
Kinetics and intracellular compartmentalization of HTLV1 gene expression
Upload
independentCategory
view
1download
0
JOURNAL OF VIROLOGY, Oct. 2009, p. 9983–9992 Vol. 83, No. 190022-538X/09/$08.00�0 doi:10.1128/JVI.01905-08Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Genetic Characterization of Hepatitis B Virus in Peripheral BloodLeukocytes: Evidence for Selection and Compartmentalization
of Viral Variants with the Immune Escape G145R Mutation�†Sibnarayan Datta,1§ Rajesh Panigrahi,1‡ Avik Biswas,1‡ Partha K. Chandra,1§ Arup Banerjee,1¶
Pradip K. Mahapatra,2 Chinmoy K. Panda,3 Shekhar Chakrabarti,1,4 Sujit K. Bhattacharya,1,4
Kuntal Biswas,5 and Runu Chakravarty1*ICMR Virus Unit, Kolkata, India1; Department of Chemistry, Jadavpur University, Kolkata, India2;
Chittaranjan National Cancer Institute (CNCI), Kolkata, India3; National Institute of Cholera andEnteric Diseases (NICED), Kolkata, India4; and Medical College and Hospital, Kolkata, India5
Received 10 September 2008/Accepted 14 April 2009
The compartmentalization of viral variants in distinct host tissues is a frequent event in many viralinfections. Although hepatitis B virus (HBV) classically is considered hepatotropic, it has strong lymphotropicproperties as well. However, unlike other viruses, molecular evolutionary studies to characterize HBV variantsin compartments other than hepatocytes or sera have not been performed. The present work attempted tocharacterize HBV sequences from the peripheral blood leukocytes (PBL) of a large set of subjects, usingadvanced molecular biology and computational methods. The results of this study revealed the exclusivecompartmentalization of HBV subgenotype Ae/A2-specific sequences with a potent immune escape G145Rmutation in the PBL of the majority of the subjects. Interestingly, entirely different HBV genotypes/subgeno-types (C, D, or Aa/A1) were found to predominate in the sera of the same study populations. These resultssuggest that subgenotype Ae/A2 is selectively archived in the PBL, and the high prevalence of G145R indicateshigh immune pressure and high evolutionary rates of HBV DNA in the PBL. The results are analogous toavailable literature on the compartmentalization of other viruses. The present work thus provides evidence infavor of the compartment-specific abundance, evolution, and emergence of the potent immune escape mutant.These findings have important implications in the field of HBV molecular epidemiology, transmission, trans-fusion medicine, organ transplantation, and vaccination strategies.
Hepatitis B virus (HBV) is the prototype member of theHepadnaviridae family and classically has been described to behepatotropic, causing a wide range of clinical and subclinicalmanifestations of liver disease (57). Nevertheless, studies ofHBV-infected human subjects and woodchucks infected withWoodchuck hepatitis virus (WHV; an animal model of hepad-naviral infection) have reported different molecular forms ofreplicative intermediates in the lymphatic cells and have estab-lished that hepadnaviruses are strongly lymphotropic in nature(29). Moreover, the results of studies of human subjects as wellas with animal models have revealed that the life-long occultpersistence of replication- and transmission-competent virusesin lymphatic cells is a strict consequence of hepadnaviral in-fections (29).
More interestingly, in animal models, lymphatic system-re-
stricted occult hepadnaviral infection has been found to betransmissible vertically as an asymptomatic, serologically oc-cult infection exclusively confined to the lymphatic system (29).Earlier we provided evidence that occult HBV persisting in thelymphatic cells are transmissible, specifically to the PBLthrough horizontal intrafamilial modes (9). These observationsclearly indicate important immunological, pathogenic, and ep-idemiological implications of lymphatic system-restricted hep-adnaviral infections. Although the involvement of specific viralvariants has been suggested to explain this lymphatic system-restricted hepadnaviral infection and transmission (29), theclassical belief that hepatocytes are the primary target and onlyreservoir of HBV has precluded the genetic characterization ofhepadnaviruses from extrahepatic sites.
Fascinatingly, despite being classically considered a hepato-tropic virus, hepatitis C virus (HCV), belonging to the familyFlaviviridae, also shows occult persistence and lymphotropismvery similar to that of hepadnaviruses (37). Similarly to WHV,HBV, and HCV, other viruses, including HIV (human immu-nodeficiency virus), small ruminant lentivirus, and Epstein-Barr virus, also have been shown to infect and persist in dif-ferent anatomical compartments of the body in addition totheir classical target cells (38, 40, 43, 45, 50). Furthermore,recent molecular evolutionary analyses based on envelope se-quences of these viruses (e.g., HIV, HCV, small ruminantlentivirus, Epstein-Barr virus, etc.) have established clearlythat these viruses undergo selection and independent evolu-tion in diverse tissues, leading to the tissue-specific compart-
* Corresponding author. Mailing address: ICMR Virus Unit, ID &BG Hospital Campus, 57, Dr. Suresh Chandra Banerjee Rd., Pin–700010, Kolkata, India. Phone: 91 33 23537425. Fax: 91 33 23537424.E-mail: [email protected].
† Supplemental material for this article may be found at http://jvi.asm.org/.
‡ These authors contributed equally to this work.§ Present address: Tulane University Health Sciences Center, De-
partment of Pathology and Laboratory Medicine, 1430 Tulane Ave.SL-79, New Orleans, LA 70112.
¶ Present address: St. Louis University School of Medicine, Divisionof Infectious Diseases & Immunology, Edward A. Doisy ResearchCenter, St. Louis, MO 63104.
� Published ahead of print on 6 May 2009.
9983

mentalization of viral populations (38, 40, 43, 45, 50). In con-trast to other viruses, to the best of our knowledge, methodicalmolecular evolutionary studies to characterize HBV sequencesisolated from extrahepatic sites of HBV-infected subjects havenot been reported in the literature.
We hypothesized that similar to other viruses, HBV alsoundergo independent evolution in different compartments ofthe body under the influence of differential immune pressure.To examine our hypothesis, we used the most easily availablelymphatic cells, the peripheral blood leukocytes (PBL), deter-mined the HBV envelope sequences from HBV DNA isolatedfrom these cells, and performed advanced genetic, phyloge-netic, and mutational analysis. The results of this work dem-onstrate a highly compartment-specific preponderance ofHBV genetic variants in serum and PBL of the same studypopulation, providing evidence in favor of the compartmental-ization of HBV genetic variants. The results and importantimplications of these findings are discussed in this work.
MATERIALS AND METHODS
Study subjects. The blood samples analyzed in this study were selected froma repository of blood samples (stored at �80°C) collected during our previousstudies of an eastern Indian population. The samples were collected at differentcollection sites and at different times. Based on a detailed examination of thequestionnaires, which were filled out during patient interviews, and on results ofthe serological assays, samples were carefully screened for inclusion in this study.Samples from subjects admitting activities related to multiple exposures to HBV(sexual interaction with multiple partners and/or commercial sex workers, intra-venous drug abuse, etc.), alcoholism, or having markers for HCV and/or HIVcoinfection were excluded. Only samples from incidentally detected, healthy,asymptomatic subjects positive for either HBsAg or anti-HBc (considered amarker of past exposure to HBV) with normal levels of alanine aminotransferasewere included in the study. These subjects also were naïve to antiviral therapy.Finally, samples from 30 HBsAg-positive and 54-HBsAg negative, anti-HBc-positive subjects were studied in details. In addition, 10 samples with no sero-logical/molecular marker of HBV infection were included to serve as controlsduring DNA extraction and amplification steps.
This work was a part of our study of the HBV genetic variability in easternIndia and was approved by the institutional ethics committee. Informed consentwas obtained from the donors before blood drawing.
Serological tests. Commercially available enzyme-linked immunosorbent as-say-based kits were used for the detection of HBsAg (Biomerieux, Boxtel, TheNetherlands), anti-HBc (Biomerieux, Boxtel, The Netherlands), anti-HCV (Or-tho-Clinical Diagnostics, NJ), and anti-HIV (Biomerieux, Boxtel, The Nether-lands).
DNA extraction and verification and HBV DNA detection. Leukocytes wereenriched and serum HBV DNA was depleted from the leukocytes by a selectiveerythrocyte lysis/leukocyte washing method. Briefly, 3 ml whole blood was incu-bated with erythrocyte lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mMNa2EDTA, pH 7.4), followed by centrifugation and the washing of the leukocytepellets twice with phosphate-buffered saline (pH 7.4). The supernatant of the lastwash step was aspirated and stored to serve as a control for the stringency of thewashing step. The leukocyte pellets were resuspended in SE buffer (75 mM NaCl,25 mM Na2EDTA, pH 8.0) and incubated with proteinase K-sodium laurylsulfate, followed by extraction with phenol-chloroform-isoamyl alcohol (25:24:1),precipitation (3 M sodium acetate, pH 5.2, isopropanol), washing (70% ethanol),and dissolving in Tris-EDTA buffer. To verify the efficiency of the erythrocytelysis/leukocyte washing method for the depletion of serum HBV DNA, werandomly selected 11 samples that were positive for HBV DNA in either PBL orin both compartments (PBL and serum) and subjected them to DNase I diges-tion (12), followed by DNA extraction with the phenol-chloroform method.Successive amplification and multiple clonal sequence analyses showed that theerythrocyte lysis/leukocyte washing method was comparable to the enzymaticmethod in its efficiency to deplete serum HBV DNA from PBL DNA and alsoprovides cells suitable for the sensitive detection of HBV genomes (data notshown). The concentration and purity of the extracted DNA was checked spec-trometrically (Varian, Victoria, Australia), diluted appropriately, and stored at�20°C until further use. Simultaneously, DNA was extracted from 200 �l serum
and from the supernatant of the last washing step of the PBL DNA extraction bythe proteinase K, phenol-chloroform-isoamyl alcohol method and stored at�20°C until further use.
DNA extraction was verified by a PCR assay targeting the human interleu-kin-1� promoter region (primers pil-1bf [5�-TGGCATTGATCTGGTTCATC-3�] and pil-1br [5�-GTTTAGGATCTTCCCACTT-3�]), producing an ampliconof 304 bp. For the detection of HBV DNA, nested PCR assays targeting thedistalX/precore/core promoter region (3) and the surface gene region (describedbelow) were used. HBV-specific cccDNA (covalently closed circular DNA) wasdetected by a previously described heminested PCR assay (7). Strict precautionswere taken to avoid cross contamination (26), and appropriate no-templatecontrols (water), negative controls (PBL DNA extracts from HBV/HIV/HCV-seronegative healthy subjects), and positive controls (PBL DNA/serum DNAextracts from HBsAg-positive subjects and/or cloned plasmid containing HBVgenotype D genome) were included during each extraction, amplification, andcycle sequencing steps to rule out contamination.
HBV genotyping, sequencing, and cloning. First, HBV genotypes were deter-mined by a rapid and highly sensitive nested PCR-restriction fragment lengthpolymorphism (PCR-RFLP) method, which we modified from a previously de-scribed single PCR-RFLP-based method for large-scale genotyping (27). Being anested PCR-based amplification assay, our modified method was appropriate forthe genotyping of HBV in HBsAg-negative samples with low viremia. Briefly, thesurface gene region encoding the major hydrophilic loop was amplified by anested PCR assay with primers 5�-ACCCCTGCTCGTGTTACAGGC-3� (sense;positions 184 to 204) and 5�-AAAGCCAGACAGTGGGGGAAA-3� (antisense;positions 731 to 711) for the first round, as well as nested primers 5�-GACTCGGTGGTGGACTTCTCTC-3� (sense; positions 251 to 271) and 5�-TAAACTGAGCCAGGAGAAACG-3� (antisense; positions 679 to 659) to amplify a 429-bpamplicon, using a HotStart Taq polymerase (Applied Biosystems, Foster City,CA) and a thermal cycling profile, including incubation for 10 min at 95°C, 40cycles (35 cycles for the second round) at 95°C for 15 s, 55°C for 15 s, and 72°Cfor 30 s, followed by a final extension step at 72°C for 5 min. PCR products werestained by ethidium bromide and visualized under UV light (Fig. 1).
The restriction sites in the 429-bp partial surface gene amplicon were derivedfrom the original report by Lindh et al. (27) and also verified by the computa-tional analysis of 112 GenBank sequences of different HBV genotypes usingBioEdit (19). It was observed that the banding pattern of Tsp509I was sufficientto distinguish between HBV genotypes A, C, and D, which are prevalent in ourpopulation. The specificity of the method was verified by using HBsAg-positivesamples, which we genotyped previously by full-genome sequencing and phylo-genetic analysis (1). After the digestion of the 429-bp amplicon with Tsp509I (at65°C for 3 h), genotype A-specific amplicons yield three major bands (126, 109,and 90 bp), genotype C-specific amplicons yield two major bands (235 and 90bp), and genotype D-specific amplicons also yield two major bands (173 and 109bp). The RFLP genotyping patterns obtained in all of the samples were con-firmed by direct sequencing and phylogenetic analysis.
Surface gene PCR products were purified (Qiagen GmbH, Hilden, Germany)and directly sequenced using a Prism Big Dye 3.0 kit and an ABI 3100/ABI3130xl automated DNA sequencer (Applied Biosystems, Foster City, CA). In thisstudy, direct sequencing was carried out to identify sites under significant evo-lutionary pressures by virtue of analyzing multiple distinct peaks (sequencingmixtures) in a sequencing electropherogram (17, 42).
To verify the results of the direct sequencing and to scan the presence of aminor subpopulation of virus in different compartments, we randomly selectedseven samples that had HBV DNA only in the PBL compartment for multipleclonal sequence analyses. Parallel PBL and serum DNA extracts from foursubjects having HBV DNA in both compartments also were selected for multipleclonal sequence analyses. In addition, two sera DNA extracts having the sub-genotype Aa/A1 (as determined in our previous studies) also were included ascontrols during the process of cloning, sequencing, and subsequent analysis torule out cross-contamination. The PCR products were cloned using a T/A clon-ing kit (Fermentas, Glen Burnie, MD), and 10 to 15 transformed colonies fromeach sample were randomly selected, amplified, sequenced, and analyzed phy-logenetically.
Molecular evolutionary analyses. We aligned and edited the sequences usingthe BioEdit v7.0 program (19). We manually examined the direct sequenceelectropherograms and replaced the N residues in the alignment with appropri-ate ambiguous nucleotide codes by following International Union of Pure andApplied Chemistry conventions.
HBV genotypes and subgenotypes were determined by phylogenetic analysiswith HBV genotype- and subgenotype-specific reference sequences retrievedfrom GenBank according to a previous analysis (34). Pairwise evolutionarydistances were calculated using the Kimura two-parameter model for the esti-
9984 DATTA ET AL. J. VIROL.

mation of distances. Phylogenetic trees were constructed by the neighbor-joiningmethod (NJ) using MEGA v3.0 (25). The significances of the interior branchesin the NJ were tested by means of bootstrap analysis with 1,000 replicates.
We used the weighted scoring matrix in the Syn-SCAN program, implementedon the web (available at http://hivdb.stanford.edu), for the weighting of themixed bases and for calculating synonymous and nonsynonymous substitutionrates. This program accepts the ambiguous nucleotide data and analyzes it usinga model that estimates nucleotide mixes as intermediate stages of intrahost viralevolution (17). Additionally, aligned PBL- and serum-derived sequences wereanalyzed for determining compartment-specific signature sequence variations(24) using the viral epidemiology signature pattern analysis program VESPA(available at http://hiv-web.lanl.gov).
Based upon phylogenetic analyses, the pairwise analyses of genetic distances,and signature amino acid patterns, we determined the HBV surface gene vari-ability in the PBL and compared them to the surface gene sequences that weregenerated using serum HBV isolates from the same study population during ourprevious studies (1, 2, 4). Following a previous report (38), viral sequences wereconsidered to be compartmentalized when sequences from PBL and serum werefound to be phylogenetically related yet sufficiently distinct to form separateclusters in the phylogenetic tree. The phylogeny was constructed using NJ, andthe topology was analyzed with phylogeny reconstructed by the maximum-like-lihood method using the program DNAML (Phylip v3.5).
Data analysis. Student’s t test was used for the comparison of continuous data,and the �2 test or Fischer’s exact test was used for the comparison of thecategorical data, where appropriate. Two-tailed P values of �0.05 were consid-ered statistically significant. Data entry, the calculation of prevalence, the rep-resentation of data, and preliminary statistical analyses were done using MSExcel (Microsoft Corporation). The statistical analysis of the categorical data wasdone using StatCalc (EpiInfo version 6.0, Centers for Disease Control andPrevention and World Health Organization, Geneva, Switzerland).
Nucleotide sequence accession numbers. The direct sequence data generatedin this work have been deposited in GenBank (http://www.ncbi.nlm.nih.gov/)under accession numbers EU275289 to EU275349.
RESULTS
Prevalence of HBV DNA in serum and PBL. The results ofthe detection of HBV DNA in serum and PBL in subjects withHBsAg-negative and HBsAg-positive serology are presented inTable 1. Statistically there was no difference in the mean age ofthe HBsAg-positive and -negative subjects included in thisstudy (P � 0.135). As expected, the detection rate of HBVDNA in serum was significantly higher in HBsAg-positive sub-jects than in HBsAg-negative subjects (23.3 and 5.5%, respec-tively; P � 0.001). The rate of HBV DNA detection in bothserum and PBL together also was significantly higher inHBsAg-positive subjects than in HBsAg-negative subjects (30and 14.8%, respectively; P � 0.017). In contrast, the rate ofHBV DNA detection only in PBL was significantly higher inHBsAg-negative subjects than in HBsAg-positive subjects(61.1 and 40%, respectively; P � 0.004). HBV DNA was notdetected in either serum or PBL in 6.6 and 18.5% of HBsAg-positive and HBsAg-negative subjects, respectively.
For studying the molecular characteristics of PBL-relatedHBV envelope sequences encoding the major hydrophilic loopregion, 12 HBsAg-positive (designated group I) and 33 HBsAg-negative (designated group II) subjects having detectable HBVDNA exclusively in the PBL were selected and further studiedin detail. This selection criterion was used to eliminate thepossibility of the contamination of PBL DNA with HBV DNAcirculating in the serum. In addition to comparing the HBVDNA sequences in the serum and PBL within the same indi-viduals, eight HBsAg-negative subjects with HBV DNA de-tectable in both serum and PBL (designated group III) alsowere selected for detailed analysis. These samples had very lowlevels of HBV DNA in their sera (102 to 103 copies ml�1). Inthese cases, the stringent washing of the leukocyte pellets wasperformed to rule out the mixing of HBV DNA from serumand PBL, and the supernatant of the last wash step was used asa control. However, the HBsAg-positive subjects who hadHBV DNA in both their serum and PBL were not included fordetailed sequence analysis if they had very high levels of HBVDNA in their sera (106 copies ml�1); thus, the likelihood ofthe contamination of the PBL DNA with serum HBV DNAwas much higher.
HBV cccDNA-specific amplicons were detectable in 25%(3/12), 18.2% (6/33), and 25% (2/8) of PBL DNA extracts fromgroups I, II, and III, respectively.
Genotypes and their distribution in serum and PBL. Resultsof the genotype-specific RFLP patterns were typical for geno-
FIG. 1. Results of representative nested PCR amplification. Shownis a gel of the 429-bp amplicon specific for HBV surface gene region incontrol and test samples. Lanes: 1, no-template control (NTC); 2 to 6,PBL extract from HBsAg-positive subjects (test samples); 7 and 9 to12, PBL extracts from HBsAg-negative, anti-HBc positive subjects(test samples); 13, positive control (plasmid containing genotypeD-specific HBV genome); 14, negative control (water); 15, PBL DNAextract from subject completely negative for HBV serological markers(mock sample); and 8 and 16, DNA molecular size markers (pUC19/MspI-digested DNA).
TABLE 1. Prevalence of HBV DNA in serum and PBL of HBsAg-positive and -negative subjects
HBsAgGroup n Age (yr)
No. (%) of samples positivefor HBV DNA
No. (%) withno HBVDNA in
serum andPBL
In serumonly
In serumand PBL
In PBLonly
Positive 30 33.9 13.4 7 (23.3) 9 (30.0) 12 (40.0) 2 (6.6)Negative 54 29.1 15.1 3 (5.5) 8 (14.8) 33 (61.1) 10 (18.5)
VOL. 83, 2009 COMPARTMENTALIZATION OF HEPATITIS B VIRUS IN PBL 9985

type A, C, or D in all but five HBsAg-positive (group I) cases.In these five cases, the RFLP banding patterns were atypical,and the sequence electropherogram from these five subjectsshowed mixed peaks at a number of nucleotide positions (seeFig. S1 in the supplemental material). The comparison of themixed peaks in these nucleotide sequences to consensus se-quences of different HBV genotypes suggested the presence oftwo different genotypes of HBV in these extracts. Subse-quently, the cloning of the amplicons from these five cases,followed by genotype analysis, revealed the presence of geno-types A and C in one sample, while genotypes A and D weredetected in four isolates. Genotype A was detected in themajority of the clones from these subjects. Nucleotide se-quences from the rest of the amplicons from group I, group II,and group III showed clearly distinct dye peaks (see Fig. S2 inthe supplemental material), with mixed residues appearing atvery few sites. The results of the phylogenetic analyses of thesesequences were consistent with the results of the RFLP geno-type results (data not shown).
Interestingly, the preponderance of genotype A was ob-served in PBL of groups I and II (Fig. 2a, b). This selectiveprevalence of HBV genotypes in the PBL was more evident ingroup III, where genotypes D and C were prevalent in theserum (Fig. 2c), significantly contrasting (P � 0.001) to thepredominance of genotype A in the PBL (Fig. 2d). Uponcomparing the paired samples (from serum and PBL) fromeight subjects of group III, we found that in six of eight subjectsthe genotype circulating in the serum was either C or D, whilegenotype A was present invariably in the PBL of the sameindividuals, clearly indicating the specific preponderance ofgenotype A in the PBL over non-A genotypes in the serum.
Phylogenetic relatedness. In the phylogenetic tree, all of thesequences generated in the present study clustered with eithergenotype A, C, or D reference sequence clusters (Fig. 3). All ofthe genotype A sequences isolated from the PBL clusteredwith subgenotype Ae/A2 reference sequences and all of thegenotype C sequences clustered with subgenotype Cs/C1 ref-erence sequences, while genotype D was the most heteroge-neous in subgenotypes, clustering with one of the subgenotypeD1, D2, D3, and D5 reference sequences (Fig. 3).
As the occurrence of subgenotype Ae/A2 sequences was not
in agreement with the results of our previous studies usingserum HBV isolates, we aligned the PBL-specific surface genesequences with serum-isolated HBV surface gene sequencesfrom our previous studies and subjected them to phylogeneticanalysis again. Genotype A sequences from PBL and serumformed clearly distinct clusters with subgenotype Ae/A2 andAa/A1 reference sequences, respectively (Fig. 4).
The results of the analysis of multiple clones also confirmedthe results of the direct sequencing. Among the three samplesselected from group I, multiple clones from isolate PB58showed a mix of subgenotype Ae/A2- and Cs/C1-specific se-quences, isolate PB72 showed a mix of subgenotype Ae/A2-and D1-specific sequences, and isolate PB74 showed onlysubgenotype D2-specific sequences (see Fig. S3 in the supple-mental material). Of the four samples selected from group II,subgenotype Ae/A2 (isolate PB47)-, subgenotype D3 (isolatePB05)-, subgenotype Cs/C1 (isolate PB18)-, and subgenotypeAe/A2 (isolate PB24)-specific sequences were detected exclu-sively in the multiple clonal analysis (see Fig. S3 in the sup-plemental material). Of the four parallel serum and PBL iso-lates from group III samples selected for multiple clonalsequence analysis, isolate PB02 showed subgenotype Cs/C1sequences in the serum and subgenotype Ae/A2 sequences inthe PBL, isolate PB32 showed subgenotype D5 sequences inthe serum and subgenotype Ae/A2 sequences in the PBL,isolate PB35 showed subgenotype D1 sequences in the serumand subgenotype Ae/A2 sequences in the PBL, and isolatePB36 showed subgenotype D5 sequences in the serum andsubgenotype Ae/A2 sequences in the PBL (see Fig. S3 in thesupplemental material). In the four isolates from group IIIdescribed above, each of the subjects showed distinct HBVgenotypes in the serum and PBL extracts. In these cases, theHBV genotype detected in the serum was not detected even asa minor subpopulation in the PBL, or vice versa, during themultiple clonal analyses. Moreover, all of the subgenotypeAe/A2-specific sequences detected in the PBL had an adenineat nucleotide position 587 (corresponding to G145R), confirm-ing the results of the direct sequencing.
Genetic diversity and sequence characteristics of Ae/A2 se-quences from PBL. On the calculation of the intergroup ge-netic distances, we found that the subgenotype Ae/A2 se-quences found in group I were the most divergent, followed bysequences from groups III and II. Furthermore, the average ofsynonymous and nonsynonymous substitution distances andthe ratio of synonymous and nonsynonymous substitution rates(dS/dN) also were found to correlate with the genetic distances(Table 2), signifying the higher rate of evolutionary pressure inthe PBL of HBsAg-positive subjects compared to that of theHBsAg-negative subjects.
When the reference sequences from GenBank were ana-lyzed based on the subgenotypes Aa/A1 and Ae/A2 for nucle-otides 341 to 660 (the region analyzed in this study) of thesurface gene, four positions were found to be phylogeneticallyinformative in differentiating these two subgenotypes of A.Nucleotides 505C, 514C, 616A, and 619T represent subgenotypeAa/A1 strains from Asia, nucleotides 505T, 514C, 616A, and619T represent subgenotype Aa/A1 strains from sub-SaharanAfrica, and nucleotides 505T, 514A, 616G, and 619C were spe-cific for subgenotype Ae/A2 sequences that are prevalent innorthwestern Europe. In all of the present genotype A samples
FIG. 2. Distribution of HBV genotypes in different groups: (a)group I, PBL; (b) group II, PBL; (c) group III, serum; and (d) groupIII, PBL.
9986 DATTA ET AL. J. VIROL.

amplified from PBL, these four informative sites had basesspecific for subgenotype Ae/A2. In addition, all of the presentgenotype Ae/A2 sequences had two additional nucleotide sub-stitutions, 358C and 587A, which were found to be coevolvingsites.
The comparison of deduced amino acid sequence revealedthat the four specific nucleotides that differed between sub-genotype Aa/A1 and Ae/A2 sequences (nucleotides 505, 514,616, and 619) were synonymous by nature. For the two otherspecific substitutions in the present Ae/A2 samples, the substi-tution at position 358 was found to be the wobble base of thecodon encoding residue 68 of HBsAg. On the other hand, thenucleotide at position 587 is the first base of the codon spec-ifying the amino acid at position 145 of HBsAg, and the mu-tation 587A results in a potent immune escape substitutionfrom glycine to arginine (G145R).
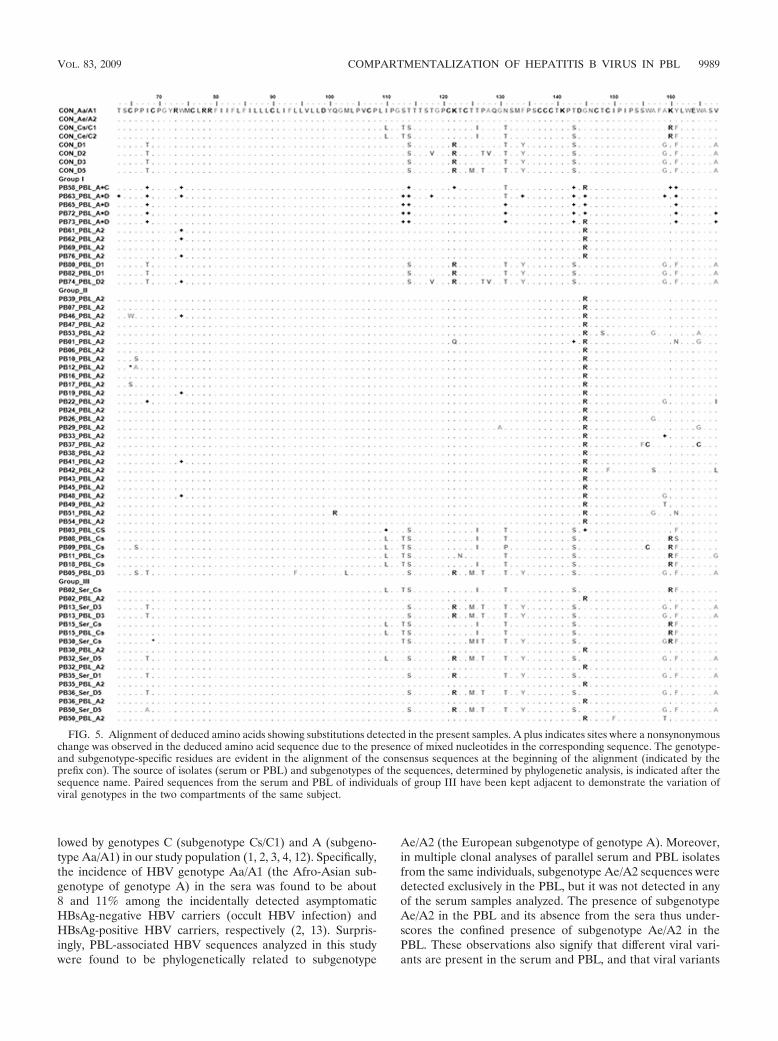
Amino acid substitutions. The alignments of deduced aminoacids and substitutions in the present samples are presented inFig. 5. Analyses of the sequences showing mixed nucleotides
revealed the changes to be mostly synonymous or genotype/serotype specific. Apart from the type-specific changes, inother sequences the substitution T125M was observed mostlyin subgenotype D3 isolates and in one Cs isolate, and it waspresent with a P127T substitution. Interestingly, the G145Rmutation was present invariably in all of the genotype A (sub-genotype Ae/A2) isolates or the isolates showing mixed geno-types (group I isolates) and was found to be a PBL-specificHBsAg signature residue in VESPA. This mutation also wasdetected in one genotype C isolate from group II. Overall, theprevalence of determinant “a” mutations was low, except thespecific association of G145R with genotype A isolates fromthe PBL. Remarkably, this mutation was not detected in any ofthe isolates from the serum in the present study.
DISCUSSION
The present study enlightens interesting and important fac-ets of HBV lymphotropism, and it supports and extends earlier
FIG. 3. Circle tree showing the phylogenetic relatedness of HBV sequences isolated from serum (f) and PBL (●) to genotype/subgenotypereference sequences retrieved from GenBank. Genotypes and subgenotypes are indicated at the corresponding nodes.
VOL. 83, 2009 COMPARTMENTALIZATION OF HEPATITIS B VIRUS IN PBL 9987

observations that occult HBV DNA persistence in the lym-phatic cells is an invariable outcome of HBV infection in themajority of subjects (7, 29, 53). Besides this persistence,cccDNA also was detected in the PBL, which suggests activeHBV replication and transcription in these cells (60). How-ever, due to the extremely small numbers of cccDNA mole-cules in the cells (60), the detection of this molecule has re-mained a challenge even in the era of sensitive PCR-basedmethods. Notably, in a previous study based on single-roundPCR, Kock et al. (23) could not detect cccDNA in the PBL,and they concluded that the detection of viral nucleic acids inthe PBL is attributable to adhered serum HBV particles (23).However, using a more sensitive PCR assay, we detected
cccDNA in the PBL of subjects who lacked HBV DNA in theirserum, in contrast to the conclusions of Kock et al. (23).
The frequent detection of HBV DNA in the PBL of HBsAg-negative anti-HBc-positive individuals is concordant with ear-lier studies of WHV (29). This phenomenon has been sug-gested to indicate low-rate viral replication and transcription inthe PBL, which is necessary for the periodic stimulation ofimmune cells, creating a negative feedback loop for keepingthe virus under strict control throughout life (29, 36). Thus,contrary to the classical belief of sterilizing immunity, the life-long trace persistence of viruses in immunologically privilegedsites seems to be common, at least in hepadnaviral infections.
In this study, we selected the HBV surface genetic regionencoding the dominant B-cell epitope (the a determinant) formolecular evolutionary analysis. A strong anti-HBs humoralresponse is induced against this epitope; thus, this region has ahigh rate of evolution (49). Analogous genetic regions of theenvelope gene frequently have been targeted for similar stud-ies in other viruses (28, 38, 40, 43, 45, 50). Although the HBVx gene region shows considerable variability, the frequent lossand low genetic variability of this HBV region in our studypopulation (3, 12) restricts its use for the robust molecularevolutionary analysis of HBV. We highlighted earlier that theHBV surface genetic region is an ideal candidate for molecularevolutionary analysis studies (11).
Interestingly, in analyzing different HBV genetic regionsfrom serum isolates, we have documented the prevalence ofHBV genotype D (subgenotypes D1, D2, D3, and D5), fol-
FIG. 4. Unrooted NJ tree of surface gene sequences from 223 serum isolates from our previous studies, 53 PBL isolates from the present study,and 79 reference sequences retrieved from GenBank. Taxon names and identical sequences have been removed to maintain the clarity of the figure.The numbers on the branches denote bootstrap values (percentages) after NJ analysis, and the asterisks signify the level of statistical significanceof the branch in the maximum-likelihood analysis (�, P � 0.001; ��, 0.05 � P 0.001). The subgenotypes Aa/A1 and Ae/A2 clusters are borderedby ellipses. Black circles indicate sequences isolated from PBL, and gray rounds indicate sequences isolated from serum.
TABLE 2. Sd, Nd, and dS/dN calculated for HBV genotype A(subgenotype Ae/A2) sequences isolated from the
PBL in this studya
Group Distanceb Sd Nd dS/dN
I (HBsAg positive, HBV DNAonly in PBL)
4.58 0.02 4.86 2.79 6.24
II (HBsAg negative, HBV DNAonly in PBL)
1.30 0.01 2.29 1.02 2.43
III (HBsAg negative, HBV DNAin PBL and serum)
1.39 0.01 2.73 0.83 3.76
a Average of synonymous substitutions (Sd), nonsynonymous substitution (Nd)distances, and ratio of synonymous and nonsynonymous substitution rates (dS/dN) were calculated for the HBV genotype A (subgenotype Ae/A2) sequencesisolated from the PBL in this study.
b Genetic distances are expressed as means standard deviations.
9988 DATTA ET AL. J. VIROL.
lowed by genotypes C (subgenotype Cs/C1) and A (subgeno-type Aa/A1) in our study population (1, 2, 3, 4, 12). Specifically,the incidence of HBV genotype Aa/A1 (the Afro-Asian sub-genotype of genotype A) in the sera was found to be about8 and 11% among the incidentally detected asymptomaticHBsAg-negative HBV carriers (occult HBV infection) andHBsAg-positive HBV carriers, respectively (2, 13). Surpris-ingly, PBL-associated HBV sequences analyzed in this studywere found to be phylogenetically related to subgenotype
Ae/A2 (the European subgenotype of genotype A). Moreover,in multiple clonal analyses of parallel serum and PBL isolatesfrom the same individuals, subgenotype Ae/A2 sequences weredetected exclusively in the PBL, but it was not detected in anyof the serum samples analyzed. The presence of subgenotypeAe/A2 in the PBL and its absence from the sera thus under-scores the confined presence of subgenotype Ae/A2 in thePBL. These observations also signify that different viral vari-ants are present in the serum and PBL, and that viral variants
FIG. 5. Alignment of deduced amino acids showing substitutions detected in the present samples. A plus indicates sites where a nonsynonymouschange was observed in the deduced amino acid sequence due to the presence of mixed nucleotides in the corresponding sequence. The genotype-and subgenotype-specific residues are evident in the alignment of the consensus sequences at the beginning of the alignment (indicated by theprefix con). The source of isolates (serum or PBL) and subgenotypes of the sequences, determined by phylogenetic analysis, is indicated after thesequence name. Paired sequences from the serum and PBL of individuals of group III have been kept adjacent to demonstrate the variation ofviral genotypes in the two compartments of the same subject.
VOL. 83, 2009 COMPARTMENTALIZATION OF HEPATITIS B VIRUS IN PBL 9989

present in the PBL are not detectable in the sera. Earlier, twocase studies of liver transplant patients also indicated thatdifferent HBV strains exist in PBL and in serum and that PBLacts as an important source of immune escape variants (5, 51).The results of the present study confirm and extend earlierstudies.
The detection of subgenotype Ae/A2 sequences with specificimmune escape variant G145R across individuals in this studyalso was surprising. However, the possible cross-contaminationbetween samples or reagents was ruled out by including ap-propriate controls during all of the experimental steps andaptly following the PCR recommendations of Kwok and Higu-chi (26). Moreover, subgenotype Ae/A2 has not been detectedin any of the group III serum HBV isolates in this study,despite the fact that they were extracted and amplified simul-taneously with the PBL samples. In addition, a number of ourother studies focused on serum HBV isolates, and in parallelor after this study, we have detected diverse HBV subgeno-types, namely, Aa/A1, Cs/C1, D1, D2, D3, and D5 (1, 2, 4, 10,12, 14), but subgenotype Ae/A2 with G145R was not detected,regardless of the fact that all of the studies were performedusing the same experimental procedures, reagent sets, andlaboratory facilities. These facts exclude the possibility that thedetection of subgenotype Ae/A2, specifically in the PBL, acrossindividuals in the present study was due to cross-contami-nation.
From the perspective of molecular epidemiology, the pres-ence of European subgenotype Ae/A2 in the sera of the Indianpopulation was quite anticipated due to India’s long history ofEuropean links (52). However, until now, studies of serumHBV isolates from different parts of India could not detect thissubgenotype in the sera of the Indian population (1, 4, 16, 20,34). Nevertheless, for the first time we reveal that subgenotypeAe/A2 sequences remain present in the leukocytes in our pop-ulation. This observation is remarkably similar to the archivingof the fittest ancestral HIV sequences in different cellular oranatomical compartments independently of the viral genotypesdominant in the serum, a phenomenon defined as cellular oranatomical memory (6). In this study, we could not detectsubgenotype Ae/A2 even after the clonal analysis of serumHBV isolates (the assay was sensitive enough to detect viralsubpopulations as low as 8 to 10%), but it might be possiblethat subgenotype Ae/A2 circulates in a very small proportionof the sera that remains untraceable, either due to the sensi-tivity of the assay used or the predominating effect of othergenotypes in the sera, similarly to the phenomenon previouslyexplained in the case of HCV (32).
Recently, molecular evolutionary analyses of the hypervari-able regions of the envelope sequences in different virusesclearly have demonstrated the independent evolution, selec-tion, and emergence of phylogenetically related, yet distinct,viral variants in different anatomical compartments of thesame individuals (33, 44, 45, 50, 58). Moreover, the compart-ment-specific predominance of particular HCV genotypes inthe leukocytes while showing different genotype in the serum,even across individuals, also has been reported recently (15).Additionally, compartment-specific signature substitutions(such as a drug resistance substitution at position 308 of HIVgp160 in the cerebrospinal fluid-derived sequence) across in-dividuals also have been well documented (39). Taking these
results together, the detection of HBV subgenotype Ae/A2with the immune escape substitution G145R in the PBL acrossindividuals in this study supports earlier observations suggest-ing that similarities in the major histocompatibility complex-restricted host immune pressure across individuals of a specificpopulation leads to the emergence of similar viral variantsacross individuals (39, 41).
It has been demonstrated that a strong antiviral humoralimmune response against HBsAg expedites the emergence ofG145R mutants (48). The high prevalence of G145R in thePBL in this study suggests that PBL-associated HBV DNA areunder strong anti-HBs-specific humoral pressures. G145R isthe most widely studied HBV immune escape mutation thatemerges naturally or in response to vaccination or anti-HBsimmunoglobulin G therapy (49) and can decrease the ability ofneutralizing anti-HBs antibodies (8, 59). Interestingly, G145Rdoes not cause any detrimental effect on virus morphogenesisand the stability of viral mRNAs, DNA, and antigens (21, 22).Moreover, HBV variants with G145R are infectious from cellto cell within a host and also are transmissible between hosts(9, 56). Thus, the selection of G145R seems to be an efficientstrategy of the PBL-associated viruses to escape the anti-HBshumoral response without compromising its critical properties.The emergence, selection, persistence, and transmission of theacquired advantageous phenotype (G145R in this case) signi-fies the Lamarkian mechanism of the evolution of advanta-geous viral variants consistently following Darwinian principlesand functioning at the molecular level, supporting the viralreplicative homeostasis hypothesis of Sallie (46).
We have detected the compartmentalization of HBV sub-genotype Ae/A2 in the PBL. This strain is well known tocirculate in the sera/hepatocytes of European populations, butit has not been reported previously in Indian populations (34,47). Thus, we could not identify a completely new genotype ofHBV in the PBL, which is phylogenetically distinct from pre-viously characterized HBV genotypes/subgenotypes (34, 47),supporting a recent observation that there exist no distinctlymphotropic variants of WHV that cannot infect the hepato-cytes (31). This observation suggests that among the differentHBV genotypes/subgenotypes, a particular one can persist inthe sera/hepatocytes of one population, while the same maypersist in the lymphatic cells in another population, indicatingthe significance of population-specific host immune factors inHBV compartmentalization. Recently, Mulrooney-Cousinsand Michalak (31) repetitively passaged wild-type WHV inlymphocyte cultures 13 times but could not distinguish anyspecific lymphotropic variant, aside from a few random muta-tions. Interestingly, the random mutations accumulated during13 repeated passages in lymphocyte cultures reverted to wild-type sequences in just a single passage of the virus in theanimals. These observations clearly highlight the role of hostimmune pressure and signify that the selection and evolutionof viral variants are more relevant in the context of immunepressure operating in vivo (28, 43) instead of in vitro cellcultures (18, 54).
HBV genotypes/subgenotypes are genetically related stableviral variants, having evolved under specific immune selectionpressures in distinct geoethnic populations (47), and the ge-netic diversity of HBV in the sera of different geoethnic pop-ulations worldwide has been studied (34, 47). Recent studies
9990 DATTA ET AL. J. VIROL.

also have shown the differential clinical/virological significanceof different HBV genotypes (13, 34, 47). The present studyoffers further evidence in favor of the differential clinical im-plications of HBV genetic variability in terms of the evolutionof immune escape variants and compartmentalization. How-ever, taking into account the key role of host-specific immunefactors in evolution and the selection of HBV genotypes (47,59), it is quite rational to anticipate different HBV genotypeswith different immune escape variants in the PBL of differentgeoethnic populations. It is thus of great clinical and scientificsubstance to investigate the genetic diversity of HBV in thePBL and other extrahepatic sites, in different populations, toenlighten the more fascinating facets of HBV epidemiology,pathogenesis, and evolution.
Using the WHV model of hepadnaviral infection, Michalakand colleagues have established that the lymphatic system actsas an important reservoir of transmission-competent occulthepadnaviral infection that invariably infects the lymphaticsystem through parenteral or vertical routes but rarely infectsliver (29, 30). In addition, we previously have shown that PBL-confined HBV infection also is capable of being transmittedexclusively to the PBL through nonparenteral, horizontal in-trafamilial routes (9). Collectively, these observations providea possible explanation for the compartment-specific dissemi-nation and maintenance of PBL-specific HBV variants while itis absent from the sera of our study population. These obser-vations signify the importance of leukocytes and encouragefurther investigation into other anatomic compartments to cor-relate the observed differences in the transmission modesamong different populations where different HBV genotypesprevail (34, 47).
Recently, HBV infection has been found to be significantlyassociated with patients with non-Hodgkin’s lymphoma, andthe oncogenic and lymphotropic properties of HBV have beensuggested to contribute to the disease (35, 55). These obser-vations indicate that the persistence of replicating virus hasimportant implications in the induction of disorders that ear-lier were not considered to be related to hepadnaviral infec-tion, and thus the further investigation of the pathogenic as-pects of the extrahepatic persistence of HBV is extremelyimportant.
In conclusion, the complete recognition of HBV strains thatinfect an individual is important to illuminate the essentials ofHBV infection, including virus transmission, compartmental-ization, persistence, and the development of extrahepatic man-ifestations, all of which still are poorly understood. To date,most of the data published on the genetic characterization ofHBV were based on virus circulating in the sera or in thehepatocytes. Moreover, the studies of vaccine efficacy or anti-virals have been performed mostly on isolates from sera andhepatocytes, but the present study clearly shows that the evo-lution of HBV in extrahepatic sites is important in the emer-gence of immune/vaccine escape variants and thus is clinicallyimportant. This is the first study of this kind to analyze thegenetic diversity and evolution of HBV in the peripheral bloodleukocytes, which opens an important field of investigation forextrahepatic HBV divergence. The findings of this study haveextremely important implications in the field of transfusionmedicine, transplantation, response to therapy, the emergenceof vaccine escape mutants, and HBV transmission that are
imperative for efficiently managing and designing strategies toreduce the burden of HBV infections.
ACKNOWLEDGMENTS
This research was supported by grants from Indian Council of Med-ical Research (ICMR), New Delhi, India. S.D. was supported by aresearch fellowship from University Grants Commission (UGC), NewDelhi, India.
We thank all of the medical officers of different hospitals, medicalcolleges, and blood banks for their help in sample collection. We thankTommy F. Liu of Stanford University HIV Drug Resistance Databasefor help with using Syn-SCAN program. We thank Tapan Chakrabortyand Sreekanta Deb for excellent technical assistance.
REFERENCES
1. Banerjee, A., F. Kurbanov, S. Datta, P. K. Chandra, Y. Tanaka, M.Mizokami, and R. Chakravarty. 2006. Phylogenetic relatedness and geneticdiversity of hepatitis B virus isolates in eastern India. J. Med. Virol. 78:1164–1174.
2. Banerjee, A., P. K. Chandra, S. Datta, A. Biswas, P Bhattacharya, S.Chakraborty, S. Chakrabarti, S. K. Bhattacharya, and R. Chakravarty.2007. Frequency and significance of hepatitis B virus surface gene variantcirculating among antiHBc only individuals in eastern India. J. Clin. Virol.40:312–317.
3. Banerjee, A., S. Banerjee, A. Chowdhury, A. Santra, S. Chowdhury, S. Roy-chowdhury, C. K. Panda, S. K. Bhattacharya, and R. Chakravarty. 2005.Nucleic acid sequence analysis of BCP/precore/core region of hepatitis Bvirus isolated from chronic carriers of the virus from Kolkata, eastern India:low frequency of mutation in the precore region. Intervirology 48:389–399.
4. Banerjee, A., S. Datta, P. K. Chandra, S. Roychowdhury, C. K. Panda, andR. Chakravarty. 2006. Distribution of hepatitis B virus genotypes: phyloge-netic analysis and virological characteristics of genotype C circulating amongHBV carriers in Kolkata, eastern India. World J. Gastroenterol. 12:5964–5971.
5. Brind, A., J. Jiang, D. Samuel, M. Gigou, C. Feray, C. Brechot, and D.Kremsdorf. 1997. Evidence for selecth of hepatitis B mutants after livertransplantation through peripheral blood mononuclear cell infection.J. Hepatol. 26:228–235.
6. Briones, C., E. Domingo, and C. M. Paris. 2003. Memory in retroviralquasispecies: experimental evidence and theoretical model for human im-munodeficiency virus. J. Mol. Biol. 331:213–229.
7. Cabrerizo, M., J. Bartolome, C. Caramelo, G. Barril, and V. Carreno. 2000.Molecular analysis of hepatitis B virus DNA in serum and peripheral bloodmononuclear cells from hepatitis B surface antigen-negative cases. Hepatol-ogy 32:116–123.
8. Carman, W. 1997. The clinical significance of surface antigen variants ofhepatitis B virus. J. Viral. Hepat. 4:11–20.
9. Chakravarty, R., M. Neogi, S. Roychowdhury, and C. K. Panda. 2002. Pres-ence of hepatitis B virus surface antigen mutant G145R DNA in the periph-eral blood leukocytes of the family members of an asymptomatic carrier andevidence of its horizontal transmission. Virus. Res. 90:133–141.
10. Chandra, P. K., A. Banerjee, S. Datta, and R. Chakravarty. 2007. G1862Tmutation among hepatitis B virus-infected individuals: association with viralgenotypes and disease outcome in Kolkata, eastern India. Intervirology 50:173–180.
11. Datta, S., A. Banerjee, P. K. Chandra, and R. Chakravarty. 2007. Selectinga genetic region for molecular analysis of hepatitis B virus transmission.J. Clin. Microbiol. 45:687–688.
12. Datta, S., A. Banerjee, P. K. Chandra, A. Biswas, R. Panigrahi, P. K.Mahapatra, C. K. Panda, S. Chakrabarti, S. K. Bhattacharya, and R.Chakravarty. 2008. Analysis of hepatitis B virus X gene phylogeny, geneticvariability and its impact on pathogenesis: implications in eastern IndianHBV carriers. Virology 382:190–198.
13. Datta, S., A. Biswas, P. K. Chandra, A. Banerjee, R. Panigrahi, P. K.Mahapatra, S. Chakrabarti, C. K. Panda, and R. Chakravarty. 2008. Mo-lecular epidemiology and clinical significance of hepatitis B virus genotypes,core promoter and precore mutation in eastern India. Intervirology 51:275–284.
14. Datta, S., P. K. Chandra, A. Banerjee, R. Chakravarty, K. M. Murhekar, andM. V. Murhekar. 2007. Predominance of hepatitis B virus genotype C amongKarens, the old settlers of Andaman and Nicobar Islands, India. Arch. Virol.152:1223–1228.
15. Di Liberto, G., A. M. Roque-Afonso, R. Kara, D. Ducoulombier, G. Fallot, D.Samuel, and C. Feray. 2006. Clinical and therapeutic implications of hepa-titis C virus compartmentalization. Gastroenterology 131:76–84.
16. Gandhe, S. S., M. S. Chadha, and V. A. Arankalle. 2003. Hepatitis B virusgenotypes and serotypes in western India: lack of clinical significance.J. Med. Virol. 63:324–330.
VOL. 83, 2009 COMPARTMENTALIZATION OF HEPATITIS B VIRUS IN PBL 9991

17. Gonzales, M. J., J. M. Dugan, and R. W. Shafer. 2002. Synonymous-nonsynonymous mutation rates between sequences containing ambiguous nu-cleotides (Syn-Scan). Bioinformatics 18:886–887.
18. Gummuluru, S., C. M. Kinsey, and M. Emerman. 2000. An in vitro rapidturnover assay for human immunodeficiency virus type 1 replication selectsfor cell-to-cell spread of virus. J. Virol. 74:10882–10891.
19. Hall, T. A. 1999. BioEdit: a user-friendly biological sequence alignmenteditor and analysis program for Windows 95/98/NT. Nucleic Acids Symp.Ser. 41:95–98.
20. Hasegawa, I., Y. Tanaka, A. Kramvis, T. Kato, F. Sugauchi, S. K. Acharya,E. Orito, R. Ueda, M. C. Kew, and M. Mizokami. 2004. Novel hepatitis Bvirus genotype A subtyping assay that distinguishes subtype Aa from Ae andits application in epidemiological studies. J. Virol. 78:7575–7581.
21. Jammeh, S., H. C. Thomas, and P. Karayiannis. 2007. Replicative compe-tence of the T131I, K141E, and G145R surface variants of hepatitis B virus.J. Infect. Dis. 196:1010–1013.
22. Kalinina, T., A. Iwanski, H. Will, and M. Sterneck. 2003. Deficiency in virionsecretion and decreased stability of the hepatitis B virus immune escapemutant G145R. Hepatology 38:1274–1281.
23. Kock, J., L. Theilmann, P. Galle, and H. J. Schlicht. 1996. Hepatitis B virusnucleic acids associated with human peripheral blood mononuclear cells donot originate from replicating virus. Hepatology 23:405–413.
24. Korber, B., and G. Myers. 1992. Signature pattern analysis: a method forassessing viral sequence relatedness. AIDS. Res. Hum. Retrovir. 8:1549–1560.
25. Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: integrated software formolecular evolutionary genetics analysis and sequence alignment. Brief.Bioinform. 5:150–163.
26. Kwok, S., and R. Higuchi. 1989. Avoiding false positives with PCR. Nature339:237–238.
27. Lindh, M., A. S. Andersson, and A. Gusdal. 1997. Genotypes, nt 1858variants, and geographic origin of hepatitis B virus—large-scale analysisusing a new genotyping method. J. Infect. Dis. 175:1285–1293.
28. McGrath, K., N. G. Hoffman, W. Resch, J. A. E. Nelson, and R. Swanstrom.2001. Using HIV-1 sequence variability to explore virus biology. Virus Res.76:137–160.
29. Michalak, T. I. 2000. Occult persistence and lymphotropism of hepadnaviralinfections: insights from the woodchuck viral hepatitis model. Immunol. Rev.174:98–111.
30. Michalak, T. I., P. M. Mulrooney, and C. S. Coffin. 2004. Low doses ofhepadnavirus induce infection of the lymphatic system that does not engagethe liver. J. Virol. 78:1730–1738.
31. Mulrooney-Cousins, P. M., and T. I. Michalak. 2008. Repeated passage ofwild-type woodchuck hepatitis virus in lymphoid cells does not generate celltype-specific variants or alter virus infectivity. J. Virol. 82:7540–7550.
32. Nakajima, N., N. Hijikata, H. Yoshikura, and Y. K. Shimizu. 1996. Charac-terization of long-term cultures of hepatitis C virus. J. Virol. 70:3325–3329.
33. Navas, S., J. Martin, J. A. Quiroga, I. Castillo, and V. Carreno. 1998.Genetic diversity and tissue compartmentalization of the hepatitis C virusgenome in blood mononuclear cells, liver, and serum from chronic hepatitisC patients. J. Virol. 72:1640–1646.
34. Norder, H., A. M. Courouce, P. Coursaget, J. M. Echevarria, S. D. Lee, I. K.Mushahwar, B. H. Robertson, S. Locarnini, and L. O. Magnius. 2004. Ge-netic diversity of hepatitis B virus strains derived worldwide: genotypes,subgenotypes, and HBsAg subtypes. Intervirology 47:289–309.
35. Park, S. C., S. H. Jeong, J. Kim, C. J. Han, Y. C. Kim, K. S. Choi, J. H. Cho,M. Lee, H. H. Jung, S. S. Ki, Y. H. Chang, S. S. Lee, Y. H. Park, and K. H.Lee. 2008. High prevalence of hepatitis B virus infection in patients withB-cell non-Hodgkin’s lymphoma in Korea. J. Med. Virol. 80:960–966.
36. Penna, A., M. Artini, A. Cavalli, M. Levero, A. Bertoletti, M. Pilli, F. V.Chisari, B. Rehermann, G. Del Prete, F. Fiaccadori, and C. Ferrari. 1996.Long-lasting memory T cell responses following self-limited acute hepatitisB. J. Clin. Investig. 98:1185–1194.
37. Pham, T. N. Q., and T. I. Michalak. 2008. Occult persistence and lympho-tropism of hepatitis C virus infection. World J. Gastroenterol. 14:2789–2793.
38. Philpott, S., H. Burger, C. Tsoukas, B. Foley, K. Anastos, C. Kitchen, and B.Weiser. 2005. Human immunodeficiency virus type 1 genomic RNA se-quences in the female genital tract and blood: compartmentalization andintrapatient recombination. J. Virol. 79:353–363.
39. Pillai, S. K., S. L. K. Pond, Y. Liu, B. M. Good, M. C. Strain, R. J. Ellis, S.Letendre, D. M. Smith, H. F. Gunthard, I. Grant, T. D. Marcotte, J. A.
McCutchan, D. D. Richman, and J. K. Wong. 2006. Genetic attributes ofcerebrospinal fluid-derived HIV-1 env. Brain 129:1872–1883.
40. Pisoni, G., P. Moroni, L. Turin, and G. Bertoni. 2007. Compartmentalizationof small ruminant lentivirus between blood and colostrum in infected goats.Virology 369:119–130.
41. Pond, S. L. K., S. D. W. Frost, Z. Grossman, M. B. Gravenor, D. D. Richman,and A. J. Leigh Brown. 2006. Adaptation to different human populations byHIV-1 revealed by codon-based analyses. PLoS Comput. Biol. 2:530–538.
42. Poon, A. F. Y., S. L. K. Pond, P. Bennett, D. D. Richman, A. J. Leigh Brown,and S. D. W. Frost. 2007. Adaptation to human populations is revealed bywithin-host polymorphisms in HIV-1 and hepatitis C virus. PLoS Pathog.3:409–417.
43. Richman, D. D., S. J. Little, D. M. Smith, T. Wrin, C. Petropoulos, and J. K.Wong. 2004. HIV evolution and escape. Trans. Am. Clin. Climatol. Assoc.115:289–303.
44. Ritola, K., K. Robertson, S. A. Fiscus, C. Hall, and R. Swanstrom. 2005.Increased human immunodeficiency virus type 1 (HIV-1) env compartmen-talization in the presence of HIV-1-associated dementia. J. Virol. 79:10830–10834.
45. Roque-Afonso, A. M., D. Ducoulombier, G. Di Liberto, R. Kara, M. Gigou,E. Dussaix, D. Samuel, and C. Feray. 2005. Compartmentalization of hep-atitis C virus genotypes between serum and peripheral blood mononuclearcells. J. Virol. 79:6349–6357.
46. Sallie, R. 2005. Replicative homeostasis: a fundamental mechanism mediat-ing selective viral replication and escape mutation. Virol. J. 2:10.
47. Schaefer, S. 2005. Hepatitis B virus: significance of genotypes. J. Viral.Hepat. 12:111–124.
48. Schilling, R., S. Ijaz, M. Davidoff, J. Y. Lee, S. Locarnini, R. Williams, andN. V. Naoumov. 2003. Endocytosis of hepatitis B immune globulin intohepatocytes inhibits the secretion of hepatitis B virus surface antigen andvirions. J. Virol. 77:8882–8892.
49. Sheldon, J., and V. Soriano. 2008. Hepatitis B virus escape mutants inducedby antiviral therapy. J. Antimicrob. Chemother. 61:766–768.
50. Sitki-Green, D., M. Covington, and N. Raab-Traub. 2003. Compartmental-ization and transmission of multiple Epstein-Barr virus strains in asymptom-atic carriers. J. Virol. 77:1840–1847.
51. Tai, D. I., Z. J. Chung, C. L. Chen, and H. L. Eng. 2001. Reappearance ofHBsAg with compartmentalized different HBV strains in allograft versusPBMC of the recipient. J. Gastroenterol. 36:200–205.
52. Thakur, V., R. C. Guptan, S. N. Kazim, V. Malhotra, and S. K. Sarin. 2002.Profile, spectrum and significance of HBV genotypes in chronic liver diseasepatients in the Indian subcontinent. J. Gastroenterol. Hepatol. 2:165–170,2002.
53. Torii, N., K. Hasegawa, R. Joh, and R. Hayashi. 2003. Configuration andreplication competence of hepatitis B virus DNA in peripheral blood mono-nuclear cells from chronic hepatitis B patients and patients who have recov-ered from acute self-limited hepatitis. Hepatol. Res. 25:234–243.
54. Voronin, Y., B. Chohan, M. Emerman, and J. Overbaugh. 2007. Primaryisolates of human immunodeficiency virus type 1 are usually dominated bythe major variants found in blood. J. Virol. 81:10232–10241.
55. Wang, F., R. Xu, B. Han, Y. Shi, H. Luo, W. Jiang, T. Lin, H. Huang, Z. Xia,and Z. Guan. 2007. High incidence of hepatitis B virus infection in B-cellsubtype non-Hodgkin lymphoma compared with other cancers. Cancer 109:1360–1364.
56. Waters, J. A., M. Kennedy, P. Voett, P. Hauser, J. Petre, W. Carman, andH. C. Thomas. 1992. Loss of the common “a” determinant of hepatitis Bsurface antigen by a vaccine-induced escape mutant. J. Clin. Investig. 90:2543–2547.
57. Wieland, S. F., and F. V. Chisari. 2005. Stealth and cunning: hepatitis B andhepatitis C viruses. J. Virol. 79:9369–9380.
58. Zhang, L., L. Rowe, T. He, C. Chung, J. Yu, W. Yu, A. Talal, M. Markowitz,and D. D. Ho. 2002. Compartmentalization of surface envelope glycoproteinof human immunodeficiency virus type 1 during acute and chronic infection.J. Virol. 76:9465–9473.
59. Zhang, S. Y., H. X. Gu, D. Li, S. F. Yang, Z. H. Zhong, X. K. Li, and X. Jin.2006. Association of leukocyte antigen polymorphism with hepatitis B virusinfection and genotypes. Jpn. J. Infect. Dis. 59:353–357.
60. Zhang, Y. Y., B. H. Zhang, D. Theele, S. Litwin, E. Toll, and J. Summers.2003. Single-cell analysis of covalently closed circular DNA copy numbers ina hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 100:12372–12377.
9992 DATTA ET AL. J. VIROL.
Copyright © 2022 FDOKUMEN




















