Investigation of protein conformation and interactions with salts via X-ray absorption spectroscopy
6
Investigation of protein conformation and interactions with salts via X-ray absorption spectroscopy Craig P. Schwartz a,b , Janel S. Uejio a,b , Andrew M. Duffin a,b , Alice H. England a,b , Daniel N. Kelly a , David Prendergast c , and Richard J. Saykally a,b,1 a Department of Chemistry, University of California, Berkeley, CA 94720; b Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720; and c Molecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720 Edited by Michael L. Klein, Temple University, Philadelphia, PA, and approved June 30, 2010 (received for review May 11, 2010) Nitrogen K-edge spectra of aqueous triglycine were measured using liquid microjets, and the effects of Hofmeister-active salts on the spectra were observed. Spectra simulated using density functional theory, sampled from room temperature classical molecular dynamics trajectories, capture all major features in the measured spectra. The spectrum of triglycine in water is quite similar to that in the presence of chaotropic sodium bromide (and other halides), which raises the solubility of proteins. However, a new feature is found when kosmotropic Na 2 SO 3 , which lowers solubility, is present; this feature results from excitations of the nitrogen atom in the terminal amino group of triglycine. Both direct interactions between this salt and the protonated amino terminus, as well as cor- responding changes in the conformational dynamics of the system, contribute to this new feature. These molecular measurements support a different mechanism for the Hofmeister effect than has previously been suggested based on thermodynamic measure- ments. It is also shown that near edge X-ray absorption fine struc- ture (NEXAFS) is sensitive to strong direct interaction between certain salts and charged peptides. However, by investigating the sensitivity of NEXAFS to the extreme structural differences between model β-sheets and α-helices, we conclude that this technique is re- latively insensitive to secondary structure of peptides and proteins. X-ray absorption near edge structure ∣ molecular dynamics ∣ density functional theory ∣ eXcited electron and Core Hole O ver 100 years ago Hofmeister discovered that adding salts to egg white protein could alter its solubility (1). Different con- centrations of salt would cause the protein to precipitate out of solution with the amount specifically related to the ion identities. This ordering of salt-protein interactions has since become known as the “Hofmeister series” (2), and the associated “Hof- meister effects” now extend to many different phenomena, including protein denaturing, optical rotation of sugars, and bacterial growth rates (3). It is known that anions have a stronger effect on protein solubility than do cations and the general anion ordering is CO 3 2− > SO 4 2− > SO 3 2− > Cl − > Br − > I − > SCN − : The species on the left are generally referred to as “kosmo- tropes,” or “structure makers,” and the species on the right are generally referred to as “chaotropes,” or “structure breakers” (3, 4). Originally, these terms derived from the entropies of hydration and referred to making and breaking the water struc- ture, which was believed to be the cause of the Hofmeister effects. However, this model of ascribing Hofmeister effects to bulk water changes has come under criticism, given more recent experimen- tal findings (3). It is now believed that interactions either directly between a protein and the ions or interactions mediated by a single solvation shell underlie the effects (3, 5). Some groups have recently presented measurements to support the original conten- tion that Hofmeister effects resulted from changes in the bulk water structure, but their models treated water as a two-state system, which has come under intense recent criticism (6, 7). It has also been proposed that absolute free energies of hydra- tion also underlie Hofmeister effects (8, 9), contending that if absolute hydration-free energies of anionic and cationic groups match, then ion pairing will occur, signifying strong ion-protein interactions (10). Cremer and coworkers have performed a series of studies wherein protein analogues were combined with various amounts of salt, causing the lower critical solution temperature (LCST)—the critical temperature below which the mixture is miscible in all portions—to change as a function of salt concen- tration and identity (3, 11–13). They observed that kosmotropes lower the LCST more than do chaotropes. Based on fitting of their measurements combined with thermodynamic arguments, they argued that the way the salts affect the LCST is dependent upon the Hofmeister activity of the anion. Chaotropes (such as Br − ) were believed to directly interact with the protein analogue near the nitrogen sites, as well as to interact with the exposed hydrophobic surfaces. Kosmotropes, on the other hand, were believed to polarize the water interacting with the nitrogen- and oxygen-containing groups as well as to interact with hydropho- bic groups. However, these conclusions were based on light scat- tering measurements and thermodynamic data, so the interactions had to be inferred, rather than being observed directly. Herein, we show evidence that suggests kosmotropes directly interact with the nitrogen backbone of peptides based on a molecular probe, casting doubt on the proposed mechanism behind the thermody- namic data. Here, we investigate triglycine as a model peptide via near edge X-ray absorption fine structure (NEXAFS) experiments and theoretical methods, seeking to further characterize the interactions of proteins with selected salts. Nitrogen K-edge (ex- citation from the N 1s orbital) spectra of triglycine in aqueous solution were measured, without cosolutes, with chaotropic coso- lute Na 2 SO 3 , and with cosolute NaBr, seeking to quantify observed differences in the spectra of triglycine due to the presence of the salts. NEXAFS probes unoccupied antibonding and Rydberg states, which are highly sensitive to intermolecular interactions (14–16). This follows similar recent studies using X-ray spectroscopy to characterize interactions between ions and small biomolecules (16, 17). There are now several methods available for obtaining core-level spectra of liquids; we use small diameter jets of water solutions (∼30 microns) with windowless coupling to a synchro- tron beamline. This approach is particularly useful for studying complex molecules, because the high velocity and continuous renewal of the liquid jet minimizes spectral contributions due to sample damage (18). The chemical information that can be extracted from such measurements is limited in many respects by the accuracy of theoretical methods for computing core-level Author contributions: R.J.S. designed research; C.P.S., J.S.U., A.M.D., A.H.E., and D.N.K. performed research; C.P.S. and D.P. analyzed data; and C.P.S. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1 To whom correspondence should be addressed. E-mail: [email protected]. 14008–14013 ∣ PNAS ∣ August 10, 2010 ∣ vol. 107 ∣ no. 32 www.pnas.org/cgi/doi/10.1073/pnas.1006435107
Transcript of Investigation of protein conformation and interactions with salts via X-ray absorption spectroscopy

Investigation of protein conformation and interactionswith salts via X-ray absorption spectroscopyCraig P. Schwartza,b, Janel S. Uejioa,b, Andrew M. Duffina,b, Alice H. Englanda,b, Daniel N. Kellya,David Prendergastc, and Richard J. Saykallya,b,1
aDepartment of Chemistry, University of California, Berkeley, CA 94720; bChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley,CA 94720; and cMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720
Edited by Michael L. Klein, Temple University, Philadelphia, PA, and approved June 30, 2010 (received for review May 11, 2010)
NitrogenK-edge spectraof aqueous triglycineweremeasuredusingliquid microjets, and the effects of Hofmeister-active salts on thespectra were observed. Spectra simulated using density functionaltheory, sampled from room temperature classical moleculardynamics trajectories, capture all major features in the measuredspectra. The spectrum of triglycine in water is quite similar to thatin the presence of chaotropic sodium bromide (and other halides),which raises the solubility of proteins. However, a new feature isfound when kosmotropic Na2SO3, which lowers solubility, ispresent; this feature results from excitations of the nitrogen atomin the terminal amino group of triglycine. Both direct interactionsbetween this salt and theprotonatedamino terminus, aswell as cor-responding changes in the conformational dynamics of the system,contribute to this new feature. These molecular measurementssupport a different mechanism for the Hofmeister effect than haspreviously been suggested based on thermodynamic measure-ments. It is also shown that near edge X-ray absorption fine struc-ture (NEXAFS) is sensitive to strong direct interaction betweencertain salts and charged peptides. However, by investigating thesensitivityofNEXAFS to theextreme structural differencesbetweenmodel β-sheets and α-helices, we conclude that this technique is re-latively insensitive to secondary structure of peptides and proteins.
X-ray absorption near edge structure ∣ molecular dynamics ∣density functional theory ∣ eXcited electron and Core Hole
Over 100 years ago Hofmeister discovered that adding salts toegg white protein could alter its solubility (1). Different con-
centrations of salt would cause the protein to precipitate out ofsolution with the amount specifically related to the ion identities.This ordering of salt-protein interactions has since becomeknown as the “Hofmeister series” (2), and the associated “Hof-meister effects” now extend to many different phenomena,including protein denaturing, optical rotation of sugars, andbacterial growth rates (3). It is known that anions have a strongereffect on protein solubility than do cations and the general anionordering is
CO32− > SO4
2− > SO32− > Cl− > Br− > I− > SCN−:
The species on the left are generally referred to as “kosmo-tropes,” or “structure makers,” and the species on the rightare generally referred to as “chaotropes,” or “structure breakers”(3, 4). Originally, these terms derived from the entropies ofhydration and referred to making and breaking the water struc-ture, which was believed to be the cause of the Hofmeister effects.However, this model of ascribing Hofmeister effects to bulk waterchanges has come under criticism, given more recent experimen-tal findings (3). It is now believed that interactions either directlybetween a protein and the ions or interactions mediated by asingle solvation shell underlie the effects (3, 5). Some groups haverecently presented measurements to support the original conten-tion that Hofmeister effects resulted from changes in the bulkwater structure, but their models treated water as a two-statesystem, which has come under intense recent criticism (6, 7).
It has also been proposed that absolute free energies of hydra-tion also underlie Hofmeister effects (8, 9), contending that ifabsolute hydration-free energies of anionic and cationic groupsmatch, then ion pairing will occur, signifying strong ion-proteininteractions (10). Cremer and coworkers have performed a seriesof studies wherein protein analogues were combined with variousamounts of salt, causing the lower critical solution temperature(LCST)—the critical temperature below which the mixture ismiscible in all portions—to change as a function of salt concen-tration and identity (3, 11–13). They observed that kosmotropeslower the LCST more than do chaotropes. Based on fitting oftheir measurements combined with thermodynamic arguments,they argued that the way the salts affect the LCST is dependentupon the Hofmeister activity of the anion. Chaotropes (such asBr−) were believed to directly interact with the protein analoguenear the nitrogen sites, as well as to interact with the exposedhydrophobic surfaces. Kosmotropes, on the other hand, werebelieved to polarize the water interacting with the nitrogen-and oxygen-containing groups as well as to interact with hydropho-bic groups. However, these conclusions were based on light scat-teringmeasurements and thermodynamic data, so the interactionshad to be inferred, rather than being observed directly. Herein,we show evidence that suggests kosmotropes directly interact withthe nitrogen backbone of peptides based on a molecular probe,casting doubt on the proposed mechanism behind the thermody-namic data.
Here, we investigate triglycine as a model peptide via nearedge X-ray absorption fine structure (NEXAFS) experimentsand theoretical methods, seeking to further characterize theinteractions of proteins with selected salts. Nitrogen K-edge (ex-citation from the N 1s orbital) spectra of triglycine in aqueoussolution were measured, without cosolutes, with chaotropic coso-lute Na2SO3, and with cosolute NaBr, seeking to quantifyobserved differences in the spectra of triglycine due to thepresence of the salts. NEXAFS probes unoccupied antibondingand Rydberg states, which are highly sensitive to intermolecularinteractions (14–16). This follows similar recent studies usingX-ray spectroscopy to characterize interactions between ionsand small biomolecules (16, 17).
There are now several methods available for obtainingcore-level spectra of liquids; we use small diameter jets of watersolutions (∼30 microns) with windowless coupling to a synchro-tron beamline. This approach is particularly useful for studyingcomplex molecules, because the high velocity and continuousrenewal of the liquid jet minimizes spectral contributions dueto sample damage (18). The chemical information that can beextracted from such measurements is limited in many respectsby the accuracy of theoretical methods for computing core-level
Author contributions: R.J.S. designed research; C.P.S., J.S.U., A.M.D., A.H.E., and D.N.K.performed research; C.P.S. and D.P. analyzed data; and C.P.S. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.1To whom correspondence should be addressed. E-mail: [email protected].
14008–14013 ∣ PNAS ∣ August 10, 2010 ∣ vol. 107 ∣ no. 32 www.pnas.org/cgi/doi/10.1073/pnas.1006435107
spectra. Here, we use the recently developed eXcited electronand Core Hole (XCH) method, which employs density functionaltheory (DFT) to accurately calculate NEXAFS spectra (19, 20).We simulated the atomic configurations of triglycine with andwithout additional salts in aqueous solution at 300 K usingclassical molecular dynamics. Sampling a trajectory in our DFTsimulation captures the impact of nuclear motion on the calcu-lated spectrum (21). This technique has been used previously tointerpret the spectra of solvated amino acids and the prototypicalaromatic molecule, pyrrole in aqueous solutions (22, 23). Toexplore possible spectral signatures of secondary structure, wealso used rigid structural models of polyalanine in both anantiparallel β-sheet and an α-helix using structural parametersfrom the literature (24) to calculate the respective N-edgeX-ray absorption spectra.
There have been many studies of proteins and amino acids insolid form using NEXAFS; every naturally occurring individualamino acid has had its core-level spectra characterized at the C,N, and O K edges (25), and a variety of polypeptides and proteinspectra have been measured under similar conditions (26–28).These data have been used to predict (using a building-blockapproach) the corresponding spectrum of a protein based on itsamino acid composition with reasonable success (28). Based onsuch studies, it has been suggested that the difference in π�-reso-nances between a model dipeptide and a protein could be due tospectral differences between an α-helix and a planar structure (26,27). In this work, we show preliminary evidence that the N K-edgespectrum will be relatively unchanged in these conformations.
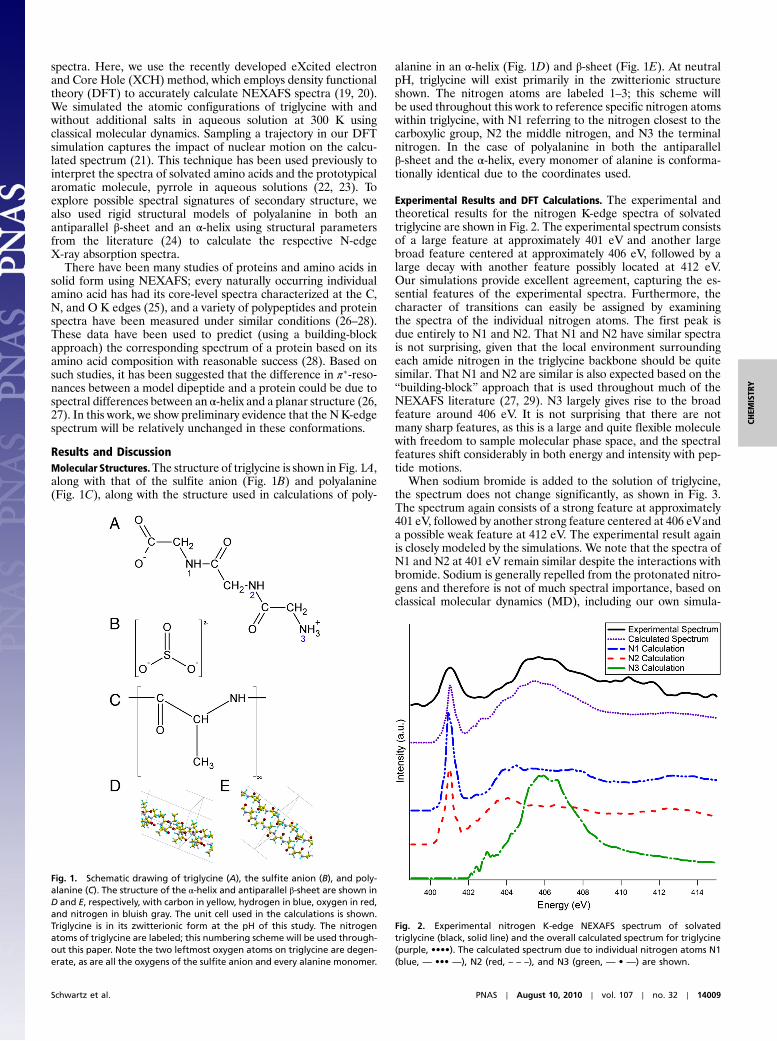
Results and DiscussionMolecular Structures.The structure of triglycine is shown in Fig. 1A,along with that of the sulfite anion (Fig. 1B) and polyalanine(Fig. 1C), along with the structure used in calculations of poly-
alanine in an α-helix (Fig. 1D) and β-sheet (Fig. 1E). At neutralpH, triglycine will exist primarily in the zwitterionic structureshown. The nitrogen atoms are labeled 1–3; this scheme willbe used throughout this work to reference specific nitrogen atomswithin triglycine, with N1 referring to the nitrogen closest to thecarboxylic group, N2 the middle nitrogen, and N3 the terminalnitrogen. In the case of polyalanine in both the antiparallelβ-sheet and the α-helix, every monomer of alanine is conforma-tionally identical due to the coordinates used.
Experimental Results and DFT Calculations. The experimental andtheoretical results for the nitrogen K-edge spectra of solvatedtriglycine are shown in Fig. 2. The experimental spectrum consistsof a large feature at approximately 401 eV and another largebroad feature centered at approximately 406 eV, followed by alarge decay with another feature possibly located at 412 eV.Our simulations provide excellent agreement, capturing the es-sential features of the experimental spectra. Furthermore, thecharacter of transitions can easily be assigned by examiningthe spectra of the individual nitrogen atoms. The first peak isdue entirely to N1 and N2. That N1 and N2 have similar spectrais not surprising, given that the local environment surroundingeach amide nitrogen in the triglycine backbone should be quitesimilar. That N1 and N2 are similar is also expected based on the“building-block” approach that is used throughout much of theNEXAFS literature (27, 29). N3 largely gives rise to the broadfeature around 406 eV. It is not surprising that there are notmany sharp features, as this is a large and quite flexible moleculewith freedom to sample molecular phase space, and the spectralfeatures shift considerably in both energy and intensity with pep-tide motions.
When sodium bromide is added to the solution of triglycine,the spectrum does not change significantly, as shown in Fig. 3.The spectrum again consists of a strong feature at approximately401 eV, followed by another strong feature centered at 406 eVanda possible weak feature at 412 eV. The experimental result againis closely modeled by the simulations. We note that the spectra ofN1 and N2 at 401 eV remain similar despite the interactions withbromide. Sodium is generally repelled from the protonated nitro-gens and therefore is not of much spectral importance, based onclassical molecular dynamics (MD), including our own simula-
Fig. 1. Schematic drawing of triglycine (A), the sulfite anion (B), and poly-alanine (C). The structure of the α-helix and antiparallel β-sheet are shown inD and E, respectively, with carbon in yellow, hydrogen in blue, oxygen in red,and nitrogen in bluish gray. The unit cell used in the calculations is shown.Triglycine is in its zwitterionic form at the pH of this study. The nitrogenatoms of triglycine are labeled; this numbering scheme will be used through-out this paper. Note the two leftmost oxygen atoms on triglycine are degen-erate, as are all the oxygens of the sulfite anion and every alanine monomer.
Fig. 2. Experimental nitrogen K-edge NEXAFS spectrum of solvatedtriglycine (black, solid line) and the overall calculated spectrum for triglycine(purple, ••••). The calculated spectrum due to individual nitrogen atoms N1(blue, — ••• —), N2 (red, – – –), and N3 (green, — • —) are shown.
Schwartz et al. PNAS ∣ August 10, 2010 ∣ vol. 107 ∣ no. 32 ∣ 14009
CHEM
ISTR
Y
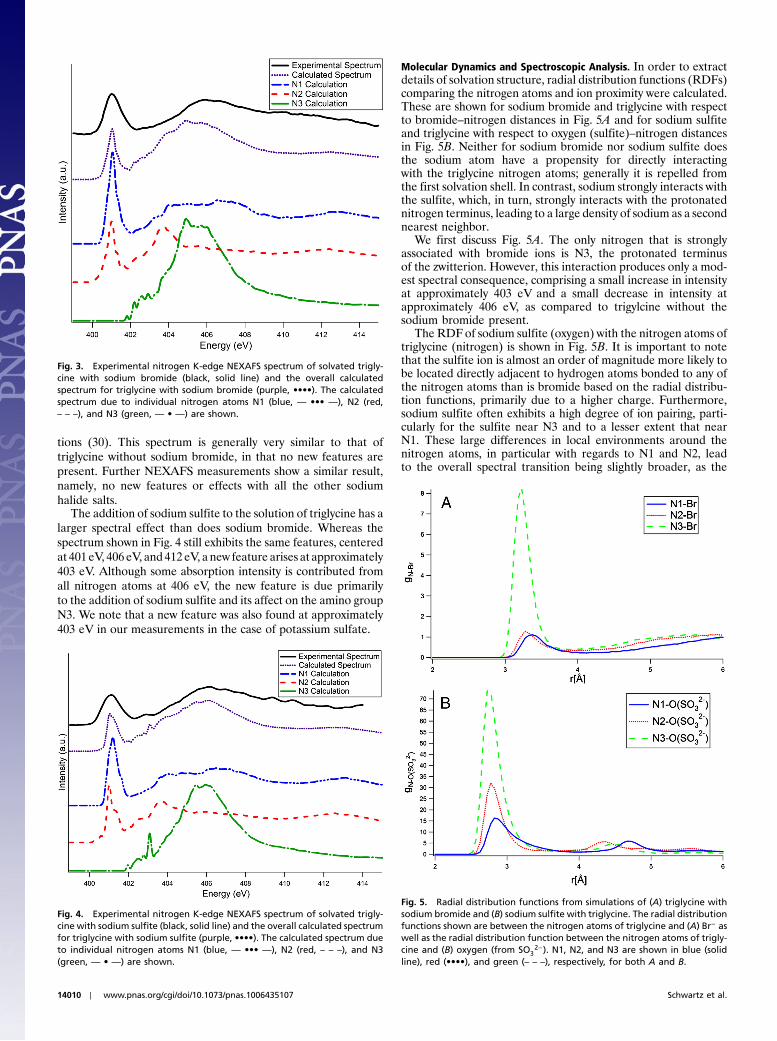
tions (30). This spectrum is generally very similar to that oftriglycine without sodium bromide, in that no new features arepresent. Further NEXAFS measurements show a similar result,namely, no new features or effects with all the other sodiumhalide salts.
The addition of sodium sulfite to the solution of triglycine has alarger spectral effect than does sodium bromide. Whereas thespectrum shown in Fig. 4 still exhibits the same features, centeredat 401 eV, 406 eV, and 412 eV, a new feature arises at approximately403 eV. Although some absorption intensity is contributed fromall nitrogen atoms at 406 eV, the new feature is due primarilyto the addition of sodium sulfite and its affect on the amino groupN3. We note that a new feature was also found at approximately403 eV in our measurements in the case of potassium sulfate.
Molecular Dynamics and Spectroscopic Analysis. In order to extractdetails of solvation structure, radial distribution functions (RDFs)comparing the nitrogen atoms and ion proximity were calculated.These are shown for sodium bromide and triglycine with respectto bromide–nitrogen distances in Fig. 5A and for sodium sulfiteand triglycine with respect to oxygen (sulfite)–nitrogen distancesin Fig. 5B. Neither for sodium bromide nor sodium sulfite doesthe sodium atom have a propensity for directly interactingwith the triglycine nitrogen atoms; generally it is repelled fromthe first solvation shell. In contrast, sodium strongly interacts withthe sulfite, which, in turn, strongly interacts with the protonatednitrogen terminus, leading to a large density of sodium as a secondnearest neighbor.
We first discuss Fig. 5A. The only nitrogen that is stronglyassociated with bromide ions is N3, the protonated terminusof the zwitterion. However, this interaction produces only a mod-est spectral consequence, comprising a small increase in intensityat approximately 403 eV and a small decrease in intensity atapproximately 406 eV, as compared to trigylcine without thesodium bromide present.
The RDFof sodium sulfite (oxygen) with the nitrogen atoms oftriglycine (nitrogen) is shown in Fig. 5B. It is important to notethat the sulfite ion is almost an order of magnitude more likely tobe located directly adjacent to hydrogen atoms bonded to any ofthe nitrogen atoms than is bromide based on the radial distribu-tion functions, primarily due to a higher charge. Furthermore,sodium sulfite often exhibits a high degree of ion pairing, parti-cularly for the sulfite near N3 and to a lesser extent that nearN1. These large differences in local environments around thenitrogen atoms, in particular with regards to N1 and N2, leadto the overall spectral transition being slightly broader, as the
Fig. 3. Experimental nitrogen K-edge NEXAFS spectrum of solvated trigly-cine with sodium bromide (black, solid line) and the overall calculatedspectrum for triglycine with sodium bromide (purple, ••••). The calculatedspectrum due to individual nitrogen atoms N1 (blue, — ••• —), N2 (red,– – –), and N3 (green, — • —) are shown.
Fig. 4. Experimental nitrogen K-edge NEXAFS spectrum of solvated trigly-cine with sodium sulfite (black, solid line) and the overall calculated spectrumfor triglycine with sodium sulfite (purple, ••••). The calculated spectrum dueto individual nitrogen atoms N1 (blue, — ••• —), N2 (red, – – –), and N3(green, — • —) are shown.
Fig. 5. Radial distribution functions from simulations of (A) triglycine withsodium bromide and (B) sodium sulfite with triglycine. The radial distributionfunctions shown are between the nitrogen atoms of triglycine and (A) Br− aswell as the radial distribution function between the nitrogen atoms of trigly-cine and (B) oxygen (from SO3
2−). N1, N2, and N3 are shown in blue (solidline), red (••••), and green (– – –), respectively, for both A and B.
14010 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1006435107 Schwartz et al.

1s → lowest unoccupied molecular orbital (LUMO) transitionis no longer energetically degenerate for N1 and N2. The 1s →LUMO transition shifts in energy as the local environment shiftsaround the N2 atom; this is particularly related to the position ofthe sulfite and the N-H bond length. We note that there are otherfactors that contribute, but a single cause cannot be isolated atthe present time. The new peak at 403 eV appears to be causedprimarily by the N3 group interacting with the sulfite ion. A largeintensity at that energy is not found in all of the calculated spec-tra, but only for a fraction of the calculated snapshots. However,many snapshots have similar 1s → LUMO transition energies(i.e., the absolute energy of many transitions is approximately403 eV). This implies a similar local environment for a varietyof snapshots, indicating that sulfite induces a certain amountof local structure in or around triglycine. If the environmentwas shifting constantly, one would not expect this peak to appear.
Although the RDFs characterize the distance between trigly-cine and ions, they provide no dynamical information. In order todetermine structural variations, the rms fitted backbone of trigly-cine from our simulations is shown in Fig. 6, with the standarddeviation in the rms fitting corresponding to the spheroid size.It is important to note that for all of these structures, the back-
bones lie roughly in a plane; i.e., they are not twisted. However,the sodium sulfite solution exhibits a different conformation oftriglycine than when sodium bromide is present. Furthermore,the rms deviation increases when triglycine is simulated witheither no salt present or with sodium bromide, as compared tosodium sulfite; this implies the existence of less conformationalvariations in the presence of sodium sulfite. This is in agreementwith the traditional interpretation of Hofmeister effects, namely,that kosmotropic solvent molecules such as SO3
2− will make itharder for a protein to bend to form a cavity (the protein becomesless conformationally flexible) (3). The chaotropic sodium bro-mide induces a very slight increase in the conformational flexibil-ity of triglycine, as compared to the peptide without cosolute, inagreement with the Hofmeister series for solvent cavity formation(3). Although this overall protein conformational flexibility maybe important for phenomena related to the Hofmeister series,NEXAFS appears to have limited sensitivity to this effect, basedon our calculations. Conformational flexibility can still be impor-tant with regard to peak width, however.
The reason for this limited spectroscopic sensitivity can beelucidated by examining the states probed in the experiment.Example states are shown for the LUMOþ 1 in Fig. 7. As seenin this image, the state is fairly well localized on a given segmentof the polypeptide chain. When N1 is excited, the excitation is notdelocalized very far along the backbone, although it does reachthe carbon of the carboxylic group. When N2 is excited, the casethat would be most representative of the middle of a large pro-tein, the state is almost entirely localized on N2 and atoms atmost two bonds away. There is no significant off-chain mixing.The state is extremely localized. The N3 excited state mixes morethan any other nitrogen, with the LUMOþ 1 delocalized allthe way back up the chain of trigylcine to N2. It also mixes withneighboring water molecules, and for that reason, N3 is likely tobe much more sensitive to its surroundings than are the othernitrogen molecules.
The applicability of our findings to the general elucidation ofHofmeister effects appears to be indirect, in particular, with re-gard to the restriction of conformational flexibility, as the kosmo-tropic sulfite ion is largely interacting with the protonated(zwitterionic) nitrogen group, wherein the anion pairs with so-dium ions, causing the carboxylic acid group to loop aroundand interact with the sodium. In other words, the present confor-mational findings are probably specific to short oligopeptides;however, one could envision such salts affecting proteins whenmultiple charged groups are in close spatial proximity, as will of-ten be the case in actual proteins. We note that it was recentlypredicted that iodide and bromide ions would not directly inter-act with the protein backbone, but rather would instead interactwith hydrophobic regions (30); we have here experimentally andtheoretically confirmed that bromide does not strongly interactwith a hydrophilic polypeptide. The calculations in the previouswork indicate that the destabilizing effect of bromide in the Hof-
Fig. 6. Result of rms fitting the backbone of triglycine to the averagestructure for a given MD simulation. The carbon atoms are in blue, andthe nitrogen atoms are in purple. The results correspond to the simulationsof (A) triglycine, (B) triglycine and sodium bromide, and (C) triglycine andsodium sulfite. The standard deviation of the fit for each atom correspondsto the size of each atom. Note how similar A and B are as compared to C andthe often larger size of the spheroids in A and B.
Fig. 7. Isosurfaces (orange and green) are shown for the LUMOþ 1 of theexcited state of triglycine solvated with sodium sulfite and water, corre-sponding to 10%of the total integrated value. Many of thewaters have beenremoved for clarity. The images show an excitation on N1, N2, and N3 fromleft to right. Despite being a delocalized state, the state does not extendthroughout the entire molecule.
Schwartz et al. PNAS ∣ August 10, 2010 ∣ vol. 107 ∣ no. 32 ∣ 14011
CHEM
ISTR
Y
meister effect is due to attraction to hydrophobic regions of theprotein.
Previous studies have addressed the thermodynamics of theHofmeister series, but molecular level insights have been muchharder to gain. Cremer and coworkers have identified systematicthermodynamic dependencies of the LCST based on specificanion identity (3, 12, 13). Their conclusion—that the more kos-motropic anions would interact with the protein via mediatingwaters and that chaotropic anions would interact with a proteindirectly—is refuted by the results presented here. In order fortheir conclusion to be correct, the interaction would have tonot affect the Hofmeister series in any way, a seemingly unlikely,though possible, outcome. However, it is important to note thattheir measurements were taken at salt concentrations that weresignificantly higher than those studied here, and it is possible thatdifferent effects occur as concentration approaches the solubilitylimit. Although this therefore does not prove that the mechanismproposed on thermodynamic arguments by Cremer and cowor-kers is incorrect, it should emphasize the difficulty of trying toinfer molecular phenomena from measurements that are inher-ently macroscopic, and should cast serious doubt on their mole-cular level interpretation.
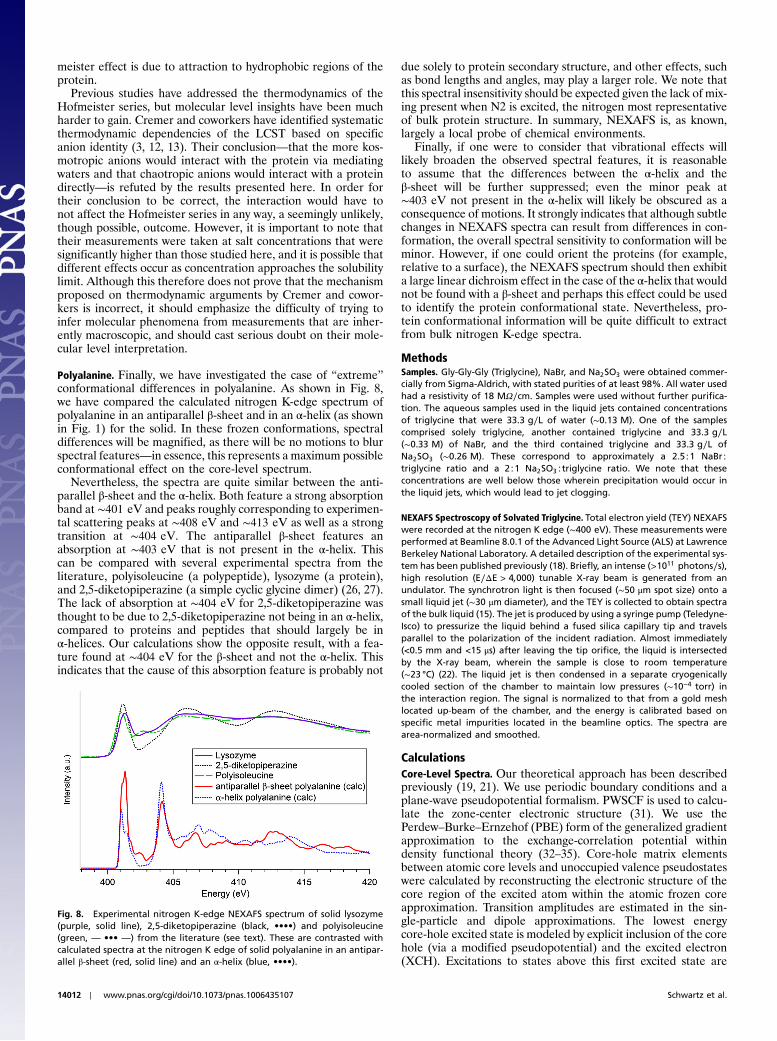
Polyalanine. Finally, we have investigated the case of “extreme”conformational differences in polyalanine. As shown in Fig. 8,we have compared the calculated nitrogen K-edge spectrum ofpolyalanine in an antiparallel β-sheet and in an α-helix (as shownin Fig. 1) for the solid. In these frozen conformations, spectraldifferences will be magnified, as there will be no motions to blurspectral features—in essence, this represents a maximum possibleconformational effect on the core-level spectrum.
Nevertheless, the spectra are quite similar between the anti-parallel β-sheet and the α-helix. Both feature a strong absorptionband at ∼401 eV and peaks roughly corresponding to experimen-tal scattering peaks at ∼408 eV and ∼413 eV as well as a strongtransition at ∼404 eV. The antiparallel β-sheet features anabsorption at ∼403 eV that is not present in the α-helix. Thiscan be compared with several experimental spectra from theliterature, polyisoleucine (a polypeptide), lysozyme (a protein),and 2,5-diketopiperazine (a simple cyclic glycine dimer) (26, 27).The lack of absorption at ∼404 eV for 2,5-diketopiperazine wasthought to be due to 2,5-diketopiperazine not being in an α-helix,compared to proteins and peptides that should largely be inα-helices. Our calculations show the opposite result, with a fea-ture found at ∼404 eV for the β-sheet and not the α-helix. Thisindicates that the cause of this absorption feature is probably not
due solely to protein secondary structure, and other effects, suchas bond lengths and angles, may play a larger role. We note thatthis spectral insensitivity should be expected given the lack of mix-ing present when N2 is excited, the nitrogen most representativeof bulk protein structure. In summary, NEXAFS is, as known,largely a local probe of chemical environments.
Finally, if one were to consider that vibrational effects willlikely broaden the observed spectral features, it is reasonableto assume that the differences between the α-helix and theβ-sheet will be further suppressed; even the minor peak at∼403 eV not present in the α-helix will likely be obscured as aconsequence of motions. It strongly indicates that although subtlechanges in NEXAFS spectra can result from differences in con-formation, the overall spectral sensitivity to conformation will beminor. However, if one could orient the proteins (for example,relative to a surface), the NEXAFS spectrum should then exhibita large linear dichroism effect in the case of the α-helix that wouldnot be found with a β-sheet and perhaps this effect could be usedto identify the protein conformational state. Nevertheless, pro-tein conformational information will be quite difficult to extractfrom bulk nitrogen K-edge spectra.
MethodsSamples. Gly-Gly-Gly (Triglycine), NaBr, and Na2SO3 were obtained commer-cially from Sigma-Aldrich, with stated purities of at least 98%. All water usedhad a resistivity of 18 MΩ∕cm. Samples were used without further purifica-tion. The aqueous samples used in the liquid jets contained concentrationsof triglycine that were 33.3 g∕L of water (∼0.13 M). One of the samplescomprised solely triglycine, another contained triglycine and 33.3 g∕L(∼0.33 M) of NaBr, and the third contained triglycine and 33.3 g∕L ofNa2SO3 (∼0.26 M). These correspond to approximately a 2.5∶1 NaBr∶triglycine ratio and a 2∶1 Na2SO3∶triglycine ratio. We note that theseconcentrations are well below those wherein precipitation would occur inthe liquid jets, which would lead to jet clogging.
NEXAFS Spectroscopy of Solvated Triglycine. Total electron yield (TEY) NEXAFSwere recorded at the nitrogen K edge (∼400 eV). These measurements wereperformed at Beamline 8.0.1 of the Advanced Light Source (ALS) at LawrenceBerkeley National Laboratory. A detailed description of the experimental sys-tem has been published previously (18). Briefly, an intense (>1011 photons∕s),high resolution (E∕ΔE > 4;000) tunable X-ray beam is generated from anundulator. The synchrotron light is then focused (∼50 μm spot size) onto asmall liquid jet (∼30 μm diameter), and the TEY is collected to obtain spectraof the bulk liquid (15). The jet is produced by using a syringe pump (Teledyne-Isco) to pressurize the liquid behind a fused silica capillary tip and travelsparallel to the polarization of the incident radiation. Almost immediately(<0.5 mm and <15 μs) after leaving the tip orifice, the liquid is intersectedby the X-ray beam, wherein the sample is close to room temperature(∼23 °C) (22). The liquid jet is then condensed in a separate cryogenicallycooled section of the chamber to maintain low pressures (∼10−4 torr) inthe interaction region. The signal is normalized to that from a gold meshlocated up-beam of the chamber, and the energy is calibrated based onspecific metal impurities located in the beamline optics. The spectra arearea-normalized and smoothed.
CalculationsCore-Level Spectra. Our theoretical approach has been describedpreviously (19, 21). We use periodic boundary conditions and aplane-wave pseudopotential formalism. PWSCF is used to calcu-late the zone-center electronic structure (31). We use thePerdew–Burke–Ernzehof (PBE) form of the generalized gradientapproximation to the exchange-correlation potential withindensity functional theory (32–35). Core-hole matrix elementsbetween atomic core levels and unoccupied valence pseudostateswere calculated by reconstructing the electronic structure of thecore region of the excited atom within the atomic frozen coreapproximation. Transition amplitudes are estimated in the sin-gle-particle and dipole approximations. The lowest energycore-hole excited state is modeled by explicit inclusion of the corehole (via a modified pseudopotential) and the excited electron(XCH). Excitations to states above this first excited state are
Fig. 8. Experimental nitrogen K-edge NEXAFS spectrum of solid lysozyme(purple, solid line), 2,5-diketopiperazine (black, ••••) and polyisoleucine(green, — ••• —) from the literature (see text). These are contrasted withcalculated spectra at the nitrogen K edge of solid polyalanine in an antipar-allel β-sheet (red, solid line) and an α-helix (blue, ••••).
14012 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1006435107 Schwartz et al.

approximated using the unoccupied Kohn–Sham eigenstatescomputed from the XCH self-consistent potential (19, 21).The spectra were aligned to experiment. These calculations wereperformed on the Franklin supercomputer at National EnergyResearch Scientific Computing Center (NERSC). All spectrawere stretched along the energy axis by 25% to improve agree-ment with measurements, due to known bandwidth underestima-tion within DFT (22, 23).
Numerical Broadening.All calculated spectra are numerically broa-dened using Gaussians of 0.2 eV full width at half maximum.Others often resort to ad-hoc energy-dependent broadeningschemes (36). We use this relatively small and uniform broaden-ing with the aim of simulating and distinguishing electronic andvibrational effects explicitly, thereby arriving at a predictivecomputational approach.
Solvated Triglycine MD Simulations. We have modeled the nucleardegrees of freedom in these systems using MD performed at300 K using a Langevin thermostat, with the generalized AMBERforce field (GAFF) (37). The resulting distribution of nuclearcoordinates was sampled at 40-ps intervals to minimize correla-tion between snapshots. This was done for 100 configurations oneach nitrogen (therefore, three calculations per configuration)(37, 38). These coordinates are subsequently used in DFTcalculations, which were by far the most significant part of thecomputational cost. The system sizes were approximately4;000 Å3 and contained approximately 120 water molecules. Inone case the triglycine was simulated with water, in another case
6 Naþ and Br− ions were added to the triglycine water simulation,and in the final case 4 Naþ ions and 2 SO3
2− ions were added tothe triglycine water simulation. The Naþ and Br− were parame-terized with the default parameters in AMBER, and SO3
2− wasparameterized using the GAFF (37). We note that this is higherthan the experimental concentration, but this was done to empha-size any effects that may exist. For statistical purposes, these MDsimulations were continued for a total of 20 ns and sampled every50 fs. These simulations were used for the RDFs and the rmspositions shown later. The rms positions of the atoms weredetermined by rms fitting the backbone of the triglycine structureto the average structure, with the average structure first rms fittedto the first coordinate set to remove rotational effects. The resultswere similar if rotational effects were not removed initially.
Spectrum of Polyalanine.The coordinates of polyalanine in both anideal α-helix and an ideal antiparallel β-sheet were taken from theliterature (24). Because all of the nitrogen atoms are effectivelydegenerate and motions were not treated, only a single NEXAFSspectrum was calculated for both the α-helix and antiparallelβ-sheet at the nitrogen K edge.
ACKNOWLEDGMENTS. We thank Wanli Yang for excellent user support ofbeamline 8.0.1. This work was supported by the Director, Office of BasicEnergy Sciences, Office of Science, U.S. Department of Energy (DOE) underContract DE-AC02-05CH11231 through the Lawrence Berkeley NationalLaboratory Chemical Sciences Division, the Molecular Foundry, and theAdvanced Light Source. Computational resources were provided by NERSC,a DOE Advanced Scientific Computing Research User Facility.
1. Hofmeister F (1888) Zur Lehre von der Wirkung der Salze. Arch Exp Pathol Pharmakol24:247–260.
2. Kunz W, Henle J, Ninham BW (2004) ‘Zur Lehre von der Wirkung der Salze’ (about thescience of the effect of salts): Franz Hofmeister’s historical papers. Curr Opin ColloidInterface Sci 9:19–37.
3. Zhang YJ, Cremer PS (2006) Interactions between macromolecules and ions: TheHofmeister series. Curr Opin Chem Biol 10:658–663.
4. Luo EP, Chai YQ, Yuan R (2007) Unsymmetrical tetradentate Schiff bases copper (II)complex as neutral carrier for thiocyanate-selective electrode. Anal Lett 40:369–386.
5. Batchelor JD, Olteanu A, Tripathy A, Pielak GJ (2004) Impact of protein denaturantsand stabilizers on water structure. J Am Chem Soc 126:1958–1961.
6. Smith JD, et al. (2005) Unified description of temperature-dependent hydrogen-bondrearrangements in liquid water. Proc Natl Acad Sci USA 102:14171–14174.
7. Nucci NV, Vanderkooi JM (2008) Effects of salts of the Hofmeister series on thehydrogen bond network of water. J Mol Liq 143:160–170.
8. Hess B, van der Vegt NFA (2009) Cation specific binding with protein surface charges.Proc Natl Acad Sci USA 106:13296–13300.
9. Collins KD (2006) Ion hydration: Implications for cellular function, polyelectrolytes,and protein crystallization. Biophys Chem 119:271–281.
10. Mason PE, et al. (2009) Specificity of ion-protein interactions: Complementary andcompetitive effects of tetrapropylammonium, guanidinium, sulfate, and chloride ions.J Phys Chem B 113:3227–3234.
11. Cho YH, et al. (2008) Effects of Hofmeister anions on the phase transition temperatureof elastin-like polypeptides. J Phys Chem B 112:13765–13771.
12. Zhang Y, et al. (2007) Effects of Hofmeister anions on the LCSTof PNIPAM as a functionof molecular weight. J Phys Chem C 111:8916–8924.
13. Zhang YJ, Furyk S, Bergbreiter DE, Cremer PS (2005) Specific ion effects on thewater solubility of macromolecules: PNIPAM and the Hofmeister series. J Am ChemSoc 127:14505–14510.
14. Aziz EF, Ottosson N, Faubel M, Hertel IV, Winter B (2008) Interaction between liquidwater and hydroxide revealed by core-hole de-excitation. Nature 455:89–91.
15. Wilson KR, et al. (2001) X-ray spectroscopy of liquid water microjets. J Phys Chem B105:3346–3349.
16. Uejio JS, et al. (2008) Characterization of selective binding of alkali cations withcarboxylate by x-ray absorption spectroscopy of liquid microjets. Proc Natl Acad SciUSA 105:6809–6812.
17. Aziz EF, et al. (2008) Cation-specific interactions with carboxylate in amino acid andacetate aqueous solutions: X-ray absorption and ab initio calculations. J Phys Chem B112:12567–12570.
18. Wilson KR, et al. (2004) Investigation of volatile liquid surfaces by synchrotron x-rayspectroscopy of liquid microjets. Rev Sci Instrum 75:725–736.
19. Uejio JS, Schwartz CP, Saykally RJ, Prendergast D (2008) Effects of vibrationalmotion on core-level spectra of prototype organic molecules. Chem Phys Lett467:195–199.
20. Prendergast D, Galli G (2006) X-ray absorption spectra of water from first principlescalculations. Phys Rev Lett 96:215502.
21. Schwartz CP, Uejio JS, Saykally RJ, Prendergast D (2009) On the importance ofnuclear quantum motions in near edge x-ray absorption fine structure spectroscopyof molecules. J Chem Phys 130:184109.
22. Schwartz CP, et al. (2009) Auto-oligomerization and hydration of pyrrole revealed byx-ray absorption spectroscopy. J Chem Phys 131:114509.
23. Uejio JS, et al. (2010) J Phys Chem B 114:4702–4709.24. Vener MV, Egorova AN, Fomin DP, Tsirelson VG (2009) Hierarchy of the non-covalent
interactions in the alanine-based secondary structures. DFT study of the frequencyshifts and electron-density features. J Phys Org Chem 22:177–185.
25. Zubavichus Y, Shaporenko A, Grunze M, Zharnikov M (2005) Innershell absorptionspectroscopy of amino acids at all relevant absorption edges. J Phys Chem A 109:6998–7000.
26. Zubavichus Y, Shaporenko A, Grunze M, Zharnikov M (2007) NEXAFS spectroscopy ofhomopolypeptides at all relevant absorption edges: Polyisoleucine, polytyrosine, andpolyhistidine. J Phys Chem B 111:9803–9807.
27. Zubavichus Y, Shaporenko A, Grunze M, Zharnikov M (2008) Is X-ray absorptionspectroscopy sensitive to the amino acid composition of functional proteins? J PhysChem B 112:4478–4480.
28. Stewart-Ornstein J, et al. (2007) Using intrinsic X-ray absorption spectral differences toidentify and map peptides and proteins. J Phys Chem B 111:7691–7699.
29. Stöhr J (1992) NEXAFS Spectroscopy (Springer, Berlin) p 403.30. Heyda J, Vincent JC, Tobias DJ, Dzubiella J, Jungwirth P (2010) Ion specificity at the
peptide bond: Molecular dynamics simulations of N-methylacetamide in aqueous saltsolutions. J Phys Chem B 114:1213–1220.
31. Baroni S, Corso AD, Gironcoli SD, Giannozzi P (2008) PWSCF v4.0.2.32. Hohenberg P, KohnW (1964) Inhomogeneous electron gas. Phys Rev B 136:B864–B871.33. Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation
effects. Phys Rev 140:A1133–A1138.34. Perdew JP, et al. (1992) Atoms, molecules, solids, and surfaces—applications of
the generalized gradient approximation for exchange and correlation. Phys Rev B46:6671–6687.
35. Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation madesimple. Phys Rev Lett 77:3865–3868.
36. Naslund LA, et al. (2005) X-ray absorption spectroscopy study of the hydrogen bondnetwork in the bulk water of aqueous solutions. J Phys Chem A 109:5995–6002.
37. Case DA, et al. (2006) AMBER 9. (University of California, San Francisco) v9.0.38. Wang JM, WangW, Kollman PA, Case DA (2006) Automatic atom type and bond type
perception in molecular mechanical calculations. J Mol Graph Model 25:247–260.
Schwartz et al. PNAS ∣ August 10, 2010 ∣ vol. 107 ∣ no. 32 ∣ 14013
CHEM
ISTR
Y