Marked microevolution of a unique Mycobacterium tuberculosis strain in 17 years of ongoing...
10
Marked Microevolution of a Unique Mycobacterium tuberculosis Strain in 17 Years of Ongoing Transmission in a High Risk Population Carolina Mehaffy 1,2 * ¤ , Jennifer L. Guthrie 1 , David C. Alexander 3 , Rebecca Stuart 4 , Elizabeth Rea 4 , Frances B. Jamieson 1,2 1 Public Health Ontario, Toronto, Canada, 2 University of Toronto, Toronto, Canada, 3 Saskatchewan Disease Control Laboratory, Regina, Canada, 4 Toronto Public Health, Toronto, Canada Abstract The transmission and persistence of Mycobacterium tuberculosis within high risk populations is a threat to tuberculosis (TB) control. In the current study, we used whole genome sequencing (WGS) to decipher the transmission dynamics and microevolution of M. tuberculosis ON-A, an endemic strain responsible for an ongoing outbreak of TB in an urban homeless/ under-housed population. Sixty-one M. tuberculosis isolates representing 57 TB cases from 1997 to 2013 were subjected to WGS. Sequencing data was integrated with available epidemiological information and analyzed to determine how the M. tuberculosis ON-A strain has evolved during almost two decades of active transmission. WGS offers higher discriminatory power than traditional genotyping techniques, dividing the M. tuberculosis ON-A strain into 6 sub-clusters, each defined by unique single nucleotide polymorphism profiles. One sub-cluster, designated ON-A NM (Natural Mutant; 26 isolates from 24 cases) was also defined by a large, 15 kb genomic deletion. WGS analysis reveals the existence of multiple transmission chains within the same population/setting. Our results help validate the utility of WGS as a powerful tool for identifying genomic changes and adaptation of M. tuberculosis. Citation: Mehaffy C, Guthrie JL, Alexander DC, Stuart R, Rea E, et al. (2014) Marked Microevolution of a Unique Mycobacterium tuberculosis Strain in 17 Years of Ongoing Transmission in a High Risk Population. PLoS ONE 9(11): e112928. doi:10.1371/journal.pone.0112928 Editor: Igor Mokrousov, St. Petersburg Pasteur Institute, Russian Federation Received June 11, 2014; Accepted August 22, 2014; Published November 18, 2014 Copyright: ß 2014 Mehaffy et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Data Availability: The authors confirm that, for approved reasons, some access restrictions apply to the data underlying the findings. All the sequencing data associated with this manuscript has now been released by NCBI. The accession number for the entire study is: SRP046976. Epidemiological data from Public Health Ontario may be available for researchers who meet the criteria for access to confidential data as determined by Public Health Ontario policies and Public Health Ontario Ethics Review Board. For requests, please contact Dr. Frances Jamieson, 81 Resources Road, Toronto, ON M9P 3T1, [email protected]. Funding: This study was supported by a grant-in-aid from The Lung Association/Ontario Thoracic Society (http://www.on.lung.ca) (CM, FBJ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Competing Interests: The authors have declared that no competing interests exist. * Email: [email protected] ¤ Current address: Colorado State University, Fort Collins, Colorado, United States of America Introduction Globally, tuberculosis (TB) is an important cause of morbidity and mortality. World-wide, M. tuberculosis is responsible for more than 1 million deaths per year. In low incidence countries, homeless/under-housed individuals represent one of the groups at greater risk for TB infection and disease [1–5]. TB outbreaks within homeless settings have been documented throughout North America [6–9]. One endemic strain, designated Ontario A (ON-A), has been circulating since at least 1997 in the urban homeless/under- housed population of Toronto, Canada and has been responsible for TB outbreaks in 2001 and 2004 with new cases identified every year [5,10]. Genotyping is an essential component of epidemio- logical investigations. However, the M. tuberculosis ON-A isolates are defined by a unique combined spoligotype and 24-locus MIRU-VNTR (24-MIRU) profile, while IS6110 RFLP generates pseudo-clusters among these strains [10]. Although several reports have highlighted the epidemiological value of whole genome sequencing (WGS) over traditional genotyping techniques [11–17], presently only a few studies have evaluated the utility of WGS to study whole genome changes of M. tuberculosis during long-term continuous active transmission [15]. We evaluated the usefulness of WGS to retrospectively validate and identify transmission events associated with TB cases due to M. tuberculosis ON-A over the last 17 years. We used a phylogenetic analysis based on single nucleotide polymorphisms (SNP) to portray the microevolution of this M. tuberculosis strain during almost two decades of on-going transmission in a high risk population. Our analysis revealed the presence of six independent transmission chains and the presence of an ON-A natural mutant, defined by a large genomic deletion that most likely emerged during the first ON-A TB outbreak in 2001. Methods M. tuberculosis clinical isolates M. tuberculosis isolates were obtained from clinical specimens routinely received at the Public Health Ontario Laboratories for TB diagnosis. All available isolates from 1997–2013 with PLOS ONE | www.plosone.org 1 November 2014 | Volume 9 | Issue 11 | e112928
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Marked microevolution of a unique Mycobacterium tuberculosis strain in 17 years of ongoing...

Marked Microevolution of a Unique Mycobacteriumtuberculosis Strain in 17 Years of Ongoing Transmissionin a High Risk PopulationCarolina Mehaffy1,2*¤, Jennifer L. Guthrie1, David C. Alexander3, Rebecca Stuart4, Elizabeth Rea4,
Frances B. Jamieson1,2
1 Public Health Ontario, Toronto, Canada, 2 University of Toronto, Toronto, Canada, 3 Saskatchewan Disease Control Laboratory, Regina, Canada, 4 Toronto Public Health,
Toronto, Canada
Abstract
The transmission and persistence of Mycobacterium tuberculosis within high risk populations is a threat to tuberculosis (TB)control. In the current study, we used whole genome sequencing (WGS) to decipher the transmission dynamics andmicroevolution of M. tuberculosis ON-A, an endemic strain responsible for an ongoing outbreak of TB in an urban homeless/under-housed population. Sixty-one M. tuberculosis isolates representing 57 TB cases from 1997 to 2013 were subjected toWGS. Sequencing data was integrated with available epidemiological information and analyzed to determine how the M.tuberculosis ON-A strain has evolved during almost two decades of active transmission. WGS offers higher discriminatorypower than traditional genotyping techniques, dividing the M. tuberculosis ON-A strain into 6 sub-clusters, each defined byunique single nucleotide polymorphism profiles. One sub-cluster, designated ON-ANM (Natural Mutant; 26 isolates from 24cases) was also defined by a large, 15 kb genomic deletion. WGS analysis reveals the existence of multiple transmissionchains within the same population/setting. Our results help validate the utility of WGS as a powerful tool for identifyinggenomic changes and adaptation of M. tuberculosis.
Citation: Mehaffy C, Guthrie JL, Alexander DC, Stuart R, Rea E, et al. (2014) Marked Microevolution of a Unique Mycobacterium tuberculosis Strain in 17 Years ofOngoing Transmission in a High Risk Population. PLoS ONE 9(11): e112928. doi:10.1371/journal.pone.0112928
Editor: Igor Mokrousov, St. Petersburg Pasteur Institute, Russian Federation
Received June 11, 2014; Accepted August 22, 2014; Published November 18, 2014
Copyright: � 2014 Mehaffy et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability: The authors confirm that, for approved reasons, some access restrictions apply to the data underlying the findings. All the sequencing dataassociated with this manuscript has now been released by NCBI. The accession number for the entire study is: SRP046976. Epidemiological data from PublicHealth Ontario may be available for researchers who meet the criteria for access to confidential data as determined by Public Health Ontario policies and PublicHealth Ontario Ethics Review Board. For requests, please contact Dr. Frances Jamieson, 81 Resources Road, Toronto, ON M9P 3T1, [email protected].
Funding: This study was supported by a grant-in-aid from The Lung Association/Ontario Thoracic Society (http://www.on.lung.ca) (CM, FBJ). The funders had norole in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: The authors have declared that no competing interests exist.
* Email: [email protected]
¤ Current address: Colorado State University, Fort Collins, Colorado, United States of America
Introduction
Globally, tuberculosis (TB) is an important cause of morbidity
and mortality. World-wide, M. tuberculosis is responsible for more
than 1 million deaths per year. In low incidence countries,
homeless/under-housed individuals represent one of the groups at
greater risk for TB infection and disease [1–5]. TB outbreaks
within homeless settings have been documented throughout North
America [6–9].
One endemic strain, designated Ontario A (ON-A), has been
circulating since at least 1997 in the urban homeless/under-
housed population of Toronto, Canada and has been responsible
for TB outbreaks in 2001 and 2004 with new cases identified every
year [5,10]. Genotyping is an essential component of epidemio-
logical investigations. However, the M. tuberculosis ON-A isolates
are defined by a unique combined spoligotype and 24-locus
MIRU-VNTR (24-MIRU) profile, while IS6110 RFLP generates
pseudo-clusters among these strains [10].
Although several reports have highlighted the epidemiological
value of whole genome sequencing (WGS) over traditional
genotyping techniques [11–17], presently only a few studies have
evaluated the utility of WGS to study whole genome changes of M.tuberculosis during long-term continuous active transmission [15].
We evaluated the usefulness of WGS to retrospectively validate
and identify transmission events associated with TB cases due to
M. tuberculosis ON-A over the last 17 years. We used a
phylogenetic analysis based on single nucleotide polymorphisms
(SNP) to portray the microevolution of this M. tuberculosis strain
during almost two decades of on-going transmission in a high risk
population. Our analysis revealed the presence of six independent
transmission chains and the presence of an ON-A natural mutant,
defined by a large genomic deletion that most likely emerged
during the first ON-A TB outbreak in 2001.
Methods
M. tuberculosis clinical isolatesM. tuberculosis isolates were obtained from clinical specimens
routinely received at the Public Health Ontario Laboratories for
TB diagnosis. All available isolates from 1997–2013 with
PLOS ONE | www.plosone.org 1 November 2014 | Volume 9 | Issue 11 | e112928

genotypes consistent with the ON-A strain [6,18] were selected.
All isolates were susceptible to all first line drugs.
The work described in this manuscript relates directly to
improvement of routine TB surveillance and outbreak manage-
ment, therefore research ethics board (REB) approval was not
required.
DNA extractionGenomic DNA (gDNA) was extracted as previously described
[19] with minor modifications [20].
Genotyping24-locus MIRU-VNTR [21], spoligotyping [22] and IS6110
RFLP [23] were performed using standard methods, and data
were analyzed with BioNumerics v6.1 (Applied Maths, St-Martin
Latem, Belgium). RFLP patterns were compared as previously
described [10].
Whole genome sequencingDNA was prepared for sequencing as described elsewhere [20].
Illumina paired-end reads were trimmed using quality scores and
then aligned to M. tuberculosis H37Rv reference genome
(NC_000962.2) using the CLC Genomics workbench (v.6.0.2)
software. For 5 of the 61 isolates, quality and/or quantity of the
DNA were not suitable for WGS and therefore these were not
included in the analysis.
Accuracy of WGS assembly and analysis workflow was assessed
by sequencing the H37Rv reference strain that is used in our
clinical lab (Material S1).
Variant callingSingle nucleotide polymorphisms (SNPs) and small insertion-
deletion (indel) events were identified using a probabilistic variant
detection with cutoffs of a minimum read depth of 20X and a
variant frequency of at least 75. Indels were not considered for any
further analyses. SNPs were further filtered by removing positions
associated with PE, PPE and PE_PGRS gene families which have
been previously shown to represent false positives and due to their
high variation are not suited for phylogenetic analysis [17]. SNPs
unique to any of the fifty-six high quality whole genome sequences
were manually inspected in each individual alignment for accuracy
and all ambiguous results were discarded.
Phylogenetic analysisA concatemer of the SNPs was generated and then used to
reconstruct the phylogeny of ON-A using SplitsTree v.4 software
[24] and the BioNJ algorithm [25]. Trees were then re-constructed
using the Equal Angle algorithm [26] with equal-daylight and box
opening optimization [27] available in SplitsTree v.4 (Figure 1).
Demographic and clinical dataDemographic characteristics and clinical information for each
TB case was obtained from Ontario’s integrated Public Health
Information System (iPHIS) as well as responsible Public Health
Units (Table 1). This information is routinely recorded by Public
Health Units for all laboratory and clinically confirmed TB cases.
Data was anonymized and all personal information removed from
the final data set. Time lapse between onset of symptoms and
diagnosis as well as treatment start date were not available for most
cases and therefore were not included in this study.
Social network visualizationKnown epidemiological/social connections identified during
routine public health contact investigation (i.e roommates, close
friends, used same drop-in, etc.) among forty-seven ON-A cases
were available in iPHIS. The igraph [28] package of R (v3.0.2)
was used to generate the analysis. Transmission events were
defined based on the genomic and epidemiological information
(i.e. SNP pattern, contact information, year of diagnosis and
infectiousness based on smear and chest x-ray results).
Results
In this study we performed the whole genome sequencing of 61
M. tuberculosis isolates identified during routine genotyping as
members of a large cluster denominated ON-A and spanning 17
years (1998–2013). The 61 isolates corresponded to 57 patients,
most of whom were homeless/under-housed individuals (75.5%).
The epidemiological and molecular typing characteristics of this
cluster up to 2008 have been published elsewhere [5,6]. Table 1
summarizes the demographic characteristics of all 57 patients.
Isolates in this cluster are characterized by a highly similar
24MIRU and spoligotype patterns while RFLP analysis of the
ON-A strain results into three pseudo-clusters defined by RFLP
types A, B and C. The main 24MIRU pattern has not been
reported in the international MIRU database (http://www.miru-
vntrplus.org/MIRU/index.faces) and personal communication
with the corresponding health authorities of the other Canadian
provinces indicates that this genotype is unique to Toronto’s inner
city population.
Of the 61 M. tuberculosis isolates, WGS was successfully
performed in 56 isolates, corresponding to 53 TB cases and further
sub-divided the large ON-A group into 6 different sub-clusters
(SC1-6) (Figure 1 and 2).
Figure 3 combines the RFLP and SNP-clustering results for all
53 cases with final WGS data. Most isolates belong to RFLP type
A (n = 34) followed by RFLP B (n = 5) and RFLP C (n = 3). Nine
isolates were not clustered by RFLP and 1 presented a mixed
pattern corresponding to an infection with strain ON-A and strain
ON-B, which is also commonly found in the homeless/under-
housed population. Figure 3 illustrates the lack of correlation
between RFLP and SNP-clustering with the exception of RFLP
type B which corresponded to SC4.
Sub-cluster classification was based on SNP analysis using as
reference, the genome of laboratory strain H37Rv. In summary,
722 SNPs were identified in ON-A isolates when compared with
H37Rv. Of these, 641 SNPs, including 333 (52%) non-synony-
mous, 224 (35%) synonymous, and 84 (13%) non-coding SNPs,
were conserved in all sequenced isolates and only served to
differentiate H37Rv from the ON-A strain. The remaining 81
SNPs were variable among the sequenced isolates and were used
to identify sub-cluster associations.
Most isolates belonged to the most recently emerged sub-cluster
SC6 (n = 24), followed by SC4 (n = 10), SC1 (n = 8), SC5 (n = 5)
and SC2 (n = 2). One isolate was not placed in any of the 6 sub-
clusters.
Of the 81 SNPs, 2 were present in the majority of ON-A
isolates, except for most isolates in SC1. One SNP was only
present in SC-6 and one was present in both SC4 and SC5
isolates. The remaining 77 SNPs were either sub-cluster associated
(i.e. present in two or more isolates) (17/77) or singletons (60/77).
Sub-clusters, SNPs and transmission eventsPhylogenetic analysis identified 6 ON-A sub-clusters (Figure 1).
One isolate did not group with any of the identified sub-clusters.
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 2 November 2014 | Volume 9 | Issue 11 | e112928

Phylogenetic analysis and temporal association of isolates from the
ON-A strain suggests the independent emergence of SC1–SC5
prior to 1997 and subsequent clonal expansion of sub-clusters
SC3, SC4 and SC5 (Figures 1 and 2). Contrary to this, SC6
appears to have emerged in 2001 during the first TB outbreak
associated with ON-A.
SNP patterns of isolates belonging to SC1 group suggest they
represent infections acquired from a common ancestor(s) prior to
1997 and most likely represent re-activation of latent TB infection
rather than immediate secondary cases. The exceptions were two
isolates, (WT24-07 and WT27-08) with SNP patterns identical to
the source isolate WT23-07. The two individuals, from whom the
isolates WT23-07 and WT24-07 were obtained, lived in the same
rooming house, and both used the same drop-in location which
was associated with the two ON-A TB outbreaks. Interestingly, the
individual associated with isolate WT27-08 was diagnosed in a
different jurisdiction north of Toronto, but is clearly related to the
other two isolates and demonstrates the high mobility of this
population.
Contrary to SC1, all other sub-clusters had a clear source case.
In group SC2, isolate WT5-00 gained 4 SNPs in approximately 2
years after infection from WT3-98. In group SC3, the source case
(WT07-01) resulted in two other primary TB cases in the same
year and a later reactivation case in 2004.
In SC4, the source case (WT17-04) resulted in secondary cases
in shelter workers (both in 2004). Five additional cases with an
identical SNP pattern as the suspected source case were also
identified in subsequent years (2005–2010). In addition, the source
was also genetically related to a case in 2006 with 1 SNP gained
and another case in 2010 with 4 additional SNPs. It is not possible
to determine if the case in 2010 (WT29-10) originated from the
source case or from one of the five cases with identical SNP
pattern to the source. However, the large number of SNPs suggests
that WT29-10 corresponds to a re-activation from an infection
acquired in 2004 or 2005. This is also supported by epidemio-
logical data that indicates WT29-10 may have shared a common
workplace with WT17-04 at the time this source case was ill.
24-MIRU, spoligotype and RFLP indicated that A/B-01
represented a mixed infection with two strains commonly found
in this population (ON-A and ON-B) [6] and therefore its WGS
data was not included in the phylogenetic analysis. However,
manual inspection of all 81 variable SNP positions in the genome
alignment generated for this isolate demonstrated the presence of
all SC5 SNPs, allowing us to include the A/B-01 case within the
SC5 group.
The two suspected source cases in group SC5 (WT6-01 and A/
B-01) resulted in two subsequent cases in 2004 and 2007, both of
which have identical SNP patterns to the suspected source(s). In
addition, these two cases were the most likely source for the
emergence of SC6 in 2001.
It is possible that the high bacteria burden present in WT6_01
or the A/B-01 mixed case (smear 2+ and 3+ respectively)
contributed to the emergence of SC6. Both patients also had
abnormal chest x-rays and together with their smear results
Figure 1. ON-A Phylogenetic tree constructed in Splitstree ver. 4. All 722 polymorphic sites were included. Phylogenetic tree was built usingBioNJ algorithm and then filtered for parsimony-informative sites.doi:10.1371/journal.pone.0112928.g001
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 3 November 2014 | Volume 9 | Issue 11 | e112928
suggest they were highly infectious. The chest x-ray of the mixed
A/B case also showed cavitation. Social network analysis
demonstrated the large number of contacts shared by these two
patients, including individuals in both ON-AWT and ON-ANM
groups (Figure S1).
The SC6 group was very homogeneous. SNPs within this group
were mostly singletons and represented temporal acquisition of
substitutions in single isolates. Nine SC6 isolates (37.5%) had no
accumulation of SNPs in an 8 year time frame (Figure 2).
Because of the high number of isolates with identical SNP
patterns in SC6, it was not possible to determine the exact
transmission chain in this group. Multiple control measures to
reduce spread of TB in Toronto’s homeless shelters were
implemented as a consequence of the outbreaks in 2001 and
2004. This provides support to the idea that most cases after 2005
correspond to reactivation of infections acquired during one of the
two TB outbreaks. The only exception is case NM17-08 which is
an individual outside of the high risk group for which contact
investigations indicate the source case for that individual was
NM15-06. For the remaining cases, the most likely index case was
NM7-01 who was highly infectious, with smear +3 and abnormal
chest x-ray.
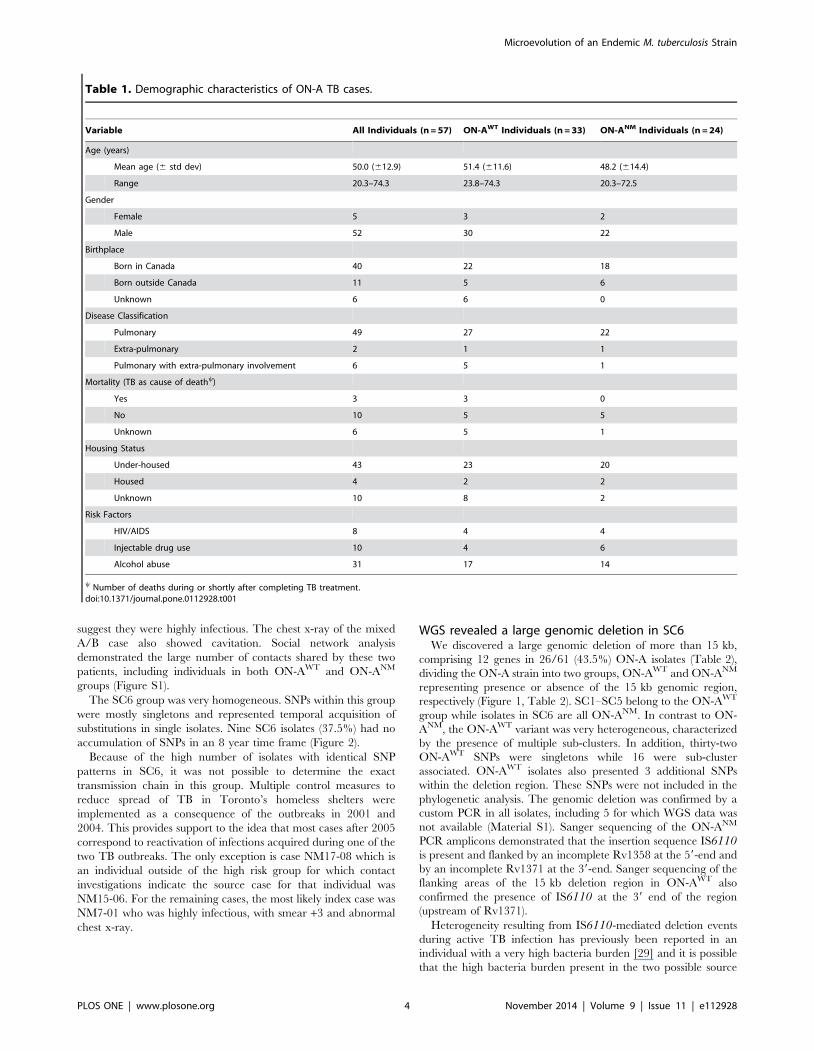
WGS revealed a large genomic deletion in SC6We discovered a large genomic deletion of more than 15 kb,
comprising 12 genes in 26/61 (43.5%) ON-A isolates (Table 2),
dividing the ON-A strain into two groups, ON-AWT and ON-ANM
representing presence or absence of the 15 kb genomic region,
respectively (Figure 1, Table 2). SC1–SC5 belong to the ON-AWT
group while isolates in SC6 are all ON-ANM. In contrast to ON-
ANM, the ON-AWT variant was very heterogeneous, characterized
by the presence of multiple sub-clusters. In addition, thirty-two
ON-AWT SNPs were singletons while 16 were sub-cluster
associated. ON-AWT isolates also presented 3 additional SNPs
within the deletion region. These SNPs were not included in the
phylogenetic analysis. The genomic deletion was confirmed by a
custom PCR in all isolates, including 5 for which WGS data was
not available (Material S1). Sanger sequencing of the ON-ANM
PCR amplicons demonstrated that the insertion sequence IS6110is present and flanked by an incomplete Rv1358 at the 59-end and
by an incomplete Rv1371 at the 39-end. Sanger sequencing of the
flanking areas of the 15 kb deletion region in ON-AWT also
confirmed the presence of IS6110 at the 39 end of the region
(upstream of Rv1371).
Heterogeneity resulting from IS6110-mediated deletion events
during active TB infection has previously been reported in an
individual with a very high bacteria burden [29] and it is possible
that the high bacteria burden present in the two possible source
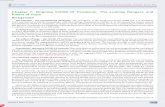
Table 1. Demographic characteristics of ON-A TB cases.
Variable All Individuals (n = 57) ON-AWT Individuals (n = 33) ON-ANM Individuals (n = 24)
Age (years)
Mean age (6 std dev) 50.0 (612.9) 51.4 (611.6) 48.2 (614.4)
Range 20.3–74.3 23.8–74.3 20.3–72.5
Gender
Female 5 3 2
Male 52 30 22
Birthplace
Born in Canada 40 22 18
Born outside Canada 11 5 6
Unknown 6 6 0
Disease Classification
Pulmonary 49 27 22
Extra-pulmonary 2 1 1
Pulmonary with extra-pulmonary involvement 6 5 1
Mortality (TB as cause of deathy)
Yes 3 3 0
No 10 5 5
Unknown 6 5 1
Housing Status
Under-housed 43 23 20
Housed 4 2 2
Unknown 10 8 2
Risk Factors
HIV/AIDS 8 4 4
Injectable drug use 10 4 6
Alcohol abuse 31 17 14
y Number of deaths during or shortly after completing TB treatment.doi:10.1371/journal.pone.0112928.t001
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 4 November 2014 | Volume 9 | Issue 11 | e112928

cases (WT6-01 and A/B-01) as described previously could have
contributed to the emergence of the deletion and origin of the ON-
ANM variant.
Discussion
Microevolution and adaptation events of M. tuberculosisduring active TB transmission
We analyzed a cluster of M. tuberculosis isolates (ON-A)
associated with on-going TB transmission in a large urban setting
and its inner-city homeless/under-housed population. In order to
evaluate the evolution of this M. tuberculosis strain during a 17
year period, we performed WGS analysis for all available ON-A
isolates. We discovered a large genomic deletion (.15 kb) in a
subset of ON-A isolates that divided the ON-A strain into two
strain variants (ON-AWT and ON-ANM). This large genomic
deletion, which is present in 43% of the ON-A isolates
demonstrates the presence of two distinct groups independent of
the RFLP pattern.
Our phylogenetic and epidemiological analyses suggest this
deletion emerged in 2000 during the first TB outbreak caused by
ON-A.
The 15 kb deletion comprises a cluster of 12 genes, of which at
least 5 code for regulatory proteins. Regulatory proteins govern
the expression of clusters of genes involved in specific molecular
pathways and therefore are ultimately responsible for the ability of
the cell to adapt and survive in new environments. Two of the
deleted genes identified in the ON-ANM correspond to regulators
Figure 2. Transmission events of ON-A TB cases based on WGS information and epidemiologic analysis. Colored solid circles representTB isolates that are identical to the suspected primary case (0 SNPs). Open circles represent related cases, separated by black solid dots representingSNPs acquired over time. Triangles represent TB cases outside of the homeless/under-housed group. Isolates from the same TB patient arerepresented by circles with dots in their background: WT25-08 and WT26-08; NM2-01 and NM3-01; and NM17-08 and NM19-08. *Isolates that hadmore than one passage prior to WGS.doi:10.1371/journal.pone.0112928.g002
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 5 November 2014 | Volume 9 | Issue 11 | e112928

of the Sigma-factor F (SigF). Sigma factors bind to RNA
polymerase and alter its promoter preference resulting in subsets
of genes that are differentially expressed upon environmental
stresses to which a particular sigma factor responds. M.tuberculosis SigF is induced by nutrient starvation [30] and it is
involved in the late stages of the disease [31]. One of the anti-sigF
deleted genes (Rv1364c) strongly interacts with sigF and also with
the sigF antagonist RsbW [32]. Other regulatory genes identified
in the 15 kb deletion are the LuxR family regulator (Rv1358)
which is also regulated by SigF [33], a signal transduction
regulatory gene (Rv1359) and a conserved gene (Rv1366) in which
we identified a domain representative of RelA-SpoT superfamily,
responsible for the regulation of ppGpp concentration. ppGpp is a
modified nucleotide used by bacteria as intracellular messengers
that respond to different environmental stresses [34,35]. In M.tuberculosis, ppGpp responses are associated with virulence and
long-term survival [35,36].
The presence of the insertion sequence IS6110 at the site of the
deletion suggests this event was IS6110 mediated. Reports
elsewhere have shown that IS6110 mediated deletion events are
common in M. tuberculosis [29,37,38] and even though the
identification of a large genomic deletion in this highly related M.tuberculosis strain is very interesting, it should not be surprising
given the high IS6110 mediated mutation rate (transposition/
generation) [39]. It has been proposed that although deleterious
IS6110 mediated mutations may occur frequently, these mutants
are rapidly purged from the population by purifying selection [39].
Contrary to this, the 15 kb deletion seems to have emerged during
the first TB outbreak in 2001; and since then the ON-ANM has
established itself among the homeless/under-housed population in
Toronto.
Other examples of non-deleterious deletions exist. An M.tuberculosis isolate from the CAS family bearing a chromosomal
deletion, was responsible for a large TB outbreak in Leicester, UK
and was shown to have a lower inflammatory phenotype and
higher intracellular growth similar to the hypervirulent strain
HN878 [40]. An IS6110-mediated deletion was also reported
elsewhere in an individual with disseminated TB and a high
bacterial burden [29]. The deleted isogenic strain in this individual
was only present in the lymph nodes and although transmission
events were not identified, it is clear that the deletion did not
impair the bacilli to replicate and cause disease.
Although the physiological effect of the ON-ANM 15 kb deletion
is presently unknown, based on the information available for the
deleted genes, five of which have some role in gene regulation
under environmental stresses; this deletion may have a direct
impact on several molecular pathways and could enhance the
capacity of this variant to spread and cause disease.
Conversely, the 15 kb deletion observed in ON-ANM is in a
hotspot position for IS6110 transposition events [41,42] and the
genes located in this region may not be essential for the bacteria to
transmit and cause disease. This would support the theory that the
ON-ANM was randomly fixed in the homeless/under-housed
population, possibly supported by higher transmission rates in
congregate setting such as shelters and a high prevalence of co-
morbidities). It is possible that a weakened immune system allows
for the appearance of potentially ‘‘unfit’’ mutations or large
deletion events such as those reported here.
Studies focusing on fitness assessment and mining of the
proteome and transcriptome of ON-A variants to determine the
role of the deleted genes in the physiology and virulence of M.tuberculosis are currently underway.
WGS as tool to determine TB transmission dynamicsAlthough RFLP, MIRU-VNTR and spoligotype are all
powerful genotyping techniques, they cannot establish chronology
of infection and lack resolution in long-term transmission events
[43–46]. In contrast WGS has the potential for resolving the
transmission dynamics of TB infection and when complemented
with epidemiological data, it is an excellent tool to identify
Figure 3. Dendrogram of IS6110 RFLP showing the 15 kbdeletion distribution. Branches indicate the clusters with identicalRFLP patterns. Colored squares represent each of the six sub-clustersidentified by WGS. Information regarding the presence or absence ofthe 15 kb deletion for each isolate is shown as ‘‘deleted’’ or ‘‘intact’’.doi:10.1371/journal.pone.0112928.g003
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 6 November 2014 | Volume 9 | Issue 11 | e112928
individual transmission events, and its use is very valuable in
highly mobile communities such as the homeless/under-housed.
In addition to providing relevant and detailed information that
can be used to construct and/or validate the transmission
dynamics of a given strain, WGS also provides important
information regarding SNPs, indels and larger genomic structural
variants that can provide insights into the physiological charac-
teristics of M. tuberculosis as a whole as well as particular
characteristics of the strain of interest.
Recent genomics studies have highlighted the benefits of whole
genome sequencing over traditional genotyping to uncover the
transmission dynamics and molecular-guided TB control and
surveillance [11,12,14–17]. For instance, Gardy et al. [12]
demonstrated the usefulness of overlapping WGS data and social
network analyses during outbreak investigations. The authors were
able to determine transmission dynamics of 32 M. tuberculosisoutbreak isolates in a period of two and half years. Similar to our
findings, their SNP analysis revealed two lineages suggesting not
one, but two simultaneous chains of transmission. The SNP calling
algorithm in Gardy’s study did not include filtering out PE/PPE
genes. This resulted in a higher genetic diversity than expected,
but it is remarkable the similar findings with our study vis-a-vis the
identification of multiple strain variants and clusters with otherwise
identical RFLP and MIRU-VNTR patterns in a high risk
population. Unlike the study by Gardy et al. [12] in which the
two strain variants diverged and circulated in the community well
before the outbreak, the ON-AWT variant appears to have evolved
during the 2001 TB outbreak and the emergent ON-ANM was
rapidly fixed in Toronto’s homeless/under-housed population.
Most WGS studies related to TB transmission dynamics have
been focused on TB outbreaks in short time frames (1–30 months)
[11–14,16]. In all cases WGS represented an invaluable tool to
either support or complement directionality of transmission based
on epidemiological contact studies. A few long-term studies have
also been conducted. Roetzer et al. [17] performed a prospective
study to evaluate the correlation of WGS analyses with contact
tracing data. Eighty-six M. tuberculosis isolates from a 13 year
time frame were sequenced. WGS analysis confirmed the clonal
expansion of an outbreak strain that was initially seen in a specific
social setting in Hamburg, Germany, but slowly spreading outside
of this particular setting [17]. In contrast to our findings, SNPs
were the primary source of genome evolution during transmission
and all small deletions and insertions detected were constant in all
isolates.
Walker et al. [15] also confirmed the high power and resolution
provided by WGS and determined the genomic diversity between
patients in MIRU-VNTR community clusters in a period
spanning 1–12 years.
Here, we studied the dynamics of TB disease over 17 years of
on-going transmission in a homeless/under-housed population.
Previous studies have demonstrated that large deletions and
hundreds of polymorphic sites can exist in isolates with identical
IS6110 and MIRU-VNTR patterns [15]. As expected, we
confirmed that clusters obtained by IS6110 RFLP typing were
not in agreement with the WGS-based division of ON-A isolates
into two major groups based on the 15 kb large genomic deletion).
In addition, we identified 81 SNPs that are variable within the
ON-A strain and further subdivide the strain into 6 distinct sub-
clusters.
The large heterogeneity observed in our study group resulted in
an average of 1.5 SNPs/case (81 SNPs/55 M. tuberculosis isolates)
is slightly larger than those reported in the majority of M.tuberculosis-WGS studies which ranged from 0–1.3. However, in
those studies, the larger variation rates were associated to
community clusters (as opposed to household groups) spanning
more than 10 yr of transmission [13,15,17]. Only two studies have
Table 2. Genes present in the 15 kb deleted region.
Deletedgene Function Annotations Reference
Rv1358 Probable transcriptionalregulatory protein
LuxR family signature, expressionregulated by SigF; deleted in someclinical isolates
[33,49]
Rv1359 Probable signaltransductionregulatory protein
Adenylate cyclase, family 3(Sensory pathways)
Rv1360 Probable oxidoreductase Expressed during GP infections;mRNA down-regulated in starvation
[50,51]
Rv1361 PPE19 - Unknown Expressed during GP infections;mRNA down-regulated instarvation; regulated by PhoP
[50–52]
Rv1362c Unknown Expressed during GP infections;membrane protein
[50]
Rv1363c Unknown None
Rv1364c Possible sigma factor Fregulatory protein
May be a capreomycin target;responds to heat stress
[53]
Rv1365 Anti-anti sigma factor F Regulated by Redox potential
Rv1366 Unknown Homology to RelA-SpoTsuperfamily (ppGpp synthesis)
Rv1367c Possibly involved in cellwall metabolism
Homology to PBPs, B-lactamases
Rv1368 Lipoprotein None
Rv1371 Unknown Probably conserved membrane protein
doi:10.1371/journal.pone.0112928.t002
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 7 November 2014 | Volume 9 | Issue 11 | e112928

shown larger variability. Cluster 9 in the study reported in Walker
et al. (2013) [15] which was associated with substance abuse and
presented an average of 2 SNPs/case in a 9 yr span; and the large
cluster in Gardy et al. (2011) with a rate of more than 5 SNPs/case
likely due to the inclusion of hypervariable PPE/PE genes [12].
Our rate of 1.5 SNPs/case may be a result from the long time
span as well as the likely possibility of on-going transmission prior
to 1997 and thus potential for missed links/cases. Although
information regarding delay in diagnosis and adherence to
treatment was not available, these are conditions likely to be
encountered in a high risk setting such as the homeless/under-
housed and could increase the possibility of bacilli evolution. For
instance, SNP patterns of isolates belonging to SC1 group suggest
they all represent infections acquired from a common ancestor
prior to 1997 and most likely represent re-activation of latent TB
infection with independently gained substitutions during latency
ranging from 3–7 SNPs per event.
This hypothesis is supported by a study of TB latency in
macaques that suggests M. tuberculosis mutation rate is fairly
similar in active and latent TB, with the rate in latent TB being
slightly higher [47]. In contrast, a recent study of latency in
humans suggests a lower mutation rate during latency when
compared to active TB [48]. However, this study only included
two subjects with a long period (.10 yr) of latent infection, and as
the authors suggested, it is possible that a broader spectrum of
mutation rates exists during latent TB.
Certainly, our study supports this idea, as we were able to show
both high heterogeneity as observed in SC1, but also several cases
with a low SNP fixation (0–2 SNPs/yr) in the other ON-A groups
particularly SC4–SC6 in which latency periods of more than 5
years and zero SNP changes were observed. For instance, in the
ON-ANM (SC6), some of the isolates obtained in 2008 did not
present any additional SNPs when compared to isolates obtained
in 2001. Similarly, group SC4 of ON-AWT was represented by
identical isolates from 2004 to 2010 and SC5 included isolates with
identical SNP pattern from 2001 to 2007.
Although most SC1 cases are probably reactivations, we
strongly believe that the high heterogeneity is not only due to
latency but also to missing links within the group. This could be
due to unrecognized/undiagnosed TB cases, TB cases ultimately
diagnosed outside the province of Ontario, or to TB cases
diagnosed prior to 1997 for which no isolates are available. Gaps
in the PHOL genotyping database are another factor. Our
laboratory implemented IS6110 RFLP in 1997 but, until the
introduction of a semi-automated MIRU-VNTR and spoligotyp-
ing program in 2007, Ontario did not have universal genotyping
and only a subset of strains were analyzed or archived. Currently,
the PHOL database includes patterns for .4000 isolates, but data
for hundreds of strains obtained between 1997 and 2007 were
never obtained.
In summary, the large number of singletons observed in our
study, and the absence of progenitors for some SCs, are most likely
due to gaps in our strain collection and genotyping database.
However, SNPs present in some isolates may represent substitu-
tions that originated during latency. In group SC6, the large
majority of cases resulted from the presence of a super-spreader
(NM7-01), and this suggestion is supported by clinical findings (e.g.
smear 3+ and abnormal chest x-rays) consistent with a highly
infectious state. Although it is possible that the high bacterial
burden in this case could have contributed to the emergence and
spread of isolates with differences of 2–4 SNPs, the large
heterogeneity observed in these isolates is also compatible with a
more plausible explanation such as mutations originating after
secondary infection and long latency periods.
Conclusion
We performed a large retrospective and longitudinal study
which resulted in the characterization of the microevolution of a
unique M. tuberculosis strain associated with a high-risk group
population. Despite a clustered genotype pattern based on the
combination of 24-MIRU and spoligotype, we identified a large
genomic deletion in nearly half of the sequenced isolates and
were able to identify the emergence of this deletion event to
have occurred during the first TB outbreak caused by ON-A in
2001. Even though loss of large genomic regions is a major
source of variation in M. tuberculosis [49], to the best of our
knowledge, this is the first study in which identification of such a
region was pinpointed during active TB transmission. Further-
more, we identified a larger than expected heterogeneity
resulting from the microevolution of ON-A in 17 years of
transmission and further delineated this strain into 6 distinct
sub-clusters.
WGS has been proposed as a ‘‘gold standard’’ for strain typing
in M. tuberculosis [12,13,15]. Our study confirms the value of
WGS to determine transmission dynamics and isolate relatedness
in a large cluster of on-going TB transmission extending over
many years in a high risk population. Nonetheless, the large
number of shared contacts between TB cases, the mobility of
homeless/under-housed individuals, and TB latency contributed
to the complexity of determining individual events of TB
transmission in this population.
The peculiarities of our study cohort are noticeable: high risk
cases, on-going transmission despite control measures, high
mobility of cases and likely missing links. All of these factors could
be associated with a higher than average microevolution dynamic
resulting in high variability and multiple transmission chains. Our
intention is not to make general conclusions adapted to other
transmission environments but to decipher the high clonal
complexity and microevolution rates that could be expected in a
transmission event in a complex population.
Supporting Information
Figure S1 Social network analysis of ON-A subjects.Social network analysis was performed using R statistical software
(v3.0.2) with the igraph package. Each large circle represents a
single individual colored by their sub-cluster as determined by
WGS SNP analysis. Grey lines represent common contacts
between study individuals and thick black lines represent direct
epidemiological/social-connections between study individuals.
(PDF)
Material S1 Supplementary methods and results sec-tion. The supplementary methods describe the PCR amplifica-
tion of regions flanking the 15 kb deletion. The supplementary
results describe the WGS results of our laboratory strain H37Rv
and how these results were use to evaluate the accuracy of our
SNP calling algorithm. Table S1 in Material S1 shows the primers
used for PCR amplification of regions flanking the 15 kb deletion.
(DOCX)
Acknowledgments
We would like to thank the Public Health Ontario TB and Mycobacter-
iology Laboratory and research staff, responsible for the initial isolation,
cultivation, and susceptibility testing of the clinical isolates used in the
study, deletion-PCR and genotyping.
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 8 November 2014 | Volume 9 | Issue 11 | e112928

Author Contributions
Conceived and designed the experiments: CM FBJ DCA. Performed the
experiments: CM JLG. Analyzed the data: CM DCA JLG RS ER.
Contributed reagents/materials/analysis tools: CM FBJ. Contributed to
the writing of the manuscript: CM. Review and edit the manuscript: JLG
DCA RS ER FBJ.
References
1. Feske ML, Teeter LD, Musser JM, Graviss EA (2013) Counting the homeless: a
previously incalculable tuberculosis risk and its social determinants. Am J Public
Health 103: 839–848. doi:10.2105/AJPH.2012.300973.
2. Haddad MB, Wilson TW, Ijaz K, Marks SM, Moore M (2005) Tuberculosis and
homelessness in the United States, 1994–2003. JAMA J Am Med Assoc 293:
2762–2766. doi:10.1001/jama.293.22.2762.
3. Bamrah S, Yelk Woodruff RS, Powell K, Ghosh S, Kammerer JS, et al. (2013)
Tuberculosis among the homeless, United States, 1994–2010. Int J Tuberc
Lung Dis Off J Int Union Tuberc Lung Dis 17: 1414–1419. doi:10.5588/
ijtld.13.0270.
4. McAdam JM, Bucher SJ, Brickner PW, Vincent RL, Lascher S (2009) Latent
tuberculosis and active tuberculosis disease rates among the homeless, New
York, New York, USA, 1992–2006. Emerg Infect Dis 15: 1109–1111.
doi:10.3201/eid1507.080410.
5. Khan K, Rea E, McDermaid C, Stuart R, Chambers C, et al. (2011) Active
tuberculosis among homeless persons, Toronto, Ontario, Canada, 1998–2007.
Emerg Infect Dis 17: 357–365. doi:10.3201/eid1703.100833.
6. Adam HJ, Guthrie JL, Bolotin S, Alexander DC, Stuart R, et al. (2010)
Genotypic characterization of tuberculosis transmission within Toronto’s under-
housed population, 1997–2008. Int J Tuberc Lung Dis Off J Int Union Tuberc
Lung Dis 14: 1350–1353.
7. Lofy KH, McElroy PD, Lake L, Cowan LS, Diem LA, et al. (2006) Outbreak of
tuberculosis in a homeless population involving multiple sites of transmission.
Int J Tuberc Lung Dis Off J Int Union Tuberc Lung Dis 10: 683–689.
8. Centers for Disease Control and Prevention (CDC) (2012) Tuberculosis outbreak
associated with a homeless shelter - Kane County, Illinois, 2007–2011. MMWR
Morb Mortal Wkly Rep 61: 186–189.
9. Centers for Disease Control and Prevention (CDC) (2013) Notes from the Field:
Outbreak of Tuberculosis Associated with a Newly Identified Mycobacterium
tuberculosis Genotype - New York City, 2010–2013. MMWR Morb Mortal
Wkly Rep 62: 904.
10. Alexander DC, Guthrie JL, Pyskir D, Maki A, Kurepina N, et al. (2009)
Mycobacterium tuberculosis in Ontario, Canada: Insights from IS6110
restriction fragment length polymorphism and mycobacterial interspersed
repetitive-unit-variable-number tandem-repeat genotyping. J Clin Microbiol
47: 2651–2654. doi:10.1128/JCM.01946-08.
11. Bryant JM, Schurch AC, van Deutekom H, Harris SR, de Beer JL, et al. (2013)
Inferring patient to patient transmission of Mycobacterium tuberculosis from
whole genome sequencing data. BMC Infect Dis 13: 110. doi:10.1186/1471-
2334-13-110.
12. Gardy JL, Johnston JC, Sui SJH, Cook VJ, Shah L, et al. (2011) Whole-Genome
Sequencing and Social-Network Analysis of a Tuberculosis Outbreak.
N Engl J Med 364: 730–739. doi:10.1056/NEJMoa1003176.
13. Kato-Maeda M, Ho C, Passarelli B, Banaei N, Grinsdale J, et al. (2013) Use of
whole genome sequencing to determine the microevolution of Mycobacterium
tuberculosis during an outbreak. PloS One 8: e58235. doi:10.1371/journal.
pone.0058235.
14. Schurch AC, Kremer K, Daviena O, Kiers A, Boeree MJ, et al. (2010) High-
resolution typing by integration of genome sequencing data in a large
tuberculosis cluster. J Clin Microbiol 48: 3403–3406. doi:10.1128/
JCM.00370-10.
15. Walker TM, Ip CLC, Harrell RH, Evans JT, Kapatai G, et al. (2013) Whole-
genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a
retrospective observational study. Lancet Infect Dis 13: 137–146. doi:10.1016/
S1473-3099(12)70277-3.
16. Torok ME, Reuter S, Bryant J, Koser CU, Stinchcombe SV, et al. (2013) Rapid
whole-genome sequencing for investigation of a suspected tuberculosis outbreak.
J Clin Microbiol 51: 611–614. doi:10.1128/JCM.02279-12.
17. Roetzer A, Diel R, Kohl TA, Ruckert C, Nubel U, et al. (2013) Whole Genome
Sequencing versus Traditional Genotyping for Investigation of a Mycobacterium
tuberculosis Outbreak: A Longitudinal Molecular Epidemiological Study. PLoS
Med 10: e1001387. doi:10.1371/journal.pmed.1001387.
18. Alexander DC, Guthrie JL, Pyskir D, Maki A, Kurepina N, et al. (2009)
Mycobacterium tuberculosis in Ontario, Canada: Insights from IS6110
Restriction Fragment Length Polymorphism and Mycobacterial Interspersed
Repetitive-Unit-Variable-Number Tandem-Repeat Genotyping. J Clin Micro-
biol 47: 2651–2654. doi:10.1128/JCM.01946-08.
19. Van Soolingen D, Hermans PW, de Haas PE, Soll DR, van Embden JD (1991)
Occurrence and stability of insertion sequences in Mycobacterium tuberculosis
complex strains: evaluation of an insertion sequence-dependent DNA polymor-
phism as a tool in the epidemiology of tuberculosis. J Clin Microbiol 29: 2578–
2586.
20. Jamieson FB, Guthrie JL, Neemuchwala A, Lastovetska O, Melano RG, et al.
(2014) Profiling of rpoB Mutations and MICs to Rifampicin and Rifabutin in
Mycobacterium tuberculosis. J Clin Microbiol. doi:10.1128/JCM.00691-14.
21. Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rusch-Gerdes S, et al.
(2006) Proposal for standardization of optimized mycobacterial interspersed
repetitive unit-variable-number tandem repeat typing of Mycobacterium
tuberculosis. J Clin Microbiol 44: 4498–4510. doi:10.1128/JCM.01392-06.
22. Cowan LS, Diem L, Brake MC, Crawford JT (2004) Transfer of a
Mycobacterium tuberculosis genotyping method, Spoligotyping, from a reverse
line-blot hybridization, membrane-based assay to the Luminex multianalyte
profiling system. J Clin Microbiol 42: 474–477.
23. Van Embden JD, Cave MD, Crawford JT, Dale JW, Eisenach KD, et al. (1993)
Strain identification of Mycobacterium tuberculosis by DNA fingerprinting:
recommendations for a standardized methodology. J Clin Microbiol 31: 406–
409.
24. Huson DH, Bryant D (2006) Application of phylogenetic networks in
evolutionary studies. Mol Biol Evol 23: 254–267. doi:10.1093/molbev/msj030.
25. Gascuel O (1997) BIONJ: an improved version of the NJ algorithm based on a
simple model of sequence data. Mol Biol Evol 14: 685–695.
26. Dress AWM, Huson DH (2004) Constructing splits graphs. IEEEACM Trans
Comput Biol Bioinforma IEEE ACM 1: 109–115. doi:10.1109/TCBB.2004.27.
27. Gambette P, Huson DH (2008) Improved layout of phylogenetic networks.
IEEEACM Trans Comput Biol Bioinforma IEEE ACM 5: 472–479.
doi:10.1109/tcbb.2007.1046.
28. Csardi G, Nepusz T (2006) The igraph software package for complex network
research. InterJournal Complex Systems: 1695.
29. Sampson SL, Richardson M, Van Helden PD, Warren RM (2004) IS6110-
mediated deletion polymorphism in isogenic strains of Mycobacterium
tuberculosis. J Clin Microbiol 42: 895–898.
30. Chen P, Ruiz RE, Li Q, Silver RF, Bishai WR (2000) Construction and
characterization of a Mycobacterium tuberculosis mutant lacking the alternate
sigma factor gene, sigF. Infect Immun 68: 5575–5580.
31. Geiman DE, Kaushal D, Ko C, Tyagi S, Manabe YC, et al. (2004) Attenuation
of late-stage disease in mice infected by the Mycobacterium tuberculosis mutant
lacking the SigF alternate sigma factor and identification of SigF-dependent
genes by microarray analysis. Infect Immun 72: 1733–1745.
32. Parida BK, Douglas T, Nino C, Dhandayuthapani S (2005) Interactions of anti-
sigma factor antagonists of Mycobacterium tuberculosis in the yeast two-hybrid
system. Tuberculosis 85: 347–355. doi:10.1016/j.tube.2005.08.001.
33. Hartkoorn RC, Sala C, Uplekar S, Busso P, Rougemont J, et al. (2012) Genome-
Wide Definition of the SigF Regulon in Mycobacterium tuberculosis. J Bacteriol
194: 2001–2009. doi:10.1128/JB.06692-11.
34. Pesavento C, Hengge R (2009) Bacterial nucleotide-based second messengers.
Curr Opin Microbiol 12: 170–176. doi:10.1016/j.mib.2009.01.007.
35. Klinkenberg LG, Lee J, Bishai WR, Karakousis PC (2010) The Stringent
Response Is Required for Full Virulence of Mycobacterium tuberculosis in
Guinea Pigs. J Infect Dis 202: 1397–1404. doi:10.1086/656524.
36. Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, et al. (2000) The
stringent response of Mycobacterium tuberculosis is required for long-term
survival. J Bacteriol 182: 4889–4898.
37. Fang Z, Doig C, Kenna DT, Smittipat N, Palittapongarnpim P, et al. (1999)
IS6110-mediated deletions of wild-type chromosomes of Mycobacterium
tuberculosis. J Bacteriol 181: 1014–1020.
38. Ho TB, Robertson BD, Taylor GM, Shaw RJ, Young DB (2000) Comparison of
Mycobacterium tuberculosis genomes reveals frequent deletions in a 20 kb
variable region in clinical isolates. Yeast Chichester Engl 17: 272–282.
doi:10.1002/1097-0061(200012)17:4,272::AID-YEA48.3.0.CO;2-2.
39. Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, et al. (2013) The
Role of Selection in Shaping Diversity of Natural M. tuberculosis Populations.
PLoS Pathog 9: e1003543. doi:10.1371/journal.ppat.1003543.
40. Newton SM, Smith RJ, Wilkinson KA, Nicol MP, Garton NJ, et al. (2006) A
deletion defining a common Asian lineage of Mycobacterium tuberculosis
associates with immune subversion. Proc Natl Acad Sci 103: 15594–15598.
doi:10.1073/pnas.0604283103.
41. Yesilkaya H, Dale JW, Strachan NJC, Forbes KJ (2005) Natural transposon
mutagenesis of clinical isolates of Mycobacterium tuberculosis: how many genes
does a pathogen need? J Bacteriol 187: 6726–6732. doi:10.1128/
JB.187.19.6726-6732.2005.
42. Reyes A, Sandoval A, Cubillos-Ruiz A, Varley KE, Hernandez-Neuta I, et al.
(2012) IS-seq: a novel high throughput survey of in vivo IS6110 transposition in
multiple Mycobacterium tuberculosis genomes. BMC Genomics 13: 249.
doi:10.1186/1471-2164-13-249.
43. Benedetti A, Menzies D, Behr MA, Schwartzman K, Jin Y (2010) How close is
close enough? Exploring matching criteria in the estimation of recent
transmission of tuberculosis. Am J Epidemiol 172: 318–326. doi:10.1093/aje/
kwq124.
44. De Boer AS, Kremer K, Borgdorff MW, de Haas PE, Heersma HF, et al. (2000)
Genetic heterogeneity in Mycobacterium tuberculosis isolates reflected in
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 9 November 2014 | Volume 9 | Issue 11 | e112928

IS6110 restriction fragment length polymorphism patterns as low-intensity
bands. J Clin Microbiol 38: 4478–4484.45. Glynn JR, Vynnycky E, Fine PE (1999) Influence of sampling on estimates of
clustering and recent transmission of Mycobacterium tuberculosis derived from
DNA fingerprinting techniques. Am J Epidemiol 149: 366–371.46. Schurch AC, Kremer K, Hendriks ACA, Freyee B, McEvoy CRE, et al. (2011)
SNP/RD typing of Mycobacterium tuberculosis Beijing strains reveals local andworldwide disseminated clonal complexes. PloS One 6: e28365. doi:10.1371/
journal.pone.0028365.
47. Ford C, Yusim K, Ioerger T, Feng S, Chase M, et al. (2012) Mycobacteriumtuberculosis – Heterogeneity revealed through whole genome sequencing.
Tuberculosis 92: 194–201. doi:10.1016/j.tube.2011.11.003.48. Colangeli R, Arcus VL, Cursons RT, Ruthe A, Karalus N, et al. (2014) Whole
Genome Sequencing of Mycobacterium tuberculosis Reveals Slow Growth andLow Mutation Rates during Latent Infections in Humans. PLoS ONE 9:
e91024. doi:10.1371/journal.pone.0091024.
49. Tsolaki AG, Hirsh AE, DeRiemer K, Enciso JA, Wong MZ, et al. (2004)Functional and evolutionary genomics of Mycobacterium tuberculosis: Insights
from genomic deletions in 100 strains. Proc Natl Acad Sci 101: 4865–4870.
doi:10.1073/pnas.0305634101.
50. Kruh NA, Troudt J, Izzo A, Prenni J, Dobos KM (2010) Portrait of a Pathogen:
The Mycobacterium tuberculosis Proteome In Vivo. PLoS ONE 5: e13938.
doi:10.1371/journal.pone.0013938.
51. Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K (2002) Evaluation of a
nutrient starvation model of Mycobacterium tuberculosis persistence by gene
and protein expression profiling. Mol Microbiol 43: 717–731.
52. Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, et al. (2006) The
Mycobacterium tuberculosis PhoPR two-component system regulates genes
essential for virulence and complex lipid biosynthesis. Mol Microbiol 60: 312–
330. doi:10.1111/j.1365-2958.2006.05102.x.
53. Arnvig KB, Comas I, Thomson NR, Houghton J, Boshoff HI, et al. (2011)
Sequence-Based Analysis Uncovers an Abundance of Non-Coding RNA in the
Total Transcriptome of Mycobacterium tuberculosis. PLoS Pathog 7: e1002342.
doi:10.1371/journal.ppat.1002342.
Microevolution of an Endemic M. tuberculosis Strain
PLOS ONE | www.plosone.org 10 November 2014 | Volume 9 | Issue 11 | e112928