
pbl talasemia 24
32
TALASEMIA Junita Karla Taneo 10 2008 161 Fakultas Kedokteran UKRIDA Semester VI Universitas Kristen Krida Wacana Jakarta Jl.Arjuna Utara No.6 Jakarta Barat 11510 [email protected] Abstrak Talasemia merupakan penyakit herediter pada manusia. Terjadinya penyakit ini akibat perkawinan pasangan yang carrier talasemia. Oleh karena sampai saat ini belum ada pengobatan yang pasti unutk talasemia, maka pencegahan harrus dilaksanakan. Caranya dapat dnegan menyaring penduduk yang menjadi carrier dan memberikan penjelasan kepada penduduk yang mempunyai resiko sebelum mereka mempunyai anak yang akan mengidap talasemia. Dalam hal ini penyuluhan akan talasemia ini perlu dilakukan agar orangtua mengerti dan dapat mengurangi ataupun mencegah terjadinya perkawinan antara carrier talasemia. Peenjelasan penyakit ini meloputi bagaiman terjadinya dan akibatnya bagi anak dan bagaiman juga cara pencegahannya.
-
Upload
vitaparamithateken -
Category
Documents
-
view
37 -
download
0
description
Makalah
Transcript of pbl talasemia 24

TALASEMIA
Junita Karla Taneo
10 2008 161
Fakultas Kedokteran UKRIDA Semester VI
Universitas Kristen Krida Wacana Jakarta
Jl.Arjuna Utara No.6 Jakarta Barat 11510
Abstrak
Talasemia merupakan penyakit herediter pada manusia. Terjadinya penyakit ini akibat
perkawinan pasangan yang carrier talasemia. Oleh karena sampai saat ini belum ada
pengobatan yang pasti unutk talasemia, maka pencegahan harrus dilaksanakan. Caranya
dapat dnegan menyaring penduduk yang menjadi carrier dan memberikan penjelasan
kepada penduduk yang mempunyai resiko sebelum mereka mempunyai anak yang akan
mengidap talasemia. Dalam hal ini penyuluhan akan talasemia ini perlu dilakukan agar
orangtua mengerti dan dapat mengurangi ataupun mencegah terjadinya perkawinan
antara carrier talasemia. Peenjelasan penyakit ini meloputi bagaiman terjadinya dan
akibatnya bagi anak dan bagaiman juga cara pencegahannya.

BAB I
Pendahuluan
Talasemia adalah sekumpulan heterogonus penyakit akibat dari gangguan sintesis hemoglobin
yang diwariskan secra autosom resesif.
Talasemia juga merupakan sindroma kelainan herediter darah yang paling sering terjadi di dunia.
Talasemia merupakan suatu penyakit darah yang ditandai dengan berkurang atau ketiadaan
produksi dari hemoglobin normal. Talasemia biasanya terjadi di daerah-daerah dimana terjadi
endemik malaria, khususnya malaria yang disebabkan oleh Plasmodium falciparum
Darah terdiri dari plasma yang berupa cairan, sel darah merah (eritrosit), sel darah putih
(leukosit), dan keping darah (trombosit). Leukosit berfungsi untuk melindungi tubuh terhadap
infeksi, dan trombosit berfungsi untuk mekanisme pembekuan darah. Eritrosit membawa satu
protein yang disebut hemoglobin yang berfungsi untuk mengikat oksigen di paru-paru,
membawanya ke peredaran darah, dan melepaskannya ke sel dan jaringan tubuh.
Molekul hemoglobin terdapat pada semua eritrosit dan menjadi penyebab dari merahnya warna
darah manusia. Hemoglobin terdiri dari haem (suatu kompleks yang terdiri dari zat besi) dan
berbagai macam globin ( rantai protein yang ada di sekeliling kompleks heme).6
Pada talasemia terjadi kelainan pada gen-gen yang mengatur pembentukan dari rantai globin
sehingga produksinya terganggu. Gangguan dari pembentukan rantai globin ini akan
mengakibatkan kerusakan pada sel darah merah yang pada akhirnya akan menimbulkan
pecahnya sel darah tersebut.4
BAB II

ISI
Anamnesis
Keluhan utama karena anemia: Pucat, gangguan nafsu makan, gangguan tumbuh kembang dan
perut membesar karena pembesaran lien dan hati. Pada umumnya keluhan ini mulai terlihat pada
anak usia 6 bulan.
Tanyakan kepada pasien ataupun keluarganya mengenai identitas pasien,. Perlu ditanyakan juga
pekerjaan orang tua untuk menunjang pengobatan nantinya.
Tanyakan keluhan yang dialaminya, pada kasus mengeluh pucat dan distensi abdomen. Lalu
tanyakan juga riwayat penyakit.
Tanyakan juga riwayat keluarga.6
Pemeriksaan
a. Fisik
1. Pucat
2. Bentuk muka mongoloid (facies Cooley)
3. Dapat ditemukan ikterus
4. Gangguan pertumbuhan- anak lebih pendek dari pertumbuhan normal seusianya.
5. Splenomegali dan hepatomegali yang menyebabkan perut membesar6
b. Laboratorium
Anemia biasanya berat, dengan kadar Hb berkisar 3-9 g/dL
Eritrosit memperlihatkan anisositosis, poikilositosis, dan hipokromia berat. Sering
ditemuakn sel target dan tear drop cell. Normoblas (eritrosit berinti) banyak
dijumpai pasca splenoktomid.
Gambaran sumsum tulang memperlihatkan eritropoiesis yang hiperaktif sebanding
dengan anemianya. Diagnosis ditegakkan dengan pemeriksaan elektroforesis Hb.

Pada talasemia beta ( ) β kadar HbF bervariasi 10-90%, sedangkan dalam keadaan
normal <1%.
Pada pemeriksaan laboratorium, penderita thalassemia berat :
1. penurunan Hemoglobin (2-5 g/dl), hematokrit, eritrosit, MCV, MCHC, MCH.
2. Kenaikan retikulosit.
3. Pada anak-anak, jumlah hemoglobin normal adalah 10 – 16 gr/dl, jumlah AL adalah
9000-12.000/ l, AL pada anak-anak adalah 200.000-400.000 / l darah, danμ μ
Hematokrit pada anak-anak adalah 33-38 volume %.5
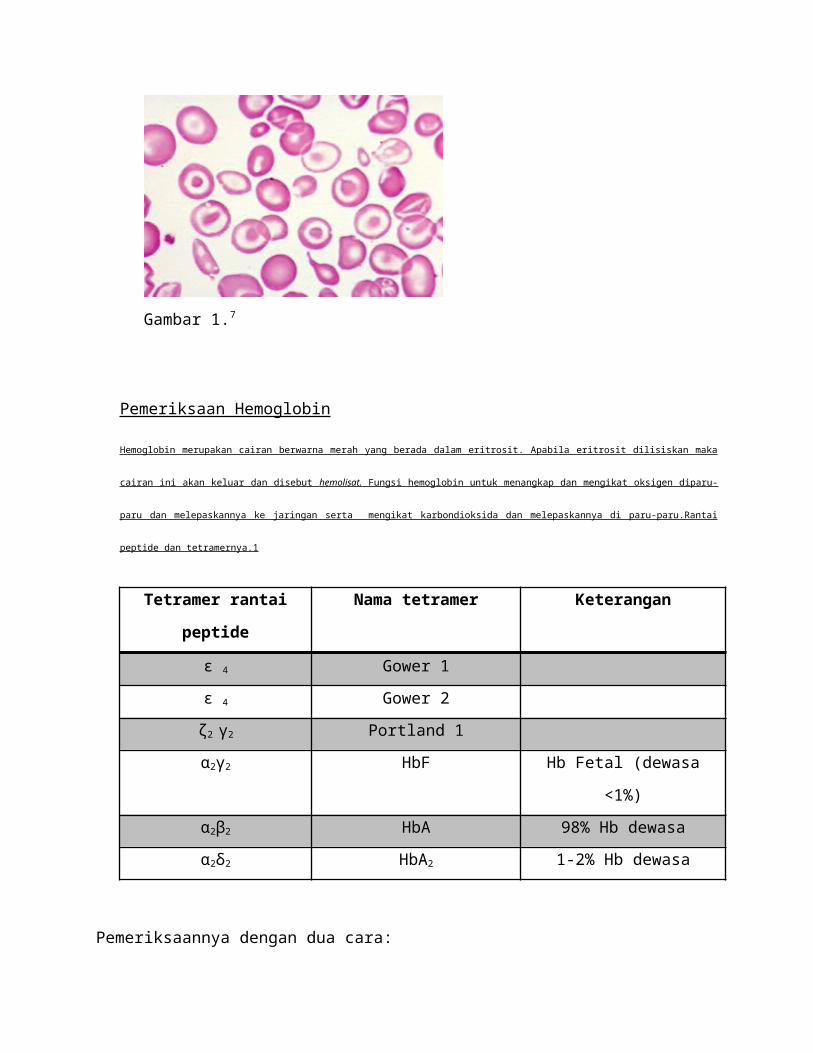
a. Pemeriksaan Hematologi
Sediaan Hapus Darah Tepi
Tujuannya: untuk evaluasi morfologi sel darah tepi (eritrosit,leukosit, trombosit),
memperkirakan jumlah leukosit dan trombosit serta mengidentifikasi parasit.
Bahan pemeriksaan :darah vena atau kapiler
Pelaporan sediaan hapus darah tepi meliputi:
1. Eritrosi : ukuran, bentuk dan warna
Normalnya: sel berbentuk bulat, tepi rata, sitoplasma merah dan pucat pada bagian
tengah. Inti sel tidak ada.
2. Leukosit: 5.000-10.000/uL
3. Trombosit: 4-8 trombosit/100 eritrosit1
Gambar 1.7
Pemeriksaan Hemoglobin

Hemoglobin merupakan cairan berwarna merah yang berada dalam eritrosit. Apabila eritrosit dilisiskan maka cairan ini akan keluar dan disebut hemolisat.
Fungsi hemoglobin untuk menangkap dan mengikat oksigen diparu-paru dan melepaskannya ke jaringan serta mengikat karbondioksida dan melepaskannya di
paru-paru.Rantai peptide dan tetramernya.1
Tetramer rantai peptide Nama tetramer Keterangan
ε 4 Gower 1
ε 4 Gower 2
ζ2 γ2 Portland 1
α2γ2 HbF Hb Fetal (dewasa <1%)
α2β2 HbA 98% Hb dewasa
α2δ2 HbA2 1-2% Hb dewasa
Pemeriksaannya dengan dua cara:
1. Sahli
Prinsipnya: Hb + HCl 0,1% Hematin asam (berwarna coklat)
2. Fotoelektrik
Prinsipnya: hemoglobin oleh K3Fe(CN)6 akan diubah menjadi methemoglobin yang
kemudian oleh KCN akan diubah menjadi Heoglobinsianiida (HiCN).1
Rumusnya:
Pemeriksaan Hematokrit
Tujuannya: mengukur volume erotrosit dalam 100ml darah, biasanya dinyatakan dalam
% (persen).
Prinsi pemeriksaan: mendapat endapan maksimal dari sel-sel darah dengan memutar
darah menggunakan mesiin sentrifus.1
1. Cara Makro (Wintrobe)
Tiga data yang dapat di nilai:
Hematokrit pria 40-48%, wanita 37-43%
Tebal Buffy coat: 0,5-1mm
indeks ikterus: 4-7 unit/satuan1
Pemeriksaan Hitung Sel Darah Tepi
Hitung sel darah adalah suatu pemeriksaan untuk menentukan jumlah sel dalam tiap mikroliter
sel darah. Pemeriksaann ini dapat dilakukan secara manual ataupun otometik. Prinsip
pemeriksaan dengan cara manual aalah dengan melakukan pengenceran darah dengan suatu
larutan. Larutan yang digunakan: formal sitrat, Hayem, gower. Alat yang dipakai Pipet Sahli
ataupun Pipet Thoma. Kamar hitung yang di gunakan, kamar hitung Improved Neubauer1
1. Hitung Eritrosit.
Nilai rujukan : wanita: 4.00-5.20x106/uL atau 4.00-5.2x109/liter
Pria :4.50-5.90x106/uL atau 4.50-5.9x109/liter
2. Hitung Retikulosit
Nilai rujukan: relative: 0.5-1.5% atau 5-15 per seribu
Absolute : (25.000 – 75.000)/ uL darah
Hb Hematokrit Erirosit
Wanita 12-15 g/dL 42-53 % 4,6-6,2 juta/uL
Pria 14-17 g/dL 38-46 % 4,2-5,2 juta/uL
Anak-anak(3-13 thn) 10-14,5 g/dL 31-43 % 3,8-5,8 juta/uL
Pemeriksaan Nilai Eritrosit Rata-rata (NER)/Mean Corpuscular Values (MCV)
Pemeriksaan NER terdiri dari 3 pemeriksaan yaitu:
Hemoglobin Eritrosit Rata-rata/Mean Corpuscular Hemoglobin (MCH)
Konsentrasi Hemoglobin Eritrosit Rata-rata/ Mean Corpuscular Hemoglobin
Concetratiaon (MCHC)
Untuk pemeriksaan ini diperlukan hasil data pemeriksaan kadar Hb (gr/dl), dan nilai hematokrit
(%), dan Hitung eritrosit (juta/uL). Pemeriksaan kadar hemoglobin dilakukan dengan cara

fotoelektrik dan tidak dianjurkan dengan cara sahli. Pemeriksaan eritrosit dengan cara otomatik
atau manual. Pemeriksaan nilai hemtokrit dapat dilakukan dengan cara makro atau mikro.
Pemeriksaan ini diperoleh dengan mengguanakan rumus:1
1.
2.
3.
b. Pemeriksaan Sumsum tulang
Merupakan pemeriksaan khusus dan bukan pemeriksaan laboratorium rutin. Pemeriksaan ini
bertujuan untuk mendapatkan gambaran tentang hematopoesisyang terjadi dalam sumsum
tulang. Oleh karena itu, untuk melakukan pemeriksan ini diperlukan beberapa data yang telah
diperoleh sebelum melakukan pemeriksaan. Data tersebut meliputi riwayat penyakit, hasil
pemeriksaan fisik, data pemeriksaan hematologi lengkap, hasil pemeriksaan laboratorium
lain yang berhubungan atau yang abnormal (pemeriksaan kimia darah, urinalisasi), serta
data-data lain misalnya radiologi,biopsy,dll.1,4
c. TIBC, Saturasi Transferin, dan Hemosiderin Sumsum Tulang
TIBC atau kapasitas mengikat besi total merupakan suatu pengukuran untuk mengukur
kapasitas transferin serum mengikat besi. Pengambilan darah unutk pemeriksaan ini
sebaiknya pada pagi hari setelah puasa 12 jam dan eksklusi suplemen besi selama 12-
24 jam. Kemampuan total transferin mengikat besi diukur dari mengukur besi total
yang terikat dan pemeriksaan TIBC ini tidak mengukur kadar transferin. Rentang
normal untuk TIBC pada orang dewasa adalah 240-360 µg/dl, dan cenderung akan
berkurang seiring bertambahnya usia sampai 250 µg/dl pada orang dengan usia di atas
70 tahun. TIBC meningkat pada defisiensi besi dan kehamilan, tetapi mungkin normal
atau rendah pada penyakit kronis dan malnutrisi .

Saturasi transferin menggambarkan perbandingan antara besi serum yang ada dengan
TIBC dalam bentuk persentase. Saturasi transferin normalnya 20-50%, ini memiliki
pola diurnal, tinggi pada pagi hari dan rendah pada siang dan sore hari. Persentase
saturasi rendah pada defisiensi besi dan penyakit kronis dan tinggi pada anemia
sideroblastik, keracunan besi, serta hemolisis intravascular dan hemokromatosis.
Pemeriksaan sumsum tulang untuk melihat kadar cadangan besi untuk proses
eritropoesis.1,4
d. Besi serum, protoporfirin eritrosit, ferritin serum
Pemeriksaan besi serum dan ferritin serum untuk melihat ada/tidaknya besi dan
cadangannya dalam tubuh. Dan protoporfirin eritrosit untuk menentukan
pembentukan heme dimana besi akan diikat oleh protoporfirin.1,4
e. Analisis DNA
Analisis DNA digunakan untuk mengetahui adanya mutasi pada gen yang memproduksi
rantai alpha dan beta. Pemeriksaan ini merupakan tes yang paling efektif untuk mendiagnosa
keadaan karier pada talasemia. 1,6,7
f. Pemeriksaan Radiologis
Gambaran radiologis tulang akan memperlihatkan medulla yang lebar, korteks tipis dan
trabekula kasar. Tulang tengkorak memperlihatkan diploe dan pada anak besar kadang-
kadang terlihat brush appearance. Sering pula ditemukan gangguan pneumatisasi rongga
sinus paranasalis. 7
Working Diagnosis
Talasemia
Differential Diagnosa
Anemia Defisiensi Besi
Gejala Umum Anemia
Gejala umum anemia yang disebut sebagai sindrom anemia (anemic syndrome) dijumpai
pada anemia defisiensi besi apabila kadar hemoglobin turun dibawah 7-8d/dl. Gejala ini

berupa badan lemas, lesu, cepat lelah, mata kunang-kunang, serta telinga yang berdenging.
Pada anemia defisiensi besi karena penurunan kadar hemoglobin yang terjadi secara
perlahan-lahan seringkali syndroma anemia tidak terlalu menyolok dibandingkan dengan
anemia yang lainyang penurunan kadar hemoglobinnya lebih cepat terjadi, oleh karena
mekanisme kompensasi tubuh dapat berjalan dengan baik. Pada pemeriksaan fisik dijumpai
pasien yang pucat, terutama pada konjungtiva dan jaringan di bawah kuku.2,3
Gejala Khas Defisiensi Besi
Gejala yang khas dijumpai pada defisiensi besi, tetapi tidak dijumpai pada anemia jenis lain
adalah:
Koilonychias: kuku sendok (spoon nail), kuku menjadi rapuh, bergaris-garis vertical dan
menjadi cekung sehingga mirip seperti sendok.
Atrofi papil lidah: permukaan lidah menjadi licin dan mengkilap karena papil lidah
menghilang.
Stomatitis angularis (cheilosis): adanya peradangan pada sudut mulut sehingga tampak
sebagai bercak berwarna pucat keputihan.
Disfagia: nyeri menelan kerena kerusakan epitel hipofaring.
Atrofi mukosa gaster sehingga menimbulkan akhloridia.
Pica : keinginan untuk memakan bahan yang tidak lazim, seperti: tanah liat, es, lem dll.2,3,5
Anemia Sideroblastik
Anemia sideroblastik adalah anemia mikrositik-hipokromik yang ditandai oleh adanya
sel-sel darah merah imatur (sideroblas) dalam sirkulasi dan sumsum tulang. Anemia
sideroblastik primer dapat terjadi akibat efek genetik pada kromosom X yang jarang
ditemukan (terutama dijumpai pada pria) atau dapat timbul secara spontan, terutama
pada orang tua. Penyebab sekunder anemia sideroblastik adalah obat-obat tertentu,
misalnya beberapa obat kemoterapi dan ingesti timah.
Anemia sideroblastik adalah anemia mikrositik hipokrom yang ditandai adanya sel-sel
darah merah abnormal (sideroblas) dalma sirkulasi dan sumsum tulang. Sideroblas
membawa besi di mitokondria bukan di molekul hemoglobin, sehingga tidak mampu
untuk mengangkut oksigen ke jaringan. Oleh sebab itu tida terjadi defisiensi besi.

Berkurangnya pengangkutan oksigen menyebabkan hipoksia. Hal ini dideteksi oleh sel-
sel ginjal penghasil eritropotein. Eritropoietin merangsang pembentukan sel-sel darah
merah baru di sum-sum tulang. Hal ini menyebabkan sumsum tulang mengalami
kongesti dan meningkatkan pembentukan sideroblas yang memperparah anemia.
Anlaisis darah
Anemia yang ditandai dengan sel mikrositik hipokrom, peningkatan besi plasma dan
kapasitas peningkatan besi normal.
Pemeriksaan sumsum tulang memperlihatkan adanya penimbunan besi, sideroblas dan
makrofak fagositik. 2,3
Hemoglobinopati
Hemoglobinopati structural
Di sini terjadi perubahan sturktur hemoglobin (kualitatif) karena substitusi satu asam
amino atau lebih pada salah satu rantai peptida hemoglobin. Hemoglobinopati yang
penting sebagian besar merupakan varian rantai Beta ( ). Pada hemoglobinopatiβ
struktural dapat ditemukan splenomegali namun tidak dapat ditemukan hepatomegali.
Contoh hemoglobinopati struktural adalah penyakit Hb C, Hb D, Hb E dan Hb SC.3,4
a. Hb C
Terdapat pada 2% kalangan kulit hitam Amerika. Pada keadaan heterozigot (Hb AC)
tidak ditemukan anemia atau penyakit, tetapi ditemukan peningkatan jumlah sel
target dalam darah tepi. Pada orang-orang homozigot (penyakit Hb CC) dapat
ditemukan anemia hemolitik dengan derajat sedang dan kadar Hb 8-11 g/dL,
retikulositosis 5-10% dan splenomegali. Darah tepi mengandung sel target dan
sferosit dalam jumlah banyak
b. Hb D
Dalam Hb D termasuk beberapa varian Hb abnormal dengan mobilitas
elektroforesis serupa dengan Hgb S, tetapi dengan sifat biokimia dan fisik yang
berbeda. Sikling tidak terjadi pada sindroma Hb D. Keadaan homozigot (Hb DD)
ditandai dengan anemia hemolitik ringan dan splenomegali.

c. Hb E
Hb E prevalen pada orang-orang dari Asia tenggara terutama Thailand. Penyakit
Hgb E homozigot ditandai dengan anemia hemolitik ringan dengan sel target nyata
serta mikrositosis dengan splenomegali sedang hingga berat. Temuan-temuan klinis
dan hematologis mirip dengan Hb C.
d. Hb SC
Jika kedua gen Hb S dan Hb C ditemukan pada orang yang sama, akan terjadi suatu
anemia dengan derajat sedang disertai splenomegali. Ditemukan episode vaso-
oklusi tetapi biasanya jarang dan ringan dibandingkan pada penyakit sel sabit.
Nekrosis apseptik dari kaput femoris kadang-kadang merupakan penyulit dan
ditemukan kerusakan retina berat. Kadar Hb rata-rata 9-10 g/dL. Sel target banyak,
tetapi sel sabit yang ireversibel jarang ditemuui dalam darah tapi. Pada
elektroforesis Hb menunjukkan campuran sama Hgb S dan Hgb C dengan sedikit
peningkatan Hgb F. Penyakt Hgb SC biasanya tidka mempengaruhi pertumbuhan
dan berhubungan dengan daya tahan yang berlanjut hingga dewasa. Krisis aplastis
dan sekuestrasi merupakan ancaman terhadap hidup.3,4
Etiologi
Genetic

Thalassemia adalah suatu kelainan darah kongenital yang disebabkan oleh menurun atau
tidak adanya sintesa salah satu atau lebih rantai globin yang berperan penting dalam
pembentukan hemoglobin. Hal ini menyebabkan kerusakan dari sel darah merah dan
hambatan produksinya. Kelainan ini diturunkan secara autosomal dari orang tua kepada
anaknya.1,2,3,4.
Gejala Klinik
Secara klinis Thalasemia dapat dibagi dalam beberapa tingkatan sesuai beratnya gejala
klinis : Mayor, Intermedia dan Minor atau troit (pembawa sifat). Batas diantara tingkatan
tersebut sering tidak jelas.
a. Thalasemia mayor (Thalasemia homozigot)
Anemia berat menjadi nyata pada umur 3 – 6 bulan setelah lahir dan tidak dapat hidup
tanpa ditransfusi. Pembesaran hati dan limpa terjadi karena penghancuran sel darah
merah berlebihan, haemopoesis ekstra modular dan kelebihan beban besi. Limpa yang
membesar meningkatkan kebutuhan darah dengan menambah penghancuran sel
darah merah dan menyebabkan pertambahan volume plasma. Perubahan pada tulang
karena hiperaktivitas sumsum merah berupa deformitas dan fraktur spontan,
terutama kasus yang tidak atau kurang mendapat transfusi darah. Deformitas tulang,
disamping mengakibatkan muka mongoloid, dapat menyebabkan pertumbuhan
berlebihan tulang prontal dan zigomaticus serta maxilla. Pertumbuhan gigi biasanya
buruk. Gejala lain yang tampak ialah anak lemah, pucat, perkembanga fisik tidak sesuai
umur, berat badan kurang, perut membuncit. Jika pasien tidak sering mendapat
transfusi darah kulit menjadi kelabu serupa dengan besi akibat penimbunan besi
dalam jaringan kulit.
b. Thalasemia intermedia
Keadaan klinisnya lebih baik dan gejala lebih ringan dari pada Thalasemia mayor,
anemia sedang (hemoglobin 7 – 10,0 g/dl) Gejala deformitas tulang, hepatomegali dan
splenomegali, eritropoesis ekstra medular dan gambaran kelebihan beban besi
nampak pada masa dewasa.
c. Thalasemia minor atau troit ( pembawa sifat)

Umumnya tidak dijumpai gejala klinis yang khas, ditandai oleh anemia mikrositik,
bentuk heterozigot tetapi tanpa anemia atau anemia ringan.3,4
Patofisiologi
Talasemia adalah penyakit keturunan dengan gejala utama pucat, perut tampak membesar karena
pembengkakakan limpa dan hati, dan apabila tidak diobati dengan baik akan terjadi perubahan
bentuk tulang muka dan warna kulit menjadi menghitam. Penyebab penyakit ini adalah
kekurangan salah satu zat pembentuk hemoglobin (Hb) sehingga produksi hemoglobin
berkurang.
Pada talasemia terjadi gangguan sintesis satu ata lebih rantai globin. Pada orang dewasa normal,
susunan Hb adalah sebagai berikut.
HbA 97% (α2 ß2)
HbA2 2-3% (α2 δ2)
Hb 1% (α2 γ2)1
Defek genetic mengakibatkan pengurangan atau peniadaan sintesis satu atau lebih rantai globin
HbA. Keadaan ini dapat mengakibatkan:
1. Pembentukan tetramer Hb berkurang sehingga terjadi anemia mikrositik hipokrom.
2. Sebagian rantai globin tidak mendapat pasangan, bebas, besifat tidak larut (insoluble) dan
tidak mampu mengikat oksigen. Akumulasi rantai globin yang bebas ini mengakibatkan
lisis eritrosit intrameduler (eritropoesis intrameduler).5
Talasemia α
Seperti yang telah diketahui sebelumnya bahwa pada bayi yang baru lahir masih terdapat
jumlah HbF( 2 2) yang masih cukup tinggi. Pada usia 20 hari sesudah kelahiran kadarα γ
HbF akan menurun dan setelah 6 bulan kadarnya akan menjadi normal seperti orang
dewasa. Selanjutnya pada masa tersebut akan terjadi konversi HbF menjadi HbA( 2 2)α β
dan HbA2 ( 2 2).α δ
Rantai globin a di produksi pada kromosom 16. Rantai globin a telah terbenntuk dari masa
embrio sampai akhir kehamilan. Apabila terjadi gangguan pembentukan rantai globin a maka

terjadi talasemia a. talasemia a sering di jumai di Asia. Nilai Mean Cell Volume (MCV), Mean
Cell Hemoglobin (MCH) dan Mean Cell Hemoglobin Conceration (MCHC) penderita talasemia
a rendah ( kadar normalnya MCV: 80-100fl, MCH: 27-34pg dan MCHC: 32-36 gr/dl). Talasemia
a terdiri dari 4 jenis yaitu : talasemia a mayor, intermedia, minor (carrier) dan silent carrier.3,5,7
1. Talasemia α mayor ( Hb-Bart’s)
Merupakan talasemia homozigot yang dijumpai di Asia Tenggara dan sedikit di beberapa
Negara di Mediterania. Talasemia a mayor disebabkan oleh kegagalan tubuh membentuk
ke empat rantai globin a pada masa fetus dan menyebabkan terbentuknya Hb-Bart’s
hydrops fetalis. Hb-Bart’s fetelis memiliki afinitas yang tinggi terhadap oksigen yang
menyebabkan penderita meninggal dalam kandungan pada usia trisemester kedua atau
ketiga dan dapat juga terjadi setelah ± 1 jam dilahirkan karena anoxia. Bayi yang lahir
dengan talasemia a mayor mengalami udem dan asites karena penumpukna cairan dalam
jaringan akibat anemia berat.3,5
2. Talasemia α intermedia (Hb H Disease)
Talasemia a intermedia di jumpai di Asia Tenggara, Mediterania beberapa Asia Tengah
dan jarang di Afrika. Talasemia a intermedia terjadi karena tidak terbentuknya 3 dari 4
globin gen α. Cirri hematologis utama di tandai dengan kelebihan rantai globin B
sehingga terbentuk homotetramer B4 atau Hb H dalam eritrosit. Penderita talasemia
intermedia mengalami pembesaran sumsum tulang yang ektopik, gallstones, jaundice,
demam, nyeri sendi, anemia kronik dengan Hb 7-10gr/dl, pembesaran limpa dan hati.3,5
3. Talasemia α Minor (Carrier)
Talasemia a minor di jumpai di Asia Tenggara, Asia Tengah dan Indian subkontinen.
Talasemia a minor terjadi karena atidak terbentuknya2 dari 4 gen a. talasemia yang di
derita di ketahui dengn pemeriksaan darah dengan di jumpai bentuk darah yang
abnormal, anemia ringan, nilai MCV 72 fl, MCH 22 pg, dan MCHC 31 gr/dl.3,5
4. Talasemia α silent carrier
Pada orang Amerika berkulit hitam (Afrika Amerika). Terjadi karena tidak terbentuknya
1 dari 4 gen a. penderita biasanya sehat dan tidak ada gangguan darah. Kadar Hb normal
baik pada wanita maupun pria.3,5

Talasemia β
Rantai globin B di produksi pada kromosom 11. Apabila terjadi gangguan pembentukan rantai
globin B maka terjadi talasemia B. talasemia B sering dijumpai di daerh laut Mediterania dan
Alfo-Karibia. Talasemia B terdiri atas 3 jenis, yaitu talasemia mayor ( Cooley’s anemia),
intermedia dan minor.3,5,7
1. Talasemia β Mayor (cooley Anemia)
Talasemia B mayor adalah talasemia B yang homozigot dan paling banyak dijumpai.
Talasemia B mayor memiliki sedikit atau tidak ada kemampuan untuk memproduksi
rantai globin B. secara patologis, penderita talasemia B mayor mengalami hemolisis yang
berat karena tidak seimbangnya produksi rantai a dan B. jumlah rantai a yang banyak
cenderung tidak stabil dan dapat menyebabkan destruksi dari dalam eritrosit. Sehingga
pada penderita ini mengalami anemia yang berat dengan Hb 2-6 gr/dl, hierplasia sumsum
tulang, zat besi berlebihan, pembesaran hati dan limpa. Selain itu, penderita talasemia
juga mnegalami keterlambatan pertumbuhan, jaundice, pembesaran maxilla, penipisan
korteks dan penebalan medulla pada tulang.3,5
2. Talasemia β intermedia
Talasemia B intermedia adalah talasemia pertengahan antara mayor dan minor. Penderita
mengalami anemia dengan Hb 6-9gr/dl, perubahan skeletal, pembesaran limpa,
meningkatnya produksi eritrosit disumsum tulang dan resopsi zat besi. Penderita
membutuhkan tranfusi darah jika di perlukan.3,5
3. Talasemia β Minor
Talasemia B minor adalah talasemia B yang heterozigot. Talasemia ini mengalami mutasi
dalam sintesa rantai globin B, dan meupakan pembawa sifat talasemia. Kadar Hb pada
penderita talasemia minor (carrier) normal baik pada pria maupun wanita.3,5
Penyebab anemia pada thalassemia bersifat primer dan sekunder. Primer adalah
berkurangnya sintesis HbA dan eritropoesis yang tidak efektif disertai penghancuran sel-
sel eritrosit intramedular. Sedangkan yang sekunder ialah karena defisiensi asam folat,
bertambahnya volume plasma intravaskular yang mengakibatkan hemodilusi, dan

destruksi eritrosit oleh sistem retikuloendotelial dalam limpa dan hati. Penelitian
biomolekular menunjukkan adanya mutasi DNA pada gen sehingga produksi rantai alfa
atau beta dari hemoglobin berkurang. Terjadinya hemosiderosis merupakan hasil
kombinasi antara transfusi berulang, peningkatan absorbsi besi dalam usus karena
eritropoesis yang tidak efektif, anemia kronis, serta proses hemolisis.7
Epideomologi
Frekuensi gen Thalassemia di Indonesia berkisar 3-10%. Berdasarkan angka ini, diperkirakan
lebih 2000 penderita baru dilahirkan setiap tahunnya di Indonesia.3,5,6
Penatalaksanaan
a. Medikamentosa
Sampai saat ini belum ditemukan cara yang dapat menyembuhkan thalassemia. Namun
terdapat beberapa terapi untuk mengurangi gejala yang ditimbulkannya:
1. Usaha untuk mencegah penumpukan besi (hemochromatosis) akibat transfusi dan
akibat patogenesis dari thalassemia dapat dilakukan dengan pemberian iron
chelator yaitu desferoksamin (desferal R) sehingga meningkatkan ekskresi besi
dalam urine. Desferal diberikan dengan infus bag atau secara subkutan.3,5
2. Pemberian asam folat 5 mg/hari secara oral untuk mencegah krisis megaloblastik.
3. Vitamin C (200 mg perhari) meningkatkan eksresi besi yang disebabkan oleh
desferioksamin.
4. Vitamin E 200-400 IU setiap hari sebagai antioksidan dapat memperpanjang umur sel
darah merah.
5. Imunisasi terhadap virus hepatitis B dan C perlu dilakukan untuk mencegah
infeksi virus tersebut melalui transfusi darah.6,7

6. Terapi definitif dengan transplantasi sumsum tulang perlu dipertimbangkan pada
setiap kasus baru dengan talasemia mayor. Transplantasi yang berhasil akan
memberikan kesembuhan permanen.
7. Secara berkala dilakukan pemantauan fungsi organ, seperti jantung, paru, hati,
endokrin termasuk kadar glukosa darah, gigi, telinga, mata, dan tulang.7
Bedah
1. Usaha untuk mengurangi proses hemolisis dengan splenektomi jika splenomegali
cukup besar dan terbukti adanya hipersplenisme sehingga membatasi gerak pasien,
menimbulkan tekanan intraabdominal yang mengganggu napas dan berisiko
mengalami ruptur. Hipersplenisme dini ditandai dengan jumlah transfusi melebihi
250 mL/kgBB dalam 1 tahun terakhir dan adanya penurunan Hb yang drastis.
Hipersplenisme lanjut ditandai oleh adanya pansitopenia. Splenektomi sebaiknya
dilakukan pada umur 5 tahun ke atas saat fungsi limpa dalam sistem imun tubuh
telah dapat diambil alih oleh organ limfoid lain.3,5,7
Splenektomi, dengan indikasi:
Limpa yang terlalu besar, sehingga membatasi gerak penderita, menimbulkan
peningkatan tekanan intraabdominal dan bahaya terjadinya rupture.
Hipersplenisme ditandai dengan peningkatan kebutuhan transfusi darah atau kebutuhan
suspensi eritrosit (PRC) melebihi 250 ml/kg berat badan dalam satu tahun.3,7
2. Cangkok sumsum tulang (CST) adalah kuratif pada penderita dan telah terbukti
keberhasilan yang meningkat, meskipun pada penderita yang telah menerima tranfusi
sangat banyak. Namun, prosedur ini membawa cukup resiko morbiditas dan mortalitas
dan biasanya hanya digunakan untuk penderita yang mempunyai saudara kandung yang
sehat yang histokompatibel.7
b. Non Medikamentosa

Atasi anemia dengan transfusi PRC (packed red cell) . Transfusi hanya diberikan bila Hb
<8g/dL. Sekali diputuskan untuk diberi transfusi darah, Hb harus selalu dipertahakan di
atas 12 g/dL tidak melebihi 15 g/dL. Bila tidak terdapat tanda gagal jantung dan Hb
sebelum transfusi di atas 5 g/dL, diberikan 10-15 mg/kgBB per satu kali pemberian
selama 2 jam atau 20 mL/kgBB dalam waktu 3-4 jam. Bila terdapat tanda gagal jantung,
pernah ada kelainan jantung, atau Hb <5 g/dL, dosis satu kali pemberian tidak boleh
lebih dari 5 ml/kgBB dengan kecepatan tidak lebih dari 2 mL/kgBB/jam. Sambil
menunggu persiapan transfusi darah diberikan oksigen dengan kecepatan 2-4/menit.
Setiap selesai pemberian satu seri transfusi, kadar Hb pasca transfusi diperiksa 30
menit setelah pemberian transfusi terakhir. Untuk mengeluarkan besi dari jaringan
tubuh diberikan chelating agents, Desferal secara im atau iv. 1,3,5,6,7
Jenis-jenis transfusi darah
a. Darah lengkap (whole blood)
Berguna untuk meningkatkan jumlah sel darah merah dan volume plasma dalam waktu
yang bersamaan, misal pada perdarahan aktif dengan kehilangan darah lebih dari 25 -35
% volume darah total.
b. Sel darah merah pekat (packed red cell)
Digunakan untuk meningkatkkan sel darah merah pada pasien yang menunjukkan gejala
anemia, misal pada pasien gagal ginjal dan keganasan.
c. Sel darah merah pekat dengan sedikit leukosit (packed red blood cell leucocyte reduced)
Digunakan untuk meningkatkan jumlah RBC pada pasien yang sering
mendapat/tergantung pada transfusi darah dan pada mereka yang mendapat reaksi
transfusi panas dan reaksi alergi yang berulang.
d. Sel darah merah pekat cuci (packed red blood cell washed)
Pada orang dewasa komponen ini dipakai untuk mencegah reaksi alergi yang berat atau
alergi yang berulang.
e. Sel darah merah pekat beku yang dicuci (packed red blood cell frozen)
Hanya digunakan untuk menyaimpan darah langka.

f. Trombosit pekat (concentrate platelets)
Diindikasikan pada kasus perdarahan karena trombositopenia atau trombositopati
congenital/didapat. Juga diindikasikan untuk mereka selama operasi atau prosedur
invasive dengan trombosit < 50.000/Ul.
g. Trombosit dengan sedikit leukosit (platelets leukocytes reduced)
Digunakan untuk pencegahan terjadinya alloimunisasi terhadap HLA, terutama pada
pasien yang menerima kemotrrapi jangka panjang.
h. Plasma segar beku (fresh frozen plasma)
Dipakai untuk pasien denagn gangguan proses pembekuan pembekuan bila tidak tersedia
faktor pembekuan pekat atau kriopresipitat, misalnya pada defisiensi faktor pembekuan
multiple. 6
Komplikasi
1. Fraktur tulang
2. Hepatosplenomegali
3. Gangguan tumbuh kembang
4. Disfungsi organ
5. Gagal jantung3,5
Pencegahan
a. Pencegahan primer
Penyuluhan sebelum perkawinan (marriage counselling) untuk mencegah perkawinan
diantara pasien Thalasemia agar tidak mendapatkan keturunan yang homozigot. Perkawinan
antara 2 hetarozigot (carrier) menghasilkan keturunan : 25 % Thalasemia (homozigot), 50 %
carrier (heterozigot) dan 25 normal.
b. Pencegahan sekunder
Pencegahan kelahiran bagi homozigot dari pasangan suami istri dengan Thalasemia
heterozigot salah satu jalan keluar adalah inseminasi buatan dengan sperma berasal dari
donor yang bebas dan Thalasemia. Kelahiran kasus homozigot terhindari, tetapi 50 % dari
anak yang lahir adalah carrier, sedangkan 50% lainnya normal. Diagnosis prenatal melalui

pemeriksaan DNA cairan amnion merupakan suatu kemajuan dan digunakan untuk
mendiagnosis kasus homozigot intra-uterin sehingga dapat dipertimbangkan tindakan
abortus.3,6
c. Edukasi:
Sampaikan kepada pasien dan keluarga mengenai kondisinya sekarang.
Beri saran agar sebelum melakukan pernikahan, cek pasangan untuk kemungkinan
thalasemia.
Hindari pemakaian obat pencetus hemolitik seperti fenasetin, klorpromazin,
penisilin, kina, dan sulfonamid.
Makan-makanan bernutrisi khususnya asupan B12 dan folic acid.6
Prognosis
Tetapi pada skenario 3 ini, terdapat gejala hepatosplenomegali yang mengindikasikan
bahwa penderita yang masih berusia 2 tahun telah sampai pada stadium berat. Dalam hal
ini, prognosisnya buruk.

BAB III
Kesimpulan
1. Dari hasil heteroanamnesis, pemeriksaan fisik, dan pemeriksaan laboratorium, anak
tersebut didiagnosa menderita thalassemia.
2. Thalassemia merupakan bagian dari hemoglobinopati yang merupakan salah satu dari
jenis anemia hemolitik.
3. Thalassemia pada anak tersebut belum pasti diketahui jenisnya. Untuk itu, perlu
dilakukan pemeriksaan lebih lanjut agar nantinya dalam penatalaksanaan penanganan
yang dilakukan dapat tepat sesuai dengan jenis thalassemianya. Akan tetapi,
kemungkinan besar thalassemia beta mayor. Hal ini dikarenakan terdapat gejala
hepatosplenomegali.
4. Penatalaksanaan pada thalassemia diberikan (desferoxamine), Vitamin C 100-250 mg
perhari, Asam folat 2-5 mg perhari, dan Vitamin E 200-400 IU (International Unit)
perhari.
5. Prognosis dari thalassemia pada umumnya baik apabila diberi penatalaksanaan yang
sesuai.
6. Di samping terapi medikamentosa, juga diberikan edukasi dan program prevensi.

Daftar Pustaka
1. Sudiono Herawati dr, Iskandar dr, Harni Edward dr, Halim SK dr, Santoso Regie dr.
Penuntun Patologi Klinik Hematologi. Cetakan III. Jakarta: SinarSurya MegaPerkasa. 2009.
Hal: 41-139.
2. Sudoyo,Setiyohadi, Alwi, et al. Ilmu Penyakit Dalam. Jilid II. Ed. IV. Jakarta: Balai
Penerbit FKUI. 2001. Hal 634-40.
3. Nelson. Ilmu Kesehatan Anak. Jakarta: EGC. 2004. Hal: 2789-793.
4. Bakta, I Made. 2006. Hematologi Klinik Ringkas. EGC: Jakarta. Hal: 19-43.
5. Robbins, Kumar Cotran. Buku Ajar Patologi. Vol.2. 2005. Jakarta: EGC. Hal: 135-39.
6. Thalassaemia. Lab Test online 2006 (sited 2007 August);1(1): Available from URL :
http://www.labtestsonline.org.uk/understanding/conditions/thalassemia.html
7. Kalpravidh RW, Wichit A, Siritanaratkul N, Fucharoen S. Effect of coenzyme Q10 as an
antioxidant in β-thalassemia/Hb E patients. BioFactors 2005 (cited 2007 August);1(1):
Available from URL : http://iospress.metapress.com/app/home/contribution.asp?
referrer=parent&backto=issue,28,32;journal,11,50;linkingpublicationresults,1:103144,1




















