i.e., osteo-differentiated
n plasma clots, chondro-differentiated in pellets, and un-, osteo- and chondro-differentiated on PVA/G scaffolds) were fixed in% neutral buffered formalin diluted in 1× PBS (0.1 M, pH 7.2)vernight at 4 ◦C, then washed in 1× PBS and stored in 70% ethanol.hole chondrogenic pellets, osteogenic plasma clots and a parts
f cell/scaffold constructs were processed for histologic proce-ures. Briefly, the samples were dehydrated with a graded seriesf ethanol aqueous solutions up to absolute ethanol (the latter for
h), clarified twice in xylene for 2 h, rinsed in liquid paraffin for h at 60 ◦C and finally paraffin-embedded. Sections were deparaf-ned in xylene and rehydrated in ethanol before being stainedr immunostained. Prior to observation, the sections were rehy-rated in ethanol, clarified in xylene and mounted in DPX mediumFluka, Buchs, Switzerland). Toluidine blue staining reveals sul-ated GAGs in violet color and was performed on chondrogenicellets. Sections were air dried and treated with 0.2% Toluidine blueSigma–Aldrich) for 10 s, washed and air dried. Alcian Blue stain-ng detects generic and sulfated GAGs in cyan color (at pH 2.5 andH 1, respectively) and was performed on chondrogenic pellets.
ections were incubated in Alcian Blue solutions kit (Bio-Optica,ilan, Italy) according to manufacturer’s instructions and coun-erstained with 0.1% nuclear fast red (Fluka). Von Kossa stainingighlights carbonate and phosphate crystals and was performed on
67 (2014) 155–168 159
DPSCs osteo-induced on 6-well plates and on fibrin clots. Sectionswere treated with 1% silver nitrate (Fluka) for 15 min at artificiallight, soaked in 0.5% pyrogallol (Fluka) for 2 min treated with 5%sodium thiosulfate (Sigma–Aldrich) for 2 min and counterstainedwith nuclear fast red.
Immunohistochemical analysis was carried out on CPs andcell/scaffold constructs. Samples were permeabilized with 0.2%Triton X-100 (Sigma–Aldrich) in 1× PBS for 10 min. Peroxidasisquenching was performed incubating the samples in a 0.6% H2O2methanol solution for 15 min in the dark. To block the aspecificbinding sites, the samples were incubated with goat serum (VectorLab, Burlingame, CA, USA) diluted 1:20 in 1× PBS at 37 ◦C for 20 min.After washing, the specimens were incubated in a moisted cham-ber overnight at 4 ◦C with mouse monoclonal anti-osteopontin(OPN) diluted 1:2000 (sc-21742, Santa Cruz Biotechnology, SantaCruz, CA, USA) and mouse monoclonal anti-Aggrecan diluted 1:50(sc-33695, Santa Cruz Biotechnology). Negative controls were per-formed incubating some sections with 0.1% BSA/1× PBS only. After24 h, the samples were incubated with goat anti-mouse biotiny-lated secondary antibodies (Vector Lab) diluted 1:200 in 1.5% goator horse serum–1× PBS solution for 60 min and then with strepta-vidin (Vectastain Elite ABC Kit Standard, Vector Lab) for 30 min.The reactions were revealed incubating the specimens in thesubstrate-chromogen solution (0.5 mg/ml, 3,3-diaminobenzidinetetrahydrochloride containing 0.02% H2O2; Amresco, Solon, OH,USA) for 5 min in the dark, counterstained with hematoxylin andobserved by a microscope (Leica Microsystems, Wetzlar, Germany).After each passage, washing in 0.01% Triton/1× PBS was performed.
2.4.7. Scanning electron microscopy (SEM) analysisAfter fixation (4% neutral buffered formalin in PBS, overnight at
4 ◦C), cell/scaffold constructs were dehydrated in a graded series ofethanol aqueous solutions up to anhydrous ethanol, dried using thecritical point method (Balzers CPD030, Oerlikon Balzers, Balzers,Liechtenstein), cross-sectioned, mounted on aluminum stumps,sputter-coated with gold (Sputter Coater Emitech K550, QuorumTechnologies Ltd., West Sussex, UK), and examined with a scanningelectron microscope (JEOL JSM-5200, JEOL Ltd., Tokyo, Japan).
2.4.8. Transmission electron microscopy (TEM) analysisTEM analysis was performed on DPSC specimens incubated
overnight with culture medium containing PLL-BNNTs at 10 �g/ml.After removal of the culture medium, cells were trypsinized,centrifuged and fixed in a solution constituted of 0.5%/4% (w/v)GTA/formaldehyde in PBS 0.1 M at pH 7.2 for 2 h at 4 ◦C. Afterwashing, the samples were post-fixed in 1% w/v OsO4 PBS 0.1 Mat pH 7.2 for 1 h, washed and dehydrated with acidified aceton-dimethylacetal (Fluka). Finally, the samples were embedded inEpon/Durcupan resin in BEEM capsules #00 (Structure Probe, WestChester, PA, USA) at 56 ◦C for 48 h. Ultra-thin sections (20–30 nmthick) were obtained with an ultramicrotome (Ultratome Nova,LKB, Bromma, Sweden) equipped with a diamond knife (Diatome,Biel/Bienne, Switzerland). The sections were placed on 200 squaremesh nickel grids, counterstained with saturated aqueous uranylacetate and lead citrate solutions and observed with a transmissionelectron microscope (Zeiss 902, Carl Zeiss, Oberkochen, Germany).Drops of diluted PLL-BNNT aqueous solution were poured on cop-per grids and observed via TEM.
2.4.9. Statistical analysisStatistical significance in quantitative analyses was evaluated
using the two-tailed t test for paired (alamarBlue® assay) orunpaired (RT-PCR, ds-DNA and GAG) data, followed by Bonferroni’scorrection. Data underwent both descriptive, i.e., mean ± standarddeviation (SD), and inferential statistics (p values).

1 icron
3
3
hdcrmtWDfiobopeeeCCttav
3
i
3
isfgca(ctsts(c
3
psm(a
3
ffFiEp
60 S. Barachini et al. / M
. Results
.1. Isolation and characterization of DPSCs
Dental pulp tissues were collected from 10 clinically extracteduman teeth. In this study, we observed that culturing humanental pulp tissue in plastic dishes led to proliferating adherentolonies of fibroblast-like cells that presented a durable prolife-ative capability in culture lasting for 4 months without evidentorphological changes. DPSCs are able to form colonies, similar
o the colonies formed by BM-MSCs, in 10–14 days of culture.hen colonies were detached and replated at defined cell density,
PSCs formed a homogenous monolayer of cells which keep thebroblast-like morphology (Fig. 1A). After studies of 10 passages,ver a period of 4 months, cell cultures reached a doublings num-er of approximately 25 PDs and showed an average doubling timef 115 h. DPSCs were characterized via flow cytometry for surfacerotein expression. We analyzed the phenotypes of all populations,ach of which corresponds to a dental pulp donor, cultivated andxpanded at different passages. As shown in Fig. 1B, almost all cellsxpressed typical mesenchymal stromal cell antigens such as CD90,D105, CD73, CD146, STRO-1, but they lacked the CD45, CD34,D31 and SSEA-4 antigens. ELISA assay were performed on cul-ure media to detect the amounts of NGF and BDNF secreted byhe DPSCs. The analyses confirmed that the expression of BDNFnd NGF was detectable in three different samples of DPSCs withalues ranging from 3 to 4 pg/cell (Fig. 1C).
.2. Differentiation of DPSCs
The DPSCs were investigated throughout a functional character-zation to assess their multipotency.
.2.1. Endothelial differentiationEndothelial differentiation was performed by culturing DPSCs
n the presence of EGM-2 for 15 days. Immunophenotype analy-is showed more than 70% of differentiated DPSCs to be positiveor CD309, and 40% to be positive for CD31 (Fig. 2A). Same anti-ens were not expressed by cells cultured under proliferatingonditions. The Ac-LDL uptake assay showed that the differenti-ted DPSCs were able to internalize Ac-LDL into their cytoplasmgreen in Fig. 2B) similarly to human umbilical vein endothelialells (HUVECs, data not shown) while undifferentiated cells failedo uptake Ac-LDL (Fig. 2C). Differentiated DPSCs formed a tube-liketructure within 12 h incubation on Matrigel (Fig. 2D), in contrasto the undifferentiated cells that were unable to form capillary-liketructures (data not shown). Gene expression analysis of PECAM1CD31) mRNAs corroborated the immunophenotypic results, thusonfirming the attainment of endothelial differentiation (Fig. 2E).
.2.2. Osteogenic differentiationThe DPSCs were induced to differentiate toward the osteoblast
henotype adding the culture medium with standard osteogenicupplements. After 21 days of culture, the deposition of calciumatrix nodules was revealed by both von Kossa and Alizarin S stain
Fig. 3A and B). Conversely, undifferentiated DPSCs did not showny positive stain for calcium deposition (Fig. 3C and D).
.2.3. Chondrogenic differentiationCPs cultured in standard conditions and CPs treated with low
requency US (namely, CPs-US) underwent histologic evaluationor cartilaginous molecule expression (i.e., GAGs and aggrecan;
ig. 4A and H). CPs-US showed higher compactness and homogene-ty than those of untreated CPs. Specifically, in CPs-US, cartilaginousCM molecules were uniformly distributed in the samples andresent to a higher extent than in CPs. Differently, in untreated67 (2014) 155–168
CPs, chondrogenic markers were often detected with zonal distri-bution. However, the investigated molecules were expressed witha similar intensity in CPs and CPs-US. In particular, generic GAGswere expressed with high intensity (+++) and uniform distribu-tion in all the samples, as shown by Alcian Blue staining at pH 2.5(Fig. 4E and F). Sulfated GAGs, typical of mature cartilage, wererevealed with strong intensity (+++) both in the CPs and CPs-US,as demonstrated by both Toluidine blue (metachromatic reaction)and Alcian Blue staining at pH 1. In the CPs-US, the positivity wasuniformly located (Fig. 4B and D), while in the CPs some areas werenegative (Fig. 4A and C). Finally, as displayed by IHC, the aggre-can marker was weakly (+) and uniformly expressed in both CPsand CPs-US samples, but in the latter with an enhanced proteinsynthesis (Fig. 4G and H).
3.3. Cell culture with biomaterial platforms
3.3.1. Human plasma clotsSignificantly reduced cell viability was detected on clots after
osteogenic induction (27.5 ± 5.5%, p < 0.05), in comparison withclots cultured in proliferation medium (40.9 ± 8.7%). Osteogenicdifferentiation was demonstrated by enhancement of mineralizedmatrix deposition, clearly evidenced through the presence of blackgranules (Fig. 3E), with respect to undifferentiated controls (Fig. 3F).The clot with the highest cell density (100,000 cells) demonstratedthe most intense mineralization.
3.3.2. PVA/G scaffoldsAs detected by alamarBlue assay, DPSCs cultured on PVA/G
scaffolds under diverse differentiative regimens were viable upto 21 culture days (Fig. 5A). Data of dye reduction percentagealong the culture times (1, 11, 21 days), as calculated following themanufacturer’s formula were: 50.12 ± 7.79%, 51.95 ± 13.99%,43.12 ± 10.11% for undifferentiated DPSCs; 50.49 ± 9.07%,40.05 ± 4.97%, 46.43 ± 5.13% for osteo-differentiated DPSCs;and 40.37 ± 9.40%, 25.61 ± 8.04%, 30.69 ± 5.62% for chondro-differentiated DPSCs. Between day 1 and day 21, no statisticallysignificant differences in cell viability were detected withinundifferentiated and osteo-differentiated groups (p > 0.05), whilechondro-differentiated samples showed a statistically signifi-cant decrease (p = 0.0002). Viability of chondro-induced samplesshowed statistically significant reductions compared to bothundifferentiated (p < 0.0001 on day 11 and p < 0.001 on day 21) andosteo-differentiated samples (p < 0.001 on day 11 and p < 0.0001 onday 21). At the endpoint, ds-DNA and GAG contents were detectedwith quantitative assays (Fig. 5B and C). Results of ds-DNAcontents were 0.31 ± 0.12, 0.26 ± 0.13 and 0.26 ± 0.64 �g/samplefor un-, osteo- and chondro-differentiated constructs, respectively.No statistically significant difference was highlighted (p > 0.05)(Fig. 5B). Findings on GAGs showed a similar trend, showingcontents of 3.35 ± 0.38 �g/sample, 3.27 ± 0.34 �g/sample and3.71 ± 0.56 �g/sample for un-, osteo- and chondro-differentiatedconstructs, respectively. On average, the highest GAG amountwas found in the chondro-induced samples; however, in any case,statistically significant differences were not detected (p > 0.05)(Fig. 5C). SEM analysis highlighted DPSCs being able to penetratein the pore network and deeply colonize the PVA scaffold in allthe types of constructs (Fig. 6A and C). Cells were well stretchedout with elongated morphology in un- and osteo-differentiatedsamples, while cells showed mildly spindle and round morphol-ogy, typical of the mature chondrocytes, in chondro-differentiatedconstructs (Fig. 6C). In un- and osteo-differentiated specimens,
immunohistochemistry revealed a widespread presence, at bothintra- and extracellular level, of the osteogenic marker OPN,with higher intensity (+++) in the osteo-differentiated than in theun-differentiated constructs (+) (Fig. 6D and E). In a similar fashion,
S. Barachini et al. / Micron 67 (2014) 155–168 161
Fig. 1. Morphological, immunological and functional characterization of DPSCs. (A) DPSCs formed a homogenous monolayer of cells that revealed typical spindle-shapedmorphology. (B) Flow cytometrical analysis of DPSC showed typical immunophenotype characterized by the expression of CD90, CD105, CD73, CD146, and STRO-1. (C) ELISAassay confirmed that DPSCs of three different samples were able to secrete BDNF and NGF neurotrophins.

162 S. Barachini et al. / Micron 67 (2014) 155–168
Fig. 2. Differentiation of DPSCs toward endothelial lineage. (A) After induction of endothelial differentiation DPSCs acquired positive stain for anti-CD309 and anti-CD31a en in Ba s foldD is ref
tmi(
3
5wpPif
ntibodies. (B) Moreover, differentiated DPSCs were able to internalize Ac-LDL (grefter 12 h incubation on Matrigel (arrows). (E) Levels of PECAM1 mRNA, reported aPSCs. (For interpretation of the references to color in this figure legend, the reader
he chondro-differentiated constructs displayed the chondrogenicarker aggrecan at intracellular level with a higher immunopos-
tivity (++) than that detected in the un-differentiated constructs−/+) (Fig. 6F and G).
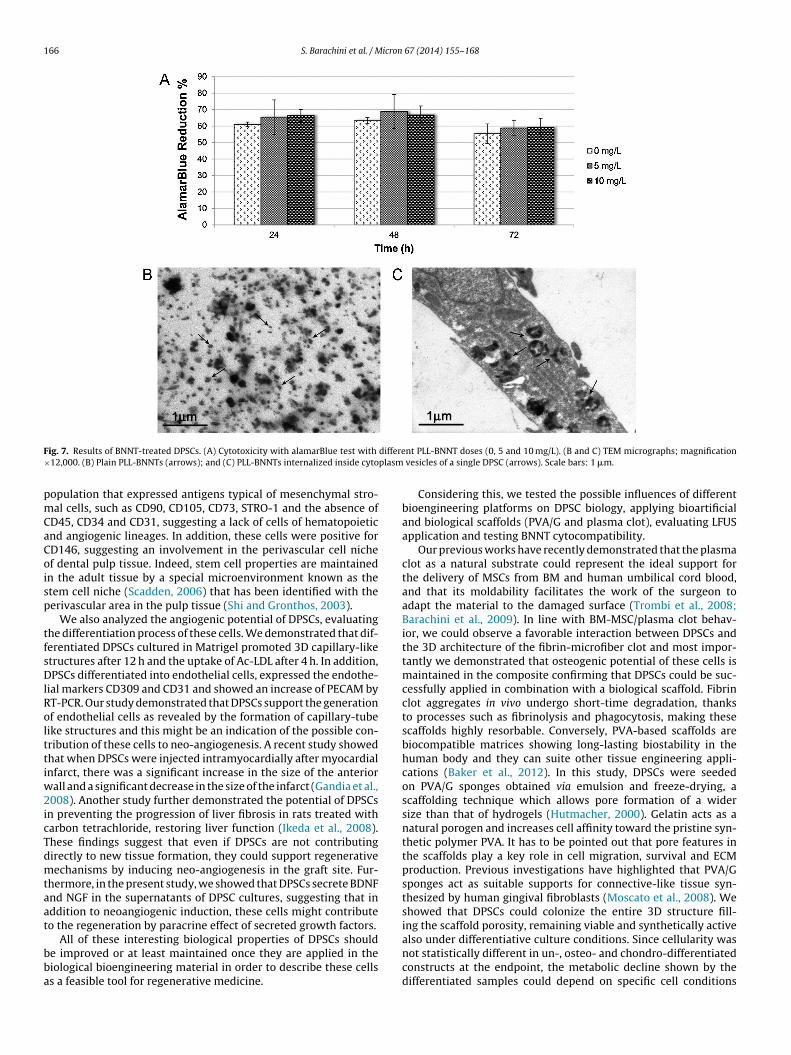
.3.3. BNNTsPLL-coated BNNTs were administrated with different doses (0,
and 10 �g/ml) to cultures of DPSCs up to 72 h, and cell viabilityas monitored using the alamarBlue test (Fig. 7A). Dye reduction
ercentage ranged between 55.36 and 63.31% in control (0 �g/mlLL-BNNTs), 58.83 and 68.87% in 5 �g/ml, and 59.21 and 66.78%n 10 �g/ml samples. Within each dose-group, and between dif-erent dose-groups at same times, cell viability did not show any) respect to (C) untreated cells, as well as (D) to form capillary tube-like structures expression change, of differentiated DPSCs resulted higher than undifferentiatederred to the web version of this article.)
statistically significant difference up to the endpoint (p > 0.05).Finally, PLL-BNNTs were observed via TEM both before and afteradministration in the culture medium (Fig. 7B and C), resultingin strongly contrasted nano-objects, which were detected as clus-ters in cytoplasm vesicles of DPSCs (Fig. 7C). The presence of suchnano-objects at nuclear level was never highlighted.
4. Discussion
Stem cell biology has emerged as a key pillar for regenerativemedicine. In order to effectively regenerate tissues via engineeredtechnologies, understanding the biology of stem cells has becomean essential basis for driving and controlling the regeneration

S. Barachini et al. / Micron 67 (2014) 155–168 163
F able
d d celld E), wh
ptSettab
satttoudta
ig. 3. Osteogenic differentiation of DPSCs. (A) Under proper stimuli, DPSCs wereetected by von Kossa staining, or (B) Alizarin S. (C and D) Conversely, untreateemonstrated in plasma clot scaffolds by the presence of black granules (arrows in
rocesses. The simple question is what are the properties of cellshat make them potentially ideal candidates for tissue engineering.erakinci and Keith (2006) indicated some of the desirable prop-rties that stem cells for transplantation should ideally possess: (i)o be easily isolated and purified, (ii) to maintain their multipoten-ial lineage capacity, (iii) to show directed differentiation, (iv) to beutologous to the patient, (v) to allow genetic modification, (vi) toe highly expanded in culture, and (vii) to be non-tumorigenic.
Nonetheless, in most cases applying multipotent cells to aurgical setting is not possible in the form of cell suspension,nd requires the use of a scaffold avoiding cell dispersion inhe surgical site. Tissue engineering is an interdisciplinary fieldhat applies the principles of engineering and life sciences tohe development of three-dimensional scaffolds able to sustainr improve the function of an applied regenerative cell pop-
lation. The scaffold’s mechanical and biochemical propertiesetermine the efficiency and avidity in which the cells can formhe composite. The ideal biomaterial should be biocompatiblend bioresorbable to support the replacement of normal tissueto differentiate toward osteogenic lineage forming mineralized nodules (arrows),s did not reveal any matrix deposition. Osteogenic potential of DPSCs were alsoile in non-osteoinductive condition (F) mineralized nodules were not detected.
without inflammation. Incompatible materials are destined for aninflammatory or foreign-body response that eventually leads torejection and/or necrosis. In addition, the degradation products,if produced, should be removed from the body at an adequaterate via metabolic pathways to keep the concentration of thesedegradation products in the tissues at a tolerable level. Further-more, the scaffold should provide an environment in which theappropriate regulation of cell behavior (e.g., adhesion, proliferation,migration, and differentiation) can occur to allow the formationof a functional tissue. Depending on their origin, biomaterials canbe divided into three classes: (i) naturally derived (e.g. plasmaclot), (ii) completely synthetic, and (iii) biomimetic materials (e.g.PVA/G). Naturally derived materials have the potential advantageof biologic recognition while synthetic polymers can be repro-duced on a large scale with controlled properties of strength,
degradation rate, and microstructure. Bioartificial materials canbe produced from blends of biologic and synthetic polymers andhave been developed to account for the advantages of both typesof biomaterials.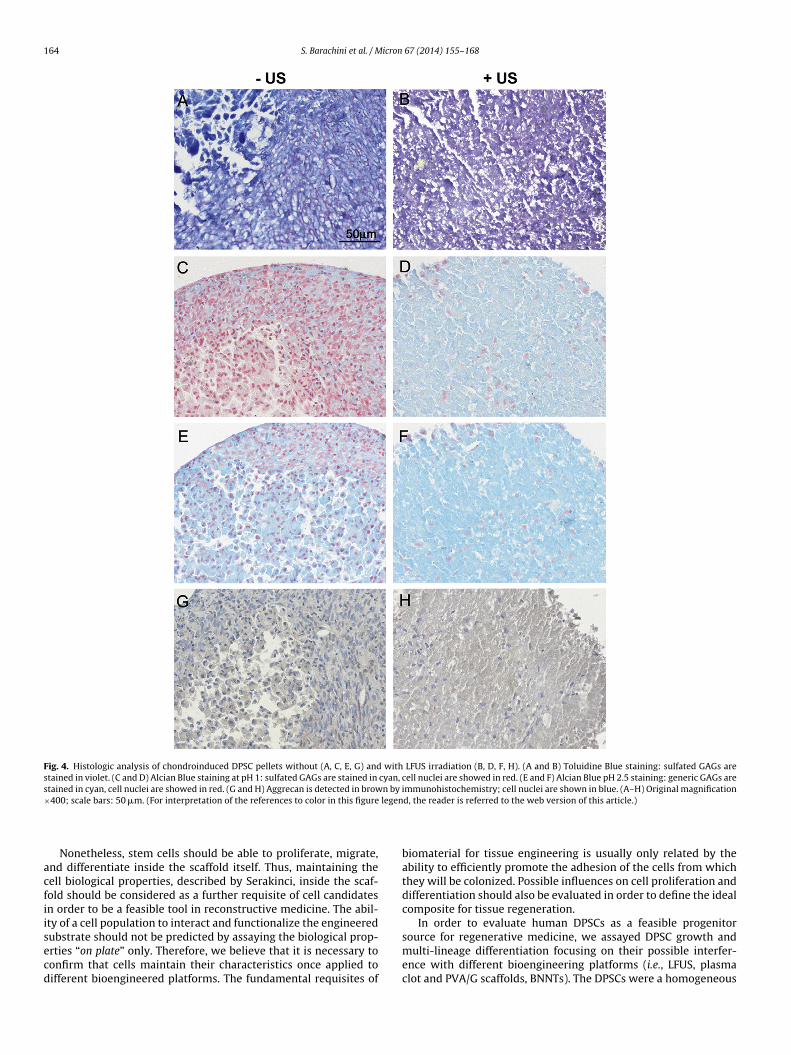
164 S. Barachini et al. / Micron 67 (2014) 155–168
Fig. 4. Histologic analysis of chondroinduced DPSC pellets without (A, C, E, G) and with LFUS irradiation (B, D, F, H). (A and B) Toluidine Blue staining: sulfated GAGs arestained in violet. (C and D) Alcian Blue staining at pH 1: sulfated GAGs are stained in cyan, cell nuclei are showed in red. (E and F) Alcian Blue pH 2.5 staining: generic GAGs arestained in cyan, cell nuclei are showed in red. (G and H) Aggrecan is detected in brown by immunohistochemistry; cell nuclei are shown in blue. (A–H) Original magnification× legen
acfiisecd
400; scale bars: 50 �m. (For interpretation of the references to color in this figure
Nonetheless, stem cells should be able to proliferate, migrate,nd differentiate inside the scaffold itself. Thus, maintaining theell biological properties, described by Serakinci, inside the scaf-old should be considered as a further requisite of cell candidatesn order to be a feasible tool in reconstructive medicine. The abil-ty of a cell population to interact and functionalize the engineered
ubstrate should not be predicted by assaying the biological prop-rties “on plate” only. Therefore, we believe that it is necessary toonfirm that cells maintain their characteristics once applied toifferent bioengineered platforms. The fundamental requisites ofd, the reader is referred to the web version of this article.)
biomaterial for tissue engineering is usually only related by theability to efficiently promote the adhesion of the cells from whichthey will be colonized. Possible influences on cell proliferation anddifferentiation should also be evaluated in order to define the idealcomposite for tissue regeneration.
In order to evaluate human DPSCs as a feasible progenitor
source for regenerative medicine, we assayed DPSC growth andmulti-lineage differentiation focusing on their possible interfer-ence with different bioengineering platforms (i.e., LFUS, plasmaclot and PVA/G scaffolds, BNNTs). The DPSCs were a homogeneous
S. Barachini et al. / Micron 67 (2014) 155–168 165
Fig. 5. Quantitative analyses on DPSC/PVA constructs. (A) Cell viability with alamarBlue test at different culture times. (B) Construct cellularity (ds-DNA content per sample)at the endpoint (21 days). (C) GAG content at the endpoint (21 days). Asterisks indicate different statistical significance of comparisons: *p < 0.01, **p < 0.001, ***p < 0.00001.
Fig. 6. SEM and immunohistochemical analysis of DPSC/PVA constructs. SEM analysis of DPSC/PVA constructs cultured (A) in standard medium, (B) in osteogenic medium(arrows: osteo-differentiated cells), (C) in chondrogenic medium (arrows: mature chondrocytes). Immunohistochemical analysis of the osteogenic and chondrogenic markers,performed on the DPSC/PVA constructs. OPN and aggrecan are displayed in brown (arrows), cell nuclei are showed in blue. OPN on the construct cultured (D) in standardmedium and (E) in osteogenic medium. Aggrecan on the construct cultured (F) in standard medium and (G) in chondrogenic medium. SC = scaffold. Original magnification×400; scale bars: 50 �m.
166 S. Barachini et al. / Micron 67 (2014) 155–168
F differe× plasm
pmCaCoisp
tfsDlRolttiw2icTdmtaat
bba
ig. 7. Results of BNNT-treated DPSCs. (A) Cytotoxicity with alamarBlue test with
12,000. (B) Plain PLL-BNNTs (arrows); and (C) PLL-BNNTs internalized inside cyto
opulation that expressed antigens typical of mesenchymal stro-al cells, such as CD90, CD105, CD73, STRO-1 and the absence of
D45, CD34 and CD31, suggesting a lack of cells of hematopoieticnd angiogenic lineages. In addition, these cells were positive forD146, suggesting an involvement in the perivascular cell nichef dental pulp tissue. Indeed, stem cell properties are maintainedn the adult tissue by a special microenvironment known as thetem cell niche (Scadden, 2006) that has been identified with theerivascular area in the pulp tissue (Shi and Gronthos, 2003).
We also analyzed the angiogenic potential of DPSCs, evaluatinghe differentiation process of these cells. We demonstrated that dif-erentiated DPSCs cultured in Matrigel promoted 3D capillary-liketructures after 12 h and the uptake of Ac-LDL after 4 h. In addition,PSCs differentiated into endothelial cells, expressed the endothe-
ial markers CD309 and CD31 and showed an increase of PECAM byT-PCR. Our study demonstrated that DPSCs support the generationf endothelial cells as revealed by the formation of capillary-tubeike structures and this might be an indication of the possible con-ribution of these cells to neo-angiogenesis. A recent study showedhat when DPSCs were injected intramyocardially after myocardialnfarct, there was a significant increase in the size of the anterior
all and a significant decrease in the size of the infarct (Gandia et al.,008). Another study further demonstrated the potential of DPSCs
n preventing the progression of liver fibrosis in rats treated witharbon tetrachloride, restoring liver function (Ikeda et al., 2008).hese findings suggest that even if DPSCs are not contributingirectly to new tissue formation, they could support regenerativeechanisms by inducing neo-angiogenesis in the graft site. Fur-
hermore, in the present study, we showed that DPSCs secrete BDNFnd NGF in the supernatants of DPSC cultures, suggesting that inddition to neoangiogenic induction, these cells might contributeo the regeneration by paracrine effect of secreted growth factors.
All of these interesting biological properties of DPSCs shoulde improved or at least maintained once they are applied in theiological bioengineering material in order to describe these cellss a feasible tool for regenerative medicine.
nt PLL-BNNT doses (0, 5 and 10 mg/L). (B and C) TEM micrographs; magnification vesicles of a single DPSC (arrows). Scale bars: 1 �m.
Considering this, we tested the possible influences of differentbioengineering platforms on DPSC biology, applying bioartificialand biological scaffolds (PVA/G and plasma clot), evaluating LFUSapplication and testing BNNT cytocompatibility.
Our previous works have recently demonstrated that the plasmaclot as a natural substrate could represent the ideal support forthe delivery of MSCs from BM and human umbilical cord blood,and that its moldability facilitates the work of the surgeon toadapt the material to the damaged surface (Trombi et al., 2008;Barachini et al., 2009). In line with BM-MSC/plasma clot behav-ior, we could observe a favorable interaction between DPSCs andthe 3D architecture of the fibrin-microfiber clot and most impor-tantly we demonstrated that osteogenic potential of these cells ismaintained in the composite confirming that DPSCs could be suc-cessfully applied in combination with a biological scaffold. Fibrinclot aggregates in vivo undergo short-time degradation, thanksto processes such as fibrinolysis and phagocytosis, making thesescaffolds highly resorbable. Conversely, PVA-based scaffolds arebiocompatible matrices showing long-lasting biostability in thehuman body and they can suite other tissue engineering appli-cations (Baker et al., 2012). In this study, DPSCs were seededon PVA/G sponges obtained via emulsion and freeze-drying, ascaffolding technique which allows pore formation of a widersize than that of hydrogels (Hutmacher, 2000). Gelatin acts as anatural porogen and increases cell affinity toward the pristine syn-thetic polymer PVA. It has to be pointed out that pore features inthe scaffolds play a key role in cell migration, survival and ECMproduction. Previous investigations have highlighted that PVA/Gsponges act as suitable supports for connective-like tissue syn-thesized by human gingival fibroblasts (Moscato et al., 2008). Weshowed that DPSCs could colonize the entire 3D structure fill-ing the scaffold porosity, remaining viable and synthetically active
also under differentiative culture conditions. Since cellularity wasnot statistically different in un-, osteo- and chondro-differentiatedconstructs at the endpoint, the metabolic decline shown by thedifferentiated samples could depend on specific cell conditions
icron
itoatspict2
dicidca2cmbmet
nsndmsptai
tadtvuaactptatplcdt
ewhdifbt
S. Barachini et al. / M
nduced by differentiation. Compared to un-differentiated con-rols, we observed enhanced expression only of early stage boner cartilage markers, such as OPN in the osteo-differentiated andggrecan in the chondro-differentiated samples. This indicated thathese specific substrates were somehow able to maintain the DPSCtemness delaying their differentiation timeline. These data sup-ort our hypothesis that some cell biological properties could be
nfluenced by the different scaffolds. Nonetheless these influencesould represent an appealing feature where a spatio-temporal con-rol of stem cell differentiation is desirable (Kinney and McDevitt,013).
Similarly to different materials, other bioengineering proce-ures could affect the biological properties of the composites,
.e. the application of US at low frequency during their in vitroulture under chondrogenic commitment. In our findings, a min-mal DPSC irradiation with LFUS resulted in a spatially uniformistribution and enhanced synthesis of GAGs and aggrecan, thusonfirming the favorable effects reported using other US sourcesnd mesenchymal stromal cell types (Lee et al., 2006; Shah et al.,013). Indeed, while cartilagineous pellets obtained by standardulture methods displayed non-homogenous cell morphology andarker distribution, CPs-US were histologically homogenous. US-
ased therapies are routinely applied in medical practice andight disclose new approaches also in the translational regen-
rative medicine of cartilage defects if combined with stem cellransplants.
Moreover, we tested DPSCs with BNNTs, a novel class ofanomaterials that has been receiving growing attention by thecientific community (Golberg et al., 2010). Similarly to carbonanotubes, BNNTs can be employed as intracellular vectors forrug and plasmid delivery, thus playing a key role in regenerativeedicine. However, owing to their chemical inertia, BNNTs have
hown a superior biocompatibility and, owing to their physicalroperties, they can also act as nanotransducers for cell stimula-ion therapies (Ciofani et al., 2013). Our assays showed that DPSCsre able to uptake BNNTs by endocytosis without suffering frommportant cytotoxical affects.
In recent years, human DPSCs have received growing atten-ion due to their characteristics in common with other MSCs, inddition to the ease of their harvesting. Teeth can be recovereduring routine dental procedures at any time during a lifespan. Ashird molar teeth are removed during standard prophylactic inter-entions and do not require additional surgical procedures, thetilization of such tissues for stem cell isolation offers an attractivelternative. In addition, owing to their immaturity, DPSCs virtu-lly possess privileged immunoregulatory properties and could beonsidered as good substitutes of BM-MSCs for many therapeu-ical uses. Moreover, in contrast to BM aspirates, human dentalulp offers several advantages: ease of harvesting, absence of riskso donors and absence of ethical problems. BM-derived MSCs aremong the most promising adult stromal cell types for regenera-ive medicine. The advantages of DPSCs include not only their highroliferation capability, but also the painless nature of stem cell col-
ection with minimal invasion. In fact, the third molars are the mostommon source of dental stromal cells, because extraction of wis-om teeth is widely performed and the teeth are usually consideredo be medical waste.
In conclusion, DPSCs appear as a feasible cellular tool to bemployed in regenerative medicine. Indeed, DPSCs are obtainableith non-invasive procedures, are easy to isolate and preserve aigh replication capability. Moreover, DPSCs show multi-lineageifferentiation, including the endothelial lineage. Finally and most
mportantly, DPSCs were highly versatile in interacting with dif-erent bioengineering strategies, making the specific combinationetween cells and a biomedical platform exploitable to target dis-inct regenerative strategies.
67 (2014) 155–168 167
Acknowledgement
The authors greatly thank Dr. Gianni Ciofani (Italian Institute ofTechnology Center for Micro-BioRobotics, Pontedera, Pisa, Italy) forproviding the PLL-BNNT samples used in this study.
References
Almushayt, A., Narayanan, K., Zaki, A.E., George, A., 2006. Dentin matrix protein 1induces cytodifferentiation of dental pulp stem cells into odontoblasts. GeneTher. 13, 611–620.
Arthur, A., Rychkov, G., Shi, S., Koblar, S.A., Gronthos, S., 2008. Adult human dentalpulp stem cells differentiate toward functionally active neurons under appro-priate environmental cues. Stem Cells 26, 1787–1795.
Alves, M.H., Jensen, B.E., Smith, A.A., Zelikin, A.N., 2011. Poly(vinyl alcohol) phys-ical hydrogels: new vista on a long serving biomaterial. Macromol. Biosci. 11,1293–1313.
Baker, M.I., Walsh, S.P., Schwartz, Z., Boyan, B.D., 2012. A review of polyvinyl alcoholand its uses in cartilage and orthopedic applications. J. Biomed. Mater. Res. B:Appl. Biomater. 100 (2), 1451–1457.
Barachini, S., Trombi, L., Danti, S., D’Alessandro, D., Battolla, B., Legitimo, A., Nesti,C., Mucci, I., D’Acunto, M., Cascone, M.G., Lazzeri, L., Mattii, L., Consolini, R.,Petrini, M., 2009. Morpho-functional characterization of human mesenchymalstem cells from umbilical cord blood for potential uses in regenerative medicine.Stem Cells Dev. 18, 293–305.
Caplan, A.I., 2007. Adult mesenchymal stem cells for tissue engineering versus regen-erative medicine. J. Cell. Physiol. 213, 341–347.
Caplan, A.I., Dennis, J.E., 2006. Mesenchymal stem cells as trophic mediators. J. Cell.Biochem. 98, 1076–1084.
Cascone, M.G., Lazzeri, L., Sparvoli, E., Scatena, M., Serino, L.P., Danti, S., 2004. Mor-phological evaluation of bioartificial hydrogels as potential tissue engineeringscaffolds. J. Mater. Sci. Mater. Med. 15, 1309–1313.
Chang, H.Y., Chi, J.T., Dudoit, S., Bondre, C., van de Rijn, M., Botstein, D., Brown, P.O.,2002. Diversity, topographic differentiation, and positional memory in humanfibroblasts. Proc. Natl. Acad. Sci. U.S.A. 99, 12877–12882.
Ciofani, G., Danti, S., 2012. Evaluation of cytocompatibility and cell response to boronnitride nanotubes. Methods Mol. Biol. 811, 193–206.
Ciofani, G., Danti, S., Genchi, G.G., Mazzolai, B., Mattoli, V., 2013. Boron nitridenanotubes: biocompatibility and potential spill-over in nanomedicine. Small 9,1672–1685.
Cook, S.D., Salkeld, S.L., Popich-Patron, L.S., Ryaby, J.P., Jones, D.G., Barrack, R.L., 2001.Improved cartilage repair after treatment with low-intensity pulsed ultrasound.Clin. Orthop. Relat. Res. 391S, S231–S243.
Cordeiro, M.M., Dong, Z., Kaneko, T., Zhang, Z., Miyazawa, M., Shi, S., et al., 2008.Dental pulp tissue engineering with stem cells from exfoliated deciduous teeth.J. Endod. 34, 962–969.
Couly, G., Creuzet, S., Bennaceur, S., Vincent, C., Le Douarin, N.M., 2002. Interactionsbetween Hox-negative cephalic neural crest cells and the foregut endodermin patterning the facial skeleton in the vertebrate head. Development 129,1061–1073.
D’Antò, V., Cantile, M., D’Armiento, M., Schiavo, G., Spagnuolo, G., Terracciano, L.,Secchione, R., Cillo, C., 2006. The HOX genes are expressed, in vivo, in humantooth germs: in vitro cAMP exposure of dental pulp cells results in paral-lel HOX network activation and neuronal differentiation. J. Cell. Biochem. 97,836–848.
D’Aquino, R., Graziano, A., Sampaolesi, M., Laino, G., Pirozzi, G., De Rosa, A., Papaccio,G., 2007. Human postnatal dental pulp cells co-differentiate into osteoblasts andendotheliocytes: a pivotal synergy leading to adult bone tissue formation. CellDeath Differ. 14, 1162–1171.
Dubruel, P., Unger, R., Vlierberghe, S.V., Cnudde, V., Jacobs, P.J., Schacht, E., Kirk-patrick, C.J., 2007. Porous gelatin hydrogels: 2. In vitro cell interaction study.Biomacromolecules 8, 338–344.
Estrela, C., Alencar, A.H., Kitten, G.T., Vencio, E.F., Gava, E., 2011. Mesenchymal stemcells in the dental tissues: perspectives for tissue regeneration. Braz. Dent. J. 22,91–98.
Gandia, C., Arminan, A., Garcia-Verdugo, J.M., Lledo, A., Ruiz, E., Minana, M.D., et al.,2008. Human dental pulp stem cells improve left ventricular function, induceangiogenesis, and reduce infarct size in rats with acute myocardial infarction.Stem Cells 26, 638–645.
Golberg, D., Bando, Y., Huang, Y., Terao, T., Mitome, M., Tang, C., Zhi, C., 2010. Boronnitride nanotubes and nanosheets. ACS Nano 4, 2979–2993.
Gronthos, S., Mankani, M., Brahim, J., Robey, P.G., Shi, S., 2000. Postnatal humandental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A.97, 13625–13630.
Gronthos, S., Brahim, J., Li, W., Fisher, L.W., Cherman, N., Boyde, A., et al., 2002. Stemcell properties of human dental pulp stem cells. J. Dent. Res. 8, 531–535.
Huang, G.T., Gronthos, S., Shi, S., 2009. Mesenchymal stem cells derived from dentaltissues vs from other sources: the biology and role in regenerative medicine. J.Dent. Res. 88, 792–806.
Hutmacher, D.W., 2000. Scaffolds in tissue engineering bone and cartilage. Bioma-terials 21, 2529–2543.
Ikeda, E., Yagi, K., Kojima, M., Yagyuu, T., Ohshima, A., Sobajima, S., et al., 2008.Multipotent cells from the human third molar: feasibility of cell-based therapyfor liver disease. Differentiation 76 (5), 495–505.

1 icron
K
K
K
K
K
L
L
L
L
M
M
M
N
N
N
68 S. Barachini et al. / M
adar, K., Kiraly, M., Porcsalmy, B., Molnar, B., Racz, G.Z., Blazsek, J., Kallo, K., Szabo,E.L., Gera, I., Gerber, G., Varga, G., 2009. Differentiation potential of stem cellsfrom human dental origin – promise for tissue engineering. J. Physiol. Pharmacol.60 (Suppl. 7), 167–175.
erkis, I., Kerkis, A., Dozortsev, D., Stukart-Parsons, G.C., Gomes Massironi, S.M.,et al., 2006. Isolation and characterization of a population of immature den-tal pulp stem cells expressing OCT-4 and other embryonic stem cell markers.Cells Tissues Organs 184, 105–116.
ern, S., Eichler, H., Toeve, J.S., Kluter, H., Bieback, K., 2006. Comparative analysis ofmesenchymal stem cells from bone marrow, umbilical cord blood, or adiposetissue. Stem Cells 24, 1294–1301.
inney, M.A., McDevitt, T.C., 2013. Emerging strategies for spatiotemporal controlof stem cell fate and morphogenesis. Trends Biotechnol. 31, 78–84.
shitiz, Park, J., Kim, P., Helen, W., Engler, A.J., Levchenko, A., Kim, D.H., 2012. Con-trol of stem cell fate and function by engineering physical microenvironments.Integr. Biol. (Camb.) 4, 1008–1018.
aino, G., D’Aquino, R., Graziano, A., Lanza, V., Carinci, F., Naro, F., et al., 2005. Anew population of human adult dental pulp stem cells: a useful source of livingautologous fibrous bone tissue (LAB). J. Bone Miner. Res. 20, 1394–1402.
aino, G., Carinci, F., Graziano, A., D’Aquino, R., Lanza, V., De Rosa, A., et al., 2006.In vitro bone production using stem cells derived from human dental pulp. J.Craniofac. Surg. 17, 511–515.
ee, H.J., Choi, B.H., Min, B.-H., Son, Y.S., Park, S.R., 2006. Low-intensity ultrasoundstimulation enhances chondrogenic differentiation in alginate culture of mes-enchymal stem cells. Artif. Organs 30, 707–715.
ivak, K.J., Schmittgen, T.D., 2001. Analysis of relative gene expression data usingreal-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25,402–408.
iura, M., Gronthos, S., Zhao, M., Lu, B., Fisher, L.W., Robey, P.G., Shi, S., 2003. SHED:stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. U.S.A.100, 5807–5812.
oens, C.B., Selleri, L., 2006. Hox cofactors in vertebrate development. Dev. Biol.291, 193–206.
oscato, S., Mattii, L., D’Alessandro, D., Cascone, M.G., Lazzeri, L., Serino, L.P., Dolfi, A.,Bernardini, N., 2008. Interaction of human gingival fibroblasts with PVA/gelatinesponges. Micron 39 (5), 569–579.
esti, C., Pardini, C., Barachini, S., D’Alessandro, D., Siciliano, G., Murri, L., Petrini,M., Vaglini, F., 2010. Human dental pulp stem cells protect mouse dopaminergicneurons against MPP+ or rotenone. Brain Res. 1367, 94–102.
osrat, I.V., Widenfalk, J., Olson, L., Norsat, C.A., 2001. Dental pulp cells produceneurotrophic factors, interact with trigeminal neurons in vitro, and rescuemotoneurons after spinal cord injury. Dev. Biol. 238, 120–132.
osrat, I.V., Smith, C.A., Mullally, P., Olson, L., Nosrat, C.A., 2004. Dental pulp cellsprovide neurotrophic support for dopaminergic neurons and differentiate into
67 (2014) 155–168
neurons in vitro; implications for tissue engineering and repair in the nervoussystem. Eur. J. Neurosci. 19, 2388–2398.
Park, H., Temenoff, J.S., Holland, T.A., Tabata, Y., Mikos, A., 2005. Delivery of TGF-beta1 and chondrocytes via injectable, biodegradable hydrogels for cartilagetissue engineering applications. J. Biomater. 26 (34), 7095–7103.
Parvizi, J., Wu, C.-C., Lewallen, D.G., Greenleaf, J.F., Bolander, M.E., 1999. Low-intensity ultrasound stimulates proteoglycan synthesis in rat chondrocytes byincreasing aggrecan gene expression. J. Orthop. Res., 12488–12494.
Paula-Silva, F.W., Ghosh, A., Silva, L.A., Kapila, Y.L., 2009. TNF-alpha pro-motes an odontoblastic phenotype in dental pulp cells. J. Dent. Res. 88,339–344.
Peters, H., Balling, R.T., 1999. Where and how to make them. Trends Genet. 15, 59–65.Picchi, J., Trombi, L., Spugnesi, L., Barachini, S., Maroni, G., Brodano, G.B., Boriani,
S., Valtieri, M., Petrini, M., Magli, M.C., 2013. HOX and TALE signatures specifyhuman stromal stem cell populations from different sources. J. Cell. Physiol. 228(4), 879–889.
Pierdomenico, L., Bonsi, L., Calvitti, M., Rondelli, D., Arpinati, M., Chirumbolo, G., et al.,2005. Multipotent mesenchymal stem cells with immunosuppressive activitycan be easily isolated from dental pulp. Transplantation 80, 836–842.
Sasaki, R., Aoki, S., Yamato, M., Uchiyama, H., Wada, K., Okano, T., Ogiuchi, H., 2008.Neurosphere generation from dental pulp of adult rat incisor. Eur. J. Neurosci.27 (3), 538–548.
Scadden, D.T., 2006. The stem-cell niche as an entity of action. Nature 441 (7097),1075–1079.
Seo, B.M., Miura, M., Gronthos, S., Bartold, P.M., Batouli, S., Brahim, J., et al., 2004.Investigation of multipotent postnatal stem cells from human periodontal liga-ment. Lancet 364, 149–155.
Serakinci, N., Keith, N.W., 2006. Therapeutic potential of adult stem cells. Eur. J.Cancer 42 (9), 1243–1246.
Shah, N., Morsi, Y., Manuelpillai, U., Barry, T., Manasseh, R., 2013. Elucidating theeffects of low-intensity ultrasound on mesenchymal stem cell proliferation andviability. J. Acoust. Soc. Am. 133 (5), 3496.
Shi, S., Gronthos, S., 2003. Perivascular niche of postnatal mesenchymal stemcells in human bone marrow and dental pulp. J. Bone Miner. Res. 8 (4),696–704.
Sonoyama, W., Liu, Y., Fang, D., Yamaza, T., Seo, B.M., Zhang, C., et al., 2006. Mes-enchymal stem cell-mediated functional tooth regeneration in swine. PloS ONE1, e79.
Trombi, L., Mattii, L., Pacini, S., D’Alessandro, D., Battolla, B., Orciuolo, E., Buda, G.,
Fazzi, R., Galimberti, S., Petrini, M., 2008. Human autologous plasma-derived clotas a biological scaffold for mesenchymal stem cells in treatment of orthopedichealing. J. Orthop. Res. 26, 176–183.Yu, J., Chen, Y., Wuhrer, R., Liu, Z., Ringe, S.P., 2005. In situ formation of BN nanotubesduring nitriding reactions. Chem. Mater. 17 (20), 5172–5176.




















