
Uji Cemaran Senyawa Sejenis Dan Titik Lebur
56
MAKALAH ANALISIS FARMASI UJI CEMARAN SENYAWA SEJENIS DAN JARAK LEBUR KELOMPOK 3 Annisa Auliyya 1406524902 Firman Mulyo Wicaksono 1406525256 Futty Dewi Nuzulia Famini 1406525262 Millatur Rodiyah 1406525470 Raissa Elvina Nanang 1406525666 Tri Amelia 1406525930 PROGRAM PROFESI APOTEKER
-
Upload
eka-wulandari -
Category
Documents
-
view
211 -
download
27
description
UJi cemaran senyawa sejenis dan titik lebur untuk analsis kualitatif pada bahan baku farmasi.
Transcript of Uji Cemaran Senyawa Sejenis Dan Titik Lebur

MAKALAH ANALISIS FARMASI
UJI CEMARAN SENYAWA SEJENIS DAN JARAK
LEBUR
KELOMPOK 3
Annisa Auliyya 1406524902
Firman Mulyo Wicaksono 1406525256
Futty Dewi Nuzulia Famini 1406525262
Millatur Rodiyah 1406525470
Raissa Elvina Nanang 1406525666
Tri Amelia 1406525930
PROGRAM PROFESI APOTEKER
FAKULTAS FARMASI
UNIVERSITAS INDONESIA
DEPOK
2014

BAB I
PENDAHULUAN
1.1. Uji Jarak Lebur
Suhu lebur atau titik lebur suatu senyawa merupakan temperatur dimana
zat padat berada dalam kesetimbangan dengan bentuk cairnya. Zat padat akan
berubah menjadi bentuk cairnya ketika molekul dari zat padat tersebut
mendapatkan energi yang cukup untuk memecah ikatan intermolekulernya. Suhu
lebur suatu zat tergantung pada struktur molekulnya.
Sementara itu, jarak lebur didefinisikan sebagai rentang temperatur atau
suhu pada saat bentuk padat tersebut mulai melebur hingga keseluruhan sampel
melebur semua. Dalam Farmakope, jarak lebur atau suhu lebur zat padat
didefinisikan sebagai rentang suhu atau suhu pada saat zat padat menyatu dan
melebur sempurna, kecuali didefinisikan lain. Alat yang digunakan untuk
penetapan titik lebur harus diperiksa ketepatan dan kebenarannya secara berkala
dengan satu atau lebih dari enam Baku Pembanding Suhu Lebur BPFI, lebih baik
digunakan satu baku yang melebur paling dekat dengan suhu lebur senyawa yang
ditetapkan seperti yang tertera pada Baku Pembanding.
Manfaat penetapan titik lebur atau jarak lebur, yaitu :
1. Suhu lebur sebagai indikator kemurnian
Suatu zat dapat dikatakan murni bila memiliki titik lebur yang sama dengan
standar zat tersebut atau jarak lebur yang sempit (1-2oC atau kurang).
Sebaliknya apabila suatu zat memiliki suhu lebur yang berbeda atau jarak
lebur yang melebar terhadap standar, maka dapat dikatakan bahwa zat
tersebut tidak murni.
2. Suhu lebur sebagai alat untuk identifikasi dan karakterisasi
Untuk mengidentifikasi dan mengkarakterisasi suatu senyawa, senyawa
tersebut harus dalam bentuk zat aktif murni dan dibandingkan dengan standar
yang memang telah terbukti kemurniannya. Apabila dua sampel memiliki
suhu lebur yang berbeda, dapat dikatakan bahwa kedua molekul sampel
tersebut berbeda baik secara struktur atau bentuk konfigurasinya. Kedua
sampel tersebut dapat diperkirakan merupakan isomer struktur. Apabila suhu

lebur antara dua sampel sama, struktur molekul kedua zat tersebut
diperkirakan sama.
Contoh alat penetapan jarak lebur yang sesuai terdiri dari:
1. Wadah gelas untuk tangas cairan dilengkapi dengan pengaduk dan diisi cairan
yang cocok. Sebagai cairan umumnya digunakan silicon cair.
2. Alat pengaduk yang sesuai
3. Termometer yang akurat
4. Kaca pembesar yang cocok.
5. Pipa kapiler berukuran panjang lebih kurang 10 cm dan diameter dalam 0,8
mm sampai 1,2 mm dengan ketebalan dinding 0,2 mm sampai 0,3 mm.
6. Sumber panas yang terkendali
Panas didapat dari api bebas atau listrik.
Cairan dalam tangas dipilih dengan melihat suhu yang dikehendaki,
tetapi umumnya digunakan parafin cair dan silikon cair yang baik untuk
rentang suhu yang lebih tinggi.
Cairan dalam tangas mempunyai kedalaman yang cukup sehingga
thermometer dapat tercelup dengan pencadang raksa tetap berada lebih
kurang 2 cm diatas dasar tangas.
Gambar 1.1. Alat Pengukuran Jarak Lebur

Gambar 1.2. Alat Penentuan Jarak Lebur
Gambar 1.3. Hasil Pengamatan penentuan Jarak Lebur
1.2. Uji Cemaran Senyawa Sejenis
Uji cemaran senyawa sejenis merupakan suatu pengujian dalam
monografi yang mengacu pada uji umum untuk menganalisis pengotor berupa
produk samping dari suatu zat aktif. Tujuan pengujian senyawa sejenis adalah

untuk mengontrol kadar produk samping saat proses sintesis dan pada
penyimpanan. Terdapat 3 metode yang digunakan untuk pengujian senyawa
sejenis :
1. Kromatografi Cair Kinerja Tinggi
A. Prinsip KCKT (Kromatografi Cair Kinerja Tinggi)
Kromatografi Cair Kinerja Tinggi (KCKT) atau High Performance Liquid
Chromatography (HPLC) merupakan suatu cara pemisahan zat yang didasarkan
pada perbedaan distribusi komponen-komponen zat yang ada pada sampel
terhadap fase gerak dan fase diam.
Instrumen KCKT
Injektor :berfungsi untuk memasukan cuplikan ke dalam kolom.
o Jenis injektor :
Aliran henti
Septum
Katup jalan kitar
Auto injektor
Pompa: untuk mengalirkan eluen kedalam kolom,pompa,segel-segel pompa
dan semua penghubung dalam sistem kromatografi harus terbuat dari bahan
yang secara kimiawi tahan terhadap fase gerak. Umumnya digunakan
gelas,baja nirkarat,teflon dan batu nilam.Tekanan minimal 103 atm.
o Jenis pompa :
Tekanan tetap
Pompa semprit
Pompa tekanan uap
Guard kolom : filter kimia untuk menahan material yang mungkin dapat
merusak atau menyumbat kolom.Berisikan fase diam yang mirip dengan
kolom
Kolom : untuk memisahkan masing-masing komponen.Kolom yang ada telah
tersedia dalam berbagai macam ukuran,kolom standar mempunyai diameter
dalam antara 4-5mm. Isi kolom harus berukuran homogen dan stabil.
Diameter partikel antara 4-7 µm, panjang kolom std 10-30 cm.

Detektor: berfungsi untuk mengidentifikasi komponen yang ada dalam eluat
dan mengukur jumlahnya.
o Sifat detektor yang ideal
Respon universal
Sensitivitas tinggi
Noisy rendah range linier dinamis
Respon tidak dipengaruhi variasi parameter
Respon terlepas dari komposisi fase gerak
Mudah digunakan dan dapat dipercaya
Tidak merusak analit
Tidak mahal
Respon stabil untuk waktu yg lama
Mampu memberikan informasi kualitatif mengenai analit
o Pengelompokan detektor KCKT berdasarkan sifat dan cara deteksi:
detektor umum: memberi respon terhadap fase gerak yang
dimodulasi dengan adanya solut.
detektor spesifik memberi respon terhadap beberapa sifat solut yang
tidak dimiliki oleh fase gerak.
detektor yang bersifat umum terhadap solute setelah fase gerak
dihilangkan dengan penguapan.
Integrator : untuk menghitung luas puncak
Fase gerak : faktor yang mempengaruhi pemisahan;variasi fase gerak sangat
beragam dalam hal kepolaran dan seletivitasnya terhadap komponen dalam
sampel;senyawa yang akan dipisahkan harus larut dalam pelarut yang
digunakan.
o Sifat eluen yang baik
Murni
Tidak bereaksi dengan kolom
Sesuai dengan detektor
Dapat melarutkan cuplikan

Selektif
Viskositas rendah
Memungkinkan dengan mudah untuk memperoleh cuplikan jika
diperlukan
Harga wajar
Dapat memisahkan zat dengan baik
Metode
Gambar 1.4. Skema Alat KCKT
Sampel yang telah dilarutkan dalam fase gerak kemudian diinjeksikan
kedalam KCKT melalui injektor, pompa akan memberi gaya pada sampel untuk
bergerak kekolom, pada kolom zat yang memiliki sifat yang sama dengan kolom
dalam hal ini polaritas zat dan kolom, zat yang bersifat polar akan tertahan pada
kolom yang bersifat polar sehingga zat yang bersifat non polar tidak tertahan dan
sebaliknya.Zat akan menuju detektor dan kemudian didapat hasil analisis berupa
kromatogram.
2. Kromatografi Lapis Tipis
Prinsip KLT (Kromatografi Lapis Tipis)
Kromatografi Lapis Tipis atau Thin Layer Chromatography (TLC)
merupakan metode pemisahan dimana yang memisahkan terdiri atas fase diam
yang ditempatkan pada penyangga berupa plat gelas, logam atau lapisan yang
cocok. Kromatografi lapis tipis termasuk kromatografi adsorpsi (serapan), dimana

fase diam digunakan zat padat yang disebut adsorben (penjerap) dan fase gerak
adalah zat cair yang disebut dengan larutan pengembang. Campuran yang akan
dipisahkan berupa larutan ditotolkan berupa bercak atau pita, kemudian plat
(lapisan) dimasukkan ke dalam bejana tertutup rapat yang berisi larutan
pengembang yang cocok (fase gerak) sehingga pemisahan terjadi selama
perambatan kapiler (pengembangan). Zat penjerap pada KLT merupakan lapisan
tipis serbuk yang dilapiskan pada lempeng kaca, plastik, atau logam secara
merata.
a. Prinsip Analisis Kualitatif
Dimana akan dibandingkan kesamaan/ kesesuaian Rf bercak zat uji dengan Rf
bercak baku pembanding dan juga spektrum serapan bercak zat uji dengan
spektrum serapan bercak baku pembanding.
b. Prinsip Analisis Kuantitatif
Dimana akan dibandingkan kesamaan/ kesesuaian Rf bercak zat uji dengan Rf
bercak baku pembanding dan juga spektrum serapan bercak zat uji dengan
spektrum serapan bercak baku pembanding.
3. Kromatografi Gas
Prinsip Kromatografi Gas
Kromatografi gas (KG) merupakan metode pemisahan dan deteksi
senyawa organik yang mudah menguap dan senyawa gas anorganik dalam suatu
campuran.Kromatografi gas dapat diotomatisasi untuk analisis sampel-sampel
padat, cair, dan gas.Prinsip kromatografi gas yaitu teknik pemisahan dimana
pembawa yang mudah menguap dan stabil terhadap suhu tinggi bermigrasi
melalui kolom yang mengandung fase diam.
Ada dua jenis kromatografi gas :
1. Kromatografi Gas Cair (KGC)
KGC menggunakan fase diam berupa cairan dengan mekanisme sorpsi-nya
yaitu partisi.

2. Kromatografi Gas Padat (KGP)
KGP menggunakan fase diam padatan dengan mekanisme sorpsi-nya yaitu
adsorpsi permukaan.
Pemisahan pada kromatografi gas didasari pada titik didih suatu senyawa
yang juga dipengaruhi oleh interaksi yang mungkin terjadi antara pembawa dan
fase diam. Fase gerak yang berupa gas akan mengelusi pembawa dari ujung
kolom lalu menghantarkannya ke detektor.
Instrumentasi
Gambar 1.5. Instrumentasi Kromatografi Gas
Bagian-bagian utama dari sebuah kromatografi gas, yaitu : gas pembawa,
pengatur kecepatan alir, ruang suntik sampel dan sampling, kolom yang
diletakkan dalam oven yang dikontrol secara termostatik, sistem deteksi dan
pencatat (detector dan recorder), serta komputer yang dilengkapi perangkat
pengolah data.
Secara singkat, suatu gas pembawa inert mengalir terus-menerus dari
sebuah tabung gas besar melalui lubang injeksi, kolom, dan detector.Kecepatan
alir dari gas pembawa secara hati-hati dikontrol untuk memastikan hasil waktu
retensi dan meminimalisasi penyimpangan atau gangguan pada detektor. Sampel

diinjeksikan, umumnya menggunakan microsyringe, melalui lubang injeksi yang
dipanaskan, kemudian sampel akan menguap dan terbawa kedalam kolom.
Sampel tersebut akan terpisahkan menjadi komponen-komponen tunggal
berdasarkan konstanta distribusinya dalam fase diam dan fase gerak. Setelah
berhasil melalui kolom, gas pembawa dan sampel akan diteruskan ke detektor.
Alat ini akan mengukur kuantitas sampel dan mengirimkan signal data menuju
sistem data atau integrator yang kemudian menghasilkan suatu kromatogram,
catatan tertulis hasil analisis kromatografi, mengintegrasi area puncak, waktu
retensi, dan kalkulasi hasil kuantitatif.
1. Gas Pembawa
Fase gerak pada KG disebut dengan gas pembawa karena tujuannya adalah
untuk membawa solut ke kolom sehingga gas pembawa tidak berpengaruh
pada selektifitas.Tujuan kedua dari fase gerak ialah untuk menghasilkan suatu
matriks yang sesuai bagi detektor untuk menganalisis komponen sampel.
Syarat dari gas pembawa, antara lain tidak reaktif; murni/kering; dan dapat
disimpan dalam tangki tekanan tinggi. Kecepatan linier dari carrier gas
menentukan efisiensi kolom. Gas yang biasa digunakan, yaitu nitrogen,
helium, argon, dan hidrogen.
2. Kecepatan Alir
Pengatur kecepatan alir penting untuk efisiensi kolom dan pengukuran analisis
kualitatif.Efisiensi kolom bergantung dari kesesuaian linieritas kecepatan alir
gas yang ditentukan oleh perubahan kecepatan alir hingga tercapainya plate
number (N) maksimum.Untuk analisis kualitatif, kecepatan alir yang konstan
menentukan waktu retensi yang dihasilkan pada kromatogram. Waktu retensi
tersebut yang kemudian akan digunakan untuk mengidentifikasi komponen-
komponen dari sampel. Sehingga, laju alir yang baik juga menentukan hasil
identifikasi senyawa yang spesifik.
3. Ruang suntik sampel
Fungsi dari ruang suntik sampel adalah untuk menghantarkan sampel ke
dalam aliran gas pembawa.Ruang suntik sampel atau lubang injeksi harus

mampu menangani berbagai bentuk sampel, baik gas, cairan, maupun padatan,
dan dengan segera dan kuantitatif diteruskan ke aliran gas pembawa. Untuk
sampel dalam bentuk gas, umumnya interaksi antara sampel gas dan cairan
pada fase diam akan menimbulkan masalah, sehingga umumnya campuran
tersebut dipanaskan hingga terbentuk gas atau diberikan tekanan hingga
terbentuk cairan. Untuk sampel dalam bentuk cairan, sebaiknya menggunakan
konsentrasi rendah dengan volume yang lebih kecil, seperti 1, 5, atau 10μL.
Sedangkan, untuk sampel dalam bentuk padatan, preparasi sampel akan lebih
mudah karena hanya melarutkan sampel tersebut dalam pelarut sesuai yang
mudah menguap.
Ruang suntik ini harus dipanaskan tersendiri (terpisah dari kolom) dan
biasanya 10-15oC lebih tinggi daripada suhu kolom maksimum. Jadi, seluruh
sampel akan menguap segera setelah sampel disuntikkan.
4. Kolom
Kolom merupakan tempat terjadinya proses pemisahan karena di dalamnya
terdapat fase diam. Ada dua jenis kolom pada KG, yaitu kolom kemas
(packing column) dan kolom kapiler (capillary column). Kolom kemas terdiri
atas fase cair yang tersebar pada permukaan penyangga yang inert yang
terdapat dalam tabung yang relative besar ( diameter 1-3 mm). Kolom kapiler
jauh lebih kecil ( 0,02 – 0.2 mm) dan dinding kapiler bertindak sebagai
penyangga lembam untuk fase diam cair. Fase diam melekat mengelilingi
dinding dalam kolom. Ada empat jenis lapisan pada kolom kapiler : WCOT (
Wall Coated Open Tube), SCOT ( Support Coated Open Tube), PLOT (
Porous Layer Open Tube), dan FSOT ( Fused Silica Open Tube).
Ketika menggambarkan suatu kolom, seseorang biasanya menyatakan panjang
kolom (dalam meter), diameter kolom ( dalam millimeter), ketebalan lapisan
fase diam ( dalam micrometer, dan jenis fase diam. Banyak bahan kimia yang
dapat dipakai sebagai fase diam, antara lain : squalen, DEGS, OV-17, dll. Fase
diam yang dipakai pada kolom kapiler dapat bersifat non polar, polar, atau
semi polar.Jenis fase diam menentukan urutan elusi komponen-komponen
dalam cairan.

Fase Diam Polaritas Golongan Sampel Suhu Maksimum
Squalen Non polar Hidrokarbon 125oC
Apiezon L Non polar Hidrokarbon, ester,
eter
300 oC
Metal silicon Non polar Steroid, pestisida,
alkaloid, ester
300 oC
Dionil ptalat Semi polar Semua jenis 170 oC
Dietilenglikolsuksinat Polar Ester 200 oC
Carbowax 20M Polar Alkohol,amina,
aromatic, keton
250 oC
Tabel 1.1. Jenis Fase Diam dan Penggunaannya
Pemisahan dengan KG didasarkan pada dua sifat senyawa yang
dipisahkan, yaitu kelarutan senyawa dalam cairan tertentu dan tekanan uap atau
keatsiriannya.Karena tekanan uap berbanding langsung dengan suhu, maka suhu
merupakan faktor yang utama pada KG.Pemisahan pada KG dapat dilakukan pada
suhu tetap yang biasanya disebut dengan pemisahan isothermal dan dapat
dilakukan menggunakan suhu yang berubah secara terkendali yang disebut dengan
pemisahan suhu terprogram.
Setelah kolom dipakai dalam jangka waktu sekian lama, kemungkinan
yang sering terjadi adalah penyumbatan kolom, sehingga mengakibatkan kinerja
kolom akan menurun. Jika hal ini terjadi, maka perlu dilakukan regenerasi untuk
mengembalikan kinerja kolom. Ada tiga cara regenerasi kolom :
a. Pemotongan kolom
Biasanya dilakukan jika terjadi penyumbatan pada ujung depan kolom.
b. Pengkondisian
Bersifat untuk memelihara kolom agar waktu hidupnya cukup lama.
c. Pencucian kolom

Untuk kolom fase terikat sebaiknya dilakukan pencucian menggunakan tangki
(tabung) pencuci yang dilakukan di luar oven.Laritan pencuci terbaik yaitu
pentana.
5. Oven (Temperatur)
Suhu kromatografi sebaiknya termostatik sehingga terjadi pemisahan yang
baik dalam waktu sesingkat mungkin dengan rentang suhu yang cukup luas.
Pengaturan suhu merupakan salah satu cara yang efektif untuk memeperbaiki
pemisahan komponen dalam campuran.
Ruang injeksi haruslah cukup panas sehingga dapat menguapkan sampel
sesegera mungkin setelah diinjeksikan supaya hasil injeksi sampel lebih
kuantitatif dan efisien.Namun, temperatur lubang injeksi haruslah serendah
mungkin dan temperatur kolom termostatik.Termperatur dari detektor
bergantung dari jenis detektor yang digunakan.Secara umum, temperatur
detektor harus cukup tinggi untuk mencegah kondensasi sampel atau cairan
dalam fase diam.
Tabel 1.2. Jenis-Jenis Detektor, Batas Deteksi, Jenis Sampel-Sampelnya, dan
Kecepatan Aliran Gas Pembawa

Apabila waktu retensi, area puncak, dan bentuk kromatogram berubah-ubah
kemungkinan terjadi dekomposisi atau modifikasi kimia bahan sampel akibat
termperatur terlalu tinggi.Sedangkan, apabila efisiensi kolom berubah
kemungkinan temperature terlalu rendah.
Jenis detektor Jenis Sampel Batas deteksi
Kecepatan Alir (ml/menit)
Gas
pembawaH2 Udara
Hantar panas Senyawa Umum 5-100 ng 15-30 - -
Ionisasi nyala Hidrokarbon 10 -100 pg 20-60 30-40 200-500
Penangkap
electron
Halogen organic,
pestisida
0,05-1 pg 30-60 - -
Nitrogen-
fosfor
Senyawa nitrogen
organik dan fosfat
organic
0,1-10 g 20-40 1-5 70-100
Fotometri
nyala (393 nm)
Senyawa-senyawa
sulfur
10-100 pg 20-40 50-70 60-80
Fotometri
nyala (526 nm)
Senyawa-senyawa
fosfor
1-10pg 20-40 120-170 100-150
Fotoionisasi Senyawa-senyawa
yang terionisasi
dengan UV
2 pg 30-40 - -
Konduktivitas
elektrolitik
Halogen, N, S 0,5 pg Cl, 2
pg S, 4 pg N
20-40 80 -
Fourier
transform-infra
red (FT-IR)
Senyawa-senyawa
organic
1000 pg 3-10 - -
Selektif masa Sesuai untuk
senyawa apapun
10 pg – 10 ng 0,5-30 - -
Emisi atom Sesuai untuk
elemen apapun
0,1 – 20 pg 60-70 - -

6. Detektor
Detektor merupakan perangkat yang diletakkan pada ujung kolom tempat
keluar fase gerak yang membawa komponen hasil pemisahan.Detektor ini
berfungsi mengubah sinyal gas pembawa dan komponen di dalamnya menjadi
sinyal elektronik, dimana sinyal elektronik ini berguna untuk analisis kualitatif
dan kuantitatif terhadap komponen-komponen yang terpisah di antara fase diam
dan fase gerak dalam bentuk suatu kromatogram.
7. Komputer (Sistem Data)
Komputer pada sistem KG berperan sebagai suatu alat pengolah data.
Informasi yang diperoleh dapat dimanfaatkan dalam analisis kualitatif, biasanya
dengan membandingkan waktu retensi sampel dalam kondisi analisis yang sama.
Sedangkan, untuk analisi kuantitatif biasanya dilakukan dengan perhitungan
relatif tinggi atau luas puncak kromatogram sampel melalui metode baku luar
(external standar) atau baku dalam (internal standar).

BAB 2
TINJAUAN PUSTAKA
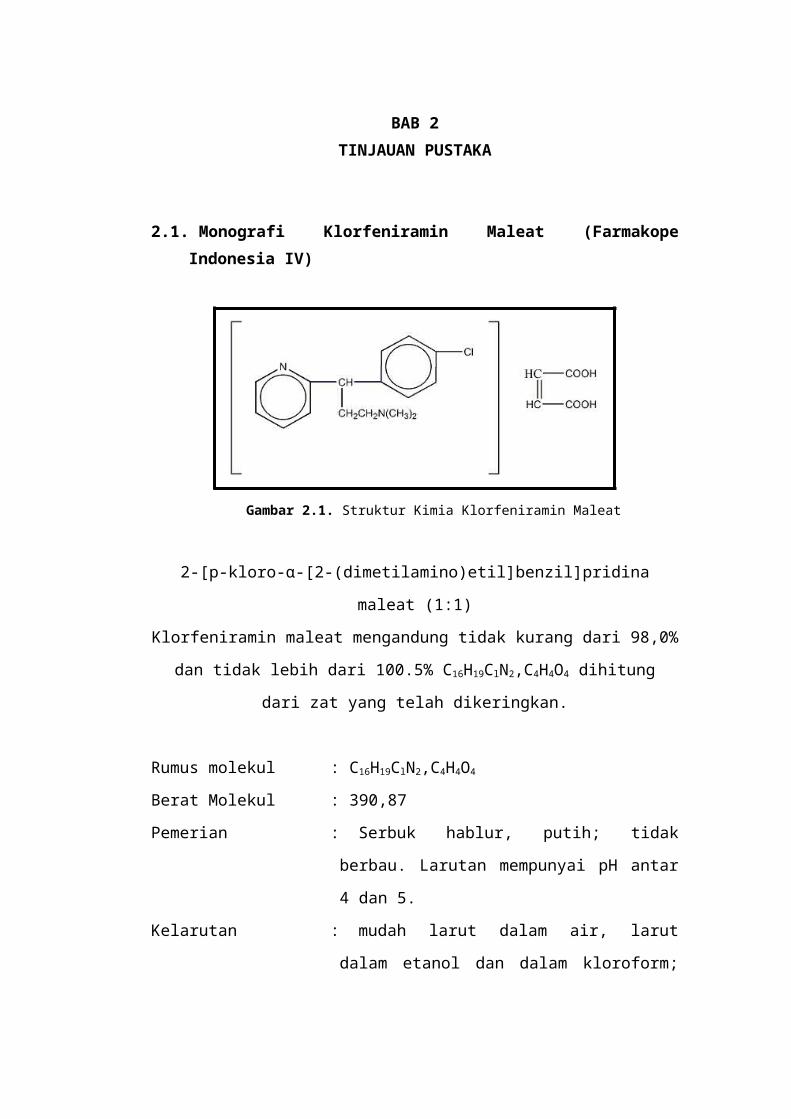
2.1. Monografi Klorfeniramin Maleat (Farmakope Indonesia IV)
Gambar 2.1. Struktur Kimia Klorfeniramin Maleat
2-[p-kloro-α-[2-(dimetilamino)etil]benzil]pridina maleat (1:1)
Klorfeniramin maleat mengandung tidak kurang dari 98,0% dan tidak lebih dari
100.5% C16H19ClN2,C4H4O4 dihitung dari zat yang telah dikeringkan.
Rumus molekul : C16H19ClN2,C4H4O4
Berat Molekul : 390,87
Pemerian : Serbuk hablur, putih; tidak berbau. Larutan
mempunyai pH antar 4 dan 5.
Kelarutan : mudah larut dalam air, larut dalam etanol dan dalam
kloroform; sukar larut dalam eter dan dalam
benzena.
Baku Pembanding : Klorfeniramin maleat BPFI; lakukan pengeringan
pada suhu 105oC selama 3 jam sebelum digunakan.
Identifikasi : Spektrum serapan infamerah zat yang didispersikan
dalam kalium bromida P menunjukkan maksimum
hanya pada panjang gelombang yang sama seperti
klorfeniramin maleat BPFI.

Susut pengeringan : Tidak lebih dari dari 0,5%, lakukan pengeringan
pada suhu 105oC selama 3 jam.
Sisa pemijaran : Tidak lebih 0,2%
Senyawa sejenis : Tidak lebih dari 2,0%.
Larutan uji : Larutkan lebih kurang 200 mg dalam
5 mL metilen klorida P.
Sistem kromatografi lakukan penetapan dengan cara
Kromatografi gas seperti yang tertera pada
kromatografi. Kromatografi gas dilengkapi dengan
detektor ionisasi nyala dan kolom kaca 1,2 m x 4
mm yang berisi bahan pengisi 3% fase diam G3
pada partikel penyangga S1AB. Pertahankan suhu
kolom, injektor, dan detektor berturut-turut pada
suhu lebih kurang 190o, 250o, dan 250o. gunakan
helium P kering sebagai gas pembawa dengan
mengatur lau aliran sehingga waktu retensi puncak
utama 4-5 menit. Lakukan kromatografi terhadap
Larutan uji, rekam luas puncak seperti yang tertera
pada prosedur. Faktor ikutan klorfeniramin maleat
tidak lebih dari 1,8.
Prosedur : suntikkan lebih kurang 1 µl Larutan uji.
Rekam kromatogram dalam eaktu tidak kurang dari
2 kali waktu retensi puncak klorfeniramin maleat
dan ukur luas puncak. Jumlah keseluruhan luas
relatif dari semua puncak kecuali puncak pelatur dan
asam maleat tidak lebih dari 2,0%.
Penetapan kadar : Timbang saksama lebih kurang 500 mg, larutkan
dalam 20 ml asam asetat glasial P, tambahkan 2 tetes
kristal violet LP dan titrasi dengan asam perklorat
0,1 N LV. Lakukan penetapan blanko.
1 asam perklorat 0,1 N setara dengan 19,54 mg
C16H19ClN2,C4H4O4.

Wadah dan penyimpanan : Dalam wadah tertutup rapat, tidak tembus cahaya.
2.2. Pengujian Jarak Lebur Klorfeniramin Maleat
Berdasarkan metode pada Farmakope Indonesia edisi IV, penentuan jarak
lebur dilakukan dengan cara sebagai berikut :
a. Sampel dimasukkan ke dalam mortir dan digerus hingga halus.
b. Setelah itu, sampel dikeringkan di atas silika.
c. Tangas dipanaskan hingga ± 10o dibawah suhu lebur yang diperkirakan
(± 120o).
d. Setelah suhu tangan mencapai ± 120o, suhu tangas air dinaikkan dengan
kecepatan 1o ± 0,5o / menit.
e. Sampel dipindahkan ke atas kertas perkamen.
f. Pipa kapiler yang salah satu ujungnya tertutup diletakkan tegak lurus di atas
sampel, kemudian diketuk-ketuk untuk memasukkan sampel ke dalam pipa
kapiler.
g. Sampel dimasukkan hingga ketinggian 2,5-3,5 mm.
h. Termometer diangkat termometer dan secepatnya tabung kapiler ditempelkan
pada termometer dengan membasahi kedua ujungnya dengan tetesan cairan
dari tangas, tinggi bahan dalam kapiler diatur hingga setinggi pencadang
raksa.
i. Termometer ditempatkan kembali dan pemanasan dilanjutkan dengan
pengadukan tetap secukupnya hingga suhu naik lebih kurang 3° di bawah dari
batas bawah jarak lebur yang diperkirakan, pemanasan dikurangi hinga suhu
naik lebih kurang 1° sampai 2° per menit.
j. Suhu pada saat kolom uji yang diamati terlepas sempurna dari dinding kapiler
dicatat sebagai permukaan melebur dan suhu pada saat sampel mencair
seluruhnya dicatat sebagai suhu lebur.
k. Kedua suhu tersebut dicatat sebagai batas jarak lebur.

2.3. Uji Jarak Lebur Zat Lain yang Menggunakan Metode yang sama
Berdasarkan Farmakope Indonesia edisi IV
1. Dietilstilbestrol
α,α’-Dietil-(E)-4,4’-stilbenediol
Gambar 2.2. Struktur Kimia Dietilstilbestrol
Jarak lebur dietilstilbestrol yaitu antara 169o dan 175o. Penetapan jarak
lebur dilakukan dengan langkah-langkah sebagai berikut :
a. Sampel dimasukkan ke dalam mortir dan digerus hingga halus.
b. Setelah itu, sampel dikeringkan di atas silika.
c. Tangas dipanaskan hingga ± 10o dibawah suhu lebur yang diperkirakan
(± 159o).
d. Setelah suhu tangan mencapai ± 159o, suhu tangas air dinaikkan dengan
kecepatan 1o ± 0,5o / menit.
e. Sampel dipindahkan ke atas kertas perkamen.
f. Pipa kapiler yang salah satu ujungnya tertutup diletakkan tegak lurus di atas
sampel, kemudian diketuk-ketuk untuk memasukkan sampel ke dalam pipa
kapiler.
g. Sampel dimasukkan hingga ketinggian 2,5-3,5 mm.
h. Termometer diangkat termometer dan secepatnya tabung kapiler ditempelkan
pada termometer dengan membasahi kedua ujungnya dengan tetesan cairan
dari tangas, tinggi bahan dalam kapiler diatur hingga setinggi pencadang
raksa.
i. Termometer ditempatkan kembali dan pemanasan dilanjutkan dengan
pengadukan tetap secukupnya hingga suhu naik lebih kurang 3° di bawah dari
batas bawah jarak lebur yang diperkirakan, pemanasan dikurangi hinga suhu
naik lebih kurang 1° sampai 2° per menit.

j. Suhu pada saat kolom uji yang diamati terlepas sempurna dari dinding kapiler
dicatat sebagai permukaan melebur dan suhu pada saat sampel mencair
seluruhnya dicatat sebagai suhu lebur.
k. Kedua suhu tersebut dicatat sebagai batas jarak lebur.
2. Isoniazid
Asam isonikotinat hidrazida
Gambar 2.3. Struktur Kimia Isoniazid
Jarak lebur isoniazid yaitu antara 170o dan 173o. Penetapan jarak lebur
dilakukan dengan langkah-langkah sebagai berikut :
a. Sampel dimasukkan ke dalam mortir dan digerus hingga halus.
b. Setelah itu, sampel dikeringkan di atas silika.
c. Tangas dipanaskan hingga ± 10o dibawah suhu lebur yang diperkirakan
(± 160o).
d. Setelah suhu tangan mencapai ± 160o, suhu tangas air dinaikkan dengan
kecepatan 1o ± 0,5o / menit.
e. Sampel dipindahkan ke atas kertas perkamen.
f. Pipa kapiler yang salah satu ujungnya tertutup diletakkan tegak lurus di atas
sampel, kemudian diketuk-ketuk untuk memasukkan sampel ke dalam pipa
kapiler.
g. Sampel dimasukkan hingga ketinggian 2,5-3,5 mm.
h. Termometer diangkat termometer dan secepatnya tabung kapiler ditempelkan
pada termometer dengan membasahi kedua ujungnya dengan tetesan cairan
dari tangas, tinggi bahan dalam kapiler diatur hingga setinggi pencadang
raksa.
i. Termometer ditempatkan kembali dan pemanasan dilanjutkan dengan
pengadukan tetap secukupnya hingga suhu naik lebih kurang 3° di bawah dari
batas bawah jarak lebur yang diperkirakan, pemanasan dikurangi hinga suhu
naik lebih kurang 1° sampai 2° per menit.
j. Suhu pada saat kolom uji yang diamati terlepas sempurna dari dinding kapiler
dicatat sebagai permukaan melebur dan suhu pada saat sampel mencair
seluruhnya dicatat sebagai suhu lebur.
k. Kedua suhu tersebut dicatat sebagai batas jarak lebur.
3. Guaifenesin
3-(o-Metoksifenoksi)-1,2-propanadiol
Gambar 2.4. Struktur Kimia Guaifenesin
Jarak lebur guaifenesin yaitu antara 78o dan 82o. Penetapan jarak lebur
dilakukan dengan langkah-langkah sebagai berikut :
a. Sampel dimasukkan ke dalam mortir dan digerus hingga halus.
b. Setelah itu, sampel dikeringkan di atas silika.
c. Tangas dipanaskan hingga ± 10o dibawah suhu lebur yang diperkirakan
(± 68o).
d. Setelah suhu tangan mencapai ± 168o, suhu tangas air dinaikkan dengan
kecepatan 1o ± 0,5o / menit.
e. Sampel dipindahkan ke atas kertas perkamen.
f. Pipa kapiler yang salah satu ujungnya tertutup diletakkan tegak lurus di atas
sampel, kemudian diketuk-ketuk untuk memasukkan sampel ke dalam pipa
kapiler.
g. Sampel dimasukkan hingga ketinggian 2,5-3,5 mm.
h. Termometer diangkat termometer dan secepatnya tabung kapiler ditempelkan
pada termometer dengan membasahi kedua ujungnya dengan tetesan cairan

dari tangas, tinggi bahan dalam kapiler diatur hingga setinggi pencadang
raksa.
i. Termometer ditempatkan kembali dan pemanasan dilanjutkan dengan
pengadukan tetap secukupnya hingga suhu naik lebih kurang 3° di bawah dari
batas bawah jarak lebur yang diperkirakan, pemanasan dikurangi hinga suhu
naik lebih kurang 1° sampai 2° per menit.
j. Suhu pada saat kolom uji yang diamati terlepas sempurna dari dinding kapiler
dicatat sebagai permukaan melebur dan suhu pada saat sampel mencair
seluruhnya dicatat sebagai suhu lebur.
k. Kedua suhu tersebut dicatat sebagai batas jarak lebur.
2.4. Uji Cemaran Senyawa Sejenis Klorfeniramin Maleat Berdasarkan
Farmakope Indonesia edisi IV
Syarat cemaran senyawa sejenis klorfeniramin maleat tidak lebih dari 2,0
%. Prosedur pengujian cemaran senyawa sejenis dengan metode kromatografi gas
adalah sebagai berikut :
1. Pembuatan larutan uji, dengan melarutkan kurang lebih 200 mg klorfeniramin
maleat dalam 5 mL metilen klorida P.
2. Sistem kromatografi :
Kromatografi gas dilengkapi dengan detektor ionisasi nyala dan kolom
kaca 1,2 m x 4 mm yang berisi bahan pengisi 3 % fase diam G3 (50%-
Fenil – 50% metilpolisiloksan) pada partikel penyangga S1AB (tanah
silika yang dicuci baik dengan asam maupun dengan basa, lalu
disilanisasikan).
Suhu kolom, injektor dan detektor berturut-turut dipertahankan pada suhu
kurang lebih 190˚C, 250˚C dan 250˚C.
Helium kering digunakan sebagai gas pembawa dengan mengatur laju
aliran sehingga waktu retensi puncak utama 4 sampai 5 menit.
3. Prosedur :
Suntikkan kurang lebih 1 µL larutan uji, lalu rekam kromatogram dalam
waktu tidak kurang dari 2 kali waktu retensi puncak klorfeniramin maleat dan
ukur luas puncak. Jumlah keseluruhan luas relatif dari semua puncak, kecuali

puncak pelarut dan asam maleat tidak lebih dari 2,0 %. Sedangkan, faktor
ikutan puncak klorfeniramin maleat tidak lebih dari 1,8.
2.5. Uji Cemaran Senyawa Sejenis Zat Lain yang Menggunakan Metode
Kromatografi Gas Berdasarkan Farmakope Indonesia edisi IV
1. Etosuksimida
2-Etil-2-metilsuksinimida
Gambar 2.5. Struktur Kimia Etosuksimida
Cemaran senyawa sejenis etosuksimida yaitu 2-Etil-2-metilsuksinat dan cemaran
lainnya tidak lebih dari 0,2%. Penetapan senyawa sejenis dilakukan dengan cara
kromatografi gas dengan langkah-langkah sebagai berikut :
a. Pembuatan larutan uji, dengan melarutkan sejumlah zat dalam kloroform P
hingga kadar menjadi 250 mg/mL.
b. Prosedur :
Suntikkan 1 µL larutan uji ke dalam kromatografi yang dilengkapi dengan
detektor ionisasi nyala dan kolom 1,8 m x 6,4 mm yang berisi fase diam 5
% G5 (3-sianopropilpolisiloksan) pada partikel penyangga S1A (tanah
silika yag telah diglukskalsinasikan dan tersilanisasi) 60 hingga 80 mesh.
Suhu injektor, kolom dan detektor dipertahankan secara berturut-turut
pada suhu 260˚C, 280˚C, dan 140˚C.
Helium P digunakan sebagai gas pembawa dengan laju alir 90 ml/menit
dan untuk udara 450 mL/menit.
Atur kepekaan alat untuk dapat mendeteksi anhidrida, biasanya 32 kali
lebih peka dari yang digunakan untuk mendeteksi etosuksimida.
Ukur luas puncak etoksuksimida dan luas puncak anhidrida atau cemaran
lain bila ada, dan lakukan koreksi untuk perbedaan dalam pengaturan
kepekaan.

Hitung jumlah dalam persen 2-etil-2-metilsuksinat anhidrida dan cemaran
lain dengan rumus :
100AB
Keterangan : A adalah jumlah luas puncak yang telah dikoreksi; B adalah jumlah
luas puncak dari etosuksimida anhidrida dan cemaran lain yang telah dikoreksi.
2. Fenfluramin HCl
Etil (α-metil-3-trifluorometilfenetil) amina
Gambar 2.6. Struktur Kimia Fenfluramin HCl
Cemaran senyawa sejenis fenfluramin HCl adalah Etil (α-metil-4-
trifluorometilfenetil) amina. Penetapan senyawa sejenis dilakukan dengan cara
kromatografi gas dengan langkah-langkah sebagai berikut :
Larutan baku dalam
Timbang sejumlah N,N-dietil-anilina P, larutkan dalam kloroform P
hingga kadar 0,01%.
Larutan baku
Timbang saksama lebih kurang 8 mg Fenfluramin HCl BPFI, larutkan
dalam 100 ml air, tambahkan 10 ml larutan KOH P 20%, ekstraksi 4 kali,
tiap kali dengan 25 ml kloroform P. saring dan uapkan kumpulan ekstrak
sampai kering dengan dialiri nitrogen P, larutkan sisa dalam 10 ml larutan
baku dalam.
Larutan uji I
Timbang saksama lebih kurang 400 mg zat uji, larutkan dalam 100 ml air,
tambahkan 10 ml larutan KOH P 20%, ekstraksi 4 kali, tiap kali dengan 25

ml kloroform P. saring dan uapkan kumpulan ekstrak sampai kering
dengan dialiri nitrogen P.
Larutan uji II
Timbang saksama lebih kurang 400 mg zat uji, larutkan dalam 100 ml air,
tambahkan 10 ml larutan KOH P 20%, ekstraksi 4 kali, tiap kali dengan 25
ml kloroform P. saring dan uapkan kumpulan ekstrak sampai kering
dengan dialiri nitrogen P, larutkan sisa dalam 10 ml larutan baku dalam.
Prosedur Uji :
Disuntikkan secara terpisah sejumlah volume sama Larutan baku, Larutan
uji I, dan Larutan uji II ke dalam KG yang dilengkapi dengan :
Detektor : Ionisasi nyala
Kolom : kolom kaca 2,75 m x 4 mm
Fase diam : senyawa polietilenglikol P (sebaiknya Carbowax 20M) dan
larutan KOH P 2% pada partikel penyangga tanah diatome cuci asam 80
mesh sampai 100 mesh
Suhu :
o Kolom :135oC
o Detektor : 200 oC
Efisiensi Kolom : Tidak kurang dari 1500 plat teoritis/meter, ditetapkan
menggunakan puncak baku dalam dalam kromatogram yang diperoleh dari
Larutan baku.
Persyaratan :
Pada kromatogram yang diperoleh dari Larutan uji I : puncak etil (α-metil-4-
trifluorometilfenetil) amina muncul segera puncak utama.
Pada kromatogram yang diperoleh dari Larutan uji II : perbandingan luas puncak
etil (α-metil-4-trifluorometilfenetil) amina terhadap puncak baku dalam tidak
lebih dari perbandingan luas puncak fenfluramin HCl terhadap puncak baku
dalam yang diperoleh dari Larutan baku.
3. Kaptopril
1-[(2S)-3-Merkapto-2-metilpropionil]-L-prolina

Gambar 2.7. Struktur Kimia Kaptopril
Cemaran senyawa sejenis kaptopril adalah Asam 3-merkapto-2-metilpropanoat.
Penetapan senyawa sejenis dilakukan dengan cara kromatografi gas dengan
langkah-langkah sebagai berikut :
Syarat: Cemaran senyawa sejenis tidak lebih dari 0,1%.
Pereaksi sililasi:
Buat larutan tert-butildimetilklorosilan dalam N-metil-N-tert-butildimetil
sililtrifluoro-asetamida (1 dalam 100).
Larutan baku internal:
Masukkan lebih kurang 0,4 mL asam 3-merkaptopropanoat ke dalam labu
ukur 10 mL, encerkan dengan metilen klorida P sampai tanda.
Larutan baku:
Timbang seksama sejumlah Garam dari Asam 3-merkapto-2-
metilpropanoat dan 1,2-Difenil-etilamin BPFI, larutkan dalam metilen
klorida P dan encerkan dengan metilen klorida P hingga kadar lebih
kurang 12 mg/mL. [Catatan : Buat segar bila hendak digunakan. Larutan
ini stabil selama lebih kurang 5 jam].
Sistem kromatografi:
Kromatografi gas dilengkapi dengan detektor ionisasi nyala, pertahankan
suhu lebih kurang 310oC dan kolom kapiler silika 15 m x 0,32 mm dilapisi
1 µm fase diam G27 (5% Fenil - 95% metilpolisiloksan) dan pemisahan
sistem injeksi dilapisi dengan wol kaca yang telah disililasi dengan
perbandingan pemisahan lebih kurang 25:1, pertahankan suhu lebih
kurang 250oC. Pertahankan suhu kolom pada 125oC selama 11 menit
setelah penyuntikan, naikkan suhu 30oC per menit hingga 300oC dan
pertahankan selama 8 menit. Gunakan helium P sebagai pembawa dan laju

aliran lebih kurang 1,7 mL/menit pada 125oC, selanjutnya laju aliran lebih
kurang 25 mL/menit.
Prosedur:
Pada 2 tabung vial yang tertutup ulir masukkan masing-masing 0,5 mL
metilen klorida P. Tambahkan 25,0 µL Larutan baku pada salah satu tabung.
Masukkan lebih kurang 100 mg kaptopril pada tabung ke dua dan campur.
Tambahkan 15,0 µL Larutan baku internal dan 0,4 mL Pereaksi sililasi pada
tiap tabung, tutup rapat tabung dengan penutup ulir dan campur hati-hati
dengan pengocok vortex. Letakkan tabung pada lempeng pemanas pada suhu
60oC selama 30 menit, angkat dan biarkan dingin.
Suntikkan 1,0 µL Larutan baku ke dalam kromatograf dan rekam luas
puncak dari larutan baku internal dan garam dari asam 3-merkapto-2-
metilpropanoat dan 1,2-difenil-etilamin (MMPA). Perbandingan simpangan
baku relatif luas puncak MMPA dan luas puncak larutan baku internal pada
penyuntikan ulang tidak lebih dari 2%. Waktu retensi relatif derivate silil dari
larutan baku internal dan derivate silil dari MMPA berturut-turut adalah lebih
kurang 0,85 dan 1,0. Dengan cara yang sama suntikkan sejumlah volume 1,0
µL Larutan uji. Hitung persentase asam 3-merkapto-2-metilpropanoat dalam
kaptopril yang digunakan dengan rumus:
( ) ( ) ( )
120,17 dan 317,45 berturut-turut adalah bobot molekul asam 3-merkapto-2-
metilpropanoat dan MMPA; C adalah kadar Garam dari asam 3-merkapto-2-
metilpropanoat dan 1,2-difeniletilamin BPFI dalam mg/mL Larutan baku; W
dalah bobot kaptopril dalam mg; Rs dan Ru berturut-turut adalah
perbandingan luas puncak asam 3-merkapto-2-metilpropanoat dan Larutan
baku internal dalam Larutan uji dan larutan baku.

BAB 3
PEMBAHASAN
3.1. Penetapan Jarak Lebur
Jarak lebur merupakan rentang temperatur pada saat bentuk padat dari
suatu zat mulai melebur hingga keseluruhan sampel melebur semua. Sehingga
secara singkat dapat dikatakan bahwa titik leleh suatu zat adalah suhu di mana
terjadi perubahan materi dari padat menjadi cair. Sebuah bahan murni meleleh
pada suhu tepat didefinisikan, karakteristik dari setiap substansi kristal dan
bergantung hanya pada tekanan (meskipun ketergantungan tekanan umumnya
dianggap tidak signifikan).
Ketika suatu padatan senyawa dipanaskan, molekul senyawa akan
menyerap energi. Makin tinggi suhu pemanasan makin banyak energi yang
diserap sehingga gerakan rotasi dan vibrasi molekul meningkat. Pada keadaan
cairan molekul masih terikat satu dengan yang lainnya tetapi sudah tidak teratur
lagi. Idealnya, penambahan panas pada saat suatu senyawa murni mencapai titik
leburnya tidak akan menyebabkan kenaikan suhu hingga seluruh padatan senyawa
melebur. Titik lebur dinyatakan dengan rentang suhu ketika padatan senyawa
mulai melebur hingga seluruh padatan melebur seluruhnya. Sehingga yang
sebenarnya dibaca pada pengukuran titik lebur adalah jarak lebur (Pasto, Johnson
& Miller, 1992; Gilbert, John & Martin, 2011).
Manfaat penetapan titik lebur atau jarak lebur, yaitu :
1. Suhu lebur sebagai indikator kemurnian
Suatu zat dapat dikatakan murni bila memiliki titik lebur yang sama dengan
standar zat tersebut atau jarak lebur yang sempit (1-2oC atau kurang).
Sebaliknya apabila suatu zat memiliki suhu lebur yang berbeda atau jarak
lebur yang melebar terhadap standar, maka dapat dikatakan bahwa zat
tersebut tidak murni.
2. Suhu lebur sebagai alat untuk identifikasi dan karakterisasi
Untuk mengidentifikasi dan mengkarakterisasi suatu senyawa, senyawa
tersebut harus dalam bentuk zat aktif murni dan dibandingkan dengan standar

yang memang telah terbukti kemurniannya. Apabila dua sampel memiliki
suhu lebur yang berbeda, dapat dikatakan bahwa kedua molekul sampel
tersebut berbeda baik secara struktur atau bentuk konfigurasinya. Kedua
sampel tersebut dapat diperkirakan merupakan isomer struktur. Apabila suhu
lebur antara dua sampel sama, struktur molekul kedua zat tersebut
diperkirakan sama.
Pada pelaksanaannya, persiapan sampel yang tidak sempurna adalah
penyebab utama hasil yang tidak akurat. Setiap sampel yang dimasukkan ke pipa
kapiler harus memenuhi beberapa syarat, yaitu :
a. Sampel harus benar-benar kering
b. Homogen
c. Dalam bentuk serbuk yang sangat halus
Persyaratan utama agar mendapatkan hasil yang baik yaitu sampel harus
berupa serbuk yang sangat halus. Hal tersebut akan menyebabkan transfer panas
ke sampel menjadi lebih efisien. Sampel yang memiliki bentuk kristal harus
digerus menggunakan mortir hingga menjadi serbuk yang sangat halus dan
homogen. Apabila sampel dalam ukuran besar diuji jarak leburnya, maka transfer
panas ke sampel menjadi tidak merata sehingga hasil yang didapatkan menjadi
tidak akurat.
Jumlah sampel yang digunakan pada pengujian jarak lebur haruslah tept.
Jumlah sampel yang terlalu banyak akan menyebabkan jarak lebur menjadi lebuh
besar karena dibutuhkan panas ekstra agar sampel tersebut mencair secara
keseluruhan. Hal tersebut menyebabkan hasil yang didapat menjadi tidak akurat.
3.2. Pembahasan Uji Cemaran Senyawa Sejenis
1. Pembahasan cemaran senyawa sejenis klorfeniramin maleat
Pada uji cemaran senyawa sejenis klorfeniramin maleat menggunakan
metode kromatografi gas. Pemilihan metilen klorida dalam pembuatan larutan uji
disebabkan karena metilen klorida bersifat semi polar sehingga dapat melarutkan

klorfeniramin maleat, serta metilen klorida memiliki titik didih yang rendah, yaitu
39,8 - 40˚C, sehingga mudah menguap.
Sistem kromatografi dalam uji ini terdiri atas fase diam yang bersifat semi
polar, yaitu fase diam G3 (50%-Fenil – 50% metilpolisiloksan). Pemilihan fase
diam disesuaikan dengan polaritas sampel, dimana sampel yang bersifat polar
menggunakan fase diam yang juga bersifat polar, dan sebaliknya. Oleh karena
klorfeniramin maleat dilarutkan dalam pelarut metilen klorida yang bersifat semi
polar, hal ini sesuai dengan fase diam yang digunakan yang juga bersifat semi
polar. Hal ini akan menyebabkan komponen-komponen dengan titik didih yang
sama dapat dipisahkan dengan adanya perbedaan koefisien partisi. Sedangkan,
detektor yang digunakan dalam kromatografi gas adalah ionisasi nyala (FID),
karena detektor ini dapat mendeteksi semua senyawa organik, dengan batas
terkecil pendeteksian 2 x 10-11 g/mL.
Setelah larutan uji dibuat, larutan tersebut diinjeksikan sebanyak 1 µL ke
dalam injektor. Suhu kolom, injektor dan detektor dijaga pada suhu 190˚C, 250˚C
dan 250˚C. Oleh karena metilen klorida memiliki titik didih yang rendah, maka
metilen klorida akan menguap terlebih dahulu, barulah disusul oleh klorfeniramin
maleat, lalu dideteksi oleh detektor. Detektor dapat menguraikan maleat yang
terkandung dalam klorfeniramin maleat menjadi asam maleat. Sehingga, hasil
kromatogram yang mungkin muncul adalah puncak metilen klorida, klorfeniramin
maleat, asam maleat, dan cemaran sejenisnya. Persyaratan cemaran senyawa
sejenis klorfeniramin maleat adalah jumlah keseluruhan luas relatif dari semua
puncak, kecuali puncak pelarut dan asam maleat tidak lebih dari 2,0 %.
2. Pembahasan cemaran senyawa sejenis etosuksimida
Pada uji cemaran senyawa sejenis etosuksimida menggunakan metode
kromatografi gas. 2-Etil-2-metilsuksinat merupakan contoh senyawa sejenis yang
sering terkandung dalam etosuksimida. Oleh karena etosuksimida merupakan zat
padat, maka untuk pengujian menggunakan kromatografi gas harus dibuat
menjadi larutan. Pemilihan kloroform dalam pembuatan larutan uji disebabkan
karena kloroform bersifat semi polar sehingga dapat melarutkan etosuksimida,

serta kloroform memiliki titik didih yang rendah, yaitu 61 - 62˚C, sehingga mudah
menguap.
Sistem kromatografi dalam uji ini terdiri atas fase diam yang bersifat semi
polar, yaitu fase diam G5 (3-sianopropilpolisiloksan). Pemilihan fase diam
disesuaikan dengan polaritas sampel, dimana larutan etosuksimida dan kloroform
bersifat semi polar, sehingga sesuai dengan fase diam yang digunakan yang juga
bersifat semi polar. Hal ini akan menyebabkan komponen-komponen dengan titik
didih yang sama dapat dipisahkan dengan adanya perbedaan koefisien partisi.
Sedangkan, detektor yang digunakan dalam kromatografi gas adalah ionisasi
nyala (FID), karena detektor ini dapat mendeteksi semua senyawa organik,
dengan batas terkecil pendeteksian 2 x 10-11 g/mL.
Setelah larutan uji dibuat, larutan tersebut diinjeksikan sebanyak 1 µL ke
dalam injektor. Suhu injektor, kolom dan detektor dijaga pada suhu 260˚C, 280˚C
dan 140˚C. Ketika larutan diinjeksikan, maka larutan kloroform yang
mengandung etosuksimida akan menguap, karena suhu dari sistem kromatografi
yang jauh lebih tinggi daripada titik didihnya. Oleh karena itu, etosuksimida dan
kloroform akan berubah menjadi gas dan dideteksi oleh detektor, sehingga
kromatogram akan muncul. Persyaratan dalam pengujian cemaran senyawa
sejenis etosuksimida adalah jumlah 2-etil-2-metilsuksinat anhidrida dan cemaran
lain tidak lebih dari 0,2%.
3. Pembahasan cemaran senyawa sejenis fenfluramin HCl
Pengujian cemaran senyawa sejenis fenfluramin HCl dilakukan
menggunakan metode kromatografi gas. Pengotor senyawa sejenis yang
terkandung dalam fenfluramin HCl yaitu etil (α-metil-4-trifluorometilfenetil)
amina. Pada prosedur, serbuk fenfluramin HCl dilarutkan ke dalam air,
ditambahkan larutan KOH P 20%, dan dilakukan ekstraksi menggunakan
kloroform sebanyak 4 kali, hal ini dilakukan untuk membantu deteksi senyawa
oleh detektor ionisasi nyala.
Detektor yang digunakan dalam pengujian iniadalah ionisasi nyala (FID).
Detektor ini dapat mendeteksi semua senyawa organik, dengan batas terkecil
pendeteksian 2 x 10-11 g/mL. Penggunaan baku dalam berupa N,N-dietil-anilina

Pditujukan agar pendeteksian lebih akurat karena digunakan perhitungan
menggunakan peak area ratio (PAR).
Larutan uji diinjeksikan sebanyak 1 µL ke dalam injektor. Suhu kolom dan
detektor dijaga pada berturut-turut pada suhu 135˚C dan 200 ˚C. Pada proses
deteksi, pelarut berupa kloroform akan menguap terlebih dahulu. Selanjutnya
pada pengujian terhadap Larutan uji I, puncak senyawa pengotor sejenis berupa
etil (α-metil-4-trifluorometilfenetil) amina akan muncul segera setelah senyawa
utama. Sedangkan pengujian terhadap Larutan Uji II memberikan perbandingan
luas puncak etil (α-metil-4-trifluorometilfenetil) amina terhadap puncak baku
dalam tidak lebih dari perbandingan luas puncak fenfluramin HCl terhadap
puncak baku dalam yang diperoleh dari Larutan baku.
4. Pembahasan cemaran senyawa sejenis kaptopril
KG merupakan teknik pemisahan yang mana solut-solut yang mudah
menguap (dan stabil terhadap panas) bermigrasi melalui kolom yang mengandung
fase diam dengan suatu kecepatan yang tergantung pada rasio distribusinya. Pada
umumnya solut akan terelusi berdasarkan pada peningkatan titik didihnya, kecuali
jika ada interaksi khusus antara solut dengan fase diam. Pemisahan pada
kromatografi gas didasarkan pada titik didih suatu senyawa dikurangi dengan
semua interaksi yang mungkin terjadi antara solut dengan fase diam. Fase gerak
yang berupa gas akan mengelusi solut dari ujung kolom lalu menghantarkannya
ke detektor. Penggunaan suhu yang meningkat (biasanya pada kisaran 50-350oC)
bertujuan untuk menjamin bahwa solut akan menguap dan karenanya akan cepat
terelusi (Riyanto, dkk., 2013:12-13).

Gambar 3.1. Diagram skematik pada KG
Fase Gerak pada KG
Fase gerak pada KG juga disebut dengan gas pembawa karena tujuan
awalnya adalah untuk membawa solut ke kolom, karenanya gas pembawa tidak
berpengaruh pada selektifitas. Gas pembawa yang digunakan adalah helium P.
Pemilihan gas pembawa tergantung pada penggunaan spesifik dan jenis detektor
yang digunakan. Detektor yang digunakan adalah ionisasi nyala (FI IV,
1995:168).
Helium merupakan tipe gas pembawa yang sering digunakan karena
memberikan efisiensi kromatografi yang lebih baik (mengurangi pelebaran pita).
Helium mempunyai viskositas yang lebih rendah sehingga menghasilkan laju alir
gas pembawa yang lebih tinggi pada efisiensi yang optimum, dan dengan
demikian waktu evaluasi menjadi lebih pendek dan analisis lebih cepat (FI IV,
1995:1013).
Setiap pemisahan dengan KG terdapat kecepatan optimum gas pembawa
yang utamanya tergantung pada diameter kolom. Kecepatan alir gas 1,7 mL/menit
untuk kolom kapiler silika dengan diameter 0,32 mm. Pada tekanan tetap,
kecepatan alir gas meningkat dengan meningkatnya suhu (sebagaimana dalam
suhu terprogram), seperti pada uji cemaran senyawa sejenis dengan metode KG
pada bahan baku obat (Kaptopril) mempunyai laju aliran lebih kurang 1,7
mL/menit pada 125oC, selanjutnya laju aliran lebih kurang 25 mL/menit (FI IV,
1995:168).

Ruang suntik sampel pada KG
Sampel yang ideal dalam kromatografi gas adalah sampel yang hanya
mengandung senyawa yang akan dipisahkan dalam kolom, dan dalam banyak hal
juga pelarut yang mudah menguap yang melarutkan sampel tersebut. Walaupun
cairan yang mudah menguap (tidak dalam larutan), tetapi kebanyakan dilarutkan
terbih dahulu dalam pelarut organik baru kemudian disuntikkan (Riyanto, dkk.,
2013:16). Ruang penyuntikan harus dipanaskan cukup tinggi (terpisah dari kolom)
agar terjadi penguapan dengan cepat, karena apabila komponen dengan tingkat
penguapan yang rendah ada dalam sampel, komponen ini akan tertinggal di ruang
suntik yang pada akhirnya akan mengurangi kinerja kolom. Namun
pemanasannya pun tidak terlalu tinggi hingga menyebabkan terjadinya peruraian
(FI IV, 1995:1012).
Pemisahan sistem injeksi dilapisi dengan wol kaca yang telah disililasi dapat
digunakan pada tempat penyuntikan dengan perbandingan pemisahan lebih
kurang 25:1, pertahankan suhu lebih kurang 250oC (FI IV, 1995:167-168). Wol
kaca yang telah disililasi digunakan untuk menangkap residu yang mudah
menguap. Jumlahnya harus sedikit mungkin untuk menghindari terjadinya
penguraian oleh permukaan yang sangat aktif (FI IV, 1995:1014). Penyuntikan
dalam KG dapat dilakukan dengan memakai alat suntik (semprit) kedap gas atau
sistem penyuntikan yang telah dirancang secara khusus. Kebanyakan penyuntikan
dilakukan dengan menggunakan alat penyuntik mikro (Riyanto, dkk., 2013:16).
Pada kolom kapiler, sampel yang diperlukan sangat sediki 1 μl. Karena
pengukuran secara akurat sulit dilakukan jika sampel yang disuntikkan terlalu
kecil (pada kolom kapiler), maka ditempuh suatu cara untuk mengecilkan ukuran
sampel setelah penyuntikan. Salah satu cara yang dilakukan adalah dengan
menggunakan teknik pemecah suntikan (split injection). Dengan menggunakan
pemecah suntikan ini, sampel yang banyaknya diketahui, disuntikkan ke dalam
aliran gas pembawa dan sebelum masuk ke kolom, gas pembawa ini dibagi
menjadi 2 aliran. Satu aliran akan masuk ke kolom dan satunya lagi akan dibuang.
Aliran relatif dalam kedua aliran ini dikendalikan dengan sejenis penghambat
seperti katup jarum pada aliran yang dibuang (Riyanto, dkk., 2013:15).

Kolom dan Fase Diam pada KG
Kolom yang digunakan pada uji cemaran senyawa sejenis dengan metode
KG pada bahan baku obat (Kaptopril) adalah Kolom Kapiler Silika 15 m x 0,32
mm. Pertahankan suhu kolom pada 125oC selama 11 menit setelah penyuntikan,
naikkan suhu 30oC per menit hingga 300oC dan pertahankan selama 8 menit (FI
IV, 1995:168).
Kolom merupakan tempat terjadinya proses pemisahan karena di dalamnya
terdapat fase diam. Kolom kapiler mempunyai dinding dalam tabung berdiameter
kecil dan dinding kapiler bertindak sebagai penyangga untuk fase diam berupa
cairan. Sifat reaktif penyangga dapat dikurangi dengan menggunakan Pereaksi
Sililasi sebelum diberi lapisan fase cair. Kolom kapiler juga disebut ”Open
tubular columns”. Fase diam melekat mengelilingi dinding dalam kolom, lapisan
yang digunakan pada kolom kapiler ini yaitu FSOT (Fused Silica Open Tube).
Ketika menggambarkan suatu kolom, seseorang biasanya menyatakan panjang
kolom (dalam meter), diameter kolom (dalam milimeter). Semakin sempit
diameter kolom, maka efisiensi pemisahan kolom semakin besar atau puncak
kromatogram yang dihasilkan semakin tajam (Riyanto, dkk., 2013:19).
Pengaturan suhu pada KG dilakukan dengan pemisahan terprogram dengan
menaikkan suhu dari suhu tertentu ke suhu tertentu yang lain dengan laju yang
diketahui dan terkendali dalam waktu tertentu. Perubahan suhu ini dapat
dilakukan secara otomatis dengan komputer. Pemisahan dengan suhu terprogram
mempunyai keuntungan, yakni mampu meningkatkan resolusi komponen-
komponen dalam suatu campuran yang mempunyai titik didih pada kisaran yang
luas. Disamping itu, pada suhu terprogram juga mampu mempercepat keseluruhan
waktu analisis, karena senyawa-senyawa dengan titik didih tinggi akan terelusi
lebih cepat (Riyanto, dkk., 2013:20-21).
Jenis fase diam akan menentukan urutan elusi komponen-komponen dalam
campuran. Fase diam yang digunakan pada uji cemaran senyawa sejenis dengan
metode KG pada bahan baku obat (Kaptopril) adalah Fase diam G27 (5% Fenil –
95% metilpolisiloksan) yang bersifat non polar dengan lapisan penyalut 1 μm (FI
IV, 1995:168 dan 1019). Semakin tipis lapisan penyalut sebagai fase diam, maka
semakin tinggi suhu operasionalnya. Untuk lapisan salut < 1 μm, suhu operasional

dapat mencapai 460ºC, sementara itu suhu minimalnya dapat mencapai - 60ºC
(Riyanto, dkk., 2013:19).
Detektor pada KG
Detektor yang digunakan pada uji cemaran senyawa sejenis dengan metode
KG pada bahan baku obat (Kaptopril) adalah Detektor Ionisasi Nyala (Flame
Ionization Detektor = FID) yang dipertahankan suhunya pada lebih kurang 310oC
untuk mencegah terjadinya kondensasi. Detektor merupakan perangkat yang
diletakkan pada ujung kolom tempat keluar fase gerak (gas pembawa) yang
membawa komponen hasil pemisahan. Detektor pada kromatografi adalah suatu
sensor elektronik yang berfungsi mengubah sinyal gas pembawa dan komponen-
komponen di dalamnya menjadi sinyal elektronik. Sinyal elektronik detektor akan
sangat berguna untuk analisis kualitatif maupun kuantitatif terhadap komponen-
komponen yang terpisah di antara fase diam dan fase gerak (FI IV, 1995:168 dan
1012).
Kromatogram yang merupakan hasil pemisahan fisik komponen-komponen
oleh KG disajikan oleh detektor sebagai deretan luas puncak terhadap waktu.
Waktu retensi tertentu dalam kromatogram dapat digunakan sebagai data
kualitatif, sedangkan luas puncak dalam kromatogram dapat dipakai sebagai data
kuantitatif yang keduanya telah dikonfirmasikan dengan senyawa baku. Detektor
ionisasi nyala ini mengukur jumlah atom karbon untuk hampir semua senyawa
organik. Disamping itu, respon detektor ionisasi nyala sangat peka, dan
mempunyai jangkauan dinamik linier yang lebar sehingga dapat diperoleh hasil
kuantitatif yang akurat (FI IV, 1995:1012-1013).
Pada dasarnya senyawa organik bila dibakar akan terurai menjadi pecahan
sederhana bermuatan positif, biasanya terdiri atas satu karbon (C+). Pecahan ini
meningkatkan daya hantar di sekitar nyala, tempat yang telah dipasang elektroda,
dan peningkatan daya hantar ini dapat diukur dengan mudah dan direkam. Sampel
yang dibawa oleh gas pembawa mengalir ke dalam nyala dan diuraikan menjadi
ion. Ion ini akan meningkatkan daya hantar dan karenanya akan meningkatkan
arus listrik yang mengalir diantara 2 elektroda (Riyanto, dkk., 2013:26).

Gambar 3.2. Diagram skematik FID
Pada pamakaian FID, ada beberapa hal yang perlu diperhatikan: pertama,
kecepatan alir O2 (udara) dan H2. Untuk memperoleh tanggapan FID yang optimal
sebaiknya kecepatan aliran H2 ± 30 ml/menit dan O2 sepuluh kalinya. Kedua
adalah bahwa suhu FID harus diatas 100oC. Hal ini bertujuan untuk mencegah
kondensasi uap air yang mengakibatkan FID berkarat atau kehilangan (menurun)
sensitivitasnya. Kalau memungkinkan pada selang waktu tertentu dengan
pertolongan mekanik, maka dapat dilakukan pembersihkan bagian atas FID
(kolektor) yang mungkin telah dilapisi berbagai macam kotoran (Riyanto, dkk.,
2013:27).

BAB 4
KESIMPULAN
1. Pengujian jarak lebur dan cemaran senyawa sejenis pada klorfeniramin
maleat dilakukan untuk memastikan kemurnin zat aktif yang digunakan.
2. Uji cemaran senyawa sejenis merupakan suatu pengujian dalam monografi
yang mengacu pada uji umum untuk menganalisis pengotor berupa produk
samping dari suatu zat aktif. Uji cemaran senyawa sejenis klorfeniramin
maleat dapat dianalisis menggunakan kromatografi gas.

DAFTAR PUSTAKA
Departemen Kesehatan Republik Indonesia. (1995). Farmakope Indonesia edisi
IV. Jakarta : Departemen Kesehatan Republik Indonesia
Gilbert, J. C. and Stephen F. (2011).Experimental Organic Chemistry: A
Miniscale and Microscale ApproachFifth Edition. Boston : Cangage Learning.
Harmita. (2006). Buku Ajar Analisis Fisikokimia. Depok : Departemen Farmasi
FMIPA UI
Pasto, D. J., Johnson, C. R., and Miller, M. J. (1992). Experiment and Techniques
in Organic Chemistry. New Jersey:Prentice Hall Inc.
Riyanto, Sugeng, Ibnu Gholib Gandjar, Sudibyo Martono, dan Endang
Lukitaningsih. (2013). Kromatografi. Yogyakarta: Fakultas Farmasi
Universitas Gadjah Mada.




















